User login
A 60-year-old man with forehead swelling
A 60-year-old man presented to our emergency department with a 4-day history of frontal headaches he described as “stinging.” He had also had a large swollen area on his forehead for the past 8 weeks.
He denied fevers, chills, nausea, vomiting, blurry vision, tinnitus, and neck pain, as well as any recent sinus infection, intransanal cocaine use, rhinorrhea, or head trauma. A month ago, he had presented to our emergency department with forehead swelling but no headaches. At that time, the swelling was thought to be an allergic reaction to lisinopril or metformin, medications he takes for hypertension and type 2 diabetes. He had been discharged home with a prescription for a course of prednisone in tapering doses, but that had failed to resolve the swelling.
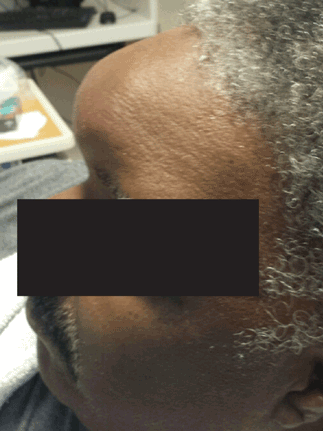
Physical examination revealed a well-circumscribed area of swelling, 3 by 4 cm, in the central forehead (Figure 1). The area was warm, erythematous, fluctuant, and tender to palpation. The nasal septum was intact and the nasal mucosa appeared pink and healthy. The remainder of the examination was unremarkable.
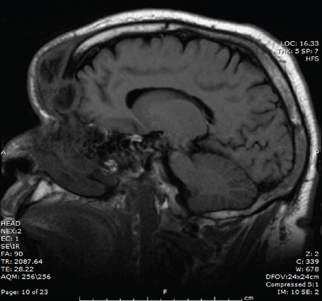
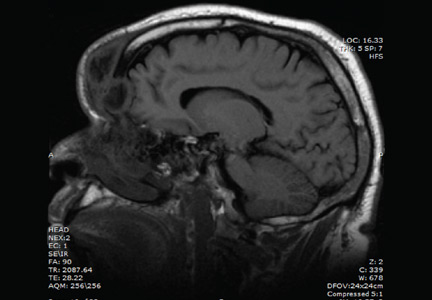
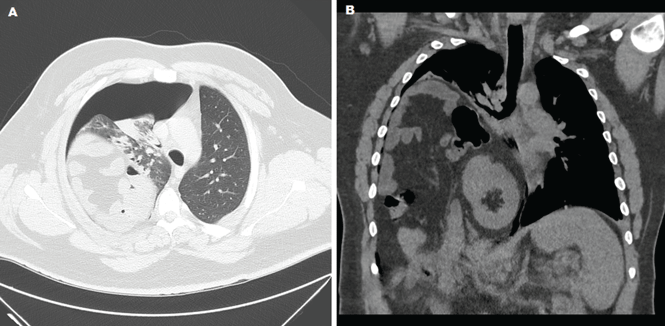
He was afebrile and hemodynamically stable. His peripheral white blood cell count was mildly elevated at 11.1 × 109. Computed tomography of the brain and sinuses revealed a fluid collection in the frontal scalp associated with erosion of the anterior frontal sinus with posterior extension and enhancement of the adjacent meninges. Magnetic resonance imaging (Figure 2) revealed similar findings. A diagnosis of Pott puffy tumor was made based on the imaging findings.
The name of this condition is misleading, as it is not a neoplasm but an infection. It requires urgent antibiotic therapy and surgical management because of the high risk of the infection spreading to the brain. Our patient was started on a broad-spectrum antibiotic regimen of intravenous vancomycin, ceftriaxone, and metronidazole pending tissue culture to identify the causative organism.
POTT PUFFY TUMOR: A BRIEF OVERVIEW
First described in 1760 by Sir Percivall Pott,1 the same English surgeon who first described tuberculosis of the spine, Pott puffy tumor is a well-demarcated area of swelling that occurs when a frontal sinus infection breaks through the anterior portion of the frontal sinus and forms an abscess between the frontal bone and periosteum with associated osteomyelitis.2 Though rare in adults (it is more common in children and adolescents),3 Pott puffy tumor is caused by conditions often encountered in internal medicine practice, such as bacterial sinusitis, head trauma, and intranasal cocaine use.
The infection can spread to the brain either directly by destruction of the posterior frontal sinus (as in our patient) or by way of the veins that drain the frontal sinus. Meningitis, epidural empyema, frontal lobe abscess, and cavernous sinus thrombosis2 have all been described. Intracranial complications are seen in nearly 100% of children and adolescents with Pott puffy tumor. The rate in adults is 30%,4,5 which is much lower but is nevertheless worrisome because patients can be initially misdiagnosed with scalp abscess,3 cellulitis, or epidermoid cyst,4 and then sent home from the emergency department or physician’s office. In a case series of 32 adult patients with Pott puffy tumor, nearly 45% were initially misdiagnosed, most often by an internist, dermatologist, ophthalmologist, or emergency room physician.4
The most common infective organisms are streptococci, staphylococci, and anaerobes,4 but Haemophilus, Aspergillus species, and invasive mucormycosis have also been described.
MANAGEMENT OPTIONS
Because of the risk of spread of the infection to the brain, rapid initiation of a broad-spectrum antibiotic is warranted in all patients with Pott puffy tumor pending results of tissue culture. Antibiotics may be necessary for at least 4 to 6 weeks to resolve osteomyelitis of the frontal bone and to decrease inflammation before surgery.6
Endoscopic sinus surgery is routinely done to drain the infected sinus and to remove or debride infected bone. Patients with intracranial extension of infection may require a combined endoscopic and neurosurgical approach.
OUTCOME
Our patient’s puffy tumor spontaneously ruptured externally on hospital day 3, and the purulent fluid was sent for culture that grew Streptococcus anginosus. His headaches improved almost immediately after this occurred. The antibiotic regimen was narrowed to ceftriaxone and metronidazole, and 1 week later he was discharged home with instructions to complete a 6-week course of antibiotics. Three weeks after he was discharged, he returned for outpatient endoscopic sinus surgery. At a follow-up visit 2 weeks after surgery, the forehead swelling had resolved, and he was well.
- Tattersall R, Tattersall R. Pott’s puffy tumor. Lancet 2002; 359:1060–1063.
- Forgie SE, Marrie TJ. Pott’s puffy tumor. Am J Med 2008; 121:1041–1042.
- Grewal HS, Dangaych NS, Esposito A. A tumor that is not a tumor but it sure can kill! Am J Case Rep 2012; 13:133–136.
- Akiyama K, Karaki M, Mori N. Evaluation of adult Pott’s puffy tumor: our five cases and 27 literature cases. Laryngoscope 2012; 122:2382–2388.
- Suwan PT, Mogal S, Chaudhary S. Pott’s puffy tumor: an uncommon clinical entity. Case Rep Pediatr 2012; 2012:386104.
- Lauria RA, Laffitte Fernandes F, Brito TP, Pereira PS, Chone CT. Extensive frontoparietal abscess: complication of frontal sinusitis (Pott’s puffy tumor). Case Rep Otolaryngol 2014; 2014:632464.
A 60-year-old man presented to our emergency department with a 4-day history of frontal headaches he described as “stinging.” He had also had a large swollen area on his forehead for the past 8 weeks.
He denied fevers, chills, nausea, vomiting, blurry vision, tinnitus, and neck pain, as well as any recent sinus infection, intransanal cocaine use, rhinorrhea, or head trauma. A month ago, he had presented to our emergency department with forehead swelling but no headaches. At that time, the swelling was thought to be an allergic reaction to lisinopril or metformin, medications he takes for hypertension and type 2 diabetes. He had been discharged home with a prescription for a course of prednisone in tapering doses, but that had failed to resolve the swelling.
Physical examination revealed a well-circumscribed area of swelling, 3 by 4 cm, in the central forehead (Figure 1). The area was warm, erythematous, fluctuant, and tender to palpation. The nasal septum was intact and the nasal mucosa appeared pink and healthy. The remainder of the examination was unremarkable.
He was afebrile and hemodynamically stable. His peripheral white blood cell count was mildly elevated at 11.1 × 109. Computed tomography of the brain and sinuses revealed a fluid collection in the frontal scalp associated with erosion of the anterior frontal sinus with posterior extension and enhancement of the adjacent meninges. Magnetic resonance imaging (Figure 2) revealed similar findings. A diagnosis of Pott puffy tumor was made based on the imaging findings.
The name of this condition is misleading, as it is not a neoplasm but an infection. It requires urgent antibiotic therapy and surgical management because of the high risk of the infection spreading to the brain. Our patient was started on a broad-spectrum antibiotic regimen of intravenous vancomycin, ceftriaxone, and metronidazole pending tissue culture to identify the causative organism.
POTT PUFFY TUMOR: A BRIEF OVERVIEW
First described in 1760 by Sir Percivall Pott,1 the same English surgeon who first described tuberculosis of the spine, Pott puffy tumor is a well-demarcated area of swelling that occurs when a frontal sinus infection breaks through the anterior portion of the frontal sinus and forms an abscess between the frontal bone and periosteum with associated osteomyelitis.2 Though rare in adults (it is more common in children and adolescents),3 Pott puffy tumor is caused by conditions often encountered in internal medicine practice, such as bacterial sinusitis, head trauma, and intranasal cocaine use.
The infection can spread to the brain either directly by destruction of the posterior frontal sinus (as in our patient) or by way of the veins that drain the frontal sinus. Meningitis, epidural empyema, frontal lobe abscess, and cavernous sinus thrombosis2 have all been described. Intracranial complications are seen in nearly 100% of children and adolescents with Pott puffy tumor. The rate in adults is 30%,4,5 which is much lower but is nevertheless worrisome because patients can be initially misdiagnosed with scalp abscess,3 cellulitis, or epidermoid cyst,4 and then sent home from the emergency department or physician’s office. In a case series of 32 adult patients with Pott puffy tumor, nearly 45% were initially misdiagnosed, most often by an internist, dermatologist, ophthalmologist, or emergency room physician.4
The most common infective organisms are streptococci, staphylococci, and anaerobes,4 but Haemophilus, Aspergillus species, and invasive mucormycosis have also been described.
MANAGEMENT OPTIONS
Because of the risk of spread of the infection to the brain, rapid initiation of a broad-spectrum antibiotic is warranted in all patients with Pott puffy tumor pending results of tissue culture. Antibiotics may be necessary for at least 4 to 6 weeks to resolve osteomyelitis of the frontal bone and to decrease inflammation before surgery.6
Endoscopic sinus surgery is routinely done to drain the infected sinus and to remove or debride infected bone. Patients with intracranial extension of infection may require a combined endoscopic and neurosurgical approach.
OUTCOME
Our patient’s puffy tumor spontaneously ruptured externally on hospital day 3, and the purulent fluid was sent for culture that grew Streptococcus anginosus. His headaches improved almost immediately after this occurred. The antibiotic regimen was narrowed to ceftriaxone and metronidazole, and 1 week later he was discharged home with instructions to complete a 6-week course of antibiotics. Three weeks after he was discharged, he returned for outpatient endoscopic sinus surgery. At a follow-up visit 2 weeks after surgery, the forehead swelling had resolved, and he was well.
A 60-year-old man presented to our emergency department with a 4-day history of frontal headaches he described as “stinging.” He had also had a large swollen area on his forehead for the past 8 weeks.
He denied fevers, chills, nausea, vomiting, blurry vision, tinnitus, and neck pain, as well as any recent sinus infection, intransanal cocaine use, rhinorrhea, or head trauma. A month ago, he had presented to our emergency department with forehead swelling but no headaches. At that time, the swelling was thought to be an allergic reaction to lisinopril or metformin, medications he takes for hypertension and type 2 diabetes. He had been discharged home with a prescription for a course of prednisone in tapering doses, but that had failed to resolve the swelling.
Physical examination revealed a well-circumscribed area of swelling, 3 by 4 cm, in the central forehead (Figure 1). The area was warm, erythematous, fluctuant, and tender to palpation. The nasal septum was intact and the nasal mucosa appeared pink and healthy. The remainder of the examination was unremarkable.
He was afebrile and hemodynamically stable. His peripheral white blood cell count was mildly elevated at 11.1 × 109. Computed tomography of the brain and sinuses revealed a fluid collection in the frontal scalp associated with erosion of the anterior frontal sinus with posterior extension and enhancement of the adjacent meninges. Magnetic resonance imaging (Figure 2) revealed similar findings. A diagnosis of Pott puffy tumor was made based on the imaging findings.
The name of this condition is misleading, as it is not a neoplasm but an infection. It requires urgent antibiotic therapy and surgical management because of the high risk of the infection spreading to the brain. Our patient was started on a broad-spectrum antibiotic regimen of intravenous vancomycin, ceftriaxone, and metronidazole pending tissue culture to identify the causative organism.
POTT PUFFY TUMOR: A BRIEF OVERVIEW
First described in 1760 by Sir Percivall Pott,1 the same English surgeon who first described tuberculosis of the spine, Pott puffy tumor is a well-demarcated area of swelling that occurs when a frontal sinus infection breaks through the anterior portion of the frontal sinus and forms an abscess between the frontal bone and periosteum with associated osteomyelitis.2 Though rare in adults (it is more common in children and adolescents),3 Pott puffy tumor is caused by conditions often encountered in internal medicine practice, such as bacterial sinusitis, head trauma, and intranasal cocaine use.
The infection can spread to the brain either directly by destruction of the posterior frontal sinus (as in our patient) or by way of the veins that drain the frontal sinus. Meningitis, epidural empyema, frontal lobe abscess, and cavernous sinus thrombosis2 have all been described. Intracranial complications are seen in nearly 100% of children and adolescents with Pott puffy tumor. The rate in adults is 30%,4,5 which is much lower but is nevertheless worrisome because patients can be initially misdiagnosed with scalp abscess,3 cellulitis, or epidermoid cyst,4 and then sent home from the emergency department or physician’s office. In a case series of 32 adult patients with Pott puffy tumor, nearly 45% were initially misdiagnosed, most often by an internist, dermatologist, ophthalmologist, or emergency room physician.4
The most common infective organisms are streptococci, staphylococci, and anaerobes,4 but Haemophilus, Aspergillus species, and invasive mucormycosis have also been described.
MANAGEMENT OPTIONS
Because of the risk of spread of the infection to the brain, rapid initiation of a broad-spectrum antibiotic is warranted in all patients with Pott puffy tumor pending results of tissue culture. Antibiotics may be necessary for at least 4 to 6 weeks to resolve osteomyelitis of the frontal bone and to decrease inflammation before surgery.6
Endoscopic sinus surgery is routinely done to drain the infected sinus and to remove or debride infected bone. Patients with intracranial extension of infection may require a combined endoscopic and neurosurgical approach.
OUTCOME
Our patient’s puffy tumor spontaneously ruptured externally on hospital day 3, and the purulent fluid was sent for culture that grew Streptococcus anginosus. His headaches improved almost immediately after this occurred. The antibiotic regimen was narrowed to ceftriaxone and metronidazole, and 1 week later he was discharged home with instructions to complete a 6-week course of antibiotics. Three weeks after he was discharged, he returned for outpatient endoscopic sinus surgery. At a follow-up visit 2 weeks after surgery, the forehead swelling had resolved, and he was well.
- Tattersall R, Tattersall R. Pott’s puffy tumor. Lancet 2002; 359:1060–1063.
- Forgie SE, Marrie TJ. Pott’s puffy tumor. Am J Med 2008; 121:1041–1042.
- Grewal HS, Dangaych NS, Esposito A. A tumor that is not a tumor but it sure can kill! Am J Case Rep 2012; 13:133–136.
- Akiyama K, Karaki M, Mori N. Evaluation of adult Pott’s puffy tumor: our five cases and 27 literature cases. Laryngoscope 2012; 122:2382–2388.
- Suwan PT, Mogal S, Chaudhary S. Pott’s puffy tumor: an uncommon clinical entity. Case Rep Pediatr 2012; 2012:386104.
- Lauria RA, Laffitte Fernandes F, Brito TP, Pereira PS, Chone CT. Extensive frontoparietal abscess: complication of frontal sinusitis (Pott’s puffy tumor). Case Rep Otolaryngol 2014; 2014:632464.
- Tattersall R, Tattersall R. Pott’s puffy tumor. Lancet 2002; 359:1060–1063.
- Forgie SE, Marrie TJ. Pott’s puffy tumor. Am J Med 2008; 121:1041–1042.
- Grewal HS, Dangaych NS, Esposito A. A tumor that is not a tumor but it sure can kill! Am J Case Rep 2012; 13:133–136.
- Akiyama K, Karaki M, Mori N. Evaluation of adult Pott’s puffy tumor: our five cases and 27 literature cases. Laryngoscope 2012; 122:2382–2388.
- Suwan PT, Mogal S, Chaudhary S. Pott’s puffy tumor: an uncommon clinical entity. Case Rep Pediatr 2012; 2012:386104.
- Lauria RA, Laffitte Fernandes F, Brito TP, Pereira PS, Chone CT. Extensive frontoparietal abscess: complication of frontal sinusitis (Pott’s puffy tumor). Case Rep Otolaryngol 2014; 2014:632464.
Eventration of the diaphragm presenting as spontaneous pneumothorax
A 25-year-old man with a 2-day history of upper respiratory tract infection presents to the emergency department with the sudden onset of right-sided back and chest pain and shortness of breath after a severe coughing fit.
He is morbidly obese, is a long-time smoker, and has had recurrent exacerbations of asthma with frequent upper respiratory tract infections. He has no history of recent trauma.
A review of systems reveals no significant impairment in exercise tolerance. He has been able to continue doing manual labor at his job as a railroad worker.
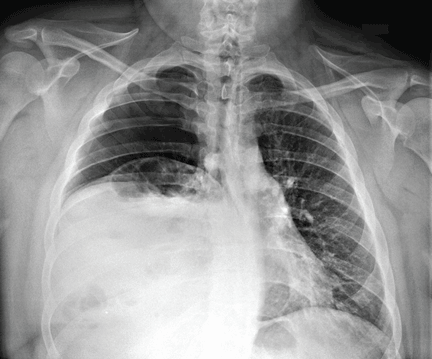
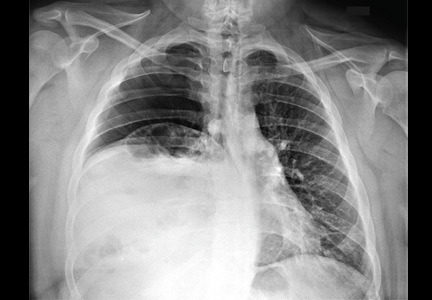
Radiography shows a large right pneumothorax and an elevated right diaphragm (Figure 1). Computed tomography (CT) (Figure 2) reveals a right anterior apical pneumothorax with hypoplastic lung and significant elevation of the right diaphragm with fat, bowel, and kidney within the right thorax. He is hemodynamically stable and shows no signs of bowel obstruction.
The physical examination is normal except for diminished breath sounds on the right side. He is diagnosed with congenital diaphragmatic hernia and spontaneous pneumothorax. A 10-F locking pigtail catheter is inserted under CT guidance, leading to complete resolution of the pneumothorax. He is discharged home the next day with a plan for elective repair of the hernia.
Two months later, he returns for scheduled right thoracotomy to repair the hernia. However, while preparing the chest cavity, the surgeon finds no diaphragmatic hernia and no intra-abdominal content—but rather, a severely elevated and thinned-out diaphragm with uninterrupted continuity. The diagnosis is changed to congenital diaphragmatic eventration, and plication of the diaphragm is performed with a series of interrupted, pledgeted polypropylene sutures.
CONGENITAL EVENTRATION OF THE DIAPHRAGM
Congenital diaphragmatic eventration is a rare developmental defect of the central, muscular portion of the diaphragm. The true prevalence is not known, but early reports identified this condition in less than 0.1% of adult.1
Symptomatic patients usually experience dyspnea secondary to ventilation-perfusion mismatch resulting from chronic atelectasis and lung hypoplasia, as well as impaired ventilation resulting from the limited caudal migration of the diaphragm.2,3 Increased susceptibility to recurrent upper respiratory tract infections and pneumonia is also a common feature.
Although rare, spontaneous pneumothorax can develop in patients such as ours, whose lengthy history of smoking and asthma predisposed him to the development of emphysema-like blebs and bullae and to subsequent rupture of blebs brought on by vigorous coughing that caused an involuntary Valsalva maneuver.4
As in our patient, distinguishing congenital diaphragmatic eventration from hernia preoperatively can be difficult with plain chest radiography. Spiral CT with multiplanar reconstruction or with magnetic resonance imaging can help establish the diagnosis.3 However, a severely attenuated diaphragm can be difficult to visualize on CT, as in our patient, leading to a presumptive diagnosis of diaphragmatic hernia. In such situations, the diagnosis of eventration can only be made intraoperatively.
Surgical repair is indicated only for patients with symptoms. Other potential causes of the symptoms should first be ruled out, however, including primary pulmonary disease, cardiac dysfunction, and morbid obesity.
- Chin EF, Lynn RB. Surgery of eventration of the diaphragm. J Thorac Surg 1956; 32:6–14.
- Ridyard JB, Stewart RM. Regional lung function in unilateral diaphragmatic paralysis. Thorax 1976; 31:438–442.
- Shen C, Che G. Congenital eventration of hemidiaphragm in an adult. Ann Thorac Surg 2012; 94:e137–e139.
- Porpodis K, Zarogoulidis P, Spyratos D, et al. Pneumothorax and asthma. J Thorac Dis 2014; 6(suppl 1):S152–S161.
A 25-year-old man with a 2-day history of upper respiratory tract infection presents to the emergency department with the sudden onset of right-sided back and chest pain and shortness of breath after a severe coughing fit.
He is morbidly obese, is a long-time smoker, and has had recurrent exacerbations of asthma with frequent upper respiratory tract infections. He has no history of recent trauma.
A review of systems reveals no significant impairment in exercise tolerance. He has been able to continue doing manual labor at his job as a railroad worker.
Radiography shows a large right pneumothorax and an elevated right diaphragm (Figure 1). Computed tomography (CT) (Figure 2) reveals a right anterior apical pneumothorax with hypoplastic lung and significant elevation of the right diaphragm with fat, bowel, and kidney within the right thorax. He is hemodynamically stable and shows no signs of bowel obstruction.
The physical examination is normal except for diminished breath sounds on the right side. He is diagnosed with congenital diaphragmatic hernia and spontaneous pneumothorax. A 10-F locking pigtail catheter is inserted under CT guidance, leading to complete resolution of the pneumothorax. He is discharged home the next day with a plan for elective repair of the hernia.
Two months later, he returns for scheduled right thoracotomy to repair the hernia. However, while preparing the chest cavity, the surgeon finds no diaphragmatic hernia and no intra-abdominal content—but rather, a severely elevated and thinned-out diaphragm with uninterrupted continuity. The diagnosis is changed to congenital diaphragmatic eventration, and plication of the diaphragm is performed with a series of interrupted, pledgeted polypropylene sutures.
CONGENITAL EVENTRATION OF THE DIAPHRAGM
Congenital diaphragmatic eventration is a rare developmental defect of the central, muscular portion of the diaphragm. The true prevalence is not known, but early reports identified this condition in less than 0.1% of adult.1
Symptomatic patients usually experience dyspnea secondary to ventilation-perfusion mismatch resulting from chronic atelectasis and lung hypoplasia, as well as impaired ventilation resulting from the limited caudal migration of the diaphragm.2,3 Increased susceptibility to recurrent upper respiratory tract infections and pneumonia is also a common feature.
Although rare, spontaneous pneumothorax can develop in patients such as ours, whose lengthy history of smoking and asthma predisposed him to the development of emphysema-like blebs and bullae and to subsequent rupture of blebs brought on by vigorous coughing that caused an involuntary Valsalva maneuver.4
As in our patient, distinguishing congenital diaphragmatic eventration from hernia preoperatively can be difficult with plain chest radiography. Spiral CT with multiplanar reconstruction or with magnetic resonance imaging can help establish the diagnosis.3 However, a severely attenuated diaphragm can be difficult to visualize on CT, as in our patient, leading to a presumptive diagnosis of diaphragmatic hernia. In such situations, the diagnosis of eventration can only be made intraoperatively.
Surgical repair is indicated only for patients with symptoms. Other potential causes of the symptoms should first be ruled out, however, including primary pulmonary disease, cardiac dysfunction, and morbid obesity.
A 25-year-old man with a 2-day history of upper respiratory tract infection presents to the emergency department with the sudden onset of right-sided back and chest pain and shortness of breath after a severe coughing fit.
He is morbidly obese, is a long-time smoker, and has had recurrent exacerbations of asthma with frequent upper respiratory tract infections. He has no history of recent trauma.
A review of systems reveals no significant impairment in exercise tolerance. He has been able to continue doing manual labor at his job as a railroad worker.
Radiography shows a large right pneumothorax and an elevated right diaphragm (Figure 1). Computed tomography (CT) (Figure 2) reveals a right anterior apical pneumothorax with hypoplastic lung and significant elevation of the right diaphragm with fat, bowel, and kidney within the right thorax. He is hemodynamically stable and shows no signs of bowel obstruction.
The physical examination is normal except for diminished breath sounds on the right side. He is diagnosed with congenital diaphragmatic hernia and spontaneous pneumothorax. A 10-F locking pigtail catheter is inserted under CT guidance, leading to complete resolution of the pneumothorax. He is discharged home the next day with a plan for elective repair of the hernia.
Two months later, he returns for scheduled right thoracotomy to repair the hernia. However, while preparing the chest cavity, the surgeon finds no diaphragmatic hernia and no intra-abdominal content—but rather, a severely elevated and thinned-out diaphragm with uninterrupted continuity. The diagnosis is changed to congenital diaphragmatic eventration, and plication of the diaphragm is performed with a series of interrupted, pledgeted polypropylene sutures.
CONGENITAL EVENTRATION OF THE DIAPHRAGM
Congenital diaphragmatic eventration is a rare developmental defect of the central, muscular portion of the diaphragm. The true prevalence is not known, but early reports identified this condition in less than 0.1% of adult.1
Symptomatic patients usually experience dyspnea secondary to ventilation-perfusion mismatch resulting from chronic atelectasis and lung hypoplasia, as well as impaired ventilation resulting from the limited caudal migration of the diaphragm.2,3 Increased susceptibility to recurrent upper respiratory tract infections and pneumonia is also a common feature.
Although rare, spontaneous pneumothorax can develop in patients such as ours, whose lengthy history of smoking and asthma predisposed him to the development of emphysema-like blebs and bullae and to subsequent rupture of blebs brought on by vigorous coughing that caused an involuntary Valsalva maneuver.4
As in our patient, distinguishing congenital diaphragmatic eventration from hernia preoperatively can be difficult with plain chest radiography. Spiral CT with multiplanar reconstruction or with magnetic resonance imaging can help establish the diagnosis.3 However, a severely attenuated diaphragm can be difficult to visualize on CT, as in our patient, leading to a presumptive diagnosis of diaphragmatic hernia. In such situations, the diagnosis of eventration can only be made intraoperatively.
Surgical repair is indicated only for patients with symptoms. Other potential causes of the symptoms should first be ruled out, however, including primary pulmonary disease, cardiac dysfunction, and morbid obesity.
- Chin EF, Lynn RB. Surgery of eventration of the diaphragm. J Thorac Surg 1956; 32:6–14.
- Ridyard JB, Stewart RM. Regional lung function in unilateral diaphragmatic paralysis. Thorax 1976; 31:438–442.
- Shen C, Che G. Congenital eventration of hemidiaphragm in an adult. Ann Thorac Surg 2012; 94:e137–e139.
- Porpodis K, Zarogoulidis P, Spyratos D, et al. Pneumothorax and asthma. J Thorac Dis 2014; 6(suppl 1):S152–S161.
- Chin EF, Lynn RB. Surgery of eventration of the diaphragm. J Thorac Surg 1956; 32:6–14.
- Ridyard JB, Stewart RM. Regional lung function in unilateral diaphragmatic paralysis. Thorax 1976; 31:438–442.
- Shen C, Che G. Congenital eventration of hemidiaphragm in an adult. Ann Thorac Surg 2012; 94:e137–e139.
- Porpodis K, Zarogoulidis P, Spyratos D, et al. Pneumothorax and asthma. J Thorac Dis 2014; 6(suppl 1):S152–S161.
Bony bumps in the mouth
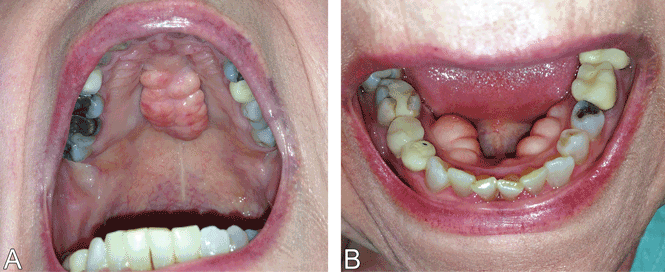
A 79-year-old woman with a long history of limited scleroderma was being evaluated in the rheumatology clinic. During routine examination of the oral cavity, masses were noted on her hard palate and on the lingual surface of both sides of the mandible (Figure 1). The masses had a bony consistency. The patient said that she had had these lumps for as long as she could remember, and that they were painless and had never caused any discomfort.
The masses were diagnosed as torus palatinus and torus mandibularis, localized benign overgrowths of cortical bone. The patient was reassured about the benign nature of these masses, and as they were asymptomatic, no further action was considered necessary.
TORUS PALATINUS AND TORUS MANDIBULARIS
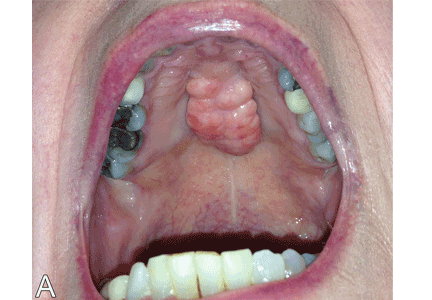
Torus palatinus and torus mandibularis are common exostoses of the mouth, ie, localized benign bony overgrowths arising from cortical bone.1 They are occasionally found incidentally during routine examination of the oral cavity. Patients should be reassured about the nonpathologic nature of this condition.
The condition is thought to be multifactorial, with causal factors including autosomal dominant inheritance, trauma, and lifestyle factors2 such as vitamin deficiency,3 a calcium-rich diet,3 fish consumption,4,5 and chewing on dry, raw, or frozen meat (as in Eskimo cultures).3 Masticatory hyperfunction and bruxism are thought to be risk factors.2,3
Epidemiologic studies indicate that oral tori are more common in women, and the prevalence varies considerably between geographic areas and ethnic groups.3 It is more common in Native Americans, Eskimos, Norwegians, and Thais.4
Torus palatinus is the most prevalent oral torus, occurring in 20% of the US population.6 It arises from the median raphe of the palatine bone and can vary in shape and size. Torus mandibularis is a protuberance arising in the premolar area of the lingual surface of the mandible.3 This form is much less common than torus palatinus, with a prevalence of 6%, and is bilateral in about 80% of cases.
Microscopic examination of tori reveal a mass of dense, lamellar, cortical bone with a small amount of fibrofatty marrow.1 An inner zone of trabecular bone may also be present.1
DIFFERENTIAL DIAGNOSIS
Oral tori must be differentiated from other growths in the mouth including fibromas, mucoceles, osteomas, osteochondromas, and osteoid osteomas.4 However, oral tori can usually be distinguished from other conditions on the basis of clinical findings alone. Biopsy may be warranted if there is doubt.4
Tori tend to grow gradually throughout life and do not have potential for malignant transformation.4 Although they are typically asymptomatic, removal is sometimes warranted for proper fitting of prostheses or for use in autogenous cortical bone grafting.5
- Neville BW, Douglas DD, Carl MA, Bouquot J. Developmental defects of the oral and maxillofacial region. In: Neville BW, Douglas DD, Carl MA, Bouquot J, eds. Oral and Maxillofacial Pathology. 3rd ed. St. Louis, MO: WB Saunders; 2009:1–53.
- Eggen S. Torus mandibularis: an estimation of the degree of genetic determination. Acta Odontol Scand 1989; 47:409–415.
- Loukas M, Hulsberg P, Tubbs RS, et al. The tori of the mouth and ear: a review. Clin Anat 2013; 26:953–960.
- Ladizinski B, Lee KC. A nodular protuberance on the hard palate. JAMA 2014; 311:1558–1559.
- García-García AS, Martínez-González JM, Gómez-Font R, Soto-Rivadeneira A, Oviedo-Roldán L. Current status of the torus palatinus and torus mandibularis. Med Oral Patol Oral Cir Bucal 2010; 15:e353–e360.
- Larheim TA, Westesson PL. Facial growth disturbances. In: Maxillofacial Imaging. Berlin/Heidelberg: Springer-Verlag, 2008:231.
A 79-year-old woman with a long history of limited scleroderma was being evaluated in the rheumatology clinic. During routine examination of the oral cavity, masses were noted on her hard palate and on the lingual surface of both sides of the mandible (Figure 1). The masses had a bony consistency. The patient said that she had had these lumps for as long as she could remember, and that they were painless and had never caused any discomfort.
The masses were diagnosed as torus palatinus and torus mandibularis, localized benign overgrowths of cortical bone. The patient was reassured about the benign nature of these masses, and as they were asymptomatic, no further action was considered necessary.
TORUS PALATINUS AND TORUS MANDIBULARIS
Torus palatinus and torus mandibularis are common exostoses of the mouth, ie, localized benign bony overgrowths arising from cortical bone.1 They are occasionally found incidentally during routine examination of the oral cavity. Patients should be reassured about the nonpathologic nature of this condition.
The condition is thought to be multifactorial, with causal factors including autosomal dominant inheritance, trauma, and lifestyle factors2 such as vitamin deficiency,3 a calcium-rich diet,3 fish consumption,4,5 and chewing on dry, raw, or frozen meat (as in Eskimo cultures).3 Masticatory hyperfunction and bruxism are thought to be risk factors.2,3
Epidemiologic studies indicate that oral tori are more common in women, and the prevalence varies considerably between geographic areas and ethnic groups.3 It is more common in Native Americans, Eskimos, Norwegians, and Thais.4
Torus palatinus is the most prevalent oral torus, occurring in 20% of the US population.6 It arises from the median raphe of the palatine bone and can vary in shape and size. Torus mandibularis is a protuberance arising in the premolar area of the lingual surface of the mandible.3 This form is much less common than torus palatinus, with a prevalence of 6%, and is bilateral in about 80% of cases.
Microscopic examination of tori reveal a mass of dense, lamellar, cortical bone with a small amount of fibrofatty marrow.1 An inner zone of trabecular bone may also be present.1
DIFFERENTIAL DIAGNOSIS
Oral tori must be differentiated from other growths in the mouth including fibromas, mucoceles, osteomas, osteochondromas, and osteoid osteomas.4 However, oral tori can usually be distinguished from other conditions on the basis of clinical findings alone. Biopsy may be warranted if there is doubt.4
Tori tend to grow gradually throughout life and do not have potential for malignant transformation.4 Although they are typically asymptomatic, removal is sometimes warranted for proper fitting of prostheses or for use in autogenous cortical bone grafting.5
A 79-year-old woman with a long history of limited scleroderma was being evaluated in the rheumatology clinic. During routine examination of the oral cavity, masses were noted on her hard palate and on the lingual surface of both sides of the mandible (Figure 1). The masses had a bony consistency. The patient said that she had had these lumps for as long as she could remember, and that they were painless and had never caused any discomfort.
The masses were diagnosed as torus palatinus and torus mandibularis, localized benign overgrowths of cortical bone. The patient was reassured about the benign nature of these masses, and as they were asymptomatic, no further action was considered necessary.
TORUS PALATINUS AND TORUS MANDIBULARIS
Torus palatinus and torus mandibularis are common exostoses of the mouth, ie, localized benign bony overgrowths arising from cortical bone.1 They are occasionally found incidentally during routine examination of the oral cavity. Patients should be reassured about the nonpathologic nature of this condition.
The condition is thought to be multifactorial, with causal factors including autosomal dominant inheritance, trauma, and lifestyle factors2 such as vitamin deficiency,3 a calcium-rich diet,3 fish consumption,4,5 and chewing on dry, raw, or frozen meat (as in Eskimo cultures).3 Masticatory hyperfunction and bruxism are thought to be risk factors.2,3
Epidemiologic studies indicate that oral tori are more common in women, and the prevalence varies considerably between geographic areas and ethnic groups.3 It is more common in Native Americans, Eskimos, Norwegians, and Thais.4
Torus palatinus is the most prevalent oral torus, occurring in 20% of the US population.6 It arises from the median raphe of the palatine bone and can vary in shape and size. Torus mandibularis is a protuberance arising in the premolar area of the lingual surface of the mandible.3 This form is much less common than torus palatinus, with a prevalence of 6%, and is bilateral in about 80% of cases.
Microscopic examination of tori reveal a mass of dense, lamellar, cortical bone with a small amount of fibrofatty marrow.1 An inner zone of trabecular bone may also be present.1
DIFFERENTIAL DIAGNOSIS
Oral tori must be differentiated from other growths in the mouth including fibromas, mucoceles, osteomas, osteochondromas, and osteoid osteomas.4 However, oral tori can usually be distinguished from other conditions on the basis of clinical findings alone. Biopsy may be warranted if there is doubt.4
Tori tend to grow gradually throughout life and do not have potential for malignant transformation.4 Although they are typically asymptomatic, removal is sometimes warranted for proper fitting of prostheses or for use in autogenous cortical bone grafting.5
- Neville BW, Douglas DD, Carl MA, Bouquot J. Developmental defects of the oral and maxillofacial region. In: Neville BW, Douglas DD, Carl MA, Bouquot J, eds. Oral and Maxillofacial Pathology. 3rd ed. St. Louis, MO: WB Saunders; 2009:1–53.
- Eggen S. Torus mandibularis: an estimation of the degree of genetic determination. Acta Odontol Scand 1989; 47:409–415.
- Loukas M, Hulsberg P, Tubbs RS, et al. The tori of the mouth and ear: a review. Clin Anat 2013; 26:953–960.
- Ladizinski B, Lee KC. A nodular protuberance on the hard palate. JAMA 2014; 311:1558–1559.
- García-García AS, Martínez-González JM, Gómez-Font R, Soto-Rivadeneira A, Oviedo-Roldán L. Current status of the torus palatinus and torus mandibularis. Med Oral Patol Oral Cir Bucal 2010; 15:e353–e360.
- Larheim TA, Westesson PL. Facial growth disturbances. In: Maxillofacial Imaging. Berlin/Heidelberg: Springer-Verlag, 2008:231.
- Neville BW, Douglas DD, Carl MA, Bouquot J. Developmental defects of the oral and maxillofacial region. In: Neville BW, Douglas DD, Carl MA, Bouquot J, eds. Oral and Maxillofacial Pathology. 3rd ed. St. Louis, MO: WB Saunders; 2009:1–53.
- Eggen S. Torus mandibularis: an estimation of the degree of genetic determination. Acta Odontol Scand 1989; 47:409–415.
- Loukas M, Hulsberg P, Tubbs RS, et al. The tori of the mouth and ear: a review. Clin Anat 2013; 26:953–960.
- Ladizinski B, Lee KC. A nodular protuberance on the hard palate. JAMA 2014; 311:1558–1559.
- García-García AS, Martínez-González JM, Gómez-Font R, Soto-Rivadeneira A, Oviedo-Roldán L. Current status of the torus palatinus and torus mandibularis. Med Oral Patol Oral Cir Bucal 2010; 15:e353–e360.
- Larheim TA, Westesson PL. Facial growth disturbances. In: Maxillofacial Imaging. Berlin/Heidelberg: Springer-Verlag, 2008:231.
An alerting sign: Enlarged cardiac silhouette
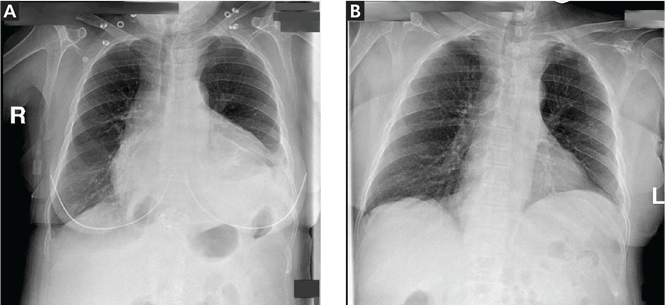
A 75-year-old woman with a history of hypertension and left-lung lobectomy for a carcinoid tumor 10 years ago presented with a 2-week history of progressive cough, dyspnea, and fatigue. Her heart rate was 159 beats per minute with an irregularly irregular rhythm, and her respiratory rate was 36 breaths per minute. Her blood pressure was 140/90 mm Hg. Examination revealed decreased breath sounds and dullness on percussion at the left lung base, jugular venous distention with a positive hepatojugular reflux sign, and an enlarged liver. Electrocardiography showed atrial fibrillation. Chest radiography (Figure 1) revealed enlargement of the cardiac silhouette, with a disproportionately increased transverse diameter, and an obscured left costophrenic angle. A radiograph taken 13 months earlier (Figure 1) had shown a normal cardiothoracic ratio.
EVALUATION OF PERICARDIAL EFFUSION
Pericardial effusion should be suspected in patients presenting with symptoms of impaired cardiac function such as fatigue, dyspnea, nausea, palpitations, lightheadedness, cough, and hoarseness. Patients may also present with chest pain, often decreased by sitting up and leaning forward and exacerbated by lying supine.
Physical examination may reveal distant heart sounds, an absent or displaced apical impulse, dullness and increased fremitus beneath the angle of the left scapula (the Ewart sign), pulsus paradoxus, and nonspecific findings such as tachycardia and hypotension. Jugular venous distention, hepatojugular reflux, and peripheral edema suggest impaired cardiac function.
A chest radiograph showing unexplained new symmetric cardiomegaly (which is often globe-shaped) without signs of pulmonary congestion1 or with a left dominant pleural effusion is an indicator of pericardial effusion, as in our patient. Pericardial fluid may be seen outlining the heart between the epicardial and mediastinal fat, posterior to the sternum in a lateral view.
Other common causes of cardiomegaly include hypertension, congestive heart failure, valvular disease, cardiomyopathy, ischemic heart disease, and pulmonary disease.
Once pericardial effusion is suspected, the next step is to confirm its presence and determine its hemodynamic significance. Transthoracic echocardiography is the imaging test of choice to confirm effusion, as it can be done rapidly and in unstable patients.2
If transthoracic echocardiography is nondiagnostic but suspicion is high, further evaluation may include transesophageal echocardiography,3 computed tomography, or magnetic resonance imaging.
MAKING THE DIAGNOSIS
Pericardial effusion can occur as part of various diseases involving the pericardium, eg, acute pericarditis, myocarditis, autoimmune disease, postmyocardial infarction, malignancy, aortic dissection, and chest trauma. It can also be associated with certain drugs.
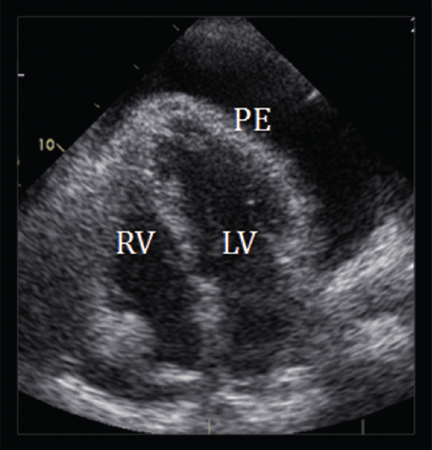
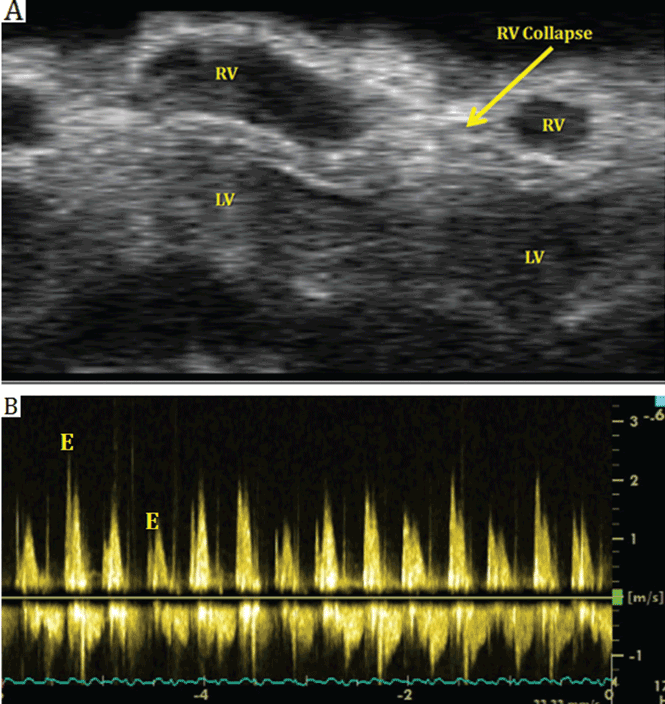
In our patient, echocardiography (Figure 2, Figure 3) demonstrated a large amount of pericardial fluid, and 820 mL of red fluid was aspirated by pericardiocentesis, resulting in relief of her respiratory symptoms. Subcostal two-dimensional echocardiography demonstrated rocking of the heart and intermittent right-ventricular collapse (watch video at www.ccjm.org). Flow cytometry demonstrated 10% kappa+ monoclonal cells. Bone marrow biopsy with immunohistochemical staining revealed infiltration by CD20+, CD5+, CD23+, and BCL1– cells, compatible with small lymphocytic lymphoma.
MALIGNANT PERICARDIAL EFFUSION
Pericardial disease can be the first manifestation of malignancy,4 more often when the patient presents with a large pericardial effusion or tamponade. Malignant tumors of the lung, breast, and esophagus—as well as lymphoma, leukemia, and melanoma—often spread to the pericardium directly or through the lymphatic vessels or bloodstream.4 In our patient, corticosteroid treatment was initiated, and echocardiography at a follow-up visit 2 months later showed no pericardial fluid.
- Khandaker MH, Espinosa RE, Nishimura RA, et al. Pericardial disease: diagnosis and management. Mayo Clin Proc 2010; 85:572–593.
- Cheitlin MD, Armstrong WF, Aurigemma GP, et al; American College of Cardiology; American Heart Association; American Society of Echocardiography. ACC/AHA/ASE 2003 guideline update for the clinical application of echocardiography: summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/ASE Committee to Update the 1997 Guidelines for the Clinical Application of Echocardiography). Circulation 2003; 108:1146–1162.
- Verhaert D, Gabriel RS, Johnston D, Lytle BW, Desai MY, Klein AL. The role of multimodality imaging in the management of pericardial disease. Circ Cardiovascular Imaging 2010; 3:333–343.
- Burazor I, Imazio M, Markel G, Adler Y. Malignant pericardial effusion. Cardiology 2013; 124:224–232.
A 75-year-old woman with a history of hypertension and left-lung lobectomy for a carcinoid tumor 10 years ago presented with a 2-week history of progressive cough, dyspnea, and fatigue. Her heart rate was 159 beats per minute with an irregularly irregular rhythm, and her respiratory rate was 36 breaths per minute. Her blood pressure was 140/90 mm Hg. Examination revealed decreased breath sounds and dullness on percussion at the left lung base, jugular venous distention with a positive hepatojugular reflux sign, and an enlarged liver. Electrocardiography showed atrial fibrillation. Chest radiography (Figure 1) revealed enlargement of the cardiac silhouette, with a disproportionately increased transverse diameter, and an obscured left costophrenic angle. A radiograph taken 13 months earlier (Figure 1) had shown a normal cardiothoracic ratio.
EVALUATION OF PERICARDIAL EFFUSION
Pericardial effusion should be suspected in patients presenting with symptoms of impaired cardiac function such as fatigue, dyspnea, nausea, palpitations, lightheadedness, cough, and hoarseness. Patients may also present with chest pain, often decreased by sitting up and leaning forward and exacerbated by lying supine.
Physical examination may reveal distant heart sounds, an absent or displaced apical impulse, dullness and increased fremitus beneath the angle of the left scapula (the Ewart sign), pulsus paradoxus, and nonspecific findings such as tachycardia and hypotension. Jugular venous distention, hepatojugular reflux, and peripheral edema suggest impaired cardiac function.
A chest radiograph showing unexplained new symmetric cardiomegaly (which is often globe-shaped) without signs of pulmonary congestion1 or with a left dominant pleural effusion is an indicator of pericardial effusion, as in our patient. Pericardial fluid may be seen outlining the heart between the epicardial and mediastinal fat, posterior to the sternum in a lateral view.
Other common causes of cardiomegaly include hypertension, congestive heart failure, valvular disease, cardiomyopathy, ischemic heart disease, and pulmonary disease.
Once pericardial effusion is suspected, the next step is to confirm its presence and determine its hemodynamic significance. Transthoracic echocardiography is the imaging test of choice to confirm effusion, as it can be done rapidly and in unstable patients.2
If transthoracic echocardiography is nondiagnostic but suspicion is high, further evaluation may include transesophageal echocardiography,3 computed tomography, or magnetic resonance imaging.
MAKING THE DIAGNOSIS
Pericardial effusion can occur as part of various diseases involving the pericardium, eg, acute pericarditis, myocarditis, autoimmune disease, postmyocardial infarction, malignancy, aortic dissection, and chest trauma. It can also be associated with certain drugs.
In our patient, echocardiography (Figure 2, Figure 3) demonstrated a large amount of pericardial fluid, and 820 mL of red fluid was aspirated by pericardiocentesis, resulting in relief of her respiratory symptoms. Subcostal two-dimensional echocardiography demonstrated rocking of the heart and intermittent right-ventricular collapse (watch video at www.ccjm.org). Flow cytometry demonstrated 10% kappa+ monoclonal cells. Bone marrow biopsy with immunohistochemical staining revealed infiltration by CD20+, CD5+, CD23+, and BCL1– cells, compatible with small lymphocytic lymphoma.
MALIGNANT PERICARDIAL EFFUSION
Pericardial disease can be the first manifestation of malignancy,4 more often when the patient presents with a large pericardial effusion or tamponade. Malignant tumors of the lung, breast, and esophagus—as well as lymphoma, leukemia, and melanoma—often spread to the pericardium directly or through the lymphatic vessels or bloodstream.4 In our patient, corticosteroid treatment was initiated, and echocardiography at a follow-up visit 2 months later showed no pericardial fluid.
A 75-year-old woman with a history of hypertension and left-lung lobectomy for a carcinoid tumor 10 years ago presented with a 2-week history of progressive cough, dyspnea, and fatigue. Her heart rate was 159 beats per minute with an irregularly irregular rhythm, and her respiratory rate was 36 breaths per minute. Her blood pressure was 140/90 mm Hg. Examination revealed decreased breath sounds and dullness on percussion at the left lung base, jugular venous distention with a positive hepatojugular reflux sign, and an enlarged liver. Electrocardiography showed atrial fibrillation. Chest radiography (Figure 1) revealed enlargement of the cardiac silhouette, with a disproportionately increased transverse diameter, and an obscured left costophrenic angle. A radiograph taken 13 months earlier (Figure 1) had shown a normal cardiothoracic ratio.
EVALUATION OF PERICARDIAL EFFUSION
Pericardial effusion should be suspected in patients presenting with symptoms of impaired cardiac function such as fatigue, dyspnea, nausea, palpitations, lightheadedness, cough, and hoarseness. Patients may also present with chest pain, often decreased by sitting up and leaning forward and exacerbated by lying supine.
Physical examination may reveal distant heart sounds, an absent or displaced apical impulse, dullness and increased fremitus beneath the angle of the left scapula (the Ewart sign), pulsus paradoxus, and nonspecific findings such as tachycardia and hypotension. Jugular venous distention, hepatojugular reflux, and peripheral edema suggest impaired cardiac function.
A chest radiograph showing unexplained new symmetric cardiomegaly (which is often globe-shaped) without signs of pulmonary congestion1 or with a left dominant pleural effusion is an indicator of pericardial effusion, as in our patient. Pericardial fluid may be seen outlining the heart between the epicardial and mediastinal fat, posterior to the sternum in a lateral view.
Other common causes of cardiomegaly include hypertension, congestive heart failure, valvular disease, cardiomyopathy, ischemic heart disease, and pulmonary disease.
Once pericardial effusion is suspected, the next step is to confirm its presence and determine its hemodynamic significance. Transthoracic echocardiography is the imaging test of choice to confirm effusion, as it can be done rapidly and in unstable patients.2
If transthoracic echocardiography is nondiagnostic but suspicion is high, further evaluation may include transesophageal echocardiography,3 computed tomography, or magnetic resonance imaging.
MAKING THE DIAGNOSIS
Pericardial effusion can occur as part of various diseases involving the pericardium, eg, acute pericarditis, myocarditis, autoimmune disease, postmyocardial infarction, malignancy, aortic dissection, and chest trauma. It can also be associated with certain drugs.
In our patient, echocardiography (Figure 2, Figure 3) demonstrated a large amount of pericardial fluid, and 820 mL of red fluid was aspirated by pericardiocentesis, resulting in relief of her respiratory symptoms. Subcostal two-dimensional echocardiography demonstrated rocking of the heart and intermittent right-ventricular collapse (watch video at www.ccjm.org). Flow cytometry demonstrated 10% kappa+ monoclonal cells. Bone marrow biopsy with immunohistochemical staining revealed infiltration by CD20+, CD5+, CD23+, and BCL1– cells, compatible with small lymphocytic lymphoma.
MALIGNANT PERICARDIAL EFFUSION
Pericardial disease can be the first manifestation of malignancy,4 more often when the patient presents with a large pericardial effusion or tamponade. Malignant tumors of the lung, breast, and esophagus—as well as lymphoma, leukemia, and melanoma—often spread to the pericardium directly or through the lymphatic vessels or bloodstream.4 In our patient, corticosteroid treatment was initiated, and echocardiography at a follow-up visit 2 months later showed no pericardial fluid.
- Khandaker MH, Espinosa RE, Nishimura RA, et al. Pericardial disease: diagnosis and management. Mayo Clin Proc 2010; 85:572–593.
- Cheitlin MD, Armstrong WF, Aurigemma GP, et al; American College of Cardiology; American Heart Association; American Society of Echocardiography. ACC/AHA/ASE 2003 guideline update for the clinical application of echocardiography: summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/ASE Committee to Update the 1997 Guidelines for the Clinical Application of Echocardiography). Circulation 2003; 108:1146–1162.
- Verhaert D, Gabriel RS, Johnston D, Lytle BW, Desai MY, Klein AL. The role of multimodality imaging in the management of pericardial disease. Circ Cardiovascular Imaging 2010; 3:333–343.
- Burazor I, Imazio M, Markel G, Adler Y. Malignant pericardial effusion. Cardiology 2013; 124:224–232.
- Khandaker MH, Espinosa RE, Nishimura RA, et al. Pericardial disease: diagnosis and management. Mayo Clin Proc 2010; 85:572–593.
- Cheitlin MD, Armstrong WF, Aurigemma GP, et al; American College of Cardiology; American Heart Association; American Society of Echocardiography. ACC/AHA/ASE 2003 guideline update for the clinical application of echocardiography: summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/ASE Committee to Update the 1997 Guidelines for the Clinical Application of Echocardiography). Circulation 2003; 108:1146–1162.
- Verhaert D, Gabriel RS, Johnston D, Lytle BW, Desai MY, Klein AL. The role of multimodality imaging in the management of pericardial disease. Circ Cardiovascular Imaging 2010; 3:333–343.
- Burazor I, Imazio M, Markel G, Adler Y. Malignant pericardial effusion. Cardiology 2013; 124:224–232.
Brown tumor of the pelvis
A 39-year-old man presented with acute left hip pain and inability to bear weight following a minor trauma. The patient had a history of polycystic kidney disease and was on dialysis. Five years ago he had undergone bilateral nephrectomy and a renal transplantation that subsequently failed.
On examination, the active and passive range of motion of the left hip were limited due to pain. His serum laboratory values were:
- Parathyroid hormone 259.7 pmol/L (reference range 1.5–9.3)
- Calcium 2.32 mmol/L (1.15–1.32)
- Phosphate 3.26 mmol/L (0.8–1.45).
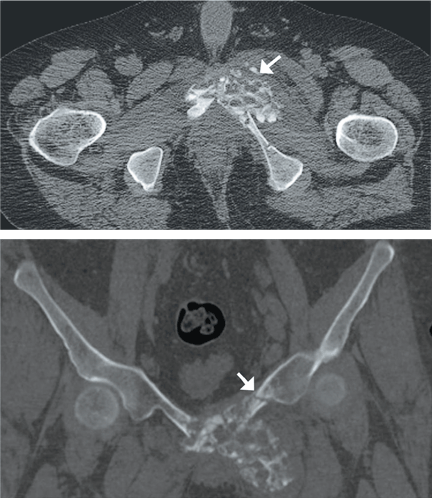
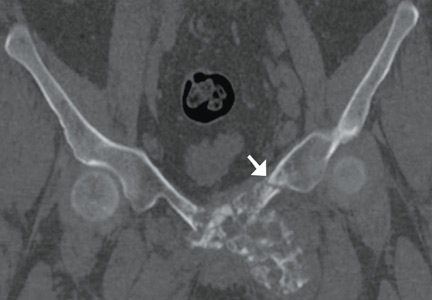
Computed tomography of the pelvis revealed an exophytic calcified lesion with multiple cystic spaces and fluid-fluid levels centered on the left pubis, extending medially into the right pubis and laterally into the left adductor muscle group. An acute pathologic fracture was documented in the left inferior pubic ramus (Figure 1). Other radiographic signs of long-standing hyperparathyroidism were present, including subperiosteal bone resorption at the radial side of the middle phalanges and the clavicle epiphysis.
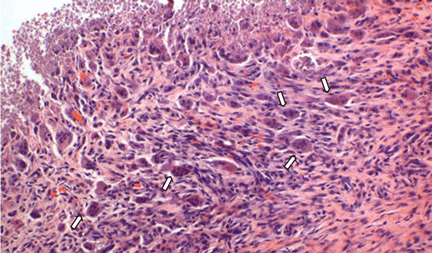
The differential diagnosis of the pelvic lesion included giant cell tumor of bone with aneurysmal bone-cyst-like changes, osteitis fibrosa cystica, and, less likely, metastatic bone disease. Biopsy of the lesion showed clusters of osteoclast-type giant cells on a background of spindle cells and fibrous stroma that in this clinical context was consistent with the diagnosis of brown tumor (Figure 2).1
BROWN TUMOR
Brown tumor has been reported in fewer than 2% of patients with primary hyperparathyroidism and in 1.5% to 1.7% of those with secondary hyperparathyroidism (ie, from chronic renal failure, malabsorption, vitamin D deficiency, or hypocalcemia).2–4 An excess of parathyroid hormone increases the number and activity of osteoclasts, which are responsible for the lytic lesions. Brown tumor is the localized form of osteitis fibrosa cystica and is the most characteristic of the many skeletal changes that accompany secondary hyperparathyroidism.
Brown tumor is named for its color, which results from hemorrhages with accumulation of hemosiderin within the vascularized fibrous tissue. The tumor most commonly affects the pelvis, ribs, long-bone shafts, clavicle, and mandible.5 Clinical symptoms are nonspecific and depend on the size and location of the lesion.
Medical management of secondary hyperparathyroidism in dialysis patients involves some combination of phosphate binders (either calcium-containing or non-calcium-containing binders), calcitriol or synthetic vitamin D analogs, and a calcimimetic. Parathyroidectomy is required if drug therapy is ineffective. Surgical excision of brown tumor should be considered in patients who have large bone defects with spontaneous fracture risk or increasing pain. Our patient declined surgical intervention.
- Davies AM, Evans N, Mangham DC, Grimer RJ. MR imaging of brown tumour with fluid-fluid levels: a report of three cases. Eur Radiol 2001; 11:1445–1449.
- Silverberg SJ, Bilezikian JP. Evaluation and management of primary hyperparathyroidism. J Clin Endocrinol Metab 1996; 81:2036–2040.
- Bohlman ME, Kim YC, Eagan J, Spees EK. Brown tumor in secondary hyperparathyroidism causing acute paraplegia. Am J Med 1986; 81:545–547.
- Demay MB, Rosenthal DI, Deshpande V. Case records of the Massachusetts General Hospital. Case 16-2008. A 46-year-old woman with bone pain. N Engl J Med 2008; 358:2266–2274.
- Perlman JS, Pletcher SD, Schmidt BL, Eisele DW. Pathology quiz case 2. Giant cell lesion (brown tumor) of the mandible, associated with primary hyperparathyroidism (HPT). Arch Otolaryngol Head Neck Surg 2004; 130:793–794.
A 39-year-old man presented with acute left hip pain and inability to bear weight following a minor trauma. The patient had a history of polycystic kidney disease and was on dialysis. Five years ago he had undergone bilateral nephrectomy and a renal transplantation that subsequently failed.
On examination, the active and passive range of motion of the left hip were limited due to pain. His serum laboratory values were:
- Parathyroid hormone 259.7 pmol/L (reference range 1.5–9.3)
- Calcium 2.32 mmol/L (1.15–1.32)
- Phosphate 3.26 mmol/L (0.8–1.45).
Computed tomography of the pelvis revealed an exophytic calcified lesion with multiple cystic spaces and fluid-fluid levels centered on the left pubis, extending medially into the right pubis and laterally into the left adductor muscle group. An acute pathologic fracture was documented in the left inferior pubic ramus (Figure 1). Other radiographic signs of long-standing hyperparathyroidism were present, including subperiosteal bone resorption at the radial side of the middle phalanges and the clavicle epiphysis.
The differential diagnosis of the pelvic lesion included giant cell tumor of bone with aneurysmal bone-cyst-like changes, osteitis fibrosa cystica, and, less likely, metastatic bone disease. Biopsy of the lesion showed clusters of osteoclast-type giant cells on a background of spindle cells and fibrous stroma that in this clinical context was consistent with the diagnosis of brown tumor (Figure 2).1
BROWN TUMOR
Brown tumor has been reported in fewer than 2% of patients with primary hyperparathyroidism and in 1.5% to 1.7% of those with secondary hyperparathyroidism (ie, from chronic renal failure, malabsorption, vitamin D deficiency, or hypocalcemia).2–4 An excess of parathyroid hormone increases the number and activity of osteoclasts, which are responsible for the lytic lesions. Brown tumor is the localized form of osteitis fibrosa cystica and is the most characteristic of the many skeletal changes that accompany secondary hyperparathyroidism.
Brown tumor is named for its color, which results from hemorrhages with accumulation of hemosiderin within the vascularized fibrous tissue. The tumor most commonly affects the pelvis, ribs, long-bone shafts, clavicle, and mandible.5 Clinical symptoms are nonspecific and depend on the size and location of the lesion.
Medical management of secondary hyperparathyroidism in dialysis patients involves some combination of phosphate binders (either calcium-containing or non-calcium-containing binders), calcitriol or synthetic vitamin D analogs, and a calcimimetic. Parathyroidectomy is required if drug therapy is ineffective. Surgical excision of brown tumor should be considered in patients who have large bone defects with spontaneous fracture risk or increasing pain. Our patient declined surgical intervention.
A 39-year-old man presented with acute left hip pain and inability to bear weight following a minor trauma. The patient had a history of polycystic kidney disease and was on dialysis. Five years ago he had undergone bilateral nephrectomy and a renal transplantation that subsequently failed.
On examination, the active and passive range of motion of the left hip were limited due to pain. His serum laboratory values were:
- Parathyroid hormone 259.7 pmol/L (reference range 1.5–9.3)
- Calcium 2.32 mmol/L (1.15–1.32)
- Phosphate 3.26 mmol/L (0.8–1.45).
Computed tomography of the pelvis revealed an exophytic calcified lesion with multiple cystic spaces and fluid-fluid levels centered on the left pubis, extending medially into the right pubis and laterally into the left adductor muscle group. An acute pathologic fracture was documented in the left inferior pubic ramus (Figure 1). Other radiographic signs of long-standing hyperparathyroidism were present, including subperiosteal bone resorption at the radial side of the middle phalanges and the clavicle epiphysis.
The differential diagnosis of the pelvic lesion included giant cell tumor of bone with aneurysmal bone-cyst-like changes, osteitis fibrosa cystica, and, less likely, metastatic bone disease. Biopsy of the lesion showed clusters of osteoclast-type giant cells on a background of spindle cells and fibrous stroma that in this clinical context was consistent with the diagnosis of brown tumor (Figure 2).1
BROWN TUMOR
Brown tumor has been reported in fewer than 2% of patients with primary hyperparathyroidism and in 1.5% to 1.7% of those with secondary hyperparathyroidism (ie, from chronic renal failure, malabsorption, vitamin D deficiency, or hypocalcemia).2–4 An excess of parathyroid hormone increases the number and activity of osteoclasts, which are responsible for the lytic lesions. Brown tumor is the localized form of osteitis fibrosa cystica and is the most characteristic of the many skeletal changes that accompany secondary hyperparathyroidism.
Brown tumor is named for its color, which results from hemorrhages with accumulation of hemosiderin within the vascularized fibrous tissue. The tumor most commonly affects the pelvis, ribs, long-bone shafts, clavicle, and mandible.5 Clinical symptoms are nonspecific and depend on the size and location of the lesion.
Medical management of secondary hyperparathyroidism in dialysis patients involves some combination of phosphate binders (either calcium-containing or non-calcium-containing binders), calcitriol or synthetic vitamin D analogs, and a calcimimetic. Parathyroidectomy is required if drug therapy is ineffective. Surgical excision of brown tumor should be considered in patients who have large bone defects with spontaneous fracture risk or increasing pain. Our patient declined surgical intervention.
- Davies AM, Evans N, Mangham DC, Grimer RJ. MR imaging of brown tumour with fluid-fluid levels: a report of three cases. Eur Radiol 2001; 11:1445–1449.
- Silverberg SJ, Bilezikian JP. Evaluation and management of primary hyperparathyroidism. J Clin Endocrinol Metab 1996; 81:2036–2040.
- Bohlman ME, Kim YC, Eagan J, Spees EK. Brown tumor in secondary hyperparathyroidism causing acute paraplegia. Am J Med 1986; 81:545–547.
- Demay MB, Rosenthal DI, Deshpande V. Case records of the Massachusetts General Hospital. Case 16-2008. A 46-year-old woman with bone pain. N Engl J Med 2008; 358:2266–2274.
- Perlman JS, Pletcher SD, Schmidt BL, Eisele DW. Pathology quiz case 2. Giant cell lesion (brown tumor) of the mandible, associated with primary hyperparathyroidism (HPT). Arch Otolaryngol Head Neck Surg 2004; 130:793–794.
- Davies AM, Evans N, Mangham DC, Grimer RJ. MR imaging of brown tumour with fluid-fluid levels: a report of three cases. Eur Radiol 2001; 11:1445–1449.
- Silverberg SJ, Bilezikian JP. Evaluation and management of primary hyperparathyroidism. J Clin Endocrinol Metab 1996; 81:2036–2040.
- Bohlman ME, Kim YC, Eagan J, Spees EK. Brown tumor in secondary hyperparathyroidism causing acute paraplegia. Am J Med 1986; 81:545–547.
- Demay MB, Rosenthal DI, Deshpande V. Case records of the Massachusetts General Hospital. Case 16-2008. A 46-year-old woman with bone pain. N Engl J Med 2008; 358:2266–2274.
- Perlman JS, Pletcher SD, Schmidt BL, Eisele DW. Pathology quiz case 2. Giant cell lesion (brown tumor) of the mandible, associated with primary hyperparathyroidism (HPT). Arch Otolaryngol Head Neck Surg 2004; 130:793–794.
The color purple
A 58-year-old man with a history of cystoprostatectomy for prostate cancer, end-stage renal disease on hemodialysis, and distal ureteral obstruction requiring bilateral nephrostomy tubes noticed that one of the nephrostomy bags looked “purple” (Figure 1). A specimen collected from one bag was reddish purple (Figure 2). The urine in the other bag was normal. The condition was diagnosed as purple urine bag syndrome.
PURPLE URINE BAG SYNDROME

Purple urine bag syndrome, a relatively rare condition that appears after 2 to 3 months of indwelling urinary catheterization, is usually asymptomatic, the only signs being the purplish urine and staining of the urinary bags and catheters. However, it should be considered a sign of underlying urinary tract infection, which can disseminate causing local complications (Fournier gangrene), systemic complications (septicemia), and death.1–3

The syndrome, first described in 1978 in children with spina bifida and urinary diversion,4 is more prevalent in women than in men, possibly because of the shorter urethra and closer proximity to the anus, which predispose women to bacterial colonization of the urinary tract. Predisposing conditions include dementia,5 female sex, increased dietary tryptophan, bacteriuria, urinary tract infection, constipation, older age, immobility, and alkaline urine.6–8
The cause of the discoloration
The purple color is from indigo and indirubin compounds in the urine, the result of the breakdown of dietary tryptophan. The color varies depending on the proportions of the two pigments.
Dietary tryptophan is broken down into indole by colonic bacteria. After reaching the portal circulation, it is excreted into the urine as indoxyl sulfate, which is broken down to indoxyl by sulfatase-producing bacteria (eg, Klebsiella pneumonia, Proteus mirabilis, Pseudomonas aeruginosa, Escherichia coli, Providencia species, Morganella morganii). Indoxyl is then oxidized to indigo and indirubin.
These compounds do not discolor the urine directly, but rather precipitate after interacting with the lining of the urinary catheter and bags, thereby imparting a purple color.1,9–13
Management
Effective initial measures are improved urinary hygiene (eg, frequent, careful changing of the urinary catheter) and management of constipation, as constipation leads to increased colonization of the intestine by bacteria that metabolize dietary tryptophan into indoxyl. Antibiotics should be given for symptomatic urinary tract infection (fever, increased urinary frequency, dysuria, abdominal pain) but not for color change alone. Coverage should be for gram-negative bacilli, although methicillin-resistant Staphylococcus aureus, which is gram-positive, has also been reported to cause purple urine bag syndrome.
In most cases, purple urine bag syndrome is benign and requires no therapy other than that mentioned above.3,13–15 However, in rare cases, immunocompromised patients (eg, people with diabetes) can develop local complications and sepsis from dissemination of bacterial infection, requiring aggressive therapy.14 Therefore, purple urine bag syndrome should be recognized as an indicator of an underlying urinary tract infection and should be treated if symptomatic. Nevertheless, the long-term prognosis is generally good.
OUR PATIENT’S MANAGEMENT
Our patient was confirmed to have urinary colonization with P aeruginosa and E coli, and alkaline urine. He underwent replacement of the nephrostomy tubes and urinary bag during his hospital stay (he was already in the hospital for another indication), but he continued to produce purple-colored urine from his right side and normal-colored urine from his left side. The unilateral involvement was likely from selective colonization of the right-sided nephrostomy tube with gram-negative bacteria.
- Kang KH, Jeong KH, Baik SK, et al. Purple urine bag syndrome: case report and literature review. Clin Nephrol 2011; 75:557–559.
- Ribeiro JP, Marcelino P, Marum S, Fernandes AP, Grilo A. Case report: purple urine bag syndrome. Crit Care 2004; 8:R137.
- Robinson J. Purple urinary bag syndrome: a harmless but alarming problem. Br J Community Nurs 2003; 8:263–266.
- Barlow GB, Dickson JAS. Purple urine bags. Lancet 1978; 1:220–221.
- Ga H, Kojima T. Purple urine bag syndrome. JAMA 2012; 307:1912–1913.
- Ishida T, Ogura S, Kawakami Y. Five cases of purple urine bag syndrome in a geriatric ward. Nihon Ronen Igakkai Zasshi 1999; 36:826–829. Japanese.
- Gautam G, Kothari A, Kumar R, Dogra PN. Purple urine bag syndrome: a rare clinical entity in patients with long term indwelling catheters. Int Urol Nephrol 2007; 39:155–156.
- Shiao CC, Weng CY, Chuang JC, Huang MS, Chen ZY. Purple urine bag syndrome: a community-based study and literature review. Nephrology (Carlton) 2008; 13:554–559.
- Chong VH. Purple urine bag syndrome: it is the urine bag and not the urine that is discolored purple. South Med J 2012; 105:446.
- Chung SD, Liao CH, Sun HD. Purple urine bag syndrome with acidic urine. Int J Infect Dis 2008; 12:526–527.
- Wu HH, Yang WC, Lin CC. Purple urine bag syndrome. Am J Med Sci 2009; 337:368.
- Achtergael W, Michielsen D, Gorus FK, Gerlo E. Indoxyl sulphate and the purple urine bag syndrome: a case report. Acta Clin Belg 2006; 61:38–41.
- Hadano Y, Shimizu T, Takada S, Inoue T, Sorano S. An update on purple urine bag syndrome. Int J Gen Med 2012; 5:707–710.
- Tasi YM, Huang MS, Yang CJ, Yeh SM, Liu CC. Purple urine bag syndrome, not always a benign process. Am J Emerg Med 2009; 27:895–897.
- Ferrara F, D’Angelo G, Costantino G. Monolateral purple urine bag syndrome in bilateral nephrostomy. Postgrad Med J 2010; 86:627.
A 58-year-old man with a history of cystoprostatectomy for prostate cancer, end-stage renal disease on hemodialysis, and distal ureteral obstruction requiring bilateral nephrostomy tubes noticed that one of the nephrostomy bags looked “purple” (Figure 1). A specimen collected from one bag was reddish purple (Figure 2). The urine in the other bag was normal. The condition was diagnosed as purple urine bag syndrome.
PURPLE URINE BAG SYNDROME
Purple urine bag syndrome, a relatively rare condition that appears after 2 to 3 months of indwelling urinary catheterization, is usually asymptomatic, the only signs being the purplish urine and staining of the urinary bags and catheters. However, it should be considered a sign of underlying urinary tract infection, which can disseminate causing local complications (Fournier gangrene), systemic complications (septicemia), and death.1–3
The syndrome, first described in 1978 in children with spina bifida and urinary diversion,4 is more prevalent in women than in men, possibly because of the shorter urethra and closer proximity to the anus, which predispose women to bacterial colonization of the urinary tract. Predisposing conditions include dementia,5 female sex, increased dietary tryptophan, bacteriuria, urinary tract infection, constipation, older age, immobility, and alkaline urine.6–8
The cause of the discoloration
The purple color is from indigo and indirubin compounds in the urine, the result of the breakdown of dietary tryptophan. The color varies depending on the proportions of the two pigments.
Dietary tryptophan is broken down into indole by colonic bacteria. After reaching the portal circulation, it is excreted into the urine as indoxyl sulfate, which is broken down to indoxyl by sulfatase-producing bacteria (eg, Klebsiella pneumonia, Proteus mirabilis, Pseudomonas aeruginosa, Escherichia coli, Providencia species, Morganella morganii). Indoxyl is then oxidized to indigo and indirubin.
These compounds do not discolor the urine directly, but rather precipitate after interacting with the lining of the urinary catheter and bags, thereby imparting a purple color.1,9–13
Management
Effective initial measures are improved urinary hygiene (eg, frequent, careful changing of the urinary catheter) and management of constipation, as constipation leads to increased colonization of the intestine by bacteria that metabolize dietary tryptophan into indoxyl. Antibiotics should be given for symptomatic urinary tract infection (fever, increased urinary frequency, dysuria, abdominal pain) but not for color change alone. Coverage should be for gram-negative bacilli, although methicillin-resistant Staphylococcus aureus, which is gram-positive, has also been reported to cause purple urine bag syndrome.
In most cases, purple urine bag syndrome is benign and requires no therapy other than that mentioned above.3,13–15 However, in rare cases, immunocompromised patients (eg, people with diabetes) can develop local complications and sepsis from dissemination of bacterial infection, requiring aggressive therapy.14 Therefore, purple urine bag syndrome should be recognized as an indicator of an underlying urinary tract infection and should be treated if symptomatic. Nevertheless, the long-term prognosis is generally good.
OUR PATIENT’S MANAGEMENT
Our patient was confirmed to have urinary colonization with P aeruginosa and E coli, and alkaline urine. He underwent replacement of the nephrostomy tubes and urinary bag during his hospital stay (he was already in the hospital for another indication), but he continued to produce purple-colored urine from his right side and normal-colored urine from his left side. The unilateral involvement was likely from selective colonization of the right-sided nephrostomy tube with gram-negative bacteria.
A 58-year-old man with a history of cystoprostatectomy for prostate cancer, end-stage renal disease on hemodialysis, and distal ureteral obstruction requiring bilateral nephrostomy tubes noticed that one of the nephrostomy bags looked “purple” (Figure 1). A specimen collected from one bag was reddish purple (Figure 2). The urine in the other bag was normal. The condition was diagnosed as purple urine bag syndrome.
PURPLE URINE BAG SYNDROME
Purple urine bag syndrome, a relatively rare condition that appears after 2 to 3 months of indwelling urinary catheterization, is usually asymptomatic, the only signs being the purplish urine and staining of the urinary bags and catheters. However, it should be considered a sign of underlying urinary tract infection, which can disseminate causing local complications (Fournier gangrene), systemic complications (septicemia), and death.1–3
The syndrome, first described in 1978 in children with spina bifida and urinary diversion,4 is more prevalent in women than in men, possibly because of the shorter urethra and closer proximity to the anus, which predispose women to bacterial colonization of the urinary tract. Predisposing conditions include dementia,5 female sex, increased dietary tryptophan, bacteriuria, urinary tract infection, constipation, older age, immobility, and alkaline urine.6–8
The cause of the discoloration
The purple color is from indigo and indirubin compounds in the urine, the result of the breakdown of dietary tryptophan. The color varies depending on the proportions of the two pigments.
Dietary tryptophan is broken down into indole by colonic bacteria. After reaching the portal circulation, it is excreted into the urine as indoxyl sulfate, which is broken down to indoxyl by sulfatase-producing bacteria (eg, Klebsiella pneumonia, Proteus mirabilis, Pseudomonas aeruginosa, Escherichia coli, Providencia species, Morganella morganii). Indoxyl is then oxidized to indigo and indirubin.
These compounds do not discolor the urine directly, but rather precipitate after interacting with the lining of the urinary catheter and bags, thereby imparting a purple color.1,9–13
Management
Effective initial measures are improved urinary hygiene (eg, frequent, careful changing of the urinary catheter) and management of constipation, as constipation leads to increased colonization of the intestine by bacteria that metabolize dietary tryptophan into indoxyl. Antibiotics should be given for symptomatic urinary tract infection (fever, increased urinary frequency, dysuria, abdominal pain) but not for color change alone. Coverage should be for gram-negative bacilli, although methicillin-resistant Staphylococcus aureus, which is gram-positive, has also been reported to cause purple urine bag syndrome.
In most cases, purple urine bag syndrome is benign and requires no therapy other than that mentioned above.3,13–15 However, in rare cases, immunocompromised patients (eg, people with diabetes) can develop local complications and sepsis from dissemination of bacterial infection, requiring aggressive therapy.14 Therefore, purple urine bag syndrome should be recognized as an indicator of an underlying urinary tract infection and should be treated if symptomatic. Nevertheless, the long-term prognosis is generally good.
OUR PATIENT’S MANAGEMENT
Our patient was confirmed to have urinary colonization with P aeruginosa and E coli, and alkaline urine. He underwent replacement of the nephrostomy tubes and urinary bag during his hospital stay (he was already in the hospital for another indication), but he continued to produce purple-colored urine from his right side and normal-colored urine from his left side. The unilateral involvement was likely from selective colonization of the right-sided nephrostomy tube with gram-negative bacteria.
- Kang KH, Jeong KH, Baik SK, et al. Purple urine bag syndrome: case report and literature review. Clin Nephrol 2011; 75:557–559.
- Ribeiro JP, Marcelino P, Marum S, Fernandes AP, Grilo A. Case report: purple urine bag syndrome. Crit Care 2004; 8:R137.
- Robinson J. Purple urinary bag syndrome: a harmless but alarming problem. Br J Community Nurs 2003; 8:263–266.
- Barlow GB, Dickson JAS. Purple urine bags. Lancet 1978; 1:220–221.
- Ga H, Kojima T. Purple urine bag syndrome. JAMA 2012; 307:1912–1913.
- Ishida T, Ogura S, Kawakami Y. Five cases of purple urine bag syndrome in a geriatric ward. Nihon Ronen Igakkai Zasshi 1999; 36:826–829. Japanese.
- Gautam G, Kothari A, Kumar R, Dogra PN. Purple urine bag syndrome: a rare clinical entity in patients with long term indwelling catheters. Int Urol Nephrol 2007; 39:155–156.
- Shiao CC, Weng CY, Chuang JC, Huang MS, Chen ZY. Purple urine bag syndrome: a community-based study and literature review. Nephrology (Carlton) 2008; 13:554–559.
- Chong VH. Purple urine bag syndrome: it is the urine bag and not the urine that is discolored purple. South Med J 2012; 105:446.
- Chung SD, Liao CH, Sun HD. Purple urine bag syndrome with acidic urine. Int J Infect Dis 2008; 12:526–527.
- Wu HH, Yang WC, Lin CC. Purple urine bag syndrome. Am J Med Sci 2009; 337:368.
- Achtergael W, Michielsen D, Gorus FK, Gerlo E. Indoxyl sulphate and the purple urine bag syndrome: a case report. Acta Clin Belg 2006; 61:38–41.
- Hadano Y, Shimizu T, Takada S, Inoue T, Sorano S. An update on purple urine bag syndrome. Int J Gen Med 2012; 5:707–710.
- Tasi YM, Huang MS, Yang CJ, Yeh SM, Liu CC. Purple urine bag syndrome, not always a benign process. Am J Emerg Med 2009; 27:895–897.
- Ferrara F, D’Angelo G, Costantino G. Monolateral purple urine bag syndrome in bilateral nephrostomy. Postgrad Med J 2010; 86:627.
- Kang KH, Jeong KH, Baik SK, et al. Purple urine bag syndrome: case report and literature review. Clin Nephrol 2011; 75:557–559.
- Ribeiro JP, Marcelino P, Marum S, Fernandes AP, Grilo A. Case report: purple urine bag syndrome. Crit Care 2004; 8:R137.
- Robinson J. Purple urinary bag syndrome: a harmless but alarming problem. Br J Community Nurs 2003; 8:263–266.
- Barlow GB, Dickson JAS. Purple urine bags. Lancet 1978; 1:220–221.
- Ga H, Kojima T. Purple urine bag syndrome. JAMA 2012; 307:1912–1913.
- Ishida T, Ogura S, Kawakami Y. Five cases of purple urine bag syndrome in a geriatric ward. Nihon Ronen Igakkai Zasshi 1999; 36:826–829. Japanese.
- Gautam G, Kothari A, Kumar R, Dogra PN. Purple urine bag syndrome: a rare clinical entity in patients with long term indwelling catheters. Int Urol Nephrol 2007; 39:155–156.
- Shiao CC, Weng CY, Chuang JC, Huang MS, Chen ZY. Purple urine bag syndrome: a community-based study and literature review. Nephrology (Carlton) 2008; 13:554–559.
- Chong VH. Purple urine bag syndrome: it is the urine bag and not the urine that is discolored purple. South Med J 2012; 105:446.
- Chung SD, Liao CH, Sun HD. Purple urine bag syndrome with acidic urine. Int J Infect Dis 2008; 12:526–527.
- Wu HH, Yang WC, Lin CC. Purple urine bag syndrome. Am J Med Sci 2009; 337:368.
- Achtergael W, Michielsen D, Gorus FK, Gerlo E. Indoxyl sulphate and the purple urine bag syndrome: a case report. Acta Clin Belg 2006; 61:38–41.
- Hadano Y, Shimizu T, Takada S, Inoue T, Sorano S. An update on purple urine bag syndrome. Int J Gen Med 2012; 5:707–710.
- Tasi YM, Huang MS, Yang CJ, Yeh SM, Liu CC. Purple urine bag syndrome, not always a benign process. Am J Emerg Med 2009; 27:895–897.
- Ferrara F, D’Angelo G, Costantino G. Monolateral purple urine bag syndrome in bilateral nephrostomy. Postgrad Med J 2010; 86:627.
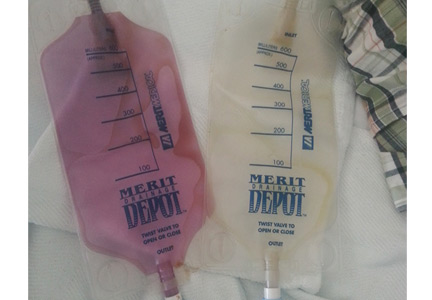
Stellate ulceration in a nonuremic patient
A 64-year-old man was admitted for extensive painful ulceration of the left lower leg (Figure 1) that occurred after a fall and that had worsened over the last 4 months.
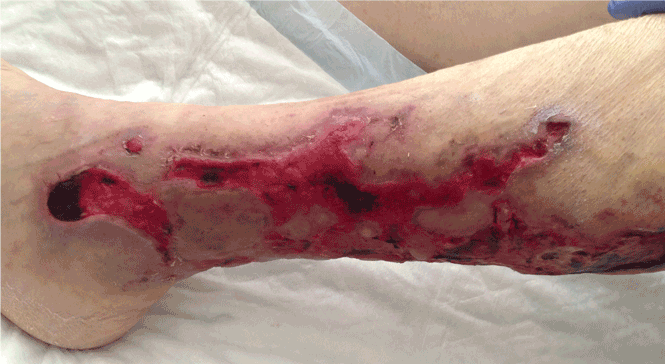
His medical history included hyperuricemia, hypertension, and type 2 diabetes mellitus. He had no known cardiac or renal disease.
Results of initial laboratory testing showed the following:
- Hemoglobin 10.9 g/dL (reference range 13.5–17.5); red blood cells were normocytic and normochromic
- White blood cell count 10.2 × 109/L (4.5–11.0)
- Neutrophil count 9.11 × 109/L (2.0–8.5)
- C-reactive protein 259 mg/L (< 5)
- Creatinine, urea, sodium, potassium, calcium, and phosphate were within normal limits.
Doppler ultrasonography of the legs showed mild diffuse atheromatous arterial disease without significant blockage of blood flow, in addition to mild bilateral venous insufficiency.
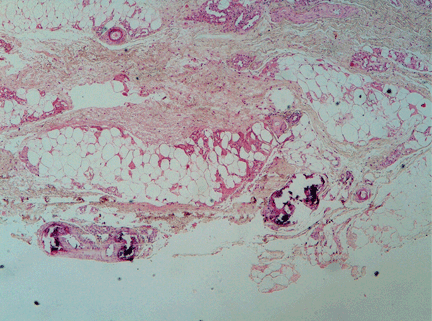
Cutaneous biopsy showed intravascular calcium deposition in the hypodermis (Figure 2) and reticular dermis, erythrocyte extravasation in the superficial dermis, and epidermal necrosis, thus establishing the diagnosis of nonuremic calciphylaxis. The vascular occlusion with spreading necrosis gave the characteristic stellate appearance.
Aside from diabetes, our patient had none of the conditions usually associated with nonuremic calciphylaxis—namely, hyperparathyroidism, previous corticosteroid therapy, warfarin use, connective tissue disease, or malignancy.
A POORLY UNDERSTOOD SMALL-VESSEL VASCULOPATHY
Calciphylaxis is a poorly understood small-vessel vasculopathy, most often associated with end-stage renal disease, with a prevalence of 1% to 4% in patients on dialysis.1 It carries a high risk of death, most often from sepsis.
The cause is still unclear, but several conditions have been implicated, including primary hyperparathyroidism, malignancies, alcoholic liver disease, connective tissue disease, and diabetes.2
Making the diagnosis may be challenging, especially in nonuremic patients. It is a rare condition, the presentation is not always typical, and it can occur with fully normal kidney function and normal indicators of calcium and phosphate metabolism.
The differential diagnosis includes:
- Vasculitis, either primary or secondary to an autoimmune disorder such as rheumatoid arthritis, systemic lupus erythematosus, or cryoglobulinemia
- Peripheral vascular disease
- Other inflammatory conditions such as pyoderma gangrenosum and panniculitis
- Infections such as cellulitis and necrotizing fasciitis
- Iatrogenic disorders such as warfarin necrosis and early-stage nephrogenic systemic fibrosis.3,4
The current approach to treatment is multidisciplinary and is based only on case reports and small case series, since no randomized prospective trial has been done. The goal is optimal control of calcium and phosphate homeostasis and correction of hypercoagulability.5 Available data4,5 support appropriate wound care and surgical debridement.4,5 Intravenous sodium thiosulfate is the most widely used medical treatment and can be given regardless of the level of renal function. Resolution rates have been greater than 90% in patients with normal renal function, whereas improvement in cutaneous ulcers and pain has been observed in 70% of hemodialysis patients.4 However, it does not reduce the associated mortality rate.4
Awareness of nonuremic calciphylaxis and a high index of suspicion are needed when any patient presents with a leg ulcer and no clear cause. It should be considered in the differential diagnosis of leg ulcer in patients with chronic renal failure even if they have risk factors for more common causes of ulcers, and even occasionally in patients such as ours without chronic kidney disease or other risk factors for this condition.
OUR PATIENT’S MANAGEMENT
The patient developed profuse diarrhea, and infection with Clostridium difficile was confirmed. Despite treatment with metronidazole and vancomycin, he died several days later. No treatment directed to calciphylaxis was ever started because of the patient’s unstable condition during the entire hospitalization.
- Van Hattem S, Bootsma AH, Thio HB. Skin manifestations of diabetes. Cleve Clin J Med 2008; 75:772–777.
- Nigwekar SU, Wolf M, Sterns RH, Hix JK. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol 2008; 3:1139–1143.
- Lee JL, Naguwa SM, Cheema G, Gershwin ME. Recognizing calcific uremic arteriolopathy in autoimmune disease: An emerging mimicker of vasculitis. Autoimmun Rev 2008; 7:638–643.
- Wollina U. Update on cutaneous calciphylaxis. Indian J Dermatol 2013; 58:87–92.
- Ross EA. Evolution of treatment strategies for calciphylaxis. Am J Nephrol 2011; 34:460–467.
A 64-year-old man was admitted for extensive painful ulceration of the left lower leg (Figure 1) that occurred after a fall and that had worsened over the last 4 months.
His medical history included hyperuricemia, hypertension, and type 2 diabetes mellitus. He had no known cardiac or renal disease.
Results of initial laboratory testing showed the following:
- Hemoglobin 10.9 g/dL (reference range 13.5–17.5); red blood cells were normocytic and normochromic
- White blood cell count 10.2 × 109/L (4.5–11.0)
- Neutrophil count 9.11 × 109/L (2.0–8.5)
- C-reactive protein 259 mg/L (< 5)
- Creatinine, urea, sodium, potassium, calcium, and phosphate were within normal limits.
Doppler ultrasonography of the legs showed mild diffuse atheromatous arterial disease without significant blockage of blood flow, in addition to mild bilateral venous insufficiency.
Cutaneous biopsy showed intravascular calcium deposition in the hypodermis (Figure 2) and reticular dermis, erythrocyte extravasation in the superficial dermis, and epidermal necrosis, thus establishing the diagnosis of nonuremic calciphylaxis. The vascular occlusion with spreading necrosis gave the characteristic stellate appearance.
Aside from diabetes, our patient had none of the conditions usually associated with nonuremic calciphylaxis—namely, hyperparathyroidism, previous corticosteroid therapy, warfarin use, connective tissue disease, or malignancy.
A POORLY UNDERSTOOD SMALL-VESSEL VASCULOPATHY
Calciphylaxis is a poorly understood small-vessel vasculopathy, most often associated with end-stage renal disease, with a prevalence of 1% to 4% in patients on dialysis.1 It carries a high risk of death, most often from sepsis.
The cause is still unclear, but several conditions have been implicated, including primary hyperparathyroidism, malignancies, alcoholic liver disease, connective tissue disease, and diabetes.2
Making the diagnosis may be challenging, especially in nonuremic patients. It is a rare condition, the presentation is not always typical, and it can occur with fully normal kidney function and normal indicators of calcium and phosphate metabolism.
The differential diagnosis includes:
- Vasculitis, either primary or secondary to an autoimmune disorder such as rheumatoid arthritis, systemic lupus erythematosus, or cryoglobulinemia
- Peripheral vascular disease
- Other inflammatory conditions such as pyoderma gangrenosum and panniculitis
- Infections such as cellulitis and necrotizing fasciitis
- Iatrogenic disorders such as warfarin necrosis and early-stage nephrogenic systemic fibrosis.3,4
The current approach to treatment is multidisciplinary and is based only on case reports and small case series, since no randomized prospective trial has been done. The goal is optimal control of calcium and phosphate homeostasis and correction of hypercoagulability.5 Available data4,5 support appropriate wound care and surgical debridement.4,5 Intravenous sodium thiosulfate is the most widely used medical treatment and can be given regardless of the level of renal function. Resolution rates have been greater than 90% in patients with normal renal function, whereas improvement in cutaneous ulcers and pain has been observed in 70% of hemodialysis patients.4 However, it does not reduce the associated mortality rate.4
Awareness of nonuremic calciphylaxis and a high index of suspicion are needed when any patient presents with a leg ulcer and no clear cause. It should be considered in the differential diagnosis of leg ulcer in patients with chronic renal failure even if they have risk factors for more common causes of ulcers, and even occasionally in patients such as ours without chronic kidney disease or other risk factors for this condition.
OUR PATIENT’S MANAGEMENT
The patient developed profuse diarrhea, and infection with Clostridium difficile was confirmed. Despite treatment with metronidazole and vancomycin, he died several days later. No treatment directed to calciphylaxis was ever started because of the patient’s unstable condition during the entire hospitalization.
A 64-year-old man was admitted for extensive painful ulceration of the left lower leg (Figure 1) that occurred after a fall and that had worsened over the last 4 months.
His medical history included hyperuricemia, hypertension, and type 2 diabetes mellitus. He had no known cardiac or renal disease.
Results of initial laboratory testing showed the following:
- Hemoglobin 10.9 g/dL (reference range 13.5–17.5); red blood cells were normocytic and normochromic
- White blood cell count 10.2 × 109/L (4.5–11.0)
- Neutrophil count 9.11 × 109/L (2.0–8.5)
- C-reactive protein 259 mg/L (< 5)
- Creatinine, urea, sodium, potassium, calcium, and phosphate were within normal limits.
Doppler ultrasonography of the legs showed mild diffuse atheromatous arterial disease without significant blockage of blood flow, in addition to mild bilateral venous insufficiency.
Cutaneous biopsy showed intravascular calcium deposition in the hypodermis (Figure 2) and reticular dermis, erythrocyte extravasation in the superficial dermis, and epidermal necrosis, thus establishing the diagnosis of nonuremic calciphylaxis. The vascular occlusion with spreading necrosis gave the characteristic stellate appearance.
Aside from diabetes, our patient had none of the conditions usually associated with nonuremic calciphylaxis—namely, hyperparathyroidism, previous corticosteroid therapy, warfarin use, connective tissue disease, or malignancy.
A POORLY UNDERSTOOD SMALL-VESSEL VASCULOPATHY
Calciphylaxis is a poorly understood small-vessel vasculopathy, most often associated with end-stage renal disease, with a prevalence of 1% to 4% in patients on dialysis.1 It carries a high risk of death, most often from sepsis.
The cause is still unclear, but several conditions have been implicated, including primary hyperparathyroidism, malignancies, alcoholic liver disease, connective tissue disease, and diabetes.2
Making the diagnosis may be challenging, especially in nonuremic patients. It is a rare condition, the presentation is not always typical, and it can occur with fully normal kidney function and normal indicators of calcium and phosphate metabolism.
The differential diagnosis includes:
- Vasculitis, either primary or secondary to an autoimmune disorder such as rheumatoid arthritis, systemic lupus erythematosus, or cryoglobulinemia
- Peripheral vascular disease
- Other inflammatory conditions such as pyoderma gangrenosum and panniculitis
- Infections such as cellulitis and necrotizing fasciitis
- Iatrogenic disorders such as warfarin necrosis and early-stage nephrogenic systemic fibrosis.3,4
The current approach to treatment is multidisciplinary and is based only on case reports and small case series, since no randomized prospective trial has been done. The goal is optimal control of calcium and phosphate homeostasis and correction of hypercoagulability.5 Available data4,5 support appropriate wound care and surgical debridement.4,5 Intravenous sodium thiosulfate is the most widely used medical treatment and can be given regardless of the level of renal function. Resolution rates have been greater than 90% in patients with normal renal function, whereas improvement in cutaneous ulcers and pain has been observed in 70% of hemodialysis patients.4 However, it does not reduce the associated mortality rate.4
Awareness of nonuremic calciphylaxis and a high index of suspicion are needed when any patient presents with a leg ulcer and no clear cause. It should be considered in the differential diagnosis of leg ulcer in patients with chronic renal failure even if they have risk factors for more common causes of ulcers, and even occasionally in patients such as ours without chronic kidney disease or other risk factors for this condition.
OUR PATIENT’S MANAGEMENT
The patient developed profuse diarrhea, and infection with Clostridium difficile was confirmed. Despite treatment with metronidazole and vancomycin, he died several days later. No treatment directed to calciphylaxis was ever started because of the patient’s unstable condition during the entire hospitalization.
- Van Hattem S, Bootsma AH, Thio HB. Skin manifestations of diabetes. Cleve Clin J Med 2008; 75:772–777.
- Nigwekar SU, Wolf M, Sterns RH, Hix JK. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol 2008; 3:1139–1143.
- Lee JL, Naguwa SM, Cheema G, Gershwin ME. Recognizing calcific uremic arteriolopathy in autoimmune disease: An emerging mimicker of vasculitis. Autoimmun Rev 2008; 7:638–643.
- Wollina U. Update on cutaneous calciphylaxis. Indian J Dermatol 2013; 58:87–92.
- Ross EA. Evolution of treatment strategies for calciphylaxis. Am J Nephrol 2011; 34:460–467.
- Van Hattem S, Bootsma AH, Thio HB. Skin manifestations of diabetes. Cleve Clin J Med 2008; 75:772–777.
- Nigwekar SU, Wolf M, Sterns RH, Hix JK. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol 2008; 3:1139–1143.
- Lee JL, Naguwa SM, Cheema G, Gershwin ME. Recognizing calcific uremic arteriolopathy in autoimmune disease: An emerging mimicker of vasculitis. Autoimmun Rev 2008; 7:638–643.
- Wollina U. Update on cutaneous calciphylaxis. Indian J Dermatol 2013; 58:87–92.
- Ross EA. Evolution of treatment strategies for calciphylaxis. Am J Nephrol 2011; 34:460–467.
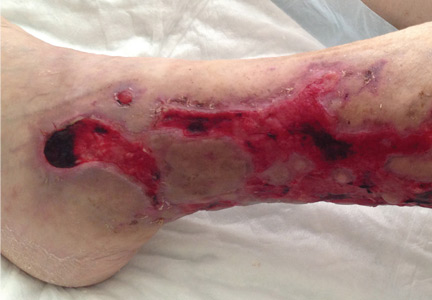
Upper-limb deep vein thrombosis in Paget-Schroetter syndrome
A 43-year-old man with no medical history presented with pain and swelling in his left arm for 2 weeks. He was a regular weight lifter, and his exercise routine included repetitive hyperextension and hyperabduction of his arms while lifting heavy weights.
He had no history of recent trauma or venous cannulation of the left arm. His family history was negative for thrombophilic disorders. Physical examination revealed a swollen and erythematous left arm and visible venous collaterals at the neck, shoulder, and chest. There was no evidence of arterial insufficiency.
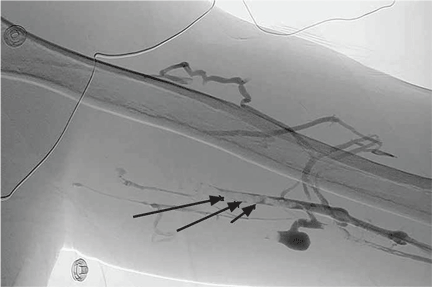
Duplex ultrasonography confirmed thrombosis of the left brachial, axillary, and subclavian veins. Further evaluation with computed tomography showed no intrathoracic mass but revealed several subsegmental pulmonary thrombi in the right lung. A screen for thrombophilia was negative. Venography confirmed complete thrombotic occlusion of the subclavian, axillary, and brachial veins (Figure 1).
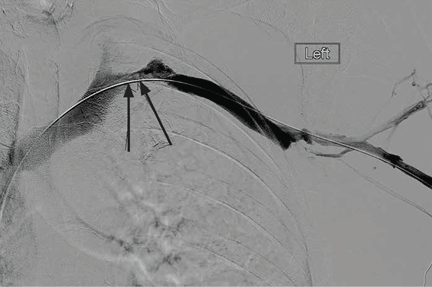
Catheter-directed thrombolysis with tissue plasminogen activator resulted in complete resolution of the thrombosis, but venography after 3 days of thrombolysis showed 50% residual stenosis of the left subclavian vein where it passes under the first rib (Figure 2). The redness and swelling had markedly improved 2 days after thrombolytic therapy. He was discharged home on rivaroxaban 20 mg daily.
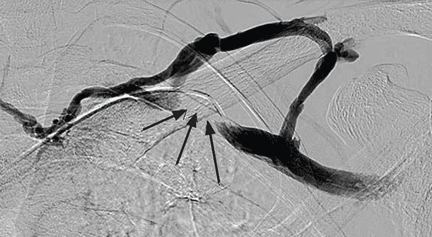
Follow-up venography 2 months later (Figure 3), with the patient performing hyperabduction of the arms, showed a patent subclavian vein with no thrombosis, but dynamic compression and occlusion of the subclavian vein where it passes the first rib. Magnetic resonance imaging (MRI) of the neck showed no cervical (ie, extra) rib and no soft-tissue abnormalities of the scalene triangle.
Following this, the patient underwent resection of the left first rib for decompression of the venous thoracic outlet, which resulted in resolution of his symptoms. He remained asymptomatic at 6-month follow-up.
PAGET-SCHROETTER SYNDROME
Paget-Schroetter syndrome, also referred to as effort-induced or effort thrombosis, is thrombosis of the axillary or subclavian vein associated with strenuous and repetitive activity of the arms. Anatomic abnormalities at the thoracic outlet—cervical rib, congenital bands, hypertrophy of scalene tendons, abnormal insertion of the costoclavicular ligament—and repetitive trauma to the endothelium of the subclavian vein are key factors in its initiation and progression.
The condition is seen primarily in young people who participate in strenuous activities such as rowing, weight lifting, and baseball pitching. It is estimated to be the cause of 40% of cases of primary upper-extremity deep vein thrombosis in the absence of an obvious risk factor or trigger such as a central venous catheter, pacemaker, port, or occult malignancy.1
A provocative test such as the Adson test or hyperabduction test during MRI or venography helps confirm thoracic outlet obstruction by demonstrating dynamic obstruction.2
TREATMENT CONSIDERATIONS
There are no universal guidelines for the treatment of Paget-Schroetter syndrome. However, the available data3–5 suggest a multimodal approach that involves early catheter-directed thrombolysis and subsequent surgical decompression of the thoracic outlet. This can restore venous patency and reduce the risk of long-term complications such as rethrombosis and postthrombotic syndrome.3–5
Surgical treatment includes resection of the first rib and division of the scalene muscles and the costoclavicular ligament. MRI with provocative testing helps guide the surgical approach. Anticoagulation therapy alone—ie, without thrombolysis and surgical decompression—is inadequate as it leads to recurrence of thrombosis and residual symptoms.6
Paget-Schroetter syndrome should not be managed the same as lower-extremity deep vein thrombosis because the cause and the exacerbating factors are different.
Unanswered questions
Because we have no data from randomized controlled trials, questions about management remain. What should be the duration of anticoagulation, especially in the absence of coexisting thrombophilia? Is thrombophilia screening useful? What is the optimal timing for starting thrombolytic therapy?
A careful history and heightened suspicion are required to make this diagnosis. If undiagnosed, it carries a risk of significant long-term morbidity and death. Dynamic obstruction during venography, in addition to MRI, can help identify an anatomic obstruction.
- Bernardi E, Pesavento R, Prandoni P. Upper extremity deep venous thrombosis. Semin Thromb Hemost 2006; 32:729–736.
- Demirbag D, Unlu E, Ozdemir F, et al. The relationship between magnetic resonance imaging findings and postural maneuver and physical examination tests in patients with thoracic outlet syndrome: results of a double-blind, controlled study. Arch Phys Med Rehabil 2007; 88:844–851.
- Alla VM, Natarajan N, Kaushik M, Warrier R, Nair CK. Paget-Schroetter syndrome: review of pathogenesis and treatment of effort thrombosis. West J Emerg Med 2010; 11:358–362.
- Molina JE, Hunter DW, Dietz CA. Paget-Schroetter syndrome treated with thrombolytics and immediate surgery. J Vasc Surg 2007; 45:328–334.
- Thompson RW. Comprehensive management of subclavian vein effort thrombosis. Semin Intervent Radiol 2012; 29:44–51.
- AbuRahma AF, Robinson PA. Effort subclavian vein thrombosis: evolution of management. J Endovasc Ther 2000; 7:302–308.
A 43-year-old man with no medical history presented with pain and swelling in his left arm for 2 weeks. He was a regular weight lifter, and his exercise routine included repetitive hyperextension and hyperabduction of his arms while lifting heavy weights.
He had no history of recent trauma or venous cannulation of the left arm. His family history was negative for thrombophilic disorders. Physical examination revealed a swollen and erythematous left arm and visible venous collaterals at the neck, shoulder, and chest. There was no evidence of arterial insufficiency.
Duplex ultrasonography confirmed thrombosis of the left brachial, axillary, and subclavian veins. Further evaluation with computed tomography showed no intrathoracic mass but revealed several subsegmental pulmonary thrombi in the right lung. A screen for thrombophilia was negative. Venography confirmed complete thrombotic occlusion of the subclavian, axillary, and brachial veins (Figure 1).
Catheter-directed thrombolysis with tissue plasminogen activator resulted in complete resolution of the thrombosis, but venography after 3 days of thrombolysis showed 50% residual stenosis of the left subclavian vein where it passes under the first rib (Figure 2). The redness and swelling had markedly improved 2 days after thrombolytic therapy. He was discharged home on rivaroxaban 20 mg daily.
Follow-up venography 2 months later (Figure 3), with the patient performing hyperabduction of the arms, showed a patent subclavian vein with no thrombosis, but dynamic compression and occlusion of the subclavian vein where it passes the first rib. Magnetic resonance imaging (MRI) of the neck showed no cervical (ie, extra) rib and no soft-tissue abnormalities of the scalene triangle.
Following this, the patient underwent resection of the left first rib for decompression of the venous thoracic outlet, which resulted in resolution of his symptoms. He remained asymptomatic at 6-month follow-up.
PAGET-SCHROETTER SYNDROME
Paget-Schroetter syndrome, also referred to as effort-induced or effort thrombosis, is thrombosis of the axillary or subclavian vein associated with strenuous and repetitive activity of the arms. Anatomic abnormalities at the thoracic outlet—cervical rib, congenital bands, hypertrophy of scalene tendons, abnormal insertion of the costoclavicular ligament—and repetitive trauma to the endothelium of the subclavian vein are key factors in its initiation and progression.
The condition is seen primarily in young people who participate in strenuous activities such as rowing, weight lifting, and baseball pitching. It is estimated to be the cause of 40% of cases of primary upper-extremity deep vein thrombosis in the absence of an obvious risk factor or trigger such as a central venous catheter, pacemaker, port, or occult malignancy.1
A provocative test such as the Adson test or hyperabduction test during MRI or venography helps confirm thoracic outlet obstruction by demonstrating dynamic obstruction.2
TREATMENT CONSIDERATIONS
There are no universal guidelines for the treatment of Paget-Schroetter syndrome. However, the available data3–5 suggest a multimodal approach that involves early catheter-directed thrombolysis and subsequent surgical decompression of the thoracic outlet. This can restore venous patency and reduce the risk of long-term complications such as rethrombosis and postthrombotic syndrome.3–5
Surgical treatment includes resection of the first rib and division of the scalene muscles and the costoclavicular ligament. MRI with provocative testing helps guide the surgical approach. Anticoagulation therapy alone—ie, without thrombolysis and surgical decompression—is inadequate as it leads to recurrence of thrombosis and residual symptoms.6
Paget-Schroetter syndrome should not be managed the same as lower-extremity deep vein thrombosis because the cause and the exacerbating factors are different.
Unanswered questions
Because we have no data from randomized controlled trials, questions about management remain. What should be the duration of anticoagulation, especially in the absence of coexisting thrombophilia? Is thrombophilia screening useful? What is the optimal timing for starting thrombolytic therapy?
A careful history and heightened suspicion are required to make this diagnosis. If undiagnosed, it carries a risk of significant long-term morbidity and death. Dynamic obstruction during venography, in addition to MRI, can help identify an anatomic obstruction.
A 43-year-old man with no medical history presented with pain and swelling in his left arm for 2 weeks. He was a regular weight lifter, and his exercise routine included repetitive hyperextension and hyperabduction of his arms while lifting heavy weights.
He had no history of recent trauma or venous cannulation of the left arm. His family history was negative for thrombophilic disorders. Physical examination revealed a swollen and erythematous left arm and visible venous collaterals at the neck, shoulder, and chest. There was no evidence of arterial insufficiency.
Duplex ultrasonography confirmed thrombosis of the left brachial, axillary, and subclavian veins. Further evaluation with computed tomography showed no intrathoracic mass but revealed several subsegmental pulmonary thrombi in the right lung. A screen for thrombophilia was negative. Venography confirmed complete thrombotic occlusion of the subclavian, axillary, and brachial veins (Figure 1).
Catheter-directed thrombolysis with tissue plasminogen activator resulted in complete resolution of the thrombosis, but venography after 3 days of thrombolysis showed 50% residual stenosis of the left subclavian vein where it passes under the first rib (Figure 2). The redness and swelling had markedly improved 2 days after thrombolytic therapy. He was discharged home on rivaroxaban 20 mg daily.
Follow-up venography 2 months later (Figure 3), with the patient performing hyperabduction of the arms, showed a patent subclavian vein with no thrombosis, but dynamic compression and occlusion of the subclavian vein where it passes the first rib. Magnetic resonance imaging (MRI) of the neck showed no cervical (ie, extra) rib and no soft-tissue abnormalities of the scalene triangle.
Following this, the patient underwent resection of the left first rib for decompression of the venous thoracic outlet, which resulted in resolution of his symptoms. He remained asymptomatic at 6-month follow-up.
PAGET-SCHROETTER SYNDROME
Paget-Schroetter syndrome, also referred to as effort-induced or effort thrombosis, is thrombosis of the axillary or subclavian vein associated with strenuous and repetitive activity of the arms. Anatomic abnormalities at the thoracic outlet—cervical rib, congenital bands, hypertrophy of scalene tendons, abnormal insertion of the costoclavicular ligament—and repetitive trauma to the endothelium of the subclavian vein are key factors in its initiation and progression.
The condition is seen primarily in young people who participate in strenuous activities such as rowing, weight lifting, and baseball pitching. It is estimated to be the cause of 40% of cases of primary upper-extremity deep vein thrombosis in the absence of an obvious risk factor or trigger such as a central venous catheter, pacemaker, port, or occult malignancy.1
A provocative test such as the Adson test or hyperabduction test during MRI or venography helps confirm thoracic outlet obstruction by demonstrating dynamic obstruction.2
TREATMENT CONSIDERATIONS
There are no universal guidelines for the treatment of Paget-Schroetter syndrome. However, the available data3–5 suggest a multimodal approach that involves early catheter-directed thrombolysis and subsequent surgical decompression of the thoracic outlet. This can restore venous patency and reduce the risk of long-term complications such as rethrombosis and postthrombotic syndrome.3–5
Surgical treatment includes resection of the first rib and division of the scalene muscles and the costoclavicular ligament. MRI with provocative testing helps guide the surgical approach. Anticoagulation therapy alone—ie, without thrombolysis and surgical decompression—is inadequate as it leads to recurrence of thrombosis and residual symptoms.6
Paget-Schroetter syndrome should not be managed the same as lower-extremity deep vein thrombosis because the cause and the exacerbating factors are different.
Unanswered questions
Because we have no data from randomized controlled trials, questions about management remain. What should be the duration of anticoagulation, especially in the absence of coexisting thrombophilia? Is thrombophilia screening useful? What is the optimal timing for starting thrombolytic therapy?
A careful history and heightened suspicion are required to make this diagnosis. If undiagnosed, it carries a risk of significant long-term morbidity and death. Dynamic obstruction during venography, in addition to MRI, can help identify an anatomic obstruction.
- Bernardi E, Pesavento R, Prandoni P. Upper extremity deep venous thrombosis. Semin Thromb Hemost 2006; 32:729–736.
- Demirbag D, Unlu E, Ozdemir F, et al. The relationship between magnetic resonance imaging findings and postural maneuver and physical examination tests in patients with thoracic outlet syndrome: results of a double-blind, controlled study. Arch Phys Med Rehabil 2007; 88:844–851.
- Alla VM, Natarajan N, Kaushik M, Warrier R, Nair CK. Paget-Schroetter syndrome: review of pathogenesis and treatment of effort thrombosis. West J Emerg Med 2010; 11:358–362.
- Molina JE, Hunter DW, Dietz CA. Paget-Schroetter syndrome treated with thrombolytics and immediate surgery. J Vasc Surg 2007; 45:328–334.
- Thompson RW. Comprehensive management of subclavian vein effort thrombosis. Semin Intervent Radiol 2012; 29:44–51.
- AbuRahma AF, Robinson PA. Effort subclavian vein thrombosis: evolution of management. J Endovasc Ther 2000; 7:302–308.
- Bernardi E, Pesavento R, Prandoni P. Upper extremity deep venous thrombosis. Semin Thromb Hemost 2006; 32:729–736.
- Demirbag D, Unlu E, Ozdemir F, et al. The relationship between magnetic resonance imaging findings and postural maneuver and physical examination tests in patients with thoracic outlet syndrome: results of a double-blind, controlled study. Arch Phys Med Rehabil 2007; 88:844–851.
- Alla VM, Natarajan N, Kaushik M, Warrier R, Nair CK. Paget-Schroetter syndrome: review of pathogenesis and treatment of effort thrombosis. West J Emerg Med 2010; 11:358–362.
- Molina JE, Hunter DW, Dietz CA. Paget-Schroetter syndrome treated with thrombolytics and immediate surgery. J Vasc Surg 2007; 45:328–334.
- Thompson RW. Comprehensive management of subclavian vein effort thrombosis. Semin Intervent Radiol 2012; 29:44–51.
- AbuRahma AF, Robinson PA. Effort subclavian vein thrombosis: evolution of management. J Endovasc Ther 2000; 7:302–308.
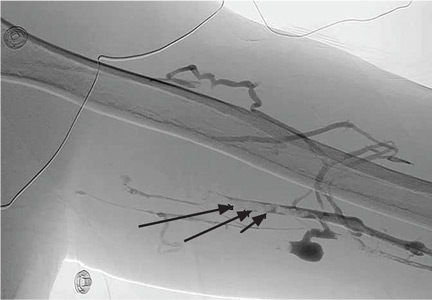
Lady Windermere syndrome: Mycobacterium of sophistication
A 75-year-old woman was referred to our pulmonary clinic with a 4-year history of intermittent episodes of persistent cough, occasionally productive of sputum, and mild exertional dyspnea. She had been treated with azithromycin for presumed community-acquired pneumonia, and her symptoms had initially improved. Subsequently, she experienced discrete, recurrent episodes of “bronchitis,” with productive cough and mild exertional dyspnea. Testing for latent tuberculosis had been negative. She reported a 10-pack-year smoking history in the remote past.
Her medical history included asthma, atrial fibrillation, gastroesophageal reflux disorder, hyperlipidemia, osteopenia, hypothyroidism, and allergic rhinitis. Her current medications were metoprolol, propafenone, and warfarin.
ABNORMALITIES ON PREVIOUS IMAGING
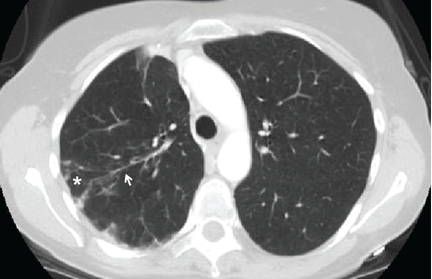
Computed tomography (CT) in April 2010 had revealed scattered linear, nodular, and “tree-in-bud” opacities involving the bilateral apices and the upper, middle, and lower lobes of the right lung, suggestive of bronchiolitis. Mild bronchiectasis had also been noted (Figure 1). Chest radiography had demonstrated signs of bronchiectasis and several scattered nodules (Figure 2). These abnormalities were still present on another CT scan in May 2013.
The patient had not undergone bronchoscopy before she was referred to our clinic.
WORKUP AT OUR CLINIC
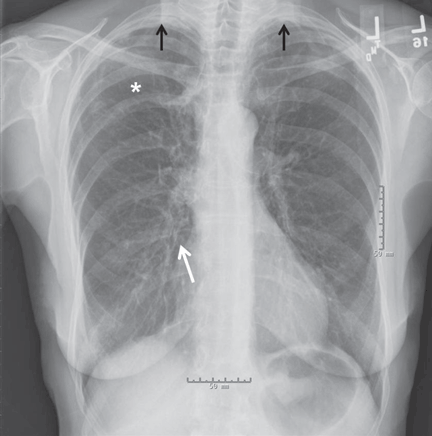
On examination, the patient was lean, with a body mass index of 20.53 kg/m2. She appeared calm, well-groomed, and well-dressed, and had a very polite manner. When she coughed, she tried to suppress it, as if she were self-conscious about it. Her heart rhythm was irregularly irregular with a normal rate.
Expectorated sputum samples were obtained. Stains for acid-fast bacilli were negative, but three cultures were positive for acid-fast bacilli consistent with Mycobacterium avium-intracellulare. Serologic studies were negative for fungal infection and immunoglobulin deficiency.
Based on her symptoms and on the findings of imaging studies and sputum culture, we arrived at the diagnosis of nontuberculous mycobacterial lung infection, specifically, Lady Windermere syndrome.
NONTUBERCULOUS MYOCOBACTERIAL LUNG INFECTION
The diagnosis of nontuberculous mycobacterial lung infection is based on respiratory symptoms, findings on imaging (eg, nodular or cavitary opacities on radiography, or multifocal bronchiectasis and multiple small nodules on CT), and a positive culture for nontuberculous mycobacterial infection in more than two specimens of expectorated sputum or in more than one specimen from bronchoalveolar lavage. Lung biopsy with tissue culture is another way to confirm the diagnosis.
LADY WINDERMERE SYNDROME
Lady Windermere syndrome was described more than 20 years ago.1 The name derives from the lead character in Oscar Wilde’s play Lady Windermere’s Fan, which satirizes the strict morals and polite manners typical of the Victorian era in Great Britain.2
The patient with Lady Windermere syndrome is typically a thin, lean, well-mannered elderly woman who voluntarily suppresses her cough out of politeness. Suppression of the cough is thought to predispose to lung infection by allowing secretions to collect in the airways, especially in the right middle lobe, which has the longest and narrowest of the lobar bronchi.3,4
Symptoms of Lady Windermere syndrome include cough, sputum production, and fatigue similar to that of acute or chronic bronchitis. Dyspnea, fever, and hemoptysis are less common.5 The differential diagnosis for these symptoms is broad and includes asthma, chronic obstructive pulmonary disease, gastroesophageal reflux disease, pneumonia, bronchiectasis, cystic fibrosis, interstitial lung disease, postnasal drip, lung cancer, and heart failure.
A prospective cohort study by Kim et al6 yielded descriptions of typical patients with Lady Windermere syndrome. Patients were tall and lean, tended to have scoliosis, and more commonly had pectus excavatum or mitral valve prolapse; 95% were women, 91% were white, and the average age was 60. The morphologic features are thought to contribute to impaired clearance of airway secretions by altered mechanics during coughing.
HALLMARKS ON IMAGING
Kim et al6 reported that the most common findings on lung imaging in nontuberculous mycobacterial infection were bronchiectasis involving the right middle lobe (90%), nodules involving the right lower lobe (73%) and right middle lobe (71%), and, less commonly, a cavitary infiltrate involving the right upper lobe (17%) or right middle lobe (10%).
Key findings on imaging in Lady Windermere syndrome include opacities and “cylindrical bronchiectasis” predominantly involving the right middle lobe or lingula.5 Bronchiolar inflammation in response to nontuberculous mycobacterial infection may cause a nodular appearance, often progressing to a tree-in-bud appearance on CT.
Other diagnostic considerations for tree-in-bud appearance on CT include fungal, viral, or other bacterial infection, aspiration pneumonitis, inhalation of a foreign substance, cystic fibrosis, rheumatoid arthritis, SjÖgren syndrome, bronchiolitis obliterans, and neoplastic disease.
CURRENT TREATMENT OPTIONS
Treatment of nontuberculous mycobacterial lung infection, including Lady Windermere syndrome, is not necessary in every case, given the variability in clinical symptoms and in disease progression. Patients with progressive symptoms or radiographic changes should be considered candidates for treatment.
Management is directed at the underlying infection. M avium-intracellulare is ubiquitous in the environment, including in soil and water, and it has been reported as the most common pathogen in nontuberculous mycobacterial lung infection.7
Nodular-bronchiectatic nontuberculous mycobacterial lung disease typically progresses more slowly than fibrocavitary disease. For patients with nodular-bronchiectatic disease, follow-up over months or years may be needed before clinical or radiographic changes become apparent.
When treatment is indicated for nodular-bronchiectatic nontuberculous mycobacterial lung infection, it should include a macrolide antibiotic, ethambutol, and rifampin.7,8 Monotherapy with a macrolide is not recommended because of the risk of macrolide resistance. Addition of an aminoglycoside may be considered when treating fibrocavitary disease or widespread nodular bronchiectatic disease.
Management of bronchiectasis, when present, includes chest physiotherapy, pulmonary hygiene therapy, and awareness of the predisposition for nonmycobacterial lung infection. The decision to prescribe antimicrobials should take into consideration the risks and benefits for each patient.
Because treatment involves multidrug regimens, drug interactions and adverse effects need to be considered and monitored, especially in elderly patients, who may already be taking multiple medications. Treatment should be continued until a patient has negative sputum cultures for acid-fast bacilli while on therapy, for 1 year.
- Reich JM, Johnson RE. Mycobacterium avium complex pulmonary disease presenting as an isolated lingular or middle lobe pattern. The Lady Windermere syndrome. Chest 1992; 101:1605–1609.
- Kasthoori JJ, Liam CK, Wastie ML. Lady Windermere syndrome: an inappropriate eponym for an increasingly important condition. Singapore Med J 2008; 49:e47–e49.
- Dhillon SS, Watanakunakorn C. Lady Windermere syndrome: middle lobe bronchiectasis and mycobacterium avium complex infection due to voluntary cough suppression. Clin Infect Dis 2000; 30:572–575.
- Reich JM. Pathogenesis of Lady Windermere syndrome. Scand J Infect Dis 2012; 44:1–2.
- Glassroth J. Pulmonary disease due to nontuberculous mycobacteria. Chest 2008; 133:243–251.
- Kim RD, Greenberg DE, Ehrmantraut ME, et al. Pulmonary nontuberculous mycobacterial disease: prospective study of a distinct preexisting syndrome. Am J Respir Crit Care Med 2008; 178:1066–1074.
- Griffith DE, Aksamit T, Brown-Elliott BA, et al; ATS Mycobacterial Diseases Subcommittee; American Thoracic Society; Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med 2007; 175:367–416.
- Mason RJ, Broaddus VC, Martin T, et al, editors. Murray and Nadel’s Textbook of Respiratory Medicine. 5th ed. Philadelphia, PA: Saunders; 2010.
A 75-year-old woman was referred to our pulmonary clinic with a 4-year history of intermittent episodes of persistent cough, occasionally productive of sputum, and mild exertional dyspnea. She had been treated with azithromycin for presumed community-acquired pneumonia, and her symptoms had initially improved. Subsequently, she experienced discrete, recurrent episodes of “bronchitis,” with productive cough and mild exertional dyspnea. Testing for latent tuberculosis had been negative. She reported a 10-pack-year smoking history in the remote past.
Her medical history included asthma, atrial fibrillation, gastroesophageal reflux disorder, hyperlipidemia, osteopenia, hypothyroidism, and allergic rhinitis. Her current medications were metoprolol, propafenone, and warfarin.
ABNORMALITIES ON PREVIOUS IMAGING
Computed tomography (CT) in April 2010 had revealed scattered linear, nodular, and “tree-in-bud” opacities involving the bilateral apices and the upper, middle, and lower lobes of the right lung, suggestive of bronchiolitis. Mild bronchiectasis had also been noted (Figure 1). Chest radiography had demonstrated signs of bronchiectasis and several scattered nodules (Figure 2). These abnormalities were still present on another CT scan in May 2013.
The patient had not undergone bronchoscopy before she was referred to our clinic.
WORKUP AT OUR CLINIC
On examination, the patient was lean, with a body mass index of 20.53 kg/m2. She appeared calm, well-groomed, and well-dressed, and had a very polite manner. When she coughed, she tried to suppress it, as if she were self-conscious about it. Her heart rhythm was irregularly irregular with a normal rate.
Expectorated sputum samples were obtained. Stains for acid-fast bacilli were negative, but three cultures were positive for acid-fast bacilli consistent with Mycobacterium avium-intracellulare. Serologic studies were negative for fungal infection and immunoglobulin deficiency.
Based on her symptoms and on the findings of imaging studies and sputum culture, we arrived at the diagnosis of nontuberculous mycobacterial lung infection, specifically, Lady Windermere syndrome.
NONTUBERCULOUS MYOCOBACTERIAL LUNG INFECTION
The diagnosis of nontuberculous mycobacterial lung infection is based on respiratory symptoms, findings on imaging (eg, nodular or cavitary opacities on radiography, or multifocal bronchiectasis and multiple small nodules on CT), and a positive culture for nontuberculous mycobacterial infection in more than two specimens of expectorated sputum or in more than one specimen from bronchoalveolar lavage. Lung biopsy with tissue culture is another way to confirm the diagnosis.
LADY WINDERMERE SYNDROME
Lady Windermere syndrome was described more than 20 years ago.1 The name derives from the lead character in Oscar Wilde’s play Lady Windermere’s Fan, which satirizes the strict morals and polite manners typical of the Victorian era in Great Britain.2
The patient with Lady Windermere syndrome is typically a thin, lean, well-mannered elderly woman who voluntarily suppresses her cough out of politeness. Suppression of the cough is thought to predispose to lung infection by allowing secretions to collect in the airways, especially in the right middle lobe, which has the longest and narrowest of the lobar bronchi.3,4
Symptoms of Lady Windermere syndrome include cough, sputum production, and fatigue similar to that of acute or chronic bronchitis. Dyspnea, fever, and hemoptysis are less common.5 The differential diagnosis for these symptoms is broad and includes asthma, chronic obstructive pulmonary disease, gastroesophageal reflux disease, pneumonia, bronchiectasis, cystic fibrosis, interstitial lung disease, postnasal drip, lung cancer, and heart failure.
A prospective cohort study by Kim et al6 yielded descriptions of typical patients with Lady Windermere syndrome. Patients were tall and lean, tended to have scoliosis, and more commonly had pectus excavatum or mitral valve prolapse; 95% were women, 91% were white, and the average age was 60. The morphologic features are thought to contribute to impaired clearance of airway secretions by altered mechanics during coughing.
HALLMARKS ON IMAGING
Kim et al6 reported that the most common findings on lung imaging in nontuberculous mycobacterial infection were bronchiectasis involving the right middle lobe (90%), nodules involving the right lower lobe (73%) and right middle lobe (71%), and, less commonly, a cavitary infiltrate involving the right upper lobe (17%) or right middle lobe (10%).
Key findings on imaging in Lady Windermere syndrome include opacities and “cylindrical bronchiectasis” predominantly involving the right middle lobe or lingula.5 Bronchiolar inflammation in response to nontuberculous mycobacterial infection may cause a nodular appearance, often progressing to a tree-in-bud appearance on CT.
Other diagnostic considerations for tree-in-bud appearance on CT include fungal, viral, or other bacterial infection, aspiration pneumonitis, inhalation of a foreign substance, cystic fibrosis, rheumatoid arthritis, SjÖgren syndrome, bronchiolitis obliterans, and neoplastic disease.
CURRENT TREATMENT OPTIONS
Treatment of nontuberculous mycobacterial lung infection, including Lady Windermere syndrome, is not necessary in every case, given the variability in clinical symptoms and in disease progression. Patients with progressive symptoms or radiographic changes should be considered candidates for treatment.
Management is directed at the underlying infection. M avium-intracellulare is ubiquitous in the environment, including in soil and water, and it has been reported as the most common pathogen in nontuberculous mycobacterial lung infection.7
Nodular-bronchiectatic nontuberculous mycobacterial lung disease typically progresses more slowly than fibrocavitary disease. For patients with nodular-bronchiectatic disease, follow-up over months or years may be needed before clinical or radiographic changes become apparent.
When treatment is indicated for nodular-bronchiectatic nontuberculous mycobacterial lung infection, it should include a macrolide antibiotic, ethambutol, and rifampin.7,8 Monotherapy with a macrolide is not recommended because of the risk of macrolide resistance. Addition of an aminoglycoside may be considered when treating fibrocavitary disease or widespread nodular bronchiectatic disease.
Management of bronchiectasis, when present, includes chest physiotherapy, pulmonary hygiene therapy, and awareness of the predisposition for nonmycobacterial lung infection. The decision to prescribe antimicrobials should take into consideration the risks and benefits for each patient.
Because treatment involves multidrug regimens, drug interactions and adverse effects need to be considered and monitored, especially in elderly patients, who may already be taking multiple medications. Treatment should be continued until a patient has negative sputum cultures for acid-fast bacilli while on therapy, for 1 year.
A 75-year-old woman was referred to our pulmonary clinic with a 4-year history of intermittent episodes of persistent cough, occasionally productive of sputum, and mild exertional dyspnea. She had been treated with azithromycin for presumed community-acquired pneumonia, and her symptoms had initially improved. Subsequently, she experienced discrete, recurrent episodes of “bronchitis,” with productive cough and mild exertional dyspnea. Testing for latent tuberculosis had been negative. She reported a 10-pack-year smoking history in the remote past.
Her medical history included asthma, atrial fibrillation, gastroesophageal reflux disorder, hyperlipidemia, osteopenia, hypothyroidism, and allergic rhinitis. Her current medications were metoprolol, propafenone, and warfarin.
ABNORMALITIES ON PREVIOUS IMAGING
Computed tomography (CT) in April 2010 had revealed scattered linear, nodular, and “tree-in-bud” opacities involving the bilateral apices and the upper, middle, and lower lobes of the right lung, suggestive of bronchiolitis. Mild bronchiectasis had also been noted (Figure 1). Chest radiography had demonstrated signs of bronchiectasis and several scattered nodules (Figure 2). These abnormalities were still present on another CT scan in May 2013.
The patient had not undergone bronchoscopy before she was referred to our clinic.
WORKUP AT OUR CLINIC
On examination, the patient was lean, with a body mass index of 20.53 kg/m2. She appeared calm, well-groomed, and well-dressed, and had a very polite manner. When she coughed, she tried to suppress it, as if she were self-conscious about it. Her heart rhythm was irregularly irregular with a normal rate.
Expectorated sputum samples were obtained. Stains for acid-fast bacilli were negative, but three cultures were positive for acid-fast bacilli consistent with Mycobacterium avium-intracellulare. Serologic studies were negative for fungal infection and immunoglobulin deficiency.
Based on her symptoms and on the findings of imaging studies and sputum culture, we arrived at the diagnosis of nontuberculous mycobacterial lung infection, specifically, Lady Windermere syndrome.
NONTUBERCULOUS MYOCOBACTERIAL LUNG INFECTION
The diagnosis of nontuberculous mycobacterial lung infection is based on respiratory symptoms, findings on imaging (eg, nodular or cavitary opacities on radiography, or multifocal bronchiectasis and multiple small nodules on CT), and a positive culture for nontuberculous mycobacterial infection in more than two specimens of expectorated sputum or in more than one specimen from bronchoalveolar lavage. Lung biopsy with tissue culture is another way to confirm the diagnosis.
LADY WINDERMERE SYNDROME
Lady Windermere syndrome was described more than 20 years ago.1 The name derives from the lead character in Oscar Wilde’s play Lady Windermere’s Fan, which satirizes the strict morals and polite manners typical of the Victorian era in Great Britain.2
The patient with Lady Windermere syndrome is typically a thin, lean, well-mannered elderly woman who voluntarily suppresses her cough out of politeness. Suppression of the cough is thought to predispose to lung infection by allowing secretions to collect in the airways, especially in the right middle lobe, which has the longest and narrowest of the lobar bronchi.3,4
Symptoms of Lady Windermere syndrome include cough, sputum production, and fatigue similar to that of acute or chronic bronchitis. Dyspnea, fever, and hemoptysis are less common.5 The differential diagnosis for these symptoms is broad and includes asthma, chronic obstructive pulmonary disease, gastroesophageal reflux disease, pneumonia, bronchiectasis, cystic fibrosis, interstitial lung disease, postnasal drip, lung cancer, and heart failure.
A prospective cohort study by Kim et al6 yielded descriptions of typical patients with Lady Windermere syndrome. Patients were tall and lean, tended to have scoliosis, and more commonly had pectus excavatum or mitral valve prolapse; 95% were women, 91% were white, and the average age was 60. The morphologic features are thought to contribute to impaired clearance of airway secretions by altered mechanics during coughing.
HALLMARKS ON IMAGING
Kim et al6 reported that the most common findings on lung imaging in nontuberculous mycobacterial infection were bronchiectasis involving the right middle lobe (90%), nodules involving the right lower lobe (73%) and right middle lobe (71%), and, less commonly, a cavitary infiltrate involving the right upper lobe (17%) or right middle lobe (10%).
Key findings on imaging in Lady Windermere syndrome include opacities and “cylindrical bronchiectasis” predominantly involving the right middle lobe or lingula.5 Bronchiolar inflammation in response to nontuberculous mycobacterial infection may cause a nodular appearance, often progressing to a tree-in-bud appearance on CT.
Other diagnostic considerations for tree-in-bud appearance on CT include fungal, viral, or other bacterial infection, aspiration pneumonitis, inhalation of a foreign substance, cystic fibrosis, rheumatoid arthritis, SjÖgren syndrome, bronchiolitis obliterans, and neoplastic disease.
CURRENT TREATMENT OPTIONS
Treatment of nontuberculous mycobacterial lung infection, including Lady Windermere syndrome, is not necessary in every case, given the variability in clinical symptoms and in disease progression. Patients with progressive symptoms or radiographic changes should be considered candidates for treatment.
Management is directed at the underlying infection. M avium-intracellulare is ubiquitous in the environment, including in soil and water, and it has been reported as the most common pathogen in nontuberculous mycobacterial lung infection.7
Nodular-bronchiectatic nontuberculous mycobacterial lung disease typically progresses more slowly than fibrocavitary disease. For patients with nodular-bronchiectatic disease, follow-up over months or years may be needed before clinical or radiographic changes become apparent.
When treatment is indicated for nodular-bronchiectatic nontuberculous mycobacterial lung infection, it should include a macrolide antibiotic, ethambutol, and rifampin.7,8 Monotherapy with a macrolide is not recommended because of the risk of macrolide resistance. Addition of an aminoglycoside may be considered when treating fibrocavitary disease or widespread nodular bronchiectatic disease.
Management of bronchiectasis, when present, includes chest physiotherapy, pulmonary hygiene therapy, and awareness of the predisposition for nonmycobacterial lung infection. The decision to prescribe antimicrobials should take into consideration the risks and benefits for each patient.
Because treatment involves multidrug regimens, drug interactions and adverse effects need to be considered and monitored, especially in elderly patients, who may already be taking multiple medications. Treatment should be continued until a patient has negative sputum cultures for acid-fast bacilli while on therapy, for 1 year.
- Reich JM, Johnson RE. Mycobacterium avium complex pulmonary disease presenting as an isolated lingular or middle lobe pattern. The Lady Windermere syndrome. Chest 1992; 101:1605–1609.
- Kasthoori JJ, Liam CK, Wastie ML. Lady Windermere syndrome: an inappropriate eponym for an increasingly important condition. Singapore Med J 2008; 49:e47–e49.
- Dhillon SS, Watanakunakorn C. Lady Windermere syndrome: middle lobe bronchiectasis and mycobacterium avium complex infection due to voluntary cough suppression. Clin Infect Dis 2000; 30:572–575.
- Reich JM. Pathogenesis of Lady Windermere syndrome. Scand J Infect Dis 2012; 44:1–2.
- Glassroth J. Pulmonary disease due to nontuberculous mycobacteria. Chest 2008; 133:243–251.
- Kim RD, Greenberg DE, Ehrmantraut ME, et al. Pulmonary nontuberculous mycobacterial disease: prospective study of a distinct preexisting syndrome. Am J Respir Crit Care Med 2008; 178:1066–1074.
- Griffith DE, Aksamit T, Brown-Elliott BA, et al; ATS Mycobacterial Diseases Subcommittee; American Thoracic Society; Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med 2007; 175:367–416.
- Mason RJ, Broaddus VC, Martin T, et al, editors. Murray and Nadel’s Textbook of Respiratory Medicine. 5th ed. Philadelphia, PA: Saunders; 2010.
- Reich JM, Johnson RE. Mycobacterium avium complex pulmonary disease presenting as an isolated lingular or middle lobe pattern. The Lady Windermere syndrome. Chest 1992; 101:1605–1609.
- Kasthoori JJ, Liam CK, Wastie ML. Lady Windermere syndrome: an inappropriate eponym for an increasingly important condition. Singapore Med J 2008; 49:e47–e49.
- Dhillon SS, Watanakunakorn C. Lady Windermere syndrome: middle lobe bronchiectasis and mycobacterium avium complex infection due to voluntary cough suppression. Clin Infect Dis 2000; 30:572–575.
- Reich JM. Pathogenesis of Lady Windermere syndrome. Scand J Infect Dis 2012; 44:1–2.
- Glassroth J. Pulmonary disease due to nontuberculous mycobacteria. Chest 2008; 133:243–251.
- Kim RD, Greenberg DE, Ehrmantraut ME, et al. Pulmonary nontuberculous mycobacterial disease: prospective study of a distinct preexisting syndrome. Am J Respir Crit Care Med 2008; 178:1066–1074.
- Griffith DE, Aksamit T, Brown-Elliott BA, et al; ATS Mycobacterial Diseases Subcommittee; American Thoracic Society; Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med 2007; 175:367–416.
- Mason RJ, Broaddus VC, Martin T, et al, editors. Murray and Nadel’s Textbook of Respiratory Medicine. 5th ed. Philadelphia, PA: Saunders; 2010.
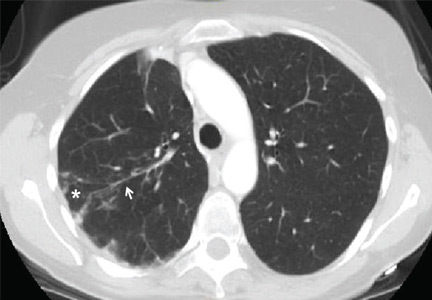
Herpes zoster triplex
A 77-year-old man presented with a 4-day history of painful eruptions on the left chest, right lower groin, and left thigh. He had been taking oral prednisolone 16 mg daily for interstitial pneumonia for 5 years. Ten days earlier, he had started to feel a stinging pain in these areas, but without eruptions.
Physical examination showed several grouped erythematous vesicles in the T2 dermatome of the left chest, L1 dermatome of the right groin, and L2 dermatome of the left upper anterior thigh (Figure 1).
Based on the presentation and a Tzanck smear of the lesions, a diagnosis of preherpetic neuralgia with herpes zoster triplex was made. The patient received intravenous acyclovir 750 mg/day for 7 days and continued to take the prednisolone. The lesions improved within 1 month, leaving scarring but no postherpetic neuralgia.
PREHERPETIC NEURALGIA
Herpes zoster usually occurs unilaterally in a single dermatome, with dermatomal pain appearing before the rash.1 Preherpetic neuralgia may be misdiagnosed as myocardial infarction or renal colic, especially in a case of zoster sine herpete.
Making the diagnosis of preherpetic neuralgia in our patient was difficult because it occurred simultaneously in three dermatomes. At first, his symptoms were suspected of being a recurrence of his past illnesses, including aortic dissection, gallstones, and diverticulitis. Anti-varicella-zoster virus immunoglobulin (Ig) M antibody was not detected, and the IgG antibody titer did not increase.
It has been suggested that cellular immunity is more important than humoral immunity for the surveillance and control of reactivations of herpes viruses. Risk factors for reactivation are increasing age, cancer, acquired immunodeficiency syndrome, and immunosuppressive medications.2,3 In addition, varicella-zoster virus can cause atypical lesions, including recurrent chickenpox, single-dermatomal herpes zoster with scattered rash, and herpes zoster in multiple dermatomes.4
Clinical suspicion for herpes zoster is important in the differential diagnosis of acute pain of uncertain origin, even if it occurs in multiple dermatomes in an immunocompromised patient.
- James WD, Berger TG, Elston DM. Andrew’s Diseases of the Skin. Clinical Dermatology, 10th ed. Philadelphia, PA: WB Saunders; 2006.
- Vu AQ, Radonich MA, Heald PW. Herpes zoster in seven disparate dermatomes (zoster multiplex): report of a case and review of literature. J Am Acad Dermatol 1999; 40:868–869.
- Failla V, Jacques J, Castronovo C, Nikkels AF. Herpes zoster in patients treated with biologicals. Dermatology 2012; 224:251–256.
- Kennedy PG, Steiner I. A molecular and cellular model to explain the differences in reactivation from latency by herpes simplex and varicella-zoster viruses. Neuropathol Appl Neurobiol 1994; 20:368–374.
A 77-year-old man presented with a 4-day history of painful eruptions on the left chest, right lower groin, and left thigh. He had been taking oral prednisolone 16 mg daily for interstitial pneumonia for 5 years. Ten days earlier, he had started to feel a stinging pain in these areas, but without eruptions.
Physical examination showed several grouped erythematous vesicles in the T2 dermatome of the left chest, L1 dermatome of the right groin, and L2 dermatome of the left upper anterior thigh (Figure 1).
Based on the presentation and a Tzanck smear of the lesions, a diagnosis of preherpetic neuralgia with herpes zoster triplex was made. The patient received intravenous acyclovir 750 mg/day for 7 days and continued to take the prednisolone. The lesions improved within 1 month, leaving scarring but no postherpetic neuralgia.
PREHERPETIC NEURALGIA
Herpes zoster usually occurs unilaterally in a single dermatome, with dermatomal pain appearing before the rash.1 Preherpetic neuralgia may be misdiagnosed as myocardial infarction or renal colic, especially in a case of zoster sine herpete.
Making the diagnosis of preherpetic neuralgia in our patient was difficult because it occurred simultaneously in three dermatomes. At first, his symptoms were suspected of being a recurrence of his past illnesses, including aortic dissection, gallstones, and diverticulitis. Anti-varicella-zoster virus immunoglobulin (Ig) M antibody was not detected, and the IgG antibody titer did not increase.
It has been suggested that cellular immunity is more important than humoral immunity for the surveillance and control of reactivations of herpes viruses. Risk factors for reactivation are increasing age, cancer, acquired immunodeficiency syndrome, and immunosuppressive medications.2,3 In addition, varicella-zoster virus can cause atypical lesions, including recurrent chickenpox, single-dermatomal herpes zoster with scattered rash, and herpes zoster in multiple dermatomes.4
Clinical suspicion for herpes zoster is important in the differential diagnosis of acute pain of uncertain origin, even if it occurs in multiple dermatomes in an immunocompromised patient.
A 77-year-old man presented with a 4-day history of painful eruptions on the left chest, right lower groin, and left thigh. He had been taking oral prednisolone 16 mg daily for interstitial pneumonia for 5 years. Ten days earlier, he had started to feel a stinging pain in these areas, but without eruptions.
Physical examination showed several grouped erythematous vesicles in the T2 dermatome of the left chest, L1 dermatome of the right groin, and L2 dermatome of the left upper anterior thigh (Figure 1).
Based on the presentation and a Tzanck smear of the lesions, a diagnosis of preherpetic neuralgia with herpes zoster triplex was made. The patient received intravenous acyclovir 750 mg/day for 7 days and continued to take the prednisolone. The lesions improved within 1 month, leaving scarring but no postherpetic neuralgia.
PREHERPETIC NEURALGIA
Herpes zoster usually occurs unilaterally in a single dermatome, with dermatomal pain appearing before the rash.1 Preherpetic neuralgia may be misdiagnosed as myocardial infarction or renal colic, especially in a case of zoster sine herpete.
Making the diagnosis of preherpetic neuralgia in our patient was difficult because it occurred simultaneously in three dermatomes. At first, his symptoms were suspected of being a recurrence of his past illnesses, including aortic dissection, gallstones, and diverticulitis. Anti-varicella-zoster virus immunoglobulin (Ig) M antibody was not detected, and the IgG antibody titer did not increase.
It has been suggested that cellular immunity is more important than humoral immunity for the surveillance and control of reactivations of herpes viruses. Risk factors for reactivation are increasing age, cancer, acquired immunodeficiency syndrome, and immunosuppressive medications.2,3 In addition, varicella-zoster virus can cause atypical lesions, including recurrent chickenpox, single-dermatomal herpes zoster with scattered rash, and herpes zoster in multiple dermatomes.4
Clinical suspicion for herpes zoster is important in the differential diagnosis of acute pain of uncertain origin, even if it occurs in multiple dermatomes in an immunocompromised patient.
- James WD, Berger TG, Elston DM. Andrew’s Diseases of the Skin. Clinical Dermatology, 10th ed. Philadelphia, PA: WB Saunders; 2006.
- Vu AQ, Radonich MA, Heald PW. Herpes zoster in seven disparate dermatomes (zoster multiplex): report of a case and review of literature. J Am Acad Dermatol 1999; 40:868–869.
- Failla V, Jacques J, Castronovo C, Nikkels AF. Herpes zoster in patients treated with biologicals. Dermatology 2012; 224:251–256.
- Kennedy PG, Steiner I. A molecular and cellular model to explain the differences in reactivation from latency by herpes simplex and varicella-zoster viruses. Neuropathol Appl Neurobiol 1994; 20:368–374.
- James WD, Berger TG, Elston DM. Andrew’s Diseases of the Skin. Clinical Dermatology, 10th ed. Philadelphia, PA: WB Saunders; 2006.
- Vu AQ, Radonich MA, Heald PW. Herpes zoster in seven disparate dermatomes (zoster multiplex): report of a case and review of literature. J Am Acad Dermatol 1999; 40:868–869.
- Failla V, Jacques J, Castronovo C, Nikkels AF. Herpes zoster in patients treated with biologicals. Dermatology 2012; 224:251–256.
- Kennedy PG, Steiner I. A molecular and cellular model to explain the differences in reactivation from latency by herpes simplex and varicella-zoster viruses. Neuropathol Appl Neurobiol 1994; 20:368–374.