User login
Intravenous immunoglobulin controls dermatomyositis in phase 3 trial
Nearly 50% achieve moderate improvement or better
The first multinational, phase 3, placebo-controlled trial conducted with intravenous immunoglobulin therapy (IVIg) for dermatomyositis has confirmed significant efficacy and acceptable safety, according to data presented at the opening plenary abstract session of the annual European Congress of Rheumatology.
At the week 16 evaluation of the trial, called ProDERM, the response rates were 78.7% and 43.8% (P = .0008) for active therapy and placebo, respectively, reported Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh.
ProDERM is a “much-awaited study,” according to session moderator Hendrik Schulze-Koops, MD, PhD, of the division of rheumatology and clinical immunology at Ludwig Maximilian University of Munich (Germany). He was not involved in the study.
“We all have been doing what we have been doing,” Dr. Schulze-Koops said, referring to the use of IVIg for the control of dermatomyositis, “but we had no evidence for support.”
This statement could apply not only to IVIg, which has long been listed among treatment options by the Myositis Association despite the absence of controlled studies, but also to most immunosuppressive therapies and other options used for this challenging disease.
The proprietary IVIg employed in this study, Octagam 10%, has been approved in the United States for the treatment of chronic immune thrombocytopenic purpura. Its manufacturer, Octagam, plans to file a supplemental new drug application with the Food and Drug Administration for the treatment of dermatomyositis. The agent is already approved for dermatomyositis by the European Medicines Agency, according to Dr. Aggarwal.
Multiple response criteria favor IVIg
In the trial, 95 patients with dermatomyositis were randomized to 2 g/kg of IVIg (Octagam 10%) or placebo administered every 4 weeks. In a subsequent open-label extension study in which patients on placebo were switched to active therapy, the same every-4-week treatment schedule was used. The patients’ mean age was 53; 75% were women, and 92% were White.
The primary endpoint was at least minimal improvement on 2016 ACR/EULAR (American College of Rheumatology/European Alliance of Associations for Rheumatology) myositis response criteria, defined as a 20-point or greater gain in the Total Improvement Score (TIS) and no clinical worsening at two consecutive visits. But IVIg also provided a large relative benefit over placebo using more rigorous definitions of improvement. For moderate improvement, defined as at least a 40-point TIS improvement, there was a 45.2% relative advantage for IVIg over placebo (68.1% vs. 22.9%; P < .0001). For major improvement, defined as at least a 60-point TIS improvement, the relative advantage was 23.6% (31.9% vs. 8.3%; P < .0062).
At 16 weeks, the mean TIS score was more than twice as high in those receiving IVIg than in those randomized to placebo (48.4 vs. 21.6). At that point, an open-label extension was initiated. Those in the IVIg group were permitted to remain on therapy for an additional 24 weeks if they had not worsened in the blinded phase.
The mean TIS score in the IVIg group continued to rise during the extension phase. By 12 weeks in this phase, it reached 54.0. Over the same period, mean TIS scores climbed steeply among the placebo-treated patients who had switched to active therapy, reaching 44.4.
At the end of 24 weeks of the extension trial, when patients initiated on IVIg had been on active therapy for 40 weeks, the mean TIS score advantage of starting on IVIg rather than placebo was relatively modest (55.4 vs. 51.1).
Benefit is significant for skin and muscle
Changes in the two major components of dermatomyositis were tracked individually. For skin symptoms, patients were evaluated with the Cutaneous Dermatomyositis Disease Areas and Severity Index (CDASI). For muscle involvement, symptoms were evaluated with the 8-item Manual Muscle Testing (MMT-8) tool.
“The effects of IVIg on the muscle and the skin were both highly statistically significant,” Dr. Aggarwal reported. He said the CDASI score was reduced by almost half at the end of 16 weeks among those treated with IVIg relative to those treated with placebo. Improvement in MMT-8 scores were also clinically as well as statistically significant.
The IVIg therapy was well tolerated. The most common adverse effects in this study, like those reported with IVIg when used to treat other diseases, were headache, pyrexia, and nausea, but Dr. Aggarwal reported that these were generally mild.
Serious adverse events, particularly thromboembolism, did occur over the course of the study, but the rate of events was only slightly higher in the group receiving active therapy (5.8% vs. 4.2%).
Patients who entered the study were permitted to remain on most immunosuppressive therapies, such as methotrexate, mycophenolate, tacrolimus, and glucocorticoids. Dr. Aggarwal said that the majority of patients were taking a glucocorticoid and at least one nonglucocorticoid immunosuppressant.
Effect on associated conditions is planned
The data from this trial have not yet been analyzed for the impact of IVIg on conditions that occur frequently in association with dermatomyositis, such as interstitial lung disease (ILD) and dysphagia, but Dr. Aggarwal reported that there are plans to do so. Although severe ILD was a trial exclusion, the presence of mild to moderate ILD and dysphagia were evaluated at baseline, so the impact of treatment can be assessed.
There are also plans to evaluate how the presence or absence of myositis-specific antibodies, which were also evaluated at baseline, affected response to IVIg.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Dr. Schulze-Koops reported no relevant potential conflicts of interest.
Nearly 50% achieve moderate improvement or better
Nearly 50% achieve moderate improvement or better
The first multinational, phase 3, placebo-controlled trial conducted with intravenous immunoglobulin therapy (IVIg) for dermatomyositis has confirmed significant efficacy and acceptable safety, according to data presented at the opening plenary abstract session of the annual European Congress of Rheumatology.
At the week 16 evaluation of the trial, called ProDERM, the response rates were 78.7% and 43.8% (P = .0008) for active therapy and placebo, respectively, reported Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh.
ProDERM is a “much-awaited study,” according to session moderator Hendrik Schulze-Koops, MD, PhD, of the division of rheumatology and clinical immunology at Ludwig Maximilian University of Munich (Germany). He was not involved in the study.
“We all have been doing what we have been doing,” Dr. Schulze-Koops said, referring to the use of IVIg for the control of dermatomyositis, “but we had no evidence for support.”
This statement could apply not only to IVIg, which has long been listed among treatment options by the Myositis Association despite the absence of controlled studies, but also to most immunosuppressive therapies and other options used for this challenging disease.
The proprietary IVIg employed in this study, Octagam 10%, has been approved in the United States for the treatment of chronic immune thrombocytopenic purpura. Its manufacturer, Octagam, plans to file a supplemental new drug application with the Food and Drug Administration for the treatment of dermatomyositis. The agent is already approved for dermatomyositis by the European Medicines Agency, according to Dr. Aggarwal.
Multiple response criteria favor IVIg
In the trial, 95 patients with dermatomyositis were randomized to 2 g/kg of IVIg (Octagam 10%) or placebo administered every 4 weeks. In a subsequent open-label extension study in which patients on placebo were switched to active therapy, the same every-4-week treatment schedule was used. The patients’ mean age was 53; 75% were women, and 92% were White.
The primary endpoint was at least minimal improvement on 2016 ACR/EULAR (American College of Rheumatology/European Alliance of Associations for Rheumatology) myositis response criteria, defined as a 20-point or greater gain in the Total Improvement Score (TIS) and no clinical worsening at two consecutive visits. But IVIg also provided a large relative benefit over placebo using more rigorous definitions of improvement. For moderate improvement, defined as at least a 40-point TIS improvement, there was a 45.2% relative advantage for IVIg over placebo (68.1% vs. 22.9%; P < .0001). For major improvement, defined as at least a 60-point TIS improvement, the relative advantage was 23.6% (31.9% vs. 8.3%; P < .0062).
At 16 weeks, the mean TIS score was more than twice as high in those receiving IVIg than in those randomized to placebo (48.4 vs. 21.6). At that point, an open-label extension was initiated. Those in the IVIg group were permitted to remain on therapy for an additional 24 weeks if they had not worsened in the blinded phase.
The mean TIS score in the IVIg group continued to rise during the extension phase. By 12 weeks in this phase, it reached 54.0. Over the same period, mean TIS scores climbed steeply among the placebo-treated patients who had switched to active therapy, reaching 44.4.
At the end of 24 weeks of the extension trial, when patients initiated on IVIg had been on active therapy for 40 weeks, the mean TIS score advantage of starting on IVIg rather than placebo was relatively modest (55.4 vs. 51.1).
Benefit is significant for skin and muscle
Changes in the two major components of dermatomyositis were tracked individually. For skin symptoms, patients were evaluated with the Cutaneous Dermatomyositis Disease Areas and Severity Index (CDASI). For muscle involvement, symptoms were evaluated with the 8-item Manual Muscle Testing (MMT-8) tool.
“The effects of IVIg on the muscle and the skin were both highly statistically significant,” Dr. Aggarwal reported. He said the CDASI score was reduced by almost half at the end of 16 weeks among those treated with IVIg relative to those treated with placebo. Improvement in MMT-8 scores were also clinically as well as statistically significant.
The IVIg therapy was well tolerated. The most common adverse effects in this study, like those reported with IVIg when used to treat other diseases, were headache, pyrexia, and nausea, but Dr. Aggarwal reported that these were generally mild.
Serious adverse events, particularly thromboembolism, did occur over the course of the study, but the rate of events was only slightly higher in the group receiving active therapy (5.8% vs. 4.2%).
Patients who entered the study were permitted to remain on most immunosuppressive therapies, such as methotrexate, mycophenolate, tacrolimus, and glucocorticoids. Dr. Aggarwal said that the majority of patients were taking a glucocorticoid and at least one nonglucocorticoid immunosuppressant.
Effect on associated conditions is planned
The data from this trial have not yet been analyzed for the impact of IVIg on conditions that occur frequently in association with dermatomyositis, such as interstitial lung disease (ILD) and dysphagia, but Dr. Aggarwal reported that there are plans to do so. Although severe ILD was a trial exclusion, the presence of mild to moderate ILD and dysphagia were evaluated at baseline, so the impact of treatment can be assessed.
There are also plans to evaluate how the presence or absence of myositis-specific antibodies, which were also evaluated at baseline, affected response to IVIg.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Dr. Schulze-Koops reported no relevant potential conflicts of interest.
The first multinational, phase 3, placebo-controlled trial conducted with intravenous immunoglobulin therapy (IVIg) for dermatomyositis has confirmed significant efficacy and acceptable safety, according to data presented at the opening plenary abstract session of the annual European Congress of Rheumatology.
At the week 16 evaluation of the trial, called ProDERM, the response rates were 78.7% and 43.8% (P = .0008) for active therapy and placebo, respectively, reported Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh.
ProDERM is a “much-awaited study,” according to session moderator Hendrik Schulze-Koops, MD, PhD, of the division of rheumatology and clinical immunology at Ludwig Maximilian University of Munich (Germany). He was not involved in the study.
“We all have been doing what we have been doing,” Dr. Schulze-Koops said, referring to the use of IVIg for the control of dermatomyositis, “but we had no evidence for support.”
This statement could apply not only to IVIg, which has long been listed among treatment options by the Myositis Association despite the absence of controlled studies, but also to most immunosuppressive therapies and other options used for this challenging disease.
The proprietary IVIg employed in this study, Octagam 10%, has been approved in the United States for the treatment of chronic immune thrombocytopenic purpura. Its manufacturer, Octagam, plans to file a supplemental new drug application with the Food and Drug Administration for the treatment of dermatomyositis. The agent is already approved for dermatomyositis by the European Medicines Agency, according to Dr. Aggarwal.
Multiple response criteria favor IVIg
In the trial, 95 patients with dermatomyositis were randomized to 2 g/kg of IVIg (Octagam 10%) or placebo administered every 4 weeks. In a subsequent open-label extension study in which patients on placebo were switched to active therapy, the same every-4-week treatment schedule was used. The patients’ mean age was 53; 75% were women, and 92% were White.
The primary endpoint was at least minimal improvement on 2016 ACR/EULAR (American College of Rheumatology/European Alliance of Associations for Rheumatology) myositis response criteria, defined as a 20-point or greater gain in the Total Improvement Score (TIS) and no clinical worsening at two consecutive visits. But IVIg also provided a large relative benefit over placebo using more rigorous definitions of improvement. For moderate improvement, defined as at least a 40-point TIS improvement, there was a 45.2% relative advantage for IVIg over placebo (68.1% vs. 22.9%; P < .0001). For major improvement, defined as at least a 60-point TIS improvement, the relative advantage was 23.6% (31.9% vs. 8.3%; P < .0062).
At 16 weeks, the mean TIS score was more than twice as high in those receiving IVIg than in those randomized to placebo (48.4 vs. 21.6). At that point, an open-label extension was initiated. Those in the IVIg group were permitted to remain on therapy for an additional 24 weeks if they had not worsened in the blinded phase.
The mean TIS score in the IVIg group continued to rise during the extension phase. By 12 weeks in this phase, it reached 54.0. Over the same period, mean TIS scores climbed steeply among the placebo-treated patients who had switched to active therapy, reaching 44.4.
At the end of 24 weeks of the extension trial, when patients initiated on IVIg had been on active therapy for 40 weeks, the mean TIS score advantage of starting on IVIg rather than placebo was relatively modest (55.4 vs. 51.1).
Benefit is significant for skin and muscle
Changes in the two major components of dermatomyositis were tracked individually. For skin symptoms, patients were evaluated with the Cutaneous Dermatomyositis Disease Areas and Severity Index (CDASI). For muscle involvement, symptoms were evaluated with the 8-item Manual Muscle Testing (MMT-8) tool.
“The effects of IVIg on the muscle and the skin were both highly statistically significant,” Dr. Aggarwal reported. He said the CDASI score was reduced by almost half at the end of 16 weeks among those treated with IVIg relative to those treated with placebo. Improvement in MMT-8 scores were also clinically as well as statistically significant.
The IVIg therapy was well tolerated. The most common adverse effects in this study, like those reported with IVIg when used to treat other diseases, were headache, pyrexia, and nausea, but Dr. Aggarwal reported that these were generally mild.
Serious adverse events, particularly thromboembolism, did occur over the course of the study, but the rate of events was only slightly higher in the group receiving active therapy (5.8% vs. 4.2%).
Patients who entered the study were permitted to remain on most immunosuppressive therapies, such as methotrexate, mycophenolate, tacrolimus, and glucocorticoids. Dr. Aggarwal said that the majority of patients were taking a glucocorticoid and at least one nonglucocorticoid immunosuppressant.
Effect on associated conditions is planned
The data from this trial have not yet been analyzed for the impact of IVIg on conditions that occur frequently in association with dermatomyositis, such as interstitial lung disease (ILD) and dysphagia, but Dr. Aggarwal reported that there are plans to do so. Although severe ILD was a trial exclusion, the presence of mild to moderate ILD and dysphagia were evaluated at baseline, so the impact of treatment can be assessed.
There are also plans to evaluate how the presence or absence of myositis-specific antibodies, which were also evaluated at baseline, affected response to IVIg.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Dr. Schulze-Koops reported no relevant potential conflicts of interest.
FROM THE EULAR 2021 CONGRESS
Sickle cell disease: Epidemiological change in bacterial infections
Among children with sickle cell disease who have not undergone hematopoietic stem cell transplant, Salmonella is now the leading cause of invasive bacterial infection (IBI), according to a new retrospective study (BACT-SPRING) conducted in Europe. Streptococcus pneumoniae was the second most common source of infection, marking a shift from years past, when S. pneumoniae was the most common source. The epidemiology of IBI in Europe has been altered by adoption of prophylaxis and the introduction of the pneumococcal conjugated vaccine (PCV13) in 2009.
Previous studies of IBI have been single center with small sample sizes, and few have been conducted since 2016, said Jean Gaschignard, MD, PhD, during his presentation of the study at the annual meeting of the European Society for Paediatric Infectious Diseases, held virtually this year.
Dr. Gaschignard is head of pediatrics at Groupe Hospitalier Nord Essonne in Longjumeau, France.
The study produced some unexpected results. “We were surprised,” said Dr. Gaschignard, by results indicating that not all children aged under 10 years were undergoing prophylaxis. Instead, the figures were closer to 80% or 90%. Among children over 10, the rate of prophylaxis varies between countries. “Our study is a clue to discuss again the indications for the age limit for prophylaxis against pneumococcus,” said Dr. Gauschignard, during the question-and-answer session following his talk.

The data give clinicians an updated picture of the epidemiology in this population following introduction of the PCV13 vaccine. “It was very important to have new data on microbiology after this implementation,” said Marie Rohr, MD, who is a fellow in pediatric infectious diseases at the University Hospitals of Geneva. Dr. Rohr moderated the session where the study was presented.
Dr. Rohr noted the shift from the dominant cause of IBI after the introduction of the PCV10/13 vaccine, from S. pneumoniae to Salmonella. The researchers also found a preponderance of bacteremia and osteoarticular infections. “The mortality and morbidity are still considerable despite infection preventive measures,” said Dr. Rohr.
The results should also prompt a second look at prevention strategies. “Even if the antibiotic prophylaxis is prescribed for a large [proportion of children with sickle cell disease] under 10 years old, the median age of invasive bacterial infection is 7 years old. This calls into question systematic antibiotic prophylaxis and case-control studies are needed to evaluate this and possibly modify antibiotic prophylaxis recommendations in the future,” said Dr. Rohr.
The BACT-SPRING study was conducted between Jan. 1, 2014, and Dec. 31, 2019, using online data. It included 217 IBI episodes from 26 centers in five European countries. Just over half were from France, while about a quarter occurred in Spain. Other countries included Belgium, Portugal, and Great Britain. Participants were younger than 18 and had an IBI confirmed by bacterial culture or PCR from normally sterile fluid.
Thirty-eight episodes occurred in children who had undergone hematopoietic stem cell transplantation (HSCT), and 179 in children who had not undergone HSCT. The presentation focused exclusively on the latter group.
Among episodes in children without HSCT, the mean age was 7. Forty-eight patients had a history of acute chest syndrome, 47 had a history of ICU admission, 29 had a history of IBI, and 27 had a history of acute splenic sequestration. Thirteen underwent a splenectomy. Almost half of children had none of these characteristics, while about one-fourth had two or more.
In the HSCT group, 141 children were on prophylaxis at the time of the infection; 74 were on hydroxyurea, and 36 were currently or previously on a transfusion program. Sixty-eight cases were primary bacteremia and 55 were osteoarticular. Other syndromes included pneumonia empyema (n = 18), and meningitis (n = 17), among others. In 44 cases, the isolated bacteria was Salmonella, followed by S. pneumoniae in 32 cases. Escherichia coli accounted for 22. Haemophilus influenza was identified in six episodes, and group A Streptococcus in three.
The study is the first large European epidemiologic study investigating IBI in children with sickle cell disease, and one of its strengths was the strict inclusion criteria. However, it was limited by its retrospective nature.
Dr. Gaschignard and Dr. Rohr have no relevant financial disclosures.
Among children with sickle cell disease who have not undergone hematopoietic stem cell transplant, Salmonella is now the leading cause of invasive bacterial infection (IBI), according to a new retrospective study (BACT-SPRING) conducted in Europe. Streptococcus pneumoniae was the second most common source of infection, marking a shift from years past, when S. pneumoniae was the most common source. The epidemiology of IBI in Europe has been altered by adoption of prophylaxis and the introduction of the pneumococcal conjugated vaccine (PCV13) in 2009.
Previous studies of IBI have been single center with small sample sizes, and few have been conducted since 2016, said Jean Gaschignard, MD, PhD, during his presentation of the study at the annual meeting of the European Society for Paediatric Infectious Diseases, held virtually this year.
Dr. Gaschignard is head of pediatrics at Groupe Hospitalier Nord Essonne in Longjumeau, France.
The study produced some unexpected results. “We were surprised,” said Dr. Gaschignard, by results indicating that not all children aged under 10 years were undergoing prophylaxis. Instead, the figures were closer to 80% or 90%. Among children over 10, the rate of prophylaxis varies between countries. “Our study is a clue to discuss again the indications for the age limit for prophylaxis against pneumococcus,” said Dr. Gauschignard, during the question-and-answer session following his talk.
The data give clinicians an updated picture of the epidemiology in this population following introduction of the PCV13 vaccine. “It was very important to have new data on microbiology after this implementation,” said Marie Rohr, MD, who is a fellow in pediatric infectious diseases at the University Hospitals of Geneva. Dr. Rohr moderated the session where the study was presented.
Dr. Rohr noted the shift from the dominant cause of IBI after the introduction of the PCV10/13 vaccine, from S. pneumoniae to Salmonella. The researchers also found a preponderance of bacteremia and osteoarticular infections. “The mortality and morbidity are still considerable despite infection preventive measures,” said Dr. Rohr.
The results should also prompt a second look at prevention strategies. “Even if the antibiotic prophylaxis is prescribed for a large [proportion of children with sickle cell disease] under 10 years old, the median age of invasive bacterial infection is 7 years old. This calls into question systematic antibiotic prophylaxis and case-control studies are needed to evaluate this and possibly modify antibiotic prophylaxis recommendations in the future,” said Dr. Rohr.
The BACT-SPRING study was conducted between Jan. 1, 2014, and Dec. 31, 2019, using online data. It included 217 IBI episodes from 26 centers in five European countries. Just over half were from France, while about a quarter occurred in Spain. Other countries included Belgium, Portugal, and Great Britain. Participants were younger than 18 and had an IBI confirmed by bacterial culture or PCR from normally sterile fluid.
Thirty-eight episodes occurred in children who had undergone hematopoietic stem cell transplantation (HSCT), and 179 in children who had not undergone HSCT. The presentation focused exclusively on the latter group.
Among episodes in children without HSCT, the mean age was 7. Forty-eight patients had a history of acute chest syndrome, 47 had a history of ICU admission, 29 had a history of IBI, and 27 had a history of acute splenic sequestration. Thirteen underwent a splenectomy. Almost half of children had none of these characteristics, while about one-fourth had two or more.
In the HSCT group, 141 children were on prophylaxis at the time of the infection; 74 were on hydroxyurea, and 36 were currently or previously on a transfusion program. Sixty-eight cases were primary bacteremia and 55 were osteoarticular. Other syndromes included pneumonia empyema (n = 18), and meningitis (n = 17), among others. In 44 cases, the isolated bacteria was Salmonella, followed by S. pneumoniae in 32 cases. Escherichia coli accounted for 22. Haemophilus influenza was identified in six episodes, and group A Streptococcus in three.
The study is the first large European epidemiologic study investigating IBI in children with sickle cell disease, and one of its strengths was the strict inclusion criteria. However, it was limited by its retrospective nature.
Dr. Gaschignard and Dr. Rohr have no relevant financial disclosures.
Among children with sickle cell disease who have not undergone hematopoietic stem cell transplant, Salmonella is now the leading cause of invasive bacterial infection (IBI), according to a new retrospective study (BACT-SPRING) conducted in Europe. Streptococcus pneumoniae was the second most common source of infection, marking a shift from years past, when S. pneumoniae was the most common source. The epidemiology of IBI in Europe has been altered by adoption of prophylaxis and the introduction of the pneumococcal conjugated vaccine (PCV13) in 2009.
Previous studies of IBI have been single center with small sample sizes, and few have been conducted since 2016, said Jean Gaschignard, MD, PhD, during his presentation of the study at the annual meeting of the European Society for Paediatric Infectious Diseases, held virtually this year.
Dr. Gaschignard is head of pediatrics at Groupe Hospitalier Nord Essonne in Longjumeau, France.
The study produced some unexpected results. “We were surprised,” said Dr. Gaschignard, by results indicating that not all children aged under 10 years were undergoing prophylaxis. Instead, the figures were closer to 80% or 90%. Among children over 10, the rate of prophylaxis varies between countries. “Our study is a clue to discuss again the indications for the age limit for prophylaxis against pneumococcus,” said Dr. Gauschignard, during the question-and-answer session following his talk.
The data give clinicians an updated picture of the epidemiology in this population following introduction of the PCV13 vaccine. “It was very important to have new data on microbiology after this implementation,” said Marie Rohr, MD, who is a fellow in pediatric infectious diseases at the University Hospitals of Geneva. Dr. Rohr moderated the session where the study was presented.
Dr. Rohr noted the shift from the dominant cause of IBI after the introduction of the PCV10/13 vaccine, from S. pneumoniae to Salmonella. The researchers also found a preponderance of bacteremia and osteoarticular infections. “The mortality and morbidity are still considerable despite infection preventive measures,” said Dr. Rohr.
The results should also prompt a second look at prevention strategies. “Even if the antibiotic prophylaxis is prescribed for a large [proportion of children with sickle cell disease] under 10 years old, the median age of invasive bacterial infection is 7 years old. This calls into question systematic antibiotic prophylaxis and case-control studies are needed to evaluate this and possibly modify antibiotic prophylaxis recommendations in the future,” said Dr. Rohr.
The BACT-SPRING study was conducted between Jan. 1, 2014, and Dec. 31, 2019, using online data. It included 217 IBI episodes from 26 centers in five European countries. Just over half were from France, while about a quarter occurred in Spain. Other countries included Belgium, Portugal, and Great Britain. Participants were younger than 18 and had an IBI confirmed by bacterial culture or PCR from normally sterile fluid.
Thirty-eight episodes occurred in children who had undergone hematopoietic stem cell transplantation (HSCT), and 179 in children who had not undergone HSCT. The presentation focused exclusively on the latter group.
Among episodes in children without HSCT, the mean age was 7. Forty-eight patients had a history of acute chest syndrome, 47 had a history of ICU admission, 29 had a history of IBI, and 27 had a history of acute splenic sequestration. Thirteen underwent a splenectomy. Almost half of children had none of these characteristics, while about one-fourth had two or more.
In the HSCT group, 141 children were on prophylaxis at the time of the infection; 74 were on hydroxyurea, and 36 were currently or previously on a transfusion program. Sixty-eight cases were primary bacteremia and 55 were osteoarticular. Other syndromes included pneumonia empyema (n = 18), and meningitis (n = 17), among others. In 44 cases, the isolated bacteria was Salmonella, followed by S. pneumoniae in 32 cases. Escherichia coli accounted for 22. Haemophilus influenza was identified in six episodes, and group A Streptococcus in three.
The study is the first large European epidemiologic study investigating IBI in children with sickle cell disease, and one of its strengths was the strict inclusion criteria. However, it was limited by its retrospective nature.
Dr. Gaschignard and Dr. Rohr have no relevant financial disclosures.
FROM ESPID 2021
Sustained long-term benefit of gene therapy for SMA
, new long-term follow-up data show. At a median of 5.2 years since receiving the approved therapeutic dose, onasemnogene abeparvovec provided “sustained, durable efficacy, with all patients alive and without the need for permanent ventilation,” reported Jerry Mendell, MD, with the Center for Gene Therapy, Nationwide Children’s Hospital, Columbus, Ohio, and colleagues.
The study was published online May 17 in JAMA Neurology.
Single infusion
SMA is a rare genetic disease that can lead to paralysis, breathing difficulty, and death. The disorder is caused by a mutation in the survival motor neuron 1 (SMN1) gene, which encodes the SMN protein critical for maintenance and function of motor neurons.
In 2019, Zolgensma was approved in the United States for children with SMA and younger than 2 years of age.
Zolgensma is an adeno-associated virus vector-based gene therapy that addresses the genetic root cause of SMA by replacing the defective or missing SMN1 gene to halt disease progression. A single, one-time intravenous infusion results in expression of the SMN protein motor neurons, which improves chances of survival, as well as muscle movement and function.
In the phase 1 START study, 15 infants with SMA type 1 were treated with either a low or therapeutic dose of Zolgensma at Nationwide Children’s Hospital between 2014 and 2017.
The START long-term follow-up study (START LTFU) is an ongoing, observational study assessing safety and durability of response over 15 years in 13 of the infants; three infants received the low dose and 10 received the approved high dose.
Prior to baseline, four patients (40%) in the therapeutic dose cohort required noninvasive ventilatory support, and six (60%) did not require regular ventilatory support, which did not change in long-term follow-up.
All 10 patients who received the therapeutic dose remained alive and without the need for permanent ventilation up to 6.2 years after dosing, Dr. Mendell and colleagues report.
These patients also maintained previously acquired motor milestones. Two patients attained the new milestone of “standing with assistance” without the use of nusinersen (Spinraza, Biogen).
Serious adverse events occurred in eight patients (62%), none of which resulted in study discontinuation or death. The most common serious adverse events were related to the underlying SMA disease process and included acute respiratory failure (31%), pneumonia (31%), dehydration (23%), respiratory distress (15%), and bronchiolitis (15%).
Importantly, the investigators noted, no new safety signals or “adverse events of special interest” emerged during follow-up, including liver function enzyme elevations, new incidences of malignancy or hematologic disorders, and new incidences or exacerbations of existing neurologic or autoimmune disorders.
The investigators acknowledged that this follow-up study is limited by the small sample size of the patient population and confounded by treatment with nusinersen in several patients. “However, given that the two patients who acquired the new motor milestone of standing with assistance did not receive nusinersen at any time, this benefit can be attributed solely to onasemnogene abeparvovec,” Dr. Mendell and colleagues said.
The study was supported by Novartis Gene Therapies. Dr. Mendell and several co-investigators have disclosed financial relationships with the company.
A version of this article first appeared on Medscape.com.
, new long-term follow-up data show. At a median of 5.2 years since receiving the approved therapeutic dose, onasemnogene abeparvovec provided “sustained, durable efficacy, with all patients alive and without the need for permanent ventilation,” reported Jerry Mendell, MD, with the Center for Gene Therapy, Nationwide Children’s Hospital, Columbus, Ohio, and colleagues.
The study was published online May 17 in JAMA Neurology.
Single infusion
SMA is a rare genetic disease that can lead to paralysis, breathing difficulty, and death. The disorder is caused by a mutation in the survival motor neuron 1 (SMN1) gene, which encodes the SMN protein critical for maintenance and function of motor neurons.
In 2019, Zolgensma was approved in the United States for children with SMA and younger than 2 years of age.
Zolgensma is an adeno-associated virus vector-based gene therapy that addresses the genetic root cause of SMA by replacing the defective or missing SMN1 gene to halt disease progression. A single, one-time intravenous infusion results in expression of the SMN protein motor neurons, which improves chances of survival, as well as muscle movement and function.
In the phase 1 START study, 15 infants with SMA type 1 were treated with either a low or therapeutic dose of Zolgensma at Nationwide Children’s Hospital between 2014 and 2017.
The START long-term follow-up study (START LTFU) is an ongoing, observational study assessing safety and durability of response over 15 years in 13 of the infants; three infants received the low dose and 10 received the approved high dose.
Prior to baseline, four patients (40%) in the therapeutic dose cohort required noninvasive ventilatory support, and six (60%) did not require regular ventilatory support, which did not change in long-term follow-up.
All 10 patients who received the therapeutic dose remained alive and without the need for permanent ventilation up to 6.2 years after dosing, Dr. Mendell and colleagues report.
These patients also maintained previously acquired motor milestones. Two patients attained the new milestone of “standing with assistance” without the use of nusinersen (Spinraza, Biogen).
Serious adverse events occurred in eight patients (62%), none of which resulted in study discontinuation or death. The most common serious adverse events were related to the underlying SMA disease process and included acute respiratory failure (31%), pneumonia (31%), dehydration (23%), respiratory distress (15%), and bronchiolitis (15%).
Importantly, the investigators noted, no new safety signals or “adverse events of special interest” emerged during follow-up, including liver function enzyme elevations, new incidences of malignancy or hematologic disorders, and new incidences or exacerbations of existing neurologic or autoimmune disorders.
The investigators acknowledged that this follow-up study is limited by the small sample size of the patient population and confounded by treatment with nusinersen in several patients. “However, given that the two patients who acquired the new motor milestone of standing with assistance did not receive nusinersen at any time, this benefit can be attributed solely to onasemnogene abeparvovec,” Dr. Mendell and colleagues said.
The study was supported by Novartis Gene Therapies. Dr. Mendell and several co-investigators have disclosed financial relationships with the company.
A version of this article first appeared on Medscape.com.
, new long-term follow-up data show. At a median of 5.2 years since receiving the approved therapeutic dose, onasemnogene abeparvovec provided “sustained, durable efficacy, with all patients alive and without the need for permanent ventilation,” reported Jerry Mendell, MD, with the Center for Gene Therapy, Nationwide Children’s Hospital, Columbus, Ohio, and colleagues.
The study was published online May 17 in JAMA Neurology.
Single infusion
SMA is a rare genetic disease that can lead to paralysis, breathing difficulty, and death. The disorder is caused by a mutation in the survival motor neuron 1 (SMN1) gene, which encodes the SMN protein critical for maintenance and function of motor neurons.
In 2019, Zolgensma was approved in the United States for children with SMA and younger than 2 years of age.
Zolgensma is an adeno-associated virus vector-based gene therapy that addresses the genetic root cause of SMA by replacing the defective or missing SMN1 gene to halt disease progression. A single, one-time intravenous infusion results in expression of the SMN protein motor neurons, which improves chances of survival, as well as muscle movement and function.
In the phase 1 START study, 15 infants with SMA type 1 were treated with either a low or therapeutic dose of Zolgensma at Nationwide Children’s Hospital between 2014 and 2017.
The START long-term follow-up study (START LTFU) is an ongoing, observational study assessing safety and durability of response over 15 years in 13 of the infants; three infants received the low dose and 10 received the approved high dose.
Prior to baseline, four patients (40%) in the therapeutic dose cohort required noninvasive ventilatory support, and six (60%) did not require regular ventilatory support, which did not change in long-term follow-up.
All 10 patients who received the therapeutic dose remained alive and without the need for permanent ventilation up to 6.2 years after dosing, Dr. Mendell and colleagues report.
These patients also maintained previously acquired motor milestones. Two patients attained the new milestone of “standing with assistance” without the use of nusinersen (Spinraza, Biogen).
Serious adverse events occurred in eight patients (62%), none of which resulted in study discontinuation or death. The most common serious adverse events were related to the underlying SMA disease process and included acute respiratory failure (31%), pneumonia (31%), dehydration (23%), respiratory distress (15%), and bronchiolitis (15%).
Importantly, the investigators noted, no new safety signals or “adverse events of special interest” emerged during follow-up, including liver function enzyme elevations, new incidences of malignancy or hematologic disorders, and new incidences or exacerbations of existing neurologic or autoimmune disorders.
The investigators acknowledged that this follow-up study is limited by the small sample size of the patient population and confounded by treatment with nusinersen in several patients. “However, given that the two patients who acquired the new motor milestone of standing with assistance did not receive nusinersen at any time, this benefit can be attributed solely to onasemnogene abeparvovec,” Dr. Mendell and colleagues said.
The study was supported by Novartis Gene Therapies. Dr. Mendell and several co-investigators have disclosed financial relationships with the company.
A version of this article first appeared on Medscape.com.
Two treatments show early promise for hypothalamic obesity
Two different agents showed potential for safely treating patients with hypothalamic obesity in two pilot studies with small numbers of patients.
One study prospectively randomized 21 adults with acquired hypothalamic obesity to treatment with placebo or Tesomet, a compound that combines the novel monoamine reuptake inhibitor tesofensine with metoprolol, a beta-blocker added to protect against adverse effects from tesofensine on heart rate and cardiac contractility. After 24 weeks of treatment, people on tesofensine/metoprolol had significant weight loss, compared with controls, while showing good tolerance with no significant effects on heart rate, blood pressure, or heart rhythm, Ulla Feldt-Rasmussen, MD, DMSc, reported at the annual meeting of the Endocrine Society.
The second report reviewed 18 children and adolescents with either acquired or genetic hypothalamic obesity who received open-label treatment with dextroamphetamine for an average of 20 months, and overall patients safely lost an average of 0.43 in their body mass index (BMI) standard deviation score, reported Jiska van Schaik, MD, in a separate talk at the meeting.
‘A supplement for lost satiety’
Patients with hypothalamic obesity face a dual problem from hypothalamic dysfunction that’s addressed by tesofensine, the weight-loss agent in Tesomet that increases hypothalamic levels of dopamine, serotonin, and noradrenaline by blocking reuptake, and thereby dulls appetite and food craving while also increasing fat metabolism, explained Dr. Feldt-Rasmussen, a professor of medical endocrinology at the University of Denmark and Rigshospitalet in Copenhagen. No treatment currently has regulatory approval for treating any form of hypothalamic obesity.
Tesofensine works as “a supplement for lost satiety, and satiety is what is lost” in patients with hypothalamic obesity as well in patients as Prader-Willi syndrome, the two disorders for which tesofensine/metoprolol is currently undergoing testing. “That’s the rationale, and it seems to work,” she declared during her talk. The formulation contains 0.5 mg tesofensine and 50 mg metoprolol administered orally once daily.
The study, run at Rigshospitalet, randomized 21 patients aged 18-75 years and with a BMI of at least 27 kg/m2who all had acquired hypothalamic obesity secondary to hypothalamic damage following cancer treatment. Patients averaged about 45 years of age, three-quarters were women, and their average BMI was about 37, with 90% having a BMI of at least 30.
The study’s design calls for 48-week follow-up; Dr. Feldt-Rasmussen presented the interim results after 24 weeks, with 18 of the 21 enrolled patients remaining in the study through 24 weeks. Three patients dropped out because of adverse events: one in the placebo arm, and two who received tesofensine/metoprolol.
Weight dropped by an average of 6.6 kg from baseline among the 11 patients who completed 24 weeks on tesofensine/metoprolol treatment, compared with no average change from baseline among the seven patients who completed the study on placebo, a significant difference. The researchers measured a validated, composite satiety score every 4 weeks, and found significantly more improvement among patients on tesofensine/metoprolol than in those on placebo during the study’s first half, but subsequently average scores among the actively treated patients fell to the same level of modest improvement as in the placebo patients.
Despite this, average weight loss in the patients on tesofensine/metoprolol steadily increased throughout the full 24 weeks.
Safety measures of diastolic blood pressure, heart rate, and corrected QT interval showed no significant between-group difference. Systolic pressure showed a transient average rise of 4 mm Hg above baseline in the tesofensine/metoprolol group, compared with a small dip in the control patients, but by 24 weeks average systolic blood pressure had reverted closer to baseline levels in both subgroups and showed no significant between-group difference. Two patients on tesofensine/metoprolol developed serious adverse events. In one patient these were not treatment related. The other patient developed anxiety after 8 weeks that was possibly treatment related but remained on treatment. Other adverse effects on tesofensine/metoprolol included dizziness, sleep disorder, and dry mouth, but all of these were mild and patients were willing to tolerate them to achieve their weight loss, Dr. Feldt-Rasmussen said.
Repurposing an ADHD treatment
Dextroamphetamine increases satiety and boosts resting energy expenditure, and is a common treatment for attention deficit hyperactivity disorder. Dr. van Schaik and coauthors reviewed 13 children and adolescents with acquired hypothalamic obesity and 5 with genetic hypothalamic obesity who received the treatment at either of two Dutch hospitals during 2014-2020. All 18 patients went on dextroamphetamine after other interventions had failed to produce improvement, said Dr. van Schaik, a researcher at University Medical Center and Wilhelmina Children’s Hospital in Utrecht, the Netherlands. The patients averaged about 13 years of age.
In addition to an overall effect on weight across all 18 subjects, the researchers found they could subdivide the full cohort into 10 responders (56%), 4 (22%) with weight stabilization on treatment, and 4 nonresponders (22%) who continued to gain weight despite treatment. The 10 responding patients had an average drop in their BMI standard deviation score of 0.91. All 10 responders had acquired hypothalamic obesity, and they averaged a 12.5 percentage point rise in their resting energy expenditure level, compared with baseline, while on treatment. The four whose weight stabilized on treatment included three patients with genetic hypothalamic obesity. The four nonresponders split into two with acquired hypothalamic obesity and two with the genetic form.
Thirteen patients (72%) had improvements in hyperphagia, energy, and behavior, and no patient had a serious adverse effect. One patient stopped treatment after 1 month because of elevated blood pressure.
“Dextroamphetamine may be promising, especially for acquired hypothalamic obesity,” Dr. van Schaik concluded, adding that prospective, controlled assessments are needed, and that a healthy lifestyle is the foundation of hypothalamic obesity treatment.
The Tesomet study was sponsored by Saniona, the company developing Tesomet. Dr Feldt-Rasmussen is an advisor to Saniona, and some of the coauthors on the study are Saniona employees. Dr. van Schaik had no disclosures.
Two different agents showed potential for safely treating patients with hypothalamic obesity in two pilot studies with small numbers of patients.
One study prospectively randomized 21 adults with acquired hypothalamic obesity to treatment with placebo or Tesomet, a compound that combines the novel monoamine reuptake inhibitor tesofensine with metoprolol, a beta-blocker added to protect against adverse effects from tesofensine on heart rate and cardiac contractility. After 24 weeks of treatment, people on tesofensine/metoprolol had significant weight loss, compared with controls, while showing good tolerance with no significant effects on heart rate, blood pressure, or heart rhythm, Ulla Feldt-Rasmussen, MD, DMSc, reported at the annual meeting of the Endocrine Society.
The second report reviewed 18 children and adolescents with either acquired or genetic hypothalamic obesity who received open-label treatment with dextroamphetamine for an average of 20 months, and overall patients safely lost an average of 0.43 in their body mass index (BMI) standard deviation score, reported Jiska van Schaik, MD, in a separate talk at the meeting.
‘A supplement for lost satiety’
Patients with hypothalamic obesity face a dual problem from hypothalamic dysfunction that’s addressed by tesofensine, the weight-loss agent in Tesomet that increases hypothalamic levels of dopamine, serotonin, and noradrenaline by blocking reuptake, and thereby dulls appetite and food craving while also increasing fat metabolism, explained Dr. Feldt-Rasmussen, a professor of medical endocrinology at the University of Denmark and Rigshospitalet in Copenhagen. No treatment currently has regulatory approval for treating any form of hypothalamic obesity.
Tesofensine works as “a supplement for lost satiety, and satiety is what is lost” in patients with hypothalamic obesity as well in patients as Prader-Willi syndrome, the two disorders for which tesofensine/metoprolol is currently undergoing testing. “That’s the rationale, and it seems to work,” she declared during her talk. The formulation contains 0.5 mg tesofensine and 50 mg metoprolol administered orally once daily.
The study, run at Rigshospitalet, randomized 21 patients aged 18-75 years and with a BMI of at least 27 kg/m2who all had acquired hypothalamic obesity secondary to hypothalamic damage following cancer treatment. Patients averaged about 45 years of age, three-quarters were women, and their average BMI was about 37, with 90% having a BMI of at least 30.
The study’s design calls for 48-week follow-up; Dr. Feldt-Rasmussen presented the interim results after 24 weeks, with 18 of the 21 enrolled patients remaining in the study through 24 weeks. Three patients dropped out because of adverse events: one in the placebo arm, and two who received tesofensine/metoprolol.
Weight dropped by an average of 6.6 kg from baseline among the 11 patients who completed 24 weeks on tesofensine/metoprolol treatment, compared with no average change from baseline among the seven patients who completed the study on placebo, a significant difference. The researchers measured a validated, composite satiety score every 4 weeks, and found significantly more improvement among patients on tesofensine/metoprolol than in those on placebo during the study’s first half, but subsequently average scores among the actively treated patients fell to the same level of modest improvement as in the placebo patients.
Despite this, average weight loss in the patients on tesofensine/metoprolol steadily increased throughout the full 24 weeks.
Safety measures of diastolic blood pressure, heart rate, and corrected QT interval showed no significant between-group difference. Systolic pressure showed a transient average rise of 4 mm Hg above baseline in the tesofensine/metoprolol group, compared with a small dip in the control patients, but by 24 weeks average systolic blood pressure had reverted closer to baseline levels in both subgroups and showed no significant between-group difference. Two patients on tesofensine/metoprolol developed serious adverse events. In one patient these were not treatment related. The other patient developed anxiety after 8 weeks that was possibly treatment related but remained on treatment. Other adverse effects on tesofensine/metoprolol included dizziness, sleep disorder, and dry mouth, but all of these were mild and patients were willing to tolerate them to achieve their weight loss, Dr. Feldt-Rasmussen said.
Repurposing an ADHD treatment
Dextroamphetamine increases satiety and boosts resting energy expenditure, and is a common treatment for attention deficit hyperactivity disorder. Dr. van Schaik and coauthors reviewed 13 children and adolescents with acquired hypothalamic obesity and 5 with genetic hypothalamic obesity who received the treatment at either of two Dutch hospitals during 2014-2020. All 18 patients went on dextroamphetamine after other interventions had failed to produce improvement, said Dr. van Schaik, a researcher at University Medical Center and Wilhelmina Children’s Hospital in Utrecht, the Netherlands. The patients averaged about 13 years of age.
In addition to an overall effect on weight across all 18 subjects, the researchers found they could subdivide the full cohort into 10 responders (56%), 4 (22%) with weight stabilization on treatment, and 4 nonresponders (22%) who continued to gain weight despite treatment. The 10 responding patients had an average drop in their BMI standard deviation score of 0.91. All 10 responders had acquired hypothalamic obesity, and they averaged a 12.5 percentage point rise in their resting energy expenditure level, compared with baseline, while on treatment. The four whose weight stabilized on treatment included three patients with genetic hypothalamic obesity. The four nonresponders split into two with acquired hypothalamic obesity and two with the genetic form.
Thirteen patients (72%) had improvements in hyperphagia, energy, and behavior, and no patient had a serious adverse effect. One patient stopped treatment after 1 month because of elevated blood pressure.
“Dextroamphetamine may be promising, especially for acquired hypothalamic obesity,” Dr. van Schaik concluded, adding that prospective, controlled assessments are needed, and that a healthy lifestyle is the foundation of hypothalamic obesity treatment.
The Tesomet study was sponsored by Saniona, the company developing Tesomet. Dr Feldt-Rasmussen is an advisor to Saniona, and some of the coauthors on the study are Saniona employees. Dr. van Schaik had no disclosures.
Two different agents showed potential for safely treating patients with hypothalamic obesity in two pilot studies with small numbers of patients.
One study prospectively randomized 21 adults with acquired hypothalamic obesity to treatment with placebo or Tesomet, a compound that combines the novel monoamine reuptake inhibitor tesofensine with metoprolol, a beta-blocker added to protect against adverse effects from tesofensine on heart rate and cardiac contractility. After 24 weeks of treatment, people on tesofensine/metoprolol had significant weight loss, compared with controls, while showing good tolerance with no significant effects on heart rate, blood pressure, or heart rhythm, Ulla Feldt-Rasmussen, MD, DMSc, reported at the annual meeting of the Endocrine Society.
The second report reviewed 18 children and adolescents with either acquired or genetic hypothalamic obesity who received open-label treatment with dextroamphetamine for an average of 20 months, and overall patients safely lost an average of 0.43 in their body mass index (BMI) standard deviation score, reported Jiska van Schaik, MD, in a separate talk at the meeting.
‘A supplement for lost satiety’
Patients with hypothalamic obesity face a dual problem from hypothalamic dysfunction that’s addressed by tesofensine, the weight-loss agent in Tesomet that increases hypothalamic levels of dopamine, serotonin, and noradrenaline by blocking reuptake, and thereby dulls appetite and food craving while also increasing fat metabolism, explained Dr. Feldt-Rasmussen, a professor of medical endocrinology at the University of Denmark and Rigshospitalet in Copenhagen. No treatment currently has regulatory approval for treating any form of hypothalamic obesity.
Tesofensine works as “a supplement for lost satiety, and satiety is what is lost” in patients with hypothalamic obesity as well in patients as Prader-Willi syndrome, the two disorders for which tesofensine/metoprolol is currently undergoing testing. “That’s the rationale, and it seems to work,” she declared during her talk. The formulation contains 0.5 mg tesofensine and 50 mg metoprolol administered orally once daily.
The study, run at Rigshospitalet, randomized 21 patients aged 18-75 years and with a BMI of at least 27 kg/m2who all had acquired hypothalamic obesity secondary to hypothalamic damage following cancer treatment. Patients averaged about 45 years of age, three-quarters were women, and their average BMI was about 37, with 90% having a BMI of at least 30.
The study’s design calls for 48-week follow-up; Dr. Feldt-Rasmussen presented the interim results after 24 weeks, with 18 of the 21 enrolled patients remaining in the study through 24 weeks. Three patients dropped out because of adverse events: one in the placebo arm, and two who received tesofensine/metoprolol.
Weight dropped by an average of 6.6 kg from baseline among the 11 patients who completed 24 weeks on tesofensine/metoprolol treatment, compared with no average change from baseline among the seven patients who completed the study on placebo, a significant difference. The researchers measured a validated, composite satiety score every 4 weeks, and found significantly more improvement among patients on tesofensine/metoprolol than in those on placebo during the study’s first half, but subsequently average scores among the actively treated patients fell to the same level of modest improvement as in the placebo patients.
Despite this, average weight loss in the patients on tesofensine/metoprolol steadily increased throughout the full 24 weeks.
Safety measures of diastolic blood pressure, heart rate, and corrected QT interval showed no significant between-group difference. Systolic pressure showed a transient average rise of 4 mm Hg above baseline in the tesofensine/metoprolol group, compared with a small dip in the control patients, but by 24 weeks average systolic blood pressure had reverted closer to baseline levels in both subgroups and showed no significant between-group difference. Two patients on tesofensine/metoprolol developed serious adverse events. In one patient these were not treatment related. The other patient developed anxiety after 8 weeks that was possibly treatment related but remained on treatment. Other adverse effects on tesofensine/metoprolol included dizziness, sleep disorder, and dry mouth, but all of these were mild and patients were willing to tolerate them to achieve their weight loss, Dr. Feldt-Rasmussen said.
Repurposing an ADHD treatment
Dextroamphetamine increases satiety and boosts resting energy expenditure, and is a common treatment for attention deficit hyperactivity disorder. Dr. van Schaik and coauthors reviewed 13 children and adolescents with acquired hypothalamic obesity and 5 with genetic hypothalamic obesity who received the treatment at either of two Dutch hospitals during 2014-2020. All 18 patients went on dextroamphetamine after other interventions had failed to produce improvement, said Dr. van Schaik, a researcher at University Medical Center and Wilhelmina Children’s Hospital in Utrecht, the Netherlands. The patients averaged about 13 years of age.
In addition to an overall effect on weight across all 18 subjects, the researchers found they could subdivide the full cohort into 10 responders (56%), 4 (22%) with weight stabilization on treatment, and 4 nonresponders (22%) who continued to gain weight despite treatment. The 10 responding patients had an average drop in their BMI standard deviation score of 0.91. All 10 responders had acquired hypothalamic obesity, and they averaged a 12.5 percentage point rise in their resting energy expenditure level, compared with baseline, while on treatment. The four whose weight stabilized on treatment included three patients with genetic hypothalamic obesity. The four nonresponders split into two with acquired hypothalamic obesity and two with the genetic form.
Thirteen patients (72%) had improvements in hyperphagia, energy, and behavior, and no patient had a serious adverse effect. One patient stopped treatment after 1 month because of elevated blood pressure.
“Dextroamphetamine may be promising, especially for acquired hypothalamic obesity,” Dr. van Schaik concluded, adding that prospective, controlled assessments are needed, and that a healthy lifestyle is the foundation of hypothalamic obesity treatment.
The Tesomet study was sponsored by Saniona, the company developing Tesomet. Dr Feldt-Rasmussen is an advisor to Saniona, and some of the coauthors on the study are Saniona employees. Dr. van Schaik had no disclosures.
FROM ENDO 2021
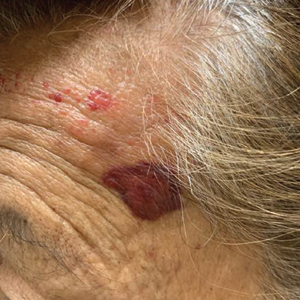
Mycosis Fungoides in Black Patients: Time for a Better Look
Recent advances in the immunopathogenesis and therapy of cutaneous T-cell lymphoma (CTCL) have shown great promise for the care of patients with mycosis fungoides (MF) and Sézary syndrome (SS).1-3 Research into the tumor microenvironment, microbiome, and molecular genetics may yield further information as we strive to develop MF/SS therapy from the bench to the bedside.3 Although progress has been made on multiple fronts in MF, some important—particularly epidemiologic and clinical—questions remain unanswered.
Racial disparities are well known to exist in CTCLs, particularly MF and SS.4-7 The incidence of MF and SS in the United States is higher in African American/Black patients than in White patients4; in addition, MF has an earlier age at onset in Black patients compared with White patients.4,5 Gender disparities also exist, with relatively more Black females than males affected with MF4-6; in particular, early-onset MF (ie, <40 years of age) is more common in Black females than Black males.6,7 According to Surveillance, Epidemiology, and End Results (SEER) data4 and the US National Cancer Database,5 African American/Black patients with MF have worse outcomes compared with other races (shorter overall survival and higher mortality) and also exhibit higher stages of disease at presentation (stage IIb or higher).5 Black race also was found to be a predictor of poor overall survival after accounting for disease characteristics, socioeconomicfactors, and types of treatment. The factors responsible for these racial disparities remain unclear.
A fortuitous collision of interests and technology may have helped to shed light on some of the reasons for these racial disparities in MF. Nearly 2 decades ago, high-quality, whole-body digital cutaneous photography was implemented by the Dermatology Service at Memorial Sloan Kettering Cancer Center Dermatology Service (New York, New York).8 Although the standardized 20-pose positioning images initially were used for the follow-up evaluation of patients with multiple nevi and melanomas, we incorporated the same photography technique into our multidisciplinary Cutaneous Lymphoma Clinic at Memorial Sloan Kettering Cancer Center. The multiplicity and clinical heterogeneity of MF lesions is well known, as is the fact that individual MF lesions may develop, respond to therapy, or change independently of other lesions in a given patient. We regularly reviewed these digital images with patients during their visits to assess treatment responses, discussed the need for changes in therapy in the face of progressive disease, and provided encouragement and positive reinforcement for those who improved with time-consuming regimens (eg, phototherapy).
Ultimately, as we became more familiar with looking at images in skin of color, we recognized different clinical features among our Black patients. In the literature, hypopigmented MF is a variant that typically is characterized by CD8+-predominant T cells and is seen more frequently in dark-skinned patients.9 In contrast, hyperpigmented MF has been considered a relatively rare presentation of MF.10 However, using only clinical and demographic information, we were able to identify 2 very different prognostic groups: those with hypopigmented lesions and those with only hyperpigmented and/or erythematous skin lesions.11 In our retrospective review of 157 African American/Black MF patients at our institution—122 with early-stage and 35 with late-stage MF—45% of patients had hypopigmented lesions vs 52% with hyperpigmented and/or erythematous lesions but no hypopigmentation. Those with hypopigmentation had superior outcomes, with better overall survival (P=.002) and progression-free survival (P=.014). In addition, more than 80% of patients who progressed or died from disease had hyperpigmented and/or erythematous lesions without hypopigmentation.11
Sometimes we have to go backward to go forward. Going from the bedside to the bench in our Black MF/SS patients—initially through the clinical recognition of prognostically different lesions, and then through clinicopathologic correlation with immunophenotyping and molecular studies—should provide important clues. Further investigation of Black patients who share similar pigmentary phenotypes of MF also may shed light on the pathogenetic mechanisms responsible for these prognostically significant skin findings. Through these efforts, we hope to identify higher-risk patients, which ultimately will lead to earlier intervention, more effective therapeutic regimens, and improved outcomes.
- Durgin JS, Weiner DM, Wysocka M, et al. The immunopathogenesis and immunotherapy of cutaneous T cell lymphoma: pathways and targets for immune restoration and tumor eradication. J Am Acad Dermatol. 2021;84:587-595.
- Weiner DM, Durgin JS, Wysocka M, et al. The immunopathogenesis and immunotherapy of cutaneous T cell lymphoma: current and future approaches. J Am Acad Dermatol. 2021;84:597-604.
- Quaglino P, Fava P, Pileri A, et al. Phenotypical markers, molecular mutations, and immune microenvironment as targets for new treatments in patients with mycosis fungoides and/or Sézary syndrome. J Invest Dermatol. 2021;141:484-495.
- Nath SK, Yu JB, Wilson LD. Poorer prognosis of African-American patients with mycosis fungoides: an analysis of the SEER dataset, 1988 to 2008. Clin Lymphoma Myeloma Leuk. 2014;14:419-423.
- Su C, Nguyen KA, Bai HX, et al. Racial disparity in mycosis fungoides: an analysis of 4495 cases from the US National Cancer Database. J Am Acad Dermatol. 2017;77:497-502.
- Balagula Y, Dusza SW, Zampella J, et al. Early-onset mycosis fungoides among African American women: a single-institution study. J Am Acad Dermatol. 2014;71:597-598.
- Virmani P, Levin L, Myskowski PL, et al. Clinical outcome and prognosis of young patients with mycosis fungoides. Pediatr Dermatol. 2017;34:547-553.
- Halpern AC, Marghoob AA, Bialoglow TW, et al. Standardized positioning of patients (poses) for whole body cutaneous photography. J Am Acad Dermatol. 2003;49:593-598.
- Rodney IJ, Kindred C, Angra K, et al. Hypopigmented mycosis fungoides: a retrospective clinicohistopathologic study. J Eur Acad Dermatol Venereol. 2017;31:808-814.
- Kondo M, Igawa K, Munetsugu T, et al. Increasing numbers of mast cells in skin lesions of hyperpigmented mycosis fungoides with large-cell transformation. Ann Dermatol. 2016;28:115-116.
- Geller S, Lebowitz E, Pulitzer MP, et al. Outcomes and prognostic factors in African American and Black patients with mycosis fungoides/Sézary syndrome: retrospective analysis of 157 patients from a referral cancer center. J Am Acad Dermatol. 2020;83:430-439.
Recent advances in the immunopathogenesis and therapy of cutaneous T-cell lymphoma (CTCL) have shown great promise for the care of patients with mycosis fungoides (MF) and Sézary syndrome (SS).1-3 Research into the tumor microenvironment, microbiome, and molecular genetics may yield further information as we strive to develop MF/SS therapy from the bench to the bedside.3 Although progress has been made on multiple fronts in MF, some important—particularly epidemiologic and clinical—questions remain unanswered.
Racial disparities are well known to exist in CTCLs, particularly MF and SS.4-7 The incidence of MF and SS in the United States is higher in African American/Black patients than in White patients4; in addition, MF has an earlier age at onset in Black patients compared with White patients.4,5 Gender disparities also exist, with relatively more Black females than males affected with MF4-6; in particular, early-onset MF (ie, <40 years of age) is more common in Black females than Black males.6,7 According to Surveillance, Epidemiology, and End Results (SEER) data4 and the US National Cancer Database,5 African American/Black patients with MF have worse outcomes compared with other races (shorter overall survival and higher mortality) and also exhibit higher stages of disease at presentation (stage IIb or higher).5 Black race also was found to be a predictor of poor overall survival after accounting for disease characteristics, socioeconomicfactors, and types of treatment. The factors responsible for these racial disparities remain unclear.
A fortuitous collision of interests and technology may have helped to shed light on some of the reasons for these racial disparities in MF. Nearly 2 decades ago, high-quality, whole-body digital cutaneous photography was implemented by the Dermatology Service at Memorial Sloan Kettering Cancer Center Dermatology Service (New York, New York).8 Although the standardized 20-pose positioning images initially were used for the follow-up evaluation of patients with multiple nevi and melanomas, we incorporated the same photography technique into our multidisciplinary Cutaneous Lymphoma Clinic at Memorial Sloan Kettering Cancer Center. The multiplicity and clinical heterogeneity of MF lesions is well known, as is the fact that individual MF lesions may develop, respond to therapy, or change independently of other lesions in a given patient. We regularly reviewed these digital images with patients during their visits to assess treatment responses, discussed the need for changes in therapy in the face of progressive disease, and provided encouragement and positive reinforcement for those who improved with time-consuming regimens (eg, phototherapy).
Ultimately, as we became more familiar with looking at images in skin of color, we recognized different clinical features among our Black patients. In the literature, hypopigmented MF is a variant that typically is characterized by CD8+-predominant T cells and is seen more frequently in dark-skinned patients.9 In contrast, hyperpigmented MF has been considered a relatively rare presentation of MF.10 However, using only clinical and demographic information, we were able to identify 2 very different prognostic groups: those with hypopigmented lesions and those with only hyperpigmented and/or erythematous skin lesions.11 In our retrospective review of 157 African American/Black MF patients at our institution—122 with early-stage and 35 with late-stage MF—45% of patients had hypopigmented lesions vs 52% with hyperpigmented and/or erythematous lesions but no hypopigmentation. Those with hypopigmentation had superior outcomes, with better overall survival (P=.002) and progression-free survival (P=.014). In addition, more than 80% of patients who progressed or died from disease had hyperpigmented and/or erythematous lesions without hypopigmentation.11
Sometimes we have to go backward to go forward. Going from the bedside to the bench in our Black MF/SS patients—initially through the clinical recognition of prognostically different lesions, and then through clinicopathologic correlation with immunophenotyping and molecular studies—should provide important clues. Further investigation of Black patients who share similar pigmentary phenotypes of MF also may shed light on the pathogenetic mechanisms responsible for these prognostically significant skin findings. Through these efforts, we hope to identify higher-risk patients, which ultimately will lead to earlier intervention, more effective therapeutic regimens, and improved outcomes.
Recent advances in the immunopathogenesis and therapy of cutaneous T-cell lymphoma (CTCL) have shown great promise for the care of patients with mycosis fungoides (MF) and Sézary syndrome (SS).1-3 Research into the tumor microenvironment, microbiome, and molecular genetics may yield further information as we strive to develop MF/SS therapy from the bench to the bedside.3 Although progress has been made on multiple fronts in MF, some important—particularly epidemiologic and clinical—questions remain unanswered.
Racial disparities are well known to exist in CTCLs, particularly MF and SS.4-7 The incidence of MF and SS in the United States is higher in African American/Black patients than in White patients4; in addition, MF has an earlier age at onset in Black patients compared with White patients.4,5 Gender disparities also exist, with relatively more Black females than males affected with MF4-6; in particular, early-onset MF (ie, <40 years of age) is more common in Black females than Black males.6,7 According to Surveillance, Epidemiology, and End Results (SEER) data4 and the US National Cancer Database,5 African American/Black patients with MF have worse outcomes compared with other races (shorter overall survival and higher mortality) and also exhibit higher stages of disease at presentation (stage IIb or higher).5 Black race also was found to be a predictor of poor overall survival after accounting for disease characteristics, socioeconomicfactors, and types of treatment. The factors responsible for these racial disparities remain unclear.
A fortuitous collision of interests and technology may have helped to shed light on some of the reasons for these racial disparities in MF. Nearly 2 decades ago, high-quality, whole-body digital cutaneous photography was implemented by the Dermatology Service at Memorial Sloan Kettering Cancer Center Dermatology Service (New York, New York).8 Although the standardized 20-pose positioning images initially were used for the follow-up evaluation of patients with multiple nevi and melanomas, we incorporated the same photography technique into our multidisciplinary Cutaneous Lymphoma Clinic at Memorial Sloan Kettering Cancer Center. The multiplicity and clinical heterogeneity of MF lesions is well known, as is the fact that individual MF lesions may develop, respond to therapy, or change independently of other lesions in a given patient. We regularly reviewed these digital images with patients during their visits to assess treatment responses, discussed the need for changes in therapy in the face of progressive disease, and provided encouragement and positive reinforcement for those who improved with time-consuming regimens (eg, phototherapy).
Ultimately, as we became more familiar with looking at images in skin of color, we recognized different clinical features among our Black patients. In the literature, hypopigmented MF is a variant that typically is characterized by CD8+-predominant T cells and is seen more frequently in dark-skinned patients.9 In contrast, hyperpigmented MF has been considered a relatively rare presentation of MF.10 However, using only clinical and demographic information, we were able to identify 2 very different prognostic groups: those with hypopigmented lesions and those with only hyperpigmented and/or erythematous skin lesions.11 In our retrospective review of 157 African American/Black MF patients at our institution—122 with early-stage and 35 with late-stage MF—45% of patients had hypopigmented lesions vs 52% with hyperpigmented and/or erythematous lesions but no hypopigmentation. Those with hypopigmentation had superior outcomes, with better overall survival (P=.002) and progression-free survival (P=.014). In addition, more than 80% of patients who progressed or died from disease had hyperpigmented and/or erythematous lesions without hypopigmentation.11
Sometimes we have to go backward to go forward. Going from the bedside to the bench in our Black MF/SS patients—initially through the clinical recognition of prognostically different lesions, and then through clinicopathologic correlation with immunophenotyping and molecular studies—should provide important clues. Further investigation of Black patients who share similar pigmentary phenotypes of MF also may shed light on the pathogenetic mechanisms responsible for these prognostically significant skin findings. Through these efforts, we hope to identify higher-risk patients, which ultimately will lead to earlier intervention, more effective therapeutic regimens, and improved outcomes.
- Durgin JS, Weiner DM, Wysocka M, et al. The immunopathogenesis and immunotherapy of cutaneous T cell lymphoma: pathways and targets for immune restoration and tumor eradication. J Am Acad Dermatol. 2021;84:587-595.
- Weiner DM, Durgin JS, Wysocka M, et al. The immunopathogenesis and immunotherapy of cutaneous T cell lymphoma: current and future approaches. J Am Acad Dermatol. 2021;84:597-604.
- Quaglino P, Fava P, Pileri A, et al. Phenotypical markers, molecular mutations, and immune microenvironment as targets for new treatments in patients with mycosis fungoides and/or Sézary syndrome. J Invest Dermatol. 2021;141:484-495.
- Nath SK, Yu JB, Wilson LD. Poorer prognosis of African-American patients with mycosis fungoides: an analysis of the SEER dataset, 1988 to 2008. Clin Lymphoma Myeloma Leuk. 2014;14:419-423.
- Su C, Nguyen KA, Bai HX, et al. Racial disparity in mycosis fungoides: an analysis of 4495 cases from the US National Cancer Database. J Am Acad Dermatol. 2017;77:497-502.
- Balagula Y, Dusza SW, Zampella J, et al. Early-onset mycosis fungoides among African American women: a single-institution study. J Am Acad Dermatol. 2014;71:597-598.
- Virmani P, Levin L, Myskowski PL, et al. Clinical outcome and prognosis of young patients with mycosis fungoides. Pediatr Dermatol. 2017;34:547-553.
- Halpern AC, Marghoob AA, Bialoglow TW, et al. Standardized positioning of patients (poses) for whole body cutaneous photography. J Am Acad Dermatol. 2003;49:593-598.
- Rodney IJ, Kindred C, Angra K, et al. Hypopigmented mycosis fungoides: a retrospective clinicohistopathologic study. J Eur Acad Dermatol Venereol. 2017;31:808-814.
- Kondo M, Igawa K, Munetsugu T, et al. Increasing numbers of mast cells in skin lesions of hyperpigmented mycosis fungoides with large-cell transformation. Ann Dermatol. 2016;28:115-116.
- Geller S, Lebowitz E, Pulitzer MP, et al. Outcomes and prognostic factors in African American and Black patients with mycosis fungoides/Sézary syndrome: retrospective analysis of 157 patients from a referral cancer center. J Am Acad Dermatol. 2020;83:430-439.
- Durgin JS, Weiner DM, Wysocka M, et al. The immunopathogenesis and immunotherapy of cutaneous T cell lymphoma: pathways and targets for immune restoration and tumor eradication. J Am Acad Dermatol. 2021;84:587-595.
- Weiner DM, Durgin JS, Wysocka M, et al. The immunopathogenesis and immunotherapy of cutaneous T cell lymphoma: current and future approaches. J Am Acad Dermatol. 2021;84:597-604.
- Quaglino P, Fava P, Pileri A, et al. Phenotypical markers, molecular mutations, and immune microenvironment as targets for new treatments in patients with mycosis fungoides and/or Sézary syndrome. J Invest Dermatol. 2021;141:484-495.
- Nath SK, Yu JB, Wilson LD. Poorer prognosis of African-American patients with mycosis fungoides: an analysis of the SEER dataset, 1988 to 2008. Clin Lymphoma Myeloma Leuk. 2014;14:419-423.
- Su C, Nguyen KA, Bai HX, et al. Racial disparity in mycosis fungoides: an analysis of 4495 cases from the US National Cancer Database. J Am Acad Dermatol. 2017;77:497-502.
- Balagula Y, Dusza SW, Zampella J, et al. Early-onset mycosis fungoides among African American women: a single-institution study. J Am Acad Dermatol. 2014;71:597-598.
- Virmani P, Levin L, Myskowski PL, et al. Clinical outcome and prognosis of young patients with mycosis fungoides. Pediatr Dermatol. 2017;34:547-553.
- Halpern AC, Marghoob AA, Bialoglow TW, et al. Standardized positioning of patients (poses) for whole body cutaneous photography. J Am Acad Dermatol. 2003;49:593-598.
- Rodney IJ, Kindred C, Angra K, et al. Hypopigmented mycosis fungoides: a retrospective clinicohistopathologic study. J Eur Acad Dermatol Venereol. 2017;31:808-814.
- Kondo M, Igawa K, Munetsugu T, et al. Increasing numbers of mast cells in skin lesions of hyperpigmented mycosis fungoides with large-cell transformation. Ann Dermatol. 2016;28:115-116.
- Geller S, Lebowitz E, Pulitzer MP, et al. Outcomes and prognostic factors in African American and Black patients with mycosis fungoides/Sézary syndrome: retrospective analysis of 157 patients from a referral cancer center. J Am Acad Dermatol. 2020;83:430-439.
Rituximab’s serious infection risk in ANCA-vasculitis allayed by antibiotic use
The serious infection risk associated with rituximab treatment for antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) is high but can be offset by co-prescribing co-trimoxazole, data from a single-center, retrospective study reaffirm.
Over the course of a 3-year study period, 14 (28%) of 50 patients with AAV treated with rituximab experienced at latest one severe infection defined as a grade 3 or higher event. The incidence of severe infections was 15.4 per 100 person-years.
However, a lower rate of infections was seen in patients who had been co-prescribed co-trimoxazole (trimethoprim and sulfamethoxazole), Francesco Dernie, a fifth-year medical student at the University of Oxford (England), reported at the British Society for Rheumatology annual conference.
“In the case of rituximab, the depletion of B cells and associated immune suppression is a double-edged sword, allowing effective disease control, but also leaving the body vulnerable to opportunistic and severe infections,” Mr. Dernie said at the meeting.
Of the patients who developed a severe infection on rituximab, just 7% had been treated with co-trimoxazole. In comparison, 44% of those who did not get a severe infection had received co-trimoxazole. Multivariate analysis confirmed that co-trimoxazole use was an influencing factor, with an odds ratio (OR) of 0.096 (95% confidence interval, 0.009–0.996; P = .05).
Another finding was that patients with low immunoglobulin G levels (less than 6 g/L) were more likely to develop a severe infection than were those with higher IgG levels. Indeed, the OR for hypogammaglobulinemia and the risk for infection was 8.782 (95% CI, 1.19–64.6; P = .033).
“Our results support the monitoring of IgG levels to identify patients who may be more susceptible to infection, as well as the prescription of prophylactic co-trimoxazole to reduce overall severe infection risk,” Mr. Dernie and associates concluded in their abstract.
It’s a “really important message around co-trimoxazole,” observed Neil Basu, MBChB, a clinical senior lecturer and honorary consultant at the Institute of Infection, Immunity & Inflammation, University of Glasgow (Scotland).
“It still frustrates me when I see that patients haven’t received that while receiving rituximab. Of course, co-trimoxazole can have its problems,” said Dr. Basu, who was not involved in the study. “It’s not uncommon for patients to develop reactions or be intolerant to the drug.”
Raashid Luqmani, DM, a senior coauthor of the work and professor of rheumatology at the Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Science, University of Oxford, said: “The tolerance of co-trimoxazole has been remarkably good in this cohort.” If there was a problem with using co-trimoxazole, then “our standard would be to go with trimethoprim alone as the next in line and follow that with inhaled pentamidine. So, it’s kind of following what we would all generally do,” Dr. Luqmani said.

These data add further support for coprescribing antibiotic treatment with rituximab, he suggested.
“Worry about infection, worry about it a lot; not just worry about it, do something about it,” Dr. Luqmani said, and co-trimoxazole “is probably an effective means to do something about it.”
Study details
To look at the characteristics of and risk factors for serious infections associated with rituximab use in AAV, Mr. Dernie and associates retrospectively examined the electronic records of patients who had been treated between August 2016 and August 2019. Follow-up was until August 2020.
Of the 50 patients identified, nearly half (48%) were men. The average age was 60 years, ranging from 25 to 90 years. Most (n = 36; 72%) patients had a diagnosis of granulomatosis with polyangiitis, while another 2 (4%) had microscopic polyangiitis, 1 (2%) had eosinophilic granulomatosis with polyangiitis, and 11 (22%) had an overlapping type of vasculitis or undefined AAV.
Of the 18 severe infection events recorded, most (56%) involved the respiratory tract. Less than one-third (28%) were sepsis or neutropenic sepsis events, and there was one case each (6%) of cellulitis, complicated urinary tract infection, and recurrent wound infection.
There were “small numbers of individual comorbidities that were not sufficient to enter into our regression analysis,” Mr. Dernie noted. “It’s likely that comorbid conditions such as COPD [chronic obstructive pulmonary disease] also contribute to an individual’s risk of developing severe infections, and thus should factor into their individualized management.”
Mr. Dernie acknowledged in discussion: “One of the limitations of the study was we just looked at patients in a time when they were receiving rituximab, so they may have historically been exposed to other treatment options.” However, he added, “they weren’t having any other major DMARDs or immunosuppressive treatments at the time.”
Dr. Luqmani observed: “If you look at Francesco’s data on the hypogammaglobulinemia at the start of rituximab, that probably gives you a good idea of just how immunosuppressed these patients were already before we got to this point.”
Dr. Luqmani added: “I suspect that’s in keeping with a lot of other centers that have started using rituximab an awful lot for patients who previously had episodes of vasculitis treated with other disease-modifying therapies, particularly cyclophosphamide.”
But for how long should co-trimoxazole be given after the last rituximab dose? asked the chair of the session, Richard Watts, DM, of Norwich (England) Medical School. These data are purely observational, so it’s not possible to say, Mr. Dernie noted: “The patients that we included as having co-trimoxazole seem to be on it more or less consistently, permanently,” he said.
What about the best dose? “It’s a tricky one,” Dr. Luqmani said, as “we not only use co-trimoxazole for prophylaxis, but we often also want to use it for treatment of the vasculitis itself.”
It’s very likely that there was a mix of patients in the analysis that had received co-trimoxazole as either a treatment or prophylaxis, which means different doses, he said.
“It might be interesting to know whether there was a difference” between doses used and the prevention of infection, added Dr. Luqmani, “but I suspect the numbers are too small to tell.”
Mr. Dernie, Dr. Luqmani, and the other coauthors had no disclosures.
The serious infection risk associated with rituximab treatment for antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) is high but can be offset by co-prescribing co-trimoxazole, data from a single-center, retrospective study reaffirm.
Over the course of a 3-year study period, 14 (28%) of 50 patients with AAV treated with rituximab experienced at latest one severe infection defined as a grade 3 or higher event. The incidence of severe infections was 15.4 per 100 person-years.
However, a lower rate of infections was seen in patients who had been co-prescribed co-trimoxazole (trimethoprim and sulfamethoxazole), Francesco Dernie, a fifth-year medical student at the University of Oxford (England), reported at the British Society for Rheumatology annual conference.
“In the case of rituximab, the depletion of B cells and associated immune suppression is a double-edged sword, allowing effective disease control, but also leaving the body vulnerable to opportunistic and severe infections,” Mr. Dernie said at the meeting.
Of the patients who developed a severe infection on rituximab, just 7% had been treated with co-trimoxazole. In comparison, 44% of those who did not get a severe infection had received co-trimoxazole. Multivariate analysis confirmed that co-trimoxazole use was an influencing factor, with an odds ratio (OR) of 0.096 (95% confidence interval, 0.009–0.996; P = .05).
Another finding was that patients with low immunoglobulin G levels (less than 6 g/L) were more likely to develop a severe infection than were those with higher IgG levels. Indeed, the OR for hypogammaglobulinemia and the risk for infection was 8.782 (95% CI, 1.19–64.6; P = .033).
“Our results support the monitoring of IgG levels to identify patients who may be more susceptible to infection, as well as the prescription of prophylactic co-trimoxazole to reduce overall severe infection risk,” Mr. Dernie and associates concluded in their abstract.
It’s a “really important message around co-trimoxazole,” observed Neil Basu, MBChB, a clinical senior lecturer and honorary consultant at the Institute of Infection, Immunity & Inflammation, University of Glasgow (Scotland).
“It still frustrates me when I see that patients haven’t received that while receiving rituximab. Of course, co-trimoxazole can have its problems,” said Dr. Basu, who was not involved in the study. “It’s not uncommon for patients to develop reactions or be intolerant to the drug.”
Raashid Luqmani, DM, a senior coauthor of the work and professor of rheumatology at the Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Science, University of Oxford, said: “The tolerance of co-trimoxazole has been remarkably good in this cohort.” If there was a problem with using co-trimoxazole, then “our standard would be to go with trimethoprim alone as the next in line and follow that with inhaled pentamidine. So, it’s kind of following what we would all generally do,” Dr. Luqmani said.
These data add further support for coprescribing antibiotic treatment with rituximab, he suggested.
“Worry about infection, worry about it a lot; not just worry about it, do something about it,” Dr. Luqmani said, and co-trimoxazole “is probably an effective means to do something about it.”
Study details
To look at the characteristics of and risk factors for serious infections associated with rituximab use in AAV, Mr. Dernie and associates retrospectively examined the electronic records of patients who had been treated between August 2016 and August 2019. Follow-up was until August 2020.
Of the 50 patients identified, nearly half (48%) were men. The average age was 60 years, ranging from 25 to 90 years. Most (n = 36; 72%) patients had a diagnosis of granulomatosis with polyangiitis, while another 2 (4%) had microscopic polyangiitis, 1 (2%) had eosinophilic granulomatosis with polyangiitis, and 11 (22%) had an overlapping type of vasculitis or undefined AAV.
Of the 18 severe infection events recorded, most (56%) involved the respiratory tract. Less than one-third (28%) were sepsis or neutropenic sepsis events, and there was one case each (6%) of cellulitis, complicated urinary tract infection, and recurrent wound infection.
There were “small numbers of individual comorbidities that were not sufficient to enter into our regression analysis,” Mr. Dernie noted. “It’s likely that comorbid conditions such as COPD [chronic obstructive pulmonary disease] also contribute to an individual’s risk of developing severe infections, and thus should factor into their individualized management.”
Mr. Dernie acknowledged in discussion: “One of the limitations of the study was we just looked at patients in a time when they were receiving rituximab, so they may have historically been exposed to other treatment options.” However, he added, “they weren’t having any other major DMARDs or immunosuppressive treatments at the time.”
Dr. Luqmani observed: “If you look at Francesco’s data on the hypogammaglobulinemia at the start of rituximab, that probably gives you a good idea of just how immunosuppressed these patients were already before we got to this point.”
Dr. Luqmani added: “I suspect that’s in keeping with a lot of other centers that have started using rituximab an awful lot for patients who previously had episodes of vasculitis treated with other disease-modifying therapies, particularly cyclophosphamide.”
But for how long should co-trimoxazole be given after the last rituximab dose? asked the chair of the session, Richard Watts, DM, of Norwich (England) Medical School. These data are purely observational, so it’s not possible to say, Mr. Dernie noted: “The patients that we included as having co-trimoxazole seem to be on it more or less consistently, permanently,” he said.
What about the best dose? “It’s a tricky one,” Dr. Luqmani said, as “we not only use co-trimoxazole for prophylaxis, but we often also want to use it for treatment of the vasculitis itself.”
It’s very likely that there was a mix of patients in the analysis that had received co-trimoxazole as either a treatment or prophylaxis, which means different doses, he said.
“It might be interesting to know whether there was a difference” between doses used and the prevention of infection, added Dr. Luqmani, “but I suspect the numbers are too small to tell.”
Mr. Dernie, Dr. Luqmani, and the other coauthors had no disclosures.
The serious infection risk associated with rituximab treatment for antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) is high but can be offset by co-prescribing co-trimoxazole, data from a single-center, retrospective study reaffirm.
Over the course of a 3-year study period, 14 (28%) of 50 patients with AAV treated with rituximab experienced at latest one severe infection defined as a grade 3 or higher event. The incidence of severe infections was 15.4 per 100 person-years.
However, a lower rate of infections was seen in patients who had been co-prescribed co-trimoxazole (trimethoprim and sulfamethoxazole), Francesco Dernie, a fifth-year medical student at the University of Oxford (England), reported at the British Society for Rheumatology annual conference.
“In the case of rituximab, the depletion of B cells and associated immune suppression is a double-edged sword, allowing effective disease control, but also leaving the body vulnerable to opportunistic and severe infections,” Mr. Dernie said at the meeting.
Of the patients who developed a severe infection on rituximab, just 7% had been treated with co-trimoxazole. In comparison, 44% of those who did not get a severe infection had received co-trimoxazole. Multivariate analysis confirmed that co-trimoxazole use was an influencing factor, with an odds ratio (OR) of 0.096 (95% confidence interval, 0.009–0.996; P = .05).
Another finding was that patients with low immunoglobulin G levels (less than 6 g/L) were more likely to develop a severe infection than were those with higher IgG levels. Indeed, the OR for hypogammaglobulinemia and the risk for infection was 8.782 (95% CI, 1.19–64.6; P = .033).
“Our results support the monitoring of IgG levels to identify patients who may be more susceptible to infection, as well as the prescription of prophylactic co-trimoxazole to reduce overall severe infection risk,” Mr. Dernie and associates concluded in their abstract.
It’s a “really important message around co-trimoxazole,” observed Neil Basu, MBChB, a clinical senior lecturer and honorary consultant at the Institute of Infection, Immunity & Inflammation, University of Glasgow (Scotland).
“It still frustrates me when I see that patients haven’t received that while receiving rituximab. Of course, co-trimoxazole can have its problems,” said Dr. Basu, who was not involved in the study. “It’s not uncommon for patients to develop reactions or be intolerant to the drug.”
Raashid Luqmani, DM, a senior coauthor of the work and professor of rheumatology at the Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Science, University of Oxford, said: “The tolerance of co-trimoxazole has been remarkably good in this cohort.” If there was a problem with using co-trimoxazole, then “our standard would be to go with trimethoprim alone as the next in line and follow that with inhaled pentamidine. So, it’s kind of following what we would all generally do,” Dr. Luqmani said.
These data add further support for coprescribing antibiotic treatment with rituximab, he suggested.
“Worry about infection, worry about it a lot; not just worry about it, do something about it,” Dr. Luqmani said, and co-trimoxazole “is probably an effective means to do something about it.”
Study details
To look at the characteristics of and risk factors for serious infections associated with rituximab use in AAV, Mr. Dernie and associates retrospectively examined the electronic records of patients who had been treated between August 2016 and August 2019. Follow-up was until August 2020.
Of the 50 patients identified, nearly half (48%) were men. The average age was 60 years, ranging from 25 to 90 years. Most (n = 36; 72%) patients had a diagnosis of granulomatosis with polyangiitis, while another 2 (4%) had microscopic polyangiitis, 1 (2%) had eosinophilic granulomatosis with polyangiitis, and 11 (22%) had an overlapping type of vasculitis or undefined AAV.
Of the 18 severe infection events recorded, most (56%) involved the respiratory tract. Less than one-third (28%) were sepsis or neutropenic sepsis events, and there was one case each (6%) of cellulitis, complicated urinary tract infection, and recurrent wound infection.
There were “small numbers of individual comorbidities that were not sufficient to enter into our regression analysis,” Mr. Dernie noted. “It’s likely that comorbid conditions such as COPD [chronic obstructive pulmonary disease] also contribute to an individual’s risk of developing severe infections, and thus should factor into their individualized management.”
Mr. Dernie acknowledged in discussion: “One of the limitations of the study was we just looked at patients in a time when they were receiving rituximab, so they may have historically been exposed to other treatment options.” However, he added, “they weren’t having any other major DMARDs or immunosuppressive treatments at the time.”
Dr. Luqmani observed: “If you look at Francesco’s data on the hypogammaglobulinemia at the start of rituximab, that probably gives you a good idea of just how immunosuppressed these patients were already before we got to this point.”
Dr. Luqmani added: “I suspect that’s in keeping with a lot of other centers that have started using rituximab an awful lot for patients who previously had episodes of vasculitis treated with other disease-modifying therapies, particularly cyclophosphamide.”
But for how long should co-trimoxazole be given after the last rituximab dose? asked the chair of the session, Richard Watts, DM, of Norwich (England) Medical School. These data are purely observational, so it’s not possible to say, Mr. Dernie noted: “The patients that we included as having co-trimoxazole seem to be on it more or less consistently, permanently,” he said.
What about the best dose? “It’s a tricky one,” Dr. Luqmani said, as “we not only use co-trimoxazole for prophylaxis, but we often also want to use it for treatment of the vasculitis itself.”
It’s very likely that there was a mix of patients in the analysis that had received co-trimoxazole as either a treatment or prophylaxis, which means different doses, he said.
“It might be interesting to know whether there was a difference” between doses used and the prevention of infection, added Dr. Luqmani, “but I suspect the numbers are too small to tell.”
Mr. Dernie, Dr. Luqmani, and the other coauthors had no disclosures.
FROM BSR 2021
FDA panel narrowly backs avacopan approval
A panel of federal advisers on May 6 lent support to the ChemoCentryx bid for approval of avacopan for a rare and serious autoimmune condition. But they also flagged concerns about both the evidence supporting claims of a benefit for this experimental drug and its safety.

At a meeting of the Food and Drug Administration’s Arthritis Advisory Committee, panelists voted 10-8 on a question of whether the risk-benefit profile of avacopan is adequate to support approval.
ChemoCentryx is seeking approval of avacopan for antineutrophil cytoplasmic autoantibody (ANCA)–associated vasculitis in the subtypes of granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA).
Regardless of their vote on this approval question, the panelists shared an interest in avacopan’s potential to reduce glucocorticoid use among some patients with ANCA-associated vasculitis, also called AAV. Mara L. Becker, MD, MSCE, the chair of the FDA’s panel, was among the panelists who said they reluctantly voted no.

“It pains me because I really want more steroid-sparing” medicines, said Dr. Becker of Duke University, Durham, N.C., who cited a need to gather more data on avacopan.
Margrit Wiesendanger, MD, PhD, of the Icahn School of Medicine at Mount Sinai, New York, who was among the panelists voting yes, spoke of a need for caution if the FDA approves avacopan.
“Judicious use of this new medication will be warranted and perhaps additional guidance could be given to rheumatologists to help them decide for whom this medication is best,” she said.
Panelists had spoken earlier of avacopan as a possible alternative medicine for people with AAV who have conditions that make glucocorticoids riskier for them, such as those who have diabetes.
Close votes on safety profile, efficacy
The panel also voted 10-8 on a question about whether the safety profile of avacopan is adequate to support approval of avacopan for the treatment of adult patients with AAV.
In addition, the panel voted 9-9 on a question about whether efficacy data support approval of avacopan for the treatment of adult patients with AAV.
The FDA considers the recommendations of its advisory panels, but is not bound by them.
The FDA staff clearly expressed the view that ChemoCentryx fell short with the evidence presented for avacopan approval. Shares of San Carlos, Calif.–based ChemoCentryx dropped sharply from a May 3 closing price of $48.82 to a May 4 closing price of $26.63 after the FDA released the staff’s review of avacopan.
In a briefing prepared for the meeting, FDA staff detailed concerns about the evidence ChemoCentryx is using to seek approval. While acknowledging a need for new treatments for AAV as a rare condition, FDA staff honed in on what they described flaws in the testing of this experimental medicine, which is a small-molecule antagonist of the receptor of C5a, an end product of the complement cascade that acts as a potent neutrophil chemoattractant and agonist.
The FDA usually requires two phase 3 studies for approval of a new medicine but will do so with a single trial in cases of exceptional need, the agency staff said. But in these cases, the bar rises for the evidence provided from that single trial.
Difficulties in interpretation of complex study design
In the case of avacopan, though, the data from the key avacopan trial, Study CL010_168, known as ADVOCATE, there were substantial uncertainties around the phase 3 study design and results, raising questions about the adequacy of this single trial to inform the benefit-risk assessment.
In the briefing document, the FDA staff noted that it had “communicated many of the concerns” about ChemoCentryx’s research earlier to the company.
“Complexities of the study design, as detailed in the briefing document, raise questions about the interpretability of the data to define a clinically meaningful benefit of avacopan and its role in the management of AAV,” the FDA staff wrote.
“We acknowledge that AAV is a rare and serious disease associated with high morbidity and increased mortality. It is also a disease with high unmet need for new therapies. However, FDA wants to ensure that new products have a defined context of use, i.e., how a product would be used, and a favorable benefit-risk assessment for patients,” the staff added.
In addition, there were differences in the assessments performed by investigators and the adjudication committee, most frequently related to the attribution of persistent vasculitis, the FDA staff noted.
Statistical analyses of the primary endpoint using investigators’ estimates “resulted in more conservative estimates of treatment effect, e.g., statistical significance for superiority would no longer be demonstrated,” the FDA staff noted. “While the prespecified analysis used the Adjudicator assessments, the assessment based on the Investigators, experienced in management of vasculitis, may better reflect real-world use.”
Imbalances in use of glucocorticoids and maintenance therapy
Also among the complications in assessing the ADVOCATE trial data were the glucocorticoids taken by patients in the study, the FDA staff said.
In the avacopan arm of the trial, 86% of patients received non–study-supplied glucocorticoids. In addition, more avacopan‐treated patients experienced adverse events and serious adverse events within the hepatobiliary system leading to discontinuation.
Subgroups given different treatments represented another challenge in interpreting ADVOCATE results for the FDA staff.
At week 26, the proportion of patients in disease remission in the avacopan group (72.3%) was noninferior to the prednisone group (70.1%), the FDA staff said in the briefing document.
But at week 52, a disparity was observed between subgroups that had received rituximab and cyclophosphamide (intravenous and oral) induction treatment. The estimated risk difference for disease remission at week 52 was 15.0% (95% CI, 2.2%-27.7%) in the subgroup receiving induction with rituximab and 3.3% (95% CI, –14.8% to 21.4%) in the cyclophosphamide plus maintenance azathioprine subgroup, the agency’s staff said.
“Based on the data, there is no evidence of clinically meaningful treatment effect in the cyclophosphamide induction subgroup,” the FDA staff wrote. “Further, the treatment comparison in the complementary rituximab induction subgroup may not be considered meaningful because these patients did not receive maintenance therapy, i.e., due to undertreating of patients, the effect observed in the rituximab subgroup may not represent a clinically meaningful treatment effect, compared to standard of care.”

Rachel L. Glaser, MD, clinical team leader in FDA’s division of rheumatology and transplant medicine, reiterated these concerns to the advisory committee at the May 6 meeting.
“Throughout the development program, FDA advised the applicant that a noninferiority comparison would not be sufficient to show that avacopan can replaced glucocorticoids as it would be difficult to establish whether avacopan is effective or whether an effect was due to the rituximab or cyclophosphamide administered to both treatment arms,” she said.
In its briefing for the meeting, ChemoCentryx noted the limits of treatments now available for AAV. It also emphasized the toll of the condition, ranging from skin manifestations to glomerulonephritis to life-threatening pulmonary hemorrhage. If untreated, 80% of patients with GPA or MPA die within 2 years of disease onset, ChemoCentryx said in its briefing materials for the meeting.
The side effects of glucocorticoids were well known to the FDA panelists and the ChemoCentryx presenters. Witnesses at an open public hearing told their own stories of depression, anxiety, and irritability caused by these medicines.

During the ChemoCentryx presentation, a presenter for the company, Peter Merkel, MD, MPH, of the University of Pennsylvania, Philadelphia, said avacopan would provide patients with AAV with an alternative allowing them “to go on a much lower glucocorticoids regimen.”
A similar view was presented in a February 2021 editorial in the New England Journal of Medicine, titled “Avacopan – Time to Replace Glucocorticoids?” Written by Kenneth J. Warrington, MD, of the Mayo Clinic, Rochester, Minn., the opinion article called the ADVOCATE trial “a milestone in the treatment of ANCA-associated vasculitis; complement inhibition with avacopan has glucocorticoid-sparing effects and results in superior disease control.”
Dr. Warrington reported no conflicts in connection with his editorial nor payments from ChemoCentryx. He did report grants from other firms such as Eli Lilly.
Julia Lewis, MD, of Vanderbilt University, Nashville, Tenn., was among the more skeptical members of the FDA panel. She was among the “nays” in all three voting questions put to the panel. Still, she said there were signs of “clinically meaningful benefit” in the data presented, but noted that the nonstudy use of glucocorticoids made it difficult to interpret the ADVOCATE results.
Dr. Lewis noted that the FDA usually requires two studies for a drug approval, particularly with a compound not yet cleared for any use. While ANCA-associated vasculitis is rare, it would be possible to recruit patients for another trial of avacopan, adding to the results reported already for avacopan from ADVOCATE, she said.
“Were there to be another study, this would certainly be a supportive study and maybe qualify as two studies,” she said.
A panel of federal advisers on May 6 lent support to the ChemoCentryx bid for approval of avacopan for a rare and serious autoimmune condition. But they also flagged concerns about both the evidence supporting claims of a benefit for this experimental drug and its safety.
At a meeting of the Food and Drug Administration’s Arthritis Advisory Committee, panelists voted 10-8 on a question of whether the risk-benefit profile of avacopan is adequate to support approval.
ChemoCentryx is seeking approval of avacopan for antineutrophil cytoplasmic autoantibody (ANCA)–associated vasculitis in the subtypes of granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA).
Regardless of their vote on this approval question, the panelists shared an interest in avacopan’s potential to reduce glucocorticoid use among some patients with ANCA-associated vasculitis, also called AAV. Mara L. Becker, MD, MSCE, the chair of the FDA’s panel, was among the panelists who said they reluctantly voted no.
“It pains me because I really want more steroid-sparing” medicines, said Dr. Becker of Duke University, Durham, N.C., who cited a need to gather more data on avacopan.
Margrit Wiesendanger, MD, PhD, of the Icahn School of Medicine at Mount Sinai, New York, who was among the panelists voting yes, spoke of a need for caution if the FDA approves avacopan.
“Judicious use of this new medication will be warranted and perhaps additional guidance could be given to rheumatologists to help them decide for whom this medication is best,” she said.
Panelists had spoken earlier of avacopan as a possible alternative medicine for people with AAV who have conditions that make glucocorticoids riskier for them, such as those who have diabetes.
Close votes on safety profile, efficacy
The panel also voted 10-8 on a question about whether the safety profile of avacopan is adequate to support approval of avacopan for the treatment of adult patients with AAV.
In addition, the panel voted 9-9 on a question about whether efficacy data support approval of avacopan for the treatment of adult patients with AAV.
The FDA considers the recommendations of its advisory panels, but is not bound by them.
The FDA staff clearly expressed the view that ChemoCentryx fell short with the evidence presented for avacopan approval. Shares of San Carlos, Calif.–based ChemoCentryx dropped sharply from a May 3 closing price of $48.82 to a May 4 closing price of $26.63 after the FDA released the staff’s review of avacopan.
In a briefing prepared for the meeting, FDA staff detailed concerns about the evidence ChemoCentryx is using to seek approval. While acknowledging a need for new treatments for AAV as a rare condition, FDA staff honed in on what they described flaws in the testing of this experimental medicine, which is a small-molecule antagonist of the receptor of C5a, an end product of the complement cascade that acts as a potent neutrophil chemoattractant and agonist.
The FDA usually requires two phase 3 studies for approval of a new medicine but will do so with a single trial in cases of exceptional need, the agency staff said. But in these cases, the bar rises for the evidence provided from that single trial.
Difficulties in interpretation of complex study design
In the case of avacopan, though, the data from the key avacopan trial, Study CL010_168, known as ADVOCATE, there were substantial uncertainties around the phase 3 study design and results, raising questions about the adequacy of this single trial to inform the benefit-risk assessment.
In the briefing document, the FDA staff noted that it had “communicated many of the concerns” about ChemoCentryx’s research earlier to the company.
“Complexities of the study design, as detailed in the briefing document, raise questions about the interpretability of the data to define a clinically meaningful benefit of avacopan and its role in the management of AAV,” the FDA staff wrote.
“We acknowledge that AAV is a rare and serious disease associated with high morbidity and increased mortality. It is also a disease with high unmet need for new therapies. However, FDA wants to ensure that new products have a defined context of use, i.e., how a product would be used, and a favorable benefit-risk assessment for patients,” the staff added.
In addition, there were differences in the assessments performed by investigators and the adjudication committee, most frequently related to the attribution of persistent vasculitis, the FDA staff noted.
Statistical analyses of the primary endpoint using investigators’ estimates “resulted in more conservative estimates of treatment effect, e.g., statistical significance for superiority would no longer be demonstrated,” the FDA staff noted. “While the prespecified analysis used the Adjudicator assessments, the assessment based on the Investigators, experienced in management of vasculitis, may better reflect real-world use.”
Imbalances in use of glucocorticoids and maintenance therapy
Also among the complications in assessing the ADVOCATE trial data were the glucocorticoids taken by patients in the study, the FDA staff said.
In the avacopan arm of the trial, 86% of patients received non–study-supplied glucocorticoids. In addition, more avacopan‐treated patients experienced adverse events and serious adverse events within the hepatobiliary system leading to discontinuation.
Subgroups given different treatments represented another challenge in interpreting ADVOCATE results for the FDA staff.
At week 26, the proportion of patients in disease remission in the avacopan group (72.3%) was noninferior to the prednisone group (70.1%), the FDA staff said in the briefing document.
But at week 52, a disparity was observed between subgroups that had received rituximab and cyclophosphamide (intravenous and oral) induction treatment. The estimated risk difference for disease remission at week 52 was 15.0% (95% CI, 2.2%-27.7%) in the subgroup receiving induction with rituximab and 3.3% (95% CI, –14.8% to 21.4%) in the cyclophosphamide plus maintenance azathioprine subgroup, the agency’s staff said.
“Based on the data, there is no evidence of clinically meaningful treatment effect in the cyclophosphamide induction subgroup,” the FDA staff wrote. “Further, the treatment comparison in the complementary rituximab induction subgroup may not be considered meaningful because these patients did not receive maintenance therapy, i.e., due to undertreating of patients, the effect observed in the rituximab subgroup may not represent a clinically meaningful treatment effect, compared to standard of care.”
Rachel L. Glaser, MD, clinical team leader in FDA’s division of rheumatology and transplant medicine, reiterated these concerns to the advisory committee at the May 6 meeting.
“Throughout the development program, FDA advised the applicant that a noninferiority comparison would not be sufficient to show that avacopan can replaced glucocorticoids as it would be difficult to establish whether avacopan is effective or whether an effect was due to the rituximab or cyclophosphamide administered to both treatment arms,” she said.
In its briefing for the meeting, ChemoCentryx noted the limits of treatments now available for AAV. It also emphasized the toll of the condition, ranging from skin manifestations to glomerulonephritis to life-threatening pulmonary hemorrhage. If untreated, 80% of patients with GPA or MPA die within 2 years of disease onset, ChemoCentryx said in its briefing materials for the meeting.
The side effects of glucocorticoids were well known to the FDA panelists and the ChemoCentryx presenters. Witnesses at an open public hearing told their own stories of depression, anxiety, and irritability caused by these medicines.
During the ChemoCentryx presentation, a presenter for the company, Peter Merkel, MD, MPH, of the University of Pennsylvania, Philadelphia, said avacopan would provide patients with AAV with an alternative allowing them “to go on a much lower glucocorticoids regimen.”
A similar view was presented in a February 2021 editorial in the New England Journal of Medicine, titled “Avacopan – Time to Replace Glucocorticoids?” Written by Kenneth J. Warrington, MD, of the Mayo Clinic, Rochester, Minn., the opinion article called the ADVOCATE trial “a milestone in the treatment of ANCA-associated vasculitis; complement inhibition with avacopan has glucocorticoid-sparing effects and results in superior disease control.”
Dr. Warrington reported no conflicts in connection with his editorial nor payments from ChemoCentryx. He did report grants from other firms such as Eli Lilly.
Julia Lewis, MD, of Vanderbilt University, Nashville, Tenn., was among the more skeptical members of the FDA panel. She was among the “nays” in all three voting questions put to the panel. Still, she said there were signs of “clinically meaningful benefit” in the data presented, but noted that the nonstudy use of glucocorticoids made it difficult to interpret the ADVOCATE results.
Dr. Lewis noted that the FDA usually requires two studies for a drug approval, particularly with a compound not yet cleared for any use. While ANCA-associated vasculitis is rare, it would be possible to recruit patients for another trial of avacopan, adding to the results reported already for avacopan from ADVOCATE, she said.
“Were there to be another study, this would certainly be a supportive study and maybe qualify as two studies,” she said.
A panel of federal advisers on May 6 lent support to the ChemoCentryx bid for approval of avacopan for a rare and serious autoimmune condition. But they also flagged concerns about both the evidence supporting claims of a benefit for this experimental drug and its safety.
At a meeting of the Food and Drug Administration’s Arthritis Advisory Committee, panelists voted 10-8 on a question of whether the risk-benefit profile of avacopan is adequate to support approval.
ChemoCentryx is seeking approval of avacopan for antineutrophil cytoplasmic autoantibody (ANCA)–associated vasculitis in the subtypes of granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA).
Regardless of their vote on this approval question, the panelists shared an interest in avacopan’s potential to reduce glucocorticoid use among some patients with ANCA-associated vasculitis, also called AAV. Mara L. Becker, MD, MSCE, the chair of the FDA’s panel, was among the panelists who said they reluctantly voted no.
“It pains me because I really want more steroid-sparing” medicines, said Dr. Becker of Duke University, Durham, N.C., who cited a need to gather more data on avacopan.
Margrit Wiesendanger, MD, PhD, of the Icahn School of Medicine at Mount Sinai, New York, who was among the panelists voting yes, spoke of a need for caution if the FDA approves avacopan.
“Judicious use of this new medication will be warranted and perhaps additional guidance could be given to rheumatologists to help them decide for whom this medication is best,” she said.
Panelists had spoken earlier of avacopan as a possible alternative medicine for people with AAV who have conditions that make glucocorticoids riskier for them, such as those who have diabetes.
Close votes on safety profile, efficacy
The panel also voted 10-8 on a question about whether the safety profile of avacopan is adequate to support approval of avacopan for the treatment of adult patients with AAV.
In addition, the panel voted 9-9 on a question about whether efficacy data support approval of avacopan for the treatment of adult patients with AAV.
The FDA considers the recommendations of its advisory panels, but is not bound by them.
The FDA staff clearly expressed the view that ChemoCentryx fell short with the evidence presented for avacopan approval. Shares of San Carlos, Calif.–based ChemoCentryx dropped sharply from a May 3 closing price of $48.82 to a May 4 closing price of $26.63 after the FDA released the staff’s review of avacopan.
In a briefing prepared for the meeting, FDA staff detailed concerns about the evidence ChemoCentryx is using to seek approval. While acknowledging a need for new treatments for AAV as a rare condition, FDA staff honed in on what they described flaws in the testing of this experimental medicine, which is a small-molecule antagonist of the receptor of C5a, an end product of the complement cascade that acts as a potent neutrophil chemoattractant and agonist.
The FDA usually requires two phase 3 studies for approval of a new medicine but will do so with a single trial in cases of exceptional need, the agency staff said. But in these cases, the bar rises for the evidence provided from that single trial.
Difficulties in interpretation of complex study design
In the case of avacopan, though, the data from the key avacopan trial, Study CL010_168, known as ADVOCATE, there were substantial uncertainties around the phase 3 study design and results, raising questions about the adequacy of this single trial to inform the benefit-risk assessment.
In the briefing document, the FDA staff noted that it had “communicated many of the concerns” about ChemoCentryx’s research earlier to the company.
“Complexities of the study design, as detailed in the briefing document, raise questions about the interpretability of the data to define a clinically meaningful benefit of avacopan and its role in the management of AAV,” the FDA staff wrote.
“We acknowledge that AAV is a rare and serious disease associated with high morbidity and increased mortality. It is also a disease with high unmet need for new therapies. However, FDA wants to ensure that new products have a defined context of use, i.e., how a product would be used, and a favorable benefit-risk assessment for patients,” the staff added.
In addition, there were differences in the assessments performed by investigators and the adjudication committee, most frequently related to the attribution of persistent vasculitis, the FDA staff noted.
Statistical analyses of the primary endpoint using investigators’ estimates “resulted in more conservative estimates of treatment effect, e.g., statistical significance for superiority would no longer be demonstrated,” the FDA staff noted. “While the prespecified analysis used the Adjudicator assessments, the assessment based on the Investigators, experienced in management of vasculitis, may better reflect real-world use.”
Imbalances in use of glucocorticoids and maintenance therapy
Also among the complications in assessing the ADVOCATE trial data were the glucocorticoids taken by patients in the study, the FDA staff said.
In the avacopan arm of the trial, 86% of patients received non–study-supplied glucocorticoids. In addition, more avacopan‐treated patients experienced adverse events and serious adverse events within the hepatobiliary system leading to discontinuation.
Subgroups given different treatments represented another challenge in interpreting ADVOCATE results for the FDA staff.
At week 26, the proportion of patients in disease remission in the avacopan group (72.3%) was noninferior to the prednisone group (70.1%), the FDA staff said in the briefing document.
But at week 52, a disparity was observed between subgroups that had received rituximab and cyclophosphamide (intravenous and oral) induction treatment. The estimated risk difference for disease remission at week 52 was 15.0% (95% CI, 2.2%-27.7%) in the subgroup receiving induction with rituximab and 3.3% (95% CI, –14.8% to 21.4%) in the cyclophosphamide plus maintenance azathioprine subgroup, the agency’s staff said.
“Based on the data, there is no evidence of clinically meaningful treatment effect in the cyclophosphamide induction subgroup,” the FDA staff wrote. “Further, the treatment comparison in the complementary rituximab induction subgroup may not be considered meaningful because these patients did not receive maintenance therapy, i.e., due to undertreating of patients, the effect observed in the rituximab subgroup may not represent a clinically meaningful treatment effect, compared to standard of care.”
Rachel L. Glaser, MD, clinical team leader in FDA’s division of rheumatology and transplant medicine, reiterated these concerns to the advisory committee at the May 6 meeting.
“Throughout the development program, FDA advised the applicant that a noninferiority comparison would not be sufficient to show that avacopan can replaced glucocorticoids as it would be difficult to establish whether avacopan is effective or whether an effect was due to the rituximab or cyclophosphamide administered to both treatment arms,” she said.
In its briefing for the meeting, ChemoCentryx noted the limits of treatments now available for AAV. It also emphasized the toll of the condition, ranging from skin manifestations to glomerulonephritis to life-threatening pulmonary hemorrhage. If untreated, 80% of patients with GPA or MPA die within 2 years of disease onset, ChemoCentryx said in its briefing materials for the meeting.
The side effects of glucocorticoids were well known to the FDA panelists and the ChemoCentryx presenters. Witnesses at an open public hearing told their own stories of depression, anxiety, and irritability caused by these medicines.
During the ChemoCentryx presentation, a presenter for the company, Peter Merkel, MD, MPH, of the University of Pennsylvania, Philadelphia, said avacopan would provide patients with AAV with an alternative allowing them “to go on a much lower glucocorticoids regimen.”
A similar view was presented in a February 2021 editorial in the New England Journal of Medicine, titled “Avacopan – Time to Replace Glucocorticoids?” Written by Kenneth J. Warrington, MD, of the Mayo Clinic, Rochester, Minn., the opinion article called the ADVOCATE trial “a milestone in the treatment of ANCA-associated vasculitis; complement inhibition with avacopan has glucocorticoid-sparing effects and results in superior disease control.”
Dr. Warrington reported no conflicts in connection with his editorial nor payments from ChemoCentryx. He did report grants from other firms such as Eli Lilly.
Julia Lewis, MD, of Vanderbilt University, Nashville, Tenn., was among the more skeptical members of the FDA panel. She was among the “nays” in all three voting questions put to the panel. Still, she said there were signs of “clinically meaningful benefit” in the data presented, but noted that the nonstudy use of glucocorticoids made it difficult to interpret the ADVOCATE results.
Dr. Lewis noted that the FDA usually requires two studies for a drug approval, particularly with a compound not yet cleared for any use. While ANCA-associated vasculitis is rare, it would be possible to recruit patients for another trial of avacopan, adding to the results reported already for avacopan from ADVOCATE, she said.
“Were there to be another study, this would certainly be a supportive study and maybe qualify as two studies,” she said.
Gene therapy shows promise for Sanfilippo syndrome
. Most of the benefit from the treatment came in patients who began treatment at younger age, but comparisons to natural history controls showed profound improvement among many recipients, some of whom attained normal developmental trajectories.
The study was presented at the American Academy of Neurology’s 2021 annual meeting by Kevin Flanigan, MD, an attending neurologist at Nationwide Children’s Hospital in Columbus, Ohio. He highlighted the improved developmental outcomes. “There’s been nothing shown to change the cognitive pathway of the disease. This is the first time it’s been seen as a treatment effect,” Dr. Flanigan said during a follow-up Q&A session.
The therapy was delivered using an adeno-associated virus-9 (AAV-9) vector, which led one questioner to ask about potential safety concerns, since AAV-associated risks date back to the death of Jesse Gelsinger in 1999. “There is concern about AAV therapies related to immune responses to potentially complement-mediated activation and thrombocytopenic syndrome, which has led to clinical holds on some other AAV-9 products related to muscular dystrophies. We’ve not seen signals of anything reminiscent of that, and we’re at AAV-9 dosages that are quite similar to what’s been used elsewhere in the field,” said Dr. Flanigan.
The results have him optimistic about the therapy. “I do think if it continues to be increasing divergent from the natural history, it will be questionable as to whether a subsequent trial will be necessary for this. That’s a decision for the [Food and Drug Administration] and the company to decide. Each observation point that goes by, each patient treated, and each time we get more data, I get more and more confident. It’s really gratifying to watch,” said Dr. Flanigan.
The study confirms the potential of gene replacement therapy autosomal recessive conditions, according to Nicholas Johnson, MD, associate professor of neurology at Virginia Commonwealth University, Richmond, as well as a fellow of the American Academy of Neurology. “Where the genetic problem is loss of gene function, the ability to replace that gene using a viral approach is going to be transformative across the board for many of these different conditions, including Sanfilippo syndrome,” said Dr. Johnson, who attended the session but was not involved in the research.
Toxicity could remain an issue, even in the absence of AAV-based safety concerns. “The rate limiting step in terms of gene replacement therapy development likely relates to the ability to provide those therapies to larger adults, because many approaches are weight based and it’s unclear what the upper limit of toxicity would be for adults,” said Dr. Johnson.
Transpher A study results
Dr. Flanigan presented results from Transpher A, a phase 1/2 clinical trial that has enrolled 20 patients to date in three cohorts: Cohort 1, with 3 patients, received 5 x 1,012 vg/kg, and had a mean follow-up of 58 months; cohort 2, with 3 patients, received 1 x 1,013 vg/kg, and had a mean follow-up of 49 months; and cohort 3, with 14 patients, received 3 x 1,013 vg/kg, with a mean follow-up of 24 months. Included patients ranged from birth to age 2, or older than age 2 with a development quotient of 60 or higher on the Bayley Scale.
Dr. Flanigan showed a plot of developmental progress compared with natural history controls, which showed that patients treated before age 2 or with a developmental quotient of 60 or higher had improved outcomes compared to other patients in the high dose cohort. They continued to show normal developmental progression at 30-36 months post treatment, at a time when the natural history data suggested they would suffer cognitive decline. Two years after administration, this group had cerebral spinal fluid levels of heparan sulfate that fell below the lower limit of detection. Patients in the high-dose cohort had normalized CSF levels of GM2 and GM3 gangliosides, and there were reductions in plasma heparan sulfate and urinary glycosaminoglycans. There was also a sustained decrease in liver volume.
The highest dose group was originally given to older patients, and most were similar to the natural history cohort, though some did stabilize. “More compellingly, patients (in the high-dose group) who were treated younger actually showed continued increase in development. One individual follows the normal development quotient line, and we would say that these are really quite distinct from what we typically see in patients,” said Dr. Flanigan.
The treatment was well tolerated. There were no deaths or treatment-related serious adverse events, and no clinically-significant adverse events within the first 5 years of follow-up.
The study was funded by Abeona Therapeutics. Dr. Flanigan has been on advisory boards for Apic Bio and 4D Molecular Therapeutics, consulted for Encoded Therapeutics, and has received royalties from Audentes Therapeutics. Dr. Flanigan has received funding from and been a consultant for Avidity.
. Most of the benefit from the treatment came in patients who began treatment at younger age, but comparisons to natural history controls showed profound improvement among many recipients, some of whom attained normal developmental trajectories.
The study was presented at the American Academy of Neurology’s 2021 annual meeting by Kevin Flanigan, MD, an attending neurologist at Nationwide Children’s Hospital in Columbus, Ohio. He highlighted the improved developmental outcomes. “There’s been nothing shown to change the cognitive pathway of the disease. This is the first time it’s been seen as a treatment effect,” Dr. Flanigan said during a follow-up Q&A session.
The therapy was delivered using an adeno-associated virus-9 (AAV-9) vector, which led one questioner to ask about potential safety concerns, since AAV-associated risks date back to the death of Jesse Gelsinger in 1999. “There is concern about AAV therapies related to immune responses to potentially complement-mediated activation and thrombocytopenic syndrome, which has led to clinical holds on some other AAV-9 products related to muscular dystrophies. We’ve not seen signals of anything reminiscent of that, and we’re at AAV-9 dosages that are quite similar to what’s been used elsewhere in the field,” said Dr. Flanigan.
The results have him optimistic about the therapy. “I do think if it continues to be increasing divergent from the natural history, it will be questionable as to whether a subsequent trial will be necessary for this. That’s a decision for the [Food and Drug Administration] and the company to decide. Each observation point that goes by, each patient treated, and each time we get more data, I get more and more confident. It’s really gratifying to watch,” said Dr. Flanigan.
The study confirms the potential of gene replacement therapy autosomal recessive conditions, according to Nicholas Johnson, MD, associate professor of neurology at Virginia Commonwealth University, Richmond, as well as a fellow of the American Academy of Neurology. “Where the genetic problem is loss of gene function, the ability to replace that gene using a viral approach is going to be transformative across the board for many of these different conditions, including Sanfilippo syndrome,” said Dr. Johnson, who attended the session but was not involved in the research.
Toxicity could remain an issue, even in the absence of AAV-based safety concerns. “The rate limiting step in terms of gene replacement therapy development likely relates to the ability to provide those therapies to larger adults, because many approaches are weight based and it’s unclear what the upper limit of toxicity would be for adults,” said Dr. Johnson.
Transpher A study results
Dr. Flanigan presented results from Transpher A, a phase 1/2 clinical trial that has enrolled 20 patients to date in three cohorts: Cohort 1, with 3 patients, received 5 x 1,012 vg/kg, and had a mean follow-up of 58 months; cohort 2, with 3 patients, received 1 x 1,013 vg/kg, and had a mean follow-up of 49 months; and cohort 3, with 14 patients, received 3 x 1,013 vg/kg, with a mean follow-up of 24 months. Included patients ranged from birth to age 2, or older than age 2 with a development quotient of 60 or higher on the Bayley Scale.
Dr. Flanigan showed a plot of developmental progress compared with natural history controls, which showed that patients treated before age 2 or with a developmental quotient of 60 or higher had improved outcomes compared to other patients in the high dose cohort. They continued to show normal developmental progression at 30-36 months post treatment, at a time when the natural history data suggested they would suffer cognitive decline. Two years after administration, this group had cerebral spinal fluid levels of heparan sulfate that fell below the lower limit of detection. Patients in the high-dose cohort had normalized CSF levels of GM2 and GM3 gangliosides, and there were reductions in plasma heparan sulfate and urinary glycosaminoglycans. There was also a sustained decrease in liver volume.
The highest dose group was originally given to older patients, and most were similar to the natural history cohort, though some did stabilize. “More compellingly, patients (in the high-dose group) who were treated younger actually showed continued increase in development. One individual follows the normal development quotient line, and we would say that these are really quite distinct from what we typically see in patients,” said Dr. Flanigan.
The treatment was well tolerated. There were no deaths or treatment-related serious adverse events, and no clinically-significant adverse events within the first 5 years of follow-up.
The study was funded by Abeona Therapeutics. Dr. Flanigan has been on advisory boards for Apic Bio and 4D Molecular Therapeutics, consulted for Encoded Therapeutics, and has received royalties from Audentes Therapeutics. Dr. Flanigan has received funding from and been a consultant for Avidity.
. Most of the benefit from the treatment came in patients who began treatment at younger age, but comparisons to natural history controls showed profound improvement among many recipients, some of whom attained normal developmental trajectories.
The study was presented at the American Academy of Neurology’s 2021 annual meeting by Kevin Flanigan, MD, an attending neurologist at Nationwide Children’s Hospital in Columbus, Ohio. He highlighted the improved developmental outcomes. “There’s been nothing shown to change the cognitive pathway of the disease. This is the first time it’s been seen as a treatment effect,” Dr. Flanigan said during a follow-up Q&A session.
The therapy was delivered using an adeno-associated virus-9 (AAV-9) vector, which led one questioner to ask about potential safety concerns, since AAV-associated risks date back to the death of Jesse Gelsinger in 1999. “There is concern about AAV therapies related to immune responses to potentially complement-mediated activation and thrombocytopenic syndrome, which has led to clinical holds on some other AAV-9 products related to muscular dystrophies. We’ve not seen signals of anything reminiscent of that, and we’re at AAV-9 dosages that are quite similar to what’s been used elsewhere in the field,” said Dr. Flanigan.
The results have him optimistic about the therapy. “I do think if it continues to be increasing divergent from the natural history, it will be questionable as to whether a subsequent trial will be necessary for this. That’s a decision for the [Food and Drug Administration] and the company to decide. Each observation point that goes by, each patient treated, and each time we get more data, I get more and more confident. It’s really gratifying to watch,” said Dr. Flanigan.
The study confirms the potential of gene replacement therapy autosomal recessive conditions, according to Nicholas Johnson, MD, associate professor of neurology at Virginia Commonwealth University, Richmond, as well as a fellow of the American Academy of Neurology. “Where the genetic problem is loss of gene function, the ability to replace that gene using a viral approach is going to be transformative across the board for many of these different conditions, including Sanfilippo syndrome,” said Dr. Johnson, who attended the session but was not involved in the research.
Toxicity could remain an issue, even in the absence of AAV-based safety concerns. “The rate limiting step in terms of gene replacement therapy development likely relates to the ability to provide those therapies to larger adults, because many approaches are weight based and it’s unclear what the upper limit of toxicity would be for adults,” said Dr. Johnson.
Transpher A study results
Dr. Flanigan presented results from Transpher A, a phase 1/2 clinical trial that has enrolled 20 patients to date in three cohorts: Cohort 1, with 3 patients, received 5 x 1,012 vg/kg, and had a mean follow-up of 58 months; cohort 2, with 3 patients, received 1 x 1,013 vg/kg, and had a mean follow-up of 49 months; and cohort 3, with 14 patients, received 3 x 1,013 vg/kg, with a mean follow-up of 24 months. Included patients ranged from birth to age 2, or older than age 2 with a development quotient of 60 or higher on the Bayley Scale.
Dr. Flanigan showed a plot of developmental progress compared with natural history controls, which showed that patients treated before age 2 or with a developmental quotient of 60 or higher had improved outcomes compared to other patients in the high dose cohort. They continued to show normal developmental progression at 30-36 months post treatment, at a time when the natural history data suggested they would suffer cognitive decline. Two years after administration, this group had cerebral spinal fluid levels of heparan sulfate that fell below the lower limit of detection. Patients in the high-dose cohort had normalized CSF levels of GM2 and GM3 gangliosides, and there were reductions in plasma heparan sulfate and urinary glycosaminoglycans. There was also a sustained decrease in liver volume.
The highest dose group was originally given to older patients, and most were similar to the natural history cohort, though some did stabilize. “More compellingly, patients (in the high-dose group) who were treated younger actually showed continued increase in development. One individual follows the normal development quotient line, and we would say that these are really quite distinct from what we typically see in patients,” said Dr. Flanigan.
The treatment was well tolerated. There were no deaths or treatment-related serious adverse events, and no clinically-significant adverse events within the first 5 years of follow-up.
The study was funded by Abeona Therapeutics. Dr. Flanigan has been on advisory boards for Apic Bio and 4D Molecular Therapeutics, consulted for Encoded Therapeutics, and has received royalties from Audentes Therapeutics. Dr. Flanigan has received funding from and been a consultant for Avidity.
FROM AAN 2021
Rare Neurological Disease Special Report

Read the issue by clicking on the cover image or HERE
--
This 2021 issue is our seventh annual Rare Neurological Disease Special Report. It comes at a time when the global pandemic is still very much a part of our everyday lives. While the COVID-19 crisis is the polar opposite of a rare disease, it may offer some insights into the rare disease community. Collectively, we faced a disease no one knew much about—an untreatable condition that sent us into isolation, changed our world view, and altered our daily lives in ways we never imagined. We experienced, many of us for the first time, the fear and helplessness of a medical situation that was potentially fatal. Isn’t that what patients with rare diseases and their families face every day? Fear of the unknown; the desperate search for answers; isolation from a world and, all too often, a medical community that does not understand your situation; the terror of suspecting that time may not be on your side, and the valiant effort to press on despite desperate odds—these are things the rare disease community know all too well.
In publishing the Rare Neurological Disease Special Report, our goal has always been to educate the medical community on conditions they may rarely see and offer some hope to those who battle a rare neurological disease. I hope you enjoy reading our seventh annual issue.
—Glenn S. Williams, vice president, group editor, Neurology Reviews and MDedge Neurology
Read the issue by clicking on the cover image or HERE
--
This 2021 issue is our seventh annual Rare Neurological Disease Special Report. It comes at a time when the global pandemic is still very much a part of our everyday lives. While the COVID-19 crisis is the polar opposite of a rare disease, it may offer some insights into the rare disease community. Collectively, we faced a disease no one knew much about—an untreatable condition that sent us into isolation, changed our world view, and altered our daily lives in ways we never imagined. We experienced, many of us for the first time, the fear and helplessness of a medical situation that was potentially fatal. Isn’t that what patients with rare diseases and their families face every day? Fear of the unknown; the desperate search for answers; isolation from a world and, all too often, a medical community that does not understand your situation; the terror of suspecting that time may not be on your side, and the valiant effort to press on despite desperate odds—these are things the rare disease community know all too well.
In publishing the Rare Neurological Disease Special Report, our goal has always been to educate the medical community on conditions they may rarely see and offer some hope to those who battle a rare neurological disease. I hope you enjoy reading our seventh annual issue.
—Glenn S. Williams, vice president, group editor, Neurology Reviews and MDedge Neurology
Read the issue by clicking on the cover image or HERE
--
This 2021 issue is our seventh annual Rare Neurological Disease Special Report. It comes at a time when the global pandemic is still very much a part of our everyday lives. While the COVID-19 crisis is the polar opposite of a rare disease, it may offer some insights into the rare disease community. Collectively, we faced a disease no one knew much about—an untreatable condition that sent us into isolation, changed our world view, and altered our daily lives in ways we never imagined. We experienced, many of us for the first time, the fear and helplessness of a medical situation that was potentially fatal. Isn’t that what patients with rare diseases and their families face every day? Fear of the unknown; the desperate search for answers; isolation from a world and, all too often, a medical community that does not understand your situation; the terror of suspecting that time may not be on your side, and the valiant effort to press on despite desperate odds—these are things the rare disease community know all too well.
In publishing the Rare Neurological Disease Special Report, our goal has always been to educate the medical community on conditions they may rarely see and offer some hope to those who battle a rare neurological disease. I hope you enjoy reading our seventh annual issue.
—Glenn S. Williams, vice president, group editor, Neurology Reviews and MDedge Neurology

Asymptomatic Hemorrhagic Lesions in an Anemic Woman
The Diagnosis: Bullous Amyloidosis
A punch biopsy from the left temple showed deposits of amorphous eosinophilic material at the tips of dermal papillae and in the papillary dermis with hemorrhage present (Figure 1). A diagnosis of amyloidosis was confirmed on the biopsy of the skin bulla. The low κ/λ light chain ratio and M-spike with notably elevated free λ light chains in both serum and urine were consistent with a λ light chain primary systemic amyloidosis. The patient was seen by hematology and oncology. A bone marrow biopsy demonstrated that 15% to 20% of the clonal-cell population was λ light chain restricted. Eosinophilic extracellular deposits found in the adjacent soft tissue and bone marrow space were confirmed as amyloid with apple green birefringence under polarized light on Congo red stain and metachromatic staining with crystal violet. The patient ultimately was diagnosed with λ light chain multiple myeloma and primary systemic amyloidosis.
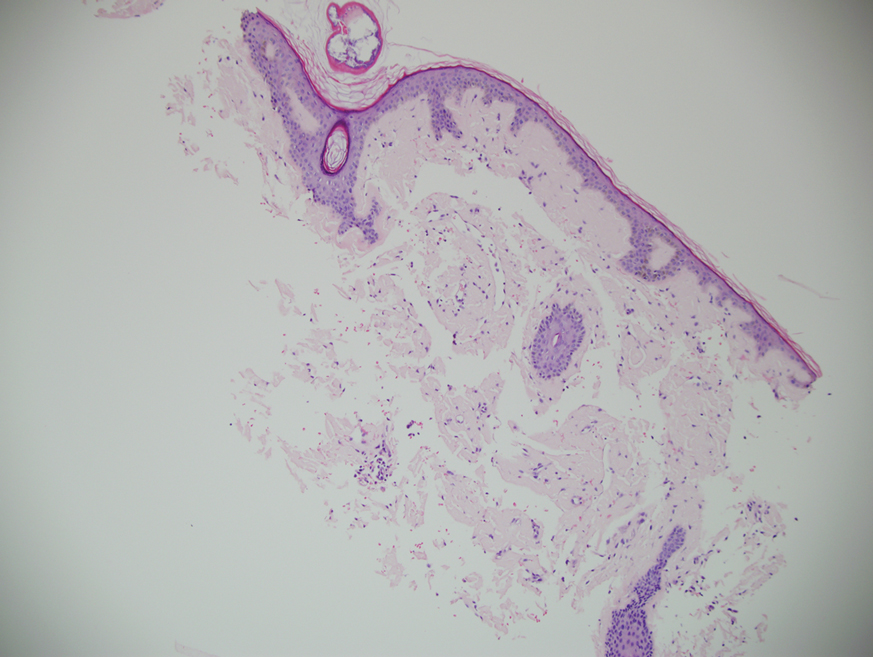
Our patient was treated with a combination therapy of bortezomib, cyclophosphamide, and dexamethasone on 21-day cycles, with bortezomib on days 1, 4, 8, and 11. She had received 3 cycles of chemotherapy before developing diarrhea, hypotension, acute on chronic heart failure, and acute renal failure requiring hospitalization. She had several related complications due to amyloid light chain (AL) amyloidosis and subsequently died 16 days after her initial hospitalization from complications of methicillin-resistant Staphylococcus aureus bacteremia and septic shock.
Amyloidosis is the pathologic deposition of abnormal protein in the extracellular space of any tissue. Various soluble precursor proteins can make up amyloid, and these proteins polymerize into insoluble fibrils that damage the surrounding parenchyma. The clinical presentation of amyloidosis varies depending on the affected tissue as well as the constituent protein. The amyloidoses are divided into localized cutaneous, primary systemic, and secondary systemic variants. The initial distinction in amyloidosis is determining whether it is skin limited or systemic. Localized cutaneous amyloidosis comprises 30% to 40% of all amyloidosis cases and is further divided into 3 main subtypes: macular, lichen, and nodular amyloidosis.1 Macular and lichen amyloidosis are composed of keratin derivatives and typically are induced by patients when rubbing or scratching the skin. Histologically, macular and lichen amyloidosis are restricted to the superficial papillary dermis.1 Nodular amyloidosis is composed of λ or κ light chain immunoglobulins, which are produced by cutaneous infiltrates of monoclonal plasma cells. Histologically, nodular amyloidosis is characterized by a diffuse dermal infiltrate of amorphous eosinophilic material.1 Primary systemic amyloidosis is associated with an underlying plasma cell dyscrasia, and unlike secondary keratinocyte-derived amyloid, it can involve internal organs. Similar to nodular amyloidosis, primary systemic amyloidosis is composed of AL proteins, and it is histologically similar to nodular amyloidosis.1
Primary systemic AL amyloidosis commonly affects individuals aged 50 to 60 years. Males and females are equally affected. Macroglossia and periorbital purpura are some of the pathognomonic presentations in AL amyloidosis. The major cause of death in these patients is cardiac and renal involvement. Renal involvement commonly presents as nephrotic syndrome, and cardiac involvement can present as a restrictive cardiomyopathy with dyspnea. Other symptoms include edema, hepatosplenomegaly, bleeding diathesis, and carpal tunnel syndrome.2 An evaluation for AL amyloidosis should include a complete review of systems and physical examination with studies such as complete blood cell count, comprehensive metabolic panel, serum and urine protein electrophoresis and immunofixation, and electrocardiogram.
Cutaneous involvement in AL amyloidosis most commonly includes yellowish waxy papules, nodules, and plaques but also can include purpura and petechiae.2 Bullous amyloidosis, as seen in our patient, is a rare cutaneous presentation of AL amyloidosis that usually is negative for the Nikolsky sign (Figure 2). Bullae form due to weakness in amyloid-laden dermal connective tissue.3 Eighty-eight percent of cases of bullous amyloidosis have systemic involvement.1 Some cases have reported a familial linkage, suggesting there might be a genetic component to the disease.4 A PubMed search of articles indexed for MEDLINE using the terms bullous amyloidosis, bullous, amyloidosis, and amyloid revealed fewer than 35 cases of bullous amyloidosis in the English-language literature.5 Bullae can be located intradermally or subepidermally and commonly are hemorrhagic but also can be translucent, tender, and tense.
A study of electron microscopy in a patient with systemic bullous amyloidosis demonstrated amyloid and keratinocyte protrusions that perforated the dermis through the spaces in the lamina densa. The study concluded that the disintegration of the lamina densa and expansion of the intercellular spaces between keratinocytes were the causes of skin fragility as well as fluid exudation.5 Trauma or friction to the skin are local precipitating factors for blister formation in bullous amyloidosis.
Bullae can become apparent at any stage of AL amyloidosis, but they generally increase in size and number over time and are most common in intertriginous areas. Bullous amyloid lesions, especially those located in intertriginous areas, can have secondary impetiginization.6 In many cases, patients who present with bullous amyloidosis ultimately will be diagnosed with multiple myeloma or another plasma cell dyscrasia. In AL amyloidosis, only 10% to 15% of cases meet criteria for multiple myeloma, whereas 80% or more patients have a monoclonal gammopathy of undetermined significance.7
The prognosis of cutaneous amyloidosis depends on the extent of organ involvement and response to treatment. Treatment is aimed at eliminating clonal plasma cell populations to decrease the production of light chains, thereby decreasing protein burden and amyloid progression. Historically, treatment options included cytotoxic chemotherapy such as oral melphalan and dexamethasone, followed by hematopoietic stem cell transplant. More recent treatment options include bortezomib, thalidomide, pomalidomide, and lenalidomide.8 Our patient received a regimen of bortezomib, cyclophosphamide, and dexamethasone that is used for patients with extensive multiple myeloma.
The differential diagnosis in our patient included bullous drug eruption, which should be considered if the bullae are reoccurring at the same location and in association with the administration of a culprit drug. Bullous pemphigoid is preceded by pruritus, and biopsy demonstrates subepidermal bullae with associated eosinophilic infiltrate. Epidermolysis bullosa acquisita can present with milia and a linear pattern along the basement membrane zone with direct immunofluorescence. Traumatic purpura usually present with the classic shape and hue of an ecchymosis, and the patient will have a history of trauma.
Cutaneous involvement of amyloidosis can be an early clue to the diagnosis of plasma cell dyscrasia. Early diagnosis and treatment can portend a better prognosis and prevent progression to renal or cardiac disease.
- Heaton J, Steinhoff N, Wanner B, et al. A review of primary cutaneous amyloidosis. J Am Osteopath Coll Dermatol. doi:10.1007/springerreference_42272
- Ventarola DJ, Schuster MW, Cohen JA, et al. JAAD grand rounds quiz. bullae and nodules on the legs of a 57-year-old woman. J Am Acad Dermatol. 2014;71:1035-1037.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Suranagi VV, Siddramappa B, Bannur HB, et al. Bullous variant of familial biphasic lichen amyloidosis: a unique combination of three rare presentations. Indian J Dermatol. 2015;60:105.
- Antúnez-Lay A, Jaque A, González S. Hemorrhagic bullous skin lesions. Int J Dermatol. 2017;56:145-147.
- Reddy K, Hoda S, Penstein A, et al. Bullous amyloidosis complicated by cellulitis and sepsis: a case report. Arch Dermatol. 2011;147:126-127.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
The Diagnosis: Bullous Amyloidosis
A punch biopsy from the left temple showed deposits of amorphous eosinophilic material at the tips of dermal papillae and in the papillary dermis with hemorrhage present (Figure 1). A diagnosis of amyloidosis was confirmed on the biopsy of the skin bulla. The low κ/λ light chain ratio and M-spike with notably elevated free λ light chains in both serum and urine were consistent with a λ light chain primary systemic amyloidosis. The patient was seen by hematology and oncology. A bone marrow biopsy demonstrated that 15% to 20% of the clonal-cell population was λ light chain restricted. Eosinophilic extracellular deposits found in the adjacent soft tissue and bone marrow space were confirmed as amyloid with apple green birefringence under polarized light on Congo red stain and metachromatic staining with crystal violet. The patient ultimately was diagnosed with λ light chain multiple myeloma and primary systemic amyloidosis.
Our patient was treated with a combination therapy of bortezomib, cyclophosphamide, and dexamethasone on 21-day cycles, with bortezomib on days 1, 4, 8, and 11. She had received 3 cycles of chemotherapy before developing diarrhea, hypotension, acute on chronic heart failure, and acute renal failure requiring hospitalization. She had several related complications due to amyloid light chain (AL) amyloidosis and subsequently died 16 days after her initial hospitalization from complications of methicillin-resistant Staphylococcus aureus bacteremia and septic shock.
Amyloidosis is the pathologic deposition of abnormal protein in the extracellular space of any tissue. Various soluble precursor proteins can make up amyloid, and these proteins polymerize into insoluble fibrils that damage the surrounding parenchyma. The clinical presentation of amyloidosis varies depending on the affected tissue as well as the constituent protein. The amyloidoses are divided into localized cutaneous, primary systemic, and secondary systemic variants. The initial distinction in amyloidosis is determining whether it is skin limited or systemic. Localized cutaneous amyloidosis comprises 30% to 40% of all amyloidosis cases and is further divided into 3 main subtypes: macular, lichen, and nodular amyloidosis.1 Macular and lichen amyloidosis are composed of keratin derivatives and typically are induced by patients when rubbing or scratching the skin. Histologically, macular and lichen amyloidosis are restricted to the superficial papillary dermis.1 Nodular amyloidosis is composed of λ or κ light chain immunoglobulins, which are produced by cutaneous infiltrates of monoclonal plasma cells. Histologically, nodular amyloidosis is characterized by a diffuse dermal infiltrate of amorphous eosinophilic material.1 Primary systemic amyloidosis is associated with an underlying plasma cell dyscrasia, and unlike secondary keratinocyte-derived amyloid, it can involve internal organs. Similar to nodular amyloidosis, primary systemic amyloidosis is composed of AL proteins, and it is histologically similar to nodular amyloidosis.1
Primary systemic AL amyloidosis commonly affects individuals aged 50 to 60 years. Males and females are equally affected. Macroglossia and periorbital purpura are some of the pathognomonic presentations in AL amyloidosis. The major cause of death in these patients is cardiac and renal involvement. Renal involvement commonly presents as nephrotic syndrome, and cardiac involvement can present as a restrictive cardiomyopathy with dyspnea. Other symptoms include edema, hepatosplenomegaly, bleeding diathesis, and carpal tunnel syndrome.2 An evaluation for AL amyloidosis should include a complete review of systems and physical examination with studies such as complete blood cell count, comprehensive metabolic panel, serum and urine protein electrophoresis and immunofixation, and electrocardiogram.
Cutaneous involvement in AL amyloidosis most commonly includes yellowish waxy papules, nodules, and plaques but also can include purpura and petechiae.2 Bullous amyloidosis, as seen in our patient, is a rare cutaneous presentation of AL amyloidosis that usually is negative for the Nikolsky sign (Figure 2). Bullae form due to weakness in amyloid-laden dermal connective tissue.3 Eighty-eight percent of cases of bullous amyloidosis have systemic involvement.1 Some cases have reported a familial linkage, suggesting there might be a genetic component to the disease.4 A PubMed search of articles indexed for MEDLINE using the terms bullous amyloidosis, bullous, amyloidosis, and amyloid revealed fewer than 35 cases of bullous amyloidosis in the English-language literature.5 Bullae can be located intradermally or subepidermally and commonly are hemorrhagic but also can be translucent, tender, and tense.
A study of electron microscopy in a patient with systemic bullous amyloidosis demonstrated amyloid and keratinocyte protrusions that perforated the dermis through the spaces in the lamina densa. The study concluded that the disintegration of the lamina densa and expansion of the intercellular spaces between keratinocytes were the causes of skin fragility as well as fluid exudation.5 Trauma or friction to the skin are local precipitating factors for blister formation in bullous amyloidosis.
Bullae can become apparent at any stage of AL amyloidosis, but they generally increase in size and number over time and are most common in intertriginous areas. Bullous amyloid lesions, especially those located in intertriginous areas, can have secondary impetiginization.6 In many cases, patients who present with bullous amyloidosis ultimately will be diagnosed with multiple myeloma or another plasma cell dyscrasia. In AL amyloidosis, only 10% to 15% of cases meet criteria for multiple myeloma, whereas 80% or more patients have a monoclonal gammopathy of undetermined significance.7
The prognosis of cutaneous amyloidosis depends on the extent of organ involvement and response to treatment. Treatment is aimed at eliminating clonal plasma cell populations to decrease the production of light chains, thereby decreasing protein burden and amyloid progression. Historically, treatment options included cytotoxic chemotherapy such as oral melphalan and dexamethasone, followed by hematopoietic stem cell transplant. More recent treatment options include bortezomib, thalidomide, pomalidomide, and lenalidomide.8 Our patient received a regimen of bortezomib, cyclophosphamide, and dexamethasone that is used for patients with extensive multiple myeloma.
The differential diagnosis in our patient included bullous drug eruption, which should be considered if the bullae are reoccurring at the same location and in association with the administration of a culprit drug. Bullous pemphigoid is preceded by pruritus, and biopsy demonstrates subepidermal bullae with associated eosinophilic infiltrate. Epidermolysis bullosa acquisita can present with milia and a linear pattern along the basement membrane zone with direct immunofluorescence. Traumatic purpura usually present with the classic shape and hue of an ecchymosis, and the patient will have a history of trauma.
Cutaneous involvement of amyloidosis can be an early clue to the diagnosis of plasma cell dyscrasia. Early diagnosis and treatment can portend a better prognosis and prevent progression to renal or cardiac disease.
The Diagnosis: Bullous Amyloidosis
A punch biopsy from the left temple showed deposits of amorphous eosinophilic material at the tips of dermal papillae and in the papillary dermis with hemorrhage present (Figure 1). A diagnosis of amyloidosis was confirmed on the biopsy of the skin bulla. The low κ/λ light chain ratio and M-spike with notably elevated free λ light chains in both serum and urine were consistent with a λ light chain primary systemic amyloidosis. The patient was seen by hematology and oncology. A bone marrow biopsy demonstrated that 15% to 20% of the clonal-cell population was λ light chain restricted. Eosinophilic extracellular deposits found in the adjacent soft tissue and bone marrow space were confirmed as amyloid with apple green birefringence under polarized light on Congo red stain and metachromatic staining with crystal violet. The patient ultimately was diagnosed with λ light chain multiple myeloma and primary systemic amyloidosis.
Our patient was treated with a combination therapy of bortezomib, cyclophosphamide, and dexamethasone on 21-day cycles, with bortezomib on days 1, 4, 8, and 11. She had received 3 cycles of chemotherapy before developing diarrhea, hypotension, acute on chronic heart failure, and acute renal failure requiring hospitalization. She had several related complications due to amyloid light chain (AL) amyloidosis and subsequently died 16 days after her initial hospitalization from complications of methicillin-resistant Staphylococcus aureus bacteremia and septic shock.
Amyloidosis is the pathologic deposition of abnormal protein in the extracellular space of any tissue. Various soluble precursor proteins can make up amyloid, and these proteins polymerize into insoluble fibrils that damage the surrounding parenchyma. The clinical presentation of amyloidosis varies depending on the affected tissue as well as the constituent protein. The amyloidoses are divided into localized cutaneous, primary systemic, and secondary systemic variants. The initial distinction in amyloidosis is determining whether it is skin limited or systemic. Localized cutaneous amyloidosis comprises 30% to 40% of all amyloidosis cases and is further divided into 3 main subtypes: macular, lichen, and nodular amyloidosis.1 Macular and lichen amyloidosis are composed of keratin derivatives and typically are induced by patients when rubbing or scratching the skin. Histologically, macular and lichen amyloidosis are restricted to the superficial papillary dermis.1 Nodular amyloidosis is composed of λ or κ light chain immunoglobulins, which are produced by cutaneous infiltrates of monoclonal plasma cells. Histologically, nodular amyloidosis is characterized by a diffuse dermal infiltrate of amorphous eosinophilic material.1 Primary systemic amyloidosis is associated with an underlying plasma cell dyscrasia, and unlike secondary keratinocyte-derived amyloid, it can involve internal organs. Similar to nodular amyloidosis, primary systemic amyloidosis is composed of AL proteins, and it is histologically similar to nodular amyloidosis.1
Primary systemic AL amyloidosis commonly affects individuals aged 50 to 60 years. Males and females are equally affected. Macroglossia and periorbital purpura are some of the pathognomonic presentations in AL amyloidosis. The major cause of death in these patients is cardiac and renal involvement. Renal involvement commonly presents as nephrotic syndrome, and cardiac involvement can present as a restrictive cardiomyopathy with dyspnea. Other symptoms include edema, hepatosplenomegaly, bleeding diathesis, and carpal tunnel syndrome.2 An evaluation for AL amyloidosis should include a complete review of systems and physical examination with studies such as complete blood cell count, comprehensive metabolic panel, serum and urine protein electrophoresis and immunofixation, and electrocardiogram.
Cutaneous involvement in AL amyloidosis most commonly includes yellowish waxy papules, nodules, and plaques but also can include purpura and petechiae.2 Bullous amyloidosis, as seen in our patient, is a rare cutaneous presentation of AL amyloidosis that usually is negative for the Nikolsky sign (Figure 2). Bullae form due to weakness in amyloid-laden dermal connective tissue.3 Eighty-eight percent of cases of bullous amyloidosis have systemic involvement.1 Some cases have reported a familial linkage, suggesting there might be a genetic component to the disease.4 A PubMed search of articles indexed for MEDLINE using the terms bullous amyloidosis, bullous, amyloidosis, and amyloid revealed fewer than 35 cases of bullous amyloidosis in the English-language literature.5 Bullae can be located intradermally or subepidermally and commonly are hemorrhagic but also can be translucent, tender, and tense.
A study of electron microscopy in a patient with systemic bullous amyloidosis demonstrated amyloid and keratinocyte protrusions that perforated the dermis through the spaces in the lamina densa. The study concluded that the disintegration of the lamina densa and expansion of the intercellular spaces between keratinocytes were the causes of skin fragility as well as fluid exudation.5 Trauma or friction to the skin are local precipitating factors for blister formation in bullous amyloidosis.
Bullae can become apparent at any stage of AL amyloidosis, but they generally increase in size and number over time and are most common in intertriginous areas. Bullous amyloid lesions, especially those located in intertriginous areas, can have secondary impetiginization.6 In many cases, patients who present with bullous amyloidosis ultimately will be diagnosed with multiple myeloma or another plasma cell dyscrasia. In AL amyloidosis, only 10% to 15% of cases meet criteria for multiple myeloma, whereas 80% or more patients have a monoclonal gammopathy of undetermined significance.7
The prognosis of cutaneous amyloidosis depends on the extent of organ involvement and response to treatment. Treatment is aimed at eliminating clonal plasma cell populations to decrease the production of light chains, thereby decreasing protein burden and amyloid progression. Historically, treatment options included cytotoxic chemotherapy such as oral melphalan and dexamethasone, followed by hematopoietic stem cell transplant. More recent treatment options include bortezomib, thalidomide, pomalidomide, and lenalidomide.8 Our patient received a regimen of bortezomib, cyclophosphamide, and dexamethasone that is used for patients with extensive multiple myeloma.
The differential diagnosis in our patient included bullous drug eruption, which should be considered if the bullae are reoccurring at the same location and in association with the administration of a culprit drug. Bullous pemphigoid is preceded by pruritus, and biopsy demonstrates subepidermal bullae with associated eosinophilic infiltrate. Epidermolysis bullosa acquisita can present with milia and a linear pattern along the basement membrane zone with direct immunofluorescence. Traumatic purpura usually present with the classic shape and hue of an ecchymosis, and the patient will have a history of trauma.
Cutaneous involvement of amyloidosis can be an early clue to the diagnosis of plasma cell dyscrasia. Early diagnosis and treatment can portend a better prognosis and prevent progression to renal or cardiac disease.
- Heaton J, Steinhoff N, Wanner B, et al. A review of primary cutaneous amyloidosis. J Am Osteopath Coll Dermatol. doi:10.1007/springerreference_42272
- Ventarola DJ, Schuster MW, Cohen JA, et al. JAAD grand rounds quiz. bullae and nodules on the legs of a 57-year-old woman. J Am Acad Dermatol. 2014;71:1035-1037.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Suranagi VV, Siddramappa B, Bannur HB, et al. Bullous variant of familial biphasic lichen amyloidosis: a unique combination of three rare presentations. Indian J Dermatol. 2015;60:105.
- Antúnez-Lay A, Jaque A, González S. Hemorrhagic bullous skin lesions. Int J Dermatol. 2017;56:145-147.
- Reddy K, Hoda S, Penstein A, et al. Bullous amyloidosis complicated by cellulitis and sepsis: a case report. Arch Dermatol. 2011;147:126-127.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
- Heaton J, Steinhoff N, Wanner B, et al. A review of primary cutaneous amyloidosis. J Am Osteopath Coll Dermatol. doi:10.1007/springerreference_42272
- Ventarola DJ, Schuster MW, Cohen JA, et al. JAAD grand rounds quiz. bullae and nodules on the legs of a 57-year-old woman. J Am Acad Dermatol. 2014;71:1035-1037.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Suranagi VV, Siddramappa B, Bannur HB, et al. Bullous variant of familial biphasic lichen amyloidosis: a unique combination of three rare presentations. Indian J Dermatol. 2015;60:105.
- Antúnez-Lay A, Jaque A, González S. Hemorrhagic bullous skin lesions. Int J Dermatol. 2017;56:145-147.
- Reddy K, Hoda S, Penstein A, et al. Bullous amyloidosis complicated by cellulitis and sepsis: a case report. Arch Dermatol. 2011;147:126-127.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
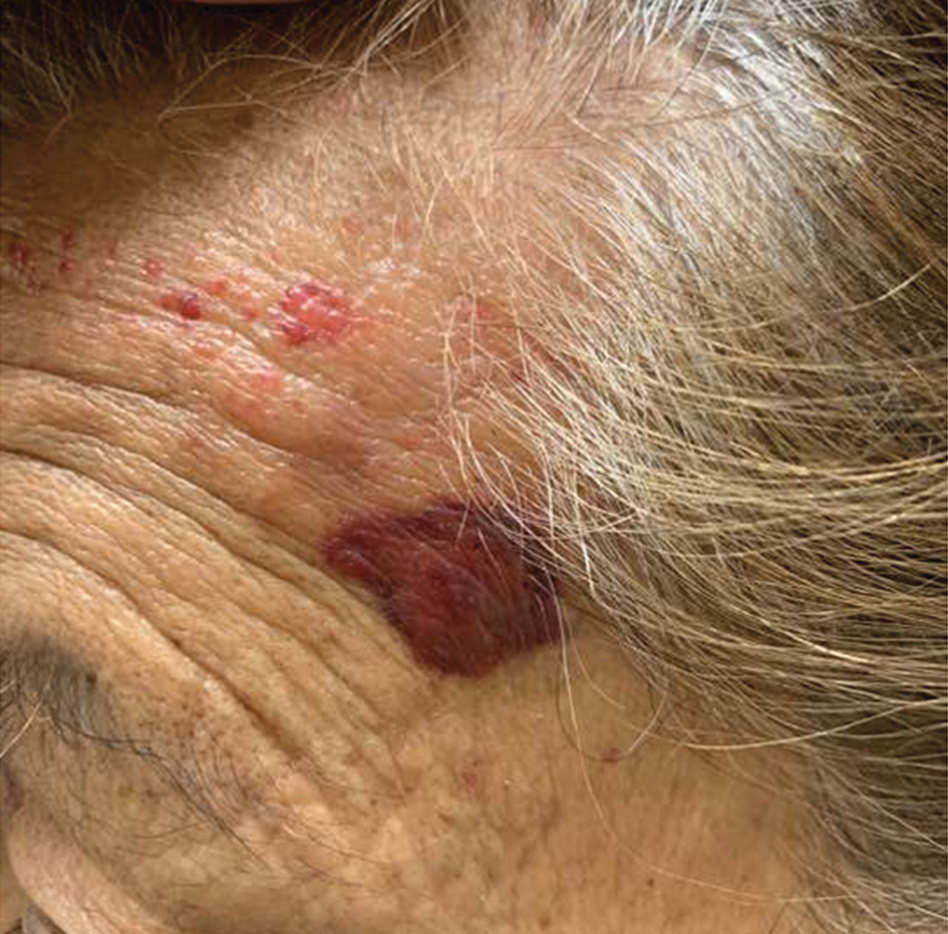
A 67-year-old woman with a medical history of type 2 diabetes mellitus, unspecified leukocytosis, and anemia presented to the dermatology clinic with asymptomatic hemorrhagic bullae on the face, chest, and tongue, as well as a large, tender, tense, hemorrhagic bulla on the groin of 3 to 4 months’ duration. A review of systems was negative for fever, chills, night sweats, malaise, shortness of breath, and dyspnea on exertion. A complete blood cell count showed mild leukocytosis, anemia, and thrombocytopenia. Her creatinine level was slightly elevated. Chest computed tomography showed early pulmonary fibrosis and coronary artery calcification. An echocardiogram showed diastolic dysfunction with moderate left ventricle thickening. A serum and urine electrophoresis demonstrated elevated free λ light chains with an M-spike. A punch biopsy was performed.