User login
Time to Get a Move On
Hospitalists might want to incorporate questions about how much time a patient spends sitting into their diagnostic interview—then consider the same question for themselves.
A study published this spring in Medicine & Science in Sports & Exercise reports that time spent watching TV or riding in a car “were significant cardiovascular disease (CVD) mortality predictors” (Med Sci Sports Exerc. 2010;42(5):879-885). The research gained traction in medical publications and was highlighted in The New York Times. But for one of the researchers at the University of South Carolina, more work is needed.
“This is sort of a new way of looking at the equation,” says Steven Hooker, PhD, director of the university's Prevention Research Center. “We really don’t have any formal guidelines or recommendations on limiting sedentary behavior, although I think at some point in time we’ll get to that stage.”
Until then, hospitalists are in a prime position to determine a patient’s lifestyle—sedentary or active—via routine checklist questions they ask upon admission. Although some HM groups already ask questions about how often a patient exercises, Dr. Hooker suspects only a few groups ask how often a patient breaks up long periods of sitting.
Further, he suggests that while there are no standard recommendations, hospitalists would serve their patients well by incorporating helpful hints in discharge instructions or admission interviews.
“Encourage a person to build in standing or slight walking breaks in a daily routine, recommend they stand periodically while attending long meetings or during long periods of travel,” Dr. Hooker says. “Common sense reigns here.”
On the flip side, hospitalists would do well to remember that while their workday might include a high level of light-intensity activity, they face the same pitfalls as their patients: commuting, sitting in meetings, long periods of time in front of the computer, TV, or PlayStation.
“We have to get the public looking at physical activity and physical inactivity as two completely separate things,” says study first author Tatiana Warren, MS, a doctoral student in the department of exercise science at the University of South Carolina's Arnold School of Public Health. “We have to continue to do more research and get the word out.”
Hospitalists might want to incorporate questions about how much time a patient spends sitting into their diagnostic interview—then consider the same question for themselves.
A study published this spring in Medicine & Science in Sports & Exercise reports that time spent watching TV or riding in a car “were significant cardiovascular disease (CVD) mortality predictors” (Med Sci Sports Exerc. 2010;42(5):879-885). The research gained traction in medical publications and was highlighted in The New York Times. But for one of the researchers at the University of South Carolina, more work is needed.
“This is sort of a new way of looking at the equation,” says Steven Hooker, PhD, director of the university's Prevention Research Center. “We really don’t have any formal guidelines or recommendations on limiting sedentary behavior, although I think at some point in time we’ll get to that stage.”
Until then, hospitalists are in a prime position to determine a patient’s lifestyle—sedentary or active—via routine checklist questions they ask upon admission. Although some HM groups already ask questions about how often a patient exercises, Dr. Hooker suspects only a few groups ask how often a patient breaks up long periods of sitting.
Further, he suggests that while there are no standard recommendations, hospitalists would serve their patients well by incorporating helpful hints in discharge instructions or admission interviews.
“Encourage a person to build in standing or slight walking breaks in a daily routine, recommend they stand periodically while attending long meetings or during long periods of travel,” Dr. Hooker says. “Common sense reigns here.”
On the flip side, hospitalists would do well to remember that while their workday might include a high level of light-intensity activity, they face the same pitfalls as their patients: commuting, sitting in meetings, long periods of time in front of the computer, TV, or PlayStation.
“We have to get the public looking at physical activity and physical inactivity as two completely separate things,” says study first author Tatiana Warren, MS, a doctoral student in the department of exercise science at the University of South Carolina's Arnold School of Public Health. “We have to continue to do more research and get the word out.”
Hospitalists might want to incorporate questions about how much time a patient spends sitting into their diagnostic interview—then consider the same question for themselves.
A study published this spring in Medicine & Science in Sports & Exercise reports that time spent watching TV or riding in a car “were significant cardiovascular disease (CVD) mortality predictors” (Med Sci Sports Exerc. 2010;42(5):879-885). The research gained traction in medical publications and was highlighted in The New York Times. But for one of the researchers at the University of South Carolina, more work is needed.
“This is sort of a new way of looking at the equation,” says Steven Hooker, PhD, director of the university's Prevention Research Center. “We really don’t have any formal guidelines or recommendations on limiting sedentary behavior, although I think at some point in time we’ll get to that stage.”
Until then, hospitalists are in a prime position to determine a patient’s lifestyle—sedentary or active—via routine checklist questions they ask upon admission. Although some HM groups already ask questions about how often a patient exercises, Dr. Hooker suspects only a few groups ask how often a patient breaks up long periods of sitting.
Further, he suggests that while there are no standard recommendations, hospitalists would serve their patients well by incorporating helpful hints in discharge instructions or admission interviews.
“Encourage a person to build in standing or slight walking breaks in a daily routine, recommend they stand periodically while attending long meetings or during long periods of travel,” Dr. Hooker says. “Common sense reigns here.”
On the flip side, hospitalists would do well to remember that while their workday might include a high level of light-intensity activity, they face the same pitfalls as their patients: commuting, sitting in meetings, long periods of time in front of the computer, TV, or PlayStation.
“We have to get the public looking at physical activity and physical inactivity as two completely separate things,” says study first author Tatiana Warren, MS, a doctoral student in the department of exercise science at the University of South Carolina's Arnold School of Public Health. “We have to continue to do more research and get the word out.”
N.C. Hospital Names Hospitalist Its Physician of the Year
Suzanne Wilson, MD, is a self-proclaimed “outdoor-sy girl”—so much so that when the opportunity arose to work as a hospitalist in the golf mecca of Pinehurst, N.C., she didn’t think twice.
“My husband and I wanted to live somewhere we wanted to be,” says Dr. Wilson, a hospitalist for 10 years.
Pinehurst is home to one of the world’s most elegant and famous golf resorts, but it was at Moore Regional Hospital that Dr. Wilson was recently named Physician of the Year. She is Moore Regional’s first hospitalist to receive the honor.
The award process begins with the nurses, who are in charge of nominations. An independent board “reads the stories” about the nominees and their outstanding care, then chooses the winner.
Although Dr. Wilson says she was “stunned” when she was told she received the award, her colleagues were not. “Dr. Wilson is a consistent professional. It’s the way she relates to patients; her manner is very comfortable and easy going, and her ability to communicate with them—it’s like going home,” says Katherine Marsh, an oncology nurse at Moore Regional.
Dan Barnes, MD, director of the 23-hospitalist program at Moore Regional, says Dr. Wilson’s extensive interaction with the hospital’s nurses has helped improve the relationship between doctors and other staff.
“She is a true advocate for quality improvement and is always willing to help with that,” Dr. Barnes says, citing a soon-to-be-live Computerized Physician Order Entry (CPOE) initiative at the hospital, a project for which Dr. Wilson serves as officer.
Dr. Wilson’s focus on forging relationships between varying medical staff can be traced back to her early exposure to medicine. She grew up in southern Indiana with an RN mother and medical photographer father. She says that combination made for “interesting dinner conversations” as a child.
“We have to keep asking ourselves, ‘How can we do this better?'” Dr. Wilson says. “It’s all about the patient.”
Suzanne Wilson, MD, is a self-proclaimed “outdoor-sy girl”—so much so that when the opportunity arose to work as a hospitalist in the golf mecca of Pinehurst, N.C., she didn’t think twice.
“My husband and I wanted to live somewhere we wanted to be,” says Dr. Wilson, a hospitalist for 10 years.
Pinehurst is home to one of the world’s most elegant and famous golf resorts, but it was at Moore Regional Hospital that Dr. Wilson was recently named Physician of the Year. She is Moore Regional’s first hospitalist to receive the honor.
The award process begins with the nurses, who are in charge of nominations. An independent board “reads the stories” about the nominees and their outstanding care, then chooses the winner.
Although Dr. Wilson says she was “stunned” when she was told she received the award, her colleagues were not. “Dr. Wilson is a consistent professional. It’s the way she relates to patients; her manner is very comfortable and easy going, and her ability to communicate with them—it’s like going home,” says Katherine Marsh, an oncology nurse at Moore Regional.
Dan Barnes, MD, director of the 23-hospitalist program at Moore Regional, says Dr. Wilson’s extensive interaction with the hospital’s nurses has helped improve the relationship between doctors and other staff.
“She is a true advocate for quality improvement and is always willing to help with that,” Dr. Barnes says, citing a soon-to-be-live Computerized Physician Order Entry (CPOE) initiative at the hospital, a project for which Dr. Wilson serves as officer.
Dr. Wilson’s focus on forging relationships between varying medical staff can be traced back to her early exposure to medicine. She grew up in southern Indiana with an RN mother and medical photographer father. She says that combination made for “interesting dinner conversations” as a child.
“We have to keep asking ourselves, ‘How can we do this better?'” Dr. Wilson says. “It’s all about the patient.”
Suzanne Wilson, MD, is a self-proclaimed “outdoor-sy girl”—so much so that when the opportunity arose to work as a hospitalist in the golf mecca of Pinehurst, N.C., she didn’t think twice.
“My husband and I wanted to live somewhere we wanted to be,” says Dr. Wilson, a hospitalist for 10 years.
Pinehurst is home to one of the world’s most elegant and famous golf resorts, but it was at Moore Regional Hospital that Dr. Wilson was recently named Physician of the Year. She is Moore Regional’s first hospitalist to receive the honor.
The award process begins with the nurses, who are in charge of nominations. An independent board “reads the stories” about the nominees and their outstanding care, then chooses the winner.
Although Dr. Wilson says she was “stunned” when she was told she received the award, her colleagues were not. “Dr. Wilson is a consistent professional. It’s the way she relates to patients; her manner is very comfortable and easy going, and her ability to communicate with them—it’s like going home,” says Katherine Marsh, an oncology nurse at Moore Regional.
Dan Barnes, MD, director of the 23-hospitalist program at Moore Regional, says Dr. Wilson’s extensive interaction with the hospital’s nurses has helped improve the relationship between doctors and other staff.
“She is a true advocate for quality improvement and is always willing to help with that,” Dr. Barnes says, citing a soon-to-be-live Computerized Physician Order Entry (CPOE) initiative at the hospital, a project for which Dr. Wilson serves as officer.
Dr. Wilson’s focus on forging relationships between varying medical staff can be traced back to her early exposure to medicine. She grew up in southern Indiana with an RN mother and medical photographer father. She says that combination made for “interesting dinner conversations” as a child.
“We have to keep asking ourselves, ‘How can we do this better?'” Dr. Wilson says. “It’s all about the patient.”
Innovators Descend on Annual Pediatric HM Conference
More than 400 people attended the Pediatric Hospital Medicine annual conference July 22-25 in Minneapolis. The annual meeting is the premier networking and educational event for pediatric hospitalists and is sponsored by the American Academy of Pediatrics (AAP), SHM, and the Academic Pediatric Association (APA).
Innovation and improvement were popular topics throughout the conference. Keynote speaker George Buckley, CEO of manufacturing and technology conglomerate 3M, spoke about inspiring innovation, and a large percentage of the sessions and posters had quality-improvement (QI) themes. Experts from Cincinnati Children’s Hospital, led by Steve Muething, MD, assistant vice president of patient safety, and Shannon Phillips, MD, MPH, Cleveland Clinic’s patient safety officer, guided several popular sessions on QI.
A major innovation announced at the conference was the planned launch of a journal of pediatric hospital medicine, which will be sponsored by the AAP. (Update 09.14.2010--The journal has yet to officially announce an editor).
Research presentations have continued to increase in this young field, and the meeting was full of poster and platform presentations in the areas of clinical research, QI, educational research, and health services research. Vineeta Mittal, MD, of the University of Texas Southwestern and Children’s Medical Center in Dallas presented research on family-centered rounds, which was recently published in Pediatrics and picked up by the National Association of Children’s Hospitals (NACHRI) for dissemination.1 Patrick Brady, MD, of Cincinnati Children’s Hospital presented his research on short- versus long-course IV therapy for pediatric urinary tract infections, also published in Pediatrics.2
Other buzzed-about sessions included Vanderbilt University pediatric hospitalist Dr. Paul Hain’s ambitious attempt to create a PHM performance dashboard, and a case of “situational” epilepsy presented by Dr. Lisa Zaoutis of CHOP.
As in years past, the hottest ticket was for the luncheon presentation of the “Top Articles in Pediatric Hospital Medicine,” paneled this year by Drs. John Pope, Kris Rehm, and Brian Alverson. Raj Srivastava, MD, of Primary Children’s Medical Center in Salt Lake City and chairperson of the Pediatric Research in Inpatient Settings network, announced that the network had been awarded major grant funding.
Dan Rauch, MD, chair of the AAP’s Section on Hospital Medicine, dropped the biggest bombshell of all: He announced that the American Board of Pediatrics will support the development of pediatric HM as a full-fledged subspecialty in the near future. TH
Dr. Ralston is associate professor of pediatrics and chief of the division of inpatient pediatrics at the University of Texas Health Science Center in San Antonio, and medical director of inpatient services at Christus Santa Rosa Children’s Hospital.
References
- Mittal VS, Sigrest T, Ottolini MC, et al. Family-centered rounds on pediatric wards: a PRIS network survey of U.S. and Canadian hospitalists. Pediatrics. 2010;126(1):37-43.
- Brady PW, Conway PH, Goudie A. Length of intravenous antibiotic therapy and treatment failure in infants with urinary track infections. Pediatrics. 2010;126(2):196-203.
More than 400 people attended the Pediatric Hospital Medicine annual conference July 22-25 in Minneapolis. The annual meeting is the premier networking and educational event for pediatric hospitalists and is sponsored by the American Academy of Pediatrics (AAP), SHM, and the Academic Pediatric Association (APA).
Innovation and improvement were popular topics throughout the conference. Keynote speaker George Buckley, CEO of manufacturing and technology conglomerate 3M, spoke about inspiring innovation, and a large percentage of the sessions and posters had quality-improvement (QI) themes. Experts from Cincinnati Children’s Hospital, led by Steve Muething, MD, assistant vice president of patient safety, and Shannon Phillips, MD, MPH, Cleveland Clinic’s patient safety officer, guided several popular sessions on QI.
A major innovation announced at the conference was the planned launch of a journal of pediatric hospital medicine, which will be sponsored by the AAP. (Update 09.14.2010--The journal has yet to officially announce an editor).
Research presentations have continued to increase in this young field, and the meeting was full of poster and platform presentations in the areas of clinical research, QI, educational research, and health services research. Vineeta Mittal, MD, of the University of Texas Southwestern and Children’s Medical Center in Dallas presented research on family-centered rounds, which was recently published in Pediatrics and picked up by the National Association of Children’s Hospitals (NACHRI) for dissemination.1 Patrick Brady, MD, of Cincinnati Children’s Hospital presented his research on short- versus long-course IV therapy for pediatric urinary tract infections, also published in Pediatrics.2
Other buzzed-about sessions included Vanderbilt University pediatric hospitalist Dr. Paul Hain’s ambitious attempt to create a PHM performance dashboard, and a case of “situational” epilepsy presented by Dr. Lisa Zaoutis of CHOP.
As in years past, the hottest ticket was for the luncheon presentation of the “Top Articles in Pediatric Hospital Medicine,” paneled this year by Drs. John Pope, Kris Rehm, and Brian Alverson. Raj Srivastava, MD, of Primary Children’s Medical Center in Salt Lake City and chairperson of the Pediatric Research in Inpatient Settings network, announced that the network had been awarded major grant funding.
Dan Rauch, MD, chair of the AAP’s Section on Hospital Medicine, dropped the biggest bombshell of all: He announced that the American Board of Pediatrics will support the development of pediatric HM as a full-fledged subspecialty in the near future. TH
Dr. Ralston is associate professor of pediatrics and chief of the division of inpatient pediatrics at the University of Texas Health Science Center in San Antonio, and medical director of inpatient services at Christus Santa Rosa Children’s Hospital.
References
- Mittal VS, Sigrest T, Ottolini MC, et al. Family-centered rounds on pediatric wards: a PRIS network survey of U.S. and Canadian hospitalists. Pediatrics. 2010;126(1):37-43.
- Brady PW, Conway PH, Goudie A. Length of intravenous antibiotic therapy and treatment failure in infants with urinary track infections. Pediatrics. 2010;126(2):196-203.
More than 400 people attended the Pediatric Hospital Medicine annual conference July 22-25 in Minneapolis. The annual meeting is the premier networking and educational event for pediatric hospitalists and is sponsored by the American Academy of Pediatrics (AAP), SHM, and the Academic Pediatric Association (APA).
Innovation and improvement were popular topics throughout the conference. Keynote speaker George Buckley, CEO of manufacturing and technology conglomerate 3M, spoke about inspiring innovation, and a large percentage of the sessions and posters had quality-improvement (QI) themes. Experts from Cincinnati Children’s Hospital, led by Steve Muething, MD, assistant vice president of patient safety, and Shannon Phillips, MD, MPH, Cleveland Clinic’s patient safety officer, guided several popular sessions on QI.
A major innovation announced at the conference was the planned launch of a journal of pediatric hospital medicine, which will be sponsored by the AAP. (Update 09.14.2010--The journal has yet to officially announce an editor).
Research presentations have continued to increase in this young field, and the meeting was full of poster and platform presentations in the areas of clinical research, QI, educational research, and health services research. Vineeta Mittal, MD, of the University of Texas Southwestern and Children’s Medical Center in Dallas presented research on family-centered rounds, which was recently published in Pediatrics and picked up by the National Association of Children’s Hospitals (NACHRI) for dissemination.1 Patrick Brady, MD, of Cincinnati Children’s Hospital presented his research on short- versus long-course IV therapy for pediatric urinary tract infections, also published in Pediatrics.2
Other buzzed-about sessions included Vanderbilt University pediatric hospitalist Dr. Paul Hain’s ambitious attempt to create a PHM performance dashboard, and a case of “situational” epilepsy presented by Dr. Lisa Zaoutis of CHOP.
As in years past, the hottest ticket was for the luncheon presentation of the “Top Articles in Pediatric Hospital Medicine,” paneled this year by Drs. John Pope, Kris Rehm, and Brian Alverson. Raj Srivastava, MD, of Primary Children’s Medical Center in Salt Lake City and chairperson of the Pediatric Research in Inpatient Settings network, announced that the network had been awarded major grant funding.
Dan Rauch, MD, chair of the AAP’s Section on Hospital Medicine, dropped the biggest bombshell of all: He announced that the American Board of Pediatrics will support the development of pediatric HM as a full-fledged subspecialty in the near future. TH
Dr. Ralston is associate professor of pediatrics and chief of the division of inpatient pediatrics at the University of Texas Health Science Center in San Antonio, and medical director of inpatient services at Christus Santa Rosa Children’s Hospital.
References
- Mittal VS, Sigrest T, Ottolini MC, et al. Family-centered rounds on pediatric wards: a PRIS network survey of U.S. and Canadian hospitalists. Pediatrics. 2010;126(1):37-43.
- Brady PW, Conway PH, Goudie A. Length of intravenous antibiotic therapy and treatment failure in infants with urinary track infections. Pediatrics. 2010;126(2):196-203.
Hospitalist/Intensivist Model Lowers Costs, Maintains Quality of Care
As the field of HM continues to mature, branch out, and is called upon to lead in the care of a larger cross-section of hospitalized patients, it is only natural that this includes the critically ill patient. Hospitalists already care for—and are the attending of record for—this patient population in most U.S. hospitals. It is my position that a technically proficient hospitalist service, which is facility-exclusive and offers 24/7 coverage, is able to offer the same quality of care as an intensivist group. An important feature of this model is the inclusion and “buy in” from community pulmonologists in order to provide backup and consultative assistance when warranted.
Our program at Westside Regional Medical Center in Plantation, Fla., has made great strides as we continue to integrate this model in the hospital. We are actively tracking ICU length of stay and throughput, incidence of ventilator-associated pneumonia (VAP), central-line infection rates, and ICU mortality.
I believe that a clinically competent and aggressive HM service is able to drive down costs and generate revenue by establishing clinically beneficial quality-improvement (QI) protocols; drive down ICU length of stay; provide effective and timely procedural services; and incur a lower cost burden (i.e., hospitalists cost less than intensivists). And I believe all of these benefits are available without sacrificing quality or patient care.
Leadership from medical staff and administration is imperative to establish the appropriate vision and drive toward hospitalist/intensivist implementation. Finding the right supporting physicians who bring excitement and energy is equally as important. Establishing expectations for skill sets, as well as the opportunity and mechanism by which these skill sets might be acquired and refined, is a must. The following technical skills should be required of hospitalist/intensivists:
- Ultrasound-guided central line insertion;
- PICC line insertion;
- Endotracheal intubation;
- Advanced airway management;
- Thoracostomy tube insertion;
- Arterial-line insertion;
- Transvenous pacing wire insertion;
- Lumbar puncture;
- Thoracentesis; and
- Paracentesis.
An important starting point is the identification of skill sets for each hospitalist. Once this information is ascertained, the next step is to understand what the credentialing requirements for the individual procedures are. This usually consists of a certain number of “logged” cases, which must be put forward for review by the medical staff leadership. Most physicians completing residency are required to keep a procedural log where cases are documented. Any deficiencies within the log can be supplemented by establishing a practice log where proctored cases are documented until the recommended number of cases are completed and put forward for credentialing.
Obtaining buy-in from the medical staff is important. They can serve as allies in many areas, specifically as proctors in the credentialing process. The key to successful interface is in awakening them to the beneficial impact a service such as this can have on patients and on the lifestyle of providers.
As an example, before our group started the hybrid model at Westside, the nursing staff would call anesthesia to evaluate patients for endotracheal intubation. This system took anesthesia away from its OR cases, causing delays and frustration. After a conversation, the anesthesia director realized the benefits that would come with assisting the hospitalists in becoming more proficient with intubations. This same scenario has been true in our experience with ED physicians, cardiothoracic surgeons (chest tubes), and so on.
Other resources for hospitalists include the National Procedure Institute, which offers CME credit and certification toward “Hospitalist Procedures.” Additionally, difficult airway or advanced airway courses provide certification.
Hospitalists have long been called on to provide emergency services for unstable patients via rapid response or codes. In many facilities hospitalists serve as the lead physicians in the management of critically ill patients. Our hospitalist model serves as a great launching pad for the development and evolution of this new breed of physician.
There exists no clinical evidence to assert inferiority between the care provided by an in-house, 24/7 hospitalist group with assistance from pulmonary medicine versus an intensivist group. It is my belief that if the appropriate infrastructure, fostered skill sets, pulmonologist partnership, and QI protocols are implemented, there will be no measurable difference in scope of care or outcomes.
The inpatient management of critically ill and unstable patients continues to be a significant and important subgroup of hospital patient populations. As patients continue to live longer with debilitating chronic diseases, the fallout from decompensation can be devastating. Many facilities have hospitalists leading the charge in the care of these patients. It is undeniable that the next evolution in HM will require a more proactive inpatient physician, with both the clinical and technical acumen to manage all patients across the hospital spectrum.
Ulises A. Perez, MD,
medical director, hospitalist division,
Westside Regional Medical Center, Plantation, Fla.,
Kendall Regional Medical Center, Miami
As the field of HM continues to mature, branch out, and is called upon to lead in the care of a larger cross-section of hospitalized patients, it is only natural that this includes the critically ill patient. Hospitalists already care for—and are the attending of record for—this patient population in most U.S. hospitals. It is my position that a technically proficient hospitalist service, which is facility-exclusive and offers 24/7 coverage, is able to offer the same quality of care as an intensivist group. An important feature of this model is the inclusion and “buy in” from community pulmonologists in order to provide backup and consultative assistance when warranted.
Our program at Westside Regional Medical Center in Plantation, Fla., has made great strides as we continue to integrate this model in the hospital. We are actively tracking ICU length of stay and throughput, incidence of ventilator-associated pneumonia (VAP), central-line infection rates, and ICU mortality.
I believe that a clinically competent and aggressive HM service is able to drive down costs and generate revenue by establishing clinically beneficial quality-improvement (QI) protocols; drive down ICU length of stay; provide effective and timely procedural services; and incur a lower cost burden (i.e., hospitalists cost less than intensivists). And I believe all of these benefits are available without sacrificing quality or patient care.
Leadership from medical staff and administration is imperative to establish the appropriate vision and drive toward hospitalist/intensivist implementation. Finding the right supporting physicians who bring excitement and energy is equally as important. Establishing expectations for skill sets, as well as the opportunity and mechanism by which these skill sets might be acquired and refined, is a must. The following technical skills should be required of hospitalist/intensivists:
- Ultrasound-guided central line insertion;
- PICC line insertion;
- Endotracheal intubation;
- Advanced airway management;
- Thoracostomy tube insertion;
- Arterial-line insertion;
- Transvenous pacing wire insertion;
- Lumbar puncture;
- Thoracentesis; and
- Paracentesis.
An important starting point is the identification of skill sets for each hospitalist. Once this information is ascertained, the next step is to understand what the credentialing requirements for the individual procedures are. This usually consists of a certain number of “logged” cases, which must be put forward for review by the medical staff leadership. Most physicians completing residency are required to keep a procedural log where cases are documented. Any deficiencies within the log can be supplemented by establishing a practice log where proctored cases are documented until the recommended number of cases are completed and put forward for credentialing.
Obtaining buy-in from the medical staff is important. They can serve as allies in many areas, specifically as proctors in the credentialing process. The key to successful interface is in awakening them to the beneficial impact a service such as this can have on patients and on the lifestyle of providers.
As an example, before our group started the hybrid model at Westside, the nursing staff would call anesthesia to evaluate patients for endotracheal intubation. This system took anesthesia away from its OR cases, causing delays and frustration. After a conversation, the anesthesia director realized the benefits that would come with assisting the hospitalists in becoming more proficient with intubations. This same scenario has been true in our experience with ED physicians, cardiothoracic surgeons (chest tubes), and so on.
Other resources for hospitalists include the National Procedure Institute, which offers CME credit and certification toward “Hospitalist Procedures.” Additionally, difficult airway or advanced airway courses provide certification.
Hospitalists have long been called on to provide emergency services for unstable patients via rapid response or codes. In many facilities hospitalists serve as the lead physicians in the management of critically ill patients. Our hospitalist model serves as a great launching pad for the development and evolution of this new breed of physician.
There exists no clinical evidence to assert inferiority between the care provided by an in-house, 24/7 hospitalist group with assistance from pulmonary medicine versus an intensivist group. It is my belief that if the appropriate infrastructure, fostered skill sets, pulmonologist partnership, and QI protocols are implemented, there will be no measurable difference in scope of care or outcomes.
The inpatient management of critically ill and unstable patients continues to be a significant and important subgroup of hospital patient populations. As patients continue to live longer with debilitating chronic diseases, the fallout from decompensation can be devastating. Many facilities have hospitalists leading the charge in the care of these patients. It is undeniable that the next evolution in HM will require a more proactive inpatient physician, with both the clinical and technical acumen to manage all patients across the hospital spectrum.
Ulises A. Perez, MD,
medical director, hospitalist division,
Westside Regional Medical Center, Plantation, Fla.,
Kendall Regional Medical Center, Miami
As the field of HM continues to mature, branch out, and is called upon to lead in the care of a larger cross-section of hospitalized patients, it is only natural that this includes the critically ill patient. Hospitalists already care for—and are the attending of record for—this patient population in most U.S. hospitals. It is my position that a technically proficient hospitalist service, which is facility-exclusive and offers 24/7 coverage, is able to offer the same quality of care as an intensivist group. An important feature of this model is the inclusion and “buy in” from community pulmonologists in order to provide backup and consultative assistance when warranted.
Our program at Westside Regional Medical Center in Plantation, Fla., has made great strides as we continue to integrate this model in the hospital. We are actively tracking ICU length of stay and throughput, incidence of ventilator-associated pneumonia (VAP), central-line infection rates, and ICU mortality.
I believe that a clinically competent and aggressive HM service is able to drive down costs and generate revenue by establishing clinically beneficial quality-improvement (QI) protocols; drive down ICU length of stay; provide effective and timely procedural services; and incur a lower cost burden (i.e., hospitalists cost less than intensivists). And I believe all of these benefits are available without sacrificing quality or patient care.
Leadership from medical staff and administration is imperative to establish the appropriate vision and drive toward hospitalist/intensivist implementation. Finding the right supporting physicians who bring excitement and energy is equally as important. Establishing expectations for skill sets, as well as the opportunity and mechanism by which these skill sets might be acquired and refined, is a must. The following technical skills should be required of hospitalist/intensivists:
- Ultrasound-guided central line insertion;
- PICC line insertion;
- Endotracheal intubation;
- Advanced airway management;
- Thoracostomy tube insertion;
- Arterial-line insertion;
- Transvenous pacing wire insertion;
- Lumbar puncture;
- Thoracentesis; and
- Paracentesis.
An important starting point is the identification of skill sets for each hospitalist. Once this information is ascertained, the next step is to understand what the credentialing requirements for the individual procedures are. This usually consists of a certain number of “logged” cases, which must be put forward for review by the medical staff leadership. Most physicians completing residency are required to keep a procedural log where cases are documented. Any deficiencies within the log can be supplemented by establishing a practice log where proctored cases are documented until the recommended number of cases are completed and put forward for credentialing.
Obtaining buy-in from the medical staff is important. They can serve as allies in many areas, specifically as proctors in the credentialing process. The key to successful interface is in awakening them to the beneficial impact a service such as this can have on patients and on the lifestyle of providers.
As an example, before our group started the hybrid model at Westside, the nursing staff would call anesthesia to evaluate patients for endotracheal intubation. This system took anesthesia away from its OR cases, causing delays and frustration. After a conversation, the anesthesia director realized the benefits that would come with assisting the hospitalists in becoming more proficient with intubations. This same scenario has been true in our experience with ED physicians, cardiothoracic surgeons (chest tubes), and so on.
Other resources for hospitalists include the National Procedure Institute, which offers CME credit and certification toward “Hospitalist Procedures.” Additionally, difficult airway or advanced airway courses provide certification.
Hospitalists have long been called on to provide emergency services for unstable patients via rapid response or codes. In many facilities hospitalists serve as the lead physicians in the management of critically ill patients. Our hospitalist model serves as a great launching pad for the development and evolution of this new breed of physician.
There exists no clinical evidence to assert inferiority between the care provided by an in-house, 24/7 hospitalist group with assistance from pulmonary medicine versus an intensivist group. It is my belief that if the appropriate infrastructure, fostered skill sets, pulmonologist partnership, and QI protocols are implemented, there will be no measurable difference in scope of care or outcomes.
The inpatient management of critically ill and unstable patients continues to be a significant and important subgroup of hospital patient populations. As patients continue to live longer with debilitating chronic diseases, the fallout from decompensation can be devastating. Many facilities have hospitalists leading the charge in the care of these patients. It is undeniable that the next evolution in HM will require a more proactive inpatient physician, with both the clinical and technical acumen to manage all patients across the hospital spectrum.
Ulises A. Perez, MD,
medical director, hospitalist division,
Westside Regional Medical Center, Plantation, Fla.,
Kendall Regional Medical Center, Miami
SHM+MGMA = Better Survey
As HM continues to grow, the need for clear and accurate data about the specialty will only become more intense. Hospitalists and HM group leaders use survey information to better understand how they compare to other practices across the country, in terms of size and practice characteristics, as well as compensation and productivity.
Increasingly, healthcare executives are turning to survey data—either independently or via their hospitalist group leaders—to get a grasp on the best practices in the industry.
That’s why SHM teamed up with the Medical Group Management Association (MGMA), the industry leader for professional administrators and leaders of medical group practices, to research and develop the State of Hospital Medicine: 2010 Report Based on 2009 Data.
Previously, SHM created and fielded a biannual survey, then analyzed the results independently.

—Leslie A. Flores, MHA, SHM senior advisor for practice management
“Our partnership with MGMA expands our survey population, delivers more information, and brings MGMA’s 90 years of industry credibility in the medical practice management field,” says Leslie A. Flores, MHA, SHM senior advisor for practice management. “The 2010 survey gives hospitalists and hospital administrators an unprecedented snapshot of the state of hospital medicine.”
The 2010 report will be available this month in the “Practice Resources” section of the SHM website (www.hospitalmedicine.org).
The print version will be available to SHM members for $125; for $175, members receive both the print version and the report on CD-ROM.
“This is a first-ever opportunity for hospitalists, group leaders, and healthcare executives to get the clearest picture possible of a rapidly changing industry,” Flores says. TH
Brendon Shank is a freelance writer based in Philadelphia.
SHM Adopts Strict Code for Industry Relations
A long with nearly 20 other organizations, SHM has adopted the Code for Industry Relations (http://cmss.org/) established by the Council on Medical Specialty Societies (CMSS). In an era of digital communication, SHM has created a Web area (www.hospital medicine.org/industry) to continuously update its policies toward industry, display its current partnerships, and disclose the potential conflicts of interest of its board or directors, editors, and CEO.
The message from SHM leadership: SHM is committed to being a leader in an era of transparency and disclosure.
Transparency serves an important role for medical specialty societies, says Norman B. Kahn Jr., MD, executive vice president and CEO of CMSS. The code developed by CMSS and adopted by SHM “assures in interactions with industry that the patients’ needs come first,” Dr. Kahn says. “The bottom line is that this is all about protecting the independence of societies from industry without abrogating the relationship.”
From its early days, SHM has been aware of the need to balance the responsibilities of speaking for HM and the need to disclose any potential conflicts of interests. In 2000, SHM developed its Principles of Organizational Relationships (www.hospitalmedicine.org/OrgRelationships), which have guided the society’s efforts.
The principles call for a clear, bright line that is a barrier between the support of a partner and SHM’s control of content. Among other activities, SHM has applied those principles to meetings and educational initiatives, quality-improvement (QI) projects, and publications.
The principles also were the foundation for the tough conflict-of-interest policies (www.hospital medicine.org/interestpolicies) the board approved in 2005.
Over the last decade, as HM has grown, national hospitalist leaders have become the experts on a wide range of topics and are asked to speak, write, or advise government agencies, foundations, and industry.
As SHM has developed its resources to help hospitalists improve glycemic control, reduce unnecessary DVTs and PEs, and improve the transitions of care, SHM has engaged in partnerships with government agencies, foundations, and industry as well.
“SHM’s leadership recognizes that it has a fiduciary responsibility to its members—a responsibility to provide expert direction, and necessary resources, to enable the hospitalist to ensure the best possible care of his or her patients, and to advance the quality of the hospital system,” says SHM President Jeff Wiese, MD, SFHM. “But SHM cannot do this alone, and when external partnerships are established, it is the organization’s responsibility to enter into these partnerships judiciously, and to be fully transparent to the membership with respect to the arrangements of these partnerships.
“We are confident that no other organization has a more robust disclosure policy than SHM.”
Today’s Nominations, Tomorrow’s Leaders
HM leaders aren’t born that way—they’re nominated.
SHM is accepting nominations for its board election; new members will take office in May at HM11 in Dallas.
The nomination deadline is Oct. 31. Online ballots will be available to all SHM members in late 2010. The results of the election will be announced online in early 2011.
Nominees must be SHM members in good standing. SHM members may nominate themselves or be nominated by another SHM member. Nominations must include a letter of nomination, a one-page CV, and a recent photo.
The nomination committee considers candidates based on length of SHM membership, activity as a hospitalist and SHM member, the prominence of the candidate within the specialty, and a number of other factors.
Board members serve a three-year term and normally serve on one or more committees.
“Participating in SHM’s leadership is one of the best ways to help guide the future of hospital medicine,” says Larry Wellikson, MD, SFHM, CEO of SHM. “That begins by submitting a board nomination to SHM this year.”
Chapter Updates
Chicago
The Chicago chapter met May 19 at Sullivan’s Steakhouse. Twenty-five hospitalists from the Chicago area, including hospitalists at Loyola Hospital, Lutheran General Hospital, Illinois Masonic Medical Center, Trinity Hospital, Silver Cross, and Evanston Hospital, as well as hospitalist groups like Cogent and Vista, attended the meeting.
Dr. Robin Ross of Season Hospice and Palliative Care, which sponsored the event, presented clinical pearls for end-of-life care for the busy hospitalist. The meeting also featured a town-hall discussion, with topics relevant to everyday hospitalist practice—coding, consultations, and the use of observation units. Notification of chapter elections were summated to all chapter members in July and August.
Harrisburg/South Central Pennsylvania
Thirty hospitalists representing six HM programs attended the Harrisburg/South Central Pennsylvania chapter of SHM June 9 at Passage to India in Harrisburg.
Eric Kupersmith, MD, SFHM, division head of the hospitalist program at Cooper University Hospital in Camden, N.J., led an open discussion regarding “Transition of Care and the Hospitalist’s Role.” Chapter members had the opportunity to discuss how each individual program actively seeks to decrease readmission rates. Discharge-planning specifics generated group discussion, and a handful of hospitalists offered testimonials about what is working in their practices.
The meeting was sponsored by Merck.
A BOOST for all seasons: Discharge improvement resources now available year round
When Project BOOST (Better Outcomes for Older Adults through Safer Transitions), SHM’s groundbreaking program to reduce readmissions, first began in 2008, hospital sites applied to participate in a yearlong program of one-on-one mentorships, regular sessions to share best practices, and a resource toolkit.
Since then, Project BOOST has grown and evolved. Some BOOST iterations now include third parties, such as the University of Michigan, Blue Cross/Blue Shield of Michigan, and the California HealthCare Foundation. SHM also recently introduced a nationwide, tuition-based version of the BOOST initiative.
Now, SHM is announcing that new resources are available to all hospitals, regardless of their participation in Project BOOST, all year. Some resources were previously available only to Project BOOST participants; others are brand-new materials available to any hospital or hospitalist trying to reduce unplanned readmissions to their hospital.
“No matter where you are in the hospital, you have an opportunity to improve discharge,” says Tina Budnitz, MPH, senior advisor for quality improvement. “The response to Project BOOST has been overwhelmingly positive. That’s why we’re so excited to make these new materials available to anyone responsible for lowering readmissions.”
Individuals can download the Project BOOST implementation course, a new training program specifically designed to help nurses use the proven “teachback” method, and the Project BOOST patient PASS form.
Budnitz and Project BOOST organizers also plan to launch supplemental products for self-implementers, including access to Project BOOST e-mail listservs, data centers, and webinars.
Prepare Now for Flu Season
With most of the country still enjoying warm weather, it’s easy to forget that flu season is right around the corner. Hospitals can either be part of the solution—or part of the problem.
Compared to friends and family outside of the hospital, patients in the hospital are especially at risk. The chances of contracting the flu are higher, given decreased immune responses and increased proximity to caregivers and other potentially infected patients. Plus, the impact of the flu on healthcare providers can be significantly more severe.
By now, most hospitals have protocols for preparing for flu season and isolating infected patients, says Danielle Scheurer, MD, MSc, SFHM, assistant professor of medicine at Harvard Medical School in Boston and director of general medical service at Brigham and Women’s Hospitalist Service. Hospitalists play a special role within those protocols, she adds.
“Hospitalists can be instrumental in preventing the spread of flu within the hospital by having a low threshold for diagnostic testing of patients, immediate isolation of those patients, and strict adherence to infection-control measures in those with suspected influenza,” Dr. Scheurer says.
Dr. Scheurer also emphasizes the fact that flu prevention doesn’t end with better clinical practices. Patient education is key.
“Hospitalists can be vital in educating patients about how to avoid symptomatic contacts,” she says, “and how to advocate for themselves in insisting that all care providers use strict handwashing protocols to avoid transmitting influenza among patients.” TH
As HM continues to grow, the need for clear and accurate data about the specialty will only become more intense. Hospitalists and HM group leaders use survey information to better understand how they compare to other practices across the country, in terms of size and practice characteristics, as well as compensation and productivity.
Increasingly, healthcare executives are turning to survey data—either independently or via their hospitalist group leaders—to get a grasp on the best practices in the industry.
That’s why SHM teamed up with the Medical Group Management Association (MGMA), the industry leader for professional administrators and leaders of medical group practices, to research and develop the State of Hospital Medicine: 2010 Report Based on 2009 Data.
Previously, SHM created and fielded a biannual survey, then analyzed the results independently.
—Leslie A. Flores, MHA, SHM senior advisor for practice management
“Our partnership with MGMA expands our survey population, delivers more information, and brings MGMA’s 90 years of industry credibility in the medical practice management field,” says Leslie A. Flores, MHA, SHM senior advisor for practice management. “The 2010 survey gives hospitalists and hospital administrators an unprecedented snapshot of the state of hospital medicine.”
The 2010 report will be available this month in the “Practice Resources” section of the SHM website (www.hospitalmedicine.org).
The print version will be available to SHM members for $125; for $175, members receive both the print version and the report on CD-ROM.
“This is a first-ever opportunity for hospitalists, group leaders, and healthcare executives to get the clearest picture possible of a rapidly changing industry,” Flores says. TH
Brendon Shank is a freelance writer based in Philadelphia.
SHM Adopts Strict Code for Industry Relations
A long with nearly 20 other organizations, SHM has adopted the Code for Industry Relations (http://cmss.org/) established by the Council on Medical Specialty Societies (CMSS). In an era of digital communication, SHM has created a Web area (www.hospital medicine.org/industry) to continuously update its policies toward industry, display its current partnerships, and disclose the potential conflicts of interest of its board or directors, editors, and CEO.
The message from SHM leadership: SHM is committed to being a leader in an era of transparency and disclosure.
Transparency serves an important role for medical specialty societies, says Norman B. Kahn Jr., MD, executive vice president and CEO of CMSS. The code developed by CMSS and adopted by SHM “assures in interactions with industry that the patients’ needs come first,” Dr. Kahn says. “The bottom line is that this is all about protecting the independence of societies from industry without abrogating the relationship.”
From its early days, SHM has been aware of the need to balance the responsibilities of speaking for HM and the need to disclose any potential conflicts of interests. In 2000, SHM developed its Principles of Organizational Relationships (www.hospitalmedicine.org/OrgRelationships), which have guided the society’s efforts.
The principles call for a clear, bright line that is a barrier between the support of a partner and SHM’s control of content. Among other activities, SHM has applied those principles to meetings and educational initiatives, quality-improvement (QI) projects, and publications.
The principles also were the foundation for the tough conflict-of-interest policies (www.hospital medicine.org/interestpolicies) the board approved in 2005.
Over the last decade, as HM has grown, national hospitalist leaders have become the experts on a wide range of topics and are asked to speak, write, or advise government agencies, foundations, and industry.
As SHM has developed its resources to help hospitalists improve glycemic control, reduce unnecessary DVTs and PEs, and improve the transitions of care, SHM has engaged in partnerships with government agencies, foundations, and industry as well.
“SHM’s leadership recognizes that it has a fiduciary responsibility to its members—a responsibility to provide expert direction, and necessary resources, to enable the hospitalist to ensure the best possible care of his or her patients, and to advance the quality of the hospital system,” says SHM President Jeff Wiese, MD, SFHM. “But SHM cannot do this alone, and when external partnerships are established, it is the organization’s responsibility to enter into these partnerships judiciously, and to be fully transparent to the membership with respect to the arrangements of these partnerships.
“We are confident that no other organization has a more robust disclosure policy than SHM.”
Today’s Nominations, Tomorrow’s Leaders
HM leaders aren’t born that way—they’re nominated.
SHM is accepting nominations for its board election; new members will take office in May at HM11 in Dallas.
The nomination deadline is Oct. 31. Online ballots will be available to all SHM members in late 2010. The results of the election will be announced online in early 2011.
Nominees must be SHM members in good standing. SHM members may nominate themselves or be nominated by another SHM member. Nominations must include a letter of nomination, a one-page CV, and a recent photo.
The nomination committee considers candidates based on length of SHM membership, activity as a hospitalist and SHM member, the prominence of the candidate within the specialty, and a number of other factors.
Board members serve a three-year term and normally serve on one or more committees.
“Participating in SHM’s leadership is one of the best ways to help guide the future of hospital medicine,” says Larry Wellikson, MD, SFHM, CEO of SHM. “That begins by submitting a board nomination to SHM this year.”
Chapter Updates
Chicago
The Chicago chapter met May 19 at Sullivan’s Steakhouse. Twenty-five hospitalists from the Chicago area, including hospitalists at Loyola Hospital, Lutheran General Hospital, Illinois Masonic Medical Center, Trinity Hospital, Silver Cross, and Evanston Hospital, as well as hospitalist groups like Cogent and Vista, attended the meeting.
Dr. Robin Ross of Season Hospice and Palliative Care, which sponsored the event, presented clinical pearls for end-of-life care for the busy hospitalist. The meeting also featured a town-hall discussion, with topics relevant to everyday hospitalist practice—coding, consultations, and the use of observation units. Notification of chapter elections were summated to all chapter members in July and August.
Harrisburg/South Central Pennsylvania
Thirty hospitalists representing six HM programs attended the Harrisburg/South Central Pennsylvania chapter of SHM June 9 at Passage to India in Harrisburg.
Eric Kupersmith, MD, SFHM, division head of the hospitalist program at Cooper University Hospital in Camden, N.J., led an open discussion regarding “Transition of Care and the Hospitalist’s Role.” Chapter members had the opportunity to discuss how each individual program actively seeks to decrease readmission rates. Discharge-planning specifics generated group discussion, and a handful of hospitalists offered testimonials about what is working in their practices.
The meeting was sponsored by Merck.
A BOOST for all seasons: Discharge improvement resources now available year round
When Project BOOST (Better Outcomes for Older Adults through Safer Transitions), SHM’s groundbreaking program to reduce readmissions, first began in 2008, hospital sites applied to participate in a yearlong program of one-on-one mentorships, regular sessions to share best practices, and a resource toolkit.
Since then, Project BOOST has grown and evolved. Some BOOST iterations now include third parties, such as the University of Michigan, Blue Cross/Blue Shield of Michigan, and the California HealthCare Foundation. SHM also recently introduced a nationwide, tuition-based version of the BOOST initiative.
Now, SHM is announcing that new resources are available to all hospitals, regardless of their participation in Project BOOST, all year. Some resources were previously available only to Project BOOST participants; others are brand-new materials available to any hospital or hospitalist trying to reduce unplanned readmissions to their hospital.
“No matter where you are in the hospital, you have an opportunity to improve discharge,” says Tina Budnitz, MPH, senior advisor for quality improvement. “The response to Project BOOST has been overwhelmingly positive. That’s why we’re so excited to make these new materials available to anyone responsible for lowering readmissions.”
Individuals can download the Project BOOST implementation course, a new training program specifically designed to help nurses use the proven “teachback” method, and the Project BOOST patient PASS form.
Budnitz and Project BOOST organizers also plan to launch supplemental products for self-implementers, including access to Project BOOST e-mail listservs, data centers, and webinars.
Prepare Now for Flu Season
With most of the country still enjoying warm weather, it’s easy to forget that flu season is right around the corner. Hospitals can either be part of the solution—or part of the problem.
Compared to friends and family outside of the hospital, patients in the hospital are especially at risk. The chances of contracting the flu are higher, given decreased immune responses and increased proximity to caregivers and other potentially infected patients. Plus, the impact of the flu on healthcare providers can be significantly more severe.
By now, most hospitals have protocols for preparing for flu season and isolating infected patients, says Danielle Scheurer, MD, MSc, SFHM, assistant professor of medicine at Harvard Medical School in Boston and director of general medical service at Brigham and Women’s Hospitalist Service. Hospitalists play a special role within those protocols, she adds.
“Hospitalists can be instrumental in preventing the spread of flu within the hospital by having a low threshold for diagnostic testing of patients, immediate isolation of those patients, and strict adherence to infection-control measures in those with suspected influenza,” Dr. Scheurer says.
Dr. Scheurer also emphasizes the fact that flu prevention doesn’t end with better clinical practices. Patient education is key.
“Hospitalists can be vital in educating patients about how to avoid symptomatic contacts,” she says, “and how to advocate for themselves in insisting that all care providers use strict handwashing protocols to avoid transmitting influenza among patients.” TH
As HM continues to grow, the need for clear and accurate data about the specialty will only become more intense. Hospitalists and HM group leaders use survey information to better understand how they compare to other practices across the country, in terms of size and practice characteristics, as well as compensation and productivity.
Increasingly, healthcare executives are turning to survey data—either independently or via their hospitalist group leaders—to get a grasp on the best practices in the industry.
That’s why SHM teamed up with the Medical Group Management Association (MGMA), the industry leader for professional administrators and leaders of medical group practices, to research and develop the State of Hospital Medicine: 2010 Report Based on 2009 Data.
Previously, SHM created and fielded a biannual survey, then analyzed the results independently.
—Leslie A. Flores, MHA, SHM senior advisor for practice management
“Our partnership with MGMA expands our survey population, delivers more information, and brings MGMA’s 90 years of industry credibility in the medical practice management field,” says Leslie A. Flores, MHA, SHM senior advisor for practice management. “The 2010 survey gives hospitalists and hospital administrators an unprecedented snapshot of the state of hospital medicine.”
The 2010 report will be available this month in the “Practice Resources” section of the SHM website (www.hospitalmedicine.org).
The print version will be available to SHM members for $125; for $175, members receive both the print version and the report on CD-ROM.
“This is a first-ever opportunity for hospitalists, group leaders, and healthcare executives to get the clearest picture possible of a rapidly changing industry,” Flores says. TH
Brendon Shank is a freelance writer based in Philadelphia.
SHM Adopts Strict Code for Industry Relations
A long with nearly 20 other organizations, SHM has adopted the Code for Industry Relations (http://cmss.org/) established by the Council on Medical Specialty Societies (CMSS). In an era of digital communication, SHM has created a Web area (www.hospital medicine.org/industry) to continuously update its policies toward industry, display its current partnerships, and disclose the potential conflicts of interest of its board or directors, editors, and CEO.
The message from SHM leadership: SHM is committed to being a leader in an era of transparency and disclosure.
Transparency serves an important role for medical specialty societies, says Norman B. Kahn Jr., MD, executive vice president and CEO of CMSS. The code developed by CMSS and adopted by SHM “assures in interactions with industry that the patients’ needs come first,” Dr. Kahn says. “The bottom line is that this is all about protecting the independence of societies from industry without abrogating the relationship.”
From its early days, SHM has been aware of the need to balance the responsibilities of speaking for HM and the need to disclose any potential conflicts of interests. In 2000, SHM developed its Principles of Organizational Relationships (www.hospitalmedicine.org/OrgRelationships), which have guided the society’s efforts.
The principles call for a clear, bright line that is a barrier between the support of a partner and SHM’s control of content. Among other activities, SHM has applied those principles to meetings and educational initiatives, quality-improvement (QI) projects, and publications.
The principles also were the foundation for the tough conflict-of-interest policies (www.hospital medicine.org/interestpolicies) the board approved in 2005.
Over the last decade, as HM has grown, national hospitalist leaders have become the experts on a wide range of topics and are asked to speak, write, or advise government agencies, foundations, and industry.
As SHM has developed its resources to help hospitalists improve glycemic control, reduce unnecessary DVTs and PEs, and improve the transitions of care, SHM has engaged in partnerships with government agencies, foundations, and industry as well.
“SHM’s leadership recognizes that it has a fiduciary responsibility to its members—a responsibility to provide expert direction, and necessary resources, to enable the hospitalist to ensure the best possible care of his or her patients, and to advance the quality of the hospital system,” says SHM President Jeff Wiese, MD, SFHM. “But SHM cannot do this alone, and when external partnerships are established, it is the organization’s responsibility to enter into these partnerships judiciously, and to be fully transparent to the membership with respect to the arrangements of these partnerships.
“We are confident that no other organization has a more robust disclosure policy than SHM.”
Today’s Nominations, Tomorrow’s Leaders
HM leaders aren’t born that way—they’re nominated.
SHM is accepting nominations for its board election; new members will take office in May at HM11 in Dallas.
The nomination deadline is Oct. 31. Online ballots will be available to all SHM members in late 2010. The results of the election will be announced online in early 2011.
Nominees must be SHM members in good standing. SHM members may nominate themselves or be nominated by another SHM member. Nominations must include a letter of nomination, a one-page CV, and a recent photo.
The nomination committee considers candidates based on length of SHM membership, activity as a hospitalist and SHM member, the prominence of the candidate within the specialty, and a number of other factors.
Board members serve a three-year term and normally serve on one or more committees.
“Participating in SHM’s leadership is one of the best ways to help guide the future of hospital medicine,” says Larry Wellikson, MD, SFHM, CEO of SHM. “That begins by submitting a board nomination to SHM this year.”
Chapter Updates
Chicago
The Chicago chapter met May 19 at Sullivan’s Steakhouse. Twenty-five hospitalists from the Chicago area, including hospitalists at Loyola Hospital, Lutheran General Hospital, Illinois Masonic Medical Center, Trinity Hospital, Silver Cross, and Evanston Hospital, as well as hospitalist groups like Cogent and Vista, attended the meeting.
Dr. Robin Ross of Season Hospice and Palliative Care, which sponsored the event, presented clinical pearls for end-of-life care for the busy hospitalist. The meeting also featured a town-hall discussion, with topics relevant to everyday hospitalist practice—coding, consultations, and the use of observation units. Notification of chapter elections were summated to all chapter members in July and August.
Harrisburg/South Central Pennsylvania
Thirty hospitalists representing six HM programs attended the Harrisburg/South Central Pennsylvania chapter of SHM June 9 at Passage to India in Harrisburg.
Eric Kupersmith, MD, SFHM, division head of the hospitalist program at Cooper University Hospital in Camden, N.J., led an open discussion regarding “Transition of Care and the Hospitalist’s Role.” Chapter members had the opportunity to discuss how each individual program actively seeks to decrease readmission rates. Discharge-planning specifics generated group discussion, and a handful of hospitalists offered testimonials about what is working in their practices.
The meeting was sponsored by Merck.
A BOOST for all seasons: Discharge improvement resources now available year round
When Project BOOST (Better Outcomes for Older Adults through Safer Transitions), SHM’s groundbreaking program to reduce readmissions, first began in 2008, hospital sites applied to participate in a yearlong program of one-on-one mentorships, regular sessions to share best practices, and a resource toolkit.
Since then, Project BOOST has grown and evolved. Some BOOST iterations now include third parties, such as the University of Michigan, Blue Cross/Blue Shield of Michigan, and the California HealthCare Foundation. SHM also recently introduced a nationwide, tuition-based version of the BOOST initiative.
Now, SHM is announcing that new resources are available to all hospitals, regardless of their participation in Project BOOST, all year. Some resources were previously available only to Project BOOST participants; others are brand-new materials available to any hospital or hospitalist trying to reduce unplanned readmissions to their hospital.
“No matter where you are in the hospital, you have an opportunity to improve discharge,” says Tina Budnitz, MPH, senior advisor for quality improvement. “The response to Project BOOST has been overwhelmingly positive. That’s why we’re so excited to make these new materials available to anyone responsible for lowering readmissions.”
Individuals can download the Project BOOST implementation course, a new training program specifically designed to help nurses use the proven “teachback” method, and the Project BOOST patient PASS form.
Budnitz and Project BOOST organizers also plan to launch supplemental products for self-implementers, including access to Project BOOST e-mail listservs, data centers, and webinars.
Prepare Now for Flu Season
With most of the country still enjoying warm weather, it’s easy to forget that flu season is right around the corner. Hospitals can either be part of the solution—or part of the problem.
Compared to friends and family outside of the hospital, patients in the hospital are especially at risk. The chances of contracting the flu are higher, given decreased immune responses and increased proximity to caregivers and other potentially infected patients. Plus, the impact of the flu on healthcare providers can be significantly more severe.
By now, most hospitals have protocols for preparing for flu season and isolating infected patients, says Danielle Scheurer, MD, MSc, SFHM, assistant professor of medicine at Harvard Medical School in Boston and director of general medical service at Brigham and Women’s Hospitalist Service. Hospitalists play a special role within those protocols, she adds.
“Hospitalists can be instrumental in preventing the spread of flu within the hospital by having a low threshold for diagnostic testing of patients, immediate isolation of those patients, and strict adherence to infection-control measures in those with suspected influenza,” Dr. Scheurer says.
Dr. Scheurer also emphasizes the fact that flu prevention doesn’t end with better clinical practices. Patient education is key.
“Hospitalists can be vital in educating patients about how to avoid symptomatic contacts,” she says, “and how to advocate for themselves in insisting that all care providers use strict handwashing protocols to avoid transmitting influenza among patients.” TH
In the Literature
In This Edition
Literature at a Glance
A guide to this month’s studies
- Antibiotics after drainage of uncomplicated skin abscesses
- Clopidogrel vs. combined aspirin-dipyridamole for acute ischemic stroke
- BNP-guided therapy in chronic heart failure outpatients
- Cognitive decline and dementia after hospitalization
- Clopidogrel delays up risks for DES implantation patients
- Clinical score identifies prolonged length of stay
- Time to therapy reduces mortality in sepsis patients
- PEEP associated with lower mortality for ARDS patients
Antibiotics Might Be Unnecessary after Drainage of Uncomplicated Skin Abscesses
Clinical question: Does trimethoprim/sulfamethoxazole (TMP/SMX) treatment after drainage of a skin abscess reduce treatment failure at seven days or development of new lesions at 30 days?
Background: Community ac-quired methicillin-resistant Staphylococcus aureus (MRSA) skin abscesses are increasing in frequency. The benefit of antibiotic treatment after incision and drainage is not clear, as there is a high cure rate without antibiotics.
Study design: Multicenter, double-blinded, randomized, placebo-controlled trial.
Setting: Four military EDs treating civilians and military patients.
Synopsis: The study enrolled a convenience sample of 220 patients, each of whom presented to EDs with uncomplicated skin abscesses from November 2007 to June 2009. Abscesses were drained in the ED, then patients were randomized to either placebo or to TMP/SMX (two DS tablets twice daily) for seven days. Re-evaluation for wound checks occurred at two days and seven days.
Treatment failure at seven days, defined as worsening infection, new lesions, or absence of clinical improvement, occurred in 26% of placebo patients and 17% of patients in the treatment arm, a nonsignificant difference (P=0.12). Fewer patients in the treatment arm had new lesions at 30 days (28% vs. 9%, P=0.02). MRSA was cultured from 53% of patients overall; all samples were sensitive to TMP/SMX.
The study was limited by the fact that only 69% of patients were evaluated at 30 days.
Bottom line: TMP/SMX treatment of uncomplicated skin abscess after drainage in EDs does not decrease treatment failure at seven days, but might decrease the development of new lesions.
Citation: Schmitz GR, Bruner D, Pitotti R, et al. Randomized controlled trial of trimethoprim-sulfamethoxazole for uncomplicated skin abscesses in patients at risk for community-associated methicillin-resistant Staphylococcus aureus infection [published online ahead of print March 29, 2010]. Ann Emerg Med. doi:10.1016/j.annemerg med.2010.03.002.
Clopidogrel and Combined Aspirin-Dipyridamole Have Similar Safety and Efficacy Profiles for Acute Ischemic Stroke
Clinical question: What is the efficacy and safety of combined aspirin and extended-release dipyridamole (Asp/ER-DP) compared to clopidogrel in patients with acute ischemic stroke?
Background: Long-term antiplatelet therapy is effective at reducing recurrence after ischemic stroke. However, the relative safety and efficacy of Asp/ER-DP or clopidogrel is not known in patients with acute ischemic stroke.
Study design: Randomized, controlled trial.
Setting: A multicenter trial involving 695 sites in 35 countries.
Synopsis: This post-hoc subgroup analysis of the PRoFESS (Prevention Regimen for Effectively Avoiding Second Strokes) trial assessed the relative safety and efficacy of Asp/ER-DP versus clopidogrel administered within 72 hours of stroke onset in 1,360 patients. The primary endpoint was functional outcome at 30 days.
Secondary outcomes included symptomatic hemorrhagic transformation of the infarct, cerebral edema, recurrent stroke, myocardial infarction (MI), composite vascular events (combination of nonfatal stroke, nonfatal MI, and vascular death), death, cognition, bleeding, and serious adverse events studied at seven, 30, and 90 days.
Combined death or dependency did not differ between treatment groups. Nonsignificant trends to reduced recurrence and vascular events were present with Asp/ER-DP. Rates of death, major bleeding, and serious adverse events did not differ between treatment groups.
Bottom line: Either clopidogrel or combined aspirin and extended-release dipyridamole can be used to treat acute ischemic stroke, with similar outcomes and safety profiles.
Citation: Bath PM, Cotton D, Martin RH, et al. Effect of combined aspirin and extended-release dipyridamole versus clopidogrel on functional outcome and recurrence in acute, mild ischemic stroke: PRoFESS subgroup analysis. Stroke. 2010;41(4):732-738.
BNP-Guided Therapy Reduces All-Cause Mortality in Outpatients with Chronic Heart Failure
Clinical question: Is there a clinical benefit in using B-type natriuretic peptide (BNP) to guide adjustment of proven medications in chronic heart failure?
Background: BNP is secreted by the heart in response to increased volume. It has been shown to be useful in the diagnosis of decompensated heart failure, and it can be decreased by treatment with proven heart failure medications. It is unclear if this effect provides clinical benefit on mortality and hospitalization.
Study design: Meta-analysis of prospective randomized controlled trials.
Setting: Eight studies involving 1,726 patients, published internationally from 2005-2009.
Synopsis: Study sizes ranged from 41 to 499 patients, with three- to 24-month follow-up. Patients had New York Heart Association (NYHA) class II or greater heart failure, with ejection fractions <50%.
All-cause mortality was significantly lower in BNP-guided therapy compared with clinical-guided therapy (RR=0.76; 95% CI, 0.63-0.91; P=0.003), specifically in patients younger than 75 years old (RR=0.52; 95% CI, 0.33-0.82; P=0.005).
A proposed mechanism for this result was a statistically significant increase in adjustment of most heart failure medications for BNP-guided therapy compared with clinical-guided therapy (75% vs. 58%, P<0.001 in diuretics; 49.6% vs. 30.9%, P<0.001 in ACE inhibitors or Angiotensin II receptor blockers (ARBs); and 51.1% vs. 41.6%, P=0.02 in beta-blockers) and a higher percentage reaching target doses in the BNP-guided therapy group. However, there was no significant decrease in all-cause hospitalization or survival free of hospitalization.
The study limitations include: Hospitalization for heart failure was not meta-analyzed, the pooled data were weighted toward one study, and BNP-guided titration parameters varied across studies.
Bottom line: BNP-guided therapy reduces all-cause mortality in chronic heart failure patients younger than 75 years old, but not all-cause hospitalization or survival free of hospitalization.
Citation: Porapakkham P, Porapakkham P, Zimmet H, Billah B, Krum H. B-type natriuretic peptide-guided heart failure therapy: A meta-analysis. Arch Intern Med. 2010;170(6):507-514.
Hospitalization Is Associated with Cognitive Decline and Subsequent Risk for Dementia in the Elderly
Clinical question: Is critical illness in patients 65 and older associated with long-term cognitive impairment, and does it affect the incidence of dementia?
Background: There is literature suggesting that survivors of critical illness suffer long-term cognitive impairment, but premorbid measures of cognitive function have not been researched. No studies have evaluated the risk of incident dementia among this patient population.
Study design: Prospective cohort study.
Setting: Group Health Cooperative in Seattle.
Synopsis: This study analyzed data from 2,929 community-dwelling adults older than 65 without baseline dementia. From 1994 to 2007, the individuals were screened with the Cognitive Abilities Screening Instrument (CASI) at follow-up visits every two years. CASI scores lower than 86 (out of 100) led to an examination for dementia; the diagnosis of dementia was an outcome measure. Scores were adjusted for baseline cognitive scores, age, and other risk factors.
For patients following acute-care hospitalization, adjusted CASI scores were 1.01 points lower on average than for those not hospitalized. For patients following critical-illness hospitalization, scores were 2.14 points lower. The dementia rate was 14.6 cases per 1,000 person-years among patients not hospitalized, and 33.6 among those admitted for noncritical illness.
As suspected, hospitalization might be a marker for cognitive decline in the elderly after adjusting for premorbid CASI scores and comorbid illness. Some factors in acute illness—and moreso in critical illness—might be causally related to cognitive decline.
Bottom line: In elderly patients without dementia at baseline, hospitalization for acute care and critical illness increases the likelihood of cognitive decline compared with patients who were not hospitalized. Only noncritical-illness hospitalization was not associated with the development of dementia.
Citation: Ehlenbach WJ, Hough CL, Crane PK, et al. Association between acute care and critical illness hospitalization and cognitive function in older adults. JAMA. 2010;303(8): 763-770.
Increased Risk of Death and Myocardial Infarction in Patients Who Delay Filling Clopidogrel Prescription after Drug-Eluting Stent Implantation
Clinical question: Is there an increased risk of death or myocardial infarction (MI) in patients with recent drug-eluting stent (DES) implantation who delayed filling their clopidogrel prescription compared with those who filled their prescription on the day of hospital discharge?
Background: Filling an initial prescription of clopidogrel on the day of discharge is important after DES implantation, as prior studies suggest that lack of thienopyridine therapy is a risk factor for early stent thrombosis.
Study design: Retrospective cohort study.
Setting: Three large, integrated healthcare systems.
Synopsis: The cohort included 7,042 patients discharged after DES implantation. Filling of a clopidogrel prescription was based on pharmacy dispensing data. Primary analysis divided patients based on whether they filled the prescription on the day of discharge or any time after discharge. Secondary analysis further characterized delays as >1 day, >3 days, or >5 days after discharge.
One in 6 patients delayed filling the initial prescription. Patients with any degree of delay had significantly higher death and MI rates during follow-up (14.2% vs. 7.9%, P<0.001), as well as an increased risk of death/MI (hazard ratio 1.53; 95% CI, 1.25-1.87). Factors associated with a delay in filling clopidogrel included older age, prior MI, diabetes, renal dysfunction, prior revascularization, cardiogenic shock, in-hospital bleeding, and use of clopidogrel upon admission.
The study was limited in that data were based on pharmacy records, and that patients might have received medication at discharge or outside the healthcare system.
Bottom line: The delay in filling a clopidogrel prescription is associated with an increased risk of death and MI in patients with recent DES implantation.
Citation: Ho PM, Tsai TT, Maddox TM, et al. Delays in filling clopidogrel prescription after hospital discharge and adverse outcomes after drug-eluting stent implantation: implications for transitions of care. Circ Cardiovasc Qual Outcomes. 2010;3(3):261-266.
Predicting Length of Stay after Stroke
Clinical question: Does a clinical score accurately predict prolonged length of stay after stroke?
Background: Stroke is a costly health problem, and length of stay is the most prominent factor contributing to the high costs. The factors leading to prolonged length of stay are varied, and there are no established tools to predict length of stay.
Study design: Prospective cohort study.
Setting: All 28 Israeli hospitals that admit stroke patients.
Synopsis: All patients admitted to Israeli hospitals during established two-month periods in 2004 (1,700 patients) and 2007 (1,648 patients) were included in the National Acute Stroke Israeli Survey (NASIS), and served as the derivation and validation cohort for development of a Prolonged Length of Stay (PLOS) score.
Using the 2004 data, investigators identified stroke severity using the National Institutes of Health Stroke Scale (NIHSS), history of congestive heart failure (CHF), history of atrial fibrillation, decreased level of consciousness on presentation, and intracerebral hemorrhage (as opposed to ischemic stroke) as predictors of prolonged length of stay. Four of these factors were expressed as dichotomous variables, whereas the stroke severity by NIHSS class was incorporated as a range; all were incorporated into a PLOS score.
Higher PLOS score correlated with longer length of stay. In the derivation cohort, 22% of patients with a PLOS score of 0 had a prolonged length of stay, whereas 85% of patients with PLOS scores of 6 or 7 had a prolonged length of stay. In the validation cohort, the corresponding figures were 19% and 72%.
Bottom line: Use of a simple score can predict risk of prolonged length of stay after stroke.
Citation: Koton S, Bornstein NM, Tsabari R, Tanne D, NASIS Investigators. Derivation and validation of the prolonged length of stay score in acute stroke patients. Neurology. 2010;74(19);1511-1516.
Earlier Administration of Appropriate Antimicrobials Decreases Mortality in Patients with Severe Sepsis and Septic Shock
Clinical question: Is the timing of antimicrobial administration an important determinant of survival in patients diagnosed with severe sepsis and septic shock?
Background: Severe sepsis and septic shock are associated with a 25% to 50% mortality rate. Early goal-directed therapy has been shown to increase survival in these patients. Antimicrobial treatment is a mainstay of this therapy, but the most effective timing of this treatment remains unclear.
Study design: Retrospective, single-center cohort study.
Setting: ED at an academic tertiary-care center.
Synopsis: Two hundred sixty-one patients in the ED in 2005-2006 presenting with severe sepsis or septic shock were enrolled in the hospital’s early goal-directed therapy (EGDT) algorithm, either at triage or later during their ED stay. Labs showed 56.7% of patients were culture-positive, with the most common sources being respiratory (30.6%), genitourinary (22.8%), and gastrointestinal (19.7%).
All patients received antibiotics and were stratified in one-hour intervals by the following categories: time from triage to antibiotics; time from qualification for EGDT to antibiotics; time from triage to appropriate antibiotics; and time from qualification for EGDT to appropriate antibiotics.
Total in-hospital mortality was 31% (35.1% for culture-positive patients vs. 25.7% for culture-negative patients, P=0.11). A significant decrease in mortality was only found when appropriate antibiotics were administered within one hour of triage, or within one hour of qualification for EGDT (OR=0.30; 95% CI, 0.11-0.83; P=0.02, and OR=0.50; 95% CI, 0.27-0.92; P=0.03, respectively).
Study limitations included the single-center site and small sample size.
Bottom line: In patients with severe sepsis and septic shock, initiating appropriate antimicrobial therapy within one hour of triage or entry into goal-directed therapy significantly reduces mortality.
Citation: Gaieski DF, Mikkelsen ME, Band RA, et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit Care Med. 2010;38(4):1045-1053.
Treatment with Higher Levels of Positive End-Expiratory Pressure Has Limited Affect on Hospital Survival
Clinical question: Is treatment with higher versus lower levels of positive end-expiratory pressure (PEEP) associated with improved hospital survival?
Background: In the management of patients with acute lung injury or acute respiratory distress syndrome (ARDS), a fundamental goal is to protect the lungs from ventilation-induced injury, but the optimal PEEP level has not been established.
Study design: Systematic review and meta-analysis.
Setting: N/A.
Synopsis: Three randomized-controlled trials eligible for this review included 2,299 critically ill adults with acute lung injury, as defined by the American-European Consensus Conference. The meta-analysis compared higher and lower PEEP levels with a mean difference of at least 3 cm H2O, incorporated a target tidal volume of less than 8 mL/kg of predicted body weight in both ventilation strategies, and provided patient follow-up until death or for at least 20 days.
This review demonstrated no statistically significant difference in hospital mortality between the groups. However, in patients with ARDS, higher levels of PEEP were associated with a relative reduction in mortality of 10%. This is supported by a recent cohort study in patients with acute lung injury or ARDS, which showed that the effect of PEEP on lung recruitment was associated with the proportion of potentially recruitable lung, as determined by computed tomography.
Since patients with ARDS have more pulmonary edema than those with acute lung injury without ARDS, the former have greater recruitability, and thus might benefit more from higher levels of PEEP.
Bottom line: Higher levels of PEEP might be associated with lower hospital mortality in patients with ARDS, but such a benefit is unlikely in patients with less severe lung injuries, and could actually be harmful.
Citation: Briel M, Meade M, Mercat A, et al. Higher vs lower positive end-expiratory pressure in patients with acute lung injury and acute respiratory distress syndrome: systematic review and meta-analysis. JAMA. 2010;303(9):865-873. TH
PEDIATRIC HM LITERATURE
By Mark Shen, MD
High-Performing State Healthcare Systems Have Higher Children’s Hospital Readmission Rates

Clinical question: What is the relationship between a hospital’s readmission rate and performance of the surrounding healthcare system?
Background: Hospital readmission rates might be influenced by factors related to the specific patient and hospital care, as well as such external factors as the performance of the surrounding healthcare system. Traditionally, readmission rates are thought to most accurately reflect the quality of hospital care; however, the relative contributions of patient, hospital, and external factors to hospital readmission rates have not been delineated.
Study design: Multilevel cohort study.
Setting: Thirty-nine children’s hospitals in 24 states.
Synopsis: The Pediatric Health Information System (PHIS) administrative database was sampled for the 2005 calendar year to review discharges from 39 participating children’s hospitals. Patients 2 to 18 years were included, and out of a total of 198,422 patients, 32,196 were readmitted within 365 days of discharge.
The Commonwealth Fund’s 2008 State Variations in Child Health System Performance ranking was used to define the state-level health system performance. Higher readmission rates correlated with higher-ranked state child health systems after adjustment for patient-level characteristics.
This surprising result calls into question the often-assumed link between hospital readmission rates and poor systems of care. However, despite the strength of its large sample size, this study’s macro-level view of the healthcare system might be too crude to truly define the external factors that play a role in readmission. State-level healthcare rankings might not accurately reflect the healthcare ecosystem surrounding each children’s hospital, and children’s hospitals do not care for the majority of children in the U.S.
Bottom line: Children’s hospital readmissions correlate with higher state child health system performance.
Citation: Feudtner C, Pati S, Goodman DM, et al. State-level child health system performance and the likelihood of readmission to children’s hospitals. J Pediatr. 2010;157(1):98-102.
In This Edition
Literature at a Glance
A guide to this month’s studies
- Antibiotics after drainage of uncomplicated skin abscesses
- Clopidogrel vs. combined aspirin-dipyridamole for acute ischemic stroke
- BNP-guided therapy in chronic heart failure outpatients
- Cognitive decline and dementia after hospitalization
- Clopidogrel delays up risks for DES implantation patients
- Clinical score identifies prolonged length of stay
- Time to therapy reduces mortality in sepsis patients
- PEEP associated with lower mortality for ARDS patients
Antibiotics Might Be Unnecessary after Drainage of Uncomplicated Skin Abscesses
Clinical question: Does trimethoprim/sulfamethoxazole (TMP/SMX) treatment after drainage of a skin abscess reduce treatment failure at seven days or development of new lesions at 30 days?
Background: Community ac-quired methicillin-resistant Staphylococcus aureus (MRSA) skin abscesses are increasing in frequency. The benefit of antibiotic treatment after incision and drainage is not clear, as there is a high cure rate without antibiotics.
Study design: Multicenter, double-blinded, randomized, placebo-controlled trial.
Setting: Four military EDs treating civilians and military patients.
Synopsis: The study enrolled a convenience sample of 220 patients, each of whom presented to EDs with uncomplicated skin abscesses from November 2007 to June 2009. Abscesses were drained in the ED, then patients were randomized to either placebo or to TMP/SMX (two DS tablets twice daily) for seven days. Re-evaluation for wound checks occurred at two days and seven days.
Treatment failure at seven days, defined as worsening infection, new lesions, or absence of clinical improvement, occurred in 26% of placebo patients and 17% of patients in the treatment arm, a nonsignificant difference (P=0.12). Fewer patients in the treatment arm had new lesions at 30 days (28% vs. 9%, P=0.02). MRSA was cultured from 53% of patients overall; all samples were sensitive to TMP/SMX.
The study was limited by the fact that only 69% of patients were evaluated at 30 days.
Bottom line: TMP/SMX treatment of uncomplicated skin abscess after drainage in EDs does not decrease treatment failure at seven days, but might decrease the development of new lesions.
Citation: Schmitz GR, Bruner D, Pitotti R, et al. Randomized controlled trial of trimethoprim-sulfamethoxazole for uncomplicated skin abscesses in patients at risk for community-associated methicillin-resistant Staphylococcus aureus infection [published online ahead of print March 29, 2010]. Ann Emerg Med. doi:10.1016/j.annemerg med.2010.03.002.
Clopidogrel and Combined Aspirin-Dipyridamole Have Similar Safety and Efficacy Profiles for Acute Ischemic Stroke
Clinical question: What is the efficacy and safety of combined aspirin and extended-release dipyridamole (Asp/ER-DP) compared to clopidogrel in patients with acute ischemic stroke?
Background: Long-term antiplatelet therapy is effective at reducing recurrence after ischemic stroke. However, the relative safety and efficacy of Asp/ER-DP or clopidogrel is not known in patients with acute ischemic stroke.
Study design: Randomized, controlled trial.
Setting: A multicenter trial involving 695 sites in 35 countries.
Synopsis: This post-hoc subgroup analysis of the PRoFESS (Prevention Regimen for Effectively Avoiding Second Strokes) trial assessed the relative safety and efficacy of Asp/ER-DP versus clopidogrel administered within 72 hours of stroke onset in 1,360 patients. The primary endpoint was functional outcome at 30 days.
Secondary outcomes included symptomatic hemorrhagic transformation of the infarct, cerebral edema, recurrent stroke, myocardial infarction (MI), composite vascular events (combination of nonfatal stroke, nonfatal MI, and vascular death), death, cognition, bleeding, and serious adverse events studied at seven, 30, and 90 days.
Combined death or dependency did not differ between treatment groups. Nonsignificant trends to reduced recurrence and vascular events were present with Asp/ER-DP. Rates of death, major bleeding, and serious adverse events did not differ between treatment groups.
Bottom line: Either clopidogrel or combined aspirin and extended-release dipyridamole can be used to treat acute ischemic stroke, with similar outcomes and safety profiles.
Citation: Bath PM, Cotton D, Martin RH, et al. Effect of combined aspirin and extended-release dipyridamole versus clopidogrel on functional outcome and recurrence in acute, mild ischemic stroke: PRoFESS subgroup analysis. Stroke. 2010;41(4):732-738.
BNP-Guided Therapy Reduces All-Cause Mortality in Outpatients with Chronic Heart Failure
Clinical question: Is there a clinical benefit in using B-type natriuretic peptide (BNP) to guide adjustment of proven medications in chronic heart failure?
Background: BNP is secreted by the heart in response to increased volume. It has been shown to be useful in the diagnosis of decompensated heart failure, and it can be decreased by treatment with proven heart failure medications. It is unclear if this effect provides clinical benefit on mortality and hospitalization.
Study design: Meta-analysis of prospective randomized controlled trials.
Setting: Eight studies involving 1,726 patients, published internationally from 2005-2009.
Synopsis: Study sizes ranged from 41 to 499 patients, with three- to 24-month follow-up. Patients had New York Heart Association (NYHA) class II or greater heart failure, with ejection fractions <50%.
All-cause mortality was significantly lower in BNP-guided therapy compared with clinical-guided therapy (RR=0.76; 95% CI, 0.63-0.91; P=0.003), specifically in patients younger than 75 years old (RR=0.52; 95% CI, 0.33-0.82; P=0.005).
A proposed mechanism for this result was a statistically significant increase in adjustment of most heart failure medications for BNP-guided therapy compared with clinical-guided therapy (75% vs. 58%, P<0.001 in diuretics; 49.6% vs. 30.9%, P<0.001 in ACE inhibitors or Angiotensin II receptor blockers (ARBs); and 51.1% vs. 41.6%, P=0.02 in beta-blockers) and a higher percentage reaching target doses in the BNP-guided therapy group. However, there was no significant decrease in all-cause hospitalization or survival free of hospitalization.
The study limitations include: Hospitalization for heart failure was not meta-analyzed, the pooled data were weighted toward one study, and BNP-guided titration parameters varied across studies.
Bottom line: BNP-guided therapy reduces all-cause mortality in chronic heart failure patients younger than 75 years old, but not all-cause hospitalization or survival free of hospitalization.
Citation: Porapakkham P, Porapakkham P, Zimmet H, Billah B, Krum H. B-type natriuretic peptide-guided heart failure therapy: A meta-analysis. Arch Intern Med. 2010;170(6):507-514.
Hospitalization Is Associated with Cognitive Decline and Subsequent Risk for Dementia in the Elderly
Clinical question: Is critical illness in patients 65 and older associated with long-term cognitive impairment, and does it affect the incidence of dementia?
Background: There is literature suggesting that survivors of critical illness suffer long-term cognitive impairment, but premorbid measures of cognitive function have not been researched. No studies have evaluated the risk of incident dementia among this patient population.
Study design: Prospective cohort study.
Setting: Group Health Cooperative in Seattle.
Synopsis: This study analyzed data from 2,929 community-dwelling adults older than 65 without baseline dementia. From 1994 to 2007, the individuals were screened with the Cognitive Abilities Screening Instrument (CASI) at follow-up visits every two years. CASI scores lower than 86 (out of 100) led to an examination for dementia; the diagnosis of dementia was an outcome measure. Scores were adjusted for baseline cognitive scores, age, and other risk factors.
For patients following acute-care hospitalization, adjusted CASI scores were 1.01 points lower on average than for those not hospitalized. For patients following critical-illness hospitalization, scores were 2.14 points lower. The dementia rate was 14.6 cases per 1,000 person-years among patients not hospitalized, and 33.6 among those admitted for noncritical illness.
As suspected, hospitalization might be a marker for cognitive decline in the elderly after adjusting for premorbid CASI scores and comorbid illness. Some factors in acute illness—and moreso in critical illness—might be causally related to cognitive decline.
Bottom line: In elderly patients without dementia at baseline, hospitalization for acute care and critical illness increases the likelihood of cognitive decline compared with patients who were not hospitalized. Only noncritical-illness hospitalization was not associated with the development of dementia.
Citation: Ehlenbach WJ, Hough CL, Crane PK, et al. Association between acute care and critical illness hospitalization and cognitive function in older adults. JAMA. 2010;303(8): 763-770.
Increased Risk of Death and Myocardial Infarction in Patients Who Delay Filling Clopidogrel Prescription after Drug-Eluting Stent Implantation
Clinical question: Is there an increased risk of death or myocardial infarction (MI) in patients with recent drug-eluting stent (DES) implantation who delayed filling their clopidogrel prescription compared with those who filled their prescription on the day of hospital discharge?
Background: Filling an initial prescription of clopidogrel on the day of discharge is important after DES implantation, as prior studies suggest that lack of thienopyridine therapy is a risk factor for early stent thrombosis.
Study design: Retrospective cohort study.
Setting: Three large, integrated healthcare systems.
Synopsis: The cohort included 7,042 patients discharged after DES implantation. Filling of a clopidogrel prescription was based on pharmacy dispensing data. Primary analysis divided patients based on whether they filled the prescription on the day of discharge or any time after discharge. Secondary analysis further characterized delays as >1 day, >3 days, or >5 days after discharge.
One in 6 patients delayed filling the initial prescription. Patients with any degree of delay had significantly higher death and MI rates during follow-up (14.2% vs. 7.9%, P<0.001), as well as an increased risk of death/MI (hazard ratio 1.53; 95% CI, 1.25-1.87). Factors associated with a delay in filling clopidogrel included older age, prior MI, diabetes, renal dysfunction, prior revascularization, cardiogenic shock, in-hospital bleeding, and use of clopidogrel upon admission.
The study was limited in that data were based on pharmacy records, and that patients might have received medication at discharge or outside the healthcare system.
Bottom line: The delay in filling a clopidogrel prescription is associated with an increased risk of death and MI in patients with recent DES implantation.
Citation: Ho PM, Tsai TT, Maddox TM, et al. Delays in filling clopidogrel prescription after hospital discharge and adverse outcomes after drug-eluting stent implantation: implications for transitions of care. Circ Cardiovasc Qual Outcomes. 2010;3(3):261-266.
Predicting Length of Stay after Stroke
Clinical question: Does a clinical score accurately predict prolonged length of stay after stroke?
Background: Stroke is a costly health problem, and length of stay is the most prominent factor contributing to the high costs. The factors leading to prolonged length of stay are varied, and there are no established tools to predict length of stay.
Study design: Prospective cohort study.
Setting: All 28 Israeli hospitals that admit stroke patients.
Synopsis: All patients admitted to Israeli hospitals during established two-month periods in 2004 (1,700 patients) and 2007 (1,648 patients) were included in the National Acute Stroke Israeli Survey (NASIS), and served as the derivation and validation cohort for development of a Prolonged Length of Stay (PLOS) score.
Using the 2004 data, investigators identified stroke severity using the National Institutes of Health Stroke Scale (NIHSS), history of congestive heart failure (CHF), history of atrial fibrillation, decreased level of consciousness on presentation, and intracerebral hemorrhage (as opposed to ischemic stroke) as predictors of prolonged length of stay. Four of these factors were expressed as dichotomous variables, whereas the stroke severity by NIHSS class was incorporated as a range; all were incorporated into a PLOS score.
Higher PLOS score correlated with longer length of stay. In the derivation cohort, 22% of patients with a PLOS score of 0 had a prolonged length of stay, whereas 85% of patients with PLOS scores of 6 or 7 had a prolonged length of stay. In the validation cohort, the corresponding figures were 19% and 72%.
Bottom line: Use of a simple score can predict risk of prolonged length of stay after stroke.
Citation: Koton S, Bornstein NM, Tsabari R, Tanne D, NASIS Investigators. Derivation and validation of the prolonged length of stay score in acute stroke patients. Neurology. 2010;74(19);1511-1516.
Earlier Administration of Appropriate Antimicrobials Decreases Mortality in Patients with Severe Sepsis and Septic Shock
Clinical question: Is the timing of antimicrobial administration an important determinant of survival in patients diagnosed with severe sepsis and septic shock?
Background: Severe sepsis and septic shock are associated with a 25% to 50% mortality rate. Early goal-directed therapy has been shown to increase survival in these patients. Antimicrobial treatment is a mainstay of this therapy, but the most effective timing of this treatment remains unclear.
Study design: Retrospective, single-center cohort study.
Setting: ED at an academic tertiary-care center.
Synopsis: Two hundred sixty-one patients in the ED in 2005-2006 presenting with severe sepsis or septic shock were enrolled in the hospital’s early goal-directed therapy (EGDT) algorithm, either at triage or later during their ED stay. Labs showed 56.7% of patients were culture-positive, with the most common sources being respiratory (30.6%), genitourinary (22.8%), and gastrointestinal (19.7%).
All patients received antibiotics and were stratified in one-hour intervals by the following categories: time from triage to antibiotics; time from qualification for EGDT to antibiotics; time from triage to appropriate antibiotics; and time from qualification for EGDT to appropriate antibiotics.
Total in-hospital mortality was 31% (35.1% for culture-positive patients vs. 25.7% for culture-negative patients, P=0.11). A significant decrease in mortality was only found when appropriate antibiotics were administered within one hour of triage, or within one hour of qualification for EGDT (OR=0.30; 95% CI, 0.11-0.83; P=0.02, and OR=0.50; 95% CI, 0.27-0.92; P=0.03, respectively).
Study limitations included the single-center site and small sample size.
Bottom line: In patients with severe sepsis and septic shock, initiating appropriate antimicrobial therapy within one hour of triage or entry into goal-directed therapy significantly reduces mortality.
Citation: Gaieski DF, Mikkelsen ME, Band RA, et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit Care Med. 2010;38(4):1045-1053.
Treatment with Higher Levels of Positive End-Expiratory Pressure Has Limited Affect on Hospital Survival
Clinical question: Is treatment with higher versus lower levels of positive end-expiratory pressure (PEEP) associated with improved hospital survival?
Background: In the management of patients with acute lung injury or acute respiratory distress syndrome (ARDS), a fundamental goal is to protect the lungs from ventilation-induced injury, but the optimal PEEP level has not been established.
Study design: Systematic review and meta-analysis.
Setting: N/A.
Synopsis: Three randomized-controlled trials eligible for this review included 2,299 critically ill adults with acute lung injury, as defined by the American-European Consensus Conference. The meta-analysis compared higher and lower PEEP levels with a mean difference of at least 3 cm H2O, incorporated a target tidal volume of less than 8 mL/kg of predicted body weight in both ventilation strategies, and provided patient follow-up until death or for at least 20 days.
This review demonstrated no statistically significant difference in hospital mortality between the groups. However, in patients with ARDS, higher levels of PEEP were associated with a relative reduction in mortality of 10%. This is supported by a recent cohort study in patients with acute lung injury or ARDS, which showed that the effect of PEEP on lung recruitment was associated with the proportion of potentially recruitable lung, as determined by computed tomography.
Since patients with ARDS have more pulmonary edema than those with acute lung injury without ARDS, the former have greater recruitability, and thus might benefit more from higher levels of PEEP.
Bottom line: Higher levels of PEEP might be associated with lower hospital mortality in patients with ARDS, but such a benefit is unlikely in patients with less severe lung injuries, and could actually be harmful.
Citation: Briel M, Meade M, Mercat A, et al. Higher vs lower positive end-expiratory pressure in patients with acute lung injury and acute respiratory distress syndrome: systematic review and meta-analysis. JAMA. 2010;303(9):865-873. TH
PEDIATRIC HM LITERATURE
By Mark Shen, MD
High-Performing State Healthcare Systems Have Higher Children’s Hospital Readmission Rates
Clinical question: What is the relationship between a hospital’s readmission rate and performance of the surrounding healthcare system?
Background: Hospital readmission rates might be influenced by factors related to the specific patient and hospital care, as well as such external factors as the performance of the surrounding healthcare system. Traditionally, readmission rates are thought to most accurately reflect the quality of hospital care; however, the relative contributions of patient, hospital, and external factors to hospital readmission rates have not been delineated.
Study design: Multilevel cohort study.
Setting: Thirty-nine children’s hospitals in 24 states.
Synopsis: The Pediatric Health Information System (PHIS) administrative database was sampled for the 2005 calendar year to review discharges from 39 participating children’s hospitals. Patients 2 to 18 years were included, and out of a total of 198,422 patients, 32,196 were readmitted within 365 days of discharge.
The Commonwealth Fund’s 2008 State Variations in Child Health System Performance ranking was used to define the state-level health system performance. Higher readmission rates correlated with higher-ranked state child health systems after adjustment for patient-level characteristics.
This surprising result calls into question the often-assumed link between hospital readmission rates and poor systems of care. However, despite the strength of its large sample size, this study’s macro-level view of the healthcare system might be too crude to truly define the external factors that play a role in readmission. State-level healthcare rankings might not accurately reflect the healthcare ecosystem surrounding each children’s hospital, and children’s hospitals do not care for the majority of children in the U.S.
Bottom line: Children’s hospital readmissions correlate with higher state child health system performance.
Citation: Feudtner C, Pati S, Goodman DM, et al. State-level child health system performance and the likelihood of readmission to children’s hospitals. J Pediatr. 2010;157(1):98-102.
In This Edition
Literature at a Glance
A guide to this month’s studies
- Antibiotics after drainage of uncomplicated skin abscesses
- Clopidogrel vs. combined aspirin-dipyridamole for acute ischemic stroke
- BNP-guided therapy in chronic heart failure outpatients
- Cognitive decline and dementia after hospitalization
- Clopidogrel delays up risks for DES implantation patients
- Clinical score identifies prolonged length of stay
- Time to therapy reduces mortality in sepsis patients
- PEEP associated with lower mortality for ARDS patients
Antibiotics Might Be Unnecessary after Drainage of Uncomplicated Skin Abscesses
Clinical question: Does trimethoprim/sulfamethoxazole (TMP/SMX) treatment after drainage of a skin abscess reduce treatment failure at seven days or development of new lesions at 30 days?
Background: Community ac-quired methicillin-resistant Staphylococcus aureus (MRSA) skin abscesses are increasing in frequency. The benefit of antibiotic treatment after incision and drainage is not clear, as there is a high cure rate without antibiotics.
Study design: Multicenter, double-blinded, randomized, placebo-controlled trial.
Setting: Four military EDs treating civilians and military patients.
Synopsis: The study enrolled a convenience sample of 220 patients, each of whom presented to EDs with uncomplicated skin abscesses from November 2007 to June 2009. Abscesses were drained in the ED, then patients were randomized to either placebo or to TMP/SMX (two DS tablets twice daily) for seven days. Re-evaluation for wound checks occurred at two days and seven days.
Treatment failure at seven days, defined as worsening infection, new lesions, or absence of clinical improvement, occurred in 26% of placebo patients and 17% of patients in the treatment arm, a nonsignificant difference (P=0.12). Fewer patients in the treatment arm had new lesions at 30 days (28% vs. 9%, P=0.02). MRSA was cultured from 53% of patients overall; all samples were sensitive to TMP/SMX.
The study was limited by the fact that only 69% of patients were evaluated at 30 days.
Bottom line: TMP/SMX treatment of uncomplicated skin abscess after drainage in EDs does not decrease treatment failure at seven days, but might decrease the development of new lesions.
Citation: Schmitz GR, Bruner D, Pitotti R, et al. Randomized controlled trial of trimethoprim-sulfamethoxazole for uncomplicated skin abscesses in patients at risk for community-associated methicillin-resistant Staphylococcus aureus infection [published online ahead of print March 29, 2010]. Ann Emerg Med. doi:10.1016/j.annemerg med.2010.03.002.
Clopidogrel and Combined Aspirin-Dipyridamole Have Similar Safety and Efficacy Profiles for Acute Ischemic Stroke
Clinical question: What is the efficacy and safety of combined aspirin and extended-release dipyridamole (Asp/ER-DP) compared to clopidogrel in patients with acute ischemic stroke?
Background: Long-term antiplatelet therapy is effective at reducing recurrence after ischemic stroke. However, the relative safety and efficacy of Asp/ER-DP or clopidogrel is not known in patients with acute ischemic stroke.
Study design: Randomized, controlled trial.
Setting: A multicenter trial involving 695 sites in 35 countries.
Synopsis: This post-hoc subgroup analysis of the PRoFESS (Prevention Regimen for Effectively Avoiding Second Strokes) trial assessed the relative safety and efficacy of Asp/ER-DP versus clopidogrel administered within 72 hours of stroke onset in 1,360 patients. The primary endpoint was functional outcome at 30 days.
Secondary outcomes included symptomatic hemorrhagic transformation of the infarct, cerebral edema, recurrent stroke, myocardial infarction (MI), composite vascular events (combination of nonfatal stroke, nonfatal MI, and vascular death), death, cognition, bleeding, and serious adverse events studied at seven, 30, and 90 days.
Combined death or dependency did not differ between treatment groups. Nonsignificant trends to reduced recurrence and vascular events were present with Asp/ER-DP. Rates of death, major bleeding, and serious adverse events did not differ between treatment groups.
Bottom line: Either clopidogrel or combined aspirin and extended-release dipyridamole can be used to treat acute ischemic stroke, with similar outcomes and safety profiles.
Citation: Bath PM, Cotton D, Martin RH, et al. Effect of combined aspirin and extended-release dipyridamole versus clopidogrel on functional outcome and recurrence in acute, mild ischemic stroke: PRoFESS subgroup analysis. Stroke. 2010;41(4):732-738.
BNP-Guided Therapy Reduces All-Cause Mortality in Outpatients with Chronic Heart Failure
Clinical question: Is there a clinical benefit in using B-type natriuretic peptide (BNP) to guide adjustment of proven medications in chronic heart failure?
Background: BNP is secreted by the heart in response to increased volume. It has been shown to be useful in the diagnosis of decompensated heart failure, and it can be decreased by treatment with proven heart failure medications. It is unclear if this effect provides clinical benefit on mortality and hospitalization.
Study design: Meta-analysis of prospective randomized controlled trials.
Setting: Eight studies involving 1,726 patients, published internationally from 2005-2009.
Synopsis: Study sizes ranged from 41 to 499 patients, with three- to 24-month follow-up. Patients had New York Heart Association (NYHA) class II or greater heart failure, with ejection fractions <50%.
All-cause mortality was significantly lower in BNP-guided therapy compared with clinical-guided therapy (RR=0.76; 95% CI, 0.63-0.91; P=0.003), specifically in patients younger than 75 years old (RR=0.52; 95% CI, 0.33-0.82; P=0.005).
A proposed mechanism for this result was a statistically significant increase in adjustment of most heart failure medications for BNP-guided therapy compared with clinical-guided therapy (75% vs. 58%, P<0.001 in diuretics; 49.6% vs. 30.9%, P<0.001 in ACE inhibitors or Angiotensin II receptor blockers (ARBs); and 51.1% vs. 41.6%, P=0.02 in beta-blockers) and a higher percentage reaching target doses in the BNP-guided therapy group. However, there was no significant decrease in all-cause hospitalization or survival free of hospitalization.
The study limitations include: Hospitalization for heart failure was not meta-analyzed, the pooled data were weighted toward one study, and BNP-guided titration parameters varied across studies.
Bottom line: BNP-guided therapy reduces all-cause mortality in chronic heart failure patients younger than 75 years old, but not all-cause hospitalization or survival free of hospitalization.
Citation: Porapakkham P, Porapakkham P, Zimmet H, Billah B, Krum H. B-type natriuretic peptide-guided heart failure therapy: A meta-analysis. Arch Intern Med. 2010;170(6):507-514.
Hospitalization Is Associated with Cognitive Decline and Subsequent Risk for Dementia in the Elderly
Clinical question: Is critical illness in patients 65 and older associated with long-term cognitive impairment, and does it affect the incidence of dementia?
Background: There is literature suggesting that survivors of critical illness suffer long-term cognitive impairment, but premorbid measures of cognitive function have not been researched. No studies have evaluated the risk of incident dementia among this patient population.
Study design: Prospective cohort study.
Setting: Group Health Cooperative in Seattle.
Synopsis: This study analyzed data from 2,929 community-dwelling adults older than 65 without baseline dementia. From 1994 to 2007, the individuals were screened with the Cognitive Abilities Screening Instrument (CASI) at follow-up visits every two years. CASI scores lower than 86 (out of 100) led to an examination for dementia; the diagnosis of dementia was an outcome measure. Scores were adjusted for baseline cognitive scores, age, and other risk factors.
For patients following acute-care hospitalization, adjusted CASI scores were 1.01 points lower on average than for those not hospitalized. For patients following critical-illness hospitalization, scores were 2.14 points lower. The dementia rate was 14.6 cases per 1,000 person-years among patients not hospitalized, and 33.6 among those admitted for noncritical illness.
As suspected, hospitalization might be a marker for cognitive decline in the elderly after adjusting for premorbid CASI scores and comorbid illness. Some factors in acute illness—and moreso in critical illness—might be causally related to cognitive decline.
Bottom line: In elderly patients without dementia at baseline, hospitalization for acute care and critical illness increases the likelihood of cognitive decline compared with patients who were not hospitalized. Only noncritical-illness hospitalization was not associated with the development of dementia.
Citation: Ehlenbach WJ, Hough CL, Crane PK, et al. Association between acute care and critical illness hospitalization and cognitive function in older adults. JAMA. 2010;303(8): 763-770.
Increased Risk of Death and Myocardial Infarction in Patients Who Delay Filling Clopidogrel Prescription after Drug-Eluting Stent Implantation
Clinical question: Is there an increased risk of death or myocardial infarction (MI) in patients with recent drug-eluting stent (DES) implantation who delayed filling their clopidogrel prescription compared with those who filled their prescription on the day of hospital discharge?
Background: Filling an initial prescription of clopidogrel on the day of discharge is important after DES implantation, as prior studies suggest that lack of thienopyridine therapy is a risk factor for early stent thrombosis.
Study design: Retrospective cohort study.
Setting: Three large, integrated healthcare systems.
Synopsis: The cohort included 7,042 patients discharged after DES implantation. Filling of a clopidogrel prescription was based on pharmacy dispensing data. Primary analysis divided patients based on whether they filled the prescription on the day of discharge or any time after discharge. Secondary analysis further characterized delays as >1 day, >3 days, or >5 days after discharge.
One in 6 patients delayed filling the initial prescription. Patients with any degree of delay had significantly higher death and MI rates during follow-up (14.2% vs. 7.9%, P<0.001), as well as an increased risk of death/MI (hazard ratio 1.53; 95% CI, 1.25-1.87). Factors associated with a delay in filling clopidogrel included older age, prior MI, diabetes, renal dysfunction, prior revascularization, cardiogenic shock, in-hospital bleeding, and use of clopidogrel upon admission.
The study was limited in that data were based on pharmacy records, and that patients might have received medication at discharge or outside the healthcare system.
Bottom line: The delay in filling a clopidogrel prescription is associated with an increased risk of death and MI in patients with recent DES implantation.
Citation: Ho PM, Tsai TT, Maddox TM, et al. Delays in filling clopidogrel prescription after hospital discharge and adverse outcomes after drug-eluting stent implantation: implications for transitions of care. Circ Cardiovasc Qual Outcomes. 2010;3(3):261-266.
Predicting Length of Stay after Stroke
Clinical question: Does a clinical score accurately predict prolonged length of stay after stroke?
Background: Stroke is a costly health problem, and length of stay is the most prominent factor contributing to the high costs. The factors leading to prolonged length of stay are varied, and there are no established tools to predict length of stay.
Study design: Prospective cohort study.
Setting: All 28 Israeli hospitals that admit stroke patients.
Synopsis: All patients admitted to Israeli hospitals during established two-month periods in 2004 (1,700 patients) and 2007 (1,648 patients) were included in the National Acute Stroke Israeli Survey (NASIS), and served as the derivation and validation cohort for development of a Prolonged Length of Stay (PLOS) score.
Using the 2004 data, investigators identified stroke severity using the National Institutes of Health Stroke Scale (NIHSS), history of congestive heart failure (CHF), history of atrial fibrillation, decreased level of consciousness on presentation, and intracerebral hemorrhage (as opposed to ischemic stroke) as predictors of prolonged length of stay. Four of these factors were expressed as dichotomous variables, whereas the stroke severity by NIHSS class was incorporated as a range; all were incorporated into a PLOS score.
Higher PLOS score correlated with longer length of stay. In the derivation cohort, 22% of patients with a PLOS score of 0 had a prolonged length of stay, whereas 85% of patients with PLOS scores of 6 or 7 had a prolonged length of stay. In the validation cohort, the corresponding figures were 19% and 72%.
Bottom line: Use of a simple score can predict risk of prolonged length of stay after stroke.
Citation: Koton S, Bornstein NM, Tsabari R, Tanne D, NASIS Investigators. Derivation and validation of the prolonged length of stay score in acute stroke patients. Neurology. 2010;74(19);1511-1516.
Earlier Administration of Appropriate Antimicrobials Decreases Mortality in Patients with Severe Sepsis and Septic Shock
Clinical question: Is the timing of antimicrobial administration an important determinant of survival in patients diagnosed with severe sepsis and septic shock?
Background: Severe sepsis and septic shock are associated with a 25% to 50% mortality rate. Early goal-directed therapy has been shown to increase survival in these patients. Antimicrobial treatment is a mainstay of this therapy, but the most effective timing of this treatment remains unclear.
Study design: Retrospective, single-center cohort study.
Setting: ED at an academic tertiary-care center.
Synopsis: Two hundred sixty-one patients in the ED in 2005-2006 presenting with severe sepsis or septic shock were enrolled in the hospital’s early goal-directed therapy (EGDT) algorithm, either at triage or later during their ED stay. Labs showed 56.7% of patients were culture-positive, with the most common sources being respiratory (30.6%), genitourinary (22.8%), and gastrointestinal (19.7%).
All patients received antibiotics and were stratified in one-hour intervals by the following categories: time from triage to antibiotics; time from qualification for EGDT to antibiotics; time from triage to appropriate antibiotics; and time from qualification for EGDT to appropriate antibiotics.
Total in-hospital mortality was 31% (35.1% for culture-positive patients vs. 25.7% for culture-negative patients, P=0.11). A significant decrease in mortality was only found when appropriate antibiotics were administered within one hour of triage, or within one hour of qualification for EGDT (OR=0.30; 95% CI, 0.11-0.83; P=0.02, and OR=0.50; 95% CI, 0.27-0.92; P=0.03, respectively).
Study limitations included the single-center site and small sample size.
Bottom line: In patients with severe sepsis and septic shock, initiating appropriate antimicrobial therapy within one hour of triage or entry into goal-directed therapy significantly reduces mortality.
Citation: Gaieski DF, Mikkelsen ME, Band RA, et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit Care Med. 2010;38(4):1045-1053.
Treatment with Higher Levels of Positive End-Expiratory Pressure Has Limited Affect on Hospital Survival
Clinical question: Is treatment with higher versus lower levels of positive end-expiratory pressure (PEEP) associated with improved hospital survival?
Background: In the management of patients with acute lung injury or acute respiratory distress syndrome (ARDS), a fundamental goal is to protect the lungs from ventilation-induced injury, but the optimal PEEP level has not been established.
Study design: Systematic review and meta-analysis.
Setting: N/A.
Synopsis: Three randomized-controlled trials eligible for this review included 2,299 critically ill adults with acute lung injury, as defined by the American-European Consensus Conference. The meta-analysis compared higher and lower PEEP levels with a mean difference of at least 3 cm H2O, incorporated a target tidal volume of less than 8 mL/kg of predicted body weight in both ventilation strategies, and provided patient follow-up until death or for at least 20 days.
This review demonstrated no statistically significant difference in hospital mortality between the groups. However, in patients with ARDS, higher levels of PEEP were associated with a relative reduction in mortality of 10%. This is supported by a recent cohort study in patients with acute lung injury or ARDS, which showed that the effect of PEEP on lung recruitment was associated with the proportion of potentially recruitable lung, as determined by computed tomography.
Since patients with ARDS have more pulmonary edema than those with acute lung injury without ARDS, the former have greater recruitability, and thus might benefit more from higher levels of PEEP.
Bottom line: Higher levels of PEEP might be associated with lower hospital mortality in patients with ARDS, but such a benefit is unlikely in patients with less severe lung injuries, and could actually be harmful.
Citation: Briel M, Meade M, Mercat A, et al. Higher vs lower positive end-expiratory pressure in patients with acute lung injury and acute respiratory distress syndrome: systematic review and meta-analysis. JAMA. 2010;303(9):865-873. TH
PEDIATRIC HM LITERATURE
By Mark Shen, MD
High-Performing State Healthcare Systems Have Higher Children’s Hospital Readmission Rates
Clinical question: What is the relationship between a hospital’s readmission rate and performance of the surrounding healthcare system?
Background: Hospital readmission rates might be influenced by factors related to the specific patient and hospital care, as well as such external factors as the performance of the surrounding healthcare system. Traditionally, readmission rates are thought to most accurately reflect the quality of hospital care; however, the relative contributions of patient, hospital, and external factors to hospital readmission rates have not been delineated.
Study design: Multilevel cohort study.
Setting: Thirty-nine children’s hospitals in 24 states.
Synopsis: The Pediatric Health Information System (PHIS) administrative database was sampled for the 2005 calendar year to review discharges from 39 participating children’s hospitals. Patients 2 to 18 years were included, and out of a total of 198,422 patients, 32,196 were readmitted within 365 days of discharge.
The Commonwealth Fund’s 2008 State Variations in Child Health System Performance ranking was used to define the state-level health system performance. Higher readmission rates correlated with higher-ranked state child health systems after adjustment for patient-level characteristics.
This surprising result calls into question the often-assumed link between hospital readmission rates and poor systems of care. However, despite the strength of its large sample size, this study’s macro-level view of the healthcare system might be too crude to truly define the external factors that play a role in readmission. State-level healthcare rankings might not accurately reflect the healthcare ecosystem surrounding each children’s hospital, and children’s hospitals do not care for the majority of children in the U.S.
Bottom line: Children’s hospital readmissions correlate with higher state child health system performance.
Citation: Feudtner C, Pati S, Goodman DM, et al. State-level child health system performance and the likelihood of readmission to children’s hospitals. J Pediatr. 2010;157(1):98-102.
Market Watch
New Generics
- Desloratadine tablets (generic Clarinex)1
- Didanosine delayed-release capsules (Generic Videx EC)2
New Indications, Dosage Forms, and Recommendations
- Ganciclovir ophthalmic gel 0.15% (Zirgan) has been approved by the FDA for treating acute herpetic keratitis.3 The recommended dose is one drop in the affected eye five times daily until the ulcer heals, then one drop three times daily for seven more days. The most common side effects in clinical trials were blurred vision, eye irritation, punctate keratitis, and conjunctival hyperemia. It will be available in a 5-g tube.
- Immune globulin intravenous 10% liquid (human) (Privigen) has received an updated approval from the FDA, which allows for room temperature storage throughout its entire 36-month shelf life.4 The agent is used to treat patients with primary immunodeficiency disorders.
- Miconazole buccal tablets (Oravig) have been approved by the FDA for treating oropharyngeal candidiasis in adults and children 16 years of age and older. It is the first, and currently the only local, buccal prescription formulation of miconazole.5 The buccal tablet was developed to adhere to the gum. It should not be crushed, chewed, or swallowed. The most common adverse effects in clinical trials were diarrhea, nausea, headache, dysgeusia, upper abdominal pain, and vomiting. It is recommended to monitor patients with a history of hypersensitivity to azoles, as there is no information regarding cross-reactivity between miconazole and other azole agents.
- A supplemental new drug application (sNDA) has been submitted to the FDA for naltrexone extended-release injectable suspension (Vivitrol) for treating opioid dependence.6 It is administered as a once-monthly intramuscular injection and currently is approved by the FDA for treating alcohol dependence.
- Oxycodone controlled-release (OxyContin) has been approved by the FDA in a new, abuse-deterrent formulation.7
- Pancrelipase delayed-release capsules (Pancreaze) joins Creon (Abbott Labs) and Zenpep (Eurand) as the third pancreatic enzyme product (PEP) to be approved by the FDA for treating exocrine pancreatic insufficiency.8
- Pramipexole extended-release tablets (Mirapex ER) have been approved by the FDA as a once-daily treatment for the signs and symptoms of idiopathic Parkinson’s disease (early and late).9
- The active ingredient in the vaccine Diamyd, rhGAD65, has received orphan drug status for treating Type 1 diabetes mellitus (T1DM) with residual beta cell function.10,11 This agent is in Phase 3 clinical trials and is being investigated to determine whether it can stop or slow the autoimmune destruction of insulin-producing beta cell function. The DiaPrevent study is enrolling patients. In Phase 2 studies, the agent preserved remaining beta cell function in adolescents and children recently diagnosed with T1DM.
- Warfarin genetic diagnostic: Machaon Diagnostics has received FDA approval for an array-based diagnostic technology that detects genetic variation and could aid in determining an accurate initial warfarin dose.12 At least 40% of Americans have at least one genetic variation involved in warfarin metabolism, which can cause a more than fivefold disparity in the weekly warfarin dose. This test can be used to more accurately determine dosing for warfarin-treated patients.
Pipeline
- The NDA for DM-1796 (gabapentin extended-release tablet) has been submitted to the FDA for treatment of postherpetic neuralgia.13 It is a once-daily, extended-release formulation of gabapentin.
- The “quad” combination of elvitegravir, cobicistat (formerly GS 9350), emtricitabine, and tenofovir disoproxil fumarate in a fixed-dose single tablet is currently in Phase 3 clinical trials for treatment of HIV.14
- FTY720 is an investigational oral immune modulator agent for treating relapsing-remitting multiple sclerosis (RR-MS).15 The NDA for this agent was submitted in December 2009; the FDA granted it a priority review in February. Two-year data from the FREEDOMS trial showed that FTY720 reduced annual relapse rates by 62%, compared with treatment-naive patients. For patients that had received prior treatments, the annual relapse rate was reduced by 44%. At two years, FTY720 delayed disability progression by 30% for patients treated with 0.5 mg, compared with placebo. The serious infection rate was comparable in the different “immune modulator” treatment groups.
Product Removal
Inhalers containing ozone-depleting chlorofluorocarbons (CFCs) are continuing to be phased out.16 These agents are used to treat asthma and COPD, and alternate products that do not contain CFCs are available. Some pharmacies might be depleting stock after the “last-sale date.” The affected products and their phase-out dates are:
- Tilade (nedocromil): June 14, 2010;
- Alupent (metaproterenol): June 14, 2010;
- Aerobid (flunisolide): June 30, 2010;
- Azmacort (triamcinolone): Dec. 31, 2010;
- Intal (cromolyn): Dec. 31, 2010;
- Combivent (albuterol/ipratropium): December 31, 2013; and
- Maxair (pirbuterol) autohaler: December 31, 2013. TH
Michele B. Kaufman, PharmD, BSc, RPh, is a freelance medical writer based in New York City and a clinical pharmacist at New York Downtown Hospital.
References
- Orange Book: Approved drug products with therapeutic equivalence evaluations. U.S. Food and Drug Administration website. Available at: www.accessdata.fda.gov/scripts/cder/ob/docs/obdetail.cfm?Appl_No=078357&TABLE1=OB_Rx. Accessed April 27, 2010.
- Mylan announces approval under PEPFAR for generic version of Videx EC HIV treatment. Medical News Today website. Available at: www.medicalnewstoday.com/articles/186273.php. Accessed April 27, 2010.
- Sirion Therapeutics announces availability of Zirgan (ganciclovir ophthalmic gel) 0.15% for ocular herpes. PR Newswire website. Available at: www.prnewswire.com/news-releases/sirion-therapeutics-announces-availability-of-zirgantm-ganciclovir-ophthalmic-gel-015-for-ocular-herpes-92084614.html. Accessed April 27, 2010.
- CSL Behring receives FDA approval to extend shelf life for Privigen from 24 to 36 months. CSL Behring website. Available at: www.cslbehring-us.com/s1/cs/enus/1154272074489/news/1255923905944/prdetail.htm. Accessed April 27, 2010.
- FDA approves Oravig (miconazole) buccal tablets for treatment of oropharyngeal candidiasis. PAR Pharmaceuticals website. Available at: investors.parpharm.com/phoenix.zhtml?c=81806&p=irol-newsArticle&ID=1413993&highlight=. Accessed April 27, 2010.
- Alkermes submits supplemental new drug application for Vivitrol for the treatment of opioid dependence. Medical News Today website. Available at: www.medicalnewstoday.com/articles/185456.php. Accessed April 27, 2010.
- FDA approves reformulated oxycontin. Contract Pharma website. www.contractpharma.com/news/2010/04/07/fda_approves_reformulated_oxycontin. Accessed April 27, 2010.
- Gansz Bobo E. FDA approves pancreatic enzyme product, Pancreaze. FDA website. Available at: www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm208135.htm. Accessed April 27, 2010.
- Once-daily Mirapex ER now approved by FDA for both early and advanced Parkinson’s disease. Medical News Today website. Available at: www.medicalnewstoday.com/printerfriendlynews.php?newsid=183272. Accessed April 27, 2010.
- DiaPrevent diabetes research. DiaPrevent website. Available at: www.diaprevent.diamyd.com/. Accessed April 27, 2010.
- Diamyd granted orphan drug designation in the US. Diamyd website. Available at: www.diamyd.com/docs/pressClip.aspx?section=investor&ClipID=479460. Accessed April 27, 2010.
- Same-day genetic testing service available for safer warfarin dosing. Monthly Prescribing Reference website. Available at: www.empr.com/same-day-genetic-testing-service-available-for-safer-warfarin-dosing/article/167586/. Accessed April 27, 2010.
- NDA submitted for DM-1796 for postherpetic neuralgia (PHN). Monthly Prescribing Reference website. Available at: www.empr.com/nda-submitted-for-dm-1796-for-postherpetic-neuralgia-phn/article/167056/. Accessed April 26, 2010.
- Gilead initiates Phase III clinical program evaluating single-table, once-daily “quad” regimen for HIV. Gilead website. Available at: www.gilead.com/pr_1411934. Accessed April 27, 2010.
- Novartis investigational multiple sclerosis therapy Gilenia (FTY720) shown to reduce relapse rates regardless of treatment history. Drugs.com website. Available at: www.drugs.com/clinical_trials/novartis-investigational-multiple-sclerosis-therapy-gilenia-fty720-shown-reduce-relapse-rates-9139.html. Accessed April 27, 2010.
- Inhalers containing CFCs being eliminated. Pharamacist eLink website. Available at: www.pharmacistelink.com/index.php/Drugs-and-Treatment/Inhalers-containing-CFC-s-being-eliminated.html. Accessed April 27, 2010.
New Generics
- Desloratadine tablets (generic Clarinex)1
- Didanosine delayed-release capsules (Generic Videx EC)2
New Indications, Dosage Forms, and Recommendations
- Ganciclovir ophthalmic gel 0.15% (Zirgan) has been approved by the FDA for treating acute herpetic keratitis.3 The recommended dose is one drop in the affected eye five times daily until the ulcer heals, then one drop three times daily for seven more days. The most common side effects in clinical trials were blurred vision, eye irritation, punctate keratitis, and conjunctival hyperemia. It will be available in a 5-g tube.
- Immune globulin intravenous 10% liquid (human) (Privigen) has received an updated approval from the FDA, which allows for room temperature storage throughout its entire 36-month shelf life.4 The agent is used to treat patients with primary immunodeficiency disorders.
- Miconazole buccal tablets (Oravig) have been approved by the FDA for treating oropharyngeal candidiasis in adults and children 16 years of age and older. It is the first, and currently the only local, buccal prescription formulation of miconazole.5 The buccal tablet was developed to adhere to the gum. It should not be crushed, chewed, or swallowed. The most common adverse effects in clinical trials were diarrhea, nausea, headache, dysgeusia, upper abdominal pain, and vomiting. It is recommended to monitor patients with a history of hypersensitivity to azoles, as there is no information regarding cross-reactivity between miconazole and other azole agents.
- A supplemental new drug application (sNDA) has been submitted to the FDA for naltrexone extended-release injectable suspension (Vivitrol) for treating opioid dependence.6 It is administered as a once-monthly intramuscular injection and currently is approved by the FDA for treating alcohol dependence.
- Oxycodone controlled-release (OxyContin) has been approved by the FDA in a new, abuse-deterrent formulation.7
- Pancrelipase delayed-release capsules (Pancreaze) joins Creon (Abbott Labs) and Zenpep (Eurand) as the third pancreatic enzyme product (PEP) to be approved by the FDA for treating exocrine pancreatic insufficiency.8
- Pramipexole extended-release tablets (Mirapex ER) have been approved by the FDA as a once-daily treatment for the signs and symptoms of idiopathic Parkinson’s disease (early and late).9
- The active ingredient in the vaccine Diamyd, rhGAD65, has received orphan drug status for treating Type 1 diabetes mellitus (T1DM) with residual beta cell function.10,11 This agent is in Phase 3 clinical trials and is being investigated to determine whether it can stop or slow the autoimmune destruction of insulin-producing beta cell function. The DiaPrevent study is enrolling patients. In Phase 2 studies, the agent preserved remaining beta cell function in adolescents and children recently diagnosed with T1DM.
- Warfarin genetic diagnostic: Machaon Diagnostics has received FDA approval for an array-based diagnostic technology that detects genetic variation and could aid in determining an accurate initial warfarin dose.12 At least 40% of Americans have at least one genetic variation involved in warfarin metabolism, which can cause a more than fivefold disparity in the weekly warfarin dose. This test can be used to more accurately determine dosing for warfarin-treated patients.
Pipeline
- The NDA for DM-1796 (gabapentin extended-release tablet) has been submitted to the FDA for treatment of postherpetic neuralgia.13 It is a once-daily, extended-release formulation of gabapentin.
- The “quad” combination of elvitegravir, cobicistat (formerly GS 9350), emtricitabine, and tenofovir disoproxil fumarate in a fixed-dose single tablet is currently in Phase 3 clinical trials for treatment of HIV.14
- FTY720 is an investigational oral immune modulator agent for treating relapsing-remitting multiple sclerosis (RR-MS).15 The NDA for this agent was submitted in December 2009; the FDA granted it a priority review in February. Two-year data from the FREEDOMS trial showed that FTY720 reduced annual relapse rates by 62%, compared with treatment-naive patients. For patients that had received prior treatments, the annual relapse rate was reduced by 44%. At two years, FTY720 delayed disability progression by 30% for patients treated with 0.5 mg, compared with placebo. The serious infection rate was comparable in the different “immune modulator” treatment groups.
Product Removal
Inhalers containing ozone-depleting chlorofluorocarbons (CFCs) are continuing to be phased out.16 These agents are used to treat asthma and COPD, and alternate products that do not contain CFCs are available. Some pharmacies might be depleting stock after the “last-sale date.” The affected products and their phase-out dates are:
- Tilade (nedocromil): June 14, 2010;
- Alupent (metaproterenol): June 14, 2010;
- Aerobid (flunisolide): June 30, 2010;
- Azmacort (triamcinolone): Dec. 31, 2010;
- Intal (cromolyn): Dec. 31, 2010;
- Combivent (albuterol/ipratropium): December 31, 2013; and
- Maxair (pirbuterol) autohaler: December 31, 2013. TH
Michele B. Kaufman, PharmD, BSc, RPh, is a freelance medical writer based in New York City and a clinical pharmacist at New York Downtown Hospital.
References
- Orange Book: Approved drug products with therapeutic equivalence evaluations. U.S. Food and Drug Administration website. Available at: www.accessdata.fda.gov/scripts/cder/ob/docs/obdetail.cfm?Appl_No=078357&TABLE1=OB_Rx. Accessed April 27, 2010.
- Mylan announces approval under PEPFAR for generic version of Videx EC HIV treatment. Medical News Today website. Available at: www.medicalnewstoday.com/articles/186273.php. Accessed April 27, 2010.
- Sirion Therapeutics announces availability of Zirgan (ganciclovir ophthalmic gel) 0.15% for ocular herpes. PR Newswire website. Available at: www.prnewswire.com/news-releases/sirion-therapeutics-announces-availability-of-zirgantm-ganciclovir-ophthalmic-gel-015-for-ocular-herpes-92084614.html. Accessed April 27, 2010.
- CSL Behring receives FDA approval to extend shelf life for Privigen from 24 to 36 months. CSL Behring website. Available at: www.cslbehring-us.com/s1/cs/enus/1154272074489/news/1255923905944/prdetail.htm. Accessed April 27, 2010.
- FDA approves Oravig (miconazole) buccal tablets for treatment of oropharyngeal candidiasis. PAR Pharmaceuticals website. Available at: investors.parpharm.com/phoenix.zhtml?c=81806&p=irol-newsArticle&ID=1413993&highlight=. Accessed April 27, 2010.
- Alkermes submits supplemental new drug application for Vivitrol for the treatment of opioid dependence. Medical News Today website. Available at: www.medicalnewstoday.com/articles/185456.php. Accessed April 27, 2010.
- FDA approves reformulated oxycontin. Contract Pharma website. www.contractpharma.com/news/2010/04/07/fda_approves_reformulated_oxycontin. Accessed April 27, 2010.
- Gansz Bobo E. FDA approves pancreatic enzyme product, Pancreaze. FDA website. Available at: www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm208135.htm. Accessed April 27, 2010.
- Once-daily Mirapex ER now approved by FDA for both early and advanced Parkinson’s disease. Medical News Today website. Available at: www.medicalnewstoday.com/printerfriendlynews.php?newsid=183272. Accessed April 27, 2010.
- DiaPrevent diabetes research. DiaPrevent website. Available at: www.diaprevent.diamyd.com/. Accessed April 27, 2010.
- Diamyd granted orphan drug designation in the US. Diamyd website. Available at: www.diamyd.com/docs/pressClip.aspx?section=investor&ClipID=479460. Accessed April 27, 2010.
- Same-day genetic testing service available for safer warfarin dosing. Monthly Prescribing Reference website. Available at: www.empr.com/same-day-genetic-testing-service-available-for-safer-warfarin-dosing/article/167586/. Accessed April 27, 2010.
- NDA submitted for DM-1796 for postherpetic neuralgia (PHN). Monthly Prescribing Reference website. Available at: www.empr.com/nda-submitted-for-dm-1796-for-postherpetic-neuralgia-phn/article/167056/. Accessed April 26, 2010.
- Gilead initiates Phase III clinical program evaluating single-table, once-daily “quad” regimen for HIV. Gilead website. Available at: www.gilead.com/pr_1411934. Accessed April 27, 2010.
- Novartis investigational multiple sclerosis therapy Gilenia (FTY720) shown to reduce relapse rates regardless of treatment history. Drugs.com website. Available at: www.drugs.com/clinical_trials/novartis-investigational-multiple-sclerosis-therapy-gilenia-fty720-shown-reduce-relapse-rates-9139.html. Accessed April 27, 2010.
- Inhalers containing CFCs being eliminated. Pharamacist eLink website. Available at: www.pharmacistelink.com/index.php/Drugs-and-Treatment/Inhalers-containing-CFC-s-being-eliminated.html. Accessed April 27, 2010.
New Generics
- Desloratadine tablets (generic Clarinex)1
- Didanosine delayed-release capsules (Generic Videx EC)2
New Indications, Dosage Forms, and Recommendations
- Ganciclovir ophthalmic gel 0.15% (Zirgan) has been approved by the FDA for treating acute herpetic keratitis.3 The recommended dose is one drop in the affected eye five times daily until the ulcer heals, then one drop three times daily for seven more days. The most common side effects in clinical trials were blurred vision, eye irritation, punctate keratitis, and conjunctival hyperemia. It will be available in a 5-g tube.
- Immune globulin intravenous 10% liquid (human) (Privigen) has received an updated approval from the FDA, which allows for room temperature storage throughout its entire 36-month shelf life.4 The agent is used to treat patients with primary immunodeficiency disorders.
- Miconazole buccal tablets (Oravig) have been approved by the FDA for treating oropharyngeal candidiasis in adults and children 16 years of age and older. It is the first, and currently the only local, buccal prescription formulation of miconazole.5 The buccal tablet was developed to adhere to the gum. It should not be crushed, chewed, or swallowed. The most common adverse effects in clinical trials were diarrhea, nausea, headache, dysgeusia, upper abdominal pain, and vomiting. It is recommended to monitor patients with a history of hypersensitivity to azoles, as there is no information regarding cross-reactivity between miconazole and other azole agents.
- A supplemental new drug application (sNDA) has been submitted to the FDA for naltrexone extended-release injectable suspension (Vivitrol) for treating opioid dependence.6 It is administered as a once-monthly intramuscular injection and currently is approved by the FDA for treating alcohol dependence.
- Oxycodone controlled-release (OxyContin) has been approved by the FDA in a new, abuse-deterrent formulation.7
- Pancrelipase delayed-release capsules (Pancreaze) joins Creon (Abbott Labs) and Zenpep (Eurand) as the third pancreatic enzyme product (PEP) to be approved by the FDA for treating exocrine pancreatic insufficiency.8
- Pramipexole extended-release tablets (Mirapex ER) have been approved by the FDA as a once-daily treatment for the signs and symptoms of idiopathic Parkinson’s disease (early and late).9
- The active ingredient in the vaccine Diamyd, rhGAD65, has received orphan drug status for treating Type 1 diabetes mellitus (T1DM) with residual beta cell function.10,11 This agent is in Phase 3 clinical trials and is being investigated to determine whether it can stop or slow the autoimmune destruction of insulin-producing beta cell function. The DiaPrevent study is enrolling patients. In Phase 2 studies, the agent preserved remaining beta cell function in adolescents and children recently diagnosed with T1DM.
- Warfarin genetic diagnostic: Machaon Diagnostics has received FDA approval for an array-based diagnostic technology that detects genetic variation and could aid in determining an accurate initial warfarin dose.12 At least 40% of Americans have at least one genetic variation involved in warfarin metabolism, which can cause a more than fivefold disparity in the weekly warfarin dose. This test can be used to more accurately determine dosing for warfarin-treated patients.
Pipeline
- The NDA for DM-1796 (gabapentin extended-release tablet) has been submitted to the FDA for treatment of postherpetic neuralgia.13 It is a once-daily, extended-release formulation of gabapentin.
- The “quad” combination of elvitegravir, cobicistat (formerly GS 9350), emtricitabine, and tenofovir disoproxil fumarate in a fixed-dose single tablet is currently in Phase 3 clinical trials for treatment of HIV.14
- FTY720 is an investigational oral immune modulator agent for treating relapsing-remitting multiple sclerosis (RR-MS).15 The NDA for this agent was submitted in December 2009; the FDA granted it a priority review in February. Two-year data from the FREEDOMS trial showed that FTY720 reduced annual relapse rates by 62%, compared with treatment-naive patients. For patients that had received prior treatments, the annual relapse rate was reduced by 44%. At two years, FTY720 delayed disability progression by 30% for patients treated with 0.5 mg, compared with placebo. The serious infection rate was comparable in the different “immune modulator” treatment groups.
Product Removal
Inhalers containing ozone-depleting chlorofluorocarbons (CFCs) are continuing to be phased out.16 These agents are used to treat asthma and COPD, and alternate products that do not contain CFCs are available. Some pharmacies might be depleting stock after the “last-sale date.” The affected products and their phase-out dates are:
- Tilade (nedocromil): June 14, 2010;
- Alupent (metaproterenol): June 14, 2010;
- Aerobid (flunisolide): June 30, 2010;
- Azmacort (triamcinolone): Dec. 31, 2010;
- Intal (cromolyn): Dec. 31, 2010;
- Combivent (albuterol/ipratropium): December 31, 2013; and
- Maxair (pirbuterol) autohaler: December 31, 2013. TH
Michele B. Kaufman, PharmD, BSc, RPh, is a freelance medical writer based in New York City and a clinical pharmacist at New York Downtown Hospital.
References
- Orange Book: Approved drug products with therapeutic equivalence evaluations. U.S. Food and Drug Administration website. Available at: www.accessdata.fda.gov/scripts/cder/ob/docs/obdetail.cfm?Appl_No=078357&TABLE1=OB_Rx. Accessed April 27, 2010.
- Mylan announces approval under PEPFAR for generic version of Videx EC HIV treatment. Medical News Today website. Available at: www.medicalnewstoday.com/articles/186273.php. Accessed April 27, 2010.
- Sirion Therapeutics announces availability of Zirgan (ganciclovir ophthalmic gel) 0.15% for ocular herpes. PR Newswire website. Available at: www.prnewswire.com/news-releases/sirion-therapeutics-announces-availability-of-zirgantm-ganciclovir-ophthalmic-gel-015-for-ocular-herpes-92084614.html. Accessed April 27, 2010.
- CSL Behring receives FDA approval to extend shelf life for Privigen from 24 to 36 months. CSL Behring website. Available at: www.cslbehring-us.com/s1/cs/enus/1154272074489/news/1255923905944/prdetail.htm. Accessed April 27, 2010.
- FDA approves Oravig (miconazole) buccal tablets for treatment of oropharyngeal candidiasis. PAR Pharmaceuticals website. Available at: investors.parpharm.com/phoenix.zhtml?c=81806&p=irol-newsArticle&ID=1413993&highlight=. Accessed April 27, 2010.
- Alkermes submits supplemental new drug application for Vivitrol for the treatment of opioid dependence. Medical News Today website. Available at: www.medicalnewstoday.com/articles/185456.php. Accessed April 27, 2010.
- FDA approves reformulated oxycontin. Contract Pharma website. www.contractpharma.com/news/2010/04/07/fda_approves_reformulated_oxycontin. Accessed April 27, 2010.
- Gansz Bobo E. FDA approves pancreatic enzyme product, Pancreaze. FDA website. Available at: www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm208135.htm. Accessed April 27, 2010.
- Once-daily Mirapex ER now approved by FDA for both early and advanced Parkinson’s disease. Medical News Today website. Available at: www.medicalnewstoday.com/printerfriendlynews.php?newsid=183272. Accessed April 27, 2010.
- DiaPrevent diabetes research. DiaPrevent website. Available at: www.diaprevent.diamyd.com/. Accessed April 27, 2010.
- Diamyd granted orphan drug designation in the US. Diamyd website. Available at: www.diamyd.com/docs/pressClip.aspx?section=investor&ClipID=479460. Accessed April 27, 2010.
- Same-day genetic testing service available for safer warfarin dosing. Monthly Prescribing Reference website. Available at: www.empr.com/same-day-genetic-testing-service-available-for-safer-warfarin-dosing/article/167586/. Accessed April 27, 2010.
- NDA submitted for DM-1796 for postherpetic neuralgia (PHN). Monthly Prescribing Reference website. Available at: www.empr.com/nda-submitted-for-dm-1796-for-postherpetic-neuralgia-phn/article/167056/. Accessed April 26, 2010.
- Gilead initiates Phase III clinical program evaluating single-table, once-daily “quad” regimen for HIV. Gilead website. Available at: www.gilead.com/pr_1411934. Accessed April 27, 2010.
- Novartis investigational multiple sclerosis therapy Gilenia (FTY720) shown to reduce relapse rates regardless of treatment history. Drugs.com website. Available at: www.drugs.com/clinical_trials/novartis-investigational-multiple-sclerosis-therapy-gilenia-fty720-shown-reduce-relapse-rates-9139.html. Accessed April 27, 2010.
- Inhalers containing CFCs being eliminated. Pharamacist eLink website. Available at: www.pharmacistelink.com/index.php/Drugs-and-Treatment/Inhalers-containing-CFC-s-being-eliminated.html. Accessed April 27, 2010.
What Are the Chances a Hospitalized Patient Will Survive In-Hospital Arrest?
Case
A 66-year-old woman with metastatic, non-small-cell carcinoma of the lung, chronic obstructive pulmonary disease (COPD), and hypertension presents with progressive shortness of breath and back pain. Her vital signs are normal, with the exception of tachypnea and an oxygen saturation of 84% on room air. A CT scan shows marked progression of her disease and new metastases to her spine. You begin a discussion about advance directives and code status. During the exchange, the patient asks for guidance regarding resuscitation. How can you best answer her questions about the likelihood of surviving an in-hospital arrest?
Background
Discussion regarding resuscitation status is a challenge for most hospitalists. The absence of an established relationship, limited time, patient emotion, and difficulty applying general scientific data to a single patient coalesce into a complex interaction. Further complicating matters, patients frequently have unrealistic expectations and overestimate their chance of survival.
Experience has shown that many patients pursue what physicians consider inappropriately aggressive resuscitation measures. Before you have an informed discussion about cardiopulmonary resuscitation (CPR) outcomes, patients tend to overestimate their likelihood of survival.1 In 2009, Kaldjian and colleagues found that patients’ initial mean prediction of post-arrest survival was 60.4%, compared with the actual mean of approximately 17%.2,3 Furthermore, nearly half of the patients who initially expressed a desire to receive CPR in the event of cardiac arrest opted to change their code status after they were informed of the actual survival estimates.1,2
Patient autonomy and the law, as defined by the 1990 Patient Self-Determination Act, require that physicians share responsibility with patients in making prospective resuscitation decisions.4 Shared decision-making necessitates a basic discussion on admission within the context of the patient’s prognosis and previously expressed wishes. It might simply include an acknowledgment of a previously completed advance directive. A more complex discussion might require in-depth conversation to address patient performance status, prognosis of acute and chronic illnesses, and education about the typical resuscitation procedures. After listening to the patient’s perspective, the admitting physician can provide input and an interpretation of available data regarding the patient’s likelihood of surviving an in-hospital arrest.
Review of the Data
In the past 40 years, the overall survival rates for cardiac arrest have changed little. Despite numerous advances made in the delivery of medical care, on average, only 17% of all adult arrest patients survive to hospital discharge.3 A variety of factors influence this overall survival rate, both pre-arrest and intra-arrest. Clinical experience allows most physicians to sense what probability a patient has for survival and quality of life following a cardiac arrest. However, anecdotal evidence alone does not provide a patient and their family with the information necessary to make an informed decision regarding code status.
Numerous studies have investigated the patient factors that might influence how likely one is to survive a cardiac arrest. Researchers have paid particular attention to such factors as age, race, presence or absence of a cancer diagnosis, and associated comorbidities. Not surprisingly, older age has been shown to be significantly associated with a lower likelihood of survival to discharge following cardiac arrest.5,6
Ehlenbach and colleagues examined medical data from 433,985 Medicare patients 65 and older who underwent in-hospital CPR.5 Both older age and prior residence in a skilled nursing facility were found to be associated with poorer survival rates.5 Although neither study was able to define an upper-age cutoff for certain peri-arrest mortality, age affects overall survival likelihood in an inverse fashion, with those 85 and older having only a 6% chance of surviving to hospital discharge (see Figure 1, p. 18).1,5,6
The degree of comorbid illness can be used to help predict mortality following cardiac arrest. Review of data from the National Registry of Cardiopulmonary Resuscitation (NRCPR) identified particular comorbidities that portend poor post-arrest prognosis.6 In general, the more pre-existing comorbidities a patient has, the less likely they are to survive.6 The presence of hepatic insufficiency, acute stroke, immunodeficiency, renal failure, or dialysis were associated with lower survival rates (see Figure 2, right).6,7
Poor performance status on admission, defined as severe disability, coma, or vegetative state, was predictive of worse outcomes.6 Understandably, patients with hypotension and those who required vasopressors or mechanical ventilation also tended to have lower post-arrest survival rates.6
The presence of a cancer diagnosis is another prognostic factor of interest when considering the chances of surviving an arrest. Classically, CPR was thought to be a futile intervention in this patient population. Specific characteristics within this subset of patients have been shown to influence prognosis, and multiple studies have confirmed that cancer patients generally do worse after an arrest with an overall survival rate of only 6.2%.8 Survival rates tend to be lower in patients with metastatic disease, hematologic malignancies, a history of stem cell transplant, those who arrest within an ICU, and inpatients whose cardiac arrest was anticipated.8,9
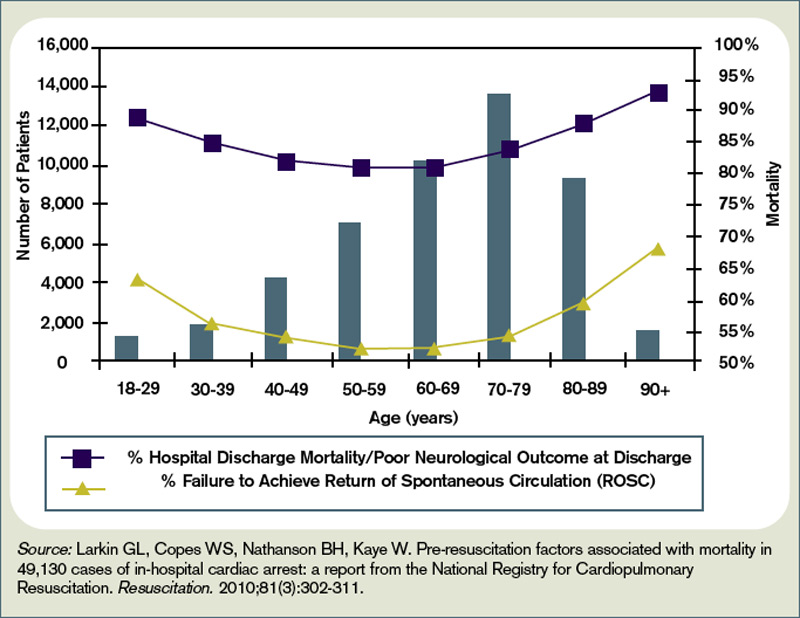
In fact, cancer patients whose hospital course followed a path of gradual deterioration showed a 0% survival rate.9 In patients with metastatic disease, poor performance status prior to arrest appeared to account for their particularly poor survival odds (this supports the intuitive, rule-of-thumb that sicker cancer patients have worse outcomes).8
Growing evidence suggests the probability of post-arrest survival is not equal between racial groups. Specifically, black or nonwhite race is associated with higher utilization of CPR and lower survival rates (see Figure 3, right).10 Among Medicare patients, Ehlenbach and colleagues found that black and nonwhite patients were much more likely to undergo CPR, presumably as a result of being less likely to opt for DNR status.5,10 Although this could account for the differences seen in survival rates among these populations, these findings also raise concerns about the possibility of racial disparities in medical care. A subsequent cohort study also suggested that blacks and nonwhites were less likely to survive following cardiac arrest.10 However, adjusted analysis revealed that these differences were strongly associated with the medical center at which these patients received care. Therefore, although being nonwhite does portend worse outcomes following an arrest, the increased risk is likely attributed to the fact that many of these patients receive care at hospitals that have poorer overall CPR performance measures.5,10
Survival is not the only outcome measure patients need to take into account when deciding whether to undergo CPR. Quality of life following resuscitation also warrants consideration. Interestingly, research has shown that neurologic outcomes among the majority of cardiac arrest survivors are generally good.3
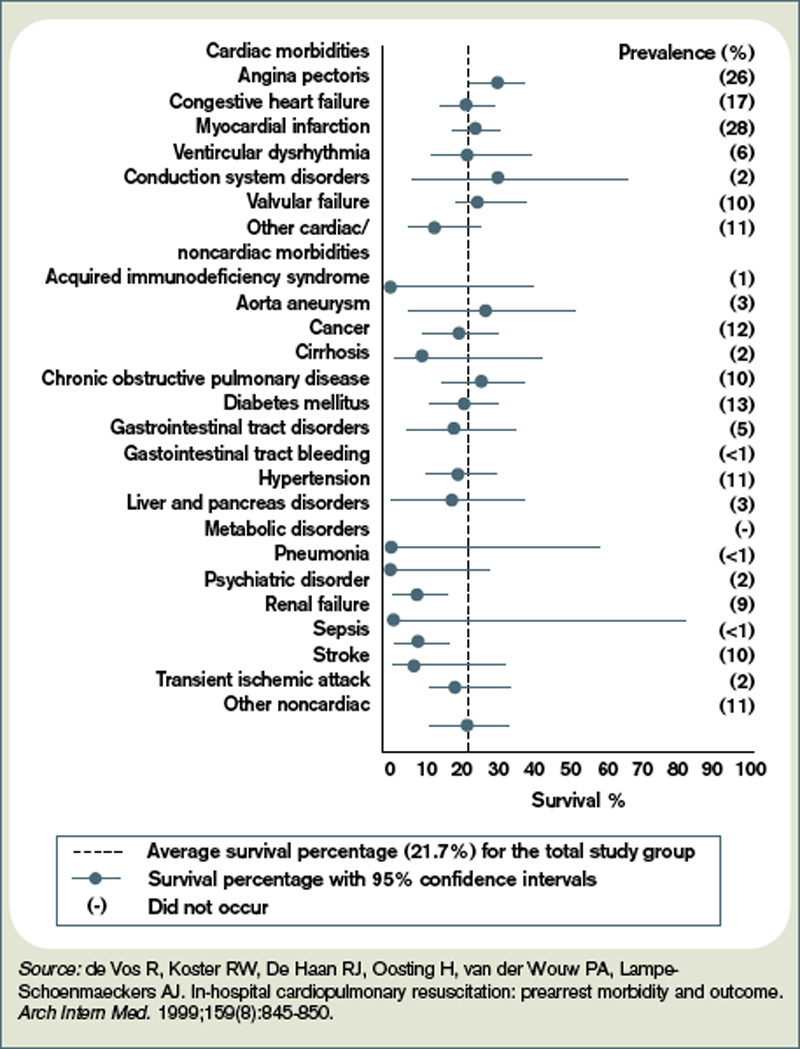
Approximately 86% of survivors with intact pre-arrest cerebral performance maintain it on discharge, and only a minority of survivors are eventually declared brain-dead.3 Still, there are certain peri-arrest factors that pose risk for poorer neurologic and functional outcomes. For arrest from a shockable rhythm, time to defibrillation is a key determinant.11 In patients for whom time to defibrillation is greater than two minutes, there is significantly higher risk of permanent disability following cardiac arrest.11
In the event of coma following resuscitation, particular clinical findings can be used to accurately predict poor outcome.12 The absence of pupillary reflexes, corneal reflexes, or absent or extensor motor responses three days after arrest are poor prognostic indicators.12 As a general rule, if a patient does not awaken within three days, neurologic and functional impairment can be expected.12 For those patients who do survive to hospital discharge, more than 50% ultimately will be able to be discharged home.3
However, nearly a quarter will need to be newly placed in a rehabilitation or skilled nursing facility.3
Back to the Case
The patient was admitted with hypoxia secondary to both progressive lung malignancy and COPD exacerbation. She had no advanced directives, so the admitting hospitalist, in collaboration with her oncologist, had a detailed discussion regarding her understanding of her disease progression, prognosis, and goals for her remaining time. Her questions regarding survivability of cardiac arrest were answered directly with an estimate of 5% to 10%, based on her age, comorbidities, and the presence of advanced malignancy.
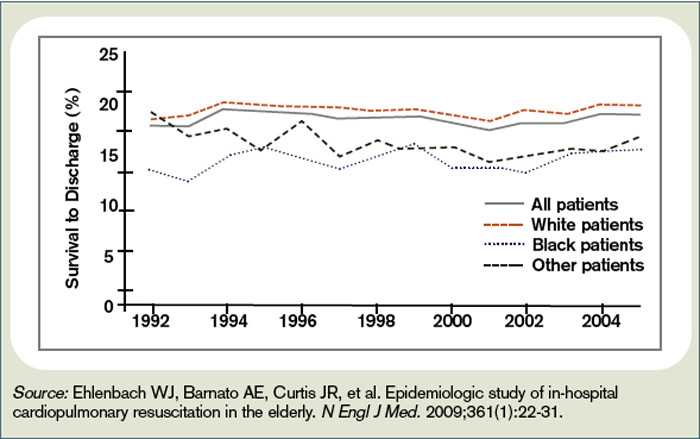
After hearing this information, the patient responded, “I still want everything done.” The hospitalist acknowledged her feelings of wanting to fight on, but asked her to think about what “everything” meant to her. After taking some additional time to reflect with friends and family, the patient was clear that she wanted to continue disease-focused therapies, but did not want to be resuscitated in the event of cardiac or pulmonary arrest.
Eventually, her hypoxia improved with antibiotics, steroids, and bronchodilators. She was discharged home with follow-up in the oncology clinic for additional chemotherapy and palliative radiation.
Bottom Line
For hospitalized adults, the average survival rate to discharge after cardiac arrest is about 17%. Many factors lower a patient’s chance of survival, including advanced age, performance status, malignancy, and presence of multiple comorbidities. TH
Dr. Neagle and Dr. Wachsberg are hospitalists and instructors in the division of hospital medicine at Northwestern University Medical Center in Chicago.
References
- Murphy DJ, Burrows D, Santali S, et al. The influence of the probability of survival on patients’ preferences regarding cardiopulmonary resuscitation. N Engl J Med. 1994;330(8):545-549.
- Kaldjian LC, Erekson ZD, Haberle TH, et al. Code status discussions and goals of care among hospitalised adults. J Med Ethics. 2009;35(6):338-342.
- Peberdy MA, Kaye W, Ornato JP, et al. Cardiopulmonary resuscitation of adults in the hospital: a report of 14720 cardiac arrests from the National Registry of Cardiopulmonary Resuscitation. Resuscitation. 2003;58(3):297-308.
- La Puma J, Orentlicher D, Moss RJ. Advance directives on admission. Clinical implications and analysis of the Patient Self-Determination Act of 1990. JAMA. 1991;266(3):402-405.
- Ehlenbach WJ, Barnato AE, Curtis JR, et al. Epidemiologic study of in-hospital cardiopulmonary resuscitation in the elderly. N Engl J Med. 2009;361(1):22-31.
- Larkin GL, Copes WS, Nathanson BH, Kaye W. Pre-resuscitation factors associated with mortality in 49,130 cases of in-hospital cardiac arrest: a report from the National Registry for Cardiopulmonary Resuscitation. Resuscitation. 2010;81(3):302-311.
- de Vos R, Koster RW, De Haan RJ, Oosting H, van der Wouw PA, Lampe-Schoenmaeckers AJ. In-hospital cardiopulmonary resuscitation: prearrest morbidity and outcome. Arch Intern Med. 1999;159(8):845-850.
- Reisfield GM, Wallace SK, Munsell MF, Webb FJ, Alvarez ER, Wilson GR. Survival in cancer patients undergoing in-hospital cardiopulmonary resuscitation: a meta-analysis. Resuscitation. 2006;71(2):152-160.
- Ewer MS, Kish SK, Martin CG, Price KJ, Feeley TW. Characteristics of cardiac arrest in cancer patients as a predictor of survival after cardiopulmonary resuscitation. Cancer. 2001;92(7):1905-1912.
- Chan PS, Nichol G, Krumholz HM, et al. Racial differences in survival after in-hospital cardiac arrest. JAMA. 2009;302(11):1195-1201.
- Chan PS, Krumholz HM, Nichol G, Nallamothu BK. Delayed time to defibrillation after in-hospital cardiac arrest. N Engl J Med. 2008;358(1):9-17.
- Wijdicks EF, Hijdra A, Young GB, Bassetti CL, Wiebe S. Practice parameter: prediction of outcome in comatose survivors after cardiopulmonary resuscitation (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2006;67(2):203-210.
Case
A 66-year-old woman with metastatic, non-small-cell carcinoma of the lung, chronic obstructive pulmonary disease (COPD), and hypertension presents with progressive shortness of breath and back pain. Her vital signs are normal, with the exception of tachypnea and an oxygen saturation of 84% on room air. A CT scan shows marked progression of her disease and new metastases to her spine. You begin a discussion about advance directives and code status. During the exchange, the patient asks for guidance regarding resuscitation. How can you best answer her questions about the likelihood of surviving an in-hospital arrest?
Background
Discussion regarding resuscitation status is a challenge for most hospitalists. The absence of an established relationship, limited time, patient emotion, and difficulty applying general scientific data to a single patient coalesce into a complex interaction. Further complicating matters, patients frequently have unrealistic expectations and overestimate their chance of survival.
Experience has shown that many patients pursue what physicians consider inappropriately aggressive resuscitation measures. Before you have an informed discussion about cardiopulmonary resuscitation (CPR) outcomes, patients tend to overestimate their likelihood of survival.1 In 2009, Kaldjian and colleagues found that patients’ initial mean prediction of post-arrest survival was 60.4%, compared with the actual mean of approximately 17%.2,3 Furthermore, nearly half of the patients who initially expressed a desire to receive CPR in the event of cardiac arrest opted to change their code status after they were informed of the actual survival estimates.1,2
Patient autonomy and the law, as defined by the 1990 Patient Self-Determination Act, require that physicians share responsibility with patients in making prospective resuscitation decisions.4 Shared decision-making necessitates a basic discussion on admission within the context of the patient’s prognosis and previously expressed wishes. It might simply include an acknowledgment of a previously completed advance directive. A more complex discussion might require in-depth conversation to address patient performance status, prognosis of acute and chronic illnesses, and education about the typical resuscitation procedures. After listening to the patient’s perspective, the admitting physician can provide input and an interpretation of available data regarding the patient’s likelihood of surviving an in-hospital arrest.
Review of the Data
In the past 40 years, the overall survival rates for cardiac arrest have changed little. Despite numerous advances made in the delivery of medical care, on average, only 17% of all adult arrest patients survive to hospital discharge.3 A variety of factors influence this overall survival rate, both pre-arrest and intra-arrest. Clinical experience allows most physicians to sense what probability a patient has for survival and quality of life following a cardiac arrest. However, anecdotal evidence alone does not provide a patient and their family with the information necessary to make an informed decision regarding code status.
Numerous studies have investigated the patient factors that might influence how likely one is to survive a cardiac arrest. Researchers have paid particular attention to such factors as age, race, presence or absence of a cancer diagnosis, and associated comorbidities. Not surprisingly, older age has been shown to be significantly associated with a lower likelihood of survival to discharge following cardiac arrest.5,6
Ehlenbach and colleagues examined medical data from 433,985 Medicare patients 65 and older who underwent in-hospital CPR.5 Both older age and prior residence in a skilled nursing facility were found to be associated with poorer survival rates.5 Although neither study was able to define an upper-age cutoff for certain peri-arrest mortality, age affects overall survival likelihood in an inverse fashion, with those 85 and older having only a 6% chance of surviving to hospital discharge (see Figure 1, p. 18).1,5,6
The degree of comorbid illness can be used to help predict mortality following cardiac arrest. Review of data from the National Registry of Cardiopulmonary Resuscitation (NRCPR) identified particular comorbidities that portend poor post-arrest prognosis.6 In general, the more pre-existing comorbidities a patient has, the less likely they are to survive.6 The presence of hepatic insufficiency, acute stroke, immunodeficiency, renal failure, or dialysis were associated with lower survival rates (see Figure 2, right).6,7
Poor performance status on admission, defined as severe disability, coma, or vegetative state, was predictive of worse outcomes.6 Understandably, patients with hypotension and those who required vasopressors or mechanical ventilation also tended to have lower post-arrest survival rates.6
The presence of a cancer diagnosis is another prognostic factor of interest when considering the chances of surviving an arrest. Classically, CPR was thought to be a futile intervention in this patient population. Specific characteristics within this subset of patients have been shown to influence prognosis, and multiple studies have confirmed that cancer patients generally do worse after an arrest with an overall survival rate of only 6.2%.8 Survival rates tend to be lower in patients with metastatic disease, hematologic malignancies, a history of stem cell transplant, those who arrest within an ICU, and inpatients whose cardiac arrest was anticipated.8,9
In fact, cancer patients whose hospital course followed a path of gradual deterioration showed a 0% survival rate.9 In patients with metastatic disease, poor performance status prior to arrest appeared to account for their particularly poor survival odds (this supports the intuitive, rule-of-thumb that sicker cancer patients have worse outcomes).8
Growing evidence suggests the probability of post-arrest survival is not equal between racial groups. Specifically, black or nonwhite race is associated with higher utilization of CPR and lower survival rates (see Figure 3, right).10 Among Medicare patients, Ehlenbach and colleagues found that black and nonwhite patients were much more likely to undergo CPR, presumably as a result of being less likely to opt for DNR status.5,10 Although this could account for the differences seen in survival rates among these populations, these findings also raise concerns about the possibility of racial disparities in medical care. A subsequent cohort study also suggested that blacks and nonwhites were less likely to survive following cardiac arrest.10 However, adjusted analysis revealed that these differences were strongly associated with the medical center at which these patients received care. Therefore, although being nonwhite does portend worse outcomes following an arrest, the increased risk is likely attributed to the fact that many of these patients receive care at hospitals that have poorer overall CPR performance measures.5,10
Survival is not the only outcome measure patients need to take into account when deciding whether to undergo CPR. Quality of life following resuscitation also warrants consideration. Interestingly, research has shown that neurologic outcomes among the majority of cardiac arrest survivors are generally good.3
Approximately 86% of survivors with intact pre-arrest cerebral performance maintain it on discharge, and only a minority of survivors are eventually declared brain-dead.3 Still, there are certain peri-arrest factors that pose risk for poorer neurologic and functional outcomes. For arrest from a shockable rhythm, time to defibrillation is a key determinant.11 In patients for whom time to defibrillation is greater than two minutes, there is significantly higher risk of permanent disability following cardiac arrest.11
In the event of coma following resuscitation, particular clinical findings can be used to accurately predict poor outcome.12 The absence of pupillary reflexes, corneal reflexes, or absent or extensor motor responses three days after arrest are poor prognostic indicators.12 As a general rule, if a patient does not awaken within three days, neurologic and functional impairment can be expected.12 For those patients who do survive to hospital discharge, more than 50% ultimately will be able to be discharged home.3
However, nearly a quarter will need to be newly placed in a rehabilitation or skilled nursing facility.3
Back to the Case
The patient was admitted with hypoxia secondary to both progressive lung malignancy and COPD exacerbation. She had no advanced directives, so the admitting hospitalist, in collaboration with her oncologist, had a detailed discussion regarding her understanding of her disease progression, prognosis, and goals for her remaining time. Her questions regarding survivability of cardiac arrest were answered directly with an estimate of 5% to 10%, based on her age, comorbidities, and the presence of advanced malignancy.
After hearing this information, the patient responded, “I still want everything done.” The hospitalist acknowledged her feelings of wanting to fight on, but asked her to think about what “everything” meant to her. After taking some additional time to reflect with friends and family, the patient was clear that she wanted to continue disease-focused therapies, but did not want to be resuscitated in the event of cardiac or pulmonary arrest.
Eventually, her hypoxia improved with antibiotics, steroids, and bronchodilators. She was discharged home with follow-up in the oncology clinic for additional chemotherapy and palliative radiation.
Bottom Line
For hospitalized adults, the average survival rate to discharge after cardiac arrest is about 17%. Many factors lower a patient’s chance of survival, including advanced age, performance status, malignancy, and presence of multiple comorbidities. TH
Dr. Neagle and Dr. Wachsberg are hospitalists and instructors in the division of hospital medicine at Northwestern University Medical Center in Chicago.
References
- Murphy DJ, Burrows D, Santali S, et al. The influence of the probability of survival on patients’ preferences regarding cardiopulmonary resuscitation. N Engl J Med. 1994;330(8):545-549.
- Kaldjian LC, Erekson ZD, Haberle TH, et al. Code status discussions and goals of care among hospitalised adults. J Med Ethics. 2009;35(6):338-342.
- Peberdy MA, Kaye W, Ornato JP, et al. Cardiopulmonary resuscitation of adults in the hospital: a report of 14720 cardiac arrests from the National Registry of Cardiopulmonary Resuscitation. Resuscitation. 2003;58(3):297-308.
- La Puma J, Orentlicher D, Moss RJ. Advance directives on admission. Clinical implications and analysis of the Patient Self-Determination Act of 1990. JAMA. 1991;266(3):402-405.
- Ehlenbach WJ, Barnato AE, Curtis JR, et al. Epidemiologic study of in-hospital cardiopulmonary resuscitation in the elderly. N Engl J Med. 2009;361(1):22-31.
- Larkin GL, Copes WS, Nathanson BH, Kaye W. Pre-resuscitation factors associated with mortality in 49,130 cases of in-hospital cardiac arrest: a report from the National Registry for Cardiopulmonary Resuscitation. Resuscitation. 2010;81(3):302-311.
- de Vos R, Koster RW, De Haan RJ, Oosting H, van der Wouw PA, Lampe-Schoenmaeckers AJ. In-hospital cardiopulmonary resuscitation: prearrest morbidity and outcome. Arch Intern Med. 1999;159(8):845-850.
- Reisfield GM, Wallace SK, Munsell MF, Webb FJ, Alvarez ER, Wilson GR. Survival in cancer patients undergoing in-hospital cardiopulmonary resuscitation: a meta-analysis. Resuscitation. 2006;71(2):152-160.
- Ewer MS, Kish SK, Martin CG, Price KJ, Feeley TW. Characteristics of cardiac arrest in cancer patients as a predictor of survival after cardiopulmonary resuscitation. Cancer. 2001;92(7):1905-1912.
- Chan PS, Nichol G, Krumholz HM, et al. Racial differences in survival after in-hospital cardiac arrest. JAMA. 2009;302(11):1195-1201.
- Chan PS, Krumholz HM, Nichol G, Nallamothu BK. Delayed time to defibrillation after in-hospital cardiac arrest. N Engl J Med. 2008;358(1):9-17.
- Wijdicks EF, Hijdra A, Young GB, Bassetti CL, Wiebe S. Practice parameter: prediction of outcome in comatose survivors after cardiopulmonary resuscitation (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2006;67(2):203-210.
Case
A 66-year-old woman with metastatic, non-small-cell carcinoma of the lung, chronic obstructive pulmonary disease (COPD), and hypertension presents with progressive shortness of breath and back pain. Her vital signs are normal, with the exception of tachypnea and an oxygen saturation of 84% on room air. A CT scan shows marked progression of her disease and new metastases to her spine. You begin a discussion about advance directives and code status. During the exchange, the patient asks for guidance regarding resuscitation. How can you best answer her questions about the likelihood of surviving an in-hospital arrest?
Background
Discussion regarding resuscitation status is a challenge for most hospitalists. The absence of an established relationship, limited time, patient emotion, and difficulty applying general scientific data to a single patient coalesce into a complex interaction. Further complicating matters, patients frequently have unrealistic expectations and overestimate their chance of survival.
Experience has shown that many patients pursue what physicians consider inappropriately aggressive resuscitation measures. Before you have an informed discussion about cardiopulmonary resuscitation (CPR) outcomes, patients tend to overestimate their likelihood of survival.1 In 2009, Kaldjian and colleagues found that patients’ initial mean prediction of post-arrest survival was 60.4%, compared with the actual mean of approximately 17%.2,3 Furthermore, nearly half of the patients who initially expressed a desire to receive CPR in the event of cardiac arrest opted to change their code status after they were informed of the actual survival estimates.1,2
Patient autonomy and the law, as defined by the 1990 Patient Self-Determination Act, require that physicians share responsibility with patients in making prospective resuscitation decisions.4 Shared decision-making necessitates a basic discussion on admission within the context of the patient’s prognosis and previously expressed wishes. It might simply include an acknowledgment of a previously completed advance directive. A more complex discussion might require in-depth conversation to address patient performance status, prognosis of acute and chronic illnesses, and education about the typical resuscitation procedures. After listening to the patient’s perspective, the admitting physician can provide input and an interpretation of available data regarding the patient’s likelihood of surviving an in-hospital arrest.
Review of the Data
In the past 40 years, the overall survival rates for cardiac arrest have changed little. Despite numerous advances made in the delivery of medical care, on average, only 17% of all adult arrest patients survive to hospital discharge.3 A variety of factors influence this overall survival rate, both pre-arrest and intra-arrest. Clinical experience allows most physicians to sense what probability a patient has for survival and quality of life following a cardiac arrest. However, anecdotal evidence alone does not provide a patient and their family with the information necessary to make an informed decision regarding code status.
Numerous studies have investigated the patient factors that might influence how likely one is to survive a cardiac arrest. Researchers have paid particular attention to such factors as age, race, presence or absence of a cancer diagnosis, and associated comorbidities. Not surprisingly, older age has been shown to be significantly associated with a lower likelihood of survival to discharge following cardiac arrest.5,6
Ehlenbach and colleagues examined medical data from 433,985 Medicare patients 65 and older who underwent in-hospital CPR.5 Both older age and prior residence in a skilled nursing facility were found to be associated with poorer survival rates.5 Although neither study was able to define an upper-age cutoff for certain peri-arrest mortality, age affects overall survival likelihood in an inverse fashion, with those 85 and older having only a 6% chance of surviving to hospital discharge (see Figure 1, p. 18).1,5,6
The degree of comorbid illness can be used to help predict mortality following cardiac arrest. Review of data from the National Registry of Cardiopulmonary Resuscitation (NRCPR) identified particular comorbidities that portend poor post-arrest prognosis.6 In general, the more pre-existing comorbidities a patient has, the less likely they are to survive.6 The presence of hepatic insufficiency, acute stroke, immunodeficiency, renal failure, or dialysis were associated with lower survival rates (see Figure 2, right).6,7
Poor performance status on admission, defined as severe disability, coma, or vegetative state, was predictive of worse outcomes.6 Understandably, patients with hypotension and those who required vasopressors or mechanical ventilation also tended to have lower post-arrest survival rates.6
The presence of a cancer diagnosis is another prognostic factor of interest when considering the chances of surviving an arrest. Classically, CPR was thought to be a futile intervention in this patient population. Specific characteristics within this subset of patients have been shown to influence prognosis, and multiple studies have confirmed that cancer patients generally do worse after an arrest with an overall survival rate of only 6.2%.8 Survival rates tend to be lower in patients with metastatic disease, hematologic malignancies, a history of stem cell transplant, those who arrest within an ICU, and inpatients whose cardiac arrest was anticipated.8,9
In fact, cancer patients whose hospital course followed a path of gradual deterioration showed a 0% survival rate.9 In patients with metastatic disease, poor performance status prior to arrest appeared to account for their particularly poor survival odds (this supports the intuitive, rule-of-thumb that sicker cancer patients have worse outcomes).8
Growing evidence suggests the probability of post-arrest survival is not equal between racial groups. Specifically, black or nonwhite race is associated with higher utilization of CPR and lower survival rates (see Figure 3, right).10 Among Medicare patients, Ehlenbach and colleagues found that black and nonwhite patients were much more likely to undergo CPR, presumably as a result of being less likely to opt for DNR status.5,10 Although this could account for the differences seen in survival rates among these populations, these findings also raise concerns about the possibility of racial disparities in medical care. A subsequent cohort study also suggested that blacks and nonwhites were less likely to survive following cardiac arrest.10 However, adjusted analysis revealed that these differences were strongly associated with the medical center at which these patients received care. Therefore, although being nonwhite does portend worse outcomes following an arrest, the increased risk is likely attributed to the fact that many of these patients receive care at hospitals that have poorer overall CPR performance measures.5,10
Survival is not the only outcome measure patients need to take into account when deciding whether to undergo CPR. Quality of life following resuscitation also warrants consideration. Interestingly, research has shown that neurologic outcomes among the majority of cardiac arrest survivors are generally good.3
Approximately 86% of survivors with intact pre-arrest cerebral performance maintain it on discharge, and only a minority of survivors are eventually declared brain-dead.3 Still, there are certain peri-arrest factors that pose risk for poorer neurologic and functional outcomes. For arrest from a shockable rhythm, time to defibrillation is a key determinant.11 In patients for whom time to defibrillation is greater than two minutes, there is significantly higher risk of permanent disability following cardiac arrest.11
In the event of coma following resuscitation, particular clinical findings can be used to accurately predict poor outcome.12 The absence of pupillary reflexes, corneal reflexes, or absent or extensor motor responses three days after arrest are poor prognostic indicators.12 As a general rule, if a patient does not awaken within three days, neurologic and functional impairment can be expected.12 For those patients who do survive to hospital discharge, more than 50% ultimately will be able to be discharged home.3
However, nearly a quarter will need to be newly placed in a rehabilitation or skilled nursing facility.3
Back to the Case
The patient was admitted with hypoxia secondary to both progressive lung malignancy and COPD exacerbation. She had no advanced directives, so the admitting hospitalist, in collaboration with her oncologist, had a detailed discussion regarding her understanding of her disease progression, prognosis, and goals for her remaining time. Her questions regarding survivability of cardiac arrest were answered directly with an estimate of 5% to 10%, based on her age, comorbidities, and the presence of advanced malignancy.
After hearing this information, the patient responded, “I still want everything done.” The hospitalist acknowledged her feelings of wanting to fight on, but asked her to think about what “everything” meant to her. After taking some additional time to reflect with friends and family, the patient was clear that she wanted to continue disease-focused therapies, but did not want to be resuscitated in the event of cardiac or pulmonary arrest.
Eventually, her hypoxia improved with antibiotics, steroids, and bronchodilators. She was discharged home with follow-up in the oncology clinic for additional chemotherapy and palliative radiation.
Bottom Line
For hospitalized adults, the average survival rate to discharge after cardiac arrest is about 17%. Many factors lower a patient’s chance of survival, including advanced age, performance status, malignancy, and presence of multiple comorbidities. TH
Dr. Neagle and Dr. Wachsberg are hospitalists and instructors in the division of hospital medicine at Northwestern University Medical Center in Chicago.
References
- Murphy DJ, Burrows D, Santali S, et al. The influence of the probability of survival on patients’ preferences regarding cardiopulmonary resuscitation. N Engl J Med. 1994;330(8):545-549.
- Kaldjian LC, Erekson ZD, Haberle TH, et al. Code status discussions and goals of care among hospitalised adults. J Med Ethics. 2009;35(6):338-342.
- Peberdy MA, Kaye W, Ornato JP, et al. Cardiopulmonary resuscitation of adults in the hospital: a report of 14720 cardiac arrests from the National Registry of Cardiopulmonary Resuscitation. Resuscitation. 2003;58(3):297-308.
- La Puma J, Orentlicher D, Moss RJ. Advance directives on admission. Clinical implications and analysis of the Patient Self-Determination Act of 1990. JAMA. 1991;266(3):402-405.
- Ehlenbach WJ, Barnato AE, Curtis JR, et al. Epidemiologic study of in-hospital cardiopulmonary resuscitation in the elderly. N Engl J Med. 2009;361(1):22-31.
- Larkin GL, Copes WS, Nathanson BH, Kaye W. Pre-resuscitation factors associated with mortality in 49,130 cases of in-hospital cardiac arrest: a report from the National Registry for Cardiopulmonary Resuscitation. Resuscitation. 2010;81(3):302-311.
- de Vos R, Koster RW, De Haan RJ, Oosting H, van der Wouw PA, Lampe-Schoenmaeckers AJ. In-hospital cardiopulmonary resuscitation: prearrest morbidity and outcome. Arch Intern Med. 1999;159(8):845-850.
- Reisfield GM, Wallace SK, Munsell MF, Webb FJ, Alvarez ER, Wilson GR. Survival in cancer patients undergoing in-hospital cardiopulmonary resuscitation: a meta-analysis. Resuscitation. 2006;71(2):152-160.
- Ewer MS, Kish SK, Martin CG, Price KJ, Feeley TW. Characteristics of cardiac arrest in cancer patients as a predictor of survival after cardiopulmonary resuscitation. Cancer. 2001;92(7):1905-1912.
- Chan PS, Nichol G, Krumholz HM, et al. Racial differences in survival after in-hospital cardiac arrest. JAMA. 2009;302(11):1195-1201.
- Chan PS, Krumholz HM, Nichol G, Nallamothu BK. Delayed time to defibrillation after in-hospital cardiac arrest. N Engl J Med. 2008;358(1):9-17.
- Wijdicks EF, Hijdra A, Young GB, Bassetti CL, Wiebe S. Practice parameter: prediction of outcome in comatose survivors after cardiopulmonary resuscitation (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2006;67(2):203-210.
Productivity Capacity

The mark of any great society is balance—balance between the production realized today and the preservation of “production capacity” to ensure the same or greater production in the future. HM is not exempt from this fundamental tenet. What we do now in the way of advancing quality, efficiency, and patient safety will matter little if our contributions are not sustained by the generation that follows us.
It is tempting to think that the issue of how we train residents is germane only to universities, but the reality is that it affects us all. There are 126 “university” medical school programs, but there are 384 residency programs, most of which are within community-based hospitals. The result is that most hospitalists encounter resident physicians in some capacity, and all hospitalists will encounter the results of residency training when they welcome a new recruit to their ranks.
The education and socialization of our residents will define the character of the hospitalists of the future. But the “residency” in which most of us trained does not exist anymore: The duty-hours changes and additional training requirements have dramatically changed the landscape of residency training in the past 10 years, and another series of sea changes is underway. As with all things HM, we again have a choice: Be reactive, wait for the dust to clear, and then lament the results, or be proactive and see this change for what it is—an opportunity to improve healthcare quality now, and in the future.
The ACGME
HM felt the impact of the first wave of duty-hours restrictions beginning in 2003, as many training programs opted to employ hospitalists to provide the coverage that could no longer be maintained by residents working under tighter admission caps and duty-hour restrictions. In doing so, hospitalists have provided a valuable service in preserving the integrity of training environments and fidelity to the Accreditation Council for Graduate Medical Education (ACGME) regulations (more than 85% of training programs have hospitalists working in their systems). But the model of hospitalists working solely as “resident-extenders” is not sustainable.
First, hospitalists who work solely on nonteaching services are at great risk of burning out, especially if the distribution of patients has been manipulated such that the more interesting patients are funneled away from the hospitalist’s service to the teaching service. Second, there is a risk in perception: In models in which the hospitalist is solely the “overflow cap coverage” or the night-float physician (i.e., the resident-extender), residents come to see hospitalists as the “PGY-4, 5, 6 …” physicians—that is, the physician who becomes a resident for life. The result is a serious pipeline issue for us, as the most talented resident physicians are unlikely to forego subspecialty training for a career in HM if hospitalists are perceived as perpetual residents.
The solution is simple: The hospitalist’s role in training environments has to be more than merely solving admission cap or duty-hour issues. It is fine for hospitalists to operate nonteaching services, but the hospitalist also has to be a part of the fulfillment that comes with overseeing teaching services. Further, residents have to see the hospitalist career for what it actually is: Academic or not, HM is much more than merely clinical service. HM is about the value-added services of system interventions to improve quality and patient safety; it is about developing a career as a systems architect. Getting the best and brightest residents to choose HM as a career is contingent upon residents seeing hospitalists in the training environment who are happy and fulfilled in the execution of this career goal.
The hospitalist’s plight was helped substantially on June 23, when ACGME released for comment the revised Common Program Requirements (www.acgme.org). The duty-hours changes are unlikely to substantially alter hospitalists’ lives; the only significant change was a limitation on intern shift durations to fewer than 16 hours in a row (upper-level residents still operate under the 24+6 hour rule, with increased flexibility to stay longer by volition). But the interesting part of the new requirements is an augmented focus on teaching residents transitions-of-care skills, improving direct supervision of residents, and constructing educational systems that minimize handoffs.
There is no specialty that is as suited as HM for fulfilling these unique (and, as of yet, unmet) requirements. Transitions, quality, being present on the hospital wards … this is what we do. And requiring instruction in transitions and quality is an unprecedented leverage point for HM to advance the quality of future physicians. How great it would be to attend HM20 and realize that the attendees had already learned the “Quality 101” lessons (i.e., those we are currently teaching at our annual meeting) as part of their residency? Freed from the need to do basic quality sessions, the content of the annual meeting could escalate to even higher-level principles that would result in substantial and sustainable quality improvement (QI).
MedPAC and GME Funding
Simultaneous with the ACGME changes are changes at the Medicare Payment Advisory Committee (MedPAC), the advisory organization responsible for recommending changes in the distribution of Centers for Medicare and Medicaid Services (CMS) funds to support graduate medical education. CMS is the primary funding agent for residency training. Each hospital receives direct medical expenditures to cover a resident’s salary and benefits. Each hospital has a pre-set per-resident allotment, or PRA. This number varies by hospital, but the average is $100,000 per resident. CMS reimburses the hospital a percentage of this number based upon the percentage of hospital days occupied by Medicare patients (e.g., 35% Medicare days=$35,000 per resident).
The hospital also receives indirect medical expenditures, or IME. IME is not a distinct payment to the hospital, but rather an “inflator” of the clinical-care payments the hospital receives from CMS. IME is paid to the hospital under the presumption that a typical training facility incurs greater cost due to higher patient severity, a higher indigent care percentage, and has higher resource utilization due to residents’ excessive testing, etc. The final presumption is that support is needed for the educational infrastructure (i.e., supervision and teaching).
IME is not inconsequential to a hospital; depending upon the payor mix, a 200-bed hospital might have from $4 million to $8 million in annual IME payments. CMS’ total IME payments to hospitals is more than $6 billion a year. Each hospital’s IME revenue can be found at www.graham-center.org/online/graham/home/tools-resources/data-tables/dt001-gme-2007.html.
The game-changing event occurred in April, when MedPAC announced its intent to reassess the mechanisms of IME funding, with a vision of IME funding eventually being linked to a hospital’s training programs’ ability to demonstrate substantial improvement in quality and patient safety. And here is the leverage point that is a unique opportunity for hospitalists in the training environment. For many hospitalists, especially if employed directly by the hospital, there is little financial incentive to engaging on a teaching service. The ACGME caps limit the service size, and this in turn limits the possible RVUs. Up until now, asking the hospital to compensate for teaching time (i.e., EVUs) was a pipe dream. But the linking of IME funding to quality outcomes (and quality instruction to residents) could change all of that.
If you put the two together: ACGME calling for instruction in quality and transitions, plus MedPAC calling for payments linked to resident outcomes in quality and patient safety, you have one inescapable conclusion—the residency of the future will hinge upon having supervisors with the necessary expertise to ensure that residents participate in, and understand the principles of, patient safety and quality as a part of the residency curriculum. And the people who can ensure that goal are likely to be in a position to warrant compensation for doing so.
Who is better to do this than the hospitalist?
SHM’s Proactive Strategy
This is the opportune time for HM to advance its stature as a profession and to ensure its future via a pipeline of residents adequately training in quality and patient safety. But it is not enough to merely wish for this to happen. There are real barriers that have kept hospitalists from being more intimately involved in physician training, the first of which is age.
HM is a young specialty (the average hospitalist is 37; the average HM leader is 41), and its youth makes it hard to compete with older subspecialists/generalists who have more experience in education. But deficits in experience can be compensated by additional training.
The Academic Hospitalist Academy (AHA)—cosponsored by SHM, the Society of General Internal Medicine (SGIM), and the Association of Chiefs and Leaders of General Internal Medicine (ACLGIM)—is the key to the strategy of catching up quickly. The academy will convene this month outside of Atlanta, and it is very important that each training facility think about sending one of its hospitalists to receive the advanced training in education necessary to compensate for not having years of experience in medical education. Academy details are available at http://academichospitalist.org.
SHM’s initiatives on this front do not stop with the academy. Over the past three months, Kevin O’Leary, MD, and his Quality Improvement Education Committee have been furiously building a “Quality and Patient Safety” curriculum, with a target audience of new hospitalists and resident physicians. The vision is to create a Web-based, interactive curriculum that teaches resident physicians the basics of quality and patient safety, design projects with their colleagues (under the supervision of their hospitalist mentor), and track their data to see real-time results.
Unlike other curricula on the market, the SHM Quality Curriculum for residents will be dynamic, requiring participating institutions commit to SHM’s modus operandi of mentored implementation by sponsoring a hospitalist to receive the training necessary to put the curriculum in motion. To this end, SHM has collaborated with the Alliance for Internal Medicine (AIM) in co-sponsoring the Quality Academy, with a focus on how to teach quality and patient safety. Jen Meyers, MD, FHM, and Jeff Glasheen, MD, SFHM, will be leading the team responsible for the development of this Quality Training Course, which should emerge in the fall of 2011.
As this project proceeds, Paul Grant, MD, chair of the Early Career Hospitalist Committee, and Cheryl O’Malley, MD, chair of the Pipeline Committee, will provide counsel. Both of these groups will continue efforts to improve the process by which residents transition from residency to HM practice, and supporting young physicians with distance mentoring.
The SHM vision of our production capacity is simple: Bring in the best and brightest hospitalists who are interested in teaching quality and patient safety, train them in the fundamentals of medical education, provide them with an “off the net” curriculum for how to teach quality, then return them to their respective training environments to coach residents on the principles of quality.
Training programs that invest in this vision will reap the rewards of fidelity to the new ACGME requirements. Hospitals that support such a vision will receive assurances, should MedPAC’s recommendation come to fruition, that DME and IME funding is secure. Hospitalists investing in this vision will find a fulfilling career in quality education.
And all of us will find assurances that, for as good as things are right now for HM, the future will be even better. TH
Dr. Wiese is president of SHM.
The mark of any great society is balance—balance between the production realized today and the preservation of “production capacity” to ensure the same or greater production in the future. HM is not exempt from this fundamental tenet. What we do now in the way of advancing quality, efficiency, and patient safety will matter little if our contributions are not sustained by the generation that follows us.
It is tempting to think that the issue of how we train residents is germane only to universities, but the reality is that it affects us all. There are 126 “university” medical school programs, but there are 384 residency programs, most of which are within community-based hospitals. The result is that most hospitalists encounter resident physicians in some capacity, and all hospitalists will encounter the results of residency training when they welcome a new recruit to their ranks.
The education and socialization of our residents will define the character of the hospitalists of the future. But the “residency” in which most of us trained does not exist anymore: The duty-hours changes and additional training requirements have dramatically changed the landscape of residency training in the past 10 years, and another series of sea changes is underway. As with all things HM, we again have a choice: Be reactive, wait for the dust to clear, and then lament the results, or be proactive and see this change for what it is—an opportunity to improve healthcare quality now, and in the future.
The ACGME
HM felt the impact of the first wave of duty-hours restrictions beginning in 2003, as many training programs opted to employ hospitalists to provide the coverage that could no longer be maintained by residents working under tighter admission caps and duty-hour restrictions. In doing so, hospitalists have provided a valuable service in preserving the integrity of training environments and fidelity to the Accreditation Council for Graduate Medical Education (ACGME) regulations (more than 85% of training programs have hospitalists working in their systems). But the model of hospitalists working solely as “resident-extenders” is not sustainable.
First, hospitalists who work solely on nonteaching services are at great risk of burning out, especially if the distribution of patients has been manipulated such that the more interesting patients are funneled away from the hospitalist’s service to the teaching service. Second, there is a risk in perception: In models in which the hospitalist is solely the “overflow cap coverage” or the night-float physician (i.e., the resident-extender), residents come to see hospitalists as the “PGY-4, 5, 6 …” physicians—that is, the physician who becomes a resident for life. The result is a serious pipeline issue for us, as the most talented resident physicians are unlikely to forego subspecialty training for a career in HM if hospitalists are perceived as perpetual residents.
The solution is simple: The hospitalist’s role in training environments has to be more than merely solving admission cap or duty-hour issues. It is fine for hospitalists to operate nonteaching services, but the hospitalist also has to be a part of the fulfillment that comes with overseeing teaching services. Further, residents have to see the hospitalist career for what it actually is: Academic or not, HM is much more than merely clinical service. HM is about the value-added services of system interventions to improve quality and patient safety; it is about developing a career as a systems architect. Getting the best and brightest residents to choose HM as a career is contingent upon residents seeing hospitalists in the training environment who are happy and fulfilled in the execution of this career goal.
The hospitalist’s plight was helped substantially on June 23, when ACGME released for comment the revised Common Program Requirements (www.acgme.org). The duty-hours changes are unlikely to substantially alter hospitalists’ lives; the only significant change was a limitation on intern shift durations to fewer than 16 hours in a row (upper-level residents still operate under the 24+6 hour rule, with increased flexibility to stay longer by volition). But the interesting part of the new requirements is an augmented focus on teaching residents transitions-of-care skills, improving direct supervision of residents, and constructing educational systems that minimize handoffs.
There is no specialty that is as suited as HM for fulfilling these unique (and, as of yet, unmet) requirements. Transitions, quality, being present on the hospital wards … this is what we do. And requiring instruction in transitions and quality is an unprecedented leverage point for HM to advance the quality of future physicians. How great it would be to attend HM20 and realize that the attendees had already learned the “Quality 101” lessons (i.e., those we are currently teaching at our annual meeting) as part of their residency? Freed from the need to do basic quality sessions, the content of the annual meeting could escalate to even higher-level principles that would result in substantial and sustainable quality improvement (QI).
MedPAC and GME Funding
Simultaneous with the ACGME changes are changes at the Medicare Payment Advisory Committee (MedPAC), the advisory organization responsible for recommending changes in the distribution of Centers for Medicare and Medicaid Services (CMS) funds to support graduate medical education. CMS is the primary funding agent for residency training. Each hospital receives direct medical expenditures to cover a resident’s salary and benefits. Each hospital has a pre-set per-resident allotment, or PRA. This number varies by hospital, but the average is $100,000 per resident. CMS reimburses the hospital a percentage of this number based upon the percentage of hospital days occupied by Medicare patients (e.g., 35% Medicare days=$35,000 per resident).
The hospital also receives indirect medical expenditures, or IME. IME is not a distinct payment to the hospital, but rather an “inflator” of the clinical-care payments the hospital receives from CMS. IME is paid to the hospital under the presumption that a typical training facility incurs greater cost due to higher patient severity, a higher indigent care percentage, and has higher resource utilization due to residents’ excessive testing, etc. The final presumption is that support is needed for the educational infrastructure (i.e., supervision and teaching).
IME is not inconsequential to a hospital; depending upon the payor mix, a 200-bed hospital might have from $4 million to $8 million in annual IME payments. CMS’ total IME payments to hospitals is more than $6 billion a year. Each hospital’s IME revenue can be found at www.graham-center.org/online/graham/home/tools-resources/data-tables/dt001-gme-2007.html.
The game-changing event occurred in April, when MedPAC announced its intent to reassess the mechanisms of IME funding, with a vision of IME funding eventually being linked to a hospital’s training programs’ ability to demonstrate substantial improvement in quality and patient safety. And here is the leverage point that is a unique opportunity for hospitalists in the training environment. For many hospitalists, especially if employed directly by the hospital, there is little financial incentive to engaging on a teaching service. The ACGME caps limit the service size, and this in turn limits the possible RVUs. Up until now, asking the hospital to compensate for teaching time (i.e., EVUs) was a pipe dream. But the linking of IME funding to quality outcomes (and quality instruction to residents) could change all of that.
If you put the two together: ACGME calling for instruction in quality and transitions, plus MedPAC calling for payments linked to resident outcomes in quality and patient safety, you have one inescapable conclusion—the residency of the future will hinge upon having supervisors with the necessary expertise to ensure that residents participate in, and understand the principles of, patient safety and quality as a part of the residency curriculum. And the people who can ensure that goal are likely to be in a position to warrant compensation for doing so.
Who is better to do this than the hospitalist?
SHM’s Proactive Strategy
This is the opportune time for HM to advance its stature as a profession and to ensure its future via a pipeline of residents adequately training in quality and patient safety. But it is not enough to merely wish for this to happen. There are real barriers that have kept hospitalists from being more intimately involved in physician training, the first of which is age.
HM is a young specialty (the average hospitalist is 37; the average HM leader is 41), and its youth makes it hard to compete with older subspecialists/generalists who have more experience in education. But deficits in experience can be compensated by additional training.
The Academic Hospitalist Academy (AHA)—cosponsored by SHM, the Society of General Internal Medicine (SGIM), and the Association of Chiefs and Leaders of General Internal Medicine (ACLGIM)—is the key to the strategy of catching up quickly. The academy will convene this month outside of Atlanta, and it is very important that each training facility think about sending one of its hospitalists to receive the advanced training in education necessary to compensate for not having years of experience in medical education. Academy details are available at http://academichospitalist.org.
SHM’s initiatives on this front do not stop with the academy. Over the past three months, Kevin O’Leary, MD, and his Quality Improvement Education Committee have been furiously building a “Quality and Patient Safety” curriculum, with a target audience of new hospitalists and resident physicians. The vision is to create a Web-based, interactive curriculum that teaches resident physicians the basics of quality and patient safety, design projects with their colleagues (under the supervision of their hospitalist mentor), and track their data to see real-time results.
Unlike other curricula on the market, the SHM Quality Curriculum for residents will be dynamic, requiring participating institutions commit to SHM’s modus operandi of mentored implementation by sponsoring a hospitalist to receive the training necessary to put the curriculum in motion. To this end, SHM has collaborated with the Alliance for Internal Medicine (AIM) in co-sponsoring the Quality Academy, with a focus on how to teach quality and patient safety. Jen Meyers, MD, FHM, and Jeff Glasheen, MD, SFHM, will be leading the team responsible for the development of this Quality Training Course, which should emerge in the fall of 2011.
As this project proceeds, Paul Grant, MD, chair of the Early Career Hospitalist Committee, and Cheryl O’Malley, MD, chair of the Pipeline Committee, will provide counsel. Both of these groups will continue efforts to improve the process by which residents transition from residency to HM practice, and supporting young physicians with distance mentoring.
The SHM vision of our production capacity is simple: Bring in the best and brightest hospitalists who are interested in teaching quality and patient safety, train them in the fundamentals of medical education, provide them with an “off the net” curriculum for how to teach quality, then return them to their respective training environments to coach residents on the principles of quality.
Training programs that invest in this vision will reap the rewards of fidelity to the new ACGME requirements. Hospitals that support such a vision will receive assurances, should MedPAC’s recommendation come to fruition, that DME and IME funding is secure. Hospitalists investing in this vision will find a fulfilling career in quality education.
And all of us will find assurances that, for as good as things are right now for HM, the future will be even better. TH
Dr. Wiese is president of SHM.
The mark of any great society is balance—balance between the production realized today and the preservation of “production capacity” to ensure the same or greater production in the future. HM is not exempt from this fundamental tenet. What we do now in the way of advancing quality, efficiency, and patient safety will matter little if our contributions are not sustained by the generation that follows us.
It is tempting to think that the issue of how we train residents is germane only to universities, but the reality is that it affects us all. There are 126 “university” medical school programs, but there are 384 residency programs, most of which are within community-based hospitals. The result is that most hospitalists encounter resident physicians in some capacity, and all hospitalists will encounter the results of residency training when they welcome a new recruit to their ranks.
The education and socialization of our residents will define the character of the hospitalists of the future. But the “residency” in which most of us trained does not exist anymore: The duty-hours changes and additional training requirements have dramatically changed the landscape of residency training in the past 10 years, and another series of sea changes is underway. As with all things HM, we again have a choice: Be reactive, wait for the dust to clear, and then lament the results, or be proactive and see this change for what it is—an opportunity to improve healthcare quality now, and in the future.
The ACGME
HM felt the impact of the first wave of duty-hours restrictions beginning in 2003, as many training programs opted to employ hospitalists to provide the coverage that could no longer be maintained by residents working under tighter admission caps and duty-hour restrictions. In doing so, hospitalists have provided a valuable service in preserving the integrity of training environments and fidelity to the Accreditation Council for Graduate Medical Education (ACGME) regulations (more than 85% of training programs have hospitalists working in their systems). But the model of hospitalists working solely as “resident-extenders” is not sustainable.
First, hospitalists who work solely on nonteaching services are at great risk of burning out, especially if the distribution of patients has been manipulated such that the more interesting patients are funneled away from the hospitalist’s service to the teaching service. Second, there is a risk in perception: In models in which the hospitalist is solely the “overflow cap coverage” or the night-float physician (i.e., the resident-extender), residents come to see hospitalists as the “PGY-4, 5, 6 …” physicians—that is, the physician who becomes a resident for life. The result is a serious pipeline issue for us, as the most talented resident physicians are unlikely to forego subspecialty training for a career in HM if hospitalists are perceived as perpetual residents.
The solution is simple: The hospitalist’s role in training environments has to be more than merely solving admission cap or duty-hour issues. It is fine for hospitalists to operate nonteaching services, but the hospitalist also has to be a part of the fulfillment that comes with overseeing teaching services. Further, residents have to see the hospitalist career for what it actually is: Academic or not, HM is much more than merely clinical service. HM is about the value-added services of system interventions to improve quality and patient safety; it is about developing a career as a systems architect. Getting the best and brightest residents to choose HM as a career is contingent upon residents seeing hospitalists in the training environment who are happy and fulfilled in the execution of this career goal.
The hospitalist’s plight was helped substantially on June 23, when ACGME released for comment the revised Common Program Requirements (www.acgme.org). The duty-hours changes are unlikely to substantially alter hospitalists’ lives; the only significant change was a limitation on intern shift durations to fewer than 16 hours in a row (upper-level residents still operate under the 24+6 hour rule, with increased flexibility to stay longer by volition). But the interesting part of the new requirements is an augmented focus on teaching residents transitions-of-care skills, improving direct supervision of residents, and constructing educational systems that minimize handoffs.
There is no specialty that is as suited as HM for fulfilling these unique (and, as of yet, unmet) requirements. Transitions, quality, being present on the hospital wards … this is what we do. And requiring instruction in transitions and quality is an unprecedented leverage point for HM to advance the quality of future physicians. How great it would be to attend HM20 and realize that the attendees had already learned the “Quality 101” lessons (i.e., those we are currently teaching at our annual meeting) as part of their residency? Freed from the need to do basic quality sessions, the content of the annual meeting could escalate to even higher-level principles that would result in substantial and sustainable quality improvement (QI).
MedPAC and GME Funding
Simultaneous with the ACGME changes are changes at the Medicare Payment Advisory Committee (MedPAC), the advisory organization responsible for recommending changes in the distribution of Centers for Medicare and Medicaid Services (CMS) funds to support graduate medical education. CMS is the primary funding agent for residency training. Each hospital receives direct medical expenditures to cover a resident’s salary and benefits. Each hospital has a pre-set per-resident allotment, or PRA. This number varies by hospital, but the average is $100,000 per resident. CMS reimburses the hospital a percentage of this number based upon the percentage of hospital days occupied by Medicare patients (e.g., 35% Medicare days=$35,000 per resident).
The hospital also receives indirect medical expenditures, or IME. IME is not a distinct payment to the hospital, but rather an “inflator” of the clinical-care payments the hospital receives from CMS. IME is paid to the hospital under the presumption that a typical training facility incurs greater cost due to higher patient severity, a higher indigent care percentage, and has higher resource utilization due to residents’ excessive testing, etc. The final presumption is that support is needed for the educational infrastructure (i.e., supervision and teaching).
IME is not inconsequential to a hospital; depending upon the payor mix, a 200-bed hospital might have from $4 million to $8 million in annual IME payments. CMS’ total IME payments to hospitals is more than $6 billion a year. Each hospital’s IME revenue can be found at www.graham-center.org/online/graham/home/tools-resources/data-tables/dt001-gme-2007.html.
The game-changing event occurred in April, when MedPAC announced its intent to reassess the mechanisms of IME funding, with a vision of IME funding eventually being linked to a hospital’s training programs’ ability to demonstrate substantial improvement in quality and patient safety. And here is the leverage point that is a unique opportunity for hospitalists in the training environment. For many hospitalists, especially if employed directly by the hospital, there is little financial incentive to engaging on a teaching service. The ACGME caps limit the service size, and this in turn limits the possible RVUs. Up until now, asking the hospital to compensate for teaching time (i.e., EVUs) was a pipe dream. But the linking of IME funding to quality outcomes (and quality instruction to residents) could change all of that.
If you put the two together: ACGME calling for instruction in quality and transitions, plus MedPAC calling for payments linked to resident outcomes in quality and patient safety, you have one inescapable conclusion—the residency of the future will hinge upon having supervisors with the necessary expertise to ensure that residents participate in, and understand the principles of, patient safety and quality as a part of the residency curriculum. And the people who can ensure that goal are likely to be in a position to warrant compensation for doing so.
Who is better to do this than the hospitalist?
SHM’s Proactive Strategy
This is the opportune time for HM to advance its stature as a profession and to ensure its future via a pipeline of residents adequately training in quality and patient safety. But it is not enough to merely wish for this to happen. There are real barriers that have kept hospitalists from being more intimately involved in physician training, the first of which is age.
HM is a young specialty (the average hospitalist is 37; the average HM leader is 41), and its youth makes it hard to compete with older subspecialists/generalists who have more experience in education. But deficits in experience can be compensated by additional training.
The Academic Hospitalist Academy (AHA)—cosponsored by SHM, the Society of General Internal Medicine (SGIM), and the Association of Chiefs and Leaders of General Internal Medicine (ACLGIM)—is the key to the strategy of catching up quickly. The academy will convene this month outside of Atlanta, and it is very important that each training facility think about sending one of its hospitalists to receive the advanced training in education necessary to compensate for not having years of experience in medical education. Academy details are available at http://academichospitalist.org.
SHM’s initiatives on this front do not stop with the academy. Over the past three months, Kevin O’Leary, MD, and his Quality Improvement Education Committee have been furiously building a “Quality and Patient Safety” curriculum, with a target audience of new hospitalists and resident physicians. The vision is to create a Web-based, interactive curriculum that teaches resident physicians the basics of quality and patient safety, design projects with their colleagues (under the supervision of their hospitalist mentor), and track their data to see real-time results.
Unlike other curricula on the market, the SHM Quality Curriculum for residents will be dynamic, requiring participating institutions commit to SHM’s modus operandi of mentored implementation by sponsoring a hospitalist to receive the training necessary to put the curriculum in motion. To this end, SHM has collaborated with the Alliance for Internal Medicine (AIM) in co-sponsoring the Quality Academy, with a focus on how to teach quality and patient safety. Jen Meyers, MD, FHM, and Jeff Glasheen, MD, SFHM, will be leading the team responsible for the development of this Quality Training Course, which should emerge in the fall of 2011.
As this project proceeds, Paul Grant, MD, chair of the Early Career Hospitalist Committee, and Cheryl O’Malley, MD, chair of the Pipeline Committee, will provide counsel. Both of these groups will continue efforts to improve the process by which residents transition from residency to HM practice, and supporting young physicians with distance mentoring.
The SHM vision of our production capacity is simple: Bring in the best and brightest hospitalists who are interested in teaching quality and patient safety, train them in the fundamentals of medical education, provide them with an “off the net” curriculum for how to teach quality, then return them to their respective training environments to coach residents on the principles of quality.
Training programs that invest in this vision will reap the rewards of fidelity to the new ACGME requirements. Hospitals that support such a vision will receive assurances, should MedPAC’s recommendation come to fruition, that DME and IME funding is secure. Hospitalists investing in this vision will find a fulfilling career in quality education.
And all of us will find assurances that, for as good as things are right now for HM, the future will be even better. TH
Dr. Wiese is president of SHM.
The Devil & the Details

Act I: The Negotiation
(A barren academic office, dimly lit, the pall of difficult negotiations afloat, backlit like dust in the air. Seated, under a strangely intense incandescent bulb, a man, who looks eerily like a good-looking version of me, sits uncomfortably adjusting himself in his seat. His eyes constrict on his counterpart, a miserly sort peering out from behind wire-rim glasses and a shock of hair improbably combed over from ear to ear. The tension crests.)
GOOD-LOOKING ME
(Voice cracking)
I’ve come to ask for a raise for our hospitalist group.
MISER
(Adjusts his clip-on tie)
We just gave you a raise in 2004.
GOOD-LOOKING ME
(Smiles uncomfortably)
That was very gracious, sir, but I think the numbers support another.
MISER
(Incredulous look at his watch)
But your work RVUs are thousands below what I’d like to see.
GOOD-LOOKING ME
(Dabs bead of sweat away from chiseled chin)
That’s because you’ve set your benchmark thousands above a reasonable number.
MISER
(Voice flitting with child-like condescension)
But those are the numbers my finance guy gave me. It’s the benchmark.
(Blackout and end of Act I.)
Mutual Agreement
Tony Award-winning stuff for sure—and based on a true story! In fact, this scene no doubt plays out annually for those of you unfortunate enough to have to negotiate with hospital executives for programmatic support. To be fair, hospital administrators deserve to know that they are getting what they pay for. Thus, the concepts of a benchmark are reasonable. The problem lies in setting mutually-agreed-upon standards.
Act II: Disbelief and Confusion
GOOD-LOOKING ME
(Unsteadily hands document to Miser)
Sir, I’ve highlighted the national benchmarks for you to see. Column four of this 2007-2008 SHM survey clearly shows that the average academic hospitalist should make $168,800 and achieve 2,813 work RVUs. We achieve the latter benchmark but are severely underpaid.
MISER
(Produces a folded cocktail napkin from his shirt pocket)
But look at this: My executive-friends-at-other-medical-centers-who-overwork-and-underpay-their-hospitalists benchmark shows that you should be well over 4,500 work RVUs. And besides, the SHM numbers are skewed; it’s a survey of hospitalists done by a group that represents hospitalists. I don’t believe them.
GOOD-LOOKING ME
(Eyes averted, adopts a tone of trepidation)
But sir, with all due respect, don’t your numbers reflect a survey of hospital administrators who might have a bias toward more expected productivity? Which benchmark should we believe?
(Blackout and end of Act II.)
A New HM Benchmark Arises
It’s all about the benchmark you choose to believe. For years, the best source of data regarding hospitalist compensation and productivity was that published every other year by SHM. It is a fair, but unfounded, concern that these data might tilt toward the benefit of hospitalists. Likewise, the hospital administrator I work most closely with (who, for the record, reads this publication and IS NOT miserly, has a FULL HEAD of hair, and is, for innumerable reasons, a TRULY GREAT man) will produce benchmarks from organizations like the Association of American Medical Colleges (AAMC) or the University HealthSystems Consortium (UHC), all of which show surprisingly disparate numbers dripping with a similar tilt toward the medical center.
Thus, the importance of the 2010 SHM/MGMA report. The Medical Group Management Association (MGMA) consists of administrators and leaders of medical group practices. Since 1926, they’ve been providing accurate, independent data on physician practice metrics. For most hospital administrators, it is the benchmark. The problem is that in the past, MGMA has struggled to identify hospitalists; the MGMA data were always underpowered and, therefore, suspect.
Enter SHM and its large database of HM groups. What has resulted in the new survey rivals those old commercials in which a person walking with a piece of chocolate slams into another with a jar of peanut butter, resulting in the creation of the Reese’s Peanut Butter Cup (apologies to those readers under the age of 35).
Act III: No Raise; Children Go Hungry
GOOD-LOOKING ME
(Unsheathing haloed document from his portfolio)
Perhaps we could agree to use these new SHM/MGMA numbers as our benchmark. It includes data from more than 440 HM groups and 4,200 hospitalists. And it appears to be fair and balanced.
MISER
(Eyes alight, peering through a shroud of compromise)
MGMA, huh? Let’s take a look. Hmmm. Well. But wait—this says the average hospitalist makes $215,000! That’s outrageous.
GOOD-LOOKING ME
(Smugly retorts)
Yes, sir, we are severely underpaid.
MISER
(Reading; a weasel-like countenance overtakes his face)
Let me take a closer look at this. Aha! Here it is. You see, this only included community hospitalist practices. You will be getting no raise!
(Blackout and end of Act III.)
A Cautionary Tale
Alas, the miser is right. It’s not always what the data say but also what they don’t say.
The one snag with the new data is that it only included a handful of academic HM groups (only 1% of respondents). In fact, the survey actively instructed academic HM practices to not complete the survey. Rather, we academic types were instructed to await the MGMA survey of academic practices completed every fall to be reported early next year.
This is emblematic of the need to dig deep when interpreting these data. As tempting as it is to use a sound bite or two of these data to your advantage, the truth lies in the details. It’s easy to say that all hospitalists should make $215,000, see 2,229 encounters, and achieve 4,107 wRVUs annually.
However, just as there is no average hospitalist, there are no average numbers. There are just too many variables (e.g., practice ownership, geography, group size, night coverage, staffing model, compensation structure) to say definitively what an individual hospitalist should look like or achieve. Rather, these numbers should be used as a guide, adapted to each individual situation.
Act IV: See You This Spring
(Standing, Good-Looking Me shakes his foe’s shriveled claw of a hand while looking him intensely in the eye—a look that says, “I’ll see you this spring.” In his rival’s eyes, the Miser sees his future—a future that involves another meeting, more practice-appropriate data, and a dusting off of his checkbook.)
(Blackout and end of Act IV.) TH
Dr. Glasheen is associate professor of medicine at the University of Colorado Denver, where he serves as director of the Hospital Medicine Program and the Hospitalist Training Program, and as associate program director of the Internal Medicine Residency Program.
Act I: The Negotiation
(A barren academic office, dimly lit, the pall of difficult negotiations afloat, backlit like dust in the air. Seated, under a strangely intense incandescent bulb, a man, who looks eerily like a good-looking version of me, sits uncomfortably adjusting himself in his seat. His eyes constrict on his counterpart, a miserly sort peering out from behind wire-rim glasses and a shock of hair improbably combed over from ear to ear. The tension crests.)
GOOD-LOOKING ME
(Voice cracking)
I’ve come to ask for a raise for our hospitalist group.
MISER
(Adjusts his clip-on tie)
We just gave you a raise in 2004.
GOOD-LOOKING ME
(Smiles uncomfortably)
That was very gracious, sir, but I think the numbers support another.
MISER
(Incredulous look at his watch)
But your work RVUs are thousands below what I’d like to see.
GOOD-LOOKING ME
(Dabs bead of sweat away from chiseled chin)
That’s because you’ve set your benchmark thousands above a reasonable number.
MISER
(Voice flitting with child-like condescension)
But those are the numbers my finance guy gave me. It’s the benchmark.
(Blackout and end of Act I.)
Mutual Agreement
Tony Award-winning stuff for sure—and based on a true story! In fact, this scene no doubt plays out annually for those of you unfortunate enough to have to negotiate with hospital executives for programmatic support. To be fair, hospital administrators deserve to know that they are getting what they pay for. Thus, the concepts of a benchmark are reasonable. The problem lies in setting mutually-agreed-upon standards.
Act II: Disbelief and Confusion
GOOD-LOOKING ME
(Unsteadily hands document to Miser)
Sir, I’ve highlighted the national benchmarks for you to see. Column four of this 2007-2008 SHM survey clearly shows that the average academic hospitalist should make $168,800 and achieve 2,813 work RVUs. We achieve the latter benchmark but are severely underpaid.
MISER
(Produces a folded cocktail napkin from his shirt pocket)
But look at this: My executive-friends-at-other-medical-centers-who-overwork-and-underpay-their-hospitalists benchmark shows that you should be well over 4,500 work RVUs. And besides, the SHM numbers are skewed; it’s a survey of hospitalists done by a group that represents hospitalists. I don’t believe them.
GOOD-LOOKING ME
(Eyes averted, adopts a tone of trepidation)
But sir, with all due respect, don’t your numbers reflect a survey of hospital administrators who might have a bias toward more expected productivity? Which benchmark should we believe?
(Blackout and end of Act II.)
A New HM Benchmark Arises
It’s all about the benchmark you choose to believe. For years, the best source of data regarding hospitalist compensation and productivity was that published every other year by SHM. It is a fair, but unfounded, concern that these data might tilt toward the benefit of hospitalists. Likewise, the hospital administrator I work most closely with (who, for the record, reads this publication and IS NOT miserly, has a FULL HEAD of hair, and is, for innumerable reasons, a TRULY GREAT man) will produce benchmarks from organizations like the Association of American Medical Colleges (AAMC) or the University HealthSystems Consortium (UHC), all of which show surprisingly disparate numbers dripping with a similar tilt toward the medical center.
Thus, the importance of the 2010 SHM/MGMA report. The Medical Group Management Association (MGMA) consists of administrators and leaders of medical group practices. Since 1926, they’ve been providing accurate, independent data on physician practice metrics. For most hospital administrators, it is the benchmark. The problem is that in the past, MGMA has struggled to identify hospitalists; the MGMA data were always underpowered and, therefore, suspect.
Enter SHM and its large database of HM groups. What has resulted in the new survey rivals those old commercials in which a person walking with a piece of chocolate slams into another with a jar of peanut butter, resulting in the creation of the Reese’s Peanut Butter Cup (apologies to those readers under the age of 35).
Act III: No Raise; Children Go Hungry
GOOD-LOOKING ME
(Unsheathing haloed document from his portfolio)
Perhaps we could agree to use these new SHM/MGMA numbers as our benchmark. It includes data from more than 440 HM groups and 4,200 hospitalists. And it appears to be fair and balanced.
MISER
(Eyes alight, peering through a shroud of compromise)
MGMA, huh? Let’s take a look. Hmmm. Well. But wait—this says the average hospitalist makes $215,000! That’s outrageous.
GOOD-LOOKING ME
(Smugly retorts)
Yes, sir, we are severely underpaid.
MISER
(Reading; a weasel-like countenance overtakes his face)
Let me take a closer look at this. Aha! Here it is. You see, this only included community hospitalist practices. You will be getting no raise!
(Blackout and end of Act III.)
A Cautionary Tale
Alas, the miser is right. It’s not always what the data say but also what they don’t say.
The one snag with the new data is that it only included a handful of academic HM groups (only 1% of respondents). In fact, the survey actively instructed academic HM practices to not complete the survey. Rather, we academic types were instructed to await the MGMA survey of academic practices completed every fall to be reported early next year.
This is emblematic of the need to dig deep when interpreting these data. As tempting as it is to use a sound bite or two of these data to your advantage, the truth lies in the details. It’s easy to say that all hospitalists should make $215,000, see 2,229 encounters, and achieve 4,107 wRVUs annually.
However, just as there is no average hospitalist, there are no average numbers. There are just too many variables (e.g., practice ownership, geography, group size, night coverage, staffing model, compensation structure) to say definitively what an individual hospitalist should look like or achieve. Rather, these numbers should be used as a guide, adapted to each individual situation.
Act IV: See You This Spring
(Standing, Good-Looking Me shakes his foe’s shriveled claw of a hand while looking him intensely in the eye—a look that says, “I’ll see you this spring.” In his rival’s eyes, the Miser sees his future—a future that involves another meeting, more practice-appropriate data, and a dusting off of his checkbook.)
(Blackout and end of Act IV.) TH
Dr. Glasheen is associate professor of medicine at the University of Colorado Denver, where he serves as director of the Hospital Medicine Program and the Hospitalist Training Program, and as associate program director of the Internal Medicine Residency Program.
Act I: The Negotiation
(A barren academic office, dimly lit, the pall of difficult negotiations afloat, backlit like dust in the air. Seated, under a strangely intense incandescent bulb, a man, who looks eerily like a good-looking version of me, sits uncomfortably adjusting himself in his seat. His eyes constrict on his counterpart, a miserly sort peering out from behind wire-rim glasses and a shock of hair improbably combed over from ear to ear. The tension crests.)
GOOD-LOOKING ME
(Voice cracking)
I’ve come to ask for a raise for our hospitalist group.
MISER
(Adjusts his clip-on tie)
We just gave you a raise in 2004.
GOOD-LOOKING ME
(Smiles uncomfortably)
That was very gracious, sir, but I think the numbers support another.
MISER
(Incredulous look at his watch)
But your work RVUs are thousands below what I’d like to see.
GOOD-LOOKING ME
(Dabs bead of sweat away from chiseled chin)
That’s because you’ve set your benchmark thousands above a reasonable number.
MISER
(Voice flitting with child-like condescension)
But those are the numbers my finance guy gave me. It’s the benchmark.
(Blackout and end of Act I.)
Mutual Agreement
Tony Award-winning stuff for sure—and based on a true story! In fact, this scene no doubt plays out annually for those of you unfortunate enough to have to negotiate with hospital executives for programmatic support. To be fair, hospital administrators deserve to know that they are getting what they pay for. Thus, the concepts of a benchmark are reasonable. The problem lies in setting mutually-agreed-upon standards.
Act II: Disbelief and Confusion
GOOD-LOOKING ME
(Unsteadily hands document to Miser)
Sir, I’ve highlighted the national benchmarks for you to see. Column four of this 2007-2008 SHM survey clearly shows that the average academic hospitalist should make $168,800 and achieve 2,813 work RVUs. We achieve the latter benchmark but are severely underpaid.
MISER
(Produces a folded cocktail napkin from his shirt pocket)
But look at this: My executive-friends-at-other-medical-centers-who-overwork-and-underpay-their-hospitalists benchmark shows that you should be well over 4,500 work RVUs. And besides, the SHM numbers are skewed; it’s a survey of hospitalists done by a group that represents hospitalists. I don’t believe them.
GOOD-LOOKING ME
(Eyes averted, adopts a tone of trepidation)
But sir, with all due respect, don’t your numbers reflect a survey of hospital administrators who might have a bias toward more expected productivity? Which benchmark should we believe?
(Blackout and end of Act II.)
A New HM Benchmark Arises
It’s all about the benchmark you choose to believe. For years, the best source of data regarding hospitalist compensation and productivity was that published every other year by SHM. It is a fair, but unfounded, concern that these data might tilt toward the benefit of hospitalists. Likewise, the hospital administrator I work most closely with (who, for the record, reads this publication and IS NOT miserly, has a FULL HEAD of hair, and is, for innumerable reasons, a TRULY GREAT man) will produce benchmarks from organizations like the Association of American Medical Colleges (AAMC) or the University HealthSystems Consortium (UHC), all of which show surprisingly disparate numbers dripping with a similar tilt toward the medical center.
Thus, the importance of the 2010 SHM/MGMA report. The Medical Group Management Association (MGMA) consists of administrators and leaders of medical group practices. Since 1926, they’ve been providing accurate, independent data on physician practice metrics. For most hospital administrators, it is the benchmark. The problem is that in the past, MGMA has struggled to identify hospitalists; the MGMA data were always underpowered and, therefore, suspect.
Enter SHM and its large database of HM groups. What has resulted in the new survey rivals those old commercials in which a person walking with a piece of chocolate slams into another with a jar of peanut butter, resulting in the creation of the Reese’s Peanut Butter Cup (apologies to those readers under the age of 35).
Act III: No Raise; Children Go Hungry
GOOD-LOOKING ME
(Unsheathing haloed document from his portfolio)
Perhaps we could agree to use these new SHM/MGMA numbers as our benchmark. It includes data from more than 440 HM groups and 4,200 hospitalists. And it appears to be fair and balanced.
MISER
(Eyes alight, peering through a shroud of compromise)
MGMA, huh? Let’s take a look. Hmmm. Well. But wait—this says the average hospitalist makes $215,000! That’s outrageous.
GOOD-LOOKING ME
(Smugly retorts)
Yes, sir, we are severely underpaid.
MISER
(Reading; a weasel-like countenance overtakes his face)
Let me take a closer look at this. Aha! Here it is. You see, this only included community hospitalist practices. You will be getting no raise!
(Blackout and end of Act III.)
A Cautionary Tale
Alas, the miser is right. It’s not always what the data say but also what they don’t say.
The one snag with the new data is that it only included a handful of academic HM groups (only 1% of respondents). In fact, the survey actively instructed academic HM practices to not complete the survey. Rather, we academic types were instructed to await the MGMA survey of academic practices completed every fall to be reported early next year.
This is emblematic of the need to dig deep when interpreting these data. As tempting as it is to use a sound bite or two of these data to your advantage, the truth lies in the details. It’s easy to say that all hospitalists should make $215,000, see 2,229 encounters, and achieve 4,107 wRVUs annually.
However, just as there is no average hospitalist, there are no average numbers. There are just too many variables (e.g., practice ownership, geography, group size, night coverage, staffing model, compensation structure) to say definitively what an individual hospitalist should look like or achieve. Rather, these numbers should be used as a guide, adapted to each individual situation.
Act IV: See You This Spring
(Standing, Good-Looking Me shakes his foe’s shriveled claw of a hand while looking him intensely in the eye—a look that says, “I’ll see you this spring.” In his rival’s eyes, the Miser sees his future—a future that involves another meeting, more practice-appropriate data, and a dusting off of his checkbook.)
(Blackout and end of Act IV.) TH
Dr. Glasheen is associate professor of medicine at the University of Colorado Denver, where he serves as director of the Hospital Medicine Program and the Hospitalist Training Program, and as associate program director of the Internal Medicine Residency Program.