User login
EADV: Vismodegib treatment breaks don’t hurt efficacy
COPENHAGEN – Treatment breaks due to adverse events in patients taking vismodegib for advanced basal cell carcinoma don’t appear to compromise the oral hedgehog pathway inhibitor’s efficacy; in fact, they might even enhance it, according to a prespecified interim analysis of the STEVIE trial.
STEVIE is an ongoing phase II, long-term, open-label international study designed primarily to assess the safety of vismodegib (Erivedge) in a situation similar to routine clinical practice. Efficacy and impact on quality of life are secondary endpoints. Although STEVIE has enrolled 1,227 patients, a prespecified interim analysis was conducted in the first 499 followed for at least 12 months, of whom 468 had locally advanced basal cell carcinoma (BCC) and 31 had metastatic BCC, explained Dr. Johan Hansson, an oncologist at the Karolinska Institute in Stockholm.

The drug was dosed at 150 mg once daily continuously in 28-day cycles until disease progression, intolerable toxicity, or study withdrawal. Safety follow-up was conducted at 1, 3, 5, 9, and 12 months. In an earlier report, the complete and partial response rates were 34% and 33%, respectively, in patients with locally advanced BCC, and 7% and 31% in those with metastatic disease (Lancet Oncol. 2015 Jun;16[6]:729-36).
Dr. Hansson presented new data on efficacy outcomes broken down according to treatment breaks, as well as quality of life results, at the annual congress of the European Academy of Dermatology and Venereology.
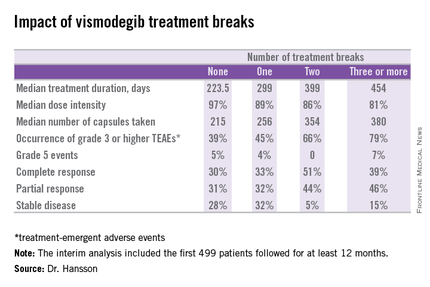
Twenty-six percent of patients had one or more treatment breaks. Seventy-six patients had one, 41 had two, and 14 had three or more. The median duration of the breaks was 22 days. The two most frequent reasons for treatment breaks were intolerable adverse events in 53% of cases, and lesser adverse events in 23%.
Close to 100% of STEVIE participants had treatment-emergent adverse events. The most common were muscle spasms, alopecia, altered sense of smell, and weight loss.
Although the number of patients with treatment breaks was relatively small, the response rates were higher in patients with more treatment breaks. So was median treatment duration as well as the median number of capsules taken.
Median progression-free survival was 19.8 months in patients with no treatment breaks, was 19.0 months in those with one, and hasn’t yet been reached in patients with two or more breaks.
In interpreting these findings, Dr. Hansson said, “We have to remember that although intriguing, these are tentative results from an exploratory analysis of subgroups in an ongoing study and should be interpreted with caution.”
The oncologist added, however, based upon these promising results he and his coinvestigators plan to look further into the concept of deliberate intermittent dosing of vismodegib.
Quality of life was assessed using the Skindex-16 questionnaire at baseline, again after two and seven 28-day cycles of vismodegib, and at 12 months. Three domains were examined: emotion, function, and symptoms.
A clinically meaningful improvement – defined as a 10-point or greater reduction from baseline – was seen in the emotion domain at all time points in patients with locally advanced BCC, with median improvements of 14.3 points after two cycles and 23.8 points after seven cycles and at the 12-month mark. Clinically meaningful improvement in symptom scores on the Skindex-16 were noted in patients aged 65 and older, in women, and in those with BCCs in locations other than the head or neck. However, no clinically meaningful improvement in the domain of function was seen at any time in patients with locally advanced BCC.
Patients with metastatic BCC didn’t show significant improvement in any of the three quality of life domains at any time point, added Dr. Hansson.
The STEVIE trial is sponsored by F. Hoffmann–La Roche/Genentech. Dr. Hansson reported receiving research grants from and serving as a consultant to Bristol-Myers Squibb, GlaxoSmithKline, Merck, Novartis, and Roche.
COPENHAGEN – Treatment breaks due to adverse events in patients taking vismodegib for advanced basal cell carcinoma don’t appear to compromise the oral hedgehog pathway inhibitor’s efficacy; in fact, they might even enhance it, according to a prespecified interim analysis of the STEVIE trial.
STEVIE is an ongoing phase II, long-term, open-label international study designed primarily to assess the safety of vismodegib (Erivedge) in a situation similar to routine clinical practice. Efficacy and impact on quality of life are secondary endpoints. Although STEVIE has enrolled 1,227 patients, a prespecified interim analysis was conducted in the first 499 followed for at least 12 months, of whom 468 had locally advanced basal cell carcinoma (BCC) and 31 had metastatic BCC, explained Dr. Johan Hansson, an oncologist at the Karolinska Institute in Stockholm.
The drug was dosed at 150 mg once daily continuously in 28-day cycles until disease progression, intolerable toxicity, or study withdrawal. Safety follow-up was conducted at 1, 3, 5, 9, and 12 months. In an earlier report, the complete and partial response rates were 34% and 33%, respectively, in patients with locally advanced BCC, and 7% and 31% in those with metastatic disease (Lancet Oncol. 2015 Jun;16[6]:729-36).
Dr. Hansson presented new data on efficacy outcomes broken down according to treatment breaks, as well as quality of life results, at the annual congress of the European Academy of Dermatology and Venereology.
Twenty-six percent of patients had one or more treatment breaks. Seventy-six patients had one, 41 had two, and 14 had three or more. The median duration of the breaks was 22 days. The two most frequent reasons for treatment breaks were intolerable adverse events in 53% of cases, and lesser adverse events in 23%.
Close to 100% of STEVIE participants had treatment-emergent adverse events. The most common were muscle spasms, alopecia, altered sense of smell, and weight loss.
Although the number of patients with treatment breaks was relatively small, the response rates were higher in patients with more treatment breaks. So was median treatment duration as well as the median number of capsules taken.
Median progression-free survival was 19.8 months in patients with no treatment breaks, was 19.0 months in those with one, and hasn’t yet been reached in patients with two or more breaks.
In interpreting these findings, Dr. Hansson said, “We have to remember that although intriguing, these are tentative results from an exploratory analysis of subgroups in an ongoing study and should be interpreted with caution.”
The oncologist added, however, based upon these promising results he and his coinvestigators plan to look further into the concept of deliberate intermittent dosing of vismodegib.
Quality of life was assessed using the Skindex-16 questionnaire at baseline, again after two and seven 28-day cycles of vismodegib, and at 12 months. Three domains were examined: emotion, function, and symptoms.
A clinically meaningful improvement – defined as a 10-point or greater reduction from baseline – was seen in the emotion domain at all time points in patients with locally advanced BCC, with median improvements of 14.3 points after two cycles and 23.8 points after seven cycles and at the 12-month mark. Clinically meaningful improvement in symptom scores on the Skindex-16 were noted in patients aged 65 and older, in women, and in those with BCCs in locations other than the head or neck. However, no clinically meaningful improvement in the domain of function was seen at any time in patients with locally advanced BCC.
Patients with metastatic BCC didn’t show significant improvement in any of the three quality of life domains at any time point, added Dr. Hansson.
The STEVIE trial is sponsored by F. Hoffmann–La Roche/Genentech. Dr. Hansson reported receiving research grants from and serving as a consultant to Bristol-Myers Squibb, GlaxoSmithKline, Merck, Novartis, and Roche.
COPENHAGEN – Treatment breaks due to adverse events in patients taking vismodegib for advanced basal cell carcinoma don’t appear to compromise the oral hedgehog pathway inhibitor’s efficacy; in fact, they might even enhance it, according to a prespecified interim analysis of the STEVIE trial.
STEVIE is an ongoing phase II, long-term, open-label international study designed primarily to assess the safety of vismodegib (Erivedge) in a situation similar to routine clinical practice. Efficacy and impact on quality of life are secondary endpoints. Although STEVIE has enrolled 1,227 patients, a prespecified interim analysis was conducted in the first 499 followed for at least 12 months, of whom 468 had locally advanced basal cell carcinoma (BCC) and 31 had metastatic BCC, explained Dr. Johan Hansson, an oncologist at the Karolinska Institute in Stockholm.
The drug was dosed at 150 mg once daily continuously in 28-day cycles until disease progression, intolerable toxicity, or study withdrawal. Safety follow-up was conducted at 1, 3, 5, 9, and 12 months. In an earlier report, the complete and partial response rates were 34% and 33%, respectively, in patients with locally advanced BCC, and 7% and 31% in those with metastatic disease (Lancet Oncol. 2015 Jun;16[6]:729-36).
Dr. Hansson presented new data on efficacy outcomes broken down according to treatment breaks, as well as quality of life results, at the annual congress of the European Academy of Dermatology and Venereology.
Twenty-six percent of patients had one or more treatment breaks. Seventy-six patients had one, 41 had two, and 14 had three or more. The median duration of the breaks was 22 days. The two most frequent reasons for treatment breaks were intolerable adverse events in 53% of cases, and lesser adverse events in 23%.
Close to 100% of STEVIE participants had treatment-emergent adverse events. The most common were muscle spasms, alopecia, altered sense of smell, and weight loss.
Although the number of patients with treatment breaks was relatively small, the response rates were higher in patients with more treatment breaks. So was median treatment duration as well as the median number of capsules taken.
Median progression-free survival was 19.8 months in patients with no treatment breaks, was 19.0 months in those with one, and hasn’t yet been reached in patients with two or more breaks.
In interpreting these findings, Dr. Hansson said, “We have to remember that although intriguing, these are tentative results from an exploratory analysis of subgroups in an ongoing study and should be interpreted with caution.”
The oncologist added, however, based upon these promising results he and his coinvestigators plan to look further into the concept of deliberate intermittent dosing of vismodegib.
Quality of life was assessed using the Skindex-16 questionnaire at baseline, again after two and seven 28-day cycles of vismodegib, and at 12 months. Three domains were examined: emotion, function, and symptoms.
A clinically meaningful improvement – defined as a 10-point or greater reduction from baseline – was seen in the emotion domain at all time points in patients with locally advanced BCC, with median improvements of 14.3 points after two cycles and 23.8 points after seven cycles and at the 12-month mark. Clinically meaningful improvement in symptom scores on the Skindex-16 were noted in patients aged 65 and older, in women, and in those with BCCs in locations other than the head or neck. However, no clinically meaningful improvement in the domain of function was seen at any time in patients with locally advanced BCC.
Patients with metastatic BCC didn’t show significant improvement in any of the three quality of life domains at any time point, added Dr. Hansson.
The STEVIE trial is sponsored by F. Hoffmann–La Roche/Genentech. Dr. Hansson reported receiving research grants from and serving as a consultant to Bristol-Myers Squibb, GlaxoSmithKline, Merck, Novartis, and Roche.
AT THE EADV CONGRESS
Key clinical point: Treatment breaks due to adverse events in patients taking vismodegib for advanced basal cell carcinoma don’t compromise efficacy.
Major finding: The complete response rate to vismodegib in patients with advanced BCC was intriguingly higher in those with more treatment breaks due to adverse events.
Data source: A prespecified interim analysis of the first 499 patients with advanced BCC enrolled in STEVIE, a large ongoing phase II, long-term, open-label international safety study of vismodegib.
Disclosures: The STEVIE trial is sponsored by F. Hoffmann–La Roche/Genentech. The presenter reported receiving research grants from and serving as a consultant to Bristol-Myers Squibb, GlaxoSmithKline, Merck, Novartis, and Roche.

Cancer prevention field riding high into the new year
The new year has us all looking forward and the cancer prevention community is no exception.
In a special report entitled “Transforming Cancer Prevention through Precision Medicine and Immune-Oncology,” a team of experts offer a brief look at what we can expect in the near future for cancer prevention research, including a Pre-Cancer Genome Atlas (PCGA), and highlight some of the recent advances shaping their optimism.
“Just as precision therapy and immunotherapy are transforming cancer treatment, precision medicine and immunoprevention approaches are being translated to the clinic and showing great promise. We stand at the edge of a new frontier that will include comprehensively characterizing the molecular and cellular events that drive premalignant progression (e.g. PCGA),” Dr. Scott M. Lippman, director of the University of California San Diego Moores Cancer Center, and his coauthors wrote (Cancer Prev Res. 2016;9:2-10).
The report details some of the clinical firsts in 2015 including genomic studies suggesting that clonal hematopoiesis is a premalignant state for blood cancer, the first precision medicine trial in cancer prevention (EPOC) reporting that loss of heterozygosity can predict which patients with premalignant mouth lesions are most likely to develop oral cancer, and the U.S. Preventive Services Task Force recommending low-dose aspirin for colorectal cancer prevention based on age and risk.
Randomized trials have also suggested that a single dose of human papillomavirus vaccine can provide durable protection against HPV infection. Tumor biology studies established new chemoprevention for familial adenomatous polyposis syndrome and universal tumor screening guidelines based on DNA mismatch repair mutations and microsatellite instability for colorectal cancer in patients with Lynch syndrome.
Further, remarkable advances have been made in liquid biopsy technology, high-throughput functional screening, and computational biology methods and algorithms that “provide unprecedented opportunities to interrogate the biology of premalignancy...” they noted.
In an American Association for Cancer Research blog post, Dr. Lippman acknowledges that not everyone is the same page when it comes to the underlying principles of cancer prevention.
A “contentious” paper published at the start of 2015 suggested that variations in cancer risk are due to random mutations or what might otherwise be called bad luck. The new year was heralded in by a second paper, however, that came to roughly the opposite conclusion or that most cancers are preventable.
In February, an AACR Cancer Prevention Summit will bring together various stakeholders to discuss the current state of cancer prevention and to identify top priorities and research directions for the field, he noted.
The authors acknowledged grant support from the National Institutes of Health/National Cancer Institute.
The new year has us all looking forward and the cancer prevention community is no exception.
In a special report entitled “Transforming Cancer Prevention through Precision Medicine and Immune-Oncology,” a team of experts offer a brief look at what we can expect in the near future for cancer prevention research, including a Pre-Cancer Genome Atlas (PCGA), and highlight some of the recent advances shaping their optimism.
“Just as precision therapy and immunotherapy are transforming cancer treatment, precision medicine and immunoprevention approaches are being translated to the clinic and showing great promise. We stand at the edge of a new frontier that will include comprehensively characterizing the molecular and cellular events that drive premalignant progression (e.g. PCGA),” Dr. Scott M. Lippman, director of the University of California San Diego Moores Cancer Center, and his coauthors wrote (Cancer Prev Res. 2016;9:2-10).
The report details some of the clinical firsts in 2015 including genomic studies suggesting that clonal hematopoiesis is a premalignant state for blood cancer, the first precision medicine trial in cancer prevention (EPOC) reporting that loss of heterozygosity can predict which patients with premalignant mouth lesions are most likely to develop oral cancer, and the U.S. Preventive Services Task Force recommending low-dose aspirin for colorectal cancer prevention based on age and risk.
Randomized trials have also suggested that a single dose of human papillomavirus vaccine can provide durable protection against HPV infection. Tumor biology studies established new chemoprevention for familial adenomatous polyposis syndrome and universal tumor screening guidelines based on DNA mismatch repair mutations and microsatellite instability for colorectal cancer in patients with Lynch syndrome.
Further, remarkable advances have been made in liquid biopsy technology, high-throughput functional screening, and computational biology methods and algorithms that “provide unprecedented opportunities to interrogate the biology of premalignancy...” they noted.
In an American Association for Cancer Research blog post, Dr. Lippman acknowledges that not everyone is the same page when it comes to the underlying principles of cancer prevention.
A “contentious” paper published at the start of 2015 suggested that variations in cancer risk are due to random mutations or what might otherwise be called bad luck. The new year was heralded in by a second paper, however, that came to roughly the opposite conclusion or that most cancers are preventable.
In February, an AACR Cancer Prevention Summit will bring together various stakeholders to discuss the current state of cancer prevention and to identify top priorities and research directions for the field, he noted.
The authors acknowledged grant support from the National Institutes of Health/National Cancer Institute.
The new year has us all looking forward and the cancer prevention community is no exception.
In a special report entitled “Transforming Cancer Prevention through Precision Medicine and Immune-Oncology,” a team of experts offer a brief look at what we can expect in the near future for cancer prevention research, including a Pre-Cancer Genome Atlas (PCGA), and highlight some of the recent advances shaping their optimism.
“Just as precision therapy and immunotherapy are transforming cancer treatment, precision medicine and immunoprevention approaches are being translated to the clinic and showing great promise. We stand at the edge of a new frontier that will include comprehensively characterizing the molecular and cellular events that drive premalignant progression (e.g. PCGA),” Dr. Scott M. Lippman, director of the University of California San Diego Moores Cancer Center, and his coauthors wrote (Cancer Prev Res. 2016;9:2-10).
The report details some of the clinical firsts in 2015 including genomic studies suggesting that clonal hematopoiesis is a premalignant state for blood cancer, the first precision medicine trial in cancer prevention (EPOC) reporting that loss of heterozygosity can predict which patients with premalignant mouth lesions are most likely to develop oral cancer, and the U.S. Preventive Services Task Force recommending low-dose aspirin for colorectal cancer prevention based on age and risk.
Randomized trials have also suggested that a single dose of human papillomavirus vaccine can provide durable protection against HPV infection. Tumor biology studies established new chemoprevention for familial adenomatous polyposis syndrome and universal tumor screening guidelines based on DNA mismatch repair mutations and microsatellite instability for colorectal cancer in patients with Lynch syndrome.
Further, remarkable advances have been made in liquid biopsy technology, high-throughput functional screening, and computational biology methods and algorithms that “provide unprecedented opportunities to interrogate the biology of premalignancy...” they noted.
In an American Association for Cancer Research blog post, Dr. Lippman acknowledges that not everyone is the same page when it comes to the underlying principles of cancer prevention.
A “contentious” paper published at the start of 2015 suggested that variations in cancer risk are due to random mutations or what might otherwise be called bad luck. The new year was heralded in by a second paper, however, that came to roughly the opposite conclusion or that most cancers are preventable.
In February, an AACR Cancer Prevention Summit will bring together various stakeholders to discuss the current state of cancer prevention and to identify top priorities and research directions for the field, he noted.
The authors acknowledged grant support from the National Institutes of Health/National Cancer Institute.
FROM CANCER PREVENTION RESEARCH
T-VEC: Advancing the Fight Against Melanoma

Following a phase III, open-label trial conducted by Andtbacka et al (J Clin Oncol. 2015;33:2780-2788), the US Food and Drug Administration recently approved the first oncolytic immunotherapy talimogene laherparepvec (T-VEC) for the treatment of unresectable cutaneous, subcutaneous, and nodal lesions in patients with advanced melanoma (stage IIIB/C–stage IV) following initial surgery.
A group of 436 patients with injectable melanomas (melanomas that are accessible via a percutaneous injection) that were not surgically resectable were randomly assigned (2:1) to treatment with intralesional T-VEC or subcutaneous granulocyte macrophage colony-stimulating factor (GM-CSF). The primary endpoint of the study was durable response rate (DRR), defined as objective response lasting continuously for 6 months or longer. Secondary endpoints included overall survival (OS) and overall response rate.
Talimogene laherparepvec was shown to extend DRRs compared to GM-CSF. Durable response rates were significantly higher with T-VEC (16.3%; 95% confidence interval [CI], 12.1%–20.5%) versus GM-CSF (2.1%; 95% CI, 0%–4.5%)(odds ratio, 8.9; P<.001).
In the OS analysis, a 4.4-month extension with T-VEC was observed; however, this was not deemed to be statistically significant (P=.051). The median OS was 23.3 months (95% CI, 19.5–29.6 months) with T-VEC and 18.9 months (95% CI, 16.0–23.7 months) with GM-CSF (hazard ratio, 0.79; 95% CI, 0.62–1.00; P=.051). Overall response rate also was higher in the T-VEC arm (26.4%; 95% CI, 21.4%–31.5%) versus GM-CSF (5.7%; 95% CI, 1.9%–9.5%).
Talimogene laherparepvec is a herpes simplex virus type 1–derived oncolytic immunotherapy designed to replicate within tumors and produce GM-CSF, which enhances systemic antitumor immune responses and induces tumor lysis.
In this study, T-VEC efficacy was greatest in patients with stage IIIB, IIIC, or IVM1a melanomas and in patients with treatment-naive disease. Differences in DRRs in patients with stage IIIB/C melanomas were 33% in the T-VEC group versus 0% for patients treated with GM-CSF alone. In the stage IVM1a group, DRR was 16% with T-VEC versus 2% with GM-CSF. The difference between both treatments was smaller in more advanced melanomas (IVM1b group, 3% vs 4%; IVM1c, 7% vs 3%). In the first-line treatment, the DRR with T-VEC was 24% versus 0% with GM-CSF. In the second-line and beyond, the DRR with T-VEC was 10% compared to 4% for GM-CSF.
The main adverse events seen in this study were fatigue, chills, and pyrexia. Serious adverse events occurred in 25.7% and 13.4% of participants in the T-VEC and GM-CSF arms, respectively, with disease progression (3.1% vs 1.6%) and cellulitis (2.4% vs 0.8%) being the most common. Six immune-mediated events occurred in the T-VEC group compared to 3 in the GM-CSF group.
Twelve patient deaths occurred within 30 days of the last dose of T-VEC; 9 were associated with progressive disease and the other 3 were associated with myocardial infarction, cardiac arrest, and sepsis, respectively. Four patient deaths were reported in the GM-CSF arm within the same 30 days.
What’s the Issue?
Immunotherapy represents a promising treatment option for metastatic melanoma. These promising results along with the US Food and Drug Administration’s approval of T-VEC will lead to further studies of the uses of T-VEC in combination with other therapies, including a phase I/II study to assess T-VEC in combination with ipilimumab for unresected melanomas (NCT01740297) and a phase III study of T-VEC in combination with pembrolizumab for unresected melanomas (NCT02263508). It is important for dermatologists to be familiar with the new frontier of melanoma treatments. How will these new immunotherapies affect your treatment of melanoma?
Following a phase III, open-label trial conducted by Andtbacka et al (J Clin Oncol. 2015;33:2780-2788), the US Food and Drug Administration recently approved the first oncolytic immunotherapy talimogene laherparepvec (T-VEC) for the treatment of unresectable cutaneous, subcutaneous, and nodal lesions in patients with advanced melanoma (stage IIIB/C–stage IV) following initial surgery.
A group of 436 patients with injectable melanomas (melanomas that are accessible via a percutaneous injection) that were not surgically resectable were randomly assigned (2:1) to treatment with intralesional T-VEC or subcutaneous granulocyte macrophage colony-stimulating factor (GM-CSF). The primary endpoint of the study was durable response rate (DRR), defined as objective response lasting continuously for 6 months or longer. Secondary endpoints included overall survival (OS) and overall response rate.
Talimogene laherparepvec was shown to extend DRRs compared to GM-CSF. Durable response rates were significantly higher with T-VEC (16.3%; 95% confidence interval [CI], 12.1%–20.5%) versus GM-CSF (2.1%; 95% CI, 0%–4.5%)(odds ratio, 8.9; P<.001).
In the OS analysis, a 4.4-month extension with T-VEC was observed; however, this was not deemed to be statistically significant (P=.051). The median OS was 23.3 months (95% CI, 19.5–29.6 months) with T-VEC and 18.9 months (95% CI, 16.0–23.7 months) with GM-CSF (hazard ratio, 0.79; 95% CI, 0.62–1.00; P=.051). Overall response rate also was higher in the T-VEC arm (26.4%; 95% CI, 21.4%–31.5%) versus GM-CSF (5.7%; 95% CI, 1.9%–9.5%).
Talimogene laherparepvec is a herpes simplex virus type 1–derived oncolytic immunotherapy designed to replicate within tumors and produce GM-CSF, which enhances systemic antitumor immune responses and induces tumor lysis.
In this study, T-VEC efficacy was greatest in patients with stage IIIB, IIIC, or IVM1a melanomas and in patients with treatment-naive disease. Differences in DRRs in patients with stage IIIB/C melanomas were 33% in the T-VEC group versus 0% for patients treated with GM-CSF alone. In the stage IVM1a group, DRR was 16% with T-VEC versus 2% with GM-CSF. The difference between both treatments was smaller in more advanced melanomas (IVM1b group, 3% vs 4%; IVM1c, 7% vs 3%). In the first-line treatment, the DRR with T-VEC was 24% versus 0% with GM-CSF. In the second-line and beyond, the DRR with T-VEC was 10% compared to 4% for GM-CSF.
The main adverse events seen in this study were fatigue, chills, and pyrexia. Serious adverse events occurred in 25.7% and 13.4% of participants in the T-VEC and GM-CSF arms, respectively, with disease progression (3.1% vs 1.6%) and cellulitis (2.4% vs 0.8%) being the most common. Six immune-mediated events occurred in the T-VEC group compared to 3 in the GM-CSF group.
Twelve patient deaths occurred within 30 days of the last dose of T-VEC; 9 were associated with progressive disease and the other 3 were associated with myocardial infarction, cardiac arrest, and sepsis, respectively. Four patient deaths were reported in the GM-CSF arm within the same 30 days.
What’s the Issue?
Immunotherapy represents a promising treatment option for metastatic melanoma. These promising results along with the US Food and Drug Administration’s approval of T-VEC will lead to further studies of the uses of T-VEC in combination with other therapies, including a phase I/II study to assess T-VEC in combination with ipilimumab for unresected melanomas (NCT01740297) and a phase III study of T-VEC in combination with pembrolizumab for unresected melanomas (NCT02263508). It is important for dermatologists to be familiar with the new frontier of melanoma treatments. How will these new immunotherapies affect your treatment of melanoma?
Following a phase III, open-label trial conducted by Andtbacka et al (J Clin Oncol. 2015;33:2780-2788), the US Food and Drug Administration recently approved the first oncolytic immunotherapy talimogene laherparepvec (T-VEC) for the treatment of unresectable cutaneous, subcutaneous, and nodal lesions in patients with advanced melanoma (stage IIIB/C–stage IV) following initial surgery.
A group of 436 patients with injectable melanomas (melanomas that are accessible via a percutaneous injection) that were not surgically resectable were randomly assigned (2:1) to treatment with intralesional T-VEC or subcutaneous granulocyte macrophage colony-stimulating factor (GM-CSF). The primary endpoint of the study was durable response rate (DRR), defined as objective response lasting continuously for 6 months or longer. Secondary endpoints included overall survival (OS) and overall response rate.
Talimogene laherparepvec was shown to extend DRRs compared to GM-CSF. Durable response rates were significantly higher with T-VEC (16.3%; 95% confidence interval [CI], 12.1%–20.5%) versus GM-CSF (2.1%; 95% CI, 0%–4.5%)(odds ratio, 8.9; P<.001).
In the OS analysis, a 4.4-month extension with T-VEC was observed; however, this was not deemed to be statistically significant (P=.051). The median OS was 23.3 months (95% CI, 19.5–29.6 months) with T-VEC and 18.9 months (95% CI, 16.0–23.7 months) with GM-CSF (hazard ratio, 0.79; 95% CI, 0.62–1.00; P=.051). Overall response rate also was higher in the T-VEC arm (26.4%; 95% CI, 21.4%–31.5%) versus GM-CSF (5.7%; 95% CI, 1.9%–9.5%).
Talimogene laherparepvec is a herpes simplex virus type 1–derived oncolytic immunotherapy designed to replicate within tumors and produce GM-CSF, which enhances systemic antitumor immune responses and induces tumor lysis.
In this study, T-VEC efficacy was greatest in patients with stage IIIB, IIIC, or IVM1a melanomas and in patients with treatment-naive disease. Differences in DRRs in patients with stage IIIB/C melanomas were 33% in the T-VEC group versus 0% for patients treated with GM-CSF alone. In the stage IVM1a group, DRR was 16% with T-VEC versus 2% with GM-CSF. The difference between both treatments was smaller in more advanced melanomas (IVM1b group, 3% vs 4%; IVM1c, 7% vs 3%). In the first-line treatment, the DRR with T-VEC was 24% versus 0% with GM-CSF. In the second-line and beyond, the DRR with T-VEC was 10% compared to 4% for GM-CSF.
The main adverse events seen in this study were fatigue, chills, and pyrexia. Serious adverse events occurred in 25.7% and 13.4% of participants in the T-VEC and GM-CSF arms, respectively, with disease progression (3.1% vs 1.6%) and cellulitis (2.4% vs 0.8%) being the most common. Six immune-mediated events occurred in the T-VEC group compared to 3 in the GM-CSF group.
Twelve patient deaths occurred within 30 days of the last dose of T-VEC; 9 were associated with progressive disease and the other 3 were associated with myocardial infarction, cardiac arrest, and sepsis, respectively. Four patient deaths were reported in the GM-CSF arm within the same 30 days.
What’s the Issue?
Immunotherapy represents a promising treatment option for metastatic melanoma. These promising results along with the US Food and Drug Administration’s approval of T-VEC will lead to further studies of the uses of T-VEC in combination with other therapies, including a phase I/II study to assess T-VEC in combination with ipilimumab for unresected melanomas (NCT01740297) and a phase III study of T-VEC in combination with pembrolizumab for unresected melanomas (NCT02263508). It is important for dermatologists to be familiar with the new frontier of melanoma treatments. How will these new immunotherapies affect your treatment of melanoma?

FDA approves pembrolizumab as first-line advanced melanoma therapy
Pembrolizumab, the programmed death receptor-1 (PD-1)–blocking antibody, is now approved as a first-line treatment of unresectable or metastatic melanoma, based on the results of the KEYNOTE-006 study.
This is the second approval for an advanced melanoma indication for pembrolizumab (Keytruda), according to a statement issued by Merck, the manufacturer. Pembrolizumab was first approved in 2014 for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor.
The FDA-approved dose is 2 mg/kg every 3 weeks.
In KEYNOTE-006, a phase III study that compared pembrolizumab to ipilimumab (Yervoy), also a PD-1–blocking antibody, in 834 patients with unresectable or metastatic melanoma with progression of disease, overall survival and progression-free survival were significantly increased among those treated with pembrolizumab, compared with ipilimumab.
The results were reported at the annual meeting of the American Association for Cancer Research in April, and published simultaneously in the New England Journal of Medicine (2015; 372:2521-32).
Pembrolizumab, the programmed death receptor-1 (PD-1)–blocking antibody, is now approved as a first-line treatment of unresectable or metastatic melanoma, based on the results of the KEYNOTE-006 study.
This is the second approval for an advanced melanoma indication for pembrolizumab (Keytruda), according to a statement issued by Merck, the manufacturer. Pembrolizumab was first approved in 2014 for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor.
The FDA-approved dose is 2 mg/kg every 3 weeks.
In KEYNOTE-006, a phase III study that compared pembrolizumab to ipilimumab (Yervoy), also a PD-1–blocking antibody, in 834 patients with unresectable or metastatic melanoma with progression of disease, overall survival and progression-free survival were significantly increased among those treated with pembrolizumab, compared with ipilimumab.
The results were reported at the annual meeting of the American Association for Cancer Research in April, and published simultaneously in the New England Journal of Medicine (2015; 372:2521-32).
Pembrolizumab, the programmed death receptor-1 (PD-1)–blocking antibody, is now approved as a first-line treatment of unresectable or metastatic melanoma, based on the results of the KEYNOTE-006 study.
This is the second approval for an advanced melanoma indication for pembrolizumab (Keytruda), according to a statement issued by Merck, the manufacturer. Pembrolizumab was first approved in 2014 for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor.
The FDA-approved dose is 2 mg/kg every 3 weeks.
In KEYNOTE-006, a phase III study that compared pembrolizumab to ipilimumab (Yervoy), also a PD-1–blocking antibody, in 834 patients with unresectable or metastatic melanoma with progression of disease, overall survival and progression-free survival were significantly increased among those treated with pembrolizumab, compared with ipilimumab.
The results were reported at the annual meeting of the American Association for Cancer Research in April, and published simultaneously in the New England Journal of Medicine (2015; 372:2521-32).
FDA proposes ban on indoor tanning for minors
The Food and Drug Administration wants to prohibit indoor tanning for minors and is seeking stricter regulations to enhance the safety of indoor tanning and use of sunlamp products for all users.
“Today’s action is intended to help protect young people from a known and preventable cause of skin cancer and other harms. Individuals under 18 years are at greatest risk of the adverse health consequences of indoor tanning,” said acting FDA Commissioner Stephen Ostroff in a news release outlining the two proposed rules.

Currently, 1.6 million minors tan indoors yearly, according to data from the 2013 National Youth Risk Behavior Survey, cited by the FDA. Although many states require parental consent before minors can use tanning facilities, a national standard is lacking.
An additional measure in the first proposed rule requires adults who use commercial tanning beds to sign a “risk acknowledgment certification” statement before the first tanning session and every 6 months thereafter. These restrictions are appropriate, the FDA statement said, because of the increased risk of skin cancer, notably melanoma, and other risks associated with indoor tanning. According to the proposed rule, some research has found that “doses of UVA radiation emitted by high-power sunlamp products may be up to 10-15 times higher than that of the midday sun, resulting in an intense amount of exposure that does not exist in nature.”
The second proposed ruleis aimed at tanning facilities and sunlamp manufacturers, and contains measures to bring about safer use of tanning devices. These rules would set limits on the amount of light allowed through protective eyewear when tanning, make an emergency “off” switch mandatory on tanning beds, improve warning signs and tanning bulb labeling, and prohibit device modifications without FDA recertification.
According to the FDA, these regulations would affect more than 30,000 tanning salons, health clubs, and spas that offer indoor tanning. “These proposed rules are meant to help adults make their decisions based on truthful information and to ensure manufacturers and tanning facilities take additional steps to improve the safety of these devices,” Dr. Ostroff said.
The rules and opportunities to comment are available starting Monday, Dec. 21 at www.regulations.gov. Public comments will be open for 90 days.
On Twitter @karioakes
The Food and Drug Administration wants to prohibit indoor tanning for minors and is seeking stricter regulations to enhance the safety of indoor tanning and use of sunlamp products for all users.
“Today’s action is intended to help protect young people from a known and preventable cause of skin cancer and other harms. Individuals under 18 years are at greatest risk of the adverse health consequences of indoor tanning,” said acting FDA Commissioner Stephen Ostroff in a news release outlining the two proposed rules.
Currently, 1.6 million minors tan indoors yearly, according to data from the 2013 National Youth Risk Behavior Survey, cited by the FDA. Although many states require parental consent before minors can use tanning facilities, a national standard is lacking.
An additional measure in the first proposed rule requires adults who use commercial tanning beds to sign a “risk acknowledgment certification” statement before the first tanning session and every 6 months thereafter. These restrictions are appropriate, the FDA statement said, because of the increased risk of skin cancer, notably melanoma, and other risks associated with indoor tanning. According to the proposed rule, some research has found that “doses of UVA radiation emitted by high-power sunlamp products may be up to 10-15 times higher than that of the midday sun, resulting in an intense amount of exposure that does not exist in nature.”
The second proposed ruleis aimed at tanning facilities and sunlamp manufacturers, and contains measures to bring about safer use of tanning devices. These rules would set limits on the amount of light allowed through protective eyewear when tanning, make an emergency “off” switch mandatory on tanning beds, improve warning signs and tanning bulb labeling, and prohibit device modifications without FDA recertification.
According to the FDA, these regulations would affect more than 30,000 tanning salons, health clubs, and spas that offer indoor tanning. “These proposed rules are meant to help adults make their decisions based on truthful information and to ensure manufacturers and tanning facilities take additional steps to improve the safety of these devices,” Dr. Ostroff said.
The rules and opportunities to comment are available starting Monday, Dec. 21 at www.regulations.gov. Public comments will be open for 90 days.
On Twitter @karioakes
The Food and Drug Administration wants to prohibit indoor tanning for minors and is seeking stricter regulations to enhance the safety of indoor tanning and use of sunlamp products for all users.
“Today’s action is intended to help protect young people from a known and preventable cause of skin cancer and other harms. Individuals under 18 years are at greatest risk of the adverse health consequences of indoor tanning,” said acting FDA Commissioner Stephen Ostroff in a news release outlining the two proposed rules.
Currently, 1.6 million minors tan indoors yearly, according to data from the 2013 National Youth Risk Behavior Survey, cited by the FDA. Although many states require parental consent before minors can use tanning facilities, a national standard is lacking.
An additional measure in the first proposed rule requires adults who use commercial tanning beds to sign a “risk acknowledgment certification” statement before the first tanning session and every 6 months thereafter. These restrictions are appropriate, the FDA statement said, because of the increased risk of skin cancer, notably melanoma, and other risks associated with indoor tanning. According to the proposed rule, some research has found that “doses of UVA radiation emitted by high-power sunlamp products may be up to 10-15 times higher than that of the midday sun, resulting in an intense amount of exposure that does not exist in nature.”
The second proposed ruleis aimed at tanning facilities and sunlamp manufacturers, and contains measures to bring about safer use of tanning devices. These rules would set limits on the amount of light allowed through protective eyewear when tanning, make an emergency “off” switch mandatory on tanning beds, improve warning signs and tanning bulb labeling, and prohibit device modifications without FDA recertification.
According to the FDA, these regulations would affect more than 30,000 tanning salons, health clubs, and spas that offer indoor tanning. “These proposed rules are meant to help adults make their decisions based on truthful information and to ensure manufacturers and tanning facilities take additional steps to improve the safety of these devices,” Dr. Ostroff said.
The rules and opportunities to comment are available starting Monday, Dec. 21 at www.regulations.gov. Public comments will be open for 90 days.
On Twitter @karioakes

Entering an era of intelligent combination therapy in cancer
The past few decades have witnessed unprecedented advances in our understanding of the molecular underpinnings of cancer. Although indiscriminately cytotoxic therapies like chemo- and radiation therapy remain standard of care for many cancer types, more precise targeted therapies and immune-boosting immunotherapies have added to our arsenal and afforded considerable survival gains. Despite those advances, we are still no closer to a cure, particularly for the most aggressive and insidious cancers that progress rapidly or go undiagnosed until advanced stages of disease. The substantial genetic diversity of tumors and universal nature of drug resistance present the most formidable and enduring challenges to effective cancer treatment.
Click on the PDF icon at the top of this introduction to read the full article.
The past few decades have witnessed unprecedented advances in our understanding of the molecular underpinnings of cancer. Although indiscriminately cytotoxic therapies like chemo- and radiation therapy remain standard of care for many cancer types, more precise targeted therapies and immune-boosting immunotherapies have added to our arsenal and afforded considerable survival gains. Despite those advances, we are still no closer to a cure, particularly for the most aggressive and insidious cancers that progress rapidly or go undiagnosed until advanced stages of disease. The substantial genetic diversity of tumors and universal nature of drug resistance present the most formidable and enduring challenges to effective cancer treatment.
Click on the PDF icon at the top of this introduction to read the full article.
The past few decades have witnessed unprecedented advances in our understanding of the molecular underpinnings of cancer. Although indiscriminately cytotoxic therapies like chemo- and radiation therapy remain standard of care for many cancer types, more precise targeted therapies and immune-boosting immunotherapies have added to our arsenal and afforded considerable survival gains. Despite those advances, we are still no closer to a cure, particularly for the most aggressive and insidious cancers that progress rapidly or go undiagnosed until advanced stages of disease. The substantial genetic diversity of tumors and universal nature of drug resistance present the most formidable and enduring challenges to effective cancer treatment.
Click on the PDF icon at the top of this introduction to read the full article.
Oncology 2015: new therapies and new transitions toward value-based cancer care
The past year has been an exciting one for new oncology and hematology drug approvals and the continued evolution of our oncology delivery system toward high quality and value. In all, at press time in mid-November, the US Food and Drug Administration (FDA) had approved or granted expanded indications for 24 drugs, compared with 19 in the 2 preceding years. Of those 24 approvals, 7 were accelerated and 6 were expanded approvals, and 3 alone were for the immunotherapeutic drug, nivolumab – 2 for non-small-cell lung cancer (NSCLC) and 1 for metastatic melanoma.
Click on the PDF icon at the top of this introduction to read the full article.
The past year has been an exciting one for new oncology and hematology drug approvals and the continued evolution of our oncology delivery system toward high quality and value. In all, at press time in mid-November, the US Food and Drug Administration (FDA) had approved or granted expanded indications for 24 drugs, compared with 19 in the 2 preceding years. Of those 24 approvals, 7 were accelerated and 6 were expanded approvals, and 3 alone were for the immunotherapeutic drug, nivolumab – 2 for non-small-cell lung cancer (NSCLC) and 1 for metastatic melanoma.
Click on the PDF icon at the top of this introduction to read the full article.
The past year has been an exciting one for new oncology and hematology drug approvals and the continued evolution of our oncology delivery system toward high quality and value. In all, at press time in mid-November, the US Food and Drug Administration (FDA) had approved or granted expanded indications for 24 drugs, compared with 19 in the 2 preceding years. Of those 24 approvals, 7 were accelerated and 6 were expanded approvals, and 3 alone were for the immunotherapeutic drug, nivolumab – 2 for non-small-cell lung cancer (NSCLC) and 1 for metastatic melanoma.
Click on the PDF icon at the top of this introduction to read the full article.
FDA approves treatment for chemotherapy ODs, life-threatening toxicities
Uridine triacetate, a pyrimidine analogue, has been approved for the emergency treatment of fluorouracil or capecitabine overdoses in adults and children, and for patients who develop “certain severe or life-threatening toxicities within 4 days of receiving” these treatments, the Food and Drug Administration announced on Dec. 11.

“Today’s approval is a first-of-its-kind therapy that can potentially save lives following overdose or life-threatening toxicity from these chemotherapy agents,” Dr. Richard Pazdur, director of the office of hematology and oncology products in the FDA’s Center for Drug Evaluation and Research, said in the FDA statement. It will be marketed as Vistogard by Wellstat Therapeutics.
Uridine comes in an oral granule formulation that can be mixed into soft foods or, when necessary, administered via a nasogastric or gastrostomy tube, the prescribing information states. The indication is for use after an overdose “regardless of the presence of symptoms,” and for treating “early-onset, severe, or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of fluorouracil or capecitabine administration,” according to the prescribing information.
Uridine blocks cell damage and cell death caused by fluorouracil chemotherapy, according to the statement, which adds that it is up to the patient’s health care provider to “determine when he or she should return to the prescribed chemotherapy after treatment with Vistogard.”
Uridine was evaluated in two studies of 135 adults and children with cancer, treated with uridine for a fluorouracil or capecitabine overdose, or for early-onset, unusually severe or life-threatening toxicities within 96 hours after receiving fluorouracil (not because of an overdose). Among those treated for an overdose, 97% were alive 30 days after treatment, and among those treated for early-onset severe or life-threatening toxicity, 89% were alive 30 days after treatment. In addition, 33% of the patients resumed chemotherapy within 30 days, according to the FDA statement. Diarrhea, vomiting, and nausea were the most common adverse events associated with treatment.
Uridine was granted orphan drug, priority review, and fast track designations.
Uridine triacetate, a pyrimidine analogue, has been approved for the emergency treatment of fluorouracil or capecitabine overdoses in adults and children, and for patients who develop “certain severe or life-threatening toxicities within 4 days of receiving” these treatments, the Food and Drug Administration announced on Dec. 11.
“Today’s approval is a first-of-its-kind therapy that can potentially save lives following overdose or life-threatening toxicity from these chemotherapy agents,” Dr. Richard Pazdur, director of the office of hematology and oncology products in the FDA’s Center for Drug Evaluation and Research, said in the FDA statement. It will be marketed as Vistogard by Wellstat Therapeutics.
Uridine comes in an oral granule formulation that can be mixed into soft foods or, when necessary, administered via a nasogastric or gastrostomy tube, the prescribing information states. The indication is for use after an overdose “regardless of the presence of symptoms,” and for treating “early-onset, severe, or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of fluorouracil or capecitabine administration,” according to the prescribing information.
Uridine blocks cell damage and cell death caused by fluorouracil chemotherapy, according to the statement, which adds that it is up to the patient’s health care provider to “determine when he or she should return to the prescribed chemotherapy after treatment with Vistogard.”
Uridine was evaluated in two studies of 135 adults and children with cancer, treated with uridine for a fluorouracil or capecitabine overdose, or for early-onset, unusually severe or life-threatening toxicities within 96 hours after receiving fluorouracil (not because of an overdose). Among those treated for an overdose, 97% were alive 30 days after treatment, and among those treated for early-onset severe or life-threatening toxicity, 89% were alive 30 days after treatment. In addition, 33% of the patients resumed chemotherapy within 30 days, according to the FDA statement. Diarrhea, vomiting, and nausea were the most common adverse events associated with treatment.
Uridine was granted orphan drug, priority review, and fast track designations.
Uridine triacetate, a pyrimidine analogue, has been approved for the emergency treatment of fluorouracil or capecitabine overdoses in adults and children, and for patients who develop “certain severe or life-threatening toxicities within 4 days of receiving” these treatments, the Food and Drug Administration announced on Dec. 11.
“Today’s approval is a first-of-its-kind therapy that can potentially save lives following overdose or life-threatening toxicity from these chemotherapy agents,” Dr. Richard Pazdur, director of the office of hematology and oncology products in the FDA’s Center for Drug Evaluation and Research, said in the FDA statement. It will be marketed as Vistogard by Wellstat Therapeutics.
Uridine comes in an oral granule formulation that can be mixed into soft foods or, when necessary, administered via a nasogastric or gastrostomy tube, the prescribing information states. The indication is for use after an overdose “regardless of the presence of symptoms,” and for treating “early-onset, severe, or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of fluorouracil or capecitabine administration,” according to the prescribing information.
Uridine blocks cell damage and cell death caused by fluorouracil chemotherapy, according to the statement, which adds that it is up to the patient’s health care provider to “determine when he or she should return to the prescribed chemotherapy after treatment with Vistogard.”
Uridine was evaluated in two studies of 135 adults and children with cancer, treated with uridine for a fluorouracil or capecitabine overdose, or for early-onset, unusually severe or life-threatening toxicities within 96 hours after receiving fluorouracil (not because of an overdose). Among those treated for an overdose, 97% were alive 30 days after treatment, and among those treated for early-onset severe or life-threatening toxicity, 89% were alive 30 days after treatment. In addition, 33% of the patients resumed chemotherapy within 30 days, according to the FDA statement. Diarrhea, vomiting, and nausea were the most common adverse events associated with treatment.
Uridine was granted orphan drug, priority review, and fast track designations.
Documentation for Mohs Surgery
In 2013, the Centers for Medicare and Medicaid Services (CMS) issued a guidance to reduce reimbursement issues for Mohs micrographic surgery (MMS).1 One crucial question that remains is when and if these documentation guidelines will be formally implemented. The guidelines outlined by the CMS currently are regarded as suggestions until Medicare contractors adopt them into the local coverage determinations (LCDs).
Key Documentation Guidelines
To reduce MMS reimbursement issues, documentation in the patient’s medical record should support the medical necessity of the procedure and reflect the number and anatomic locations of specimens taken and the reason for the procedure should be clearly communicated. The specific tumor type also should be approved for treatment with MMS in the respective LCD.
Nonphysician providers are not authorized by Medicare to perform MMS. To ensure proper coding, both surgery and pathology must be performed by a single physician and should be supported by documentation in the patient’s medical record (eg, relevant chart notes should be made under the provider’s signature). These documentation guidelines are not new but are included in the CMS guidance to reiterate their importance in reducing MMS reimbursement issues.
Per customary clinical practice, the CMS guidance specifies that MMS documentation should include gross description of the tissue removed, including the location, number, and size of the lesions, as well as how many specimens were removed for each stage. However, the guidance diverges from routine MMS documentation requirements in its emphasis on providing a histologic description of the tissue removed. The guidance suggests that the depth of tumor invasion, pathologic pattern, cell morphology (which is not typically specified for skin cancers), and, if present, the existence of perineural invasion or scar tissue should be documented. If these features are constant across stages, they only need to be noted for the first stage.
Adapting Guidelines for Clinical Practice
The CMS guidance may create some conundrums for physicians regarding MMS documentation; for instance, if a tumor is cleared in one stage, as is often the case, no tumor will be seen on glass slides prepared to assess tissue margins during the procedure and therefore documentation of characteristics like depth and pattern will be impossible. Similarly, cell morphology is not a feature that usually is relevant for most squamous and basal cell carcinomas, although it may be useful in certain unusual instances, such as in cases of rare tumors with particular histologic features that may influence management and/or prognosis. When in doubt regarding the appropriate documentation method for MMS, the surgeon should use his or her best judgment based on clinical experience rather than simply following guidelines that may not be applicable.
Final Thoughts
The CMS guidance serves as a reminder of the documentation requirements for MMS and extends current practice by suggesting a detailed microscopic description of the removed tissue. The American Academy of Dermatology has developed a guide to help Mohs surgeons provide the necessary documentation without creating cumbersome chart notes.2 Mohs surgeons should consult the most recent version of the LCD that applies to their geographic area to determine if the new documentation guidelines have been adopted.
- Centers for Medicare and Medicaid Services. Guidance to Reduce Mohs Surgery Reimbursement Issues. Bethesda, MD: Centers for Medicare and Medicaid Services, US Department of Health and Human Services; 2013. MLN Matters SE1318.
- Position statement on documentation of frozen section specimens during Mohs micrographic surgery. American Academy of Dermatology Web site. https://www.aad.org/forms/policies/Uploads/PS/PS%20-%20Documentation%20of%20Frozen%20Section%20Specimens%20during%20Mohs%20Micrographic%20Surgery.pdf. Accessed November 30, 2015.
In 2013, the Centers for Medicare and Medicaid Services (CMS) issued a guidance to reduce reimbursement issues for Mohs micrographic surgery (MMS).1 One crucial question that remains is when and if these documentation guidelines will be formally implemented. The guidelines outlined by the CMS currently are regarded as suggestions until Medicare contractors adopt them into the local coverage determinations (LCDs).
Key Documentation Guidelines
To reduce MMS reimbursement issues, documentation in the patient’s medical record should support the medical necessity of the procedure and reflect the number and anatomic locations of specimens taken and the reason for the procedure should be clearly communicated. The specific tumor type also should be approved for treatment with MMS in the respective LCD.
Nonphysician providers are not authorized by Medicare to perform MMS. To ensure proper coding, both surgery and pathology must be performed by a single physician and should be supported by documentation in the patient’s medical record (eg, relevant chart notes should be made under the provider’s signature). These documentation guidelines are not new but are included in the CMS guidance to reiterate their importance in reducing MMS reimbursement issues.
Per customary clinical practice, the CMS guidance specifies that MMS documentation should include gross description of the tissue removed, including the location, number, and size of the lesions, as well as how many specimens were removed for each stage. However, the guidance diverges from routine MMS documentation requirements in its emphasis on providing a histologic description of the tissue removed. The guidance suggests that the depth of tumor invasion, pathologic pattern, cell morphology (which is not typically specified for skin cancers), and, if present, the existence of perineural invasion or scar tissue should be documented. If these features are constant across stages, they only need to be noted for the first stage.
Adapting Guidelines for Clinical Practice
The CMS guidance may create some conundrums for physicians regarding MMS documentation; for instance, if a tumor is cleared in one stage, as is often the case, no tumor will be seen on glass slides prepared to assess tissue margins during the procedure and therefore documentation of characteristics like depth and pattern will be impossible. Similarly, cell morphology is not a feature that usually is relevant for most squamous and basal cell carcinomas, although it may be useful in certain unusual instances, such as in cases of rare tumors with particular histologic features that may influence management and/or prognosis. When in doubt regarding the appropriate documentation method for MMS, the surgeon should use his or her best judgment based on clinical experience rather than simply following guidelines that may not be applicable.
Final Thoughts
The CMS guidance serves as a reminder of the documentation requirements for MMS and extends current practice by suggesting a detailed microscopic description of the removed tissue. The American Academy of Dermatology has developed a guide to help Mohs surgeons provide the necessary documentation without creating cumbersome chart notes.2 Mohs surgeons should consult the most recent version of the LCD that applies to their geographic area to determine if the new documentation guidelines have been adopted.
In 2013, the Centers for Medicare and Medicaid Services (CMS) issued a guidance to reduce reimbursement issues for Mohs micrographic surgery (MMS).1 One crucial question that remains is when and if these documentation guidelines will be formally implemented. The guidelines outlined by the CMS currently are regarded as suggestions until Medicare contractors adopt them into the local coverage determinations (LCDs).
Key Documentation Guidelines
To reduce MMS reimbursement issues, documentation in the patient’s medical record should support the medical necessity of the procedure and reflect the number and anatomic locations of specimens taken and the reason for the procedure should be clearly communicated. The specific tumor type also should be approved for treatment with MMS in the respective LCD.
Nonphysician providers are not authorized by Medicare to perform MMS. To ensure proper coding, both surgery and pathology must be performed by a single physician and should be supported by documentation in the patient’s medical record (eg, relevant chart notes should be made under the provider’s signature). These documentation guidelines are not new but are included in the CMS guidance to reiterate their importance in reducing MMS reimbursement issues.
Per customary clinical practice, the CMS guidance specifies that MMS documentation should include gross description of the tissue removed, including the location, number, and size of the lesions, as well as how many specimens were removed for each stage. However, the guidance diverges from routine MMS documentation requirements in its emphasis on providing a histologic description of the tissue removed. The guidance suggests that the depth of tumor invasion, pathologic pattern, cell morphology (which is not typically specified for skin cancers), and, if present, the existence of perineural invasion or scar tissue should be documented. If these features are constant across stages, they only need to be noted for the first stage.
Adapting Guidelines for Clinical Practice
The CMS guidance may create some conundrums for physicians regarding MMS documentation; for instance, if a tumor is cleared in one stage, as is often the case, no tumor will be seen on glass slides prepared to assess tissue margins during the procedure and therefore documentation of characteristics like depth and pattern will be impossible. Similarly, cell morphology is not a feature that usually is relevant for most squamous and basal cell carcinomas, although it may be useful in certain unusual instances, such as in cases of rare tumors with particular histologic features that may influence management and/or prognosis. When in doubt regarding the appropriate documentation method for MMS, the surgeon should use his or her best judgment based on clinical experience rather than simply following guidelines that may not be applicable.
Final Thoughts
The CMS guidance serves as a reminder of the documentation requirements for MMS and extends current practice by suggesting a detailed microscopic description of the removed tissue. The American Academy of Dermatology has developed a guide to help Mohs surgeons provide the necessary documentation without creating cumbersome chart notes.2 Mohs surgeons should consult the most recent version of the LCD that applies to their geographic area to determine if the new documentation guidelines have been adopted.
- Centers for Medicare and Medicaid Services. Guidance to Reduce Mohs Surgery Reimbursement Issues. Bethesda, MD: Centers for Medicare and Medicaid Services, US Department of Health and Human Services; 2013. MLN Matters SE1318.
- Position statement on documentation of frozen section specimens during Mohs micrographic surgery. American Academy of Dermatology Web site. https://www.aad.org/forms/policies/Uploads/PS/PS%20-%20Documentation%20of%20Frozen%20Section%20Specimens%20during%20Mohs%20Micrographic%20Surgery.pdf. Accessed November 30, 2015.
- Centers for Medicare and Medicaid Services. Guidance to Reduce Mohs Surgery Reimbursement Issues. Bethesda, MD: Centers for Medicare and Medicaid Services, US Department of Health and Human Services; 2013. MLN Matters SE1318.
- Position statement on documentation of frozen section specimens during Mohs micrographic surgery. American Academy of Dermatology Web site. https://www.aad.org/forms/policies/Uploads/PS/PS%20-%20Documentation%20of%20Frozen%20Section%20Specimens%20during%20Mohs%20Micrographic%20Surgery.pdf. Accessed November 30, 2015.

EADV: Focus on non-UV triggers of melanoma
COPENHAGEN – Sunlight is recognized as the 800-pound gorilla of melanoma risk, with roughly 65% of all melanomas worldwide attributable to exposure to UV radiation. But what about the other 35%?
About 10% of melanomas are due to an inherited germline mutation, and new germline mutations are being discovered all the time. However, the incidence of melanoma has been rising steadily in industrialized countries since the 1950s, and genetic predisposition can’t explain that phenomenon. Environmental and lifestyle factors are likely to be involved. Indeed, melanomas arising from epigenetic modifications by extrinsic environmental and lifestyle factors other than UV exposure are drawing increasing research attention because of the potential for preventing the malignancy through avoidance of these risk factors, Dr. Veronique del Marmol said at the annual congress of the European Academy of Dermatology and Venereology.

One promising avenue of investigation focuses on microRNAs (miRs) as environmental drivers of melanoma risk. The miRs are important posttranscriptional regulators of gene expression. In particular, overexpression of miR-21 has been shown to accompany the transition of benign melanocytes into melanoma. miR-21 affects numerous genes critical to oncogenesis, including genes promoting sustained proliferation, angiogenesis, evasion from apoptosis, genetic instability, oxidative stress, and invasion and metastasis, as detailed recently [J Transl Med. 2015 Jun 27;13:202] by Dr. Bodo C. Melnik of the department of dermatology, environmental medicine, and health theory, University of Osnabrück (Germany).
Translational work by Dr. Melnik and others has shown that miR-21 expression is upregulated by several elements that investigators have long suspected are associated with an increased risk of melanoma, including smoking, air pollution, polychlorinated biphenyls and other noxious chemicals, chronic inflammation, a high-fat/high-sugar diet, and a sedentary lifestyle. Even cow’s milk has generated suspicion, since bovine miR-21 is identical to human miR-21, noted Dr. del Marmol, head of the department of dermatology at Erasmus Hospital in Brussels and chair of the pan-European Euromelanoma project.
Two noteworthy factors that have generated interest recently are coffee consumption, shown in a large observational study to protect against melanoma, and sildenafil (Viagra), which was shown in an analysis of 25,848 physicians enrolled in the Health Professionals’ Follow-Up Study to be associated with a 2.24-fold increased risk of subsequently developing melanoma during prospective follow-up (JAMA Intern Med. 2014 Jun;174[6]:964-70).
An association between increased risk of melanoma and the use of sildenafil or other phosphodiesterase-5 (PDE-5) inhibitors commonly used by men with erectile dysfunction is biologically plausible, according to Dr. del Marmol. Activation of oncogenic BRAF is a hallmark of 40%-60% of melanomas, and BRAF activation, like sildenafil use, downregulates phosphodiesterase-5A, which increases the invasiveness of melanoma cells.
The coffee connection was identified through an analysis of food frequency questionnaires completed by 447,357 non-Hispanic whites enrolled in the NIH-AARP Diet and Health Study, a prospective study developed at the National Cancer Institute.
During 4.3 million person-years of follow-up, 2,904 individuals were diagnosed with malignant melanoma. In a multivariate analysis, quaffing 4 or more cups of caffeinated coffee daily was associated with a 25% reduction in the risk of developing melanoma. Lesser consumption had no significant impact on melanoma risk (J Natl Cancer Inst. 2015 Jan 20;107[2]: pii: dju421. doi: 10.1093/jnci/dju421].
COPENHAGEN – Sunlight is recognized as the 800-pound gorilla of melanoma risk, with roughly 65% of all melanomas worldwide attributable to exposure to UV radiation. But what about the other 35%?
About 10% of melanomas are due to an inherited germline mutation, and new germline mutations are being discovered all the time. However, the incidence of melanoma has been rising steadily in industrialized countries since the 1950s, and genetic predisposition can’t explain that phenomenon. Environmental and lifestyle factors are likely to be involved. Indeed, melanomas arising from epigenetic modifications by extrinsic environmental and lifestyle factors other than UV exposure are drawing increasing research attention because of the potential for preventing the malignancy through avoidance of these risk factors, Dr. Veronique del Marmol said at the annual congress of the European Academy of Dermatology and Venereology.
One promising avenue of investigation focuses on microRNAs (miRs) as environmental drivers of melanoma risk. The miRs are important posttranscriptional regulators of gene expression. In particular, overexpression of miR-21 has been shown to accompany the transition of benign melanocytes into melanoma. miR-21 affects numerous genes critical to oncogenesis, including genes promoting sustained proliferation, angiogenesis, evasion from apoptosis, genetic instability, oxidative stress, and invasion and metastasis, as detailed recently [J Transl Med. 2015 Jun 27;13:202] by Dr. Bodo C. Melnik of the department of dermatology, environmental medicine, and health theory, University of Osnabrück (Germany).
Translational work by Dr. Melnik and others has shown that miR-21 expression is upregulated by several elements that investigators have long suspected are associated with an increased risk of melanoma, including smoking, air pollution, polychlorinated biphenyls and other noxious chemicals, chronic inflammation, a high-fat/high-sugar diet, and a sedentary lifestyle. Even cow’s milk has generated suspicion, since bovine miR-21 is identical to human miR-21, noted Dr. del Marmol, head of the department of dermatology at Erasmus Hospital in Brussels and chair of the pan-European Euromelanoma project.
Two noteworthy factors that have generated interest recently are coffee consumption, shown in a large observational study to protect against melanoma, and sildenafil (Viagra), which was shown in an analysis of 25,848 physicians enrolled in the Health Professionals’ Follow-Up Study to be associated with a 2.24-fold increased risk of subsequently developing melanoma during prospective follow-up (JAMA Intern Med. 2014 Jun;174[6]:964-70).
An association between increased risk of melanoma and the use of sildenafil or other phosphodiesterase-5 (PDE-5) inhibitors commonly used by men with erectile dysfunction is biologically plausible, according to Dr. del Marmol. Activation of oncogenic BRAF is a hallmark of 40%-60% of melanomas, and BRAF activation, like sildenafil use, downregulates phosphodiesterase-5A, which increases the invasiveness of melanoma cells.
The coffee connection was identified through an analysis of food frequency questionnaires completed by 447,357 non-Hispanic whites enrolled in the NIH-AARP Diet and Health Study, a prospective study developed at the National Cancer Institute.
During 4.3 million person-years of follow-up, 2,904 individuals were diagnosed with malignant melanoma. In a multivariate analysis, quaffing 4 or more cups of caffeinated coffee daily was associated with a 25% reduction in the risk of developing melanoma. Lesser consumption had no significant impact on melanoma risk (J Natl Cancer Inst. 2015 Jan 20;107[2]: pii: dju421. doi: 10.1093/jnci/dju421].
COPENHAGEN – Sunlight is recognized as the 800-pound gorilla of melanoma risk, with roughly 65% of all melanomas worldwide attributable to exposure to UV radiation. But what about the other 35%?
About 10% of melanomas are due to an inherited germline mutation, and new germline mutations are being discovered all the time. However, the incidence of melanoma has been rising steadily in industrialized countries since the 1950s, and genetic predisposition can’t explain that phenomenon. Environmental and lifestyle factors are likely to be involved. Indeed, melanomas arising from epigenetic modifications by extrinsic environmental and lifestyle factors other than UV exposure are drawing increasing research attention because of the potential for preventing the malignancy through avoidance of these risk factors, Dr. Veronique del Marmol said at the annual congress of the European Academy of Dermatology and Venereology.
One promising avenue of investigation focuses on microRNAs (miRs) as environmental drivers of melanoma risk. The miRs are important posttranscriptional regulators of gene expression. In particular, overexpression of miR-21 has been shown to accompany the transition of benign melanocytes into melanoma. miR-21 affects numerous genes critical to oncogenesis, including genes promoting sustained proliferation, angiogenesis, evasion from apoptosis, genetic instability, oxidative stress, and invasion and metastasis, as detailed recently [J Transl Med. 2015 Jun 27;13:202] by Dr. Bodo C. Melnik of the department of dermatology, environmental medicine, and health theory, University of Osnabrück (Germany).
Translational work by Dr. Melnik and others has shown that miR-21 expression is upregulated by several elements that investigators have long suspected are associated with an increased risk of melanoma, including smoking, air pollution, polychlorinated biphenyls and other noxious chemicals, chronic inflammation, a high-fat/high-sugar diet, and a sedentary lifestyle. Even cow’s milk has generated suspicion, since bovine miR-21 is identical to human miR-21, noted Dr. del Marmol, head of the department of dermatology at Erasmus Hospital in Brussels and chair of the pan-European Euromelanoma project.
Two noteworthy factors that have generated interest recently are coffee consumption, shown in a large observational study to protect against melanoma, and sildenafil (Viagra), which was shown in an analysis of 25,848 physicians enrolled in the Health Professionals’ Follow-Up Study to be associated with a 2.24-fold increased risk of subsequently developing melanoma during prospective follow-up (JAMA Intern Med. 2014 Jun;174[6]:964-70).
An association between increased risk of melanoma and the use of sildenafil or other phosphodiesterase-5 (PDE-5) inhibitors commonly used by men with erectile dysfunction is biologically plausible, according to Dr. del Marmol. Activation of oncogenic BRAF is a hallmark of 40%-60% of melanomas, and BRAF activation, like sildenafil use, downregulates phosphodiesterase-5A, which increases the invasiveness of melanoma cells.
The coffee connection was identified through an analysis of food frequency questionnaires completed by 447,357 non-Hispanic whites enrolled in the NIH-AARP Diet and Health Study, a prospective study developed at the National Cancer Institute.
During 4.3 million person-years of follow-up, 2,904 individuals were diagnosed with malignant melanoma. In a multivariate analysis, quaffing 4 or more cups of caffeinated coffee daily was associated with a 25% reduction in the risk of developing melanoma. Lesser consumption had no significant impact on melanoma risk (J Natl Cancer Inst. 2015 Jan 20;107[2]: pii: dju421. doi: 10.1093/jnci/dju421].
EXPERT ANALYSIS FROM THE EADV CONGRESS
