User login
Noninvasive Ventilation Improves Outcomes for COPD Inpatients
Clinical question: Do patients hospitalized with acute COPD exacerbations have improved outcomes with noninvasive ventilation (NIV) compared to those treated with invasive mechanical ventilation (IMV)?
Background: Previous studies have shown that in select patients, NIV has a mortality benefit over IMV for acute COPD exacerbations requiring hospitalization. NIV may also decrease complication rates and reduce length of stay; however, the previous prospective studies have been small.
Study design: Retrospective cohort study.
Setting: 420 structurally and geographically diverse U.S. hospitals.
Synopsis: Using the Premier Healthcare Informatics database, this study looked at 25,628 patients over 40 years old who were hospitalized with COPD exacerbations. Compared with patients who were initially treated with IMV, patients treated with NIV demonstrated lower mortality rates with an odds ratio of 0.54, lower risk of hospital-acquired pneumonia with an odds ratio of 0.53, and a 32% cost reduction. They also had shorter lengths of stay.
This was a retrospective study using a limited data set, and the authors did not have access to potentially confounding factors between the two groups, including vital signs and blood gases. Additionally, the advantages of NIV were attenuated among patients with pneumonia present on admission, patients with high burden of comorbid diseases, and patients older than 85 years.
Bottom line: Treatment of acute COPD exacerbations with NIV is associated with lower mortality, lower costs, and shorter length of stay as compared with IMV.
Citation: Lindenauer PK, Stefan MS, Shieh MS, Pekow PS, Rothberg MB, Hill NS. Outcomes associated with invasive and noninvasive ventilation among patients hospitalized with exacerbations of chronic obstructive pulmonary disease. JAMA Intern Med. 2014;174(12):1982-1993.
Clinical question: Do patients hospitalized with acute COPD exacerbations have improved outcomes with noninvasive ventilation (NIV) compared to those treated with invasive mechanical ventilation (IMV)?
Background: Previous studies have shown that in select patients, NIV has a mortality benefit over IMV for acute COPD exacerbations requiring hospitalization. NIV may also decrease complication rates and reduce length of stay; however, the previous prospective studies have been small.
Study design: Retrospective cohort study.
Setting: 420 structurally and geographically diverse U.S. hospitals.
Synopsis: Using the Premier Healthcare Informatics database, this study looked at 25,628 patients over 40 years old who were hospitalized with COPD exacerbations. Compared with patients who were initially treated with IMV, patients treated with NIV demonstrated lower mortality rates with an odds ratio of 0.54, lower risk of hospital-acquired pneumonia with an odds ratio of 0.53, and a 32% cost reduction. They also had shorter lengths of stay.
This was a retrospective study using a limited data set, and the authors did not have access to potentially confounding factors between the two groups, including vital signs and blood gases. Additionally, the advantages of NIV were attenuated among patients with pneumonia present on admission, patients with high burden of comorbid diseases, and patients older than 85 years.
Bottom line: Treatment of acute COPD exacerbations with NIV is associated with lower mortality, lower costs, and shorter length of stay as compared with IMV.
Citation: Lindenauer PK, Stefan MS, Shieh MS, Pekow PS, Rothberg MB, Hill NS. Outcomes associated with invasive and noninvasive ventilation among patients hospitalized with exacerbations of chronic obstructive pulmonary disease. JAMA Intern Med. 2014;174(12):1982-1993.
Clinical question: Do patients hospitalized with acute COPD exacerbations have improved outcomes with noninvasive ventilation (NIV) compared to those treated with invasive mechanical ventilation (IMV)?
Background: Previous studies have shown that in select patients, NIV has a mortality benefit over IMV for acute COPD exacerbations requiring hospitalization. NIV may also decrease complication rates and reduce length of stay; however, the previous prospective studies have been small.
Study design: Retrospective cohort study.
Setting: 420 structurally and geographically diverse U.S. hospitals.
Synopsis: Using the Premier Healthcare Informatics database, this study looked at 25,628 patients over 40 years old who were hospitalized with COPD exacerbations. Compared with patients who were initially treated with IMV, patients treated with NIV demonstrated lower mortality rates with an odds ratio of 0.54, lower risk of hospital-acquired pneumonia with an odds ratio of 0.53, and a 32% cost reduction. They also had shorter lengths of stay.
This was a retrospective study using a limited data set, and the authors did not have access to potentially confounding factors between the two groups, including vital signs and blood gases. Additionally, the advantages of NIV were attenuated among patients with pneumonia present on admission, patients with high burden of comorbid diseases, and patients older than 85 years.
Bottom line: Treatment of acute COPD exacerbations with NIV is associated with lower mortality, lower costs, and shorter length of stay as compared with IMV.
Citation: Lindenauer PK, Stefan MS, Shieh MS, Pekow PS, Rothberg MB, Hill NS. Outcomes associated with invasive and noninvasive ventilation among patients hospitalized with exacerbations of chronic obstructive pulmonary disease. JAMA Intern Med. 2014;174(12):1982-1993.
Bova Risk Model Predicts 30-Day Pulmonary Embolism-Related Complications
Clinical question: Can the Bova risk model stratify patients with acute PE into stages of increasing risk for 30-day pulmonary embolism (PE)-related complications?
Background: The Bova score is based on four variables assessed at the time of PE diagnosis: heart rate, systolic blood pressure, cardiac troponin, and a marker of right ventricular (RV) dysfunction. In the original study, the Bova risk model was derived from 2,874 normotensive patients with PE. This study performed a retrospective validation of this model on a different cohort of patients.
Study design: Retrospective cohort study.
Setting: Academic urban ED in Madrid, Spain.
Synopsis: Investigators included 1,083 patients with normotensive PE, and the Bova risk score classified 80% into class I, 15% into class II, and 5% into class III—correlating 30-day PE-related complication rates were 4.4%, 18%, and 42%, respectively. When dichotomized into low risk (class I and II) versus intermediate to high risk (class III), the model had a specificity of 97%, a positive predictive value of 42%, and a positive likelihood ratio of 7.9 for predicting 30-day PE-related complications.
The existing risk assessment models, the pulmonary embolism severity index (PESI) and the simplified PESI (sPESI), have been extensively validated but were specifically developed to identity patients with low risk for mortality. The Bova risk model could be used in a stepwise fashion, with the PESI or sPESI model, to further assess intermediate-risk patients.
This model was derived and validated at one single center, so the results may not be generalizable. Additionally, the variables were collected prospectively, but this validation analysis was performed retrospectively.
Bottom line: The Bova risk model accurately stratifies patients with normotensive PE into stages of increasing risk for developing 30-day PE-related complications.
Citation: Fernández C, Bova C, Sanchez O, et al. Validation of a model for identification of patients at intermediate to high risk for complications associated with acute symptomatic pulmonary embolism [published online ahead of print January 29, 2015]. Chest.
Clinical question: Can the Bova risk model stratify patients with acute PE into stages of increasing risk for 30-day pulmonary embolism (PE)-related complications?
Background: The Bova score is based on four variables assessed at the time of PE diagnosis: heart rate, systolic blood pressure, cardiac troponin, and a marker of right ventricular (RV) dysfunction. In the original study, the Bova risk model was derived from 2,874 normotensive patients with PE. This study performed a retrospective validation of this model on a different cohort of patients.
Study design: Retrospective cohort study.
Setting: Academic urban ED in Madrid, Spain.
Synopsis: Investigators included 1,083 patients with normotensive PE, and the Bova risk score classified 80% into class I, 15% into class II, and 5% into class III—correlating 30-day PE-related complication rates were 4.4%, 18%, and 42%, respectively. When dichotomized into low risk (class I and II) versus intermediate to high risk (class III), the model had a specificity of 97%, a positive predictive value of 42%, and a positive likelihood ratio of 7.9 for predicting 30-day PE-related complications.
The existing risk assessment models, the pulmonary embolism severity index (PESI) and the simplified PESI (sPESI), have been extensively validated but were specifically developed to identity patients with low risk for mortality. The Bova risk model could be used in a stepwise fashion, with the PESI or sPESI model, to further assess intermediate-risk patients.
This model was derived and validated at one single center, so the results may not be generalizable. Additionally, the variables were collected prospectively, but this validation analysis was performed retrospectively.
Bottom line: The Bova risk model accurately stratifies patients with normotensive PE into stages of increasing risk for developing 30-day PE-related complications.
Citation: Fernández C, Bova C, Sanchez O, et al. Validation of a model for identification of patients at intermediate to high risk for complications associated with acute symptomatic pulmonary embolism [published online ahead of print January 29, 2015]. Chest.
Clinical question: Can the Bova risk model stratify patients with acute PE into stages of increasing risk for 30-day pulmonary embolism (PE)-related complications?
Background: The Bova score is based on four variables assessed at the time of PE diagnosis: heart rate, systolic blood pressure, cardiac troponin, and a marker of right ventricular (RV) dysfunction. In the original study, the Bova risk model was derived from 2,874 normotensive patients with PE. This study performed a retrospective validation of this model on a different cohort of patients.
Study design: Retrospective cohort study.
Setting: Academic urban ED in Madrid, Spain.
Synopsis: Investigators included 1,083 patients with normotensive PE, and the Bova risk score classified 80% into class I, 15% into class II, and 5% into class III—correlating 30-day PE-related complication rates were 4.4%, 18%, and 42%, respectively. When dichotomized into low risk (class I and II) versus intermediate to high risk (class III), the model had a specificity of 97%, a positive predictive value of 42%, and a positive likelihood ratio of 7.9 for predicting 30-day PE-related complications.
The existing risk assessment models, the pulmonary embolism severity index (PESI) and the simplified PESI (sPESI), have been extensively validated but were specifically developed to identity patients with low risk for mortality. The Bova risk model could be used in a stepwise fashion, with the PESI or sPESI model, to further assess intermediate-risk patients.
This model was derived and validated at one single center, so the results may not be generalizable. Additionally, the variables were collected prospectively, but this validation analysis was performed retrospectively.
Bottom line: The Bova risk model accurately stratifies patients with normotensive PE into stages of increasing risk for developing 30-day PE-related complications.
Citation: Fernández C, Bova C, Sanchez O, et al. Validation of a model for identification of patients at intermediate to high risk for complications associated with acute symptomatic pulmonary embolism [published online ahead of print January 29, 2015]. Chest.
Intracranial Bleeding Risk for Head Injury Patients on Warfarin
Clinical question: Do minor and minimal head injuries in patients on warfarin lead to significant intracranial bleed?
Background: Warfarin use is common, and many of these patients sustain minor and minimal head injuries. When presenting to the ED, these patients pose a clinical dilemma regarding whether to obtain neuroimaging and/or admit.
Study design: Retrospective cohort study.
Setting: Two urban tertiary care EDs in Ottawa, Canada, over a two-year period.
Synopsis: Using the Canadian National Ambulatory Care Reporting System database and the associated coding data, 259 patients were identified that fit the inclusion criteria GCS ≥13 and INR >1.5. This study showed that the rate of intracranial bleeds in this group of patients was high (15.9%); for minor and minimal head injury groups, the rate was 21.9% and 4.8%, respectively. Additionally, loss of consciousness was associated with higher rates of intracranial bleeding.
The risk of intracranial bleed after a head injury while on warfarin is considerably high, particularly for those patients with minor head injury (21.9%), which is about three times the rate previously reported. Hospitalists evaluating these patients should consider obtaining neuroimaging.
Nonetheless, these rates may be overestimating the true prevalence due to the following: 1) Coding data may overlook minor and minimal head injuries in the presence of more serious injuries, and 2) patients with minimal head injuries may not seek medical care.
Bottom line: Patients sustaining minor head injury while on warfarin have a high rate of intracranial bleed.
Reference: Alrajhi KN, Perry JJ, Forster AJ. Intracranial bleeds after minor and minimal head injury in patients on warfarin. J Emer Med. 2015;48(2):137-142.
Clinical question: Do minor and minimal head injuries in patients on warfarin lead to significant intracranial bleed?
Background: Warfarin use is common, and many of these patients sustain minor and minimal head injuries. When presenting to the ED, these patients pose a clinical dilemma regarding whether to obtain neuroimaging and/or admit.
Study design: Retrospective cohort study.
Setting: Two urban tertiary care EDs in Ottawa, Canada, over a two-year period.
Synopsis: Using the Canadian National Ambulatory Care Reporting System database and the associated coding data, 259 patients were identified that fit the inclusion criteria GCS ≥13 and INR >1.5. This study showed that the rate of intracranial bleeds in this group of patients was high (15.9%); for minor and minimal head injury groups, the rate was 21.9% and 4.8%, respectively. Additionally, loss of consciousness was associated with higher rates of intracranial bleeding.
The risk of intracranial bleed after a head injury while on warfarin is considerably high, particularly for those patients with minor head injury (21.9%), which is about three times the rate previously reported. Hospitalists evaluating these patients should consider obtaining neuroimaging.
Nonetheless, these rates may be overestimating the true prevalence due to the following: 1) Coding data may overlook minor and minimal head injuries in the presence of more serious injuries, and 2) patients with minimal head injuries may not seek medical care.
Bottom line: Patients sustaining minor head injury while on warfarin have a high rate of intracranial bleed.
Reference: Alrajhi KN, Perry JJ, Forster AJ. Intracranial bleeds after minor and minimal head injury in patients on warfarin. J Emer Med. 2015;48(2):137-142.
Clinical question: Do minor and minimal head injuries in patients on warfarin lead to significant intracranial bleed?
Background: Warfarin use is common, and many of these patients sustain minor and minimal head injuries. When presenting to the ED, these patients pose a clinical dilemma regarding whether to obtain neuroimaging and/or admit.
Study design: Retrospective cohort study.
Setting: Two urban tertiary care EDs in Ottawa, Canada, over a two-year period.
Synopsis: Using the Canadian National Ambulatory Care Reporting System database and the associated coding data, 259 patients were identified that fit the inclusion criteria GCS ≥13 and INR >1.5. This study showed that the rate of intracranial bleeds in this group of patients was high (15.9%); for minor and minimal head injury groups, the rate was 21.9% and 4.8%, respectively. Additionally, loss of consciousness was associated with higher rates of intracranial bleeding.
The risk of intracranial bleed after a head injury while on warfarin is considerably high, particularly for those patients with minor head injury (21.9%), which is about three times the rate previously reported. Hospitalists evaluating these patients should consider obtaining neuroimaging.
Nonetheless, these rates may be overestimating the true prevalence due to the following: 1) Coding data may overlook minor and minimal head injuries in the presence of more serious injuries, and 2) patients with minimal head injuries may not seek medical care.
Bottom line: Patients sustaining minor head injury while on warfarin have a high rate of intracranial bleed.
Reference: Alrajhi KN, Perry JJ, Forster AJ. Intracranial bleeds after minor and minimal head injury in patients on warfarin. J Emer Med. 2015;48(2):137-142.
Enriched Nutritional Formulas Help Heal Pressure Ulcers
Clinical question: Does a high-calorie, high-protein formula enriched with supplements of arginine, zinc, and antioxidants improve pressure ulcer healing?
Background: Malnutrition is thought to be a major factor in the development and poor healing of pressure ulcers. Trials evaluating whether or not the addition of antioxidants, arginine, and zinc to nutritional formulas improves pressure ulcer healing have been small and inconsistent.
Study design: Multicenter, randomized, controlled, blinded trial.
Setting: Long-term care facilities and patients receiving home care services.
Synopsis: Two hundred patients with stage II, III, or IV pressure ulcers receiving standardized wound care were randomly assigned to a control formula or an experimental formula enriched with arginine, zinc, and antioxidants. At eight weeks, the experimental formula group had an 18.7% (CI, 5.7% to 31.8%, P=0.017) mean reduction in pressure ulcer size compared with the control formula group, although both groups showed efficacy in wound healing.
Nutrition is an important part of wound healing and should be incorporated into the plan of care for the hospitalized patient with pressure ulcers. Hospitalists should be mindful that this study was conducted in non-acute settings, with a chronically ill patient population; more research needs to be done to investigate the effect of these specific immune-modulating nutritional supplements in acutely ill hospitalized patients, given the inconclusive safety profile of certain nutrients such as arginine in severe sepsis.
Bottom line: Enhanced nutritional support with an oral nutritional formula enriched with arginine, zinc, and antioxidants improves pressure ulcer healing in malnourished patients already receiving standard wound care.
Citation: Cereda E, Klersy C, Serioli M, Crespi A, D’Andrea F, OligoElement Sore Trial Study Group. A nutritional formula enriched with arginine, zinc, and antioxidants for the healing of pressure ulcers: a randomized trial. Ann Intern Med. 2015;162(3):167-174.
Clinical question: Does a high-calorie, high-protein formula enriched with supplements of arginine, zinc, and antioxidants improve pressure ulcer healing?
Background: Malnutrition is thought to be a major factor in the development and poor healing of pressure ulcers. Trials evaluating whether or not the addition of antioxidants, arginine, and zinc to nutritional formulas improves pressure ulcer healing have been small and inconsistent.
Study design: Multicenter, randomized, controlled, blinded trial.
Setting: Long-term care facilities and patients receiving home care services.
Synopsis: Two hundred patients with stage II, III, or IV pressure ulcers receiving standardized wound care were randomly assigned to a control formula or an experimental formula enriched with arginine, zinc, and antioxidants. At eight weeks, the experimental formula group had an 18.7% (CI, 5.7% to 31.8%, P=0.017) mean reduction in pressure ulcer size compared with the control formula group, although both groups showed efficacy in wound healing.
Nutrition is an important part of wound healing and should be incorporated into the plan of care for the hospitalized patient with pressure ulcers. Hospitalists should be mindful that this study was conducted in non-acute settings, with a chronically ill patient population; more research needs to be done to investigate the effect of these specific immune-modulating nutritional supplements in acutely ill hospitalized patients, given the inconclusive safety profile of certain nutrients such as arginine in severe sepsis.
Bottom line: Enhanced nutritional support with an oral nutritional formula enriched with arginine, zinc, and antioxidants improves pressure ulcer healing in malnourished patients already receiving standard wound care.
Citation: Cereda E, Klersy C, Serioli M, Crespi A, D’Andrea F, OligoElement Sore Trial Study Group. A nutritional formula enriched with arginine, zinc, and antioxidants for the healing of pressure ulcers: a randomized trial. Ann Intern Med. 2015;162(3):167-174.
Clinical question: Does a high-calorie, high-protein formula enriched with supplements of arginine, zinc, and antioxidants improve pressure ulcer healing?
Background: Malnutrition is thought to be a major factor in the development and poor healing of pressure ulcers. Trials evaluating whether or not the addition of antioxidants, arginine, and zinc to nutritional formulas improves pressure ulcer healing have been small and inconsistent.
Study design: Multicenter, randomized, controlled, blinded trial.
Setting: Long-term care facilities and patients receiving home care services.
Synopsis: Two hundred patients with stage II, III, or IV pressure ulcers receiving standardized wound care were randomly assigned to a control formula or an experimental formula enriched with arginine, zinc, and antioxidants. At eight weeks, the experimental formula group had an 18.7% (CI, 5.7% to 31.8%, P=0.017) mean reduction in pressure ulcer size compared with the control formula group, although both groups showed efficacy in wound healing.
Nutrition is an important part of wound healing and should be incorporated into the plan of care for the hospitalized patient with pressure ulcers. Hospitalists should be mindful that this study was conducted in non-acute settings, with a chronically ill patient population; more research needs to be done to investigate the effect of these specific immune-modulating nutritional supplements in acutely ill hospitalized patients, given the inconclusive safety profile of certain nutrients such as arginine in severe sepsis.
Bottom line: Enhanced nutritional support with an oral nutritional formula enriched with arginine, zinc, and antioxidants improves pressure ulcer healing in malnourished patients already receiving standard wound care.
Citation: Cereda E, Klersy C, Serioli M, Crespi A, D’Andrea F, OligoElement Sore Trial Study Group. A nutritional formula enriched with arginine, zinc, and antioxidants for the healing of pressure ulcers: a randomized trial. Ann Intern Med. 2015;162(3):167-174.
High-Volume Hospitals Have Higher Readmission Rates
Clinical question: Is there an association between hospital volume and hospital readmission rates?
Background: There is an established association between high patient volume and reduced complications or mortality after surgical procedures; however, readmission represents a different type of quality metric than mortality or complications. Studies on the association between hospital patient volume and readmission rates have been controversial.
Study design: Retrospective, cross-sectional study.
Setting: Acute care hospitals.
Synopsis: The study included 6,916,644 admissions to 4,651 hospitals, where patients were assigned to one of five cohorts: medicine, surgery/gynecology, cardiorespiratory, cardiovascular, and neurology. The hospital with the highest volume group had a hospital-wide mean standardized readmission rate of 15.9%, while the hospital with the lowest volume group had a readmission rate of 14.7%. This was a 1.2 percentage point absolute difference between the two hospitals (95% confidence interval 0.9 to 1.5). This trend continued when specialty cohorts were examined, with the exception of the procedure-heavy cardiovascular cohort.
Results showed a trend toward decreased readmission rates in lower-volume hospitals; however, it is unclear why this trend exists. Possible reasons include different patient populations and different practitioner-to-patient ratios in low-volume hospitals.
Limitations of this study are the inclusion of only patients 65 years and older and the fact that all admissions per patient were included, which may bias the results against hospitals with many frequently admitted patients.
Bottom line: Hospitals with high patient volumes are associated with higher readmission rates, except in procedure-heavy patient groups.
Citation: Horwitz LI, Lin Z, Herrin J, et al.Association of hospital volume with readmission rates: a retrospective cross-sectional study. BMJ. 2015;350:h447.
Clinical question: Is there an association between hospital volume and hospital readmission rates?
Background: There is an established association between high patient volume and reduced complications or mortality after surgical procedures; however, readmission represents a different type of quality metric than mortality or complications. Studies on the association between hospital patient volume and readmission rates have been controversial.
Study design: Retrospective, cross-sectional study.
Setting: Acute care hospitals.
Synopsis: The study included 6,916,644 admissions to 4,651 hospitals, where patients were assigned to one of five cohorts: medicine, surgery/gynecology, cardiorespiratory, cardiovascular, and neurology. The hospital with the highest volume group had a hospital-wide mean standardized readmission rate of 15.9%, while the hospital with the lowest volume group had a readmission rate of 14.7%. This was a 1.2 percentage point absolute difference between the two hospitals (95% confidence interval 0.9 to 1.5). This trend continued when specialty cohorts were examined, with the exception of the procedure-heavy cardiovascular cohort.
Results showed a trend toward decreased readmission rates in lower-volume hospitals; however, it is unclear why this trend exists. Possible reasons include different patient populations and different practitioner-to-patient ratios in low-volume hospitals.
Limitations of this study are the inclusion of only patients 65 years and older and the fact that all admissions per patient were included, which may bias the results against hospitals with many frequently admitted patients.
Bottom line: Hospitals with high patient volumes are associated with higher readmission rates, except in procedure-heavy patient groups.
Citation: Horwitz LI, Lin Z, Herrin J, et al.Association of hospital volume with readmission rates: a retrospective cross-sectional study. BMJ. 2015;350:h447.
Clinical question: Is there an association between hospital volume and hospital readmission rates?
Background: There is an established association between high patient volume and reduced complications or mortality after surgical procedures; however, readmission represents a different type of quality metric than mortality or complications. Studies on the association between hospital patient volume and readmission rates have been controversial.
Study design: Retrospective, cross-sectional study.
Setting: Acute care hospitals.
Synopsis: The study included 6,916,644 admissions to 4,651 hospitals, where patients were assigned to one of five cohorts: medicine, surgery/gynecology, cardiorespiratory, cardiovascular, and neurology. The hospital with the highest volume group had a hospital-wide mean standardized readmission rate of 15.9%, while the hospital with the lowest volume group had a readmission rate of 14.7%. This was a 1.2 percentage point absolute difference between the two hospitals (95% confidence interval 0.9 to 1.5). This trend continued when specialty cohorts were examined, with the exception of the procedure-heavy cardiovascular cohort.
Results showed a trend toward decreased readmission rates in lower-volume hospitals; however, it is unclear why this trend exists. Possible reasons include different patient populations and different practitioner-to-patient ratios in low-volume hospitals.
Limitations of this study are the inclusion of only patients 65 years and older and the fact that all admissions per patient were included, which may bias the results against hospitals with many frequently admitted patients.
Bottom line: Hospitals with high patient volumes are associated with higher readmission rates, except in procedure-heavy patient groups.
Citation: Horwitz LI, Lin Z, Herrin J, et al.Association of hospital volume with readmission rates: a retrospective cross-sectional study. BMJ. 2015;350:h447.
Arthroscopic Treatment of Tibial Spine Malunion With Resorbable Screws
Anterior tibial spine fractures are rare, occurring with an incidence of 3 per 100,000 per year.1,2 Historically, this fracture has occurred more frequently in children,3-5 and was considered a condition of skeletal immaturity and the pediatric equivalent of an anterior cruciate ligament (ACL) rupture.6 However, recent literature indicates that this fracture is more common in the adult population than previously thought.7 The tibial spine is an attachment point for the ACL and an avulsion may produce ACL laxity,8 predisposing to further symptomatic laxity and premature osteoarthritis. Nearly 40% of these fractures are associated with concomitant injuries to surrounding structures.9
Meyers and McKeever10,11 originally classified these fractures into 3 groups on the basis of displacement. Type I fractures present with no significant displacement of the anterior margin, type II involve displacement and are hinged, while type III have complete displacement.10,11 More recently, a type IV fracture has been added, involving comminution of the displaced fragment. Nondisplaced fractures are commonly treated with immobilization in varying degrees of extension; this allows the femoral condyles to compress and to reduce the fracture while arthroscopic or open reduction is the preferred method for displaced fractures of the tibial spine.2,4,8,10
We report the case of an 11-year-old boy with a tibial spine fracture that failed conservative management. He developed a subsequent malunion with impingement anteriorly of the tibial spine on the notch, and residual instability of the ACL. The patient’s parents provided written informed consent for print and electronic publication of this case report.
Case Report
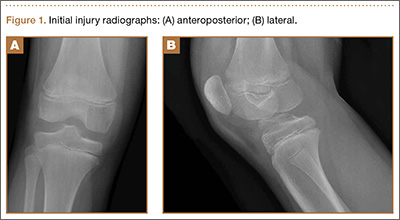
An 11-year-old Caucasian boy was referred to our office for evaluation of right knee injury. He sustained the injury approximately 3 months earlier, and it was determined that he had a tibial spine fracture. Conservative management with immobilization in extension and activity modification was undertaken; however, he was referred for further evaluation because of healing in a malreduced position and residual ACL laxity. Physical examination showed a grade 2A Lachman test (contralateral limb with negative Lachman examination), negative McMurray test, and pain with forced hyperextension; range-of-motion examination showed lack of the terminal 5º of extension. Magnetic resonance and computed tomography imaging from an outside facility showed a skeletally immature individual with a large tibial spine fracture that had healed in a malunited position with the fragment extended on a posterior hinge, creating a large prominence anteriorly (Figures 1A, 1B). Magnetic resonance imaging showed that the ACL fibers were likely to remain intact but would lack appropriate tension secondary to the displacement of the tibial insertion.
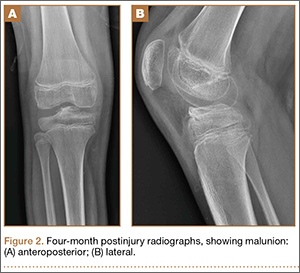
Because of healing in a displaced position, lack of terminal extension, ACL laxity, and subjective complaints of pain, we discussed surgery with the patient and his parents (Figures 2A, 2B). Four months after the initial injury, the patient underwent surgery for a right tibial spine malunion arthroscopic takedown and repair, as well as an intraoperative evaluation of the ACL. Standard arthroscopy was performed, using anterolateral and anteromedial arthroscopic portals, and an accessory medial peripatellar portal. During surgery, a large prominence was noted in the region of the anterior tibial spine (Figure 3A). The ACL fibers maintained a slack position secondary to the elevation of the tibial insertion point, and intraoperative Lachman examination showed anterior translation of the tibia on the femur as the slack was removed from the ACL. During surgery, impingement of the anterior tibial spine along the femoral notch was shown to be significant by taking the knee into near-full extension (Figure 3B). A cam-like effect was noted at the time of impingement with the posterior soft tissues relaxing to accommodate slight further extension.
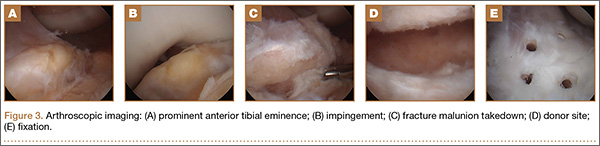
Based on these findings, we chose to take down the malunited fracture and repair it (Figure 3C). PDS suture (Ethicon, Somerville, New Jersey) was temporarily placed along the intermeniscal ligament and anterior horns of the medial and lateral menisci, using a system of spinal needles to facilitate suture passage. Surgical clamps were hung from the suture to provide traction on the sutures throughout the case, allowing the intermeniscal ligament and menisci to recede anteriorly to improve working space and aid in preventing iatrogenic injury. These sutures were removed at the conclusion of the case. Using a combination of curettes, elevator, and small shaver, we were able to meticulously remove interposed malunited callus to allow for mobilization of the displaced fragment. After removal of the excess bone formation, a typical donor site was created, allowing the displaced spine fragment to be hinged into appropriate alignment (Figure 3D). We were able to maintain a posterior cortical hinge to facilitate this process.
Then, we placed Kirschner wires (K-wires) across the fracture in an antegrade fashion, anterior to the trochlea and notch, using an accessory medial peripatellar starting point percutaneously, under direct visualization to avoid iatrogenic chondral injury. The tibial spine fragment was temporarily maintained in a reduced position with an arthroscopic probe and pinned in place with two 0.062-in K-wires. The fracture was stabilized with 8 resorbable 1.6-mm poly-L-lactic/polyglycolic acid (PLLA/PGA) nails, in varying lengths from 18 mm to 22 mm. Excellent fixation was obtained, and range of motion was tested from 0º to 80º, without movement of the fracture site (Figure 3E). Fluoroscopy with multi-axial views verified adequate fixation and reduction. Further, we examined and noted a taut ACL after fixation. The patient was placed in a long leg cast for 3 weeks at 30º, based upon intraoperative determination of the position of least tension on the fracture fragment.
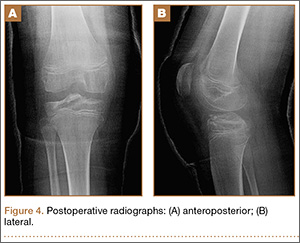
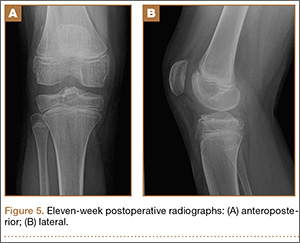
At 3-week follow-up, the patient was progressing well and transitioned from a long leg cast to a hinged knee brace, to allow for early range of motion. Radiographs showed appropriate alignment of the tibial spine fracture with no significant loss of fixation (Figures 4A, 4B). Physical therapy was initiated between 0º and 30º, and flexion was progressively increased over the course of the first 3 weeks. Active and active-assist, closed-chain activities were maintained. Seven weeks postoperatively, the patient displayed continued clinical progression. Radiographs showed interval healing with slight lucency over the anterolateral aspect of the fracture fragment, likely related to the early resorptive process of healing. Physical examination showed movement between 0º and 120º, stable Lachman test, and stable anterior drawer. Crutches were discontinued and hinged knee brace was converted to an ACL brace. By the 11th week, motion had increased to 140º, and radiographs continued to show acceptable alignment and healing (Figures 5A, 5B). The patient was released to return to play as tolerated; however, an ACL brace was recommended during his initial return to provide additional support.
Discussion
In this report, we present an approach for arthroscopic reduction of a malunited tibial spine fracture using resorbable PLLA/PGA nails. The number of polyglycolic nails employed is individualized per case, dependent on the surface area and the quality of the bone within the fractured fragment. Preoperative templating allows for measurements from the fractured fragment to the level of the proximal tibial physis. Based on these measurements, nails are chosen to maximize fixation length and avoid the physis. Despite studies that have examined the effect of transphyseal K-wire pinning or drilling on subsequent growth, there is no consensus about optimal technique. Experiments in animal models indicate that drill injuries destroying less than 8% to 9% of the physis do not impact total bone growth.12,13 Further, temporary crossing of the physeal plate for internal fixation of dislocated joint injuries has not been shown to result in bone bridging or growth disturbance.14,15
Each nail is 1.6 mm in diameter, leaving a small footprint. The nails are used judiciously to provide effective stabilization of the fragment and to maintain a cost-conscious approach. An accessory superomedial peripatellar portal allows an appropriate angle for nail placement. This portal allows access to all regions of the fractured fragment, while an anteromedial and anterolateral portal are used as working and camera portals, respectively. Nails are placed to provide an axis perpendicular to the fracture line to allow appropriate compression. By virtue of the shape of the typical fragment in a tibial spine fracture, the nails vary in insertion angle.
The occurrence of anterior tibial spine fractures is rare, and while several techniques have been described to repair this fracture, there remains a great deal of uncertainty regarding the best course of treatment. A review of the literature finds arthroscopic and open approaches, as well as techniques employing K-wire fixation, metal screw fixation, staple fixation, absorbable fixation, and fixation with sutures passed through the tibial tunnel.16-18
Avulsion fractures of the tibial eminence were treated with open fixation until McLennan8 first reported the benefits of reduction with an arthroscope. Open reduction and internal fixation provide the benefit of direct visualization,9 while arthroscopic reduction offers decreased morbidity and an accelerated recovery of knee functions,8 despite the fact that a higher rate of range-of-motion deficits were seen in patients treated arthroscopically.19 We feel that with proper early rehabilitation to achieve range of motion, the risk of this can be minimal.
Various arthroscopic approaches that improve the accuracy of the reduction and decrease surgical invasiveness have been described. Suture and screw fixation are among the most common methods, and both have resulted in positive outcomes.20-24 Suture fixation of the tibial eminence is technically demanding but offers secure fixation without the need for follow-up hardware removal. Screw fixation results in secure fixation; however, numerous hardware-related issues may necessitate removal. Furthermore, in skeletally immature patients, screw fixation may disturb the growth plate if it crosses an open physis.9
Hunter and Willis25 retrospectively reviewed patients with tibial eminence fractures treated with either screw or suture fixation and found a 44% reoperation rate in the screw-fixation group. Removal was often recommended as a result of hardware-related issues. There was a 13% reoperation rate in the suture-fixation group, which resulted largely from stiffness.25 In a recent review, Gans and colleagues19 reviewed 6 publications comparing screw and suture fixation of tibial eminence fractures and found 82.4% of screw patients had laxity on both the anterior drawer and Lachman tests, compared with 18.8% in the suture-fixation group. This study also noted a slightly higher rate of arthrofibrosis in patients treated with suture fixation.19 Biomechanical studies indicate that suture fixation imparts greater strength under cyclic-loading conditions;26 however, there does not appear to be a difference in ultimate force required for fixation failure.27
Ultimately, both suture and screw fixation result in secure methods of fixation; however, there are often greater issues with screw fixation because of the persistent hardware. Metal has been the most popular method for fracture fixation, and while biodegradable materials have been alluring, adverse tissue reactions have slowed implementation. However, these implants have become increasingly sophisticated, thereby reducing disadvantages.28 Previous biodegradable devices were often composed of a single polymer, and many caused adverse reactions by degrading too quickly or provided no real advantages because they degraded too slowly.29 As the number of polymers approved for internal use and surgical applications continues to rise, so too will the benefits of employing this technology. Furthermore, by including multiple polymers in these implants, one is better able to control the degradation rate, limiting the tissue response.
In this study, we employed PLLA/PGA nails. Studies of PGA implants indicate this molecule degrades at a fast rate resulting in adverse tissue reactions. Adverse reactions in studies of PLLA implants are less frequent because of their slower rate of degradation.29,30 Combining these monomers results in appropriate strength and a controlled degradation rate, reducing the likelihood of adverse reactions. Furthermore, numerous studies have reported that inflammatory responses in children are rare and mild in nature.31,32 Absorbable implants have displayed efficacy in numerous orthopedic settings33-36 and are beneficial in procedures that are not suitable for repeated surgeries, such as reconstruction of the ACL.37 There is some concern about the use of absorbable implants in synovial joints. Polyglycolic acid use in synovial joints may cause foreign-body reactions and may increase the risk of intra-articular dissemination of polymeric debris;38 however, use of a multipolymer construct decreases the likelihood of this occurrence.
Polyglycolic nails confer the advantage over nonresorbable screw fixation because further procedure for hardware removal is not required. Although suture fixation has proved to be beneficial over nonresorbable screw fixation, implantation of resorbable nails appears to have several advantages. In Dr. Estes’ experience, placement of resorbable screws through an accessory superomedial portal is far less technically demanding than placement of suture through the fracture fragment. Further, as sutures are passed from the extra-articular to the intra-articular region of the joint, capsular layers of the knee may inadvertently be bound up in the fixation, predisposing to arthrofibrosis.
At the same time, biodegradable devices are often more costly than alternative forms of treatment; however, a true cost-to-benefit analysis requires consideration of other factors. One of the benefits of biodegradable hardware is that there is no need for follow-up hardware removal. Reports have indicated that up to 91% of patients thought that hardware removal was the most negative aspect of metal implants.39 It is estimated that if the removal rate for metallic implants is higher than 19% to 54%, resorbable implants would be more cost-effective.40 The cost of sutures and screws is variable, however; they are invariably less expensive than biodegradable nails. A study of fracture patients determined that biodegradable implants were cheaper on average after considering the cost of implant removal.40 Ultimately, the hardware choice depends on numerous factors, including surgeon’s discretion; however, biodegradable hardware should not be discounted for financial reasons because the difference in cost is likely negligible.
Conclusion
The approach described in this report offers efficient and secure fixation with resorbable hardware without a reduction in range of motion. Resorbable implants may prove beneficial in the treatment of tibial eminence fractures by offering robust fixation without the concerns associated with permanent hardware.
1. Hargrove R, Parsons S, Payne R. Anterior tibial spine fracture – an easy fracture to miss. Accid Emerg Nurs. 2004;12(3):173-175.
2. Aderinto J, Walmsley P, Keating JF. Fractures of the tibial spine: epidemiology and outcome. Knee. 2008;15(3):164-167.
3. Driessen MJ, Winkelman PA. Fractures of the intercondylar eminence of the tibia in childhood. Neth J Surg. 1984;36(3):69-72.
4. Zaricznyj B. Avulsion fracture of the tibial eminence: treatment by open reduction and pinning. J Bone Joint Surg Am. 1977;59(8):1111-1114.
5. Molander ML, Wallin G, Wikstad I. Fracture of the intercondylar eminence of the tibia: a review of 35 patients. J Bone Joint Surg Br. 1981;63(1):89-91.
6. Kieser DC, Gwynne-Jones D, Dreyer S. Displaced tibial intercondylar eminence fractures. J Orthop Surg. 2011;19(3):292-296.
7. Ishibashi Y, Tsuda E, Sasaki T, Toh S. Magnetic resonance imaging AIDS in detecting concomitant injuries in patients with tibial spine fractures. Clin Orthop. 2005;(434):207-212.
8. McLennan JG. The role of arthroscopic surgery in the treatment of fractures of the intercondylar eminence of the tibia. J Bone Joint Surg Br. 1982;64(4):477-480.
9. Lafrance RM, Giordano B, Goldblatt J, Voloshin I, Maloney M. Pediatric tibial eminence fractures: evaluation and management. J Am Acad Orthop Surg. 2010;18(7):395-405.
10. Meyers MH, McKeever FM. Fracture of the intercondylar eminence of the tibia. J Bone Joint Surg Am. 1959;41(2):209-220.
11. Meyers MH, McKeever FM. Fracture of the intercondylar eminence of the tibia. J Bone Joint Surg Am. 1970;52(8):1677-1684.
12. Garcés GL, Mugica-Garay I, López-González Coviella N, Guerado E. Growth-plate modifications after drilling. J Pediatr Orthop. 1994;14(2):225-228.
13. Janarv PM, Wikström B, Hirsch G. The influence of transphyseal drilling and tendon grafting on bone growth: an experimental study in the rabbit. J Pediatr Orthop. 1998;18(2):149-154.
14. Boelitz R, Dallek M, Meenen NM, Jungbluth KH. Reaction of the epiphyseal groove to groove-crossing bore-wire osteosynthesis. Results of a histomorphologic small animal study. Unfallchirurgie. 1994;20(3):131-137.
15. Yung PS, Lam CY, Ng BK, Lam TP, Cheng JC. Percutaneous transphyseal intramedullary Kirschner wire pinning: a safe and effective procedure for treatment of displaced diaphyseal forearm fracture in children. J Pediatr Orthop. 2004;24(1):7-12.
16. Bong MR, Romero A, Kubiak E, et al. Suture versus screw fixation of displaced tibial eminence fractures: a biomechanical comparison. Arthroscopy. 2005;21(10):1172-1176.
17. Vega JR, Irribarra LA, Baar AK, Iñiguez M, Salgado M, Gana N. Arthroscopic fixation of displaced tibial eminence fractures: a new growth plate-sparing method. Arthroscopy. 2008;24(11):1239-1243.
18. Shepley RW. Arthroscopic treatment of type III tibial spine fractures using absorbable fixation. Orthopedics. 2004;27(7):767-769.
19. Gans I, Baldwin KD, Ganley TJ. Treatment and management outcomes of tibial eminence fractures in pediatric patients: a systematic review. Am J Sports Med. 2013;42(7):1743-1750.
20. Delcogliano A, Chiossi S, Caporaso A, Menghi A, Rinonapoli G. Tibial intercondylar eminence fractures in adults: arthroscopic treatment. Knee Surg Sports Traumatol Arthrosc. 2003;11(4):255-259.
21. Mulhall KJ, Dowdall J, Grannell M, McCabe JP. Tibial spine fractures: an analysis of outcome in surgically treated type III injuries. Injury. 1999;30(4):289-292.
22. Geissler WB, Matthews DE. Arthroscopic suture fixation of displaced tibial eminence fractures. Orthopedics. 1993;16(3):331-333.
23. Mah JY, Otsuka NY, McLean J. An arthroscopic technique for the reduction and fixation of tibial-eminence fractures. J Pediatr Orthop. 1996;16(1):119-121.
24. Reynders P, Reynders K, Broos P. Pediatric and adolescent tibial eminence fractures: arthroscopic cannulated screw fixation. J Trauma. 2002;53(1):49-54.
25. Hunter RE, Willis JA. Arthroscopic fixation of avulsion fractures of the tibial eminence: technique and outcome. Arthroscopy. 2004;20(2):113-121.
26. Eggers AK, Becker C, Weimann A, et al. Biomechanical evaluation of different fixation methods for tibial eminence fractures. Am J Sports Med. 2007;35(3):404-410.
27. Mahar AT, Duncan D, Oka R, Lowry A, Gillingham B, Chambers H. Biomechanical comparison of four different fixation techniques for pediatric tibial eminence avulsion fractures. J Pediatr Orthop. 2008;28(2):159-162.
28. Toro C, Robiony M, Zerman N, Politi M. Resorbable plates in maxillary fixation. A 5-year experience. Minerva Stomatol. 2005;54(4):199-206.
29. Andriano KP, Pohjonen T, Törmälä P. Processing and characterization of absorbable polylactide polymers for use in surgical implants. J Appl Biomater.1994;5(2):133-140.
30. Böstman O, Pihlajamäki H. Clinical biocompatibility of biodegradable orthopaedic implants for internal fixation: a review. Biomaterials. 2000;21(24):2615-2621.
31. Rokkanen PU, Böstman O, Hirvensalo E, et al. Bioabsorbable fixation in orthopaedic surgery and traumatology. Biomaterials. 2000;21(24):2607-2613.
32. Athanasiou KA, Niederauer GG, Agrawal CM. Sterilization, toxicity, biocompatibility and clinical applications of polylactic acid/polyglycolic acid copolymers. Biomaterials. 1996;17(2):93-102.
33. Li ZH, Yu AX, Guo XP, Qi BW, Zhou M, Wang WY. Absorbable implants versus metal implants for the treatment of ankle fractures: A meta-analysis. Exp Ther Med. 2013;5(5):1531-1537.
34. Singh G, Mohammad S, Chak RK, Lepcha N, Singh N, Malkunje LR. Bio-resorbable plates as effective implant in paediatric mandibular fracture. J Maxillofac Oral Surg. 2012;11(4):400-406.
35. Sakamoto Y, Shimizu Y, Nagasao T, Kishi K. Combined use of resorbable poly-L-lactic acid-polyglycolic acid implant and bone cement for treating large orbital floor fractures. J Plast Reconstr Aesthet Surg. 2014;67(3):e88-e90.
36. Benz G, Kallieris D, Seeböck T, McIntosh A, Daum R. Bioresorbable pins and screws in paediatric traumatology. Eur J Pediatr Surg. 1994;4(2):103-107.
37. Gaweda K, Walawski J, Weglowski R, Krzyzanowski W. Comparison of bioabsorbable interference screws and posts for distal fixation in anterior cruciate ligament reconstruction. Int Orthop. 2009;33(1):123-127.
38. Böstman OM. Osteoarthritis of the ankle after foreign-body reaction to absorbable pins and screws: a three- to nine-year follow-up study. J Bone Joint Surg Br. 1998;80(2):333-338.
39. Mittal R, Morley J, Dinopoulos H, Drakoulakis EG, Vermani E, Giannoudis PV. Use of bio-resorbable implants for stabilisation of distal radius fractures: the United Kingdom patients’ perspective. Injury. 2005;36(2):333-338.
40. Böstman OM. Metallic or absorbable fracture fixation devices. A cost minimization analysis. Clin Orthop. 1996;(329):233-239.
Anterior tibial spine fractures are rare, occurring with an incidence of 3 per 100,000 per year.1,2 Historically, this fracture has occurred more frequently in children,3-5 and was considered a condition of skeletal immaturity and the pediatric equivalent of an anterior cruciate ligament (ACL) rupture.6 However, recent literature indicates that this fracture is more common in the adult population than previously thought.7 The tibial spine is an attachment point for the ACL and an avulsion may produce ACL laxity,8 predisposing to further symptomatic laxity and premature osteoarthritis. Nearly 40% of these fractures are associated with concomitant injuries to surrounding structures.9
Meyers and McKeever10,11 originally classified these fractures into 3 groups on the basis of displacement. Type I fractures present with no significant displacement of the anterior margin, type II involve displacement and are hinged, while type III have complete displacement.10,11 More recently, a type IV fracture has been added, involving comminution of the displaced fragment. Nondisplaced fractures are commonly treated with immobilization in varying degrees of extension; this allows the femoral condyles to compress and to reduce the fracture while arthroscopic or open reduction is the preferred method for displaced fractures of the tibial spine.2,4,8,10
We report the case of an 11-year-old boy with a tibial spine fracture that failed conservative management. He developed a subsequent malunion with impingement anteriorly of the tibial spine on the notch, and residual instability of the ACL. The patient’s parents provided written informed consent for print and electronic publication of this case report.
Case Report
An 11-year-old Caucasian boy was referred to our office for evaluation of right knee injury. He sustained the injury approximately 3 months earlier, and it was determined that he had a tibial spine fracture. Conservative management with immobilization in extension and activity modification was undertaken; however, he was referred for further evaluation because of healing in a malreduced position and residual ACL laxity. Physical examination showed a grade 2A Lachman test (contralateral limb with negative Lachman examination), negative McMurray test, and pain with forced hyperextension; range-of-motion examination showed lack of the terminal 5º of extension. Magnetic resonance and computed tomography imaging from an outside facility showed a skeletally immature individual with a large tibial spine fracture that had healed in a malunited position with the fragment extended on a posterior hinge, creating a large prominence anteriorly (Figures 1A, 1B). Magnetic resonance imaging showed that the ACL fibers were likely to remain intact but would lack appropriate tension secondary to the displacement of the tibial insertion.
Because of healing in a displaced position, lack of terminal extension, ACL laxity, and subjective complaints of pain, we discussed surgery with the patient and his parents (Figures 2A, 2B). Four months after the initial injury, the patient underwent surgery for a right tibial spine malunion arthroscopic takedown and repair, as well as an intraoperative evaluation of the ACL. Standard arthroscopy was performed, using anterolateral and anteromedial arthroscopic portals, and an accessory medial peripatellar portal. During surgery, a large prominence was noted in the region of the anterior tibial spine (Figure 3A). The ACL fibers maintained a slack position secondary to the elevation of the tibial insertion point, and intraoperative Lachman examination showed anterior translation of the tibia on the femur as the slack was removed from the ACL. During surgery, impingement of the anterior tibial spine along the femoral notch was shown to be significant by taking the knee into near-full extension (Figure 3B). A cam-like effect was noted at the time of impingement with the posterior soft tissues relaxing to accommodate slight further extension.
Based on these findings, we chose to take down the malunited fracture and repair it (Figure 3C). PDS suture (Ethicon, Somerville, New Jersey) was temporarily placed along the intermeniscal ligament and anterior horns of the medial and lateral menisci, using a system of spinal needles to facilitate suture passage. Surgical clamps were hung from the suture to provide traction on the sutures throughout the case, allowing the intermeniscal ligament and menisci to recede anteriorly to improve working space and aid in preventing iatrogenic injury. These sutures were removed at the conclusion of the case. Using a combination of curettes, elevator, and small shaver, we were able to meticulously remove interposed malunited callus to allow for mobilization of the displaced fragment. After removal of the excess bone formation, a typical donor site was created, allowing the displaced spine fragment to be hinged into appropriate alignment (Figure 3D). We were able to maintain a posterior cortical hinge to facilitate this process.
Then, we placed Kirschner wires (K-wires) across the fracture in an antegrade fashion, anterior to the trochlea and notch, using an accessory medial peripatellar starting point percutaneously, under direct visualization to avoid iatrogenic chondral injury. The tibial spine fragment was temporarily maintained in a reduced position with an arthroscopic probe and pinned in place with two 0.062-in K-wires. The fracture was stabilized with 8 resorbable 1.6-mm poly-L-lactic/polyglycolic acid (PLLA/PGA) nails, in varying lengths from 18 mm to 22 mm. Excellent fixation was obtained, and range of motion was tested from 0º to 80º, without movement of the fracture site (Figure 3E). Fluoroscopy with multi-axial views verified adequate fixation and reduction. Further, we examined and noted a taut ACL after fixation. The patient was placed in a long leg cast for 3 weeks at 30º, based upon intraoperative determination of the position of least tension on the fracture fragment.
At 3-week follow-up, the patient was progressing well and transitioned from a long leg cast to a hinged knee brace, to allow for early range of motion. Radiographs showed appropriate alignment of the tibial spine fracture with no significant loss of fixation (Figures 4A, 4B). Physical therapy was initiated between 0º and 30º, and flexion was progressively increased over the course of the first 3 weeks. Active and active-assist, closed-chain activities were maintained. Seven weeks postoperatively, the patient displayed continued clinical progression. Radiographs showed interval healing with slight lucency over the anterolateral aspect of the fracture fragment, likely related to the early resorptive process of healing. Physical examination showed movement between 0º and 120º, stable Lachman test, and stable anterior drawer. Crutches were discontinued and hinged knee brace was converted to an ACL brace. By the 11th week, motion had increased to 140º, and radiographs continued to show acceptable alignment and healing (Figures 5A, 5B). The patient was released to return to play as tolerated; however, an ACL brace was recommended during his initial return to provide additional support.
Discussion
In this report, we present an approach for arthroscopic reduction of a malunited tibial spine fracture using resorbable PLLA/PGA nails. The number of polyglycolic nails employed is individualized per case, dependent on the surface area and the quality of the bone within the fractured fragment. Preoperative templating allows for measurements from the fractured fragment to the level of the proximal tibial physis. Based on these measurements, nails are chosen to maximize fixation length and avoid the physis. Despite studies that have examined the effect of transphyseal K-wire pinning or drilling on subsequent growth, there is no consensus about optimal technique. Experiments in animal models indicate that drill injuries destroying less than 8% to 9% of the physis do not impact total bone growth.12,13 Further, temporary crossing of the physeal plate for internal fixation of dislocated joint injuries has not been shown to result in bone bridging or growth disturbance.14,15
Each nail is 1.6 mm in diameter, leaving a small footprint. The nails are used judiciously to provide effective stabilization of the fragment and to maintain a cost-conscious approach. An accessory superomedial peripatellar portal allows an appropriate angle for nail placement. This portal allows access to all regions of the fractured fragment, while an anteromedial and anterolateral portal are used as working and camera portals, respectively. Nails are placed to provide an axis perpendicular to the fracture line to allow appropriate compression. By virtue of the shape of the typical fragment in a tibial spine fracture, the nails vary in insertion angle.
The occurrence of anterior tibial spine fractures is rare, and while several techniques have been described to repair this fracture, there remains a great deal of uncertainty regarding the best course of treatment. A review of the literature finds arthroscopic and open approaches, as well as techniques employing K-wire fixation, metal screw fixation, staple fixation, absorbable fixation, and fixation with sutures passed through the tibial tunnel.16-18
Avulsion fractures of the tibial eminence were treated with open fixation until McLennan8 first reported the benefits of reduction with an arthroscope. Open reduction and internal fixation provide the benefit of direct visualization,9 while arthroscopic reduction offers decreased morbidity and an accelerated recovery of knee functions,8 despite the fact that a higher rate of range-of-motion deficits were seen in patients treated arthroscopically.19 We feel that with proper early rehabilitation to achieve range of motion, the risk of this can be minimal.
Various arthroscopic approaches that improve the accuracy of the reduction and decrease surgical invasiveness have been described. Suture and screw fixation are among the most common methods, and both have resulted in positive outcomes.20-24 Suture fixation of the tibial eminence is technically demanding but offers secure fixation without the need for follow-up hardware removal. Screw fixation results in secure fixation; however, numerous hardware-related issues may necessitate removal. Furthermore, in skeletally immature patients, screw fixation may disturb the growth plate if it crosses an open physis.9
Hunter and Willis25 retrospectively reviewed patients with tibial eminence fractures treated with either screw or suture fixation and found a 44% reoperation rate in the screw-fixation group. Removal was often recommended as a result of hardware-related issues. There was a 13% reoperation rate in the suture-fixation group, which resulted largely from stiffness.25 In a recent review, Gans and colleagues19 reviewed 6 publications comparing screw and suture fixation of tibial eminence fractures and found 82.4% of screw patients had laxity on both the anterior drawer and Lachman tests, compared with 18.8% in the suture-fixation group. This study also noted a slightly higher rate of arthrofibrosis in patients treated with suture fixation.19 Biomechanical studies indicate that suture fixation imparts greater strength under cyclic-loading conditions;26 however, there does not appear to be a difference in ultimate force required for fixation failure.27
Ultimately, both suture and screw fixation result in secure methods of fixation; however, there are often greater issues with screw fixation because of the persistent hardware. Metal has been the most popular method for fracture fixation, and while biodegradable materials have been alluring, adverse tissue reactions have slowed implementation. However, these implants have become increasingly sophisticated, thereby reducing disadvantages.28 Previous biodegradable devices were often composed of a single polymer, and many caused adverse reactions by degrading too quickly or provided no real advantages because they degraded too slowly.29 As the number of polymers approved for internal use and surgical applications continues to rise, so too will the benefits of employing this technology. Furthermore, by including multiple polymers in these implants, one is better able to control the degradation rate, limiting the tissue response.
In this study, we employed PLLA/PGA nails. Studies of PGA implants indicate this molecule degrades at a fast rate resulting in adverse tissue reactions. Adverse reactions in studies of PLLA implants are less frequent because of their slower rate of degradation.29,30 Combining these monomers results in appropriate strength and a controlled degradation rate, reducing the likelihood of adverse reactions. Furthermore, numerous studies have reported that inflammatory responses in children are rare and mild in nature.31,32 Absorbable implants have displayed efficacy in numerous orthopedic settings33-36 and are beneficial in procedures that are not suitable for repeated surgeries, such as reconstruction of the ACL.37 There is some concern about the use of absorbable implants in synovial joints. Polyglycolic acid use in synovial joints may cause foreign-body reactions and may increase the risk of intra-articular dissemination of polymeric debris;38 however, use of a multipolymer construct decreases the likelihood of this occurrence.
Polyglycolic nails confer the advantage over nonresorbable screw fixation because further procedure for hardware removal is not required. Although suture fixation has proved to be beneficial over nonresorbable screw fixation, implantation of resorbable nails appears to have several advantages. In Dr. Estes’ experience, placement of resorbable screws through an accessory superomedial portal is far less technically demanding than placement of suture through the fracture fragment. Further, as sutures are passed from the extra-articular to the intra-articular region of the joint, capsular layers of the knee may inadvertently be bound up in the fixation, predisposing to arthrofibrosis.
At the same time, biodegradable devices are often more costly than alternative forms of treatment; however, a true cost-to-benefit analysis requires consideration of other factors. One of the benefits of biodegradable hardware is that there is no need for follow-up hardware removal. Reports have indicated that up to 91% of patients thought that hardware removal was the most negative aspect of metal implants.39 It is estimated that if the removal rate for metallic implants is higher than 19% to 54%, resorbable implants would be more cost-effective.40 The cost of sutures and screws is variable, however; they are invariably less expensive than biodegradable nails. A study of fracture patients determined that biodegradable implants were cheaper on average after considering the cost of implant removal.40 Ultimately, the hardware choice depends on numerous factors, including surgeon’s discretion; however, biodegradable hardware should not be discounted for financial reasons because the difference in cost is likely negligible.
Conclusion
The approach described in this report offers efficient and secure fixation with resorbable hardware without a reduction in range of motion. Resorbable implants may prove beneficial in the treatment of tibial eminence fractures by offering robust fixation without the concerns associated with permanent hardware.
Anterior tibial spine fractures are rare, occurring with an incidence of 3 per 100,000 per year.1,2 Historically, this fracture has occurred more frequently in children,3-5 and was considered a condition of skeletal immaturity and the pediatric equivalent of an anterior cruciate ligament (ACL) rupture.6 However, recent literature indicates that this fracture is more common in the adult population than previously thought.7 The tibial spine is an attachment point for the ACL and an avulsion may produce ACL laxity,8 predisposing to further symptomatic laxity and premature osteoarthritis. Nearly 40% of these fractures are associated with concomitant injuries to surrounding structures.9
Meyers and McKeever10,11 originally classified these fractures into 3 groups on the basis of displacement. Type I fractures present with no significant displacement of the anterior margin, type II involve displacement and are hinged, while type III have complete displacement.10,11 More recently, a type IV fracture has been added, involving comminution of the displaced fragment. Nondisplaced fractures are commonly treated with immobilization in varying degrees of extension; this allows the femoral condyles to compress and to reduce the fracture while arthroscopic or open reduction is the preferred method for displaced fractures of the tibial spine.2,4,8,10
We report the case of an 11-year-old boy with a tibial spine fracture that failed conservative management. He developed a subsequent malunion with impingement anteriorly of the tibial spine on the notch, and residual instability of the ACL. The patient’s parents provided written informed consent for print and electronic publication of this case report.
Case Report
An 11-year-old Caucasian boy was referred to our office for evaluation of right knee injury. He sustained the injury approximately 3 months earlier, and it was determined that he had a tibial spine fracture. Conservative management with immobilization in extension and activity modification was undertaken; however, he was referred for further evaluation because of healing in a malreduced position and residual ACL laxity. Physical examination showed a grade 2A Lachman test (contralateral limb with negative Lachman examination), negative McMurray test, and pain with forced hyperextension; range-of-motion examination showed lack of the terminal 5º of extension. Magnetic resonance and computed tomography imaging from an outside facility showed a skeletally immature individual with a large tibial spine fracture that had healed in a malunited position with the fragment extended on a posterior hinge, creating a large prominence anteriorly (Figures 1A, 1B). Magnetic resonance imaging showed that the ACL fibers were likely to remain intact but would lack appropriate tension secondary to the displacement of the tibial insertion.
Because of healing in a displaced position, lack of terminal extension, ACL laxity, and subjective complaints of pain, we discussed surgery with the patient and his parents (Figures 2A, 2B). Four months after the initial injury, the patient underwent surgery for a right tibial spine malunion arthroscopic takedown and repair, as well as an intraoperative evaluation of the ACL. Standard arthroscopy was performed, using anterolateral and anteromedial arthroscopic portals, and an accessory medial peripatellar portal. During surgery, a large prominence was noted in the region of the anterior tibial spine (Figure 3A). The ACL fibers maintained a slack position secondary to the elevation of the tibial insertion point, and intraoperative Lachman examination showed anterior translation of the tibia on the femur as the slack was removed from the ACL. During surgery, impingement of the anterior tibial spine along the femoral notch was shown to be significant by taking the knee into near-full extension (Figure 3B). A cam-like effect was noted at the time of impingement with the posterior soft tissues relaxing to accommodate slight further extension.
Based on these findings, we chose to take down the malunited fracture and repair it (Figure 3C). PDS suture (Ethicon, Somerville, New Jersey) was temporarily placed along the intermeniscal ligament and anterior horns of the medial and lateral menisci, using a system of spinal needles to facilitate suture passage. Surgical clamps were hung from the suture to provide traction on the sutures throughout the case, allowing the intermeniscal ligament and menisci to recede anteriorly to improve working space and aid in preventing iatrogenic injury. These sutures were removed at the conclusion of the case. Using a combination of curettes, elevator, and small shaver, we were able to meticulously remove interposed malunited callus to allow for mobilization of the displaced fragment. After removal of the excess bone formation, a typical donor site was created, allowing the displaced spine fragment to be hinged into appropriate alignment (Figure 3D). We were able to maintain a posterior cortical hinge to facilitate this process.
Then, we placed Kirschner wires (K-wires) across the fracture in an antegrade fashion, anterior to the trochlea and notch, using an accessory medial peripatellar starting point percutaneously, under direct visualization to avoid iatrogenic chondral injury. The tibial spine fragment was temporarily maintained in a reduced position with an arthroscopic probe and pinned in place with two 0.062-in K-wires. The fracture was stabilized with 8 resorbable 1.6-mm poly-L-lactic/polyglycolic acid (PLLA/PGA) nails, in varying lengths from 18 mm to 22 mm. Excellent fixation was obtained, and range of motion was tested from 0º to 80º, without movement of the fracture site (Figure 3E). Fluoroscopy with multi-axial views verified adequate fixation and reduction. Further, we examined and noted a taut ACL after fixation. The patient was placed in a long leg cast for 3 weeks at 30º, based upon intraoperative determination of the position of least tension on the fracture fragment.
At 3-week follow-up, the patient was progressing well and transitioned from a long leg cast to a hinged knee brace, to allow for early range of motion. Radiographs showed appropriate alignment of the tibial spine fracture with no significant loss of fixation (Figures 4A, 4B). Physical therapy was initiated between 0º and 30º, and flexion was progressively increased over the course of the first 3 weeks. Active and active-assist, closed-chain activities were maintained. Seven weeks postoperatively, the patient displayed continued clinical progression. Radiographs showed interval healing with slight lucency over the anterolateral aspect of the fracture fragment, likely related to the early resorptive process of healing. Physical examination showed movement between 0º and 120º, stable Lachman test, and stable anterior drawer. Crutches were discontinued and hinged knee brace was converted to an ACL brace. By the 11th week, motion had increased to 140º, and radiographs continued to show acceptable alignment and healing (Figures 5A, 5B). The patient was released to return to play as tolerated; however, an ACL brace was recommended during his initial return to provide additional support.
Discussion
In this report, we present an approach for arthroscopic reduction of a malunited tibial spine fracture using resorbable PLLA/PGA nails. The number of polyglycolic nails employed is individualized per case, dependent on the surface area and the quality of the bone within the fractured fragment. Preoperative templating allows for measurements from the fractured fragment to the level of the proximal tibial physis. Based on these measurements, nails are chosen to maximize fixation length and avoid the physis. Despite studies that have examined the effect of transphyseal K-wire pinning or drilling on subsequent growth, there is no consensus about optimal technique. Experiments in animal models indicate that drill injuries destroying less than 8% to 9% of the physis do not impact total bone growth.12,13 Further, temporary crossing of the physeal plate for internal fixation of dislocated joint injuries has not been shown to result in bone bridging or growth disturbance.14,15
Each nail is 1.6 mm in diameter, leaving a small footprint. The nails are used judiciously to provide effective stabilization of the fragment and to maintain a cost-conscious approach. An accessory superomedial peripatellar portal allows an appropriate angle for nail placement. This portal allows access to all regions of the fractured fragment, while an anteromedial and anterolateral portal are used as working and camera portals, respectively. Nails are placed to provide an axis perpendicular to the fracture line to allow appropriate compression. By virtue of the shape of the typical fragment in a tibial spine fracture, the nails vary in insertion angle.
The occurrence of anterior tibial spine fractures is rare, and while several techniques have been described to repair this fracture, there remains a great deal of uncertainty regarding the best course of treatment. A review of the literature finds arthroscopic and open approaches, as well as techniques employing K-wire fixation, metal screw fixation, staple fixation, absorbable fixation, and fixation with sutures passed through the tibial tunnel.16-18
Avulsion fractures of the tibial eminence were treated with open fixation until McLennan8 first reported the benefits of reduction with an arthroscope. Open reduction and internal fixation provide the benefit of direct visualization,9 while arthroscopic reduction offers decreased morbidity and an accelerated recovery of knee functions,8 despite the fact that a higher rate of range-of-motion deficits were seen in patients treated arthroscopically.19 We feel that with proper early rehabilitation to achieve range of motion, the risk of this can be minimal.
Various arthroscopic approaches that improve the accuracy of the reduction and decrease surgical invasiveness have been described. Suture and screw fixation are among the most common methods, and both have resulted in positive outcomes.20-24 Suture fixation of the tibial eminence is technically demanding but offers secure fixation without the need for follow-up hardware removal. Screw fixation results in secure fixation; however, numerous hardware-related issues may necessitate removal. Furthermore, in skeletally immature patients, screw fixation may disturb the growth plate if it crosses an open physis.9
Hunter and Willis25 retrospectively reviewed patients with tibial eminence fractures treated with either screw or suture fixation and found a 44% reoperation rate in the screw-fixation group. Removal was often recommended as a result of hardware-related issues. There was a 13% reoperation rate in the suture-fixation group, which resulted largely from stiffness.25 In a recent review, Gans and colleagues19 reviewed 6 publications comparing screw and suture fixation of tibial eminence fractures and found 82.4% of screw patients had laxity on both the anterior drawer and Lachman tests, compared with 18.8% in the suture-fixation group. This study also noted a slightly higher rate of arthrofibrosis in patients treated with suture fixation.19 Biomechanical studies indicate that suture fixation imparts greater strength under cyclic-loading conditions;26 however, there does not appear to be a difference in ultimate force required for fixation failure.27
Ultimately, both suture and screw fixation result in secure methods of fixation; however, there are often greater issues with screw fixation because of the persistent hardware. Metal has been the most popular method for fracture fixation, and while biodegradable materials have been alluring, adverse tissue reactions have slowed implementation. However, these implants have become increasingly sophisticated, thereby reducing disadvantages.28 Previous biodegradable devices were often composed of a single polymer, and many caused adverse reactions by degrading too quickly or provided no real advantages because they degraded too slowly.29 As the number of polymers approved for internal use and surgical applications continues to rise, so too will the benefits of employing this technology. Furthermore, by including multiple polymers in these implants, one is better able to control the degradation rate, limiting the tissue response.
In this study, we employed PLLA/PGA nails. Studies of PGA implants indicate this molecule degrades at a fast rate resulting in adverse tissue reactions. Adverse reactions in studies of PLLA implants are less frequent because of their slower rate of degradation.29,30 Combining these monomers results in appropriate strength and a controlled degradation rate, reducing the likelihood of adverse reactions. Furthermore, numerous studies have reported that inflammatory responses in children are rare and mild in nature.31,32 Absorbable implants have displayed efficacy in numerous orthopedic settings33-36 and are beneficial in procedures that are not suitable for repeated surgeries, such as reconstruction of the ACL.37 There is some concern about the use of absorbable implants in synovial joints. Polyglycolic acid use in synovial joints may cause foreign-body reactions and may increase the risk of intra-articular dissemination of polymeric debris;38 however, use of a multipolymer construct decreases the likelihood of this occurrence.
Polyglycolic nails confer the advantage over nonresorbable screw fixation because further procedure for hardware removal is not required. Although suture fixation has proved to be beneficial over nonresorbable screw fixation, implantation of resorbable nails appears to have several advantages. In Dr. Estes’ experience, placement of resorbable screws through an accessory superomedial portal is far less technically demanding than placement of suture through the fracture fragment. Further, as sutures are passed from the extra-articular to the intra-articular region of the joint, capsular layers of the knee may inadvertently be bound up in the fixation, predisposing to arthrofibrosis.
At the same time, biodegradable devices are often more costly than alternative forms of treatment; however, a true cost-to-benefit analysis requires consideration of other factors. One of the benefits of biodegradable hardware is that there is no need for follow-up hardware removal. Reports have indicated that up to 91% of patients thought that hardware removal was the most negative aspect of metal implants.39 It is estimated that if the removal rate for metallic implants is higher than 19% to 54%, resorbable implants would be more cost-effective.40 The cost of sutures and screws is variable, however; they are invariably less expensive than biodegradable nails. A study of fracture patients determined that biodegradable implants were cheaper on average after considering the cost of implant removal.40 Ultimately, the hardware choice depends on numerous factors, including surgeon’s discretion; however, biodegradable hardware should not be discounted for financial reasons because the difference in cost is likely negligible.
Conclusion
The approach described in this report offers efficient and secure fixation with resorbable hardware without a reduction in range of motion. Resorbable implants may prove beneficial in the treatment of tibial eminence fractures by offering robust fixation without the concerns associated with permanent hardware.
1. Hargrove R, Parsons S, Payne R. Anterior tibial spine fracture – an easy fracture to miss. Accid Emerg Nurs. 2004;12(3):173-175.
2. Aderinto J, Walmsley P, Keating JF. Fractures of the tibial spine: epidemiology and outcome. Knee. 2008;15(3):164-167.
3. Driessen MJ, Winkelman PA. Fractures of the intercondylar eminence of the tibia in childhood. Neth J Surg. 1984;36(3):69-72.
4. Zaricznyj B. Avulsion fracture of the tibial eminence: treatment by open reduction and pinning. J Bone Joint Surg Am. 1977;59(8):1111-1114.
5. Molander ML, Wallin G, Wikstad I. Fracture of the intercondylar eminence of the tibia: a review of 35 patients. J Bone Joint Surg Br. 1981;63(1):89-91.
6. Kieser DC, Gwynne-Jones D, Dreyer S. Displaced tibial intercondylar eminence fractures. J Orthop Surg. 2011;19(3):292-296.
7. Ishibashi Y, Tsuda E, Sasaki T, Toh S. Magnetic resonance imaging AIDS in detecting concomitant injuries in patients with tibial spine fractures. Clin Orthop. 2005;(434):207-212.
8. McLennan JG. The role of arthroscopic surgery in the treatment of fractures of the intercondylar eminence of the tibia. J Bone Joint Surg Br. 1982;64(4):477-480.
9. Lafrance RM, Giordano B, Goldblatt J, Voloshin I, Maloney M. Pediatric tibial eminence fractures: evaluation and management. J Am Acad Orthop Surg. 2010;18(7):395-405.
10. Meyers MH, McKeever FM. Fracture of the intercondylar eminence of the tibia. J Bone Joint Surg Am. 1959;41(2):209-220.
11. Meyers MH, McKeever FM. Fracture of the intercondylar eminence of the tibia. J Bone Joint Surg Am. 1970;52(8):1677-1684.
12. Garcés GL, Mugica-Garay I, López-González Coviella N, Guerado E. Growth-plate modifications after drilling. J Pediatr Orthop. 1994;14(2):225-228.
13. Janarv PM, Wikström B, Hirsch G. The influence of transphyseal drilling and tendon grafting on bone growth: an experimental study in the rabbit. J Pediatr Orthop. 1998;18(2):149-154.
14. Boelitz R, Dallek M, Meenen NM, Jungbluth KH. Reaction of the epiphyseal groove to groove-crossing bore-wire osteosynthesis. Results of a histomorphologic small animal study. Unfallchirurgie. 1994;20(3):131-137.
15. Yung PS, Lam CY, Ng BK, Lam TP, Cheng JC. Percutaneous transphyseal intramedullary Kirschner wire pinning: a safe and effective procedure for treatment of displaced diaphyseal forearm fracture in children. J Pediatr Orthop. 2004;24(1):7-12.
16. Bong MR, Romero A, Kubiak E, et al. Suture versus screw fixation of displaced tibial eminence fractures: a biomechanical comparison. Arthroscopy. 2005;21(10):1172-1176.
17. Vega JR, Irribarra LA, Baar AK, Iñiguez M, Salgado M, Gana N. Arthroscopic fixation of displaced tibial eminence fractures: a new growth plate-sparing method. Arthroscopy. 2008;24(11):1239-1243.
18. Shepley RW. Arthroscopic treatment of type III tibial spine fractures using absorbable fixation. Orthopedics. 2004;27(7):767-769.
19. Gans I, Baldwin KD, Ganley TJ. Treatment and management outcomes of tibial eminence fractures in pediatric patients: a systematic review. Am J Sports Med. 2013;42(7):1743-1750.
20. Delcogliano A, Chiossi S, Caporaso A, Menghi A, Rinonapoli G. Tibial intercondylar eminence fractures in adults: arthroscopic treatment. Knee Surg Sports Traumatol Arthrosc. 2003;11(4):255-259.
21. Mulhall KJ, Dowdall J, Grannell M, McCabe JP. Tibial spine fractures: an analysis of outcome in surgically treated type III injuries. Injury. 1999;30(4):289-292.
22. Geissler WB, Matthews DE. Arthroscopic suture fixation of displaced tibial eminence fractures. Orthopedics. 1993;16(3):331-333.
23. Mah JY, Otsuka NY, McLean J. An arthroscopic technique for the reduction and fixation of tibial-eminence fractures. J Pediatr Orthop. 1996;16(1):119-121.
24. Reynders P, Reynders K, Broos P. Pediatric and adolescent tibial eminence fractures: arthroscopic cannulated screw fixation. J Trauma. 2002;53(1):49-54.
25. Hunter RE, Willis JA. Arthroscopic fixation of avulsion fractures of the tibial eminence: technique and outcome. Arthroscopy. 2004;20(2):113-121.
26. Eggers AK, Becker C, Weimann A, et al. Biomechanical evaluation of different fixation methods for tibial eminence fractures. Am J Sports Med. 2007;35(3):404-410.
27. Mahar AT, Duncan D, Oka R, Lowry A, Gillingham B, Chambers H. Biomechanical comparison of four different fixation techniques for pediatric tibial eminence avulsion fractures. J Pediatr Orthop. 2008;28(2):159-162.
28. Toro C, Robiony M, Zerman N, Politi M. Resorbable plates in maxillary fixation. A 5-year experience. Minerva Stomatol. 2005;54(4):199-206.
29. Andriano KP, Pohjonen T, Törmälä P. Processing and characterization of absorbable polylactide polymers for use in surgical implants. J Appl Biomater.1994;5(2):133-140.
30. Böstman O, Pihlajamäki H. Clinical biocompatibility of biodegradable orthopaedic implants for internal fixation: a review. Biomaterials. 2000;21(24):2615-2621.
31. Rokkanen PU, Böstman O, Hirvensalo E, et al. Bioabsorbable fixation in orthopaedic surgery and traumatology. Biomaterials. 2000;21(24):2607-2613.
32. Athanasiou KA, Niederauer GG, Agrawal CM. Sterilization, toxicity, biocompatibility and clinical applications of polylactic acid/polyglycolic acid copolymers. Biomaterials. 1996;17(2):93-102.
33. Li ZH, Yu AX, Guo XP, Qi BW, Zhou M, Wang WY. Absorbable implants versus metal implants for the treatment of ankle fractures: A meta-analysis. Exp Ther Med. 2013;5(5):1531-1537.
34. Singh G, Mohammad S, Chak RK, Lepcha N, Singh N, Malkunje LR. Bio-resorbable plates as effective implant in paediatric mandibular fracture. J Maxillofac Oral Surg. 2012;11(4):400-406.
35. Sakamoto Y, Shimizu Y, Nagasao T, Kishi K. Combined use of resorbable poly-L-lactic acid-polyglycolic acid implant and bone cement for treating large orbital floor fractures. J Plast Reconstr Aesthet Surg. 2014;67(3):e88-e90.
36. Benz G, Kallieris D, Seeböck T, McIntosh A, Daum R. Bioresorbable pins and screws in paediatric traumatology. Eur J Pediatr Surg. 1994;4(2):103-107.
37. Gaweda K, Walawski J, Weglowski R, Krzyzanowski W. Comparison of bioabsorbable interference screws and posts for distal fixation in anterior cruciate ligament reconstruction. Int Orthop. 2009;33(1):123-127.
38. Böstman OM. Osteoarthritis of the ankle after foreign-body reaction to absorbable pins and screws: a three- to nine-year follow-up study. J Bone Joint Surg Br. 1998;80(2):333-338.
39. Mittal R, Morley J, Dinopoulos H, Drakoulakis EG, Vermani E, Giannoudis PV. Use of bio-resorbable implants for stabilisation of distal radius fractures: the United Kingdom patients’ perspective. Injury. 2005;36(2):333-338.
40. Böstman OM. Metallic or absorbable fracture fixation devices. A cost minimization analysis. Clin Orthop. 1996;(329):233-239.
1. Hargrove R, Parsons S, Payne R. Anterior tibial spine fracture – an easy fracture to miss. Accid Emerg Nurs. 2004;12(3):173-175.
2. Aderinto J, Walmsley P, Keating JF. Fractures of the tibial spine: epidemiology and outcome. Knee. 2008;15(3):164-167.
3. Driessen MJ, Winkelman PA. Fractures of the intercondylar eminence of the tibia in childhood. Neth J Surg. 1984;36(3):69-72.
4. Zaricznyj B. Avulsion fracture of the tibial eminence: treatment by open reduction and pinning. J Bone Joint Surg Am. 1977;59(8):1111-1114.
5. Molander ML, Wallin G, Wikstad I. Fracture of the intercondylar eminence of the tibia: a review of 35 patients. J Bone Joint Surg Br. 1981;63(1):89-91.
6. Kieser DC, Gwynne-Jones D, Dreyer S. Displaced tibial intercondylar eminence fractures. J Orthop Surg. 2011;19(3):292-296.
7. Ishibashi Y, Tsuda E, Sasaki T, Toh S. Magnetic resonance imaging AIDS in detecting concomitant injuries in patients with tibial spine fractures. Clin Orthop. 2005;(434):207-212.
8. McLennan JG. The role of arthroscopic surgery in the treatment of fractures of the intercondylar eminence of the tibia. J Bone Joint Surg Br. 1982;64(4):477-480.
9. Lafrance RM, Giordano B, Goldblatt J, Voloshin I, Maloney M. Pediatric tibial eminence fractures: evaluation and management. J Am Acad Orthop Surg. 2010;18(7):395-405.
10. Meyers MH, McKeever FM. Fracture of the intercondylar eminence of the tibia. J Bone Joint Surg Am. 1959;41(2):209-220.
11. Meyers MH, McKeever FM. Fracture of the intercondylar eminence of the tibia. J Bone Joint Surg Am. 1970;52(8):1677-1684.
12. Garcés GL, Mugica-Garay I, López-González Coviella N, Guerado E. Growth-plate modifications after drilling. J Pediatr Orthop. 1994;14(2):225-228.
13. Janarv PM, Wikström B, Hirsch G. The influence of transphyseal drilling and tendon grafting on bone growth: an experimental study in the rabbit. J Pediatr Orthop. 1998;18(2):149-154.
14. Boelitz R, Dallek M, Meenen NM, Jungbluth KH. Reaction of the epiphyseal groove to groove-crossing bore-wire osteosynthesis. Results of a histomorphologic small animal study. Unfallchirurgie. 1994;20(3):131-137.
15. Yung PS, Lam CY, Ng BK, Lam TP, Cheng JC. Percutaneous transphyseal intramedullary Kirschner wire pinning: a safe and effective procedure for treatment of displaced diaphyseal forearm fracture in children. J Pediatr Orthop. 2004;24(1):7-12.
16. Bong MR, Romero A, Kubiak E, et al. Suture versus screw fixation of displaced tibial eminence fractures: a biomechanical comparison. Arthroscopy. 2005;21(10):1172-1176.
17. Vega JR, Irribarra LA, Baar AK, Iñiguez M, Salgado M, Gana N. Arthroscopic fixation of displaced tibial eminence fractures: a new growth plate-sparing method. Arthroscopy. 2008;24(11):1239-1243.
18. Shepley RW. Arthroscopic treatment of type III tibial spine fractures using absorbable fixation. Orthopedics. 2004;27(7):767-769.
19. Gans I, Baldwin KD, Ganley TJ. Treatment and management outcomes of tibial eminence fractures in pediatric patients: a systematic review. Am J Sports Med. 2013;42(7):1743-1750.
20. Delcogliano A, Chiossi S, Caporaso A, Menghi A, Rinonapoli G. Tibial intercondylar eminence fractures in adults: arthroscopic treatment. Knee Surg Sports Traumatol Arthrosc. 2003;11(4):255-259.
21. Mulhall KJ, Dowdall J, Grannell M, McCabe JP. Tibial spine fractures: an analysis of outcome in surgically treated type III injuries. Injury. 1999;30(4):289-292.
22. Geissler WB, Matthews DE. Arthroscopic suture fixation of displaced tibial eminence fractures. Orthopedics. 1993;16(3):331-333.
23. Mah JY, Otsuka NY, McLean J. An arthroscopic technique for the reduction and fixation of tibial-eminence fractures. J Pediatr Orthop. 1996;16(1):119-121.
24. Reynders P, Reynders K, Broos P. Pediatric and adolescent tibial eminence fractures: arthroscopic cannulated screw fixation. J Trauma. 2002;53(1):49-54.
25. Hunter RE, Willis JA. Arthroscopic fixation of avulsion fractures of the tibial eminence: technique and outcome. Arthroscopy. 2004;20(2):113-121.
26. Eggers AK, Becker C, Weimann A, et al. Biomechanical evaluation of different fixation methods for tibial eminence fractures. Am J Sports Med. 2007;35(3):404-410.
27. Mahar AT, Duncan D, Oka R, Lowry A, Gillingham B, Chambers H. Biomechanical comparison of four different fixation techniques for pediatric tibial eminence avulsion fractures. J Pediatr Orthop. 2008;28(2):159-162.
28. Toro C, Robiony M, Zerman N, Politi M. Resorbable plates in maxillary fixation. A 5-year experience. Minerva Stomatol. 2005;54(4):199-206.
29. Andriano KP, Pohjonen T, Törmälä P. Processing and characterization of absorbable polylactide polymers for use in surgical implants. J Appl Biomater.1994;5(2):133-140.
30. Böstman O, Pihlajamäki H. Clinical biocompatibility of biodegradable orthopaedic implants for internal fixation: a review. Biomaterials. 2000;21(24):2615-2621.
31. Rokkanen PU, Böstman O, Hirvensalo E, et al. Bioabsorbable fixation in orthopaedic surgery and traumatology. Biomaterials. 2000;21(24):2607-2613.
32. Athanasiou KA, Niederauer GG, Agrawal CM. Sterilization, toxicity, biocompatibility and clinical applications of polylactic acid/polyglycolic acid copolymers. Biomaterials. 1996;17(2):93-102.
33. Li ZH, Yu AX, Guo XP, Qi BW, Zhou M, Wang WY. Absorbable implants versus metal implants for the treatment of ankle fractures: A meta-analysis. Exp Ther Med. 2013;5(5):1531-1537.
34. Singh G, Mohammad S, Chak RK, Lepcha N, Singh N, Malkunje LR. Bio-resorbable plates as effective implant in paediatric mandibular fracture. J Maxillofac Oral Surg. 2012;11(4):400-406.
35. Sakamoto Y, Shimizu Y, Nagasao T, Kishi K. Combined use of resorbable poly-L-lactic acid-polyglycolic acid implant and bone cement for treating large orbital floor fractures. J Plast Reconstr Aesthet Surg. 2014;67(3):e88-e90.
36. Benz G, Kallieris D, Seeböck T, McIntosh A, Daum R. Bioresorbable pins and screws in paediatric traumatology. Eur J Pediatr Surg. 1994;4(2):103-107.
37. Gaweda K, Walawski J, Weglowski R, Krzyzanowski W. Comparison of bioabsorbable interference screws and posts for distal fixation in anterior cruciate ligament reconstruction. Int Orthop. 2009;33(1):123-127.
38. Böstman OM. Osteoarthritis of the ankle after foreign-body reaction to absorbable pins and screws: a three- to nine-year follow-up study. J Bone Joint Surg Br. 1998;80(2):333-338.
39. Mittal R, Morley J, Dinopoulos H, Drakoulakis EG, Vermani E, Giannoudis PV. Use of bio-resorbable implants for stabilisation of distal radius fractures: the United Kingdom patients’ perspective. Injury. 2005;36(2):333-338.
40. Böstman OM. Metallic or absorbable fracture fixation devices. A cost minimization analysis. Clin Orthop. 1996;(329):233-239.

Hospital Testing Overuse Done to Reassure Patients, Families
Clinical Question: What is the extent of, and factors associated with, testing overuse in U.S. hospitals for pre-operative evaluation and syncope.
Background: Little is known about the extent and drivers of overuse by hospitalists.
Study design: Two vignettes (pre-operative evaluation and syncope) were mailed to hospitalists. They were asked to identify what most hospitalists at their institution would recommend and “the most likely primary driver of the hospitalist’s decision.”
Setting: Random selection of hospitalists from SHM member database and SHM national meeting attendees.
Synopsis: Investigators mailed 1,753 surveys and received a 68% response rate. For the pre-operative evaluation vignette, 52% of hospitalists reported overuse of pre-operative testing. When a family member was a physician and requested further testing, overuse increased significantly to 65%. For the syncope vignette, any choice involving admission was considered overuse.
Eighty-two percent of respondents reported overuse; when the wife was a lawyer or requested further testing, overuse remained the same. Overuse in both cases was more frequent due to a hospitalist’s desire to reassure patients or themselves, rather than a belief that it was clinically indicated (pre-operative evaluation, 63% vs. 37%; syncope, 69% vs. 31%, P<0.001).
The survey responses do not necessarily represent actual clinical choices, and the hospitalist sample may not be representative of all hospitalists; however, this study shows that efforts to reduce overuse in hospitals need to move beyond financial incentives and/or informing providers of evidence-based recommendations.
Bottom line: A survey of hospitalists showed substantial overuse in two common clinical situations, syncope and pre-operative evaluation, mostly driven by a desire to reassure patients, families, or themselves.
Citation: Kachalia A, Berg A, Fagerlin A, et al. Overuse of testing in preoperative evaluation and syncope: a survey of hospitalists. Ann Intern Med. 2015;162(2):100-108.
Clinical Question: What is the extent of, and factors associated with, testing overuse in U.S. hospitals for pre-operative evaluation and syncope.
Background: Little is known about the extent and drivers of overuse by hospitalists.
Study design: Two vignettes (pre-operative evaluation and syncope) were mailed to hospitalists. They were asked to identify what most hospitalists at their institution would recommend and “the most likely primary driver of the hospitalist’s decision.”
Setting: Random selection of hospitalists from SHM member database and SHM national meeting attendees.
Synopsis: Investigators mailed 1,753 surveys and received a 68% response rate. For the pre-operative evaluation vignette, 52% of hospitalists reported overuse of pre-operative testing. When a family member was a physician and requested further testing, overuse increased significantly to 65%. For the syncope vignette, any choice involving admission was considered overuse.
Eighty-two percent of respondents reported overuse; when the wife was a lawyer or requested further testing, overuse remained the same. Overuse in both cases was more frequent due to a hospitalist’s desire to reassure patients or themselves, rather than a belief that it was clinically indicated (pre-operative evaluation, 63% vs. 37%; syncope, 69% vs. 31%, P<0.001).
The survey responses do not necessarily represent actual clinical choices, and the hospitalist sample may not be representative of all hospitalists; however, this study shows that efforts to reduce overuse in hospitals need to move beyond financial incentives and/or informing providers of evidence-based recommendations.
Bottom line: A survey of hospitalists showed substantial overuse in two common clinical situations, syncope and pre-operative evaluation, mostly driven by a desire to reassure patients, families, or themselves.
Citation: Kachalia A, Berg A, Fagerlin A, et al. Overuse of testing in preoperative evaluation and syncope: a survey of hospitalists. Ann Intern Med. 2015;162(2):100-108.
Clinical Question: What is the extent of, and factors associated with, testing overuse in U.S. hospitals for pre-operative evaluation and syncope.
Background: Little is known about the extent and drivers of overuse by hospitalists.
Study design: Two vignettes (pre-operative evaluation and syncope) were mailed to hospitalists. They were asked to identify what most hospitalists at their institution would recommend and “the most likely primary driver of the hospitalist’s decision.”
Setting: Random selection of hospitalists from SHM member database and SHM national meeting attendees.
Synopsis: Investigators mailed 1,753 surveys and received a 68% response rate. For the pre-operative evaluation vignette, 52% of hospitalists reported overuse of pre-operative testing. When a family member was a physician and requested further testing, overuse increased significantly to 65%. For the syncope vignette, any choice involving admission was considered overuse.
Eighty-two percent of respondents reported overuse; when the wife was a lawyer or requested further testing, overuse remained the same. Overuse in both cases was more frequent due to a hospitalist’s desire to reassure patients or themselves, rather than a belief that it was clinically indicated (pre-operative evaluation, 63% vs. 37%; syncope, 69% vs. 31%, P<0.001).
The survey responses do not necessarily represent actual clinical choices, and the hospitalist sample may not be representative of all hospitalists; however, this study shows that efforts to reduce overuse in hospitals need to move beyond financial incentives and/or informing providers of evidence-based recommendations.
Bottom line: A survey of hospitalists showed substantial overuse in two common clinical situations, syncope and pre-operative evaluation, mostly driven by a desire to reassure patients, families, or themselves.
Citation: Kachalia A, Berg A, Fagerlin A, et al. Overuse of testing in preoperative evaluation and syncope: a survey of hospitalists. Ann Intern Med. 2015;162(2):100-108.
Functional Impairment Boosts Readmission for Medicare Seniors
Clinical question: Is functional impairment associated with an increased risk of 30-day readmission?
Background: Many Medicare seniors suffer from some level of impairment in functional status, which, in turn, has been linked to high healthcare utilization. Studies that examine the role of functional impairment with readmission rates are limited.
Study design: Prospective, cohort study.
Setting: Seniors enrolled in the Health and Retirement Study (HRS) with Medicare hospitalizations from Jan. 1, 2000, to Dec. 31, 2010.
Synopsis: The primary outcome was readmissions within 30 days of discharge. Activities of daily living (ADL) scale and instrumental ADL were used as measures of functional impairment.
Overall, 48.3% of patients had preadmission functional impairments with a readmission rate of 15.5%. There was a progressive increase in the adjusted risk of readmission as the degree of functional impairment increased: 13.5% with no functional impairment, 14.3% with difficulty in one or more instrumental ADLs (OR 1.06; 95% CI 0.94-1.20), 14.4% with difficulty in one or more ADLs (OR 1.08; 95% CI 0.96-1.21), 16.5% with dependency in one or two ADLs (OR, 1.26; 95% CI 1.11-1.44), and 18.2% with dependency in three or more ADLs (OR 1.42; 95% CI 1.20-1.69).
This observation was more pronounced in patients admitted for heart failure, MI, and pneumonia (16.9% readmission rate for no impairment vs. 25.7% dependency in three or more ADLs, OR 1.70; 95% CI 1.04-2.78).
Although the study is limited by reliance on survey data and Medicare claim data, functional status may be an important variable in calculating readmission risk and a potential target for intervention.
Bottom line: Functional impairment is associated with an increased risk of 30-day readmission, especially in patients admitted for heart failure, MI, and pneumonia.
Citation: Greysen SR, Stijacic Cenzer I, Auerbach AD, Covinsky KE. Functional impairment and hospital readmission in Medicare seniors. JAMA Intern Med. 2015;175(4):559-565.
Clinical question: Is functional impairment associated with an increased risk of 30-day readmission?
Background: Many Medicare seniors suffer from some level of impairment in functional status, which, in turn, has been linked to high healthcare utilization. Studies that examine the role of functional impairment with readmission rates are limited.
Study design: Prospective, cohort study.
Setting: Seniors enrolled in the Health and Retirement Study (HRS) with Medicare hospitalizations from Jan. 1, 2000, to Dec. 31, 2010.
Synopsis: The primary outcome was readmissions within 30 days of discharge. Activities of daily living (ADL) scale and instrumental ADL were used as measures of functional impairment.
Overall, 48.3% of patients had preadmission functional impairments with a readmission rate of 15.5%. There was a progressive increase in the adjusted risk of readmission as the degree of functional impairment increased: 13.5% with no functional impairment, 14.3% with difficulty in one or more instrumental ADLs (OR 1.06; 95% CI 0.94-1.20), 14.4% with difficulty in one or more ADLs (OR 1.08; 95% CI 0.96-1.21), 16.5% with dependency in one or two ADLs (OR, 1.26; 95% CI 1.11-1.44), and 18.2% with dependency in three or more ADLs (OR 1.42; 95% CI 1.20-1.69).
This observation was more pronounced in patients admitted for heart failure, MI, and pneumonia (16.9% readmission rate for no impairment vs. 25.7% dependency in three or more ADLs, OR 1.70; 95% CI 1.04-2.78).
Although the study is limited by reliance on survey data and Medicare claim data, functional status may be an important variable in calculating readmission risk and a potential target for intervention.
Bottom line: Functional impairment is associated with an increased risk of 30-day readmission, especially in patients admitted for heart failure, MI, and pneumonia.
Citation: Greysen SR, Stijacic Cenzer I, Auerbach AD, Covinsky KE. Functional impairment and hospital readmission in Medicare seniors. JAMA Intern Med. 2015;175(4):559-565.
Clinical question: Is functional impairment associated with an increased risk of 30-day readmission?
Background: Many Medicare seniors suffer from some level of impairment in functional status, which, in turn, has been linked to high healthcare utilization. Studies that examine the role of functional impairment with readmission rates are limited.
Study design: Prospective, cohort study.
Setting: Seniors enrolled in the Health and Retirement Study (HRS) with Medicare hospitalizations from Jan. 1, 2000, to Dec. 31, 2010.
Synopsis: The primary outcome was readmissions within 30 days of discharge. Activities of daily living (ADL) scale and instrumental ADL were used as measures of functional impairment.
Overall, 48.3% of patients had preadmission functional impairments with a readmission rate of 15.5%. There was a progressive increase in the adjusted risk of readmission as the degree of functional impairment increased: 13.5% with no functional impairment, 14.3% with difficulty in one or more instrumental ADLs (OR 1.06; 95% CI 0.94-1.20), 14.4% with difficulty in one or more ADLs (OR 1.08; 95% CI 0.96-1.21), 16.5% with dependency in one or two ADLs (OR, 1.26; 95% CI 1.11-1.44), and 18.2% with dependency in three or more ADLs (OR 1.42; 95% CI 1.20-1.69).
This observation was more pronounced in patients admitted for heart failure, MI, and pneumonia (16.9% readmission rate for no impairment vs. 25.7% dependency in three or more ADLs, OR 1.70; 95% CI 1.04-2.78).
Although the study is limited by reliance on survey data and Medicare claim data, functional status may be an important variable in calculating readmission risk and a potential target for intervention.
Bottom line: Functional impairment is associated with an increased risk of 30-day readmission, especially in patients admitted for heart failure, MI, and pneumonia.
Citation: Greysen SR, Stijacic Cenzer I, Auerbach AD, Covinsky KE. Functional impairment and hospital readmission in Medicare seniors. JAMA Intern Med. 2015;175(4):559-565.
Delirium, Falls Reduced by Nonpharmacological Intervention
Clinical question: Are multicomponent, nonpharmacological interventions effective in decreasing delirium and falls?
Background: Delirium is prevalent among elderly hospitalized patients and is associated with increased morbidity, length of stay, healthcare costs, and risk of institutionalization. Multicomponent nonpharmacologic interventions have been used to prevent incident delirium in the elderly, but data regarding their effectiveness and impact on preventing poor outcomes are lacking.
Study design: Systematic literature review and meta-analysis.
Setting: Review of medical databases from Jan. 1, 1999, to Dec. 31, 2013.
Synopsis: Fourteen studies were included involving 4,267 elderly patients from 12 acute medical and surgical sites from around the world. There was a 53% reduction in delirium incidence associated with multicomponent, nonpharmacological interventions (OR, 0.47; 95% CI, 0.38-0.58). The odds of falling were 62% lower among intervention patients compared with controls (2.79 vs. 7.05 falls per 1,000 patient-days). The intervention group also showed a decrease in length of stay, with a mean difference of -0.16 (95% CI, -0.97 to 0.64) days and a 5% lower chance of institutionalization (95% CI, 0.71 to 1.26); however, the differences were not statistically significant.
Although the small number and heterogeneity of the studies included limited the analysis, the use of nonpharmacologic interventions appears to be a low-risk, low-cost strategy to prevent delirium. The challenge for the hospitalist in developing a nonpharmacological protocol is to determine which interventions to include; the study did not look at which interventions were most effective.
Bottom line: The use of multicomponent nonpharmacological interventions in older patients can lower the risk of delirium and falls.
Citation: Hshieh TT, Yue J, Oh E, et al. Effectiveness of multicomponent nonpharmacological delirium interventions: a meta-analysis. JAMA Intern Med. 2015;175(4):512-520.
Clinical question: Are multicomponent, nonpharmacological interventions effective in decreasing delirium and falls?
Background: Delirium is prevalent among elderly hospitalized patients and is associated with increased morbidity, length of stay, healthcare costs, and risk of institutionalization. Multicomponent nonpharmacologic interventions have been used to prevent incident delirium in the elderly, but data regarding their effectiveness and impact on preventing poor outcomes are lacking.
Study design: Systematic literature review and meta-analysis.
Setting: Review of medical databases from Jan. 1, 1999, to Dec. 31, 2013.
Synopsis: Fourteen studies were included involving 4,267 elderly patients from 12 acute medical and surgical sites from around the world. There was a 53% reduction in delirium incidence associated with multicomponent, nonpharmacological interventions (OR, 0.47; 95% CI, 0.38-0.58). The odds of falling were 62% lower among intervention patients compared with controls (2.79 vs. 7.05 falls per 1,000 patient-days). The intervention group also showed a decrease in length of stay, with a mean difference of -0.16 (95% CI, -0.97 to 0.64) days and a 5% lower chance of institutionalization (95% CI, 0.71 to 1.26); however, the differences were not statistically significant.
Although the small number and heterogeneity of the studies included limited the analysis, the use of nonpharmacologic interventions appears to be a low-risk, low-cost strategy to prevent delirium. The challenge for the hospitalist in developing a nonpharmacological protocol is to determine which interventions to include; the study did not look at which interventions were most effective.
Bottom line: The use of multicomponent nonpharmacological interventions in older patients can lower the risk of delirium and falls.
Citation: Hshieh TT, Yue J, Oh E, et al. Effectiveness of multicomponent nonpharmacological delirium interventions: a meta-analysis. JAMA Intern Med. 2015;175(4):512-520.
Clinical question: Are multicomponent, nonpharmacological interventions effective in decreasing delirium and falls?
Background: Delirium is prevalent among elderly hospitalized patients and is associated with increased morbidity, length of stay, healthcare costs, and risk of institutionalization. Multicomponent nonpharmacologic interventions have been used to prevent incident delirium in the elderly, but data regarding their effectiveness and impact on preventing poor outcomes are lacking.
Study design: Systematic literature review and meta-analysis.
Setting: Review of medical databases from Jan. 1, 1999, to Dec. 31, 2013.
Synopsis: Fourteen studies were included involving 4,267 elderly patients from 12 acute medical and surgical sites from around the world. There was a 53% reduction in delirium incidence associated with multicomponent, nonpharmacological interventions (OR, 0.47; 95% CI, 0.38-0.58). The odds of falling were 62% lower among intervention patients compared with controls (2.79 vs. 7.05 falls per 1,000 patient-days). The intervention group also showed a decrease in length of stay, with a mean difference of -0.16 (95% CI, -0.97 to 0.64) days and a 5% lower chance of institutionalization (95% CI, 0.71 to 1.26); however, the differences were not statistically significant.
Although the small number and heterogeneity of the studies included limited the analysis, the use of nonpharmacologic interventions appears to be a low-risk, low-cost strategy to prevent delirium. The challenge for the hospitalist in developing a nonpharmacological protocol is to determine which interventions to include; the study did not look at which interventions were most effective.
Bottom line: The use of multicomponent nonpharmacological interventions in older patients can lower the risk of delirium and falls.
Citation: Hshieh TT, Yue J, Oh E, et al. Effectiveness of multicomponent nonpharmacological delirium interventions: a meta-analysis. JAMA Intern Med. 2015;175(4):512-520.
Bridging Anticoagulation for Patients with Atrial Fibrillation
Clinical question: Is bridging anticoagulation for procedures associated with a higher bleeding risk and increased adverse outcomes compared to no bridging?
Background: Practice guidelines have been published to determine when, how, and on whom to bridge anticoagulation for procedures; however, uncertainty remains as to whether or not bridging changes outcomes.
Study design: Prospective, observational study.
Setting: Outcomes Registry for Better Informed treatment of Atrial Fibrillation (ORBIT-AF) study.
Synopsis: Investigators included 10,132 patients who were 18 years and older, with a baseline EKG documenting atrial fibrillation (Afib) and undergoing procedures. Interruptions of oral anticoagulation for a procedure, as well as the use and type of bridging method, were recorded. Six hundred sixty-five patients (24%) used bridging anticoagulation (73% low molecular weight heparin, 15% unfractionated heparin) prior to a procedure. Bridged patients were more likely to have had a mechanical valve replacement (9.6% vs. 2.4%, P<0.0001) and prior stroke (22% vs. 15%, P=0.0003).
Multivariate adjusted analysis showed that bridged patients, compared with non-bridged patients, had higher rates of bleeding (5.0% vs. 1.3%, adjusted odds ratio (OR) 3.84, P<0.0001) and an increased risk for adverse events, including the composite of myocardial infarction (MI), bleeding, stroke or systemic embolism, hospitalization, or death within 30 days (OR 1.94, 95% CI 1.38-271, P=0.0001). Rates of CHADS2 ≥2 or CHA2DS2-VASc score ≥2 were similar between bridged and nonbridged patients.
These results are observational and, therefore, a causal relationship cannot be established; however, the Effectiveness of Bridging Anticoagulation for Surgery (BRIDGE) study will give us more insight and answers.
Bottom line: Bridging anticoagulation prior to procedures is associated with a higher risk of bleeding and adverse outcomes.
Citation: Steinberg BA, Peterson ED, Kim S, et al. Use and outcomes associated with bridging during anticoagulation interruptions in patients with atrial fibrillation: Findings from the Outcomes Registry for Better Informed Treatment of Atrial Fibrillation (ORBIT-AF). Circulation. 2015;131(5):488-494.
Clinical question: Is bridging anticoagulation for procedures associated with a higher bleeding risk and increased adverse outcomes compared to no bridging?
Background: Practice guidelines have been published to determine when, how, and on whom to bridge anticoagulation for procedures; however, uncertainty remains as to whether or not bridging changes outcomes.
Study design: Prospective, observational study.
Setting: Outcomes Registry for Better Informed treatment of Atrial Fibrillation (ORBIT-AF) study.
Synopsis: Investigators included 10,132 patients who were 18 years and older, with a baseline EKG documenting atrial fibrillation (Afib) and undergoing procedures. Interruptions of oral anticoagulation for a procedure, as well as the use and type of bridging method, were recorded. Six hundred sixty-five patients (24%) used bridging anticoagulation (73% low molecular weight heparin, 15% unfractionated heparin) prior to a procedure. Bridged patients were more likely to have had a mechanical valve replacement (9.6% vs. 2.4%, P<0.0001) and prior stroke (22% vs. 15%, P=0.0003).
Multivariate adjusted analysis showed that bridged patients, compared with non-bridged patients, had higher rates of bleeding (5.0% vs. 1.3%, adjusted odds ratio (OR) 3.84, P<0.0001) and an increased risk for adverse events, including the composite of myocardial infarction (MI), bleeding, stroke or systemic embolism, hospitalization, or death within 30 days (OR 1.94, 95% CI 1.38-271, P=0.0001). Rates of CHADS2 ≥2 or CHA2DS2-VASc score ≥2 were similar between bridged and nonbridged patients.
These results are observational and, therefore, a causal relationship cannot be established; however, the Effectiveness of Bridging Anticoagulation for Surgery (BRIDGE) study will give us more insight and answers.
Bottom line: Bridging anticoagulation prior to procedures is associated with a higher risk of bleeding and adverse outcomes.
Citation: Steinberg BA, Peterson ED, Kim S, et al. Use and outcomes associated with bridging during anticoagulation interruptions in patients with atrial fibrillation: Findings from the Outcomes Registry for Better Informed Treatment of Atrial Fibrillation (ORBIT-AF). Circulation. 2015;131(5):488-494.
Clinical question: Is bridging anticoagulation for procedures associated with a higher bleeding risk and increased adverse outcomes compared to no bridging?
Background: Practice guidelines have been published to determine when, how, and on whom to bridge anticoagulation for procedures; however, uncertainty remains as to whether or not bridging changes outcomes.
Study design: Prospective, observational study.
Setting: Outcomes Registry for Better Informed treatment of Atrial Fibrillation (ORBIT-AF) study.
Synopsis: Investigators included 10,132 patients who were 18 years and older, with a baseline EKG documenting atrial fibrillation (Afib) and undergoing procedures. Interruptions of oral anticoagulation for a procedure, as well as the use and type of bridging method, were recorded. Six hundred sixty-five patients (24%) used bridging anticoagulation (73% low molecular weight heparin, 15% unfractionated heparin) prior to a procedure. Bridged patients were more likely to have had a mechanical valve replacement (9.6% vs. 2.4%, P<0.0001) and prior stroke (22% vs. 15%, P=0.0003).
Multivariate adjusted analysis showed that bridged patients, compared with non-bridged patients, had higher rates of bleeding (5.0% vs. 1.3%, adjusted odds ratio (OR) 3.84, P<0.0001) and an increased risk for adverse events, including the composite of myocardial infarction (MI), bleeding, stroke or systemic embolism, hospitalization, or death within 30 days (OR 1.94, 95% CI 1.38-271, P=0.0001). Rates of CHADS2 ≥2 or CHA2DS2-VASc score ≥2 were similar between bridged and nonbridged patients.
These results are observational and, therefore, a causal relationship cannot be established; however, the Effectiveness of Bridging Anticoagulation for Surgery (BRIDGE) study will give us more insight and answers.
Bottom line: Bridging anticoagulation prior to procedures is associated with a higher risk of bleeding and adverse outcomes.
Citation: Steinberg BA, Peterson ED, Kim S, et al. Use and outcomes associated with bridging during anticoagulation interruptions in patients with atrial fibrillation: Findings from the Outcomes Registry for Better Informed Treatment of Atrial Fibrillation (ORBIT-AF). Circulation. 2015;131(5):488-494.