User login
Wegener's Granulomatosis
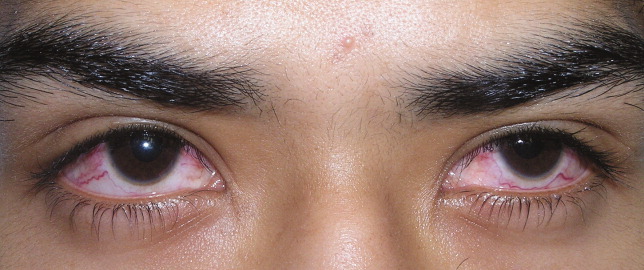
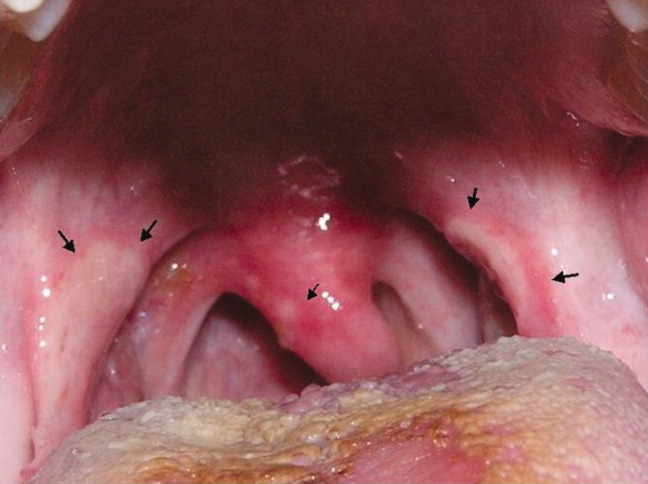
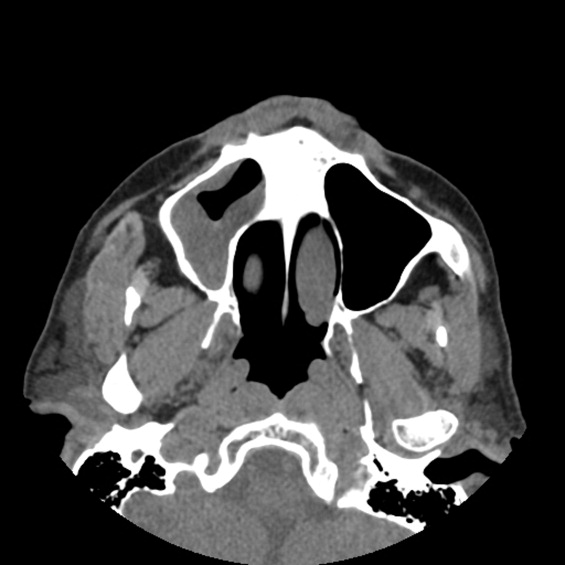
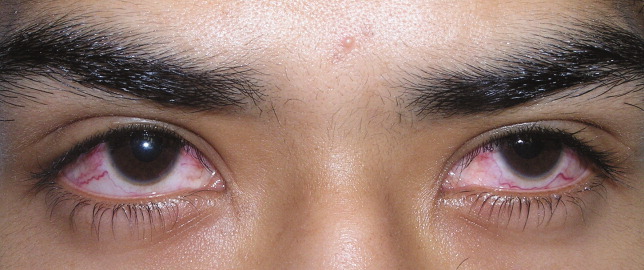
A previously healthy 21‐year‐old man presented with 6 weeks of low‐grade fever, sore throat, red eyes, and hematuria. Physical examination revealed episcleral injection consistent with episcleritis (Figure 1), oral ulcers (Figure 2, black arrows), diffuse fine crackles on chest auscultation and testicular tenderness. Laboratory workup was significant for leukocytosis (14,000 cell/mL), hematuria with red blood cell (RBC) casts and serum creatinine level of 2.1 mg/dL, which subsequently rose rapidly to 4.1 mg/dL. Test for cytoplasmic‐stainingantineutrophil cytoplasmic antibody (C‐ANCA) was positive. Antiproteinase 3 (PR3) antibodies were also positive. Chest x‐ray showed bilateral pulmonary opacities and sinus computed tomography (CT) scan showed mucosal thickening of the sinuses consistent with sinusitis (Figure 3). Renal biopsy revealed segmental necrotizing glomerulonephritis that was pauci‐immune on immunofluorescence staining. The patient was diagnosed with Wegener's granulomatosis with rapidly progressive glomerulonephritis. He was treated with intravenous corticosteroids, cyclophosphamide, and trimethoprim‐sulfamethoxazole. The patient's symptoms and acute renal failure resolved with this medical regimen.
A previously healthy 21‐year‐old man presented with 6 weeks of low‐grade fever, sore throat, red eyes, and hematuria. Physical examination revealed episcleral injection consistent with episcleritis (Figure 1), oral ulcers (Figure 2, black arrows), diffuse fine crackles on chest auscultation and testicular tenderness. Laboratory workup was significant for leukocytosis (14,000 cell/mL), hematuria with red blood cell (RBC) casts and serum creatinine level of 2.1 mg/dL, which subsequently rose rapidly to 4.1 mg/dL. Test for cytoplasmic‐stainingantineutrophil cytoplasmic antibody (C‐ANCA) was positive. Antiproteinase 3 (PR3) antibodies were also positive. Chest x‐ray showed bilateral pulmonary opacities and sinus computed tomography (CT) scan showed mucosal thickening of the sinuses consistent with sinusitis (Figure 3). Renal biopsy revealed segmental necrotizing glomerulonephritis that was pauci‐immune on immunofluorescence staining. The patient was diagnosed with Wegener's granulomatosis with rapidly progressive glomerulonephritis. He was treated with intravenous corticosteroids, cyclophosphamide, and trimethoprim‐sulfamethoxazole. The patient's symptoms and acute renal failure resolved with this medical regimen.
A previously healthy 21‐year‐old man presented with 6 weeks of low‐grade fever, sore throat, red eyes, and hematuria. Physical examination revealed episcleral injection consistent with episcleritis (Figure 1), oral ulcers (Figure 2, black arrows), diffuse fine crackles on chest auscultation and testicular tenderness. Laboratory workup was significant for leukocytosis (14,000 cell/mL), hematuria with red blood cell (RBC) casts and serum creatinine level of 2.1 mg/dL, which subsequently rose rapidly to 4.1 mg/dL. Test for cytoplasmic‐stainingantineutrophil cytoplasmic antibody (C‐ANCA) was positive. Antiproteinase 3 (PR3) antibodies were also positive. Chest x‐ray showed bilateral pulmonary opacities and sinus computed tomography (CT) scan showed mucosal thickening of the sinuses consistent with sinusitis (Figure 3). Renal biopsy revealed segmental necrotizing glomerulonephritis that was pauci‐immune on immunofluorescence staining. The patient was diagnosed with Wegener's granulomatosis with rapidly progressive glomerulonephritis. He was treated with intravenous corticosteroids, cyclophosphamide, and trimethoprim‐sulfamethoxazole. The patient's symptoms and acute renal failure resolved with this medical regimen.
Pulmonary Artery Dissection
A 51‐year‐old African American woman with medical history of essential hypertension and chronic obstructive pulmonary disease (COPD) presented to the hospital with chest pain and shortness of breath. The chest pain was retrosternal and radiated to the back. It lasted for about an hour and resolved without any intervention. After some time, she again felt discomfort in the chest, which was a constant and dull ache.
She had similar episodes of chest pain 1 week prior, although less severe in intensity, for which she went to an outside hospital before coming to our hospital. Acute coronary syndrome was ruled out with serial cardiac enzymes measurements. An exercise stress test was also performed at that time, which failed to show any stress‐induced ischemia.
Her medications included lisinopril for hypertension and aspirin, which had been started 1 week prior to admission. She gave a 10‐pack‐year history of smoking tobacco. Family history was significant for hypertension in her father and coronary artery disease in her mother at the age of 58 years. A review of systems was negative for fever, cough, orthopnea, wheezing, palpitations, nausea, vomiting, recent surgery, or any significant trauma.
Assessment
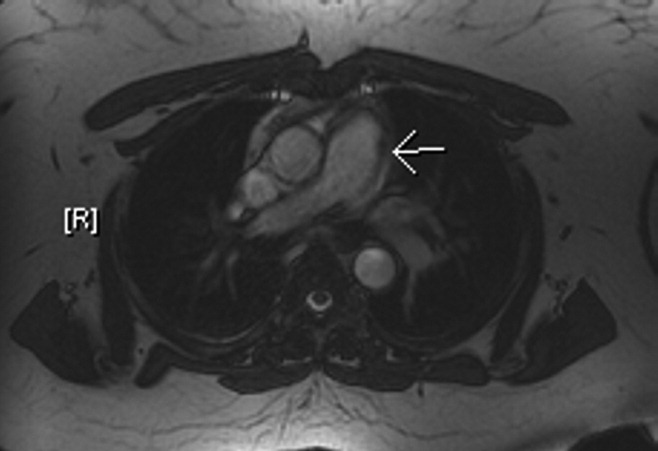
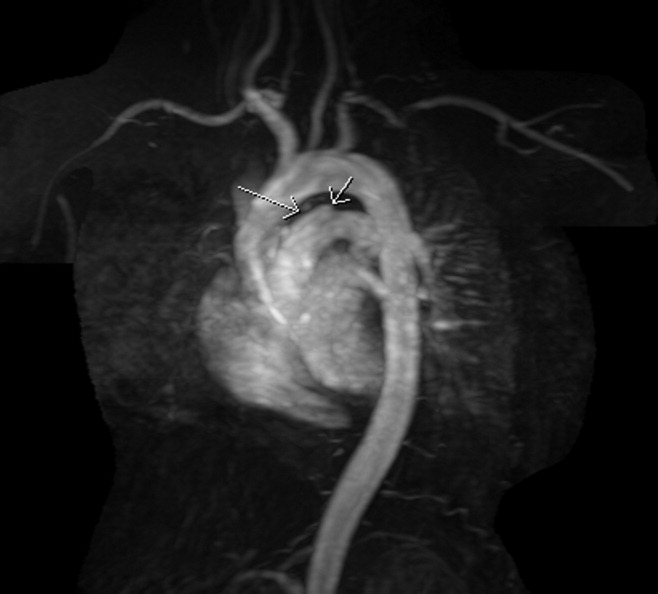
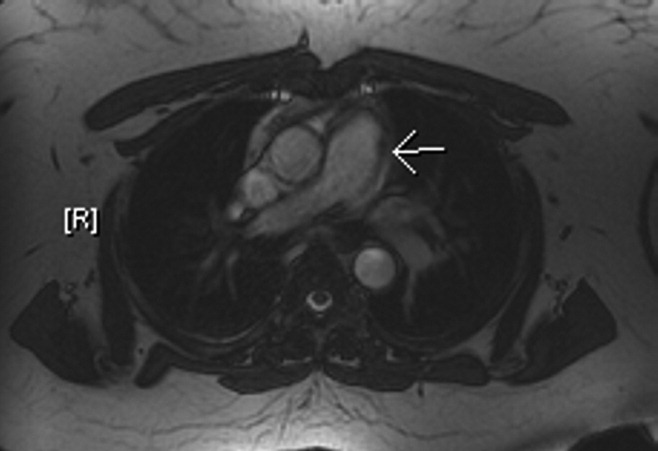
The patient's physical examination was only remarkable for a blood pressure (BP) of 181/100 mm Hg. She did not have Marfanoid features, hyperflexible joints, or easy bruisability. Laboratory tests, including complete blood count, comprehensive metabolic panel, and cardiac enzymes, were within normal limits. A contrast‐enhanced computed tomography (CT) of the chest showed a linear hypodense area in the left lateral aspect of the main pulmonary trunk, which suggested an intimal dissection of the main pulmonary artery. Magnetic resonance angiography/emmaging (MRA/MRI) confirmed dissection of the main pulmonary artery extending into the proximal left pulmonary artery and associated with a 12 8 mm2 aneurysm (Figures 1 and 2). The entry site of dissection was located in the main pulmonary artery just after its origin and the exit site was located in the left pulmonary artery 5 mm distal to the bifurcation of main pulmonary artery. The pulmonary artery diameter at the dissection was 27 mm.
Diagnosis
To investigate possible etiologies, a transthoracic echocardiogram with Doppler was done to look for pulmonary hypertension. The echocardiogram showed normal pulmonary artery pressure with normal right ventricular systolic pressure. There was no evidence of pericardial effusion or structural cardiac abnormality on echocardiogram. Further investigations including work up for connective tissue diseases and infectious etiologies (Table 1) were normal.
| Variable | Reference Range | Patient's Result |
|---|---|---|
| ||
| ANA | Negative | Negative |
| C3 complement level | 88201 | 175 |
| C4 complement level | 1647 | 49 |
| RF | 20 | 20 |
| Anti‐centromere Ab | Negative | Negative |
| Anti‐Scl 70 Ab | Negative | Negative |
| Anti‐smooth muscle Ab | Negative | Negative |
| Anti‐mitochondrial Ab | Negative | Negative |
| Anti‐parietal cell Ab | Negative | Negative |
| TB skin test | 5 mm | |
| RPR | Negative | Negative |
According to Shilkin et al.,1 Helmbrecht first reported pulmonary artery dissection (PAD) in 1842. PAD is very rare and is usually diagnosed at autopsy. There are 71 other cases of PAD reported in the English literature, of which 16 cases are in living patients.217 Unlike aortic dissection, which is fairly common, the reentry circuit for PAD is formed by the rupture of the free wall of the pulmonary artery leading to hemopericardium, cardiac tamponade, and sudden death.2, 8, 9 There is wide variation in age of incidence, ranging from 26 to 85 years of age, with a slightly higher prevalence in females (male‐to‐female ratio 1:1.2).1, 2 The main pulmonary artery is usually involved, with or without involvement of its branches. Isolated left and right pulmonary artery involvement is seen in 6% and 4% of cases, respectively.2
Pulmonary hypertension, either primary or secondary (collagen vascular diseases, COPD, congenital heart diseases, etc.), is the most common underlying etiology. Other less common, but well‐described etiologies include: Marfan's syndrome, instrumentation of pulmonary artery, tuberculosis, syphilis, pregnancy, idiopathic cystic medial necrosis, and amyloidosis.2, 8
As noted earlier, underlying pulmonary hypertension is usually a major risk factor for PAD. More than 75% of the patients have underlying acute or chronic pulmonary hypertension.2 Our patient had COPD without pulmonary hypertension. Despite extensive investigation, no underlying cause of her pulmonary dissection was identified. The differential diagnosis includes cryptogenic cystic medionecrosis; however, because the patient refused surgery the diagnosis remains unknown. As in our case, idiopathic PAD is extremely rare, and only 4 other cases have been described in the literature.2 Underlying etiologies should always be ruled out to identify correctable causes such as congenital abnormalities of the heart leading to pulmonary hypertension.
Chest pain is a very common presenting complaint in the emergency department. Although rare, PAD should be suspected in a patient with retrosternal chest pain when other common causes of chest pain are excluded. Some of the more suggestive findings are the presence of a new diastolic murmur, a wide mediastinum on chest x‐ray, and CT scan of chest showing an intimal flap.2, 8 CT scan of the chest is an acceptable imaging modality to diagnose PAD.18 According to Neimatallah et al.,18 there are only 5 cases in the literature reported with detailed CT scans demonstrating PAD. If the diagnosis remains uncertain, it should be confirmed by MRI/MRA.16 Transthoracic echocardiography can be used for diagnosis and follow‐up of conservatively managed patients with PAD.3, 8, 19 The echocardiographic findings suggestive of PAD include direct or indirect evidence of pulmonary artery hypertension, with a large main pulmonary artery and an intimal flap across the pulmonary trunk.
Management
No consensus strategy is available for the management of PAD because of the rarity of this condition. In general, operative repair is the treatment of choice for PAD.2, 8, 9, 11 There are 16 cases diagnosed in living patients, out of which 6 were managed medically, 8 were managed surgically, and management was not discussed in 2 of the case reports (Table 2). In these case reports, medically managed patients were treated with oxygen, vasodilators (nitrates, angiotensin‐converting enzyme [ACE] inhibitors, dihydropyridine calcium channel blockers, sildenafil), diuretics and beta‐blockers. These patients did well on follow‐up, ranging from 3 weeks to 4 years, except for 1 who died on day 4 in the intensive care unit (ICU).
| Case Report | Etiology of PAD | Management | Outcome |
|---|---|---|---|
| |||
| Janus et al.3 | Balloon valvuloplasty for pulmonary stenosis | Medical (beta blocker) | Stable during 4 years of follow‐up |
| Khattar et al.8 | Secondary PH from COPD | Medical (diuretics, ACE inhibitor) | Stable during 1 year of follow‐up |
| Lobato et al.9 | Aortic valve replacement | Medical (vasodilators, diuretics) | Stable during 3 weeks of follow‐up |
| Smalcelj et al.10 | Primary PH | Medical (Sildenafil) | Stable during 8 months of follow‐up |
| Song and Kolecki11 | Secondary PH from VSD, Eisenmenger's syndrome | Medical (Nitroprusside) | Patient died on day 4 of admission |
| Steurer et al.15 | Primary PH | Medical (ACE inhibitor, CCB) | Stable during 1 year of follow‐up |
| Wuyts et al.4 | Secondary PH from VSD | Surgical (heart lung transplant) | Follow‐up not mentioned |
| Sakamaki et. al.5 | Primary PH | Surgical (reanastomosis) | Stable during 37 months of follow‐up |
| Westaby et al.7 | Secondary PH from VSD, Eisenmenger's syndrome | Surgical (vascular prosthesis) | Follow‐up not discussed, stable on discharge on tenth day |
| Senbaklavaci et al.12 | Primary PH | Surgical | Stable during 10 months of follow‐up |
| Inayama et al.2 | PH secondary to pulmonary thrombosis | Surgical | Follow‐up not discussed, stable at discharge |
| Wunderbaldinger et al.13 | Primary PH | Surgical | Follow‐up not discussed |
| Lopez‐Candales et al.14 | Secondary PH from partially corrected pulmonary stenosis | Surgical | Follow‐up not discussed, stable on discharge at 1 week |
| Khatchatourian and Vala17 | Associated with aortic dissection | Surgical | Stable during 3 months of follow‐up |
| Rosenson and Sutton6 | Secondary PH from mitral stenosis | Management not discussed | Follow‐up not discussed |
| Stern et al.16 | Secondary PH from hypersensitivity pneumonitis | Management not discussed | Follow‐up not discussed |
Conservative management may be tried in patients who are hemodynamically stable and do not have pericardial effusion.2, 9 The aim of conservative management is to decrease right ventricular preload and afterload. Preload reduction can be dangerous in patients with PAD and should be done in the intensive care setting as this can lead to profound hypotension. Nitrates for preload reduction should be used cautiously in patients taking sildenafil or similar agents for erectile dysfunction or pulmonary artery hypertension because of significant risk of cardiovascular collapse. The American Heart Association and American College of Cardiology both recommend that there should be a time gap of at least 24 hours between the last dose of sildenafil and the first dose of nitrates. Conservatively managed patients should be followed with interval CT scans2, 9, 18 or echocardiography.3, 19 In addition, the underlying etiology should always be investigated to predict prognosis and recommend future management strategies.
The patient was offered surgical repair but she declined. She was managed conservatively with nitrates and beta‐blockers and was pain free within 24 hours. Her BP was brought down to a systolic BP range of 130140 mm Hg. A repeat CT scan of the chest at 1‐month follow up was unchanged. The patient was doing well at 6‐month follow‐up.
Conclusions
PAD is an extremely rare cause of chest pain and a rare antemortem diagnosis. It is usually associated with underlying pulmonary hypertension. This case describes a patient with PAD in the absence of pulmonary hypertension. The patient was managed with conservative medical therapy and did well at 6‐month follow‐up. There are a total of 6 other case reports of patients with PAD managed conservatively, out of which 5 patients did well at follow‐up and 1 patient died. More case reports and longer follow‐up are needed to assess the effectiveness of conservative medical therapy in patients with PAD. To our knowledge, this is the first case report of idiopathic PAD diagnosed in a living patient and managed conservatively. This case also highlights better prognosis for patients with PAD without underlying pulmonary hypertension.
- ,,.Dissecting aneurysm of the pulmonary artery.J. Pathol.1969;98;25–29.
- ,,.Pulmonary artery dissection in patients without underlying pulmonary hypertension.Histopathology.2001;38:435–442.
- ,,,,,.Pulmonary artery dissection: a rare complication of pulmonary balloon valvuloplasty diagnosed 11 years after the procedure.J Am Soc Echocardiogr.2006;19:1191,e1195–e1198.
- ,,,,.Extensive dissection of the pulmonary artery treated with combined heart‐lung transplantation.J Thorac Cardiovasc Surg.2006;132:205–206.
- ,,, et al.Pulmonary artery dissection complicating lung transplantation for primary pulmonary hypertension.Ann Thorac Surg.2006;81:360–362.
- ,.Dissecting aneurysm of the pulmonary trunk in mitral stenosis.Am J Cardiol.1986;58:1140–1141.
- ,,.Pulmonary‐artery dissection in patients with Eisenmenger's syndrome.N Engl J Med.2007;356:2110–2112.
- ,,,.Pulmonary artery dissection: an emerging cardiovascular complication in surviving patients with chronic pulmonary hypertension.Heart.2005;91:142–145.
- ,,, et al.Pulmonary artery dissection and conservative medical management.Int J Cardiol.2007;119:e25–e26.
- ,,,,,.Giant, dissecting, high‐pressure pulmonary artery aneurysm: case report of a 1‐year natural course.Tex Heart Inst J.2005;32:589–594.
- ,.A case of pulmonary artery dissection diagnosed in the Emergency Department.J Emerg Med.2002;23:155–159.
- ,,, et al.Rupture and dissection in pulmonary artery aneurysms: incidence, cause, and treatment—review and case report.J Thorac Cardiovasc Surg.2001;121:1006–1008.
- ,,,,,.Acute pulmonary trunk dissection in a patient with primary pulmonary hypertension.J Comput Assist Tomogr.2000;24:92–95.
- ,,,,.Pulmonary artery aneurysm: review and case report.Clin Cardiol.1995;18:738–740.
- ,,,,,.Dissecting aneurysm of the pulmonary artery with pulmonary hypertension.Am Rev Respir Dis.1990;142:1219–1221.
- ,,,,.Pulmonary artery dissection: MR findings.J Comput Assist Tomogr.1992;16:481–483.
- ,.Images in cardiovascular medicine. Acute type I aortic dissection with concomitant pulmonary artery dissection.Circulation.2005;112:e313–314.
- ,,,.CT findings of pulmonary artery dissection.Br J Radiol.2007;80:e61–e63.
- ,.Pulmonary artery dissection: echocardiographic findings and diagnosis.Echocardiography.2003;20:375–377.
A 51‐year‐old African American woman with medical history of essential hypertension and chronic obstructive pulmonary disease (COPD) presented to the hospital with chest pain and shortness of breath. The chest pain was retrosternal and radiated to the back. It lasted for about an hour and resolved without any intervention. After some time, she again felt discomfort in the chest, which was a constant and dull ache.
She had similar episodes of chest pain 1 week prior, although less severe in intensity, for which she went to an outside hospital before coming to our hospital. Acute coronary syndrome was ruled out with serial cardiac enzymes measurements. An exercise stress test was also performed at that time, which failed to show any stress‐induced ischemia.
Her medications included lisinopril for hypertension and aspirin, which had been started 1 week prior to admission. She gave a 10‐pack‐year history of smoking tobacco. Family history was significant for hypertension in her father and coronary artery disease in her mother at the age of 58 years. A review of systems was negative for fever, cough, orthopnea, wheezing, palpitations, nausea, vomiting, recent surgery, or any significant trauma.
Assessment
The patient's physical examination was only remarkable for a blood pressure (BP) of 181/100 mm Hg. She did not have Marfanoid features, hyperflexible joints, or easy bruisability. Laboratory tests, including complete blood count, comprehensive metabolic panel, and cardiac enzymes, were within normal limits. A contrast‐enhanced computed tomography (CT) of the chest showed a linear hypodense area in the left lateral aspect of the main pulmonary trunk, which suggested an intimal dissection of the main pulmonary artery. Magnetic resonance angiography/emmaging (MRA/MRI) confirmed dissection of the main pulmonary artery extending into the proximal left pulmonary artery and associated with a 12 8 mm2 aneurysm (Figures 1 and 2). The entry site of dissection was located in the main pulmonary artery just after its origin and the exit site was located in the left pulmonary artery 5 mm distal to the bifurcation of main pulmonary artery. The pulmonary artery diameter at the dissection was 27 mm.
Diagnosis
To investigate possible etiologies, a transthoracic echocardiogram with Doppler was done to look for pulmonary hypertension. The echocardiogram showed normal pulmonary artery pressure with normal right ventricular systolic pressure. There was no evidence of pericardial effusion or structural cardiac abnormality on echocardiogram. Further investigations including work up for connective tissue diseases and infectious etiologies (Table 1) were normal.
| Variable | Reference Range | Patient's Result |
|---|---|---|
| ||
| ANA | Negative | Negative |
| C3 complement level | 88201 | 175 |
| C4 complement level | 1647 | 49 |
| RF | 20 | 20 |
| Anti‐centromere Ab | Negative | Negative |
| Anti‐Scl 70 Ab | Negative | Negative |
| Anti‐smooth muscle Ab | Negative | Negative |
| Anti‐mitochondrial Ab | Negative | Negative |
| Anti‐parietal cell Ab | Negative | Negative |
| TB skin test | 5 mm | |
| RPR | Negative | Negative |
According to Shilkin et al.,1 Helmbrecht first reported pulmonary artery dissection (PAD) in 1842. PAD is very rare and is usually diagnosed at autopsy. There are 71 other cases of PAD reported in the English literature, of which 16 cases are in living patients.217 Unlike aortic dissection, which is fairly common, the reentry circuit for PAD is formed by the rupture of the free wall of the pulmonary artery leading to hemopericardium, cardiac tamponade, and sudden death.2, 8, 9 There is wide variation in age of incidence, ranging from 26 to 85 years of age, with a slightly higher prevalence in females (male‐to‐female ratio 1:1.2).1, 2 The main pulmonary artery is usually involved, with or without involvement of its branches. Isolated left and right pulmonary artery involvement is seen in 6% and 4% of cases, respectively.2
Pulmonary hypertension, either primary or secondary (collagen vascular diseases, COPD, congenital heart diseases, etc.), is the most common underlying etiology. Other less common, but well‐described etiologies include: Marfan's syndrome, instrumentation of pulmonary artery, tuberculosis, syphilis, pregnancy, idiopathic cystic medial necrosis, and amyloidosis.2, 8
As noted earlier, underlying pulmonary hypertension is usually a major risk factor for PAD. More than 75% of the patients have underlying acute or chronic pulmonary hypertension.2 Our patient had COPD without pulmonary hypertension. Despite extensive investigation, no underlying cause of her pulmonary dissection was identified. The differential diagnosis includes cryptogenic cystic medionecrosis; however, because the patient refused surgery the diagnosis remains unknown. As in our case, idiopathic PAD is extremely rare, and only 4 other cases have been described in the literature.2 Underlying etiologies should always be ruled out to identify correctable causes such as congenital abnormalities of the heart leading to pulmonary hypertension.
Chest pain is a very common presenting complaint in the emergency department. Although rare, PAD should be suspected in a patient with retrosternal chest pain when other common causes of chest pain are excluded. Some of the more suggestive findings are the presence of a new diastolic murmur, a wide mediastinum on chest x‐ray, and CT scan of chest showing an intimal flap.2, 8 CT scan of the chest is an acceptable imaging modality to diagnose PAD.18 According to Neimatallah et al.,18 there are only 5 cases in the literature reported with detailed CT scans demonstrating PAD. If the diagnosis remains uncertain, it should be confirmed by MRI/MRA.16 Transthoracic echocardiography can be used for diagnosis and follow‐up of conservatively managed patients with PAD.3, 8, 19 The echocardiographic findings suggestive of PAD include direct or indirect evidence of pulmonary artery hypertension, with a large main pulmonary artery and an intimal flap across the pulmonary trunk.
Management
No consensus strategy is available for the management of PAD because of the rarity of this condition. In general, operative repair is the treatment of choice for PAD.2, 8, 9, 11 There are 16 cases diagnosed in living patients, out of which 6 were managed medically, 8 were managed surgically, and management was not discussed in 2 of the case reports (Table 2). In these case reports, medically managed patients were treated with oxygen, vasodilators (nitrates, angiotensin‐converting enzyme [ACE] inhibitors, dihydropyridine calcium channel blockers, sildenafil), diuretics and beta‐blockers. These patients did well on follow‐up, ranging from 3 weeks to 4 years, except for 1 who died on day 4 in the intensive care unit (ICU).
| Case Report | Etiology of PAD | Management | Outcome |
|---|---|---|---|
| |||
| Janus et al.3 | Balloon valvuloplasty for pulmonary stenosis | Medical (beta blocker) | Stable during 4 years of follow‐up |
| Khattar et al.8 | Secondary PH from COPD | Medical (diuretics, ACE inhibitor) | Stable during 1 year of follow‐up |
| Lobato et al.9 | Aortic valve replacement | Medical (vasodilators, diuretics) | Stable during 3 weeks of follow‐up |
| Smalcelj et al.10 | Primary PH | Medical (Sildenafil) | Stable during 8 months of follow‐up |
| Song and Kolecki11 | Secondary PH from VSD, Eisenmenger's syndrome | Medical (Nitroprusside) | Patient died on day 4 of admission |
| Steurer et al.15 | Primary PH | Medical (ACE inhibitor, CCB) | Stable during 1 year of follow‐up |
| Wuyts et al.4 | Secondary PH from VSD | Surgical (heart lung transplant) | Follow‐up not mentioned |
| Sakamaki et. al.5 | Primary PH | Surgical (reanastomosis) | Stable during 37 months of follow‐up |
| Westaby et al.7 | Secondary PH from VSD, Eisenmenger's syndrome | Surgical (vascular prosthesis) | Follow‐up not discussed, stable on discharge on tenth day |
| Senbaklavaci et al.12 | Primary PH | Surgical | Stable during 10 months of follow‐up |
| Inayama et al.2 | PH secondary to pulmonary thrombosis | Surgical | Follow‐up not discussed, stable at discharge |
| Wunderbaldinger et al.13 | Primary PH | Surgical | Follow‐up not discussed |
| Lopez‐Candales et al.14 | Secondary PH from partially corrected pulmonary stenosis | Surgical | Follow‐up not discussed, stable on discharge at 1 week |
| Khatchatourian and Vala17 | Associated with aortic dissection | Surgical | Stable during 3 months of follow‐up |
| Rosenson and Sutton6 | Secondary PH from mitral stenosis | Management not discussed | Follow‐up not discussed |
| Stern et al.16 | Secondary PH from hypersensitivity pneumonitis | Management not discussed | Follow‐up not discussed |
Conservative management may be tried in patients who are hemodynamically stable and do not have pericardial effusion.2, 9 The aim of conservative management is to decrease right ventricular preload and afterload. Preload reduction can be dangerous in patients with PAD and should be done in the intensive care setting as this can lead to profound hypotension. Nitrates for preload reduction should be used cautiously in patients taking sildenafil or similar agents for erectile dysfunction or pulmonary artery hypertension because of significant risk of cardiovascular collapse. The American Heart Association and American College of Cardiology both recommend that there should be a time gap of at least 24 hours between the last dose of sildenafil and the first dose of nitrates. Conservatively managed patients should be followed with interval CT scans2, 9, 18 or echocardiography.3, 19 In addition, the underlying etiology should always be investigated to predict prognosis and recommend future management strategies.
The patient was offered surgical repair but she declined. She was managed conservatively with nitrates and beta‐blockers and was pain free within 24 hours. Her BP was brought down to a systolic BP range of 130140 mm Hg. A repeat CT scan of the chest at 1‐month follow up was unchanged. The patient was doing well at 6‐month follow‐up.
Conclusions
PAD is an extremely rare cause of chest pain and a rare antemortem diagnosis. It is usually associated with underlying pulmonary hypertension. This case describes a patient with PAD in the absence of pulmonary hypertension. The patient was managed with conservative medical therapy and did well at 6‐month follow‐up. There are a total of 6 other case reports of patients with PAD managed conservatively, out of which 5 patients did well at follow‐up and 1 patient died. More case reports and longer follow‐up are needed to assess the effectiveness of conservative medical therapy in patients with PAD. To our knowledge, this is the first case report of idiopathic PAD diagnosed in a living patient and managed conservatively. This case also highlights better prognosis for patients with PAD without underlying pulmonary hypertension.
A 51‐year‐old African American woman with medical history of essential hypertension and chronic obstructive pulmonary disease (COPD) presented to the hospital with chest pain and shortness of breath. The chest pain was retrosternal and radiated to the back. It lasted for about an hour and resolved without any intervention. After some time, she again felt discomfort in the chest, which was a constant and dull ache.
She had similar episodes of chest pain 1 week prior, although less severe in intensity, for which she went to an outside hospital before coming to our hospital. Acute coronary syndrome was ruled out with serial cardiac enzymes measurements. An exercise stress test was also performed at that time, which failed to show any stress‐induced ischemia.
Her medications included lisinopril for hypertension and aspirin, which had been started 1 week prior to admission. She gave a 10‐pack‐year history of smoking tobacco. Family history was significant for hypertension in her father and coronary artery disease in her mother at the age of 58 years. A review of systems was negative for fever, cough, orthopnea, wheezing, palpitations, nausea, vomiting, recent surgery, or any significant trauma.
Assessment
The patient's physical examination was only remarkable for a blood pressure (BP) of 181/100 mm Hg. She did not have Marfanoid features, hyperflexible joints, or easy bruisability. Laboratory tests, including complete blood count, comprehensive metabolic panel, and cardiac enzymes, were within normal limits. A contrast‐enhanced computed tomography (CT) of the chest showed a linear hypodense area in the left lateral aspect of the main pulmonary trunk, which suggested an intimal dissection of the main pulmonary artery. Magnetic resonance angiography/emmaging (MRA/MRI) confirmed dissection of the main pulmonary artery extending into the proximal left pulmonary artery and associated with a 12 8 mm2 aneurysm (Figures 1 and 2). The entry site of dissection was located in the main pulmonary artery just after its origin and the exit site was located in the left pulmonary artery 5 mm distal to the bifurcation of main pulmonary artery. The pulmonary artery diameter at the dissection was 27 mm.
Diagnosis
To investigate possible etiologies, a transthoracic echocardiogram with Doppler was done to look for pulmonary hypertension. The echocardiogram showed normal pulmonary artery pressure with normal right ventricular systolic pressure. There was no evidence of pericardial effusion or structural cardiac abnormality on echocardiogram. Further investigations including work up for connective tissue diseases and infectious etiologies (Table 1) were normal.
| Variable | Reference Range | Patient's Result |
|---|---|---|
| ||
| ANA | Negative | Negative |
| C3 complement level | 88201 | 175 |
| C4 complement level | 1647 | 49 |
| RF | 20 | 20 |
| Anti‐centromere Ab | Negative | Negative |
| Anti‐Scl 70 Ab | Negative | Negative |
| Anti‐smooth muscle Ab | Negative | Negative |
| Anti‐mitochondrial Ab | Negative | Negative |
| Anti‐parietal cell Ab | Negative | Negative |
| TB skin test | 5 mm | |
| RPR | Negative | Negative |
According to Shilkin et al.,1 Helmbrecht first reported pulmonary artery dissection (PAD) in 1842. PAD is very rare and is usually diagnosed at autopsy. There are 71 other cases of PAD reported in the English literature, of which 16 cases are in living patients.217 Unlike aortic dissection, which is fairly common, the reentry circuit for PAD is formed by the rupture of the free wall of the pulmonary artery leading to hemopericardium, cardiac tamponade, and sudden death.2, 8, 9 There is wide variation in age of incidence, ranging from 26 to 85 years of age, with a slightly higher prevalence in females (male‐to‐female ratio 1:1.2).1, 2 The main pulmonary artery is usually involved, with or without involvement of its branches. Isolated left and right pulmonary artery involvement is seen in 6% and 4% of cases, respectively.2
Pulmonary hypertension, either primary or secondary (collagen vascular diseases, COPD, congenital heart diseases, etc.), is the most common underlying etiology. Other less common, but well‐described etiologies include: Marfan's syndrome, instrumentation of pulmonary artery, tuberculosis, syphilis, pregnancy, idiopathic cystic medial necrosis, and amyloidosis.2, 8
As noted earlier, underlying pulmonary hypertension is usually a major risk factor for PAD. More than 75% of the patients have underlying acute or chronic pulmonary hypertension.2 Our patient had COPD without pulmonary hypertension. Despite extensive investigation, no underlying cause of her pulmonary dissection was identified. The differential diagnosis includes cryptogenic cystic medionecrosis; however, because the patient refused surgery the diagnosis remains unknown. As in our case, idiopathic PAD is extremely rare, and only 4 other cases have been described in the literature.2 Underlying etiologies should always be ruled out to identify correctable causes such as congenital abnormalities of the heart leading to pulmonary hypertension.
Chest pain is a very common presenting complaint in the emergency department. Although rare, PAD should be suspected in a patient with retrosternal chest pain when other common causes of chest pain are excluded. Some of the more suggestive findings are the presence of a new diastolic murmur, a wide mediastinum on chest x‐ray, and CT scan of chest showing an intimal flap.2, 8 CT scan of the chest is an acceptable imaging modality to diagnose PAD.18 According to Neimatallah et al.,18 there are only 5 cases in the literature reported with detailed CT scans demonstrating PAD. If the diagnosis remains uncertain, it should be confirmed by MRI/MRA.16 Transthoracic echocardiography can be used for diagnosis and follow‐up of conservatively managed patients with PAD.3, 8, 19 The echocardiographic findings suggestive of PAD include direct or indirect evidence of pulmonary artery hypertension, with a large main pulmonary artery and an intimal flap across the pulmonary trunk.
Management
No consensus strategy is available for the management of PAD because of the rarity of this condition. In general, operative repair is the treatment of choice for PAD.2, 8, 9, 11 There are 16 cases diagnosed in living patients, out of which 6 were managed medically, 8 were managed surgically, and management was not discussed in 2 of the case reports (Table 2). In these case reports, medically managed patients were treated with oxygen, vasodilators (nitrates, angiotensin‐converting enzyme [ACE] inhibitors, dihydropyridine calcium channel blockers, sildenafil), diuretics and beta‐blockers. These patients did well on follow‐up, ranging from 3 weeks to 4 years, except for 1 who died on day 4 in the intensive care unit (ICU).
| Case Report | Etiology of PAD | Management | Outcome |
|---|---|---|---|
| |||
| Janus et al.3 | Balloon valvuloplasty for pulmonary stenosis | Medical (beta blocker) | Stable during 4 years of follow‐up |
| Khattar et al.8 | Secondary PH from COPD | Medical (diuretics, ACE inhibitor) | Stable during 1 year of follow‐up |
| Lobato et al.9 | Aortic valve replacement | Medical (vasodilators, diuretics) | Stable during 3 weeks of follow‐up |
| Smalcelj et al.10 | Primary PH | Medical (Sildenafil) | Stable during 8 months of follow‐up |
| Song and Kolecki11 | Secondary PH from VSD, Eisenmenger's syndrome | Medical (Nitroprusside) | Patient died on day 4 of admission |
| Steurer et al.15 | Primary PH | Medical (ACE inhibitor, CCB) | Stable during 1 year of follow‐up |
| Wuyts et al.4 | Secondary PH from VSD | Surgical (heart lung transplant) | Follow‐up not mentioned |
| Sakamaki et. al.5 | Primary PH | Surgical (reanastomosis) | Stable during 37 months of follow‐up |
| Westaby et al.7 | Secondary PH from VSD, Eisenmenger's syndrome | Surgical (vascular prosthesis) | Follow‐up not discussed, stable on discharge on tenth day |
| Senbaklavaci et al.12 | Primary PH | Surgical | Stable during 10 months of follow‐up |
| Inayama et al.2 | PH secondary to pulmonary thrombosis | Surgical | Follow‐up not discussed, stable at discharge |
| Wunderbaldinger et al.13 | Primary PH | Surgical | Follow‐up not discussed |
| Lopez‐Candales et al.14 | Secondary PH from partially corrected pulmonary stenosis | Surgical | Follow‐up not discussed, stable on discharge at 1 week |
| Khatchatourian and Vala17 | Associated with aortic dissection | Surgical | Stable during 3 months of follow‐up |
| Rosenson and Sutton6 | Secondary PH from mitral stenosis | Management not discussed | Follow‐up not discussed |
| Stern et al.16 | Secondary PH from hypersensitivity pneumonitis | Management not discussed | Follow‐up not discussed |
Conservative management may be tried in patients who are hemodynamically stable and do not have pericardial effusion.2, 9 The aim of conservative management is to decrease right ventricular preload and afterload. Preload reduction can be dangerous in patients with PAD and should be done in the intensive care setting as this can lead to profound hypotension. Nitrates for preload reduction should be used cautiously in patients taking sildenafil or similar agents for erectile dysfunction or pulmonary artery hypertension because of significant risk of cardiovascular collapse. The American Heart Association and American College of Cardiology both recommend that there should be a time gap of at least 24 hours between the last dose of sildenafil and the first dose of nitrates. Conservatively managed patients should be followed with interval CT scans2, 9, 18 or echocardiography.3, 19 In addition, the underlying etiology should always be investigated to predict prognosis and recommend future management strategies.
The patient was offered surgical repair but she declined. She was managed conservatively with nitrates and beta‐blockers and was pain free within 24 hours. Her BP was brought down to a systolic BP range of 130140 mm Hg. A repeat CT scan of the chest at 1‐month follow up was unchanged. The patient was doing well at 6‐month follow‐up.
Conclusions
PAD is an extremely rare cause of chest pain and a rare antemortem diagnosis. It is usually associated with underlying pulmonary hypertension. This case describes a patient with PAD in the absence of pulmonary hypertension. The patient was managed with conservative medical therapy and did well at 6‐month follow‐up. There are a total of 6 other case reports of patients with PAD managed conservatively, out of which 5 patients did well at follow‐up and 1 patient died. More case reports and longer follow‐up are needed to assess the effectiveness of conservative medical therapy in patients with PAD. To our knowledge, this is the first case report of idiopathic PAD diagnosed in a living patient and managed conservatively. This case also highlights better prognosis for patients with PAD without underlying pulmonary hypertension.
- ,,.Dissecting aneurysm of the pulmonary artery.J. Pathol.1969;98;25–29.
- ,,.Pulmonary artery dissection in patients without underlying pulmonary hypertension.Histopathology.2001;38:435–442.
- ,,,,,.Pulmonary artery dissection: a rare complication of pulmonary balloon valvuloplasty diagnosed 11 years after the procedure.J Am Soc Echocardiogr.2006;19:1191,e1195–e1198.
- ,,,,.Extensive dissection of the pulmonary artery treated with combined heart‐lung transplantation.J Thorac Cardiovasc Surg.2006;132:205–206.
- ,,, et al.Pulmonary artery dissection complicating lung transplantation for primary pulmonary hypertension.Ann Thorac Surg.2006;81:360–362.
- ,.Dissecting aneurysm of the pulmonary trunk in mitral stenosis.Am J Cardiol.1986;58:1140–1141.
- ,,.Pulmonary‐artery dissection in patients with Eisenmenger's syndrome.N Engl J Med.2007;356:2110–2112.
- ,,,.Pulmonary artery dissection: an emerging cardiovascular complication in surviving patients with chronic pulmonary hypertension.Heart.2005;91:142–145.
- ,,, et al.Pulmonary artery dissection and conservative medical management.Int J Cardiol.2007;119:e25–e26.
- ,,,,,.Giant, dissecting, high‐pressure pulmonary artery aneurysm: case report of a 1‐year natural course.Tex Heart Inst J.2005;32:589–594.
- ,.A case of pulmonary artery dissection diagnosed in the Emergency Department.J Emerg Med.2002;23:155–159.
- ,,, et al.Rupture and dissection in pulmonary artery aneurysms: incidence, cause, and treatment—review and case report.J Thorac Cardiovasc Surg.2001;121:1006–1008.
- ,,,,,.Acute pulmonary trunk dissection in a patient with primary pulmonary hypertension.J Comput Assist Tomogr.2000;24:92–95.
- ,,,,.Pulmonary artery aneurysm: review and case report.Clin Cardiol.1995;18:738–740.
- ,,,,,.Dissecting aneurysm of the pulmonary artery with pulmonary hypertension.Am Rev Respir Dis.1990;142:1219–1221.
- ,,,,.Pulmonary artery dissection: MR findings.J Comput Assist Tomogr.1992;16:481–483.
- ,.Images in cardiovascular medicine. Acute type I aortic dissection with concomitant pulmonary artery dissection.Circulation.2005;112:e313–314.
- ,,,.CT findings of pulmonary artery dissection.Br J Radiol.2007;80:e61–e63.
- ,.Pulmonary artery dissection: echocardiographic findings and diagnosis.Echocardiography.2003;20:375–377.
- ,,.Dissecting aneurysm of the pulmonary artery.J. Pathol.1969;98;25–29.
- ,,.Pulmonary artery dissection in patients without underlying pulmonary hypertension.Histopathology.2001;38:435–442.
- ,,,,,.Pulmonary artery dissection: a rare complication of pulmonary balloon valvuloplasty diagnosed 11 years after the procedure.J Am Soc Echocardiogr.2006;19:1191,e1195–e1198.
- ,,,,.Extensive dissection of the pulmonary artery treated with combined heart‐lung transplantation.J Thorac Cardiovasc Surg.2006;132:205–206.
- ,,, et al.Pulmonary artery dissection complicating lung transplantation for primary pulmonary hypertension.Ann Thorac Surg.2006;81:360–362.
- ,.Dissecting aneurysm of the pulmonary trunk in mitral stenosis.Am J Cardiol.1986;58:1140–1141.
- ,,.Pulmonary‐artery dissection in patients with Eisenmenger's syndrome.N Engl J Med.2007;356:2110–2112.
- ,,,.Pulmonary artery dissection: an emerging cardiovascular complication in surviving patients with chronic pulmonary hypertension.Heart.2005;91:142–145.
- ,,, et al.Pulmonary artery dissection and conservative medical management.Int J Cardiol.2007;119:e25–e26.
- ,,,,,.Giant, dissecting, high‐pressure pulmonary artery aneurysm: case report of a 1‐year natural course.Tex Heart Inst J.2005;32:589–594.
- ,.A case of pulmonary artery dissection diagnosed in the Emergency Department.J Emerg Med.2002;23:155–159.
- ,,, et al.Rupture and dissection in pulmonary artery aneurysms: incidence, cause, and treatment—review and case report.J Thorac Cardiovasc Surg.2001;121:1006–1008.
- ,,,,,.Acute pulmonary trunk dissection in a patient with primary pulmonary hypertension.J Comput Assist Tomogr.2000;24:92–95.
- ,,,,.Pulmonary artery aneurysm: review and case report.Clin Cardiol.1995;18:738–740.
- ,,,,,.Dissecting aneurysm of the pulmonary artery with pulmonary hypertension.Am Rev Respir Dis.1990;142:1219–1221.
- ,,,,.Pulmonary artery dissection: MR findings.J Comput Assist Tomogr.1992;16:481–483.
- ,.Images in cardiovascular medicine. Acute type I aortic dissection with concomitant pulmonary artery dissection.Circulation.2005;112:e313–314.
- ,,,.CT findings of pulmonary artery dissection.Br J Radiol.2007;80:e61–e63.
- ,.Pulmonary artery dissection: echocardiographic findings and diagnosis.Echocardiography.2003;20:375–377.
Hospitals and Recession
With the United States mired in its most severe recession in decades, stories of hospital struggles have emerged. Beaumont Hospital, located near the headquarters of major automakers and several assembly plants outside Detroit, recently cut hundreds of jobs and put major construction on indefinite hold.1 The CEO of Boston's Beth Israel Deaconess Medical Center made an agreement with employees to take large cuts in pay and vacation time to prevent laying off 10% of the staff.2 The University of Chicago Medical Center made plans to limit the number of emergency room beds, thereby decreasing low‐reimbursing emergency admissions while making beds available for higher‐paying elective hospitalizations.3
What is surprising about these stories is that hospitals have long been considered recession‐proof. Yet, with one‐half of US hospitals having reduced their staff to balance their budgets4 and with hospitals' financial margins falling dramatically,5 economic struggles are now a widespread problem.
Furthermore, it is difficult to determine if hospitals' clinical care has been damaged by the recession. The measurement of hospital quality is new and still under‐developed: there is virtually no reliable information on hospital quality from previous recessions, and even now it will be difficult to assess quality in real time.
Critics of waste and excess in the US health care system may see tough economic times as a Darwinian proving ground for hospitals, through which efficiency will improve and poor performers will close their doors. But more likely, hospital cutbacks will risk the quality and safety of health care delivery. For reasons of both public health and fiscal impact on communities, state and federal leaders may need to watch these trends closely to design and to be ready to implement potential government remedies for hospitals' fiscal woes.
In this commentary, we describe how hospitals have fared historically during recessions, how this recession could have different effectsfirst fiscally, then clinically, and we examine policy options to mitigate these untoward effects.
Decades of Recession‐Proof Hospitals
During the Great Depression, hospital insolvency was a national problem that prompted federal and state aid. Keeping hospitals alive was a critical policy goal and proved central to the early development of health insurance that focused on payment for hospital care.6
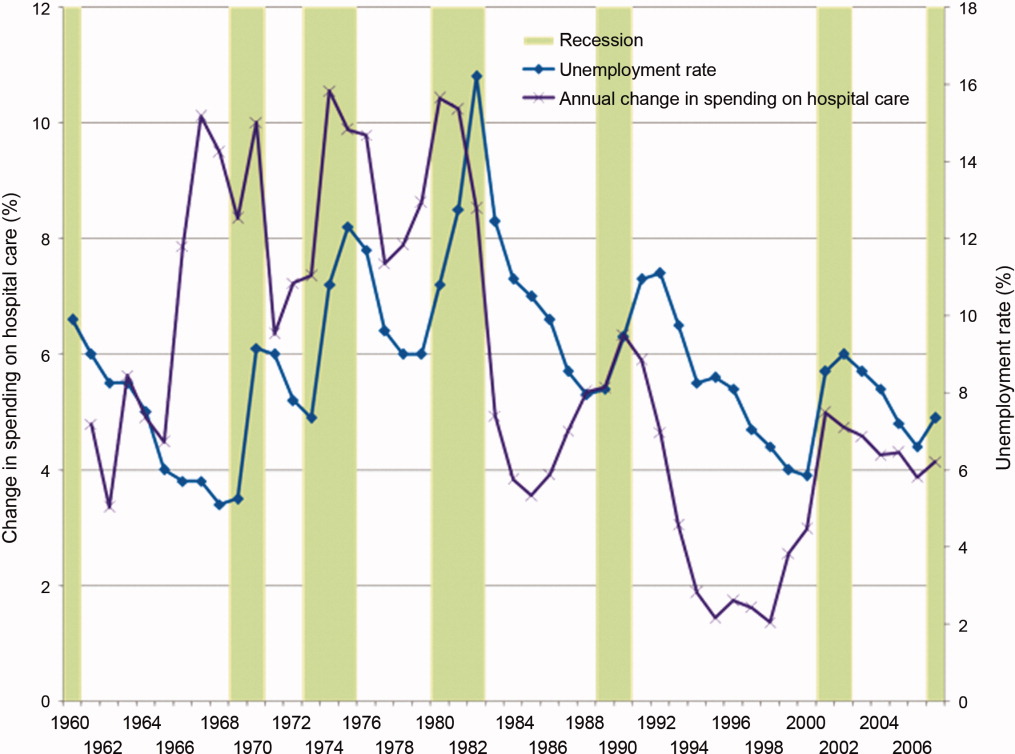
Since WWII, growth in America's hospitals has been only loosely related to national macroeconomic trends, with other changes like technological innovations and the advent of managed care far more influential to hospital finances. In fact, during recessions, hospital care spending growth often escalates in tandem with worsening unemployment (Figure 1). One explanation for this phenomenon is that economic pressures lead to declining primary care utilization, with adverse consequences for individuals' health.7
Hospitals' Current Fiscal Vulnerability
However, the current recession is the worst in 70 years. Every method of income generation available to hospitals appears at risk, including reimbursement per discharge (70% of hospitals report moderate or significant increases in uncompensated care), number of inpatient admissions (over one‐half report a moderate or significant decrease), difficulty obtaining bonds (60% report at least significant problems), and charitable donations.4 Over 50% of US hospitals had negative margins in the fourth quarter of 2008, though there has been some improvement since that time.8
Future hospital stability concerns remain. Growth in revenue per discharge is still below the norm.5 Because employment lags a recovering economy, further reimbursement decreases are possible from increasing proportions of patients with low‐reimbursing insurers or no coverage at all, decreasing payment rates from all payers, and decreasing elective care. The lower‐reimbursing payers, like state Medicaid programs, are experiencing increased enrollment as Americans lose their jobs and their better‐paying, employer‐sponsored private insurance.9 There's also evidence that reimbursement rates are declining from both Medicare and private insurers,10 which threatens the fragile cost‐shift through which hospitals have long used private insurance reimbursement to subsidize government reimbursements.11
Hospitals' specific financial challenges will likely vary across markets. The authors' state of Michigan has been hit particularly long and hard by the current recession. Unemployment rates exceeding 11% are expected to cause dramatic losses in private health insurance.9 Patients' increasing need with decreasing ability to pay will make markets in the deepest recession particularly vulnerable.
Hospital Quality and Safety at Risk?
The effect of the recession on the quality of hospital care is less clear. Until the 1990s, hospital quality was essentially assumed and virtually unmeasured. Even now, measuring hospital quality is difficult and rarely timely. Medicare data often take 1 to 2 years to become publicly available for analysis. Reports by trade organizations like the American Hospital Association are up‐to‐date but have conflicts of interest and are less rigorous. The most timely measures of hospitals' distressflawed as they may bewill come from the hospitals themselves, just like reports of economic woe from other businesses and government agencies during challenging economic times.
However, since the publication of the 1999 report To Err is Human,12 major improvements in hospital quality and safety have transformed the delivery of inpatient care. These improvements have taken the form of simple interventions like nationally consistent medical abbreviations, management initiatives like Six Sigma, and technological advances including computerized health records.
Nonetheless, during this recession and recovery, slashed hospital budgets may slow or even stop the momentum towards further improvements in quality and safety. Frontline care delivery could be at risk. Understaffed and under financed hospitals are rarely safe. Dissatisfaction and layoffs hurt the interactions between employees and patients. Robust nurse‐to‐patient ratios which have proven vital to patients' hospital outcomes could be at risk.13 Admittedly, recession‐induced threats to quality and safety are conjectures on our part: unfortunately, no recession measures of hospitals' specific spending on staffing, technology, or process improvements exist.
However, there are many small, evidence‐based changes that could improve hospital safety dramatically in the near future. Michigan's Keystone ICU Initiative showed that systematic interventions in routine care delivery could reduce the risk of catheter‐related bloodstream infections, which currently are implicated in the death of 28,000 Americans per year, to nearly zero.14 The Institute for Healthcare Improvement's 100,000 Lives Campaign also illustrated that dramatic improvements in hospital‐related mortality can occur with fairly focused interventions. In the month after discharge, more than one‐quarter of all hospitalized patients go to an emergency room or need to be rehospitalized. This rate can be cut by 30% by inserting a nurse discharge advocate into the discharge process.15 Instituting a simple safety checklist before surgery decreased surgery‐related mortality and complications by over one‐third.16
Such interventions are effective, reasonable, and widely accessible. Over the long‐term, many may even be cost‐saving. But, importantly, they all require an institutional investment in start‐up money and an organizational will to change how things have been done. In a period of recession with severe cost‐cutting, and a recovery period of cautious spending, this may not be possible.
A Possible Stimulus: Investing in Quality Initiatives at Fiscally Vulnerable Hospitals
It is not enough to keep hospitals' doors open in a recession. Hospitals must continue to improve the quality and safety of the care they delivervital for their future patients and also for their communities who depend on them as anchors of health systems. We believe there is a need for a new, federally supported alignment of hospital finance and hospital quality that can limit damage to hospitals, help community employment, and improve patient safety.
Timely, structural quality measures could speed the introduction of functional value‐based purchasing, promote hospital safety, and help local economies at the same time. There are many simple structural measures that could be examined, such as development of discharge coordinators, promoting effective nurse‐to‐patient ratios, and encouraging health information technology (IT). Importantly, this would not duplicate efforts already underway to promote quality with process measures. With effective financial monitoring in real time, these measures could focus on high‐risk, fiscally disadvantaged hospitals.
To its credit, the Obama administration has already reached out to support hospitals, although aid has not been targeted specifically to hospitals in the most dire financial circumstances. Along with support for Medicaid and community health centers to improve primary care during the recession, the administration has provided a $268 million increase in Disproportionate Share Hospital payments towards hospitals that care for vulnerable patients, an increase of about 3%.17 Concurrently, the Centers for Medicare and Medicaid Services are implementing a value‐based purchasing program that starts with a 5% withhold in reimbursement that institutions need to earn back through a combination of mortality, process, and patient satisfaction metrics.18 The administration also reserved $19 billion to promote improvement of health IT for American medicine.19
Using health IT investment to help hospitals is an appealing concept, but for many institutions the infrastructure required to make that transition directly competes with other patient needs, including bedside patient care. IT investments have large initial costs, at a time when bank loans are difficult to acquire and few organizations can make expensive capital improvements. In fact, one‐quarter of hospitals report scaling back health IT investments that they had already started, in spite of the stimulus funds available.4
Instead, the administration may have more influence on improving care delivery by focusing on connecting hospital safety with hospital financial stability, by appropriating stimulus funds to center on quality and safety programs like those described above. Here is how: a hospital that would receive stimulus money for employing nurse discharge advocates would preserve employment while advancing patient safety, as would a hospital that retains a nurse‐to‐patient ratio above a specified threshold. By focusing on measures of structural quality, the government could improve care in ways that are easy to measure and maximize local economic stimulus without difficult outcomes assessment, insurance reform, or duplicating process measure efforts. There could even be an innovation differential (ie, payment/reward) for hospitals that improve quality while holding flat or lowering overall costs.
Equally important is to use this national financial crisis as an opportunity to improve monitoring of hospital quality. While quality assessment of hospitals is difficult, increased federal awareness of local medical need, hospital financial stability, and government awareness of emergency services overcrowding, nurse‐to‐patient ratios, and IT utilization are all valuable and easy to measure.
None of these quality‐focused fiscal interventions would be guaranteed to prevent hospital closure. Especially in small population centers, hospital closures can affect an entire community's financial growth and clinical safety net,20 while leaving hundreds or even thousands unemployed. Hospital closure should be assessed by state and federal government officials in these larger terms, perhaps even encouraging closure when appropriate, and helping prevent it when necessary.
Conclusion
Hospitals, as complex pieces of America's health care system, are central to communities' safety and economic growth. While national health coverage reform, as currently being discussed in Washington, would make hospital infrastructure less sensitive to macroeconomic changes, major reform would not come fast enough if hospitals start closing. While the worst of the recession may be over, recovery and the continuing rise in unemployment is a tenuous lifeline for hospitals on the financial brink.
We are not arguing against all hospital layoffs, or even closures. Indeed, this recession is a lean time for most industries and is likely to lead to closures for hospitals that cannot compete on efficiency or quality. But a hospital closure is a major event for a community and should not be permitted to occur without thorough consideration of alternatives. Current data on hospitals' financial status and clinical safety are limited, potentially biased, and not timely enough for this rapidly changing economic crisis. Therefore, state and federal government officials should assess whether hospitals would be eligible not just for possible emergency loans, but for linking loans to quality of care and community need. In so doing, this difficult time could be an opportunity to help hospitals improve their care, rather than watching it diminish.
- Michigan's Health Care Safety Net: In Jeopardy.2009.
- .Final budget decisions.Running A Hospital. Vol 2009.Boston, MA;2009.
- .Doctors Plan to Limit Beds in ER.Wall Street Journal.2009.
- The Impact of the Economic Crisis on Health Services for Patients and Communities.Washington, DC2009.
- ,.Hospital Operational and Financial Performance Improving.Ann Arbor, MI:Thomson Reuters Center for Healthcare Improvement.2009.
- .The Social Transformation of American Medicine.New York, NY:Basic Books;1983.
- AAFP.Patient Care during the 2008‐2009 Recession – Online Survey.Leawood, KS:AAFP.2009.
- The Impact of the Economic Crisis on Health Services for Patients and Communities.Washington, D.C.:American Hospital Association.2009.
- The economic downturn and its impact on hospitals. American Hospital Association Trendwatch.2009.
- ,,.The Current Recession and U.S. Hospitals:Center for Healthcare Improvement.2009.
- ,,.The cost‐shift payment ‘hydraulic’: foundation, history, and implications.Health Aff (Millwood).2006;25(1):22–33.
- ,.To Err Is Human: Building a Safer Health System.Washington, DC:National Academy Press;1999.
- ,,,,.Nurse‐staffing levels and the quality of care in hospitals.N Engl J Med.2002;346(22):1715–1722.
- ,,, et al.An intervention to decrease catheter‐related bloodstream infections in the ICU.N Engl J Med.2006;355(26):2725–2732.
- ,,, et al.A reengineered hospital discharge program to decrease rehospitalization: a randomized trial.Ann Intern Med.2009;150(3):178–187.
- ,,, et al.A surgical safety checklist to reduce morbidity and mortality in a global population.N Engl J Med.2009;360(5):491–499.
- Disproportionate Share Hospital (DSH). Available at: http://www.hhs. gov/recovery/cms/dsh.html. Accessed December 2009.
- ,,.Measuring outcomes and efficiency in medicare value‐based purchasing.Health Aff (Millwood).2009;28(2):w251–w261.
- .Stimulating the adoption of health information technology.N Engl J Med.2009;360(15):1477–1479.
- ,,,.The effect of rural hospital closures on community economic health.Health Serv Res.2006;41(2):467–485.
With the United States mired in its most severe recession in decades, stories of hospital struggles have emerged. Beaumont Hospital, located near the headquarters of major automakers and several assembly plants outside Detroit, recently cut hundreds of jobs and put major construction on indefinite hold.1 The CEO of Boston's Beth Israel Deaconess Medical Center made an agreement with employees to take large cuts in pay and vacation time to prevent laying off 10% of the staff.2 The University of Chicago Medical Center made plans to limit the number of emergency room beds, thereby decreasing low‐reimbursing emergency admissions while making beds available for higher‐paying elective hospitalizations.3
What is surprising about these stories is that hospitals have long been considered recession‐proof. Yet, with one‐half of US hospitals having reduced their staff to balance their budgets4 and with hospitals' financial margins falling dramatically,5 economic struggles are now a widespread problem.
Furthermore, it is difficult to determine if hospitals' clinical care has been damaged by the recession. The measurement of hospital quality is new and still under‐developed: there is virtually no reliable information on hospital quality from previous recessions, and even now it will be difficult to assess quality in real time.
Critics of waste and excess in the US health care system may see tough economic times as a Darwinian proving ground for hospitals, through which efficiency will improve and poor performers will close their doors. But more likely, hospital cutbacks will risk the quality and safety of health care delivery. For reasons of both public health and fiscal impact on communities, state and federal leaders may need to watch these trends closely to design and to be ready to implement potential government remedies for hospitals' fiscal woes.
In this commentary, we describe how hospitals have fared historically during recessions, how this recession could have different effectsfirst fiscally, then clinically, and we examine policy options to mitigate these untoward effects.
Decades of Recession‐Proof Hospitals
During the Great Depression, hospital insolvency was a national problem that prompted federal and state aid. Keeping hospitals alive was a critical policy goal and proved central to the early development of health insurance that focused on payment for hospital care.6
Since WWII, growth in America's hospitals has been only loosely related to national macroeconomic trends, with other changes like technological innovations and the advent of managed care far more influential to hospital finances. In fact, during recessions, hospital care spending growth often escalates in tandem with worsening unemployment (Figure 1). One explanation for this phenomenon is that economic pressures lead to declining primary care utilization, with adverse consequences for individuals' health.7
Hospitals' Current Fiscal Vulnerability
However, the current recession is the worst in 70 years. Every method of income generation available to hospitals appears at risk, including reimbursement per discharge (70% of hospitals report moderate or significant increases in uncompensated care), number of inpatient admissions (over one‐half report a moderate or significant decrease), difficulty obtaining bonds (60% report at least significant problems), and charitable donations.4 Over 50% of US hospitals had negative margins in the fourth quarter of 2008, though there has been some improvement since that time.8
Future hospital stability concerns remain. Growth in revenue per discharge is still below the norm.5 Because employment lags a recovering economy, further reimbursement decreases are possible from increasing proportions of patients with low‐reimbursing insurers or no coverage at all, decreasing payment rates from all payers, and decreasing elective care. The lower‐reimbursing payers, like state Medicaid programs, are experiencing increased enrollment as Americans lose their jobs and their better‐paying, employer‐sponsored private insurance.9 There's also evidence that reimbursement rates are declining from both Medicare and private insurers,10 which threatens the fragile cost‐shift through which hospitals have long used private insurance reimbursement to subsidize government reimbursements.11
Hospitals' specific financial challenges will likely vary across markets. The authors' state of Michigan has been hit particularly long and hard by the current recession. Unemployment rates exceeding 11% are expected to cause dramatic losses in private health insurance.9 Patients' increasing need with decreasing ability to pay will make markets in the deepest recession particularly vulnerable.
Hospital Quality and Safety at Risk?
The effect of the recession on the quality of hospital care is less clear. Until the 1990s, hospital quality was essentially assumed and virtually unmeasured. Even now, measuring hospital quality is difficult and rarely timely. Medicare data often take 1 to 2 years to become publicly available for analysis. Reports by trade organizations like the American Hospital Association are up‐to‐date but have conflicts of interest and are less rigorous. The most timely measures of hospitals' distressflawed as they may bewill come from the hospitals themselves, just like reports of economic woe from other businesses and government agencies during challenging economic times.
However, since the publication of the 1999 report To Err is Human,12 major improvements in hospital quality and safety have transformed the delivery of inpatient care. These improvements have taken the form of simple interventions like nationally consistent medical abbreviations, management initiatives like Six Sigma, and technological advances including computerized health records.
Nonetheless, during this recession and recovery, slashed hospital budgets may slow or even stop the momentum towards further improvements in quality and safety. Frontline care delivery could be at risk. Understaffed and under financed hospitals are rarely safe. Dissatisfaction and layoffs hurt the interactions between employees and patients. Robust nurse‐to‐patient ratios which have proven vital to patients' hospital outcomes could be at risk.13 Admittedly, recession‐induced threats to quality and safety are conjectures on our part: unfortunately, no recession measures of hospitals' specific spending on staffing, technology, or process improvements exist.
However, there are many small, evidence‐based changes that could improve hospital safety dramatically in the near future. Michigan's Keystone ICU Initiative showed that systematic interventions in routine care delivery could reduce the risk of catheter‐related bloodstream infections, which currently are implicated in the death of 28,000 Americans per year, to nearly zero.14 The Institute for Healthcare Improvement's 100,000 Lives Campaign also illustrated that dramatic improvements in hospital‐related mortality can occur with fairly focused interventions. In the month after discharge, more than one‐quarter of all hospitalized patients go to an emergency room or need to be rehospitalized. This rate can be cut by 30% by inserting a nurse discharge advocate into the discharge process.15 Instituting a simple safety checklist before surgery decreased surgery‐related mortality and complications by over one‐third.16
Such interventions are effective, reasonable, and widely accessible. Over the long‐term, many may even be cost‐saving. But, importantly, they all require an institutional investment in start‐up money and an organizational will to change how things have been done. In a period of recession with severe cost‐cutting, and a recovery period of cautious spending, this may not be possible.
A Possible Stimulus: Investing in Quality Initiatives at Fiscally Vulnerable Hospitals
It is not enough to keep hospitals' doors open in a recession. Hospitals must continue to improve the quality and safety of the care they delivervital for their future patients and also for their communities who depend on them as anchors of health systems. We believe there is a need for a new, federally supported alignment of hospital finance and hospital quality that can limit damage to hospitals, help community employment, and improve patient safety.
Timely, structural quality measures could speed the introduction of functional value‐based purchasing, promote hospital safety, and help local economies at the same time. There are many simple structural measures that could be examined, such as development of discharge coordinators, promoting effective nurse‐to‐patient ratios, and encouraging health information technology (IT). Importantly, this would not duplicate efforts already underway to promote quality with process measures. With effective financial monitoring in real time, these measures could focus on high‐risk, fiscally disadvantaged hospitals.
To its credit, the Obama administration has already reached out to support hospitals, although aid has not been targeted specifically to hospitals in the most dire financial circumstances. Along with support for Medicaid and community health centers to improve primary care during the recession, the administration has provided a $268 million increase in Disproportionate Share Hospital payments towards hospitals that care for vulnerable patients, an increase of about 3%.17 Concurrently, the Centers for Medicare and Medicaid Services are implementing a value‐based purchasing program that starts with a 5% withhold in reimbursement that institutions need to earn back through a combination of mortality, process, and patient satisfaction metrics.18 The administration also reserved $19 billion to promote improvement of health IT for American medicine.19
Using health IT investment to help hospitals is an appealing concept, but for many institutions the infrastructure required to make that transition directly competes with other patient needs, including bedside patient care. IT investments have large initial costs, at a time when bank loans are difficult to acquire and few organizations can make expensive capital improvements. In fact, one‐quarter of hospitals report scaling back health IT investments that they had already started, in spite of the stimulus funds available.4
Instead, the administration may have more influence on improving care delivery by focusing on connecting hospital safety with hospital financial stability, by appropriating stimulus funds to center on quality and safety programs like those described above. Here is how: a hospital that would receive stimulus money for employing nurse discharge advocates would preserve employment while advancing patient safety, as would a hospital that retains a nurse‐to‐patient ratio above a specified threshold. By focusing on measures of structural quality, the government could improve care in ways that are easy to measure and maximize local economic stimulus without difficult outcomes assessment, insurance reform, or duplicating process measure efforts. There could even be an innovation differential (ie, payment/reward) for hospitals that improve quality while holding flat or lowering overall costs.
Equally important is to use this national financial crisis as an opportunity to improve monitoring of hospital quality. While quality assessment of hospitals is difficult, increased federal awareness of local medical need, hospital financial stability, and government awareness of emergency services overcrowding, nurse‐to‐patient ratios, and IT utilization are all valuable and easy to measure.
None of these quality‐focused fiscal interventions would be guaranteed to prevent hospital closure. Especially in small population centers, hospital closures can affect an entire community's financial growth and clinical safety net,20 while leaving hundreds or even thousands unemployed. Hospital closure should be assessed by state and federal government officials in these larger terms, perhaps even encouraging closure when appropriate, and helping prevent it when necessary.
Conclusion
Hospitals, as complex pieces of America's health care system, are central to communities' safety and economic growth. While national health coverage reform, as currently being discussed in Washington, would make hospital infrastructure less sensitive to macroeconomic changes, major reform would not come fast enough if hospitals start closing. While the worst of the recession may be over, recovery and the continuing rise in unemployment is a tenuous lifeline for hospitals on the financial brink.
We are not arguing against all hospital layoffs, or even closures. Indeed, this recession is a lean time for most industries and is likely to lead to closures for hospitals that cannot compete on efficiency or quality. But a hospital closure is a major event for a community and should not be permitted to occur without thorough consideration of alternatives. Current data on hospitals' financial status and clinical safety are limited, potentially biased, and not timely enough for this rapidly changing economic crisis. Therefore, state and federal government officials should assess whether hospitals would be eligible not just for possible emergency loans, but for linking loans to quality of care and community need. In so doing, this difficult time could be an opportunity to help hospitals improve their care, rather than watching it diminish.
With the United States mired in its most severe recession in decades, stories of hospital struggles have emerged. Beaumont Hospital, located near the headquarters of major automakers and several assembly plants outside Detroit, recently cut hundreds of jobs and put major construction on indefinite hold.1 The CEO of Boston's Beth Israel Deaconess Medical Center made an agreement with employees to take large cuts in pay and vacation time to prevent laying off 10% of the staff.2 The University of Chicago Medical Center made plans to limit the number of emergency room beds, thereby decreasing low‐reimbursing emergency admissions while making beds available for higher‐paying elective hospitalizations.3
What is surprising about these stories is that hospitals have long been considered recession‐proof. Yet, with one‐half of US hospitals having reduced their staff to balance their budgets4 and with hospitals' financial margins falling dramatically,5 economic struggles are now a widespread problem.
Furthermore, it is difficult to determine if hospitals' clinical care has been damaged by the recession. The measurement of hospital quality is new and still under‐developed: there is virtually no reliable information on hospital quality from previous recessions, and even now it will be difficult to assess quality in real time.
Critics of waste and excess in the US health care system may see tough economic times as a Darwinian proving ground for hospitals, through which efficiency will improve and poor performers will close their doors. But more likely, hospital cutbacks will risk the quality and safety of health care delivery. For reasons of both public health and fiscal impact on communities, state and federal leaders may need to watch these trends closely to design and to be ready to implement potential government remedies for hospitals' fiscal woes.
In this commentary, we describe how hospitals have fared historically during recessions, how this recession could have different effectsfirst fiscally, then clinically, and we examine policy options to mitigate these untoward effects.
Decades of Recession‐Proof Hospitals
During the Great Depression, hospital insolvency was a national problem that prompted federal and state aid. Keeping hospitals alive was a critical policy goal and proved central to the early development of health insurance that focused on payment for hospital care.6
Since WWII, growth in America's hospitals has been only loosely related to national macroeconomic trends, with other changes like technological innovations and the advent of managed care far more influential to hospital finances. In fact, during recessions, hospital care spending growth often escalates in tandem with worsening unemployment (Figure 1). One explanation for this phenomenon is that economic pressures lead to declining primary care utilization, with adverse consequences for individuals' health.7
Hospitals' Current Fiscal Vulnerability
However, the current recession is the worst in 70 years. Every method of income generation available to hospitals appears at risk, including reimbursement per discharge (70% of hospitals report moderate or significant increases in uncompensated care), number of inpatient admissions (over one‐half report a moderate or significant decrease), difficulty obtaining bonds (60% report at least significant problems), and charitable donations.4 Over 50% of US hospitals had negative margins in the fourth quarter of 2008, though there has been some improvement since that time.8
Future hospital stability concerns remain. Growth in revenue per discharge is still below the norm.5 Because employment lags a recovering economy, further reimbursement decreases are possible from increasing proportions of patients with low‐reimbursing insurers or no coverage at all, decreasing payment rates from all payers, and decreasing elective care. The lower‐reimbursing payers, like state Medicaid programs, are experiencing increased enrollment as Americans lose their jobs and their better‐paying, employer‐sponsored private insurance.9 There's also evidence that reimbursement rates are declining from both Medicare and private insurers,10 which threatens the fragile cost‐shift through which hospitals have long used private insurance reimbursement to subsidize government reimbursements.11
Hospitals' specific financial challenges will likely vary across markets. The authors' state of Michigan has been hit particularly long and hard by the current recession. Unemployment rates exceeding 11% are expected to cause dramatic losses in private health insurance.9 Patients' increasing need with decreasing ability to pay will make markets in the deepest recession particularly vulnerable.
Hospital Quality and Safety at Risk?
The effect of the recession on the quality of hospital care is less clear. Until the 1990s, hospital quality was essentially assumed and virtually unmeasured. Even now, measuring hospital quality is difficult and rarely timely. Medicare data often take 1 to 2 years to become publicly available for analysis. Reports by trade organizations like the American Hospital Association are up‐to‐date but have conflicts of interest and are less rigorous. The most timely measures of hospitals' distressflawed as they may bewill come from the hospitals themselves, just like reports of economic woe from other businesses and government agencies during challenging economic times.
However, since the publication of the 1999 report To Err is Human,12 major improvements in hospital quality and safety have transformed the delivery of inpatient care. These improvements have taken the form of simple interventions like nationally consistent medical abbreviations, management initiatives like Six Sigma, and technological advances including computerized health records.
Nonetheless, during this recession and recovery, slashed hospital budgets may slow or even stop the momentum towards further improvements in quality and safety. Frontline care delivery could be at risk. Understaffed and under financed hospitals are rarely safe. Dissatisfaction and layoffs hurt the interactions between employees and patients. Robust nurse‐to‐patient ratios which have proven vital to patients' hospital outcomes could be at risk.13 Admittedly, recession‐induced threats to quality and safety are conjectures on our part: unfortunately, no recession measures of hospitals' specific spending on staffing, technology, or process improvements exist.
However, there are many small, evidence‐based changes that could improve hospital safety dramatically in the near future. Michigan's Keystone ICU Initiative showed that systematic interventions in routine care delivery could reduce the risk of catheter‐related bloodstream infections, which currently are implicated in the death of 28,000 Americans per year, to nearly zero.14 The Institute for Healthcare Improvement's 100,000 Lives Campaign also illustrated that dramatic improvements in hospital‐related mortality can occur with fairly focused interventions. In the month after discharge, more than one‐quarter of all hospitalized patients go to an emergency room or need to be rehospitalized. This rate can be cut by 30% by inserting a nurse discharge advocate into the discharge process.15 Instituting a simple safety checklist before surgery decreased surgery‐related mortality and complications by over one‐third.16
Such interventions are effective, reasonable, and widely accessible. Over the long‐term, many may even be cost‐saving. But, importantly, they all require an institutional investment in start‐up money and an organizational will to change how things have been done. In a period of recession with severe cost‐cutting, and a recovery period of cautious spending, this may not be possible.
A Possible Stimulus: Investing in Quality Initiatives at Fiscally Vulnerable Hospitals
It is not enough to keep hospitals' doors open in a recession. Hospitals must continue to improve the quality and safety of the care they delivervital for their future patients and also for their communities who depend on them as anchors of health systems. We believe there is a need for a new, federally supported alignment of hospital finance and hospital quality that can limit damage to hospitals, help community employment, and improve patient safety.
Timely, structural quality measures could speed the introduction of functional value‐based purchasing, promote hospital safety, and help local economies at the same time. There are many simple structural measures that could be examined, such as development of discharge coordinators, promoting effective nurse‐to‐patient ratios, and encouraging health information technology (IT). Importantly, this would not duplicate efforts already underway to promote quality with process measures. With effective financial monitoring in real time, these measures could focus on high‐risk, fiscally disadvantaged hospitals.
To its credit, the Obama administration has already reached out to support hospitals, although aid has not been targeted specifically to hospitals in the most dire financial circumstances. Along with support for Medicaid and community health centers to improve primary care during the recession, the administration has provided a $268 million increase in Disproportionate Share Hospital payments towards hospitals that care for vulnerable patients, an increase of about 3%.17 Concurrently, the Centers for Medicare and Medicaid Services are implementing a value‐based purchasing program that starts with a 5% withhold in reimbursement that institutions need to earn back through a combination of mortality, process, and patient satisfaction metrics.18 The administration also reserved $19 billion to promote improvement of health IT for American medicine.19
Using health IT investment to help hospitals is an appealing concept, but for many institutions the infrastructure required to make that transition directly competes with other patient needs, including bedside patient care. IT investments have large initial costs, at a time when bank loans are difficult to acquire and few organizations can make expensive capital improvements. In fact, one‐quarter of hospitals report scaling back health IT investments that they had already started, in spite of the stimulus funds available.4
Instead, the administration may have more influence on improving care delivery by focusing on connecting hospital safety with hospital financial stability, by appropriating stimulus funds to center on quality and safety programs like those described above. Here is how: a hospital that would receive stimulus money for employing nurse discharge advocates would preserve employment while advancing patient safety, as would a hospital that retains a nurse‐to‐patient ratio above a specified threshold. By focusing on measures of structural quality, the government could improve care in ways that are easy to measure and maximize local economic stimulus without difficult outcomes assessment, insurance reform, or duplicating process measure efforts. There could even be an innovation differential (ie, payment/reward) for hospitals that improve quality while holding flat or lowering overall costs.
Equally important is to use this national financial crisis as an opportunity to improve monitoring of hospital quality. While quality assessment of hospitals is difficult, increased federal awareness of local medical need, hospital financial stability, and government awareness of emergency services overcrowding, nurse‐to‐patient ratios, and IT utilization are all valuable and easy to measure.
None of these quality‐focused fiscal interventions would be guaranteed to prevent hospital closure. Especially in small population centers, hospital closures can affect an entire community's financial growth and clinical safety net,20 while leaving hundreds or even thousands unemployed. Hospital closure should be assessed by state and federal government officials in these larger terms, perhaps even encouraging closure when appropriate, and helping prevent it when necessary.
Conclusion
Hospitals, as complex pieces of America's health care system, are central to communities' safety and economic growth. While national health coverage reform, as currently being discussed in Washington, would make hospital infrastructure less sensitive to macroeconomic changes, major reform would not come fast enough if hospitals start closing. While the worst of the recession may be over, recovery and the continuing rise in unemployment is a tenuous lifeline for hospitals on the financial brink.
We are not arguing against all hospital layoffs, or even closures. Indeed, this recession is a lean time for most industries and is likely to lead to closures for hospitals that cannot compete on efficiency or quality. But a hospital closure is a major event for a community and should not be permitted to occur without thorough consideration of alternatives. Current data on hospitals' financial status and clinical safety are limited, potentially biased, and not timely enough for this rapidly changing economic crisis. Therefore, state and federal government officials should assess whether hospitals would be eligible not just for possible emergency loans, but for linking loans to quality of care and community need. In so doing, this difficult time could be an opportunity to help hospitals improve their care, rather than watching it diminish.
- Michigan's Health Care Safety Net: In Jeopardy.2009.
- .Final budget decisions.Running A Hospital. Vol 2009.Boston, MA;2009.
- .Doctors Plan to Limit Beds in ER.Wall Street Journal.2009.
- The Impact of the Economic Crisis on Health Services for Patients and Communities.Washington, DC2009.
- ,.Hospital Operational and Financial Performance Improving.Ann Arbor, MI:Thomson Reuters Center for Healthcare Improvement.2009.
- .The Social Transformation of American Medicine.New York, NY:Basic Books;1983.
- AAFP.Patient Care during the 2008‐2009 Recession – Online Survey.Leawood, KS:AAFP.2009.
- The Impact of the Economic Crisis on Health Services for Patients and Communities.Washington, D.C.:American Hospital Association.2009.
- The economic downturn and its impact on hospitals. American Hospital Association Trendwatch.2009.
- ,,.The Current Recession and U.S. Hospitals:Center for Healthcare Improvement.2009.
- ,,.The cost‐shift payment ‘hydraulic’: foundation, history, and implications.Health Aff (Millwood).2006;25(1):22–33.
- ,.To Err Is Human: Building a Safer Health System.Washington, DC:National Academy Press;1999.
- ,,,,.Nurse‐staffing levels and the quality of care in hospitals.N Engl J Med.2002;346(22):1715–1722.
- ,,, et al.An intervention to decrease catheter‐related bloodstream infections in the ICU.N Engl J Med.2006;355(26):2725–2732.
- ,,, et al.A reengineered hospital discharge program to decrease rehospitalization: a randomized trial.Ann Intern Med.2009;150(3):178–187.
- ,,, et al.A surgical safety checklist to reduce morbidity and mortality in a global population.N Engl J Med.2009;360(5):491–499.
- Disproportionate Share Hospital (DSH). Available at: http://www.hhs. gov/recovery/cms/dsh.html. Accessed December 2009.
- ,,.Measuring outcomes and efficiency in medicare value‐based purchasing.Health Aff (Millwood).2009;28(2):w251–w261.
- .Stimulating the adoption of health information technology.N Engl J Med.2009;360(15):1477–1479.
- ,,,.The effect of rural hospital closures on community economic health.Health Serv Res.2006;41(2):467–485.
- Michigan's Health Care Safety Net: In Jeopardy.2009.
- .Final budget decisions.Running A Hospital. Vol 2009.Boston, MA;2009.
- .Doctors Plan to Limit Beds in ER.Wall Street Journal.2009.
- The Impact of the Economic Crisis on Health Services for Patients and Communities.Washington, DC2009.
- ,.Hospital Operational and Financial Performance Improving.Ann Arbor, MI:Thomson Reuters Center for Healthcare Improvement.2009.
- .The Social Transformation of American Medicine.New York, NY:Basic Books;1983.
- AAFP.Patient Care during the 2008‐2009 Recession – Online Survey.Leawood, KS:AAFP.2009.
- The Impact of the Economic Crisis on Health Services for Patients and Communities.Washington, D.C.:American Hospital Association.2009.
- The economic downturn and its impact on hospitals. American Hospital Association Trendwatch.2009.
- ,,.The Current Recession and U.S. Hospitals:Center for Healthcare Improvement.2009.
- ,,.The cost‐shift payment ‘hydraulic’: foundation, history, and implications.Health Aff (Millwood).2006;25(1):22–33.
- ,.To Err Is Human: Building a Safer Health System.Washington, DC:National Academy Press;1999.
- ,,,,.Nurse‐staffing levels and the quality of care in hospitals.N Engl J Med.2002;346(22):1715–1722.
- ,,, et al.An intervention to decrease catheter‐related bloodstream infections in the ICU.N Engl J Med.2006;355(26):2725–2732.
- ,,, et al.A reengineered hospital discharge program to decrease rehospitalization: a randomized trial.Ann Intern Med.2009;150(3):178–187.
- ,,, et al.A surgical safety checklist to reduce morbidity and mortality in a global population.N Engl J Med.2009;360(5):491–499.
- Disproportionate Share Hospital (DSH). Available at: http://www.hhs. gov/recovery/cms/dsh.html. Accessed December 2009.
- ,,.Measuring outcomes and efficiency in medicare value‐based purchasing.Health Aff (Millwood).2009;28(2):w251–w261.
- .Stimulating the adoption of health information technology.N Engl J Med.2009;360(15):1477–1479.
- ,,,.The effect of rural hospital closures on community economic health.Health Serv Res.2006;41(2):467–485.
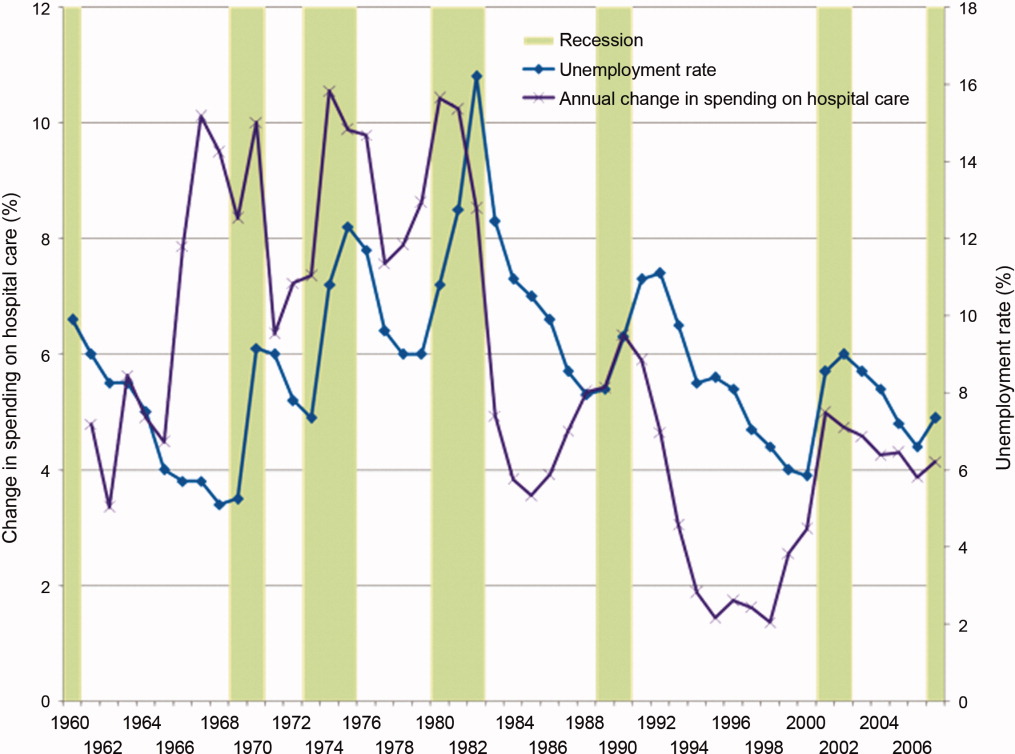
New Resident Regulations on the Horizon
The Accreditation Council for Graduate Medical Education (ACGME) task force is close to offering revised standards for medical resident work hours—a decision that could significantly change the landscape for academic hospitalist programs.
While no date has been set for the unveiling, a May 4 letter written by ACGME CEO Thomas Nasca, MD, MACP, says “the work of the task force is nearly complete.” Many expect the rules will be offered as a draft for public comment in the coming weeks. If approved, the new regulations would probably take effect in July 2011. ACGME formed the task force more than a year ago as the prescribed five-year update to the landmark 2003 duty-hour standards.
Medical experts say the new rules will in many ways mirror the recommendations of the Institute of Medicine’s 2008 report “Resident Duty Hours: Enhancing Sleep, Supervision and Safety.” The oft-quoted report recommended residents only treat patients for up to 16 hours during their shift, down from the current recommendation of 24 hours. It also suggests residents take an uninterrupted five hours for a continuous sleep period between 10 p.m. and 8 a.m.
Many HM physicians expect the new ACGME rules will include a 60-hour workweek cap, part of a growing trend to try to balance the educational requirements of medical school with the need to expose residents to practical experience. Dr. Nasca gave no hint as to what ACGME’s recommendations will be.
In an article in this month’s The Hospitalist, academic and community hospitalists say they have been keeping on eye on how the newest rules will change their playing fields: Will a wave of academics flee teaching hospitals, as additional clinical duties become an intrusion? Will teaching hospitals face financial pressure as they struggle to replace the lower-cost labor force that residents represent? And—perhaps most importantly from a medical perspective—will graduate trainees be as prepared as their predecessors when they enter practice?
“Hospitalists will always be involved in teaching—it will never go away,” says Julia Wright, MD, FHM, a member of Team Hospitalist and clinical associate professor of medicine and director of hospital medicine at the University of Wisconsin School of Medicine and Public Health in Madison. “But it will be a very different balance, a different kind of feel.”
The Accreditation Council for Graduate Medical Education (ACGME) task force is close to offering revised standards for medical resident work hours—a decision that could significantly change the landscape for academic hospitalist programs.
While no date has been set for the unveiling, a May 4 letter written by ACGME CEO Thomas Nasca, MD, MACP, says “the work of the task force is nearly complete.” Many expect the rules will be offered as a draft for public comment in the coming weeks. If approved, the new regulations would probably take effect in July 2011. ACGME formed the task force more than a year ago as the prescribed five-year update to the landmark 2003 duty-hour standards.
Medical experts say the new rules will in many ways mirror the recommendations of the Institute of Medicine’s 2008 report “Resident Duty Hours: Enhancing Sleep, Supervision and Safety.” The oft-quoted report recommended residents only treat patients for up to 16 hours during their shift, down from the current recommendation of 24 hours. It also suggests residents take an uninterrupted five hours for a continuous sleep period between 10 p.m. and 8 a.m.
Many HM physicians expect the new ACGME rules will include a 60-hour workweek cap, part of a growing trend to try to balance the educational requirements of medical school with the need to expose residents to practical experience. Dr. Nasca gave no hint as to what ACGME’s recommendations will be.
In an article in this month’s The Hospitalist, academic and community hospitalists say they have been keeping on eye on how the newest rules will change their playing fields: Will a wave of academics flee teaching hospitals, as additional clinical duties become an intrusion? Will teaching hospitals face financial pressure as they struggle to replace the lower-cost labor force that residents represent? And—perhaps most importantly from a medical perspective—will graduate trainees be as prepared as their predecessors when they enter practice?
“Hospitalists will always be involved in teaching—it will never go away,” says Julia Wright, MD, FHM, a member of Team Hospitalist and clinical associate professor of medicine and director of hospital medicine at the University of Wisconsin School of Medicine and Public Health in Madison. “But it will be a very different balance, a different kind of feel.”
The Accreditation Council for Graduate Medical Education (ACGME) task force is close to offering revised standards for medical resident work hours—a decision that could significantly change the landscape for academic hospitalist programs.
While no date has been set for the unveiling, a May 4 letter written by ACGME CEO Thomas Nasca, MD, MACP, says “the work of the task force is nearly complete.” Many expect the rules will be offered as a draft for public comment in the coming weeks. If approved, the new regulations would probably take effect in July 2011. ACGME formed the task force more than a year ago as the prescribed five-year update to the landmark 2003 duty-hour standards.
Medical experts say the new rules will in many ways mirror the recommendations of the Institute of Medicine’s 2008 report “Resident Duty Hours: Enhancing Sleep, Supervision and Safety.” The oft-quoted report recommended residents only treat patients for up to 16 hours during their shift, down from the current recommendation of 24 hours. It also suggests residents take an uninterrupted five hours for a continuous sleep period between 10 p.m. and 8 a.m.
Many HM physicians expect the new ACGME rules will include a 60-hour workweek cap, part of a growing trend to try to balance the educational requirements of medical school with the need to expose residents to practical experience. Dr. Nasca gave no hint as to what ACGME’s recommendations will be.
In an article in this month’s The Hospitalist, academic and community hospitalists say they have been keeping on eye on how the newest rules will change their playing fields: Will a wave of academics flee teaching hospitals, as additional clinical duties become an intrusion? Will teaching hospitals face financial pressure as they struggle to replace the lower-cost labor force that residents represent? And—perhaps most importantly from a medical perspective—will graduate trainees be as prepared as their predecessors when they enter practice?
“Hospitalists will always be involved in teaching—it will never go away,” says Julia Wright, MD, FHM, a member of Team Hospitalist and clinical associate professor of medicine and director of hospital medicine at the University of Wisconsin School of Medicine and Public Health in Madison. “But it will be a very different balance, a different kind of feel.”
In the Literature: Research You Need to Know
Clinical question: Is recombinant tissue-type plasminogen activator (rt-PA) at 50 mg/2 hr as effective and safe as 100 mg/2 hr for acute pulmonary thromboembolism (PTE)?
Background: The U.S. Food and Drug Administration approved a 100 mg/2 hr dose of rt-PA, which has been recommended as the standard regimen for PTE. Lower doses potentially have less bleeding but their clinical efficacy in PTE has not yet been evaluated. If efficacious, rt-PA at 50 mg/2 hr used for treating acute MI might prove to be a better regimen for acute PTE.
Study design: Prospective, randomized, open-label, multicenter trial.
Setting: Multiple centers in China.
Synopsis: 118 patients with PTE, with either hemodynamic instability or anatomically massive obstruction, were assigned to receive rt-PA at 50 mg/2 hr (n=65) or 100 mg/2 hr (n=53) and followed for 14 days. Clinical efficacy as serially measured by improvement in pulmonary artery pressure and right ventricular function on echocardiogram, lung perfusion on V/Q scan, and pulmonary artery obstruction by CTPA was not significantly different between the two groups.
Though mortality was not significantly different between both groups (three in the high-dose group and one in the low-dose group), there was one fatal ICH in the high-dose group. As can be expected, total bleeding prevalence (major and minor) was lower in the 50-mg group (17% vs. 32%, p=0.084), especially in patients with body weight <65 kgs or BMI <24 kg/m2.
The fact that two-thirds of the patients had only anatomically massive PTE without any hemodynamic instability limits the extrapolation of the efficacy of low-dose rt-PA because heparin alone is generally used in these cases. Also, patients with body weight >100 kg may need the higher dose but were not evaluated adequately in this study.
Bottom line: A lower-dose regimen of 50 mg/2 hr of rt-PA is as efficacious as 100 mg/2 hr in treatment of PTE but offers a better safety profile in patients with weight <65kg.
Citation: Wang C, Zhai Z, Yang Y, et al. Efficacy and safety of low dose recombinant tissue-type plasminogen activator for the treatment of acute pulmonary thromboembolism: a randomized, multicenter, controlled trial. Chest. 2010;137(2):254-262.
Clinical question: Is recombinant tissue-type plasminogen activator (rt-PA) at 50 mg/2 hr as effective and safe as 100 mg/2 hr for acute pulmonary thromboembolism (PTE)?
Background: The U.S. Food and Drug Administration approved a 100 mg/2 hr dose of rt-PA, which has been recommended as the standard regimen for PTE. Lower doses potentially have less bleeding but their clinical efficacy in PTE has not yet been evaluated. If efficacious, rt-PA at 50 mg/2 hr used for treating acute MI might prove to be a better regimen for acute PTE.
Study design: Prospective, randomized, open-label, multicenter trial.
Setting: Multiple centers in China.
Synopsis: 118 patients with PTE, with either hemodynamic instability or anatomically massive obstruction, were assigned to receive rt-PA at 50 mg/2 hr (n=65) or 100 mg/2 hr (n=53) and followed for 14 days. Clinical efficacy as serially measured by improvement in pulmonary artery pressure and right ventricular function on echocardiogram, lung perfusion on V/Q scan, and pulmonary artery obstruction by CTPA was not significantly different between the two groups.
Though mortality was not significantly different between both groups (three in the high-dose group and one in the low-dose group), there was one fatal ICH in the high-dose group. As can be expected, total bleeding prevalence (major and minor) was lower in the 50-mg group (17% vs. 32%, p=0.084), especially in patients with body weight <65 kgs or BMI <24 kg/m2.
The fact that two-thirds of the patients had only anatomically massive PTE without any hemodynamic instability limits the extrapolation of the efficacy of low-dose rt-PA because heparin alone is generally used in these cases. Also, patients with body weight >100 kg may need the higher dose but were not evaluated adequately in this study.
Bottom line: A lower-dose regimen of 50 mg/2 hr of rt-PA is as efficacious as 100 mg/2 hr in treatment of PTE but offers a better safety profile in patients with weight <65kg.
Citation: Wang C, Zhai Z, Yang Y, et al. Efficacy and safety of low dose recombinant tissue-type plasminogen activator for the treatment of acute pulmonary thromboembolism: a randomized, multicenter, controlled trial. Chest. 2010;137(2):254-262.
Clinical question: Is recombinant tissue-type plasminogen activator (rt-PA) at 50 mg/2 hr as effective and safe as 100 mg/2 hr for acute pulmonary thromboembolism (PTE)?
Background: The U.S. Food and Drug Administration approved a 100 mg/2 hr dose of rt-PA, which has been recommended as the standard regimen for PTE. Lower doses potentially have less bleeding but their clinical efficacy in PTE has not yet been evaluated. If efficacious, rt-PA at 50 mg/2 hr used for treating acute MI might prove to be a better regimen for acute PTE.
Study design: Prospective, randomized, open-label, multicenter trial.
Setting: Multiple centers in China.
Synopsis: 118 patients with PTE, with either hemodynamic instability or anatomically massive obstruction, were assigned to receive rt-PA at 50 mg/2 hr (n=65) or 100 mg/2 hr (n=53) and followed for 14 days. Clinical efficacy as serially measured by improvement in pulmonary artery pressure and right ventricular function on echocardiogram, lung perfusion on V/Q scan, and pulmonary artery obstruction by CTPA was not significantly different between the two groups.
Though mortality was not significantly different between both groups (three in the high-dose group and one in the low-dose group), there was one fatal ICH in the high-dose group. As can be expected, total bleeding prevalence (major and minor) was lower in the 50-mg group (17% vs. 32%, p=0.084), especially in patients with body weight <65 kgs or BMI <24 kg/m2.
The fact that two-thirds of the patients had only anatomically massive PTE without any hemodynamic instability limits the extrapolation of the efficacy of low-dose rt-PA because heparin alone is generally used in these cases. Also, patients with body weight >100 kg may need the higher dose but were not evaluated adequately in this study.
Bottom line: A lower-dose regimen of 50 mg/2 hr of rt-PA is as efficacious as 100 mg/2 hr in treatment of PTE but offers a better safety profile in patients with weight <65kg.
Citation: Wang C, Zhai Z, Yang Y, et al. Efficacy and safety of low dose recombinant tissue-type plasminogen activator for the treatment of acute pulmonary thromboembolism: a randomized, multicenter, controlled trial. Chest. 2010;137(2):254-262.
ONLINE EXCLUSIVE: Workforce Readiness

In the battle over how to best cope with the changing landscape of medical training, a key front could be job descriptions. That’s right: job descriptions.
“It behooves a program to define what and who they are, and follow it through in the hiring process,” says Ken Simone, DO, FHM, president of Hospitalist and Practice Solutions in Veazie, Maine, and author of the recently published Hospitalist Recruitment and Retention: Building a Hospital Medicine Program (Hoboken, N.J.: Wiley-Blackwell, 2010).
Dr. Simone, a member of Team Hospitalist, notes that the way any successful program—private, academic, or community—hires and keeps quality staff is to attract like-minded physicians.
For younger physicians, he says a major factor in job selection is the role of mentoring. And in the wake of new and potentially continuing changes to the training that residents are allowed to experience, that training role becomes even more important. “The good news is we can basically mold this person and we can support this person, so they become the provider we want them to be, as opposed to an experienced doctor who has habits they won’t change,” Dr. Simone says.
—Ken Simone, DO, FHM, president, Hospitalist and Practice Solutions, Veazie, Maine
And while much of the discussion about residency regulations focuses on potential downsides, Dr. Simone quickly points out that post-graduate physicians often are well versed in technology, evidence-based protocols, and other modern techniques that older physicians are reticent to adopt.
“Although we recognize that recruiting is challenging, that’s not been something that has prohibited [HM] from agreeing to be the solution here,” says Shaun Frost, MD, FACP, FHM, regional director for Cogent Healthcare in St. Paul, Minn., and an SHM board member. “With education, training, and mentoring, young hospitalists fresh out of their residencies can learn to practice as efficiently as so-called 20th-century residents.”
Richard Quinn is a freelance writer based in New Jersey.
In the battle over how to best cope with the changing landscape of medical training, a key front could be job descriptions. That’s right: job descriptions.
“It behooves a program to define what and who they are, and follow it through in the hiring process,” says Ken Simone, DO, FHM, president of Hospitalist and Practice Solutions in Veazie, Maine, and author of the recently published Hospitalist Recruitment and Retention: Building a Hospital Medicine Program (Hoboken, N.J.: Wiley-Blackwell, 2010).
Dr. Simone, a member of Team Hospitalist, notes that the way any successful program—private, academic, or community—hires and keeps quality staff is to attract like-minded physicians.
For younger physicians, he says a major factor in job selection is the role of mentoring. And in the wake of new and potentially continuing changes to the training that residents are allowed to experience, that training role becomes even more important. “The good news is we can basically mold this person and we can support this person, so they become the provider we want them to be, as opposed to an experienced doctor who has habits they won’t change,” Dr. Simone says.
—Ken Simone, DO, FHM, president, Hospitalist and Practice Solutions, Veazie, Maine
And while much of the discussion about residency regulations focuses on potential downsides, Dr. Simone quickly points out that post-graduate physicians often are well versed in technology, evidence-based protocols, and other modern techniques that older physicians are reticent to adopt.
“Although we recognize that recruiting is challenging, that’s not been something that has prohibited [HM] from agreeing to be the solution here,” says Shaun Frost, MD, FACP, FHM, regional director for Cogent Healthcare in St. Paul, Minn., and an SHM board member. “With education, training, and mentoring, young hospitalists fresh out of their residencies can learn to practice as efficiently as so-called 20th-century residents.”
Richard Quinn is a freelance writer based in New Jersey.
In the battle over how to best cope with the changing landscape of medical training, a key front could be job descriptions. That’s right: job descriptions.
“It behooves a program to define what and who they are, and follow it through in the hiring process,” says Ken Simone, DO, FHM, president of Hospitalist and Practice Solutions in Veazie, Maine, and author of the recently published Hospitalist Recruitment and Retention: Building a Hospital Medicine Program (Hoboken, N.J.: Wiley-Blackwell, 2010).
Dr. Simone, a member of Team Hospitalist, notes that the way any successful program—private, academic, or community—hires and keeps quality staff is to attract like-minded physicians.
For younger physicians, he says a major factor in job selection is the role of mentoring. And in the wake of new and potentially continuing changes to the training that residents are allowed to experience, that training role becomes even more important. “The good news is we can basically mold this person and we can support this person, so they become the provider we want them to be, as opposed to an experienced doctor who has habits they won’t change,” Dr. Simone says.
—Ken Simone, DO, FHM, president, Hospitalist and Practice Solutions, Veazie, Maine
And while much of the discussion about residency regulations focuses on potential downsides, Dr. Simone quickly points out that post-graduate physicians often are well versed in technology, evidence-based protocols, and other modern techniques that older physicians are reticent to adopt.
“Although we recognize that recruiting is challenging, that’s not been something that has prohibited [HM] from agreeing to be the solution here,” says Shaun Frost, MD, FACP, FHM, regional director for Cogent Healthcare in St. Paul, Minn., and an SHM board member. “With education, training, and mentoring, young hospitalists fresh out of their residencies can learn to practice as efficiently as so-called 20th-century residents.”
Richard Quinn is a freelance writer based in New Jersey.
ONLINE EXCLUSIVE: Audio interview with ABIM Learning Session Director Julius Yang, MD
Focused Practice in Hospital Medicine Worth the Additional Cost

Focused Practice in Hospital Medicine Worth the Additional Cost
Why are we being required to fork over an extra $380 for the Focused Practice in Hospital Medicine MOC? This feels like the icing on the cake of already a major ripoff.
Dr. Ragan
Grass Valley, Calif.
Dr. Hospitalist responds: Thank you for your frank reaction to the much-anticipated American Board of Internal Medicine (ABIM) Focused Practice in Hospital Medicine (FPHM) Maintenance of Certification (MOC) program. As you noted, an additional fee is required to participate in this recertification program.
To my knowledge, any and all fees associated with recertification are paid to ABIM. No other organization benefits from the added cost, so your question might be more appropriately addressed to ABIM (see “Focused Practice in Hospital Medicine,” May 2010, p. 1). But because you asked the question, I am happy to respond with my thoughts.
Participation in the FPHM MOC program is not mandatory. I am not aware of any organization that is requiring hospitalists to participate. I don’t expect that your lack of participation will affect your ability to obtain hospital privileges. Like any new MOC program, I would expect some up-front administrative costs associated with developing and administering the practice-improvement modules and the secure examination.
It’s up to you and others to decide whether this added recognition is worth the cost. I can tell you that I have made the decision to participate. I fully expect to be part of the inaugural class of ABIM diplomates with this added recognition by the end of the year.
What went into my own decision to participate? I can tell you that I am a practicing hospitalist who makes a salary typical of most hospitalists. I am frugal with my money and certainly do not view the added cost as an insignificant amount of money. Like most hospitalists, I am not only busy with my professional life, but I have plenty of family commitments as well.
I expect the exam will be rigorous, and the requirements of the practice-improvement modules will be demanding. I would not want it any other way. In the fast-changing healthcare environment, I believe that hospitalists will be challenged to think about what it means to care for a hospitalized patient. To succeed in the future, hospitalists will be expected to not only participate, but also lead QI efforts at their institutions. The FPHM MOC will distinguish me as a hospitalist with added qualifications in the field of QI.
So how about it, Dr. Ragan? Will you join me?
What Certification Requirements Should a Hospitalist Program Have for Its Physicians?
I hope you can help me with some questions I have concerning starting a hospitalist program at my medical center. Are there certain requirements (e.g., board certification in internal medicine, ACLS, etc.) that need to be met, or is that up to the facility? The physician interested in the position is board-certified in infectious disease. Any direction you can give me on this would be greatly appreciated.
Marisa Sellers,
Medical Staff Coordinator,
Hartselle Medical Center,
Hartselle, Ala.
Dr. Hospitalist responds: Congratulations on your medical center’s decision to establish a hospitalist program. Over the past decade, HM has been the fastest-growing field in all of American medicine. The majority of the country’s acute-care hospitals have hospitalists on staff.
Approximately 85% of the country’s hospitalists received training in internal medicine. Most of the other hospitalists received training in pediatrics or family medicine. While most hospitalists are general internists, some also have additional subspecialty training, which seems to be the case of the physician at your medical center. As you know, different medical facilities have different requirements of their medical staff. At the acute-care hospital where I work clinically, maintenance of board certification is required of all medical staff. I know that is not the case for all hospitals, yet I’m not aware of any hospitals with hospitalist-specific medical staff requirements.
Most of the hospitalists who are internists will be either board-eligible or board-certified with the American Board of Internal Medicine (ABIM). You should be aware that ABIM has developed a new program, the Recognition of Focused Practice (RFP) in Hospital Medicine. As part of this maintenance of certification (MOC) program, ABIM diplomates will have the opportunity to take the first ABIM Hospital Medicine examination in October. For more information about this exam, ABIM’s rationale for recognizing a focused practice in HM, and any other questions about this program, please visit the ABIM Web site at www.abim.org/news/news/focused-
practice-hospital-medicine-qa.aspx.
I have heard from hospitalists trained as family physicians who are interested in RFP as hospitalists. It is my understanding that the American Board of Family Medicine is studying the ABIM program and working to develop a similar program for hospitalists with family medicine board certifications.
Regarding your question about hospitalists and the American Heart Association’s advanced cardiac life support (ACLS) training and certification: While I think it is a great idea for hospitalists to receive this training and maintain this certification, I am not aware of any mandate for hospitalists to be uniformly ACLS-certified. I think this is an issue the medical staff at your medical center will have to decide; basically, what is in the best interests of your patients?
Focused Practice in Hospital Medicine Worth the Additional Cost
Why are we being required to fork over an extra $380 for the Focused Practice in Hospital Medicine MOC? This feels like the icing on the cake of already a major ripoff.
Dr. Ragan
Grass Valley, Calif.
Dr. Hospitalist responds: Thank you for your frank reaction to the much-anticipated American Board of Internal Medicine (ABIM) Focused Practice in Hospital Medicine (FPHM) Maintenance of Certification (MOC) program. As you noted, an additional fee is required to participate in this recertification program.
To my knowledge, any and all fees associated with recertification are paid to ABIM. No other organization benefits from the added cost, so your question might be more appropriately addressed to ABIM (see “Focused Practice in Hospital Medicine,” May 2010, p. 1). But because you asked the question, I am happy to respond with my thoughts.
Participation in the FPHM MOC program is not mandatory. I am not aware of any organization that is requiring hospitalists to participate. I don’t expect that your lack of participation will affect your ability to obtain hospital privileges. Like any new MOC program, I would expect some up-front administrative costs associated with developing and administering the practice-improvement modules and the secure examination.
It’s up to you and others to decide whether this added recognition is worth the cost. I can tell you that I have made the decision to participate. I fully expect to be part of the inaugural class of ABIM diplomates with this added recognition by the end of the year.
What went into my own decision to participate? I can tell you that I am a practicing hospitalist who makes a salary typical of most hospitalists. I am frugal with my money and certainly do not view the added cost as an insignificant amount of money. Like most hospitalists, I am not only busy with my professional life, but I have plenty of family commitments as well.
I expect the exam will be rigorous, and the requirements of the practice-improvement modules will be demanding. I would not want it any other way. In the fast-changing healthcare environment, I believe that hospitalists will be challenged to think about what it means to care for a hospitalized patient. To succeed in the future, hospitalists will be expected to not only participate, but also lead QI efforts at their institutions. The FPHM MOC will distinguish me as a hospitalist with added qualifications in the field of QI.
So how about it, Dr. Ragan? Will you join me?
What Certification Requirements Should a Hospitalist Program Have for Its Physicians?
I hope you can help me with some questions I have concerning starting a hospitalist program at my medical center. Are there certain requirements (e.g., board certification in internal medicine, ACLS, etc.) that need to be met, or is that up to the facility? The physician interested in the position is board-certified in infectious disease. Any direction you can give me on this would be greatly appreciated.
Marisa Sellers,
Medical Staff Coordinator,
Hartselle Medical Center,
Hartselle, Ala.
Dr. Hospitalist responds: Congratulations on your medical center’s decision to establish a hospitalist program. Over the past decade, HM has been the fastest-growing field in all of American medicine. The majority of the country’s acute-care hospitals have hospitalists on staff.
Approximately 85% of the country’s hospitalists received training in internal medicine. Most of the other hospitalists received training in pediatrics or family medicine. While most hospitalists are general internists, some also have additional subspecialty training, which seems to be the case of the physician at your medical center. As you know, different medical facilities have different requirements of their medical staff. At the acute-care hospital where I work clinically, maintenance of board certification is required of all medical staff. I know that is not the case for all hospitals, yet I’m not aware of any hospitals with hospitalist-specific medical staff requirements.
Most of the hospitalists who are internists will be either board-eligible or board-certified with the American Board of Internal Medicine (ABIM). You should be aware that ABIM has developed a new program, the Recognition of Focused Practice (RFP) in Hospital Medicine. As part of this maintenance of certification (MOC) program, ABIM diplomates will have the opportunity to take the first ABIM Hospital Medicine examination in October. For more information about this exam, ABIM’s rationale for recognizing a focused practice in HM, and any other questions about this program, please visit the ABIM Web site at www.abim.org/news/news/focused-
practice-hospital-medicine-qa.aspx.
I have heard from hospitalists trained as family physicians who are interested in RFP as hospitalists. It is my understanding that the American Board of Family Medicine is studying the ABIM program and working to develop a similar program for hospitalists with family medicine board certifications.
Regarding your question about hospitalists and the American Heart Association’s advanced cardiac life support (ACLS) training and certification: While I think it is a great idea for hospitalists to receive this training and maintain this certification, I am not aware of any mandate for hospitalists to be uniformly ACLS-certified. I think this is an issue the medical staff at your medical center will have to decide; basically, what is in the best interests of your patients?
Focused Practice in Hospital Medicine Worth the Additional Cost
Why are we being required to fork over an extra $380 for the Focused Practice in Hospital Medicine MOC? This feels like the icing on the cake of already a major ripoff.
Dr. Ragan
Grass Valley, Calif.
Dr. Hospitalist responds: Thank you for your frank reaction to the much-anticipated American Board of Internal Medicine (ABIM) Focused Practice in Hospital Medicine (FPHM) Maintenance of Certification (MOC) program. As you noted, an additional fee is required to participate in this recertification program.
To my knowledge, any and all fees associated with recertification are paid to ABIM. No other organization benefits from the added cost, so your question might be more appropriately addressed to ABIM (see “Focused Practice in Hospital Medicine,” May 2010, p. 1). But because you asked the question, I am happy to respond with my thoughts.
Participation in the FPHM MOC program is not mandatory. I am not aware of any organization that is requiring hospitalists to participate. I don’t expect that your lack of participation will affect your ability to obtain hospital privileges. Like any new MOC program, I would expect some up-front administrative costs associated with developing and administering the practice-improvement modules and the secure examination.
It’s up to you and others to decide whether this added recognition is worth the cost. I can tell you that I have made the decision to participate. I fully expect to be part of the inaugural class of ABIM diplomates with this added recognition by the end of the year.
What went into my own decision to participate? I can tell you that I am a practicing hospitalist who makes a salary typical of most hospitalists. I am frugal with my money and certainly do not view the added cost as an insignificant amount of money. Like most hospitalists, I am not only busy with my professional life, but I have plenty of family commitments as well.
I expect the exam will be rigorous, and the requirements of the practice-improvement modules will be demanding. I would not want it any other way. In the fast-changing healthcare environment, I believe that hospitalists will be challenged to think about what it means to care for a hospitalized patient. To succeed in the future, hospitalists will be expected to not only participate, but also lead QI efforts at their institutions. The FPHM MOC will distinguish me as a hospitalist with added qualifications in the field of QI.
So how about it, Dr. Ragan? Will you join me?
What Certification Requirements Should a Hospitalist Program Have for Its Physicians?
I hope you can help me with some questions I have concerning starting a hospitalist program at my medical center. Are there certain requirements (e.g., board certification in internal medicine, ACLS, etc.) that need to be met, or is that up to the facility? The physician interested in the position is board-certified in infectious disease. Any direction you can give me on this would be greatly appreciated.
Marisa Sellers,
Medical Staff Coordinator,
Hartselle Medical Center,
Hartselle, Ala.
Dr. Hospitalist responds: Congratulations on your medical center’s decision to establish a hospitalist program. Over the past decade, HM has been the fastest-growing field in all of American medicine. The majority of the country’s acute-care hospitals have hospitalists on staff.
Approximately 85% of the country’s hospitalists received training in internal medicine. Most of the other hospitalists received training in pediatrics or family medicine. While most hospitalists are general internists, some also have additional subspecialty training, which seems to be the case of the physician at your medical center. As you know, different medical facilities have different requirements of their medical staff. At the acute-care hospital where I work clinically, maintenance of board certification is required of all medical staff. I know that is not the case for all hospitals, yet I’m not aware of any hospitals with hospitalist-specific medical staff requirements.
Most of the hospitalists who are internists will be either board-eligible or board-certified with the American Board of Internal Medicine (ABIM). You should be aware that ABIM has developed a new program, the Recognition of Focused Practice (RFP) in Hospital Medicine. As part of this maintenance of certification (MOC) program, ABIM diplomates will have the opportunity to take the first ABIM Hospital Medicine examination in October. For more information about this exam, ABIM’s rationale for recognizing a focused practice in HM, and any other questions about this program, please visit the ABIM Web site at www.abim.org/news/news/focused-
practice-hospital-medicine-qa.aspx.
I have heard from hospitalists trained as family physicians who are interested in RFP as hospitalists. It is my understanding that the American Board of Family Medicine is studying the ABIM program and working to develop a similar program for hospitalists with family medicine board certifications.
Regarding your question about hospitalists and the American Heart Association’s advanced cardiac life support (ACLS) training and certification: While I think it is a great idea for hospitalists to receive this training and maintain this certification, I am not aware of any mandate for hospitalists to be uniformly ACLS-certified. I think this is an issue the medical staff at your medical center will have to decide; basically, what is in the best interests of your patients?
To Vary Is Human …

Just a few years ago, if I had been asked to comment on variation in healthcare, I would have said it needed a fundraising event for awareness, or even a respected celebrity patron—maybe Sandra Bullock decrying unnecessary variation on Oprah. Fortunately, more socially influential forces evolved. In a relatively short (from a cultural perspective) span of time, variation has emerged to become standard water-cooler talk amongst physicians and politicians alike.
Although the first analysis of medical variation surfaced in 1938, it wasn’t until Wennberg and Gittelsohn’s seminal paper that our collective medical consciousness emerged.1 Wennberg noted that if his children had simply gone to school in the neighboring district of Stowe, Vt., they would have had a 70% chance of having a tonsillectomy, as opposed to a 20% chance in their chosen district of Waterbury. Decades later, that work is the foundation of the Dartmouth Atlas project, which has turned its lenses toward unexplained variation in the costs of healthcare.
Meanwhile, in a parallel—nonmedical—universe, two engineer-statisticians were busy refining quality-control theory in the late 1930s. Shewhart developed the PDCA (plan-do-check-act) cycle, and Deming took it to Japan, revolutionizing that country’s manufacturing industry. They recognized that unwarranted variations were key quality constraints in any process, and that sustained improvements in outcomes could be attained only through careful analysis and control of this variation.
Hospital Variation and Application
Following the Institute of Medicine’s landmark report a decade ago, these fields of study explicitly converged, and variation began to emerge as a key player in healthcare quality discussions. Sprinkle in a few more ingredients—such as the looming cliff that is Medicare insolvency, a failing economy, and Atul Gawande’s uncloaking of McAllen, Texas—and the transformation of the Kool-Aid is now complete.
A fortunate (or unfortunate, depending on your perspective) byproduct of these analyses has been that physicians are at the sharp end of the most important yet variable decisions in medicine. Why are doctors so different in their practices? The short answer is that we’re human; the long answer is that, well, we’re human.
In complex settings, the literature on medical decision-making tells us that we humans simply are not wired to process more than three to five different options at any one time. Even rocket scientists might disagree if they regularly encountered large boluses of clinical data in the face of an ever-exploding body of knowledge. When we dissect the more straightforward daily decisions, the complexity of the human persona then becomes an overlay with as much variability and heterogeneity as our own genetic makeup.
The Simple Life
The late John Eisenberg, in a book titled Doctors’ Decisions and the Cost of Medical Care, lists a dizzying array of reasons behind physician decision-making: experience, risk tolerance, practice style, incentives, and concept of social good, to name a few.2 Each of these domains could be a unique area of study—just for each individual human practitioner. In an era of genomic medicine, the strongest predictor of the phenotypic quality of care might simply be the genotype of the physician.
This is not a revelation for anyone who has ever questioned another physician’s care. My guess is that it’s been less than a week for most of us. After all, we’re hospitalists, perfectly perched as second-tier providers to judge other physicians’ care. We are air-traffic control for doctors’ decisions, and it’s quite a scene: thousands of independent physicians practicing on isolated islands. Like “outside EDs,” some of these habitats appear quite a bit more aboriginal and remote than others. Now, I will admit that I’ve often dreamed of practicing on an isolated, single-palm-tree island. Armed with only a coconut (my patient) and evidence-based medicine, this would be an overdue retreat from the chaotic morass of illogical (i.e., different from my own) medical decisions.
But it is exactly this reaction that provides clues to our current state. No one prepared us for the fact that healthcare delivery is a social science, so frustration and avoidance are merely natural reflections of our immaturity. If we did receive any coaching, it tended to be of the Monday-morning-quarterback school, autocratic and self-serving in nature. We were trained to critique only the finer details of scientific “fact,” not humans in context. How, then, are we to improve our care when we can barely handle the variation?
Advanced Concepts
Adapting a Darwinian perspective, we might hunt out the highly developed and advanced tribes in our midst. One such tribe is pediatric oncology. For decades, almost all variation in pediatric oncology has been controlled through treatment protocols tailored to the particular risk factors of the patient, not the physician. Although this ostensibly improves quality of care, it has had an even greater impact on learning and eventual outcomes.
For this reason and this reason alone, if your 18-year-old child develops leukemia, you probably want to send them to a pediatric oncologist rather than an adult oncologist.7 Survival rates are better because pediatric oncologists have been able to rapidly learn from the enrollment of almost all patients into trials with standardized treatment protocols. By collecting data on a limited number of options and sharing information across practices, true rapid-cycle improvement has materialized.
The key here is not the degree of standardization or the creation of large-scale research networks. It is the extent to which independent practitioners are able to sacrifice their individual beliefs in order to partner for the greater good. Growth and learning do not occur in isolation. A team-based approach, in the setting of standardization and measurement, will accelerate the pace of our evolution. Think about this the next time you feel like throwing a coconut at the infectious-disease consultant who dares cross your island of practice. For if it is human to vary, then only through collaboration may we truly divine. TH
Dr. Shen is The Hospitalist’s pediatric editor. Read his monthly review of pediatric research in our “In the Literature” section (see p. 16).
References
- Wennberg J, Gittelsohn. Small area variations in health care delivery. Science. 1973:182(117):1102-1108.
- Eisenberg JM. Doctors’ Decisions and the Cost of Medical Care: The Reasons for Doctor’s Practice Patterns and Ways to Change Them. Chicago: Health Administration Press; 1986.
Just a few years ago, if I had been asked to comment on variation in healthcare, I would have said it needed a fundraising event for awareness, or even a respected celebrity patron—maybe Sandra Bullock decrying unnecessary variation on Oprah. Fortunately, more socially influential forces evolved. In a relatively short (from a cultural perspective) span of time, variation has emerged to become standard water-cooler talk amongst physicians and politicians alike.
Although the first analysis of medical variation surfaced in 1938, it wasn’t until Wennberg and Gittelsohn’s seminal paper that our collective medical consciousness emerged.1 Wennberg noted that if his children had simply gone to school in the neighboring district of Stowe, Vt., they would have had a 70% chance of having a tonsillectomy, as opposed to a 20% chance in their chosen district of Waterbury. Decades later, that work is the foundation of the Dartmouth Atlas project, which has turned its lenses toward unexplained variation in the costs of healthcare.
Meanwhile, in a parallel—nonmedical—universe, two engineer-statisticians were busy refining quality-control theory in the late 1930s. Shewhart developed the PDCA (plan-do-check-act) cycle, and Deming took it to Japan, revolutionizing that country’s manufacturing industry. They recognized that unwarranted variations were key quality constraints in any process, and that sustained improvements in outcomes could be attained only through careful analysis and control of this variation.
Hospital Variation and Application
Following the Institute of Medicine’s landmark report a decade ago, these fields of study explicitly converged, and variation began to emerge as a key player in healthcare quality discussions. Sprinkle in a few more ingredients—such as the looming cliff that is Medicare insolvency, a failing economy, and Atul Gawande’s uncloaking of McAllen, Texas—and the transformation of the Kool-Aid is now complete.
A fortunate (or unfortunate, depending on your perspective) byproduct of these analyses has been that physicians are at the sharp end of the most important yet variable decisions in medicine. Why are doctors so different in their practices? The short answer is that we’re human; the long answer is that, well, we’re human.
In complex settings, the literature on medical decision-making tells us that we humans simply are not wired to process more than three to five different options at any one time. Even rocket scientists might disagree if they regularly encountered large boluses of clinical data in the face of an ever-exploding body of knowledge. When we dissect the more straightforward daily decisions, the complexity of the human persona then becomes an overlay with as much variability and heterogeneity as our own genetic makeup.
The Simple Life
The late John Eisenberg, in a book titled Doctors’ Decisions and the Cost of Medical Care, lists a dizzying array of reasons behind physician decision-making: experience, risk tolerance, practice style, incentives, and concept of social good, to name a few.2 Each of these domains could be a unique area of study—just for each individual human practitioner. In an era of genomic medicine, the strongest predictor of the phenotypic quality of care might simply be the genotype of the physician.
This is not a revelation for anyone who has ever questioned another physician’s care. My guess is that it’s been less than a week for most of us. After all, we’re hospitalists, perfectly perched as second-tier providers to judge other physicians’ care. We are air-traffic control for doctors’ decisions, and it’s quite a scene: thousands of independent physicians practicing on isolated islands. Like “outside EDs,” some of these habitats appear quite a bit more aboriginal and remote than others. Now, I will admit that I’ve often dreamed of practicing on an isolated, single-palm-tree island. Armed with only a coconut (my patient) and evidence-based medicine, this would be an overdue retreat from the chaotic morass of illogical (i.e., different from my own) medical decisions.
But it is exactly this reaction that provides clues to our current state. No one prepared us for the fact that healthcare delivery is a social science, so frustration and avoidance are merely natural reflections of our immaturity. If we did receive any coaching, it tended to be of the Monday-morning-quarterback school, autocratic and self-serving in nature. We were trained to critique only the finer details of scientific “fact,” not humans in context. How, then, are we to improve our care when we can barely handle the variation?
Advanced Concepts
Adapting a Darwinian perspective, we might hunt out the highly developed and advanced tribes in our midst. One such tribe is pediatric oncology. For decades, almost all variation in pediatric oncology has been controlled through treatment protocols tailored to the particular risk factors of the patient, not the physician. Although this ostensibly improves quality of care, it has had an even greater impact on learning and eventual outcomes.
For this reason and this reason alone, if your 18-year-old child develops leukemia, you probably want to send them to a pediatric oncologist rather than an adult oncologist.7 Survival rates are better because pediatric oncologists have been able to rapidly learn from the enrollment of almost all patients into trials with standardized treatment protocols. By collecting data on a limited number of options and sharing information across practices, true rapid-cycle improvement has materialized.
The key here is not the degree of standardization or the creation of large-scale research networks. It is the extent to which independent practitioners are able to sacrifice their individual beliefs in order to partner for the greater good. Growth and learning do not occur in isolation. A team-based approach, in the setting of standardization and measurement, will accelerate the pace of our evolution. Think about this the next time you feel like throwing a coconut at the infectious-disease consultant who dares cross your island of practice. For if it is human to vary, then only through collaboration may we truly divine. TH
Dr. Shen is The Hospitalist’s pediatric editor. Read his monthly review of pediatric research in our “In the Literature” section (see p. 16).
References
- Wennberg J, Gittelsohn. Small area variations in health care delivery. Science. 1973:182(117):1102-1108.
- Eisenberg JM. Doctors’ Decisions and the Cost of Medical Care: The Reasons for Doctor’s Practice Patterns and Ways to Change Them. Chicago: Health Administration Press; 1986.
Just a few years ago, if I had been asked to comment on variation in healthcare, I would have said it needed a fundraising event for awareness, or even a respected celebrity patron—maybe Sandra Bullock decrying unnecessary variation on Oprah. Fortunately, more socially influential forces evolved. In a relatively short (from a cultural perspective) span of time, variation has emerged to become standard water-cooler talk amongst physicians and politicians alike.
Although the first analysis of medical variation surfaced in 1938, it wasn’t until Wennberg and Gittelsohn’s seminal paper that our collective medical consciousness emerged.1 Wennberg noted that if his children had simply gone to school in the neighboring district of Stowe, Vt., they would have had a 70% chance of having a tonsillectomy, as opposed to a 20% chance in their chosen district of Waterbury. Decades later, that work is the foundation of the Dartmouth Atlas project, which has turned its lenses toward unexplained variation in the costs of healthcare.
Meanwhile, in a parallel—nonmedical—universe, two engineer-statisticians were busy refining quality-control theory in the late 1930s. Shewhart developed the PDCA (plan-do-check-act) cycle, and Deming took it to Japan, revolutionizing that country’s manufacturing industry. They recognized that unwarranted variations were key quality constraints in any process, and that sustained improvements in outcomes could be attained only through careful analysis and control of this variation.
Hospital Variation and Application
Following the Institute of Medicine’s landmark report a decade ago, these fields of study explicitly converged, and variation began to emerge as a key player in healthcare quality discussions. Sprinkle in a few more ingredients—such as the looming cliff that is Medicare insolvency, a failing economy, and Atul Gawande’s uncloaking of McAllen, Texas—and the transformation of the Kool-Aid is now complete.
A fortunate (or unfortunate, depending on your perspective) byproduct of these analyses has been that physicians are at the sharp end of the most important yet variable decisions in medicine. Why are doctors so different in their practices? The short answer is that we’re human; the long answer is that, well, we’re human.
In complex settings, the literature on medical decision-making tells us that we humans simply are not wired to process more than three to five different options at any one time. Even rocket scientists might disagree if they regularly encountered large boluses of clinical data in the face of an ever-exploding body of knowledge. When we dissect the more straightforward daily decisions, the complexity of the human persona then becomes an overlay with as much variability and heterogeneity as our own genetic makeup.
The Simple Life
The late John Eisenberg, in a book titled Doctors’ Decisions and the Cost of Medical Care, lists a dizzying array of reasons behind physician decision-making: experience, risk tolerance, practice style, incentives, and concept of social good, to name a few.2 Each of these domains could be a unique area of study—just for each individual human practitioner. In an era of genomic medicine, the strongest predictor of the phenotypic quality of care might simply be the genotype of the physician.
This is not a revelation for anyone who has ever questioned another physician’s care. My guess is that it’s been less than a week for most of us. After all, we’re hospitalists, perfectly perched as second-tier providers to judge other physicians’ care. We are air-traffic control for doctors’ decisions, and it’s quite a scene: thousands of independent physicians practicing on isolated islands. Like “outside EDs,” some of these habitats appear quite a bit more aboriginal and remote than others. Now, I will admit that I’ve often dreamed of practicing on an isolated, single-palm-tree island. Armed with only a coconut (my patient) and evidence-based medicine, this would be an overdue retreat from the chaotic morass of illogical (i.e., different from my own) medical decisions.
But it is exactly this reaction that provides clues to our current state. No one prepared us for the fact that healthcare delivery is a social science, so frustration and avoidance are merely natural reflections of our immaturity. If we did receive any coaching, it tended to be of the Monday-morning-quarterback school, autocratic and self-serving in nature. We were trained to critique only the finer details of scientific “fact,” not humans in context. How, then, are we to improve our care when we can barely handle the variation?
Advanced Concepts
Adapting a Darwinian perspective, we might hunt out the highly developed and advanced tribes in our midst. One such tribe is pediatric oncology. For decades, almost all variation in pediatric oncology has been controlled through treatment protocols tailored to the particular risk factors of the patient, not the physician. Although this ostensibly improves quality of care, it has had an even greater impact on learning and eventual outcomes.
For this reason and this reason alone, if your 18-year-old child develops leukemia, you probably want to send them to a pediatric oncologist rather than an adult oncologist.7 Survival rates are better because pediatric oncologists have been able to rapidly learn from the enrollment of almost all patients into trials with standardized treatment protocols. By collecting data on a limited number of options and sharing information across practices, true rapid-cycle improvement has materialized.
The key here is not the degree of standardization or the creation of large-scale research networks. It is the extent to which independent practitioners are able to sacrifice their individual beliefs in order to partner for the greater good. Growth and learning do not occur in isolation. A team-based approach, in the setting of standardization and measurement, will accelerate the pace of our evolution. Think about this the next time you feel like throwing a coconut at the infectious-disease consultant who dares cross your island of practice. For if it is human to vary, then only through collaboration may we truly divine. TH
Dr. Shen is The Hospitalist’s pediatric editor. Read his monthly review of pediatric research in our “In the Literature” section (see p. 16).
References
- Wennberg J, Gittelsohn. Small area variations in health care delivery. Science. 1973:182(117):1102-1108.
- Eisenberg JM. Doctors’ Decisions and the Cost of Medical Care: The Reasons for Doctor’s Practice Patterns and Ways to Change Them. Chicago: Health Administration Press; 1986.
Square Peg, Square Hole

I encounter a lot of hospitalists who complain that the other doctors at their hospital think of hospitalists as second-class citizens, as sort of like career residents. HM program directors need to make sure that is not the case for the hospitalists in their practice.
SHM has worked with the AMA’s Organized Medical Staff Section to assess the perception of hospitalists by primary-care physicians (PCPs) and hospitalists themselves. When asked in a 2009 survey, “Do you agree or disagree that hospitalists are respected members of the medical staff at a hospital?” only 3 out of 4 respondents agreed or highly agreed. That percentage is up slightly from the same survey conducted in 2007, and we don’t have data regarding how the responses would have been different if the question had been asked about other specialties. But I still find it concerning that about 25% of PCPs and hospitalists don’t see hospitalists as respected members of a medical staff. (If you are wondering, there wasn’t much of a difference between how hospitalists and PCPs answered the question.)
Use First Names
In the 1980s, I left residency and entered private practice as a hospitalist in a nonteaching, suburban hospital. I had a really hard time calling other doctors by their first names, especially the highly regarded senior internist who was my former roommate’s dad. He had always been Dr. McCollough to me, and I insisted calling him “Doctor” until we had been peers on the same medical staff for about a year.
Finally, in a somewhat annoyed voice, he told me I had to start calling him “Bob,” and that I should call all the doctors by their first names. It took a while, but using first names began to feel normal. Looking back on it, I think Dr. McCollough Bob taught me an important lesson about fitting in.
So make sure the hospitalists in your group call other doctors by their first names, too.
Dress the Part
I’ve come to believe that there are a number of things some hospitalists do to sabotage their own interest in being respected by the medical staff at their hospital. To my surprise, I’ve worked with a number of hospitalist groups in which most dress and act like residents, then complain that other doctors at their hospital treat them like residents. I think the way we dress, especially early in our careers, is a pretty big deal. If you’re similar in age to residents, then you’ll sure look like a resident if you dress like them. So don’t wear scrubs and Skechers unless all of the doctors in your hospital wear scrubs and Skechers.
The best advice is to dress the way the respected doctors dress. Follow the lead on things like neckties, dresses, and the white coat (the latter is almost unheard of at my hospital unless it is used to cover up scrubs). Fortunately, few doctors dress formally anymore (e.g., suit, and tie or sport coat for men). Emerging research might push all of us toward shedding ties, long sleeves, and the white coat before long.
Of course, you should keep in mind the way patients would like to see you dress. You can find information about patient expectations through a simple Internet search or by asking the person in charge of patient satisfaction at your hospital.
Seek Social Connections
Just like the issue of dress, I’ve encountered a number of hospitalist groups that have a habit of sneaking into the physician lunchroom, grabbing food in a “to go” container, and heading back to their office to eat together. These hospitalists are missing a valuable opportunity to enjoy social conversation with physicians of all specialties. If your hospital has a physician lunch room that is crowded with doctors, take advantage of the opportunity to build social networks.
You don’t need to eat there every day. (For a number of years, I enjoyed having lunch with the social workers in our main cafeteria.) But you should eat there more frequently than sneaking back to your office to eat only with other hospitalists. (If you don’t have time for lunch, then we need to talk about workload and efficiency issues.)
Look for other opportunities to make connections with other doctors through service on hospital committees, participation in social events at the hospital, or speaking at grand rounds. Although any single activity might not have significant impact, if you do these things regularly, you will form better relationships and be less likely to be or feel “dumped on,” and if it does happen, you’re in a much better position to address it if the dumping doctor is a friend.
Leadership Positions
Work to ensure a member of your group always sits on the medical staff executive committee, and seek out leadership positions like chief of medicine or chief of staff. Don’t simply assume you are too young or too inexperienced. Your hospital really needs the leadership of doctors who have a broad view of hospital operations and medical staff affairs. Few doctors have a broader view than hospitalists.
And if you have an interest in medical staff leadership, think about whether you’d like to serve as your hospital’s chief medical officer (aka vice president of medical affairs). All of these activities are important ways to influence what happens at your hospital, but aside from that, they are an excellent way to build relationships and gain respect from throughout the medical staff.
Worthwhile Effort
Ensuring that the hospitalists in your group feel respected and valued by other doctors and everyone they work with is important. Don’t make the mistake of thinking that working on this is just about stroking hospitalists’ egos.
I coauthored a 2001 research study on hospitalist burnout that failed to show a correlation between workload and burnout, but the study found that things like poor occupational solidarity are associated with burnout.1
Feeling like you fit in and are a respected member of your peer group (medical staff) is important and worth working on diligently. TH
Dr. Nelson has been a practicing hospitalist since 1988 and is cofounder and past president of SHM. He is a principal in Nelson Flores Hospital Medicine Consultants, a national hospitalist practice management consulting firm (www.nelsonflores.com). He is course co-director and faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official position of SHM.
Reference
- Hoff T, Whitcomb WF, Nelson JR. Thriving and surviving in a new medical career: the case of hospitalist physicians. Jrl Health Social Behavior. 2001;43:72-91.
I encounter a lot of hospitalists who complain that the other doctors at their hospital think of hospitalists as second-class citizens, as sort of like career residents. HM program directors need to make sure that is not the case for the hospitalists in their practice.
SHM has worked with the AMA’s Organized Medical Staff Section to assess the perception of hospitalists by primary-care physicians (PCPs) and hospitalists themselves. When asked in a 2009 survey, “Do you agree or disagree that hospitalists are respected members of the medical staff at a hospital?” only 3 out of 4 respondents agreed or highly agreed. That percentage is up slightly from the same survey conducted in 2007, and we don’t have data regarding how the responses would have been different if the question had been asked about other specialties. But I still find it concerning that about 25% of PCPs and hospitalists don’t see hospitalists as respected members of a medical staff. (If you are wondering, there wasn’t much of a difference between how hospitalists and PCPs answered the question.)
Use First Names
In the 1980s, I left residency and entered private practice as a hospitalist in a nonteaching, suburban hospital. I had a really hard time calling other doctors by their first names, especially the highly regarded senior internist who was my former roommate’s dad. He had always been Dr. McCollough to me, and I insisted calling him “Doctor” until we had been peers on the same medical staff for about a year.
Finally, in a somewhat annoyed voice, he told me I had to start calling him “Bob,” and that I should call all the doctors by their first names. It took a while, but using first names began to feel normal. Looking back on it, I think Dr. McCollough Bob taught me an important lesson about fitting in.
So make sure the hospitalists in your group call other doctors by their first names, too.
Dress the Part
I’ve come to believe that there are a number of things some hospitalists do to sabotage their own interest in being respected by the medical staff at their hospital. To my surprise, I’ve worked with a number of hospitalist groups in which most dress and act like residents, then complain that other doctors at their hospital treat them like residents. I think the way we dress, especially early in our careers, is a pretty big deal. If you’re similar in age to residents, then you’ll sure look like a resident if you dress like them. So don’t wear scrubs and Skechers unless all of the doctors in your hospital wear scrubs and Skechers.
The best advice is to dress the way the respected doctors dress. Follow the lead on things like neckties, dresses, and the white coat (the latter is almost unheard of at my hospital unless it is used to cover up scrubs). Fortunately, few doctors dress formally anymore (e.g., suit, and tie or sport coat for men). Emerging research might push all of us toward shedding ties, long sleeves, and the white coat before long.
Of course, you should keep in mind the way patients would like to see you dress. You can find information about patient expectations through a simple Internet search or by asking the person in charge of patient satisfaction at your hospital.
Seek Social Connections
Just like the issue of dress, I’ve encountered a number of hospitalist groups that have a habit of sneaking into the physician lunchroom, grabbing food in a “to go” container, and heading back to their office to eat together. These hospitalists are missing a valuable opportunity to enjoy social conversation with physicians of all specialties. If your hospital has a physician lunch room that is crowded with doctors, take advantage of the opportunity to build social networks.
You don’t need to eat there every day. (For a number of years, I enjoyed having lunch with the social workers in our main cafeteria.) But you should eat there more frequently than sneaking back to your office to eat only with other hospitalists. (If you don’t have time for lunch, then we need to talk about workload and efficiency issues.)
Look for other opportunities to make connections with other doctors through service on hospital committees, participation in social events at the hospital, or speaking at grand rounds. Although any single activity might not have significant impact, if you do these things regularly, you will form better relationships and be less likely to be or feel “dumped on,” and if it does happen, you’re in a much better position to address it if the dumping doctor is a friend.
Leadership Positions
Work to ensure a member of your group always sits on the medical staff executive committee, and seek out leadership positions like chief of medicine or chief of staff. Don’t simply assume you are too young or too inexperienced. Your hospital really needs the leadership of doctors who have a broad view of hospital operations and medical staff affairs. Few doctors have a broader view than hospitalists.
And if you have an interest in medical staff leadership, think about whether you’d like to serve as your hospital’s chief medical officer (aka vice president of medical affairs). All of these activities are important ways to influence what happens at your hospital, but aside from that, they are an excellent way to build relationships and gain respect from throughout the medical staff.
Worthwhile Effort
Ensuring that the hospitalists in your group feel respected and valued by other doctors and everyone they work with is important. Don’t make the mistake of thinking that working on this is just about stroking hospitalists’ egos.
I coauthored a 2001 research study on hospitalist burnout that failed to show a correlation between workload and burnout, but the study found that things like poor occupational solidarity are associated with burnout.1
Feeling like you fit in and are a respected member of your peer group (medical staff) is important and worth working on diligently. TH
Dr. Nelson has been a practicing hospitalist since 1988 and is cofounder and past president of SHM. He is a principal in Nelson Flores Hospital Medicine Consultants, a national hospitalist practice management consulting firm (www.nelsonflores.com). He is course co-director and faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official position of SHM.
Reference
- Hoff T, Whitcomb WF, Nelson JR. Thriving and surviving in a new medical career: the case of hospitalist physicians. Jrl Health Social Behavior. 2001;43:72-91.
I encounter a lot of hospitalists who complain that the other doctors at their hospital think of hospitalists as second-class citizens, as sort of like career residents. HM program directors need to make sure that is not the case for the hospitalists in their practice.
SHM has worked with the AMA’s Organized Medical Staff Section to assess the perception of hospitalists by primary-care physicians (PCPs) and hospitalists themselves. When asked in a 2009 survey, “Do you agree or disagree that hospitalists are respected members of the medical staff at a hospital?” only 3 out of 4 respondents agreed or highly agreed. That percentage is up slightly from the same survey conducted in 2007, and we don’t have data regarding how the responses would have been different if the question had been asked about other specialties. But I still find it concerning that about 25% of PCPs and hospitalists don’t see hospitalists as respected members of a medical staff. (If you are wondering, there wasn’t much of a difference between how hospitalists and PCPs answered the question.)
Use First Names
In the 1980s, I left residency and entered private practice as a hospitalist in a nonteaching, suburban hospital. I had a really hard time calling other doctors by their first names, especially the highly regarded senior internist who was my former roommate’s dad. He had always been Dr. McCollough to me, and I insisted calling him “Doctor” until we had been peers on the same medical staff for about a year.
Finally, in a somewhat annoyed voice, he told me I had to start calling him “Bob,” and that I should call all the doctors by their first names. It took a while, but using first names began to feel normal. Looking back on it, I think Dr. McCollough Bob taught me an important lesson about fitting in.
So make sure the hospitalists in your group call other doctors by their first names, too.
Dress the Part
I’ve come to believe that there are a number of things some hospitalists do to sabotage their own interest in being respected by the medical staff at their hospital. To my surprise, I’ve worked with a number of hospitalist groups in which most dress and act like residents, then complain that other doctors at their hospital treat them like residents. I think the way we dress, especially early in our careers, is a pretty big deal. If you’re similar in age to residents, then you’ll sure look like a resident if you dress like them. So don’t wear scrubs and Skechers unless all of the doctors in your hospital wear scrubs and Skechers.
The best advice is to dress the way the respected doctors dress. Follow the lead on things like neckties, dresses, and the white coat (the latter is almost unheard of at my hospital unless it is used to cover up scrubs). Fortunately, few doctors dress formally anymore (e.g., suit, and tie or sport coat for men). Emerging research might push all of us toward shedding ties, long sleeves, and the white coat before long.
Of course, you should keep in mind the way patients would like to see you dress. You can find information about patient expectations through a simple Internet search or by asking the person in charge of patient satisfaction at your hospital.
Seek Social Connections
Just like the issue of dress, I’ve encountered a number of hospitalist groups that have a habit of sneaking into the physician lunchroom, grabbing food in a “to go” container, and heading back to their office to eat together. These hospitalists are missing a valuable opportunity to enjoy social conversation with physicians of all specialties. If your hospital has a physician lunch room that is crowded with doctors, take advantage of the opportunity to build social networks.
You don’t need to eat there every day. (For a number of years, I enjoyed having lunch with the social workers in our main cafeteria.) But you should eat there more frequently than sneaking back to your office to eat only with other hospitalists. (If you don’t have time for lunch, then we need to talk about workload and efficiency issues.)
Look for other opportunities to make connections with other doctors through service on hospital committees, participation in social events at the hospital, or speaking at grand rounds. Although any single activity might not have significant impact, if you do these things regularly, you will form better relationships and be less likely to be or feel “dumped on,” and if it does happen, you’re in a much better position to address it if the dumping doctor is a friend.
Leadership Positions
Work to ensure a member of your group always sits on the medical staff executive committee, and seek out leadership positions like chief of medicine or chief of staff. Don’t simply assume you are too young or too inexperienced. Your hospital really needs the leadership of doctors who have a broad view of hospital operations and medical staff affairs. Few doctors have a broader view than hospitalists.
And if you have an interest in medical staff leadership, think about whether you’d like to serve as your hospital’s chief medical officer (aka vice president of medical affairs). All of these activities are important ways to influence what happens at your hospital, but aside from that, they are an excellent way to build relationships and gain respect from throughout the medical staff.
Worthwhile Effort
Ensuring that the hospitalists in your group feel respected and valued by other doctors and everyone they work with is important. Don’t make the mistake of thinking that working on this is just about stroking hospitalists’ egos.
I coauthored a 2001 research study on hospitalist burnout that failed to show a correlation between workload and burnout, but the study found that things like poor occupational solidarity are associated with burnout.1
Feeling like you fit in and are a respected member of your peer group (medical staff) is important and worth working on diligently. TH
Dr. Nelson has been a practicing hospitalist since 1988 and is cofounder and past president of SHM. He is a principal in Nelson Flores Hospital Medicine Consultants, a national hospitalist practice management consulting firm (www.nelsonflores.com). He is course co-director and faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official position of SHM.
Reference
- Hoff T, Whitcomb WF, Nelson JR. Thriving and surviving in a new medical career: the case of hospitalist physicians. Jrl Health Social Behavior. 2001;43:72-91.