User login
Occult satellite metastasis of an auricular melanoma
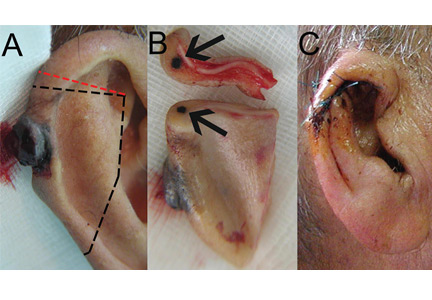
A 90-year-old man presented to our clinic with a dark, exophytic, hemorrhagic mass on the helix of his right auricle (Figure 1A). He had first noticed the lesion 6 months before.
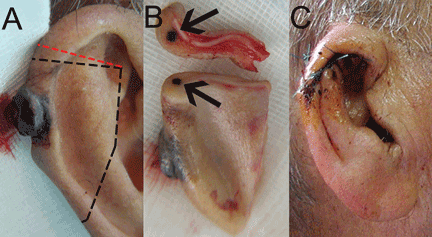
Evaluation of the lesion with the standard ABCDE criteria (Asymmetry, Border irregularity, Color variation, Diameter > 6 mm, Evolution/elevation) raised our suspicion of melanoma.1 We performed a wide, full-thickness, auricular wedge resection, which revealed a second dark lesion in the subcutaneous tissue of the upper border of the resected specimen. The rest of the second lesion was evident on the corresponding location of the edge of the remaining auricle (Figure 1B). Thus, we excised an additional strip of auricular tissue. The aesthetic result of the auricular reconstruction was quite good (Figure 1C).
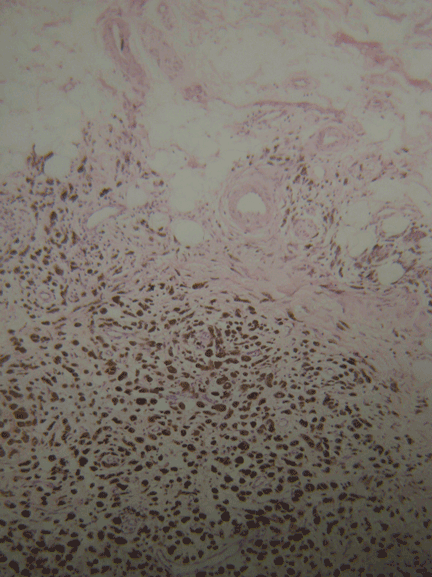
Histopathologic study confirmed cutaneous melanoma and showed the second lesion to be a satellite melanoma metastasis (Figure 2). The patient refused to undergo staging investigations for lymph node and distant metastases. He died 1 year later of ischemic stroke.
IN-TRANSIT AND SATELLITE METASTASES
Melanoma is highly metastatic. In addition to regional lymph node and distant metastases, patients may develop in-transit metastases and satellite metastases.
In-transit metastases grow more than 2 cm away from the primary tumor but not beyond the regional lymph node basin. Satellite lesions are found within 2 cm of the primary melanoma.
As seen in our patient, satellite metastases are not always cutaneous and evident. This is also true of in-transit melanoma lesions. They can also be located in subcutaneous tissue, making them difficult to detect. The presence of satellite lesions is a sign of aggressive disease and requires a thorough evaluation for metastases.2
- Thomas L, Tranchand P, Berard F, Secchi T, Colin C, Moulin G. Semiological value of ABCDE criteria in the diagnosis of cutaneous pigmented tumors. Dermatology 1998; 197:11–17.
- Homsi J, Kashani-Sabet M, Messina JL, Daud A. Cutaneous melanoma: prognostic factors. Cancer Control 2005; 12:223–229.
A 90-year-old man presented to our clinic with a dark, exophytic, hemorrhagic mass on the helix of his right auricle (Figure 1A). He had first noticed the lesion 6 months before.
Evaluation of the lesion with the standard ABCDE criteria (Asymmetry, Border irregularity, Color variation, Diameter > 6 mm, Evolution/elevation) raised our suspicion of melanoma.1 We performed a wide, full-thickness, auricular wedge resection, which revealed a second dark lesion in the subcutaneous tissue of the upper border of the resected specimen. The rest of the second lesion was evident on the corresponding location of the edge of the remaining auricle (Figure 1B). Thus, we excised an additional strip of auricular tissue. The aesthetic result of the auricular reconstruction was quite good (Figure 1C).
Histopathologic study confirmed cutaneous melanoma and showed the second lesion to be a satellite melanoma metastasis (Figure 2). The patient refused to undergo staging investigations for lymph node and distant metastases. He died 1 year later of ischemic stroke.
IN-TRANSIT AND SATELLITE METASTASES
Melanoma is highly metastatic. In addition to regional lymph node and distant metastases, patients may develop in-transit metastases and satellite metastases.
In-transit metastases grow more than 2 cm away from the primary tumor but not beyond the regional lymph node basin. Satellite lesions are found within 2 cm of the primary melanoma.
As seen in our patient, satellite metastases are not always cutaneous and evident. This is also true of in-transit melanoma lesions. They can also be located in subcutaneous tissue, making them difficult to detect. The presence of satellite lesions is a sign of aggressive disease and requires a thorough evaluation for metastases.2
A 90-year-old man presented to our clinic with a dark, exophytic, hemorrhagic mass on the helix of his right auricle (Figure 1A). He had first noticed the lesion 6 months before.
Evaluation of the lesion with the standard ABCDE criteria (Asymmetry, Border irregularity, Color variation, Diameter > 6 mm, Evolution/elevation) raised our suspicion of melanoma.1 We performed a wide, full-thickness, auricular wedge resection, which revealed a second dark lesion in the subcutaneous tissue of the upper border of the resected specimen. The rest of the second lesion was evident on the corresponding location of the edge of the remaining auricle (Figure 1B). Thus, we excised an additional strip of auricular tissue. The aesthetic result of the auricular reconstruction was quite good (Figure 1C).
Histopathologic study confirmed cutaneous melanoma and showed the second lesion to be a satellite melanoma metastasis (Figure 2). The patient refused to undergo staging investigations for lymph node and distant metastases. He died 1 year later of ischemic stroke.
IN-TRANSIT AND SATELLITE METASTASES
Melanoma is highly metastatic. In addition to regional lymph node and distant metastases, patients may develop in-transit metastases and satellite metastases.
In-transit metastases grow more than 2 cm away from the primary tumor but not beyond the regional lymph node basin. Satellite lesions are found within 2 cm of the primary melanoma.
As seen in our patient, satellite metastases are not always cutaneous and evident. This is also true of in-transit melanoma lesions. They can also be located in subcutaneous tissue, making them difficult to detect. The presence of satellite lesions is a sign of aggressive disease and requires a thorough evaluation for metastases.2
- Thomas L, Tranchand P, Berard F, Secchi T, Colin C, Moulin G. Semiological value of ABCDE criteria in the diagnosis of cutaneous pigmented tumors. Dermatology 1998; 197:11–17.
- Homsi J, Kashani-Sabet M, Messina JL, Daud A. Cutaneous melanoma: prognostic factors. Cancer Control 2005; 12:223–229.
- Thomas L, Tranchand P, Berard F, Secchi T, Colin C, Moulin G. Semiological value of ABCDE criteria in the diagnosis of cutaneous pigmented tumors. Dermatology 1998; 197:11–17.
- Homsi J, Kashani-Sabet M, Messina JL, Daud A. Cutaneous melanoma: prognostic factors. Cancer Control 2005; 12:223–229.
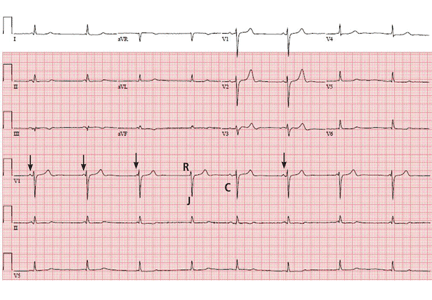
Respiratory artifact: A second vital sign on the electrocardiogram
A 57-year-old man hospitalized for treatment of multilobar pneumonia was noted to have a rapid, irregular heart rate on telemetry. He was hypoxemic and appeared to be in respiratory distress. A 12-lead electrocardiogram (ECG) demonstrated atrial fibrillation with rapid ventricular response, as well as what looked like distinct and regular P waves dissociated from the QRS complexes at a rate of about 44/min (Figure 1). What is the explanation and clinical significance of this curious finding?
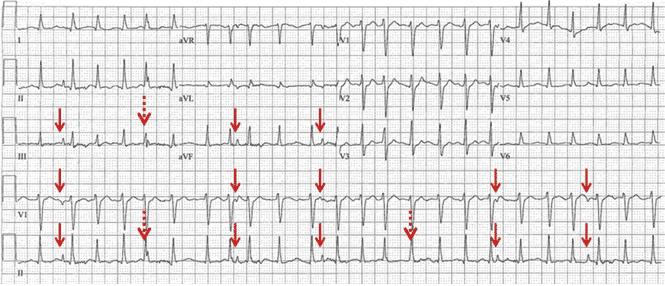
What appear to be dissociated P waves actually represent respiratory artifact.1–3 The sharp deflections mimicking P waves signify the tonic initiation of inspiratory effort; the subsequent brief periods of low-amplitude, high-frequency micro-oscillations represent surface electrical activity associated with the increased force of the accessory muscles of respiration.1–3
Surface electromyography noninvasively measures muscle activity using electrodes placed on the skin overlying the muscle.4 Using simultaneously recorded mechanical respiratory waveform tracings, we have previously demonstrated that the repetitive pseudo-P waves followed by micro-oscillations have a close temporal relationship with the inspiratory phase of respiration.3 The presence of respiratory artifact indicates a high-risk state frequently necessitating ventilation support.
In addition, when present, respiratory artifact can be viewed as the “second vital sign” on the ECG, the first vital sign being the heart rate. The respiratory rate can be approximated by counting the number of respiratory artifacts in a 10-second recording and multiplying it by 6. A more accurate rate assessment is achieved by measuring 1 or more respiratory artifact cycles in millimeters and then dividing that number into 1,500 or its multiples.3 Based on these calculations, the respiratory rate in this patient was 44/min.
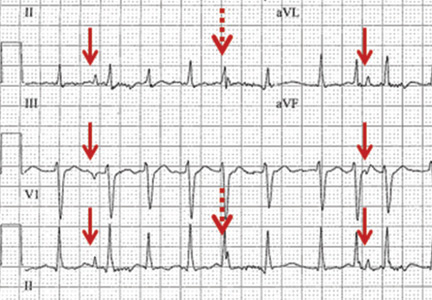
The presence of two atrial rhythms on the same ECG, one not disturbing the other, is consistent with the diagnosis of atrial dissociation.5 Atrial dissociation is a common finding in cardiac transplant recipients in whom the transplantation was performed using atrio-atrial anastomosis.6 Most other cases of apparent atrial dissociation described in the old cardiology and critical care literature probably represented unrecognized respiratory artifact.7,8
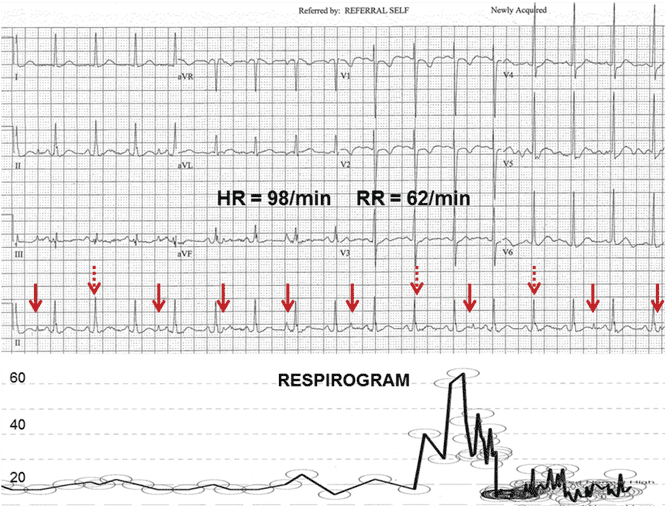
An ECG from a different patient (Figure 2) demonstrates rapid respiratory artifact that raised awareness of severe respiratory failure. The respiratory rate calculated from spacing of the pseudo-P waves is 62/min, confirmed by simultaneous respirography.
A FREQUENT FINDING IN SICK HOSPITALIZED PATIENTS
Respiratory artifact is a frequent finding in sick hospitalized patients.3 Most commonly, it manifests as repetitive micro-oscillations.3 Pseudo-P waves, as in this 57-year-old patient, are less often observed; but if their origin is not recognized, the interpretation of the ECG can become puzzling.1–3,7,8
Respiratory artifact is a marker of increased work of breathing and a strong indicator of significant cardiopulmonary compromise. Improvement in the patient’s cardiac or respiratory condition is typically associated with a decrease in the rate or complete elimination of respiratory artifact.3
Recognition of rapid respiratory artifact is less important in critical care units, where patients’ vital signs and cardiorespiratory status are carefully observed. However, in hospital settings where respiratory rate and oxygen saturation are not continuously monitored, recognizing rapid respiratory artifact can help raise awareness of the possibility of severe respiratory distress.
- Higgins TG, Phillips JH Jr, Sumner RG. Atrial dissociation: an electrocardiographic artifact produced by the accessory muscles of respiration. Am J Cardiol 1966; 18:132–139.
- Cheriex EC, Brugada P, Wellens HJ. Pseudo-atrial dissociation: a respiratory artifact. Eur Heart J 1986; 7:357–359.
- Littmann L, Rennyson SL, Wall BP, Parker JM. Significance of respiratory artifact in the electrocardiogram. Am J Cardiol 2008; 102:1090–1096.
- Pullman SL, Goodin DS, Marquinez AI, Tabbal S, Rubin M. Clinical utility of surface EMG: report of the therapeutics and technology assessment subcommittee of the American Academy of Neurology. Neurology 2000; 55:171–177.
- Chung EK. A reappraisal of atrial dissociation. Am J Cardiol 1971; 28:111–117.
- Stinson EB, Schroeder JS, Griepp RB, Shumway NE, Dong E Jr. Observations on the behavior of recipient atria after cardiac transplantation in man. Am J Cardiol 1972; 30:615–622.
- Cohen J, Scherf D. Complete interatrial and intra-atrial block (atrial dissociation). Am Heart J 1965; 70:23–34.
- Chung KY, Walsh TJ, Massie E. A review of atrial dissociation, with illustrative cases and critical discussion. Am J Med Sci 1965; 250:72–78.
A 57-year-old man hospitalized for treatment of multilobar pneumonia was noted to have a rapid, irregular heart rate on telemetry. He was hypoxemic and appeared to be in respiratory distress. A 12-lead electrocardiogram (ECG) demonstrated atrial fibrillation with rapid ventricular response, as well as what looked like distinct and regular P waves dissociated from the QRS complexes at a rate of about 44/min (Figure 1). What is the explanation and clinical significance of this curious finding?
What appear to be dissociated P waves actually represent respiratory artifact.1–3 The sharp deflections mimicking P waves signify the tonic initiation of inspiratory effort; the subsequent brief periods of low-amplitude, high-frequency micro-oscillations represent surface electrical activity associated with the increased force of the accessory muscles of respiration.1–3
Surface electromyography noninvasively measures muscle activity using electrodes placed on the skin overlying the muscle.4 Using simultaneously recorded mechanical respiratory waveform tracings, we have previously demonstrated that the repetitive pseudo-P waves followed by micro-oscillations have a close temporal relationship with the inspiratory phase of respiration.3 The presence of respiratory artifact indicates a high-risk state frequently necessitating ventilation support.
In addition, when present, respiratory artifact can be viewed as the “second vital sign” on the ECG, the first vital sign being the heart rate. The respiratory rate can be approximated by counting the number of respiratory artifacts in a 10-second recording and multiplying it by 6. A more accurate rate assessment is achieved by measuring 1 or more respiratory artifact cycles in millimeters and then dividing that number into 1,500 or its multiples.3 Based on these calculations, the respiratory rate in this patient was 44/min.
The presence of two atrial rhythms on the same ECG, one not disturbing the other, is consistent with the diagnosis of atrial dissociation.5 Atrial dissociation is a common finding in cardiac transplant recipients in whom the transplantation was performed using atrio-atrial anastomosis.6 Most other cases of apparent atrial dissociation described in the old cardiology and critical care literature probably represented unrecognized respiratory artifact.7,8
An ECG from a different patient (Figure 2) demonstrates rapid respiratory artifact that raised awareness of severe respiratory failure. The respiratory rate calculated from spacing of the pseudo-P waves is 62/min, confirmed by simultaneous respirography.
A FREQUENT FINDING IN SICK HOSPITALIZED PATIENTS
Respiratory artifact is a frequent finding in sick hospitalized patients.3 Most commonly, it manifests as repetitive micro-oscillations.3 Pseudo-P waves, as in this 57-year-old patient, are less often observed; but if their origin is not recognized, the interpretation of the ECG can become puzzling.1–3,7,8
Respiratory artifact is a marker of increased work of breathing and a strong indicator of significant cardiopulmonary compromise. Improvement in the patient’s cardiac or respiratory condition is typically associated with a decrease in the rate or complete elimination of respiratory artifact.3
Recognition of rapid respiratory artifact is less important in critical care units, where patients’ vital signs and cardiorespiratory status are carefully observed. However, in hospital settings where respiratory rate and oxygen saturation are not continuously monitored, recognizing rapid respiratory artifact can help raise awareness of the possibility of severe respiratory distress.
A 57-year-old man hospitalized for treatment of multilobar pneumonia was noted to have a rapid, irregular heart rate on telemetry. He was hypoxemic and appeared to be in respiratory distress. A 12-lead electrocardiogram (ECG) demonstrated atrial fibrillation with rapid ventricular response, as well as what looked like distinct and regular P waves dissociated from the QRS complexes at a rate of about 44/min (Figure 1). What is the explanation and clinical significance of this curious finding?
What appear to be dissociated P waves actually represent respiratory artifact.1–3 The sharp deflections mimicking P waves signify the tonic initiation of inspiratory effort; the subsequent brief periods of low-amplitude, high-frequency micro-oscillations represent surface electrical activity associated with the increased force of the accessory muscles of respiration.1–3
Surface electromyography noninvasively measures muscle activity using electrodes placed on the skin overlying the muscle.4 Using simultaneously recorded mechanical respiratory waveform tracings, we have previously demonstrated that the repetitive pseudo-P waves followed by micro-oscillations have a close temporal relationship with the inspiratory phase of respiration.3 The presence of respiratory artifact indicates a high-risk state frequently necessitating ventilation support.
In addition, when present, respiratory artifact can be viewed as the “second vital sign” on the ECG, the first vital sign being the heart rate. The respiratory rate can be approximated by counting the number of respiratory artifacts in a 10-second recording and multiplying it by 6. A more accurate rate assessment is achieved by measuring 1 or more respiratory artifact cycles in millimeters and then dividing that number into 1,500 or its multiples.3 Based on these calculations, the respiratory rate in this patient was 44/min.
The presence of two atrial rhythms on the same ECG, one not disturbing the other, is consistent with the diagnosis of atrial dissociation.5 Atrial dissociation is a common finding in cardiac transplant recipients in whom the transplantation was performed using atrio-atrial anastomosis.6 Most other cases of apparent atrial dissociation described in the old cardiology and critical care literature probably represented unrecognized respiratory artifact.7,8
An ECG from a different patient (Figure 2) demonstrates rapid respiratory artifact that raised awareness of severe respiratory failure. The respiratory rate calculated from spacing of the pseudo-P waves is 62/min, confirmed by simultaneous respirography.
A FREQUENT FINDING IN SICK HOSPITALIZED PATIENTS
Respiratory artifact is a frequent finding in sick hospitalized patients.3 Most commonly, it manifests as repetitive micro-oscillations.3 Pseudo-P waves, as in this 57-year-old patient, are less often observed; but if their origin is not recognized, the interpretation of the ECG can become puzzling.1–3,7,8
Respiratory artifact is a marker of increased work of breathing and a strong indicator of significant cardiopulmonary compromise. Improvement in the patient’s cardiac or respiratory condition is typically associated with a decrease in the rate or complete elimination of respiratory artifact.3
Recognition of rapid respiratory artifact is less important in critical care units, where patients’ vital signs and cardiorespiratory status are carefully observed. However, in hospital settings where respiratory rate and oxygen saturation are not continuously monitored, recognizing rapid respiratory artifact can help raise awareness of the possibility of severe respiratory distress.
- Higgins TG, Phillips JH Jr, Sumner RG. Atrial dissociation: an electrocardiographic artifact produced by the accessory muscles of respiration. Am J Cardiol 1966; 18:132–139.
- Cheriex EC, Brugada P, Wellens HJ. Pseudo-atrial dissociation: a respiratory artifact. Eur Heart J 1986; 7:357–359.
- Littmann L, Rennyson SL, Wall BP, Parker JM. Significance of respiratory artifact in the electrocardiogram. Am J Cardiol 2008; 102:1090–1096.
- Pullman SL, Goodin DS, Marquinez AI, Tabbal S, Rubin M. Clinical utility of surface EMG: report of the therapeutics and technology assessment subcommittee of the American Academy of Neurology. Neurology 2000; 55:171–177.
- Chung EK. A reappraisal of atrial dissociation. Am J Cardiol 1971; 28:111–117.
- Stinson EB, Schroeder JS, Griepp RB, Shumway NE, Dong E Jr. Observations on the behavior of recipient atria after cardiac transplantation in man. Am J Cardiol 1972; 30:615–622.
- Cohen J, Scherf D. Complete interatrial and intra-atrial block (atrial dissociation). Am Heart J 1965; 70:23–34.
- Chung KY, Walsh TJ, Massie E. A review of atrial dissociation, with illustrative cases and critical discussion. Am J Med Sci 1965; 250:72–78.
- Higgins TG, Phillips JH Jr, Sumner RG. Atrial dissociation: an electrocardiographic artifact produced by the accessory muscles of respiration. Am J Cardiol 1966; 18:132–139.
- Cheriex EC, Brugada P, Wellens HJ. Pseudo-atrial dissociation: a respiratory artifact. Eur Heart J 1986; 7:357–359.
- Littmann L, Rennyson SL, Wall BP, Parker JM. Significance of respiratory artifact in the electrocardiogram. Am J Cardiol 2008; 102:1090–1096.
- Pullman SL, Goodin DS, Marquinez AI, Tabbal S, Rubin M. Clinical utility of surface EMG: report of the therapeutics and technology assessment subcommittee of the American Academy of Neurology. Neurology 2000; 55:171–177.
- Chung EK. A reappraisal of atrial dissociation. Am J Cardiol 1971; 28:111–117.
- Stinson EB, Schroeder JS, Griepp RB, Shumway NE, Dong E Jr. Observations on the behavior of recipient atria after cardiac transplantation in man. Am J Cardiol 1972; 30:615–622.
- Cohen J, Scherf D. Complete interatrial and intra-atrial block (atrial dissociation). Am Heart J 1965; 70:23–34.
- Chung KY, Walsh TJ, Massie E. A review of atrial dissociation, with illustrative cases and critical discussion. Am J Med Sci 1965; 250:72–78.
Parvovirus mimicking acute HIV infection
A 25-year-old Jamaican man presented to the emergency department for evaluation of a rash on his face, back, and hands (Figure 1). He recalled a puncture injury after handling garbage at work. He denied recent travel, blood transfusions, or sick contacts. He was not aware of any recent arthropod bites. All standard vaccines including a tetanus booster were up to date.
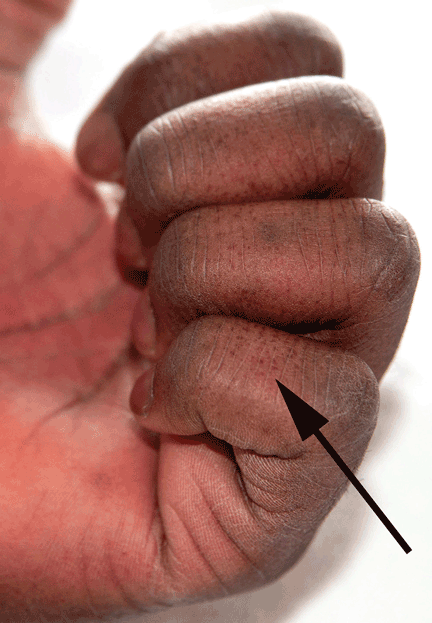
Examination revealed edema of the hands and uvula. He was discharged with diphenhydramine and a short course of a systemic corticosteroid for presumed contact dermatitis.
He returned 4 days later with new symptoms, including sore throat, fever with a temperature of 103°F (39.4°C), oropharyngeal pain, dysphagia, dysuria, and purpura on the hands, abdomen, and legs. He was admitted to the hospital.

Examination revealed bright red confluent erythema of both legs extending to the lower abdomen, with petechiae on the palms (Figure 2), soles, toes, and fingers. Several small scrotal ulcers with well-defined borders were noted. Oral examination revealed white-yellow adherent plaques on the tongue and similar small ulcers on the lower lip and soft and hard palates.
A complete blood cell count revealed absolute lymphopenia, with a white blood cell count of 0.64 × 109/L (reference range 1.0–4.8) and a neutrophil percentage of 78.9% (39%–68%). Other values were within normal limits, with a red blood cell count of 5.3 × 1012/L (3.9–5.5) and a platelet count of 161 × 109/L (150–350). Serum liver enzymes were also within normal limits.
Treatment with intravenous fluids and intramuscular penicillin G was started empirically, pending a workup for infectious disease. Tests for syphilis immunoglobulin G (IgG), streptococci, anti-streptolysin O, Epstein-Barr virus, and human immunodeficiency virus (HIV) 1 and 2 were negative. The scrotal ulcers were swabbed, and culture and direct fluorescent antibody testing for cytomegalovirus and herpes simplex virus were negative. Urine testing for gonococcal and chlamydial infection was negative.
On the fifth day of hospitalization, the patient’s condition was improving, but there was still no definitive diagnosis. Consultation with the inpatient dermatology team prompted testing for parvovirus B19 infection, based on the gloves-and-socks distribution of the purpura. Testing revealed a slightly elevated parvovirus B19 IgG titer (2.61) and a significantly elevated parvovirus B19 IgM titer (12.74), which confirmed acute parvovirus infection.
The patient’s condition improved over several days with fluid administration, and he was discharged in good condition. He returned 1 week later for a follow-up appointment, at which time only superficial desquamation was noted in the areas previously affected by purpura.
PARVOVIRUS B19: NOT ONLY IN CHILDREN
Parvovirus B19 is responsible for the common childhood viral exanthem known as fifth disease.1 However, although much less common, the virus can also affect young adults, precipitating a dermatosis referred to as gloves-and-socks syndrome characterized by purpura on the hands and feet,2,3 and with a higher incidence in the spring and summer.1
Although papular-purpuric gloves-and- socks syndrome is characterized by purpura on the hands and feet, the cheeks, oral mucosa, inner thighs, buttocks, and genitalia are affected in about 50% of patients.2 In one report, in two-thirds of adult patients the presentation was caused by parvovirus B19 infection,4 but the syndrome has also been associated with Epstein-Barr virus, cytomegalovirus, human herpesvirus types 6 and 7, hepatitis B virus, rubella virus, and varicella zoster virus.4
Parvovirus B19 infection is commonly associated with systemic manifestations such as fever, fatigue, and lymphadenopathy, as well as swelling of the lips, cutaneous and mucosal ulcerations, polyarthritis, and petechiae involving the hard palate, the soft palate, or both.1
The syndrome is self-limited and resolves within 1 to 2 weeks.1
THE DIAGNOSTIC CHALLENGE
The differential diagnosis of the syndrome’s gloves-and-socks presentation includes hand-foot-mouth disease, erythema multiforme, Henoch-Schönlein purpura, and Kawasaki disease,4 in addition to viral exanthems and sexually transmitted diseases. Our patient’s fever, rash, and absolute lymphopenia focused attention on possible HIV infection, which caused the patient significant anxiety while awaiting the results of HIV testing. Heightened awareness of the cutaneous presentation of parvovirus B19 infection can help avoid unnecessary hospitalization and patient anxiety.
- Smith PT, Landry ML, Carey H, Krasnoff J, Cooney E. Papular-purpuric gloves and socks syndrome associated with acute parvovirus B19 infection: case report and review. Clin Infect Dis 1998; 27:164–168.
- Harms M, Feldmann R, Saurat JH. Papular-purpuric “gloves and socks” syndrome. J Am Acad Dermatol 1990; 23:850–854.
- Bagot M, Revuz J. Papular-purpuric “gloves and socks” syndrome: primary infection with parvovirus B19? J Am Acad Dermatol 1991; 25:341–342.
- Gutermuth J, Nadas K, Zirbs M, et al. Papular-purpuric gloves and socks syndrome. Lancet 2011; 378:198.
A 25-year-old Jamaican man presented to the emergency department for evaluation of a rash on his face, back, and hands (Figure 1). He recalled a puncture injury after handling garbage at work. He denied recent travel, blood transfusions, or sick contacts. He was not aware of any recent arthropod bites. All standard vaccines including a tetanus booster were up to date.
Examination revealed edema of the hands and uvula. He was discharged with diphenhydramine and a short course of a systemic corticosteroid for presumed contact dermatitis.
He returned 4 days later with new symptoms, including sore throat, fever with a temperature of 103°F (39.4°C), oropharyngeal pain, dysphagia, dysuria, and purpura on the hands, abdomen, and legs. He was admitted to the hospital.
Examination revealed bright red confluent erythema of both legs extending to the lower abdomen, with petechiae on the palms (Figure 2), soles, toes, and fingers. Several small scrotal ulcers with well-defined borders were noted. Oral examination revealed white-yellow adherent plaques on the tongue and similar small ulcers on the lower lip and soft and hard palates.
A complete blood cell count revealed absolute lymphopenia, with a white blood cell count of 0.64 × 109/L (reference range 1.0–4.8) and a neutrophil percentage of 78.9% (39%–68%). Other values were within normal limits, with a red blood cell count of 5.3 × 1012/L (3.9–5.5) and a platelet count of 161 × 109/L (150–350). Serum liver enzymes were also within normal limits.
Treatment with intravenous fluids and intramuscular penicillin G was started empirically, pending a workup for infectious disease. Tests for syphilis immunoglobulin G (IgG), streptococci, anti-streptolysin O, Epstein-Barr virus, and human immunodeficiency virus (HIV) 1 and 2 were negative. The scrotal ulcers were swabbed, and culture and direct fluorescent antibody testing for cytomegalovirus and herpes simplex virus were negative. Urine testing for gonococcal and chlamydial infection was negative.
On the fifth day of hospitalization, the patient’s condition was improving, but there was still no definitive diagnosis. Consultation with the inpatient dermatology team prompted testing for parvovirus B19 infection, based on the gloves-and-socks distribution of the purpura. Testing revealed a slightly elevated parvovirus B19 IgG titer (2.61) and a significantly elevated parvovirus B19 IgM titer (12.74), which confirmed acute parvovirus infection.
The patient’s condition improved over several days with fluid administration, and he was discharged in good condition. He returned 1 week later for a follow-up appointment, at which time only superficial desquamation was noted in the areas previously affected by purpura.
PARVOVIRUS B19: NOT ONLY IN CHILDREN
Parvovirus B19 is responsible for the common childhood viral exanthem known as fifth disease.1 However, although much less common, the virus can also affect young adults, precipitating a dermatosis referred to as gloves-and-socks syndrome characterized by purpura on the hands and feet,2,3 and with a higher incidence in the spring and summer.1
Although papular-purpuric gloves-and- socks syndrome is characterized by purpura on the hands and feet, the cheeks, oral mucosa, inner thighs, buttocks, and genitalia are affected in about 50% of patients.2 In one report, in two-thirds of adult patients the presentation was caused by parvovirus B19 infection,4 but the syndrome has also been associated with Epstein-Barr virus, cytomegalovirus, human herpesvirus types 6 and 7, hepatitis B virus, rubella virus, and varicella zoster virus.4
Parvovirus B19 infection is commonly associated with systemic manifestations such as fever, fatigue, and lymphadenopathy, as well as swelling of the lips, cutaneous and mucosal ulcerations, polyarthritis, and petechiae involving the hard palate, the soft palate, or both.1
The syndrome is self-limited and resolves within 1 to 2 weeks.1
THE DIAGNOSTIC CHALLENGE
The differential diagnosis of the syndrome’s gloves-and-socks presentation includes hand-foot-mouth disease, erythema multiforme, Henoch-Schönlein purpura, and Kawasaki disease,4 in addition to viral exanthems and sexually transmitted diseases. Our patient’s fever, rash, and absolute lymphopenia focused attention on possible HIV infection, which caused the patient significant anxiety while awaiting the results of HIV testing. Heightened awareness of the cutaneous presentation of parvovirus B19 infection can help avoid unnecessary hospitalization and patient anxiety.
A 25-year-old Jamaican man presented to the emergency department for evaluation of a rash on his face, back, and hands (Figure 1). He recalled a puncture injury after handling garbage at work. He denied recent travel, blood transfusions, or sick contacts. He was not aware of any recent arthropod bites. All standard vaccines including a tetanus booster were up to date.
Examination revealed edema of the hands and uvula. He was discharged with diphenhydramine and a short course of a systemic corticosteroid for presumed contact dermatitis.
He returned 4 days later with new symptoms, including sore throat, fever with a temperature of 103°F (39.4°C), oropharyngeal pain, dysphagia, dysuria, and purpura on the hands, abdomen, and legs. He was admitted to the hospital.
Examination revealed bright red confluent erythema of both legs extending to the lower abdomen, with petechiae on the palms (Figure 2), soles, toes, and fingers. Several small scrotal ulcers with well-defined borders were noted. Oral examination revealed white-yellow adherent plaques on the tongue and similar small ulcers on the lower lip and soft and hard palates.
A complete blood cell count revealed absolute lymphopenia, with a white blood cell count of 0.64 × 109/L (reference range 1.0–4.8) and a neutrophil percentage of 78.9% (39%–68%). Other values were within normal limits, with a red blood cell count of 5.3 × 1012/L (3.9–5.5) and a platelet count of 161 × 109/L (150–350). Serum liver enzymes were also within normal limits.
Treatment with intravenous fluids and intramuscular penicillin G was started empirically, pending a workup for infectious disease. Tests for syphilis immunoglobulin G (IgG), streptococci, anti-streptolysin O, Epstein-Barr virus, and human immunodeficiency virus (HIV) 1 and 2 were negative. The scrotal ulcers were swabbed, and culture and direct fluorescent antibody testing for cytomegalovirus and herpes simplex virus were negative. Urine testing for gonococcal and chlamydial infection was negative.
On the fifth day of hospitalization, the patient’s condition was improving, but there was still no definitive diagnosis. Consultation with the inpatient dermatology team prompted testing for parvovirus B19 infection, based on the gloves-and-socks distribution of the purpura. Testing revealed a slightly elevated parvovirus B19 IgG titer (2.61) and a significantly elevated parvovirus B19 IgM titer (12.74), which confirmed acute parvovirus infection.
The patient’s condition improved over several days with fluid administration, and he was discharged in good condition. He returned 1 week later for a follow-up appointment, at which time only superficial desquamation was noted in the areas previously affected by purpura.
PARVOVIRUS B19: NOT ONLY IN CHILDREN
Parvovirus B19 is responsible for the common childhood viral exanthem known as fifth disease.1 However, although much less common, the virus can also affect young adults, precipitating a dermatosis referred to as gloves-and-socks syndrome characterized by purpura on the hands and feet,2,3 and with a higher incidence in the spring and summer.1
Although papular-purpuric gloves-and- socks syndrome is characterized by purpura on the hands and feet, the cheeks, oral mucosa, inner thighs, buttocks, and genitalia are affected in about 50% of patients.2 In one report, in two-thirds of adult patients the presentation was caused by parvovirus B19 infection,4 but the syndrome has also been associated with Epstein-Barr virus, cytomegalovirus, human herpesvirus types 6 and 7, hepatitis B virus, rubella virus, and varicella zoster virus.4
Parvovirus B19 infection is commonly associated with systemic manifestations such as fever, fatigue, and lymphadenopathy, as well as swelling of the lips, cutaneous and mucosal ulcerations, polyarthritis, and petechiae involving the hard palate, the soft palate, or both.1
The syndrome is self-limited and resolves within 1 to 2 weeks.1
THE DIAGNOSTIC CHALLENGE
The differential diagnosis of the syndrome’s gloves-and-socks presentation includes hand-foot-mouth disease, erythema multiforme, Henoch-Schönlein purpura, and Kawasaki disease,4 in addition to viral exanthems and sexually transmitted diseases. Our patient’s fever, rash, and absolute lymphopenia focused attention on possible HIV infection, which caused the patient significant anxiety while awaiting the results of HIV testing. Heightened awareness of the cutaneous presentation of parvovirus B19 infection can help avoid unnecessary hospitalization and patient anxiety.
- Smith PT, Landry ML, Carey H, Krasnoff J, Cooney E. Papular-purpuric gloves and socks syndrome associated with acute parvovirus B19 infection: case report and review. Clin Infect Dis 1998; 27:164–168.
- Harms M, Feldmann R, Saurat JH. Papular-purpuric “gloves and socks” syndrome. J Am Acad Dermatol 1990; 23:850–854.
- Bagot M, Revuz J. Papular-purpuric “gloves and socks” syndrome: primary infection with parvovirus B19? J Am Acad Dermatol 1991; 25:341–342.
- Gutermuth J, Nadas K, Zirbs M, et al. Papular-purpuric gloves and socks syndrome. Lancet 2011; 378:198.
- Smith PT, Landry ML, Carey H, Krasnoff J, Cooney E. Papular-purpuric gloves and socks syndrome associated with acute parvovirus B19 infection: case report and review. Clin Infect Dis 1998; 27:164–168.
- Harms M, Feldmann R, Saurat JH. Papular-purpuric “gloves and socks” syndrome. J Am Acad Dermatol 1990; 23:850–854.
- Bagot M, Revuz J. Papular-purpuric “gloves and socks” syndrome: primary infection with parvovirus B19? J Am Acad Dermatol 1991; 25:341–342.
- Gutermuth J, Nadas K, Zirbs M, et al. Papular-purpuric gloves and socks syndrome. Lancet 2011; 378:198.
An unusual cause of vitamin B12 and iron deficiency
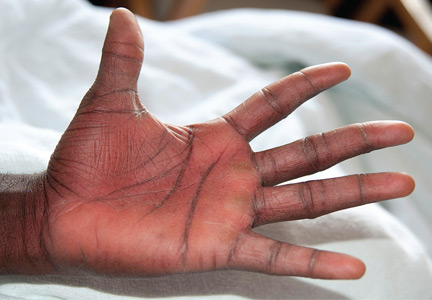
A 76-year-old woman visiting from Ethiopia presented for further evaluation of concomitant iron and vitamin B12 deficiency anemia that had developed over the previous 6 months. During that time, she had complained of ongoing fatigue and increasing paresthesias in the hands and feet.
At presentation, her hemoglobin concentration was 7.8 g/dL (reference range 11.5–15), with a mean corpuscular volume of 81.8 fL (81.5–97.0). These values were down from her baseline hemoglobin of 12 g/dL and corpuscular volume of 85.8 recorded more than 1 year ago. Serum studies showed an iron concentration of 21 µg/dL (37–170), ferritin 3 ng/mL (10–107), and percent saturation of transferrin 5% (20%–55%). Also noted was a low vitamin B12 level of 108 pg/mL (180–1,241 pg/mL). She had no overt signs of gastrointestinal blood loss. She did not report altered bowel habits or use of nonsteroidal anti-inflammatory medications.
Given her country of origin, she was sent for initial stool testing for ova and parasites, which was unrevealing.
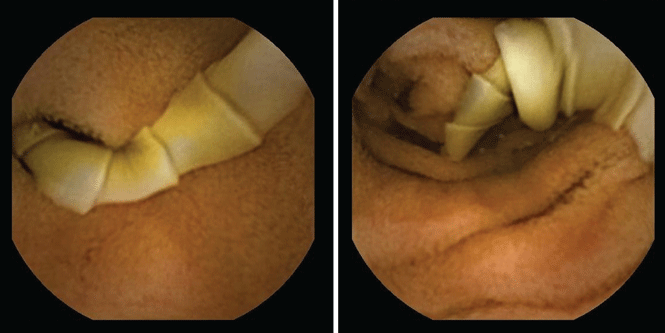
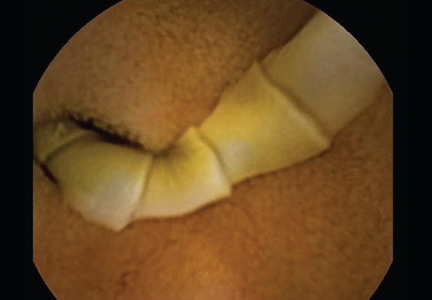
She underwent esophagogastroduodenoscopy and colonoscopy, which revealed no underlying cause of her iron deficiency or vitamin B12 insufficiency. But further evaluation with capsule endoscopy showed evidence of a tapeworm in the distal duodenum (Figure 1).
She was given praziquantel in a single oral dose of 10 mg/kg. Repeat stool culture 1 month later showed no evidence of tapeworm infection, and at follow-up 3 months later, her hemoglobin had recovered to 13.2 g/dL with a corpuscular volume of 87.6 fL and no residual vitamin B12 or iron deficiency. She reported complete resolution of fatigue and of paresthesias of the hands and feet.
DIPHYLLOBOTHRIUM LATUM
The appearance on capsule endoscopy indicated Diphyllobothrium latum as the likely parasite. This tapeworm is acquired by ingesting undercooked or raw fish. Infection is most common in Northern Europe but has been reported in Africa.1
As it grows, the tapeworm develops chains of segments and can reach a length of 1 to 15 meters.1 In humans, it typically resides in the small intestine. Most patients are asymptomatic or have moderate nonspecific symptoms such as abdominal pain and diarrhea. A key differentiating aspect of D latum infection is vitamin B12 deficiency caused by consumption of the vitamin by the parasite, as well as by parasite-mediated dissociation of the vitamin B12-intrinsic factor complex, thus making the vitamin unavailable to the host.
Up to 40% of people infected with D latum develop low levels of vitamin B12, and 2% develop symptomatic megaloblastic anemia.2 Iron deficiency anemia is uncommon but has been reported.3 In our patient, the concomitant iron deficiency was probably secondary to involvement of the duodenum, where a significant amount of dietary iron is absorbed.
The diagnosis is typically established by stool testing for ova and parasites. When stool samples do not reveal a cause of the symptoms, as in this patient, endoscopy can be used. Capsule endoscopy has not been widely used in the diagnosis of intestinal helminth infection, although reports exist describing the use of capsule endoscopy to detect intestinal parasites. Notably, as in this case, intestinal parasite infection is occasionally found during investigations of anemia and vitamin deficiencies of unknown cause.4
As in our patient, treatment of infection with this species of tapeworm typically involves a single oral dose of praziquantel; this off-label use has been shown to lead to resolution of symptoms in nearly all patients treated.5
- Schantz PM. Tapeworms (cestodiasis). Gastroenterol Clin North Am 1996; 25:637–653.
- Scholz T, Garcia HH, Kuchta R, Wicht B. Update on the human broad tapeworm (genus Diphyllobothrium), including clinical relevance. Clin Microbiol Rev 2009; 22:146–160,
- Stanciu C, Trifan A, Singeap AM, Sfarti C, Cojocariu C, Luca M. Diphyllobothrium latum identified by capsule endoscopy—an unusual cause of iron-deficiency anemia. J Gastrointestin Liver Dis 2009; 18:142.
- Soga K, Handa O, Yamada M, et al. In vivo imaging of intestinal helminths by capsule endoscopy. Parasitol Int 2014; 63:221–228.
- Drugs for Parasitic Infections. 3rd edition. Treatment guidelines from the Medical Letter 2010. The Medical Letter, Inc., New Rochelle, NY.
A 76-year-old woman visiting from Ethiopia presented for further evaluation of concomitant iron and vitamin B12 deficiency anemia that had developed over the previous 6 months. During that time, she had complained of ongoing fatigue and increasing paresthesias in the hands and feet.
At presentation, her hemoglobin concentration was 7.8 g/dL (reference range 11.5–15), with a mean corpuscular volume of 81.8 fL (81.5–97.0). These values were down from her baseline hemoglobin of 12 g/dL and corpuscular volume of 85.8 recorded more than 1 year ago. Serum studies showed an iron concentration of 21 µg/dL (37–170), ferritin 3 ng/mL (10–107), and percent saturation of transferrin 5% (20%–55%). Also noted was a low vitamin B12 level of 108 pg/mL (180–1,241 pg/mL). She had no overt signs of gastrointestinal blood loss. She did not report altered bowel habits or use of nonsteroidal anti-inflammatory medications.
Given her country of origin, she was sent for initial stool testing for ova and parasites, which was unrevealing.
She underwent esophagogastroduodenoscopy and colonoscopy, which revealed no underlying cause of her iron deficiency or vitamin B12 insufficiency. But further evaluation with capsule endoscopy showed evidence of a tapeworm in the distal duodenum (Figure 1).
She was given praziquantel in a single oral dose of 10 mg/kg. Repeat stool culture 1 month later showed no evidence of tapeworm infection, and at follow-up 3 months later, her hemoglobin had recovered to 13.2 g/dL with a corpuscular volume of 87.6 fL and no residual vitamin B12 or iron deficiency. She reported complete resolution of fatigue and of paresthesias of the hands and feet.
DIPHYLLOBOTHRIUM LATUM
The appearance on capsule endoscopy indicated Diphyllobothrium latum as the likely parasite. This tapeworm is acquired by ingesting undercooked or raw fish. Infection is most common in Northern Europe but has been reported in Africa.1
As it grows, the tapeworm develops chains of segments and can reach a length of 1 to 15 meters.1 In humans, it typically resides in the small intestine. Most patients are asymptomatic or have moderate nonspecific symptoms such as abdominal pain and diarrhea. A key differentiating aspect of D latum infection is vitamin B12 deficiency caused by consumption of the vitamin by the parasite, as well as by parasite-mediated dissociation of the vitamin B12-intrinsic factor complex, thus making the vitamin unavailable to the host.
Up to 40% of people infected with D latum develop low levels of vitamin B12, and 2% develop symptomatic megaloblastic anemia.2 Iron deficiency anemia is uncommon but has been reported.3 In our patient, the concomitant iron deficiency was probably secondary to involvement of the duodenum, where a significant amount of dietary iron is absorbed.
The diagnosis is typically established by stool testing for ova and parasites. When stool samples do not reveal a cause of the symptoms, as in this patient, endoscopy can be used. Capsule endoscopy has not been widely used in the diagnosis of intestinal helminth infection, although reports exist describing the use of capsule endoscopy to detect intestinal parasites. Notably, as in this case, intestinal parasite infection is occasionally found during investigations of anemia and vitamin deficiencies of unknown cause.4
As in our patient, treatment of infection with this species of tapeworm typically involves a single oral dose of praziquantel; this off-label use has been shown to lead to resolution of symptoms in nearly all patients treated.5
A 76-year-old woman visiting from Ethiopia presented for further evaluation of concomitant iron and vitamin B12 deficiency anemia that had developed over the previous 6 months. During that time, she had complained of ongoing fatigue and increasing paresthesias in the hands and feet.
At presentation, her hemoglobin concentration was 7.8 g/dL (reference range 11.5–15), with a mean corpuscular volume of 81.8 fL (81.5–97.0). These values were down from her baseline hemoglobin of 12 g/dL and corpuscular volume of 85.8 recorded more than 1 year ago. Serum studies showed an iron concentration of 21 µg/dL (37–170), ferritin 3 ng/mL (10–107), and percent saturation of transferrin 5% (20%–55%). Also noted was a low vitamin B12 level of 108 pg/mL (180–1,241 pg/mL). She had no overt signs of gastrointestinal blood loss. She did not report altered bowel habits or use of nonsteroidal anti-inflammatory medications.
Given her country of origin, she was sent for initial stool testing for ova and parasites, which was unrevealing.
She underwent esophagogastroduodenoscopy and colonoscopy, which revealed no underlying cause of her iron deficiency or vitamin B12 insufficiency. But further evaluation with capsule endoscopy showed evidence of a tapeworm in the distal duodenum (Figure 1).
She was given praziquantel in a single oral dose of 10 mg/kg. Repeat stool culture 1 month later showed no evidence of tapeworm infection, and at follow-up 3 months later, her hemoglobin had recovered to 13.2 g/dL with a corpuscular volume of 87.6 fL and no residual vitamin B12 or iron deficiency. She reported complete resolution of fatigue and of paresthesias of the hands and feet.
DIPHYLLOBOTHRIUM LATUM
The appearance on capsule endoscopy indicated Diphyllobothrium latum as the likely parasite. This tapeworm is acquired by ingesting undercooked or raw fish. Infection is most common in Northern Europe but has been reported in Africa.1
As it grows, the tapeworm develops chains of segments and can reach a length of 1 to 15 meters.1 In humans, it typically resides in the small intestine. Most patients are asymptomatic or have moderate nonspecific symptoms such as abdominal pain and diarrhea. A key differentiating aspect of D latum infection is vitamin B12 deficiency caused by consumption of the vitamin by the parasite, as well as by parasite-mediated dissociation of the vitamin B12-intrinsic factor complex, thus making the vitamin unavailable to the host.
Up to 40% of people infected with D latum develop low levels of vitamin B12, and 2% develop symptomatic megaloblastic anemia.2 Iron deficiency anemia is uncommon but has been reported.3 In our patient, the concomitant iron deficiency was probably secondary to involvement of the duodenum, where a significant amount of dietary iron is absorbed.
The diagnosis is typically established by stool testing for ova and parasites. When stool samples do not reveal a cause of the symptoms, as in this patient, endoscopy can be used. Capsule endoscopy has not been widely used in the diagnosis of intestinal helminth infection, although reports exist describing the use of capsule endoscopy to detect intestinal parasites. Notably, as in this case, intestinal parasite infection is occasionally found during investigations of anemia and vitamin deficiencies of unknown cause.4
As in our patient, treatment of infection with this species of tapeworm typically involves a single oral dose of praziquantel; this off-label use has been shown to lead to resolution of symptoms in nearly all patients treated.5
- Schantz PM. Tapeworms (cestodiasis). Gastroenterol Clin North Am 1996; 25:637–653.
- Scholz T, Garcia HH, Kuchta R, Wicht B. Update on the human broad tapeworm (genus Diphyllobothrium), including clinical relevance. Clin Microbiol Rev 2009; 22:146–160,
- Stanciu C, Trifan A, Singeap AM, Sfarti C, Cojocariu C, Luca M. Diphyllobothrium latum identified by capsule endoscopy—an unusual cause of iron-deficiency anemia. J Gastrointestin Liver Dis 2009; 18:142.
- Soga K, Handa O, Yamada M, et al. In vivo imaging of intestinal helminths by capsule endoscopy. Parasitol Int 2014; 63:221–228.
- Drugs for Parasitic Infections. 3rd edition. Treatment guidelines from the Medical Letter 2010. The Medical Letter, Inc., New Rochelle, NY.
- Schantz PM. Tapeworms (cestodiasis). Gastroenterol Clin North Am 1996; 25:637–653.
- Scholz T, Garcia HH, Kuchta R, Wicht B. Update on the human broad tapeworm (genus Diphyllobothrium), including clinical relevance. Clin Microbiol Rev 2009; 22:146–160,
- Stanciu C, Trifan A, Singeap AM, Sfarti C, Cojocariu C, Luca M. Diphyllobothrium latum identified by capsule endoscopy—an unusual cause of iron-deficiency anemia. J Gastrointestin Liver Dis 2009; 18:142.
- Soga K, Handa O, Yamada M, et al. In vivo imaging of intestinal helminths by capsule endoscopy. Parasitol Int 2014; 63:221–228.
- Drugs for Parasitic Infections. 3rd edition. Treatment guidelines from the Medical Letter 2010. The Medical Letter, Inc., New Rochelle, NY.
Umbilical hernia in a patient with cirrhosis
A 62-year-old man was admitted to the intensive care unit with esophageal variceal bleeding. He had a long history of alcohol abuse with secondary cirrhosis, with a Child-Pugh score of 11 on a scale of 15 (class C—the most severe) at presentation. He also had a history of uncomplicated umbilical hernia, 6 cm in diameter without overlying trophic skin alterations.
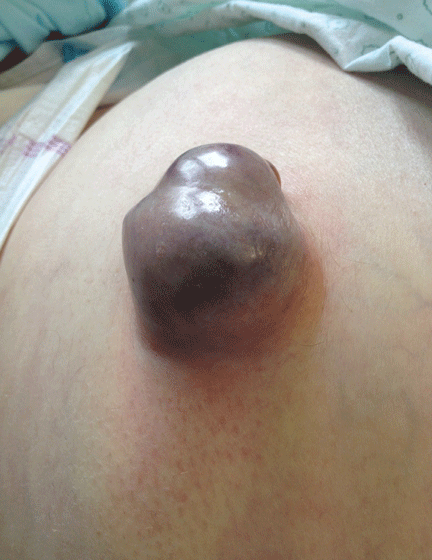
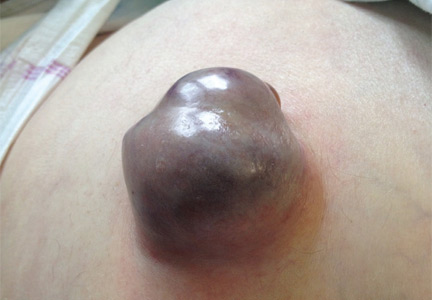
Treatment with somatostatin, endoscopic band ligation, and prophylactic antibiotics was initiated for the variceal bleeding. The next day, he was transferred to the hepatology floor. His condition stabilized during the next week, but then he abruptly became diaphoretic and less talkative. Physical examination revealed a painful and irreducible umbilical hernia (Figure 1). He was rushed for umbilical hernia repair with resection of a necrotic segment of small bowel. His recovery after surgery was uneventful, and he was eventually discharged.
UMBILICAL HERNIA AND CIRRHOSIS
Umbilical hernia is common in cirrhotic patients suffering from ascites, with a prevalence up to 20%, which is 10 times higher than in the general population.1 Ascites is the major predisposing factor since it causes muscle wasting and increases intra-abdominal pressure.
A unique feature of cirrhosis is low physiologic reserve, which increases the risk of death from complications of umbilical hernia and makes the patient more vulnerable to perioperative complications during repair. Because of the high operative risk, umbilical hernia repair has traditionally been reserved for the most complicated cases, such as strangulation of the bowel or rupture of the skin with leakage of ascitic fluid.2,3 Many patients are thus managed conservatively, with watchful waiting.
However, the natural course of umbilical hernia tends toward complications (eg, bowel incarceration, rupture of the overlying skin), which necessitate urgent repair.4 The risk of death with hernia repair in this urgent setting is seven times higher than for elective hernia repair in cirrhotic patients.5 More recent data indicate that elective repair in patients with well-compensated cirrhosis carries complication and mortality rates similar to those in noncirrhotic patients.5–8 Therefore, patients who should undergo umbilical hernia repair are not only those with complicated umbilical hernia (strangulation or ascites leak), but also those with well-compensated cirrhosis at risk of complications.
Factors that pose a particularly high risk of complications of repair are large hernia (> 5 cm), hernia associated with pain, intermittent incarceration, and trophic alterations of the overlying skin.1 In these patients, elective repair should be considered if hepatic function is preserved, if ascites is well managed (sodium restriction, diuretics, and sometimes even preoperative transjugular intrahepatic portosystemic shunt placement), and if the patient is not expected to undergo liver transplantation in the near future. If liver transplantation is anticipated in the short term, umbilical hernia can be managed concomitantly. Management of ascites after umbilical hernia repair is essential for prevention of recurrence.
- Dokmak S, Aussilhou B, Belghiti J. Umbilical hernias and cirrhose. J Visc Surg 2012; 149(suppl 5):e32–e39.
- Baron HC. Umbilical hernia secondary to cirrhosis of the liver. Complications of surgical correction. N Engl J Med 1960; 263:824–828.
- Hansen JB, Thulstrup AM, Vilstup H, Sørensen HT. Danish nationwide cohort study of postoperative death in patients with liver cirrhosis undergoing hernia repair. Br J Surg 2002; 89:805–806.
- Marsman HA, Heisterkamp J, Halm JA, Tilanus HW, Metselaar HJ, Kazemier G. Management in patients with liver cirrhosis and an umbilical hernia. Surgery 2007; 142:372–375.
- Carbonell AM, Wolfe LG, DeMaria EJ. Poor outcomes in cirrhosis-associated hernia repair: a nationwide cohort study of 32,033 patients. Hernia 2005; 9:353–357.
- Eker HH, van Ramshorst GH, de Goede B, et al. A prospective study on elective umbilical hernia repair in patients with liver cirrhosis and ascites. Surgery 2011; 150:542–546.
- Gray SH, Vick CC, Graham LA, Finan KR, Neumayer LA, Hawn MT. Umbilical herniorrhapy in cirrhosis: improved outcomes with elective repair. J Gastrointest Surg 2008; 12:675–681.
- McKay A, Dixon E, Bathe O, Sutherland F. Umbilical hernia repair in the presence of cirrhosis and ascites: results of a survey and review of the literature. Hernia 2009; 13:461–468.
A 62-year-old man was admitted to the intensive care unit with esophageal variceal bleeding. He had a long history of alcohol abuse with secondary cirrhosis, with a Child-Pugh score of 11 on a scale of 15 (class C—the most severe) at presentation. He also had a history of uncomplicated umbilical hernia, 6 cm in diameter without overlying trophic skin alterations.
Treatment with somatostatin, endoscopic band ligation, and prophylactic antibiotics was initiated for the variceal bleeding. The next day, he was transferred to the hepatology floor. His condition stabilized during the next week, but then he abruptly became diaphoretic and less talkative. Physical examination revealed a painful and irreducible umbilical hernia (Figure 1). He was rushed for umbilical hernia repair with resection of a necrotic segment of small bowel. His recovery after surgery was uneventful, and he was eventually discharged.
UMBILICAL HERNIA AND CIRRHOSIS
Umbilical hernia is common in cirrhotic patients suffering from ascites, with a prevalence up to 20%, which is 10 times higher than in the general population.1 Ascites is the major predisposing factor since it causes muscle wasting and increases intra-abdominal pressure.
A unique feature of cirrhosis is low physiologic reserve, which increases the risk of death from complications of umbilical hernia and makes the patient more vulnerable to perioperative complications during repair. Because of the high operative risk, umbilical hernia repair has traditionally been reserved for the most complicated cases, such as strangulation of the bowel or rupture of the skin with leakage of ascitic fluid.2,3 Many patients are thus managed conservatively, with watchful waiting.
However, the natural course of umbilical hernia tends toward complications (eg, bowel incarceration, rupture of the overlying skin), which necessitate urgent repair.4 The risk of death with hernia repair in this urgent setting is seven times higher than for elective hernia repair in cirrhotic patients.5 More recent data indicate that elective repair in patients with well-compensated cirrhosis carries complication and mortality rates similar to those in noncirrhotic patients.5–8 Therefore, patients who should undergo umbilical hernia repair are not only those with complicated umbilical hernia (strangulation or ascites leak), but also those with well-compensated cirrhosis at risk of complications.
Factors that pose a particularly high risk of complications of repair are large hernia (> 5 cm), hernia associated with pain, intermittent incarceration, and trophic alterations of the overlying skin.1 In these patients, elective repair should be considered if hepatic function is preserved, if ascites is well managed (sodium restriction, diuretics, and sometimes even preoperative transjugular intrahepatic portosystemic shunt placement), and if the patient is not expected to undergo liver transplantation in the near future. If liver transplantation is anticipated in the short term, umbilical hernia can be managed concomitantly. Management of ascites after umbilical hernia repair is essential for prevention of recurrence.
A 62-year-old man was admitted to the intensive care unit with esophageal variceal bleeding. He had a long history of alcohol abuse with secondary cirrhosis, with a Child-Pugh score of 11 on a scale of 15 (class C—the most severe) at presentation. He also had a history of uncomplicated umbilical hernia, 6 cm in diameter without overlying trophic skin alterations.
Treatment with somatostatin, endoscopic band ligation, and prophylactic antibiotics was initiated for the variceal bleeding. The next day, he was transferred to the hepatology floor. His condition stabilized during the next week, but then he abruptly became diaphoretic and less talkative. Physical examination revealed a painful and irreducible umbilical hernia (Figure 1). He was rushed for umbilical hernia repair with resection of a necrotic segment of small bowel. His recovery after surgery was uneventful, and he was eventually discharged.
UMBILICAL HERNIA AND CIRRHOSIS
Umbilical hernia is common in cirrhotic patients suffering from ascites, with a prevalence up to 20%, which is 10 times higher than in the general population.1 Ascites is the major predisposing factor since it causes muscle wasting and increases intra-abdominal pressure.
A unique feature of cirrhosis is low physiologic reserve, which increases the risk of death from complications of umbilical hernia and makes the patient more vulnerable to perioperative complications during repair. Because of the high operative risk, umbilical hernia repair has traditionally been reserved for the most complicated cases, such as strangulation of the bowel or rupture of the skin with leakage of ascitic fluid.2,3 Many patients are thus managed conservatively, with watchful waiting.
However, the natural course of umbilical hernia tends toward complications (eg, bowel incarceration, rupture of the overlying skin), which necessitate urgent repair.4 The risk of death with hernia repair in this urgent setting is seven times higher than for elective hernia repair in cirrhotic patients.5 More recent data indicate that elective repair in patients with well-compensated cirrhosis carries complication and mortality rates similar to those in noncirrhotic patients.5–8 Therefore, patients who should undergo umbilical hernia repair are not only those with complicated umbilical hernia (strangulation or ascites leak), but also those with well-compensated cirrhosis at risk of complications.
Factors that pose a particularly high risk of complications of repair are large hernia (> 5 cm), hernia associated with pain, intermittent incarceration, and trophic alterations of the overlying skin.1 In these patients, elective repair should be considered if hepatic function is preserved, if ascites is well managed (sodium restriction, diuretics, and sometimes even preoperative transjugular intrahepatic portosystemic shunt placement), and if the patient is not expected to undergo liver transplantation in the near future. If liver transplantation is anticipated in the short term, umbilical hernia can be managed concomitantly. Management of ascites after umbilical hernia repair is essential for prevention of recurrence.
- Dokmak S, Aussilhou B, Belghiti J. Umbilical hernias and cirrhose. J Visc Surg 2012; 149(suppl 5):e32–e39.
- Baron HC. Umbilical hernia secondary to cirrhosis of the liver. Complications of surgical correction. N Engl J Med 1960; 263:824–828.
- Hansen JB, Thulstrup AM, Vilstup H, Sørensen HT. Danish nationwide cohort study of postoperative death in patients with liver cirrhosis undergoing hernia repair. Br J Surg 2002; 89:805–806.
- Marsman HA, Heisterkamp J, Halm JA, Tilanus HW, Metselaar HJ, Kazemier G. Management in patients with liver cirrhosis and an umbilical hernia. Surgery 2007; 142:372–375.
- Carbonell AM, Wolfe LG, DeMaria EJ. Poor outcomes in cirrhosis-associated hernia repair: a nationwide cohort study of 32,033 patients. Hernia 2005; 9:353–357.
- Eker HH, van Ramshorst GH, de Goede B, et al. A prospective study on elective umbilical hernia repair in patients with liver cirrhosis and ascites. Surgery 2011; 150:542–546.
- Gray SH, Vick CC, Graham LA, Finan KR, Neumayer LA, Hawn MT. Umbilical herniorrhapy in cirrhosis: improved outcomes with elective repair. J Gastrointest Surg 2008; 12:675–681.
- McKay A, Dixon E, Bathe O, Sutherland F. Umbilical hernia repair in the presence of cirrhosis and ascites: results of a survey and review of the literature. Hernia 2009; 13:461–468.
- Dokmak S, Aussilhou B, Belghiti J. Umbilical hernias and cirrhose. J Visc Surg 2012; 149(suppl 5):e32–e39.
- Baron HC. Umbilical hernia secondary to cirrhosis of the liver. Complications of surgical correction. N Engl J Med 1960; 263:824–828.
- Hansen JB, Thulstrup AM, Vilstup H, Sørensen HT. Danish nationwide cohort study of postoperative death in patients with liver cirrhosis undergoing hernia repair. Br J Surg 2002; 89:805–806.
- Marsman HA, Heisterkamp J, Halm JA, Tilanus HW, Metselaar HJ, Kazemier G. Management in patients with liver cirrhosis and an umbilical hernia. Surgery 2007; 142:372–375.
- Carbonell AM, Wolfe LG, DeMaria EJ. Poor outcomes in cirrhosis-associated hernia repair: a nationwide cohort study of 32,033 patients. Hernia 2005; 9:353–357.
- Eker HH, van Ramshorst GH, de Goede B, et al. A prospective study on elective umbilical hernia repair in patients with liver cirrhosis and ascites. Surgery 2011; 150:542–546.
- Gray SH, Vick CC, Graham LA, Finan KR, Neumayer LA, Hawn MT. Umbilical herniorrhapy in cirrhosis: improved outcomes with elective repair. J Gastrointest Surg 2008; 12:675–681.
- McKay A, Dixon E, Bathe O, Sutherland F. Umbilical hernia repair in the presence of cirrhosis and ascites: results of a survey and review of the literature. Hernia 2009; 13:461–468.
Electrocardiographic changes in amitriptyline overdose
A 49-year-old woman with a history of depression, bipolar disorder, and chronic back pain was brought to the emergency department unresponsive after having taken an unknown quantity of amitriptyline tablets.
On arrival, she was comatose, with a score of 3 (the lowest possible score) on the 15-point Glasgow Coma Scale. Her blood pressure was 65/22 mm Hg, heart rate 121 beats per minute, respiratory rate 14 per minute, and oxygen saturation 88% on room air. The rest of the initial physical examination was normal.
She was immediately intubated, put on mechanical ventilation, and given an infusion of a 1-L bolus of normal saline and 50 mmol (1 mmol/kg) of sodium bicarbonate. Norepinephrine infusion was started. Gastric lavage was not done.
Results of initial laboratory testing showed a serum potassium of 2.9 mmol/L (reference range 3.5–5.0) and a serum magnesium of 1.6 mmol/L (1.7–2.6), which were corrected with infusion of 60 mmol of potassium chloride and 2 g of magnesium sulfate. The serum amitriptyline measurement was ordered at the time of her presentation to the emergency department.
Arterial blood gas analysis showed:
- pH 7.15 (normal range 7.35–7.45)
- Paco2 66 mm Hg (34–46)
- Pao2 229 mm Hg (85–95)
- Bicarbonate 22 mmol/L (22–26).
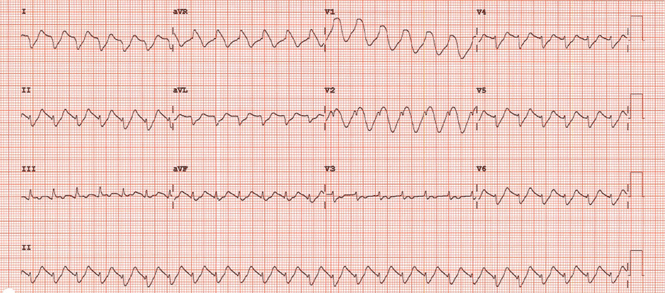
The initial electrocardiogram (ECG) (Figure 1) showed regular wide-complex tachycardia with no definite right or left bundle branch block morphology, no discernible P waves, a QRS duration of 198 msec, right axis deviation, and no Brugada criteria to suggest ventricular tachycardia.
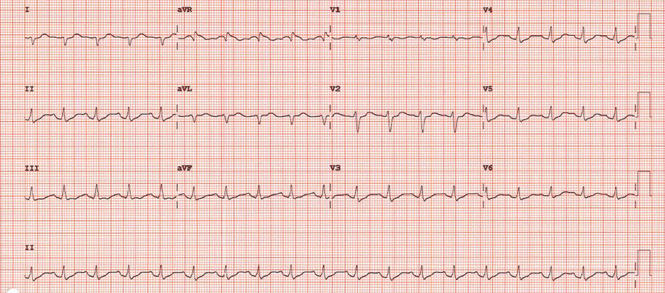
She remained hypotensive, with regular wide-complex tachycardia on the ECG. She was given an additional 1-L bolus of normal saline and 100 mmol (2 mmol/kg) of sodium bicarbonate, and within 1 minute the wide-complex tachycardia resolved to narrow-complex sinus tachycardia (Figure 2). At this point, an infusion of 150 mmol/L of sodium bicarbonate in dextrose 5% in water was started, with serial ECGs to monitor the QRS duration and serial arterial blood gas monitoring to maintain the pH between 7.45 and 7.55.
TRANSFER TO THE ICU
She was then transferred to the intensive care unit (ICU), where she remained for 2 weeks. While in the ICU, she had a single recurrence of wide-complex tachycardia that resolved immediately with an infusion of 100 mmol of sodium bicarbonate. A urine toxicology screen was negative, and the serum amitriptyline measurement, returned from the laboratory 48 hours after her initial presentation, was 594 ng/mL (reference range 100–250 ng/mL). She was eventually weaned off the norepinephrine infusion after 20 hours, the sodium bicarbonate infusion was discontinued after 4 days, and she was taken off mechanical ventilation after 10 days. Also during her ICU stay, she had seizures on day 3 and developed aspiration pneumonia.
From the ICU, she was transferred to a regular floor, where she stayed for another week and then was transferred to a rehabilitation center. This patient was known to have clinical depression and to have attempted suicide once before. She had recently been under additional psychosocial stresses, which likely prompted this second attempt.
She reportedly had no neurologic or cardiovascular sequelae after her discharge from the hospital.
AMITRIPTYLINE OVERDOSE
Amitriptyline causes a relatively high number of fatal overdoses, at 34 per 1 million prescriptions.1 Death is usually from hypotension and ventricular arrhythmia caused by blockage of cardiac fast sodium channels leading to disturbances of cardiac conduction such as wide-complex tachycardia.
Other manifestations of amitriptyline overdose include seizures, sedation, and anticholinergic toxicity from variable blockade of gamma-aminobutyric acid receptors, histamine 1 receptors, and alpha receptors.2
Of the various changes on ECG described with amitriptyline overdose, sinus tachycardia is the most common. A QRS duration greater than 100 msec, right to extreme-right axis deviation with negative QRS complexes in leads I and aVL, and an R-wave amplitude greater than 3 mm in lead aVR are indications for sodium bicarbonate infusion, especially in hemodynamically unstable patients.3 Sodium bicarbonate increases the serum concentration of sodium and thereby overcomes the sodium channel blockade. It also alkalinizes the serum, favoring an electrically neutral form of amitriptyline that binds less to receptors and binds more to alpha-1-acid glycoprotein, decreasing the fraction of free drug available for toxicity.4
In patients with amitriptyline overdose, wide-complex tachycardia and hypotension refractory to sodium bicarbonate infusion can be treated with lidocaine, magnesium sulfate, direct-current cardioversion, and lipid resuscitation.5,6 Treatment with class IA, IC, and III antiarrhythmics is contraindicated, as they block sodium channels and thus can worsen conduction disturbances.
- Henry JA, Alexander CA, Sener EK. Relative mortality from overdose of antidepressants. BMJ 1995; 310:221–224.
- Shannon M, Merola J, Lovejoy FH Jr. Hypotension in severe tricyclic antidepressant overdose. Am J Emerg Med 1988; 6:439–442.
- Liebelt EL, Francis PD, Woolf AD. ECG lead aVR versus QRS interval in predicting seizures and arrhythmias in acute tricyclic antidepressant toxicity. Ann Emerg Med 1995; 26:195–201.
- Sayniuk BI, Jhamandas V. Mechanism of reversal of toxic effects of amitriptyline on cardiac Purkinje fibres by sodium bicarbonate. J Pharmacol Exp Ther 1984; 231:387.
- Kiberd MB, Minor SF. Lipid therapy for the treatment of a refractory amitriptyline overdose. CJEM 2012; 14:193–197.
- Harvey M, Cave G. Case report: successful lipid resuscitation in multidrug overdose with predominant tricyclic antidepressant toxidrome. Int J Emerg Med 2012; 5:8.
A 49-year-old woman with a history of depression, bipolar disorder, and chronic back pain was brought to the emergency department unresponsive after having taken an unknown quantity of amitriptyline tablets.
On arrival, she was comatose, with a score of 3 (the lowest possible score) on the 15-point Glasgow Coma Scale. Her blood pressure was 65/22 mm Hg, heart rate 121 beats per minute, respiratory rate 14 per minute, and oxygen saturation 88% on room air. The rest of the initial physical examination was normal.
She was immediately intubated, put on mechanical ventilation, and given an infusion of a 1-L bolus of normal saline and 50 mmol (1 mmol/kg) of sodium bicarbonate. Norepinephrine infusion was started. Gastric lavage was not done.
Results of initial laboratory testing showed a serum potassium of 2.9 mmol/L (reference range 3.5–5.0) and a serum magnesium of 1.6 mmol/L (1.7–2.6), which were corrected with infusion of 60 mmol of potassium chloride and 2 g of magnesium sulfate. The serum amitriptyline measurement was ordered at the time of her presentation to the emergency department.
Arterial blood gas analysis showed:
- pH 7.15 (normal range 7.35–7.45)
- Paco2 66 mm Hg (34–46)
- Pao2 229 mm Hg (85–95)
- Bicarbonate 22 mmol/L (22–26).
The initial electrocardiogram (ECG) (Figure 1) showed regular wide-complex tachycardia with no definite right or left bundle branch block morphology, no discernible P waves, a QRS duration of 198 msec, right axis deviation, and no Brugada criteria to suggest ventricular tachycardia.
She remained hypotensive, with regular wide-complex tachycardia on the ECG. She was given an additional 1-L bolus of normal saline and 100 mmol (2 mmol/kg) of sodium bicarbonate, and within 1 minute the wide-complex tachycardia resolved to narrow-complex sinus tachycardia (Figure 2). At this point, an infusion of 150 mmol/L of sodium bicarbonate in dextrose 5% in water was started, with serial ECGs to monitor the QRS duration and serial arterial blood gas monitoring to maintain the pH between 7.45 and 7.55.
TRANSFER TO THE ICU
She was then transferred to the intensive care unit (ICU), where she remained for 2 weeks. While in the ICU, she had a single recurrence of wide-complex tachycardia that resolved immediately with an infusion of 100 mmol of sodium bicarbonate. A urine toxicology screen was negative, and the serum amitriptyline measurement, returned from the laboratory 48 hours after her initial presentation, was 594 ng/mL (reference range 100–250 ng/mL). She was eventually weaned off the norepinephrine infusion after 20 hours, the sodium bicarbonate infusion was discontinued after 4 days, and she was taken off mechanical ventilation after 10 days. Also during her ICU stay, she had seizures on day 3 and developed aspiration pneumonia.
From the ICU, she was transferred to a regular floor, where she stayed for another week and then was transferred to a rehabilitation center. This patient was known to have clinical depression and to have attempted suicide once before. She had recently been under additional psychosocial stresses, which likely prompted this second attempt.
She reportedly had no neurologic or cardiovascular sequelae after her discharge from the hospital.
AMITRIPTYLINE OVERDOSE
Amitriptyline causes a relatively high number of fatal overdoses, at 34 per 1 million prescriptions.1 Death is usually from hypotension and ventricular arrhythmia caused by blockage of cardiac fast sodium channels leading to disturbances of cardiac conduction such as wide-complex tachycardia.
Other manifestations of amitriptyline overdose include seizures, sedation, and anticholinergic toxicity from variable blockade of gamma-aminobutyric acid receptors, histamine 1 receptors, and alpha receptors.2
Of the various changes on ECG described with amitriptyline overdose, sinus tachycardia is the most common. A QRS duration greater than 100 msec, right to extreme-right axis deviation with negative QRS complexes in leads I and aVL, and an R-wave amplitude greater than 3 mm in lead aVR are indications for sodium bicarbonate infusion, especially in hemodynamically unstable patients.3 Sodium bicarbonate increases the serum concentration of sodium and thereby overcomes the sodium channel blockade. It also alkalinizes the serum, favoring an electrically neutral form of amitriptyline that binds less to receptors and binds more to alpha-1-acid glycoprotein, decreasing the fraction of free drug available for toxicity.4
In patients with amitriptyline overdose, wide-complex tachycardia and hypotension refractory to sodium bicarbonate infusion can be treated with lidocaine, magnesium sulfate, direct-current cardioversion, and lipid resuscitation.5,6 Treatment with class IA, IC, and III antiarrhythmics is contraindicated, as they block sodium channels and thus can worsen conduction disturbances.
A 49-year-old woman with a history of depression, bipolar disorder, and chronic back pain was brought to the emergency department unresponsive after having taken an unknown quantity of amitriptyline tablets.
On arrival, she was comatose, with a score of 3 (the lowest possible score) on the 15-point Glasgow Coma Scale. Her blood pressure was 65/22 mm Hg, heart rate 121 beats per minute, respiratory rate 14 per minute, and oxygen saturation 88% on room air. The rest of the initial physical examination was normal.
She was immediately intubated, put on mechanical ventilation, and given an infusion of a 1-L bolus of normal saline and 50 mmol (1 mmol/kg) of sodium bicarbonate. Norepinephrine infusion was started. Gastric lavage was not done.
Results of initial laboratory testing showed a serum potassium of 2.9 mmol/L (reference range 3.5–5.0) and a serum magnesium of 1.6 mmol/L (1.7–2.6), which were corrected with infusion of 60 mmol of potassium chloride and 2 g of magnesium sulfate. The serum amitriptyline measurement was ordered at the time of her presentation to the emergency department.
Arterial blood gas analysis showed:
- pH 7.15 (normal range 7.35–7.45)
- Paco2 66 mm Hg (34–46)
- Pao2 229 mm Hg (85–95)
- Bicarbonate 22 mmol/L (22–26).
The initial electrocardiogram (ECG) (Figure 1) showed regular wide-complex tachycardia with no definite right or left bundle branch block morphology, no discernible P waves, a QRS duration of 198 msec, right axis deviation, and no Brugada criteria to suggest ventricular tachycardia.
She remained hypotensive, with regular wide-complex tachycardia on the ECG. She was given an additional 1-L bolus of normal saline and 100 mmol (2 mmol/kg) of sodium bicarbonate, and within 1 minute the wide-complex tachycardia resolved to narrow-complex sinus tachycardia (Figure 2). At this point, an infusion of 150 mmol/L of sodium bicarbonate in dextrose 5% in water was started, with serial ECGs to monitor the QRS duration and serial arterial blood gas monitoring to maintain the pH between 7.45 and 7.55.
TRANSFER TO THE ICU
She was then transferred to the intensive care unit (ICU), where she remained for 2 weeks. While in the ICU, she had a single recurrence of wide-complex tachycardia that resolved immediately with an infusion of 100 mmol of sodium bicarbonate. A urine toxicology screen was negative, and the serum amitriptyline measurement, returned from the laboratory 48 hours after her initial presentation, was 594 ng/mL (reference range 100–250 ng/mL). She was eventually weaned off the norepinephrine infusion after 20 hours, the sodium bicarbonate infusion was discontinued after 4 days, and she was taken off mechanical ventilation after 10 days. Also during her ICU stay, she had seizures on day 3 and developed aspiration pneumonia.
From the ICU, she was transferred to a regular floor, where she stayed for another week and then was transferred to a rehabilitation center. This patient was known to have clinical depression and to have attempted suicide once before. She had recently been under additional psychosocial stresses, which likely prompted this second attempt.
She reportedly had no neurologic or cardiovascular sequelae after her discharge from the hospital.
AMITRIPTYLINE OVERDOSE
Amitriptyline causes a relatively high number of fatal overdoses, at 34 per 1 million prescriptions.1 Death is usually from hypotension and ventricular arrhythmia caused by blockage of cardiac fast sodium channels leading to disturbances of cardiac conduction such as wide-complex tachycardia.
Other manifestations of amitriptyline overdose include seizures, sedation, and anticholinergic toxicity from variable blockade of gamma-aminobutyric acid receptors, histamine 1 receptors, and alpha receptors.2
Of the various changes on ECG described with amitriptyline overdose, sinus tachycardia is the most common. A QRS duration greater than 100 msec, right to extreme-right axis deviation with negative QRS complexes in leads I and aVL, and an R-wave amplitude greater than 3 mm in lead aVR are indications for sodium bicarbonate infusion, especially in hemodynamically unstable patients.3 Sodium bicarbonate increases the serum concentration of sodium and thereby overcomes the sodium channel blockade. It also alkalinizes the serum, favoring an electrically neutral form of amitriptyline that binds less to receptors and binds more to alpha-1-acid glycoprotein, decreasing the fraction of free drug available for toxicity.4
In patients with amitriptyline overdose, wide-complex tachycardia and hypotension refractory to sodium bicarbonate infusion can be treated with lidocaine, magnesium sulfate, direct-current cardioversion, and lipid resuscitation.5,6 Treatment with class IA, IC, and III antiarrhythmics is contraindicated, as they block sodium channels and thus can worsen conduction disturbances.
- Henry JA, Alexander CA, Sener EK. Relative mortality from overdose of antidepressants. BMJ 1995; 310:221–224.
- Shannon M, Merola J, Lovejoy FH Jr. Hypotension in severe tricyclic antidepressant overdose. Am J Emerg Med 1988; 6:439–442.
- Liebelt EL, Francis PD, Woolf AD. ECG lead aVR versus QRS interval in predicting seizures and arrhythmias in acute tricyclic antidepressant toxicity. Ann Emerg Med 1995; 26:195–201.
- Sayniuk BI, Jhamandas V. Mechanism of reversal of toxic effects of amitriptyline on cardiac Purkinje fibres by sodium bicarbonate. J Pharmacol Exp Ther 1984; 231:387.
- Kiberd MB, Minor SF. Lipid therapy for the treatment of a refractory amitriptyline overdose. CJEM 2012; 14:193–197.
- Harvey M, Cave G. Case report: successful lipid resuscitation in multidrug overdose with predominant tricyclic antidepressant toxidrome. Int J Emerg Med 2012; 5:8.
- Henry JA, Alexander CA, Sener EK. Relative mortality from overdose of antidepressants. BMJ 1995; 310:221–224.
- Shannon M, Merola J, Lovejoy FH Jr. Hypotension in severe tricyclic antidepressant overdose. Am J Emerg Med 1988; 6:439–442.
- Liebelt EL, Francis PD, Woolf AD. ECG lead aVR versus QRS interval in predicting seizures and arrhythmias in acute tricyclic antidepressant toxicity. Ann Emerg Med 1995; 26:195–201.
- Sayniuk BI, Jhamandas V. Mechanism of reversal of toxic effects of amitriptyline on cardiac Purkinje fibres by sodium bicarbonate. J Pharmacol Exp Ther 1984; 231:387.
- Kiberd MB, Minor SF. Lipid therapy for the treatment of a refractory amitriptyline overdose. CJEM 2012; 14:193–197.
- Harvey M, Cave G. Case report: successful lipid resuscitation in multidrug overdose with predominant tricyclic antidepressant toxidrome. Int J Emerg Med 2012; 5:8.
Corneal opacities in a man with chronic kidney disease
A 40-year-old man with end-stage renal disease on intermittent hemodialysis presented to the emergency department with a 1-week history of pain affecting his left lower back, left flank, and left lower abdomen, diagnosed as zoster prodrome.
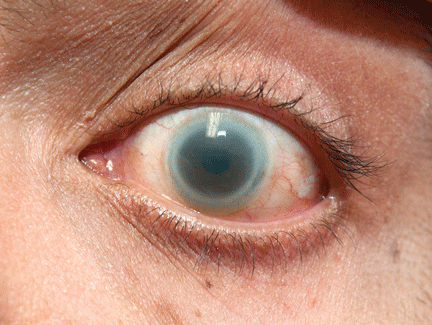
Of note, both corneas were cloudy, most severely in the limbus (Figure 1). His visual acuity and findings on funduscopic examination were normal.
CORNEAL OPACITY
The finding of corneal opacity should prompt an immediate ophthalmologic evaluation by the internist as well as an ophthalmologist. The initial examination should include visual acuity testing; gross examination with the naked eye; penlight examination of the pupil, conjunctiva, and anterior chamber; funduscopic examination to at least confirm a red reflex; and fluorescein examination of the cornea. Fluorescein testing is done last, as the dye may interfere with the other initial tests.1
A number of causes of opacity
A number of conditions can cause corneal opacity. Several genetic conditions can cause developmental anomalies of the cornea, leading to corneal defects present at birth.2 Causes of secondary corneal opacity in early infancy include infections such as herpes, iatrogenic injury during amniocentesis or forceps delivery, and infantile congenital glaucoma.2
Later in life, causes of corneal opacity include cataract, glaucoma, chemical exposure, foreign body injury, irradiation, infection (eg, syphilis, herpes, chlamydia), endophthalmitis, and metabolic genetic disorders such as Fabry disease, trisomy 18 syndrome, and lecithin-cholesterol acyltransferase (LCAT) deficiency.3
LCAT DEFICIENCY
LCAT is a key protein in reverse transport of cholesterol from the systemic circulation to the liver for excretion into the bile. Its deficiency results in low serum concentrations of high-density lipoprotein cholesterol (HDL-C).4 About 80 different mutations in the LCAT gene have been linked to LCAT deficiency.5
LCAT deficiency varies in severity. Patients with complete deficiency can have nearly undetectable levels of HDL-C, eruptive xanthoma, hepatosplenomegaly, and premature coronary artery disease (ie, by age 40).5,6 Features of coronary atherosclerosis can be lacking in patients with partial deficiency.
Regardless of the degree of LCAT deficiency, most patients have corneal opacification that is most severe near the limbus (thus, the term “fish eye syndrome”) and anemia.7 Although corneal opacification presents early in life and persists, it does not seem to affect vision.5 The anemia is associated with enhanced fractional clearance of red blood cells secondary to hypersplenism.8
LCAT deficiency and the kidneys
LCAT deficiency has its most devastating effect on the kidney. Renal disease begins early in life with mild proteinuria and microscopic hematuria. With increasing age, renal function deteriorates and proteinuria and hematuria worsen.9
Renal biopsy study may reveal foam cells in the glomerular tufts, arterioles with thickened intima and narrowed lumens, and subendothelial deposits of lipids in the renal arteries and arterioles.10 Some studies have suggested that kidney disease is most likely initiated by lipid deposition or cellular uptake of lipoproteins in the glomerular basement membrane, mesangium, and capillary subendothelium.
Treatment
There are few treatment options for patients with LCAT deficiency. Control of hypertension, if present, may halt or slow renal deterioration.5 Many patients eventually require dialysis, and some undergo renal transplantation, but the renal disease can recur.
OUR PATIENT
Our patient had a known diagnosis of LCAT deficiency. Five years before this presentation at our emergency department, he developed malignant hypertension, followed shortly by renal disease. Over the next 4 years, his kidney function deteriorated, culminating in the need for dialysis; his corneal opacities manifested and gradually worsened; and after extensive studies including kidney biopsies, he was finally diagnosed with LCAT deficiency.
He also exhibited a chronically low level of HDL-C (2 to 5 mg/dL) and significant coronary artery disease. Although unrelated, his zoster pain was treated with renally dosed acyclovir and gabapentin. He never demonstrated the characteristic rash, and his pain improved significantly within 5 days of treatment.
- Knox KA, McIntee J. Nurse management of corneal abrasion. Br J Nurs 1995; 4:440–460.
- Nischal KK. Congenital corneal opacities—a surgical approach to nomenclature and classification. Eye (Lond) 2007; 21:1326–1337.
- Chiapella AP, Rosenthal AR. One year in an eye casualty clinic. Br J Ophthalmol 1985; 69:865–870.
- Rosenson RS, Brewer HB Jr, Davidson WS, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation 2012; 125:1905–1919.
- Roshan B, Ganda OP, Desilva R, et al. Homozygous lecithin:cholesterol acyltransferase (LCAT) deficiency due to a new loss of function mutation and review of the literature. J Clin Lipidol 2011; 5:493–499.
- Kuivenhoven JA, van Voorst tot Voorst EJ, Wiebusch H, et al. A unique genetic and biochemical presentation of fish-eye disease. J Clin Invest 1995; 96:2783–2791.
- Palmiero PM, Sbeity Z, Liebmann J, Ritch R. In vivo imaging of the cornea in a patient with lecithin-cholesterol acyltransferase deficiency. Cornea 2009; 28:1061–1064.
- Norum KR, Gjone E. Familial serum-cholesterol esterification failure. A new inborn error of metabolism. Biochim Biophys Acta 1967; 144:698–700.
- Gjone E, Norum KR. Familial serum cholesterol ester deficiency. Clinical study of a patient with a new syndrome. Acta Med Scand 1968; 183:107–112.
- Lager DJ, Rosenberg BF, Shapiro H, Bernstein J. Lecithin cholesterol acyltransferase deficiency: ultrastructural examination of sequential renal biopsies. Mod Pathol 1991; 4:331–335.
A 40-year-old man with end-stage renal disease on intermittent hemodialysis presented to the emergency department with a 1-week history of pain affecting his left lower back, left flank, and left lower abdomen, diagnosed as zoster prodrome.
Of note, both corneas were cloudy, most severely in the limbus (Figure 1). His visual acuity and findings on funduscopic examination were normal.
CORNEAL OPACITY
The finding of corneal opacity should prompt an immediate ophthalmologic evaluation by the internist as well as an ophthalmologist. The initial examination should include visual acuity testing; gross examination with the naked eye; penlight examination of the pupil, conjunctiva, and anterior chamber; funduscopic examination to at least confirm a red reflex; and fluorescein examination of the cornea. Fluorescein testing is done last, as the dye may interfere with the other initial tests.1
A number of causes of opacity
A number of conditions can cause corneal opacity. Several genetic conditions can cause developmental anomalies of the cornea, leading to corneal defects present at birth.2 Causes of secondary corneal opacity in early infancy include infections such as herpes, iatrogenic injury during amniocentesis or forceps delivery, and infantile congenital glaucoma.2
Later in life, causes of corneal opacity include cataract, glaucoma, chemical exposure, foreign body injury, irradiation, infection (eg, syphilis, herpes, chlamydia), endophthalmitis, and metabolic genetic disorders such as Fabry disease, trisomy 18 syndrome, and lecithin-cholesterol acyltransferase (LCAT) deficiency.3
LCAT DEFICIENCY
LCAT is a key protein in reverse transport of cholesterol from the systemic circulation to the liver for excretion into the bile. Its deficiency results in low serum concentrations of high-density lipoprotein cholesterol (HDL-C).4 About 80 different mutations in the LCAT gene have been linked to LCAT deficiency.5
LCAT deficiency varies in severity. Patients with complete deficiency can have nearly undetectable levels of HDL-C, eruptive xanthoma, hepatosplenomegaly, and premature coronary artery disease (ie, by age 40).5,6 Features of coronary atherosclerosis can be lacking in patients with partial deficiency.
Regardless of the degree of LCAT deficiency, most patients have corneal opacification that is most severe near the limbus (thus, the term “fish eye syndrome”) and anemia.7 Although corneal opacification presents early in life and persists, it does not seem to affect vision.5 The anemia is associated with enhanced fractional clearance of red blood cells secondary to hypersplenism.8
LCAT deficiency and the kidneys
LCAT deficiency has its most devastating effect on the kidney. Renal disease begins early in life with mild proteinuria and microscopic hematuria. With increasing age, renal function deteriorates and proteinuria and hematuria worsen.9
Renal biopsy study may reveal foam cells in the glomerular tufts, arterioles with thickened intima and narrowed lumens, and subendothelial deposits of lipids in the renal arteries and arterioles.10 Some studies have suggested that kidney disease is most likely initiated by lipid deposition or cellular uptake of lipoproteins in the glomerular basement membrane, mesangium, and capillary subendothelium.
Treatment
There are few treatment options for patients with LCAT deficiency. Control of hypertension, if present, may halt or slow renal deterioration.5 Many patients eventually require dialysis, and some undergo renal transplantation, but the renal disease can recur.
OUR PATIENT
Our patient had a known diagnosis of LCAT deficiency. Five years before this presentation at our emergency department, he developed malignant hypertension, followed shortly by renal disease. Over the next 4 years, his kidney function deteriorated, culminating in the need for dialysis; his corneal opacities manifested and gradually worsened; and after extensive studies including kidney biopsies, he was finally diagnosed with LCAT deficiency.
He also exhibited a chronically low level of HDL-C (2 to 5 mg/dL) and significant coronary artery disease. Although unrelated, his zoster pain was treated with renally dosed acyclovir and gabapentin. He never demonstrated the characteristic rash, and his pain improved significantly within 5 days of treatment.
A 40-year-old man with end-stage renal disease on intermittent hemodialysis presented to the emergency department with a 1-week history of pain affecting his left lower back, left flank, and left lower abdomen, diagnosed as zoster prodrome.
Of note, both corneas were cloudy, most severely in the limbus (Figure 1). His visual acuity and findings on funduscopic examination were normal.
CORNEAL OPACITY
The finding of corneal opacity should prompt an immediate ophthalmologic evaluation by the internist as well as an ophthalmologist. The initial examination should include visual acuity testing; gross examination with the naked eye; penlight examination of the pupil, conjunctiva, and anterior chamber; funduscopic examination to at least confirm a red reflex; and fluorescein examination of the cornea. Fluorescein testing is done last, as the dye may interfere with the other initial tests.1
A number of causes of opacity
A number of conditions can cause corneal opacity. Several genetic conditions can cause developmental anomalies of the cornea, leading to corneal defects present at birth.2 Causes of secondary corneal opacity in early infancy include infections such as herpes, iatrogenic injury during amniocentesis or forceps delivery, and infantile congenital glaucoma.2
Later in life, causes of corneal opacity include cataract, glaucoma, chemical exposure, foreign body injury, irradiation, infection (eg, syphilis, herpes, chlamydia), endophthalmitis, and metabolic genetic disorders such as Fabry disease, trisomy 18 syndrome, and lecithin-cholesterol acyltransferase (LCAT) deficiency.3
LCAT DEFICIENCY
LCAT is a key protein in reverse transport of cholesterol from the systemic circulation to the liver for excretion into the bile. Its deficiency results in low serum concentrations of high-density lipoprotein cholesterol (HDL-C).4 About 80 different mutations in the LCAT gene have been linked to LCAT deficiency.5
LCAT deficiency varies in severity. Patients with complete deficiency can have nearly undetectable levels of HDL-C, eruptive xanthoma, hepatosplenomegaly, and premature coronary artery disease (ie, by age 40).5,6 Features of coronary atherosclerosis can be lacking in patients with partial deficiency.
Regardless of the degree of LCAT deficiency, most patients have corneal opacification that is most severe near the limbus (thus, the term “fish eye syndrome”) and anemia.7 Although corneal opacification presents early in life and persists, it does not seem to affect vision.5 The anemia is associated with enhanced fractional clearance of red blood cells secondary to hypersplenism.8
LCAT deficiency and the kidneys
LCAT deficiency has its most devastating effect on the kidney. Renal disease begins early in life with mild proteinuria and microscopic hematuria. With increasing age, renal function deteriorates and proteinuria and hematuria worsen.9
Renal biopsy study may reveal foam cells in the glomerular tufts, arterioles with thickened intima and narrowed lumens, and subendothelial deposits of lipids in the renal arteries and arterioles.10 Some studies have suggested that kidney disease is most likely initiated by lipid deposition or cellular uptake of lipoproteins in the glomerular basement membrane, mesangium, and capillary subendothelium.
Treatment
There are few treatment options for patients with LCAT deficiency. Control of hypertension, if present, may halt or slow renal deterioration.5 Many patients eventually require dialysis, and some undergo renal transplantation, but the renal disease can recur.
OUR PATIENT
Our patient had a known diagnosis of LCAT deficiency. Five years before this presentation at our emergency department, he developed malignant hypertension, followed shortly by renal disease. Over the next 4 years, his kidney function deteriorated, culminating in the need for dialysis; his corneal opacities manifested and gradually worsened; and after extensive studies including kidney biopsies, he was finally diagnosed with LCAT deficiency.
He also exhibited a chronically low level of HDL-C (2 to 5 mg/dL) and significant coronary artery disease. Although unrelated, his zoster pain was treated with renally dosed acyclovir and gabapentin. He never demonstrated the characteristic rash, and his pain improved significantly within 5 days of treatment.
- Knox KA, McIntee J. Nurse management of corneal abrasion. Br J Nurs 1995; 4:440–460.
- Nischal KK. Congenital corneal opacities—a surgical approach to nomenclature and classification. Eye (Lond) 2007; 21:1326–1337.
- Chiapella AP, Rosenthal AR. One year in an eye casualty clinic. Br J Ophthalmol 1985; 69:865–870.
- Rosenson RS, Brewer HB Jr, Davidson WS, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation 2012; 125:1905–1919.
- Roshan B, Ganda OP, Desilva R, et al. Homozygous lecithin:cholesterol acyltransferase (LCAT) deficiency due to a new loss of function mutation and review of the literature. J Clin Lipidol 2011; 5:493–499.
- Kuivenhoven JA, van Voorst tot Voorst EJ, Wiebusch H, et al. A unique genetic and biochemical presentation of fish-eye disease. J Clin Invest 1995; 96:2783–2791.
- Palmiero PM, Sbeity Z, Liebmann J, Ritch R. In vivo imaging of the cornea in a patient with lecithin-cholesterol acyltransferase deficiency. Cornea 2009; 28:1061–1064.
- Norum KR, Gjone E. Familial serum-cholesterol esterification failure. A new inborn error of metabolism. Biochim Biophys Acta 1967; 144:698–700.
- Gjone E, Norum KR. Familial serum cholesterol ester deficiency. Clinical study of a patient with a new syndrome. Acta Med Scand 1968; 183:107–112.
- Lager DJ, Rosenberg BF, Shapiro H, Bernstein J. Lecithin cholesterol acyltransferase deficiency: ultrastructural examination of sequential renal biopsies. Mod Pathol 1991; 4:331–335.
- Knox KA, McIntee J. Nurse management of corneal abrasion. Br J Nurs 1995; 4:440–460.
- Nischal KK. Congenital corneal opacities—a surgical approach to nomenclature and classification. Eye (Lond) 2007; 21:1326–1337.
- Chiapella AP, Rosenthal AR. One year in an eye casualty clinic. Br J Ophthalmol 1985; 69:865–870.
- Rosenson RS, Brewer HB Jr, Davidson WS, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation 2012; 125:1905–1919.
- Roshan B, Ganda OP, Desilva R, et al. Homozygous lecithin:cholesterol acyltransferase (LCAT) deficiency due to a new loss of function mutation and review of the literature. J Clin Lipidol 2011; 5:493–499.
- Kuivenhoven JA, van Voorst tot Voorst EJ, Wiebusch H, et al. A unique genetic and biochemical presentation of fish-eye disease. J Clin Invest 1995; 96:2783–2791.
- Palmiero PM, Sbeity Z, Liebmann J, Ritch R. In vivo imaging of the cornea in a patient with lecithin-cholesterol acyltransferase deficiency. Cornea 2009; 28:1061–1064.
- Norum KR, Gjone E. Familial serum-cholesterol esterification failure. A new inborn error of metabolism. Biochim Biophys Acta 1967; 144:698–700.
- Gjone E, Norum KR. Familial serum cholesterol ester deficiency. Clinical study of a patient with a new syndrome. Acta Med Scand 1968; 183:107–112.
- Lager DJ, Rosenberg BF, Shapiro H, Bernstein J. Lecithin cholesterol acyltransferase deficiency: ultrastructural examination of sequential renal biopsies. Mod Pathol 1991; 4:331–335.
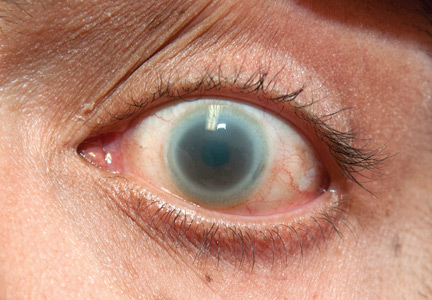
Persistent back pain in a young woman
A 24-year-old woman from northern India came to our medical center because of lower back pain for the past 2 years. The pain was initially a dull, continuous ache and did not radiate. She had no fever, night sweats, weight loss, or other constitutional symptoms.
In addition, she had seen her local practitioner 1 year earlier because of burning during urination and occasional frequency. She had been found to have an 8-mm calculus in the lower calyx of the left kidney, for which she underwent two sessions of shock-wave lithotripsy, but she did not pass any stone fragments. Because her back pain continued, she sought medical treatment at our center.
On evaluation at our facility, she was found to have paraspinal muscle spasm and scoliosis. Her gait was antalgic. Sensations were normal over both lower limbs in all dermatomes. Power was grade 5 throughout, and deep tendon reflexes were normal. The straight-leg-raising test was positive for reproducible pain in the lower back and sciatic pain radiating down the back of both legs.
Laboratory testing showed that her hemoglobin was low at 9.7 g/dL (reference range 11.5–15.5), but the rest of the complete blood cell count was within normal limits. C-reactive protein was elevated at 70.7 mg/L (reference range < 6 mg/L). An enzyme-linked immunosorbent assay was negative for human immunodeficiency virus (HIV).
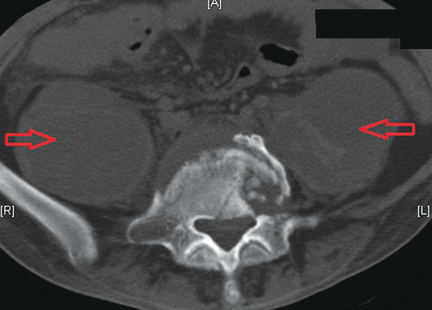
Nonenhanced computed tomography of the abdomen revealed destruction of vertebral body end plates and disks from the L2 lower end plate to the L5 superior end plate. The left transverse processes of the L3, L4, and L5 vertebral bodies were also destroyed. The scan also revealed bilateral psoas abscesses larger than 10 by 10 cm (Figure 1), with the right side larger than the left, and confirmed a stone in the left lower renal calyx (Figure 2).
She underwent bilateral ultrasonographically guided drainage of the abscesses. Culture of the thick pus that was aspirated grew Mycobacterium tuberculosis. Tuberculosis therapy was started with isoniazid, rifampicin, pyrazinamide, and ethambutol. Her condition improved rapidly over the next 2 to 3 months. She completed 18 months of tuberculosis therapy.
Because her spine was stable, with no collapse of vertebrae, she did not require orthopedic intervention.
SPINAL TUBERCULOSIS
Spinal tuberculosis, or Pott disease, is still a common cause of back pain in areas where the infection is rampant, such as northern India.1Mycobacterium infections continue to be a problem, especially coexisting with HIV infection.2,3 In fact, the World Health Organization and the United States Agency for International Development have referred to this as a twin epidemic.4
Early diagnosis and prompt treatment can prevent or minimize spinal deformity and permanent neurologic disability.5 Rapidly progressive and significant neurologic involvement requires surgical management. On the other hand, Patil et al6 reported a series of 50 cases in which early radiologic evidence of spinal cord compression from tuberculosis was managed nonoperatively.
When evaluating back pain, symptoms that should ring the alarm include weight loss, constitutional symptoms, no change in pain status after 6 weeks of treatment with a nonsteroidal anti-inflammatory drug, pain at night or at rest, and neurologic symptoms. Our patient had no relief of pain and thus sought treatment.
An important take-home message is that small, nonobstructive renal calculi almost never cause back pain, and when incidentally detected, as in this patient, should not be considered the cause of back pain.
- McLain RF, Isada C. Spinal tuberculosis deserves a place on the radar screen. Cleve Clin J Med 2004; 71:537–549.
- Vermund SH, Yamamoto N. Co-infection with human immunodeficiency virus and tuberculosis in Asia. Tuberculosis (Edinb) 2007; 87(suppl 1):S18–S25.
- Candy S, Chang G, Andronikou S. Acute myelopathy or cauda equina syndrome in HIV-positive adults in a tuberculosis endemic setting: MRI, clinical, and pathologic findings. AJNR Am J Neuroradiol 2014; 35:1634–1641.
- USAID. The twin epidemics: HIV and TB co-infection. www.usaid.gov/news-information/fact-sheets/twin-epidemics-hiv-and-tb-co-infection. Accessed April 30, 2015.
- Jain AK. Tuberculosis of the spine: a fresh look at an old disease. J Bone Joint Surg Br 2010; 92:905–913.
- Patil SS, Mohite S, Varma R, Bhojraj SY, Nene AM. Non-surgical management of cord compression in tuberculosis: a series of surprises. Asian Spine J 2014; 8:315–321.
A 24-year-old woman from northern India came to our medical center because of lower back pain for the past 2 years. The pain was initially a dull, continuous ache and did not radiate. She had no fever, night sweats, weight loss, or other constitutional symptoms.
In addition, she had seen her local practitioner 1 year earlier because of burning during urination and occasional frequency. She had been found to have an 8-mm calculus in the lower calyx of the left kidney, for which she underwent two sessions of shock-wave lithotripsy, but she did not pass any stone fragments. Because her back pain continued, she sought medical treatment at our center.
On evaluation at our facility, she was found to have paraspinal muscle spasm and scoliosis. Her gait was antalgic. Sensations were normal over both lower limbs in all dermatomes. Power was grade 5 throughout, and deep tendon reflexes were normal. The straight-leg-raising test was positive for reproducible pain in the lower back and sciatic pain radiating down the back of both legs.
Laboratory testing showed that her hemoglobin was low at 9.7 g/dL (reference range 11.5–15.5), but the rest of the complete blood cell count was within normal limits. C-reactive protein was elevated at 70.7 mg/L (reference range < 6 mg/L). An enzyme-linked immunosorbent assay was negative for human immunodeficiency virus (HIV).
Nonenhanced computed tomography of the abdomen revealed destruction of vertebral body end plates and disks from the L2 lower end plate to the L5 superior end plate. The left transverse processes of the L3, L4, and L5 vertebral bodies were also destroyed. The scan also revealed bilateral psoas abscesses larger than 10 by 10 cm (Figure 1), with the right side larger than the left, and confirmed a stone in the left lower renal calyx (Figure 2).
She underwent bilateral ultrasonographically guided drainage of the abscesses. Culture of the thick pus that was aspirated grew Mycobacterium tuberculosis. Tuberculosis therapy was started with isoniazid, rifampicin, pyrazinamide, and ethambutol. Her condition improved rapidly over the next 2 to 3 months. She completed 18 months of tuberculosis therapy.
Because her spine was stable, with no collapse of vertebrae, she did not require orthopedic intervention.
SPINAL TUBERCULOSIS
Spinal tuberculosis, or Pott disease, is still a common cause of back pain in areas where the infection is rampant, such as northern India.1Mycobacterium infections continue to be a problem, especially coexisting with HIV infection.2,3 In fact, the World Health Organization and the United States Agency for International Development have referred to this as a twin epidemic.4
Early diagnosis and prompt treatment can prevent or minimize spinal deformity and permanent neurologic disability.5 Rapidly progressive and significant neurologic involvement requires surgical management. On the other hand, Patil et al6 reported a series of 50 cases in which early radiologic evidence of spinal cord compression from tuberculosis was managed nonoperatively.
When evaluating back pain, symptoms that should ring the alarm include weight loss, constitutional symptoms, no change in pain status after 6 weeks of treatment with a nonsteroidal anti-inflammatory drug, pain at night or at rest, and neurologic symptoms. Our patient had no relief of pain and thus sought treatment.
An important take-home message is that small, nonobstructive renal calculi almost never cause back pain, and when incidentally detected, as in this patient, should not be considered the cause of back pain.
A 24-year-old woman from northern India came to our medical center because of lower back pain for the past 2 years. The pain was initially a dull, continuous ache and did not radiate. She had no fever, night sweats, weight loss, or other constitutional symptoms.
In addition, she had seen her local practitioner 1 year earlier because of burning during urination and occasional frequency. She had been found to have an 8-mm calculus in the lower calyx of the left kidney, for which she underwent two sessions of shock-wave lithotripsy, but she did not pass any stone fragments. Because her back pain continued, she sought medical treatment at our center.
On evaluation at our facility, she was found to have paraspinal muscle spasm and scoliosis. Her gait was antalgic. Sensations were normal over both lower limbs in all dermatomes. Power was grade 5 throughout, and deep tendon reflexes were normal. The straight-leg-raising test was positive for reproducible pain in the lower back and sciatic pain radiating down the back of both legs.
Laboratory testing showed that her hemoglobin was low at 9.7 g/dL (reference range 11.5–15.5), but the rest of the complete blood cell count was within normal limits. C-reactive protein was elevated at 70.7 mg/L (reference range < 6 mg/L). An enzyme-linked immunosorbent assay was negative for human immunodeficiency virus (HIV).
Nonenhanced computed tomography of the abdomen revealed destruction of vertebral body end plates and disks from the L2 lower end plate to the L5 superior end plate. The left transverse processes of the L3, L4, and L5 vertebral bodies were also destroyed. The scan also revealed bilateral psoas abscesses larger than 10 by 10 cm (Figure 1), with the right side larger than the left, and confirmed a stone in the left lower renal calyx (Figure 2).
She underwent bilateral ultrasonographically guided drainage of the abscesses. Culture of the thick pus that was aspirated grew Mycobacterium tuberculosis. Tuberculosis therapy was started with isoniazid, rifampicin, pyrazinamide, and ethambutol. Her condition improved rapidly over the next 2 to 3 months. She completed 18 months of tuberculosis therapy.
Because her spine was stable, with no collapse of vertebrae, she did not require orthopedic intervention.
SPINAL TUBERCULOSIS
Spinal tuberculosis, or Pott disease, is still a common cause of back pain in areas where the infection is rampant, such as northern India.1Mycobacterium infections continue to be a problem, especially coexisting with HIV infection.2,3 In fact, the World Health Organization and the United States Agency for International Development have referred to this as a twin epidemic.4
Early diagnosis and prompt treatment can prevent or minimize spinal deformity and permanent neurologic disability.5 Rapidly progressive and significant neurologic involvement requires surgical management. On the other hand, Patil et al6 reported a series of 50 cases in which early radiologic evidence of spinal cord compression from tuberculosis was managed nonoperatively.
When evaluating back pain, symptoms that should ring the alarm include weight loss, constitutional symptoms, no change in pain status after 6 weeks of treatment with a nonsteroidal anti-inflammatory drug, pain at night or at rest, and neurologic symptoms. Our patient had no relief of pain and thus sought treatment.
An important take-home message is that small, nonobstructive renal calculi almost never cause back pain, and when incidentally detected, as in this patient, should not be considered the cause of back pain.
- McLain RF, Isada C. Spinal tuberculosis deserves a place on the radar screen. Cleve Clin J Med 2004; 71:537–549.
- Vermund SH, Yamamoto N. Co-infection with human immunodeficiency virus and tuberculosis in Asia. Tuberculosis (Edinb) 2007; 87(suppl 1):S18–S25.
- Candy S, Chang G, Andronikou S. Acute myelopathy or cauda equina syndrome in HIV-positive adults in a tuberculosis endemic setting: MRI, clinical, and pathologic findings. AJNR Am J Neuroradiol 2014; 35:1634–1641.
- USAID. The twin epidemics: HIV and TB co-infection. www.usaid.gov/news-information/fact-sheets/twin-epidemics-hiv-and-tb-co-infection. Accessed April 30, 2015.
- Jain AK. Tuberculosis of the spine: a fresh look at an old disease. J Bone Joint Surg Br 2010; 92:905–913.
- Patil SS, Mohite S, Varma R, Bhojraj SY, Nene AM. Non-surgical management of cord compression in tuberculosis: a series of surprises. Asian Spine J 2014; 8:315–321.
- McLain RF, Isada C. Spinal tuberculosis deserves a place on the radar screen. Cleve Clin J Med 2004; 71:537–549.
- Vermund SH, Yamamoto N. Co-infection with human immunodeficiency virus and tuberculosis in Asia. Tuberculosis (Edinb) 2007; 87(suppl 1):S18–S25.
- Candy S, Chang G, Andronikou S. Acute myelopathy or cauda equina syndrome in HIV-positive adults in a tuberculosis endemic setting: MRI, clinical, and pathologic findings. AJNR Am J Neuroradiol 2014; 35:1634–1641.
- USAID. The twin epidemics: HIV and TB co-infection. www.usaid.gov/news-information/fact-sheets/twin-epidemics-hiv-and-tb-co-infection. Accessed April 30, 2015.
- Jain AK. Tuberculosis of the spine: a fresh look at an old disease. J Bone Joint Surg Br 2010; 92:905–913.
- Patil SS, Mohite S, Varma R, Bhojraj SY, Nene AM. Non-surgical management of cord compression in tuberculosis: a series of surprises. Asian Spine J 2014; 8:315–321.
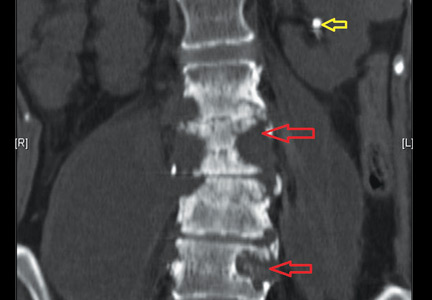
Morphea-like plaques, induration of the extremities, and eosinophilia
A 52-year-old woman who had been diagnosed with morphea 1 year before was referred to our department for evaluation of a 6-week history of myalgia and limited flexion and extension of the wrists and elbows. She had a history of hypertension, which was controlled with enalapril 10 mg daily. She had no fever, weight loss, dyspnea, dysphagia, Raynaud phenomenon, or other symptoms, and she had not traveled recently.
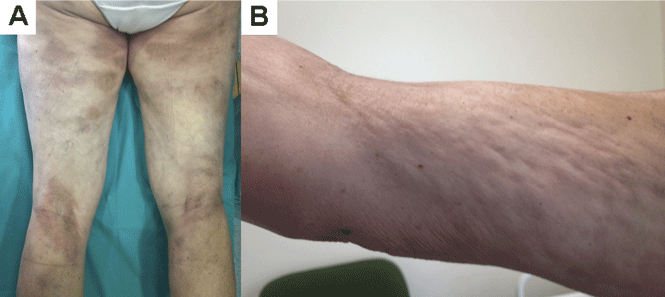
On physical examination, she had multiple dark-brown plaques on the legs (Figure 1), induration of both forearms, puckering of the skin (peau d’orange) of both arms (Figure 1), and marked limitation of motion of the wrists (Figure 2). Sclerodactyly was absent, and the rest of the examination was unremarkable. Laboratory tests showed an elevated serum C-reactive protein (1.97 mg/dL, reference range 0–0.5) and a white blood cell count of 9.59 × 109/L, with 25.2% eosinophils (reference range 0%–7%). Antinuclear antibodies were negative.
Because the patient refused full-thickness biopsy of the skin (ie, down to superficial muscle), magnetic resonance imaging (MRI) of the right arm was ordered. T1-weighted fat-saturated imaging with contrast showed marked contrast enhancement along all muscle fascia. This finding, along with the patient’s presentation and the results of laboratory testing, indicated a diagnosis of eosinophilic fasciitis.
Prednisone was prescribed in an initial dose of 1 mg/kg/day and was subsequently tapered, and this brought a partial response. Methotrexate (15 mg weekly) was added 1 month later. Follow-up visits at 3, 6, and 12 months were scheduled. At 12 months, levels of markers of inflammation were normal, the eosinophilia had completely resolved (< 0.125 × 109/L, < 2%), joint mobility had improved (Figure 2), and the morphea-like lesions had lightened somewhat. MRI repeated 6 months after the start of methotrexate showed complete resolution of the fascia infiltration.
EOSINOPHILIC FASCIITIS
Eosinophilic fasciitis, or Shulman syndrome, is a rare localized fibrosing disorder of the fascia characterized in its early phase by symmetrical limb or trunk erythema and swelling, and later by progressive thickening and induration of the dermis and subcutaneous fascia.1
The cause is unknown, but triggers have been suggested such as vigorous exercise or trauma, Borrelia infection, and drugs (statins, phenytoin, l-tryptophan). It has also been associated with hematologic disease. In our patient, none of these was present.2,3
There are no international diagnostic criteria for eosinophilic fasciitis. Rather, the diagnosis is suspected in a patient with eosinophilia and characteristic cutaneous features, such as pitting edema, induration, hyperpigmentation, and a peau d’orange aspect caused by inflammation and fibrosis of the fascia.1,2 These features have been reported in up to 90% of patients at the time of diagnosis.1–3 Myalgia, muscle weakness, and articular involvement including inflammatory arthralgia and joint contracture, as in our patient, are also common.1,2,4,5 Raynaud phenomenon in these patients is typically absent. Furthermore, pulmonary, cardiac, and renal involvement has also been reported, but this is very infrequent, and if it is present, other diseases such as Churg-Strauss vasculitis and hypereosinophilic syndrome should be excluded.1,2,4
The presence of thickened fascia and inflammatory infiltrates composed of lymphocytes or eosinophils (or both) on histologic examination of the skin-to-muscle biopsy remains the gold standard for diagnosis.2,4 However, when biopsy is nondiagnostic or is not possible, imaging with MRI or ultrasonography has been helpful.6,7 Muscle MRI is considered the best morphologic procedure for diagnosis, as it can specifically indicate the optimal location for biopsy in atypical cases (eg, fasciitis without skin changes and when eosinophilia is absent); it is also used to monitor the response to therapy.7,8 Although nonspecific, muscle MRI typically shows a markedly increased signal intensity within the fascia. And gadolinium administration shows marked fascia enhancement in the acute phase of the disease.1,4,6–8
MORPHEA PREDICTS POOR OUTCOME
Morphea has been reported to be present in 19% to 41% of patients with eosinophilic fasciitis due to dermis infiltration.2,4,5 Although rare, morphea plaques may also be present before the onset of eosinophilic fasciitis.3,4 Morphea has been described as predictive of poor outcome and residual fibrosis, and it has been associated with eosinophilic fasciitis resistant to usual therapies.3,5
Treatment of eosinophilic fasciitis is largely empirical because evidence from controlled trials is lacking. Corticosteroids remain the standard therapy. However, in patients with morphea-like lesions, treatment with an immunosuppressive drug such as methotrexate has been reported to be useful.1,4
- Lebeaux D, Sène D. Eosinophilic fasciitis (Shulman disease). Best Pract Res Clin Rheumatol 2012; 26:449–458.
- Lakhanpal S, Ginsburg WW, Michet CJ, Doyle JA, Moore SB. Eosinophilic fasciitis: clinical spectrum and therapeutic response in 52 cases. Semin Arthritis Rheum 1988; 17:221–231.
- Moulin C, Cavailhes A, Balme B, Skowron F. Morphoea-like plaques revealing an eosinophilic (Shulman) fasciitis. Clin Exp Dermatol 2009; 34:e851–e853.
- Lebeaux D, Francès C, Barete S, et al. Eosinophilic fasciitis (Shulman disease): new insights into the therapeutic management from a series of 34 patients. Rheumatology (Oxford) 2012; 51:557–561.
- Endo Y, Tamura A, Matsushima Y, et al. Eosinophilic fasciitis: report of two cases and a systematic review of the literature dealing with clinical variables that predict outcome. Clin Rheumatol 2007; 26:1445–1451.
- Dybowski F, Neuen-Jacob E, Braun J. Eosinophilic fasciitis and myositis: use of imaging modalities for diagnosis and monitoring. Ann Rheum Dis 2008; 67:572–574.
- Moulton SJ, Kransdorf MJ, Ginsburg WW, Abril A, Persellin S. Eosinophilic fasciitis: spectrum of MRI findings. AJR Am J Roentgenol 2005; 184:975–978.
- Ronneberger M, Janka R, Schett G, Manger B. Can MRI substitute for biopsy in eosinophilic fasciitis? Ann Rheum Dis 2009; 68:1651–1652.
A 52-year-old woman who had been diagnosed with morphea 1 year before was referred to our department for evaluation of a 6-week history of myalgia and limited flexion and extension of the wrists and elbows. She had a history of hypertension, which was controlled with enalapril 10 mg daily. She had no fever, weight loss, dyspnea, dysphagia, Raynaud phenomenon, or other symptoms, and she had not traveled recently.
On physical examination, she had multiple dark-brown plaques on the legs (Figure 1), induration of both forearms, puckering of the skin (peau d’orange) of both arms (Figure 1), and marked limitation of motion of the wrists (Figure 2). Sclerodactyly was absent, and the rest of the examination was unremarkable. Laboratory tests showed an elevated serum C-reactive protein (1.97 mg/dL, reference range 0–0.5) and a white blood cell count of 9.59 × 109/L, with 25.2% eosinophils (reference range 0%–7%). Antinuclear antibodies were negative.
Because the patient refused full-thickness biopsy of the skin (ie, down to superficial muscle), magnetic resonance imaging (MRI) of the right arm was ordered. T1-weighted fat-saturated imaging with contrast showed marked contrast enhancement along all muscle fascia. This finding, along with the patient’s presentation and the results of laboratory testing, indicated a diagnosis of eosinophilic fasciitis.
Prednisone was prescribed in an initial dose of 1 mg/kg/day and was subsequently tapered, and this brought a partial response. Methotrexate (15 mg weekly) was added 1 month later. Follow-up visits at 3, 6, and 12 months were scheduled. At 12 months, levels of markers of inflammation were normal, the eosinophilia had completely resolved (< 0.125 × 109/L, < 2%), joint mobility had improved (Figure 2), and the morphea-like lesions had lightened somewhat. MRI repeated 6 months after the start of methotrexate showed complete resolution of the fascia infiltration.
EOSINOPHILIC FASCIITIS
Eosinophilic fasciitis, or Shulman syndrome, is a rare localized fibrosing disorder of the fascia characterized in its early phase by symmetrical limb or trunk erythema and swelling, and later by progressive thickening and induration of the dermis and subcutaneous fascia.1
The cause is unknown, but triggers have been suggested such as vigorous exercise or trauma, Borrelia infection, and drugs (statins, phenytoin, l-tryptophan). It has also been associated with hematologic disease. In our patient, none of these was present.2,3
There are no international diagnostic criteria for eosinophilic fasciitis. Rather, the diagnosis is suspected in a patient with eosinophilia and characteristic cutaneous features, such as pitting edema, induration, hyperpigmentation, and a peau d’orange aspect caused by inflammation and fibrosis of the fascia.1,2 These features have been reported in up to 90% of patients at the time of diagnosis.1–3 Myalgia, muscle weakness, and articular involvement including inflammatory arthralgia and joint contracture, as in our patient, are also common.1,2,4,5 Raynaud phenomenon in these patients is typically absent. Furthermore, pulmonary, cardiac, and renal involvement has also been reported, but this is very infrequent, and if it is present, other diseases such as Churg-Strauss vasculitis and hypereosinophilic syndrome should be excluded.1,2,4
The presence of thickened fascia and inflammatory infiltrates composed of lymphocytes or eosinophils (or both) on histologic examination of the skin-to-muscle biopsy remains the gold standard for diagnosis.2,4 However, when biopsy is nondiagnostic or is not possible, imaging with MRI or ultrasonography has been helpful.6,7 Muscle MRI is considered the best morphologic procedure for diagnosis, as it can specifically indicate the optimal location for biopsy in atypical cases (eg, fasciitis without skin changes and when eosinophilia is absent); it is also used to monitor the response to therapy.7,8 Although nonspecific, muscle MRI typically shows a markedly increased signal intensity within the fascia. And gadolinium administration shows marked fascia enhancement in the acute phase of the disease.1,4,6–8
MORPHEA PREDICTS POOR OUTCOME
Morphea has been reported to be present in 19% to 41% of patients with eosinophilic fasciitis due to dermis infiltration.2,4,5 Although rare, morphea plaques may also be present before the onset of eosinophilic fasciitis.3,4 Morphea has been described as predictive of poor outcome and residual fibrosis, and it has been associated with eosinophilic fasciitis resistant to usual therapies.3,5
Treatment of eosinophilic fasciitis is largely empirical because evidence from controlled trials is lacking. Corticosteroids remain the standard therapy. However, in patients with morphea-like lesions, treatment with an immunosuppressive drug such as methotrexate has been reported to be useful.1,4
A 52-year-old woman who had been diagnosed with morphea 1 year before was referred to our department for evaluation of a 6-week history of myalgia and limited flexion and extension of the wrists and elbows. She had a history of hypertension, which was controlled with enalapril 10 mg daily. She had no fever, weight loss, dyspnea, dysphagia, Raynaud phenomenon, or other symptoms, and she had not traveled recently.
On physical examination, she had multiple dark-brown plaques on the legs (Figure 1), induration of both forearms, puckering of the skin (peau d’orange) of both arms (Figure 1), and marked limitation of motion of the wrists (Figure 2). Sclerodactyly was absent, and the rest of the examination was unremarkable. Laboratory tests showed an elevated serum C-reactive protein (1.97 mg/dL, reference range 0–0.5) and a white blood cell count of 9.59 × 109/L, with 25.2% eosinophils (reference range 0%–7%). Antinuclear antibodies were negative.
Because the patient refused full-thickness biopsy of the skin (ie, down to superficial muscle), magnetic resonance imaging (MRI) of the right arm was ordered. T1-weighted fat-saturated imaging with contrast showed marked contrast enhancement along all muscle fascia. This finding, along with the patient’s presentation and the results of laboratory testing, indicated a diagnosis of eosinophilic fasciitis.
Prednisone was prescribed in an initial dose of 1 mg/kg/day and was subsequently tapered, and this brought a partial response. Methotrexate (15 mg weekly) was added 1 month later. Follow-up visits at 3, 6, and 12 months were scheduled. At 12 months, levels of markers of inflammation were normal, the eosinophilia had completely resolved (< 0.125 × 109/L, < 2%), joint mobility had improved (Figure 2), and the morphea-like lesions had lightened somewhat. MRI repeated 6 months after the start of methotrexate showed complete resolution of the fascia infiltration.
EOSINOPHILIC FASCIITIS
Eosinophilic fasciitis, or Shulman syndrome, is a rare localized fibrosing disorder of the fascia characterized in its early phase by symmetrical limb or trunk erythema and swelling, and later by progressive thickening and induration of the dermis and subcutaneous fascia.1
The cause is unknown, but triggers have been suggested such as vigorous exercise or trauma, Borrelia infection, and drugs (statins, phenytoin, l-tryptophan). It has also been associated with hematologic disease. In our patient, none of these was present.2,3
There are no international diagnostic criteria for eosinophilic fasciitis. Rather, the diagnosis is suspected in a patient with eosinophilia and characteristic cutaneous features, such as pitting edema, induration, hyperpigmentation, and a peau d’orange aspect caused by inflammation and fibrosis of the fascia.1,2 These features have been reported in up to 90% of patients at the time of diagnosis.1–3 Myalgia, muscle weakness, and articular involvement including inflammatory arthralgia and joint contracture, as in our patient, are also common.1,2,4,5 Raynaud phenomenon in these patients is typically absent. Furthermore, pulmonary, cardiac, and renal involvement has also been reported, but this is very infrequent, and if it is present, other diseases such as Churg-Strauss vasculitis and hypereosinophilic syndrome should be excluded.1,2,4
The presence of thickened fascia and inflammatory infiltrates composed of lymphocytes or eosinophils (or both) on histologic examination of the skin-to-muscle biopsy remains the gold standard for diagnosis.2,4 However, when biopsy is nondiagnostic or is not possible, imaging with MRI or ultrasonography has been helpful.6,7 Muscle MRI is considered the best morphologic procedure for diagnosis, as it can specifically indicate the optimal location for biopsy in atypical cases (eg, fasciitis without skin changes and when eosinophilia is absent); it is also used to monitor the response to therapy.7,8 Although nonspecific, muscle MRI typically shows a markedly increased signal intensity within the fascia. And gadolinium administration shows marked fascia enhancement in the acute phase of the disease.1,4,6–8
MORPHEA PREDICTS POOR OUTCOME
Morphea has been reported to be present in 19% to 41% of patients with eosinophilic fasciitis due to dermis infiltration.2,4,5 Although rare, morphea plaques may also be present before the onset of eosinophilic fasciitis.3,4 Morphea has been described as predictive of poor outcome and residual fibrosis, and it has been associated with eosinophilic fasciitis resistant to usual therapies.3,5
Treatment of eosinophilic fasciitis is largely empirical because evidence from controlled trials is lacking. Corticosteroids remain the standard therapy. However, in patients with morphea-like lesions, treatment with an immunosuppressive drug such as methotrexate has been reported to be useful.1,4
- Lebeaux D, Sène D. Eosinophilic fasciitis (Shulman disease). Best Pract Res Clin Rheumatol 2012; 26:449–458.
- Lakhanpal S, Ginsburg WW, Michet CJ, Doyle JA, Moore SB. Eosinophilic fasciitis: clinical spectrum and therapeutic response in 52 cases. Semin Arthritis Rheum 1988; 17:221–231.
- Moulin C, Cavailhes A, Balme B, Skowron F. Morphoea-like plaques revealing an eosinophilic (Shulman) fasciitis. Clin Exp Dermatol 2009; 34:e851–e853.
- Lebeaux D, Francès C, Barete S, et al. Eosinophilic fasciitis (Shulman disease): new insights into the therapeutic management from a series of 34 patients. Rheumatology (Oxford) 2012; 51:557–561.
- Endo Y, Tamura A, Matsushima Y, et al. Eosinophilic fasciitis: report of two cases and a systematic review of the literature dealing with clinical variables that predict outcome. Clin Rheumatol 2007; 26:1445–1451.
- Dybowski F, Neuen-Jacob E, Braun J. Eosinophilic fasciitis and myositis: use of imaging modalities for diagnosis and monitoring. Ann Rheum Dis 2008; 67:572–574.
- Moulton SJ, Kransdorf MJ, Ginsburg WW, Abril A, Persellin S. Eosinophilic fasciitis: spectrum of MRI findings. AJR Am J Roentgenol 2005; 184:975–978.
- Ronneberger M, Janka R, Schett G, Manger B. Can MRI substitute for biopsy in eosinophilic fasciitis? Ann Rheum Dis 2009; 68:1651–1652.
- Lebeaux D, Sène D. Eosinophilic fasciitis (Shulman disease). Best Pract Res Clin Rheumatol 2012; 26:449–458.
- Lakhanpal S, Ginsburg WW, Michet CJ, Doyle JA, Moore SB. Eosinophilic fasciitis: clinical spectrum and therapeutic response in 52 cases. Semin Arthritis Rheum 1988; 17:221–231.
- Moulin C, Cavailhes A, Balme B, Skowron F. Morphoea-like plaques revealing an eosinophilic (Shulman) fasciitis. Clin Exp Dermatol 2009; 34:e851–e853.
- Lebeaux D, Francès C, Barete S, et al. Eosinophilic fasciitis (Shulman disease): new insights into the therapeutic management from a series of 34 patients. Rheumatology (Oxford) 2012; 51:557–561.
- Endo Y, Tamura A, Matsushima Y, et al. Eosinophilic fasciitis: report of two cases and a systematic review of the literature dealing with clinical variables that predict outcome. Clin Rheumatol 2007; 26:1445–1451.
- Dybowski F, Neuen-Jacob E, Braun J. Eosinophilic fasciitis and myositis: use of imaging modalities for diagnosis and monitoring. Ann Rheum Dis 2008; 67:572–574.
- Moulton SJ, Kransdorf MJ, Ginsburg WW, Abril A, Persellin S. Eosinophilic fasciitis: spectrum of MRI findings. AJR Am J Roentgenol 2005; 184:975–978.
- Ronneberger M, Janka R, Schett G, Manger B. Can MRI substitute for biopsy in eosinophilic fasciitis? Ann Rheum Dis 2009; 68:1651–1652.

Light-headedness and bradycardia in a 72-year-old woman
A 72-year-old woman came to the emergency department because of persistent light-headedness. Her medical history included end-stage renal disease, hypertension, peripheral vascular disease, and diabetes mellitus. She said she had experienced similar symptoms before, but they had gone away.
She reported no visual changes, no loss of consciousness, and no history of seizures, syncope, chest pain, palpitations, or diaphoresis. She was not taking a beta-blocker, calcium channel blocker, or digoxin.
Her blood pressure was 75/44 mm Hg, heart rate 44 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 97% while receiving oxygen at 3 L per minute. An electrolyte panel was normal except for an elevated creatinine level secondary to end-stage renal disease.
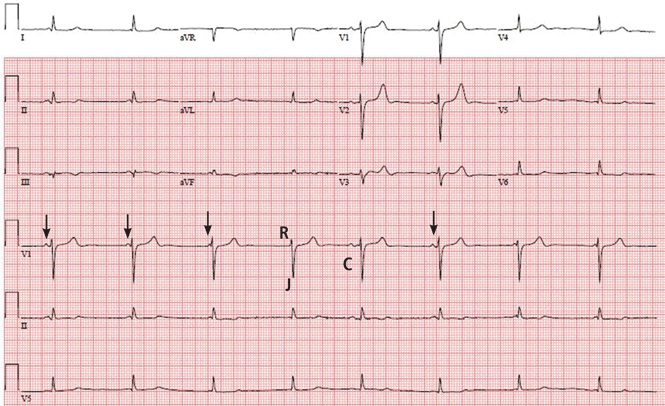
In view of her symptoms and bradycardia, she was admitted to the hospital. The initial electrocardiogram (Figure 1) showed an atrial rate of approximately 46 beats per minute, a ventricular rate of approximately 48 beats per minute, and a P wave in the refractory period caused by a junction impulse.
These findings pointed to atrioventricular (AV) dissociation, a term commonly applied to arrhythmias in which the atria and ventricles are rhythmically detached.
ATRIOVENTRICULAR DISSOCIATION
AV dissociation is often used interchangeably with complete heart block, but this is incorrect1; though complete heart block is a form of AV dissociation, not all AV dissociation is complete heart block. In complete heart block, there is no rhythmic relationship between the atria and ventricles, as they beat independently with no influence on each other. On the other hand, when a “blockade” is created by the physiologic refractory period of the atria (sinus node or atrial ectopic focus) and ventricles, interference dissociation can result.2 In this condition, when the ventricles are not in a refractory period, an atrial impulse may be conducted through the AV node, resulting in an atrial-driven beat. Simply put, a P wave has the potential to be conducted in AV dissociation if there is an opportunity, but in complete heart block it does not.1
AV dissociation is a secondary manifestation of a primary disorder or rhythm disturbance. In general, any rhythm that competes against an atrial impulse and inhibits its conduction through the AV node can cause AV dissociation. Common examples include junctional escape or accelerated rhythms, premature ventricular beats or ventricular tachycardia, and accelerated idioventricular rhythms. It also can be caused by drugs (eg, digoxin) or an increase in vagal tone.2
In normal myocardium, the sinus node has a higher impulse rate than the subordinate pacemaker (AV node or ventricular pacemaker). Generally, the atrial rate is higher than the ventricular rate in complete heart block, whereas in AV dissociation the ventricular rate is higher than the atrial rate.3
Thus, AV dissociation can result from one of the following mechanisms4:
- Slowing of the dominant pacemaker (sinus or atrial pacemaker)
- Acceleration or overtaking of the sinus node (or atrial focus) by a subordinate pacemaker (eg, a junctional or ventricular pacemaker)
- A block within the AV node that prevents an impulse generated by the dominant pacemaker (sinus or atrial focus) from crossing the AV node
- A combination of these mechanisms.
Another form of AV dissociation is isorhythmic dissociation. In this subtype, atrial and ventricular impulses occur at the same rate. This type of dissociation is most commonly confused with third-degree (or complete) heart block. It may be difficult to distinguish one from the other, but at higher sinus (or atrial) rates the difference becomes obvious—properly timed P waves may be conducted through the AV node in isorhythmic dissociation.1
The prevalence of AV dissociation is thought to be 0.48% to 0.68%,3 but it could be more common since it is underdiagnosed.5
Treatment should be directed at the primary disorder.4 The need for a pacemaker depends on the condition causing the AV dissociation. In conditions that slow the sinus node, such as increased vagal tone, patients may benefit from medications that decrease parasympathetic activity or increase adrenergic activity in the AV node (eg, isoproterenol, atropine).6
OUR PATIENT
Our patient’s electrocardiogram showed interference dissociation from competing junctional rhythms. Possibly, she had sinus node disease, explaining why the sinus node was not the dominant pacemaker. She had symptomatic hypotension, requiring dopamine for pressure support. She was started on intravenous isoproterenol, which eventually restored sinus rhythm.
During the same hospitalization, she was diagnosed with osteomyelitis of the left foot, without bacteremia. She was treated for her infection and later received a pacemaker. She was discharged to a rehabilitation facility.
TAKE-AWAY POINTS
- When an occasional impulse is conducted through the AV node, AV dissociation is most likely interference dissociation.
- AV dissociations are often confused with complete heart block.
- In AV dissociation, the ventricular rate is higher than the atrial rate.
- Complete heart block is a form of AV dissociation, but not all AV dissociation is complete heart block.
- AV dissociation can be caused by three main mechanisms or by a combination of them.
- AV dissociation is secondary to a primary rhythm disorder.
- Adrenergic drugs may help to correct the AV dissociation, but not always completely.
- Goldberger AL. Atrioventricular conduction abnormalities: delays, blocks, and dissociation syndromes. In: Goldberger AL, Goldberger ZD, Shvilkin A, eds. Clinical Electrocardiography: A Simplified Approach. 8th ed. Philadelphia, PA: Elsevier/Saunders; 2012:159–169.
- Wang K, Benditt DG. AV dissociation, an inevitable response. Ann Noninvasive Electrocardiol 2011; 16:227–231.
- Harrigan RA, Perron AD, Brady WJ. Atrioventricular dissociation. Am J Emerg Med 2001; 19:218–222.
- Jeffrey O, Zipes DP. Specific arrhythmias: diagnosis and treatment. In: Bonow RO, Mann DL, Zipes DP, Libby P, eds. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2011.
- Singh GD, Wong GB, Southard JA, Amsterdam EA. Food for thought: atrioventricular dissociation. Am J Med 2013; 126:1050–1053.
- Vavetsi S, Nikolaou N, Tsarouhas K, et al. Consecutive administration of atropine and isoproterenol for the evaluation of asymptomatic sinus bradycardia. Europace 2008; 10:1176–1181.
A 72-year-old woman came to the emergency department because of persistent light-headedness. Her medical history included end-stage renal disease, hypertension, peripheral vascular disease, and diabetes mellitus. She said she had experienced similar symptoms before, but they had gone away.
She reported no visual changes, no loss of consciousness, and no history of seizures, syncope, chest pain, palpitations, or diaphoresis. She was not taking a beta-blocker, calcium channel blocker, or digoxin.
Her blood pressure was 75/44 mm Hg, heart rate 44 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 97% while receiving oxygen at 3 L per minute. An electrolyte panel was normal except for an elevated creatinine level secondary to end-stage renal disease.
In view of her symptoms and bradycardia, she was admitted to the hospital. The initial electrocardiogram (Figure 1) showed an atrial rate of approximately 46 beats per minute, a ventricular rate of approximately 48 beats per minute, and a P wave in the refractory period caused by a junction impulse.
These findings pointed to atrioventricular (AV) dissociation, a term commonly applied to arrhythmias in which the atria and ventricles are rhythmically detached.
ATRIOVENTRICULAR DISSOCIATION
AV dissociation is often used interchangeably with complete heart block, but this is incorrect1; though complete heart block is a form of AV dissociation, not all AV dissociation is complete heart block. In complete heart block, there is no rhythmic relationship between the atria and ventricles, as they beat independently with no influence on each other. On the other hand, when a “blockade” is created by the physiologic refractory period of the atria (sinus node or atrial ectopic focus) and ventricles, interference dissociation can result.2 In this condition, when the ventricles are not in a refractory period, an atrial impulse may be conducted through the AV node, resulting in an atrial-driven beat. Simply put, a P wave has the potential to be conducted in AV dissociation if there is an opportunity, but in complete heart block it does not.1
AV dissociation is a secondary manifestation of a primary disorder or rhythm disturbance. In general, any rhythm that competes against an atrial impulse and inhibits its conduction through the AV node can cause AV dissociation. Common examples include junctional escape or accelerated rhythms, premature ventricular beats or ventricular tachycardia, and accelerated idioventricular rhythms. It also can be caused by drugs (eg, digoxin) or an increase in vagal tone.2
In normal myocardium, the sinus node has a higher impulse rate than the subordinate pacemaker (AV node or ventricular pacemaker). Generally, the atrial rate is higher than the ventricular rate in complete heart block, whereas in AV dissociation the ventricular rate is higher than the atrial rate.3
Thus, AV dissociation can result from one of the following mechanisms4:
- Slowing of the dominant pacemaker (sinus or atrial pacemaker)
- Acceleration or overtaking of the sinus node (or atrial focus) by a subordinate pacemaker (eg, a junctional or ventricular pacemaker)
- A block within the AV node that prevents an impulse generated by the dominant pacemaker (sinus or atrial focus) from crossing the AV node
- A combination of these mechanisms.
Another form of AV dissociation is isorhythmic dissociation. In this subtype, atrial and ventricular impulses occur at the same rate. This type of dissociation is most commonly confused with third-degree (or complete) heart block. It may be difficult to distinguish one from the other, but at higher sinus (or atrial) rates the difference becomes obvious—properly timed P waves may be conducted through the AV node in isorhythmic dissociation.1
The prevalence of AV dissociation is thought to be 0.48% to 0.68%,3 but it could be more common since it is underdiagnosed.5
Treatment should be directed at the primary disorder.4 The need for a pacemaker depends on the condition causing the AV dissociation. In conditions that slow the sinus node, such as increased vagal tone, patients may benefit from medications that decrease parasympathetic activity or increase adrenergic activity in the AV node (eg, isoproterenol, atropine).6
OUR PATIENT
Our patient’s electrocardiogram showed interference dissociation from competing junctional rhythms. Possibly, she had sinus node disease, explaining why the sinus node was not the dominant pacemaker. She had symptomatic hypotension, requiring dopamine for pressure support. She was started on intravenous isoproterenol, which eventually restored sinus rhythm.
During the same hospitalization, she was diagnosed with osteomyelitis of the left foot, without bacteremia. She was treated for her infection and later received a pacemaker. She was discharged to a rehabilitation facility.
TAKE-AWAY POINTS
- When an occasional impulse is conducted through the AV node, AV dissociation is most likely interference dissociation.
- AV dissociations are often confused with complete heart block.
- In AV dissociation, the ventricular rate is higher than the atrial rate.
- Complete heart block is a form of AV dissociation, but not all AV dissociation is complete heart block.
- AV dissociation can be caused by three main mechanisms or by a combination of them.
- AV dissociation is secondary to a primary rhythm disorder.
- Adrenergic drugs may help to correct the AV dissociation, but not always completely.
A 72-year-old woman came to the emergency department because of persistent light-headedness. Her medical history included end-stage renal disease, hypertension, peripheral vascular disease, and diabetes mellitus. She said she had experienced similar symptoms before, but they had gone away.
She reported no visual changes, no loss of consciousness, and no history of seizures, syncope, chest pain, palpitations, or diaphoresis. She was not taking a beta-blocker, calcium channel blocker, or digoxin.
Her blood pressure was 75/44 mm Hg, heart rate 44 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 97% while receiving oxygen at 3 L per minute. An electrolyte panel was normal except for an elevated creatinine level secondary to end-stage renal disease.
In view of her symptoms and bradycardia, she was admitted to the hospital. The initial electrocardiogram (Figure 1) showed an atrial rate of approximately 46 beats per minute, a ventricular rate of approximately 48 beats per minute, and a P wave in the refractory period caused by a junction impulse.
These findings pointed to atrioventricular (AV) dissociation, a term commonly applied to arrhythmias in which the atria and ventricles are rhythmically detached.
ATRIOVENTRICULAR DISSOCIATION
AV dissociation is often used interchangeably with complete heart block, but this is incorrect1; though complete heart block is a form of AV dissociation, not all AV dissociation is complete heart block. In complete heart block, there is no rhythmic relationship between the atria and ventricles, as they beat independently with no influence on each other. On the other hand, when a “blockade” is created by the physiologic refractory period of the atria (sinus node or atrial ectopic focus) and ventricles, interference dissociation can result.2 In this condition, when the ventricles are not in a refractory period, an atrial impulse may be conducted through the AV node, resulting in an atrial-driven beat. Simply put, a P wave has the potential to be conducted in AV dissociation if there is an opportunity, but in complete heart block it does not.1
AV dissociation is a secondary manifestation of a primary disorder or rhythm disturbance. In general, any rhythm that competes against an atrial impulse and inhibits its conduction through the AV node can cause AV dissociation. Common examples include junctional escape or accelerated rhythms, premature ventricular beats or ventricular tachycardia, and accelerated idioventricular rhythms. It also can be caused by drugs (eg, digoxin) or an increase in vagal tone.2
In normal myocardium, the sinus node has a higher impulse rate than the subordinate pacemaker (AV node or ventricular pacemaker). Generally, the atrial rate is higher than the ventricular rate in complete heart block, whereas in AV dissociation the ventricular rate is higher than the atrial rate.3
Thus, AV dissociation can result from one of the following mechanisms4:
- Slowing of the dominant pacemaker (sinus or atrial pacemaker)
- Acceleration or overtaking of the sinus node (or atrial focus) by a subordinate pacemaker (eg, a junctional or ventricular pacemaker)
- A block within the AV node that prevents an impulse generated by the dominant pacemaker (sinus or atrial focus) from crossing the AV node
- A combination of these mechanisms.
Another form of AV dissociation is isorhythmic dissociation. In this subtype, atrial and ventricular impulses occur at the same rate. This type of dissociation is most commonly confused with third-degree (or complete) heart block. It may be difficult to distinguish one from the other, but at higher sinus (or atrial) rates the difference becomes obvious—properly timed P waves may be conducted through the AV node in isorhythmic dissociation.1
The prevalence of AV dissociation is thought to be 0.48% to 0.68%,3 but it could be more common since it is underdiagnosed.5
Treatment should be directed at the primary disorder.4 The need for a pacemaker depends on the condition causing the AV dissociation. In conditions that slow the sinus node, such as increased vagal tone, patients may benefit from medications that decrease parasympathetic activity or increase adrenergic activity in the AV node (eg, isoproterenol, atropine).6
OUR PATIENT
Our patient’s electrocardiogram showed interference dissociation from competing junctional rhythms. Possibly, she had sinus node disease, explaining why the sinus node was not the dominant pacemaker. She had symptomatic hypotension, requiring dopamine for pressure support. She was started on intravenous isoproterenol, which eventually restored sinus rhythm.
During the same hospitalization, she was diagnosed with osteomyelitis of the left foot, without bacteremia. She was treated for her infection and later received a pacemaker. She was discharged to a rehabilitation facility.
TAKE-AWAY POINTS
- When an occasional impulse is conducted through the AV node, AV dissociation is most likely interference dissociation.
- AV dissociations are often confused with complete heart block.
- In AV dissociation, the ventricular rate is higher than the atrial rate.
- Complete heart block is a form of AV dissociation, but not all AV dissociation is complete heart block.
- AV dissociation can be caused by three main mechanisms or by a combination of them.
- AV dissociation is secondary to a primary rhythm disorder.
- Adrenergic drugs may help to correct the AV dissociation, but not always completely.
- Goldberger AL. Atrioventricular conduction abnormalities: delays, blocks, and dissociation syndromes. In: Goldberger AL, Goldberger ZD, Shvilkin A, eds. Clinical Electrocardiography: A Simplified Approach. 8th ed. Philadelphia, PA: Elsevier/Saunders; 2012:159–169.
- Wang K, Benditt DG. AV dissociation, an inevitable response. Ann Noninvasive Electrocardiol 2011; 16:227–231.
- Harrigan RA, Perron AD, Brady WJ. Atrioventricular dissociation. Am J Emerg Med 2001; 19:218–222.
- Jeffrey O, Zipes DP. Specific arrhythmias: diagnosis and treatment. In: Bonow RO, Mann DL, Zipes DP, Libby P, eds. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2011.
- Singh GD, Wong GB, Southard JA, Amsterdam EA. Food for thought: atrioventricular dissociation. Am J Med 2013; 126:1050–1053.
- Vavetsi S, Nikolaou N, Tsarouhas K, et al. Consecutive administration of atropine and isoproterenol for the evaluation of asymptomatic sinus bradycardia. Europace 2008; 10:1176–1181.
- Goldberger AL. Atrioventricular conduction abnormalities: delays, blocks, and dissociation syndromes. In: Goldberger AL, Goldberger ZD, Shvilkin A, eds. Clinical Electrocardiography: A Simplified Approach. 8th ed. Philadelphia, PA: Elsevier/Saunders; 2012:159–169.
- Wang K, Benditt DG. AV dissociation, an inevitable response. Ann Noninvasive Electrocardiol 2011; 16:227–231.
- Harrigan RA, Perron AD, Brady WJ. Atrioventricular dissociation. Am J Emerg Med 2001; 19:218–222.
- Jeffrey O, Zipes DP. Specific arrhythmias: diagnosis and treatment. In: Bonow RO, Mann DL, Zipes DP, Libby P, eds. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2011.
- Singh GD, Wong GB, Southard JA, Amsterdam EA. Food for thought: atrioventricular dissociation. Am J Med 2013; 126:1050–1053.
- Vavetsi S, Nikolaou N, Tsarouhas K, et al. Consecutive administration of atropine and isoproterenol for the evaluation of asymptomatic sinus bradycardia. Europace 2008; 10:1176–1181.