User login
Know the best specific signs for polycystic ovary syndrome
SAN FRANCISCO – Dermatologists are often on the frontline when it comes to diagnosing polycystic ovary syndrome (PCOS), which is one reason they should be up to date and aware of the changing diagnostic criteria for the condition, according to Kanade Shinkai, MD.
About one-quarter of patients who are diagnosed with PCOS are seen first by a dermatologist. That’s because skin conditions may be more concerning than reproductive issues in young women.
“Sometimes, people don’t see [irregular menstruation] as a problem,” explained Dr. Shinkai of the department of dermatology at the University of California, San Francisco. “Maybe they’re young, or they’re not trying to get pregnant. But if their hair is falling out, they see that as a problem, or if they have bad acne, or they’re becoming hirsute, they see that as a problem. So, they present to a dermatologist.”
Early recognition of PCOS is important, because many women with the condition go on to develop diabetes, impaired glucose intolerance, hyperlipidemia, hypertension, fertility problems, and obesity.

It used to be that physicians expected patients with PCOS to have menstrual irregularities, biochemical or clinical evidence of hyperandrogenism, and evidence of polycystic ovaries on ultrasound. But just two of the three are now considered enough to warrant a diagnosis.
“Our original view of the classic patient has gone away, and it’s really a heterogeneous phenotype,” Dr. Shinkai said. “Originally, it was all three [criteria], and the patient was obese, and they all had diabetes. Now, we know that’s not true. Every woman who has PCOS has her own version of PCOS.”
Dr. Shinkai’s team conducted a study of clinical markers associated with PCOS and found that some of the classic signs of PCOS may be unreliable.
“Alopecia turns out not to be a very reliable marker,” she explained. “That’s paradigm shifting, I think, because often if patients present with hair loss in a hormonal pattern, they get worked up for PCOS, and it turns out that workup is not always fruitful.” Acne can also be misleading, given its frequency in the general population.
More reliable signs include hirsutism and acanthosis nigricans; 70%-80% of women with hirsutism have PCOS, and 53% of patients with PCOS have hirsutism, most commonly on the trunk. Acanthosis nigricans occurs in 37% of PCOS patients.
“Those are the best specific signs for PCOS,” said Dr. Shinkai. “If we see those, we should probably work the patient up.”
In preparation, the patient should be off of birth control treatment for at least 4 weeks, because hormonal treatment can interfere with test results, Dr Shinkai noted.
She also recommended a transvaginal ultrasound and a free-testosterone test. Consensus statements recommend testing of 17-hydroxyprogesterone, but Dr. Shinkai said she isn’t so sure. “That’s only going to capture about 3% of your patients with cutaneous hyperandrogenism, so it’s pretty low yield,” she said.
For treatment of cutaneous symptoms of PCOS, it’s important for the patient to understand that treatment courses will last at least 6 months. “It’s not a quick fix,” said Dr. Shinkai. Oral contraceptives are a mainstay, and are often sufficient for mild hirsutism. But moderate or severe cases call for high doses of spironolactone (150-200 mg/day). She said she usually combines spironolactone with oral contraceptives, because the drug can lead to menstrual irregularities, which birth control pills can relieve.
Dr. Shinkai reported having no relevant financial disclosures.
SAN FRANCISCO – Dermatologists are often on the frontline when it comes to diagnosing polycystic ovary syndrome (PCOS), which is one reason they should be up to date and aware of the changing diagnostic criteria for the condition, according to Kanade Shinkai, MD.
About one-quarter of patients who are diagnosed with PCOS are seen first by a dermatologist. That’s because skin conditions may be more concerning than reproductive issues in young women.
“Sometimes, people don’t see [irregular menstruation] as a problem,” explained Dr. Shinkai of the department of dermatology at the University of California, San Francisco. “Maybe they’re young, or they’re not trying to get pregnant. But if their hair is falling out, they see that as a problem, or if they have bad acne, or they’re becoming hirsute, they see that as a problem. So, they present to a dermatologist.”
Early recognition of PCOS is important, because many women with the condition go on to develop diabetes, impaired glucose intolerance, hyperlipidemia, hypertension, fertility problems, and obesity.
It used to be that physicians expected patients with PCOS to have menstrual irregularities, biochemical or clinical evidence of hyperandrogenism, and evidence of polycystic ovaries on ultrasound. But just two of the three are now considered enough to warrant a diagnosis.
“Our original view of the classic patient has gone away, and it’s really a heterogeneous phenotype,” Dr. Shinkai said. “Originally, it was all three [criteria], and the patient was obese, and they all had diabetes. Now, we know that’s not true. Every woman who has PCOS has her own version of PCOS.”
Dr. Shinkai’s team conducted a study of clinical markers associated with PCOS and found that some of the classic signs of PCOS may be unreliable.
“Alopecia turns out not to be a very reliable marker,” she explained. “That’s paradigm shifting, I think, because often if patients present with hair loss in a hormonal pattern, they get worked up for PCOS, and it turns out that workup is not always fruitful.” Acne can also be misleading, given its frequency in the general population.
More reliable signs include hirsutism and acanthosis nigricans; 70%-80% of women with hirsutism have PCOS, and 53% of patients with PCOS have hirsutism, most commonly on the trunk. Acanthosis nigricans occurs in 37% of PCOS patients.
“Those are the best specific signs for PCOS,” said Dr. Shinkai. “If we see those, we should probably work the patient up.”
In preparation, the patient should be off of birth control treatment for at least 4 weeks, because hormonal treatment can interfere with test results, Dr Shinkai noted.
She also recommended a transvaginal ultrasound and a free-testosterone test. Consensus statements recommend testing of 17-hydroxyprogesterone, but Dr. Shinkai said she isn’t so sure. “That’s only going to capture about 3% of your patients with cutaneous hyperandrogenism, so it’s pretty low yield,” she said.
For treatment of cutaneous symptoms of PCOS, it’s important for the patient to understand that treatment courses will last at least 6 months. “It’s not a quick fix,” said Dr. Shinkai. Oral contraceptives are a mainstay, and are often sufficient for mild hirsutism. But moderate or severe cases call for high doses of spironolactone (150-200 mg/day). She said she usually combines spironolactone with oral contraceptives, because the drug can lead to menstrual irregularities, which birth control pills can relieve.
Dr. Shinkai reported having no relevant financial disclosures.
SAN FRANCISCO – Dermatologists are often on the frontline when it comes to diagnosing polycystic ovary syndrome (PCOS), which is one reason they should be up to date and aware of the changing diagnostic criteria for the condition, according to Kanade Shinkai, MD.
About one-quarter of patients who are diagnosed with PCOS are seen first by a dermatologist. That’s because skin conditions may be more concerning than reproductive issues in young women.
“Sometimes, people don’t see [irregular menstruation] as a problem,” explained Dr. Shinkai of the department of dermatology at the University of California, San Francisco. “Maybe they’re young, or they’re not trying to get pregnant. But if their hair is falling out, they see that as a problem, or if they have bad acne, or they’re becoming hirsute, they see that as a problem. So, they present to a dermatologist.”
Early recognition of PCOS is important, because many women with the condition go on to develop diabetes, impaired glucose intolerance, hyperlipidemia, hypertension, fertility problems, and obesity.
It used to be that physicians expected patients with PCOS to have menstrual irregularities, biochemical or clinical evidence of hyperandrogenism, and evidence of polycystic ovaries on ultrasound. But just two of the three are now considered enough to warrant a diagnosis.
“Our original view of the classic patient has gone away, and it’s really a heterogeneous phenotype,” Dr. Shinkai said. “Originally, it was all three [criteria], and the patient was obese, and they all had diabetes. Now, we know that’s not true. Every woman who has PCOS has her own version of PCOS.”
Dr. Shinkai’s team conducted a study of clinical markers associated with PCOS and found that some of the classic signs of PCOS may be unreliable.
“Alopecia turns out not to be a very reliable marker,” she explained. “That’s paradigm shifting, I think, because often if patients present with hair loss in a hormonal pattern, they get worked up for PCOS, and it turns out that workup is not always fruitful.” Acne can also be misleading, given its frequency in the general population.
More reliable signs include hirsutism and acanthosis nigricans; 70%-80% of women with hirsutism have PCOS, and 53% of patients with PCOS have hirsutism, most commonly on the trunk. Acanthosis nigricans occurs in 37% of PCOS patients.
“Those are the best specific signs for PCOS,” said Dr. Shinkai. “If we see those, we should probably work the patient up.”
In preparation, the patient should be off of birth control treatment for at least 4 weeks, because hormonal treatment can interfere with test results, Dr Shinkai noted.
She also recommended a transvaginal ultrasound and a free-testosterone test. Consensus statements recommend testing of 17-hydroxyprogesterone, but Dr. Shinkai said she isn’t so sure. “That’s only going to capture about 3% of your patients with cutaneous hyperandrogenism, so it’s pretty low yield,” she said.
For treatment of cutaneous symptoms of PCOS, it’s important for the patient to understand that treatment courses will last at least 6 months. “It’s not a quick fix,” said Dr. Shinkai. Oral contraceptives are a mainstay, and are often sufficient for mild hirsutism. But moderate or severe cases call for high doses of spironolactone (150-200 mg/day). She said she usually combines spironolactone with oral contraceptives, because the drug can lead to menstrual irregularities, which birth control pills can relieve.
Dr. Shinkai reported having no relevant financial disclosures.
AT PDA 2017
Hyperpigmented Patch on the Leg
The Diagnosis: Lichen Aureus
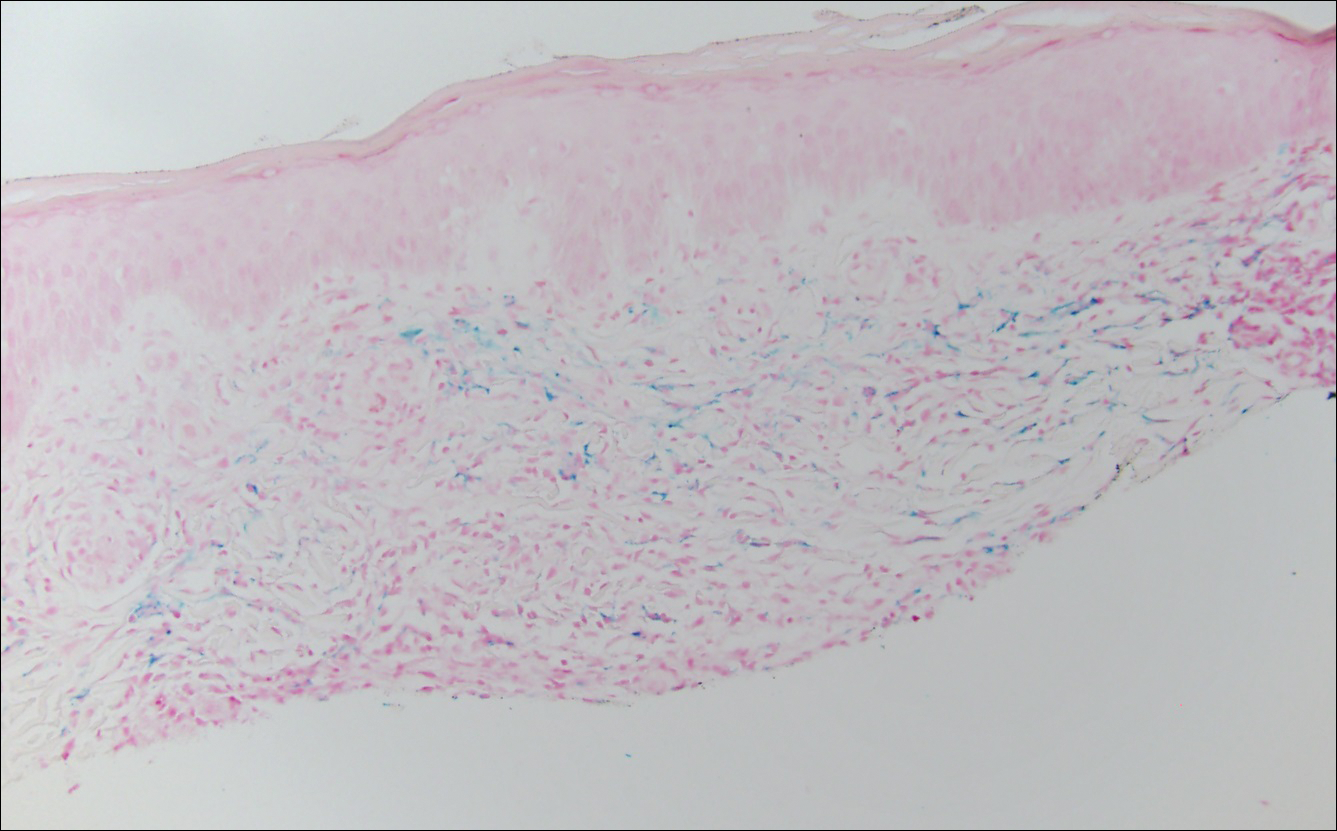
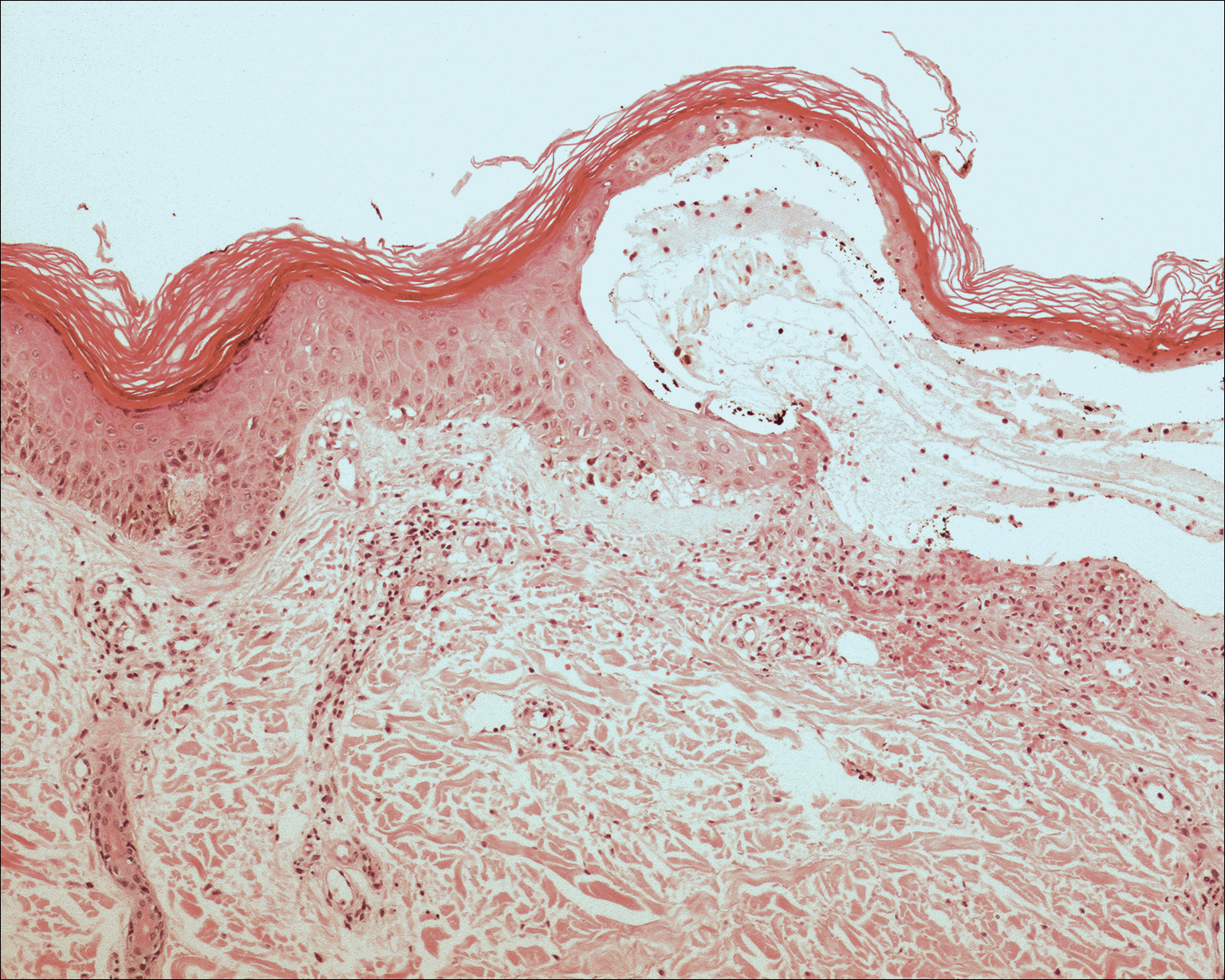
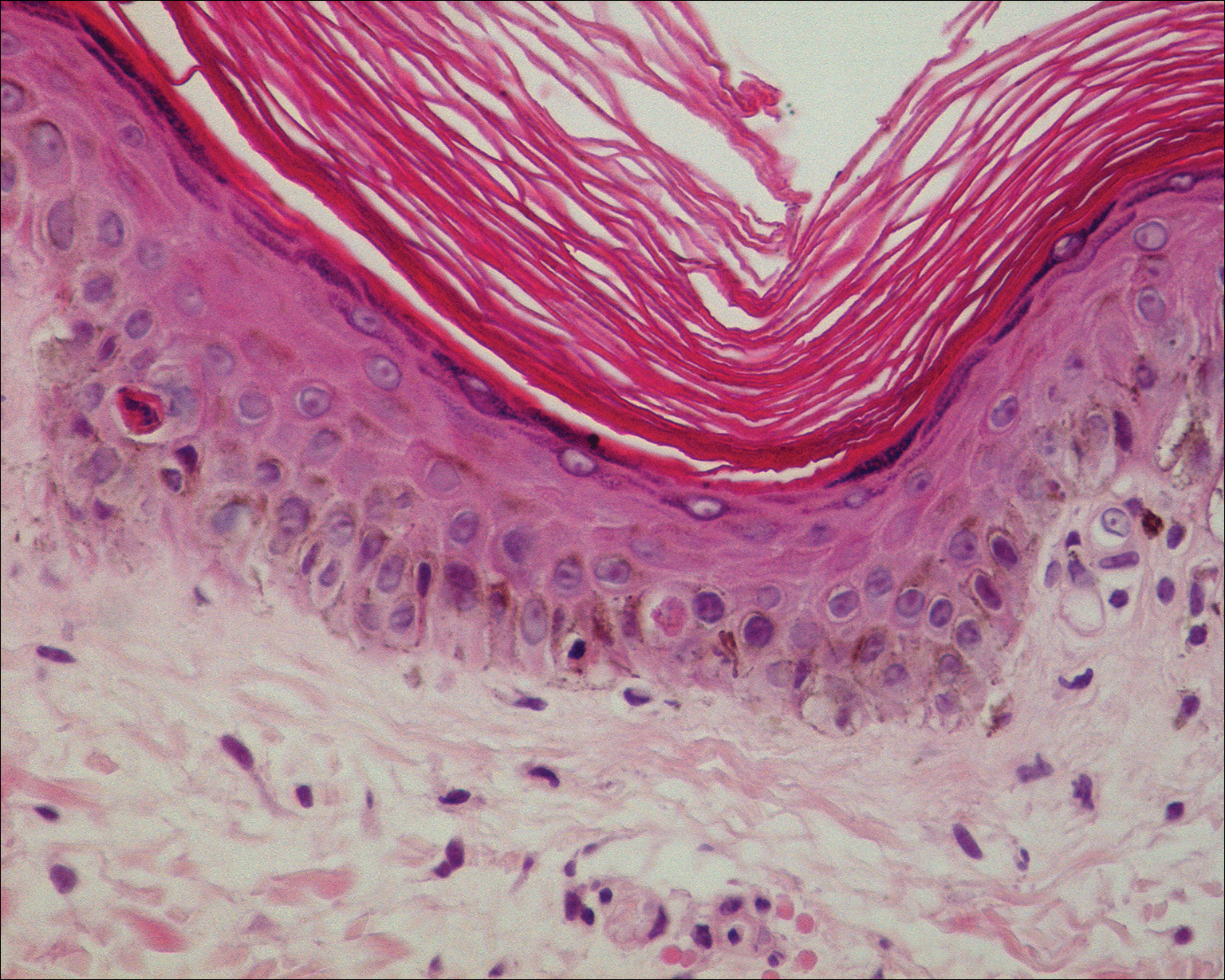
The clinicopathological findings were diagnostic of lichen aureus (LA). Microscopic examination revealed a relatively sparse, superficial, perivascular and interstitial lymphohistiocytic infiltrate with scattered siderophages in the upper dermis. Extravasation of red blood cells also was noted (Figure 1). An immunohistochemical stain for Melan-A highlighted a normal number and distribution of single melanocytes at the dermoepidermal junction with no evidence of pagetoid scatter. A Perls Prussian blue stain for iron demonstrated abundant hemosiderin in the dermis (Figure 2).
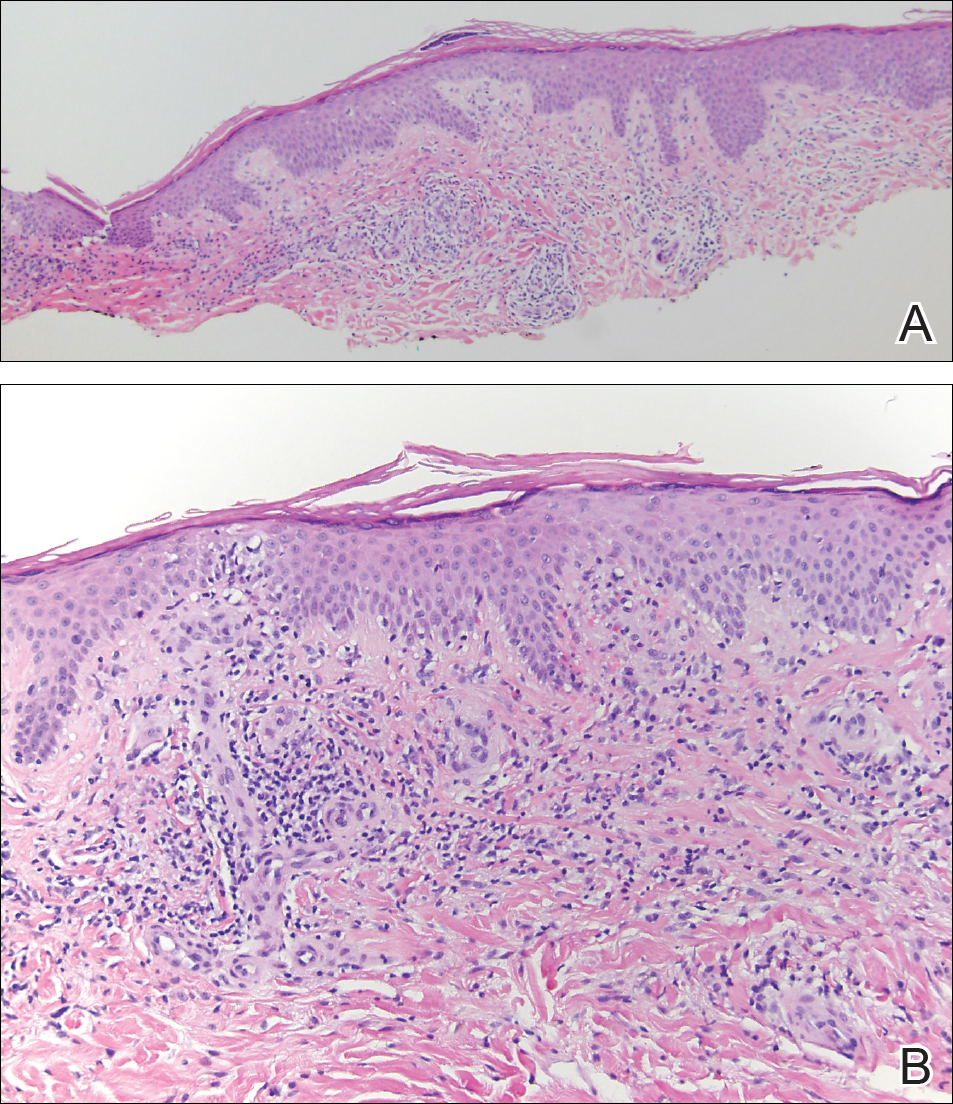
Pigmented purpuric dermatosis (PPD) describes a group of cutaneous lesions that are characterized by petechiae and pigmentary changes. These lesions most commonly present on the lower limbs; however, other sites have been reported.1 This group includes several major clinical forms such as Schamberg disease, LA, purpura annularis telangiectodes of Majocchi, eczematidlike purpura of Doucas and Kapetanakis, and lichenoid PPD of Gougerot and Blum. Lesions typically demonstrate a striking golden brown color clinically and by definition occur in the absence of platelet defects or vasculitis.1
Factors implicated in the pathogenesis of pigmented purpura include gravitational dependency, venous stasis, infection, and drugs.2 It is suggested that cellular immunity may play a role in the development of the disease based on the presence of CD4+ T lymphocytes in the infiltrate and the expression of HLA-DR by these lymphocytes and the keratinocytes.3 Lichen aureus differs in that it relates to increased intravascular pressure from an incompetent valve in an underlying perforating vein.4
Lichen aureus, also referred to as lichen purpuricus, is one major variant of PPD. The name reflects both the characteristic golden brown color and the histopathologic pattern of inflammation.1 Lichen aureus usually presents as a unilateral, asymptomatic, confined single lesion located mainly on the leg,1 though it can develop at other sites or as a localized group of lesions. Extensive lesions have been reported5 and cases with a segmental distribution have been described.6 In contrast, Schamberg disease demonstrates pinhead-sized reddish lesions giving the characteristic cayenne pepper pigmentation. These lesions coalesce to form thumbprint patches that progress proximally.1 Majocchi purpura is annular and telangiectatic, while lichenoid purpura of Gougerot and Blum presents with flat-topped, polygonal, violaceous papules that turn brown over time.
Some authors have championed a role for dermoscopy in diagnosis of LA.7 By dermoscopy, LA demonstrates a diffuse copper background reflecting the lymphohistiocytic dermal infiltrate, red dots and globules representing the extravasated red blood cells and the dilated swollen vessels, and grey dots that reflect the hemosiderin present in the dermis.8
Histologically, LA demonstrates a superficial perivascular infiltrate composed mainly of CD4+ lymphocytes surrounding the superficial capillaries. Over time, red cell extravasation leads to the formation of hemosiderin-laden macrophages, which can be highlighted with Perls Prussian blue stain. A bandlike infiltrate with thin strands of collagen separating it from the epidermis also may be noted.9
An important consideration in the differential diagnosis of PPD is mycosis fungoides (MF). Mycosis fungoides is a cutaneous T-cell lymphoma that clinically presents as a single or multiple hypopigmented or hyperpigmented patches or as erythematous scaly lesions in the patch or plaque stage. These lesions eventually may evolve into tumor stage.10 Mycosis fungoides may mimic PPD clinically and/or histopathologically, and rarely PPD also may precede MF.11 Involvement of the trunk, especially the lower abdomen and buttock region, favors a diagnosis of MF. Typically, histopathologic examination of MF demonstrates an epidermotropic lymphocytic infiltrate composed of atypical cerebriform lymphocytes overlying papillary dermal fibrosis. Although classic MF would be difficult to confuse with PPD, the atrophic lichenoid pattern of MF may show remarkable overlap with PPD.12 Such cases require clinicopathologic correlation, immunophenotyping of the epidermotropic lymphocytes, and occasionally T-cell clonality studies.
Lichen aureus is a chronic persistent disease unless the underlying incompetent perforator vessel is ligated. Various treatments have been used for other forms of pigmented purpura including topical corticosteroids, topical tacrolimus, systemic vasodilators such as prostacyclin and pentoxifylline, and phototherapy.1 Clinical follow-up is recommended for lesions that show some clinical or histopathological overlap with MF. Additional biopsies also may prove useful in establishing a definitive diagnosis in ambiguous cases.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Newton RC, Raimer SS. Pigmented purpuric eruptions. Dermatol Clin. 1985;3:165-169.
- Aiba S, Tagami H. Immunohistologic studies in Schamberg's disease. evidence for cellular immune reaction in lesional skin. Arch Dermatol. 1988;124:1058-1062.
- English J. Lichen aureus. J Am Acad Dermatol. 1985;12(2, pt 1):377-379.
- Duhra P, Tan CY. Lichen aureus. Br J Dermatol. 1986;114:395.
- Moche J, Glassman S, Modi D, et al. Segmental lichen aureus: a report of two cases treated with methylprednisolone aceponate. Australas J Dermatol. 2011;52:E15-E18.
- Zaballos P, Puig S, Malvehy J. Dermoscopy of pigmented purpuric dermatoses (lichen aureus): a useful tool for clinical diagnosis. Arch Dermatol. 2004;140:1290-1291.
- Portela PS, Melo DF, Ormiga P, et al. Dermoscopy of lichen aureus. An Bras Dermatol. 2013;88:253-255.
- Smoller BR, Kamel OW. Pigmented purpuric eruptions: immunopathologic studies supportive of a common immunophenotype. J Cutan Pathol. 1991;18:423-427.
- Jaffe ES, Harris NL, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. a progress report. Am J Clin Pathol. 1999;111(1 suppl 1):S8-S12.
- Hanna S, Walsh N, D'Intino Y, et al. Mycosis fungoides presenting as pigmented purpuric dermatitis. Pediatr Dermatol. 2006;23:350-354.
- Toro JR, Sander CA, LeBoit PE. Persistent pigmented purpuric dermatitis and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
The Diagnosis: Lichen Aureus
The clinicopathological findings were diagnostic of lichen aureus (LA). Microscopic examination revealed a relatively sparse, superficial, perivascular and interstitial lymphohistiocytic infiltrate with scattered siderophages in the upper dermis. Extravasation of red blood cells also was noted (Figure 1). An immunohistochemical stain for Melan-A highlighted a normal number and distribution of single melanocytes at the dermoepidermal junction with no evidence of pagetoid scatter. A Perls Prussian blue stain for iron demonstrated abundant hemosiderin in the dermis (Figure 2).
Pigmented purpuric dermatosis (PPD) describes a group of cutaneous lesions that are characterized by petechiae and pigmentary changes. These lesions most commonly present on the lower limbs; however, other sites have been reported.1 This group includes several major clinical forms such as Schamberg disease, LA, purpura annularis telangiectodes of Majocchi, eczematidlike purpura of Doucas and Kapetanakis, and lichenoid PPD of Gougerot and Blum. Lesions typically demonstrate a striking golden brown color clinically and by definition occur in the absence of platelet defects or vasculitis.1
Factors implicated in the pathogenesis of pigmented purpura include gravitational dependency, venous stasis, infection, and drugs.2 It is suggested that cellular immunity may play a role in the development of the disease based on the presence of CD4+ T lymphocytes in the infiltrate and the expression of HLA-DR by these lymphocytes and the keratinocytes.3 Lichen aureus differs in that it relates to increased intravascular pressure from an incompetent valve in an underlying perforating vein.4
Lichen aureus, also referred to as lichen purpuricus, is one major variant of PPD. The name reflects both the characteristic golden brown color and the histopathologic pattern of inflammation.1 Lichen aureus usually presents as a unilateral, asymptomatic, confined single lesion located mainly on the leg,1 though it can develop at other sites or as a localized group of lesions. Extensive lesions have been reported5 and cases with a segmental distribution have been described.6 In contrast, Schamberg disease demonstrates pinhead-sized reddish lesions giving the characteristic cayenne pepper pigmentation. These lesions coalesce to form thumbprint patches that progress proximally.1 Majocchi purpura is annular and telangiectatic, while lichenoid purpura of Gougerot and Blum presents with flat-topped, polygonal, violaceous papules that turn brown over time.
Some authors have championed a role for dermoscopy in diagnosis of LA.7 By dermoscopy, LA demonstrates a diffuse copper background reflecting the lymphohistiocytic dermal infiltrate, red dots and globules representing the extravasated red blood cells and the dilated swollen vessels, and grey dots that reflect the hemosiderin present in the dermis.8
Histologically, LA demonstrates a superficial perivascular infiltrate composed mainly of CD4+ lymphocytes surrounding the superficial capillaries. Over time, red cell extravasation leads to the formation of hemosiderin-laden macrophages, which can be highlighted with Perls Prussian blue stain. A bandlike infiltrate with thin strands of collagen separating it from the epidermis also may be noted.9
An important consideration in the differential diagnosis of PPD is mycosis fungoides (MF). Mycosis fungoides is a cutaneous T-cell lymphoma that clinically presents as a single or multiple hypopigmented or hyperpigmented patches or as erythematous scaly lesions in the patch or plaque stage. These lesions eventually may evolve into tumor stage.10 Mycosis fungoides may mimic PPD clinically and/or histopathologically, and rarely PPD also may precede MF.11 Involvement of the trunk, especially the lower abdomen and buttock region, favors a diagnosis of MF. Typically, histopathologic examination of MF demonstrates an epidermotropic lymphocytic infiltrate composed of atypical cerebriform lymphocytes overlying papillary dermal fibrosis. Although classic MF would be difficult to confuse with PPD, the atrophic lichenoid pattern of MF may show remarkable overlap with PPD.12 Such cases require clinicopathologic correlation, immunophenotyping of the epidermotropic lymphocytes, and occasionally T-cell clonality studies.
Lichen aureus is a chronic persistent disease unless the underlying incompetent perforator vessel is ligated. Various treatments have been used for other forms of pigmented purpura including topical corticosteroids, topical tacrolimus, systemic vasodilators such as prostacyclin and pentoxifylline, and phototherapy.1 Clinical follow-up is recommended for lesions that show some clinical or histopathological overlap with MF. Additional biopsies also may prove useful in establishing a definitive diagnosis in ambiguous cases.
The Diagnosis: Lichen Aureus
The clinicopathological findings were diagnostic of lichen aureus (LA). Microscopic examination revealed a relatively sparse, superficial, perivascular and interstitial lymphohistiocytic infiltrate with scattered siderophages in the upper dermis. Extravasation of red blood cells also was noted (Figure 1). An immunohistochemical stain for Melan-A highlighted a normal number and distribution of single melanocytes at the dermoepidermal junction with no evidence of pagetoid scatter. A Perls Prussian blue stain for iron demonstrated abundant hemosiderin in the dermis (Figure 2).
Pigmented purpuric dermatosis (PPD) describes a group of cutaneous lesions that are characterized by petechiae and pigmentary changes. These lesions most commonly present on the lower limbs; however, other sites have been reported.1 This group includes several major clinical forms such as Schamberg disease, LA, purpura annularis telangiectodes of Majocchi, eczematidlike purpura of Doucas and Kapetanakis, and lichenoid PPD of Gougerot and Blum. Lesions typically demonstrate a striking golden brown color clinically and by definition occur in the absence of platelet defects or vasculitis.1
Factors implicated in the pathogenesis of pigmented purpura include gravitational dependency, venous stasis, infection, and drugs.2 It is suggested that cellular immunity may play a role in the development of the disease based on the presence of CD4+ T lymphocytes in the infiltrate and the expression of HLA-DR by these lymphocytes and the keratinocytes.3 Lichen aureus differs in that it relates to increased intravascular pressure from an incompetent valve in an underlying perforating vein.4
Lichen aureus, also referred to as lichen purpuricus, is one major variant of PPD. The name reflects both the characteristic golden brown color and the histopathologic pattern of inflammation.1 Lichen aureus usually presents as a unilateral, asymptomatic, confined single lesion located mainly on the leg,1 though it can develop at other sites or as a localized group of lesions. Extensive lesions have been reported5 and cases with a segmental distribution have been described.6 In contrast, Schamberg disease demonstrates pinhead-sized reddish lesions giving the characteristic cayenne pepper pigmentation. These lesions coalesce to form thumbprint patches that progress proximally.1 Majocchi purpura is annular and telangiectatic, while lichenoid purpura of Gougerot and Blum presents with flat-topped, polygonal, violaceous papules that turn brown over time.
Some authors have championed a role for dermoscopy in diagnosis of LA.7 By dermoscopy, LA demonstrates a diffuse copper background reflecting the lymphohistiocytic dermal infiltrate, red dots and globules representing the extravasated red blood cells and the dilated swollen vessels, and grey dots that reflect the hemosiderin present in the dermis.8
Histologically, LA demonstrates a superficial perivascular infiltrate composed mainly of CD4+ lymphocytes surrounding the superficial capillaries. Over time, red cell extravasation leads to the formation of hemosiderin-laden macrophages, which can be highlighted with Perls Prussian blue stain. A bandlike infiltrate with thin strands of collagen separating it from the epidermis also may be noted.9
An important consideration in the differential diagnosis of PPD is mycosis fungoides (MF). Mycosis fungoides is a cutaneous T-cell lymphoma that clinically presents as a single or multiple hypopigmented or hyperpigmented patches or as erythematous scaly lesions in the patch or plaque stage. These lesions eventually may evolve into tumor stage.10 Mycosis fungoides may mimic PPD clinically and/or histopathologically, and rarely PPD also may precede MF.11 Involvement of the trunk, especially the lower abdomen and buttock region, favors a diagnosis of MF. Typically, histopathologic examination of MF demonstrates an epidermotropic lymphocytic infiltrate composed of atypical cerebriform lymphocytes overlying papillary dermal fibrosis. Although classic MF would be difficult to confuse with PPD, the atrophic lichenoid pattern of MF may show remarkable overlap with PPD.12 Such cases require clinicopathologic correlation, immunophenotyping of the epidermotropic lymphocytes, and occasionally T-cell clonality studies.
Lichen aureus is a chronic persistent disease unless the underlying incompetent perforator vessel is ligated. Various treatments have been used for other forms of pigmented purpura including topical corticosteroids, topical tacrolimus, systemic vasodilators such as prostacyclin and pentoxifylline, and phototherapy.1 Clinical follow-up is recommended for lesions that show some clinical or histopathological overlap with MF. Additional biopsies also may prove useful in establishing a definitive diagnosis in ambiguous cases.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Newton RC, Raimer SS. Pigmented purpuric eruptions. Dermatol Clin. 1985;3:165-169.
- Aiba S, Tagami H. Immunohistologic studies in Schamberg's disease. evidence for cellular immune reaction in lesional skin. Arch Dermatol. 1988;124:1058-1062.
- English J. Lichen aureus. J Am Acad Dermatol. 1985;12(2, pt 1):377-379.
- Duhra P, Tan CY. Lichen aureus. Br J Dermatol. 1986;114:395.
- Moche J, Glassman S, Modi D, et al. Segmental lichen aureus: a report of two cases treated with methylprednisolone aceponate. Australas J Dermatol. 2011;52:E15-E18.
- Zaballos P, Puig S, Malvehy J. Dermoscopy of pigmented purpuric dermatoses (lichen aureus): a useful tool for clinical diagnosis. Arch Dermatol. 2004;140:1290-1291.
- Portela PS, Melo DF, Ormiga P, et al. Dermoscopy of lichen aureus. An Bras Dermatol. 2013;88:253-255.
- Smoller BR, Kamel OW. Pigmented purpuric eruptions: immunopathologic studies supportive of a common immunophenotype. J Cutan Pathol. 1991;18:423-427.
- Jaffe ES, Harris NL, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. a progress report. Am J Clin Pathol. 1999;111(1 suppl 1):S8-S12.
- Hanna S, Walsh N, D'Intino Y, et al. Mycosis fungoides presenting as pigmented purpuric dermatitis. Pediatr Dermatol. 2006;23:350-354.
- Toro JR, Sander CA, LeBoit PE. Persistent pigmented purpuric dermatitis and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Newton RC, Raimer SS. Pigmented purpuric eruptions. Dermatol Clin. 1985;3:165-169.
- Aiba S, Tagami H. Immunohistologic studies in Schamberg's disease. evidence for cellular immune reaction in lesional skin. Arch Dermatol. 1988;124:1058-1062.
- English J. Lichen aureus. J Am Acad Dermatol. 1985;12(2, pt 1):377-379.
- Duhra P, Tan CY. Lichen aureus. Br J Dermatol. 1986;114:395.
- Moche J, Glassman S, Modi D, et al. Segmental lichen aureus: a report of two cases treated with methylprednisolone aceponate. Australas J Dermatol. 2011;52:E15-E18.
- Zaballos P, Puig S, Malvehy J. Dermoscopy of pigmented purpuric dermatoses (lichen aureus): a useful tool for clinical diagnosis. Arch Dermatol. 2004;140:1290-1291.
- Portela PS, Melo DF, Ormiga P, et al. Dermoscopy of lichen aureus. An Bras Dermatol. 2013;88:253-255.
- Smoller BR, Kamel OW. Pigmented purpuric eruptions: immunopathologic studies supportive of a common immunophenotype. J Cutan Pathol. 1991;18:423-427.
- Jaffe ES, Harris NL, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. a progress report. Am J Clin Pathol. 1999;111(1 suppl 1):S8-S12.
- Hanna S, Walsh N, D'Intino Y, et al. Mycosis fungoides presenting as pigmented purpuric dermatitis. Pediatr Dermatol. 2006;23:350-354.
- Toro JR, Sander CA, LeBoit PE. Persistent pigmented purpuric dermatitis and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
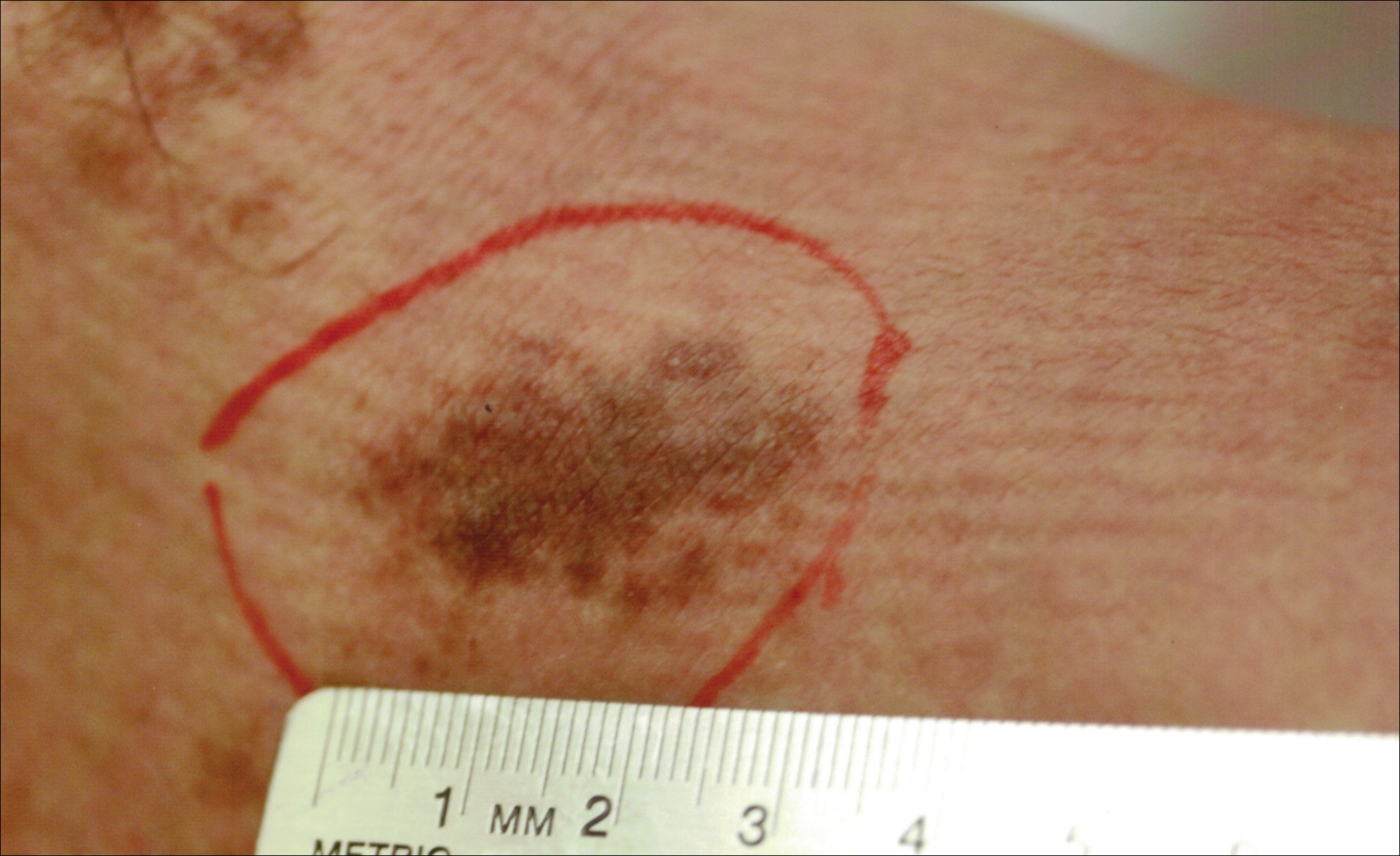
A 32-year-old man presented with an asymptomatic pigmented lesion on the left foot that developed over the course of 4 months. Physical examination revealed a 4-cm asymmetrical, deeply pigmented macule on the left foot. A shave biopsy of the lesion was performed.
Bullous Lesions in a Neonate
The Diagnosis: Incontinentia Pigmenti
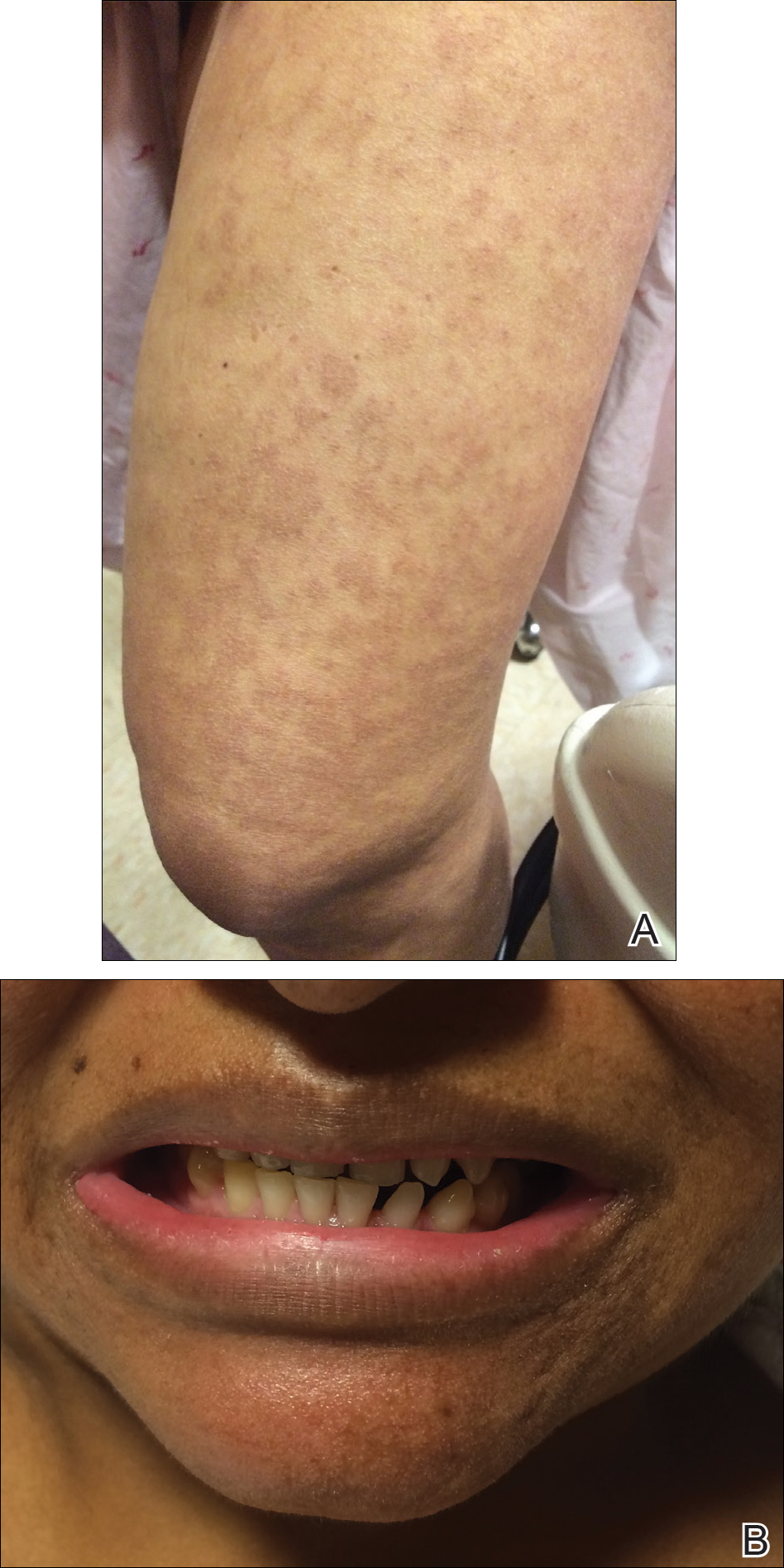
The infant's mother was noted to have diffuse hypopigmented patches over the trunk, arms, and legs (present since adolescence) with whorled cicatricial alopecia of the vertex scalp and peg-shaped teeth (Figure). Together, these findings suggested incontinentia pigmenti (IP), which the mother revealed she had been diagnosed with in childhood. The infant's characteristic lesions in the setting of her mother's diagnosed genodermatosis confirmed the diagnosis of IP.
Incontinentia pigmenti is an X-linked dominant disorder that presents with many classic dermatologic, dental, neurologic, and ophthalmologic findings. The causative mutation occurs in IKBKG/NEMO (inhibitor of κ polypeptide gene enhancer in B-cells, kinase γ/nuclear factor-κB essential modulator) gene on Xq28, disabling the resultant protein that normally protects cells from tumor necrosis factor family-induced apoptosis.1 Incontinentia pigmenti usually is lethal in males and causes an unbalanced X-inactivation in surviving female IP patients. Occurring at a rate of 1.2 per 100,000 births,2 IP typically presents in female infants with skin lesions patterned along Blaschko lines that evolve in 4 stages over a lifetime.3 Stage I, presenting in the neonatal period, manifests as vesiculobullous eruptions on the limbs and scalp. Stages II to IV vary in duration from months to years and are comprised of a verrucous stage, a hyperpigmented stage, and a hypopigmented stage, respectively.3 All stages of IP can overlap and coexist.
The vesiculobullous findings in infants with IP may be mistakenly attributed to other diseases with prominent vesicular or bullous components including herpes simplex virus, epidermolysis bullosa, and infantile acropustulosis. With neonatal herpes simplex virus infection, vesicular skin or mucocutaneous lesions occur 9 to 11 days after birth and can be confirmed by specimen culture or qualitative polymerase chain reaction, while stage I of IP appears within the first 6 to 8 weeks of life and can be present at birth.4 The hallmark of epidermolysis bullosa, caused by mutations in keratins 5 and 14, is blistering erosions of the skin in response to frictional stress,1 thus these lesions do not follow Blaschko lines. Infantile acropustulosis, a nonheritable vesiculopustular eruption of the hands and feet, rarely occurs in the immediate newborn period; it most often appears in the 3- to 6-month age range with recurrent eruptions at 3- to 4-week intervals.5 Focal dermal hypoplasia is another X-linked dominant disorder with blaschkolinear findings at birth that presents with pink or red, angular, atrophic macules, in contrast to the bullous lesions of IP.6
Incontinentia pigmenti may encompass a wide range of systemic symptoms in addition to the classic dermatologic findings. Notably, central nervous system defects are concurrent in up to 40% of IP cases, with seizures, mental retardation, and spastic paresis being the most common sequelae.7 Teeth defects, seen in 35% of patients, include delayed primary dentition and peg-shaped teeth. Many patients will experience ophthalmologic defects including vision problems (16%) and retinopathy (15%).7
The cutaneous eruptions of IP may be treated with topical corticosteroids or topical tacrolimus, and vesicles should be left intact and monitored for signs of infection.8,9 Seizures, if present, should be treated with anticonvulsants, and regular neuropsychiatric monitoring and physical rehabilitation may be warranted. Patients should be regularly monitored for retinopathy beginning at the time of diagnosis. Retinal fibrovascular proliferation is treated with xenon laser photocoagulation to reduce the high risk for retinal detachment in this population.10,11 Older and younger at-risk relatives must be evaluated by genetic testing or thorough physical examination to clarify their disease status and determine the need for additional genetic counseling.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012.
- Prevalence and incidence of rare diseases: bibliographic data. Orphanet Report Series, Rare Diseases collection. http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf. Published June 2017. Accessed July 13, 2017.
- Scheuerle AE, Ursini MV. Incontinentia pigmenti. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. Seattle, WA: University of Washington; 2015. http://www.ncbi.nlm.nih.gov/books/NBK1472/. Accessed July 25, 2017.
- James SH, Kimberlin DW. Neonatal herpes simplex virus infection. Infect Dis Clin North Am. 2015;29:391-400.
- Eichenfield LF, Frieden IJ, Mathes E, et al, eds. Neonatal and Infant Dermatology. Philadelphia, PA: Saunders; 2015.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Fusco F, Paciolla M, Conte MI, et al. Incontinentia pigmenti: report on data from 2000 to 2013. Orphanet J Rare Dis. 2014;9:93.
- Jessup CJ, Morgan SC, Cohen LM, et al. Incontinentia pigmenti: treatment of IP with topical tacrolimus. J Drugs Dermatol. 2009;8:944-946.
- Kaya TI, Tursen U, Ikizoglu G. Therapeutic use of topical corticosteroids in the vesiculobullous lesions of incontinentia pigmenti [published online June 1, 2009]. Clin Exp Dermatol. 2009;34:E611-E613.
- Nguyen JK, Brady-Mccreery KM. Laser photocoagulation in preproliferative retinopathy of incontinentia pigmenti. J AAPOS. 2001;5:258-259.
- Chen CJ, Han IC, Tian J, et al. Extended follow-up of treated and untreated retinopathy in incontinentia pigmenti: analysis of peripheral vascular changes and incidence of retinal detachment. JAMA Ophthalmol. 2015;133:542-548.
The Diagnosis: Incontinentia Pigmenti
The infant's mother was noted to have diffuse hypopigmented patches over the trunk, arms, and legs (present since adolescence) with whorled cicatricial alopecia of the vertex scalp and peg-shaped teeth (Figure). Together, these findings suggested incontinentia pigmenti (IP), which the mother revealed she had been diagnosed with in childhood. The infant's characteristic lesions in the setting of her mother's diagnosed genodermatosis confirmed the diagnosis of IP.
Incontinentia pigmenti is an X-linked dominant disorder that presents with many classic dermatologic, dental, neurologic, and ophthalmologic findings. The causative mutation occurs in IKBKG/NEMO (inhibitor of κ polypeptide gene enhancer in B-cells, kinase γ/nuclear factor-κB essential modulator) gene on Xq28, disabling the resultant protein that normally protects cells from tumor necrosis factor family-induced apoptosis.1 Incontinentia pigmenti usually is lethal in males and causes an unbalanced X-inactivation in surviving female IP patients. Occurring at a rate of 1.2 per 100,000 births,2 IP typically presents in female infants with skin lesions patterned along Blaschko lines that evolve in 4 stages over a lifetime.3 Stage I, presenting in the neonatal period, manifests as vesiculobullous eruptions on the limbs and scalp. Stages II to IV vary in duration from months to years and are comprised of a verrucous stage, a hyperpigmented stage, and a hypopigmented stage, respectively.3 All stages of IP can overlap and coexist.
The vesiculobullous findings in infants with IP may be mistakenly attributed to other diseases with prominent vesicular or bullous components including herpes simplex virus, epidermolysis bullosa, and infantile acropustulosis. With neonatal herpes simplex virus infection, vesicular skin or mucocutaneous lesions occur 9 to 11 days after birth and can be confirmed by specimen culture or qualitative polymerase chain reaction, while stage I of IP appears within the first 6 to 8 weeks of life and can be present at birth.4 The hallmark of epidermolysis bullosa, caused by mutations in keratins 5 and 14, is blistering erosions of the skin in response to frictional stress,1 thus these lesions do not follow Blaschko lines. Infantile acropustulosis, a nonheritable vesiculopustular eruption of the hands and feet, rarely occurs in the immediate newborn period; it most often appears in the 3- to 6-month age range with recurrent eruptions at 3- to 4-week intervals.5 Focal dermal hypoplasia is another X-linked dominant disorder with blaschkolinear findings at birth that presents with pink or red, angular, atrophic macules, in contrast to the bullous lesions of IP.6
Incontinentia pigmenti may encompass a wide range of systemic symptoms in addition to the classic dermatologic findings. Notably, central nervous system defects are concurrent in up to 40% of IP cases, with seizures, mental retardation, and spastic paresis being the most common sequelae.7 Teeth defects, seen in 35% of patients, include delayed primary dentition and peg-shaped teeth. Many patients will experience ophthalmologic defects including vision problems (16%) and retinopathy (15%).7
The cutaneous eruptions of IP may be treated with topical corticosteroids or topical tacrolimus, and vesicles should be left intact and monitored for signs of infection.8,9 Seizures, if present, should be treated with anticonvulsants, and regular neuropsychiatric monitoring and physical rehabilitation may be warranted. Patients should be regularly monitored for retinopathy beginning at the time of diagnosis. Retinal fibrovascular proliferation is treated with xenon laser photocoagulation to reduce the high risk for retinal detachment in this population.10,11 Older and younger at-risk relatives must be evaluated by genetic testing or thorough physical examination to clarify their disease status and determine the need for additional genetic counseling.
The Diagnosis: Incontinentia Pigmenti
The infant's mother was noted to have diffuse hypopigmented patches over the trunk, arms, and legs (present since adolescence) with whorled cicatricial alopecia of the vertex scalp and peg-shaped teeth (Figure). Together, these findings suggested incontinentia pigmenti (IP), which the mother revealed she had been diagnosed with in childhood. The infant's characteristic lesions in the setting of her mother's diagnosed genodermatosis confirmed the diagnosis of IP.
Incontinentia pigmenti is an X-linked dominant disorder that presents with many classic dermatologic, dental, neurologic, and ophthalmologic findings. The causative mutation occurs in IKBKG/NEMO (inhibitor of κ polypeptide gene enhancer in B-cells, kinase γ/nuclear factor-κB essential modulator) gene on Xq28, disabling the resultant protein that normally protects cells from tumor necrosis factor family-induced apoptosis.1 Incontinentia pigmenti usually is lethal in males and causes an unbalanced X-inactivation in surviving female IP patients. Occurring at a rate of 1.2 per 100,000 births,2 IP typically presents in female infants with skin lesions patterned along Blaschko lines that evolve in 4 stages over a lifetime.3 Stage I, presenting in the neonatal period, manifests as vesiculobullous eruptions on the limbs and scalp. Stages II to IV vary in duration from months to years and are comprised of a verrucous stage, a hyperpigmented stage, and a hypopigmented stage, respectively.3 All stages of IP can overlap and coexist.
The vesiculobullous findings in infants with IP may be mistakenly attributed to other diseases with prominent vesicular or bullous components including herpes simplex virus, epidermolysis bullosa, and infantile acropustulosis. With neonatal herpes simplex virus infection, vesicular skin or mucocutaneous lesions occur 9 to 11 days after birth and can be confirmed by specimen culture or qualitative polymerase chain reaction, while stage I of IP appears within the first 6 to 8 weeks of life and can be present at birth.4 The hallmark of epidermolysis bullosa, caused by mutations in keratins 5 and 14, is blistering erosions of the skin in response to frictional stress,1 thus these lesions do not follow Blaschko lines. Infantile acropustulosis, a nonheritable vesiculopustular eruption of the hands and feet, rarely occurs in the immediate newborn period; it most often appears in the 3- to 6-month age range with recurrent eruptions at 3- to 4-week intervals.5 Focal dermal hypoplasia is another X-linked dominant disorder with blaschkolinear findings at birth that presents with pink or red, angular, atrophic macules, in contrast to the bullous lesions of IP.6
Incontinentia pigmenti may encompass a wide range of systemic symptoms in addition to the classic dermatologic findings. Notably, central nervous system defects are concurrent in up to 40% of IP cases, with seizures, mental retardation, and spastic paresis being the most common sequelae.7 Teeth defects, seen in 35% of patients, include delayed primary dentition and peg-shaped teeth. Many patients will experience ophthalmologic defects including vision problems (16%) and retinopathy (15%).7
The cutaneous eruptions of IP may be treated with topical corticosteroids or topical tacrolimus, and vesicles should be left intact and monitored for signs of infection.8,9 Seizures, if present, should be treated with anticonvulsants, and regular neuropsychiatric monitoring and physical rehabilitation may be warranted. Patients should be regularly monitored for retinopathy beginning at the time of diagnosis. Retinal fibrovascular proliferation is treated with xenon laser photocoagulation to reduce the high risk for retinal detachment in this population.10,11 Older and younger at-risk relatives must be evaluated by genetic testing or thorough physical examination to clarify their disease status and determine the need for additional genetic counseling.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012.
- Prevalence and incidence of rare diseases: bibliographic data. Orphanet Report Series, Rare Diseases collection. http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf. Published June 2017. Accessed July 13, 2017.
- Scheuerle AE, Ursini MV. Incontinentia pigmenti. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. Seattle, WA: University of Washington; 2015. http://www.ncbi.nlm.nih.gov/books/NBK1472/. Accessed July 25, 2017.
- James SH, Kimberlin DW. Neonatal herpes simplex virus infection. Infect Dis Clin North Am. 2015;29:391-400.
- Eichenfield LF, Frieden IJ, Mathes E, et al, eds. Neonatal and Infant Dermatology. Philadelphia, PA: Saunders; 2015.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Fusco F, Paciolla M, Conte MI, et al. Incontinentia pigmenti: report on data from 2000 to 2013. Orphanet J Rare Dis. 2014;9:93.
- Jessup CJ, Morgan SC, Cohen LM, et al. Incontinentia pigmenti: treatment of IP with topical tacrolimus. J Drugs Dermatol. 2009;8:944-946.
- Kaya TI, Tursen U, Ikizoglu G. Therapeutic use of topical corticosteroids in the vesiculobullous lesions of incontinentia pigmenti [published online June 1, 2009]. Clin Exp Dermatol. 2009;34:E611-E613.
- Nguyen JK, Brady-Mccreery KM. Laser photocoagulation in preproliferative retinopathy of incontinentia pigmenti. J AAPOS. 2001;5:258-259.
- Chen CJ, Han IC, Tian J, et al. Extended follow-up of treated and untreated retinopathy in incontinentia pigmenti: analysis of peripheral vascular changes and incidence of retinal detachment. JAMA Ophthalmol. 2015;133:542-548.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012.
- Prevalence and incidence of rare diseases: bibliographic data. Orphanet Report Series, Rare Diseases collection. http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf. Published June 2017. Accessed July 13, 2017.
- Scheuerle AE, Ursini MV. Incontinentia pigmenti. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. Seattle, WA: University of Washington; 2015. http://www.ncbi.nlm.nih.gov/books/NBK1472/. Accessed July 25, 2017.
- James SH, Kimberlin DW. Neonatal herpes simplex virus infection. Infect Dis Clin North Am. 2015;29:391-400.
- Eichenfield LF, Frieden IJ, Mathes E, et al, eds. Neonatal and Infant Dermatology. Philadelphia, PA: Saunders; 2015.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Fusco F, Paciolla M, Conte MI, et al. Incontinentia pigmenti: report on data from 2000 to 2013. Orphanet J Rare Dis. 2014;9:93.
- Jessup CJ, Morgan SC, Cohen LM, et al. Incontinentia pigmenti: treatment of IP with topical tacrolimus. J Drugs Dermatol. 2009;8:944-946.
- Kaya TI, Tursen U, Ikizoglu G. Therapeutic use of topical corticosteroids in the vesiculobullous lesions of incontinentia pigmenti [published online June 1, 2009]. Clin Exp Dermatol. 2009;34:E611-E613.
- Nguyen JK, Brady-Mccreery KM. Laser photocoagulation in preproliferative retinopathy of incontinentia pigmenti. J AAPOS. 2001;5:258-259.
- Chen CJ, Han IC, Tian J, et al. Extended follow-up of treated and untreated retinopathy in incontinentia pigmenti: analysis of peripheral vascular changes and incidence of retinal detachment. JAMA Ophthalmol. 2015;133:542-548.
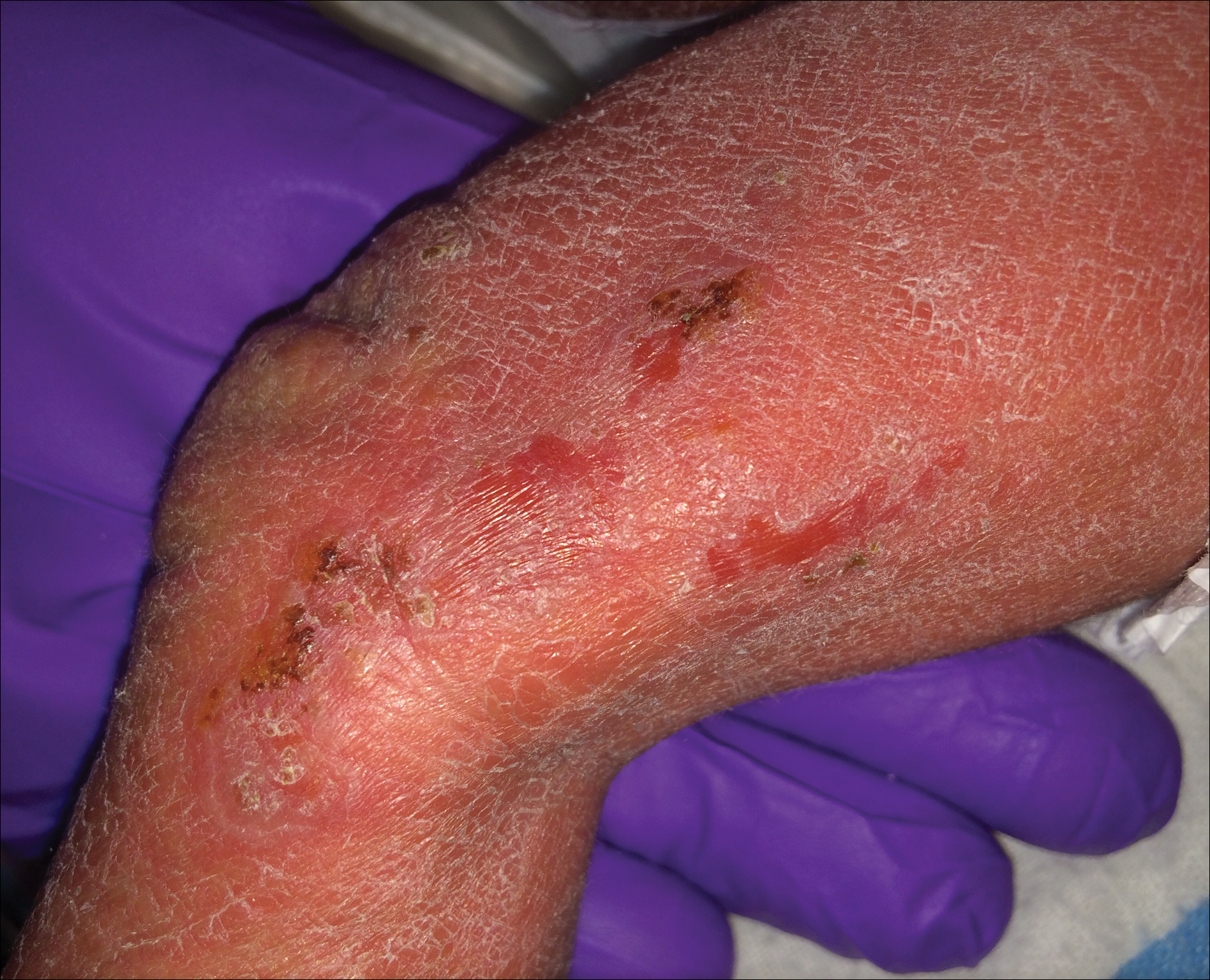
A 1-day-old Hispanic female infant was born via uncomplicated vaginal delivery at 41 weeks' gestation after a normal pregnancy. Linear plaques containing multiple ruptured vesicles and bullae following Blaschko lines were noted on the right medial thigh and anterior arm. The infant was afebrile and generally well-appearing.
Space Heater–Induced Bullous Erythema Ab Igne
To the Editor:
Erythema ab igne (EAI) is a reticular erythematous hyperpigmentation of skin repeatedly exposed to moderate heat.1 It usually is asymptomatic, though some patients report itching or burning at the site.2 Historically caused by exposure to coal stoves or open fires, EAI has become increasingly common among individuals using space heaters, heating pads, or laptop computers near bare skin.2,3 Although EAI itself is benign and usually resolves with the removal of the exposure, it remains of clinical importance because of its association with underlying chronic disease, as chronic pain often is managed with frequent heating pad or hot water bottle use.2 Additionally, accurate diagnosis is important given the future risk for malignancy, as chronic changes of EAI have been reported to lead to squamous cell carcinoma or rarely Merkel cell carcinoma.2 Erythema ab igne is not traditionally associated with the formation of bullae; however, we present a case of bullous EAI that we believe highlights the importance of including this condition in the differential diagnosis of bullous disorders.
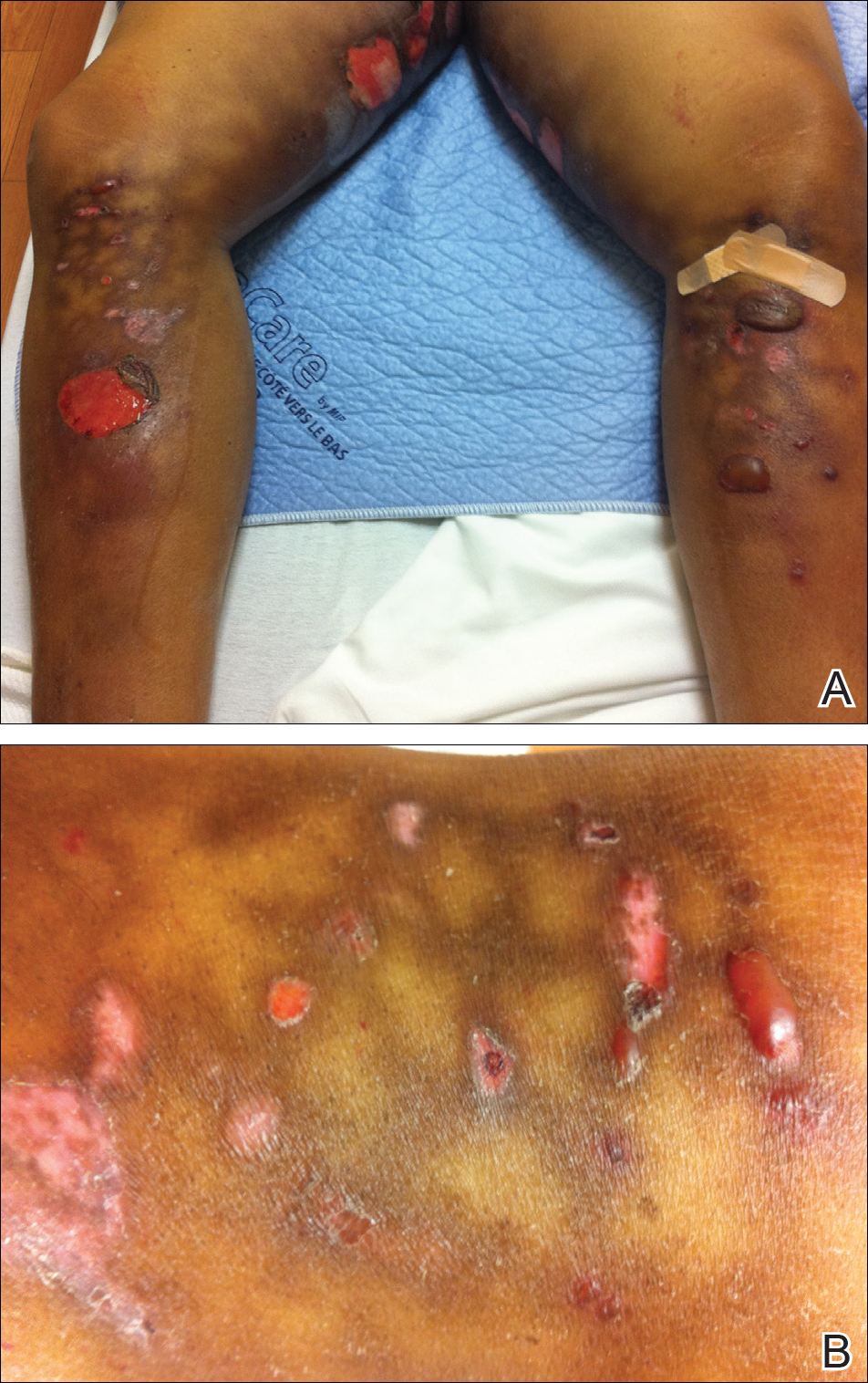
A 55-year-old man was admitted for presumed cellulitis of the bilateral legs. The patient had developed hyperpigmented discoloration of the medial surface of both legs with subsequent formation of tense bullae over the last 2 months. The dermatology department was consulted, as there was concern for bullous pemphigoid. The patient’s medical history was notable for hypertension, hyperlipidemia, diet-controlled type 2 diabetes mellitus, and hepatitis C virus with cirrhosis. The patient denied pruritus, pain, or known exposure of the legs to potential irritants prior to developing the lesions; however, with additional questioning he did report frequently sitting in front of a space heater with bare legs. Physical examination revealed multiple areas of reticulated erythematous hyperpigmentation with several overlying bullae (Figure 1). Many of the bullae were unroofed with full-thickness ulceration. Biopsies were taken for hematoxylin and eosin staining (Figure 2) and direct immunofluorescence.
Basic hematologic and metabolic laboratory test results as well as blood cultures were negative. Wound culture was positive for methicillin-resistant Staphylococcus aureus. Histologic examination showed interface dermatitis with subepidermal vesicle (Figure 2). Scattered necrotic keratinocytes were present in the adjacent epidermis, and focal subtle vacuolar alteration of the dermoepidermal junction was seen (Figure 3). Sparse perivascular mononuclear cells and scattered melanophages were present in the dermis. Direct immunofluorescence showed no diagnostic immunopathologic abnormality. Focal weak nonspecific vascular positivity for IgG and C3 was seen, but IgA and IgM were negative. Although not specific, these changes were compatible with EAI in the clinical context provided. The diagnosis of bullous EAI with superimposed staphylococcal infection was made.
Although rare, there have been reports of a bullous variant of EAI. Flanagan et al4 described 3 cases of bullous EAI with histopathology similar to our case. All 3 biopsies showed subepidermal separation with a mild perivascular dermal lymphocytic infiltrate. Direct immunofluorescence was negative in 2 cases but showed nonspecific weak patchy deposition of IgM along the dermoepidermal junction.4 Although our case was negative for IgM, there was a similar weak nonspecific distribution of IgG. Kokturk et al5 described a case of bullous EAI in a man with repeated exposure to a space heater. The lesions showed subepidermal separation of the epidermis; increased elastic fibers; dilated dermal capillaries; melanophages in the upper dermis; and a mild, superficial, perivascular-lymphocytic infiltrate. Direct immunofluorescence showed no immune deposits.5 Several earlier cases of bullae associated with EAI have been reported in the literature but were thought to be bullous lichen planus superimposed on EAI.6 Our case, which exhibited similar historical, physical, and histopathologic findings, strengthens the argument for a defined bullous variant of EAI.
- Baruchin AM. Erythema ab igne—a neglected entity? Burns. 1994;20:460-462.
- Arnold AW, Itin PH. Laptop computer−induced erythema ab igne in a child and review of the literature [published online October 4, 2010]. Pediatrics. 2010;126:E1227-E1230.
- Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162:77-78.
- Flanagan N, Watson R, Sweeney E, et al. Bullous erythema ab igne. Br J Dermatol. 1996;134:1159-1160.
- Kokturk A, Kaya TI, Baz K, et al. Bullous erythema ab igne. Dermatol Online J. 2003;9:18.
- Horio T, Imamura S. Bullous lichen planus developed on erythema ab igne. J Dermatol. 1986;13:203-207.
To the Editor:
Erythema ab igne (EAI) is a reticular erythematous hyperpigmentation of skin repeatedly exposed to moderate heat.1 It usually is asymptomatic, though some patients report itching or burning at the site.2 Historically caused by exposure to coal stoves or open fires, EAI has become increasingly common among individuals using space heaters, heating pads, or laptop computers near bare skin.2,3 Although EAI itself is benign and usually resolves with the removal of the exposure, it remains of clinical importance because of its association with underlying chronic disease, as chronic pain often is managed with frequent heating pad or hot water bottle use.2 Additionally, accurate diagnosis is important given the future risk for malignancy, as chronic changes of EAI have been reported to lead to squamous cell carcinoma or rarely Merkel cell carcinoma.2 Erythema ab igne is not traditionally associated with the formation of bullae; however, we present a case of bullous EAI that we believe highlights the importance of including this condition in the differential diagnosis of bullous disorders.
A 55-year-old man was admitted for presumed cellulitis of the bilateral legs. The patient had developed hyperpigmented discoloration of the medial surface of both legs with subsequent formation of tense bullae over the last 2 months. The dermatology department was consulted, as there was concern for bullous pemphigoid. The patient’s medical history was notable for hypertension, hyperlipidemia, diet-controlled type 2 diabetes mellitus, and hepatitis C virus with cirrhosis. The patient denied pruritus, pain, or known exposure of the legs to potential irritants prior to developing the lesions; however, with additional questioning he did report frequently sitting in front of a space heater with bare legs. Physical examination revealed multiple areas of reticulated erythematous hyperpigmentation with several overlying bullae (Figure 1). Many of the bullae were unroofed with full-thickness ulceration. Biopsies were taken for hematoxylin and eosin staining (Figure 2) and direct immunofluorescence.
Basic hematologic and metabolic laboratory test results as well as blood cultures were negative. Wound culture was positive for methicillin-resistant Staphylococcus aureus. Histologic examination showed interface dermatitis with subepidermal vesicle (Figure 2). Scattered necrotic keratinocytes were present in the adjacent epidermis, and focal subtle vacuolar alteration of the dermoepidermal junction was seen (Figure 3). Sparse perivascular mononuclear cells and scattered melanophages were present in the dermis. Direct immunofluorescence showed no diagnostic immunopathologic abnormality. Focal weak nonspecific vascular positivity for IgG and C3 was seen, but IgA and IgM were negative. Although not specific, these changes were compatible with EAI in the clinical context provided. The diagnosis of bullous EAI with superimposed staphylococcal infection was made.
Although rare, there have been reports of a bullous variant of EAI. Flanagan et al4 described 3 cases of bullous EAI with histopathology similar to our case. All 3 biopsies showed subepidermal separation with a mild perivascular dermal lymphocytic infiltrate. Direct immunofluorescence was negative in 2 cases but showed nonspecific weak patchy deposition of IgM along the dermoepidermal junction.4 Although our case was negative for IgM, there was a similar weak nonspecific distribution of IgG. Kokturk et al5 described a case of bullous EAI in a man with repeated exposure to a space heater. The lesions showed subepidermal separation of the epidermis; increased elastic fibers; dilated dermal capillaries; melanophages in the upper dermis; and a mild, superficial, perivascular-lymphocytic infiltrate. Direct immunofluorescence showed no immune deposits.5 Several earlier cases of bullae associated with EAI have been reported in the literature but were thought to be bullous lichen planus superimposed on EAI.6 Our case, which exhibited similar historical, physical, and histopathologic findings, strengthens the argument for a defined bullous variant of EAI.
To the Editor:
Erythema ab igne (EAI) is a reticular erythematous hyperpigmentation of skin repeatedly exposed to moderate heat.1 It usually is asymptomatic, though some patients report itching or burning at the site.2 Historically caused by exposure to coal stoves or open fires, EAI has become increasingly common among individuals using space heaters, heating pads, or laptop computers near bare skin.2,3 Although EAI itself is benign and usually resolves with the removal of the exposure, it remains of clinical importance because of its association with underlying chronic disease, as chronic pain often is managed with frequent heating pad or hot water bottle use.2 Additionally, accurate diagnosis is important given the future risk for malignancy, as chronic changes of EAI have been reported to lead to squamous cell carcinoma or rarely Merkel cell carcinoma.2 Erythema ab igne is not traditionally associated with the formation of bullae; however, we present a case of bullous EAI that we believe highlights the importance of including this condition in the differential diagnosis of bullous disorders.
A 55-year-old man was admitted for presumed cellulitis of the bilateral legs. The patient had developed hyperpigmented discoloration of the medial surface of both legs with subsequent formation of tense bullae over the last 2 months. The dermatology department was consulted, as there was concern for bullous pemphigoid. The patient’s medical history was notable for hypertension, hyperlipidemia, diet-controlled type 2 diabetes mellitus, and hepatitis C virus with cirrhosis. The patient denied pruritus, pain, or known exposure of the legs to potential irritants prior to developing the lesions; however, with additional questioning he did report frequently sitting in front of a space heater with bare legs. Physical examination revealed multiple areas of reticulated erythematous hyperpigmentation with several overlying bullae (Figure 1). Many of the bullae were unroofed with full-thickness ulceration. Biopsies were taken for hematoxylin and eosin staining (Figure 2) and direct immunofluorescence.
Basic hematologic and metabolic laboratory test results as well as blood cultures were negative. Wound culture was positive for methicillin-resistant Staphylococcus aureus. Histologic examination showed interface dermatitis with subepidermal vesicle (Figure 2). Scattered necrotic keratinocytes were present in the adjacent epidermis, and focal subtle vacuolar alteration of the dermoepidermal junction was seen (Figure 3). Sparse perivascular mononuclear cells and scattered melanophages were present in the dermis. Direct immunofluorescence showed no diagnostic immunopathologic abnormality. Focal weak nonspecific vascular positivity for IgG and C3 was seen, but IgA and IgM were negative. Although not specific, these changes were compatible with EAI in the clinical context provided. The diagnosis of bullous EAI with superimposed staphylococcal infection was made.
Although rare, there have been reports of a bullous variant of EAI. Flanagan et al4 described 3 cases of bullous EAI with histopathology similar to our case. All 3 biopsies showed subepidermal separation with a mild perivascular dermal lymphocytic infiltrate. Direct immunofluorescence was negative in 2 cases but showed nonspecific weak patchy deposition of IgM along the dermoepidermal junction.4 Although our case was negative for IgM, there was a similar weak nonspecific distribution of IgG. Kokturk et al5 described a case of bullous EAI in a man with repeated exposure to a space heater. The lesions showed subepidermal separation of the epidermis; increased elastic fibers; dilated dermal capillaries; melanophages in the upper dermis; and a mild, superficial, perivascular-lymphocytic infiltrate. Direct immunofluorescence showed no immune deposits.5 Several earlier cases of bullae associated with EAI have been reported in the literature but were thought to be bullous lichen planus superimposed on EAI.6 Our case, which exhibited similar historical, physical, and histopathologic findings, strengthens the argument for a defined bullous variant of EAI.
- Baruchin AM. Erythema ab igne—a neglected entity? Burns. 1994;20:460-462.
- Arnold AW, Itin PH. Laptop computer−induced erythema ab igne in a child and review of the literature [published online October 4, 2010]. Pediatrics. 2010;126:E1227-E1230.
- Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162:77-78.
- Flanagan N, Watson R, Sweeney E, et al. Bullous erythema ab igne. Br J Dermatol. 1996;134:1159-1160.
- Kokturk A, Kaya TI, Baz K, et al. Bullous erythema ab igne. Dermatol Online J. 2003;9:18.
- Horio T, Imamura S. Bullous lichen planus developed on erythema ab igne. J Dermatol. 1986;13:203-207.
- Baruchin AM. Erythema ab igne—a neglected entity? Burns. 1994;20:460-462.
- Arnold AW, Itin PH. Laptop computer−induced erythema ab igne in a child and review of the literature [published online October 4, 2010]. Pediatrics. 2010;126:E1227-E1230.
- Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162:77-78.
- Flanagan N, Watson R, Sweeney E, et al. Bullous erythema ab igne. Br J Dermatol. 1996;134:1159-1160.
- Kokturk A, Kaya TI, Baz K, et al. Bullous erythema ab igne. Dermatol Online J. 2003;9:18.
- Horio T, Imamura S. Bullous lichen planus developed on erythema ab igne. J Dermatol. 1986;13:203-207.
Practice Points
- Consider erythema ab igne (EAI) as a potential differential diagnosis in bullous eruptions.
- Space heaters, heating pads, and even laptop computers should be considered as potential causes of EAI.
Acne-associated hyperpigmentation an important consideration in patients with skin of color
NEW YORK – When treating patients with skin of color for acne, treatment goals may vary from those of patients with lighter skin, according to Andrew F. Alexis, MD.
For example, in patients with Fitzpatrick skin types V and VI, the desired treatment outcome is not only resolution of acne, but also resolution of hyperpigmentation, said Dr. Alexis, chairman of the department of dermatology at Mount Sinai St. Luke’s and Mount Sinai West, New York, N.Y.
“Postinflammatory hyperpigmentation is often the driving force for the dermatology consult” in individuals with skin of color, Dr. Alexis said at the summer meeting of the American Academy of Dermatology. “They may be just as concerned about their dark spots as underlying acne,” he noted, citing a study that he coauthored (J Clin Aesthet Dermatol. 2014 Jul;7[7]:19-31).
In the study – a survey of patients with acne to determine which treatment outcomes were most important – 41.6% of the nonwhite female patients reported that clearance of postinflammatory hyperpigmentation was the most important goal, compared with 8.4% of white female respondents (P less than .0001).

It’s important to avoid undertreating patients, especially darker-skinned patients, where ongoing subclinical inflammation may contribute to hyperpigmentation. Even in lesions that appear grossly noninflamed, biopsies may find histological evidence of inflammation, with increased T-cell infiltration of the pilosebaceous units, Dr. Alexis said.
However, there’s always a balancing act in determining how aggressively to treat patients, he added. Dermatologists have to be aware of the risk of hypertrophic scar formation in darker-skinned individuals, especially in truncal areas.
When addressing the acne, step one is to aggressively reduce acne-associated inflammation to reduce potential sequelae. This can be done with any of a number of agents, such as retinoids, benzoyl peroxide, dapsone, azelaic acid, and even intralesional corticosteroid injections, he said.
“All agents have been considered in darker skin types,” he said, noting that “retinoids are particularly important because they can also treat postinflammatory hyperpigmentation.” Tretinoin 0.1% cream and tazarotene 0.1% cream are both good choices, he added.
Adapalene in a fixed combination with benzoyl peroxide has been studied in darker-skinned patients, with no difference in tolerability or higher incidence of pigmentary sequelae than in lighter-skinned patients, he pointed out.
Dapsone 5% and 7.5% have also been studied in patients with darker skin, and both concentrations showed comparable results for safety and efficacy.
The thinking about second-line agents can shift a bit when treating acne in darker skin. For example, azelaic acid as a 20% cream or 15% gel can be a good choice, and can be helpful in treating postinflammatory hyperpigmentation, but azelaic acid is “not as good an antiacne agent as retinoids,” Dr. Alexis said.
Patients should understand that any of these choices are primarily acne-directed treatments, to be deployed over the first 3-6 months of treatment. Then, beginning at about the 3-month mark and continuing for up to a year, hyperpigmentation can be addressed. “Really emphasize the duration of treatment,” when treating hyperpigmentation, Dr. Alexis advised.
Once the acne is under control and hyperpigmentation can be assessed on its own, dermatologists can consider whether bleaching agents are appropriate. “Should they be used? If so, how?” he asked.
Bleaching agents can be effective, said Dr. Alexis, who recommends lesion-directed rather than broad-field therapy, unless there are many larger hyperpigmented macules. “The more common scenario is smaller, more distributed lesions,” he said. “Superficial chemical peels, if used with caution, can be a good adjunct,” to bleaching agents, he added.
Coming down the road are topical nitric oxide preparations, which he said are looking good for darker skin in clinical trials.
“The key to great outcomes is to initiate a combination regimen that targets inflammation and reduces hyperpigmentation,” said Dr. Alexis. Then, he advised, minimize irritation but don’t undertreat, consider adjunctive chemical peels, and above all, “set realistic timeline expectations.”
Dr. Alexis reported financial relationships with multiple pharmaceutical companies.
[email protected]
On Twitter @karioakes
NEW YORK – When treating patients with skin of color for acne, treatment goals may vary from those of patients with lighter skin, according to Andrew F. Alexis, MD.
For example, in patients with Fitzpatrick skin types V and VI, the desired treatment outcome is not only resolution of acne, but also resolution of hyperpigmentation, said Dr. Alexis, chairman of the department of dermatology at Mount Sinai St. Luke’s and Mount Sinai West, New York, N.Y.
“Postinflammatory hyperpigmentation is often the driving force for the dermatology consult” in individuals with skin of color, Dr. Alexis said at the summer meeting of the American Academy of Dermatology. “They may be just as concerned about their dark spots as underlying acne,” he noted, citing a study that he coauthored (J Clin Aesthet Dermatol. 2014 Jul;7[7]:19-31).
In the study – a survey of patients with acne to determine which treatment outcomes were most important – 41.6% of the nonwhite female patients reported that clearance of postinflammatory hyperpigmentation was the most important goal, compared with 8.4% of white female respondents (P less than .0001).
It’s important to avoid undertreating patients, especially darker-skinned patients, where ongoing subclinical inflammation may contribute to hyperpigmentation. Even in lesions that appear grossly noninflamed, biopsies may find histological evidence of inflammation, with increased T-cell infiltration of the pilosebaceous units, Dr. Alexis said.
However, there’s always a balancing act in determining how aggressively to treat patients, he added. Dermatologists have to be aware of the risk of hypertrophic scar formation in darker-skinned individuals, especially in truncal areas.
When addressing the acne, step one is to aggressively reduce acne-associated inflammation to reduce potential sequelae. This can be done with any of a number of agents, such as retinoids, benzoyl peroxide, dapsone, azelaic acid, and even intralesional corticosteroid injections, he said.
“All agents have been considered in darker skin types,” he said, noting that “retinoids are particularly important because they can also treat postinflammatory hyperpigmentation.” Tretinoin 0.1% cream and tazarotene 0.1% cream are both good choices, he added.
Adapalene in a fixed combination with benzoyl peroxide has been studied in darker-skinned patients, with no difference in tolerability or higher incidence of pigmentary sequelae than in lighter-skinned patients, he pointed out.
Dapsone 5% and 7.5% have also been studied in patients with darker skin, and both concentrations showed comparable results for safety and efficacy.
The thinking about second-line agents can shift a bit when treating acne in darker skin. For example, azelaic acid as a 20% cream or 15% gel can be a good choice, and can be helpful in treating postinflammatory hyperpigmentation, but azelaic acid is “not as good an antiacne agent as retinoids,” Dr. Alexis said.
Patients should understand that any of these choices are primarily acne-directed treatments, to be deployed over the first 3-6 months of treatment. Then, beginning at about the 3-month mark and continuing for up to a year, hyperpigmentation can be addressed. “Really emphasize the duration of treatment,” when treating hyperpigmentation, Dr. Alexis advised.
Once the acne is under control and hyperpigmentation can be assessed on its own, dermatologists can consider whether bleaching agents are appropriate. “Should they be used? If so, how?” he asked.
Bleaching agents can be effective, said Dr. Alexis, who recommends lesion-directed rather than broad-field therapy, unless there are many larger hyperpigmented macules. “The more common scenario is smaller, more distributed lesions,” he said. “Superficial chemical peels, if used with caution, can be a good adjunct,” to bleaching agents, he added.
Coming down the road are topical nitric oxide preparations, which he said are looking good for darker skin in clinical trials.
“The key to great outcomes is to initiate a combination regimen that targets inflammation and reduces hyperpigmentation,” said Dr. Alexis. Then, he advised, minimize irritation but don’t undertreat, consider adjunctive chemical peels, and above all, “set realistic timeline expectations.”
Dr. Alexis reported financial relationships with multiple pharmaceutical companies.
[email protected]
On Twitter @karioakes
NEW YORK – When treating patients with skin of color for acne, treatment goals may vary from those of patients with lighter skin, according to Andrew F. Alexis, MD.
For example, in patients with Fitzpatrick skin types V and VI, the desired treatment outcome is not only resolution of acne, but also resolution of hyperpigmentation, said Dr. Alexis, chairman of the department of dermatology at Mount Sinai St. Luke’s and Mount Sinai West, New York, N.Y.
“Postinflammatory hyperpigmentation is often the driving force for the dermatology consult” in individuals with skin of color, Dr. Alexis said at the summer meeting of the American Academy of Dermatology. “They may be just as concerned about their dark spots as underlying acne,” he noted, citing a study that he coauthored (J Clin Aesthet Dermatol. 2014 Jul;7[7]:19-31).
In the study – a survey of patients with acne to determine which treatment outcomes were most important – 41.6% of the nonwhite female patients reported that clearance of postinflammatory hyperpigmentation was the most important goal, compared with 8.4% of white female respondents (P less than .0001).
It’s important to avoid undertreating patients, especially darker-skinned patients, where ongoing subclinical inflammation may contribute to hyperpigmentation. Even in lesions that appear grossly noninflamed, biopsies may find histological evidence of inflammation, with increased T-cell infiltration of the pilosebaceous units, Dr. Alexis said.
However, there’s always a balancing act in determining how aggressively to treat patients, he added. Dermatologists have to be aware of the risk of hypertrophic scar formation in darker-skinned individuals, especially in truncal areas.
When addressing the acne, step one is to aggressively reduce acne-associated inflammation to reduce potential sequelae. This can be done with any of a number of agents, such as retinoids, benzoyl peroxide, dapsone, azelaic acid, and even intralesional corticosteroid injections, he said.
“All agents have been considered in darker skin types,” he said, noting that “retinoids are particularly important because they can also treat postinflammatory hyperpigmentation.” Tretinoin 0.1% cream and tazarotene 0.1% cream are both good choices, he added.
Adapalene in a fixed combination with benzoyl peroxide has been studied in darker-skinned patients, with no difference in tolerability or higher incidence of pigmentary sequelae than in lighter-skinned patients, he pointed out.
Dapsone 5% and 7.5% have also been studied in patients with darker skin, and both concentrations showed comparable results for safety and efficacy.
The thinking about second-line agents can shift a bit when treating acne in darker skin. For example, azelaic acid as a 20% cream or 15% gel can be a good choice, and can be helpful in treating postinflammatory hyperpigmentation, but azelaic acid is “not as good an antiacne agent as retinoids,” Dr. Alexis said.
Patients should understand that any of these choices are primarily acne-directed treatments, to be deployed over the first 3-6 months of treatment. Then, beginning at about the 3-month mark and continuing for up to a year, hyperpigmentation can be addressed. “Really emphasize the duration of treatment,” when treating hyperpigmentation, Dr. Alexis advised.
Once the acne is under control and hyperpigmentation can be assessed on its own, dermatologists can consider whether bleaching agents are appropriate. “Should they be used? If so, how?” he asked.
Bleaching agents can be effective, said Dr. Alexis, who recommends lesion-directed rather than broad-field therapy, unless there are many larger hyperpigmented macules. “The more common scenario is smaller, more distributed lesions,” he said. “Superficial chemical peels, if used with caution, can be a good adjunct,” to bleaching agents, he added.
Coming down the road are topical nitric oxide preparations, which he said are looking good for darker skin in clinical trials.
“The key to great outcomes is to initiate a combination regimen that targets inflammation and reduces hyperpigmentation,” said Dr. Alexis. Then, he advised, minimize irritation but don’t undertreat, consider adjunctive chemical peels, and above all, “set realistic timeline expectations.”
Dr. Alexis reported financial relationships with multiple pharmaceutical companies.
[email protected]
On Twitter @karioakes
EXPERT ANALYSIS FROM THE 2017 SUMMER AAD MEETING
Annular Atrophic Lichen Planus Responds to Hydroxychloroquine and Acitretin
Annular atrophic lichen planus (AALP) is a rare variant of lichen planus that was first described by Friedman and Hashimoto1 in 1991. Clinically, it combines the configuration and morphological features of both annular and atrophic lichen planus. It is a rare entity. We report a case of AALP in a 69-year-old black man. The clinical and histopathological presentation depicted the defining features of this entity with a characteristic loss of elastic fibers corresponding to central atrophy of active lesions.
Case Report
A 69-year-old black man with a history of hepatitis C virus infection and hypothyroidism presented to the dermatology clinic with a pruritic rash on the trunk, extremities, groin, and scalp of 4 months' duration. He denied any new medications, recent illnesses, or sick contacts. Physical examination demonstrated well-demarcated violaceous papules and plaques on the trunk, extensor aspect of the forearms, and thighs involving 10% of the body surface area (Figure 1A). The lesions were annular with raised borders and central depigmented atrophic scarring (Figure 1B). The examination also revealed several large hypopigmented atrophic patches and plaques in the right inguinal region and on the dorsal aspect of the penile shaft and buttocks as well as a single atrophic plaque on the scalp. No oral lesions were seen. An initial punch biopsy was consistent with a nonspecific lichenoid dermatitis (Figure 2), and the patient was prescribed triamcinolone ointment 0.1% for the trunk and extremities and tacrolimus ointment 0.1% for the groin and genital region.


The patient continued to develop new annular atrophic skin lesions over the next several months. Repeat punch biopsies of lesional and uninvolved perilesional skin from the trunk were obtained for histopathologic confirmation and special staining. Lichenoid dermatitis again was noted on the lesional biopsy, and no notable histopathologic changes were observed on the perilesional biopsy. Verhoeff-van Gieson staining for elastic fibers was performed on both biopsies, which revealed destruction of elastic fibers in the central papillary dermis and upper reticular dermis of the lesional biopsy (Figure 3A). The elastic fibers on the perilesional biopsy were preserved (Figure 3B).

The clinical presentation and histopathological findings confirmed a diagnosis of AALP. The patient was prescribed a short taper of oral prednisone, which halted further disease progression. The patient was then started on pentoxifylline and continued on tacrolimus ointment 0.1% with minimal improvement in existing lesions. These medications were discontinued after 3 months. Hydroxychloroquine 400 mg once daily was administered, which initially resulted in some thinning of the plaques on the trunk; however, further progression of the disease was noted after 3 months. Acitretin 25 mg once daily was added to his treatment regimen. Marked thinning of active lesions, hyperpigmentation, and residual scarring was noted after 2 months of combined therapy with acitretin and hydroxychloroquine (Figure 4), with continued improvement appreciable several months later.
Comment
Lichen planus is a common pruritic inflammatory disease of the skin, mucous membranes, hair follicles, and nails with a highly variable clinical pattern and disease course that typically affects the adult population.2 There are many clinical variants of lichen planus, which all demonstrate lichenoid dermatitis on histology. Annular lichen planus is an uncommon variant most commonly seen in men with asymptomatic lesions involving the axillae and groin.2 Atrophic lichen planus is another variant demonstrating atrophic papules and plaques on the trunk and extremities.3 Annular atrophic lichen planus is the rarest variant of lichen planus, incorporating features of both annular and atrophic lichen planus.
The first case of AALP involved a 56-year-old black man with a 25-year history of annular atrophic papules and plaques on the trunk and extremities.1 The second case reported by Requena et al4 in 1994 described a 65-year-old woman with characteristic lesions on the right elbow and left knee. Lipsker et al5 reported a third case in a 41-year-old man with a history of Sneddon syndrome who had lesions typical for AALP for 20 years. In all of these cases, histopathologic examination revealed a lichenoid infiltrate with thinning of the epidermis and loss of elastic fibers in the center of the active lesions.
In more recent cases of AALP, the characteristic findings primarily occurred on the trunk and extremities.6-10 Treatment with topical corticosteroids failed in most cases and some patients noted moderate improvement with tacrolimus ointment 0.1%. Sugashima and Yamamoto11 reported a unique case in 2012 of a 32-year-old woman with AALP on the lower lip. She had notable improvement with tacrolimus ointment 0.1% after 6 months.11
All of the known cases of AALP to date have occurred in adults, both male and female, presenting with a limited number of annular plaques with slightly elevated borders and depressed atrophic centers.1,3-11 Disease duration of AALP has ranged from 2 months to 25 years.11 Histopathologic findings characteristically demonstrate a lichenoid dermatitis of the raised lesional border with a flattened epidermis, loss of rete ridges, and fibrosis of dermal papillae in the lesion center.7 The elastic fibers are destroyed in the papillary dermis of the lesion center, presumably due to elastolytic activity of inflammatory cells.1 Macrophages present in the lichenoid infiltrate of acute lesions release elastases contributing to this destruction.7 Furthermore, elastic fibers appear fragmented on electron microscopy.1
The clinical course of AALP has proven to be chronic in most cases and frequently is resistant to treatment with topical corticosteroids, retinoids, phototherapy, and immunosuppressive agents.3 Treatment administered early in the disease course may provide a more favorable outcome.11 Lesions characteristically heal with scarring and hyperpigmentation. Our case displayed more extensive involvement than has previously been reported. Our patient showed minimal improvement with topical therapy; however, he demonstrated thinning and regression of active lesions after 2 months of combined treatment with hydroxychloroquine and acitretin. Our use of oral pentoxifylline, hydroxychloroquine, and acitretin has not been previously reported in the other cases of AALP we reviewed. Acitretin is the only systemic agent for lichen planus that has achieved level A evidence, as it previously was shown to be highly effective in a placebo-controlled, double-blind study of 65 patients.12
Conclusion
Annular atrophic lichen planus is a known variant of lichen planus characterized by a loss of elastic fibers in the papillary dermis in the center of active lesions. Treatment with topical corticosteroids and phototherapy frequently is ineffective. To our knowledge, there are no studies to date regarding the efficacy of systemic therapy in treatment of AALP. Hydroxychloroquine and acitretin may prove to be beneficial treatment options for resistant AALP. Additional alternative treatments continue to be explored. We encourage reporting additional cases of AALP to further characterize its clinical presentation and response to treatments.
- Friedman DB, Hashimoto K. Annular atrophic lichen planus. J Am Acad Dermatol. 1991;25:392-394.
- James WD, Berger TG, Elston DM. Lichen planus and related conditions. In: James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:213-215.
- Kim BS, Seo SH, Jang BS, et al. A case of annular atrophic lichen planus. J Eur Acad Dermatol Venereol. 2007;21:989-990.
- Requena L, Olivares M, Pique E, et al. Annular atrophic lichen planus. Dermatology. 1994;189:95-98.
- Lipsker D, Piette JC, Laporte JL, et al. Annular atrophic lichen planus and Sneddon's syndrome. Dermatology. 1997;105:402-403.
- Mseddi M, Bouassadi S, Marrakchi S, et al. Annular atrophic lichen planus. Dermatology. 2003;207:208-209.
- Morales-Callaghan A Jr, Martinez G, Aragoneses H, et al. Annular atrophic lichen planus. J Am Acad Dermatol. 2005;52:906-908.
- Ponce-Olivera RM, Tirado-Sánchez A, Montes-de-Oca-Sánchez G, et al. Annular atrophic lichen planus. Int J Dermatol. 2007;46:490-491.
- Kim JS, Kang MS, Sagong C, et al. Annular atrophic lichen planus associated with hypertrophic lichen planus. Clin Exp Dermatol. 2008;33:195-197.
- Li B, Li JH, Xiao T, et al. Annular atrophic lichen planus. Eur J Dermatol. 2010;20:842-843.
- Sugashima Y, Yamamoto T. Annular atrophic lichen planus of the lip. Dermatol Online J. 2012;18:14.
- Manousaridis I, Manousaridis K, Peitsch WK, et al. Individualizing treatment and choice of medication in lichen planus: a step by step approach. J Dtsch Dermatol Ges. 2013;11:981-991.
Annular atrophic lichen planus (AALP) is a rare variant of lichen planus that was first described by Friedman and Hashimoto1 in 1991. Clinically, it combines the configuration and morphological features of both annular and atrophic lichen planus. It is a rare entity. We report a case of AALP in a 69-year-old black man. The clinical and histopathological presentation depicted the defining features of this entity with a characteristic loss of elastic fibers corresponding to central atrophy of active lesions.
Case Report
A 69-year-old black man with a history of hepatitis C virus infection and hypothyroidism presented to the dermatology clinic with a pruritic rash on the trunk, extremities, groin, and scalp of 4 months' duration. He denied any new medications, recent illnesses, or sick contacts. Physical examination demonstrated well-demarcated violaceous papules and plaques on the trunk, extensor aspect of the forearms, and thighs involving 10% of the body surface area (Figure 1A). The lesions were annular with raised borders and central depigmented atrophic scarring (Figure 1B). The examination also revealed several large hypopigmented atrophic patches and plaques in the right inguinal region and on the dorsal aspect of the penile shaft and buttocks as well as a single atrophic plaque on the scalp. No oral lesions were seen. An initial punch biopsy was consistent with a nonspecific lichenoid dermatitis (Figure 2), and the patient was prescribed triamcinolone ointment 0.1% for the trunk and extremities and tacrolimus ointment 0.1% for the groin and genital region.
The patient continued to develop new annular atrophic skin lesions over the next several months. Repeat punch biopsies of lesional and uninvolved perilesional skin from the trunk were obtained for histopathologic confirmation and special staining. Lichenoid dermatitis again was noted on the lesional biopsy, and no notable histopathologic changes were observed on the perilesional biopsy. Verhoeff-van Gieson staining for elastic fibers was performed on both biopsies, which revealed destruction of elastic fibers in the central papillary dermis and upper reticular dermis of the lesional biopsy (Figure 3A). The elastic fibers on the perilesional biopsy were preserved (Figure 3B).
The clinical presentation and histopathological findings confirmed a diagnosis of AALP. The patient was prescribed a short taper of oral prednisone, which halted further disease progression. The patient was then started on pentoxifylline and continued on tacrolimus ointment 0.1% with minimal improvement in existing lesions. These medications were discontinued after 3 months. Hydroxychloroquine 400 mg once daily was administered, which initially resulted in some thinning of the plaques on the trunk; however, further progression of the disease was noted after 3 months. Acitretin 25 mg once daily was added to his treatment regimen. Marked thinning of active lesions, hyperpigmentation, and residual scarring was noted after 2 months of combined therapy with acitretin and hydroxychloroquine (Figure 4), with continued improvement appreciable several months later.
Comment
Lichen planus is a common pruritic inflammatory disease of the skin, mucous membranes, hair follicles, and nails with a highly variable clinical pattern and disease course that typically affects the adult population.2 There are many clinical variants of lichen planus, which all demonstrate lichenoid dermatitis on histology. Annular lichen planus is an uncommon variant most commonly seen in men with asymptomatic lesions involving the axillae and groin.2 Atrophic lichen planus is another variant demonstrating atrophic papules and plaques on the trunk and extremities.3 Annular atrophic lichen planus is the rarest variant of lichen planus, incorporating features of both annular and atrophic lichen planus.
The first case of AALP involved a 56-year-old black man with a 25-year history of annular atrophic papules and plaques on the trunk and extremities.1 The second case reported by Requena et al4 in 1994 described a 65-year-old woman with characteristic lesions on the right elbow and left knee. Lipsker et al5 reported a third case in a 41-year-old man with a history of Sneddon syndrome who had lesions typical for AALP for 20 years. In all of these cases, histopathologic examination revealed a lichenoid infiltrate with thinning of the epidermis and loss of elastic fibers in the center of the active lesions.
In more recent cases of AALP, the characteristic findings primarily occurred on the trunk and extremities.6-10 Treatment with topical corticosteroids failed in most cases and some patients noted moderate improvement with tacrolimus ointment 0.1%. Sugashima and Yamamoto11 reported a unique case in 2012 of a 32-year-old woman with AALP on the lower lip. She had notable improvement with tacrolimus ointment 0.1% after 6 months.11
All of the known cases of AALP to date have occurred in adults, both male and female, presenting with a limited number of annular plaques with slightly elevated borders and depressed atrophic centers.1,3-11 Disease duration of AALP has ranged from 2 months to 25 years.11 Histopathologic findings characteristically demonstrate a lichenoid dermatitis of the raised lesional border with a flattened epidermis, loss of rete ridges, and fibrosis of dermal papillae in the lesion center.7 The elastic fibers are destroyed in the papillary dermis of the lesion center, presumably due to elastolytic activity of inflammatory cells.1 Macrophages present in the lichenoid infiltrate of acute lesions release elastases contributing to this destruction.7 Furthermore, elastic fibers appear fragmented on electron microscopy.1
The clinical course of AALP has proven to be chronic in most cases and frequently is resistant to treatment with topical corticosteroids, retinoids, phototherapy, and immunosuppressive agents.3 Treatment administered early in the disease course may provide a more favorable outcome.11 Lesions characteristically heal with scarring and hyperpigmentation. Our case displayed more extensive involvement than has previously been reported. Our patient showed minimal improvement with topical therapy; however, he demonstrated thinning and regression of active lesions after 2 months of combined treatment with hydroxychloroquine and acitretin. Our use of oral pentoxifylline, hydroxychloroquine, and acitretin has not been previously reported in the other cases of AALP we reviewed. Acitretin is the only systemic agent for lichen planus that has achieved level A evidence, as it previously was shown to be highly effective in a placebo-controlled, double-blind study of 65 patients.12
Conclusion
Annular atrophic lichen planus is a known variant of lichen planus characterized by a loss of elastic fibers in the papillary dermis in the center of active lesions. Treatment with topical corticosteroids and phototherapy frequently is ineffective. To our knowledge, there are no studies to date regarding the efficacy of systemic therapy in treatment of AALP. Hydroxychloroquine and acitretin may prove to be beneficial treatment options for resistant AALP. Additional alternative treatments continue to be explored. We encourage reporting additional cases of AALP to further characterize its clinical presentation and response to treatments.
Annular atrophic lichen planus (AALP) is a rare variant of lichen planus that was first described by Friedman and Hashimoto1 in 1991. Clinically, it combines the configuration and morphological features of both annular and atrophic lichen planus. It is a rare entity. We report a case of AALP in a 69-year-old black man. The clinical and histopathological presentation depicted the defining features of this entity with a characteristic loss of elastic fibers corresponding to central atrophy of active lesions.
Case Report
A 69-year-old black man with a history of hepatitis C virus infection and hypothyroidism presented to the dermatology clinic with a pruritic rash on the trunk, extremities, groin, and scalp of 4 months' duration. He denied any new medications, recent illnesses, or sick contacts. Physical examination demonstrated well-demarcated violaceous papules and plaques on the trunk, extensor aspect of the forearms, and thighs involving 10% of the body surface area (Figure 1A). The lesions were annular with raised borders and central depigmented atrophic scarring (Figure 1B). The examination also revealed several large hypopigmented atrophic patches and plaques in the right inguinal region and on the dorsal aspect of the penile shaft and buttocks as well as a single atrophic plaque on the scalp. No oral lesions were seen. An initial punch biopsy was consistent with a nonspecific lichenoid dermatitis (Figure 2), and the patient was prescribed triamcinolone ointment 0.1% for the trunk and extremities and tacrolimus ointment 0.1% for the groin and genital region.
The patient continued to develop new annular atrophic skin lesions over the next several months. Repeat punch biopsies of lesional and uninvolved perilesional skin from the trunk were obtained for histopathologic confirmation and special staining. Lichenoid dermatitis again was noted on the lesional biopsy, and no notable histopathologic changes were observed on the perilesional biopsy. Verhoeff-van Gieson staining for elastic fibers was performed on both biopsies, which revealed destruction of elastic fibers in the central papillary dermis and upper reticular dermis of the lesional biopsy (Figure 3A). The elastic fibers on the perilesional biopsy were preserved (Figure 3B).
The clinical presentation and histopathological findings confirmed a diagnosis of AALP. The patient was prescribed a short taper of oral prednisone, which halted further disease progression. The patient was then started on pentoxifylline and continued on tacrolimus ointment 0.1% with minimal improvement in existing lesions. These medications were discontinued after 3 months. Hydroxychloroquine 400 mg once daily was administered, which initially resulted in some thinning of the plaques on the trunk; however, further progression of the disease was noted after 3 months. Acitretin 25 mg once daily was added to his treatment regimen. Marked thinning of active lesions, hyperpigmentation, and residual scarring was noted after 2 months of combined therapy with acitretin and hydroxychloroquine (Figure 4), with continued improvement appreciable several months later.
Comment
Lichen planus is a common pruritic inflammatory disease of the skin, mucous membranes, hair follicles, and nails with a highly variable clinical pattern and disease course that typically affects the adult population.2 There are many clinical variants of lichen planus, which all demonstrate lichenoid dermatitis on histology. Annular lichen planus is an uncommon variant most commonly seen in men with asymptomatic lesions involving the axillae and groin.2 Atrophic lichen planus is another variant demonstrating atrophic papules and plaques on the trunk and extremities.3 Annular atrophic lichen planus is the rarest variant of lichen planus, incorporating features of both annular and atrophic lichen planus.
The first case of AALP involved a 56-year-old black man with a 25-year history of annular atrophic papules and plaques on the trunk and extremities.1 The second case reported by Requena et al4 in 1994 described a 65-year-old woman with characteristic lesions on the right elbow and left knee. Lipsker et al5 reported a third case in a 41-year-old man with a history of Sneddon syndrome who had lesions typical for AALP for 20 years. In all of these cases, histopathologic examination revealed a lichenoid infiltrate with thinning of the epidermis and loss of elastic fibers in the center of the active lesions.
In more recent cases of AALP, the characteristic findings primarily occurred on the trunk and extremities.6-10 Treatment with topical corticosteroids failed in most cases and some patients noted moderate improvement with tacrolimus ointment 0.1%. Sugashima and Yamamoto11 reported a unique case in 2012 of a 32-year-old woman with AALP on the lower lip. She had notable improvement with tacrolimus ointment 0.1% after 6 months.11
All of the known cases of AALP to date have occurred in adults, both male and female, presenting with a limited number of annular plaques with slightly elevated borders and depressed atrophic centers.1,3-11 Disease duration of AALP has ranged from 2 months to 25 years.11 Histopathologic findings characteristically demonstrate a lichenoid dermatitis of the raised lesional border with a flattened epidermis, loss of rete ridges, and fibrosis of dermal papillae in the lesion center.7 The elastic fibers are destroyed in the papillary dermis of the lesion center, presumably due to elastolytic activity of inflammatory cells.1 Macrophages present in the lichenoid infiltrate of acute lesions release elastases contributing to this destruction.7 Furthermore, elastic fibers appear fragmented on electron microscopy.1
The clinical course of AALP has proven to be chronic in most cases and frequently is resistant to treatment with topical corticosteroids, retinoids, phototherapy, and immunosuppressive agents.3 Treatment administered early in the disease course may provide a more favorable outcome.11 Lesions characteristically heal with scarring and hyperpigmentation. Our case displayed more extensive involvement than has previously been reported. Our patient showed minimal improvement with topical therapy; however, he demonstrated thinning and regression of active lesions after 2 months of combined treatment with hydroxychloroquine and acitretin. Our use of oral pentoxifylline, hydroxychloroquine, and acitretin has not been previously reported in the other cases of AALP we reviewed. Acitretin is the only systemic agent for lichen planus that has achieved level A evidence, as it previously was shown to be highly effective in a placebo-controlled, double-blind study of 65 patients.12
Conclusion
Annular atrophic lichen planus is a known variant of lichen planus characterized by a loss of elastic fibers in the papillary dermis in the center of active lesions. Treatment with topical corticosteroids and phototherapy frequently is ineffective. To our knowledge, there are no studies to date regarding the efficacy of systemic therapy in treatment of AALP. Hydroxychloroquine and acitretin may prove to be beneficial treatment options for resistant AALP. Additional alternative treatments continue to be explored. We encourage reporting additional cases of AALP to further characterize its clinical presentation and response to treatments.
- Friedman DB, Hashimoto K. Annular atrophic lichen planus. J Am Acad Dermatol. 1991;25:392-394.
- James WD, Berger TG, Elston DM. Lichen planus and related conditions. In: James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:213-215.
- Kim BS, Seo SH, Jang BS, et al. A case of annular atrophic lichen planus. J Eur Acad Dermatol Venereol. 2007;21:989-990.
- Requena L, Olivares M, Pique E, et al. Annular atrophic lichen planus. Dermatology. 1994;189:95-98.
- Lipsker D, Piette JC, Laporte JL, et al. Annular atrophic lichen planus and Sneddon's syndrome. Dermatology. 1997;105:402-403.
- Mseddi M, Bouassadi S, Marrakchi S, et al. Annular atrophic lichen planus. Dermatology. 2003;207:208-209.
- Morales-Callaghan A Jr, Martinez G, Aragoneses H, et al. Annular atrophic lichen planus. J Am Acad Dermatol. 2005;52:906-908.
- Ponce-Olivera RM, Tirado-Sánchez A, Montes-de-Oca-Sánchez G, et al. Annular atrophic lichen planus. Int J Dermatol. 2007;46:490-491.
- Kim JS, Kang MS, Sagong C, et al. Annular atrophic lichen planus associated with hypertrophic lichen planus. Clin Exp Dermatol. 2008;33:195-197.
- Li B, Li JH, Xiao T, et al. Annular atrophic lichen planus. Eur J Dermatol. 2010;20:842-843.
- Sugashima Y, Yamamoto T. Annular atrophic lichen planus of the lip. Dermatol Online J. 2012;18:14.
- Manousaridis I, Manousaridis K, Peitsch WK, et al. Individualizing treatment and choice of medication in lichen planus: a step by step approach. J Dtsch Dermatol Ges. 2013;11:981-991.
- Friedman DB, Hashimoto K. Annular atrophic lichen planus. J Am Acad Dermatol. 1991;25:392-394.
- James WD, Berger TG, Elston DM. Lichen planus and related conditions. In: James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:213-215.
- Kim BS, Seo SH, Jang BS, et al. A case of annular atrophic lichen planus. J Eur Acad Dermatol Venereol. 2007;21:989-990.
- Requena L, Olivares M, Pique E, et al. Annular atrophic lichen planus. Dermatology. 1994;189:95-98.
- Lipsker D, Piette JC, Laporte JL, et al. Annular atrophic lichen planus and Sneddon's syndrome. Dermatology. 1997;105:402-403.
- Mseddi M, Bouassadi S, Marrakchi S, et al. Annular atrophic lichen planus. Dermatology. 2003;207:208-209.
- Morales-Callaghan A Jr, Martinez G, Aragoneses H, et al. Annular atrophic lichen planus. J Am Acad Dermatol. 2005;52:906-908.
- Ponce-Olivera RM, Tirado-Sánchez A, Montes-de-Oca-Sánchez G, et al. Annular atrophic lichen planus. Int J Dermatol. 2007;46:490-491.
- Kim JS, Kang MS, Sagong C, et al. Annular atrophic lichen planus associated with hypertrophic lichen planus. Clin Exp Dermatol. 2008;33:195-197.
- Li B, Li JH, Xiao T, et al. Annular atrophic lichen planus. Eur J Dermatol. 2010;20:842-843.
- Sugashima Y, Yamamoto T. Annular atrophic lichen planus of the lip. Dermatol Online J. 2012;18:14.
- Manousaridis I, Manousaridis K, Peitsch WK, et al. Individualizing treatment and choice of medication in lichen planus: a step by step approach. J Dtsch Dermatol Ges. 2013;11:981-991.
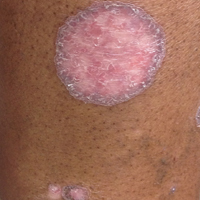
Evaluating the Clinical and Demographic Features of Extrafacial Granuloma Faciale
Granuloma faciale (GF) is a chronic benign leukocytoclastic vasculitis that can be difficult to treat. It is characterized by single or multiple, soft, well-circumscribed papules, plaques, or nodules ranging in color from red, violet, or yellow to brown that may darken with sun exposure.1 Lesions usually are smooth with follicular orifices that are accentuated, thus producing a peau d’orange appearance. Lesions generally are slow to develop and asymptomatic, though some patients report pruritus or burning.2,3 Diagnosis of GF is based on the presence of distinct histologic features. The epidermis usually is spared, with a prominent grenz zone of normal collagen separating the epidermis from a dense infiltrate of neutrophils, lymphocytes, and eosinophils. This mixed inflammatory infiltrate is seen mainly in the superficial dermis but occasionally spreads to the lower dermis and subcutaneous tissues.4
As the name implies, GF usually is confined to the face but occasionally involves extrafacial sites.5-15 The clinical characteristics of these rare extrafacial lesions are not well understood. The purpose of this study was to identify the clinical and demographic features of extrafacial GF in patients treated at Mayo Clinic (Rochester, Minnesota) during a 54-year period.
Methods
This study was approved by the Mayo institutional review board. We searched the Mayo Clinic Rochester dermatology database for all patients with a diagnosis of GF from 1959 through 2013. All histopathology slides were reviewed by a board-certified dermatologist (A.G.B.) and dermatopathologist (A.G.B.) before inclusion in this study. Histologic criteria for diagnosis of GF included the presence of a mixed inflammatory infiltrate of neutrophils, eosinophils, lymphocytes, and histiocytes in the superficial or deep dermis; a prominent grenz zone separating the uninvolved epidermis; and the presence of vascular damage, as seen by fibrin deposition in dermal blood vessels.
Medical records were reviewed for patient demographics and for history pertinent to the diagnosis of GF, including sites involved, appearance, histopathology reports, symptoms, treatments, and outcomes.
Literature Search Strategy
A computerized Ovid MEDLINE database search was undertaken to identify English-language articles concerning GF in humans using the search terms granuloma faciale with extrafacial or disseminated. To ensure that no articles were overlooked, we conducted another search for English-language articles in the Embase database (1946-2013) using the terms granuloma faciale and extrafacial or disseminated.
Statistical Analysis
Descriptive clinical and histopathologic data were summarized using means, medians, and ranges or proportions as appropriate; statistical analysis was performed using SAS software (JMP package).
Results
Ninety-six patients with a diagnosis of GF were identified, and 12 (13%) had a diagnosis of extrafacial GF. Of them, 2 patients had a diagnosis of extrafacial GF supported only by histopathology slides without accompanying clinical records and therefore were excluded from the study. Thus, 10 cases of extrafacial GF were identified from our search and were included in the study group. Clinical data for these patients are summarized in Table 1. The mean age was 58.7 years (range, 26–87 years). Six (60%) patients were male, and all patients were white. Seven patients (70%) had facial GF in addition to extrafacial GF. Six patients reported no symptoms (60%), and 4 (40%) reported pruritus, discomfort, or both associated with their GF lesions.

Extrafacial GF was diagnosed in the following anatomic locations: scalp (n=3 [30%]), posterior auricular area (n=3 [30%]), mid upper back (n=1 [10%]), right shoulder (n=1 [10%]), both ears (n=1 [10%]), right elbow (n=1 [10%]), and left infra-auricular area (n=1 [10%]). Only 1 (10%) patient had multiple extrafacial sites identified.
The lesions were characterized clinically as violet, red, and yellow to brown smooth papules, plaques, and nodules (Figure 1). Biopsies from these lesions showed a subepidermal and adnexal grenz zone; a polymorphous perivascular and periadnexal dermal infiltrate composed of neutrophils, eosinophils, lymphocytes, histiocytes, and plasma cells; and a mild subtle leukocytoclastic vasculitis with subtle mild vascular necrosis (Figure 2).
For the 9 patients who elected to undergo GF treatment, the average number of treatments attempted was 2.8 (range, 1–5). The most common method of treatment was a combination of intralesional and topical corticosteroids (n=5 [50%]). Other methods included surgery (n=3 [30%]), dapsone (n=2 [20%]), radiation therapy (n=2 [20%]), cryosurgery (n=1 [10%]), nitrogen mustard (n=1 [10%]), liquid nitrogen (n=1 [10%]), and tar shampoo and fluocinolone acetonide solution 0.01% (n=1 [10%]).
Treatment outcomes were available for 8 of 9 treated patients. Three patients (patients 7, 8, and 10) had long-term successful resolution of their lesions. Patient 7 had an extrafacial lesion that was successfully treated with intralesional and topical corticosteroids, but the facial lesions recurred. The extrafacial GF lesion in patient 8 was found adjacent to a squamous cell carcinoma and was removed with a wide surgical excision that included both lesions. Patient 10 was successfully treated with a combination of liquid nitrogen and topical corticosteroid. Patients 2 and 4 were well controlled while on dapsone; however, once the treatment was discontinued, primarily due to adverse effects, the lesions returned.
Literature Search
Our search of the English-language literature identified 20 patients with extrafacial GF (Table 2). Fifteen (75%) patients were male, which was similar to our study (6/10 [60%]). Our patient population was slightly older with a mean age of 58.7 years compared to a median age of 54 years among those identified in the literature. Additionally, 3 (30%) patients in our study had no facial lesions, as seen in classic GF, which is comparable to 8 (40%) patients identified in the literature.
Comment
Extrafacial GF primarily affects white individuals and is more prevalent in men, as demonstrated in our study. Extrafacial GF was most often found in association with facial lesions, with only 3 patients having exclusively extrafacial sites.
Data from the current study indicate that diverse modalities were used to treat extrafacial GF with variable outcomes (chronic recurrence to complete resolution). The most common first-line treatment, intralesional corticosteroid injection, was used in 5 (50%) patients but resulted in only 1 (10%) successful resolution. Other methods frequently used in our study and prior studies were surgical excision, cryotherapy, electrosurgery, and dermabrasion.1,20 These treatments do not appear to be uniformly definitive, and the ablative methods may result in scarring.1 Different laser treatments are emerging for the management of GF lesions. Prior reports of treating facial GF with argon and CO2 lasers have indicated minimized residual scarring and pigmentation.21-23 The use of pulsed dye lasers has resulted in complete clearance of facial GF lesions, without recurrence on long-term follow-up.20,24-26
The latest investigations of immunomodulatory drugs indicate these agents are promising for the management of facial GF. Eetam et al27 reported the successful use of topical tacrolimus to treat facial GF. The relatively low cost and ease of use make these topical medications a competitive alternative to currently available surgical and laser methods. The appearance of all of these novel therapeutic modalities creates the necessity for a randomized trial to establish their efficacy on extrafacial GF lesions.
The wide array of treatments reflects the recalcitrant nature of extrafacial GF lesions. Further insight into the etiology of these lesions is needed to understand their tendency to recur. The important contribution of our study is the observed
Conclusion
The findings from this study and the cases reviewed in the literature provide a unique contribution to the understanding of the clinical and demographic characteristics of extrafacial GF. The rarity of this condition is the single most important constraint of our study, reflected in the emblematic limitations of a retrospective analysis in a select group of patients. The results of analysis of data from our patients were similar to the findings reported in the English-language medical literature. Serious consideration should be given to the development of a national registry for patients with GF. A database containing the clinicopathologic features, treatments, and outcomes for patients with both facial and extrafacial manifestations of GF may be invaluable in evaluating various treatment options and increasing understanding of the etiology and epidemiology of the disease.
- Radin DA, Mehregan DR. Granuloma faciale: distribution of the lesions and review of the literature. Cutis. 2003;72:213-219.
- Dowlati B, Firooz A, Dowlati Y. Granuloma faciale: successful treatment of nine cases with a combination of cryotherapy and intralesional corticosteroid injection. Int J Dermatol. 1997;36:548-551.
- Guill MA, Aton JK. Facial granuloma responsive to dapsone therapy. Arch Dermatol. 1982;118:332-335.
- Ryan TJ. Cutaneous vasculitis. In: Champion RH, Burton JL, Burns DA, et al, eds. Rook/Wilkins/Ebling Textbook of Dermatology. 7th ed. Malden, MA: Blackwell Science; 2004.
- Castano E, Segurado A, Iglesias L, et al. Granuloma faciale entirely in an extrafacial location. Br J Dermatol. 1997;136:978-979.
- Castellano-Howard L, Fairbee SI, Hogan DJ, et al. Extrafacial granuloma faciale: report of a case and response to treatment. Cutis. 2001;67:413-415.
- Cecchi R, Paoli S, Giomi A. Granuloma faciale with extrafacial lesions. Eur J Dermatol. 2002;12:438.
- Inanir I, Alvur Y. Granuloma faciale with extrafacial lesions. Br J Dermatol. 2001;14:360-362.
- Kavanagh GM, McLaren KM, Hunter JA. Extensive extrafacial granuloma faciale of the scalp. Br J Dermatol. 1996;134:595-596.
- Marcoval J, Moreno A, Peyr J. Granuloma faciale: a clinicopathological study of 11 cases. J Am Acad Dermatol. 2004;51:269-273.
- Okun MR, Bauman L, Minor D. Granuloma faciale with lesions on the face and hand. Arch Dermatol. 1965;92:78-80.
- Roustan G, Sanchez Yus E, Salas C, et al. Granuloma faciale with extrafacial lesions. Dermatology. 1999;198:79-82.
- Rusin LJ, Dubin HV, Taylor WB. Disseminated granuloma faciale. Arch Dermatol. 1976;112:1575-1577.
- Sears JK, Gitter DG, Stone MS. Extrafacial granuloma faciale. Arch Dermatol. 1991;127:742-743.
- Zargari O. Disseminated granuloma faciale. Int J Dermatol. 2004;43:210-212.
- Lever WF, Lane CG, Downing JG, et al. Eosinophilic granuloma of the skin: report of three cases. Arch Derm Syphilol. 1948;58:430-438.
- Pedace FJ, Perry HO. Granuloma faciale: a clinical and histopathologic review. Arch Dermatol. 1966;94:387-395.
- Frost FA, Heenan PJ. Facial granuloma. Australas J Dermatol. 1984;25:121-124.
Konohana A. Extrafacial granuloma faciale. J Dermatol. 1994;21:680-682.- Ludwig E, Allam JP, Bieber T, et al. New treatment modalities for granuloma faciale. Br J Dermatol. 2003;149:634-637.
- Apfelberg DB, Druker D, Maser MR, et al. Granuloma faciale: treatment with the argon laser. Arch Dermatol. 1983;119:573-576.
- Apfelberg DB, Maser MR, Lash H, et al. Expanded role of the argon laser in plastic surgery. J Dermatol Surg Oncol. 1983;9:145-151.
- Wheeland RG, Ashley JR, Smith DA, et al. Carbon dioxide laser treatment of granuloma faciale. J Dermatol Surg Oncol. 1984;10:730-733.
- Cheung ST, Lanigan SW. Granuloma faciale treated with the pulsed-dye laser: a case series. Clin Exp Dermatol. 2005;30:373-375.
- Chatrath V, Rohrer TE. Granuloma faciale successfully treated with long-pulsed tunable dye laser. Dermatol Surg. 2002;28:527-529.
- Elston DM. Treatment of granuloma faciale with the pulsed dye laser. Cutis. 2000;65:97-98.
- Eetam I, Ertekin B, Unal I, et al. Granuloma faciale: is it a new indication for pimecrolimus? a case report. J Dermatolog Treat. 2006;17:238-240.
- Johnson WC, Higdon RS, Helwig EB. Granuloma faciale. AMA Arch Derm. 1959;79:42-52.
Granuloma faciale (GF) is a chronic benign leukocytoclastic vasculitis that can be difficult to treat. It is characterized by single or multiple, soft, well-circumscribed papules, plaques, or nodules ranging in color from red, violet, or yellow to brown that may darken with sun exposure.1 Lesions usually are smooth with follicular orifices that are accentuated, thus producing a peau d’orange appearance. Lesions generally are slow to develop and asymptomatic, though some patients report pruritus or burning.2,3 Diagnosis of GF is based on the presence of distinct histologic features. The epidermis usually is spared, with a prominent grenz zone of normal collagen separating the epidermis from a dense infiltrate of neutrophils, lymphocytes, and eosinophils. This mixed inflammatory infiltrate is seen mainly in the superficial dermis but occasionally spreads to the lower dermis and subcutaneous tissues.4
As the name implies, GF usually is confined to the face but occasionally involves extrafacial sites.5-15 The clinical characteristics of these rare extrafacial lesions are not well understood. The purpose of this study was to identify the clinical and demographic features of extrafacial GF in patients treated at Mayo Clinic (Rochester, Minnesota) during a 54-year period.
Methods
This study was approved by the Mayo institutional review board. We searched the Mayo Clinic Rochester dermatology database for all patients with a diagnosis of GF from 1959 through 2013. All histopathology slides were reviewed by a board-certified dermatologist (A.G.B.) and dermatopathologist (A.G.B.) before inclusion in this study. Histologic criteria for diagnosis of GF included the presence of a mixed inflammatory infiltrate of neutrophils, eosinophils, lymphocytes, and histiocytes in the superficial or deep dermis; a prominent grenz zone separating the uninvolved epidermis; and the presence of vascular damage, as seen by fibrin deposition in dermal blood vessels.
Medical records were reviewed for patient demographics and for history pertinent to the diagnosis of GF, including sites involved, appearance, histopathology reports, symptoms, treatments, and outcomes.
Literature Search Strategy
A computerized Ovid MEDLINE database search was undertaken to identify English-language articles concerning GF in humans using the search terms granuloma faciale with extrafacial or disseminated. To ensure that no articles were overlooked, we conducted another search for English-language articles in the Embase database (1946-2013) using the terms granuloma faciale and extrafacial or disseminated.
Statistical Analysis
Descriptive clinical and histopathologic data were summarized using means, medians, and ranges or proportions as appropriate; statistical analysis was performed using SAS software (JMP package).
Results
Ninety-six patients with a diagnosis of GF were identified, and 12 (13%) had a diagnosis of extrafacial GF. Of them, 2 patients had a diagnosis of extrafacial GF supported only by histopathology slides without accompanying clinical records and therefore were excluded from the study. Thus, 10 cases of extrafacial GF were identified from our search and were included in the study group. Clinical data for these patients are summarized in Table 1. The mean age was 58.7 years (range, 26–87 years). Six (60%) patients were male, and all patients were white. Seven patients (70%) had facial GF in addition to extrafacial GF. Six patients reported no symptoms (60%), and 4 (40%) reported pruritus, discomfort, or both associated with their GF lesions.
Extrafacial GF was diagnosed in the following anatomic locations: scalp (n=3 [30%]), posterior auricular area (n=3 [30%]), mid upper back (n=1 [10%]), right shoulder (n=1 [10%]), both ears (n=1 [10%]), right elbow (n=1 [10%]), and left infra-auricular area (n=1 [10%]). Only 1 (10%) patient had multiple extrafacial sites identified.
The lesions were characterized clinically as violet, red, and yellow to brown smooth papules, plaques, and nodules (Figure 1). Biopsies from these lesions showed a subepidermal and adnexal grenz zone; a polymorphous perivascular and periadnexal dermal infiltrate composed of neutrophils, eosinophils, lymphocytes, histiocytes, and plasma cells; and a mild subtle leukocytoclastic vasculitis with subtle mild vascular necrosis (Figure 2).
For the 9 patients who elected to undergo GF treatment, the average number of treatments attempted was 2.8 (range, 1–5). The most common method of treatment was a combination of intralesional and topical corticosteroids (n=5 [50%]). Other methods included surgery (n=3 [30%]), dapsone (n=2 [20%]), radiation therapy (n=2 [20%]), cryosurgery (n=1 [10%]), nitrogen mustard (n=1 [10%]), liquid nitrogen (n=1 [10%]), and tar shampoo and fluocinolone acetonide solution 0.01% (n=1 [10%]).
Treatment outcomes were available for 8 of 9 treated patients. Three patients (patients 7, 8, and 10) had long-term successful resolution of their lesions. Patient 7 had an extrafacial lesion that was successfully treated with intralesional and topical corticosteroids, but the facial lesions recurred. The extrafacial GF lesion in patient 8 was found adjacent to a squamous cell carcinoma and was removed with a wide surgical excision that included both lesions. Patient 10 was successfully treated with a combination of liquid nitrogen and topical corticosteroid. Patients 2 and 4 were well controlled while on dapsone; however, once the treatment was discontinued, primarily due to adverse effects, the lesions returned.
Literature Search
Our search of the English-language literature identified 20 patients with extrafacial GF (Table 2). Fifteen (75%) patients were male, which was similar to our study (6/10 [60%]). Our patient population was slightly older with a mean age of 58.7 years compared to a median age of 54 years among those identified in the literature. Additionally, 3 (30%) patients in our study had no facial lesions, as seen in classic GF, which is comparable to 8 (40%) patients identified in the literature.
Comment
Extrafacial GF primarily affects white individuals and is more prevalent in men, as demonstrated in our study. Extrafacial GF was most often found in association with facial lesions, with only 3 patients having exclusively extrafacial sites.
Data from the current study indicate that diverse modalities were used to treat extrafacial GF with variable outcomes (chronic recurrence to complete resolution). The most common first-line treatment, intralesional corticosteroid injection, was used in 5 (50%) patients but resulted in only 1 (10%) successful resolution. Other methods frequently used in our study and prior studies were surgical excision, cryotherapy, electrosurgery, and dermabrasion.1,20 These treatments do not appear to be uniformly definitive, and the ablative methods may result in scarring.1 Different laser treatments are emerging for the management of GF lesions. Prior reports of treating facial GF with argon and CO2 lasers have indicated minimized residual scarring and pigmentation.21-23 The use of pulsed dye lasers has resulted in complete clearance of facial GF lesions, without recurrence on long-term follow-up.20,24-26
The latest investigations of immunomodulatory drugs indicate these agents are promising for the management of facial GF. Eetam et al27 reported the successful use of topical tacrolimus to treat facial GF. The relatively low cost and ease of use make these topical medications a competitive alternative to currently available surgical and laser methods. The appearance of all of these novel therapeutic modalities creates the necessity for a randomized trial to establish their efficacy on extrafacial GF lesions.
The wide array of treatments reflects the recalcitrant nature of extrafacial GF lesions. Further insight into the etiology of these lesions is needed to understand their tendency to recur. The important contribution of our study is the observed
Conclusion
The findings from this study and the cases reviewed in the literature provide a unique contribution to the understanding of the clinical and demographic characteristics of extrafacial GF. The rarity of this condition is the single most important constraint of our study, reflected in the emblematic limitations of a retrospective analysis in a select group of patients. The results of analysis of data from our patients were similar to the findings reported in the English-language medical literature. Serious consideration should be given to the development of a national registry for patients with GF. A database containing the clinicopathologic features, treatments, and outcomes for patients with both facial and extrafacial manifestations of GF may be invaluable in evaluating various treatment options and increasing understanding of the etiology and epidemiology of the disease.
Granuloma faciale (GF) is a chronic benign leukocytoclastic vasculitis that can be difficult to treat. It is characterized by single or multiple, soft, well-circumscribed papules, plaques, or nodules ranging in color from red, violet, or yellow to brown that may darken with sun exposure.1 Lesions usually are smooth with follicular orifices that are accentuated, thus producing a peau d’orange appearance. Lesions generally are slow to develop and asymptomatic, though some patients report pruritus or burning.2,3 Diagnosis of GF is based on the presence of distinct histologic features. The epidermis usually is spared, with a prominent grenz zone of normal collagen separating the epidermis from a dense infiltrate of neutrophils, lymphocytes, and eosinophils. This mixed inflammatory infiltrate is seen mainly in the superficial dermis but occasionally spreads to the lower dermis and subcutaneous tissues.4
As the name implies, GF usually is confined to the face but occasionally involves extrafacial sites.5-15 The clinical characteristics of these rare extrafacial lesions are not well understood. The purpose of this study was to identify the clinical and demographic features of extrafacial GF in patients treated at Mayo Clinic (Rochester, Minnesota) during a 54-year period.
Methods
This study was approved by the Mayo institutional review board. We searched the Mayo Clinic Rochester dermatology database for all patients with a diagnosis of GF from 1959 through 2013. All histopathology slides were reviewed by a board-certified dermatologist (A.G.B.) and dermatopathologist (A.G.B.) before inclusion in this study. Histologic criteria for diagnosis of GF included the presence of a mixed inflammatory infiltrate of neutrophils, eosinophils, lymphocytes, and histiocytes in the superficial or deep dermis; a prominent grenz zone separating the uninvolved epidermis; and the presence of vascular damage, as seen by fibrin deposition in dermal blood vessels.
Medical records were reviewed for patient demographics and for history pertinent to the diagnosis of GF, including sites involved, appearance, histopathology reports, symptoms, treatments, and outcomes.
Literature Search Strategy
A computerized Ovid MEDLINE database search was undertaken to identify English-language articles concerning GF in humans using the search terms granuloma faciale with extrafacial or disseminated. To ensure that no articles were overlooked, we conducted another search for English-language articles in the Embase database (1946-2013) using the terms granuloma faciale and extrafacial or disseminated.
Statistical Analysis
Descriptive clinical and histopathologic data were summarized using means, medians, and ranges or proportions as appropriate; statistical analysis was performed using SAS software (JMP package).
Results
Ninety-six patients with a diagnosis of GF were identified, and 12 (13%) had a diagnosis of extrafacial GF. Of them, 2 patients had a diagnosis of extrafacial GF supported only by histopathology slides without accompanying clinical records and therefore were excluded from the study. Thus, 10 cases of extrafacial GF were identified from our search and were included in the study group. Clinical data for these patients are summarized in Table 1. The mean age was 58.7 years (range, 26–87 years). Six (60%) patients were male, and all patients were white. Seven patients (70%) had facial GF in addition to extrafacial GF. Six patients reported no symptoms (60%), and 4 (40%) reported pruritus, discomfort, or both associated with their GF lesions.
Extrafacial GF was diagnosed in the following anatomic locations: scalp (n=3 [30%]), posterior auricular area (n=3 [30%]), mid upper back (n=1 [10%]), right shoulder (n=1 [10%]), both ears (n=1 [10%]), right elbow (n=1 [10%]), and left infra-auricular area (n=1 [10%]). Only 1 (10%) patient had multiple extrafacial sites identified.
The lesions were characterized clinically as violet, red, and yellow to brown smooth papules, plaques, and nodules (Figure 1). Biopsies from these lesions showed a subepidermal and adnexal grenz zone; a polymorphous perivascular and periadnexal dermal infiltrate composed of neutrophils, eosinophils, lymphocytes, histiocytes, and plasma cells; and a mild subtle leukocytoclastic vasculitis with subtle mild vascular necrosis (Figure 2).
For the 9 patients who elected to undergo GF treatment, the average number of treatments attempted was 2.8 (range, 1–5). The most common method of treatment was a combination of intralesional and topical corticosteroids (n=5 [50%]). Other methods included surgery (n=3 [30%]), dapsone (n=2 [20%]), radiation therapy (n=2 [20%]), cryosurgery (n=1 [10%]), nitrogen mustard (n=1 [10%]), liquid nitrogen (n=1 [10%]), and tar shampoo and fluocinolone acetonide solution 0.01% (n=1 [10%]).
Treatment outcomes were available for 8 of 9 treated patients. Three patients (patients 7, 8, and 10) had long-term successful resolution of their lesions. Patient 7 had an extrafacial lesion that was successfully treated with intralesional and topical corticosteroids, but the facial lesions recurred. The extrafacial GF lesion in patient 8 was found adjacent to a squamous cell carcinoma and was removed with a wide surgical excision that included both lesions. Patient 10 was successfully treated with a combination of liquid nitrogen and topical corticosteroid. Patients 2 and 4 were well controlled while on dapsone; however, once the treatment was discontinued, primarily due to adverse effects, the lesions returned.
Literature Search
Our search of the English-language literature identified 20 patients with extrafacial GF (Table 2). Fifteen (75%) patients were male, which was similar to our study (6/10 [60%]). Our patient population was slightly older with a mean age of 58.7 years compared to a median age of 54 years among those identified in the literature. Additionally, 3 (30%) patients in our study had no facial lesions, as seen in classic GF, which is comparable to 8 (40%) patients identified in the literature.
Comment
Extrafacial GF primarily affects white individuals and is more prevalent in men, as demonstrated in our study. Extrafacial GF was most often found in association with facial lesions, with only 3 patients having exclusively extrafacial sites.
Data from the current study indicate that diverse modalities were used to treat extrafacial GF with variable outcomes (chronic recurrence to complete resolution). The most common first-line treatment, intralesional corticosteroid injection, was used in 5 (50%) patients but resulted in only 1 (10%) successful resolution. Other methods frequently used in our study and prior studies were surgical excision, cryotherapy, electrosurgery, and dermabrasion.1,20 These treatments do not appear to be uniformly definitive, and the ablative methods may result in scarring.1 Different laser treatments are emerging for the management of GF lesions. Prior reports of treating facial GF with argon and CO2 lasers have indicated minimized residual scarring and pigmentation.21-23 The use of pulsed dye lasers has resulted in complete clearance of facial GF lesions, without recurrence on long-term follow-up.20,24-26
The latest investigations of immunomodulatory drugs indicate these agents are promising for the management of facial GF. Eetam et al27 reported the successful use of topical tacrolimus to treat facial GF. The relatively low cost and ease of use make these topical medications a competitive alternative to currently available surgical and laser methods. The appearance of all of these novel therapeutic modalities creates the necessity for a randomized trial to establish their efficacy on extrafacial GF lesions.
The wide array of treatments reflects the recalcitrant nature of extrafacial GF lesions. Further insight into the etiology of these lesions is needed to understand their tendency to recur. The important contribution of our study is the observed
Conclusion
The findings from this study and the cases reviewed in the literature provide a unique contribution to the understanding of the clinical and demographic characteristics of extrafacial GF. The rarity of this condition is the single most important constraint of our study, reflected in the emblematic limitations of a retrospective analysis in a select group of patients. The results of analysis of data from our patients were similar to the findings reported in the English-language medical literature. Serious consideration should be given to the development of a national registry for patients with GF. A database containing the clinicopathologic features, treatments, and outcomes for patients with both facial and extrafacial manifestations of GF may be invaluable in evaluating various treatment options and increasing understanding of the etiology and epidemiology of the disease.
- Radin DA, Mehregan DR. Granuloma faciale: distribution of the lesions and review of the literature. Cutis. 2003;72:213-219.
- Dowlati B, Firooz A, Dowlati Y. Granuloma faciale: successful treatment of nine cases with a combination of cryotherapy and intralesional corticosteroid injection. Int J Dermatol. 1997;36:548-551.
- Guill MA, Aton JK. Facial granuloma responsive to dapsone therapy. Arch Dermatol. 1982;118:332-335.
- Ryan TJ. Cutaneous vasculitis. In: Champion RH, Burton JL, Burns DA, et al, eds. Rook/Wilkins/Ebling Textbook of Dermatology. 7th ed. Malden, MA: Blackwell Science; 2004.
- Castano E, Segurado A, Iglesias L, et al. Granuloma faciale entirely in an extrafacial location. Br J Dermatol. 1997;136:978-979.
- Castellano-Howard L, Fairbee SI, Hogan DJ, et al. Extrafacial granuloma faciale: report of a case and response to treatment. Cutis. 2001;67:413-415.
- Cecchi R, Paoli S, Giomi A. Granuloma faciale with extrafacial lesions. Eur J Dermatol. 2002;12:438.
- Inanir I, Alvur Y. Granuloma faciale with extrafacial lesions. Br J Dermatol. 2001;14:360-362.
- Kavanagh GM, McLaren KM, Hunter JA. Extensive extrafacial granuloma faciale of the scalp. Br J Dermatol. 1996;134:595-596.
- Marcoval J, Moreno A, Peyr J. Granuloma faciale: a clinicopathological study of 11 cases. J Am Acad Dermatol. 2004;51:269-273.
- Okun MR, Bauman L, Minor D. Granuloma faciale with lesions on the face and hand. Arch Dermatol. 1965;92:78-80.
- Roustan G, Sanchez Yus E, Salas C, et al. Granuloma faciale with extrafacial lesions. Dermatology. 1999;198:79-82.
- Rusin LJ, Dubin HV, Taylor WB. Disseminated granuloma faciale. Arch Dermatol. 1976;112:1575-1577.
- Sears JK, Gitter DG, Stone MS. Extrafacial granuloma faciale. Arch Dermatol. 1991;127:742-743.
- Zargari O. Disseminated granuloma faciale. Int J Dermatol. 2004;43:210-212.
- Lever WF, Lane CG, Downing JG, et al. Eosinophilic granuloma of the skin: report of three cases. Arch Derm Syphilol. 1948;58:430-438.
- Pedace FJ, Perry HO. Granuloma faciale: a clinical and histopathologic review. Arch Dermatol. 1966;94:387-395.
- Frost FA, Heenan PJ. Facial granuloma. Australas J Dermatol. 1984;25:121-124.
Konohana A. Extrafacial granuloma faciale. J Dermatol. 1994;21:680-682.- Ludwig E, Allam JP, Bieber T, et al. New treatment modalities for granuloma faciale. Br J Dermatol. 2003;149:634-637.
- Apfelberg DB, Druker D, Maser MR, et al. Granuloma faciale: treatment with the argon laser. Arch Dermatol. 1983;119:573-576.
- Apfelberg DB, Maser MR, Lash H, et al. Expanded role of the argon laser in plastic surgery. J Dermatol Surg Oncol. 1983;9:145-151.
- Wheeland RG, Ashley JR, Smith DA, et al. Carbon dioxide laser treatment of granuloma faciale. J Dermatol Surg Oncol. 1984;10:730-733.
- Cheung ST, Lanigan SW. Granuloma faciale treated with the pulsed-dye laser: a case series. Clin Exp Dermatol. 2005;30:373-375.
- Chatrath V, Rohrer TE. Granuloma faciale successfully treated with long-pulsed tunable dye laser. Dermatol Surg. 2002;28:527-529.
- Elston DM. Treatment of granuloma faciale with the pulsed dye laser. Cutis. 2000;65:97-98.
- Eetam I, Ertekin B, Unal I, et al. Granuloma faciale: is it a new indication for pimecrolimus? a case report. J Dermatolog Treat. 2006;17:238-240.
- Johnson WC, Higdon RS, Helwig EB. Granuloma faciale. AMA Arch Derm. 1959;79:42-52.
- Radin DA, Mehregan DR. Granuloma faciale: distribution of the lesions and review of the literature. Cutis. 2003;72:213-219.
- Dowlati B, Firooz A, Dowlati Y. Granuloma faciale: successful treatment of nine cases with a combination of cryotherapy and intralesional corticosteroid injection. Int J Dermatol. 1997;36:548-551.
- Guill MA, Aton JK. Facial granuloma responsive to dapsone therapy. Arch Dermatol. 1982;118:332-335.
- Ryan TJ. Cutaneous vasculitis. In: Champion RH, Burton JL, Burns DA, et al, eds. Rook/Wilkins/Ebling Textbook of Dermatology. 7th ed. Malden, MA: Blackwell Science; 2004.
- Castano E, Segurado A, Iglesias L, et al. Granuloma faciale entirely in an extrafacial location. Br J Dermatol. 1997;136:978-979.
- Castellano-Howard L, Fairbee SI, Hogan DJ, et al. Extrafacial granuloma faciale: report of a case and response to treatment. Cutis. 2001;67:413-415.
- Cecchi R, Paoli S, Giomi A. Granuloma faciale with extrafacial lesions. Eur J Dermatol. 2002;12:438.
- Inanir I, Alvur Y. Granuloma faciale with extrafacial lesions. Br J Dermatol. 2001;14:360-362.
- Kavanagh GM, McLaren KM, Hunter JA. Extensive extrafacial granuloma faciale of the scalp. Br J Dermatol. 1996;134:595-596.
- Marcoval J, Moreno A, Peyr J. Granuloma faciale: a clinicopathological study of 11 cases. J Am Acad Dermatol. 2004;51:269-273.
- Okun MR, Bauman L, Minor D. Granuloma faciale with lesions on the face and hand. Arch Dermatol. 1965;92:78-80.
- Roustan G, Sanchez Yus E, Salas C, et al. Granuloma faciale with extrafacial lesions. Dermatology. 1999;198:79-82.
- Rusin LJ, Dubin HV, Taylor WB. Disseminated granuloma faciale. Arch Dermatol. 1976;112:1575-1577.
- Sears JK, Gitter DG, Stone MS. Extrafacial granuloma faciale. Arch Dermatol. 1991;127:742-743.
- Zargari O. Disseminated granuloma faciale. Int J Dermatol. 2004;43:210-212.
- Lever WF, Lane CG, Downing JG, et al. Eosinophilic granuloma of the skin: report of three cases. Arch Derm Syphilol. 1948;58:430-438.
- Pedace FJ, Perry HO. Granuloma faciale: a clinical and histopathologic review. Arch Dermatol. 1966;94:387-395.
- Frost FA, Heenan PJ. Facial granuloma. Australas J Dermatol. 1984;25:121-124.
Konohana A. Extrafacial granuloma faciale. J Dermatol. 1994;21:680-682.- Ludwig E, Allam JP, Bieber T, et al. New treatment modalities for granuloma faciale. Br J Dermatol. 2003;149:634-637.
- Apfelberg DB, Druker D, Maser MR, et al. Granuloma faciale: treatment with the argon laser. Arch Dermatol. 1983;119:573-576.
- Apfelberg DB, Maser MR, Lash H, et al. Expanded role of the argon laser in plastic surgery. J Dermatol Surg Oncol. 1983;9:145-151.
- Wheeland RG, Ashley JR, Smith DA, et al. Carbon dioxide laser treatment of granuloma faciale. J Dermatol Surg Oncol. 1984;10:730-733.
- Cheung ST, Lanigan SW. Granuloma faciale treated with the pulsed-dye laser: a case series. Clin Exp Dermatol. 2005;30:373-375.
- Chatrath V, Rohrer TE. Granuloma faciale successfully treated with long-pulsed tunable dye laser. Dermatol Surg. 2002;28:527-529.
- Elston DM. Treatment of granuloma faciale with the pulsed dye laser. Cutis. 2000;65:97-98.
- Eetam I, Ertekin B, Unal I, et al. Granuloma faciale: is it a new indication for pimecrolimus? a case report. J Dermatolog Treat. 2006;17:238-240.
- Johnson WC, Higdon RS, Helwig EB. Granuloma faciale. AMA Arch Derm. 1959;79:42-52.
Practice Points
- Extrafacial lesions are rare in granuloma faciale (GF).
- Extrafacial GF should be included in the differential diagnosis of well-demarcated plaques and nodules found on the trunk or extremities.
- Diagnosis of extrafacial GF is based on the presence of distinct histologic features identical to GF.
- Granuloma faciale is a chronic benign leukocytoclastic vasculitis that can be difficult to treat.
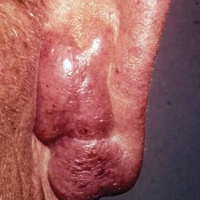
Cosmetic Corner: Dermatologists Weigh in on Redness-Reducing Products
To improve patient care and outcomes, leading dermatologists offered their recommendations on redness-reducing products. Consideration must be given to:
- Avène Antirougeurs FORT Relief Concentrate
Pierre Fabre Dermo-Cosmetique USA
“This formula has medical-grade ruscus extract to support microcirculation and soothe skin reactivity and redness, as well as soothing Avène Thermal Spring Water.” — Jeannette Graf, MD, New York, New York
- Eucerin Redness Relief
Beiersdorf Inc
“Eucerin’s Redness Relief product line has worked well for some of my patients.” — Gary Goldenberg, MD, New York, New York
- Redness Solutions Daily Protective Base Broad Spectrum SPF 15
Clinique Laboratories, LLC
“This oil-free makeup primer has a sheer green tint that camouflages redness while also protecting from UV rays.” — Shari Lipner, MD, PhD, New York, New York
Cutis invites readers to send us their recommendations. Athlete’s foot treatments, cleansing devices, and men’s products will be featured in upcoming editions of Cosmetic Corner. Please email your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on redness-reducing products. Consideration must be given to:
- Avène Antirougeurs FORT Relief Concentrate
Pierre Fabre Dermo-Cosmetique USA
“This formula has medical-grade ruscus extract to support microcirculation and soothe skin reactivity and redness, as well as soothing Avène Thermal Spring Water.” — Jeannette Graf, MD, New York, New York
- Eucerin Redness Relief
Beiersdorf Inc
“Eucerin’s Redness Relief product line has worked well for some of my patients.” — Gary Goldenberg, MD, New York, New York
- Redness Solutions Daily Protective Base Broad Spectrum SPF 15
Clinique Laboratories, LLC
“This oil-free makeup primer has a sheer green tint that camouflages redness while also protecting from UV rays.” — Shari Lipner, MD, PhD, New York, New York
Cutis invites readers to send us their recommendations. Athlete’s foot treatments, cleansing devices, and men’s products will be featured in upcoming editions of Cosmetic Corner. Please email your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on redness-reducing products. Consideration must be given to:
- Avène Antirougeurs FORT Relief Concentrate
Pierre Fabre Dermo-Cosmetique USA
“This formula has medical-grade ruscus extract to support microcirculation and soothe skin reactivity and redness, as well as soothing Avène Thermal Spring Water.” — Jeannette Graf, MD, New York, New York
- Eucerin Redness Relief
Beiersdorf Inc
“Eucerin’s Redness Relief product line has worked well for some of my patients.” — Gary Goldenberg, MD, New York, New York
- Redness Solutions Daily Protective Base Broad Spectrum SPF 15
Clinique Laboratories, LLC
“This oil-free makeup primer has a sheer green tint that camouflages redness while also protecting from UV rays.” — Shari Lipner, MD, PhD, New York, New York
Cutis invites readers to send us their recommendations. Athlete’s foot treatments, cleansing devices, and men’s products will be featured in upcoming editions of Cosmetic Corner. Please email your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.

Segmental Vitiligo–like Hypopigmentation Associated With Metastatic Melanoma
To the Editor:
Melanoma-associated hypopigmentation frequently has been reported during the disease course and can include different characteristics such as regression of the primary melanoma and/or its metastases as well as common vitiligolike hypopigmentation at sites distant from the melanoma.1,2 Among patients who present with hypopigmentation, the most common clinical presentation is hypopigmented patches in a bilateral symmetric distribution that is similar to vitiligo.1 We report a case of segmental vitiligo–like hypopigmentation associated with melanoma.
RELATED ARTICLE: Novel Melanoma Therapies and Their Side Effects
A 37-year-old man presented with achromic patches on the right side of the neck and lower face of 2 months’ duration. He had a history of melanoma (Breslow thickness, 1.37 mm; mitotic rate, 4/mm2) on the right retroauricular region that was treated by wide local excision 12 years prior; after 10 years, he began to have headaches. At that time, imaging studies including computed tomography, magnetic resonance imaging, and positron emission tomography–computed tomography revealed multiple nodules on the brain, lungs, pancreas, left scapula, and left suprarenal gland. A lung biopsy confirmed metastatic melanoma. Intr
On physical examination using a Wood lamp at the current presentation 2 months later, the achromic patches were linearly distributed on the inferior portion of the right cheek (Figure). A 2×3-cm atrophic scar was present on the right retroauricular region. No regional or distant lymph nodes were enlarged or hard on examination. Although vitiligo is diagnosed using clinical findings,3 a biopsy was performed and showed absence of melanocytes at the dermoepidermal junction (hematoxylin and eosin stain) and complete absence of melanin pigment (Fontana-Masson stain). The patient was treated with topical tacrolimus with poor improvement after 2 months.
The relationship between melanoma and vitiligolike hypopigmentation is a fascinating and controversial topic. Its association is considered to be a consequence of the immune-mediated response against antigens shared by normal melanocytes and melanoma cells.4 Vitiligolike hypopigmentation occurs in 2.8%2 of melanoma patients and is reported in metastatic disease1 as well as those undergoing immunotherapy with or without chemotherapy.5 Its development in patients with stage III or IV melanoma seems to represent an independent positive prognostic factor2 and correlates with a better therapeutic outcome in patients undergoing treatment with biotherapy.5
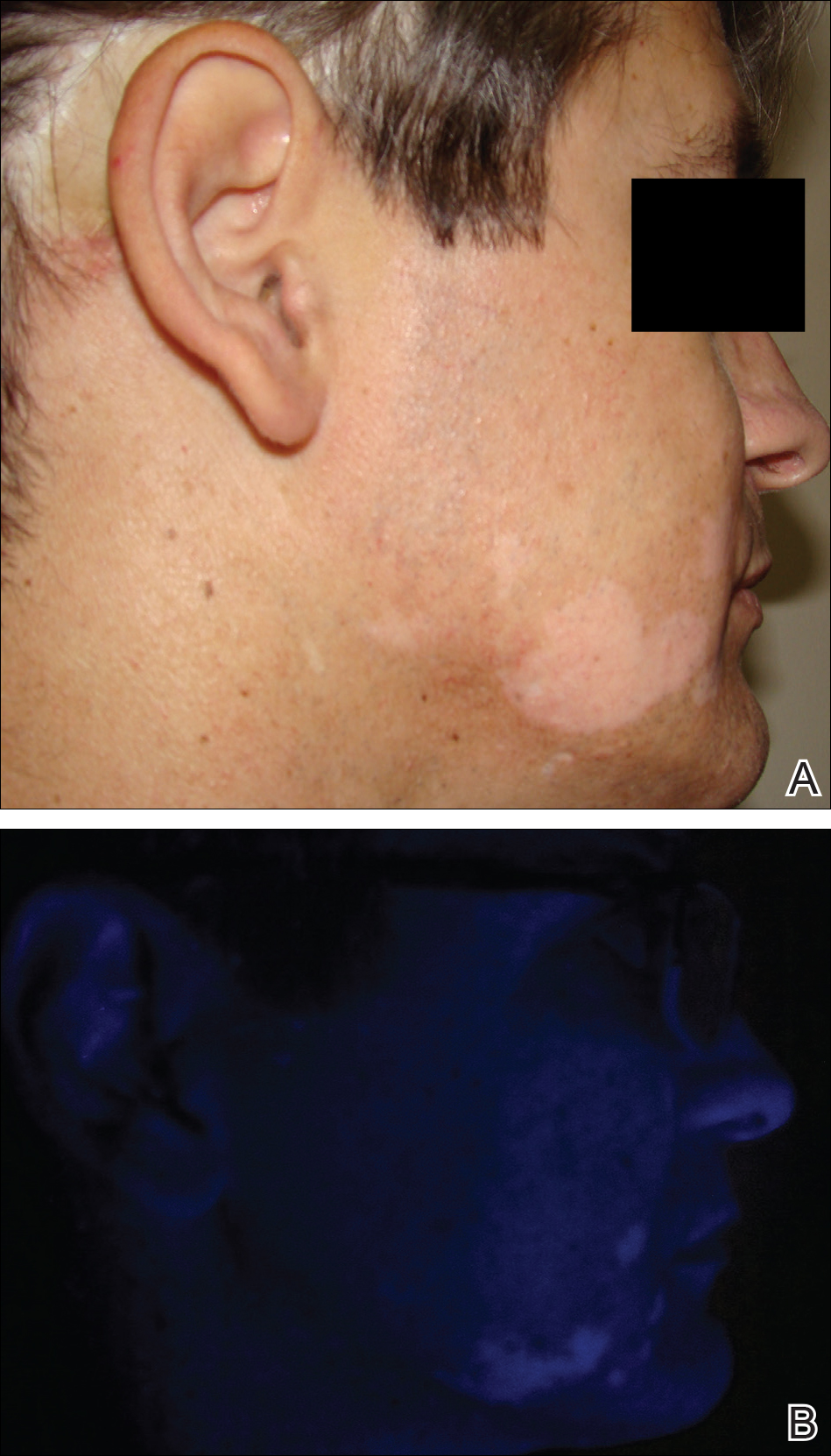
In most cases, the onset of achromic lesions follows the diagnosis of melanoma. Hypopigmentation appears on average 4.8 years after the initial diagnosis and approximately 1 to 2 years after lymph node or distant metastasis.1 In our case, it occurred 12 years after the initial diagnosis and 2 years after metastatic disease was diagnosed.
Despite having widespread metastatic melanoma, our patient only developed achromic patches on the area near the prior melanoma. However, most affected patients present with hypopigmented patches in a bilateral symmetric distribution pattern similar to common vitiligo. No correlation has been found between the hypopigmentation distribution and the location of the primary tumor.1
Because fotemustine is not likely to induce hypopigmentation, we believe that the vitiligolike hypopigmentation in our patient was related to an immune-mediated response associated with melanoma. To help explain our findings, one hypothesis considered was that cutaneous mosaicism may be involved in segmental vitiligo.6 The tumor may have triggered an immune response that affected a close susceptible area of mosaic vitiligo, leading to these clinical findings.
- Hartmann A, Bedenk C, Keikavoussi P, et al. Vitiligo and melanoma-associated hypopigmentation (MAH): shared and discriminative features. J Dtsch Dermatol Ges. 2008;6:1053-1059.
- Quaglino P, Marenco F, Osella-Abate S, et al. Vitiligo is an independent favourable prognostic factor in stage III and IV metastatic melanoma patients: results from a single-institution hospital-based observational cohort study. Ann Oncol. 2010;21:409-414.
- Taïeb A, Picardo M, VETF Members. The definition and assessment of vitiligo: a consensus report of the Vitiligo European Task Force. Pigment Cell Res. 2007;20:27-35.
- Becker JC, Guldberg P, Zeuthen J, et al. Accumulation of identical T cells in melanoma and vitiligo-like leukoderma. J Invest Dermatol. 1999;113:1033-1038.
- Boasberg PD, Hoon DS, Piro LD, et al. Enhanced survival associated with vitiligo expression during maintenance biotherapy for metastatic melanoma. J Invest Dermatol. 2006;126:2658-2663.
- Van Geel N, Speeckaert R, Melsens E, et al. The distribution pattern of segmental vitiligo: clues for somatic mosaicism. Br J Dermatol. 2013;168:56-64.
To the Editor:
Melanoma-associated hypopigmentation frequently has been reported during the disease course and can include different characteristics such as regression of the primary melanoma and/or its metastases as well as common vitiligolike hypopigmentation at sites distant from the melanoma.1,2 Among patients who present with hypopigmentation, the most common clinical presentation is hypopigmented patches in a bilateral symmetric distribution that is similar to vitiligo.1 We report a case of segmental vitiligo–like hypopigmentation associated with melanoma.
RELATED ARTICLE: Novel Melanoma Therapies and Their Side Effects
A 37-year-old man presented with achromic patches on the right side of the neck and lower face of 2 months’ duration. He had a history of melanoma (Breslow thickness, 1.37 mm; mitotic rate, 4/mm2) on the right retroauricular region that was treated by wide local excision 12 years prior; after 10 years, he began to have headaches. At that time, imaging studies including computed tomography, magnetic resonance imaging, and positron emission tomography–computed tomography revealed multiple nodules on the brain, lungs, pancreas, left scapula, and left suprarenal gland. A lung biopsy confirmed metastatic melanoma. Intr
On physical examination using a Wood lamp at the current presentation 2 months later, the achromic patches were linearly distributed on the inferior portion of the right cheek (Figure). A 2×3-cm atrophic scar was present on the right retroauricular region. No regional or distant lymph nodes were enlarged or hard on examination. Although vitiligo is diagnosed using clinical findings,3 a biopsy was performed and showed absence of melanocytes at the dermoepidermal junction (hematoxylin and eosin stain) and complete absence of melanin pigment (Fontana-Masson stain). The patient was treated with topical tacrolimus with poor improvement after 2 months.
The relationship between melanoma and vitiligolike hypopigmentation is a fascinating and controversial topic. Its association is considered to be a consequence of the immune-mediated response against antigens shared by normal melanocytes and melanoma cells.4 Vitiligolike hypopigmentation occurs in 2.8%2 of melanoma patients and is reported in metastatic disease1 as well as those undergoing immunotherapy with or without chemotherapy.5 Its development in patients with stage III or IV melanoma seems to represent an independent positive prognostic factor2 and correlates with a better therapeutic outcome in patients undergoing treatment with biotherapy.5
In most cases, the onset of achromic lesions follows the diagnosis of melanoma. Hypopigmentation appears on average 4.8 years after the initial diagnosis and approximately 1 to 2 years after lymph node or distant metastasis.1 In our case, it occurred 12 years after the initial diagnosis and 2 years after metastatic disease was diagnosed.
Despite having widespread metastatic melanoma, our patient only developed achromic patches on the area near the prior melanoma. However, most affected patients present with hypopigmented patches in a bilateral symmetric distribution pattern similar to common vitiligo. No correlation has been found between the hypopigmentation distribution and the location of the primary tumor.1
Because fotemustine is not likely to induce hypopigmentation, we believe that the vitiligolike hypopigmentation in our patient was related to an immune-mediated response associated with melanoma. To help explain our findings, one hypothesis considered was that cutaneous mosaicism may be involved in segmental vitiligo.6 The tumor may have triggered an immune response that affected a close susceptible area of mosaic vitiligo, leading to these clinical findings.
To the Editor:
Melanoma-associated hypopigmentation frequently has been reported during the disease course and can include different characteristics such as regression of the primary melanoma and/or its metastases as well as common vitiligolike hypopigmentation at sites distant from the melanoma.1,2 Among patients who present with hypopigmentation, the most common clinical presentation is hypopigmented patches in a bilateral symmetric distribution that is similar to vitiligo.1 We report a case of segmental vitiligo–like hypopigmentation associated with melanoma.
RELATED ARTICLE: Novel Melanoma Therapies and Their Side Effects
A 37-year-old man presented with achromic patches on the right side of the neck and lower face of 2 months’ duration. He had a history of melanoma (Breslow thickness, 1.37 mm; mitotic rate, 4/mm2) on the right retroauricular region that was treated by wide local excision 12 years prior; after 10 years, he began to have headaches. At that time, imaging studies including computed tomography, magnetic resonance imaging, and positron emission tomography–computed tomography revealed multiple nodules on the brain, lungs, pancreas, left scapula, and left suprarenal gland. A lung biopsy confirmed metastatic melanoma. Intr
On physical examination using a Wood lamp at the current presentation 2 months later, the achromic patches were linearly distributed on the inferior portion of the right cheek (Figure). A 2×3-cm atrophic scar was present on the right retroauricular region. No regional or distant lymph nodes were enlarged or hard on examination. Although vitiligo is diagnosed using clinical findings,3 a biopsy was performed and showed absence of melanocytes at the dermoepidermal junction (hematoxylin and eosin stain) and complete absence of melanin pigment (Fontana-Masson stain). The patient was treated with topical tacrolimus with poor improvement after 2 months.
The relationship between melanoma and vitiligolike hypopigmentation is a fascinating and controversial topic. Its association is considered to be a consequence of the immune-mediated response against antigens shared by normal melanocytes and melanoma cells.4 Vitiligolike hypopigmentation occurs in 2.8%2 of melanoma patients and is reported in metastatic disease1 as well as those undergoing immunotherapy with or without chemotherapy.5 Its development in patients with stage III or IV melanoma seems to represent an independent positive prognostic factor2 and correlates with a better therapeutic outcome in patients undergoing treatment with biotherapy.5
In most cases, the onset of achromic lesions follows the diagnosis of melanoma. Hypopigmentation appears on average 4.8 years after the initial diagnosis and approximately 1 to 2 years after lymph node or distant metastasis.1 In our case, it occurred 12 years after the initial diagnosis and 2 years after metastatic disease was diagnosed.
Despite having widespread metastatic melanoma, our patient only developed achromic patches on the area near the prior melanoma. However, most affected patients present with hypopigmented patches in a bilateral symmetric distribution pattern similar to common vitiligo. No correlation has been found between the hypopigmentation distribution and the location of the primary tumor.1
Because fotemustine is not likely to induce hypopigmentation, we believe that the vitiligolike hypopigmentation in our patient was related to an immune-mediated response associated with melanoma. To help explain our findings, one hypothesis considered was that cutaneous mosaicism may be involved in segmental vitiligo.6 The tumor may have triggered an immune response that affected a close susceptible area of mosaic vitiligo, leading to these clinical findings.
- Hartmann A, Bedenk C, Keikavoussi P, et al. Vitiligo and melanoma-associated hypopigmentation (MAH): shared and discriminative features. J Dtsch Dermatol Ges. 2008;6:1053-1059.
- Quaglino P, Marenco F, Osella-Abate S, et al. Vitiligo is an independent favourable prognostic factor in stage III and IV metastatic melanoma patients: results from a single-institution hospital-based observational cohort study. Ann Oncol. 2010;21:409-414.
- Taïeb A, Picardo M, VETF Members. The definition and assessment of vitiligo: a consensus report of the Vitiligo European Task Force. Pigment Cell Res. 2007;20:27-35.
- Becker JC, Guldberg P, Zeuthen J, et al. Accumulation of identical T cells in melanoma and vitiligo-like leukoderma. J Invest Dermatol. 1999;113:1033-1038.
- Boasberg PD, Hoon DS, Piro LD, et al. Enhanced survival associated with vitiligo expression during maintenance biotherapy for metastatic melanoma. J Invest Dermatol. 2006;126:2658-2663.
- Van Geel N, Speeckaert R, Melsens E, et al. The distribution pattern of segmental vitiligo: clues for somatic mosaicism. Br J Dermatol. 2013;168:56-64.
- Hartmann A, Bedenk C, Keikavoussi P, et al. Vitiligo and melanoma-associated hypopigmentation (MAH): shared and discriminative features. J Dtsch Dermatol Ges. 2008;6:1053-1059.
- Quaglino P, Marenco F, Osella-Abate S, et al. Vitiligo is an independent favourable prognostic factor in stage III and IV metastatic melanoma patients: results from a single-institution hospital-based observational cohort study. Ann Oncol. 2010;21:409-414.
- Taïeb A, Picardo M, VETF Members. The definition and assessment of vitiligo: a consensus report of the Vitiligo European Task Force. Pigment Cell Res. 2007;20:27-35.
- Becker JC, Guldberg P, Zeuthen J, et al. Accumulation of identical T cells in melanoma and vitiligo-like leukoderma. J Invest Dermatol. 1999;113:1033-1038.
- Boasberg PD, Hoon DS, Piro LD, et al. Enhanced survival associated with vitiligo expression during maintenance biotherapy for metastatic melanoma. J Invest Dermatol. 2006;126:2658-2663.
- Van Geel N, Speeckaert R, Melsens E, et al. The distribution pattern of segmental vitiligo: clues for somatic mosaicism. Br J Dermatol. 2013;168:56-64.
Prac
- Melanoma-associated hypopigmentation usually manifests as common vitiligo; however, little is known about the pathophysiology of segmental vitiligo–like hypopigmentation associated with melanoma.
- This case of segmental vitiligo–like hypopigmentation associated with melanoma sheds light on possible autoimmune and mosaic disease etiology.
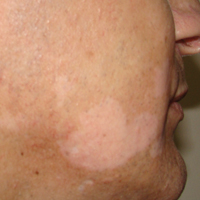
Topical JAK inhibitor showed promise in facial vitiligo
PORTLAND – Twice-daily topical therapy with the Janus kinase (JAK) inhibitor ruxolitinib led to significant improvements in facial vitiligo in a small, uncontrolled, open-label, proof-of-concept study.
Four patients with significant baseline facial involvement improved by an average of 76% on the facial Vitiligo Area Scoring Index, or VASI (95% confidence interval, 53%-99%, P = .001), Brooke Rothstein reported at the annual meeting of the Society for Investigative Dermatology. The results suggest that topical JAK inhibition might help treat facial vitiligo, while potentially sparing patients from the side effects of oral therapy, said Ms. Rothstein, a medical student at Tufts University, Boston, who conducted the study under the mentorship of David Rosmarin, MD, of the department of dermatology at Tufts.
The study included 11 patients with vitiligo affecting at least 1% of body surface area. In all, 54% were male and the average age was 52 years. Patients applied ruxolitinib 1.5% phosphate cream to affected areas twice daily for 20 weeks. The primary outcome was percent improvement in VASI from baseline, Ms. Rothstein said.
By week 20, eight (73%) patients responded to treatment. Overall VASI scores improved by 23% (95% CI, 4%-43%; P = .02) when considering all patients and affected body regions. Three of eight patients responded on the body, and one of these eight patients also improved on acral surfaces, but these improvements were modest – less than 10%, compared with baseline, which was statistically insignificant.
Adverse events were generally mild and included erythema, hyperpigmentation, and transient acne, Ms. Rothstein reported. Despite the small sample size and open-label design of this study, the findings support further studies of topical JAK inhibition in vitiligo and add to mounting evidence that targeting interferon-gamma and its associated chemokines might stimulate repigmentation of skin in affected patients, she concluded.
This study also was published online in the Journal of the American Academy of Dermatology (J Am Acad Dermatol. 2017 Apr 5. doi: 10.1016/j.jaad.2017.02.049). The work was partially supported by Incyte, manufacturer of ruxolitinib (Jakafi), which supplied the study drug and reviewed the manuscript, but did not have final approval or control over the decision to submit for publication. An Alpha Omega Alpha Carolyn L. Kuckein Student Research Fellowship also helped support the work. Ms. Rothstein and her coinvestigators reported having no financial conflicts of interest.
Ruxolitinib, in a tablet formulation, is approved by the Food and Drug Administration for treating myelofibrosis and polycythemia vera.
PORTLAND – Twice-daily topical therapy with the Janus kinase (JAK) inhibitor ruxolitinib led to significant improvements in facial vitiligo in a small, uncontrolled, open-label, proof-of-concept study.
Four patients with significant baseline facial involvement improved by an average of 76% on the facial Vitiligo Area Scoring Index, or VASI (95% confidence interval, 53%-99%, P = .001), Brooke Rothstein reported at the annual meeting of the Society for Investigative Dermatology. The results suggest that topical JAK inhibition might help treat facial vitiligo, while potentially sparing patients from the side effects of oral therapy, said Ms. Rothstein, a medical student at Tufts University, Boston, who conducted the study under the mentorship of David Rosmarin, MD, of the department of dermatology at Tufts.
The study included 11 patients with vitiligo affecting at least 1% of body surface area. In all, 54% were male and the average age was 52 years. Patients applied ruxolitinib 1.5% phosphate cream to affected areas twice daily for 20 weeks. The primary outcome was percent improvement in VASI from baseline, Ms. Rothstein said.
By week 20, eight (73%) patients responded to treatment. Overall VASI scores improved by 23% (95% CI, 4%-43%; P = .02) when considering all patients and affected body regions. Three of eight patients responded on the body, and one of these eight patients also improved on acral surfaces, but these improvements were modest – less than 10%, compared with baseline, which was statistically insignificant.
Adverse events were generally mild and included erythema, hyperpigmentation, and transient acne, Ms. Rothstein reported. Despite the small sample size and open-label design of this study, the findings support further studies of topical JAK inhibition in vitiligo and add to mounting evidence that targeting interferon-gamma and its associated chemokines might stimulate repigmentation of skin in affected patients, she concluded.
This study also was published online in the Journal of the American Academy of Dermatology (J Am Acad Dermatol. 2017 Apr 5. doi: 10.1016/j.jaad.2017.02.049). The work was partially supported by Incyte, manufacturer of ruxolitinib (Jakafi), which supplied the study drug and reviewed the manuscript, but did not have final approval or control over the decision to submit for publication. An Alpha Omega Alpha Carolyn L. Kuckein Student Research Fellowship also helped support the work. Ms. Rothstein and her coinvestigators reported having no financial conflicts of interest.
Ruxolitinib, in a tablet formulation, is approved by the Food and Drug Administration for treating myelofibrosis and polycythemia vera.
PORTLAND – Twice-daily topical therapy with the Janus kinase (JAK) inhibitor ruxolitinib led to significant improvements in facial vitiligo in a small, uncontrolled, open-label, proof-of-concept study.
Four patients with significant baseline facial involvement improved by an average of 76% on the facial Vitiligo Area Scoring Index, or VASI (95% confidence interval, 53%-99%, P = .001), Brooke Rothstein reported at the annual meeting of the Society for Investigative Dermatology. The results suggest that topical JAK inhibition might help treat facial vitiligo, while potentially sparing patients from the side effects of oral therapy, said Ms. Rothstein, a medical student at Tufts University, Boston, who conducted the study under the mentorship of David Rosmarin, MD, of the department of dermatology at Tufts.
The study included 11 patients with vitiligo affecting at least 1% of body surface area. In all, 54% were male and the average age was 52 years. Patients applied ruxolitinib 1.5% phosphate cream to affected areas twice daily for 20 weeks. The primary outcome was percent improvement in VASI from baseline, Ms. Rothstein said.
By week 20, eight (73%) patients responded to treatment. Overall VASI scores improved by 23% (95% CI, 4%-43%; P = .02) when considering all patients and affected body regions. Three of eight patients responded on the body, and one of these eight patients also improved on acral surfaces, but these improvements were modest – less than 10%, compared with baseline, which was statistically insignificant.
Adverse events were generally mild and included erythema, hyperpigmentation, and transient acne, Ms. Rothstein reported. Despite the small sample size and open-label design of this study, the findings support further studies of topical JAK inhibition in vitiligo and add to mounting evidence that targeting interferon-gamma and its associated chemokines might stimulate repigmentation of skin in affected patients, she concluded.
This study also was published online in the Journal of the American Academy of Dermatology (J Am Acad Dermatol. 2017 Apr 5. doi: 10.1016/j.jaad.2017.02.049). The work was partially supported by Incyte, manufacturer of ruxolitinib (Jakafi), which supplied the study drug and reviewed the manuscript, but did not have final approval or control over the decision to submit for publication. An Alpha Omega Alpha Carolyn L. Kuckein Student Research Fellowship also helped support the work. Ms. Rothstein and her coinvestigators reported having no financial conflicts of interest.
Ruxolitinib, in a tablet formulation, is approved by the Food and Drug Administration for treating myelofibrosis and polycythemia vera.
AT SID 2017
Key clinical point:
Major finding: Four patients with significant facial vitiligo improved by 76% on the facial Vitiligo Area Scoring Index, from baseline (P = .001).
Data source: An uncontrolled, open-label pilot study of 11 patients with vitiligo affecting more than 1% of body surface area.
Disclosures: The work was partially supported by Incyte, manufacturer of ruxolitinib, which supplied the study drug and reviewed the manuscript, but did not have final approval or control over the decision to submit for publication. An Alpha Omega Alpha Carolyn L. Kuckein Student Research Fellowship also helped support the work. Ms. Rothstein and her coinvestigators reported having no financial conflicts of interest.