User login
Positive Outcomes Following a Multidisciplinary Approach in the Diagnosis and Prevention of Hospital Delirium
From the Department of Neurology, Cedars-Sinai Medical Center, Los Angeles, CA (Drs. Ching, Darwish, Li, Wong, Simpson, and Funk), the Department of Anesthesia, Cedars-Sinai Medical Center, Los Angeles, CA (Keith Siegel), and the Department of Psychiatry, Cedars-Sinai Medical Center, Los Angeles, CA (Dr. Bamgbose).
Objectives: To reduce the incidence and duration of delirium among patients in a hospital ward through standardized delirium screening tools and nonpharmacologic interventions. To advance nursing-focused education on delirium-prevention strategies. To measure the efficacy of the interventions with the aim of reproducing best practices.
Background: Delirium is associated with poor patient outcomes but may be preventable in a significant percentage of hospitalized patients.
Methods: Following nursing-focused education to prevent delirium, we prospectively evaluated patient care outcomes in a consecutive series of patients who were admitted to a hospital medical-surgical ward within a 25-week period. All patients who had at least 1 Confusion Assessment Method (CAM) documented by a nurse during hospitalization met our inclusion criteria (N = 353). Standards for Quality Improvement Reporting Excellence guidelines were adhered to.
Results: There were 187 patients in the control group, and 166 in the postintervention group. Compared to the control group, the postintervention group had a significant decrease in the incidence of delirium during hospitalization (14.4% vs 4.2%) and a significant decrease in the mean percentage of tested nursing shifts with 1 or more positive CAM (4.9% vs 1.1%). Significant differences in secondary outcomes between the control and postintervention groups included median length of stay (6 days vs 4 days), mean length of stay (8.5 days vs 5.9 days), and use of an indwelling urinary catheter (9.1% vs 2.4%).
Conclusion: A multimodal strategy involving nursing-focused training and nonpharmacologic interventions to address hospital delirium is associated with improved patient care outcomes and nursing confidence. Nurses play an integral role in the early recognition and prevention of hospital delirium, which directly translates to reducing burdens in both patient functionality and health care costs.
Delirium is a disorder characterized by inattention and acute changes in cognition. It is defined by the American Psychiatric Association’s fifth edition of the Diagnostic and Statistical Manual of Mental Disorders as a disturbance in attention, awareness, and cognition over hours to a few days that is not better explained by a preexisting, established, or other evolving neurocognitive disorder.1 Delirium is common yet often under-recognized among hospitalized patients, particularly in the elderly. The incidence of delirium in elderly patients on admission is estimated to be 11% to 25%, and an additional 29% to 31% of elderly patients will develop delirium during the hospitalization.2 Delirium costs the health care system an estimated $38 billion to $152 billion per year.3 It is associated with negative outcomes, such as increased new placements to nursing homes, increased mortality, increased risk of dementia, and further cognitive deterioration among patients with dementia.4-6
Despite its prevalence, delirium may be preventable in a significant percentage of hospitalized patients. Targeted intervention strategies, such as frequent reorientation, maximizing sleep, early mobilization, restricting use of psychoactive medications, and addressing hearing or vision impairment, have been demonstrated to significantly reduce the incidence of hospital delirium.7,8 To achieve these goals, we explored the use of a multimodal strategy centered on nursing education. We integrated consistent, standardized delirium screening and nonpharmacologic interventions as part of a preventative protocol to reduce the incidence of delirium in the hospital ward.
Methods
We evaluated a consecutive series of patients who were admitted to a designated hospital medical-surgical ward within a 25-week period between October 2019 and April 2020. All patients during this period who had at least 1 Confusion Assessment Method (CAM) documented by a nurse during hospitalization met our inclusion criteria. Patients who did not have a CAM documented were excluded from the analysis. Delirium was defined according to the CAM diagnostic algorithm.9
Core nursing staff regularly assigned to the ward completed a multimodal training program designed to improve recognition, documentation, and prevention of hospital delirium. Prior to the training, the nurses completed a 5-point Likert scale survey assessing their level of confidence with recognizing delirium risk factors, preventing delirium, addressing delirium, utilizing the CAM tool, and educating others about delirium. Nurses completed the same survey after the study period ended.
The training curriculum for nurses began with an online module reviewing the epidemiology and risk factors for delirium. Nurses then participated in a series of in-service training sessions led by a team of physicians, during which the CAM and nonpharmacologic delirium prevention measures were reviewed then practiced first-hand. Nursing staff attended an in-person lecture reviewing the current body of literature on delirium risk factors and effective nursing interventions. After formal training was completed, nurses were instructed to document CAM screens

Patients admitted to the hospital unit from the start of the training program (week 1) until the order set was made available (week 15) constituted our control group. The postintervention study group consisted of patients admitted for 10 weeks after the completion of the interventions (weeks 16-25). A timeline of the study events is shown in Figure 1.

Patient demographics and hospital-stay metrics determined a priori were attained via the Cedars-Sinai Enterprise Information Services core. Age, sex, medical history, and incidence of surgery with anesthesia during hospitalization were recorded. The Charlson Comorbidity Index was calculated from patients’ listed diagnoses following discharge. Primary outcomes included incidence of patients with delirium during hospitalization, percentage of tested shifts with positive CAM screens, length of hospital stay, and survival. Secondary outcomes included measures associated with delirium, including the use of chemical restraints, physical restraints, sitters, indwelling urinary catheters, and new psychiatry and neurology consults. Chemical restraints were defined as administration of a new antipsychotic medication or benzodiazepine for the specific indication of hyperactive delirium or agitation.
Statistical analysis was conducted by a statistician, using R version 3.6.3.10P values of < .05 were considered significant. Categorical variables were analyzed using Fisher’s exact test. Continuous variables were analyzed with Welch’s t-test or, for highly skewed continuous variables, with Wilcoxon rank-sum test or Mood’s median test. All patient data were anonymized and stored securely in accordance with institutional guidelines.
Our project was deemed to represent nonhuman subject research and therefore did not require Institutional Review Board (IRB) approval upon review by our institution’s IRB committee and Office of Research Compliance and Quality Improvement. Standards for Quality Improvement Reporting Excellence (SQUIRE 2.0) guidelines were adhered to (Supplementary File can be found at mdedge.com/jcomjournal).
Results
We evaluated 353 patients who met our inclusion criteria: 187 in the control group, and 166 in the postintervention group. Ten patients were readmitted to the ward after their initial discharge; only the initial admission encounters were included in our analysis. Median age, sex, median Charlson Comorbidity Index, and incidence of surgery with anesthesia during hospitalization were comparable between the control and postintervention groups and are summarized in Table 2.

In the control group, 1572 CAMs were performed, with 74 positive CAMs recorded among 27 patients with delirium. In the postintervention group, 1298 CAMs were performed, with 12 positive CAMs recorded among 7 patients with delirium (Figure 2). Primary and secondary outcomes, as well as CAM compliance measures, are summarized in Table 3.

Compared to the control group, the postintervention group had a significant decrease in the incidence of delirium during hospitalization (14.4% vs 4.2%, P = .002) and a significant decrease in the mean percentage of tested nursing shifts with 1 or more positive CAM (4.9% vs 1.1%, P = .002). Significant differences in secondary outcomes between the control and postintervention groups included median length of stay (6 days vs 4 days, P = .004), mean length of stay (8.5 days vs 5.9 days, P = .003), and use of an indwelling urinary catheter (9.1% vs 2.4%, P = .012). There was a trend towards decreased incidence of chemical restraints and psychiatry consults, which did not reach statistical significance. Differences in mortality during hospitalization, physical restraint use, and sitter use could not be assessed due to low incidence.

Compliance with nursing CAM assessments was evaluated. Compared to the control group, the postintervention group saw a significant increase in the percentage of shifts with a CAM performed (54.7% vs 69.1%, P < .001). The median and mean number of CAMs performed per patient were similar between the control and postintervention groups.
Results of nursing surveys completed before and after the training program are listed in Table 4. After training, nurses had a greater level of confidence with recognizing delirium risk factors, preventing delirium, addressing delirium, utilizing the CAM tool, and educating others about delirium.

Discussion
Our study utilized a standardized delirium assessment tool to compare patient cohorts before and after nurse-targeted training interventions on delirium recognition and prevention. Our interventions emphasized nonpharmacologic intervention strategies, which are recommended as first-line in the management of patients with delirium.11 Patients were not excluded from the analysis based on preexisting medical conditions or recent surgery with anesthesia, to allow for conditions that are representative of community hospitals. We also did not use an inclusion criterion based on age; however, the majority of our patients were greater than 70 years old, representing those at highest risk for delirium.2 Significant outcomes among patients in the postintervention group include decreased incidence of delirium, lower average length of stay, decreased indwelling urinary catheter use, and increased compliance with delirium screening by nursing staff.
While the study’s focus was primarily on delirium prevention rather than treatment, these strategies may also have conferred the benefit of reversing delirium symptoms. In addition to measuring incidence of delirium, our primary outcome of percentage of tested shifts with 1 or more positive CAM was intended to assess the overall duration in which patients had delirium during their hospitalization. The reduction in shifts with positive CAMs observed in the postintervention group is notable, given that a significant percentage of patients with hospital delirium have the potential for symptom reversibility.12
Multiple studies have shown that admitted patients who develop delirium experience prolonged hospital stays, often up to 5 to 10 days longer.12-14 The decreased incidence and duration of delirium in our postintervention group is a reasonable explanation for the observed decrease in average length of stay. Our study is in line with previously documented initiatives that show that nonpharmacologic interventions can effectively address downstream health and fiscal sequelae of hospital delirium. For example, a volunteer-based initiative named the Hospital Elder Life Program, from which elements in our order set were modeled after, demonstrated significant reductions in delirium incidence, length of stay, and health care costs.14-16 Other initiatives that focused on educational training for nurses to assess and prevent delirium have also demonstrated similar positive results.17-19 Our study provides a model for effective nursing-focused education that can be reproduced in the hospital setting.
Unlike some other studies, which identified delirium based only on physician assessments, our initiative utilized the CAM performed by floor nurses to identify delirium. While this method
Our study demonstrated an increase in the overall compliance with the CAM screening during the postintervention period, which is significant given the under-recognition of delirium by health care professionals.20 We attribute this increase to greater realized importance and a higher level of confidence from nursing staff in recognizing and addressing delirium, as supported by survey data. While the increased screening of patients should be considered a positive outcome, it also poses the possibility that the observed decrease in delirium incidence in the postintervention group was in fact due to more CAMs performed on patients without delirium. Likewise, nurses may have become more adept at recognizing true delirium, as opposed to delirium mimics, in the latter period of the study.
Perhaps the greatest limitation of our study is the variability in performing and recording CAMs, as some patients had multiple CAMs recorded while others did not have any CAMs recorded. This may have been affected in part by the increase in COVID-19 cases in our hospital towards the latter half of the study, which resulted in changes in nursing assignments as well as patient comorbidities in ways that cannot be easily quantified. Given the limited size of our patient cohorts, certain outcomes, such as the use of sitters, physical restraints, and in-hospital mortality, were unable to be assessed for changes statistically. Causative relationships between our interventions and associated outcome measures are necessarily limited in a binary comparison between control and postintervention groups.
Within these limitations, our study demonstrates promising results in core dimensions of patient care. We anticipate further quality improvement initiatives involving greater numbers of nursing staff and patients to better quantify the impact of nonpharmacologic nursing-centered interventions for preventing hospital delirium.
Conclusion
A multimodal strategy involving nursing-focused training and nonpharmacologic interventions to address hospital delirium is associated with improved patient care outcomes and nursing confidence. Nurses play an integral role in the early recognition and prevention of hospital delirium, which directly translates to reducing burdens in both patient functionality and health care costs. Education and tools to equip nurses to perform standardized delirium screening and interventions should be prioritized.
Acknowledgment: The authors thanks Olena Svetlov, NP, Oscar Abarca, Jose Chavez, and Jenita Gutierrez.
Corresponding author: Jason Ching, MD, Department of Neurology, Cedars-Sinai Medical Center, 8700 Beverly Blvd, Los Angeles, CA 90048; [email protected].
Financial disclosures: None.
Funding: This research was supported by NIH National Center for Advancing Translational Science (NCATS) UCLA CTSI Grant Number UL1TR001881.
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th edition. American Psychiatric Association; 2013.
2. Vasilevskis EE, Han JH, Hughes CG, et al. Epidemiology and risk factors for delirium across hospital settings. Best Pract Res Clin Anaesthesiol. 2012;26(3):277-287. doi:10.1016/j.bpa.2012.07003
3. Leslie DL, Marcantonio ER, Zhang Y, et al. One-year health care costs associated with delirium in the elderly population. Arch Intern Med. 2008;168(1):27-32. doi:10.1001/archinternmed.2007.4
4. McCusker J, Cole M, Abrahamowicz M, et al. Delirium predicts 12-month mortality. Arch Intern Med. 2002;162(4):457-463. doi:10.1001/archinte.162.4.457
5. Witlox J, Eurelings LS, de Jonghe JF, et al. Delirium in elderly patients and the risk of postdischarge mortality, institutionalization, and dementia: a meta-analysis. JAMA. 2010;304(4):443-451. doi:10.1001/jama.2010.1013
6. Gross AL, Jones RN, Habtemariam DA, et al. Delirium and long-term cognitive trajectory among persons with dementia. Arch Intern Med. 2012;172(17):1324-1331. doi:10.1001/archinternmed.2012.3203
7. Inouye SK. Prevention of delirium in hospitalized older patients: risk factors and targeted intervention strategies. Ann Med. 2000;32(4):257-263. doi:10.3109/07853890009011770
8. Siddiqi N, Harrison JK, Clegg A, et al. Interventions for preventing delirium in hospitalised non-ICU patients. Cochrane Database Syst Rev. 2016;3:CD005563. doi:10.1002/14651858.CD005563.pub3
9. Inouye SK, van Dyck CH, Alessi CA, et al. Clarifying confusion: the confusion assessment method. A new method for detection of delirium. Ann Intern Med. 1990;113(12):941-948. doi:10.7326/0003-4819-113-12-941
10. R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2017.
11. Fong TG, Tulebaev SR, Inouye SK. Delirium in elderly adults: diagnosis, prevention and treatment. Nat Rev Neurol. 2009;5(4):210-220. doi:10.1038/nrneurol.2009.24
12. Siddiqi N, House AO, Holmes JD. Occurrence and outcome of delirium in medical in-patients: a systematic literature review. Age Ageing. 2006;35(4):350-364. doi:10.1093/ageing/afl005
13. Ely EW, Shintani A, Truman B, et al. Delirium as a predictor of mortality in mechanically ventilated patients in the intensive care unit. JAMA. 2004;291(14):1753-1762. doi:10.1001/jama.291.14.1753
14. Chen CC, Lin MT, Tien YW, et al. Modified Hospital Elder Life Program: effects on abdominal surgery patients. J Am Coll Surg. 2011;213(2):245-252. doi:10.1016/j.jamcollsurg.2011.05.004
15. Zaubler TS, Murphy K, Rizzuto L, et al. Quality improvement and cost savings with multicomponent delirium interventions: replication of the Hospital Elder Life Program in a community hospital. Psychosomatics. 2013;54(3):219-226. doi:10.1016/j.psym.2013.01.010
16. Rubin FH, Neal K, Fenlon K, et al. Sustainability and scalability of the Hospital Elder Life Program at a community hospital. J Am Geriatr Soc. 2011;59(2):359-365. doi:10.1111/j.1532-5415.2010.03243.x
17. Milisen K, Foreman MD, Abraham IL, et al. A nurse-led interdisciplinary intervention program for delirium in elderly hip-fracture patients. J Am Geriatr Soc. 2001;49(5):523-532. doi:10.1046/j.1532-5415.2001.49109.x
18. Lundström M, Edlund A, Karlsson S, et al. A multifactorial intervention program reduces the duration of delirium, length of hospitalization, and mortality in delirious patients. J Am Geriatr Soc. 2005;53(4):622-628. doi:10.1111/j.1532-5415.2005.53210.x
19. Tabet N, Hudson S, Sweeney V, et al. An educational intervention can prevent delirium on acute medical wards. Age Ageing. 2005;34(2):152-156. doi:10.1093/ageing/afi0320. Han JH, Zimmerman EE, Cutler N, et al. Delirium in older emergency department patients: recognition, risk factors, and psychomotor subtypes. Acad Emerg Med. 2009;16(3):193-200. doi:10.1111/j.1553-2712.2008.00339.x
From the Department of Neurology, Cedars-Sinai Medical Center, Los Angeles, CA (Drs. Ching, Darwish, Li, Wong, Simpson, and Funk), the Department of Anesthesia, Cedars-Sinai Medical Center, Los Angeles, CA (Keith Siegel), and the Department of Psychiatry, Cedars-Sinai Medical Center, Los Angeles, CA (Dr. Bamgbose).
Objectives: To reduce the incidence and duration of delirium among patients in a hospital ward through standardized delirium screening tools and nonpharmacologic interventions. To advance nursing-focused education on delirium-prevention strategies. To measure the efficacy of the interventions with the aim of reproducing best practices.
Background: Delirium is associated with poor patient outcomes but may be preventable in a significant percentage of hospitalized patients.
Methods: Following nursing-focused education to prevent delirium, we prospectively evaluated patient care outcomes in a consecutive series of patients who were admitted to a hospital medical-surgical ward within a 25-week period. All patients who had at least 1 Confusion Assessment Method (CAM) documented by a nurse during hospitalization met our inclusion criteria (N = 353). Standards for Quality Improvement Reporting Excellence guidelines were adhered to.
Results: There were 187 patients in the control group, and 166 in the postintervention group. Compared to the control group, the postintervention group had a significant decrease in the incidence of delirium during hospitalization (14.4% vs 4.2%) and a significant decrease in the mean percentage of tested nursing shifts with 1 or more positive CAM (4.9% vs 1.1%). Significant differences in secondary outcomes between the control and postintervention groups included median length of stay (6 days vs 4 days), mean length of stay (8.5 days vs 5.9 days), and use of an indwelling urinary catheter (9.1% vs 2.4%).
Conclusion: A multimodal strategy involving nursing-focused training and nonpharmacologic interventions to address hospital delirium is associated with improved patient care outcomes and nursing confidence. Nurses play an integral role in the early recognition and prevention of hospital delirium, which directly translates to reducing burdens in both patient functionality and health care costs.
Delirium is a disorder characterized by inattention and acute changes in cognition. It is defined by the American Psychiatric Association’s fifth edition of the Diagnostic and Statistical Manual of Mental Disorders as a disturbance in attention, awareness, and cognition over hours to a few days that is not better explained by a preexisting, established, or other evolving neurocognitive disorder.1 Delirium is common yet often under-recognized among hospitalized patients, particularly in the elderly. The incidence of delirium in elderly patients on admission is estimated to be 11% to 25%, and an additional 29% to 31% of elderly patients will develop delirium during the hospitalization.2 Delirium costs the health care system an estimated $38 billion to $152 billion per year.3 It is associated with negative outcomes, such as increased new placements to nursing homes, increased mortality, increased risk of dementia, and further cognitive deterioration among patients with dementia.4-6
Despite its prevalence, delirium may be preventable in a significant percentage of hospitalized patients. Targeted intervention strategies, such as frequent reorientation, maximizing sleep, early mobilization, restricting use of psychoactive medications, and addressing hearing or vision impairment, have been demonstrated to significantly reduce the incidence of hospital delirium.7,8 To achieve these goals, we explored the use of a multimodal strategy centered on nursing education. We integrated consistent, standardized delirium screening and nonpharmacologic interventions as part of a preventative protocol to reduce the incidence of delirium in the hospital ward.
Methods
We evaluated a consecutive series of patients who were admitted to a designated hospital medical-surgical ward within a 25-week period between October 2019 and April 2020. All patients during this period who had at least 1 Confusion Assessment Method (CAM) documented by a nurse during hospitalization met our inclusion criteria. Patients who did not have a CAM documented were excluded from the analysis. Delirium was defined according to the CAM diagnostic algorithm.9
Core nursing staff regularly assigned to the ward completed a multimodal training program designed to improve recognition, documentation, and prevention of hospital delirium. Prior to the training, the nurses completed a 5-point Likert scale survey assessing their level of confidence with recognizing delirium risk factors, preventing delirium, addressing delirium, utilizing the CAM tool, and educating others about delirium. Nurses completed the same survey after the study period ended.
The training curriculum for nurses began with an online module reviewing the epidemiology and risk factors for delirium. Nurses then participated in a series of in-service training sessions led by a team of physicians, during which the CAM and nonpharmacologic delirium prevention measures were reviewed then practiced first-hand. Nursing staff attended an in-person lecture reviewing the current body of literature on delirium risk factors and effective nursing interventions. After formal training was completed, nurses were instructed to document CAM screens
Patients admitted to the hospital unit from the start of the training program (week 1) until the order set was made available (week 15) constituted our control group. The postintervention study group consisted of patients admitted for 10 weeks after the completion of the interventions (weeks 16-25). A timeline of the study events is shown in Figure 1.
Patient demographics and hospital-stay metrics determined a priori were attained via the Cedars-Sinai Enterprise Information Services core. Age, sex, medical history, and incidence of surgery with anesthesia during hospitalization were recorded. The Charlson Comorbidity Index was calculated from patients’ listed diagnoses following discharge. Primary outcomes included incidence of patients with delirium during hospitalization, percentage of tested shifts with positive CAM screens, length of hospital stay, and survival. Secondary outcomes included measures associated with delirium, including the use of chemical restraints, physical restraints, sitters, indwelling urinary catheters, and new psychiatry and neurology consults. Chemical restraints were defined as administration of a new antipsychotic medication or benzodiazepine for the specific indication of hyperactive delirium or agitation.
Statistical analysis was conducted by a statistician, using R version 3.6.3.10P values of < .05 were considered significant. Categorical variables were analyzed using Fisher’s exact test. Continuous variables were analyzed with Welch’s t-test or, for highly skewed continuous variables, with Wilcoxon rank-sum test or Mood’s median test. All patient data were anonymized and stored securely in accordance with institutional guidelines.
Our project was deemed to represent nonhuman subject research and therefore did not require Institutional Review Board (IRB) approval upon review by our institution’s IRB committee and Office of Research Compliance and Quality Improvement. Standards for Quality Improvement Reporting Excellence (SQUIRE 2.0) guidelines were adhered to (Supplementary File can be found at mdedge.com/jcomjournal).
Results
We evaluated 353 patients who met our inclusion criteria: 187 in the control group, and 166 in the postintervention group. Ten patients were readmitted to the ward after their initial discharge; only the initial admission encounters were included in our analysis. Median age, sex, median Charlson Comorbidity Index, and incidence of surgery with anesthesia during hospitalization were comparable between the control and postintervention groups and are summarized in Table 2.
In the control group, 1572 CAMs were performed, with 74 positive CAMs recorded among 27 patients with delirium. In the postintervention group, 1298 CAMs were performed, with 12 positive CAMs recorded among 7 patients with delirium (Figure 2). Primary and secondary outcomes, as well as CAM compliance measures, are summarized in Table 3.
Compared to the control group, the postintervention group had a significant decrease in the incidence of delirium during hospitalization (14.4% vs 4.2%, P = .002) and a significant decrease in the mean percentage of tested nursing shifts with 1 or more positive CAM (4.9% vs 1.1%, P = .002). Significant differences in secondary outcomes between the control and postintervention groups included median length of stay (6 days vs 4 days, P = .004), mean length of stay (8.5 days vs 5.9 days, P = .003), and use of an indwelling urinary catheter (9.1% vs 2.4%, P = .012). There was a trend towards decreased incidence of chemical restraints and psychiatry consults, which did not reach statistical significance. Differences in mortality during hospitalization, physical restraint use, and sitter use could not be assessed due to low incidence.
Compliance with nursing CAM assessments was evaluated. Compared to the control group, the postintervention group saw a significant increase in the percentage of shifts with a CAM performed (54.7% vs 69.1%, P < .001). The median and mean number of CAMs performed per patient were similar between the control and postintervention groups.
Results of nursing surveys completed before and after the training program are listed in Table 4. After training, nurses had a greater level of confidence with recognizing delirium risk factors, preventing delirium, addressing delirium, utilizing the CAM tool, and educating others about delirium.
Discussion
Our study utilized a standardized delirium assessment tool to compare patient cohorts before and after nurse-targeted training interventions on delirium recognition and prevention. Our interventions emphasized nonpharmacologic intervention strategies, which are recommended as first-line in the management of patients with delirium.11 Patients were not excluded from the analysis based on preexisting medical conditions or recent surgery with anesthesia, to allow for conditions that are representative of community hospitals. We also did not use an inclusion criterion based on age; however, the majority of our patients were greater than 70 years old, representing those at highest risk for delirium.2 Significant outcomes among patients in the postintervention group include decreased incidence of delirium, lower average length of stay, decreased indwelling urinary catheter use, and increased compliance with delirium screening by nursing staff.
While the study’s focus was primarily on delirium prevention rather than treatment, these strategies may also have conferred the benefit of reversing delirium symptoms. In addition to measuring incidence of delirium, our primary outcome of percentage of tested shifts with 1 or more positive CAM was intended to assess the overall duration in which patients had delirium during their hospitalization. The reduction in shifts with positive CAMs observed in the postintervention group is notable, given that a significant percentage of patients with hospital delirium have the potential for symptom reversibility.12
Multiple studies have shown that admitted patients who develop delirium experience prolonged hospital stays, often up to 5 to 10 days longer.12-14 The decreased incidence and duration of delirium in our postintervention group is a reasonable explanation for the observed decrease in average length of stay. Our study is in line with previously documented initiatives that show that nonpharmacologic interventions can effectively address downstream health and fiscal sequelae of hospital delirium. For example, a volunteer-based initiative named the Hospital Elder Life Program, from which elements in our order set were modeled after, demonstrated significant reductions in delirium incidence, length of stay, and health care costs.14-16 Other initiatives that focused on educational training for nurses to assess and prevent delirium have also demonstrated similar positive results.17-19 Our study provides a model for effective nursing-focused education that can be reproduced in the hospital setting.
Unlike some other studies, which identified delirium based only on physician assessments, our initiative utilized the CAM performed by floor nurses to identify delirium. While this method
Our study demonstrated an increase in the overall compliance with the CAM screening during the postintervention period, which is significant given the under-recognition of delirium by health care professionals.20 We attribute this increase to greater realized importance and a higher level of confidence from nursing staff in recognizing and addressing delirium, as supported by survey data. While the increased screening of patients should be considered a positive outcome, it also poses the possibility that the observed decrease in delirium incidence in the postintervention group was in fact due to more CAMs performed on patients without delirium. Likewise, nurses may have become more adept at recognizing true delirium, as opposed to delirium mimics, in the latter period of the study.
Perhaps the greatest limitation of our study is the variability in performing and recording CAMs, as some patients had multiple CAMs recorded while others did not have any CAMs recorded. This may have been affected in part by the increase in COVID-19 cases in our hospital towards the latter half of the study, which resulted in changes in nursing assignments as well as patient comorbidities in ways that cannot be easily quantified. Given the limited size of our patient cohorts, certain outcomes, such as the use of sitters, physical restraints, and in-hospital mortality, were unable to be assessed for changes statistically. Causative relationships between our interventions and associated outcome measures are necessarily limited in a binary comparison between control and postintervention groups.
Within these limitations, our study demonstrates promising results in core dimensions of patient care. We anticipate further quality improvement initiatives involving greater numbers of nursing staff and patients to better quantify the impact of nonpharmacologic nursing-centered interventions for preventing hospital delirium.
Conclusion
A multimodal strategy involving nursing-focused training and nonpharmacologic interventions to address hospital delirium is associated with improved patient care outcomes and nursing confidence. Nurses play an integral role in the early recognition and prevention of hospital delirium, which directly translates to reducing burdens in both patient functionality and health care costs. Education and tools to equip nurses to perform standardized delirium screening and interventions should be prioritized.
Acknowledgment: The authors thanks Olena Svetlov, NP, Oscar Abarca, Jose Chavez, and Jenita Gutierrez.
Corresponding author: Jason Ching, MD, Department of Neurology, Cedars-Sinai Medical Center, 8700 Beverly Blvd, Los Angeles, CA 90048; [email protected].
Financial disclosures: None.
Funding: This research was supported by NIH National Center for Advancing Translational Science (NCATS) UCLA CTSI Grant Number UL1TR001881.
From the Department of Neurology, Cedars-Sinai Medical Center, Los Angeles, CA (Drs. Ching, Darwish, Li, Wong, Simpson, and Funk), the Department of Anesthesia, Cedars-Sinai Medical Center, Los Angeles, CA (Keith Siegel), and the Department of Psychiatry, Cedars-Sinai Medical Center, Los Angeles, CA (Dr. Bamgbose).
Objectives: To reduce the incidence and duration of delirium among patients in a hospital ward through standardized delirium screening tools and nonpharmacologic interventions. To advance nursing-focused education on delirium-prevention strategies. To measure the efficacy of the interventions with the aim of reproducing best practices.
Background: Delirium is associated with poor patient outcomes but may be preventable in a significant percentage of hospitalized patients.
Methods: Following nursing-focused education to prevent delirium, we prospectively evaluated patient care outcomes in a consecutive series of patients who were admitted to a hospital medical-surgical ward within a 25-week period. All patients who had at least 1 Confusion Assessment Method (CAM) documented by a nurse during hospitalization met our inclusion criteria (N = 353). Standards for Quality Improvement Reporting Excellence guidelines were adhered to.
Results: There were 187 patients in the control group, and 166 in the postintervention group. Compared to the control group, the postintervention group had a significant decrease in the incidence of delirium during hospitalization (14.4% vs 4.2%) and a significant decrease in the mean percentage of tested nursing shifts with 1 or more positive CAM (4.9% vs 1.1%). Significant differences in secondary outcomes between the control and postintervention groups included median length of stay (6 days vs 4 days), mean length of stay (8.5 days vs 5.9 days), and use of an indwelling urinary catheter (9.1% vs 2.4%).
Conclusion: A multimodal strategy involving nursing-focused training and nonpharmacologic interventions to address hospital delirium is associated with improved patient care outcomes and nursing confidence. Nurses play an integral role in the early recognition and prevention of hospital delirium, which directly translates to reducing burdens in both patient functionality and health care costs.
Delirium is a disorder characterized by inattention and acute changes in cognition. It is defined by the American Psychiatric Association’s fifth edition of the Diagnostic and Statistical Manual of Mental Disorders as a disturbance in attention, awareness, and cognition over hours to a few days that is not better explained by a preexisting, established, or other evolving neurocognitive disorder.1 Delirium is common yet often under-recognized among hospitalized patients, particularly in the elderly. The incidence of delirium in elderly patients on admission is estimated to be 11% to 25%, and an additional 29% to 31% of elderly patients will develop delirium during the hospitalization.2 Delirium costs the health care system an estimated $38 billion to $152 billion per year.3 It is associated with negative outcomes, such as increased new placements to nursing homes, increased mortality, increased risk of dementia, and further cognitive deterioration among patients with dementia.4-6
Despite its prevalence, delirium may be preventable in a significant percentage of hospitalized patients. Targeted intervention strategies, such as frequent reorientation, maximizing sleep, early mobilization, restricting use of psychoactive medications, and addressing hearing or vision impairment, have been demonstrated to significantly reduce the incidence of hospital delirium.7,8 To achieve these goals, we explored the use of a multimodal strategy centered on nursing education. We integrated consistent, standardized delirium screening and nonpharmacologic interventions as part of a preventative protocol to reduce the incidence of delirium in the hospital ward.
Methods
We evaluated a consecutive series of patients who were admitted to a designated hospital medical-surgical ward within a 25-week period between October 2019 and April 2020. All patients during this period who had at least 1 Confusion Assessment Method (CAM) documented by a nurse during hospitalization met our inclusion criteria. Patients who did not have a CAM documented were excluded from the analysis. Delirium was defined according to the CAM diagnostic algorithm.9
Core nursing staff regularly assigned to the ward completed a multimodal training program designed to improve recognition, documentation, and prevention of hospital delirium. Prior to the training, the nurses completed a 5-point Likert scale survey assessing their level of confidence with recognizing delirium risk factors, preventing delirium, addressing delirium, utilizing the CAM tool, and educating others about delirium. Nurses completed the same survey after the study period ended.
The training curriculum for nurses began with an online module reviewing the epidemiology and risk factors for delirium. Nurses then participated in a series of in-service training sessions led by a team of physicians, during which the CAM and nonpharmacologic delirium prevention measures were reviewed then practiced first-hand. Nursing staff attended an in-person lecture reviewing the current body of literature on delirium risk factors and effective nursing interventions. After formal training was completed, nurses were instructed to document CAM screens
Patients admitted to the hospital unit from the start of the training program (week 1) until the order set was made available (week 15) constituted our control group. The postintervention study group consisted of patients admitted for 10 weeks after the completion of the interventions (weeks 16-25). A timeline of the study events is shown in Figure 1.
Patient demographics and hospital-stay metrics determined a priori were attained via the Cedars-Sinai Enterprise Information Services core. Age, sex, medical history, and incidence of surgery with anesthesia during hospitalization were recorded. The Charlson Comorbidity Index was calculated from patients’ listed diagnoses following discharge. Primary outcomes included incidence of patients with delirium during hospitalization, percentage of tested shifts with positive CAM screens, length of hospital stay, and survival. Secondary outcomes included measures associated with delirium, including the use of chemical restraints, physical restraints, sitters, indwelling urinary catheters, and new psychiatry and neurology consults. Chemical restraints were defined as administration of a new antipsychotic medication or benzodiazepine for the specific indication of hyperactive delirium or agitation.
Statistical analysis was conducted by a statistician, using R version 3.6.3.10P values of < .05 were considered significant. Categorical variables were analyzed using Fisher’s exact test. Continuous variables were analyzed with Welch’s t-test or, for highly skewed continuous variables, with Wilcoxon rank-sum test or Mood’s median test. All patient data were anonymized and stored securely in accordance with institutional guidelines.
Our project was deemed to represent nonhuman subject research and therefore did not require Institutional Review Board (IRB) approval upon review by our institution’s IRB committee and Office of Research Compliance and Quality Improvement. Standards for Quality Improvement Reporting Excellence (SQUIRE 2.0) guidelines were adhered to (Supplementary File can be found at mdedge.com/jcomjournal).
Results
We evaluated 353 patients who met our inclusion criteria: 187 in the control group, and 166 in the postintervention group. Ten patients were readmitted to the ward after their initial discharge; only the initial admission encounters were included in our analysis. Median age, sex, median Charlson Comorbidity Index, and incidence of surgery with anesthesia during hospitalization were comparable between the control and postintervention groups and are summarized in Table 2.
In the control group, 1572 CAMs were performed, with 74 positive CAMs recorded among 27 patients with delirium. In the postintervention group, 1298 CAMs were performed, with 12 positive CAMs recorded among 7 patients with delirium (Figure 2). Primary and secondary outcomes, as well as CAM compliance measures, are summarized in Table 3.
Compared to the control group, the postintervention group had a significant decrease in the incidence of delirium during hospitalization (14.4% vs 4.2%, P = .002) and a significant decrease in the mean percentage of tested nursing shifts with 1 or more positive CAM (4.9% vs 1.1%, P = .002). Significant differences in secondary outcomes between the control and postintervention groups included median length of stay (6 days vs 4 days, P = .004), mean length of stay (8.5 days vs 5.9 days, P = .003), and use of an indwelling urinary catheter (9.1% vs 2.4%, P = .012). There was a trend towards decreased incidence of chemical restraints and psychiatry consults, which did not reach statistical significance. Differences in mortality during hospitalization, physical restraint use, and sitter use could not be assessed due to low incidence.
Compliance with nursing CAM assessments was evaluated. Compared to the control group, the postintervention group saw a significant increase in the percentage of shifts with a CAM performed (54.7% vs 69.1%, P < .001). The median and mean number of CAMs performed per patient were similar between the control and postintervention groups.
Results of nursing surveys completed before and after the training program are listed in Table 4. After training, nurses had a greater level of confidence with recognizing delirium risk factors, preventing delirium, addressing delirium, utilizing the CAM tool, and educating others about delirium.
Discussion
Our study utilized a standardized delirium assessment tool to compare patient cohorts before and after nurse-targeted training interventions on delirium recognition and prevention. Our interventions emphasized nonpharmacologic intervention strategies, which are recommended as first-line in the management of patients with delirium.11 Patients were not excluded from the analysis based on preexisting medical conditions or recent surgery with anesthesia, to allow for conditions that are representative of community hospitals. We also did not use an inclusion criterion based on age; however, the majority of our patients were greater than 70 years old, representing those at highest risk for delirium.2 Significant outcomes among patients in the postintervention group include decreased incidence of delirium, lower average length of stay, decreased indwelling urinary catheter use, and increased compliance with delirium screening by nursing staff.
While the study’s focus was primarily on delirium prevention rather than treatment, these strategies may also have conferred the benefit of reversing delirium symptoms. In addition to measuring incidence of delirium, our primary outcome of percentage of tested shifts with 1 or more positive CAM was intended to assess the overall duration in which patients had delirium during their hospitalization. The reduction in shifts with positive CAMs observed in the postintervention group is notable, given that a significant percentage of patients with hospital delirium have the potential for symptom reversibility.12
Multiple studies have shown that admitted patients who develop delirium experience prolonged hospital stays, often up to 5 to 10 days longer.12-14 The decreased incidence and duration of delirium in our postintervention group is a reasonable explanation for the observed decrease in average length of stay. Our study is in line with previously documented initiatives that show that nonpharmacologic interventions can effectively address downstream health and fiscal sequelae of hospital delirium. For example, a volunteer-based initiative named the Hospital Elder Life Program, from which elements in our order set were modeled after, demonstrated significant reductions in delirium incidence, length of stay, and health care costs.14-16 Other initiatives that focused on educational training for nurses to assess and prevent delirium have also demonstrated similar positive results.17-19 Our study provides a model for effective nursing-focused education that can be reproduced in the hospital setting.
Unlike some other studies, which identified delirium based only on physician assessments, our initiative utilized the CAM performed by floor nurses to identify delirium. While this method
Our study demonstrated an increase in the overall compliance with the CAM screening during the postintervention period, which is significant given the under-recognition of delirium by health care professionals.20 We attribute this increase to greater realized importance and a higher level of confidence from nursing staff in recognizing and addressing delirium, as supported by survey data. While the increased screening of patients should be considered a positive outcome, it also poses the possibility that the observed decrease in delirium incidence in the postintervention group was in fact due to more CAMs performed on patients without delirium. Likewise, nurses may have become more adept at recognizing true delirium, as opposed to delirium mimics, in the latter period of the study.
Perhaps the greatest limitation of our study is the variability in performing and recording CAMs, as some patients had multiple CAMs recorded while others did not have any CAMs recorded. This may have been affected in part by the increase in COVID-19 cases in our hospital towards the latter half of the study, which resulted in changes in nursing assignments as well as patient comorbidities in ways that cannot be easily quantified. Given the limited size of our patient cohorts, certain outcomes, such as the use of sitters, physical restraints, and in-hospital mortality, were unable to be assessed for changes statistically. Causative relationships between our interventions and associated outcome measures are necessarily limited in a binary comparison between control and postintervention groups.
Within these limitations, our study demonstrates promising results in core dimensions of patient care. We anticipate further quality improvement initiatives involving greater numbers of nursing staff and patients to better quantify the impact of nonpharmacologic nursing-centered interventions for preventing hospital delirium.
Conclusion
A multimodal strategy involving nursing-focused training and nonpharmacologic interventions to address hospital delirium is associated with improved patient care outcomes and nursing confidence. Nurses play an integral role in the early recognition and prevention of hospital delirium, which directly translates to reducing burdens in both patient functionality and health care costs. Education and tools to equip nurses to perform standardized delirium screening and interventions should be prioritized.
Acknowledgment: The authors thanks Olena Svetlov, NP, Oscar Abarca, Jose Chavez, and Jenita Gutierrez.
Corresponding author: Jason Ching, MD, Department of Neurology, Cedars-Sinai Medical Center, 8700 Beverly Blvd, Los Angeles, CA 90048; [email protected].
Financial disclosures: None.
Funding: This research was supported by NIH National Center for Advancing Translational Science (NCATS) UCLA CTSI Grant Number UL1TR001881.
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th edition. American Psychiatric Association; 2013.
2. Vasilevskis EE, Han JH, Hughes CG, et al. Epidemiology and risk factors for delirium across hospital settings. Best Pract Res Clin Anaesthesiol. 2012;26(3):277-287. doi:10.1016/j.bpa.2012.07003
3. Leslie DL, Marcantonio ER, Zhang Y, et al. One-year health care costs associated with delirium in the elderly population. Arch Intern Med. 2008;168(1):27-32. doi:10.1001/archinternmed.2007.4
4. McCusker J, Cole M, Abrahamowicz M, et al. Delirium predicts 12-month mortality. Arch Intern Med. 2002;162(4):457-463. doi:10.1001/archinte.162.4.457
5. Witlox J, Eurelings LS, de Jonghe JF, et al. Delirium in elderly patients and the risk of postdischarge mortality, institutionalization, and dementia: a meta-analysis. JAMA. 2010;304(4):443-451. doi:10.1001/jama.2010.1013
6. Gross AL, Jones RN, Habtemariam DA, et al. Delirium and long-term cognitive trajectory among persons with dementia. Arch Intern Med. 2012;172(17):1324-1331. doi:10.1001/archinternmed.2012.3203
7. Inouye SK. Prevention of delirium in hospitalized older patients: risk factors and targeted intervention strategies. Ann Med. 2000;32(4):257-263. doi:10.3109/07853890009011770
8. Siddiqi N, Harrison JK, Clegg A, et al. Interventions for preventing delirium in hospitalised non-ICU patients. Cochrane Database Syst Rev. 2016;3:CD005563. doi:10.1002/14651858.CD005563.pub3
9. Inouye SK, van Dyck CH, Alessi CA, et al. Clarifying confusion: the confusion assessment method. A new method for detection of delirium. Ann Intern Med. 1990;113(12):941-948. doi:10.7326/0003-4819-113-12-941
10. R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2017.
11. Fong TG, Tulebaev SR, Inouye SK. Delirium in elderly adults: diagnosis, prevention and treatment. Nat Rev Neurol. 2009;5(4):210-220. doi:10.1038/nrneurol.2009.24
12. Siddiqi N, House AO, Holmes JD. Occurrence and outcome of delirium in medical in-patients: a systematic literature review. Age Ageing. 2006;35(4):350-364. doi:10.1093/ageing/afl005
13. Ely EW, Shintani A, Truman B, et al. Delirium as a predictor of mortality in mechanically ventilated patients in the intensive care unit. JAMA. 2004;291(14):1753-1762. doi:10.1001/jama.291.14.1753
14. Chen CC, Lin MT, Tien YW, et al. Modified Hospital Elder Life Program: effects on abdominal surgery patients. J Am Coll Surg. 2011;213(2):245-252. doi:10.1016/j.jamcollsurg.2011.05.004
15. Zaubler TS, Murphy K, Rizzuto L, et al. Quality improvement and cost savings with multicomponent delirium interventions: replication of the Hospital Elder Life Program in a community hospital. Psychosomatics. 2013;54(3):219-226. doi:10.1016/j.psym.2013.01.010
16. Rubin FH, Neal K, Fenlon K, et al. Sustainability and scalability of the Hospital Elder Life Program at a community hospital. J Am Geriatr Soc. 2011;59(2):359-365. doi:10.1111/j.1532-5415.2010.03243.x
17. Milisen K, Foreman MD, Abraham IL, et al. A nurse-led interdisciplinary intervention program for delirium in elderly hip-fracture patients. J Am Geriatr Soc. 2001;49(5):523-532. doi:10.1046/j.1532-5415.2001.49109.x
18. Lundström M, Edlund A, Karlsson S, et al. A multifactorial intervention program reduces the duration of delirium, length of hospitalization, and mortality in delirious patients. J Am Geriatr Soc. 2005;53(4):622-628. doi:10.1111/j.1532-5415.2005.53210.x
19. Tabet N, Hudson S, Sweeney V, et al. An educational intervention can prevent delirium on acute medical wards. Age Ageing. 2005;34(2):152-156. doi:10.1093/ageing/afi0320. Han JH, Zimmerman EE, Cutler N, et al. Delirium in older emergency department patients: recognition, risk factors, and psychomotor subtypes. Acad Emerg Med. 2009;16(3):193-200. doi:10.1111/j.1553-2712.2008.00339.x
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th edition. American Psychiatric Association; 2013.
2. Vasilevskis EE, Han JH, Hughes CG, et al. Epidemiology and risk factors for delirium across hospital settings. Best Pract Res Clin Anaesthesiol. 2012;26(3):277-287. doi:10.1016/j.bpa.2012.07003
3. Leslie DL, Marcantonio ER, Zhang Y, et al. One-year health care costs associated with delirium in the elderly population. Arch Intern Med. 2008;168(1):27-32. doi:10.1001/archinternmed.2007.4
4. McCusker J, Cole M, Abrahamowicz M, et al. Delirium predicts 12-month mortality. Arch Intern Med. 2002;162(4):457-463. doi:10.1001/archinte.162.4.457
5. Witlox J, Eurelings LS, de Jonghe JF, et al. Delirium in elderly patients and the risk of postdischarge mortality, institutionalization, and dementia: a meta-analysis. JAMA. 2010;304(4):443-451. doi:10.1001/jama.2010.1013
6. Gross AL, Jones RN, Habtemariam DA, et al. Delirium and long-term cognitive trajectory among persons with dementia. Arch Intern Med. 2012;172(17):1324-1331. doi:10.1001/archinternmed.2012.3203
7. Inouye SK. Prevention of delirium in hospitalized older patients: risk factors and targeted intervention strategies. Ann Med. 2000;32(4):257-263. doi:10.3109/07853890009011770
8. Siddiqi N, Harrison JK, Clegg A, et al. Interventions for preventing delirium in hospitalised non-ICU patients. Cochrane Database Syst Rev. 2016;3:CD005563. doi:10.1002/14651858.CD005563.pub3
9. Inouye SK, van Dyck CH, Alessi CA, et al. Clarifying confusion: the confusion assessment method. A new method for detection of delirium. Ann Intern Med. 1990;113(12):941-948. doi:10.7326/0003-4819-113-12-941
10. R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2017.
11. Fong TG, Tulebaev SR, Inouye SK. Delirium in elderly adults: diagnosis, prevention and treatment. Nat Rev Neurol. 2009;5(4):210-220. doi:10.1038/nrneurol.2009.24
12. Siddiqi N, House AO, Holmes JD. Occurrence and outcome of delirium in medical in-patients: a systematic literature review. Age Ageing. 2006;35(4):350-364. doi:10.1093/ageing/afl005
13. Ely EW, Shintani A, Truman B, et al. Delirium as a predictor of mortality in mechanically ventilated patients in the intensive care unit. JAMA. 2004;291(14):1753-1762. doi:10.1001/jama.291.14.1753
14. Chen CC, Lin MT, Tien YW, et al. Modified Hospital Elder Life Program: effects on abdominal surgery patients. J Am Coll Surg. 2011;213(2):245-252. doi:10.1016/j.jamcollsurg.2011.05.004
15. Zaubler TS, Murphy K, Rizzuto L, et al. Quality improvement and cost savings with multicomponent delirium interventions: replication of the Hospital Elder Life Program in a community hospital. Psychosomatics. 2013;54(3):219-226. doi:10.1016/j.psym.2013.01.010
16. Rubin FH, Neal K, Fenlon K, et al. Sustainability and scalability of the Hospital Elder Life Program at a community hospital. J Am Geriatr Soc. 2011;59(2):359-365. doi:10.1111/j.1532-5415.2010.03243.x
17. Milisen K, Foreman MD, Abraham IL, et al. A nurse-led interdisciplinary intervention program for delirium in elderly hip-fracture patients. J Am Geriatr Soc. 2001;49(5):523-532. doi:10.1046/j.1532-5415.2001.49109.x
18. Lundström M, Edlund A, Karlsson S, et al. A multifactorial intervention program reduces the duration of delirium, length of hospitalization, and mortality in delirious patients. J Am Geriatr Soc. 2005;53(4):622-628. doi:10.1111/j.1532-5415.2005.53210.x
19. Tabet N, Hudson S, Sweeney V, et al. An educational intervention can prevent delirium on acute medical wards. Age Ageing. 2005;34(2):152-156. doi:10.1093/ageing/afi0320. Han JH, Zimmerman EE, Cutler N, et al. Delirium in older emergency department patients: recognition, risk factors, and psychomotor subtypes. Acad Emerg Med. 2009;16(3):193-200. doi:10.1111/j.1553-2712.2008.00339.x

Mask-wearing cuts new COVID-19 cases by 53%, study says
Social distancing and handwashing were also effective at lowering the number of cases, but wearing masks was the most effective tool against the coronavirus.
“Personal and social measures, including handwashing, mask wearing, and physical distancing are effective at reducing the incidence of COVID-19,” the study authors wrote.
The research team, which included public health and infectious disease specialists in Australia, China, and the U.K., evaluated 72 studies of COVID-19 precautions during the pandemic. They later looked at eight studies that focused on handwashing, mask wearing, and physical distancing.
Among six studies that looked at mask wearing, the researchers found a 53% reduction in COVID-19 cases. In the broader analysis with additional studies, wearing a mask reduced coronavirus transmission, cases, and deaths.
In one study across 200 countries, mandatory mask wearing resulted in nearly 46% fewer negative outcomes from COVID-19. In another study in the U.S., coronavirus transmission was reduced 29% in states where masks were mandatory.
But the research team couldn’t analyze the impact of the type of face mask used, the frequency of mask wearing, or the overall compliance with wearing face masks.
Among five studies that looked at physical distancing, the researchers found a 25% reduction in the rate of COVID-19. A study in the U.S. showed a 12% decrease in coronavirus transmission, while another study in Iran reported a reduction in COVID-19 mortality.
Handwashing interventions also suggested a substantial reduction of COVID-19 cases up to 53%, the researchers wrote. But in adjusted models, the results weren’t statistically significant due to the small number of studies included.
Other studies found significant decreases related to other public health measures, such as quarantines, broad lockdowns, border closures, school closures, business closures, and travel restrictions. Still, the research team couldn’t analyze the overall effectiveness of these measures due to the different ways the studies were conducted.
The study lines up with other research conducted so far during the pandemic, the research team wrote, which indicates that wearing masks and physical distancing can reduce transmission, cases, and deaths.
That said, more studies are needed, particularly now that vaccinations are available and contagious coronavirus variants have become prevalent.
“Further research is needed to assess the effectiveness of public health measures after adequate vaccination coverage has been achieved,” they wrote.
“It is likely that further control of the COVID-19 pandemic depends not only on high vaccination coverage and its effectiveness but also on ongoing adherence to effective and sustainable public health measures,” they concluded.
A version of this article first appeared on WebMD.com.
Social distancing and handwashing were also effective at lowering the number of cases, but wearing masks was the most effective tool against the coronavirus.
“Personal and social measures, including handwashing, mask wearing, and physical distancing are effective at reducing the incidence of COVID-19,” the study authors wrote.
The research team, which included public health and infectious disease specialists in Australia, China, and the U.K., evaluated 72 studies of COVID-19 precautions during the pandemic. They later looked at eight studies that focused on handwashing, mask wearing, and physical distancing.
Among six studies that looked at mask wearing, the researchers found a 53% reduction in COVID-19 cases. In the broader analysis with additional studies, wearing a mask reduced coronavirus transmission, cases, and deaths.
In one study across 200 countries, mandatory mask wearing resulted in nearly 46% fewer negative outcomes from COVID-19. In another study in the U.S., coronavirus transmission was reduced 29% in states where masks were mandatory.
But the research team couldn’t analyze the impact of the type of face mask used, the frequency of mask wearing, or the overall compliance with wearing face masks.
Among five studies that looked at physical distancing, the researchers found a 25% reduction in the rate of COVID-19. A study in the U.S. showed a 12% decrease in coronavirus transmission, while another study in Iran reported a reduction in COVID-19 mortality.
Handwashing interventions also suggested a substantial reduction of COVID-19 cases up to 53%, the researchers wrote. But in adjusted models, the results weren’t statistically significant due to the small number of studies included.
Other studies found significant decreases related to other public health measures, such as quarantines, broad lockdowns, border closures, school closures, business closures, and travel restrictions. Still, the research team couldn’t analyze the overall effectiveness of these measures due to the different ways the studies were conducted.
The study lines up with other research conducted so far during the pandemic, the research team wrote, which indicates that wearing masks and physical distancing can reduce transmission, cases, and deaths.
That said, more studies are needed, particularly now that vaccinations are available and contagious coronavirus variants have become prevalent.
“Further research is needed to assess the effectiveness of public health measures after adequate vaccination coverage has been achieved,” they wrote.
“It is likely that further control of the COVID-19 pandemic depends not only on high vaccination coverage and its effectiveness but also on ongoing adherence to effective and sustainable public health measures,” they concluded.
A version of this article first appeared on WebMD.com.
Social distancing and handwashing were also effective at lowering the number of cases, but wearing masks was the most effective tool against the coronavirus.
“Personal and social measures, including handwashing, mask wearing, and physical distancing are effective at reducing the incidence of COVID-19,” the study authors wrote.
The research team, which included public health and infectious disease specialists in Australia, China, and the U.K., evaluated 72 studies of COVID-19 precautions during the pandemic. They later looked at eight studies that focused on handwashing, mask wearing, and physical distancing.
Among six studies that looked at mask wearing, the researchers found a 53% reduction in COVID-19 cases. In the broader analysis with additional studies, wearing a mask reduced coronavirus transmission, cases, and deaths.
In one study across 200 countries, mandatory mask wearing resulted in nearly 46% fewer negative outcomes from COVID-19. In another study in the U.S., coronavirus transmission was reduced 29% in states where masks were mandatory.
But the research team couldn’t analyze the impact of the type of face mask used, the frequency of mask wearing, or the overall compliance with wearing face masks.
Among five studies that looked at physical distancing, the researchers found a 25% reduction in the rate of COVID-19. A study in the U.S. showed a 12% decrease in coronavirus transmission, while another study in Iran reported a reduction in COVID-19 mortality.
Handwashing interventions also suggested a substantial reduction of COVID-19 cases up to 53%, the researchers wrote. But in adjusted models, the results weren’t statistically significant due to the small number of studies included.
Other studies found significant decreases related to other public health measures, such as quarantines, broad lockdowns, border closures, school closures, business closures, and travel restrictions. Still, the research team couldn’t analyze the overall effectiveness of these measures due to the different ways the studies were conducted.
The study lines up with other research conducted so far during the pandemic, the research team wrote, which indicates that wearing masks and physical distancing can reduce transmission, cases, and deaths.
That said, more studies are needed, particularly now that vaccinations are available and contagious coronavirus variants have become prevalent.
“Further research is needed to assess the effectiveness of public health measures after adequate vaccination coverage has been achieved,” they wrote.
“It is likely that further control of the COVID-19 pandemic depends not only on high vaccination coverage and its effectiveness but also on ongoing adherence to effective and sustainable public health measures,” they concluded.
A version of this article first appeared on WebMD.com.
FROM THE BMJ
Texas SB8 and the future of abortion care
Texas Senate Bill 8 (SB8) is the most extreme antiabortion legislation currently in effect in the United States. SB8 was introduced by the Texas legislature on March 11, 2021, and signed into law by Governor Greg Abbott on May 19, 2021.1 The law went into effect on September 1, 2021, despite an appeal to the US Supreme Court to block the law until the courts could weigh in on its constitutionality. The bill prohibits all abortion care in the state of Texas after cardiac activity has been identified, typically at 6 weeks’ gestational age. The majority of pregnant people may be unaware at that point that they are pregnant, particularly if their menstrual cycles are irregular.2 An estimated 85% of abortions in Texas occur after the 6-week mark, leaving millions of Texans without the constitutionally protected rights assured to them in Roe v Wade.3,4 This has and will disproportionately impact communities of color and low-income people seeking abortion care.
SB8 does not contain exceptions in case of a pregnancy that results from rape, sexual assault, or incest, but it does contain an exemption for abortion care because of a medical emergency, as approved by a physician. The physician is required to note the medical emergency in the patient’s chart, stating that the “medical emergency necessitated the abortion” and “prevented compliance” with SB8.5 In practice, this exception is so vague as to leave clinicians concerned that routine management of medical conditions and complications, as in ectopic pregnancy, places them at risk of legal action against them and their colleagues should they authorize abortion care.
In Texas, abortion restrictions are nothing new. Texas patients are already subject to a 2-trip requirement: Since 2011 they have been required to have a mandatory ultrasound in one visit and schedule a second visit, 24 hours later, for the procedure.6 As of 2003, Texas law also mandates that providers discuss with patients the medical risks, adoption alternatives, and developmental stages of the pregnancy.6 There are no medical indications for either of these laws, and their impact is to delay patient care. Unfortunately, laws such as these have been increasingly common in the past decade, with 106 abortion restrictions enacted in 2021 alone.7,8
What is different about SB8?
SB8 is unique in that it deputizes private citizens to enforce the law. This represents a major change in the antichoice movement’s tactics, as previous bills have made violations a criminal offense. SB8 allows a citizen to sue anyone associated with abortion care, with a minimum penalty of $10,000. In practice, a citizen of another state, who has no connection to the patient receiving care, can sue under this Texas law.9 Anyone “aiding and abetting a violation” can be found liable for up to 4 years after the date of care, including, for example, a ride-hailing driver called to ferry the patient to the appointment, the health care team providing abortion care, or insurance companies covering the costs of care. In addition, anyone found guilty of “aiding and abetting” a violation of the bill is responsible for all costs and attorney fees associated with the civil case.5,10
Furthermore, SB8 outlines defenses that cannot be used to preempt a finding of civil liability, including “ignorance or mistake of the law,” “belief of the law’s unconstitutionality,” and “consent of the [patient] to the abortion.”5 This additional layer of restriction makes it difficult to appeal the bill and convolutes an individual’s ability to challenge the law. The law also forbids the state (Texas), a state official, a court, or a district attorney from intervening on behalf of the law—upending typical courses of appeal. This legislation also complicates both federal and state intervention regarding SB8’s constitutionality, as the state has no role in enforcing the law as it is written.5
Continue to: What has been the response?...
What has been the response?
As expected, abortion foes reacted positively to SB8, while abortion advocates expressed outrage that the law went into effect. Many were additionally confused that the Supreme Court chose not to intervene to stay the law while the courts adjudicate its constitutionality, as is typical in other cases concerning abortion restrictions.11
In a 5-4 ruling, the US Supreme Court allowed SB8 to take effect on September 1, issuing its decision on the “Shadow Docket.” As such, a decision was handed down on an expedited timeline in response to an emergency appeal without any oral arguments or a lengthy opinion explaining the ruling.11,12 The majority delivered a brief, one-paragraph order summarizing their decision, explaining that their refusal to grant the injunction was not a commentary on the law’s constitutionality. The High Court stated that they could not initially comment on the law’s constitutionality before it went into effect, citing that per the law, the state had no role in enforcement, and at the time, no private actions had yet been brought under the law. Justice Sonia Sotomayor dissented, stating, “The Court’s order is stunning. Presented with an application to enjoin a flagrantly unconstitutional law engineered to prohibit women from exercising their constitutional rights and evade judicial scrutiny, a majority of Justices have opted to bury their heads in the sand.”13
Following the Supreme Court’s refusal to act, US Attorney General Merrick Garland commented that “the Justice Department was evaluating all options to protect the constitutional rights of women and other persons.” Just one week later, the US Department of Justice filed a lawsuit against the State of Texas, arguing that SB8 was unconstitutional under the Supremacy Clause (federal law takes precedence over state law) and the Fourteenth Amendment.14,15
On October 6, in response to the Department of Justice’s challenge, District Judge Robert Pitman issued an injunction to prevent enforcement of SB8. In a 113-page ruling, Judge Pitman explained that “a person’s right under the Constitution to choose to obtain an abortion prior to fetal viability is well established.” Judge Pittman held SB8 unconstitutional, stating, “Women have been unlawfully prevented from exercising control over their lives in ways that are protected by the Constitution... Fully aware that depriving its citizens of this right by direct state action would be flagrantly unconstitutional, the State contrived an unprecedented and transparent statutory scheme to do just that.”16
Just 48 hours after the injunction issued by Judge Pitman, the Fifth Circuit Court of Appeals overturned the injunction, and SB8 went back into effect while litigation on its constitutionality proceeded.2,17 The Fifth Circuit Court of Appeals is widely considered to be one of the most conservative courts in the country.18
On October 15, 2021, the Department of Justice appealed the Fifth Circuit Court’s decision and asked the US Supreme Court to intervene, requesting that the Court issue an emergency halt to the law.19,20 On October 22, 2021, the Court declined to halt the law but scheduled oral arguments on the case for November 1, 2021. This is a stunningly fast briefing schedule for a case of such constitutional importance.
Given the legal back-and-forth, many clinicians are not providing abortion care in Texas as the litigation unfolds. SB8 permits retroactive enforcement, mandating that those “aiding and abetting” of abortion care may be civilly liable for up to 4 years after providing the care.5
Continue to: Potential outcomes, and what comes next...
Potential outcomes, and what comes next
Since the ascension of Justice Amy Coney Barrett to the High Court, there has been a nationwide increase in antiabortion legislation. Between January and July 2021, more than 90 abortion restrictions were passed, more restrictions in any single year since Roe v Wade was decided in 1973.8 In the past decade, more than 500 laws that restrict abortion have been passed across the United States, and studies indicate that 87% to 90% of American counties today are without a single abortion provider.21,22 Abortion supporters are particularly concerned about the future of Roe v Wade, with a conservative Supreme Court set to hear the challenge to SB8 on November 1, 2021, followed by a second case from Mississippi challenging the constitutionality of a 15-week ban on abortion in Dobbs v Jackson Women’s Health Organization (read about this case in “Supreme Court Case: Dobbs v Jackson Women’s Health Organization: What you need to know,” at https://www.mdedge.com/obgyn/article/245853/practice-management/supreme-court-case-dobbs-v-jackson-womens-health).23,24
At the time of this article writing, we do not know how the Supreme Court will rule on the variety of challenges to the right to privacy. That said, advocates believe it is safe to assume that the landscape of abortion access is likely to change dramatically in the coming year.
Action items: What can you do?
It is important to remember that not only does SB8 severely limit access to safe and legal abortion but also it makes pregnancy dangerous for all pregnant people in Texas and places doubt in providers’ minds on how to manage medical care for their patients.
On the federal level, many advocates are focusing on codifying the right to choose and protecting abortion care from medically unnecessary restrictions. The Women’s Health Protection Act of 2021 (WHPA) was introduced in the House of Representatives by Rep. Judy Chu (D-CA), Lois Frankel (D-FL), Ayanna Pressley (D-MA), and Veronica Escobar (D-TX), and it passed in the US House of Representatives in a 218-211 vote.25 WHPA now awaits a vote in a deeply divided US Senate. Although WHPA has wide popular support—an estimated 61% of Americans support the legislation—its future is unclear in the Senate.26 Currently, WHPA has 48 supporters, all Democrats. You can contact your legislators via the links below to encourage them to pass WHPA. If you have friends and colleagues in states in which the Senator does not support WHPA, forward these links and encourage them to sign on:
- Equal Access to Abortion, Everywhere: https://actforwomen.org/take-action/
- Physicians for Reproductive Rights: https://secure.everyaction.com/p/MOuAyW7F3Ua-FmaGtGD4Kw2
- Center for Reproductive Rights: https://reproductiverights.org/whpa-take-action/
Many also are organizing a crowdfunding campaign to support abortion providers as well as legislative resources. Additional groups to donate specifically to SB8 efforts include27:
- Equal Access to Abortion, Everywhere: https://actforwomen.org/whpa-faqs/
- Planned Parenthood of Greater Texas, Inc: https://www.plannedparenthood.org/planned-parenthood-greater-texas/senate-bill-8
- Texas Equal Access Fund: https://secure.everyaction.com/ztEh8Qeh80-k2k1Yuo5gTw2
- ActBlue Charities: https://secure.actblue.com/donate/txfunds
Furthermore, it is more important than ever to support work within states to support abortion rights. State-specific abortion advocacy groups and their efforts include:
- Avow Foundation for Abortion Access: https://avowtexas.org/support/
- Planned Parenthood of Greater Texas, Inc: https://www.plannedparenthood.org/planned-parenthood-greater-texas
- NARAL Pro-Choice Texas: https://prochoicetexas.org/
- Texas Abortion Access Network: https://txabortionaccessnetwork.org/
- ACLU Texas. Abortion in Texas. Updated October 9, 2021. Accessed November 8, 2021. https://www.aclutx.org/en/know-your-rights/abortion-texas.
- Rummler O. The 19th explains: what to know about Texas’ abortion law. The 19th. September 1, 2021; updated October 12, 2021. Accessed November 8, 2021. https://19thnews.org/2021/09/texas-new-abortion-law-what-you-need-know/.
- Kaye J, Hearron M. Even people who oppose abortion should fear Texas’s new ban. July 19, 2021. The Washington Post. Accessed November 12, 2021. https://www.washingtonpost.com/outlook/2021/07/19/texas-sb8-abortion-lawsuits/.
- Centers for Disease Control and Prevention. CDCs abortion surveillance system FAQs. November 25, 2020. Accessed November 8, 2021. https://www.cdc.gov/reproductivehealth/data_stats/abortion.htm.
- Texas Senate Bill 8. LegiScan. Accessed November 8, 2021. https://legiscan.com/TX/text/SB8/id/2395961.
- Texas abortion laws and policies. Planned Parenthood of Greater Texas, Inc. Accessed November 8, 2021. https://www.plannedparenthood.org/planned-parenthood-greater-texas/patient-resources/texas-laws-policies.
- Nash E. For the first time ever, US states enacted more than 100 abortion restrictions in a single year. October 4, 2012. Guttmacher Institute. Accessed November 12, 2021. https://www.guttmacher.org/article/2021/10/first-time-ever-us-states-enacted-more-100-abortion-restrictions-single-year.
- Nash E, Naide S. State policy trends at midyear 2021: already the worst legislative year ever for US abortion rights. July 2021. Guttmacher Institute. Accessed November 8, 2021. https://www.guttmacher.org/article/2021/07/state-policy-trends-midyear-2021-already-worst-legislative-year-ever-us-abortion.
- ACLU. Whole Women’s Health v Jackson. Updated October 7, 2021. Accessed November 8, 2021. https://www.aclu.org/cases/whole-womans-health-v-jacksonH
- Holley P, Solomon D. Your questions about Texas’s new abortion law, answered. Texas Monthly. October 7, 2021. Accessed November 8, 2021. https://www.texasmonthly.com/news-politics/texas-abortion-law-explained/.
- Millhiser I. The staggering implications of the Supreme Court’s Texas anti-abortion ruling. Vox. September 2, 2021. Accessed November 8, 2021. https://www.vox.com/22653779/supreme-court-abortion-texas-sb8-whole-womans-health-jackson-roe-wade.
- Carter S. ACLU of Texas asks US Supreme Court to stop new abortion law. Dallas Observer. August 31, 2021. Accessed November 8, 2021. https://www.dallasobserver.com/news/aclu-of-texas-asks-us-supreme-court-to-block-new-anti-abortion-law-sb-8-12314274.
- Supreme Court of the United States. Whole Women’s Health et al v Austin Reeve Jackson, Judge, et al: On application of injunction relief. September 1, 2021. Accessed November 8, 2021. https://www.supremecourt.gov/opinions/20pdf/21a24_8759.pdf.
- Lucas R. A US judge blocks enforcement of Texas’ controversial new abortion law. NPR. October 6, 2021. Accessed November 8, 2021. https://www.npr.org/2021/10/06/1040221171/a-u-s-judge-blocks-enforcement-of-texas-controversial-new-abortion-law.
- US Department of Justice. Attorney General Merrick B. Garland delivers remarks announcing lawsuit against the state of Texas to stop unconstitutional Senate Bill 8. September 8, 2021. Accessed November 8, 2021. https://www.justice.gov/opa/speech/attorney-general-merrick-b-garland-delivers-remarks-announcing-lawsuit-against-state-0.
- Barnhart T. Texas abortion law suspended by district judge hearing Biden administration challenge. Newsweek. October 6, 2021. Accessed November 8, 2021. https://www.newsweek.com/district-court-judge-issues-injunction-texas-law-banning-abortions-after-6-weeks-1636411.
- Oxner R. Appeals court allows Texas abortion law to resume, stopping federal judge’s order to block enforcement. The Texas Tribune. October 8, 2021. Accessed November 8, 2021. https://www.texastribune.org/2021/10/08/texas-abortion-appeal/.
- Oxner R. Texas’ near-total abortion ban will remain in effect as federal appeals court agrees to hear legal challenges. The Texas Tribune. October 14, 2021. Accessed November 8, 2021. https://www.texastribune.org/2021/10/14/texas-abortion-restrictions-appeal/.
- The United States District Court for the Western District of Texas, Austin Division. September 9, 2021. Accessed November 8, 2021. https://www.justsecurity.org/wp-content/uploads/2021/09/lawsuit-doj.pdf.
- Barnes R, Marimow AE. Justice Department will ask Supreme Court to block Texas abortion law while legal fights play out. Washington Post. October 15, 2021. Accessed November 8, 2021. https://www.washingtonpost.com/politics/courts_law/doj-texas-abortion-ban-supreme-court/2021/10/15/bd5762e6-2dcc-11ec-8ef6-3ca8fe943a92_story.html.
- Nash E, Bearak J, Li N, et al. Impact of Texas’ abortion ban: a 14-fold increase in driving distance to get an abortion. Guttmacher Institute. August 4, 2021; updated September 15, 2021. Accessed November 8, 2021. https://www.guttmacher.org/article/2021/08/impact-texas-abortion-ban-14-fold-increase-driving-distance-get-abortion.
- Jones RK, Jerman J. Abortion incidence and service availability in the United States, 2014. Perspect Sex Reprod Health. 2017;49:17-27. https://doi.org/10.1363/psrh.12015. Accessed November 12, 2021.
- Center for Reproductive Rights. Jackson Women’s Health Organization v Dobbs. March 19, 2018. Accessed November 8, 2021. https://reproductiverights.org/case/jackson-womens-health-organization-v-dobbs/.
- Chung A. US Supreme Court takes up Texas abortion case, lets ban remain. Oct 22, 2021. Reuters. Accessed November 8, 2021. https://www.reuters.com/world/us/us-supreme-court-hear-challenge-texas-abortion-ban-2021-10-22/.
- Equal Access to Abortion, Everywhere. Frequently asked questions. Accessed November 8, 2021. https://actforwomen.org/whpa-faqs/.
- Center for Reproductive Rights. New poll: a solid majority of voters support the Women’s Health Protection Act (WHPA). Accessed November 8, 2021. https://reproductiverights.org/wp-content/uploads/2021/06/ME-CRR_WHPA-Release-14001-June-1.pdf.
- Pardilla A, Avila A. 20 organizations fighting the Texas abortion ban. New York Magazine. September 2, 2021. Accessed November 8, 2021. https://nymag.com/strategist/2021/09/texas-abortion-ban-2021-where-to-donate.html.
Texas Senate Bill 8 (SB8) is the most extreme antiabortion legislation currently in effect in the United States. SB8 was introduced by the Texas legislature on March 11, 2021, and signed into law by Governor Greg Abbott on May 19, 2021.1 The law went into effect on September 1, 2021, despite an appeal to the US Supreme Court to block the law until the courts could weigh in on its constitutionality. The bill prohibits all abortion care in the state of Texas after cardiac activity has been identified, typically at 6 weeks’ gestational age. The majority of pregnant people may be unaware at that point that they are pregnant, particularly if their menstrual cycles are irregular.2 An estimated 85% of abortions in Texas occur after the 6-week mark, leaving millions of Texans without the constitutionally protected rights assured to them in Roe v Wade.3,4 This has and will disproportionately impact communities of color and low-income people seeking abortion care.
SB8 does not contain exceptions in case of a pregnancy that results from rape, sexual assault, or incest, but it does contain an exemption for abortion care because of a medical emergency, as approved by a physician. The physician is required to note the medical emergency in the patient’s chart, stating that the “medical emergency necessitated the abortion” and “prevented compliance” with SB8.5 In practice, this exception is so vague as to leave clinicians concerned that routine management of medical conditions and complications, as in ectopic pregnancy, places them at risk of legal action against them and their colleagues should they authorize abortion care.
In Texas, abortion restrictions are nothing new. Texas patients are already subject to a 2-trip requirement: Since 2011 they have been required to have a mandatory ultrasound in one visit and schedule a second visit, 24 hours later, for the procedure.6 As of 2003, Texas law also mandates that providers discuss with patients the medical risks, adoption alternatives, and developmental stages of the pregnancy.6 There are no medical indications for either of these laws, and their impact is to delay patient care. Unfortunately, laws such as these have been increasingly common in the past decade, with 106 abortion restrictions enacted in 2021 alone.7,8
What is different about SB8?
SB8 is unique in that it deputizes private citizens to enforce the law. This represents a major change in the antichoice movement’s tactics, as previous bills have made violations a criminal offense. SB8 allows a citizen to sue anyone associated with abortion care, with a minimum penalty of $10,000. In practice, a citizen of another state, who has no connection to the patient receiving care, can sue under this Texas law.9 Anyone “aiding and abetting a violation” can be found liable for up to 4 years after the date of care, including, for example, a ride-hailing driver called to ferry the patient to the appointment, the health care team providing abortion care, or insurance companies covering the costs of care. In addition, anyone found guilty of “aiding and abetting” a violation of the bill is responsible for all costs and attorney fees associated with the civil case.5,10
Furthermore, SB8 outlines defenses that cannot be used to preempt a finding of civil liability, including “ignorance or mistake of the law,” “belief of the law’s unconstitutionality,” and “consent of the [patient] to the abortion.”5 This additional layer of restriction makes it difficult to appeal the bill and convolutes an individual’s ability to challenge the law. The law also forbids the state (Texas), a state official, a court, or a district attorney from intervening on behalf of the law—upending typical courses of appeal. This legislation also complicates both federal and state intervention regarding SB8’s constitutionality, as the state has no role in enforcing the law as it is written.5
Continue to: What has been the response?...
What has been the response?
As expected, abortion foes reacted positively to SB8, while abortion advocates expressed outrage that the law went into effect. Many were additionally confused that the Supreme Court chose not to intervene to stay the law while the courts adjudicate its constitutionality, as is typical in other cases concerning abortion restrictions.11
In a 5-4 ruling, the US Supreme Court allowed SB8 to take effect on September 1, issuing its decision on the “Shadow Docket.” As such, a decision was handed down on an expedited timeline in response to an emergency appeal without any oral arguments or a lengthy opinion explaining the ruling.11,12 The majority delivered a brief, one-paragraph order summarizing their decision, explaining that their refusal to grant the injunction was not a commentary on the law’s constitutionality. The High Court stated that they could not initially comment on the law’s constitutionality before it went into effect, citing that per the law, the state had no role in enforcement, and at the time, no private actions had yet been brought under the law. Justice Sonia Sotomayor dissented, stating, “The Court’s order is stunning. Presented with an application to enjoin a flagrantly unconstitutional law engineered to prohibit women from exercising their constitutional rights and evade judicial scrutiny, a majority of Justices have opted to bury their heads in the sand.”13
Following the Supreme Court’s refusal to act, US Attorney General Merrick Garland commented that “the Justice Department was evaluating all options to protect the constitutional rights of women and other persons.” Just one week later, the US Department of Justice filed a lawsuit against the State of Texas, arguing that SB8 was unconstitutional under the Supremacy Clause (federal law takes precedence over state law) and the Fourteenth Amendment.14,15
On October 6, in response to the Department of Justice’s challenge, District Judge Robert Pitman issued an injunction to prevent enforcement of SB8. In a 113-page ruling, Judge Pitman explained that “a person’s right under the Constitution to choose to obtain an abortion prior to fetal viability is well established.” Judge Pittman held SB8 unconstitutional, stating, “Women have been unlawfully prevented from exercising control over their lives in ways that are protected by the Constitution... Fully aware that depriving its citizens of this right by direct state action would be flagrantly unconstitutional, the State contrived an unprecedented and transparent statutory scheme to do just that.”16
Just 48 hours after the injunction issued by Judge Pitman, the Fifth Circuit Court of Appeals overturned the injunction, and SB8 went back into effect while litigation on its constitutionality proceeded.2,17 The Fifth Circuit Court of Appeals is widely considered to be one of the most conservative courts in the country.18
On October 15, 2021, the Department of Justice appealed the Fifth Circuit Court’s decision and asked the US Supreme Court to intervene, requesting that the Court issue an emergency halt to the law.19,20 On October 22, 2021, the Court declined to halt the law but scheduled oral arguments on the case for November 1, 2021. This is a stunningly fast briefing schedule for a case of such constitutional importance.
Given the legal back-and-forth, many clinicians are not providing abortion care in Texas as the litigation unfolds. SB8 permits retroactive enforcement, mandating that those “aiding and abetting” of abortion care may be civilly liable for up to 4 years after providing the care.5
Continue to: Potential outcomes, and what comes next...
Potential outcomes, and what comes next
Since the ascension of Justice Amy Coney Barrett to the High Court, there has been a nationwide increase in antiabortion legislation. Between January and July 2021, more than 90 abortion restrictions were passed, more restrictions in any single year since Roe v Wade was decided in 1973.8 In the past decade, more than 500 laws that restrict abortion have been passed across the United States, and studies indicate that 87% to 90% of American counties today are without a single abortion provider.21,22 Abortion supporters are particularly concerned about the future of Roe v Wade, with a conservative Supreme Court set to hear the challenge to SB8 on November 1, 2021, followed by a second case from Mississippi challenging the constitutionality of a 15-week ban on abortion in Dobbs v Jackson Women’s Health Organization (read about this case in “Supreme Court Case: Dobbs v Jackson Women’s Health Organization: What you need to know,” at https://www.mdedge.com/obgyn/article/245853/practice-management/supreme-court-case-dobbs-v-jackson-womens-health).23,24
At the time of this article writing, we do not know how the Supreme Court will rule on the variety of challenges to the right to privacy. That said, advocates believe it is safe to assume that the landscape of abortion access is likely to change dramatically in the coming year.
Action items: What can you do?
It is important to remember that not only does SB8 severely limit access to safe and legal abortion but also it makes pregnancy dangerous for all pregnant people in Texas and places doubt in providers’ minds on how to manage medical care for their patients.
On the federal level, many advocates are focusing on codifying the right to choose and protecting abortion care from medically unnecessary restrictions. The Women’s Health Protection Act of 2021 (WHPA) was introduced in the House of Representatives by Rep. Judy Chu (D-CA), Lois Frankel (D-FL), Ayanna Pressley (D-MA), and Veronica Escobar (D-TX), and it passed in the US House of Representatives in a 218-211 vote.25 WHPA now awaits a vote in a deeply divided US Senate. Although WHPA has wide popular support—an estimated 61% of Americans support the legislation—its future is unclear in the Senate.26 Currently, WHPA has 48 supporters, all Democrats. You can contact your legislators via the links below to encourage them to pass WHPA. If you have friends and colleagues in states in which the Senator does not support WHPA, forward these links and encourage them to sign on:
- Equal Access to Abortion, Everywhere: https://actforwomen.org/take-action/
- Physicians for Reproductive Rights: https://secure.everyaction.com/p/MOuAyW7F3Ua-FmaGtGD4Kw2
- Center for Reproductive Rights: https://reproductiverights.org/whpa-take-action/
Many also are organizing a crowdfunding campaign to support abortion providers as well as legislative resources. Additional groups to donate specifically to SB8 efforts include27:
- Equal Access to Abortion, Everywhere: https://actforwomen.org/whpa-faqs/
- Planned Parenthood of Greater Texas, Inc: https://www.plannedparenthood.org/planned-parenthood-greater-texas/senate-bill-8
- Texas Equal Access Fund: https://secure.everyaction.com/ztEh8Qeh80-k2k1Yuo5gTw2
- ActBlue Charities: https://secure.actblue.com/donate/txfunds
Furthermore, it is more important than ever to support work within states to support abortion rights. State-specific abortion advocacy groups and their efforts include:
- Avow Foundation for Abortion Access: https://avowtexas.org/support/
- Planned Parenthood of Greater Texas, Inc: https://www.plannedparenthood.org/planned-parenthood-greater-texas
- NARAL Pro-Choice Texas: https://prochoicetexas.org/
- Texas Abortion Access Network: https://txabortionaccessnetwork.org/
Texas Senate Bill 8 (SB8) is the most extreme antiabortion legislation currently in effect in the United States. SB8 was introduced by the Texas legislature on March 11, 2021, and signed into law by Governor Greg Abbott on May 19, 2021.1 The law went into effect on September 1, 2021, despite an appeal to the US Supreme Court to block the law until the courts could weigh in on its constitutionality. The bill prohibits all abortion care in the state of Texas after cardiac activity has been identified, typically at 6 weeks’ gestational age. The majority of pregnant people may be unaware at that point that they are pregnant, particularly if their menstrual cycles are irregular.2 An estimated 85% of abortions in Texas occur after the 6-week mark, leaving millions of Texans without the constitutionally protected rights assured to them in Roe v Wade.3,4 This has and will disproportionately impact communities of color and low-income people seeking abortion care.
SB8 does not contain exceptions in case of a pregnancy that results from rape, sexual assault, or incest, but it does contain an exemption for abortion care because of a medical emergency, as approved by a physician. The physician is required to note the medical emergency in the patient’s chart, stating that the “medical emergency necessitated the abortion” and “prevented compliance” with SB8.5 In practice, this exception is so vague as to leave clinicians concerned that routine management of medical conditions and complications, as in ectopic pregnancy, places them at risk of legal action against them and their colleagues should they authorize abortion care.
In Texas, abortion restrictions are nothing new. Texas patients are already subject to a 2-trip requirement: Since 2011 they have been required to have a mandatory ultrasound in one visit and schedule a second visit, 24 hours later, for the procedure.6 As of 2003, Texas law also mandates that providers discuss with patients the medical risks, adoption alternatives, and developmental stages of the pregnancy.6 There are no medical indications for either of these laws, and their impact is to delay patient care. Unfortunately, laws such as these have been increasingly common in the past decade, with 106 abortion restrictions enacted in 2021 alone.7,8
What is different about SB8?
SB8 is unique in that it deputizes private citizens to enforce the law. This represents a major change in the antichoice movement’s tactics, as previous bills have made violations a criminal offense. SB8 allows a citizen to sue anyone associated with abortion care, with a minimum penalty of $10,000. In practice, a citizen of another state, who has no connection to the patient receiving care, can sue under this Texas law.9 Anyone “aiding and abetting a violation” can be found liable for up to 4 years after the date of care, including, for example, a ride-hailing driver called to ferry the patient to the appointment, the health care team providing abortion care, or insurance companies covering the costs of care. In addition, anyone found guilty of “aiding and abetting” a violation of the bill is responsible for all costs and attorney fees associated with the civil case.5,10
Furthermore, SB8 outlines defenses that cannot be used to preempt a finding of civil liability, including “ignorance or mistake of the law,” “belief of the law’s unconstitutionality,” and “consent of the [patient] to the abortion.”5 This additional layer of restriction makes it difficult to appeal the bill and convolutes an individual’s ability to challenge the law. The law also forbids the state (Texas), a state official, a court, or a district attorney from intervening on behalf of the law—upending typical courses of appeal. This legislation also complicates both federal and state intervention regarding SB8’s constitutionality, as the state has no role in enforcing the law as it is written.5
Continue to: What has been the response?...
What has been the response?
As expected, abortion foes reacted positively to SB8, while abortion advocates expressed outrage that the law went into effect. Many were additionally confused that the Supreme Court chose not to intervene to stay the law while the courts adjudicate its constitutionality, as is typical in other cases concerning abortion restrictions.11
In a 5-4 ruling, the US Supreme Court allowed SB8 to take effect on September 1, issuing its decision on the “Shadow Docket.” As such, a decision was handed down on an expedited timeline in response to an emergency appeal without any oral arguments or a lengthy opinion explaining the ruling.11,12 The majority delivered a brief, one-paragraph order summarizing their decision, explaining that their refusal to grant the injunction was not a commentary on the law’s constitutionality. The High Court stated that they could not initially comment on the law’s constitutionality before it went into effect, citing that per the law, the state had no role in enforcement, and at the time, no private actions had yet been brought under the law. Justice Sonia Sotomayor dissented, stating, “The Court’s order is stunning. Presented with an application to enjoin a flagrantly unconstitutional law engineered to prohibit women from exercising their constitutional rights and evade judicial scrutiny, a majority of Justices have opted to bury their heads in the sand.”13
Following the Supreme Court’s refusal to act, US Attorney General Merrick Garland commented that “the Justice Department was evaluating all options to protect the constitutional rights of women and other persons.” Just one week later, the US Department of Justice filed a lawsuit against the State of Texas, arguing that SB8 was unconstitutional under the Supremacy Clause (federal law takes precedence over state law) and the Fourteenth Amendment.14,15
On October 6, in response to the Department of Justice’s challenge, District Judge Robert Pitman issued an injunction to prevent enforcement of SB8. In a 113-page ruling, Judge Pitman explained that “a person’s right under the Constitution to choose to obtain an abortion prior to fetal viability is well established.” Judge Pittman held SB8 unconstitutional, stating, “Women have been unlawfully prevented from exercising control over their lives in ways that are protected by the Constitution... Fully aware that depriving its citizens of this right by direct state action would be flagrantly unconstitutional, the State contrived an unprecedented and transparent statutory scheme to do just that.”16
Just 48 hours after the injunction issued by Judge Pitman, the Fifth Circuit Court of Appeals overturned the injunction, and SB8 went back into effect while litigation on its constitutionality proceeded.2,17 The Fifth Circuit Court of Appeals is widely considered to be one of the most conservative courts in the country.18
On October 15, 2021, the Department of Justice appealed the Fifth Circuit Court’s decision and asked the US Supreme Court to intervene, requesting that the Court issue an emergency halt to the law.19,20 On October 22, 2021, the Court declined to halt the law but scheduled oral arguments on the case for November 1, 2021. This is a stunningly fast briefing schedule for a case of such constitutional importance.
Given the legal back-and-forth, many clinicians are not providing abortion care in Texas as the litigation unfolds. SB8 permits retroactive enforcement, mandating that those “aiding and abetting” of abortion care may be civilly liable for up to 4 years after providing the care.5
Continue to: Potential outcomes, and what comes next...
Potential outcomes, and what comes next
Since the ascension of Justice Amy Coney Barrett to the High Court, there has been a nationwide increase in antiabortion legislation. Between January and July 2021, more than 90 abortion restrictions were passed, more restrictions in any single year since Roe v Wade was decided in 1973.8 In the past decade, more than 500 laws that restrict abortion have been passed across the United States, and studies indicate that 87% to 90% of American counties today are without a single abortion provider.21,22 Abortion supporters are particularly concerned about the future of Roe v Wade, with a conservative Supreme Court set to hear the challenge to SB8 on November 1, 2021, followed by a second case from Mississippi challenging the constitutionality of a 15-week ban on abortion in Dobbs v Jackson Women’s Health Organization (read about this case in “Supreme Court Case: Dobbs v Jackson Women’s Health Organization: What you need to know,” at https://www.mdedge.com/obgyn/article/245853/practice-management/supreme-court-case-dobbs-v-jackson-womens-health).23,24
At the time of this article writing, we do not know how the Supreme Court will rule on the variety of challenges to the right to privacy. That said, advocates believe it is safe to assume that the landscape of abortion access is likely to change dramatically in the coming year.
Action items: What can you do?
It is important to remember that not only does SB8 severely limit access to safe and legal abortion but also it makes pregnancy dangerous for all pregnant people in Texas and places doubt in providers’ minds on how to manage medical care for their patients.
On the federal level, many advocates are focusing on codifying the right to choose and protecting abortion care from medically unnecessary restrictions. The Women’s Health Protection Act of 2021 (WHPA) was introduced in the House of Representatives by Rep. Judy Chu (D-CA), Lois Frankel (D-FL), Ayanna Pressley (D-MA), and Veronica Escobar (D-TX), and it passed in the US House of Representatives in a 218-211 vote.25 WHPA now awaits a vote in a deeply divided US Senate. Although WHPA has wide popular support—an estimated 61% of Americans support the legislation—its future is unclear in the Senate.26 Currently, WHPA has 48 supporters, all Democrats. You can contact your legislators via the links below to encourage them to pass WHPA. If you have friends and colleagues in states in which the Senator does not support WHPA, forward these links and encourage them to sign on:
- Equal Access to Abortion, Everywhere: https://actforwomen.org/take-action/
- Physicians for Reproductive Rights: https://secure.everyaction.com/p/MOuAyW7F3Ua-FmaGtGD4Kw2
- Center for Reproductive Rights: https://reproductiverights.org/whpa-take-action/
Many also are organizing a crowdfunding campaign to support abortion providers as well as legislative resources. Additional groups to donate specifically to SB8 efforts include27:
- Equal Access to Abortion, Everywhere: https://actforwomen.org/whpa-faqs/
- Planned Parenthood of Greater Texas, Inc: https://www.plannedparenthood.org/planned-parenthood-greater-texas/senate-bill-8
- Texas Equal Access Fund: https://secure.everyaction.com/ztEh8Qeh80-k2k1Yuo5gTw2
- ActBlue Charities: https://secure.actblue.com/donate/txfunds
Furthermore, it is more important than ever to support work within states to support abortion rights. State-specific abortion advocacy groups and their efforts include:
- Avow Foundation for Abortion Access: https://avowtexas.org/support/
- Planned Parenthood of Greater Texas, Inc: https://www.plannedparenthood.org/planned-parenthood-greater-texas
- NARAL Pro-Choice Texas: https://prochoicetexas.org/
- Texas Abortion Access Network: https://txabortionaccessnetwork.org/
- ACLU Texas. Abortion in Texas. Updated October 9, 2021. Accessed November 8, 2021. https://www.aclutx.org/en/know-your-rights/abortion-texas.
- Rummler O. The 19th explains: what to know about Texas’ abortion law. The 19th. September 1, 2021; updated October 12, 2021. Accessed November 8, 2021. https://19thnews.org/2021/09/texas-new-abortion-law-what-you-need-know/.
- Kaye J, Hearron M. Even people who oppose abortion should fear Texas’s new ban. July 19, 2021. The Washington Post. Accessed November 12, 2021. https://www.washingtonpost.com/outlook/2021/07/19/texas-sb8-abortion-lawsuits/.
- Centers for Disease Control and Prevention. CDCs abortion surveillance system FAQs. November 25, 2020. Accessed November 8, 2021. https://www.cdc.gov/reproductivehealth/data_stats/abortion.htm.
- Texas Senate Bill 8. LegiScan. Accessed November 8, 2021. https://legiscan.com/TX/text/SB8/id/2395961.
- Texas abortion laws and policies. Planned Parenthood of Greater Texas, Inc. Accessed November 8, 2021. https://www.plannedparenthood.org/planned-parenthood-greater-texas/patient-resources/texas-laws-policies.
- Nash E. For the first time ever, US states enacted more than 100 abortion restrictions in a single year. October 4, 2012. Guttmacher Institute. Accessed November 12, 2021. https://www.guttmacher.org/article/2021/10/first-time-ever-us-states-enacted-more-100-abortion-restrictions-single-year.
- Nash E, Naide S. State policy trends at midyear 2021: already the worst legislative year ever for US abortion rights. July 2021. Guttmacher Institute. Accessed November 8, 2021. https://www.guttmacher.org/article/2021/07/state-policy-trends-midyear-2021-already-worst-legislative-year-ever-us-abortion.
- ACLU. Whole Women’s Health v Jackson. Updated October 7, 2021. Accessed November 8, 2021. https://www.aclu.org/cases/whole-womans-health-v-jacksonH
- Holley P, Solomon D. Your questions about Texas’s new abortion law, answered. Texas Monthly. October 7, 2021. Accessed November 8, 2021. https://www.texasmonthly.com/news-politics/texas-abortion-law-explained/.
- Millhiser I. The staggering implications of the Supreme Court’s Texas anti-abortion ruling. Vox. September 2, 2021. Accessed November 8, 2021. https://www.vox.com/22653779/supreme-court-abortion-texas-sb8-whole-womans-health-jackson-roe-wade.
- Carter S. ACLU of Texas asks US Supreme Court to stop new abortion law. Dallas Observer. August 31, 2021. Accessed November 8, 2021. https://www.dallasobserver.com/news/aclu-of-texas-asks-us-supreme-court-to-block-new-anti-abortion-law-sb-8-12314274.
- Supreme Court of the United States. Whole Women’s Health et al v Austin Reeve Jackson, Judge, et al: On application of injunction relief. September 1, 2021. Accessed November 8, 2021. https://www.supremecourt.gov/opinions/20pdf/21a24_8759.pdf.
- Lucas R. A US judge blocks enforcement of Texas’ controversial new abortion law. NPR. October 6, 2021. Accessed November 8, 2021. https://www.npr.org/2021/10/06/1040221171/a-u-s-judge-blocks-enforcement-of-texas-controversial-new-abortion-law.
- US Department of Justice. Attorney General Merrick B. Garland delivers remarks announcing lawsuit against the state of Texas to stop unconstitutional Senate Bill 8. September 8, 2021. Accessed November 8, 2021. https://www.justice.gov/opa/speech/attorney-general-merrick-b-garland-delivers-remarks-announcing-lawsuit-against-state-0.
- Barnhart T. Texas abortion law suspended by district judge hearing Biden administration challenge. Newsweek. October 6, 2021. Accessed November 8, 2021. https://www.newsweek.com/district-court-judge-issues-injunction-texas-law-banning-abortions-after-6-weeks-1636411.
- Oxner R. Appeals court allows Texas abortion law to resume, stopping federal judge’s order to block enforcement. The Texas Tribune. October 8, 2021. Accessed November 8, 2021. https://www.texastribune.org/2021/10/08/texas-abortion-appeal/.
- Oxner R. Texas’ near-total abortion ban will remain in effect as federal appeals court agrees to hear legal challenges. The Texas Tribune. October 14, 2021. Accessed November 8, 2021. https://www.texastribune.org/2021/10/14/texas-abortion-restrictions-appeal/.
- The United States District Court for the Western District of Texas, Austin Division. September 9, 2021. Accessed November 8, 2021. https://www.justsecurity.org/wp-content/uploads/2021/09/lawsuit-doj.pdf.
- Barnes R, Marimow AE. Justice Department will ask Supreme Court to block Texas abortion law while legal fights play out. Washington Post. October 15, 2021. Accessed November 8, 2021. https://www.washingtonpost.com/politics/courts_law/doj-texas-abortion-ban-supreme-court/2021/10/15/bd5762e6-2dcc-11ec-8ef6-3ca8fe943a92_story.html.
- Nash E, Bearak J, Li N, et al. Impact of Texas’ abortion ban: a 14-fold increase in driving distance to get an abortion. Guttmacher Institute. August 4, 2021; updated September 15, 2021. Accessed November 8, 2021. https://www.guttmacher.org/article/2021/08/impact-texas-abortion-ban-14-fold-increase-driving-distance-get-abortion.
- Jones RK, Jerman J. Abortion incidence and service availability in the United States, 2014. Perspect Sex Reprod Health. 2017;49:17-27. https://doi.org/10.1363/psrh.12015. Accessed November 12, 2021.
- Center for Reproductive Rights. Jackson Women’s Health Organization v Dobbs. March 19, 2018. Accessed November 8, 2021. https://reproductiverights.org/case/jackson-womens-health-organization-v-dobbs/.
- Chung A. US Supreme Court takes up Texas abortion case, lets ban remain. Oct 22, 2021. Reuters. Accessed November 8, 2021. https://www.reuters.com/world/us/us-supreme-court-hear-challenge-texas-abortion-ban-2021-10-22/.
- Equal Access to Abortion, Everywhere. Frequently asked questions. Accessed November 8, 2021. https://actforwomen.org/whpa-faqs/.
- Center for Reproductive Rights. New poll: a solid majority of voters support the Women’s Health Protection Act (WHPA). Accessed November 8, 2021. https://reproductiverights.org/wp-content/uploads/2021/06/ME-CRR_WHPA-Release-14001-June-1.pdf.
- Pardilla A, Avila A. 20 organizations fighting the Texas abortion ban. New York Magazine. September 2, 2021. Accessed November 8, 2021. https://nymag.com/strategist/2021/09/texas-abortion-ban-2021-where-to-donate.html.
- ACLU Texas. Abortion in Texas. Updated October 9, 2021. Accessed November 8, 2021. https://www.aclutx.org/en/know-your-rights/abortion-texas.
- Rummler O. The 19th explains: what to know about Texas’ abortion law. The 19th. September 1, 2021; updated October 12, 2021. Accessed November 8, 2021. https://19thnews.org/2021/09/texas-new-abortion-law-what-you-need-know/.
- Kaye J, Hearron M. Even people who oppose abortion should fear Texas’s new ban. July 19, 2021. The Washington Post. Accessed November 12, 2021. https://www.washingtonpost.com/outlook/2021/07/19/texas-sb8-abortion-lawsuits/.
- Centers for Disease Control and Prevention. CDCs abortion surveillance system FAQs. November 25, 2020. Accessed November 8, 2021. https://www.cdc.gov/reproductivehealth/data_stats/abortion.htm.
- Texas Senate Bill 8. LegiScan. Accessed November 8, 2021. https://legiscan.com/TX/text/SB8/id/2395961.
- Texas abortion laws and policies. Planned Parenthood of Greater Texas, Inc. Accessed November 8, 2021. https://www.plannedparenthood.org/planned-parenthood-greater-texas/patient-resources/texas-laws-policies.
- Nash E. For the first time ever, US states enacted more than 100 abortion restrictions in a single year. October 4, 2012. Guttmacher Institute. Accessed November 12, 2021. https://www.guttmacher.org/article/2021/10/first-time-ever-us-states-enacted-more-100-abortion-restrictions-single-year.
- Nash E, Naide S. State policy trends at midyear 2021: already the worst legislative year ever for US abortion rights. July 2021. Guttmacher Institute. Accessed November 8, 2021. https://www.guttmacher.org/article/2021/07/state-policy-trends-midyear-2021-already-worst-legislative-year-ever-us-abortion.
- ACLU. Whole Women’s Health v Jackson. Updated October 7, 2021. Accessed November 8, 2021. https://www.aclu.org/cases/whole-womans-health-v-jacksonH
- Holley P, Solomon D. Your questions about Texas’s new abortion law, answered. Texas Monthly. October 7, 2021. Accessed November 8, 2021. https://www.texasmonthly.com/news-politics/texas-abortion-law-explained/.
- Millhiser I. The staggering implications of the Supreme Court’s Texas anti-abortion ruling. Vox. September 2, 2021. Accessed November 8, 2021. https://www.vox.com/22653779/supreme-court-abortion-texas-sb8-whole-womans-health-jackson-roe-wade.
- Carter S. ACLU of Texas asks US Supreme Court to stop new abortion law. Dallas Observer. August 31, 2021. Accessed November 8, 2021. https://www.dallasobserver.com/news/aclu-of-texas-asks-us-supreme-court-to-block-new-anti-abortion-law-sb-8-12314274.
- Supreme Court of the United States. Whole Women’s Health et al v Austin Reeve Jackson, Judge, et al: On application of injunction relief. September 1, 2021. Accessed November 8, 2021. https://www.supremecourt.gov/opinions/20pdf/21a24_8759.pdf.
- Lucas R. A US judge blocks enforcement of Texas’ controversial new abortion law. NPR. October 6, 2021. Accessed November 8, 2021. https://www.npr.org/2021/10/06/1040221171/a-u-s-judge-blocks-enforcement-of-texas-controversial-new-abortion-law.
- US Department of Justice. Attorney General Merrick B. Garland delivers remarks announcing lawsuit against the state of Texas to stop unconstitutional Senate Bill 8. September 8, 2021. Accessed November 8, 2021. https://www.justice.gov/opa/speech/attorney-general-merrick-b-garland-delivers-remarks-announcing-lawsuit-against-state-0.
- Barnhart T. Texas abortion law suspended by district judge hearing Biden administration challenge. Newsweek. October 6, 2021. Accessed November 8, 2021. https://www.newsweek.com/district-court-judge-issues-injunction-texas-law-banning-abortions-after-6-weeks-1636411.
- Oxner R. Appeals court allows Texas abortion law to resume, stopping federal judge’s order to block enforcement. The Texas Tribune. October 8, 2021. Accessed November 8, 2021. https://www.texastribune.org/2021/10/08/texas-abortion-appeal/.
- Oxner R. Texas’ near-total abortion ban will remain in effect as federal appeals court agrees to hear legal challenges. The Texas Tribune. October 14, 2021. Accessed November 8, 2021. https://www.texastribune.org/2021/10/14/texas-abortion-restrictions-appeal/.
- The United States District Court for the Western District of Texas, Austin Division. September 9, 2021. Accessed November 8, 2021. https://www.justsecurity.org/wp-content/uploads/2021/09/lawsuit-doj.pdf.
- Barnes R, Marimow AE. Justice Department will ask Supreme Court to block Texas abortion law while legal fights play out. Washington Post. October 15, 2021. Accessed November 8, 2021. https://www.washingtonpost.com/politics/courts_law/doj-texas-abortion-ban-supreme-court/2021/10/15/bd5762e6-2dcc-11ec-8ef6-3ca8fe943a92_story.html.
- Nash E, Bearak J, Li N, et al. Impact of Texas’ abortion ban: a 14-fold increase in driving distance to get an abortion. Guttmacher Institute. August 4, 2021; updated September 15, 2021. Accessed November 8, 2021. https://www.guttmacher.org/article/2021/08/impact-texas-abortion-ban-14-fold-increase-driving-distance-get-abortion.
- Jones RK, Jerman J. Abortion incidence and service availability in the United States, 2014. Perspect Sex Reprod Health. 2017;49:17-27. https://doi.org/10.1363/psrh.12015. Accessed November 12, 2021.
- Center for Reproductive Rights. Jackson Women’s Health Organization v Dobbs. March 19, 2018. Accessed November 8, 2021. https://reproductiverights.org/case/jackson-womens-health-organization-v-dobbs/.
- Chung A. US Supreme Court takes up Texas abortion case, lets ban remain. Oct 22, 2021. Reuters. Accessed November 8, 2021. https://www.reuters.com/world/us/us-supreme-court-hear-challenge-texas-abortion-ban-2021-10-22/.
- Equal Access to Abortion, Everywhere. Frequently asked questions. Accessed November 8, 2021. https://actforwomen.org/whpa-faqs/.
- Center for Reproductive Rights. New poll: a solid majority of voters support the Women’s Health Protection Act (WHPA). Accessed November 8, 2021. https://reproductiverights.org/wp-content/uploads/2021/06/ME-CRR_WHPA-Release-14001-June-1.pdf.
- Pardilla A, Avila A. 20 organizations fighting the Texas abortion ban. New York Magazine. September 2, 2021. Accessed November 8, 2021. https://nymag.com/strategist/2021/09/texas-abortion-ban-2021-where-to-donate.html.
Whistleblowers will play key role in enforcing workplace vaccine mandate
goes into effect in January.
The Occupational Safety and Health Administration (OSHA) doesn’t have enough workplace safety inspectors to cover the nation, the Associated Press reported, so the agency will count on people within organizations to identify violations.
“There is no army of OSHA inspectors that is going to be knocking on employers’ doors or even calling them,” Debbie Berkowitz, a former OSHA chief of staff who is a fellow at Georgetown University, told the news service.
“They’re going to rely on workers and their union representatives to file complaints where the company is totally flouting the law,” she said.
Last week, OSHA published the details of the Biden administration’s vaccine mandate. Companies with more than 100 employees must require their workers to get vaccinated or undergo weekly testing. Companies that don’t comply could face fines of $14,000 for each “serious” violation. Repeat violators could face 10 times that amount.
Employees who are concerned about workplace safety, unvaccinated co-workers, or people not being tested as required may report their employers, according to Reuters.
Jim Frederick, the acting chief for OSHA, told reporters that the agency will focus on job sites “where workers need assistance to have a safe and healthy workplace.”
“That typically comes through in the form of a complaint,” he said.
OSHA has jurisdiction in 29 states, the AP reported. OSHA is tasked with addressing violations of the Occupational Safety and Health Act of 1970, which is meant to create safe workplaces, and the agency has updated its guidance about COVID-19 safety in the workplace throughout this year.
Other states, such as California and Michigan, have their own workplace safety agencies, which will have until February to adopt their own version of a vaccine mandate, according to the AP.
OSHA and state counterparts will be tasked with enforcing the mandate, and their agencies are already short-staffed. About 1,850 inspectors will oversee 130 million workers at 8 million job sites.
OSHA has encouraged workers to first report complaints to employers “if possible.” Otherwise, employees can file a confidential safety complaint with OSHA or file a case through a representative, such as a lawyer or union leader, the AP reported.
But workplace experts have voiced caution about the potential risks of reporting. Whistleblowers tend to face retaliation and OSHA can’t always offer protection in these cases.
“Technically, the law says that companies can’t retaliate against a worker for raising a health and safety issue or filing an OSHA complaint or even reporting an injury,” Ms. Berkowitz said. “But retaliation is rampant.”
OSHA has some jurisdiction to pursue employers who punish workers for reporting unsafe working conditions, the AP reported. Last month, the agency sued a luxury car dealer in Texas for firing an employee who warned co-workers about potential coronavirus hazards.
But at the same time, Ms. Berkowitz and the National Employment Law Project found that OSHA dismissed more than half of the COVID-related complaints of retaliation that it received from whistleblowers. About 2% of complaints were resolved during a 5-month period last year, according to their report.
As the vaccine mandate deadline approaches, most companies are expected to comply, experts told the AP. Some employers wanted to require the shot but didn’t want to create their own rule, and others have said they’ll follow OSHA regulations as they always do.
“Most employers, they’re law abiding,” David Michaels, a former OSHA chief who is a public health professor at George Washington University, told the AP.
“They’re trying to make sure that they meet the requirements of every law and regulation,” he said. “Now OSHA will follow up. They’ll respond to complaints. They’ll do spot checks. They’ll issue citations and fines, and they’ll make a big deal of those.”
A version of this article first appeared on WebMD.com.
goes into effect in January.
The Occupational Safety and Health Administration (OSHA) doesn’t have enough workplace safety inspectors to cover the nation, the Associated Press reported, so the agency will count on people within organizations to identify violations.
“There is no army of OSHA inspectors that is going to be knocking on employers’ doors or even calling them,” Debbie Berkowitz, a former OSHA chief of staff who is a fellow at Georgetown University, told the news service.
“They’re going to rely on workers and their union representatives to file complaints where the company is totally flouting the law,” she said.
Last week, OSHA published the details of the Biden administration’s vaccine mandate. Companies with more than 100 employees must require their workers to get vaccinated or undergo weekly testing. Companies that don’t comply could face fines of $14,000 for each “serious” violation. Repeat violators could face 10 times that amount.
Employees who are concerned about workplace safety, unvaccinated co-workers, or people not being tested as required may report their employers, according to Reuters.
Jim Frederick, the acting chief for OSHA, told reporters that the agency will focus on job sites “where workers need assistance to have a safe and healthy workplace.”
“That typically comes through in the form of a complaint,” he said.
OSHA has jurisdiction in 29 states, the AP reported. OSHA is tasked with addressing violations of the Occupational Safety and Health Act of 1970, which is meant to create safe workplaces, and the agency has updated its guidance about COVID-19 safety in the workplace throughout this year.
Other states, such as California and Michigan, have their own workplace safety agencies, which will have until February to adopt their own version of a vaccine mandate, according to the AP.
OSHA and state counterparts will be tasked with enforcing the mandate, and their agencies are already short-staffed. About 1,850 inspectors will oversee 130 million workers at 8 million job sites.
OSHA has encouraged workers to first report complaints to employers “if possible.” Otherwise, employees can file a confidential safety complaint with OSHA or file a case through a representative, such as a lawyer or union leader, the AP reported.
But workplace experts have voiced caution about the potential risks of reporting. Whistleblowers tend to face retaliation and OSHA can’t always offer protection in these cases.
“Technically, the law says that companies can’t retaliate against a worker for raising a health and safety issue or filing an OSHA complaint or even reporting an injury,” Ms. Berkowitz said. “But retaliation is rampant.”
OSHA has some jurisdiction to pursue employers who punish workers for reporting unsafe working conditions, the AP reported. Last month, the agency sued a luxury car dealer in Texas for firing an employee who warned co-workers about potential coronavirus hazards.
But at the same time, Ms. Berkowitz and the National Employment Law Project found that OSHA dismissed more than half of the COVID-related complaints of retaliation that it received from whistleblowers. About 2% of complaints were resolved during a 5-month period last year, according to their report.
As the vaccine mandate deadline approaches, most companies are expected to comply, experts told the AP. Some employers wanted to require the shot but didn’t want to create their own rule, and others have said they’ll follow OSHA regulations as they always do.
“Most employers, they’re law abiding,” David Michaels, a former OSHA chief who is a public health professor at George Washington University, told the AP.
“They’re trying to make sure that they meet the requirements of every law and regulation,” he said. “Now OSHA will follow up. They’ll respond to complaints. They’ll do spot checks. They’ll issue citations and fines, and they’ll make a big deal of those.”
A version of this article first appeared on WebMD.com.
goes into effect in January.
The Occupational Safety and Health Administration (OSHA) doesn’t have enough workplace safety inspectors to cover the nation, the Associated Press reported, so the agency will count on people within organizations to identify violations.
“There is no army of OSHA inspectors that is going to be knocking on employers’ doors or even calling them,” Debbie Berkowitz, a former OSHA chief of staff who is a fellow at Georgetown University, told the news service.
“They’re going to rely on workers and their union representatives to file complaints where the company is totally flouting the law,” she said.
Last week, OSHA published the details of the Biden administration’s vaccine mandate. Companies with more than 100 employees must require their workers to get vaccinated or undergo weekly testing. Companies that don’t comply could face fines of $14,000 for each “serious” violation. Repeat violators could face 10 times that amount.
Employees who are concerned about workplace safety, unvaccinated co-workers, or people not being tested as required may report their employers, according to Reuters.
Jim Frederick, the acting chief for OSHA, told reporters that the agency will focus on job sites “where workers need assistance to have a safe and healthy workplace.”
“That typically comes through in the form of a complaint,” he said.
OSHA has jurisdiction in 29 states, the AP reported. OSHA is tasked with addressing violations of the Occupational Safety and Health Act of 1970, which is meant to create safe workplaces, and the agency has updated its guidance about COVID-19 safety in the workplace throughout this year.
Other states, such as California and Michigan, have their own workplace safety agencies, which will have until February to adopt their own version of a vaccine mandate, according to the AP.
OSHA and state counterparts will be tasked with enforcing the mandate, and their agencies are already short-staffed. About 1,850 inspectors will oversee 130 million workers at 8 million job sites.
OSHA has encouraged workers to first report complaints to employers “if possible.” Otherwise, employees can file a confidential safety complaint with OSHA or file a case through a representative, such as a lawyer or union leader, the AP reported.
But workplace experts have voiced caution about the potential risks of reporting. Whistleblowers tend to face retaliation and OSHA can’t always offer protection in these cases.
“Technically, the law says that companies can’t retaliate against a worker for raising a health and safety issue or filing an OSHA complaint or even reporting an injury,” Ms. Berkowitz said. “But retaliation is rampant.”
OSHA has some jurisdiction to pursue employers who punish workers for reporting unsafe working conditions, the AP reported. Last month, the agency sued a luxury car dealer in Texas for firing an employee who warned co-workers about potential coronavirus hazards.
But at the same time, Ms. Berkowitz and the National Employment Law Project found that OSHA dismissed more than half of the COVID-related complaints of retaliation that it received from whistleblowers. About 2% of complaints were resolved during a 5-month period last year, according to their report.
As the vaccine mandate deadline approaches, most companies are expected to comply, experts told the AP. Some employers wanted to require the shot but didn’t want to create their own rule, and others have said they’ll follow OSHA regulations as they always do.
“Most employers, they’re law abiding,” David Michaels, a former OSHA chief who is a public health professor at George Washington University, told the AP.
“They’re trying to make sure that they meet the requirements of every law and regulation,” he said. “Now OSHA will follow up. They’ll respond to complaints. They’ll do spot checks. They’ll issue citations and fines, and they’ll make a big deal of those.”
A version of this article first appeared on WebMD.com.
The Supreme Court 2020‒2021: What will affect ObGyns?
The Supreme Court’s usual processes were disrupted this term. The COVID-19 pandemic required audio hearings rather than in-person, and it resulted in a number of emergency legal appeals. As the Court began its regular sessions on October 5, 2020, there were only 8 justices—Justice Ruth Bader Ginsburg had passed away and Amy Coney Barrett had not yet been confirmed by the Senate. The Court decided many important cases this term, including dealing with the delivery of drugs to induce abortions, a Centers for Disease Control and Prevention (CDC) moratorium on housing evictions, yet another case on the Affordable Care Act, state laws concerning pharmacy benefit managers, and the Hologic and Minerva endometrial ablation systems patents. After considering these cases, we also will briefly look at other cases of general interest.
Abortion
Patient access to mifepristone
In May 2020, the American College of Obstetricians and Gynecologists (ACOG) was the named plaintiff in a lawsuit against the US Food and Drug Administration (FDA) regarding the drugs mifepristone and misoprostol that are used to induce medical abortions.1 The case was filed by the American Civil Liberties Union on behalf of ACOG and others2,3 and raised the issue of patients’ access to these medications. The basic claim of the case was that during the pandemic, the FDA’s regulation of mifepristone was unconstitutional in that they imposed an undue burden on the decision of women to have an abortion.4 (Although misoprostol is a part of the medical abortion regimen, it is not subject to special regulation and was not part of the litigation.)
The FDA regulation of mifepristone, begun in 2000 but modified since then, includes 3 elements to assure safe use:
- prescribers must have special training or certification
- the drug can be dispensed to patients only in a hospital, clinic, or medical office under the supervision of a certified health care provider (known as the “in-person dispensing requirement” because retail pharmacy or mail distribution are prohibited)
- the health care provider must review a “patient agreement form” with the patient and have the patient sign the consent form in the provider’s presence.5
The pandemic made fulfilling these requirements substantially more burdensome and difficult. The question was whether the FDA was constitutionally required to modify its regulations during a pandemic to take account of the undue burden of the regulation created by the pandemic. That is, the question was not whether the FDA could have or should have chosen to make the modification, but whether it was required to do so.
In July 2020, a federal district court in Maryland held that the FDA regulation was an unconstitutional burden on the abortion rights of women during the pandemic and issued a preliminary injunction to stop the FDA from enforcing the in-person dispensing and signature rules. The district judge applied the injunction to Maryland, but also made it a nationwide injunction. (The issue of district court nationwide injunctions is considered in, “District court ‘nationwide injunctions’”).
The FDA asked the Fourth Circuit Court of Appeals to stay the enforcement of the injunction, which the appeals court denied. The FDA then appealed to the Supreme Court, asking it to stay the injunction. In October 2020, the Court announced that it was holding the FDA’s request “in abeyance” to allow the district court to consider a motion by the FDA to dissolve or change the injunction. It gave the district court 40 days in which to act. That decision by the Court was in the “Shadow Docket” (see sidebar on page XX), so the exact vote of the Court in October is not clear, but 2 Justices (Alito and Thomas) dissented and would have stayed the injunction.6 Over the next 40 days, the district court did not withdraw its nationwide injunction.
Thus, on January 12, 2021, the case was again before the Supreme Court, which let the FDA’s regulations regarding mifepristone remain in place by lifting the district court’s injunction. Most of the justices supporting the stay did not write to explain their decision, although their dissent in the earlier cases may have served that purpose. (Maryland was permitting many kinds of activity that were more risky than visiting a clinic—indoor dining, with open hair salons, gyms, and casinos.)7 Chief Justice Roberts wrote a concurrence to indicate that, in his view, the issue was not whether the FDA’s regulations placed an undue burden on a right to an abortion generally, but that “My view is that courts owe significant deference” to the public health authorities (here meaning the FDA). Justices Sotomayor and Kagan dissented, saying that the issue was the undue burden on women, given the difficulties of the pandemic, particularly going to medical facilities during the COVID-19 pandemic.8
The injunction, sought by ACOG and others, was issued by the district court and was in effect for several months before it was dissolved by the Supreme Court. Following the change in presidential administrations, in April 2021 the FDA announced that it was going to “exercise enforcement discretion with respect to the in-person dispensing requirement…during the COVID-19 public health emergency.”9
Continue to: The Texas abortion case...
The Texas abortion case
The Court, on September 1, 2021, declined to block a Texas abortion statute from taking effect.10 This law precludes abortions after a fetal heartbeat is present at about 6 weeks of gestation. The Fifth Circuit declined to grant an injunction delaying implementation of the Texas law, and the Court did not reverse that decision.
Over the years, a variety of states have placed limitations on abortion, and those almost always have been enjoined by federal courts before they went into effect. However, the Texas statute, which undoubtedly is unconstitutional, was creatively constructed to avoid an early injunction.11 The statute does not allow state officials to enforce the new law, but rather it allows almost any private citizen to seek monetary damages from anyone performing an abortion or who “aids and abets” an abortion. Thus, it is difficult to tailor a lawsuit before this law is enforced. First, courts do not enjoin laws; they usually enjoin individuals from enforcing the law, and in this case it is difficult to know which individuals will be enforcing the laws and what their decisions might be. There also are some questions about the degree to which federal courts can enjoin state courts from deciding lawsuits under state law. For these procedural reasons, the majority of the Court found that those attacking the Texas law had not met their burden of showing that that they would win their case.
Even 3 of the dissenting justices said the defendants may be right that “existing doctrines preclude judicial intervention,” but that the consequences are such that the Court should delay the law until there is time for briefing and argument. The other 3 dissenting justices thought there would be ways of getting around the clever roadblock Texas had erected for the federal courts.
There has been some commentary that this case portends the abandonment of Roe v Wade and Casey,12 but that conclusion does not seem warranted by this case. The Court has accepted a Mississippi abortion law to be heard next term.13 In addition, the Texas statute is likely to be back in federal court once a private individual has filed a claim for money from an abortion provider (and likely even before that).
COVID-19 cases
The Supreme Court decided several cases related to COVID-19, including adjustments to election procedures, church services, and CDC eviction moratoria. As a general matter early in the pandemic, the Court deferred to government authorities, generally upholding government actions. Chief Justice Roberts emphasized the importance of the Court deferring to government officials in emergencies. As the pandemic progressed into 2021, however, the Court became less and less sympathetic to government actions that were not consistent, permitted by existing law, or reasonably necessary. For example, regulations of churches that were inconsistent with the regulation of similar organizations were struck down.14
Among the most interesting of the summer 2021 cases was the CDC eviction moratorium that essentially prohibited landlords nationwide from evicting tenants for nonpayment of rent. When the challenges to these CDC regulations first reached the Court, the moratorium was about to expire; in a 5-4 decision, the Court did not enjoin the CDC from continuing that policy. Justice Kavanaugh (the fifth vote) warned that “clear and specific congressional authorization…would be necessary to extend the moratorium past July 31.”15 Despite telling the Court that the moratorium would expire on July 31, just 3 days after the expiration and without any congressional authorization, the CDC reinstated what was practically the same moratorium.16 On August 26, the Court struck down the reinstated regulation, probably by a 6-3 margin. (Because this case arose in the “Shadow Docket,” the vote of some justices is not certain).17
Continue to: The Affordable Care Act...
The Affordable Care Act
The Affordable Care Act was challenged in the Court for the third time.18 In this term’s case, several states argued that when Congress essentially eliminated the penalty/tax for not purchasing insurance coverage, there was no longer a constitutional basis for the individual mandate. With that centerpiece gone, they claimed, the whole statute should be declared unconstitutional.
Along with many other specialty groups, ACOG joined an amicus curiae brief sponsored by the American Medical Association (AMA).19 An amicus brief is one not filed by the parties to the case, but by organizations or individuals who have information that may be of use to the Court in considering the case. Among other things, the filing of an amicus brief indicates the interest of the organization in the outcome of the case. In this case, the crux of the amicus was that even if the individual mandate currently is not constitutional, the Court should sever that provision and retain the rest of the ACA.
Despite some wild predictions about what the Court might do, it did not decide any substantive issue. Rather, it found that none of the parties to the case had “standing” to challenge the constitutionality of the ACA. Therefore, in effect, the Court dismissed the case without deciding the substantive legal issues.
Pharmacy Benefit Managers
The powerful Pharmacy Benefit Managers (PBMs) are a hidden part of the health care system; however, in recent years there has been increasing regulatory attention paid to them. Some states have begun regulating aspects of PBMs. In this term, the Court considered an Arkansas law that sought to protect local pharmacies from PBM pricing practices.20 The AMA filed an amicus brief in the case which made legal arguments, most of which had been made by the parties to the litigation.21
PBMs generally tell pharmacies how much they will reimburse the pharmacy for filling a prescription for a particular drug. In some instances, PBMs will set a reimbursement price that is lower than the wholesale price at which local pharmacies can purchase the drug. The Arkansas law prohibited PBMs in the state from reimbursing pharmacies for less than the wholesale cost the pharmacy paid for the drug.
The claim of the PBMs was that the Arkansas law violated the Employee Retirement Income Security Act (ERISA). In part, this act preempts state law that relates to fringe benefit plans. States have the authority to regulate insurance, but ERISA limits what they can do when the insurance relates to fringe benefits. The Court held that ERISA does not preempt the Arkansas law or similar state laws in other states. Because the state law was not preempted by the state law, the Arkansas regulation was upheld. The fact that this was a unanimous decision (8-0, because Justice Barrett was not on the Court when the case was heard) suggests that states may have leeway in additional regulations of PBMs, and it would not be surprising to see more of that state regulation in the future.
Continue to: Patent uncertainty...
Patent uncertainty
Csaba Truckai invented and patented the NovaSure System ablation device with a “moisture permeable” head. He sold his company and the related patents, which eventually were purchased by Hologic. Over time, Hologic added claims to the original patent. In the meantime, Truckai went on to invent another device, the Minerva Endometrial Ablation System (MEAS), which had a “moisture impermeable” head. (Note that the “Minerva Surgical, Inc.” involved in this case is not related to the company “Minerva Industries,” which some identified as a “patent troll.”)22
Hologic sued Minerva, claiming that Truckai’s second device (MEAS) infringed on its patent for the first device (NovaSure). Truckai’s defense was that the patent on NovaSure was invalid. Hologic felt that since Truckai had obtained that patent and then sold it, it was improper for him now to claim it was invalid. There is a doctrine for that: assignor estoppel—the person who sold (assigned) the patent is prevented from later claiming it was invalid. The question in this case was whether assignor estoppel is part of the patent law of the United States. It is not in the patent statutes, so it is a court-determined part of the law.
In a 5-4 decision this Term, the Court held that assignor estoppel is recognized, but that it is narrow.23 The Court identified several exceptions to assignor estoppel, notably for this case, including the situation in which the purchaser of the patent, after the purchase, returns to the Patent and Trademark Office to expand (amend) the patent’s claims. In that case, the seller could not be estopped by the amended terms of the patent. Minerva claimed that it was attacking the expanded patent that included changes made after it sold the patent. The Court, therefore, returned the case to the Federal Circuit to apply the principles it laid out about assignor estoppel.
Biotech and other fast-moving fields frequently have new technology building on slightly earlier technology. The current patent system often leaves uncertainty about who owns which part of a valid patent. This uncertainty is a drag on innovation, and the patent system is supposed to spur innovation. Assignor estoppel is likely to create additional complexity and uncertainty in some patents, which is regrettable.
Review of the Term
In addition to the other disruptions of the Term, during the first part of the Term, Amy Coney Barrett was not yet confirmed by the Senate, so there were only 8 justices until October 27. She did not participate in those cases that were heard before she joined the Court. The consensus is that the Court heard 67 cases: 57 were formally briefed and argued along with 8 summary reversals and 2 religious cases in the Shadow Docket. In my opinion, this undercounts both the number and the importance of the Shadow Docket cases, but the following data use the 67 case convention.24
The Court was unanimous in 43% of the cases, including some of the most divisive issues. That unanimity reflects very narrow decisions. There were (by conventional count) only eight 5-4 opinions (12%), an unusually low number. Justice Kavanaugh is viewed as the “median” justice. He was in the majority in 97% of all cases. Chief Justice Roberts and Justice Barrett were in the majority 91%, and Justice Gorsuch 90%. As for the other justices, they were in the majority (all cases) most of the time: Justice Alito, 83%; Justice Thomas, 81%; Justice Breyer, 76%; Justice Kagan, 75%; and Justice Sotomayor, 69%. In “divided cases” (when unanimous cases are removed), the percentages are: Justice Kavanaugh, 95%; Chief Justice Roberts and Justice Barrett, 84%; Justice Gorsuch, 82%; Justice Alito, 70%; Justice Thomas, 66%; Justice Breyer, 58%; Justice Kagan, 55%; and Justice Sotomayor, 45%.
When the term began, many Court watchers expected a relatively uninteresting term, dealing with many technical legal details. In fact, it turned out to be more interesting and important than expected, even with narrow holdings in important cases. Part of the secret of the term was that a lot of the real action was in the Shadow Docket. The end of the term is sometimes the moment when a justice announces a plan to retire. Many commentators expected Justice Breyer might announce—he has been under pressure to do so, to allow President Biden to nominate and a Democratic Senate to confirm a progressive justice. However, he did not do so. It is possible that he will announce his retirement to be effective when his successor is confirmed, but that is pure speculation.
Continue to: Next Term...
Next Term
The next term began on Monday, October 4, 2021. With the considerable current activity in the Shadow Docket, there was not much of a summer break. The coming term looks extraordinary. The headline case is an abortion case from Mississippi, Dobbs v Jackson Women’s Health Organization.25 The legal question is the constitutionality of Mississippi law that prohibits most abortions after 15 weeks of gestation. The Texas abortion law will also be back before the Court. As we saw this term, big cases may produce very narrow results, but this case has the potential for being a notable abortion decision.
In a different case the Court will decide whether a state attorney general can step in to defend an abortion law when the state health secretary does not do so.26
The Court also has accepted 3 cases dealing with reimbursement for health services. One deals with whether or not the Department of Health and Human Services can set reimbursement rates without good survey data regarding costs,27 another involves the calculation of additional payments for hospitals that serve a “disproportionate number of low-income patients,”28 and the third whether state Medicaid programs can take funds from an injured beneficiary’s tort recovery to cover future Medicaid costs.29
In other cases, the Court will review a gun control law from New York. The Court’s earlier Second Amendment cases involved guns in the home used for self-defense, but this case raises the question of whether a state can practically preclude “concealed-carry licenses.”30 Many experts believe the Court will accept a case dealing with racial preferences in college admissions, perhaps the Harvard case in which the claim is discrimination against Asian Americans.31
The ACOG mifepristone case was interesting, in part because the federal district court issued a nationwide injunction against the Americans with Disabilities Act, enforcing its rules anywhere in the country. The effect of these orders is for a single district judge to create the “law of the land,” at least until that is reviewed—which can take months. The advantage of the nationwide injunction is that it avoids having to repeatedly litigate the same issues in multiple courts around the country. The downside is that plaintiffs can seek out a nonrepresentative judge or circuit and receive an injunction that would be granted by few other circuits. In addition, a nationwide injunction can apply to specific circumstances that are not before the court issuing the injunction. In the mifepristone case, for example, 10 states requested to intervene in the ACOG case. The court rejected the request, but the nationwide injunction applied to those states.1
Although federal judges have had the authority to issue nationwide injunctions for years, they are becoming much more common. One reason is the ease of forum shopping noted earlier—organizations can cherry-pick district courts and circuits sympathetic to their views. Both left- and right-leaning organizations have learned this lesson, so left-leaning groups are likely to file in specific districts in the Ninth Circuit, and right-leaning groups to districts in the Fifth Circuit.
If the current trend of increasing nationwide injunctions continues, either the rules for the federal courts or congressional action may be required to reduce some of the abuses by both sides of the political spectrum.
The ACOG mifepristone case was interesting, in part because the federal district court issued a nationwide injunction against the Americans with Disabilities Act, enforcing its rules anywhere in the country. The effect of these orders is for a single district judge to create the “law of the land,” at least until that is reviewed—which can take months. The advantage of the nationwide injunction is that it avoids having to repeatedly litigate the same issues in multiple courts around the country. The downside is that plaintiffs can seek out a nonrepresentative judge or circuit and receive an injunction that would be granted by few other circuits. In addition, a nationwide injunction can apply to specific circumstances that are not before the court issuing the injunction. In the mifepristone case, for example, 10 states requested to intervene in the ACOG case. The court rejected the request, but the nationwide injunction applied to those states.1
Although federal judges have had the authority to issue nationwide injunctions for years, they are becoming much more common. One reason is the ease of forum shopping noted earlier—organizations can cherry-pick district courts and circuits sympathetic to their views. Both left- and right-leaning organizations have learned this lesson, so left-leaning groups are likely to file in specific districts in the Ninth Circuit, and right-leaning groups to districts in the Fifth Circuit.
If the current trend of increasing nationwide injunctions continues, either the rules for the federal courts or congressional action may be required to reduce some of the abuses by both sides of the political spectrum. Reference Am. Coll. of Obstetricians & Gynecologists v. United States FDA, 467 F. Supp. 3d 282, 284 (D. Md. 2020).
Reference
1. Am. Coll. of Obstetricians & Gynecologists v. United States FDA, 467 F. Supp. 3d 282, 284 (D. Md. 2020).
The “Shadow Docket”
The ACOG mifepristone decisions do not appear on the Supreme Court’s “Court Opinions” website.1 They appear in what has become known in recent years as “The Shadow Docket,” an informal term that includes many orders of the Court and statements of individual justices regarding some cases.2 There are hundreds of orders by the Court each Term, there is nothing particularly shadowy about any of these items—they are all publicly available on the Court’s website and later in paper format. It is, however, a little harder to find and much harder to sort through than the major opinions. In some cases, it is not possible to tell what the vote was, how each justice voted, and what the reasoning of the Court was. In a few cases it is difficult to know exactly what the Court was holding or otherwise leaves some confusion about what the law actually is.3
The part of the Shadow Docket that is most intriguing for commentators, and where the ACOG cases appear, is the “Opinions Relating to Orders.”4 These are a variety of opinions, some written by the Court and many by individual justices. It also includes the action of the Court in some cases in which there was not full briefing or oral argument. The statements by justices often are to dissent from the denial of cert of decisions of the Court. These opinions have become much more common over the years. In this past term, there were approximately 60 such opinions related to about 50 cases. In part, this relates to the number of pandemic cases that could not wait for a Court decision going through the extended ordinary process. Although the Shadow Docket has been of interest to academic observers and Court watchers for years, this year it has attracted the attention of Congress.5
References
1. Opinions of the Court. Supreme Court website. https://www.supremecourt.gov/opinions/slipopinion/20#list. Accessed October 10, 2021.
2. Baude W. Foreword: the Supreme Court’s Shadow Docket, 9 N.Y.U. J.L. & Liberty 1 (2015).
3. Vladeck SI. The Solicitor General and the Shadow Docket, 133 Harvard Law Review. 123 (2019).
4. Opinions relating to orders. Supreme Court website. https://www.supremecourt.gov/opinions/relatingtoorders/20#list. Accessed October 10, 2021.
5. The Supreme Court’s Shadow Docket: Hearing Before the Subcommittee on Courts, Intellectual Property and the Internet of the H. Committee on the Judiciary, 117th Congress (2021).
- American College of Obstetricians & Gynecologists v. United States FDA, 472 F. Supp. 3d 183 (D. Md. 2020).
- Michael Kunzelman, Doctors Sue to Block FDA Abortion Pill Rule During Pandemic, (May 29, 2020).
- ACLU, American College Of Obstetricians And Gynecologists V. U.S. Food And Drug Administration, https://www.aclu.org/cases/american-college-obstetricians-and-gynecologists-v-us-food-and-drug-administration. Updated February 12, 2021. Accessed August 27, 2021.
- Whole Woman’s Health v Hellerstedt, 579 US ___ (2016), 136 S Ct 2292.
- 2016 Clinical Review at 39, 47, 49, Opp’n Mot. PI Ex. 19, ECF No. 62-11.
- American College of Obstetricians and Gynecologists v FDA (I), decided October 8, 2020.
- October 8, 2020, dissenting opinion by Justice Alito.
- January 12, 2021, dissenting opinion by Justice Sotomayor.
- Questions and answers on Mifeprex. U.S. Food and Drug Administration website. Published April 13, 2021. https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/questions-and-answers-mifeprex. Accessed October 9, 2021.
- Whole Woman’s Health v Jackson, decided September 1, 2021.
- Texas Senate Bill 8, relating to abortion, including abortions after detection of unborn child’s heartbeat; authorizing a private civil right of action. LegiScan website. https://legiscan.com/TX/text/SB8/id/2395961. Accessed October 9, 2021.
- Planned Parenthood of Southeastern Pennsylvania v Casey, 505 U. S. 833 (1992); Roe v Wade, 410 U. S. 113 (1973).
- Dobbs v Jackson Women’s Health Organization, No. 19-1392.
- Roman Catholic Diocese of Brooklyn v Cuomo, decided November 25, 2020.
- Alabama Association of Realtors v Department of Health and Human Services, decided June 29, 2021.
- Temporary halt in residential evictions in communities with substantial or high levels of community transmission of COVID-19 to prevent the further spread of COVID-19. August 6, 2021. https://www.federalregister.gov/documents/2021/08/06/2021-16945/temporary-halt-in-residential-evictions-in-communities-with-substantial-or-high-transmission-of.
- Alabama Association of Realtors v Department of Health and Human Services, decided August 26, 2021.
- California v Texas, decided June 17, 2021.
- Brief of Amici Curiae American Medical Association, American Academy of Allergy, Asthma and Immunology, Aerospace Medical Association, American Academy of Family Physicians, American Academy of Pediatrics, American College of Cardiology, American College of Emergency Physicians, American College of Medical Genetics and Genomics, American College of Obstetricians and Gynecologists, American College of Physicians, American College of Radiation Oncology, American College of Radiology, American Psychiatric Association, American Society of Gastrointestinal Endoscopy, American Society of Hematology, American Society of Metabolic and Bariatric Surgery, Endocrine Society, GLMA: Health Professionals Advancing LGBTQ Equality, Renal Physicians Association, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology in Support of Petitioners, in California v. Texas. May 13, 2020. https://www.supremecourt.gov/DocketPDF/19/19-840/143469/20200513150051995_19-840%20Amici%20Brief%20AMA.pdf. Accessed October 9, 2021.
- Rutledge v Pharmaceutical Care Management Association, decided December 10, 2020.
- Brief of the American Medical Association, The Arkansas Medical Society, and The Litigation Center of the American Medical Association and the State Medical Societies as Amici Curiae in Support of Petitioner in Rutledge v Pharmaceutical Care Management Association. March 2, 2020. https://www.supremecourt.gov/DocketPDF/18/18-540/134670/20200302163622018_Rutledge%20v.%20PCMA%20Amicus%20Brief%20of%20AMA%20et%20al.pdf. Accessed October 9, 2021.
- Apple quietly settles patent lawsuit, promptly gets hit with another one. TechCrunch website. Published July 30, 2010. https://techcrunch.com/2010/07/30/apple-minerva-emblaze/. Accessed October 9, 2021.
- Minerva Surgical, Inc. v Hologic, Inc., decided June 29, 2021.
- Stat pack. SCOTUS Blog website. Published July 6, 2021. https://www.scotusblog.com/wp-content/uploads/2021/07/Final-Stat-Pack-7.6.21.pdf. Accessed October 9, 2021.
- Dobbs v Jackson Women’s Health Organization, No. 19-1392.
- Cameron v. EMW Women’s Surgical Center, https://www.scotusblog.com/case-files/cases/cameron-v-emw-womens-surgical-center-p-s-c/. Accessed August 28, 2021.
- American Hospital Association v Becerra, No. 20-1114.
- Becerra v Empire Health Foundation, No. 20-1312.
- Gallardo v Marstiller, No. 20-1263.
- New York State Rifle & Pistol Association Inc. v Corlett, No. 20-843.
- Students for Fair Admissions v President & Fellows of Harvard College, No. 20-1199.
The Supreme Court’s usual processes were disrupted this term. The COVID-19 pandemic required audio hearings rather than in-person, and it resulted in a number of emergency legal appeals. As the Court began its regular sessions on October 5, 2020, there were only 8 justices—Justice Ruth Bader Ginsburg had passed away and Amy Coney Barrett had not yet been confirmed by the Senate. The Court decided many important cases this term, including dealing with the delivery of drugs to induce abortions, a Centers for Disease Control and Prevention (CDC) moratorium on housing evictions, yet another case on the Affordable Care Act, state laws concerning pharmacy benefit managers, and the Hologic and Minerva endometrial ablation systems patents. After considering these cases, we also will briefly look at other cases of general interest.
Abortion
Patient access to mifepristone
In May 2020, the American College of Obstetricians and Gynecologists (ACOG) was the named plaintiff in a lawsuit against the US Food and Drug Administration (FDA) regarding the drugs mifepristone and misoprostol that are used to induce medical abortions.1 The case was filed by the American Civil Liberties Union on behalf of ACOG and others2,3 and raised the issue of patients’ access to these medications. The basic claim of the case was that during the pandemic, the FDA’s regulation of mifepristone was unconstitutional in that they imposed an undue burden on the decision of women to have an abortion.4 (Although misoprostol is a part of the medical abortion regimen, it is not subject to special regulation and was not part of the litigation.)
The FDA regulation of mifepristone, begun in 2000 but modified since then, includes 3 elements to assure safe use:
- prescribers must have special training or certification
- the drug can be dispensed to patients only in a hospital, clinic, or medical office under the supervision of a certified health care provider (known as the “in-person dispensing requirement” because retail pharmacy or mail distribution are prohibited)
- the health care provider must review a “patient agreement form” with the patient and have the patient sign the consent form in the provider’s presence.5
The pandemic made fulfilling these requirements substantially more burdensome and difficult. The question was whether the FDA was constitutionally required to modify its regulations during a pandemic to take account of the undue burden of the regulation created by the pandemic. That is, the question was not whether the FDA could have or should have chosen to make the modification, but whether it was required to do so.
In July 2020, a federal district court in Maryland held that the FDA regulation was an unconstitutional burden on the abortion rights of women during the pandemic and issued a preliminary injunction to stop the FDA from enforcing the in-person dispensing and signature rules. The district judge applied the injunction to Maryland, but also made it a nationwide injunction. (The issue of district court nationwide injunctions is considered in, “District court ‘nationwide injunctions’”).
The FDA asked the Fourth Circuit Court of Appeals to stay the enforcement of the injunction, which the appeals court denied. The FDA then appealed to the Supreme Court, asking it to stay the injunction. In October 2020, the Court announced that it was holding the FDA’s request “in abeyance” to allow the district court to consider a motion by the FDA to dissolve or change the injunction. It gave the district court 40 days in which to act. That decision by the Court was in the “Shadow Docket” (see sidebar on page XX), so the exact vote of the Court in October is not clear, but 2 Justices (Alito and Thomas) dissented and would have stayed the injunction.6 Over the next 40 days, the district court did not withdraw its nationwide injunction.
Thus, on January 12, 2021, the case was again before the Supreme Court, which let the FDA’s regulations regarding mifepristone remain in place by lifting the district court’s injunction. Most of the justices supporting the stay did not write to explain their decision, although their dissent in the earlier cases may have served that purpose. (Maryland was permitting many kinds of activity that were more risky than visiting a clinic—indoor dining, with open hair salons, gyms, and casinos.)7 Chief Justice Roberts wrote a concurrence to indicate that, in his view, the issue was not whether the FDA’s regulations placed an undue burden on a right to an abortion generally, but that “My view is that courts owe significant deference” to the public health authorities (here meaning the FDA). Justices Sotomayor and Kagan dissented, saying that the issue was the undue burden on women, given the difficulties of the pandemic, particularly going to medical facilities during the COVID-19 pandemic.8
The injunction, sought by ACOG and others, was issued by the district court and was in effect for several months before it was dissolved by the Supreme Court. Following the change in presidential administrations, in April 2021 the FDA announced that it was going to “exercise enforcement discretion with respect to the in-person dispensing requirement…during the COVID-19 public health emergency.”9
Continue to: The Texas abortion case...
The Texas abortion case
The Court, on September 1, 2021, declined to block a Texas abortion statute from taking effect.10 This law precludes abortions after a fetal heartbeat is present at about 6 weeks of gestation. The Fifth Circuit declined to grant an injunction delaying implementation of the Texas law, and the Court did not reverse that decision.
Over the years, a variety of states have placed limitations on abortion, and those almost always have been enjoined by federal courts before they went into effect. However, the Texas statute, which undoubtedly is unconstitutional, was creatively constructed to avoid an early injunction.11 The statute does not allow state officials to enforce the new law, but rather it allows almost any private citizen to seek monetary damages from anyone performing an abortion or who “aids and abets” an abortion. Thus, it is difficult to tailor a lawsuit before this law is enforced. First, courts do not enjoin laws; they usually enjoin individuals from enforcing the law, and in this case it is difficult to know which individuals will be enforcing the laws and what their decisions might be. There also are some questions about the degree to which federal courts can enjoin state courts from deciding lawsuits under state law. For these procedural reasons, the majority of the Court found that those attacking the Texas law had not met their burden of showing that that they would win their case.
Even 3 of the dissenting justices said the defendants may be right that “existing doctrines preclude judicial intervention,” but that the consequences are such that the Court should delay the law until there is time for briefing and argument. The other 3 dissenting justices thought there would be ways of getting around the clever roadblock Texas had erected for the federal courts.
There has been some commentary that this case portends the abandonment of Roe v Wade and Casey,12 but that conclusion does not seem warranted by this case. The Court has accepted a Mississippi abortion law to be heard next term.13 In addition, the Texas statute is likely to be back in federal court once a private individual has filed a claim for money from an abortion provider (and likely even before that).
COVID-19 cases
The Supreme Court decided several cases related to COVID-19, including adjustments to election procedures, church services, and CDC eviction moratoria. As a general matter early in the pandemic, the Court deferred to government authorities, generally upholding government actions. Chief Justice Roberts emphasized the importance of the Court deferring to government officials in emergencies. As the pandemic progressed into 2021, however, the Court became less and less sympathetic to government actions that were not consistent, permitted by existing law, or reasonably necessary. For example, regulations of churches that were inconsistent with the regulation of similar organizations were struck down.14
Among the most interesting of the summer 2021 cases was the CDC eviction moratorium that essentially prohibited landlords nationwide from evicting tenants for nonpayment of rent. When the challenges to these CDC regulations first reached the Court, the moratorium was about to expire; in a 5-4 decision, the Court did not enjoin the CDC from continuing that policy. Justice Kavanaugh (the fifth vote) warned that “clear and specific congressional authorization…would be necessary to extend the moratorium past July 31.”15 Despite telling the Court that the moratorium would expire on July 31, just 3 days after the expiration and without any congressional authorization, the CDC reinstated what was practically the same moratorium.16 On August 26, the Court struck down the reinstated regulation, probably by a 6-3 margin. (Because this case arose in the “Shadow Docket,” the vote of some justices is not certain).17
Continue to: The Affordable Care Act...
The Affordable Care Act
The Affordable Care Act was challenged in the Court for the third time.18 In this term’s case, several states argued that when Congress essentially eliminated the penalty/tax for not purchasing insurance coverage, there was no longer a constitutional basis for the individual mandate. With that centerpiece gone, they claimed, the whole statute should be declared unconstitutional.
Along with many other specialty groups, ACOG joined an amicus curiae brief sponsored by the American Medical Association (AMA).19 An amicus brief is one not filed by the parties to the case, but by organizations or individuals who have information that may be of use to the Court in considering the case. Among other things, the filing of an amicus brief indicates the interest of the organization in the outcome of the case. In this case, the crux of the amicus was that even if the individual mandate currently is not constitutional, the Court should sever that provision and retain the rest of the ACA.
Despite some wild predictions about what the Court might do, it did not decide any substantive issue. Rather, it found that none of the parties to the case had “standing” to challenge the constitutionality of the ACA. Therefore, in effect, the Court dismissed the case without deciding the substantive legal issues.
Pharmacy Benefit Managers
The powerful Pharmacy Benefit Managers (PBMs) are a hidden part of the health care system; however, in recent years there has been increasing regulatory attention paid to them. Some states have begun regulating aspects of PBMs. In this term, the Court considered an Arkansas law that sought to protect local pharmacies from PBM pricing practices.20 The AMA filed an amicus brief in the case which made legal arguments, most of which had been made by the parties to the litigation.21
PBMs generally tell pharmacies how much they will reimburse the pharmacy for filling a prescription for a particular drug. In some instances, PBMs will set a reimbursement price that is lower than the wholesale price at which local pharmacies can purchase the drug. The Arkansas law prohibited PBMs in the state from reimbursing pharmacies for less than the wholesale cost the pharmacy paid for the drug.
The claim of the PBMs was that the Arkansas law violated the Employee Retirement Income Security Act (ERISA). In part, this act preempts state law that relates to fringe benefit plans. States have the authority to regulate insurance, but ERISA limits what they can do when the insurance relates to fringe benefits. The Court held that ERISA does not preempt the Arkansas law or similar state laws in other states. Because the state law was not preempted by the state law, the Arkansas regulation was upheld. The fact that this was a unanimous decision (8-0, because Justice Barrett was not on the Court when the case was heard) suggests that states may have leeway in additional regulations of PBMs, and it would not be surprising to see more of that state regulation in the future.
Continue to: Patent uncertainty...
Patent uncertainty
Csaba Truckai invented and patented the NovaSure System ablation device with a “moisture permeable” head. He sold his company and the related patents, which eventually were purchased by Hologic. Over time, Hologic added claims to the original patent. In the meantime, Truckai went on to invent another device, the Minerva Endometrial Ablation System (MEAS), which had a “moisture impermeable” head. (Note that the “Minerva Surgical, Inc.” involved in this case is not related to the company “Minerva Industries,” which some identified as a “patent troll.”)22
Hologic sued Minerva, claiming that Truckai’s second device (MEAS) infringed on its patent for the first device (NovaSure). Truckai’s defense was that the patent on NovaSure was invalid. Hologic felt that since Truckai had obtained that patent and then sold it, it was improper for him now to claim it was invalid. There is a doctrine for that: assignor estoppel—the person who sold (assigned) the patent is prevented from later claiming it was invalid. The question in this case was whether assignor estoppel is part of the patent law of the United States. It is not in the patent statutes, so it is a court-determined part of the law.
In a 5-4 decision this Term, the Court held that assignor estoppel is recognized, but that it is narrow.23 The Court identified several exceptions to assignor estoppel, notably for this case, including the situation in which the purchaser of the patent, after the purchase, returns to the Patent and Trademark Office to expand (amend) the patent’s claims. In that case, the seller could not be estopped by the amended terms of the patent. Minerva claimed that it was attacking the expanded patent that included changes made after it sold the patent. The Court, therefore, returned the case to the Federal Circuit to apply the principles it laid out about assignor estoppel.
Biotech and other fast-moving fields frequently have new technology building on slightly earlier technology. The current patent system often leaves uncertainty about who owns which part of a valid patent. This uncertainty is a drag on innovation, and the patent system is supposed to spur innovation. Assignor estoppel is likely to create additional complexity and uncertainty in some patents, which is regrettable.
Review of the Term
In addition to the other disruptions of the Term, during the first part of the Term, Amy Coney Barrett was not yet confirmed by the Senate, so there were only 8 justices until October 27. She did not participate in those cases that were heard before she joined the Court. The consensus is that the Court heard 67 cases: 57 were formally briefed and argued along with 8 summary reversals and 2 religious cases in the Shadow Docket. In my opinion, this undercounts both the number and the importance of the Shadow Docket cases, but the following data use the 67 case convention.24
The Court was unanimous in 43% of the cases, including some of the most divisive issues. That unanimity reflects very narrow decisions. There were (by conventional count) only eight 5-4 opinions (12%), an unusually low number. Justice Kavanaugh is viewed as the “median” justice. He was in the majority in 97% of all cases. Chief Justice Roberts and Justice Barrett were in the majority 91%, and Justice Gorsuch 90%. As for the other justices, they were in the majority (all cases) most of the time: Justice Alito, 83%; Justice Thomas, 81%; Justice Breyer, 76%; Justice Kagan, 75%; and Justice Sotomayor, 69%. In “divided cases” (when unanimous cases are removed), the percentages are: Justice Kavanaugh, 95%; Chief Justice Roberts and Justice Barrett, 84%; Justice Gorsuch, 82%; Justice Alito, 70%; Justice Thomas, 66%; Justice Breyer, 58%; Justice Kagan, 55%; and Justice Sotomayor, 45%.
When the term began, many Court watchers expected a relatively uninteresting term, dealing with many technical legal details. In fact, it turned out to be more interesting and important than expected, even with narrow holdings in important cases. Part of the secret of the term was that a lot of the real action was in the Shadow Docket. The end of the term is sometimes the moment when a justice announces a plan to retire. Many commentators expected Justice Breyer might announce—he has been under pressure to do so, to allow President Biden to nominate and a Democratic Senate to confirm a progressive justice. However, he did not do so. It is possible that he will announce his retirement to be effective when his successor is confirmed, but that is pure speculation.
Continue to: Next Term...
Next Term
The next term began on Monday, October 4, 2021. With the considerable current activity in the Shadow Docket, there was not much of a summer break. The coming term looks extraordinary. The headline case is an abortion case from Mississippi, Dobbs v Jackson Women’s Health Organization.25 The legal question is the constitutionality of Mississippi law that prohibits most abortions after 15 weeks of gestation. The Texas abortion law will also be back before the Court. As we saw this term, big cases may produce very narrow results, but this case has the potential for being a notable abortion decision.
In a different case the Court will decide whether a state attorney general can step in to defend an abortion law when the state health secretary does not do so.26
The Court also has accepted 3 cases dealing with reimbursement for health services. One deals with whether or not the Department of Health and Human Services can set reimbursement rates without good survey data regarding costs,27 another involves the calculation of additional payments for hospitals that serve a “disproportionate number of low-income patients,”28 and the third whether state Medicaid programs can take funds from an injured beneficiary’s tort recovery to cover future Medicaid costs.29
In other cases, the Court will review a gun control law from New York. The Court’s earlier Second Amendment cases involved guns in the home used for self-defense, but this case raises the question of whether a state can practically preclude “concealed-carry licenses.”30 Many experts believe the Court will accept a case dealing with racial preferences in college admissions, perhaps the Harvard case in which the claim is discrimination against Asian Americans.31
The ACOG mifepristone case was interesting, in part because the federal district court issued a nationwide injunction against the Americans with Disabilities Act, enforcing its rules anywhere in the country. The effect of these orders is for a single district judge to create the “law of the land,” at least until that is reviewed—which can take months. The advantage of the nationwide injunction is that it avoids having to repeatedly litigate the same issues in multiple courts around the country. The downside is that plaintiffs can seek out a nonrepresentative judge or circuit and receive an injunction that would be granted by few other circuits. In addition, a nationwide injunction can apply to specific circumstances that are not before the court issuing the injunction. In the mifepristone case, for example, 10 states requested to intervene in the ACOG case. The court rejected the request, but the nationwide injunction applied to those states.1
Although federal judges have had the authority to issue nationwide injunctions for years, they are becoming much more common. One reason is the ease of forum shopping noted earlier—organizations can cherry-pick district courts and circuits sympathetic to their views. Both left- and right-leaning organizations have learned this lesson, so left-leaning groups are likely to file in specific districts in the Ninth Circuit, and right-leaning groups to districts in the Fifth Circuit.
If the current trend of increasing nationwide injunctions continues, either the rules for the federal courts or congressional action may be required to reduce some of the abuses by both sides of the political spectrum.
The ACOG mifepristone case was interesting, in part because the federal district court issued a nationwide injunction against the Americans with Disabilities Act, enforcing its rules anywhere in the country. The effect of these orders is for a single district judge to create the “law of the land,” at least until that is reviewed—which can take months. The advantage of the nationwide injunction is that it avoids having to repeatedly litigate the same issues in multiple courts around the country. The downside is that plaintiffs can seek out a nonrepresentative judge or circuit and receive an injunction that would be granted by few other circuits. In addition, a nationwide injunction can apply to specific circumstances that are not before the court issuing the injunction. In the mifepristone case, for example, 10 states requested to intervene in the ACOG case. The court rejected the request, but the nationwide injunction applied to those states.1
Although federal judges have had the authority to issue nationwide injunctions for years, they are becoming much more common. One reason is the ease of forum shopping noted earlier—organizations can cherry-pick district courts and circuits sympathetic to their views. Both left- and right-leaning organizations have learned this lesson, so left-leaning groups are likely to file in specific districts in the Ninth Circuit, and right-leaning groups to districts in the Fifth Circuit.
If the current trend of increasing nationwide injunctions continues, either the rules for the federal courts or congressional action may be required to reduce some of the abuses by both sides of the political spectrum. Reference Am. Coll. of Obstetricians & Gynecologists v. United States FDA, 467 F. Supp. 3d 282, 284 (D. Md. 2020).
Reference
1. Am. Coll. of Obstetricians & Gynecologists v. United States FDA, 467 F. Supp. 3d 282, 284 (D. Md. 2020).
The “Shadow Docket”
The ACOG mifepristone decisions do not appear on the Supreme Court’s “Court Opinions” website.1 They appear in what has become known in recent years as “The Shadow Docket,” an informal term that includes many orders of the Court and statements of individual justices regarding some cases.2 There are hundreds of orders by the Court each Term, there is nothing particularly shadowy about any of these items—they are all publicly available on the Court’s website and later in paper format. It is, however, a little harder to find and much harder to sort through than the major opinions. In some cases, it is not possible to tell what the vote was, how each justice voted, and what the reasoning of the Court was. In a few cases it is difficult to know exactly what the Court was holding or otherwise leaves some confusion about what the law actually is.3
The part of the Shadow Docket that is most intriguing for commentators, and where the ACOG cases appear, is the “Opinions Relating to Orders.”4 These are a variety of opinions, some written by the Court and many by individual justices. It also includes the action of the Court in some cases in which there was not full briefing or oral argument. The statements by justices often are to dissent from the denial of cert of decisions of the Court. These opinions have become much more common over the years. In this past term, there were approximately 60 such opinions related to about 50 cases. In part, this relates to the number of pandemic cases that could not wait for a Court decision going through the extended ordinary process. Although the Shadow Docket has been of interest to academic observers and Court watchers for years, this year it has attracted the attention of Congress.5
References
1. Opinions of the Court. Supreme Court website. https://www.supremecourt.gov/opinions/slipopinion/20#list. Accessed October 10, 2021.
2. Baude W. Foreword: the Supreme Court’s Shadow Docket, 9 N.Y.U. J.L. & Liberty 1 (2015).
3. Vladeck SI. The Solicitor General and the Shadow Docket, 133 Harvard Law Review. 123 (2019).
4. Opinions relating to orders. Supreme Court website. https://www.supremecourt.gov/opinions/relatingtoorders/20#list. Accessed October 10, 2021.
5. The Supreme Court’s Shadow Docket: Hearing Before the Subcommittee on Courts, Intellectual Property and the Internet of the H. Committee on the Judiciary, 117th Congress (2021).
The Supreme Court’s usual processes were disrupted this term. The COVID-19 pandemic required audio hearings rather than in-person, and it resulted in a number of emergency legal appeals. As the Court began its regular sessions on October 5, 2020, there were only 8 justices—Justice Ruth Bader Ginsburg had passed away and Amy Coney Barrett had not yet been confirmed by the Senate. The Court decided many important cases this term, including dealing with the delivery of drugs to induce abortions, a Centers for Disease Control and Prevention (CDC) moratorium on housing evictions, yet another case on the Affordable Care Act, state laws concerning pharmacy benefit managers, and the Hologic and Minerva endometrial ablation systems patents. After considering these cases, we also will briefly look at other cases of general interest.
Abortion
Patient access to mifepristone
In May 2020, the American College of Obstetricians and Gynecologists (ACOG) was the named plaintiff in a lawsuit against the US Food and Drug Administration (FDA) regarding the drugs mifepristone and misoprostol that are used to induce medical abortions.1 The case was filed by the American Civil Liberties Union on behalf of ACOG and others2,3 and raised the issue of patients’ access to these medications. The basic claim of the case was that during the pandemic, the FDA’s regulation of mifepristone was unconstitutional in that they imposed an undue burden on the decision of women to have an abortion.4 (Although misoprostol is a part of the medical abortion regimen, it is not subject to special regulation and was not part of the litigation.)
The FDA regulation of mifepristone, begun in 2000 but modified since then, includes 3 elements to assure safe use:
- prescribers must have special training or certification
- the drug can be dispensed to patients only in a hospital, clinic, or medical office under the supervision of a certified health care provider (known as the “in-person dispensing requirement” because retail pharmacy or mail distribution are prohibited)
- the health care provider must review a “patient agreement form” with the patient and have the patient sign the consent form in the provider’s presence.5
The pandemic made fulfilling these requirements substantially more burdensome and difficult. The question was whether the FDA was constitutionally required to modify its regulations during a pandemic to take account of the undue burden of the regulation created by the pandemic. That is, the question was not whether the FDA could have or should have chosen to make the modification, but whether it was required to do so.
In July 2020, a federal district court in Maryland held that the FDA regulation was an unconstitutional burden on the abortion rights of women during the pandemic and issued a preliminary injunction to stop the FDA from enforcing the in-person dispensing and signature rules. The district judge applied the injunction to Maryland, but also made it a nationwide injunction. (The issue of district court nationwide injunctions is considered in, “District court ‘nationwide injunctions’”).
The FDA asked the Fourth Circuit Court of Appeals to stay the enforcement of the injunction, which the appeals court denied. The FDA then appealed to the Supreme Court, asking it to stay the injunction. In October 2020, the Court announced that it was holding the FDA’s request “in abeyance” to allow the district court to consider a motion by the FDA to dissolve or change the injunction. It gave the district court 40 days in which to act. That decision by the Court was in the “Shadow Docket” (see sidebar on page XX), so the exact vote of the Court in October is not clear, but 2 Justices (Alito and Thomas) dissented and would have stayed the injunction.6 Over the next 40 days, the district court did not withdraw its nationwide injunction.
Thus, on January 12, 2021, the case was again before the Supreme Court, which let the FDA’s regulations regarding mifepristone remain in place by lifting the district court’s injunction. Most of the justices supporting the stay did not write to explain their decision, although their dissent in the earlier cases may have served that purpose. (Maryland was permitting many kinds of activity that were more risky than visiting a clinic—indoor dining, with open hair salons, gyms, and casinos.)7 Chief Justice Roberts wrote a concurrence to indicate that, in his view, the issue was not whether the FDA’s regulations placed an undue burden on a right to an abortion generally, but that “My view is that courts owe significant deference” to the public health authorities (here meaning the FDA). Justices Sotomayor and Kagan dissented, saying that the issue was the undue burden on women, given the difficulties of the pandemic, particularly going to medical facilities during the COVID-19 pandemic.8
The injunction, sought by ACOG and others, was issued by the district court and was in effect for several months before it was dissolved by the Supreme Court. Following the change in presidential administrations, in April 2021 the FDA announced that it was going to “exercise enforcement discretion with respect to the in-person dispensing requirement…during the COVID-19 public health emergency.”9
Continue to: The Texas abortion case...
The Texas abortion case
The Court, on September 1, 2021, declined to block a Texas abortion statute from taking effect.10 This law precludes abortions after a fetal heartbeat is present at about 6 weeks of gestation. The Fifth Circuit declined to grant an injunction delaying implementation of the Texas law, and the Court did not reverse that decision.
Over the years, a variety of states have placed limitations on abortion, and those almost always have been enjoined by federal courts before they went into effect. However, the Texas statute, which undoubtedly is unconstitutional, was creatively constructed to avoid an early injunction.11 The statute does not allow state officials to enforce the new law, but rather it allows almost any private citizen to seek monetary damages from anyone performing an abortion or who “aids and abets” an abortion. Thus, it is difficult to tailor a lawsuit before this law is enforced. First, courts do not enjoin laws; they usually enjoin individuals from enforcing the law, and in this case it is difficult to know which individuals will be enforcing the laws and what their decisions might be. There also are some questions about the degree to which federal courts can enjoin state courts from deciding lawsuits under state law. For these procedural reasons, the majority of the Court found that those attacking the Texas law had not met their burden of showing that that they would win their case.
Even 3 of the dissenting justices said the defendants may be right that “existing doctrines preclude judicial intervention,” but that the consequences are such that the Court should delay the law until there is time for briefing and argument. The other 3 dissenting justices thought there would be ways of getting around the clever roadblock Texas had erected for the federal courts.
There has been some commentary that this case portends the abandonment of Roe v Wade and Casey,12 but that conclusion does not seem warranted by this case. The Court has accepted a Mississippi abortion law to be heard next term.13 In addition, the Texas statute is likely to be back in federal court once a private individual has filed a claim for money from an abortion provider (and likely even before that).
COVID-19 cases
The Supreme Court decided several cases related to COVID-19, including adjustments to election procedures, church services, and CDC eviction moratoria. As a general matter early in the pandemic, the Court deferred to government authorities, generally upholding government actions. Chief Justice Roberts emphasized the importance of the Court deferring to government officials in emergencies. As the pandemic progressed into 2021, however, the Court became less and less sympathetic to government actions that were not consistent, permitted by existing law, or reasonably necessary. For example, regulations of churches that were inconsistent with the regulation of similar organizations were struck down.14
Among the most interesting of the summer 2021 cases was the CDC eviction moratorium that essentially prohibited landlords nationwide from evicting tenants for nonpayment of rent. When the challenges to these CDC regulations first reached the Court, the moratorium was about to expire; in a 5-4 decision, the Court did not enjoin the CDC from continuing that policy. Justice Kavanaugh (the fifth vote) warned that “clear and specific congressional authorization…would be necessary to extend the moratorium past July 31.”15 Despite telling the Court that the moratorium would expire on July 31, just 3 days after the expiration and without any congressional authorization, the CDC reinstated what was practically the same moratorium.16 On August 26, the Court struck down the reinstated regulation, probably by a 6-3 margin. (Because this case arose in the “Shadow Docket,” the vote of some justices is not certain).17
Continue to: The Affordable Care Act...
The Affordable Care Act
The Affordable Care Act was challenged in the Court for the third time.18 In this term’s case, several states argued that when Congress essentially eliminated the penalty/tax for not purchasing insurance coverage, there was no longer a constitutional basis for the individual mandate. With that centerpiece gone, they claimed, the whole statute should be declared unconstitutional.
Along with many other specialty groups, ACOG joined an amicus curiae brief sponsored by the American Medical Association (AMA).19 An amicus brief is one not filed by the parties to the case, but by organizations or individuals who have information that may be of use to the Court in considering the case. Among other things, the filing of an amicus brief indicates the interest of the organization in the outcome of the case. In this case, the crux of the amicus was that even if the individual mandate currently is not constitutional, the Court should sever that provision and retain the rest of the ACA.
Despite some wild predictions about what the Court might do, it did not decide any substantive issue. Rather, it found that none of the parties to the case had “standing” to challenge the constitutionality of the ACA. Therefore, in effect, the Court dismissed the case without deciding the substantive legal issues.
Pharmacy Benefit Managers
The powerful Pharmacy Benefit Managers (PBMs) are a hidden part of the health care system; however, in recent years there has been increasing regulatory attention paid to them. Some states have begun regulating aspects of PBMs. In this term, the Court considered an Arkansas law that sought to protect local pharmacies from PBM pricing practices.20 The AMA filed an amicus brief in the case which made legal arguments, most of which had been made by the parties to the litigation.21
PBMs generally tell pharmacies how much they will reimburse the pharmacy for filling a prescription for a particular drug. In some instances, PBMs will set a reimbursement price that is lower than the wholesale price at which local pharmacies can purchase the drug. The Arkansas law prohibited PBMs in the state from reimbursing pharmacies for less than the wholesale cost the pharmacy paid for the drug.
The claim of the PBMs was that the Arkansas law violated the Employee Retirement Income Security Act (ERISA). In part, this act preempts state law that relates to fringe benefit plans. States have the authority to regulate insurance, but ERISA limits what they can do when the insurance relates to fringe benefits. The Court held that ERISA does not preempt the Arkansas law or similar state laws in other states. Because the state law was not preempted by the state law, the Arkansas regulation was upheld. The fact that this was a unanimous decision (8-0, because Justice Barrett was not on the Court when the case was heard) suggests that states may have leeway in additional regulations of PBMs, and it would not be surprising to see more of that state regulation in the future.
Continue to: Patent uncertainty...
Patent uncertainty
Csaba Truckai invented and patented the NovaSure System ablation device with a “moisture permeable” head. He sold his company and the related patents, which eventually were purchased by Hologic. Over time, Hologic added claims to the original patent. In the meantime, Truckai went on to invent another device, the Minerva Endometrial Ablation System (MEAS), which had a “moisture impermeable” head. (Note that the “Minerva Surgical, Inc.” involved in this case is not related to the company “Minerva Industries,” which some identified as a “patent troll.”)22
Hologic sued Minerva, claiming that Truckai’s second device (MEAS) infringed on its patent for the first device (NovaSure). Truckai’s defense was that the patent on NovaSure was invalid. Hologic felt that since Truckai had obtained that patent and then sold it, it was improper for him now to claim it was invalid. There is a doctrine for that: assignor estoppel—the person who sold (assigned) the patent is prevented from later claiming it was invalid. The question in this case was whether assignor estoppel is part of the patent law of the United States. It is not in the patent statutes, so it is a court-determined part of the law.
In a 5-4 decision this Term, the Court held that assignor estoppel is recognized, but that it is narrow.23 The Court identified several exceptions to assignor estoppel, notably for this case, including the situation in which the purchaser of the patent, after the purchase, returns to the Patent and Trademark Office to expand (amend) the patent’s claims. In that case, the seller could not be estopped by the amended terms of the patent. Minerva claimed that it was attacking the expanded patent that included changes made after it sold the patent. The Court, therefore, returned the case to the Federal Circuit to apply the principles it laid out about assignor estoppel.
Biotech and other fast-moving fields frequently have new technology building on slightly earlier technology. The current patent system often leaves uncertainty about who owns which part of a valid patent. This uncertainty is a drag on innovation, and the patent system is supposed to spur innovation. Assignor estoppel is likely to create additional complexity and uncertainty in some patents, which is regrettable.
Review of the Term
In addition to the other disruptions of the Term, during the first part of the Term, Amy Coney Barrett was not yet confirmed by the Senate, so there were only 8 justices until October 27. She did not participate in those cases that were heard before she joined the Court. The consensus is that the Court heard 67 cases: 57 were formally briefed and argued along with 8 summary reversals and 2 religious cases in the Shadow Docket. In my opinion, this undercounts both the number and the importance of the Shadow Docket cases, but the following data use the 67 case convention.24
The Court was unanimous in 43% of the cases, including some of the most divisive issues. That unanimity reflects very narrow decisions. There were (by conventional count) only eight 5-4 opinions (12%), an unusually low number. Justice Kavanaugh is viewed as the “median” justice. He was in the majority in 97% of all cases. Chief Justice Roberts and Justice Barrett were in the majority 91%, and Justice Gorsuch 90%. As for the other justices, they were in the majority (all cases) most of the time: Justice Alito, 83%; Justice Thomas, 81%; Justice Breyer, 76%; Justice Kagan, 75%; and Justice Sotomayor, 69%. In “divided cases” (when unanimous cases are removed), the percentages are: Justice Kavanaugh, 95%; Chief Justice Roberts and Justice Barrett, 84%; Justice Gorsuch, 82%; Justice Alito, 70%; Justice Thomas, 66%; Justice Breyer, 58%; Justice Kagan, 55%; and Justice Sotomayor, 45%.
When the term began, many Court watchers expected a relatively uninteresting term, dealing with many technical legal details. In fact, it turned out to be more interesting and important than expected, even with narrow holdings in important cases. Part of the secret of the term was that a lot of the real action was in the Shadow Docket. The end of the term is sometimes the moment when a justice announces a plan to retire. Many commentators expected Justice Breyer might announce—he has been under pressure to do so, to allow President Biden to nominate and a Democratic Senate to confirm a progressive justice. However, he did not do so. It is possible that he will announce his retirement to be effective when his successor is confirmed, but that is pure speculation.
Continue to: Next Term...
Next Term
The next term began on Monday, October 4, 2021. With the considerable current activity in the Shadow Docket, there was not much of a summer break. The coming term looks extraordinary. The headline case is an abortion case from Mississippi, Dobbs v Jackson Women’s Health Organization.25 The legal question is the constitutionality of Mississippi law that prohibits most abortions after 15 weeks of gestation. The Texas abortion law will also be back before the Court. As we saw this term, big cases may produce very narrow results, but this case has the potential for being a notable abortion decision.
In a different case the Court will decide whether a state attorney general can step in to defend an abortion law when the state health secretary does not do so.26
The Court also has accepted 3 cases dealing with reimbursement for health services. One deals with whether or not the Department of Health and Human Services can set reimbursement rates without good survey data regarding costs,27 another involves the calculation of additional payments for hospitals that serve a “disproportionate number of low-income patients,”28 and the third whether state Medicaid programs can take funds from an injured beneficiary’s tort recovery to cover future Medicaid costs.29
In other cases, the Court will review a gun control law from New York. The Court’s earlier Second Amendment cases involved guns in the home used for self-defense, but this case raises the question of whether a state can practically preclude “concealed-carry licenses.”30 Many experts believe the Court will accept a case dealing with racial preferences in college admissions, perhaps the Harvard case in which the claim is discrimination against Asian Americans.31
The ACOG mifepristone case was interesting, in part because the federal district court issued a nationwide injunction against the Americans with Disabilities Act, enforcing its rules anywhere in the country. The effect of these orders is for a single district judge to create the “law of the land,” at least until that is reviewed—which can take months. The advantage of the nationwide injunction is that it avoids having to repeatedly litigate the same issues in multiple courts around the country. The downside is that plaintiffs can seek out a nonrepresentative judge or circuit and receive an injunction that would be granted by few other circuits. In addition, a nationwide injunction can apply to specific circumstances that are not before the court issuing the injunction. In the mifepristone case, for example, 10 states requested to intervene in the ACOG case. The court rejected the request, but the nationwide injunction applied to those states.1
Although federal judges have had the authority to issue nationwide injunctions for years, they are becoming much more common. One reason is the ease of forum shopping noted earlier—organizations can cherry-pick district courts and circuits sympathetic to their views. Both left- and right-leaning organizations have learned this lesson, so left-leaning groups are likely to file in specific districts in the Ninth Circuit, and right-leaning groups to districts in the Fifth Circuit.
If the current trend of increasing nationwide injunctions continues, either the rules for the federal courts or congressional action may be required to reduce some of the abuses by both sides of the political spectrum.
The ACOG mifepristone case was interesting, in part because the federal district court issued a nationwide injunction against the Americans with Disabilities Act, enforcing its rules anywhere in the country. The effect of these orders is for a single district judge to create the “law of the land,” at least until that is reviewed—which can take months. The advantage of the nationwide injunction is that it avoids having to repeatedly litigate the same issues in multiple courts around the country. The downside is that plaintiffs can seek out a nonrepresentative judge or circuit and receive an injunction that would be granted by few other circuits. In addition, a nationwide injunction can apply to specific circumstances that are not before the court issuing the injunction. In the mifepristone case, for example, 10 states requested to intervene in the ACOG case. The court rejected the request, but the nationwide injunction applied to those states.1
Although federal judges have had the authority to issue nationwide injunctions for years, they are becoming much more common. One reason is the ease of forum shopping noted earlier—organizations can cherry-pick district courts and circuits sympathetic to their views. Both left- and right-leaning organizations have learned this lesson, so left-leaning groups are likely to file in specific districts in the Ninth Circuit, and right-leaning groups to districts in the Fifth Circuit.
If the current trend of increasing nationwide injunctions continues, either the rules for the federal courts or congressional action may be required to reduce some of the abuses by both sides of the political spectrum. Reference Am. Coll. of Obstetricians & Gynecologists v. United States FDA, 467 F. Supp. 3d 282, 284 (D. Md. 2020).
Reference
1. Am. Coll. of Obstetricians & Gynecologists v. United States FDA, 467 F. Supp. 3d 282, 284 (D. Md. 2020).
The “Shadow Docket”
The ACOG mifepristone decisions do not appear on the Supreme Court’s “Court Opinions” website.1 They appear in what has become known in recent years as “The Shadow Docket,” an informal term that includes many orders of the Court and statements of individual justices regarding some cases.2 There are hundreds of orders by the Court each Term, there is nothing particularly shadowy about any of these items—they are all publicly available on the Court’s website and later in paper format. It is, however, a little harder to find and much harder to sort through than the major opinions. In some cases, it is not possible to tell what the vote was, how each justice voted, and what the reasoning of the Court was. In a few cases it is difficult to know exactly what the Court was holding or otherwise leaves some confusion about what the law actually is.3
The part of the Shadow Docket that is most intriguing for commentators, and where the ACOG cases appear, is the “Opinions Relating to Orders.”4 These are a variety of opinions, some written by the Court and many by individual justices. It also includes the action of the Court in some cases in which there was not full briefing or oral argument. The statements by justices often are to dissent from the denial of cert of decisions of the Court. These opinions have become much more common over the years. In this past term, there were approximately 60 such opinions related to about 50 cases. In part, this relates to the number of pandemic cases that could not wait for a Court decision going through the extended ordinary process. Although the Shadow Docket has been of interest to academic observers and Court watchers for years, this year it has attracted the attention of Congress.5
References
1. Opinions of the Court. Supreme Court website. https://www.supremecourt.gov/opinions/slipopinion/20#list. Accessed October 10, 2021.
2. Baude W. Foreword: the Supreme Court’s Shadow Docket, 9 N.Y.U. J.L. & Liberty 1 (2015).
3. Vladeck SI. The Solicitor General and the Shadow Docket, 133 Harvard Law Review. 123 (2019).
4. Opinions relating to orders. Supreme Court website. https://www.supremecourt.gov/opinions/relatingtoorders/20#list. Accessed October 10, 2021.
5. The Supreme Court’s Shadow Docket: Hearing Before the Subcommittee on Courts, Intellectual Property and the Internet of the H. Committee on the Judiciary, 117th Congress (2021).
- American College of Obstetricians & Gynecologists v. United States FDA, 472 F. Supp. 3d 183 (D. Md. 2020).
- Michael Kunzelman, Doctors Sue to Block FDA Abortion Pill Rule During Pandemic, (May 29, 2020).
- ACLU, American College Of Obstetricians And Gynecologists V. U.S. Food And Drug Administration, https://www.aclu.org/cases/american-college-obstetricians-and-gynecologists-v-us-food-and-drug-administration. Updated February 12, 2021. Accessed August 27, 2021.
- Whole Woman’s Health v Hellerstedt, 579 US ___ (2016), 136 S Ct 2292.
- 2016 Clinical Review at 39, 47, 49, Opp’n Mot. PI Ex. 19, ECF No. 62-11.
- American College of Obstetricians and Gynecologists v FDA (I), decided October 8, 2020.
- October 8, 2020, dissenting opinion by Justice Alito.
- January 12, 2021, dissenting opinion by Justice Sotomayor.
- Questions and answers on Mifeprex. U.S. Food and Drug Administration website. Published April 13, 2021. https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/questions-and-answers-mifeprex. Accessed October 9, 2021.
- Whole Woman’s Health v Jackson, decided September 1, 2021.
- Texas Senate Bill 8, relating to abortion, including abortions after detection of unborn child’s heartbeat; authorizing a private civil right of action. LegiScan website. https://legiscan.com/TX/text/SB8/id/2395961. Accessed October 9, 2021.
- Planned Parenthood of Southeastern Pennsylvania v Casey, 505 U. S. 833 (1992); Roe v Wade, 410 U. S. 113 (1973).
- Dobbs v Jackson Women’s Health Organization, No. 19-1392.
- Roman Catholic Diocese of Brooklyn v Cuomo, decided November 25, 2020.
- Alabama Association of Realtors v Department of Health and Human Services, decided June 29, 2021.
- Temporary halt in residential evictions in communities with substantial or high levels of community transmission of COVID-19 to prevent the further spread of COVID-19. August 6, 2021. https://www.federalregister.gov/documents/2021/08/06/2021-16945/temporary-halt-in-residential-evictions-in-communities-with-substantial-or-high-transmission-of.
- Alabama Association of Realtors v Department of Health and Human Services, decided August 26, 2021.
- California v Texas, decided June 17, 2021.
- Brief of Amici Curiae American Medical Association, American Academy of Allergy, Asthma and Immunology, Aerospace Medical Association, American Academy of Family Physicians, American Academy of Pediatrics, American College of Cardiology, American College of Emergency Physicians, American College of Medical Genetics and Genomics, American College of Obstetricians and Gynecologists, American College of Physicians, American College of Radiation Oncology, American College of Radiology, American Psychiatric Association, American Society of Gastrointestinal Endoscopy, American Society of Hematology, American Society of Metabolic and Bariatric Surgery, Endocrine Society, GLMA: Health Professionals Advancing LGBTQ Equality, Renal Physicians Association, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology in Support of Petitioners, in California v. Texas. May 13, 2020. https://www.supremecourt.gov/DocketPDF/19/19-840/143469/20200513150051995_19-840%20Amici%20Brief%20AMA.pdf. Accessed October 9, 2021.
- Rutledge v Pharmaceutical Care Management Association, decided December 10, 2020.
- Brief of the American Medical Association, The Arkansas Medical Society, and The Litigation Center of the American Medical Association and the State Medical Societies as Amici Curiae in Support of Petitioner in Rutledge v Pharmaceutical Care Management Association. March 2, 2020. https://www.supremecourt.gov/DocketPDF/18/18-540/134670/20200302163622018_Rutledge%20v.%20PCMA%20Amicus%20Brief%20of%20AMA%20et%20al.pdf. Accessed October 9, 2021.
- Apple quietly settles patent lawsuit, promptly gets hit with another one. TechCrunch website. Published July 30, 2010. https://techcrunch.com/2010/07/30/apple-minerva-emblaze/. Accessed October 9, 2021.
- Minerva Surgical, Inc. v Hologic, Inc., decided June 29, 2021.
- Stat pack. SCOTUS Blog website. Published July 6, 2021. https://www.scotusblog.com/wp-content/uploads/2021/07/Final-Stat-Pack-7.6.21.pdf. Accessed October 9, 2021.
- Dobbs v Jackson Women’s Health Organization, No. 19-1392.
- Cameron v. EMW Women’s Surgical Center, https://www.scotusblog.com/case-files/cases/cameron-v-emw-womens-surgical-center-p-s-c/. Accessed August 28, 2021.
- American Hospital Association v Becerra, No. 20-1114.
- Becerra v Empire Health Foundation, No. 20-1312.
- Gallardo v Marstiller, No. 20-1263.
- New York State Rifle & Pistol Association Inc. v Corlett, No. 20-843.
- Students for Fair Admissions v President & Fellows of Harvard College, No. 20-1199.
- American College of Obstetricians & Gynecologists v. United States FDA, 472 F. Supp. 3d 183 (D. Md. 2020).
- Michael Kunzelman, Doctors Sue to Block FDA Abortion Pill Rule During Pandemic, (May 29, 2020).
- ACLU, American College Of Obstetricians And Gynecologists V. U.S. Food And Drug Administration, https://www.aclu.org/cases/american-college-obstetricians-and-gynecologists-v-us-food-and-drug-administration. Updated February 12, 2021. Accessed August 27, 2021.
- Whole Woman’s Health v Hellerstedt, 579 US ___ (2016), 136 S Ct 2292.
- 2016 Clinical Review at 39, 47, 49, Opp’n Mot. PI Ex. 19, ECF No. 62-11.
- American College of Obstetricians and Gynecologists v FDA (I), decided October 8, 2020.
- October 8, 2020, dissenting opinion by Justice Alito.
- January 12, 2021, dissenting opinion by Justice Sotomayor.
- Questions and answers on Mifeprex. U.S. Food and Drug Administration website. Published April 13, 2021. https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/questions-and-answers-mifeprex. Accessed October 9, 2021.
- Whole Woman’s Health v Jackson, decided September 1, 2021.
- Texas Senate Bill 8, relating to abortion, including abortions after detection of unborn child’s heartbeat; authorizing a private civil right of action. LegiScan website. https://legiscan.com/TX/text/SB8/id/2395961. Accessed October 9, 2021.
- Planned Parenthood of Southeastern Pennsylvania v Casey, 505 U. S. 833 (1992); Roe v Wade, 410 U. S. 113 (1973).
- Dobbs v Jackson Women’s Health Organization, No. 19-1392.
- Roman Catholic Diocese of Brooklyn v Cuomo, decided November 25, 2020.
- Alabama Association of Realtors v Department of Health and Human Services, decided June 29, 2021.
- Temporary halt in residential evictions in communities with substantial or high levels of community transmission of COVID-19 to prevent the further spread of COVID-19. August 6, 2021. https://www.federalregister.gov/documents/2021/08/06/2021-16945/temporary-halt-in-residential-evictions-in-communities-with-substantial-or-high-transmission-of.
- Alabama Association of Realtors v Department of Health and Human Services, decided August 26, 2021.
- California v Texas, decided June 17, 2021.
- Brief of Amici Curiae American Medical Association, American Academy of Allergy, Asthma and Immunology, Aerospace Medical Association, American Academy of Family Physicians, American Academy of Pediatrics, American College of Cardiology, American College of Emergency Physicians, American College of Medical Genetics and Genomics, American College of Obstetricians and Gynecologists, American College of Physicians, American College of Radiation Oncology, American College of Radiology, American Psychiatric Association, American Society of Gastrointestinal Endoscopy, American Society of Hematology, American Society of Metabolic and Bariatric Surgery, Endocrine Society, GLMA: Health Professionals Advancing LGBTQ Equality, Renal Physicians Association, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology in Support of Petitioners, in California v. Texas. May 13, 2020. https://www.supremecourt.gov/DocketPDF/19/19-840/143469/20200513150051995_19-840%20Amici%20Brief%20AMA.pdf. Accessed October 9, 2021.
- Rutledge v Pharmaceutical Care Management Association, decided December 10, 2020.
- Brief of the American Medical Association, The Arkansas Medical Society, and The Litigation Center of the American Medical Association and the State Medical Societies as Amici Curiae in Support of Petitioner in Rutledge v Pharmaceutical Care Management Association. March 2, 2020. https://www.supremecourt.gov/DocketPDF/18/18-540/134670/20200302163622018_Rutledge%20v.%20PCMA%20Amicus%20Brief%20of%20AMA%20et%20al.pdf. Accessed October 9, 2021.
- Apple quietly settles patent lawsuit, promptly gets hit with another one. TechCrunch website. Published July 30, 2010. https://techcrunch.com/2010/07/30/apple-minerva-emblaze/. Accessed October 9, 2021.
- Minerva Surgical, Inc. v Hologic, Inc., decided June 29, 2021.
- Stat pack. SCOTUS Blog website. Published July 6, 2021. https://www.scotusblog.com/wp-content/uploads/2021/07/Final-Stat-Pack-7.6.21.pdf. Accessed October 9, 2021.
- Dobbs v Jackson Women’s Health Organization, No. 19-1392.
- Cameron v. EMW Women’s Surgical Center, https://www.scotusblog.com/case-files/cases/cameron-v-emw-womens-surgical-center-p-s-c/. Accessed August 28, 2021.
- American Hospital Association v Becerra, No. 20-1114.
- Becerra v Empire Health Foundation, No. 20-1312.
- Gallardo v Marstiller, No. 20-1263.
- New York State Rifle & Pistol Association Inc. v Corlett, No. 20-843.
- Students for Fair Admissions v President & Fellows of Harvard College, No. 20-1199.
Advocacy Update: Is Your Practice Equipped to Handle Looming Changes in Dermatopathology?
The proposed 2022 Medicare physician fee schedule and quality payment program (QPP) regulations were released on July 13, 2021.1 Final regulations are expected to be released on or around November 1, 2021, but they may be delayed. Multiple national medical organizations, including the College of American Pathologists (CAP), the American Society of Dermatopathology, the American Academy of Dermatology Association (AADA), and the American Medical Association (AMA) Physicians’ Grassroots Network all work together to engage with the Centers for Medicare & Medicaid Services (CMS) to influence these regulations. Stated advocacy priorities include protecting the value of dermatopathology services, mobilizing dermatopathologists for political action, ensuring dermatopathologists can participate in new payment models, strengthening the profession with advocacy on a state level, and conducting socioeconomic research. Is your practice aware and prepared to handle the changes coming in 2022?
The recent revisions and revaluations of the outpatient evaluation and management (E/M) codes2 resulted in a considerable redistribution of Medicare dollars in 2021, negatively impacting dermatopathologists and other specialties and services due to budget neutrality required by law (Figure). Important steps were taken to mitigate the 2021 Medicare cuts for all non–office-based dermatopathology services (eg, pathology, surgical services, emergency department).1,3 Direct engagement by the CAP, American Society of Dermatopathology, and AADA, along with the AMA Physicians’ Grassroots Network resulted in legislative action on December 27, 2020, which directed Medicare to make a 3.75% positive adjustment to the 2021 physician payments. Additionally, the CMS updated the 2021 physician conversion factor to $34.8931, a 3.3% reduction from the 2020 conversion factor rather than $32.41, or a 10.20% decrease. The 2% payment adjustment (sequestration) through December 21, 2021, also was suspended, and Congress and the Biden administration mandated delayed implementation of the inherent complexity add-on code for E/M services (G2211) until 2024.1,3
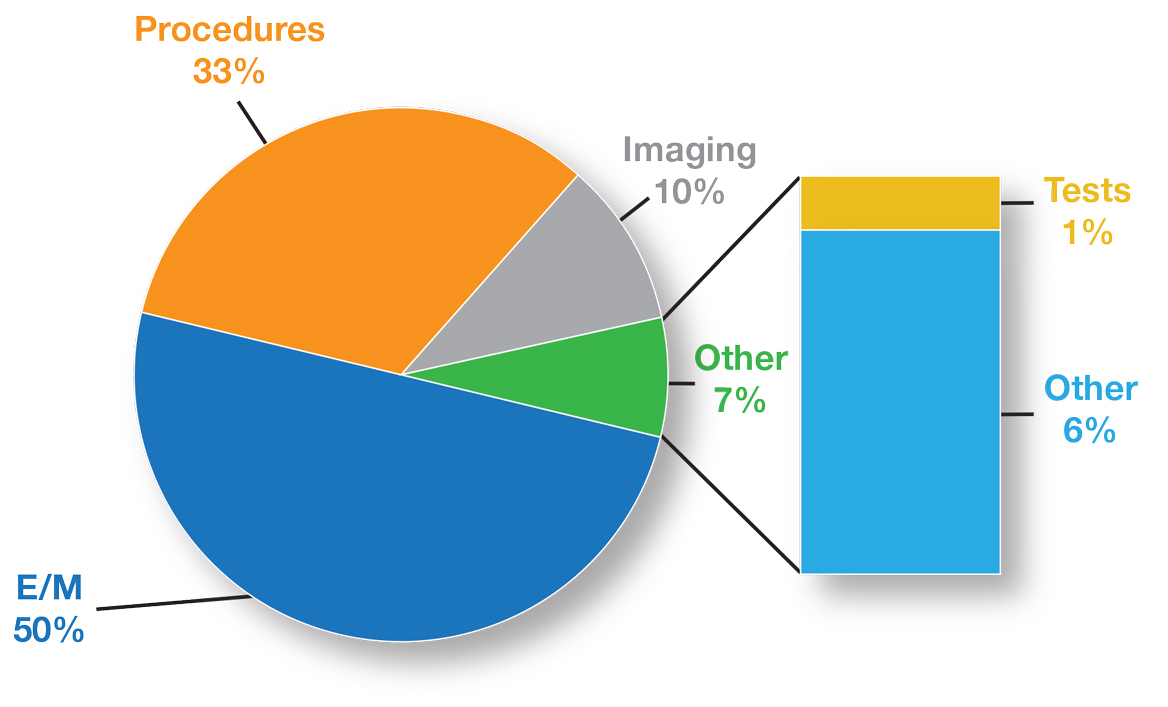
Threat of Medicare Cuts in 2022
Based on dermatopathology utilization data, the overall impact on reimbursement for 2022 represents an approximately 5% decrease from 2021 dermatopathology payments (Table 1).1,4 This represents a 3.75% cut from revaluation of E/M services, and a 1% cut due to changes in practice expense pricing. The estimated change in reimbursement for independent laboratories is a 6% decrease. Advocacy groups have been working to mitigate the 2022 cuts by engaging with Congress and urging them to act before these changes go into effect next year. Keep in mind that approximately half of all pathology Current Procedural Terminology (CPT) codes have been targeted for evaluation by the CMS since 2006.1,4
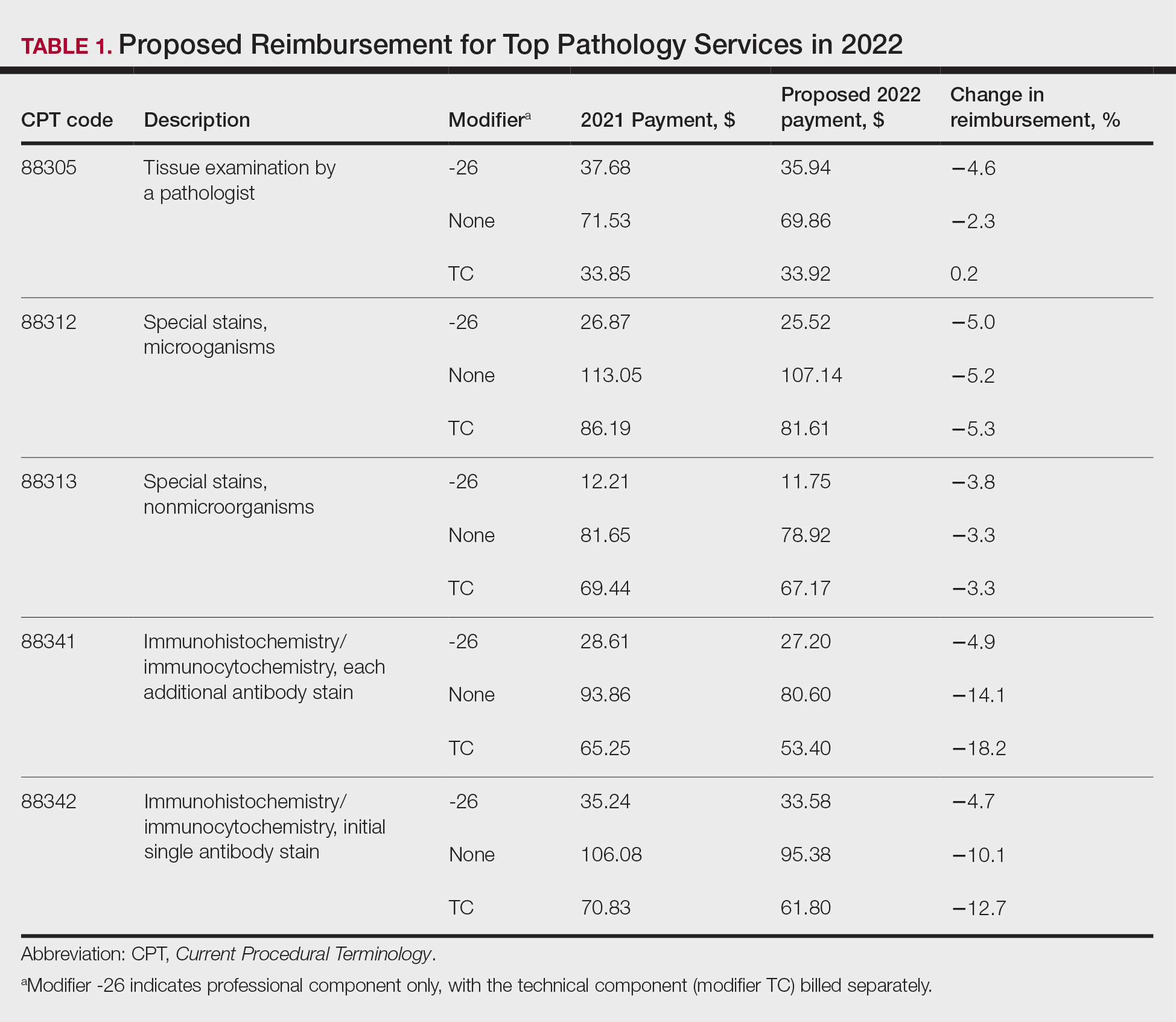
The current clinical pathology consultation services (CPT codes 80500 and 80502) previously were identified as potentially misvalued for review by the AMA Relative Value Scale Update Committee’s (RUC’s) relativity assessment workgroup.4 Consequently, the CAP worked with the AMA’s CPT Editorial Panel to delete codes 80500 and 80502, as well as to modernize and create the 4 new clinical pathology consultation codes: 80XX0, 80XX1, 80XX2, and 80XX3. Then the CAP worked with the RUC to develop physician work and practice expense values for the new clinical pathology consultation codes. Once the fee schedule is finalized, pathologists can begin using the new codes to bill these services in 2022 (Table 2).4

According to CPT, clinical pathology consultation services may be reported when the following criteria have been met: (1) the pathologist renders a clinical pathology consultation at the request of a physician or qualified health care professional at the same or another institution; (2) the pathology clinical consultation request relating to pathology and laboratory findings or other relevant clinical or diagnostic information requiring additional medical interpretative judgment is made; and (3) these codes are not reported in conjunction with codes 88321, 88323, and 88325.4
Proposed 2022 Medicare QPP Requirements
On July 13, 2021, the CMS also published its proposed 2022 QPP proposals that will take effect next year.4 According to the proposed regulation, nearly all dermatopathologists will be required to participate in Medicare’s QPP, either through advanced alternative payment models (APMs) or the Merit-based Incentive Payment System (MIPS). The CAP has long advocated for reducing MIPS reporting burdens for dermatopathologists. In this regulation, the CMS is proposing key program changes that move the program forward but also introduce additional complexities; for example, the CMS will move forward with a new participation pathway called MIPS Value Pathways (MVPs). The CMS proposed 7 specific MVPs that align with certain clinical topics; however, it will not implement these MVPs until the 2023 MIPS performance period.
In 2022, dermatopathologists who are eligible for MIPS will have to take action to avoid penalties that reduce future Medicare Part B payments for their services. Performance in MIPS in 2022 affects Medicare Part B payments in 2024 by an increase of 9% to a decrease of 9%.
In its proposed 2022 QPP regulations, the CMS proposed an increase of the performance threshold from 60 MIPS points to 75 MIPS points. It also proposed an increase of the exceptional Performance Threshold from 85 MIPS points to 89 MIPS points.
The CMS also proposed notable scoring changes for quality measures, including removing the 3-point floor for measures that can be scored against a benchmark. These measures would receive 1 to 10 points. Measures without a benchmark or that do not meet case requirements would earn 0 points, with an exception for small practices. The CMS also proposed removing bonus points for reporting additional outcomes and high-priority measures beyond the 1 that is required, as well as establishing a 5-point floor for the first 2 performance periods for new measures, which is in line with the CAP’s advocacy.
The Pathology Specialty Measure Set will remain the same as the 2021 set containing 6 quality measures, including the AADA-stewarded quality measure #440 (skin cancer: biopsy reporting time—pathologist to clinician). Although the CAP recognizes the importance of prompt turnaround of biopsy reports, it also is working with the CMS and the AADA to mitigate the operational challenges dermatopathologists encounter when using this measure.
Due to advocacy from the CAP, the CMS included a CAP-proposed improvement activity on implementation of a laboratory preparedness plan to support continued or expanded patient care during the COVID-19 pandemic or another public health emergency. This plan should address how the laboratory would maintain or expand access to improve beneficiary health outcomes and reduce health care disparities.
The CAP has actively worked with the CMS to demonstrate the need for more appropriate and alternative measures and improvement activities so that pathologists can more fully participate in MIPS.
Alternative Payment Models—For those dermatopathologists who practice in an APM, the proposed 2022 QPP makes minimal changes to the advanced APM track while adding transition time for accountable care organizations in the Medicare Shared Savings Program to report on certain quality measures and increasing flexibility related to the program’s quality performance standard.
Cures Act 2021: To Do No Harm
The 21st Century Cures Act (Cures Act) was signed into federal law in 2016. The Office of the National Coordinator for Health Information Technology (ONC) laid the groundwork for patients to have easier access to and control of their health information.5 The ONC’s final rule, which went into effect on April 5, 2021, requires that all providers make their office notes, laboratory results, and other diagnostic reports (including dermatopathology reports) available to patients as soon as the physician’s office receives an electronic copy. Penalty for noncompliance has not been determined.
There are information-blocking exceptions, but delaying access to a patient’s report so that a provider can review the result before the patient receives it is not considered an exception.6 The exceptions are situational and must be evaluated by the referring clinician or their employer. Documentation of the exception is critical. The specific facts and circumstances associated with your decision to use an exception will be important to include in your documentation. Information blocking necessary to prevent “harm” to a patient or another person requires a reasonable belief that the practice will substantially reduce the risk of harm.6
The AMA passed a resolution in June 2021 calling for changes to this rule to allow for a delay of pathology results, advocating to the Office for Civil Rights to revise the harm exception to include psychological distress.6 In August 2021, the AADA met with senior officials at the ONC also asking to revise its definition of harm, sharing examples of emotional strain that resulted from receiving results without clinical context.7 California enacted a law requiring a delay before a patient receives the result of a malignant diagnosis, giving the clinician time to contact the patient before they see their report.8
The Cures Act requirements are about patients accessing their health care information. Always consider what is best for the patient and ensure that your policies and procedures reflect this.5
Final Thoughts
It is important to learn and support advocacy priorities and efforts and to join forces to protect your practice. Physician advocacy is no longer an elective pursuit. We need to be involved and engaged through our medical societies to help patients, communities, and ourselves.
- Centers for Medicare & Medicaid Services. Calendar Year (CY) 2022 Medicare Physician Fee Schedule Proposed Rule. Published July 13, 2021. Accessed October 22, 2021. https://www.cms.gov/newsroom/fact-sheets/calendar-year-cy-2022-medicare-physician-fee-schedule-proposed-rule
- Healthcare spending and the Medicare program. Medicare Payment Advisory Commission; July 2020. Accessed October 25, 2021.http://www.medpac.gov/docs/default-source/data-book/july2020_databook_entirereport_sec.pdf
- Frieden J. 2021 Medicare fee schedule includes 10.2% cut in conversion factor. MedPage Today website. Published December 2, 2020. Accessed October 22, 2021. https://www.medpagetoday.com/practicemanagement/reimbursement/89970
- Advocacy. College of American Pathologists website. Accessed October 13, 2021. https://www.cap.org/advocacy
- ONC’s Cures Act Final Rule. The Office of the National Coordinator for Health Information Technology website. Accessed October 13, 2021. https://www.healthit.gov/curesrule/
- Nelson H. Delegates call AMA to advocate for provider info-blocking flexibility. Published June 18, 2021. Accessed October 13, 2021. https://ehrintelligence.com/news/delegates-call-ama-to-advocate-for-provider-info-blocking-flexibility
- Rosamilia LL. Immediate Pathology report release to patients—is the 21st Century Cures Act worse than the disease? American Academy of Dermatology website. Published August 25, 2021. Accessed October 22, 2021. https://www.aad.org/dw/dw-insights-and-inquiries/archive/2021/cures-act-immediate-pathology-report-release-to-patients
- Purington K, Alfreds ST, Pritts J, et al; The National Academy for State Health Policy. Electronic release of clinical laboratory results: a review of state and federal policy. Published January 2010. Accessed October 13, 2021. https://www.nashp.org/wp-content/uploads/2010/02/ElectronicLabResultsExchangePolicy.pdf
The proposed 2022 Medicare physician fee schedule and quality payment program (QPP) regulations were released on July 13, 2021.1 Final regulations are expected to be released on or around November 1, 2021, but they may be delayed. Multiple national medical organizations, including the College of American Pathologists (CAP), the American Society of Dermatopathology, the American Academy of Dermatology Association (AADA), and the American Medical Association (AMA) Physicians’ Grassroots Network all work together to engage with the Centers for Medicare & Medicaid Services (CMS) to influence these regulations. Stated advocacy priorities include protecting the value of dermatopathology services, mobilizing dermatopathologists for political action, ensuring dermatopathologists can participate in new payment models, strengthening the profession with advocacy on a state level, and conducting socioeconomic research. Is your practice aware and prepared to handle the changes coming in 2022?
The recent revisions and revaluations of the outpatient evaluation and management (E/M) codes2 resulted in a considerable redistribution of Medicare dollars in 2021, negatively impacting dermatopathologists and other specialties and services due to budget neutrality required by law (Figure). Important steps were taken to mitigate the 2021 Medicare cuts for all non–office-based dermatopathology services (eg, pathology, surgical services, emergency department).1,3 Direct engagement by the CAP, American Society of Dermatopathology, and AADA, along with the AMA Physicians’ Grassroots Network resulted in legislative action on December 27, 2020, which directed Medicare to make a 3.75% positive adjustment to the 2021 physician payments. Additionally, the CMS updated the 2021 physician conversion factor to $34.8931, a 3.3% reduction from the 2020 conversion factor rather than $32.41, or a 10.20% decrease. The 2% payment adjustment (sequestration) through December 21, 2021, also was suspended, and Congress and the Biden administration mandated delayed implementation of the inherent complexity add-on code for E/M services (G2211) until 2024.1,3
Threat of Medicare Cuts in 2022
Based on dermatopathology utilization data, the overall impact on reimbursement for 2022 represents an approximately 5% decrease from 2021 dermatopathology payments (Table 1).1,4 This represents a 3.75% cut from revaluation of E/M services, and a 1% cut due to changes in practice expense pricing. The estimated change in reimbursement for independent laboratories is a 6% decrease. Advocacy groups have been working to mitigate the 2022 cuts by engaging with Congress and urging them to act before these changes go into effect next year. Keep in mind that approximately half of all pathology Current Procedural Terminology (CPT) codes have been targeted for evaluation by the CMS since 2006.1,4
The current clinical pathology consultation services (CPT codes 80500 and 80502) previously were identified as potentially misvalued for review by the AMA Relative Value Scale Update Committee’s (RUC’s) relativity assessment workgroup.4 Consequently, the CAP worked with the AMA’s CPT Editorial Panel to delete codes 80500 and 80502, as well as to modernize and create the 4 new clinical pathology consultation codes: 80XX0, 80XX1, 80XX2, and 80XX3. Then the CAP worked with the RUC to develop physician work and practice expense values for the new clinical pathology consultation codes. Once the fee schedule is finalized, pathologists can begin using the new codes to bill these services in 2022 (Table 2).4
According to CPT, clinical pathology consultation services may be reported when the following criteria have been met: (1) the pathologist renders a clinical pathology consultation at the request of a physician or qualified health care professional at the same or another institution; (2) the pathology clinical consultation request relating to pathology and laboratory findings or other relevant clinical or diagnostic information requiring additional medical interpretative judgment is made; and (3) these codes are not reported in conjunction with codes 88321, 88323, and 88325.4
Proposed 2022 Medicare QPP Requirements
On July 13, 2021, the CMS also published its proposed 2022 QPP proposals that will take effect next year.4 According to the proposed regulation, nearly all dermatopathologists will be required to participate in Medicare’s QPP, either through advanced alternative payment models (APMs) or the Merit-based Incentive Payment System (MIPS). The CAP has long advocated for reducing MIPS reporting burdens for dermatopathologists. In this regulation, the CMS is proposing key program changes that move the program forward but also introduce additional complexities; for example, the CMS will move forward with a new participation pathway called MIPS Value Pathways (MVPs). The CMS proposed 7 specific MVPs that align with certain clinical topics; however, it will not implement these MVPs until the 2023 MIPS performance period.
In 2022, dermatopathologists who are eligible for MIPS will have to take action to avoid penalties that reduce future Medicare Part B payments for their services. Performance in MIPS in 2022 affects Medicare Part B payments in 2024 by an increase of 9% to a decrease of 9%.
In its proposed 2022 QPP regulations, the CMS proposed an increase of the performance threshold from 60 MIPS points to 75 MIPS points. It also proposed an increase of the exceptional Performance Threshold from 85 MIPS points to 89 MIPS points.
The CMS also proposed notable scoring changes for quality measures, including removing the 3-point floor for measures that can be scored against a benchmark. These measures would receive 1 to 10 points. Measures without a benchmark or that do not meet case requirements would earn 0 points, with an exception for small practices. The CMS also proposed removing bonus points for reporting additional outcomes and high-priority measures beyond the 1 that is required, as well as establishing a 5-point floor for the first 2 performance periods for new measures, which is in line with the CAP’s advocacy.
The Pathology Specialty Measure Set will remain the same as the 2021 set containing 6 quality measures, including the AADA-stewarded quality measure #440 (skin cancer: biopsy reporting time—pathologist to clinician). Although the CAP recognizes the importance of prompt turnaround of biopsy reports, it also is working with the CMS and the AADA to mitigate the operational challenges dermatopathologists encounter when using this measure.
Due to advocacy from the CAP, the CMS included a CAP-proposed improvement activity on implementation of a laboratory preparedness plan to support continued or expanded patient care during the COVID-19 pandemic or another public health emergency. This plan should address how the laboratory would maintain or expand access to improve beneficiary health outcomes and reduce health care disparities.
The CAP has actively worked with the CMS to demonstrate the need for more appropriate and alternative measures and improvement activities so that pathologists can more fully participate in MIPS.
Alternative Payment Models—For those dermatopathologists who practice in an APM, the proposed 2022 QPP makes minimal changes to the advanced APM track while adding transition time for accountable care organizations in the Medicare Shared Savings Program to report on certain quality measures and increasing flexibility related to the program’s quality performance standard.
Cures Act 2021: To Do No Harm
The 21st Century Cures Act (Cures Act) was signed into federal law in 2016. The Office of the National Coordinator for Health Information Technology (ONC) laid the groundwork for patients to have easier access to and control of their health information.5 The ONC’s final rule, which went into effect on April 5, 2021, requires that all providers make their office notes, laboratory results, and other diagnostic reports (including dermatopathology reports) available to patients as soon as the physician’s office receives an electronic copy. Penalty for noncompliance has not been determined.
There are information-blocking exceptions, but delaying access to a patient’s report so that a provider can review the result before the patient receives it is not considered an exception.6 The exceptions are situational and must be evaluated by the referring clinician or their employer. Documentation of the exception is critical. The specific facts and circumstances associated with your decision to use an exception will be important to include in your documentation. Information blocking necessary to prevent “harm” to a patient or another person requires a reasonable belief that the practice will substantially reduce the risk of harm.6
The AMA passed a resolution in June 2021 calling for changes to this rule to allow for a delay of pathology results, advocating to the Office for Civil Rights to revise the harm exception to include psychological distress.6 In August 2021, the AADA met with senior officials at the ONC also asking to revise its definition of harm, sharing examples of emotional strain that resulted from receiving results without clinical context.7 California enacted a law requiring a delay before a patient receives the result of a malignant diagnosis, giving the clinician time to contact the patient before they see their report.8
The Cures Act requirements are about patients accessing their health care information. Always consider what is best for the patient and ensure that your policies and procedures reflect this.5
Final Thoughts
It is important to learn and support advocacy priorities and efforts and to join forces to protect your practice. Physician advocacy is no longer an elective pursuit. We need to be involved and engaged through our medical societies to help patients, communities, and ourselves.
The proposed 2022 Medicare physician fee schedule and quality payment program (QPP) regulations were released on July 13, 2021.1 Final regulations are expected to be released on or around November 1, 2021, but they may be delayed. Multiple national medical organizations, including the College of American Pathologists (CAP), the American Society of Dermatopathology, the American Academy of Dermatology Association (AADA), and the American Medical Association (AMA) Physicians’ Grassroots Network all work together to engage with the Centers for Medicare & Medicaid Services (CMS) to influence these regulations. Stated advocacy priorities include protecting the value of dermatopathology services, mobilizing dermatopathologists for political action, ensuring dermatopathologists can participate in new payment models, strengthening the profession with advocacy on a state level, and conducting socioeconomic research. Is your practice aware and prepared to handle the changes coming in 2022?
The recent revisions and revaluations of the outpatient evaluation and management (E/M) codes2 resulted in a considerable redistribution of Medicare dollars in 2021, negatively impacting dermatopathologists and other specialties and services due to budget neutrality required by law (Figure). Important steps were taken to mitigate the 2021 Medicare cuts for all non–office-based dermatopathology services (eg, pathology, surgical services, emergency department).1,3 Direct engagement by the CAP, American Society of Dermatopathology, and AADA, along with the AMA Physicians’ Grassroots Network resulted in legislative action on December 27, 2020, which directed Medicare to make a 3.75% positive adjustment to the 2021 physician payments. Additionally, the CMS updated the 2021 physician conversion factor to $34.8931, a 3.3% reduction from the 2020 conversion factor rather than $32.41, or a 10.20% decrease. The 2% payment adjustment (sequestration) through December 21, 2021, also was suspended, and Congress and the Biden administration mandated delayed implementation of the inherent complexity add-on code for E/M services (G2211) until 2024.1,3
Threat of Medicare Cuts in 2022
Based on dermatopathology utilization data, the overall impact on reimbursement for 2022 represents an approximately 5% decrease from 2021 dermatopathology payments (Table 1).1,4 This represents a 3.75% cut from revaluation of E/M services, and a 1% cut due to changes in practice expense pricing. The estimated change in reimbursement for independent laboratories is a 6% decrease. Advocacy groups have been working to mitigate the 2022 cuts by engaging with Congress and urging them to act before these changes go into effect next year. Keep in mind that approximately half of all pathology Current Procedural Terminology (CPT) codes have been targeted for evaluation by the CMS since 2006.1,4
The current clinical pathology consultation services (CPT codes 80500 and 80502) previously were identified as potentially misvalued for review by the AMA Relative Value Scale Update Committee’s (RUC’s) relativity assessment workgroup.4 Consequently, the CAP worked with the AMA’s CPT Editorial Panel to delete codes 80500 and 80502, as well as to modernize and create the 4 new clinical pathology consultation codes: 80XX0, 80XX1, 80XX2, and 80XX3. Then the CAP worked with the RUC to develop physician work and practice expense values for the new clinical pathology consultation codes. Once the fee schedule is finalized, pathologists can begin using the new codes to bill these services in 2022 (Table 2).4
According to CPT, clinical pathology consultation services may be reported when the following criteria have been met: (1) the pathologist renders a clinical pathology consultation at the request of a physician or qualified health care professional at the same or another institution; (2) the pathology clinical consultation request relating to pathology and laboratory findings or other relevant clinical or diagnostic information requiring additional medical interpretative judgment is made; and (3) these codes are not reported in conjunction with codes 88321, 88323, and 88325.4
Proposed 2022 Medicare QPP Requirements
On July 13, 2021, the CMS also published its proposed 2022 QPP proposals that will take effect next year.4 According to the proposed regulation, nearly all dermatopathologists will be required to participate in Medicare’s QPP, either through advanced alternative payment models (APMs) or the Merit-based Incentive Payment System (MIPS). The CAP has long advocated for reducing MIPS reporting burdens for dermatopathologists. In this regulation, the CMS is proposing key program changes that move the program forward but also introduce additional complexities; for example, the CMS will move forward with a new participation pathway called MIPS Value Pathways (MVPs). The CMS proposed 7 specific MVPs that align with certain clinical topics; however, it will not implement these MVPs until the 2023 MIPS performance period.
In 2022, dermatopathologists who are eligible for MIPS will have to take action to avoid penalties that reduce future Medicare Part B payments for their services. Performance in MIPS in 2022 affects Medicare Part B payments in 2024 by an increase of 9% to a decrease of 9%.
In its proposed 2022 QPP regulations, the CMS proposed an increase of the performance threshold from 60 MIPS points to 75 MIPS points. It also proposed an increase of the exceptional Performance Threshold from 85 MIPS points to 89 MIPS points.
The CMS also proposed notable scoring changes for quality measures, including removing the 3-point floor for measures that can be scored against a benchmark. These measures would receive 1 to 10 points. Measures without a benchmark or that do not meet case requirements would earn 0 points, with an exception for small practices. The CMS also proposed removing bonus points for reporting additional outcomes and high-priority measures beyond the 1 that is required, as well as establishing a 5-point floor for the first 2 performance periods for new measures, which is in line with the CAP’s advocacy.
The Pathology Specialty Measure Set will remain the same as the 2021 set containing 6 quality measures, including the AADA-stewarded quality measure #440 (skin cancer: biopsy reporting time—pathologist to clinician). Although the CAP recognizes the importance of prompt turnaround of biopsy reports, it also is working with the CMS and the AADA to mitigate the operational challenges dermatopathologists encounter when using this measure.
Due to advocacy from the CAP, the CMS included a CAP-proposed improvement activity on implementation of a laboratory preparedness plan to support continued or expanded patient care during the COVID-19 pandemic or another public health emergency. This plan should address how the laboratory would maintain or expand access to improve beneficiary health outcomes and reduce health care disparities.
The CAP has actively worked with the CMS to demonstrate the need for more appropriate and alternative measures and improvement activities so that pathologists can more fully participate in MIPS.
Alternative Payment Models—For those dermatopathologists who practice in an APM, the proposed 2022 QPP makes minimal changes to the advanced APM track while adding transition time for accountable care organizations in the Medicare Shared Savings Program to report on certain quality measures and increasing flexibility related to the program’s quality performance standard.
Cures Act 2021: To Do No Harm
The 21st Century Cures Act (Cures Act) was signed into federal law in 2016. The Office of the National Coordinator for Health Information Technology (ONC) laid the groundwork for patients to have easier access to and control of their health information.5 The ONC’s final rule, which went into effect on April 5, 2021, requires that all providers make their office notes, laboratory results, and other diagnostic reports (including dermatopathology reports) available to patients as soon as the physician’s office receives an electronic copy. Penalty for noncompliance has not been determined.
There are information-blocking exceptions, but delaying access to a patient’s report so that a provider can review the result before the patient receives it is not considered an exception.6 The exceptions are situational and must be evaluated by the referring clinician or their employer. Documentation of the exception is critical. The specific facts and circumstances associated with your decision to use an exception will be important to include in your documentation. Information blocking necessary to prevent “harm” to a patient or another person requires a reasonable belief that the practice will substantially reduce the risk of harm.6
The AMA passed a resolution in June 2021 calling for changes to this rule to allow for a delay of pathology results, advocating to the Office for Civil Rights to revise the harm exception to include psychological distress.6 In August 2021, the AADA met with senior officials at the ONC also asking to revise its definition of harm, sharing examples of emotional strain that resulted from receiving results without clinical context.7 California enacted a law requiring a delay before a patient receives the result of a malignant diagnosis, giving the clinician time to contact the patient before they see their report.8
The Cures Act requirements are about patients accessing their health care information. Always consider what is best for the patient and ensure that your policies and procedures reflect this.5
Final Thoughts
It is important to learn and support advocacy priorities and efforts and to join forces to protect your practice. Physician advocacy is no longer an elective pursuit. We need to be involved and engaged through our medical societies to help patients, communities, and ourselves.
- Centers for Medicare & Medicaid Services. Calendar Year (CY) 2022 Medicare Physician Fee Schedule Proposed Rule. Published July 13, 2021. Accessed October 22, 2021. https://www.cms.gov/newsroom/fact-sheets/calendar-year-cy-2022-medicare-physician-fee-schedule-proposed-rule
- Healthcare spending and the Medicare program. Medicare Payment Advisory Commission; July 2020. Accessed October 25, 2021.http://www.medpac.gov/docs/default-source/data-book/july2020_databook_entirereport_sec.pdf
- Frieden J. 2021 Medicare fee schedule includes 10.2% cut in conversion factor. MedPage Today website. Published December 2, 2020. Accessed October 22, 2021. https://www.medpagetoday.com/practicemanagement/reimbursement/89970
- Advocacy. College of American Pathologists website. Accessed October 13, 2021. https://www.cap.org/advocacy
- ONC’s Cures Act Final Rule. The Office of the National Coordinator for Health Information Technology website. Accessed October 13, 2021. https://www.healthit.gov/curesrule/
- Nelson H. Delegates call AMA to advocate for provider info-blocking flexibility. Published June 18, 2021. Accessed October 13, 2021. https://ehrintelligence.com/news/delegates-call-ama-to-advocate-for-provider-info-blocking-flexibility
- Rosamilia LL. Immediate Pathology report release to patients—is the 21st Century Cures Act worse than the disease? American Academy of Dermatology website. Published August 25, 2021. Accessed October 22, 2021. https://www.aad.org/dw/dw-insights-and-inquiries/archive/2021/cures-act-immediate-pathology-report-release-to-patients
- Purington K, Alfreds ST, Pritts J, et al; The National Academy for State Health Policy. Electronic release of clinical laboratory results: a review of state and federal policy. Published January 2010. Accessed October 13, 2021. https://www.nashp.org/wp-content/uploads/2010/02/ElectronicLabResultsExchangePolicy.pdf
- Centers for Medicare & Medicaid Services. Calendar Year (CY) 2022 Medicare Physician Fee Schedule Proposed Rule. Published July 13, 2021. Accessed October 22, 2021. https://www.cms.gov/newsroom/fact-sheets/calendar-year-cy-2022-medicare-physician-fee-schedule-proposed-rule
- Healthcare spending and the Medicare program. Medicare Payment Advisory Commission; July 2020. Accessed October 25, 2021.http://www.medpac.gov/docs/default-source/data-book/july2020_databook_entirereport_sec.pdf
- Frieden J. 2021 Medicare fee schedule includes 10.2% cut in conversion factor. MedPage Today website. Published December 2, 2020. Accessed October 22, 2021. https://www.medpagetoday.com/practicemanagement/reimbursement/89970
- Advocacy. College of American Pathologists website. Accessed October 13, 2021. https://www.cap.org/advocacy
- ONC’s Cures Act Final Rule. The Office of the National Coordinator for Health Information Technology website. Accessed October 13, 2021. https://www.healthit.gov/curesrule/
- Nelson H. Delegates call AMA to advocate for provider info-blocking flexibility. Published June 18, 2021. Accessed October 13, 2021. https://ehrintelligence.com/news/delegates-call-ama-to-advocate-for-provider-info-blocking-flexibility
- Rosamilia LL. Immediate Pathology report release to patients—is the 21st Century Cures Act worse than the disease? American Academy of Dermatology website. Published August 25, 2021. Accessed October 22, 2021. https://www.aad.org/dw/dw-insights-and-inquiries/archive/2021/cures-act-immediate-pathology-report-release-to-patients
- Purington K, Alfreds ST, Pritts J, et al; The National Academy for State Health Policy. Electronic release of clinical laboratory results: a review of state and federal policy. Published January 2010. Accessed October 13, 2021. https://www.nashp.org/wp-content/uploads/2010/02/ElectronicLabResultsExchangePolicy.pdf
Practice Points
- A proposed 2022 fee schedule negatively impacting dermatopathology practices has been published by the Centers for Medicare & Medicaid Services (CMS) in July 2021.
- New pathology consultation codes with new payment rates proposed by CMS can be used starting January 1, 2022.
- The 21st Century Cures Act Final Rule has information blocking provisions.
FDA posts new websites on accelerated approvals for cancer drugs
, including a public list detailing cases where accelerated approvals have been rescinded for lack of evidence.
On Oct. 29, the Food and Drug Administration posted new websites detailing the status of oncology medicines given these special clearances:
- Ongoing | Cancer Accelerated Approvals
- Verified Clinical Benefit | Cancer Accelerated Approvals
- Withdrawn | Cancer Accelerated Approvals
The FDA’s cancer center also has created a web page called Project Confirm to provide more information on the way it uses accelerated approvals.
There has been increased concern about medicines cleared by accelerated approvals in recent years, culminating in an uproar over the controversial June approval of aducanumab (Aduhelm) for Alzheimer’s disease. This drew more attention to a debate already underway about how much data supports some of the indications for some cancer drugs.
Federal and state officials and advisers are putting more pressure on pharmaceutical companies to prove that medicines that are put on the market through accelerated approval do deliver meaningful benefits for patients.
In addition, earlier this month two of the top health advisers in Barack Obama’s administration proposed a new model through which Medicare could reduce payments for certain cancer drugs cleared through accelerated approvals – and even cut off reimbursements in cases where companies fail to deliver confirmatory evidence for expected benefits.
This “Pay for Drugs That Work Model” was proposed by Richard Frank, PhD, and Ezekiel Emanuel, MD, PhD, in a recent JAMA article. In their view, the FDA’s accelerated drug approval process allows for too many delays in obtaining answers as to whether medicines cleared this way provide expected benefits.
“The proposed Pay for Drugs That Work model could test a modified approach for incentivizing rapid completion of confirmatory trials to inform clinicians and patients about the true risks and benefits of new drugs and improve the value for money of cancer drugs that receive accelerated approval,” they wrote.
Excel files, regular updates
For the FDA, accelerated approvals require balancing an estimated potential benefit for people facing serious diseases (for example, cancer) against serious risks, including potentially exposing patients to costly, toxic drugs that will later be shown not to work for their conditions.
For many years, there has been significant pressure on the FDA to lean toward speedier approvals, with members of Congress, advocacy groups, and drugmakers advocating for broad use of surrogate data in deciding on clearances. The FDA posts biannual reports on its website that highlight how quickly approvals have been granted. But these biannual reports don’t provide much information on the status of accelerated-approval drugs, other than to say if they have been given full approval or withdrawn.
The newly created websites from the FDA’s oncology division appear to reflect growing public interest in knowing what standards the agency sets for confirmatory trials and what deadlines companies face to deliver evidence of significant benefit for their drugs.
The new sortable websites also include details on trials and have links to Excel files which will help researchers and others seeking to track patterns with accelerated approvals. The FDA said in an interview that it intends to update these sites when there are developments with accelerated approvals for cancer drugs, such as new clearances of this type, conversions to regular approvals, and withdrawn approvals.
Julia Beaver, MD, chief of medical oncology at the FDA’s Oncology Center of Excellence, and acting deputy director of the Office of Oncologic Diseases of the FDA’s Center for Drug Evaluation and Research, described the new websites as part of a “commitment to preserve the integrity” of the accelerated approval program.
“These new web pages will make information on our accelerated approvals more transparent,” Dr. Beaver said in an email to this news organization.
The FDA has been able to speed many medicines to market and clear additional uses for drugs already sold through the program, giving people earlier access in many cases to critical medicines, Dr. Beaver said.
More than 165 oncology indications have received accelerated approval, with almost half converted to regular approval in a median of 3 years. Less than 10% of these indications were withdrawn, Dr. Beaver said.
“Of those accelerated approvals that were converted to regular approval, many demonstrated survival advantages to patients with several types of cancer or provided meaningful therapeutic options where none previously existed,” she said.
However, Dr. Beaver also has made public the FDA’s concerns with what she and Richard Pazdur, MD, director of the Oncology Center of Excellence, have described as “dangling” accelerated approvals.
These are cases where the required trials did not end up confirming benefit for a medicine, yet the manufacturer did not move to withdraw an accelerated approval. The FDA’s cancer center has already announced that it is doing an “industry-wide evaluation of accelerated approvals in oncology in which confirmatory trials did not confirm clinical benefit.”
This stems in part from what can be called the FDA’s “growing pains” in its efforts to manage the rapidly changing landscape for these immunotherapy checkpoint inhibitors. This field of medicine has experienced an “unprecedented level of drug development” in recent years, FDA officials said in briefing materials for an Oncologic Drugs Advisory Committee (ODAC) meeting last April on dangling accelerated approvals.
A newly posted chart on withdrawn oncology accelerated approvals, posted by the FDA’s cancer division, makes it clear that the pace of these rescinded clearances has picked up. The chart lists a total 14 withdrawn indications of oncology accelerated approvals.
Six of these withdrawals happened this year.
There were two withdrawals in 2020, including the December withdrawal of nivolumab, (Opdivo) for a form of metastatic lung cancer.
Then there was a significant gap, with no withdrawals going back to 2013 (when there was one). There were two withdrawals in 2012 and three in 2011.
A version of this article first appeared on Medscape.com.
, including a public list detailing cases where accelerated approvals have been rescinded for lack of evidence.
On Oct. 29, the Food and Drug Administration posted new websites detailing the status of oncology medicines given these special clearances:
- Ongoing | Cancer Accelerated Approvals
- Verified Clinical Benefit | Cancer Accelerated Approvals
- Withdrawn | Cancer Accelerated Approvals
The FDA’s cancer center also has created a web page called Project Confirm to provide more information on the way it uses accelerated approvals.
There has been increased concern about medicines cleared by accelerated approvals in recent years, culminating in an uproar over the controversial June approval of aducanumab (Aduhelm) for Alzheimer’s disease. This drew more attention to a debate already underway about how much data supports some of the indications for some cancer drugs.
Federal and state officials and advisers are putting more pressure on pharmaceutical companies to prove that medicines that are put on the market through accelerated approval do deliver meaningful benefits for patients.
In addition, earlier this month two of the top health advisers in Barack Obama’s administration proposed a new model through which Medicare could reduce payments for certain cancer drugs cleared through accelerated approvals – and even cut off reimbursements in cases where companies fail to deliver confirmatory evidence for expected benefits.
This “Pay for Drugs That Work Model” was proposed by Richard Frank, PhD, and Ezekiel Emanuel, MD, PhD, in a recent JAMA article. In their view, the FDA’s accelerated drug approval process allows for too many delays in obtaining answers as to whether medicines cleared this way provide expected benefits.
“The proposed Pay for Drugs That Work model could test a modified approach for incentivizing rapid completion of confirmatory trials to inform clinicians and patients about the true risks and benefits of new drugs and improve the value for money of cancer drugs that receive accelerated approval,” they wrote.
Excel files, regular updates
For the FDA, accelerated approvals require balancing an estimated potential benefit for people facing serious diseases (for example, cancer) against serious risks, including potentially exposing patients to costly, toxic drugs that will later be shown not to work for their conditions.
For many years, there has been significant pressure on the FDA to lean toward speedier approvals, with members of Congress, advocacy groups, and drugmakers advocating for broad use of surrogate data in deciding on clearances. The FDA posts biannual reports on its website that highlight how quickly approvals have been granted. But these biannual reports don’t provide much information on the status of accelerated-approval drugs, other than to say if they have been given full approval or withdrawn.
The newly created websites from the FDA’s oncology division appear to reflect growing public interest in knowing what standards the agency sets for confirmatory trials and what deadlines companies face to deliver evidence of significant benefit for their drugs.
The new sortable websites also include details on trials and have links to Excel files which will help researchers and others seeking to track patterns with accelerated approvals. The FDA said in an interview that it intends to update these sites when there are developments with accelerated approvals for cancer drugs, such as new clearances of this type, conversions to regular approvals, and withdrawn approvals.
Julia Beaver, MD, chief of medical oncology at the FDA’s Oncology Center of Excellence, and acting deputy director of the Office of Oncologic Diseases of the FDA’s Center for Drug Evaluation and Research, described the new websites as part of a “commitment to preserve the integrity” of the accelerated approval program.
“These new web pages will make information on our accelerated approvals more transparent,” Dr. Beaver said in an email to this news organization.
The FDA has been able to speed many medicines to market and clear additional uses for drugs already sold through the program, giving people earlier access in many cases to critical medicines, Dr. Beaver said.
More than 165 oncology indications have received accelerated approval, with almost half converted to regular approval in a median of 3 years. Less than 10% of these indications were withdrawn, Dr. Beaver said.
“Of those accelerated approvals that were converted to regular approval, many demonstrated survival advantages to patients with several types of cancer or provided meaningful therapeutic options where none previously existed,” she said.
However, Dr. Beaver also has made public the FDA’s concerns with what she and Richard Pazdur, MD, director of the Oncology Center of Excellence, have described as “dangling” accelerated approvals.
These are cases where the required trials did not end up confirming benefit for a medicine, yet the manufacturer did not move to withdraw an accelerated approval. The FDA’s cancer center has already announced that it is doing an “industry-wide evaluation of accelerated approvals in oncology in which confirmatory trials did not confirm clinical benefit.”
This stems in part from what can be called the FDA’s “growing pains” in its efforts to manage the rapidly changing landscape for these immunotherapy checkpoint inhibitors. This field of medicine has experienced an “unprecedented level of drug development” in recent years, FDA officials said in briefing materials for an Oncologic Drugs Advisory Committee (ODAC) meeting last April on dangling accelerated approvals.
A newly posted chart on withdrawn oncology accelerated approvals, posted by the FDA’s cancer division, makes it clear that the pace of these rescinded clearances has picked up. The chart lists a total 14 withdrawn indications of oncology accelerated approvals.
Six of these withdrawals happened this year.
There were two withdrawals in 2020, including the December withdrawal of nivolumab, (Opdivo) for a form of metastatic lung cancer.
Then there was a significant gap, with no withdrawals going back to 2013 (when there was one). There were two withdrawals in 2012 and three in 2011.
A version of this article first appeared on Medscape.com.
, including a public list detailing cases where accelerated approvals have been rescinded for lack of evidence.
On Oct. 29, the Food and Drug Administration posted new websites detailing the status of oncology medicines given these special clearances:
- Ongoing | Cancer Accelerated Approvals
- Verified Clinical Benefit | Cancer Accelerated Approvals
- Withdrawn | Cancer Accelerated Approvals
The FDA’s cancer center also has created a web page called Project Confirm to provide more information on the way it uses accelerated approvals.
There has been increased concern about medicines cleared by accelerated approvals in recent years, culminating in an uproar over the controversial June approval of aducanumab (Aduhelm) for Alzheimer’s disease. This drew more attention to a debate already underway about how much data supports some of the indications for some cancer drugs.
Federal and state officials and advisers are putting more pressure on pharmaceutical companies to prove that medicines that are put on the market through accelerated approval do deliver meaningful benefits for patients.
In addition, earlier this month two of the top health advisers in Barack Obama’s administration proposed a new model through which Medicare could reduce payments for certain cancer drugs cleared through accelerated approvals – and even cut off reimbursements in cases where companies fail to deliver confirmatory evidence for expected benefits.
This “Pay for Drugs That Work Model” was proposed by Richard Frank, PhD, and Ezekiel Emanuel, MD, PhD, in a recent JAMA article. In their view, the FDA’s accelerated drug approval process allows for too many delays in obtaining answers as to whether medicines cleared this way provide expected benefits.
“The proposed Pay for Drugs That Work model could test a modified approach for incentivizing rapid completion of confirmatory trials to inform clinicians and patients about the true risks and benefits of new drugs and improve the value for money of cancer drugs that receive accelerated approval,” they wrote.
Excel files, regular updates
For the FDA, accelerated approvals require balancing an estimated potential benefit for people facing serious diseases (for example, cancer) against serious risks, including potentially exposing patients to costly, toxic drugs that will later be shown not to work for their conditions.
For many years, there has been significant pressure on the FDA to lean toward speedier approvals, with members of Congress, advocacy groups, and drugmakers advocating for broad use of surrogate data in deciding on clearances. The FDA posts biannual reports on its website that highlight how quickly approvals have been granted. But these biannual reports don’t provide much information on the status of accelerated-approval drugs, other than to say if they have been given full approval or withdrawn.
The newly created websites from the FDA’s oncology division appear to reflect growing public interest in knowing what standards the agency sets for confirmatory trials and what deadlines companies face to deliver evidence of significant benefit for their drugs.
The new sortable websites also include details on trials and have links to Excel files which will help researchers and others seeking to track patterns with accelerated approvals. The FDA said in an interview that it intends to update these sites when there are developments with accelerated approvals for cancer drugs, such as new clearances of this type, conversions to regular approvals, and withdrawn approvals.
Julia Beaver, MD, chief of medical oncology at the FDA’s Oncology Center of Excellence, and acting deputy director of the Office of Oncologic Diseases of the FDA’s Center for Drug Evaluation and Research, described the new websites as part of a “commitment to preserve the integrity” of the accelerated approval program.
“These new web pages will make information on our accelerated approvals more transparent,” Dr. Beaver said in an email to this news organization.
The FDA has been able to speed many medicines to market and clear additional uses for drugs already sold through the program, giving people earlier access in many cases to critical medicines, Dr. Beaver said.
More than 165 oncology indications have received accelerated approval, with almost half converted to regular approval in a median of 3 years. Less than 10% of these indications were withdrawn, Dr. Beaver said.
“Of those accelerated approvals that were converted to regular approval, many demonstrated survival advantages to patients with several types of cancer or provided meaningful therapeutic options where none previously existed,” she said.
However, Dr. Beaver also has made public the FDA’s concerns with what she and Richard Pazdur, MD, director of the Oncology Center of Excellence, have described as “dangling” accelerated approvals.
These are cases where the required trials did not end up confirming benefit for a medicine, yet the manufacturer did not move to withdraw an accelerated approval. The FDA’s cancer center has already announced that it is doing an “industry-wide evaluation of accelerated approvals in oncology in which confirmatory trials did not confirm clinical benefit.”
This stems in part from what can be called the FDA’s “growing pains” in its efforts to manage the rapidly changing landscape for these immunotherapy checkpoint inhibitors. This field of medicine has experienced an “unprecedented level of drug development” in recent years, FDA officials said in briefing materials for an Oncologic Drugs Advisory Committee (ODAC) meeting last April on dangling accelerated approvals.
A newly posted chart on withdrawn oncology accelerated approvals, posted by the FDA’s cancer division, makes it clear that the pace of these rescinded clearances has picked up. The chart lists a total 14 withdrawn indications of oncology accelerated approvals.
Six of these withdrawals happened this year.
There were two withdrawals in 2020, including the December withdrawal of nivolumab, (Opdivo) for a form of metastatic lung cancer.
Then there was a significant gap, with no withdrawals going back to 2013 (when there was one). There were two withdrawals in 2012 and three in 2011.
A version of this article first appeared on Medscape.com.
Can we return to the ABCs of crafting a medical record note?
Prior to 1980, medical record notes were generally hand-written, short, and to the point. Senior physicians often wrote their 3-line notes using a fountain pen in an elegant cursive. With the transition to electronic medical records, notes have become bloated with irrelevant information and frequently lack a focus on the critical clinical insights that optimize patient care. The use of smart phrases to pull vast amounts of raw data into the note is a major contributor to note bloat. The unrestrained use of the copy and paste functionality generates a sequence of cloned notes that grow in length as new information is added and little information from prior notes removed. With each subsequent clone the note often becomes less accurate, lengthier, and more difficult for a reader to understand. In one survey of 253 physicians who wrote electronic notes, 90% reported that they used the copy and paste function, with 71% reporting that use of this function caused inconsistencies within and among notes and increased the repetitive presentation of outdated information in the note.1 Although the surveyed clinicians recognized that the copy and paste function caused problems, 80% reported that they planned to continue to use the copy and paste function.1
The SOAP note
The problem-oriented SOAP note is written in the classic structure of subjective and objective information, followed by an assessment and plan.2 The structure of the SOAP note emphasizes the logical and sequential collection of data followed by data analysis, resulting in a focused assessment and plan. When notes were hand-written and short, the entire SOAP note could be viewed on one page. Like a dashboard, the eye could quickly scan each key component of the note, facilitating the simultaneous integration of all 4 components of the note, facilitating understanding of the patient’s clinical situation. When the SOAP note structure is used to create a multipage electronic note, the result is a note that often confuses rather than enlightens the reader. A 5- to 10-page SOAP note is often useless for patient care but demonstrates the ability of computer-savvy clinicians to quickly generate a note thousands of words in length.
The APSO note, a response to note bloat
When a medical record note becomes a multipage document, clinicians should consider switching from the SOAP note structure to the APSO note, where the assessment and plan are at the top of the note, and the subjective and objective information is below the assessment and plan. The APSO format permits the reader to more quickly grasp the critical thinking of the author and facilitates a focus on key points relevant to the patient’s condition. The note can be written in the SOAP format, but then the assessment and plan are brought to the top of the note. In my clinical experience fewer than 10% of clinicians are using an APSO note structure. I believe that, with a multipage note, the APSO structure improves the experience of the reader and should be more widely utilized, especially by clinicians who are prone to crafting a bloated note. In a survey of more than 3,000 clinicians, approximately two-thirds of the respondents reported that, compared with SOAP notes, APSO notes were easier and faster to read, and APSO notes made it easier to follow the clinical reasoning of the author.3
Continue to: New evaluation and management billing guidelines—An opportunity to reduce note bloat...
New evaluation and management billing guidelines—An opportunity to reduce note bloat
Previous evaluation and management federal billing guidelines emphasized documentation of a myriad of clinically irrelevant details contributing to note bloat. The new federal evaluation and management billing guidelines pivot the focus of the note to the quality and complexity of medical decision making as demonstrated in the assessment and plan.4 Prioritizing the assessment and plan as the key feature of the medical record note should help reduce the length of notes. The American College of Physicians recently recommended deleting the complete review of systems and prior histories from most notes unless relevant to medical decision making and the assessment and plan.5
The open note
The open note mandate was contained in federal regulations developed to implement the 21st Century Cures Act, which required patients to have access to the information in their medical record. In order to comply with the regulation, health systems are sending most notes and test results to the patient through the health system’s patient gateway. The open note process entered my practice through a stealthy progression, from an initial step of permitting a clinician to easily share their note with a patient to a top-down edict that all notes, except some notes that have a high risk of causing patient harm, must be sent immediately to the patient. Obviously, an open note supports “transparency,” but I am unaware of high quality evidence that open notes improve the health of a population or reduce morbidity or mortality from health problems.
The federal mandate that clinicians share their notes or risk fiscal penalties is coercive and undermines the independence of health professionals. Open notes may have many benefits, including:
- improving a patient’s comprehension and sense of control over their health issues
- increasing patient trust in their health system
- increasing the number of questions patients ask their clinician.6
Open notes may also cause unintended adverse emotional trauma to patients, especially when the note communicates “bad news.” In one study of 100 oncology patients, approximately 25% of respondents reported that reading clinical notes was emotionally difficult, and they sometimes regretted having read the note.6 One patient reported, “I think MyChart is great but in this whole cancer thing MyChart has not been a good thing.” Another patient reported, “Reading serious stuff like that is just too taxing for me to be honest with you.”6 An additional finding of the study was that patients reported their notes were written with too much medical jargon and repetition of information.
Open laboratory, pathology, and imaging data—Helpful or harmful?
A component of the open note mandate is that laboratory, pathology, and imaging data must be shared timely with patients. Some health systems incorporate a 3-day pause prior to sharing such data, in order to provide the clinical team with time to communicate with the patient before the test results are shared. Some health systems, including my health system, have engineered the open note data-sharing system to immediately share the results of most completed laboratory, pathology, and imaging studies with the patient. Immediate sharing of data may result in the patient first learning that they have a serious, life-threatening health problem, such as cancer, from their patient portal rather than from a clinician. As an example, a patient may first learn that they have metastatic cancer from a CT scan that was ordered for a benign indication.
Another example is that a patient may first learn that they have an HIV infection from their patient portal. This can be a shocking and emotionally damaging experience for the patient. For many test results, it would be best if a clinician were able to communicate the result to the patient, providing support and context to the meaning of the result, rather than sending sensitive, life-altering information directly from the laboratory or imaging department to the patient. Leaders in medical education have spent decades teaching clinicians how to communicate “bad news” in a sensitive, supportive, and effective manner. The open sharing of laboratory, pathology, and imaging data short-circuits the superior process of relying on a highly capable clinician to communicate bad news.
Continue to: Crafting the open medical record note...
Crafting the open medical record note
Building on the advice that “when life gives you lemons, make lemonade,” I have begun to pivot the purpose of my medical notes from a product useful to myself and other clinicians to a product whose primary purpose is to be helpful for the patient. The open note can facilitate building a trusting relationship with the patient. My notes are becoming a series of written conversations with the patient, emphasizing compassion and empathy. I am increasing significantly the amount of educational information in the note to help the patient understand their situation. In addition, I am replacing traditional medical terms with verbiage more appropriate in the context of a conversation with the patient, reducing the use of medical jargon. For example, I have stopped using “chief complaint” and replaced it with “health issues.” I am diligently avoiding the use of medical terms that have negative connotations, including “obese,” “psychosomatic,” “alcoholic,” and “drug addiction.” I include encouragement and positive comments in many of my notes. For example, “Ms. X is successfully managing her health issues and experiencing improved health. It is a pleasure collaborating with her on achieving optimal health.”
Can we bring sanity back to medical note writing?
The primary role of a clinician is to spend as much time as possible listening to patients, understanding their needs, and helping them achieve optimal health. There are many benefits to an electronic medical record, including legibility, accessibility, interoperability, and efficiency. However, in current practice “note bloat” undermines the potential of the electronic medical record and makes many notes ineffective to the process of advancing the patient’s health. We are competent and highly trained clinicians. We can craft notes that are simple, specific, story-driven, compassionate, and empathetic. If we return to the ABCs of note writing, focusing on accuracy, brevity, and clarity, we will make note writing and reading more rewarding and improve patient care. ●
- O’Donnell HC, Kaushal R, Barron Y, et al. Physicians’ attitudes towards copy and pasting in the electronic note writing. J Gen Intern Med. 2009;24:63-68.
- Weed LL. Medical records, patient care and medical education. Ir J Med Sci. 1964;462:271-282.
- Sieja A, Pell J, Markley K, et al. Successful implementation of APSO notes across a major health system. Am J Account Care. 2017;5:29-34.
- Barbieri RL, Levy B. Major changes in Medicare billing are planned for January 2021: some specialists fare better that others. OBG Manag. 2020;32:9, 10, 12, 14.
- State of the note summit, 2021. Medical specialty dos and don’ts. https://www.acponline.org/system/files/documents/practice-resources/business-resources/coding/state-of-the-note-summit-2021/sotn21-specialtycare.pdf. Accessed September 21, 2021.
- Kayashtha N, Pollak KI, LeBLanc TW. Open oncology notes: a qualitative study of oncology patients’ experiences reading their cancer care notes. Am Soc Clin Oncol. 2018;14:e251-e257.

Robert L. Barbieri, MD
Chair Emeritus, Department of Obstetrics and Gynecology
Interim Chief, Obstetrics
Brigham and Women’s Hospital
Kate Macy Ladd Distinguished Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Boston, Massachusetts
Dr. Barbieri reports no financial relationships relevant to this article.
Robert L. Barbieri, MD
Chair Emeritus, Department of Obstetrics and Gynecology
Interim Chief, Obstetrics
Brigham and Women’s Hospital
Kate Macy Ladd Distinguished Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Boston, Massachusetts
Dr. Barbieri reports no financial relationships relevant to this article.
Robert L. Barbieri, MD
Chair Emeritus, Department of Obstetrics and Gynecology
Interim Chief, Obstetrics
Brigham and Women’s Hospital
Kate Macy Ladd Distinguished Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Boston, Massachusetts
Dr. Barbieri reports no financial relationships relevant to this article.
Prior to 1980, medical record notes were generally hand-written, short, and to the point. Senior physicians often wrote their 3-line notes using a fountain pen in an elegant cursive. With the transition to electronic medical records, notes have become bloated with irrelevant information and frequently lack a focus on the critical clinical insights that optimize patient care. The use of smart phrases to pull vast amounts of raw data into the note is a major contributor to note bloat. The unrestrained use of the copy and paste functionality generates a sequence of cloned notes that grow in length as new information is added and little information from prior notes removed. With each subsequent clone the note often becomes less accurate, lengthier, and more difficult for a reader to understand. In one survey of 253 physicians who wrote electronic notes, 90% reported that they used the copy and paste function, with 71% reporting that use of this function caused inconsistencies within and among notes and increased the repetitive presentation of outdated information in the note.1 Although the surveyed clinicians recognized that the copy and paste function caused problems, 80% reported that they planned to continue to use the copy and paste function.1
The SOAP note
The problem-oriented SOAP note is written in the classic structure of subjective and objective information, followed by an assessment and plan.2 The structure of the SOAP note emphasizes the logical and sequential collection of data followed by data analysis, resulting in a focused assessment and plan. When notes were hand-written and short, the entire SOAP note could be viewed on one page. Like a dashboard, the eye could quickly scan each key component of the note, facilitating the simultaneous integration of all 4 components of the note, facilitating understanding of the patient’s clinical situation. When the SOAP note structure is used to create a multipage electronic note, the result is a note that often confuses rather than enlightens the reader. A 5- to 10-page SOAP note is often useless for patient care but demonstrates the ability of computer-savvy clinicians to quickly generate a note thousands of words in length.
The APSO note, a response to note bloat
When a medical record note becomes a multipage document, clinicians should consider switching from the SOAP note structure to the APSO note, where the assessment and plan are at the top of the note, and the subjective and objective information is below the assessment and plan. The APSO format permits the reader to more quickly grasp the critical thinking of the author and facilitates a focus on key points relevant to the patient’s condition. The note can be written in the SOAP format, but then the assessment and plan are brought to the top of the note. In my clinical experience fewer than 10% of clinicians are using an APSO note structure. I believe that, with a multipage note, the APSO structure improves the experience of the reader and should be more widely utilized, especially by clinicians who are prone to crafting a bloated note. In a survey of more than 3,000 clinicians, approximately two-thirds of the respondents reported that, compared with SOAP notes, APSO notes were easier and faster to read, and APSO notes made it easier to follow the clinical reasoning of the author.3
Continue to: New evaluation and management billing guidelines—An opportunity to reduce note bloat...
New evaluation and management billing guidelines—An opportunity to reduce note bloat
Previous evaluation and management federal billing guidelines emphasized documentation of a myriad of clinically irrelevant details contributing to note bloat. The new federal evaluation and management billing guidelines pivot the focus of the note to the quality and complexity of medical decision making as demonstrated in the assessment and plan.4 Prioritizing the assessment and plan as the key feature of the medical record note should help reduce the length of notes. The American College of Physicians recently recommended deleting the complete review of systems and prior histories from most notes unless relevant to medical decision making and the assessment and plan.5
The open note
The open note mandate was contained in federal regulations developed to implement the 21st Century Cures Act, which required patients to have access to the information in their medical record. In order to comply with the regulation, health systems are sending most notes and test results to the patient through the health system’s patient gateway. The open note process entered my practice through a stealthy progression, from an initial step of permitting a clinician to easily share their note with a patient to a top-down edict that all notes, except some notes that have a high risk of causing patient harm, must be sent immediately to the patient. Obviously, an open note supports “transparency,” but I am unaware of high quality evidence that open notes improve the health of a population or reduce morbidity or mortality from health problems.
The federal mandate that clinicians share their notes or risk fiscal penalties is coercive and undermines the independence of health professionals. Open notes may have many benefits, including:
- improving a patient’s comprehension and sense of control over their health issues
- increasing patient trust in their health system
- increasing the number of questions patients ask their clinician.6
Open notes may also cause unintended adverse emotional trauma to patients, especially when the note communicates “bad news.” In one study of 100 oncology patients, approximately 25% of respondents reported that reading clinical notes was emotionally difficult, and they sometimes regretted having read the note.6 One patient reported, “I think MyChart is great but in this whole cancer thing MyChart has not been a good thing.” Another patient reported, “Reading serious stuff like that is just too taxing for me to be honest with you.”6 An additional finding of the study was that patients reported their notes were written with too much medical jargon and repetition of information.
Open laboratory, pathology, and imaging data—Helpful or harmful?
A component of the open note mandate is that laboratory, pathology, and imaging data must be shared timely with patients. Some health systems incorporate a 3-day pause prior to sharing such data, in order to provide the clinical team with time to communicate with the patient before the test results are shared. Some health systems, including my health system, have engineered the open note data-sharing system to immediately share the results of most completed laboratory, pathology, and imaging studies with the patient. Immediate sharing of data may result in the patient first learning that they have a serious, life-threatening health problem, such as cancer, from their patient portal rather than from a clinician. As an example, a patient may first learn that they have metastatic cancer from a CT scan that was ordered for a benign indication.
Another example is that a patient may first learn that they have an HIV infection from their patient portal. This can be a shocking and emotionally damaging experience for the patient. For many test results, it would be best if a clinician were able to communicate the result to the patient, providing support and context to the meaning of the result, rather than sending sensitive, life-altering information directly from the laboratory or imaging department to the patient. Leaders in medical education have spent decades teaching clinicians how to communicate “bad news” in a sensitive, supportive, and effective manner. The open sharing of laboratory, pathology, and imaging data short-circuits the superior process of relying on a highly capable clinician to communicate bad news.
Continue to: Crafting the open medical record note...
Crafting the open medical record note
Building on the advice that “when life gives you lemons, make lemonade,” I have begun to pivot the purpose of my medical notes from a product useful to myself and other clinicians to a product whose primary purpose is to be helpful for the patient. The open note can facilitate building a trusting relationship with the patient. My notes are becoming a series of written conversations with the patient, emphasizing compassion and empathy. I am increasing significantly the amount of educational information in the note to help the patient understand their situation. In addition, I am replacing traditional medical terms with verbiage more appropriate in the context of a conversation with the patient, reducing the use of medical jargon. For example, I have stopped using “chief complaint” and replaced it with “health issues.” I am diligently avoiding the use of medical terms that have negative connotations, including “obese,” “psychosomatic,” “alcoholic,” and “drug addiction.” I include encouragement and positive comments in many of my notes. For example, “Ms. X is successfully managing her health issues and experiencing improved health. It is a pleasure collaborating with her on achieving optimal health.”
Can we bring sanity back to medical note writing?
The primary role of a clinician is to spend as much time as possible listening to patients, understanding their needs, and helping them achieve optimal health. There are many benefits to an electronic medical record, including legibility, accessibility, interoperability, and efficiency. However, in current practice “note bloat” undermines the potential of the electronic medical record and makes many notes ineffective to the process of advancing the patient’s health. We are competent and highly trained clinicians. We can craft notes that are simple, specific, story-driven, compassionate, and empathetic. If we return to the ABCs of note writing, focusing on accuracy, brevity, and clarity, we will make note writing and reading more rewarding and improve patient care. ●
Prior to 1980, medical record notes were generally hand-written, short, and to the point. Senior physicians often wrote their 3-line notes using a fountain pen in an elegant cursive. With the transition to electronic medical records, notes have become bloated with irrelevant information and frequently lack a focus on the critical clinical insights that optimize patient care. The use of smart phrases to pull vast amounts of raw data into the note is a major contributor to note bloat. The unrestrained use of the copy and paste functionality generates a sequence of cloned notes that grow in length as new information is added and little information from prior notes removed. With each subsequent clone the note often becomes less accurate, lengthier, and more difficult for a reader to understand. In one survey of 253 physicians who wrote electronic notes, 90% reported that they used the copy and paste function, with 71% reporting that use of this function caused inconsistencies within and among notes and increased the repetitive presentation of outdated information in the note.1 Although the surveyed clinicians recognized that the copy and paste function caused problems, 80% reported that they planned to continue to use the copy and paste function.1
The SOAP note
The problem-oriented SOAP note is written in the classic structure of subjective and objective information, followed by an assessment and plan.2 The structure of the SOAP note emphasizes the logical and sequential collection of data followed by data analysis, resulting in a focused assessment and plan. When notes were hand-written and short, the entire SOAP note could be viewed on one page. Like a dashboard, the eye could quickly scan each key component of the note, facilitating the simultaneous integration of all 4 components of the note, facilitating understanding of the patient’s clinical situation. When the SOAP note structure is used to create a multipage electronic note, the result is a note that often confuses rather than enlightens the reader. A 5- to 10-page SOAP note is often useless for patient care but demonstrates the ability of computer-savvy clinicians to quickly generate a note thousands of words in length.
The APSO note, a response to note bloat
When a medical record note becomes a multipage document, clinicians should consider switching from the SOAP note structure to the APSO note, where the assessment and plan are at the top of the note, and the subjective and objective information is below the assessment and plan. The APSO format permits the reader to more quickly grasp the critical thinking of the author and facilitates a focus on key points relevant to the patient’s condition. The note can be written in the SOAP format, but then the assessment and plan are brought to the top of the note. In my clinical experience fewer than 10% of clinicians are using an APSO note structure. I believe that, with a multipage note, the APSO structure improves the experience of the reader and should be more widely utilized, especially by clinicians who are prone to crafting a bloated note. In a survey of more than 3,000 clinicians, approximately two-thirds of the respondents reported that, compared with SOAP notes, APSO notes were easier and faster to read, and APSO notes made it easier to follow the clinical reasoning of the author.3
Continue to: New evaluation and management billing guidelines—An opportunity to reduce note bloat...
New evaluation and management billing guidelines—An opportunity to reduce note bloat
Previous evaluation and management federal billing guidelines emphasized documentation of a myriad of clinically irrelevant details contributing to note bloat. The new federal evaluation and management billing guidelines pivot the focus of the note to the quality and complexity of medical decision making as demonstrated in the assessment and plan.4 Prioritizing the assessment and plan as the key feature of the medical record note should help reduce the length of notes. The American College of Physicians recently recommended deleting the complete review of systems and prior histories from most notes unless relevant to medical decision making and the assessment and plan.5
The open note
The open note mandate was contained in federal regulations developed to implement the 21st Century Cures Act, which required patients to have access to the information in their medical record. In order to comply with the regulation, health systems are sending most notes and test results to the patient through the health system’s patient gateway. The open note process entered my practice through a stealthy progression, from an initial step of permitting a clinician to easily share their note with a patient to a top-down edict that all notes, except some notes that have a high risk of causing patient harm, must be sent immediately to the patient. Obviously, an open note supports “transparency,” but I am unaware of high quality evidence that open notes improve the health of a population or reduce morbidity or mortality from health problems.
The federal mandate that clinicians share their notes or risk fiscal penalties is coercive and undermines the independence of health professionals. Open notes may have many benefits, including:
- improving a patient’s comprehension and sense of control over their health issues
- increasing patient trust in their health system
- increasing the number of questions patients ask their clinician.6
Open notes may also cause unintended adverse emotional trauma to patients, especially when the note communicates “bad news.” In one study of 100 oncology patients, approximately 25% of respondents reported that reading clinical notes was emotionally difficult, and they sometimes regretted having read the note.6 One patient reported, “I think MyChart is great but in this whole cancer thing MyChart has not been a good thing.” Another patient reported, “Reading serious stuff like that is just too taxing for me to be honest with you.”6 An additional finding of the study was that patients reported their notes were written with too much medical jargon and repetition of information.
Open laboratory, pathology, and imaging data—Helpful or harmful?
A component of the open note mandate is that laboratory, pathology, and imaging data must be shared timely with patients. Some health systems incorporate a 3-day pause prior to sharing such data, in order to provide the clinical team with time to communicate with the patient before the test results are shared. Some health systems, including my health system, have engineered the open note data-sharing system to immediately share the results of most completed laboratory, pathology, and imaging studies with the patient. Immediate sharing of data may result in the patient first learning that they have a serious, life-threatening health problem, such as cancer, from their patient portal rather than from a clinician. As an example, a patient may first learn that they have metastatic cancer from a CT scan that was ordered for a benign indication.
Another example is that a patient may first learn that they have an HIV infection from their patient portal. This can be a shocking and emotionally damaging experience for the patient. For many test results, it would be best if a clinician were able to communicate the result to the patient, providing support and context to the meaning of the result, rather than sending sensitive, life-altering information directly from the laboratory or imaging department to the patient. Leaders in medical education have spent decades teaching clinicians how to communicate “bad news” in a sensitive, supportive, and effective manner. The open sharing of laboratory, pathology, and imaging data short-circuits the superior process of relying on a highly capable clinician to communicate bad news.
Continue to: Crafting the open medical record note...
Crafting the open medical record note
Building on the advice that “when life gives you lemons, make lemonade,” I have begun to pivot the purpose of my medical notes from a product useful to myself and other clinicians to a product whose primary purpose is to be helpful for the patient. The open note can facilitate building a trusting relationship with the patient. My notes are becoming a series of written conversations with the patient, emphasizing compassion and empathy. I am increasing significantly the amount of educational information in the note to help the patient understand their situation. In addition, I am replacing traditional medical terms with verbiage more appropriate in the context of a conversation with the patient, reducing the use of medical jargon. For example, I have stopped using “chief complaint” and replaced it with “health issues.” I am diligently avoiding the use of medical terms that have negative connotations, including “obese,” “psychosomatic,” “alcoholic,” and “drug addiction.” I include encouragement and positive comments in many of my notes. For example, “Ms. X is successfully managing her health issues and experiencing improved health. It is a pleasure collaborating with her on achieving optimal health.”
Can we bring sanity back to medical note writing?
The primary role of a clinician is to spend as much time as possible listening to patients, understanding their needs, and helping them achieve optimal health. There are many benefits to an electronic medical record, including legibility, accessibility, interoperability, and efficiency. However, in current practice “note bloat” undermines the potential of the electronic medical record and makes many notes ineffective to the process of advancing the patient’s health. We are competent and highly trained clinicians. We can craft notes that are simple, specific, story-driven, compassionate, and empathetic. If we return to the ABCs of note writing, focusing on accuracy, brevity, and clarity, we will make note writing and reading more rewarding and improve patient care. ●
- O’Donnell HC, Kaushal R, Barron Y, et al. Physicians’ attitudes towards copy and pasting in the electronic note writing. J Gen Intern Med. 2009;24:63-68.
- Weed LL. Medical records, patient care and medical education. Ir J Med Sci. 1964;462:271-282.
- Sieja A, Pell J, Markley K, et al. Successful implementation of APSO notes across a major health system. Am J Account Care. 2017;5:29-34.
- Barbieri RL, Levy B. Major changes in Medicare billing are planned for January 2021: some specialists fare better that others. OBG Manag. 2020;32:9, 10, 12, 14.
- State of the note summit, 2021. Medical specialty dos and don’ts. https://www.acponline.org/system/files/documents/practice-resources/business-resources/coding/state-of-the-note-summit-2021/sotn21-specialtycare.pdf. Accessed September 21, 2021.
- Kayashtha N, Pollak KI, LeBLanc TW. Open oncology notes: a qualitative study of oncology patients’ experiences reading their cancer care notes. Am Soc Clin Oncol. 2018;14:e251-e257.
- O’Donnell HC, Kaushal R, Barron Y, et al. Physicians’ attitudes towards copy and pasting in the electronic note writing. J Gen Intern Med. 2009;24:63-68.
- Weed LL. Medical records, patient care and medical education. Ir J Med Sci. 1964;462:271-282.
- Sieja A, Pell J, Markley K, et al. Successful implementation of APSO notes across a major health system. Am J Account Care. 2017;5:29-34.
- Barbieri RL, Levy B. Major changes in Medicare billing are planned for January 2021: some specialists fare better that others. OBG Manag. 2020;32:9, 10, 12, 14.
- State of the note summit, 2021. Medical specialty dos and don’ts. https://www.acponline.org/system/files/documents/practice-resources/business-resources/coding/state-of-the-note-summit-2021/sotn21-specialtycare.pdf. Accessed September 21, 2021.
- Kayashtha N, Pollak KI, LeBLanc TW. Open oncology notes: a qualitative study of oncology patients’ experiences reading their cancer care notes. Am Soc Clin Oncol. 2018;14:e251-e257.

Mobile Integrated Health: Reducing Chronic Obstructive Pulmonary Disease Hospitalizations Through Novel Outpatient Care Initiatives
From the Mobile Integrated Health and Emergency Medicine Department, South Shore Health, Weymouth, MA.
Objective: To develop a process through which Mobile Integrated Health (MIH) can treat patients with chronic obstructive pulmonary disease (COPD) at high risk for readmission in an outpatient setting. In turn, South Shore Hospital (SSH) looks to leverage MIH to improve hospital flow, decrease costs, and improve patient quality of life.
Methods: With the recent approval of hospital-based MIH programs in Massachusetts, SSH used MIH to target specific patient demographics in an at-home setting. Here, we describe the planning and implementation of this program for patients with COPD. Key components to success include collaboration among providers, early follow-up visits, patient education, and in-depth medical reconciliations. Analysis includes a retrospective examination of a structured COPD outpatient pathway.
Results: A total of 214 patients with COPD were treated with MIH from March 2, 2020, to August 1, 2021. Eighty-seven emergent visits were conducted, and more than 650 total visits were made. A more intensive outpatient pathway was implemented for patients deemed to be at the highest risk for readmission by pulmonary specialists.
Conclusion: This process can serve as a template for future institutions to treat patients with COPD using MIH or similar hospital-at-home services.
Keywords: Mobile Integrated Health; MIH; COPD; population health.
It is estimated that chronic obstructive pulmonary disease (COPD) affects more than 16 million Americans1 and accounts for more than 700 000 hospitalizations each year in the US.2 Thirty-day COPD readmission rates hover around 22.6%,3 and readmission within 90 days of initial discharge can jump to between 31% and 35%.4 This is the highest of any patient demographic, and more than half of these readmissions are due to COPD. To counter this, government and state entities have made nationwide efforts to encourage health systems to focus on preventing readmissions. In October 2014, the US added COPD to the active list of diseases in Medicare’s Hospital Readmissions Reduction Program (HRRP), later adding COPD to various risk-based bundle programs that hospitals may choose to opt into. These programs are designed to reduce all-cause readmissions after an acute exacerbation of COPD, as the HRRP penalizes hospitals for all-cause 30-day readmissions.3 However, what is most troubling is that, despite these efforts, readmission rates have not dropped in the past decade.5 COPD remains the third leading cause of death in America and still poses a significant burden both clinically and economically to hospitals across the country.3
A solution that is gaining traction is to encourage outpatient care initiatives and discharge pathways. Early follow-up is proven to decrease chances of readmission, and studies have shown that more than half of readmitted patients did not follow up with a primary care physician (PCP) within 30 days of their initial discharge.6 Additionally, large meta-analyses show hospital-at-home–type programs can lead to reductions in mortality, decrease costs, decrease readmissions, and increase patient satisfaction.7-9 Therefore, for more challenging patient populations with regard to readmissions and mortality, Mobile Integrated Health (MIH) may be the solution that we are looking for.
This article presents a viable process to treat patients with COPD in an outpatient setting with MIH Services. It includes an examination of what makes MIH successful as well as a closer look at a structured COPD outpatient pathway.
Methods
South Shore Hospital (SSH) is an independent, not-for-profit hospital located in Weymouth, Massachusetts. It is host to 400 beds, 100 000 annual visits to the emergency department (ED), and its own emergency medical services program. In March 2020, SSH became the first Massachusetts hospital-based program to acquire an MIH license. MIH paramedics receive 300 hours of specialized training, including time in clinical clerkships shadowing pulmonary specialists, cardiology/congestive heart failure (CHF) providers, addiction medicine specialists, home care and care progression colleagues, and wound center providers. Specialist providers become more comfortable with paramedic capabilities as a result of these clerkships, improving interactions and relationships going forward. At the time of writing, SSH MIH is staffed by 12 paramedics, 4 of whom are full time; 2 medical directors; 2 internal coordinators; and 1 registered nurse (RN). A minimum of 2 paramedics are on call each day, each with twice-daily intravenous (IV) capabilities. The first shift slot is 16 hours, from 7:00 AM to 11:00
The goal of developing MIH is to improve upon the current standard of care. For hospitals without MIH capabilities, there are limited options to treat acute exacerbations of chronic obstructive pulmonary disease (AECOPD) patients postdischarge. It is common for the only outpatient referral to be a lone PCP visit, and many patients who need more extensive treatment options don’t have access to a timely PCP follow-up or resources for alternative care. This is part of why there has been little improvement in the 21st century with regard to reducing COPD hospitalizations. As it stands, approximately 10% to 55% of all AECOPD readmissions are preventable, and more than one-fifth of patients with COPD are rehospitalized within 30 days of discharge.3 In response, MIH has been designed to provide robust care options postdischarge in the patient home, with the eventual goal of reducing preventable hospitalizations and readmissions for all patients with COPD.
Patient selection
Patients with COPD are admitted to the MIH program in 1 of 3 ways: (1) directly from the ED; (2) at discharge from inpatient care; or (3) from a SSH affiliate referral.
With option 1, the ED physician assesses patient need for MIH services and places a referral to MIH in the electronic medical record (EMR). The ED provider also specifies whether follow-up is “urgent” and sets an alternative level of priority if not. With option 2, the inpatient provider and case manager follow a similar process, first determining whether a patient is stable enough to go home with outpatient services and then if MIH would be beneficial to the patient. If the patient is discharged home, a follow-up visit by an MIH paramedic is scheduled within 48 hours. With option 3, the patient is referred to MIH by an affiliate of SSH. This can be through the patient’s PCP, their visiting nurse association (VNA) service provider, or through any SSH urgent care center. In all 3 referral processes, the patient has the option to consent into the program or refuse services. Once referred, MIH coordinators review patients on a case-by-case basis. Patients with a history of prior admissions are given preference, with the goal being to keep the frailer, older, and comorbid patients at home. Other considerations include recent admission(s), length of stay, and overall stability. Social factors considered by the team include whether the patient lives alone and has alternative home services and the patient’s total distance from the hospital. Patients with a history of violence, mental health concerns, or substance abuse go through a more extensive screening process to ensure paramedic safety.
Given their patient profile and high hospital usage rates, MIH is sometimes requested for patients with end-stage COPD. Many of these patients benefit from MIH goals-of-care conversations to ensure they understand all their options and choose an approach that fits their preferences. In these cases, MIH has been instrumental in assisting patients and families with completing Medical Orders for Life-Sustaining Treatment and health care proxy forms and transitioning patients to palliative care, hospice, advanced-illness care management programs, or other long-term care options to prevent the need for rehospitalization. The MIH team focuses heavily on providing quality end-of-life care for patients and aligning care models with patient and family goals, often finding that having these sensitive conversations in the comfort of home enables transparency and comfort not otherwise experienced by hospitalized patients.
Initial patient follow-up
For patients with COPD enrolled in the MIH program, their first patient visit is scheduled within 48 hours of discharge from the ED or inpatient hospital. In many cases, this visit can be conducted within 24 hours of returning home. Once at the patient’s home, the paramedic begins with general introductions, vital signs, and a basic physical examination. The remainder of the visit focuses on patient education and symptom recognition. The paramedic reviews the COPD action plan (Figure 1), including how to recognize the onset of a “COPD flare-up” and the appropriate response. Patients are provided with a paper copy of the action plan for future reference.
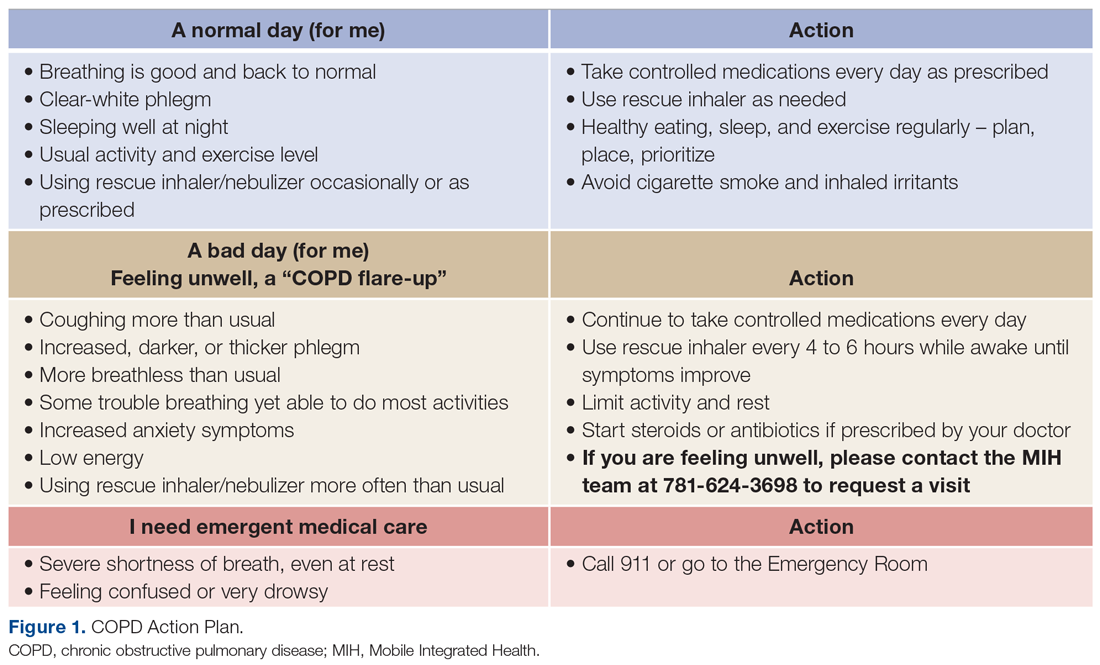
The next point of educational emphasis is the patient’s individual medication regimen. This involves differentiating between control (daily) and rescue medications, how to use oxygen tanks, and how to safely wean off of oxygen. Specific attention is given to how to use a metered-dose inhaler, as studies have found that more than half of all patients use their inhaler devices incorrectly.10
Paramedics also complete a home safety evaluation of the patient’s residence, which involves checking for tripping hazards, lighting, handrails, slippery surfaces, and general access to patient medication. If an issue cannot be resolved by the paramedic on site and is considered a safety hazard, it is reported back to the hospital team for assistance.
Finally, patients are educated on the capabilities of MIH as a program and what to expect when they reach out over the phone. Patients are given a phone number to call for both “urgent” and “nonemergent” situations. In both cases, they will be greeted by one of the MIH coordinators or nurses who assist with triaging patient symptoms, scheduling a visit, or providing other guidance. It is a point of emphasis that the patient can use MIH for more than just COPD and should call in the event of any illness or discomfort (eg, dehydration, fever) in an effort to prevent unnecessary ED visits.
Medication reconciliation
Patients with COPD often have complex medication regimens. To help alleviate any confusion, medication reconciliations are done in conjunction with every COPD patient’s initial visit. During this process, the paramedic first takes an inventory of all medications in the patient home. Common reasons for nonadherence include confusing packaging, inability to reach the pharmacy, or medication not being covered by insurance. The paramedic reconciles the updated medication regimen against the medications that are physically in the home. Once the initial review is complete, the paramedic teleconferences with a registered hospitalist pharmacist (RHP) for a more in-depth review. Over video chat, the RHP reviews each medication individually to make sure the patient understands how many times per day they take each medication, whether it is a control or rescue medication, and what times of the day to take them. The RHP will then clarify any other medication questions the patient has, assure all recent medications have been picked up from the pharmacy, and determine any barriers, such as cost or transportation.
Follow-ups and PCP involvement
At each in-person visit, paramedics coordinate with an advanced practice clinician (APC) through telehealth communication. On these video calls with a provider, the paramedic relays relevant information pertaining to patient history, vital signs, and current status. Any concerning findings, symptoms of COPD flare-ups, or recent changes in status will be discussed. The APC then speaks directly to the patient to gather additional details about their condition and any recent hospitalizations, with their primary role being to make clinical decisions on further treatment. For the COPD population, this often includes orders for the MIH paramedic to administer IV medication (ie, IV methylprednisolone or other corticosteroids), antibiotics, home nebulizers, and at-home oxygen.
Second and third follow-up paramedic visits are often less intensive. Although these visits often still involve telehealth calls to the APC, the overall focus shifts toward medication adherence, ED avoidance, and readmission avoidance. On these visits, the paramedic also checks vitals, conducts a physical examination, and completes follow-up testing or orders per the APC.
PCP involvement is critical to streamlining and transitioning patient care. Patients who are admitted to MIH without insurance or a PCP are assisted in the process of finding one. PCPs automatically receive a patient enrollment letter when their patient is seen by an MIH paramedic. Following each individual visit, paramedic and APC notes are sent to the PCP through the EMR or via fax, at which time the PCP may be consulted on patient history and/or future care decisions. After the transition back to care by their PCP, patients are still encouraged to utilize MIH if acute changes arise. If a patient is readmitted back to the hospital, MIH is automatically notified, and coordinators will assess whether there is continued need for outpatient services or areas for potential improvement.
Emergent MIH visits
While MIH visits with patients with COPD are often scheduled, MIH can also be leveraged in urgent situations to prevent the need for a patient to come to the ED or hospital. Patients with COPD are told to call MIH if they have worsening symptoms or have exhausted all methods of self-treatment without an improvement in status. In this case, a paramedic is notified and sent to the patient’s home at the earliest time possible. The paramedic then completes an assessment of the patient’s status and relays information to the MIH APC or medical director. From there, treatment decisions, such as starting the patient on an IV, using nebulizers, or doing an electrocardiogram for diagnostic purposes, are guided by the provider team with the ultimate goal of caring for the patient in the home. For our population, providing urgent care in the home has proven to be an effective way to avoid unnecessary readmissions while still ensuring high-quality patient care.
Outpatient pathway
In May 2021, select patients with COPD were given the option to participate in a more intensive MIH outpatient pathway. Pilot patients were chosen by 2 pulmonary specialists, with a focus on enrolling patients with COPD at the highest risk for readmission. Patients who opted in were followed by MIH for a total of 30 days.
The first visit was made as usual within 48 hours of discharge. Patients received education, medication reconciliation, vitals examination, home safety evaluation, and a facilitated telehealth evaluation with the APC. What differentiates the pathway from standard MIH services is that after the first visit, the follow-ups are prescheduled and more numerous. This is outlined best in Figure 2, which serves as a guideline for coordinators and paramedics in the cadence and focus of visits for each patient on the pathway. The initial 2 weeks are designed to check in on the patient in person and ensure active recovery. The latter 2 weeks are designed to ensure that the patient follows up with their care team and understands their medications and action plan going forward. Pathway patients were also monitored using a remote patient monitoring (RPM) kit. On the initial visit, paramedics set up the RPM equipment and provided a demonstration on how to use each device. Patients were issued a Bluetooth-enabled scale, blood pressure cuff, video-enabled tablet, and wearable device. The wearable device continuously recorded respiration rate, heart rate, and oxygen saturation and had fall-detection enabled. Over the course of a month, an experienced MIH nurse monitored the vitals transmitted by the wearable device and checked patient weight and blood pressure 1 to 2 times per day, utilizing these data to proactively outreach to patients if abnormalities occurred. Prior to the start of the program, the MIH nurse contacted each patient to introduce herself and notify them that they would receive a call if any vitals were unusual.
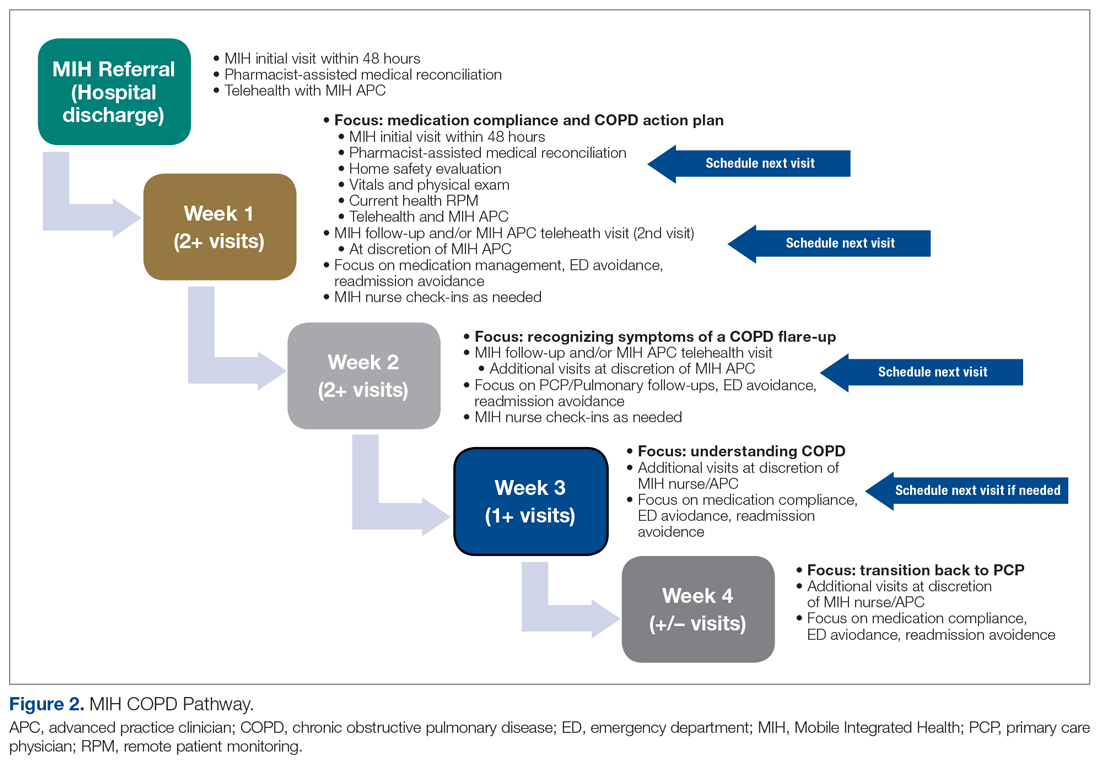
Results
MIH treated 214 patients with COPD from March 2, 2020, to August 2, 2021. In total, paramedics made more than 650 visits. Eighty-seven of these were documented as urgent visits with AECOPD, shortness of breath, cough, or wheezing as the primary concern.
In the calendar year of 2019, our institution admitted 804 patients with a primary diagnosis of COPD. In 2020, the first year with MIH, total COPD admissions decreased to 473; however, the effect of the COVID-19 pandemic cannot be discounted. At of the time of writing—219 days into 2021—253 patients with COPD have been admitted thus far (Table 1).

Pathway results
Sixteen patients were referred to the MIH COPD Discharge Pathway Pilot during May 2021. Ten patients went on to complete the entire 30-day pathway. Six did not finish the program. Three of these 6 patients were referred by a pulmonary specialist for enrollment but not ultimately referred to the pilot program by case management and therefore not enrolled. The other 3 of the 6 patients who did not complete the pilot program were enrolled but discontinued owing to noncompliance.
Of the 10 patients who completed the pathway, 3 patients were male, and 7 were female. Ages ranged from 55 to 84 years. On average, the RHP found 3.6 medication reconciliation errors per patient. One patient was readmitted within 30 days (only 3 days after the initial discharge), and 5 were readmitted within 90 days.
A retrospective analysis was conducted on patients with COPD who were not provided with MIH services and were admitted to our hospital between September 1, 2020, and March 1, 2021, for comparison. Age, sex, and other related conditions are shown in Table 2. Medication reconciliation error data were not tracked for this demographic, as they did not have an in-home medication reconciliation completed.
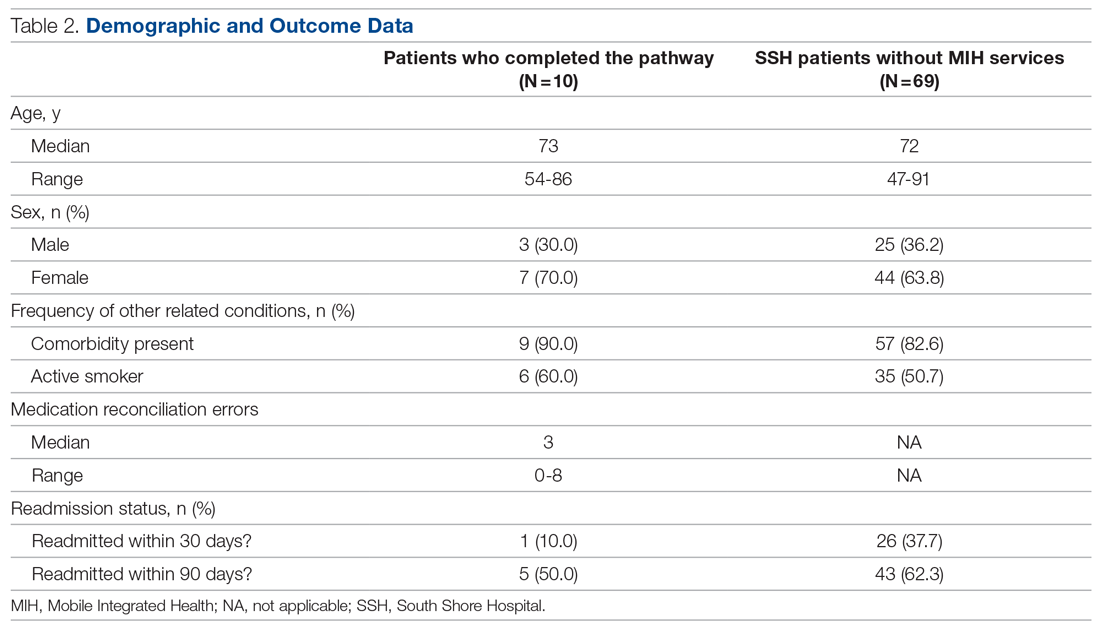
Discussion
MIH has treated 214 patients with COPD from March 2, 2020, to August 2, 2021, a 17-month period. In that same timeframe, the hospital experienced a 42% decrease in COPD admissions. Although this effect is not the sole product of MIH (specifically, COVID-19 caused a drop in all-cause hospital admissions), we believe MIH did play a small role in this reduction. Eighty-seven emergent visits were conducted for patients with a primary complaint of AECOPD, shortness of breath, cough, or wheezing. On these visits, MIH provided urgent treatment to prevent the patient returning to the ED and potentially leading to readmission.
The program’s impact extends beyond the numbers. With more than 200 patients with COPD treated at home, we improved hospital flow, shortened patients’ overall length of stay, and increased capacity in the ED and inpatient units. In addition, MIH has been able to fill in care gaps present in the current health care system by providing acute care in the home to patients who otherwise have access-to-care and transportation issues.
What made the program successful
With the COPD population prone to having complex medication regimens, medication reconciliations were critical to improving patient outcomes. During the documented medication reconciliations for pathway patients, 8 of 10 patients had medication errors identified. Some of the more common errors included incorrect inhaler usage, patient medication not arriving to the pharmacy for a week or more after discharge, prescribed medication dosages that were too high or too low, and a lack of transportation to pick up the patient’s prescription. Even more problematic is that 7 of these 8 patients required multiple interventions to correct their regimen. What was cited as most beneficial by both the paramedic and the RHP was taking time to walk through each medication individually and ensuring that the patient could recite back how often and when they should be using it. What also proved to be helpful was spending extra time on the inhalers and nebulizers. Multiple patients did not know how to use them properly and/or cited a history of struggling with them.
The MIH COPD pathway patients showed encouraging preliminary results. In the initial 30-day window, only 1 of 10 (10%) patients was readmitted, which is lower than the 37.7% rate for comparable patients who did not have MIH services. This could imply that patients with COPD respond positively to active and consistent management with predetermined points of contact. Ninety-day readmission rates jumped to 5 of 10, with 4 of these patients being readmitted multiple times. Approximately half of these readmissions were COPD related. It is important to remember that the patients being targeted by the pathway are deemed to be at very high risk of readmission. As such, one could expect that even with a successful reduction in rates, pathway patient readmission rates may be slightly elevated compared with national COPD averages.
Given the more personalized and at-home care, patients also expressed higher levels of care satisfaction. Most patients want to avoid the hospital at all costs, and MIH provides a safe and effective alternative. Patients with COPD have also relayed that the education they receive on their medication, disease, and how to use MIH has been useful. This is reflected in the volume of urgent calls that MIH receives. A patient calling MIH in place of 911 shows not only that the patient has a level of trust in the MIH team, but also that they have learned how to recognize symptoms earlier to prevent major flare-ups.
This study had several limitations. On the pilot pathway, 3 patients were removed from MIH services because of repeated noncompliance. These instances primarily involved aggression toward the paramedics, both verbal and physical, as well as refusal to allow the MIH paramedics into the home. Going forward, it will be valuable to have a screening process for pathway patients to determine likelihood of compliance. This could include speaking to the patient’s PCP or other in-hospital providers before accepting them into the program.
Remote patient monitoring also presented its challenges. Despite extensive equipment demonstrations, some patients struggled to grasp the technology. Some of the biggest problems cited were confusion operating the tablet, inability to charge the devices, and issues with connectivity. In the future, it may be useful to simplify the devices even more. Further work should also be done to evaluate the efficacy of remote patient technology in this specific setting, as studies have shown varied results with regard to RPM success. In 1 meta-analysis of 91 different published studies that took place between 2015 and 2020, approximately half of the RPM studies resulted in no change in hospital readmissions, length of stay, or ED presentations, while the other half saw improvement in these categories.11 We suspect that the greatest benefits of our work came from the patient education, trust built over time, in-home urgent evaluations, and 1-on-1 time with the paramedic.
With many people forgoing care during the pandemic, COVID-19 has also caused a downward trend in overall and non-COVID-19 admissions. In a review of more than 500 000 ED visits in Massachusetts between March 11, 2020, and September 8, 2021, there was a 32% decrease in admissions when compared with those same weeks in 2019.10 There was an even greater drop-off when it came to COPD and other respiratory-related admissions. In evaluating the impact SSH MIH has made, it is important to recognize that the pandemic contributed to reducing total COPD admissions. Adding merit to the success of MIH in contributing to the reduction in admissions is the continued downward trend in total COPD admissions year-to-date in 2021. Despite total hospital usage rates increasing at our institution over the course of this year, the overall COPD usage rates have remained lower than before.
Another limitation is that in the selection of patients, both for general MIH care and for the COPD pathway, there was room for bias. The pilot pathway was offered specifically to patients at the highest risk for readmission; however, patients were referred at the discretion of our pulmonologist care team and not selected by any standardized rubric. Additionally, MIH only operates on a 16-hour schedule. This means that patients admitted to the ED or inpatient at night may sometimes be missed and not referred to MIH for care.
The biggest caveat to the pathway results is, of course, the small sample size. With only 10 patients completing the pilot, it is impossible to come to any concrete conclusions. Such an intensive pathway requires dedicating large amounts of time and resources, which is why the pilot was small. However, considering the preliminary results, the outline given could provide a starting point for future work to evaluate a similar COPD pathway on a larger scale.
Future considerations
Risk stratification of patients is critical to achieving even further reductions in readmissions and mortality. Hospitals can get the most value from MIH by focusing on patients with COPD at the highest risk for return, and it would be valuable to explicitly define who fits into this criterion. Utilizing a tool similar to the LACE index for readmission but tailoring it to patients with COPD when admitting patients into the program would be a logical next step.
Reducing the points of patient contact could also prove valuable. Over the course of a patient’s time with MIH, they interact with an RHP, APC, paramedic, RN, and discharging hospitalist. Additionally, we found many patients had VNA services, home health aides, care managers, and/or social workers involved in their care. Some patients found this to be stressful and overwhelming, especially regarding the number of outreach calls soon after discharge.
It would also be useful to look at the impact of MIH on total COPD admissions independent of the artificial variation created by COVID-19. This may require waiting until there are higher levels of vaccination and/or finding ways to control for the potential variation. In doing so, one could look at the direct effect MIH has on COPD readmissions when compared with a control group without MIH services, which could then serve as a comparison point to the results of this study. As it stands, given the relative novelty of MIH, there are primarily only broad reviews of MIH’s effectiveness and/or impact on patient populations that have been published. Of these, only a few directly mentioned MIH in relation to COPD, and none have comparable designs that look at overall COPD hospitalization reductions post-MIH implementation. There is also little to no literature looking at the utilization of MIH in a more intensive COPD outpatient pathway.
Finally, MIH has proven to be a useful tool for our institution in many areas outside of COPD management. Specifically, MIH has been utilized as a mobile influenza and COVID-19 vaccination unit and in-home testing service and now operates both a hospital-at-home and skilled nursing facility-at-home program. Analysis of the overall needs of the system and where this valuable MIH resource would have the biggest impact will be key in future growth opportunities.
Conclusion
MIH has been an invaluable tool for our hospital, especially in light of the recent shift toward more in-home and virtual care. MIH cared for 214 patients with COPD with more than 650 visits between March 2020 and August 2021. Eighty-seven emergent COPD visits were conducted, and COPD admissions were reduced dramatically from 2019 to 2020. MIH services have improved hospital flow, allowed for earlier discharge from the ED and inpatient care, and helped improve all-cause COPD readmission rates. The importance of postdischarge care and follow-up visits for patients with COPD, especially those at higher risk for readmission, cannot be understated. We hope our experience working to improve COPD patient outcomes serves as valuable a reference point for future MIH programs.
Corresponding author: Kelly Lannutti, DO, Mobile Integrated Health and Emergency Medicine Department, South Shore Health, 55 Fogg Rd, South Weymouth, MA 02190; [email protected].
Financial disclosures: None.
1. Centers for Disease Control and Prevention. Chronic obstructive pulmonary disease (COPD). Accessed September 10, 2011. https://www.cdc.gov/copd/index.html
2. Wier LM, Elixhauser A, Pfuntner A, AuDH. Overview of Hospitalizations among Patients with COPD, 2008. Statistical Brief #106. In: Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Agency for Healthcare Research and Quality; 2011.
3. Shah T, Press,VG, Huisingh-Scheetz M, White SR. COPD Readmissions: Addressing COPD in the Era of Value-Based Health Care. Chest. 2016;150(4):916-926. doi:10.1016/j.chest.2016.05.002
4. Harries TH, Thornton H, Crichton S, et al. Hospital readmissions for COPD: a retrospective longitudinal study. NPJ Prim Care Respir Med. 2017;27(1):31. doi:10.1038/s41533-017-0028-8
5. Ford ES. Hospital discharges, readmissions, and ED visits for COPD or bronchiectasis among US adults: findings from the nationwide inpatient sample 2001-2012 and Nationwide Emergency Department Sample 2006-2011. Chest. 2015;147(4):989-998. doi:10.1378/chest.14-2146
6. Jencks SF, Williams MV, Coleman EA. Rehospitalizations among patients in the Medicare fee-for-service program. N Engl J Med. 2009;360(14):1418-1428. doi:10.1056/NEJMsa0803563
7. Shepperd S, Doll H, Angus RM, et al. Avoiding hospital admission through provision of hospital care at home: a systematic review and meta-analysis of individual patient data. CMAJ. 2009;180(2):175-182. doi:10.1503/cmaj.081491
8. Caplan GA, Sulaiman NS, Mangin DA, et al. A meta-analysis of “hospital in the home.” Med J Aust. 2012;197(9):512-519. doi:10.5694/mja12.10480
9. Portillo EC, Wilcox A, Seckel E, et al. Reducing COPD readmission rates: using a COPD care service during care transitions. Fed Pract. 2018;35(11):30-36.
10. Nourazari S, Davis SR, Granovsky R, et al. Decreased hospital admissions through emergency departments during the COVID-19 pandemic. Am J Emerg Med. 2021;42:203-210. doi:10.1016/j.ajem.2020.11.029
11. Taylor ML, Thomas EE, Snoswell CL, et al. Does remote patient monitoring reduce acute care use? A systematic review. BMJ Open. 2021;11(3):e040232. doi:10.1136/bmj/open-2020-040232
From the Mobile Integrated Health and Emergency Medicine Department, South Shore Health, Weymouth, MA.
Objective: To develop a process through which Mobile Integrated Health (MIH) can treat patients with chronic obstructive pulmonary disease (COPD) at high risk for readmission in an outpatient setting. In turn, South Shore Hospital (SSH) looks to leverage MIH to improve hospital flow, decrease costs, and improve patient quality of life.
Methods: With the recent approval of hospital-based MIH programs in Massachusetts, SSH used MIH to target specific patient demographics in an at-home setting. Here, we describe the planning and implementation of this program for patients with COPD. Key components to success include collaboration among providers, early follow-up visits, patient education, and in-depth medical reconciliations. Analysis includes a retrospective examination of a structured COPD outpatient pathway.
Results: A total of 214 patients with COPD were treated with MIH from March 2, 2020, to August 1, 2021. Eighty-seven emergent visits were conducted, and more than 650 total visits were made. A more intensive outpatient pathway was implemented for patients deemed to be at the highest risk for readmission by pulmonary specialists.
Conclusion: This process can serve as a template for future institutions to treat patients with COPD using MIH or similar hospital-at-home services.
Keywords: Mobile Integrated Health; MIH; COPD; population health.
It is estimated that chronic obstructive pulmonary disease (COPD) affects more than 16 million Americans1 and accounts for more than 700 000 hospitalizations each year in the US.2 Thirty-day COPD readmission rates hover around 22.6%,3 and readmission within 90 days of initial discharge can jump to between 31% and 35%.4 This is the highest of any patient demographic, and more than half of these readmissions are due to COPD. To counter this, government and state entities have made nationwide efforts to encourage health systems to focus on preventing readmissions. In October 2014, the US added COPD to the active list of diseases in Medicare’s Hospital Readmissions Reduction Program (HRRP), later adding COPD to various risk-based bundle programs that hospitals may choose to opt into. These programs are designed to reduce all-cause readmissions after an acute exacerbation of COPD, as the HRRP penalizes hospitals for all-cause 30-day readmissions.3 However, what is most troubling is that, despite these efforts, readmission rates have not dropped in the past decade.5 COPD remains the third leading cause of death in America and still poses a significant burden both clinically and economically to hospitals across the country.3
A solution that is gaining traction is to encourage outpatient care initiatives and discharge pathways. Early follow-up is proven to decrease chances of readmission, and studies have shown that more than half of readmitted patients did not follow up with a primary care physician (PCP) within 30 days of their initial discharge.6 Additionally, large meta-analyses show hospital-at-home–type programs can lead to reductions in mortality, decrease costs, decrease readmissions, and increase patient satisfaction.7-9 Therefore, for more challenging patient populations with regard to readmissions and mortality, Mobile Integrated Health (MIH) may be the solution that we are looking for.
This article presents a viable process to treat patients with COPD in an outpatient setting with MIH Services. It includes an examination of what makes MIH successful as well as a closer look at a structured COPD outpatient pathway.
Methods
South Shore Hospital (SSH) is an independent, not-for-profit hospital located in Weymouth, Massachusetts. It is host to 400 beds, 100 000 annual visits to the emergency department (ED), and its own emergency medical services program. In March 2020, SSH became the first Massachusetts hospital-based program to acquire an MIH license. MIH paramedics receive 300 hours of specialized training, including time in clinical clerkships shadowing pulmonary specialists, cardiology/congestive heart failure (CHF) providers, addiction medicine specialists, home care and care progression colleagues, and wound center providers. Specialist providers become more comfortable with paramedic capabilities as a result of these clerkships, improving interactions and relationships going forward. At the time of writing, SSH MIH is staffed by 12 paramedics, 4 of whom are full time; 2 medical directors; 2 internal coordinators; and 1 registered nurse (RN). A minimum of 2 paramedics are on call each day, each with twice-daily intravenous (IV) capabilities. The first shift slot is 16 hours, from 7:00 AM to 11:00
The goal of developing MIH is to improve upon the current standard of care. For hospitals without MIH capabilities, there are limited options to treat acute exacerbations of chronic obstructive pulmonary disease (AECOPD) patients postdischarge. It is common for the only outpatient referral to be a lone PCP visit, and many patients who need more extensive treatment options don’t have access to a timely PCP follow-up or resources for alternative care. This is part of why there has been little improvement in the 21st century with regard to reducing COPD hospitalizations. As it stands, approximately 10% to 55% of all AECOPD readmissions are preventable, and more than one-fifth of patients with COPD are rehospitalized within 30 days of discharge.3 In response, MIH has been designed to provide robust care options postdischarge in the patient home, with the eventual goal of reducing preventable hospitalizations and readmissions for all patients with COPD.
Patient selection
Patients with COPD are admitted to the MIH program in 1 of 3 ways: (1) directly from the ED; (2) at discharge from inpatient care; or (3) from a SSH affiliate referral.
With option 1, the ED physician assesses patient need for MIH services and places a referral to MIH in the electronic medical record (EMR). The ED provider also specifies whether follow-up is “urgent” and sets an alternative level of priority if not. With option 2, the inpatient provider and case manager follow a similar process, first determining whether a patient is stable enough to go home with outpatient services and then if MIH would be beneficial to the patient. If the patient is discharged home, a follow-up visit by an MIH paramedic is scheduled within 48 hours. With option 3, the patient is referred to MIH by an affiliate of SSH. This can be through the patient’s PCP, their visiting nurse association (VNA) service provider, or through any SSH urgent care center. In all 3 referral processes, the patient has the option to consent into the program or refuse services. Once referred, MIH coordinators review patients on a case-by-case basis. Patients with a history of prior admissions are given preference, with the goal being to keep the frailer, older, and comorbid patients at home. Other considerations include recent admission(s), length of stay, and overall stability. Social factors considered by the team include whether the patient lives alone and has alternative home services and the patient’s total distance from the hospital. Patients with a history of violence, mental health concerns, or substance abuse go through a more extensive screening process to ensure paramedic safety.
Given their patient profile and high hospital usage rates, MIH is sometimes requested for patients with end-stage COPD. Many of these patients benefit from MIH goals-of-care conversations to ensure they understand all their options and choose an approach that fits their preferences. In these cases, MIH has been instrumental in assisting patients and families with completing Medical Orders for Life-Sustaining Treatment and health care proxy forms and transitioning patients to palliative care, hospice, advanced-illness care management programs, or other long-term care options to prevent the need for rehospitalization. The MIH team focuses heavily on providing quality end-of-life care for patients and aligning care models with patient and family goals, often finding that having these sensitive conversations in the comfort of home enables transparency and comfort not otherwise experienced by hospitalized patients.
Initial patient follow-up
For patients with COPD enrolled in the MIH program, their first patient visit is scheduled within 48 hours of discharge from the ED or inpatient hospital. In many cases, this visit can be conducted within 24 hours of returning home. Once at the patient’s home, the paramedic begins with general introductions, vital signs, and a basic physical examination. The remainder of the visit focuses on patient education and symptom recognition. The paramedic reviews the COPD action plan (Figure 1), including how to recognize the onset of a “COPD flare-up” and the appropriate response. Patients are provided with a paper copy of the action plan for future reference.
The next point of educational emphasis is the patient’s individual medication regimen. This involves differentiating between control (daily) and rescue medications, how to use oxygen tanks, and how to safely wean off of oxygen. Specific attention is given to how to use a metered-dose inhaler, as studies have found that more than half of all patients use their inhaler devices incorrectly.10
Paramedics also complete a home safety evaluation of the patient’s residence, which involves checking for tripping hazards, lighting, handrails, slippery surfaces, and general access to patient medication. If an issue cannot be resolved by the paramedic on site and is considered a safety hazard, it is reported back to the hospital team for assistance.
Finally, patients are educated on the capabilities of MIH as a program and what to expect when they reach out over the phone. Patients are given a phone number to call for both “urgent” and “nonemergent” situations. In both cases, they will be greeted by one of the MIH coordinators or nurses who assist with triaging patient symptoms, scheduling a visit, or providing other guidance. It is a point of emphasis that the patient can use MIH for more than just COPD and should call in the event of any illness or discomfort (eg, dehydration, fever) in an effort to prevent unnecessary ED visits.
Medication reconciliation
Patients with COPD often have complex medication regimens. To help alleviate any confusion, medication reconciliations are done in conjunction with every COPD patient’s initial visit. During this process, the paramedic first takes an inventory of all medications in the patient home. Common reasons for nonadherence include confusing packaging, inability to reach the pharmacy, or medication not being covered by insurance. The paramedic reconciles the updated medication regimen against the medications that are physically in the home. Once the initial review is complete, the paramedic teleconferences with a registered hospitalist pharmacist (RHP) for a more in-depth review. Over video chat, the RHP reviews each medication individually to make sure the patient understands how many times per day they take each medication, whether it is a control or rescue medication, and what times of the day to take them. The RHP will then clarify any other medication questions the patient has, assure all recent medications have been picked up from the pharmacy, and determine any barriers, such as cost or transportation.
Follow-ups and PCP involvement
At each in-person visit, paramedics coordinate with an advanced practice clinician (APC) through telehealth communication. On these video calls with a provider, the paramedic relays relevant information pertaining to patient history, vital signs, and current status. Any concerning findings, symptoms of COPD flare-ups, or recent changes in status will be discussed. The APC then speaks directly to the patient to gather additional details about their condition and any recent hospitalizations, with their primary role being to make clinical decisions on further treatment. For the COPD population, this often includes orders for the MIH paramedic to administer IV medication (ie, IV methylprednisolone or other corticosteroids), antibiotics, home nebulizers, and at-home oxygen.
Second and third follow-up paramedic visits are often less intensive. Although these visits often still involve telehealth calls to the APC, the overall focus shifts toward medication adherence, ED avoidance, and readmission avoidance. On these visits, the paramedic also checks vitals, conducts a physical examination, and completes follow-up testing or orders per the APC.
PCP involvement is critical to streamlining and transitioning patient care. Patients who are admitted to MIH without insurance or a PCP are assisted in the process of finding one. PCPs automatically receive a patient enrollment letter when their patient is seen by an MIH paramedic. Following each individual visit, paramedic and APC notes are sent to the PCP through the EMR or via fax, at which time the PCP may be consulted on patient history and/or future care decisions. After the transition back to care by their PCP, patients are still encouraged to utilize MIH if acute changes arise. If a patient is readmitted back to the hospital, MIH is automatically notified, and coordinators will assess whether there is continued need for outpatient services or areas for potential improvement.
Emergent MIH visits
While MIH visits with patients with COPD are often scheduled, MIH can also be leveraged in urgent situations to prevent the need for a patient to come to the ED or hospital. Patients with COPD are told to call MIH if they have worsening symptoms or have exhausted all methods of self-treatment without an improvement in status. In this case, a paramedic is notified and sent to the patient’s home at the earliest time possible. The paramedic then completes an assessment of the patient’s status and relays information to the MIH APC or medical director. From there, treatment decisions, such as starting the patient on an IV, using nebulizers, or doing an electrocardiogram for diagnostic purposes, are guided by the provider team with the ultimate goal of caring for the patient in the home. For our population, providing urgent care in the home has proven to be an effective way to avoid unnecessary readmissions while still ensuring high-quality patient care.
Outpatient pathway
In May 2021, select patients with COPD were given the option to participate in a more intensive MIH outpatient pathway. Pilot patients were chosen by 2 pulmonary specialists, with a focus on enrolling patients with COPD at the highest risk for readmission. Patients who opted in were followed by MIH for a total of 30 days.
The first visit was made as usual within 48 hours of discharge. Patients received education, medication reconciliation, vitals examination, home safety evaluation, and a facilitated telehealth evaluation with the APC. What differentiates the pathway from standard MIH services is that after the first visit, the follow-ups are prescheduled and more numerous. This is outlined best in Figure 2, which serves as a guideline for coordinators and paramedics in the cadence and focus of visits for each patient on the pathway. The initial 2 weeks are designed to check in on the patient in person and ensure active recovery. The latter 2 weeks are designed to ensure that the patient follows up with their care team and understands their medications and action plan going forward. Pathway patients were also monitored using a remote patient monitoring (RPM) kit. On the initial visit, paramedics set up the RPM equipment and provided a demonstration on how to use each device. Patients were issued a Bluetooth-enabled scale, blood pressure cuff, video-enabled tablet, and wearable device. The wearable device continuously recorded respiration rate, heart rate, and oxygen saturation and had fall-detection enabled. Over the course of a month, an experienced MIH nurse monitored the vitals transmitted by the wearable device and checked patient weight and blood pressure 1 to 2 times per day, utilizing these data to proactively outreach to patients if abnormalities occurred. Prior to the start of the program, the MIH nurse contacted each patient to introduce herself and notify them that they would receive a call if any vitals were unusual.
Results
MIH treated 214 patients with COPD from March 2, 2020, to August 2, 2021. In total, paramedics made more than 650 visits. Eighty-seven of these were documented as urgent visits with AECOPD, shortness of breath, cough, or wheezing as the primary concern.
In the calendar year of 2019, our institution admitted 804 patients with a primary diagnosis of COPD. In 2020, the first year with MIH, total COPD admissions decreased to 473; however, the effect of the COVID-19 pandemic cannot be discounted. At of the time of writing—219 days into 2021—253 patients with COPD have been admitted thus far (Table 1).
Pathway results
Sixteen patients were referred to the MIH COPD Discharge Pathway Pilot during May 2021. Ten patients went on to complete the entire 30-day pathway. Six did not finish the program. Three of these 6 patients were referred by a pulmonary specialist for enrollment but not ultimately referred to the pilot program by case management and therefore not enrolled. The other 3 of the 6 patients who did not complete the pilot program were enrolled but discontinued owing to noncompliance.
Of the 10 patients who completed the pathway, 3 patients were male, and 7 were female. Ages ranged from 55 to 84 years. On average, the RHP found 3.6 medication reconciliation errors per patient. One patient was readmitted within 30 days (only 3 days after the initial discharge), and 5 were readmitted within 90 days.
A retrospective analysis was conducted on patients with COPD who were not provided with MIH services and were admitted to our hospital between September 1, 2020, and March 1, 2021, for comparison. Age, sex, and other related conditions are shown in Table 2. Medication reconciliation error data were not tracked for this demographic, as they did not have an in-home medication reconciliation completed.
Discussion
MIH has treated 214 patients with COPD from March 2, 2020, to August 2, 2021, a 17-month period. In that same timeframe, the hospital experienced a 42% decrease in COPD admissions. Although this effect is not the sole product of MIH (specifically, COVID-19 caused a drop in all-cause hospital admissions), we believe MIH did play a small role in this reduction. Eighty-seven emergent visits were conducted for patients with a primary complaint of AECOPD, shortness of breath, cough, or wheezing. On these visits, MIH provided urgent treatment to prevent the patient returning to the ED and potentially leading to readmission.
The program’s impact extends beyond the numbers. With more than 200 patients with COPD treated at home, we improved hospital flow, shortened patients’ overall length of stay, and increased capacity in the ED and inpatient units. In addition, MIH has been able to fill in care gaps present in the current health care system by providing acute care in the home to patients who otherwise have access-to-care and transportation issues.
What made the program successful
With the COPD population prone to having complex medication regimens, medication reconciliations were critical to improving patient outcomes. During the documented medication reconciliations for pathway patients, 8 of 10 patients had medication errors identified. Some of the more common errors included incorrect inhaler usage, patient medication not arriving to the pharmacy for a week or more after discharge, prescribed medication dosages that were too high or too low, and a lack of transportation to pick up the patient’s prescription. Even more problematic is that 7 of these 8 patients required multiple interventions to correct their regimen. What was cited as most beneficial by both the paramedic and the RHP was taking time to walk through each medication individually and ensuring that the patient could recite back how often and when they should be using it. What also proved to be helpful was spending extra time on the inhalers and nebulizers. Multiple patients did not know how to use them properly and/or cited a history of struggling with them.
The MIH COPD pathway patients showed encouraging preliminary results. In the initial 30-day window, only 1 of 10 (10%) patients was readmitted, which is lower than the 37.7% rate for comparable patients who did not have MIH services. This could imply that patients with COPD respond positively to active and consistent management with predetermined points of contact. Ninety-day readmission rates jumped to 5 of 10, with 4 of these patients being readmitted multiple times. Approximately half of these readmissions were COPD related. It is important to remember that the patients being targeted by the pathway are deemed to be at very high risk of readmission. As such, one could expect that even with a successful reduction in rates, pathway patient readmission rates may be slightly elevated compared with national COPD averages.
Given the more personalized and at-home care, patients also expressed higher levels of care satisfaction. Most patients want to avoid the hospital at all costs, and MIH provides a safe and effective alternative. Patients with COPD have also relayed that the education they receive on their medication, disease, and how to use MIH has been useful. This is reflected in the volume of urgent calls that MIH receives. A patient calling MIH in place of 911 shows not only that the patient has a level of trust in the MIH team, but also that they have learned how to recognize symptoms earlier to prevent major flare-ups.
This study had several limitations. On the pilot pathway, 3 patients were removed from MIH services because of repeated noncompliance. These instances primarily involved aggression toward the paramedics, both verbal and physical, as well as refusal to allow the MIH paramedics into the home. Going forward, it will be valuable to have a screening process for pathway patients to determine likelihood of compliance. This could include speaking to the patient’s PCP or other in-hospital providers before accepting them into the program.
Remote patient monitoring also presented its challenges. Despite extensive equipment demonstrations, some patients struggled to grasp the technology. Some of the biggest problems cited were confusion operating the tablet, inability to charge the devices, and issues with connectivity. In the future, it may be useful to simplify the devices even more. Further work should also be done to evaluate the efficacy of remote patient technology in this specific setting, as studies have shown varied results with regard to RPM success. In 1 meta-analysis of 91 different published studies that took place between 2015 and 2020, approximately half of the RPM studies resulted in no change in hospital readmissions, length of stay, or ED presentations, while the other half saw improvement in these categories.11 We suspect that the greatest benefits of our work came from the patient education, trust built over time, in-home urgent evaluations, and 1-on-1 time with the paramedic.
With many people forgoing care during the pandemic, COVID-19 has also caused a downward trend in overall and non-COVID-19 admissions. In a review of more than 500 000 ED visits in Massachusetts between March 11, 2020, and September 8, 2021, there was a 32% decrease in admissions when compared with those same weeks in 2019.10 There was an even greater drop-off when it came to COPD and other respiratory-related admissions. In evaluating the impact SSH MIH has made, it is important to recognize that the pandemic contributed to reducing total COPD admissions. Adding merit to the success of MIH in contributing to the reduction in admissions is the continued downward trend in total COPD admissions year-to-date in 2021. Despite total hospital usage rates increasing at our institution over the course of this year, the overall COPD usage rates have remained lower than before.
Another limitation is that in the selection of patients, both for general MIH care and for the COPD pathway, there was room for bias. The pilot pathway was offered specifically to patients at the highest risk for readmission; however, patients were referred at the discretion of our pulmonologist care team and not selected by any standardized rubric. Additionally, MIH only operates on a 16-hour schedule. This means that patients admitted to the ED or inpatient at night may sometimes be missed and not referred to MIH for care.
The biggest caveat to the pathway results is, of course, the small sample size. With only 10 patients completing the pilot, it is impossible to come to any concrete conclusions. Such an intensive pathway requires dedicating large amounts of time and resources, which is why the pilot was small. However, considering the preliminary results, the outline given could provide a starting point for future work to evaluate a similar COPD pathway on a larger scale.
Future considerations
Risk stratification of patients is critical to achieving even further reductions in readmissions and mortality. Hospitals can get the most value from MIH by focusing on patients with COPD at the highest risk for return, and it would be valuable to explicitly define who fits into this criterion. Utilizing a tool similar to the LACE index for readmission but tailoring it to patients with COPD when admitting patients into the program would be a logical next step.
Reducing the points of patient contact could also prove valuable. Over the course of a patient’s time with MIH, they interact with an RHP, APC, paramedic, RN, and discharging hospitalist. Additionally, we found many patients had VNA services, home health aides, care managers, and/or social workers involved in their care. Some patients found this to be stressful and overwhelming, especially regarding the number of outreach calls soon after discharge.
It would also be useful to look at the impact of MIH on total COPD admissions independent of the artificial variation created by COVID-19. This may require waiting until there are higher levels of vaccination and/or finding ways to control for the potential variation. In doing so, one could look at the direct effect MIH has on COPD readmissions when compared with a control group without MIH services, which could then serve as a comparison point to the results of this study. As it stands, given the relative novelty of MIH, there are primarily only broad reviews of MIH’s effectiveness and/or impact on patient populations that have been published. Of these, only a few directly mentioned MIH in relation to COPD, and none have comparable designs that look at overall COPD hospitalization reductions post-MIH implementation. There is also little to no literature looking at the utilization of MIH in a more intensive COPD outpatient pathway.
Finally, MIH has proven to be a useful tool for our institution in many areas outside of COPD management. Specifically, MIH has been utilized as a mobile influenza and COVID-19 vaccination unit and in-home testing service and now operates both a hospital-at-home and skilled nursing facility-at-home program. Analysis of the overall needs of the system and where this valuable MIH resource would have the biggest impact will be key in future growth opportunities.
Conclusion
MIH has been an invaluable tool for our hospital, especially in light of the recent shift toward more in-home and virtual care. MIH cared for 214 patients with COPD with more than 650 visits between March 2020 and August 2021. Eighty-seven emergent COPD visits were conducted, and COPD admissions were reduced dramatically from 2019 to 2020. MIH services have improved hospital flow, allowed for earlier discharge from the ED and inpatient care, and helped improve all-cause COPD readmission rates. The importance of postdischarge care and follow-up visits for patients with COPD, especially those at higher risk for readmission, cannot be understated. We hope our experience working to improve COPD patient outcomes serves as valuable a reference point for future MIH programs.
Corresponding author: Kelly Lannutti, DO, Mobile Integrated Health and Emergency Medicine Department, South Shore Health, 55 Fogg Rd, South Weymouth, MA 02190; [email protected].
Financial disclosures: None.
From the Mobile Integrated Health and Emergency Medicine Department, South Shore Health, Weymouth, MA.
Objective: To develop a process through which Mobile Integrated Health (MIH) can treat patients with chronic obstructive pulmonary disease (COPD) at high risk for readmission in an outpatient setting. In turn, South Shore Hospital (SSH) looks to leverage MIH to improve hospital flow, decrease costs, and improve patient quality of life.
Methods: With the recent approval of hospital-based MIH programs in Massachusetts, SSH used MIH to target specific patient demographics in an at-home setting. Here, we describe the planning and implementation of this program for patients with COPD. Key components to success include collaboration among providers, early follow-up visits, patient education, and in-depth medical reconciliations. Analysis includes a retrospective examination of a structured COPD outpatient pathway.
Results: A total of 214 patients with COPD were treated with MIH from March 2, 2020, to August 1, 2021. Eighty-seven emergent visits were conducted, and more than 650 total visits were made. A more intensive outpatient pathway was implemented for patients deemed to be at the highest risk for readmission by pulmonary specialists.
Conclusion: This process can serve as a template for future institutions to treat patients with COPD using MIH or similar hospital-at-home services.
Keywords: Mobile Integrated Health; MIH; COPD; population health.
It is estimated that chronic obstructive pulmonary disease (COPD) affects more than 16 million Americans1 and accounts for more than 700 000 hospitalizations each year in the US.2 Thirty-day COPD readmission rates hover around 22.6%,3 and readmission within 90 days of initial discharge can jump to between 31% and 35%.4 This is the highest of any patient demographic, and more than half of these readmissions are due to COPD. To counter this, government and state entities have made nationwide efforts to encourage health systems to focus on preventing readmissions. In October 2014, the US added COPD to the active list of diseases in Medicare’s Hospital Readmissions Reduction Program (HRRP), later adding COPD to various risk-based bundle programs that hospitals may choose to opt into. These programs are designed to reduce all-cause readmissions after an acute exacerbation of COPD, as the HRRP penalizes hospitals for all-cause 30-day readmissions.3 However, what is most troubling is that, despite these efforts, readmission rates have not dropped in the past decade.5 COPD remains the third leading cause of death in America and still poses a significant burden both clinically and economically to hospitals across the country.3
A solution that is gaining traction is to encourage outpatient care initiatives and discharge pathways. Early follow-up is proven to decrease chances of readmission, and studies have shown that more than half of readmitted patients did not follow up with a primary care physician (PCP) within 30 days of their initial discharge.6 Additionally, large meta-analyses show hospital-at-home–type programs can lead to reductions in mortality, decrease costs, decrease readmissions, and increase patient satisfaction.7-9 Therefore, for more challenging patient populations with regard to readmissions and mortality, Mobile Integrated Health (MIH) may be the solution that we are looking for.
This article presents a viable process to treat patients with COPD in an outpatient setting with MIH Services. It includes an examination of what makes MIH successful as well as a closer look at a structured COPD outpatient pathway.
Methods
South Shore Hospital (SSH) is an independent, not-for-profit hospital located in Weymouth, Massachusetts. It is host to 400 beds, 100 000 annual visits to the emergency department (ED), and its own emergency medical services program. In March 2020, SSH became the first Massachusetts hospital-based program to acquire an MIH license. MIH paramedics receive 300 hours of specialized training, including time in clinical clerkships shadowing pulmonary specialists, cardiology/congestive heart failure (CHF) providers, addiction medicine specialists, home care and care progression colleagues, and wound center providers. Specialist providers become more comfortable with paramedic capabilities as a result of these clerkships, improving interactions and relationships going forward. At the time of writing, SSH MIH is staffed by 12 paramedics, 4 of whom are full time; 2 medical directors; 2 internal coordinators; and 1 registered nurse (RN). A minimum of 2 paramedics are on call each day, each with twice-daily intravenous (IV) capabilities. The first shift slot is 16 hours, from 7:00 AM to 11:00
The goal of developing MIH is to improve upon the current standard of care. For hospitals without MIH capabilities, there are limited options to treat acute exacerbations of chronic obstructive pulmonary disease (AECOPD) patients postdischarge. It is common for the only outpatient referral to be a lone PCP visit, and many patients who need more extensive treatment options don’t have access to a timely PCP follow-up or resources for alternative care. This is part of why there has been little improvement in the 21st century with regard to reducing COPD hospitalizations. As it stands, approximately 10% to 55% of all AECOPD readmissions are preventable, and more than one-fifth of patients with COPD are rehospitalized within 30 days of discharge.3 In response, MIH has been designed to provide robust care options postdischarge in the patient home, with the eventual goal of reducing preventable hospitalizations and readmissions for all patients with COPD.
Patient selection
Patients with COPD are admitted to the MIH program in 1 of 3 ways: (1) directly from the ED; (2) at discharge from inpatient care; or (3) from a SSH affiliate referral.
With option 1, the ED physician assesses patient need for MIH services and places a referral to MIH in the electronic medical record (EMR). The ED provider also specifies whether follow-up is “urgent” and sets an alternative level of priority if not. With option 2, the inpatient provider and case manager follow a similar process, first determining whether a patient is stable enough to go home with outpatient services and then if MIH would be beneficial to the patient. If the patient is discharged home, a follow-up visit by an MIH paramedic is scheduled within 48 hours. With option 3, the patient is referred to MIH by an affiliate of SSH. This can be through the patient’s PCP, their visiting nurse association (VNA) service provider, or through any SSH urgent care center. In all 3 referral processes, the patient has the option to consent into the program or refuse services. Once referred, MIH coordinators review patients on a case-by-case basis. Patients with a history of prior admissions are given preference, with the goal being to keep the frailer, older, and comorbid patients at home. Other considerations include recent admission(s), length of stay, and overall stability. Social factors considered by the team include whether the patient lives alone and has alternative home services and the patient’s total distance from the hospital. Patients with a history of violence, mental health concerns, or substance abuse go through a more extensive screening process to ensure paramedic safety.
Given their patient profile and high hospital usage rates, MIH is sometimes requested for patients with end-stage COPD. Many of these patients benefit from MIH goals-of-care conversations to ensure they understand all their options and choose an approach that fits their preferences. In these cases, MIH has been instrumental in assisting patients and families with completing Medical Orders for Life-Sustaining Treatment and health care proxy forms and transitioning patients to palliative care, hospice, advanced-illness care management programs, or other long-term care options to prevent the need for rehospitalization. The MIH team focuses heavily on providing quality end-of-life care for patients and aligning care models with patient and family goals, often finding that having these sensitive conversations in the comfort of home enables transparency and comfort not otherwise experienced by hospitalized patients.
Initial patient follow-up
For patients with COPD enrolled in the MIH program, their first patient visit is scheduled within 48 hours of discharge from the ED or inpatient hospital. In many cases, this visit can be conducted within 24 hours of returning home. Once at the patient’s home, the paramedic begins with general introductions, vital signs, and a basic physical examination. The remainder of the visit focuses on patient education and symptom recognition. The paramedic reviews the COPD action plan (Figure 1), including how to recognize the onset of a “COPD flare-up” and the appropriate response. Patients are provided with a paper copy of the action plan for future reference.
The next point of educational emphasis is the patient’s individual medication regimen. This involves differentiating between control (daily) and rescue medications, how to use oxygen tanks, and how to safely wean off of oxygen. Specific attention is given to how to use a metered-dose inhaler, as studies have found that more than half of all patients use their inhaler devices incorrectly.10
Paramedics also complete a home safety evaluation of the patient’s residence, which involves checking for tripping hazards, lighting, handrails, slippery surfaces, and general access to patient medication. If an issue cannot be resolved by the paramedic on site and is considered a safety hazard, it is reported back to the hospital team for assistance.
Finally, patients are educated on the capabilities of MIH as a program and what to expect when they reach out over the phone. Patients are given a phone number to call for both “urgent” and “nonemergent” situations. In both cases, they will be greeted by one of the MIH coordinators or nurses who assist with triaging patient symptoms, scheduling a visit, or providing other guidance. It is a point of emphasis that the patient can use MIH for more than just COPD and should call in the event of any illness or discomfort (eg, dehydration, fever) in an effort to prevent unnecessary ED visits.
Medication reconciliation
Patients with COPD often have complex medication regimens. To help alleviate any confusion, medication reconciliations are done in conjunction with every COPD patient’s initial visit. During this process, the paramedic first takes an inventory of all medications in the patient home. Common reasons for nonadherence include confusing packaging, inability to reach the pharmacy, or medication not being covered by insurance. The paramedic reconciles the updated medication regimen against the medications that are physically in the home. Once the initial review is complete, the paramedic teleconferences with a registered hospitalist pharmacist (RHP) for a more in-depth review. Over video chat, the RHP reviews each medication individually to make sure the patient understands how many times per day they take each medication, whether it is a control or rescue medication, and what times of the day to take them. The RHP will then clarify any other medication questions the patient has, assure all recent medications have been picked up from the pharmacy, and determine any barriers, such as cost or transportation.
Follow-ups and PCP involvement
At each in-person visit, paramedics coordinate with an advanced practice clinician (APC) through telehealth communication. On these video calls with a provider, the paramedic relays relevant information pertaining to patient history, vital signs, and current status. Any concerning findings, symptoms of COPD flare-ups, or recent changes in status will be discussed. The APC then speaks directly to the patient to gather additional details about their condition and any recent hospitalizations, with their primary role being to make clinical decisions on further treatment. For the COPD population, this often includes orders for the MIH paramedic to administer IV medication (ie, IV methylprednisolone or other corticosteroids), antibiotics, home nebulizers, and at-home oxygen.
Second and third follow-up paramedic visits are often less intensive. Although these visits often still involve telehealth calls to the APC, the overall focus shifts toward medication adherence, ED avoidance, and readmission avoidance. On these visits, the paramedic also checks vitals, conducts a physical examination, and completes follow-up testing or orders per the APC.
PCP involvement is critical to streamlining and transitioning patient care. Patients who are admitted to MIH without insurance or a PCP are assisted in the process of finding one. PCPs automatically receive a patient enrollment letter when their patient is seen by an MIH paramedic. Following each individual visit, paramedic and APC notes are sent to the PCP through the EMR or via fax, at which time the PCP may be consulted on patient history and/or future care decisions. After the transition back to care by their PCP, patients are still encouraged to utilize MIH if acute changes arise. If a patient is readmitted back to the hospital, MIH is automatically notified, and coordinators will assess whether there is continued need for outpatient services or areas for potential improvement.
Emergent MIH visits
While MIH visits with patients with COPD are often scheduled, MIH can also be leveraged in urgent situations to prevent the need for a patient to come to the ED or hospital. Patients with COPD are told to call MIH if they have worsening symptoms or have exhausted all methods of self-treatment without an improvement in status. In this case, a paramedic is notified and sent to the patient’s home at the earliest time possible. The paramedic then completes an assessment of the patient’s status and relays information to the MIH APC or medical director. From there, treatment decisions, such as starting the patient on an IV, using nebulizers, or doing an electrocardiogram for diagnostic purposes, are guided by the provider team with the ultimate goal of caring for the patient in the home. For our population, providing urgent care in the home has proven to be an effective way to avoid unnecessary readmissions while still ensuring high-quality patient care.
Outpatient pathway
In May 2021, select patients with COPD were given the option to participate in a more intensive MIH outpatient pathway. Pilot patients were chosen by 2 pulmonary specialists, with a focus on enrolling patients with COPD at the highest risk for readmission. Patients who opted in were followed by MIH for a total of 30 days.
The first visit was made as usual within 48 hours of discharge. Patients received education, medication reconciliation, vitals examination, home safety evaluation, and a facilitated telehealth evaluation with the APC. What differentiates the pathway from standard MIH services is that after the first visit, the follow-ups are prescheduled and more numerous. This is outlined best in Figure 2, which serves as a guideline for coordinators and paramedics in the cadence and focus of visits for each patient on the pathway. The initial 2 weeks are designed to check in on the patient in person and ensure active recovery. The latter 2 weeks are designed to ensure that the patient follows up with their care team and understands their medications and action plan going forward. Pathway patients were also monitored using a remote patient monitoring (RPM) kit. On the initial visit, paramedics set up the RPM equipment and provided a demonstration on how to use each device. Patients were issued a Bluetooth-enabled scale, blood pressure cuff, video-enabled tablet, and wearable device. The wearable device continuously recorded respiration rate, heart rate, and oxygen saturation and had fall-detection enabled. Over the course of a month, an experienced MIH nurse monitored the vitals transmitted by the wearable device and checked patient weight and blood pressure 1 to 2 times per day, utilizing these data to proactively outreach to patients if abnormalities occurred. Prior to the start of the program, the MIH nurse contacted each patient to introduce herself and notify them that they would receive a call if any vitals were unusual.
Results
MIH treated 214 patients with COPD from March 2, 2020, to August 2, 2021. In total, paramedics made more than 650 visits. Eighty-seven of these were documented as urgent visits with AECOPD, shortness of breath, cough, or wheezing as the primary concern.
In the calendar year of 2019, our institution admitted 804 patients with a primary diagnosis of COPD. In 2020, the first year with MIH, total COPD admissions decreased to 473; however, the effect of the COVID-19 pandemic cannot be discounted. At of the time of writing—219 days into 2021—253 patients with COPD have been admitted thus far (Table 1).
Pathway results
Sixteen patients were referred to the MIH COPD Discharge Pathway Pilot during May 2021. Ten patients went on to complete the entire 30-day pathway. Six did not finish the program. Three of these 6 patients were referred by a pulmonary specialist for enrollment but not ultimately referred to the pilot program by case management and therefore not enrolled. The other 3 of the 6 patients who did not complete the pilot program were enrolled but discontinued owing to noncompliance.
Of the 10 patients who completed the pathway, 3 patients were male, and 7 were female. Ages ranged from 55 to 84 years. On average, the RHP found 3.6 medication reconciliation errors per patient. One patient was readmitted within 30 days (only 3 days after the initial discharge), and 5 were readmitted within 90 days.
A retrospective analysis was conducted on patients with COPD who were not provided with MIH services and were admitted to our hospital between September 1, 2020, and March 1, 2021, for comparison. Age, sex, and other related conditions are shown in Table 2. Medication reconciliation error data were not tracked for this demographic, as they did not have an in-home medication reconciliation completed.
Discussion
MIH has treated 214 patients with COPD from March 2, 2020, to August 2, 2021, a 17-month period. In that same timeframe, the hospital experienced a 42% decrease in COPD admissions. Although this effect is not the sole product of MIH (specifically, COVID-19 caused a drop in all-cause hospital admissions), we believe MIH did play a small role in this reduction. Eighty-seven emergent visits were conducted for patients with a primary complaint of AECOPD, shortness of breath, cough, or wheezing. On these visits, MIH provided urgent treatment to prevent the patient returning to the ED and potentially leading to readmission.
The program’s impact extends beyond the numbers. With more than 200 patients with COPD treated at home, we improved hospital flow, shortened patients’ overall length of stay, and increased capacity in the ED and inpatient units. In addition, MIH has been able to fill in care gaps present in the current health care system by providing acute care in the home to patients who otherwise have access-to-care and transportation issues.
What made the program successful
With the COPD population prone to having complex medication regimens, medication reconciliations were critical to improving patient outcomes. During the documented medication reconciliations for pathway patients, 8 of 10 patients had medication errors identified. Some of the more common errors included incorrect inhaler usage, patient medication not arriving to the pharmacy for a week or more after discharge, prescribed medication dosages that were too high or too low, and a lack of transportation to pick up the patient’s prescription. Even more problematic is that 7 of these 8 patients required multiple interventions to correct their regimen. What was cited as most beneficial by both the paramedic and the RHP was taking time to walk through each medication individually and ensuring that the patient could recite back how often and when they should be using it. What also proved to be helpful was spending extra time on the inhalers and nebulizers. Multiple patients did not know how to use them properly and/or cited a history of struggling with them.
The MIH COPD pathway patients showed encouraging preliminary results. In the initial 30-day window, only 1 of 10 (10%) patients was readmitted, which is lower than the 37.7% rate for comparable patients who did not have MIH services. This could imply that patients with COPD respond positively to active and consistent management with predetermined points of contact. Ninety-day readmission rates jumped to 5 of 10, with 4 of these patients being readmitted multiple times. Approximately half of these readmissions were COPD related. It is important to remember that the patients being targeted by the pathway are deemed to be at very high risk of readmission. As such, one could expect that even with a successful reduction in rates, pathway patient readmission rates may be slightly elevated compared with national COPD averages.
Given the more personalized and at-home care, patients also expressed higher levels of care satisfaction. Most patients want to avoid the hospital at all costs, and MIH provides a safe and effective alternative. Patients with COPD have also relayed that the education they receive on their medication, disease, and how to use MIH has been useful. This is reflected in the volume of urgent calls that MIH receives. A patient calling MIH in place of 911 shows not only that the patient has a level of trust in the MIH team, but also that they have learned how to recognize symptoms earlier to prevent major flare-ups.
This study had several limitations. On the pilot pathway, 3 patients were removed from MIH services because of repeated noncompliance. These instances primarily involved aggression toward the paramedics, both verbal and physical, as well as refusal to allow the MIH paramedics into the home. Going forward, it will be valuable to have a screening process for pathway patients to determine likelihood of compliance. This could include speaking to the patient’s PCP or other in-hospital providers before accepting them into the program.
Remote patient monitoring also presented its challenges. Despite extensive equipment demonstrations, some patients struggled to grasp the technology. Some of the biggest problems cited were confusion operating the tablet, inability to charge the devices, and issues with connectivity. In the future, it may be useful to simplify the devices even more. Further work should also be done to evaluate the efficacy of remote patient technology in this specific setting, as studies have shown varied results with regard to RPM success. In 1 meta-analysis of 91 different published studies that took place between 2015 and 2020, approximately half of the RPM studies resulted in no change in hospital readmissions, length of stay, or ED presentations, while the other half saw improvement in these categories.11 We suspect that the greatest benefits of our work came from the patient education, trust built over time, in-home urgent evaluations, and 1-on-1 time with the paramedic.
With many people forgoing care during the pandemic, COVID-19 has also caused a downward trend in overall and non-COVID-19 admissions. In a review of more than 500 000 ED visits in Massachusetts between March 11, 2020, and September 8, 2021, there was a 32% decrease in admissions when compared with those same weeks in 2019.10 There was an even greater drop-off when it came to COPD and other respiratory-related admissions. In evaluating the impact SSH MIH has made, it is important to recognize that the pandemic contributed to reducing total COPD admissions. Adding merit to the success of MIH in contributing to the reduction in admissions is the continued downward trend in total COPD admissions year-to-date in 2021. Despite total hospital usage rates increasing at our institution over the course of this year, the overall COPD usage rates have remained lower than before.
Another limitation is that in the selection of patients, both for general MIH care and for the COPD pathway, there was room for bias. The pilot pathway was offered specifically to patients at the highest risk for readmission; however, patients were referred at the discretion of our pulmonologist care team and not selected by any standardized rubric. Additionally, MIH only operates on a 16-hour schedule. This means that patients admitted to the ED or inpatient at night may sometimes be missed and not referred to MIH for care.
The biggest caveat to the pathway results is, of course, the small sample size. With only 10 patients completing the pilot, it is impossible to come to any concrete conclusions. Such an intensive pathway requires dedicating large amounts of time and resources, which is why the pilot was small. However, considering the preliminary results, the outline given could provide a starting point for future work to evaluate a similar COPD pathway on a larger scale.
Future considerations
Risk stratification of patients is critical to achieving even further reductions in readmissions and mortality. Hospitals can get the most value from MIH by focusing on patients with COPD at the highest risk for return, and it would be valuable to explicitly define who fits into this criterion. Utilizing a tool similar to the LACE index for readmission but tailoring it to patients with COPD when admitting patients into the program would be a logical next step.
Reducing the points of patient contact could also prove valuable. Over the course of a patient’s time with MIH, they interact with an RHP, APC, paramedic, RN, and discharging hospitalist. Additionally, we found many patients had VNA services, home health aides, care managers, and/or social workers involved in their care. Some patients found this to be stressful and overwhelming, especially regarding the number of outreach calls soon after discharge.
It would also be useful to look at the impact of MIH on total COPD admissions independent of the artificial variation created by COVID-19. This may require waiting until there are higher levels of vaccination and/or finding ways to control for the potential variation. In doing so, one could look at the direct effect MIH has on COPD readmissions when compared with a control group without MIH services, which could then serve as a comparison point to the results of this study. As it stands, given the relative novelty of MIH, there are primarily only broad reviews of MIH’s effectiveness and/or impact on patient populations that have been published. Of these, only a few directly mentioned MIH in relation to COPD, and none have comparable designs that look at overall COPD hospitalization reductions post-MIH implementation. There is also little to no literature looking at the utilization of MIH in a more intensive COPD outpatient pathway.
Finally, MIH has proven to be a useful tool for our institution in many areas outside of COPD management. Specifically, MIH has been utilized as a mobile influenza and COVID-19 vaccination unit and in-home testing service and now operates both a hospital-at-home and skilled nursing facility-at-home program. Analysis of the overall needs of the system and where this valuable MIH resource would have the biggest impact will be key in future growth opportunities.
Conclusion
MIH has been an invaluable tool for our hospital, especially in light of the recent shift toward more in-home and virtual care. MIH cared for 214 patients with COPD with more than 650 visits between March 2020 and August 2021. Eighty-seven emergent COPD visits were conducted, and COPD admissions were reduced dramatically from 2019 to 2020. MIH services have improved hospital flow, allowed for earlier discharge from the ED and inpatient care, and helped improve all-cause COPD readmission rates. The importance of postdischarge care and follow-up visits for patients with COPD, especially those at higher risk for readmission, cannot be understated. We hope our experience working to improve COPD patient outcomes serves as valuable a reference point for future MIH programs.
Corresponding author: Kelly Lannutti, DO, Mobile Integrated Health and Emergency Medicine Department, South Shore Health, 55 Fogg Rd, South Weymouth, MA 02190; [email protected].
Financial disclosures: None.
1. Centers for Disease Control and Prevention. Chronic obstructive pulmonary disease (COPD). Accessed September 10, 2011. https://www.cdc.gov/copd/index.html
2. Wier LM, Elixhauser A, Pfuntner A, AuDH. Overview of Hospitalizations among Patients with COPD, 2008. Statistical Brief #106. In: Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Agency for Healthcare Research and Quality; 2011.
3. Shah T, Press,VG, Huisingh-Scheetz M, White SR. COPD Readmissions: Addressing COPD in the Era of Value-Based Health Care. Chest. 2016;150(4):916-926. doi:10.1016/j.chest.2016.05.002
4. Harries TH, Thornton H, Crichton S, et al. Hospital readmissions for COPD: a retrospective longitudinal study. NPJ Prim Care Respir Med. 2017;27(1):31. doi:10.1038/s41533-017-0028-8
5. Ford ES. Hospital discharges, readmissions, and ED visits for COPD or bronchiectasis among US adults: findings from the nationwide inpatient sample 2001-2012 and Nationwide Emergency Department Sample 2006-2011. Chest. 2015;147(4):989-998. doi:10.1378/chest.14-2146
6. Jencks SF, Williams MV, Coleman EA. Rehospitalizations among patients in the Medicare fee-for-service program. N Engl J Med. 2009;360(14):1418-1428. doi:10.1056/NEJMsa0803563
7. Shepperd S, Doll H, Angus RM, et al. Avoiding hospital admission through provision of hospital care at home: a systematic review and meta-analysis of individual patient data. CMAJ. 2009;180(2):175-182. doi:10.1503/cmaj.081491
8. Caplan GA, Sulaiman NS, Mangin DA, et al. A meta-analysis of “hospital in the home.” Med J Aust. 2012;197(9):512-519. doi:10.5694/mja12.10480
9. Portillo EC, Wilcox A, Seckel E, et al. Reducing COPD readmission rates: using a COPD care service during care transitions. Fed Pract. 2018;35(11):30-36.
10. Nourazari S, Davis SR, Granovsky R, et al. Decreased hospital admissions through emergency departments during the COVID-19 pandemic. Am J Emerg Med. 2021;42:203-210. doi:10.1016/j.ajem.2020.11.029
11. Taylor ML, Thomas EE, Snoswell CL, et al. Does remote patient monitoring reduce acute care use? A systematic review. BMJ Open. 2021;11(3):e040232. doi:10.1136/bmj/open-2020-040232
1. Centers for Disease Control and Prevention. Chronic obstructive pulmonary disease (COPD). Accessed September 10, 2011. https://www.cdc.gov/copd/index.html
2. Wier LM, Elixhauser A, Pfuntner A, AuDH. Overview of Hospitalizations among Patients with COPD, 2008. Statistical Brief #106. In: Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Agency for Healthcare Research and Quality; 2011.
3. Shah T, Press,VG, Huisingh-Scheetz M, White SR. COPD Readmissions: Addressing COPD in the Era of Value-Based Health Care. Chest. 2016;150(4):916-926. doi:10.1016/j.chest.2016.05.002
4. Harries TH, Thornton H, Crichton S, et al. Hospital readmissions for COPD: a retrospective longitudinal study. NPJ Prim Care Respir Med. 2017;27(1):31. doi:10.1038/s41533-017-0028-8
5. Ford ES. Hospital discharges, readmissions, and ED visits for COPD or bronchiectasis among US adults: findings from the nationwide inpatient sample 2001-2012 and Nationwide Emergency Department Sample 2006-2011. Chest. 2015;147(4):989-998. doi:10.1378/chest.14-2146
6. Jencks SF, Williams MV, Coleman EA. Rehospitalizations among patients in the Medicare fee-for-service program. N Engl J Med. 2009;360(14):1418-1428. doi:10.1056/NEJMsa0803563
7. Shepperd S, Doll H, Angus RM, et al. Avoiding hospital admission through provision of hospital care at home: a systematic review and meta-analysis of individual patient data. CMAJ. 2009;180(2):175-182. doi:10.1503/cmaj.081491
8. Caplan GA, Sulaiman NS, Mangin DA, et al. A meta-analysis of “hospital in the home.” Med J Aust. 2012;197(9):512-519. doi:10.5694/mja12.10480
9. Portillo EC, Wilcox A, Seckel E, et al. Reducing COPD readmission rates: using a COPD care service during care transitions. Fed Pract. 2018;35(11):30-36.
10. Nourazari S, Davis SR, Granovsky R, et al. Decreased hospital admissions through emergency departments during the COVID-19 pandemic. Am J Emerg Med. 2021;42:203-210. doi:10.1016/j.ajem.2020.11.029
11. Taylor ML, Thomas EE, Snoswell CL, et al. Does remote patient monitoring reduce acute care use? A systematic review. BMJ Open. 2021;11(3):e040232. doi:10.1136/bmj/open-2020-040232
Improving Physicians’ Bowel Documentation on Geriatric Wards
From Sheffield Teaching Hospitals, Sheffield, UK, S5 7AU.
Objective: Constipation is widely prevalent in older adults and may result in complications such as urinary retention, delirium, and bowel obstruction. Previous studies have indicated that while the nursing staff do well in completing stool charts, doctors monitor them infrequently. This project aimed to improve the documentation of bowel movement by doctors on ward rounds to 85%, by the end of a 3-month period.
Methods: Baseline, postintervention, and sustainability data were collected from inpatient notes on weekdays on a geriatric ward in Northern General Hospital, Sheffield, UK. Posters and stickers of the poo emoji were placed on walls and in inpatient notes, respectively, as a reminder.
Results: Data on bowel activity documentation were collected from 28 patients. The baseline data showed that bowel activity was monitored daily on the ward 60.49% of the time. However, following the interventions, there was a significant increase in documentation, to 86.78%. The sustainability study showed that bowel activity was documented on the ward 56.56% of the time.
Conclusion: This study shows how a strong initial effect on behavioral change can be accomplished through simple interventions such as stickers and posters. As most wards currently still use paper notes, this is a generalizable model that other wards can trial. However, this study also shows the difficulty in maintaining behavioral change over extended periods of time.
Keywords: bowel movement; documentation; obstruction; constipation; geriatrics; incontinence; junior doctor; quality improvement.
Constipation is widely prevalent in the elderly, encountered frequently in both community and hospital medicine.1 Its estimated prevalence in adults over 84 years old is 34% for women and 25% for men, rising to up to 80% for long-term care residents.2
Chronic constipation is generally characterized by unsatisfactory defecation due to infrequent bowel emptying or difficulty with stool passage, which may lead to incomplete evacuation.2-4 Constipation in the elderly, in addition to causing abdominal pain, nausea, and reduced appetite, may result in complications such as fecal incontinence (and overflow diarrhea), urinary retention, delirium, and bowel obstruction, which may in result in life-threatening perforation.5,6 For inpatients on geriatric wards, these consequences may increase morbidity and mortality, while prolonging hospital stays, thereby also increasing exposure to hospital-acquired infections.7 Furthermore, constipation is also associated with impaired health-related quality of life.8
Management includes treating the cause, stopping contributing medications, early mobilization, diet modification, and, if all else fails, prescription laxatives. Therefore, early identification and appropriate treatment of constipation is beneficial in inpatient care, as well as when planning safe and patient-centered discharges.
Given the risks and complications of constipation in the elderly, we, a group of Foundation Year 2 (FY2) doctors in the UK Foundation Programme, decided to explore how doctors can help to recognize this condition early. Regular bowel movement documentation in patient notes on ward rounds is crucial, as it has been shown to reduce constipation-associated complications.5 However, complications from constipation can take significant amounts of time to develop and, therefore, documenting bowel movements on a daily basis is not necessary.
Based on these observations along with targets set out in previous studies,7 our aim was to improve documentation of bowel movement on ward rounds to 85% by March 2020.
Methods
Before the data collection process, a fishbone diagram was designed to identify the potential causes of poor documentation of bowel movement on geriatric wards. There were several aspects that were reviewed, including, for example, patients, health care professionals, organizational policies, procedures, and equipment. It was then decided to focus on raising awareness of the documentation of bowel movement by doctors specifically.
Retrospective data were collected from the inpatient paper notes of 28 patients on Brearley 6, a geriatric ward at the Northern General Hospital within Sheffield Teaching Hospitals (STH), on weekdays over a 3-week period. The baseline data collected included the bed number of the patient, whether or not bowel movement on initial ward round was documented, and whether it was the junior, registrar, or consultant leading the ward round. End-of-life and discharged patients were excluded (Table).
The interventions consisted of posters and stickers. Posters were displayed on Brearley 6, including the doctors’ office, nurses’ station, and around the bays where notes were kept, in order to emphasize their importance. The stickers of the poo emoji were also printed and placed at the front of each set of inpatient paper notes as a reminder for the doctor documenting on the ward round. The interventions were also introduced in the morning board meeting to ensure all staff on Brearley 6 were aware of them.
Data were collected on weekdays over a 3-week period starting 2 weeks after the interventions were put in place (Table). In order to assess that the intervention had been sustained, data were again collected 1 month later over a 2-week period (Table). Microsoft Excel (Microsoft Corporation, Redmond, Washington, USA) was used to analyze all data, and control charts were used to assess variability in the data.
Results
The baseline data showed that bowel movement was documented 60.49% of the time by doctors on the initial ward round before intervention, as illustrated in Figure 1. There was no evidence of an out-of-control process in this baseline data set.
The comparison between the preintervention and postintervention data is illustrated in Figure 1. The postintervention data, which were taken 2 weeks after intervention, showed a significant increase in the documentation of bowel movements, to 86.78%. The figure displays a number of features consistent with an out-of-control process: beyond limits (≥ 1 points beyond control limits), Zone A rule (2 out of 3 consecutive points beyond 2 standard deviations from the mean), Zone B rule (4 out of 5 consecutive points beyond 1 standard deviation from the mean), and Zone C rule (≥ 8 consecutive points on 1 side of the mean). These findings demonstrate a special cause variation in the documentation of bowel movements.
Figure 2 shows the sustainability of the intervention, which averaged 56.56% postintervention nearly 2 months later. The data returned to preintervention variability levels.
Discussion
Our project explored an important issue that was frequently encountered by department clinicians. Our team of FY2 doctors, in general, had little experience with quality improvement. We have developed our understanding and experience through planning, making, and measuring improvement.
It was challenging deciding on how to deal with the problem. A number of ways were considered to improve the paper rounding chart, but the nursing team had already planned to make changes to it. Bowel activity is mainly documented by nursing staff, but there was no specific protocol for recognizing constipation and when to inform the medical team. We decided to focus on doctors’ documentation in patient notes during the ward round, as this is where the decision regarding management of bowels is made, including interventions that could only be done by doctors, such as prescribing laxatives.
Strom et al9 have described a number of successful quality improvement interventions, and we decided to follow the authors’ guidance to implement a reminder system strategy using both posters and stickers to prompt doctors to document bowel activity. Both of these were simple, and the text on the poster was concise. The only cost incurred on the project was from printing the stickers; this totalled £2.99 (US $4.13). Individual stickers for each ward round entry were considered but not used, as it would create an additional task for doctors.
The data initially indicated that the interventions had their desired effect. However, this positive change was unsustainable, most likely suggesting that the novelty of the stickers and posters wore off at some point, leading to junior doctors no longer noticing them. Further Plan-Do-Study-Act cycles should examine the reasons why the change is difficult to sustain and implement new policies that aim to overcome them.
There were a number of limitations to this study. A patient could be discharged before data collection, which was done twice weekly. This could have resulted in missed data during the collection period. In addition, the accuracy of the documentation is dependent on nursing staff correctly recording—as well as the doctors correctly viewing—all sources of information on bowel activity. Observer bias is possible, too, as a steering group member was involved in data collection. Their awareness of the project could cause a positive skew in the data. And, unfortunately, the project came to an abrupt end because of COVID-19 cases on the ward.
We examined the daily documentation of bowel activity, which may not be necessary considering that internationally recognized constipation classifications, such as the Rome III criteria, define constipation as fewer than 3 bowel movements per week.10 However, the data collection sheet did not include patient identifiers, so it was impossible to determine whether bowel activity had been documented 3 or more times per week for each patient. This is important because a clinician may only decide to act if there is no bowel movement activity for 3 or more days.
Because our data were collected on a single geriatric ward, which had an emphasis on Parkinson’s disease, it is unclear whether our findings are generalizable to other clinical areas in STH. However, constipation is common in the elderly, so it is likely to be relevant to other wards, as more than a third of STH hospital beds are occupied by patients aged 75 years and older.11
Conclusion
Overall, our study highlights the fact that monitoring bowel activity is important on a geriatric ward. Recognizing constipation early prevents complications and delays to discharge. As mentioned earlier, our aim was achieved initially but not sustained. Therefore, future development should focus on sustainability. For example, laxative-focused ward rounds have shown to be effective at recognizing and preventing constipation by intervening early.12 Future cycles that we considered included using an electronic reminder on the hospital IT system, as the trust is aiming to introduce electronic documentation. Focus could also be placed on improving documentation in bowel charts by ward staff. This could be achieved by organizing regular educational sessions on the complications of constipation and when to inform the medical team regarding concerns.
Acknowledgments: The authors thank Dr. Jamie Kapur, Sheffield Teaching Hospitals, for his guidance and supervision, as well as our collaborators: Rachel Hallam, Claire Walker, Monisha Chakravorty, and Hamza Khan.
Corresponding author: Alexander P. Noar, BMBCh, BA, 10 Stanhope Gardens, London, N6 5TS; [email protected].
Financial disclosures: None.
1. Forootan M, Bagheri N, Darvishi M. Chronic constipation: A review of literature. Medicine (Baltimore). 2018;97:e10631. doi:10.1097/MD.00000000000.10631
2. Schuster BG, Kosar L, Kamrul R. Constipation in older adults: stepwise approach to keep things moving. Can Fam Physician. 2015;61:152-158.
3. Gray JR. What is chronic constipation? Definition and diagnosis. Can J Gastroenterol. 2011;25 (Suppl B):7B-10B.
4. American Gastroenterological Association, Bharucha AE, Dorn SD, Lembo A, Pressman A. American Gastroenterological Association medical position statement on constipation. Gastroenterology. 2013;144:211-217. doi:10.1053/j.gastro.2012.10.029
5. Maung TZ, Singh K. Regular monitoring with stool chart prevents constipation, urinary retention and delirium in elderly patients: an audit leading to clinical effectiveness, efficiency and patient centredness. Future Healthc J. 2019;6(Suppl 2):3. doi:10.7861/futurehosp.6-2s-s3
6. Mostafa SM, Bhandari S, Ritchie G, et al. Constipation and its implications in the critically ill patient. Br J Anaesth. 2003;91:815-819. doi:10.1093/bja/aeg275
7. Jackson R, Cheng P, Moreman S, et al. “The constipation conundrum”: Improving recognition of constipation on a gastroenterology ward. BMJ Qual Improv Rep. 2016;5(1):u212167.w3007. doi:10.1136/bmjquality.u212167.w3007
8. Rao S, Go JT. Update on the management of constipation in the elderly: new treatment options. Clin Interv Aging. 2010;5:163-171. doi:10.2147/cia.s8100
9. Strom KL. Quality improvement interventions: what works? J Healthc Qual. 2001;23(5):4-24. doi:10.1111/j.1945-1474.2001.tb00368.x
10. De Giorgio R, Ruggeri E, Stanghellini V, et al. Chronic constipation in the elderly: a primer for the gastroenterologist. BMC Gastroenterol. 2015;15:130. doi:10.1186/s12876-015-366-3
11. The Health Foundation. Improving the flow of older people. April 2013. Accessed August 11, 2021. https://www.england.nhs.uk/wp-content/uploads/2013/08/sheff-study.pdf
12. Linton A. Improving management of constipation in an inpatient setting using a care bundle. BMJ Qual Improv Rep. 2014;3(1):u201903.w1002. doi:10.1136/bmjquality.u201903.w1002
From Sheffield Teaching Hospitals, Sheffield, UK, S5 7AU.
Objective: Constipation is widely prevalent in older adults and may result in complications such as urinary retention, delirium, and bowel obstruction. Previous studies have indicated that while the nursing staff do well in completing stool charts, doctors monitor them infrequently. This project aimed to improve the documentation of bowel movement by doctors on ward rounds to 85%, by the end of a 3-month period.
Methods: Baseline, postintervention, and sustainability data were collected from inpatient notes on weekdays on a geriatric ward in Northern General Hospital, Sheffield, UK. Posters and stickers of the poo emoji were placed on walls and in inpatient notes, respectively, as a reminder.
Results: Data on bowel activity documentation were collected from 28 patients. The baseline data showed that bowel activity was monitored daily on the ward 60.49% of the time. However, following the interventions, there was a significant increase in documentation, to 86.78%. The sustainability study showed that bowel activity was documented on the ward 56.56% of the time.
Conclusion: This study shows how a strong initial effect on behavioral change can be accomplished through simple interventions such as stickers and posters. As most wards currently still use paper notes, this is a generalizable model that other wards can trial. However, this study also shows the difficulty in maintaining behavioral change over extended periods of time.
Keywords: bowel movement; documentation; obstruction; constipation; geriatrics; incontinence; junior doctor; quality improvement.
Constipation is widely prevalent in the elderly, encountered frequently in both community and hospital medicine.1 Its estimated prevalence in adults over 84 years old is 34% for women and 25% for men, rising to up to 80% for long-term care residents.2
Chronic constipation is generally characterized by unsatisfactory defecation due to infrequent bowel emptying or difficulty with stool passage, which may lead to incomplete evacuation.2-4 Constipation in the elderly, in addition to causing abdominal pain, nausea, and reduced appetite, may result in complications such as fecal incontinence (and overflow diarrhea), urinary retention, delirium, and bowel obstruction, which may in result in life-threatening perforation.5,6 For inpatients on geriatric wards, these consequences may increase morbidity and mortality, while prolonging hospital stays, thereby also increasing exposure to hospital-acquired infections.7 Furthermore, constipation is also associated with impaired health-related quality of life.8
Management includes treating the cause, stopping contributing medications, early mobilization, diet modification, and, if all else fails, prescription laxatives. Therefore, early identification and appropriate treatment of constipation is beneficial in inpatient care, as well as when planning safe and patient-centered discharges.
Given the risks and complications of constipation in the elderly, we, a group of Foundation Year 2 (FY2) doctors in the UK Foundation Programme, decided to explore how doctors can help to recognize this condition early. Regular bowel movement documentation in patient notes on ward rounds is crucial, as it has been shown to reduce constipation-associated complications.5 However, complications from constipation can take significant amounts of time to develop and, therefore, documenting bowel movements on a daily basis is not necessary.
Based on these observations along with targets set out in previous studies,7 our aim was to improve documentation of bowel movement on ward rounds to 85% by March 2020.
Methods
Before the data collection process, a fishbone diagram was designed to identify the potential causes of poor documentation of bowel movement on geriatric wards. There were several aspects that were reviewed, including, for example, patients, health care professionals, organizational policies, procedures, and equipment. It was then decided to focus on raising awareness of the documentation of bowel movement by doctors specifically.
Retrospective data were collected from the inpatient paper notes of 28 patients on Brearley 6, a geriatric ward at the Northern General Hospital within Sheffield Teaching Hospitals (STH), on weekdays over a 3-week period. The baseline data collected included the bed number of the patient, whether or not bowel movement on initial ward round was documented, and whether it was the junior, registrar, or consultant leading the ward round. End-of-life and discharged patients were excluded (Table).
The interventions consisted of posters and stickers. Posters were displayed on Brearley 6, including the doctors’ office, nurses’ station, and around the bays where notes were kept, in order to emphasize their importance. The stickers of the poo emoji were also printed and placed at the front of each set of inpatient paper notes as a reminder for the doctor documenting on the ward round. The interventions were also introduced in the morning board meeting to ensure all staff on Brearley 6 were aware of them.
Data were collected on weekdays over a 3-week period starting 2 weeks after the interventions were put in place (Table). In order to assess that the intervention had been sustained, data were again collected 1 month later over a 2-week period (Table). Microsoft Excel (Microsoft Corporation, Redmond, Washington, USA) was used to analyze all data, and control charts were used to assess variability in the data.
Results
The baseline data showed that bowel movement was documented 60.49% of the time by doctors on the initial ward round before intervention, as illustrated in Figure 1. There was no evidence of an out-of-control process in this baseline data set.
The comparison between the preintervention and postintervention data is illustrated in Figure 1. The postintervention data, which were taken 2 weeks after intervention, showed a significant increase in the documentation of bowel movements, to 86.78%. The figure displays a number of features consistent with an out-of-control process: beyond limits (≥ 1 points beyond control limits), Zone A rule (2 out of 3 consecutive points beyond 2 standard deviations from the mean), Zone B rule (4 out of 5 consecutive points beyond 1 standard deviation from the mean), and Zone C rule (≥ 8 consecutive points on 1 side of the mean). These findings demonstrate a special cause variation in the documentation of bowel movements.
Figure 2 shows the sustainability of the intervention, which averaged 56.56% postintervention nearly 2 months later. The data returned to preintervention variability levels.
Discussion
Our project explored an important issue that was frequently encountered by department clinicians. Our team of FY2 doctors, in general, had little experience with quality improvement. We have developed our understanding and experience through planning, making, and measuring improvement.
It was challenging deciding on how to deal with the problem. A number of ways were considered to improve the paper rounding chart, but the nursing team had already planned to make changes to it. Bowel activity is mainly documented by nursing staff, but there was no specific protocol for recognizing constipation and when to inform the medical team. We decided to focus on doctors’ documentation in patient notes during the ward round, as this is where the decision regarding management of bowels is made, including interventions that could only be done by doctors, such as prescribing laxatives.
Strom et al9 have described a number of successful quality improvement interventions, and we decided to follow the authors’ guidance to implement a reminder system strategy using both posters and stickers to prompt doctors to document bowel activity. Both of these were simple, and the text on the poster was concise. The only cost incurred on the project was from printing the stickers; this totalled £2.99 (US $4.13). Individual stickers for each ward round entry were considered but not used, as it would create an additional task for doctors.
The data initially indicated that the interventions had their desired effect. However, this positive change was unsustainable, most likely suggesting that the novelty of the stickers and posters wore off at some point, leading to junior doctors no longer noticing them. Further Plan-Do-Study-Act cycles should examine the reasons why the change is difficult to sustain and implement new policies that aim to overcome them.
There were a number of limitations to this study. A patient could be discharged before data collection, which was done twice weekly. This could have resulted in missed data during the collection period. In addition, the accuracy of the documentation is dependent on nursing staff correctly recording—as well as the doctors correctly viewing—all sources of information on bowel activity. Observer bias is possible, too, as a steering group member was involved in data collection. Their awareness of the project could cause a positive skew in the data. And, unfortunately, the project came to an abrupt end because of COVID-19 cases on the ward.
We examined the daily documentation of bowel activity, which may not be necessary considering that internationally recognized constipation classifications, such as the Rome III criteria, define constipation as fewer than 3 bowel movements per week.10 However, the data collection sheet did not include patient identifiers, so it was impossible to determine whether bowel activity had been documented 3 or more times per week for each patient. This is important because a clinician may only decide to act if there is no bowel movement activity for 3 or more days.
Because our data were collected on a single geriatric ward, which had an emphasis on Parkinson’s disease, it is unclear whether our findings are generalizable to other clinical areas in STH. However, constipation is common in the elderly, so it is likely to be relevant to other wards, as more than a third of STH hospital beds are occupied by patients aged 75 years and older.11
Conclusion
Overall, our study highlights the fact that monitoring bowel activity is important on a geriatric ward. Recognizing constipation early prevents complications and delays to discharge. As mentioned earlier, our aim was achieved initially but not sustained. Therefore, future development should focus on sustainability. For example, laxative-focused ward rounds have shown to be effective at recognizing and preventing constipation by intervening early.12 Future cycles that we considered included using an electronic reminder on the hospital IT system, as the trust is aiming to introduce electronic documentation. Focus could also be placed on improving documentation in bowel charts by ward staff. This could be achieved by organizing regular educational sessions on the complications of constipation and when to inform the medical team regarding concerns.
Acknowledgments: The authors thank Dr. Jamie Kapur, Sheffield Teaching Hospitals, for his guidance and supervision, as well as our collaborators: Rachel Hallam, Claire Walker, Monisha Chakravorty, and Hamza Khan.
Corresponding author: Alexander P. Noar, BMBCh, BA, 10 Stanhope Gardens, London, N6 5TS; [email protected].
Financial disclosures: None.
From Sheffield Teaching Hospitals, Sheffield, UK, S5 7AU.
Objective: Constipation is widely prevalent in older adults and may result in complications such as urinary retention, delirium, and bowel obstruction. Previous studies have indicated that while the nursing staff do well in completing stool charts, doctors monitor them infrequently. This project aimed to improve the documentation of bowel movement by doctors on ward rounds to 85%, by the end of a 3-month period.
Methods: Baseline, postintervention, and sustainability data were collected from inpatient notes on weekdays on a geriatric ward in Northern General Hospital, Sheffield, UK. Posters and stickers of the poo emoji were placed on walls and in inpatient notes, respectively, as a reminder.
Results: Data on bowel activity documentation were collected from 28 patients. The baseline data showed that bowel activity was monitored daily on the ward 60.49% of the time. However, following the interventions, there was a significant increase in documentation, to 86.78%. The sustainability study showed that bowel activity was documented on the ward 56.56% of the time.
Conclusion: This study shows how a strong initial effect on behavioral change can be accomplished through simple interventions such as stickers and posters. As most wards currently still use paper notes, this is a generalizable model that other wards can trial. However, this study also shows the difficulty in maintaining behavioral change over extended periods of time.
Keywords: bowel movement; documentation; obstruction; constipation; geriatrics; incontinence; junior doctor; quality improvement.
Constipation is widely prevalent in the elderly, encountered frequently in both community and hospital medicine.1 Its estimated prevalence in adults over 84 years old is 34% for women and 25% for men, rising to up to 80% for long-term care residents.2
Chronic constipation is generally characterized by unsatisfactory defecation due to infrequent bowel emptying or difficulty with stool passage, which may lead to incomplete evacuation.2-4 Constipation in the elderly, in addition to causing abdominal pain, nausea, and reduced appetite, may result in complications such as fecal incontinence (and overflow diarrhea), urinary retention, delirium, and bowel obstruction, which may in result in life-threatening perforation.5,6 For inpatients on geriatric wards, these consequences may increase morbidity and mortality, while prolonging hospital stays, thereby also increasing exposure to hospital-acquired infections.7 Furthermore, constipation is also associated with impaired health-related quality of life.8
Management includes treating the cause, stopping contributing medications, early mobilization, diet modification, and, if all else fails, prescription laxatives. Therefore, early identification and appropriate treatment of constipation is beneficial in inpatient care, as well as when planning safe and patient-centered discharges.
Given the risks and complications of constipation in the elderly, we, a group of Foundation Year 2 (FY2) doctors in the UK Foundation Programme, decided to explore how doctors can help to recognize this condition early. Regular bowel movement documentation in patient notes on ward rounds is crucial, as it has been shown to reduce constipation-associated complications.5 However, complications from constipation can take significant amounts of time to develop and, therefore, documenting bowel movements on a daily basis is not necessary.
Based on these observations along with targets set out in previous studies,7 our aim was to improve documentation of bowel movement on ward rounds to 85% by March 2020.
Methods
Before the data collection process, a fishbone diagram was designed to identify the potential causes of poor documentation of bowel movement on geriatric wards. There were several aspects that were reviewed, including, for example, patients, health care professionals, organizational policies, procedures, and equipment. It was then decided to focus on raising awareness of the documentation of bowel movement by doctors specifically.
Retrospective data were collected from the inpatient paper notes of 28 patients on Brearley 6, a geriatric ward at the Northern General Hospital within Sheffield Teaching Hospitals (STH), on weekdays over a 3-week period. The baseline data collected included the bed number of the patient, whether or not bowel movement on initial ward round was documented, and whether it was the junior, registrar, or consultant leading the ward round. End-of-life and discharged patients were excluded (Table).
The interventions consisted of posters and stickers. Posters were displayed on Brearley 6, including the doctors’ office, nurses’ station, and around the bays where notes were kept, in order to emphasize their importance. The stickers of the poo emoji were also printed and placed at the front of each set of inpatient paper notes as a reminder for the doctor documenting on the ward round. The interventions were also introduced in the morning board meeting to ensure all staff on Brearley 6 were aware of them.
Data were collected on weekdays over a 3-week period starting 2 weeks after the interventions were put in place (Table). In order to assess that the intervention had been sustained, data were again collected 1 month later over a 2-week period (Table). Microsoft Excel (Microsoft Corporation, Redmond, Washington, USA) was used to analyze all data, and control charts were used to assess variability in the data.
Results
The baseline data showed that bowel movement was documented 60.49% of the time by doctors on the initial ward round before intervention, as illustrated in Figure 1. There was no evidence of an out-of-control process in this baseline data set.
The comparison between the preintervention and postintervention data is illustrated in Figure 1. The postintervention data, which were taken 2 weeks after intervention, showed a significant increase in the documentation of bowel movements, to 86.78%. The figure displays a number of features consistent with an out-of-control process: beyond limits (≥ 1 points beyond control limits), Zone A rule (2 out of 3 consecutive points beyond 2 standard deviations from the mean), Zone B rule (4 out of 5 consecutive points beyond 1 standard deviation from the mean), and Zone C rule (≥ 8 consecutive points on 1 side of the mean). These findings demonstrate a special cause variation in the documentation of bowel movements.
Figure 2 shows the sustainability of the intervention, which averaged 56.56% postintervention nearly 2 months later. The data returned to preintervention variability levels.
Discussion
Our project explored an important issue that was frequently encountered by department clinicians. Our team of FY2 doctors, in general, had little experience with quality improvement. We have developed our understanding and experience through planning, making, and measuring improvement.
It was challenging deciding on how to deal with the problem. A number of ways were considered to improve the paper rounding chart, but the nursing team had already planned to make changes to it. Bowel activity is mainly documented by nursing staff, but there was no specific protocol for recognizing constipation and when to inform the medical team. We decided to focus on doctors’ documentation in patient notes during the ward round, as this is where the decision regarding management of bowels is made, including interventions that could only be done by doctors, such as prescribing laxatives.
Strom et al9 have described a number of successful quality improvement interventions, and we decided to follow the authors’ guidance to implement a reminder system strategy using both posters and stickers to prompt doctors to document bowel activity. Both of these were simple, and the text on the poster was concise. The only cost incurred on the project was from printing the stickers; this totalled £2.99 (US $4.13). Individual stickers for each ward round entry were considered but not used, as it would create an additional task for doctors.
The data initially indicated that the interventions had their desired effect. However, this positive change was unsustainable, most likely suggesting that the novelty of the stickers and posters wore off at some point, leading to junior doctors no longer noticing them. Further Plan-Do-Study-Act cycles should examine the reasons why the change is difficult to sustain and implement new policies that aim to overcome them.
There were a number of limitations to this study. A patient could be discharged before data collection, which was done twice weekly. This could have resulted in missed data during the collection period. In addition, the accuracy of the documentation is dependent on nursing staff correctly recording—as well as the doctors correctly viewing—all sources of information on bowel activity. Observer bias is possible, too, as a steering group member was involved in data collection. Their awareness of the project could cause a positive skew in the data. And, unfortunately, the project came to an abrupt end because of COVID-19 cases on the ward.
We examined the daily documentation of bowel activity, which may not be necessary considering that internationally recognized constipation classifications, such as the Rome III criteria, define constipation as fewer than 3 bowel movements per week.10 However, the data collection sheet did not include patient identifiers, so it was impossible to determine whether bowel activity had been documented 3 or more times per week for each patient. This is important because a clinician may only decide to act if there is no bowel movement activity for 3 or more days.
Because our data were collected on a single geriatric ward, which had an emphasis on Parkinson’s disease, it is unclear whether our findings are generalizable to other clinical areas in STH. However, constipation is common in the elderly, so it is likely to be relevant to other wards, as more than a third of STH hospital beds are occupied by patients aged 75 years and older.11
Conclusion
Overall, our study highlights the fact that monitoring bowel activity is important on a geriatric ward. Recognizing constipation early prevents complications and delays to discharge. As mentioned earlier, our aim was achieved initially but not sustained. Therefore, future development should focus on sustainability. For example, laxative-focused ward rounds have shown to be effective at recognizing and preventing constipation by intervening early.12 Future cycles that we considered included using an electronic reminder on the hospital IT system, as the trust is aiming to introduce electronic documentation. Focus could also be placed on improving documentation in bowel charts by ward staff. This could be achieved by organizing regular educational sessions on the complications of constipation and when to inform the medical team regarding concerns.
Acknowledgments: The authors thank Dr. Jamie Kapur, Sheffield Teaching Hospitals, for his guidance and supervision, as well as our collaborators: Rachel Hallam, Claire Walker, Monisha Chakravorty, and Hamza Khan.
Corresponding author: Alexander P. Noar, BMBCh, BA, 10 Stanhope Gardens, London, N6 5TS; [email protected].
Financial disclosures: None.
1. Forootan M, Bagheri N, Darvishi M. Chronic constipation: A review of literature. Medicine (Baltimore). 2018;97:e10631. doi:10.1097/MD.00000000000.10631
2. Schuster BG, Kosar L, Kamrul R. Constipation in older adults: stepwise approach to keep things moving. Can Fam Physician. 2015;61:152-158.
3. Gray JR. What is chronic constipation? Definition and diagnosis. Can J Gastroenterol. 2011;25 (Suppl B):7B-10B.
4. American Gastroenterological Association, Bharucha AE, Dorn SD, Lembo A, Pressman A. American Gastroenterological Association medical position statement on constipation. Gastroenterology. 2013;144:211-217. doi:10.1053/j.gastro.2012.10.029
5. Maung TZ, Singh K. Regular monitoring with stool chart prevents constipation, urinary retention and delirium in elderly patients: an audit leading to clinical effectiveness, efficiency and patient centredness. Future Healthc J. 2019;6(Suppl 2):3. doi:10.7861/futurehosp.6-2s-s3
6. Mostafa SM, Bhandari S, Ritchie G, et al. Constipation and its implications in the critically ill patient. Br J Anaesth. 2003;91:815-819. doi:10.1093/bja/aeg275
7. Jackson R, Cheng P, Moreman S, et al. “The constipation conundrum”: Improving recognition of constipation on a gastroenterology ward. BMJ Qual Improv Rep. 2016;5(1):u212167.w3007. doi:10.1136/bmjquality.u212167.w3007
8. Rao S, Go JT. Update on the management of constipation in the elderly: new treatment options. Clin Interv Aging. 2010;5:163-171. doi:10.2147/cia.s8100
9. Strom KL. Quality improvement interventions: what works? J Healthc Qual. 2001;23(5):4-24. doi:10.1111/j.1945-1474.2001.tb00368.x
10. De Giorgio R, Ruggeri E, Stanghellini V, et al. Chronic constipation in the elderly: a primer for the gastroenterologist. BMC Gastroenterol. 2015;15:130. doi:10.1186/s12876-015-366-3
11. The Health Foundation. Improving the flow of older people. April 2013. Accessed August 11, 2021. https://www.england.nhs.uk/wp-content/uploads/2013/08/sheff-study.pdf
12. Linton A. Improving management of constipation in an inpatient setting using a care bundle. BMJ Qual Improv Rep. 2014;3(1):u201903.w1002. doi:10.1136/bmjquality.u201903.w1002
1. Forootan M, Bagheri N, Darvishi M. Chronic constipation: A review of literature. Medicine (Baltimore). 2018;97:e10631. doi:10.1097/MD.00000000000.10631
2. Schuster BG, Kosar L, Kamrul R. Constipation in older adults: stepwise approach to keep things moving. Can Fam Physician. 2015;61:152-158.
3. Gray JR. What is chronic constipation? Definition and diagnosis. Can J Gastroenterol. 2011;25 (Suppl B):7B-10B.
4. American Gastroenterological Association, Bharucha AE, Dorn SD, Lembo A, Pressman A. American Gastroenterological Association medical position statement on constipation. Gastroenterology. 2013;144:211-217. doi:10.1053/j.gastro.2012.10.029
5. Maung TZ, Singh K. Regular monitoring with stool chart prevents constipation, urinary retention and delirium in elderly patients: an audit leading to clinical effectiveness, efficiency and patient centredness. Future Healthc J. 2019;6(Suppl 2):3. doi:10.7861/futurehosp.6-2s-s3
6. Mostafa SM, Bhandari S, Ritchie G, et al. Constipation and its implications in the critically ill patient. Br J Anaesth. 2003;91:815-819. doi:10.1093/bja/aeg275
7. Jackson R, Cheng P, Moreman S, et al. “The constipation conundrum”: Improving recognition of constipation on a gastroenterology ward. BMJ Qual Improv Rep. 2016;5(1):u212167.w3007. doi:10.1136/bmjquality.u212167.w3007
8. Rao S, Go JT. Update on the management of constipation in the elderly: new treatment options. Clin Interv Aging. 2010;5:163-171. doi:10.2147/cia.s8100
9. Strom KL. Quality improvement interventions: what works? J Healthc Qual. 2001;23(5):4-24. doi:10.1111/j.1945-1474.2001.tb00368.x
10. De Giorgio R, Ruggeri E, Stanghellini V, et al. Chronic constipation in the elderly: a primer for the gastroenterologist. BMC Gastroenterol. 2015;15:130. doi:10.1186/s12876-015-366-3
11. The Health Foundation. Improving the flow of older people. April 2013. Accessed August 11, 2021. https://www.england.nhs.uk/wp-content/uploads/2013/08/sheff-study.pdf
12. Linton A. Improving management of constipation in an inpatient setting using a care bundle. BMJ Qual Improv Rep. 2014;3(1):u201903.w1002. doi:10.1136/bmjquality.u201903.w1002