User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Abrocitinib efficacy dose-dependent, similar across AD age groups
and was comparable in patients aged 51 years and older, results from a post hoc analysis of four trials showed.

Abrocitinib (Cibinqo) is an oral, once-daily, Janus kinase 1 selective inhibitor that has shown good efficacy and safety as monotherapy or combined with topical therapy for treatment of patients with moderate to severe AD. The agent was approved in mid-December in Europe for the treatment of moderate to severe AD in adults who are candidates for systemic therapy and is currently under review by the Food and Drug Administration.
“We know that responses to, and adverse events associated with, systemic therapies may vary among patients of different ages,” Andrew F. Alexis, MD, MPH, said during a late-breaking abstract session at the Revolutionizing Atopic Dermatitis virtual symposium. “The efficacy and safety of abrocitinib monotherapy were previously evaluated in adolescent and adult subpopulations from controlled clinical trials in patients with moderate to severe AD. The objective of the current study was to assess the impact of age on short-term responses to abrocitinib treatment in patients with moderate to severe AD.”
Dr. Alexis, professor of clinical dermatology at Weill Cornell Medicine, New York, and colleagues performed a post hoc analysis across four randomized, double-blind studies that was stratified by age group: 12-17 years, 18-40 years, 41-50 years, and 51 years and older. Efficacy data were assessed separately for patients in the monotherapy pool and in the JADE COMPARE trial. The monotherapy pool included patients from one phase 2b study and two phase 3 studies who received abrocitinib 200 mg, abrocitinib 100 mg, or placebo monotherapy for 12 weeks (JADE-MONO-1 and JADE-MONO-2).
The JADE COMPARE pool included patients who received abrocitinib 200 mg, abrocitinib 100 mg, or placebo, plus medicated topical therapy for 16 weeks. Data from patients in all four trials were pooled for the analysis of treatment-emergent adverse events. Efficacy points analyzed were the Investigator Global Assessment (IGA) score of 0/1 (clear or almost clear), a 75% reduction from baseline in the Eczema Area and Severity Index (EASI-75), or Peak Pruritus Numeric Rating Scale score (PP-NRS4) at week 12 for the monotherapy pool and at week 16 for COMPARE.
In the monotherapy pool, the proportions of patients ages 12-17 years, 18-40 years, 41-50 years, and 51 years and older who achieved an IGA 0/1 response at 12 weeks were 31.3%, 40.2%, 43.8%, and 50.8% (abrocitinib 200 mg); 22%, 23.7%, 22.4%, and 40.8% (abrocitinib 100 mg); and 8.7%, 8%, 3.3%, and 10% (placebo).
In JADE COMPARE, the proportions of patients aged 18-40 years, 41-50 years, and 51 years and older who achieved an IGA 0/1 response were 50%, 53.2%, and 34.8% (abrocitinib 200 mg); 36.9%, 37.1%, and 26.1% (abrocitinib 100 mg); and 12%, 11.8%, and 16.7% (placebo) at 16 weeks. Similar trends were observed for EASI-75 and PP-NRS4 responses at 12 weeks.
Across all age groups, the most common treatment-emergent adverse events were infections/infestations and gastrointestinal effects; most cases were mild or moderate. Nausea was more frequent in the two younger age groups and was dose related: For abrocitinib 200 mg and abrocitinib 100 mg, respectively, the rates of nausea were 18.8% and 7.8% in patients aged 12-17 years; 17.1% and 6.4% in patients aged 18-40 years; and 7.1% and 3.3% in patients aged 51 and older.
“Efficacy responses in patients 51 years of age and older were comparable to those in other age groups,” concluded Dr. Alexis, vice chair for diversity and inclusion in the department of dermatology at Weill Cornell. “The safety profile was consistent across age ranges and was similar to that reported previously.”
The investigators found that treatment response to abrocitinib “in the absence or presence of medicated topical therapy was fairly consistent across age groups, showed similar dose-dependency, and importantly, did not show reduced efficacy in older adults as measured by lesional severity, extent, and itch at 4 months,” said Raj Chovatiya, MD, PhD, assistant professor of dermatology at Northwestern University, Chicago, who was asked to comment on the study.

“Furthermore, the safety profile was consistent across all adults, though notably, nausea was more common among younger age groups, highlighting an area of future investigation,” he added. “Overall, these data show that abrocitinib is associated with similar short-term responses across adulthood and underscore the importance of the JAK-STAT pathway in the underlying pathophysiology of AD in different age groups. It will be interesting to see how these data reflect the real-world setting with both short- and long-term outcomes in a heterogeneous patient population.”
In the interview, Dr. Chovatiya said, “the next frontier in personalized therapy for AD involves deeper clinical phenotyping of our patients and a better understanding of how efficacy and safety vary across patient groups.” For example, he noted, “AD in earlier versus later adulthood may be associated with different clinical signs, symptoms, comorbidities, and other measures of patient burden, and thus, may be associated with different treatment responses to systemic therapy.”
Dr. Alexis disclosed that he has served as an adviser to, or has received consulting fees from, Leo, Galderma, Pfizer, Sanofi-Regeneron, Dermavant, Beiersdorf, Valeant, L’Oréal, BMS, Bausch Health, UCB, Vyne, Arcutis, Janssen, Allergan, Almirall, AbbVie, Sol-Gel, and Amgen.
Dr. Chovatiya disclosed that he is a consultant to, a speaker for, and/or a member of the advisory board for AbbVie, Arena, Arcutis, Incyte, Pfizer, Regeneron, and Sanofi-Genzyme.
A version of this article first appeared on Medscape.com.
and was comparable in patients aged 51 years and older, results from a post hoc analysis of four trials showed.
Abrocitinib (Cibinqo) is an oral, once-daily, Janus kinase 1 selective inhibitor that has shown good efficacy and safety as monotherapy or combined with topical therapy for treatment of patients with moderate to severe AD. The agent was approved in mid-December in Europe for the treatment of moderate to severe AD in adults who are candidates for systemic therapy and is currently under review by the Food and Drug Administration.
“We know that responses to, and adverse events associated with, systemic therapies may vary among patients of different ages,” Andrew F. Alexis, MD, MPH, said during a late-breaking abstract session at the Revolutionizing Atopic Dermatitis virtual symposium. “The efficacy and safety of abrocitinib monotherapy were previously evaluated in adolescent and adult subpopulations from controlled clinical trials in patients with moderate to severe AD. The objective of the current study was to assess the impact of age on short-term responses to abrocitinib treatment in patients with moderate to severe AD.”
Dr. Alexis, professor of clinical dermatology at Weill Cornell Medicine, New York, and colleagues performed a post hoc analysis across four randomized, double-blind studies that was stratified by age group: 12-17 years, 18-40 years, 41-50 years, and 51 years and older. Efficacy data were assessed separately for patients in the monotherapy pool and in the JADE COMPARE trial. The monotherapy pool included patients from one phase 2b study and two phase 3 studies who received abrocitinib 200 mg, abrocitinib 100 mg, or placebo monotherapy for 12 weeks (JADE-MONO-1 and JADE-MONO-2).
The JADE COMPARE pool included patients who received abrocitinib 200 mg, abrocitinib 100 mg, or placebo, plus medicated topical therapy for 16 weeks. Data from patients in all four trials were pooled for the analysis of treatment-emergent adverse events. Efficacy points analyzed were the Investigator Global Assessment (IGA) score of 0/1 (clear or almost clear), a 75% reduction from baseline in the Eczema Area and Severity Index (EASI-75), or Peak Pruritus Numeric Rating Scale score (PP-NRS4) at week 12 for the monotherapy pool and at week 16 for COMPARE.
In the monotherapy pool, the proportions of patients ages 12-17 years, 18-40 years, 41-50 years, and 51 years and older who achieved an IGA 0/1 response at 12 weeks were 31.3%, 40.2%, 43.8%, and 50.8% (abrocitinib 200 mg); 22%, 23.7%, 22.4%, and 40.8% (abrocitinib 100 mg); and 8.7%, 8%, 3.3%, and 10% (placebo).
In JADE COMPARE, the proportions of patients aged 18-40 years, 41-50 years, and 51 years and older who achieved an IGA 0/1 response were 50%, 53.2%, and 34.8% (abrocitinib 200 mg); 36.9%, 37.1%, and 26.1% (abrocitinib 100 mg); and 12%, 11.8%, and 16.7% (placebo) at 16 weeks. Similar trends were observed for EASI-75 and PP-NRS4 responses at 12 weeks.
Across all age groups, the most common treatment-emergent adverse events were infections/infestations and gastrointestinal effects; most cases were mild or moderate. Nausea was more frequent in the two younger age groups and was dose related: For abrocitinib 200 mg and abrocitinib 100 mg, respectively, the rates of nausea were 18.8% and 7.8% in patients aged 12-17 years; 17.1% and 6.4% in patients aged 18-40 years; and 7.1% and 3.3% in patients aged 51 and older.
“Efficacy responses in patients 51 years of age and older were comparable to those in other age groups,” concluded Dr. Alexis, vice chair for diversity and inclusion in the department of dermatology at Weill Cornell. “The safety profile was consistent across age ranges and was similar to that reported previously.”
The investigators found that treatment response to abrocitinib “in the absence or presence of medicated topical therapy was fairly consistent across age groups, showed similar dose-dependency, and importantly, did not show reduced efficacy in older adults as measured by lesional severity, extent, and itch at 4 months,” said Raj Chovatiya, MD, PhD, assistant professor of dermatology at Northwestern University, Chicago, who was asked to comment on the study.
“Furthermore, the safety profile was consistent across all adults, though notably, nausea was more common among younger age groups, highlighting an area of future investigation,” he added. “Overall, these data show that abrocitinib is associated with similar short-term responses across adulthood and underscore the importance of the JAK-STAT pathway in the underlying pathophysiology of AD in different age groups. It will be interesting to see how these data reflect the real-world setting with both short- and long-term outcomes in a heterogeneous patient population.”
In the interview, Dr. Chovatiya said, “the next frontier in personalized therapy for AD involves deeper clinical phenotyping of our patients and a better understanding of how efficacy and safety vary across patient groups.” For example, he noted, “AD in earlier versus later adulthood may be associated with different clinical signs, symptoms, comorbidities, and other measures of patient burden, and thus, may be associated with different treatment responses to systemic therapy.”
Dr. Alexis disclosed that he has served as an adviser to, or has received consulting fees from, Leo, Galderma, Pfizer, Sanofi-Regeneron, Dermavant, Beiersdorf, Valeant, L’Oréal, BMS, Bausch Health, UCB, Vyne, Arcutis, Janssen, Allergan, Almirall, AbbVie, Sol-Gel, and Amgen.
Dr. Chovatiya disclosed that he is a consultant to, a speaker for, and/or a member of the advisory board for AbbVie, Arena, Arcutis, Incyte, Pfizer, Regeneron, and Sanofi-Genzyme.
A version of this article first appeared on Medscape.com.
and was comparable in patients aged 51 years and older, results from a post hoc analysis of four trials showed.
Abrocitinib (Cibinqo) is an oral, once-daily, Janus kinase 1 selective inhibitor that has shown good efficacy and safety as monotherapy or combined with topical therapy for treatment of patients with moderate to severe AD. The agent was approved in mid-December in Europe for the treatment of moderate to severe AD in adults who are candidates for systemic therapy and is currently under review by the Food and Drug Administration.
“We know that responses to, and adverse events associated with, systemic therapies may vary among patients of different ages,” Andrew F. Alexis, MD, MPH, said during a late-breaking abstract session at the Revolutionizing Atopic Dermatitis virtual symposium. “The efficacy and safety of abrocitinib monotherapy were previously evaluated in adolescent and adult subpopulations from controlled clinical trials in patients with moderate to severe AD. The objective of the current study was to assess the impact of age on short-term responses to abrocitinib treatment in patients with moderate to severe AD.”
Dr. Alexis, professor of clinical dermatology at Weill Cornell Medicine, New York, and colleagues performed a post hoc analysis across four randomized, double-blind studies that was stratified by age group: 12-17 years, 18-40 years, 41-50 years, and 51 years and older. Efficacy data were assessed separately for patients in the monotherapy pool and in the JADE COMPARE trial. The monotherapy pool included patients from one phase 2b study and two phase 3 studies who received abrocitinib 200 mg, abrocitinib 100 mg, or placebo monotherapy for 12 weeks (JADE-MONO-1 and JADE-MONO-2).
The JADE COMPARE pool included patients who received abrocitinib 200 mg, abrocitinib 100 mg, or placebo, plus medicated topical therapy for 16 weeks. Data from patients in all four trials were pooled for the analysis of treatment-emergent adverse events. Efficacy points analyzed were the Investigator Global Assessment (IGA) score of 0/1 (clear or almost clear), a 75% reduction from baseline in the Eczema Area and Severity Index (EASI-75), or Peak Pruritus Numeric Rating Scale score (PP-NRS4) at week 12 for the monotherapy pool and at week 16 for COMPARE.
In the monotherapy pool, the proportions of patients ages 12-17 years, 18-40 years, 41-50 years, and 51 years and older who achieved an IGA 0/1 response at 12 weeks were 31.3%, 40.2%, 43.8%, and 50.8% (abrocitinib 200 mg); 22%, 23.7%, 22.4%, and 40.8% (abrocitinib 100 mg); and 8.7%, 8%, 3.3%, and 10% (placebo).
In JADE COMPARE, the proportions of patients aged 18-40 years, 41-50 years, and 51 years and older who achieved an IGA 0/1 response were 50%, 53.2%, and 34.8% (abrocitinib 200 mg); 36.9%, 37.1%, and 26.1% (abrocitinib 100 mg); and 12%, 11.8%, and 16.7% (placebo) at 16 weeks. Similar trends were observed for EASI-75 and PP-NRS4 responses at 12 weeks.
Across all age groups, the most common treatment-emergent adverse events were infections/infestations and gastrointestinal effects; most cases were mild or moderate. Nausea was more frequent in the two younger age groups and was dose related: For abrocitinib 200 mg and abrocitinib 100 mg, respectively, the rates of nausea were 18.8% and 7.8% in patients aged 12-17 years; 17.1% and 6.4% in patients aged 18-40 years; and 7.1% and 3.3% in patients aged 51 and older.
“Efficacy responses in patients 51 years of age and older were comparable to those in other age groups,” concluded Dr. Alexis, vice chair for diversity and inclusion in the department of dermatology at Weill Cornell. “The safety profile was consistent across age ranges and was similar to that reported previously.”
The investigators found that treatment response to abrocitinib “in the absence or presence of medicated topical therapy was fairly consistent across age groups, showed similar dose-dependency, and importantly, did not show reduced efficacy in older adults as measured by lesional severity, extent, and itch at 4 months,” said Raj Chovatiya, MD, PhD, assistant professor of dermatology at Northwestern University, Chicago, who was asked to comment on the study.
“Furthermore, the safety profile was consistent across all adults, though notably, nausea was more common among younger age groups, highlighting an area of future investigation,” he added. “Overall, these data show that abrocitinib is associated with similar short-term responses across adulthood and underscore the importance of the JAK-STAT pathway in the underlying pathophysiology of AD in different age groups. It will be interesting to see how these data reflect the real-world setting with both short- and long-term outcomes in a heterogeneous patient population.”
In the interview, Dr. Chovatiya said, “the next frontier in personalized therapy for AD involves deeper clinical phenotyping of our patients and a better understanding of how efficacy and safety vary across patient groups.” For example, he noted, “AD in earlier versus later adulthood may be associated with different clinical signs, symptoms, comorbidities, and other measures of patient burden, and thus, may be associated with different treatment responses to systemic therapy.”
Dr. Alexis disclosed that he has served as an adviser to, or has received consulting fees from, Leo, Galderma, Pfizer, Sanofi-Regeneron, Dermavant, Beiersdorf, Valeant, L’Oréal, BMS, Bausch Health, UCB, Vyne, Arcutis, Janssen, Allergan, Almirall, AbbVie, Sol-Gel, and Amgen.
Dr. Chovatiya disclosed that he is a consultant to, a speaker for, and/or a member of the advisory board for AbbVie, Arena, Arcutis, Incyte, Pfizer, Regeneron, and Sanofi-Genzyme.
A version of this article first appeared on Medscape.com.
FROM REVOLUTIONIZING AD 2021
CRP elevated in adults with AD and sleep disturbance
and mortality, results from a large cohort analysis showed.
“The implications of these findings are vast,” presenting author Varsha Parthasarathy said during a late-breaking abstract session at the Revolutionizing Atopic Dermatitis virtual symposium. “Poor sleep quality is known to be associated with increased inflammatory markers such as IL-6, IL-17, and CRP, so it is interesting to see this reflected in AD patients with versus without sleep disturbance. Additionally, we know that CRP is a driver of inflammation and is strongly associated with cardiovascular complications such as heart attack and stroke. Therefore, CRP may be a useful prognostic marker in AD patients with sleep disturbances.”
To examine the comorbidity burden of sleep disorders in AD patients and associate findings with inflammatory CRP and cardiovascular comorbidities, Mr. Parthasarathy, a medical student and itch fellow in the department of dermatology at the Johns Hopkins University School of Medicine, Baltimore, and colleagues drew from TriNetX, a health care network of approximately 73 million de-identified medical records in 53 organizations. The years of study were 2015 to 2021. The researchers limited the analysis to adults with at least two instances of International Classification of Diseases, Tenth Revision (ICD-10) code L28 for AD, to capture a population with true AD. Controls were adults without AD who presented for general checkup and were matched to AD patients by age, race, and sex.
The study population consisted of 120,480 AD patients and matched controls. Their mean age was 36 years, 61% were female, and 26% were Black. Compared with controls, AD patients had an increased risk of developing general sleep disorders over the 6-year period (relative risk, 1.10), as well as obstructive sleep apnea (RR, 1.13), insomnia (RR, 1.10), hypersomnia (RR, 1.24), sleep-related movement disorders (RR, 1.36), restless legs syndrome (RR, 1.25), sleep deprivation (RR, 1.36), and unspecified sleep disorders (RR, 1.22).
To examine the association of sleep disturbance with the inflammatory biomarker CRP, the researchers measured CRP levels between these patient groups. They found a substantially higher CRP in AD patients compared with controls (21.2 mg/L vs. 7.6 mg/L, respectively; P < .0001). This finding “is suggestive of a higher level of inflammation in these patients,” Mr. Parthasarathy said. Interestingly, he added, they also found a higher CRP level in AD patients with sleep disturbances compared to AD patients without sleep disturbances (23.3 vs. 20.6 mg/L; P = .02), “also pointing to a higher inflammatory burden in AD patients whose sleep was affected.”
Compared to matched AD patients without sleep disorders, AD patients with sleep disorders were more likely to develop obesity (RR, 2.65), hyperlipidemia (RR, 2.18), type 2 diabetes (RR, 2.45), metabolic syndrome (RR, 4.16), atherosclerosis (RR, 2.42), peripheral vascular disease (RR, 2.47), stroke (RR, 2.37), venous thromboembolism (RR, 2.93), and mortality (hazard ratio, 1.24).
“There is a consequence of not treating patients with atopic dermatitis, especially those patients with sleep disturbance,” the study’s primary author, Shawn G. Kwatra, MD, associate professor of dermatology at Johns Hopkins, told this news organization. “Chronic inflammation can lead to the development of comorbidities, so it is important to offer patients early treatment to reduce their overall inflammation.” He said that he was most surprised by the degree of increased inflammation in the blood of AD as compared to healthy controls. “This likely plays a part in the development of several comorbidities,” he said.
Mr. Parthasarathy acknowledged certain limitations of the study, including the inability to infer causal relationships, as uncontrolled factors may be present. “Additionally, sampling of only patients that have had medical encounters limits the generalizability of the findings,” she said. “However, findings in this large cohort study suggest that clinicians should seek to identify sleep disorders in AD patients and screen for cardiac comorbidities secondary to inflammation in this patient population.”
“There is increased data to suggest that adults with AD, particularly those with more severe disease, may be at an increased risk of cardiovascular disease and the results from [this study] further support the concept of AD as systemic disease,” said Zelma C. Chiesa Fuxench, MD, MSCE, assistant professor of dermatology at the University of Pennsylvania, Philadelphia, who was asked to comment on the study. She cited the large population-based, retrospective design and use of two instances of ICD codes for AD to confirm diagnosis as key strengths of the research. “However, it is unclear if for each patient CRP levels were measured at one single timepoint,” Dr. Chiesa Fuxench said. “For future studies, it would be interesting to see if these levels fluctuate with time and if persistently elevated levels are associated with worse cardiovascular outcomes in this population. More data is needed to better understand the relationship better atopic dermatitis disease severity, impact on sleep, and how this relates to increased systemic inflammation and worse cardiovascular outcomes in this population.”
Dr. Kwatra disclosed support by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number K23AR077073-01A1 and previous funding by the Dermatology Foundation and Skin of Color Society. Dr. Kwatra is also an advisory board member/consultant for AbbVie, Celldex Therapeutics, Galderma, Incyte Corporation, Johnson & Johnson, Novartis Pharmaceuticals Corporation, Pfizer, Regeneron Pharmaceuticals, Sanofi, and Kiniksa Pharmaceuticals and has served as an investigator for Galderma, Pfizer, and Sanofi. Dr. Chiesa Fuxench disclosed research grants from several pharmaceutical companies for work related to AD. She has also served as a consultant for the Asthma and Allergy Foundation of America, National Eczema Association, AbbVie, Incyte Corporation, and Pfizer.
A version of this article first appeared on Medscape.com.
and mortality, results from a large cohort analysis showed.
“The implications of these findings are vast,” presenting author Varsha Parthasarathy said during a late-breaking abstract session at the Revolutionizing Atopic Dermatitis virtual symposium. “Poor sleep quality is known to be associated with increased inflammatory markers such as IL-6, IL-17, and CRP, so it is interesting to see this reflected in AD patients with versus without sleep disturbance. Additionally, we know that CRP is a driver of inflammation and is strongly associated with cardiovascular complications such as heart attack and stroke. Therefore, CRP may be a useful prognostic marker in AD patients with sleep disturbances.”
To examine the comorbidity burden of sleep disorders in AD patients and associate findings with inflammatory CRP and cardiovascular comorbidities, Mr. Parthasarathy, a medical student and itch fellow in the department of dermatology at the Johns Hopkins University School of Medicine, Baltimore, and colleagues drew from TriNetX, a health care network of approximately 73 million de-identified medical records in 53 organizations. The years of study were 2015 to 2021. The researchers limited the analysis to adults with at least two instances of International Classification of Diseases, Tenth Revision (ICD-10) code L28 for AD, to capture a population with true AD. Controls were adults without AD who presented for general checkup and were matched to AD patients by age, race, and sex.
The study population consisted of 120,480 AD patients and matched controls. Their mean age was 36 years, 61% were female, and 26% were Black. Compared with controls, AD patients had an increased risk of developing general sleep disorders over the 6-year period (relative risk, 1.10), as well as obstructive sleep apnea (RR, 1.13), insomnia (RR, 1.10), hypersomnia (RR, 1.24), sleep-related movement disorders (RR, 1.36), restless legs syndrome (RR, 1.25), sleep deprivation (RR, 1.36), and unspecified sleep disorders (RR, 1.22).
To examine the association of sleep disturbance with the inflammatory biomarker CRP, the researchers measured CRP levels between these patient groups. They found a substantially higher CRP in AD patients compared with controls (21.2 mg/L vs. 7.6 mg/L, respectively; P < .0001). This finding “is suggestive of a higher level of inflammation in these patients,” Mr. Parthasarathy said. Interestingly, he added, they also found a higher CRP level in AD patients with sleep disturbances compared to AD patients without sleep disturbances (23.3 vs. 20.6 mg/L; P = .02), “also pointing to a higher inflammatory burden in AD patients whose sleep was affected.”
Compared to matched AD patients without sleep disorders, AD patients with sleep disorders were more likely to develop obesity (RR, 2.65), hyperlipidemia (RR, 2.18), type 2 diabetes (RR, 2.45), metabolic syndrome (RR, 4.16), atherosclerosis (RR, 2.42), peripheral vascular disease (RR, 2.47), stroke (RR, 2.37), venous thromboembolism (RR, 2.93), and mortality (hazard ratio, 1.24).
“There is a consequence of not treating patients with atopic dermatitis, especially those patients with sleep disturbance,” the study’s primary author, Shawn G. Kwatra, MD, associate professor of dermatology at Johns Hopkins, told this news organization. “Chronic inflammation can lead to the development of comorbidities, so it is important to offer patients early treatment to reduce their overall inflammation.” He said that he was most surprised by the degree of increased inflammation in the blood of AD as compared to healthy controls. “This likely plays a part in the development of several comorbidities,” he said.
Mr. Parthasarathy acknowledged certain limitations of the study, including the inability to infer causal relationships, as uncontrolled factors may be present. “Additionally, sampling of only patients that have had medical encounters limits the generalizability of the findings,” she said. “However, findings in this large cohort study suggest that clinicians should seek to identify sleep disorders in AD patients and screen for cardiac comorbidities secondary to inflammation in this patient population.”
“There is increased data to suggest that adults with AD, particularly those with more severe disease, may be at an increased risk of cardiovascular disease and the results from [this study] further support the concept of AD as systemic disease,” said Zelma C. Chiesa Fuxench, MD, MSCE, assistant professor of dermatology at the University of Pennsylvania, Philadelphia, who was asked to comment on the study. She cited the large population-based, retrospective design and use of two instances of ICD codes for AD to confirm diagnosis as key strengths of the research. “However, it is unclear if for each patient CRP levels were measured at one single timepoint,” Dr. Chiesa Fuxench said. “For future studies, it would be interesting to see if these levels fluctuate with time and if persistently elevated levels are associated with worse cardiovascular outcomes in this population. More data is needed to better understand the relationship better atopic dermatitis disease severity, impact on sleep, and how this relates to increased systemic inflammation and worse cardiovascular outcomes in this population.”
Dr. Kwatra disclosed support by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number K23AR077073-01A1 and previous funding by the Dermatology Foundation and Skin of Color Society. Dr. Kwatra is also an advisory board member/consultant for AbbVie, Celldex Therapeutics, Galderma, Incyte Corporation, Johnson & Johnson, Novartis Pharmaceuticals Corporation, Pfizer, Regeneron Pharmaceuticals, Sanofi, and Kiniksa Pharmaceuticals and has served as an investigator for Galderma, Pfizer, and Sanofi. Dr. Chiesa Fuxench disclosed research grants from several pharmaceutical companies for work related to AD. She has also served as a consultant for the Asthma and Allergy Foundation of America, National Eczema Association, AbbVie, Incyte Corporation, and Pfizer.
A version of this article first appeared on Medscape.com.
and mortality, results from a large cohort analysis showed.
“The implications of these findings are vast,” presenting author Varsha Parthasarathy said during a late-breaking abstract session at the Revolutionizing Atopic Dermatitis virtual symposium. “Poor sleep quality is known to be associated with increased inflammatory markers such as IL-6, IL-17, and CRP, so it is interesting to see this reflected in AD patients with versus without sleep disturbance. Additionally, we know that CRP is a driver of inflammation and is strongly associated with cardiovascular complications such as heart attack and stroke. Therefore, CRP may be a useful prognostic marker in AD patients with sleep disturbances.”
To examine the comorbidity burden of sleep disorders in AD patients and associate findings with inflammatory CRP and cardiovascular comorbidities, Mr. Parthasarathy, a medical student and itch fellow in the department of dermatology at the Johns Hopkins University School of Medicine, Baltimore, and colleagues drew from TriNetX, a health care network of approximately 73 million de-identified medical records in 53 organizations. The years of study were 2015 to 2021. The researchers limited the analysis to adults with at least two instances of International Classification of Diseases, Tenth Revision (ICD-10) code L28 for AD, to capture a population with true AD. Controls were adults without AD who presented for general checkup and were matched to AD patients by age, race, and sex.
The study population consisted of 120,480 AD patients and matched controls. Their mean age was 36 years, 61% were female, and 26% were Black. Compared with controls, AD patients had an increased risk of developing general sleep disorders over the 6-year period (relative risk, 1.10), as well as obstructive sleep apnea (RR, 1.13), insomnia (RR, 1.10), hypersomnia (RR, 1.24), sleep-related movement disorders (RR, 1.36), restless legs syndrome (RR, 1.25), sleep deprivation (RR, 1.36), and unspecified sleep disorders (RR, 1.22).
To examine the association of sleep disturbance with the inflammatory biomarker CRP, the researchers measured CRP levels between these patient groups. They found a substantially higher CRP in AD patients compared with controls (21.2 mg/L vs. 7.6 mg/L, respectively; P < .0001). This finding “is suggestive of a higher level of inflammation in these patients,” Mr. Parthasarathy said. Interestingly, he added, they also found a higher CRP level in AD patients with sleep disturbances compared to AD patients without sleep disturbances (23.3 vs. 20.6 mg/L; P = .02), “also pointing to a higher inflammatory burden in AD patients whose sleep was affected.”
Compared to matched AD patients without sleep disorders, AD patients with sleep disorders were more likely to develop obesity (RR, 2.65), hyperlipidemia (RR, 2.18), type 2 diabetes (RR, 2.45), metabolic syndrome (RR, 4.16), atherosclerosis (RR, 2.42), peripheral vascular disease (RR, 2.47), stroke (RR, 2.37), venous thromboembolism (RR, 2.93), and mortality (hazard ratio, 1.24).
“There is a consequence of not treating patients with atopic dermatitis, especially those patients with sleep disturbance,” the study’s primary author, Shawn G. Kwatra, MD, associate professor of dermatology at Johns Hopkins, told this news organization. “Chronic inflammation can lead to the development of comorbidities, so it is important to offer patients early treatment to reduce their overall inflammation.” He said that he was most surprised by the degree of increased inflammation in the blood of AD as compared to healthy controls. “This likely plays a part in the development of several comorbidities,” he said.
Mr. Parthasarathy acknowledged certain limitations of the study, including the inability to infer causal relationships, as uncontrolled factors may be present. “Additionally, sampling of only patients that have had medical encounters limits the generalizability of the findings,” she said. “However, findings in this large cohort study suggest that clinicians should seek to identify sleep disorders in AD patients and screen for cardiac comorbidities secondary to inflammation in this patient population.”
“There is increased data to suggest that adults with AD, particularly those with more severe disease, may be at an increased risk of cardiovascular disease and the results from [this study] further support the concept of AD as systemic disease,” said Zelma C. Chiesa Fuxench, MD, MSCE, assistant professor of dermatology at the University of Pennsylvania, Philadelphia, who was asked to comment on the study. She cited the large population-based, retrospective design and use of two instances of ICD codes for AD to confirm diagnosis as key strengths of the research. “However, it is unclear if for each patient CRP levels were measured at one single timepoint,” Dr. Chiesa Fuxench said. “For future studies, it would be interesting to see if these levels fluctuate with time and if persistently elevated levels are associated with worse cardiovascular outcomes in this population. More data is needed to better understand the relationship better atopic dermatitis disease severity, impact on sleep, and how this relates to increased systemic inflammation and worse cardiovascular outcomes in this population.”
Dr. Kwatra disclosed support by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number K23AR077073-01A1 and previous funding by the Dermatology Foundation and Skin of Color Society. Dr. Kwatra is also an advisory board member/consultant for AbbVie, Celldex Therapeutics, Galderma, Incyte Corporation, Johnson & Johnson, Novartis Pharmaceuticals Corporation, Pfizer, Regeneron Pharmaceuticals, Sanofi, and Kiniksa Pharmaceuticals and has served as an investigator for Galderma, Pfizer, and Sanofi. Dr. Chiesa Fuxench disclosed research grants from several pharmaceutical companies for work related to AD. She has also served as a consultant for the Asthma and Allergy Foundation of America, National Eczema Association, AbbVie, Incyte Corporation, and Pfizer.
A version of this article first appeared on Medscape.com.
Upadacitinib (Rinvoq) gains psoriatic arthritis as second FDA-approved indication
upadacitinib (Rinvoq) for adults with psoriatic arthritis who had an inadequate response or intolerance to one or more anti-tumor necrosis factor drugs, manufacturer AbbVie announced December 14.

The approval is the second indication given by the agency for the selective Janus kinase (JAK) inhibitor upadacitinib, which was previously approved for rheumatoid arthritis (RA) in 2019.
Upadacitinib 15 mg is also approved by the European Commission for adults with RA, psoriatic arthritis, and ankylosing spondylitis. The European Commission also approved the drug for moderate to severe atopic dermatitis at both 15- and 30-mg doses for adults and at 15 mg for adolescents.
The approval is based on two phase 3 trials, SELECT-PsA 1 and SELECT-PsA 2, which together randomized more than 2,300 patients with psoriatic arthritis. In the trials, significantly more patients who took upadacitinib 15 mg met their primary endpoint of 20% improvement in American College of Rheumatology response criteria (ACR20) at week 12 (71% in SELECT-PsA 1 and 57% in SELECT-PsA 2) vs placebo (36% and 24%, respectively). Both trials also included treatment arms for upadacitinib at 30 mg, but the FDA approved only the 15-mg dose.
In the announcement, AbbVie noted that significantly higher percentages of patients treated with upadacitinib 15 mg in the SELECT-PSA 1 and 2 trials, respectively, met ACR50 (38% and 32%) and ACR70 (16% and 9%) criteria than did patients on placebo (13% and 5% for ACR50 and 2% and 1% for ACR70). Symptoms of dactylitis and enthesitis improved with upadacitinib for patients who had them at baseline.
The trials’ 12-week results also indicated that upadacitinib significantly improved physical function relative to placebo at baseline, based on the Health Assessment Questionnaire-Disability Index, as well as fatigue, according to Functional Assessment of Chronic Illness Therapy – Fatigue (FACIT-F) scores. Skin manifestations also improved during the trial, but upadacitinib has not been studied for treating plaque psoriasis.
AbbVie reported that the safety results of upadacitinib in the trials were consistent with the results seen in patients with rheumatoid arthritis, and during the trials’ 24-week placebo-controlled period, the most common adverse events reported with upadacitinib were upper respiratory tract infection and blood creatine phosphokinase elevations.
Upadacitinib comes with a boxed warning that was formally placed on the drug’s label this month after data from a postmarketing trial of the JAK inhibitor tofacitinib (Xeljanz and Xeljanz XR) in patients with RA aged 50 years and older with at least one cardiovascular risk factor showed numerically higher risks for all-cause mortality; lymphoma and other malignancies; major adverse cardiovascular events (cardiovascular death, myocardial infarction, and stroke); and thrombosis, including deep venous thrombosis, pulmonary embolism, and arterial thrombosis.
Upadacitinib also carries a boxed warning for an elevated risk of serious infection leading to hospitalization or death. In the SELECT-PsA 1 and 2 trials overall, rates of herpes zoster and herpes simplex were 1.1% and 1.4% with upadacitinib, compared with 0.8% and 1.3% with placebo.
Phase 3 trials of upadacitinib in RA, atopic dermatitis, psoriatic arthritis, axial spondyloarthritis, Crohn’s disease, ulcerative colitis, giant cell arteritis, and Takayasu arteritis are ongoing, according to AbbVie.
A version of this article first appeared on Medscape.com.
upadacitinib (Rinvoq) for adults with psoriatic arthritis who had an inadequate response or intolerance to one or more anti-tumor necrosis factor drugs, manufacturer AbbVie announced December 14.
The approval is the second indication given by the agency for the selective Janus kinase (JAK) inhibitor upadacitinib, which was previously approved for rheumatoid arthritis (RA) in 2019.
Upadacitinib 15 mg is also approved by the European Commission for adults with RA, psoriatic arthritis, and ankylosing spondylitis. The European Commission also approved the drug for moderate to severe atopic dermatitis at both 15- and 30-mg doses for adults and at 15 mg for adolescents.
The approval is based on two phase 3 trials, SELECT-PsA 1 and SELECT-PsA 2, which together randomized more than 2,300 patients with psoriatic arthritis. In the trials, significantly more patients who took upadacitinib 15 mg met their primary endpoint of 20% improvement in American College of Rheumatology response criteria (ACR20) at week 12 (71% in SELECT-PsA 1 and 57% in SELECT-PsA 2) vs placebo (36% and 24%, respectively). Both trials also included treatment arms for upadacitinib at 30 mg, but the FDA approved only the 15-mg dose.
In the announcement, AbbVie noted that significantly higher percentages of patients treated with upadacitinib 15 mg in the SELECT-PSA 1 and 2 trials, respectively, met ACR50 (38% and 32%) and ACR70 (16% and 9%) criteria than did patients on placebo (13% and 5% for ACR50 and 2% and 1% for ACR70). Symptoms of dactylitis and enthesitis improved with upadacitinib for patients who had them at baseline.
The trials’ 12-week results also indicated that upadacitinib significantly improved physical function relative to placebo at baseline, based on the Health Assessment Questionnaire-Disability Index, as well as fatigue, according to Functional Assessment of Chronic Illness Therapy – Fatigue (FACIT-F) scores. Skin manifestations also improved during the trial, but upadacitinib has not been studied for treating plaque psoriasis.
AbbVie reported that the safety results of upadacitinib in the trials were consistent with the results seen in patients with rheumatoid arthritis, and during the trials’ 24-week placebo-controlled period, the most common adverse events reported with upadacitinib were upper respiratory tract infection and blood creatine phosphokinase elevations.
Upadacitinib comes with a boxed warning that was formally placed on the drug’s label this month after data from a postmarketing trial of the JAK inhibitor tofacitinib (Xeljanz and Xeljanz XR) in patients with RA aged 50 years and older with at least one cardiovascular risk factor showed numerically higher risks for all-cause mortality; lymphoma and other malignancies; major adverse cardiovascular events (cardiovascular death, myocardial infarction, and stroke); and thrombosis, including deep venous thrombosis, pulmonary embolism, and arterial thrombosis.
Upadacitinib also carries a boxed warning for an elevated risk of serious infection leading to hospitalization or death. In the SELECT-PsA 1 and 2 trials overall, rates of herpes zoster and herpes simplex were 1.1% and 1.4% with upadacitinib, compared with 0.8% and 1.3% with placebo.
Phase 3 trials of upadacitinib in RA, atopic dermatitis, psoriatic arthritis, axial spondyloarthritis, Crohn’s disease, ulcerative colitis, giant cell arteritis, and Takayasu arteritis are ongoing, according to AbbVie.
A version of this article first appeared on Medscape.com.
upadacitinib (Rinvoq) for adults with psoriatic arthritis who had an inadequate response or intolerance to one or more anti-tumor necrosis factor drugs, manufacturer AbbVie announced December 14.
The approval is the second indication given by the agency for the selective Janus kinase (JAK) inhibitor upadacitinib, which was previously approved for rheumatoid arthritis (RA) in 2019.
Upadacitinib 15 mg is also approved by the European Commission for adults with RA, psoriatic arthritis, and ankylosing spondylitis. The European Commission also approved the drug for moderate to severe atopic dermatitis at both 15- and 30-mg doses for adults and at 15 mg for adolescents.
The approval is based on two phase 3 trials, SELECT-PsA 1 and SELECT-PsA 2, which together randomized more than 2,300 patients with psoriatic arthritis. In the trials, significantly more patients who took upadacitinib 15 mg met their primary endpoint of 20% improvement in American College of Rheumatology response criteria (ACR20) at week 12 (71% in SELECT-PsA 1 and 57% in SELECT-PsA 2) vs placebo (36% and 24%, respectively). Both trials also included treatment arms for upadacitinib at 30 mg, but the FDA approved only the 15-mg dose.
In the announcement, AbbVie noted that significantly higher percentages of patients treated with upadacitinib 15 mg in the SELECT-PSA 1 and 2 trials, respectively, met ACR50 (38% and 32%) and ACR70 (16% and 9%) criteria than did patients on placebo (13% and 5% for ACR50 and 2% and 1% for ACR70). Symptoms of dactylitis and enthesitis improved with upadacitinib for patients who had them at baseline.
The trials’ 12-week results also indicated that upadacitinib significantly improved physical function relative to placebo at baseline, based on the Health Assessment Questionnaire-Disability Index, as well as fatigue, according to Functional Assessment of Chronic Illness Therapy – Fatigue (FACIT-F) scores. Skin manifestations also improved during the trial, but upadacitinib has not been studied for treating plaque psoriasis.
AbbVie reported that the safety results of upadacitinib in the trials were consistent with the results seen in patients with rheumatoid arthritis, and during the trials’ 24-week placebo-controlled period, the most common adverse events reported with upadacitinib were upper respiratory tract infection and blood creatine phosphokinase elevations.
Upadacitinib comes with a boxed warning that was formally placed on the drug’s label this month after data from a postmarketing trial of the JAK inhibitor tofacitinib (Xeljanz and Xeljanz XR) in patients with RA aged 50 years and older with at least one cardiovascular risk factor showed numerically higher risks for all-cause mortality; lymphoma and other malignancies; major adverse cardiovascular events (cardiovascular death, myocardial infarction, and stroke); and thrombosis, including deep venous thrombosis, pulmonary embolism, and arterial thrombosis.
Upadacitinib also carries a boxed warning for an elevated risk of serious infection leading to hospitalization or death. In the SELECT-PsA 1 and 2 trials overall, rates of herpes zoster and herpes simplex were 1.1% and 1.4% with upadacitinib, compared with 0.8% and 1.3% with placebo.
Phase 3 trials of upadacitinib in RA, atopic dermatitis, psoriatic arthritis, axial spondyloarthritis, Crohn’s disease, ulcerative colitis, giant cell arteritis, and Takayasu arteritis are ongoing, according to AbbVie.
A version of this article first appeared on Medscape.com.
COVID-19 asymptomatic infection rate remains high
Based on data from a meta-analysis of 95 studies that included nearly 30,000,000 individuals, the pooled percentage of asymptomatic COVID-19 infections was 0.25% in the tested population and 40.5% among confirmed cases.
, wrote Qiuyue Ma, PhD, and colleagues of Peking University, Beijing.
In a study published in JAMA Network Open the researchers identified 44 cross-sectional studies, 41 cohort studies, seven case series, and three case series on transmission studies. A total of 74 studies were conducted in developed countries, including those in Europe, North America, and Asia. Approximately one-third (37) of the studies were conducted among health care workers or in-hospital patients, 17 among nursing home staff or residents, and 14 among community residents. In addition, 13 studies involved pregnant women, eight involved air or cruise ship travelers, and six involved close contacts of individuals with confirmed infections.
The meta-analysis included 29,776,306 tested individuals; 11,516 of them had asymptomatic infections.
Overall, the pooled percentage of asymptomatic infections among the tested population was 0.25%. In an analysis of different study populations, the percentage was higher in nursing home residents or staff (4.52%), air or cruise ship travelers (2.02%), and pregnant women (2.34%), compared against the pooled percentage.
The pooled percentage of asymptomatic infections among the confirmed population was 40.50%, and this percentage was higher in pregnant women (54.11%), air or cruise ship travelers (52.91%), and nursing home residents or staff (47.53%).
The pooled percentage in the tested population was higher than the overall percentage when the mean age of the study population was 60 years or older (3.69%). By contrast, in the confirmed population, the pooled percentage was higher than the overall percentage when the study population was younger than 20 years (60.2%) or aged 20 to 39 years (49.5%).
The researchers noted in their discussion that the varying percentage of asymptomatic individuals according to community prevalence might impact the heterogeneity of the included studies. They also noted the high number of studies conducted in nursing home populations, groups in which asymptomatic individuals were more likely to be tested.
The study findings were limited by several factors, including the potential for missed studies that were not published at the time of the meta-analysis, as well as the exclusion of studies written in Chinese, the researchers noted. Other limitations included lack of follow-up on presymptomatic and covert infections, and the focus on specific populations, factors that may limit the degree to which the results can be generalized.
However, the results highlight the need to screen for asymptomatic infections, especially in countries where COVID-19 has been better controlled, the researchers said. Management strategies for asymptomatic infections, when identified, should include isolation and contact tracing similar to strategies used with confirmed cases, they added.
More testing needed to catch cases early
“During the initial phase of [the] COVID-19 pandemic, testing was not widely available in the United States or the rest of the world,” Setu Patolia, MD, of Saint Louis University School of Medicine, Missouri, said in an interview. Much of the world still lacks access to COVID-19 testing, and early in the pandemic only severely symptomatic patients were tested, he said. “With new variants, particularly the Omicron variant, which may have mild or minimally symptomatic disease, asymptomatic carriers play an important role in propagation of the pandemic,” he explained. “It is important to know the asymptomatic carrier rate among the general population for the future control of [the] pandemic,” he added.
Dr. Patolia said he was surprised by the study finding that one in 400 people in the general population could be asymptomatic carriers of COVID-19.
“Also, nursing home patients are more at risk of complications of COVID, and I expected that they would have a higher rate of symptomatic disease as compared to [the] general population,” said Dr. Patolia. He was also surprised by the high rate of asymptomatic infections in travelers.
“Physicians should be more aware about the asymptomatic carrier rate, particularly in travelers and nursing home patients,” he noted. “Travelers carry high risk of transferring infection from one region to another region of the world, and physicians should advise them to get tested despite the absence of symptoms,” Dr. Patolia emphasized. “Similarly, once any nursing home patient has been diagnosed with COVID-19, physicians should be more careful with the rest of the nursing home patients and test them despite the absence of the symptoms,” he added.
Dr. Patolia also recommended that pregnant women wear masks to help prevent disease transmission when visiting a doctor’s office or labor unit.
Looking ahead, there is a need for cheaper at-home testing kits so that all vulnerable populations can be tested fast and frequently, Dr. Patolia said.
The study was supported by the National Natural Science Foundation of China. The researchers had no financial conflicts to disclose. Dr. Patolia has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Based on data from a meta-analysis of 95 studies that included nearly 30,000,000 individuals, the pooled percentage of asymptomatic COVID-19 infections was 0.25% in the tested population and 40.5% among confirmed cases.
, wrote Qiuyue Ma, PhD, and colleagues of Peking University, Beijing.
In a study published in JAMA Network Open the researchers identified 44 cross-sectional studies, 41 cohort studies, seven case series, and three case series on transmission studies. A total of 74 studies were conducted in developed countries, including those in Europe, North America, and Asia. Approximately one-third (37) of the studies were conducted among health care workers or in-hospital patients, 17 among nursing home staff or residents, and 14 among community residents. In addition, 13 studies involved pregnant women, eight involved air or cruise ship travelers, and six involved close contacts of individuals with confirmed infections.
The meta-analysis included 29,776,306 tested individuals; 11,516 of them had asymptomatic infections.
Overall, the pooled percentage of asymptomatic infections among the tested population was 0.25%. In an analysis of different study populations, the percentage was higher in nursing home residents or staff (4.52%), air or cruise ship travelers (2.02%), and pregnant women (2.34%), compared against the pooled percentage.
The pooled percentage of asymptomatic infections among the confirmed population was 40.50%, and this percentage was higher in pregnant women (54.11%), air or cruise ship travelers (52.91%), and nursing home residents or staff (47.53%).
The pooled percentage in the tested population was higher than the overall percentage when the mean age of the study population was 60 years or older (3.69%). By contrast, in the confirmed population, the pooled percentage was higher than the overall percentage when the study population was younger than 20 years (60.2%) or aged 20 to 39 years (49.5%).
The researchers noted in their discussion that the varying percentage of asymptomatic individuals according to community prevalence might impact the heterogeneity of the included studies. They also noted the high number of studies conducted in nursing home populations, groups in which asymptomatic individuals were more likely to be tested.
The study findings were limited by several factors, including the potential for missed studies that were not published at the time of the meta-analysis, as well as the exclusion of studies written in Chinese, the researchers noted. Other limitations included lack of follow-up on presymptomatic and covert infections, and the focus on specific populations, factors that may limit the degree to which the results can be generalized.
However, the results highlight the need to screen for asymptomatic infections, especially in countries where COVID-19 has been better controlled, the researchers said. Management strategies for asymptomatic infections, when identified, should include isolation and contact tracing similar to strategies used with confirmed cases, they added.
More testing needed to catch cases early
“During the initial phase of [the] COVID-19 pandemic, testing was not widely available in the United States or the rest of the world,” Setu Patolia, MD, of Saint Louis University School of Medicine, Missouri, said in an interview. Much of the world still lacks access to COVID-19 testing, and early in the pandemic only severely symptomatic patients were tested, he said. “With new variants, particularly the Omicron variant, which may have mild or minimally symptomatic disease, asymptomatic carriers play an important role in propagation of the pandemic,” he explained. “It is important to know the asymptomatic carrier rate among the general population for the future control of [the] pandemic,” he added.
Dr. Patolia said he was surprised by the study finding that one in 400 people in the general population could be asymptomatic carriers of COVID-19.
“Also, nursing home patients are more at risk of complications of COVID, and I expected that they would have a higher rate of symptomatic disease as compared to [the] general population,” said Dr. Patolia. He was also surprised by the high rate of asymptomatic infections in travelers.
“Physicians should be more aware about the asymptomatic carrier rate, particularly in travelers and nursing home patients,” he noted. “Travelers carry high risk of transferring infection from one region to another region of the world, and physicians should advise them to get tested despite the absence of symptoms,” Dr. Patolia emphasized. “Similarly, once any nursing home patient has been diagnosed with COVID-19, physicians should be more careful with the rest of the nursing home patients and test them despite the absence of the symptoms,” he added.
Dr. Patolia also recommended that pregnant women wear masks to help prevent disease transmission when visiting a doctor’s office or labor unit.
Looking ahead, there is a need for cheaper at-home testing kits so that all vulnerable populations can be tested fast and frequently, Dr. Patolia said.
The study was supported by the National Natural Science Foundation of China. The researchers had no financial conflicts to disclose. Dr. Patolia has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Based on data from a meta-analysis of 95 studies that included nearly 30,000,000 individuals, the pooled percentage of asymptomatic COVID-19 infections was 0.25% in the tested population and 40.5% among confirmed cases.
, wrote Qiuyue Ma, PhD, and colleagues of Peking University, Beijing.
In a study published in JAMA Network Open the researchers identified 44 cross-sectional studies, 41 cohort studies, seven case series, and three case series on transmission studies. A total of 74 studies were conducted in developed countries, including those in Europe, North America, and Asia. Approximately one-third (37) of the studies were conducted among health care workers or in-hospital patients, 17 among nursing home staff or residents, and 14 among community residents. In addition, 13 studies involved pregnant women, eight involved air or cruise ship travelers, and six involved close contacts of individuals with confirmed infections.
The meta-analysis included 29,776,306 tested individuals; 11,516 of them had asymptomatic infections.
Overall, the pooled percentage of asymptomatic infections among the tested population was 0.25%. In an analysis of different study populations, the percentage was higher in nursing home residents or staff (4.52%), air or cruise ship travelers (2.02%), and pregnant women (2.34%), compared against the pooled percentage.
The pooled percentage of asymptomatic infections among the confirmed population was 40.50%, and this percentage was higher in pregnant women (54.11%), air or cruise ship travelers (52.91%), and nursing home residents or staff (47.53%).
The pooled percentage in the tested population was higher than the overall percentage when the mean age of the study population was 60 years or older (3.69%). By contrast, in the confirmed population, the pooled percentage was higher than the overall percentage when the study population was younger than 20 years (60.2%) or aged 20 to 39 years (49.5%).
The researchers noted in their discussion that the varying percentage of asymptomatic individuals according to community prevalence might impact the heterogeneity of the included studies. They also noted the high number of studies conducted in nursing home populations, groups in which asymptomatic individuals were more likely to be tested.
The study findings were limited by several factors, including the potential for missed studies that were not published at the time of the meta-analysis, as well as the exclusion of studies written in Chinese, the researchers noted. Other limitations included lack of follow-up on presymptomatic and covert infections, and the focus on specific populations, factors that may limit the degree to which the results can be generalized.
However, the results highlight the need to screen for asymptomatic infections, especially in countries where COVID-19 has been better controlled, the researchers said. Management strategies for asymptomatic infections, when identified, should include isolation and contact tracing similar to strategies used with confirmed cases, they added.
More testing needed to catch cases early
“During the initial phase of [the] COVID-19 pandemic, testing was not widely available in the United States or the rest of the world,” Setu Patolia, MD, of Saint Louis University School of Medicine, Missouri, said in an interview. Much of the world still lacks access to COVID-19 testing, and early in the pandemic only severely symptomatic patients were tested, he said. “With new variants, particularly the Omicron variant, which may have mild or minimally symptomatic disease, asymptomatic carriers play an important role in propagation of the pandemic,” he explained. “It is important to know the asymptomatic carrier rate among the general population for the future control of [the] pandemic,” he added.
Dr. Patolia said he was surprised by the study finding that one in 400 people in the general population could be asymptomatic carriers of COVID-19.
“Also, nursing home patients are more at risk of complications of COVID, and I expected that they would have a higher rate of symptomatic disease as compared to [the] general population,” said Dr. Patolia. He was also surprised by the high rate of asymptomatic infections in travelers.
“Physicians should be more aware about the asymptomatic carrier rate, particularly in travelers and nursing home patients,” he noted. “Travelers carry high risk of transferring infection from one region to another region of the world, and physicians should advise them to get tested despite the absence of symptoms,” Dr. Patolia emphasized. “Similarly, once any nursing home patient has been diagnosed with COVID-19, physicians should be more careful with the rest of the nursing home patients and test them despite the absence of the symptoms,” he added.
Dr. Patolia also recommended that pregnant women wear masks to help prevent disease transmission when visiting a doctor’s office or labor unit.
Looking ahead, there is a need for cheaper at-home testing kits so that all vulnerable populations can be tested fast and frequently, Dr. Patolia said.
The study was supported by the National Natural Science Foundation of China. The researchers had no financial conflicts to disclose. Dr. Patolia has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Is it OK to just be satisfied?
It is possible to talk to a patient for a brief moment and just know if he or she is a satisficer or a maximizer. A “satisficer” when presented with treatment options will invariably say: “I’ll do whatever you say, Doctor.” A “maximizer,” in contrast, would like a printed copy of treatment choices, then would seek a second opinion before ultimately buying an UpToDate subscription to research treatments for him or herself.

.
This notion that we have tendencies toward maximizing or satisficing is thanks to Nobel Memorial Prize winner and all-around smart guy, Herbert A. Simon, PhD. Dr. Simon recognized that, although each person might be expected to make optimal decisions to benefit himself or herself, this is practically impossible. To do so would require an infinite amount of time and energy. He found therefore that we actually exhibit “bounded rationality;” that is, we make the best decision given the limits of time, the price of acquiring information, and even our cognitive abilities. The amount of effort we give to make a decision also depends on the situation: You might be very invested in choosing the right spouse, but not at all invested in choosing soup or salad. (Although, we all have friends who are: “Um, is there any thyme in the soup?”)
You’ll certainly recognize that people have different set points on the spectrum between being a satisficer, one who will take the first option that meets a standard, and a maximizer, one who will seek and accept only the best, even if choosing is at great cost. There are risks and benefits of each. In getting the best job, maximizers might be more successful, but satisficers seem to be happier.
How much this extends into other spheres of life is unclear. It is clear, though, that the work of choosing can come at a cost.
The psychologist Barry Schwartz, PhD, believes that, in general, having more choices leads to more anxiety, not more contentment. For example, which Christmas tree lot would you rather visit: One with hundreds of trees of half a dozen varieties? Or one with just a few trees each of Balsam and Douglas Firs? Dr. Schwartz would argue that you might waste an entire afternoon in the first lot only to bring it home and have remorse when you realize it’s a little lopsided. Or let’s say your child applied to all the Ivy League and Public Ivy schools and also threw in all the top liberal arts colleges. The anxiety of selecting the best and the terror that the “best one” might not choose him or her could be overwhelming. A key lesson is that more in life is by chance than we realize, including how straight your tree is and who gets into Princeton this year. Yet, our expectation that things will work out perfectly if only we maximize is ubiquitous. That confidence in our ability to choose correctly is, however, unwarranted. Better to do your best and know that your tree will be festive and there are many colleges which would lead to a happy life than to fret in choosing and then suffer from dashed expectations. Sometimes good enough is good enough.
Being a satisficer or maximizer is probably somewhat fixed, a personality trait, like being extroverted or conscientious. Yet, having insight can be helpful. If choosing a restaurant in Manhattan becomes an actual project for you with spreadsheets and your own statistical analysis, then go for it! Just know that if that process causes you angst and apprehension, then there is another way. Go to Eleven Madison Park, just because I say so. You might have the best dinner of your life or maybe not. At least by not choosing you’ll have the gift of time to spend picking out a tree instead.
Dr. Benabio is director of Healthcare Transformation and chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @Dermdoc on Twitter. Write to him at [email protected].
It is possible to talk to a patient for a brief moment and just know if he or she is a satisficer or a maximizer. A “satisficer” when presented with treatment options will invariably say: “I’ll do whatever you say, Doctor.” A “maximizer,” in contrast, would like a printed copy of treatment choices, then would seek a second opinion before ultimately buying an UpToDate subscription to research treatments for him or herself.
.
This notion that we have tendencies toward maximizing or satisficing is thanks to Nobel Memorial Prize winner and all-around smart guy, Herbert A. Simon, PhD. Dr. Simon recognized that, although each person might be expected to make optimal decisions to benefit himself or herself, this is practically impossible. To do so would require an infinite amount of time and energy. He found therefore that we actually exhibit “bounded rationality;” that is, we make the best decision given the limits of time, the price of acquiring information, and even our cognitive abilities. The amount of effort we give to make a decision also depends on the situation: You might be very invested in choosing the right spouse, but not at all invested in choosing soup or salad. (Although, we all have friends who are: “Um, is there any thyme in the soup?”)
You’ll certainly recognize that people have different set points on the spectrum between being a satisficer, one who will take the first option that meets a standard, and a maximizer, one who will seek and accept only the best, even if choosing is at great cost. There are risks and benefits of each. In getting the best job, maximizers might be more successful, but satisficers seem to be happier.
How much this extends into other spheres of life is unclear. It is clear, though, that the work of choosing can come at a cost.
The psychologist Barry Schwartz, PhD, believes that, in general, having more choices leads to more anxiety, not more contentment. For example, which Christmas tree lot would you rather visit: One with hundreds of trees of half a dozen varieties? Or one with just a few trees each of Balsam and Douglas Firs? Dr. Schwartz would argue that you might waste an entire afternoon in the first lot only to bring it home and have remorse when you realize it’s a little lopsided. Or let’s say your child applied to all the Ivy League and Public Ivy schools and also threw in all the top liberal arts colleges. The anxiety of selecting the best and the terror that the “best one” might not choose him or her could be overwhelming. A key lesson is that more in life is by chance than we realize, including how straight your tree is and who gets into Princeton this year. Yet, our expectation that things will work out perfectly if only we maximize is ubiquitous. That confidence in our ability to choose correctly is, however, unwarranted. Better to do your best and know that your tree will be festive and there are many colleges which would lead to a happy life than to fret in choosing and then suffer from dashed expectations. Sometimes good enough is good enough.
Being a satisficer or maximizer is probably somewhat fixed, a personality trait, like being extroverted or conscientious. Yet, having insight can be helpful. If choosing a restaurant in Manhattan becomes an actual project for you with spreadsheets and your own statistical analysis, then go for it! Just know that if that process causes you angst and apprehension, then there is another way. Go to Eleven Madison Park, just because I say so. You might have the best dinner of your life or maybe not. At least by not choosing you’ll have the gift of time to spend picking out a tree instead.
Dr. Benabio is director of Healthcare Transformation and chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @Dermdoc on Twitter. Write to him at [email protected].
It is possible to talk to a patient for a brief moment and just know if he or she is a satisficer or a maximizer. A “satisficer” when presented with treatment options will invariably say: “I’ll do whatever you say, Doctor.” A “maximizer,” in contrast, would like a printed copy of treatment choices, then would seek a second opinion before ultimately buying an UpToDate subscription to research treatments for him or herself.
.
This notion that we have tendencies toward maximizing or satisficing is thanks to Nobel Memorial Prize winner and all-around smart guy, Herbert A. Simon, PhD. Dr. Simon recognized that, although each person might be expected to make optimal decisions to benefit himself or herself, this is practically impossible. To do so would require an infinite amount of time and energy. He found therefore that we actually exhibit “bounded rationality;” that is, we make the best decision given the limits of time, the price of acquiring information, and even our cognitive abilities. The amount of effort we give to make a decision also depends on the situation: You might be very invested in choosing the right spouse, but not at all invested in choosing soup or salad. (Although, we all have friends who are: “Um, is there any thyme in the soup?”)
You’ll certainly recognize that people have different set points on the spectrum between being a satisficer, one who will take the first option that meets a standard, and a maximizer, one who will seek and accept only the best, even if choosing is at great cost. There are risks and benefits of each. In getting the best job, maximizers might be more successful, but satisficers seem to be happier.
How much this extends into other spheres of life is unclear. It is clear, though, that the work of choosing can come at a cost.
The psychologist Barry Schwartz, PhD, believes that, in general, having more choices leads to more anxiety, not more contentment. For example, which Christmas tree lot would you rather visit: One with hundreds of trees of half a dozen varieties? Or one with just a few trees each of Balsam and Douglas Firs? Dr. Schwartz would argue that you might waste an entire afternoon in the first lot only to bring it home and have remorse when you realize it’s a little lopsided. Or let’s say your child applied to all the Ivy League and Public Ivy schools and also threw in all the top liberal arts colleges. The anxiety of selecting the best and the terror that the “best one” might not choose him or her could be overwhelming. A key lesson is that more in life is by chance than we realize, including how straight your tree is and who gets into Princeton this year. Yet, our expectation that things will work out perfectly if only we maximize is ubiquitous. That confidence in our ability to choose correctly is, however, unwarranted. Better to do your best and know that your tree will be festive and there are many colleges which would lead to a happy life than to fret in choosing and then suffer from dashed expectations. Sometimes good enough is good enough.
Being a satisficer or maximizer is probably somewhat fixed, a personality trait, like being extroverted or conscientious. Yet, having insight can be helpful. If choosing a restaurant in Manhattan becomes an actual project for you with spreadsheets and your own statistical analysis, then go for it! Just know that if that process causes you angst and apprehension, then there is another way. Go to Eleven Madison Park, just because I say so. You might have the best dinner of your life or maybe not. At least by not choosing you’ll have the gift of time to spend picking out a tree instead.
Dr. Benabio is director of Healthcare Transformation and chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @Dermdoc on Twitter. Write to him at [email protected].
Physician gender pay gap isn’t news; health inequity is rampant
A recent study examined projected career earnings between the genders in a largely community-based physician population, finding a difference of about $2 million in career earnings. That a gender pay gap exists in medicine is not news – but the manner in which this study was done, the investigators’ ability to control for a number of confounding variables, and the size of the study group (over 80,000) are newsworthy.
Some of the key findings include that gender pay gaps start with your first job, and you never close the gap, even as you gain experience and efficiency. Also, the more highly remunerated your specialty, the larger the gap. The gender pay gap joins a growing list of inequities within health care. Although physician compensation is not the most important, given that nearly all physicians are well-paid, and we have much more significant inequities that lead to direct patient harm, the reasons for this discrepancy warrant further consideration.
When I was first being educated about social inequity as part of work in social determinants of health, I made the error of using “inequality” and “inequity” interchangeably. The subtle yet important difference between the two terms was quickly described to me. Inequality is a gastroenterologist getting paid more money to do a colonoscopy than a family physician. Inequity is a female gastroenterologist getting paid less than a male gastroenterologist. Global Health Europe boldly identifies that “inequity is the result of failure.” In looking at the inequity inherent in the gender pay gap, I consider what failed and why.
I’m currently making a major career change, leaving an executive leadership position to return to full-time clinical practice. There is a significant pay decrease that will accompany this change because I am in a primary care specialty. Beyond that, I am considering two employment contracts from different systems to do a similar clinical role.
One of the questions my husband asked was which will pay more over the long run. This is difficult to discern because the compensation formula each health system uses is different, even though they are based on standard national benchmarking data. It is possible that women, in general, are like I am and look for factors other than compensation to make a job decision – assuming, like I do, that it will be close enough to not matter or is generally fair. In fact, while compensation is most certainly a consideration for me, once I determined that it was likely to be in the same ballpark, I stopped comparing. Even as the sole breadwinner in our family, I take this (probably faulty) approach.
It’s time to reconsider how we pay physicians
Women may be more likely to gloss over compensation details that men evaluate and negotiate carefully. To change this, women must first take responsibility for being an active, informed, and engaged part of compensation negotiations. In addition, employers who value gender pay equity must negotiate in good faith, keeping in mind the well-described vulnerabilities in discussions about pay. Finally, male and female mentors and leaders should actively coach female physicians on how to approach these conversations with confidence and skill.
In primary care, female physicians spend, on average, about 15% more time with their patients during a visit. Despite spending as much time in clinic seeing patients per week, they see fewer patients, thereby generating less revenue. For compensation plans that are based on productivity, the extra time spent costs money. In this case, it costs the female physicians lost compensation.
The way in which women are more likely to practice medicine, which includes the amount of time they spend with patients, may affect clinical outcomes without directly increasing productivity. A 2017 study demonstrated that elderly patients had lower rates of mortality and readmission when cared for by a female rather than a male physician. These findings require health systems to critically evaluate what compensation plans value and to promote an appropriate balance between quality of care, quantity of care, and style of care.
Although I’ve seen gender pay inequity as blatant as two different salaries for physicians doing the same work – one male and one female – I think this is uncommon. Like many forms of inequity, the outputs are often related to a failed system rather than solely a series of individual failures. Making compensation formulas gender-blind is an important step – but it is only the first step, not the last. Recognizing that the structure of a compensation formula may be biased toward a style of medical practice more likely to be espoused by one gender is necessary as well.
The data, including the findings of this recent study, clearly identify the gender pay gap that exists in medicine, as it does in many other fields, and that it is not explainable solely by differences in specialties, work hours, family status, or title.
To address the inequity, it is imperative that women engage with employers and leaders to both understand and develop skills around effective and appropriate compensation negotiation. Recognizing that compensation plans, especially those built on productivity models, may fail to place adequate value on gender-specific practice styles.
Jennifer Frank is a family physician, physician leader, wife, and mother in Northeast Wisconsin.
A version of this article first appeared on Medscape.com.
A recent study examined projected career earnings between the genders in a largely community-based physician population, finding a difference of about $2 million in career earnings. That a gender pay gap exists in medicine is not news – but the manner in which this study was done, the investigators’ ability to control for a number of confounding variables, and the size of the study group (over 80,000) are newsworthy.
Some of the key findings include that gender pay gaps start with your first job, and you never close the gap, even as you gain experience and efficiency. Also, the more highly remunerated your specialty, the larger the gap. The gender pay gap joins a growing list of inequities within health care. Although physician compensation is not the most important, given that nearly all physicians are well-paid, and we have much more significant inequities that lead to direct patient harm, the reasons for this discrepancy warrant further consideration.
When I was first being educated about social inequity as part of work in social determinants of health, I made the error of using “inequality” and “inequity” interchangeably. The subtle yet important difference between the two terms was quickly described to me. Inequality is a gastroenterologist getting paid more money to do a colonoscopy than a family physician. Inequity is a female gastroenterologist getting paid less than a male gastroenterologist. Global Health Europe boldly identifies that “inequity is the result of failure.” In looking at the inequity inherent in the gender pay gap, I consider what failed and why.
I’m currently making a major career change, leaving an executive leadership position to return to full-time clinical practice. There is a significant pay decrease that will accompany this change because I am in a primary care specialty. Beyond that, I am considering two employment contracts from different systems to do a similar clinical role.
One of the questions my husband asked was which will pay more over the long run. This is difficult to discern because the compensation formula each health system uses is different, even though they are based on standard national benchmarking data. It is possible that women, in general, are like I am and look for factors other than compensation to make a job decision – assuming, like I do, that it will be close enough to not matter or is generally fair. In fact, while compensation is most certainly a consideration for me, once I determined that it was likely to be in the same ballpark, I stopped comparing. Even as the sole breadwinner in our family, I take this (probably faulty) approach.
It’s time to reconsider how we pay physicians
Women may be more likely to gloss over compensation details that men evaluate and negotiate carefully. To change this, women must first take responsibility for being an active, informed, and engaged part of compensation negotiations. In addition, employers who value gender pay equity must negotiate in good faith, keeping in mind the well-described vulnerabilities in discussions about pay. Finally, male and female mentors and leaders should actively coach female physicians on how to approach these conversations with confidence and skill.
In primary care, female physicians spend, on average, about 15% more time with their patients during a visit. Despite spending as much time in clinic seeing patients per week, they see fewer patients, thereby generating less revenue. For compensation plans that are based on productivity, the extra time spent costs money. In this case, it costs the female physicians lost compensation.
The way in which women are more likely to practice medicine, which includes the amount of time they spend with patients, may affect clinical outcomes without directly increasing productivity. A 2017 study demonstrated that elderly patients had lower rates of mortality and readmission when cared for by a female rather than a male physician. These findings require health systems to critically evaluate what compensation plans value and to promote an appropriate balance between quality of care, quantity of care, and style of care.
Although I’ve seen gender pay inequity as blatant as two different salaries for physicians doing the same work – one male and one female – I think this is uncommon. Like many forms of inequity, the outputs are often related to a failed system rather than solely a series of individual failures. Making compensation formulas gender-blind is an important step – but it is only the first step, not the last. Recognizing that the structure of a compensation formula may be biased toward a style of medical practice more likely to be espoused by one gender is necessary as well.
The data, including the findings of this recent study, clearly identify the gender pay gap that exists in medicine, as it does in many other fields, and that it is not explainable solely by differences in specialties, work hours, family status, or title.
To address the inequity, it is imperative that women engage with employers and leaders to both understand and develop skills around effective and appropriate compensation negotiation. Recognizing that compensation plans, especially those built on productivity models, may fail to place adequate value on gender-specific practice styles.
Jennifer Frank is a family physician, physician leader, wife, and mother in Northeast Wisconsin.
A version of this article first appeared on Medscape.com.
A recent study examined projected career earnings between the genders in a largely community-based physician population, finding a difference of about $2 million in career earnings. That a gender pay gap exists in medicine is not news – but the manner in which this study was done, the investigators’ ability to control for a number of confounding variables, and the size of the study group (over 80,000) are newsworthy.
Some of the key findings include that gender pay gaps start with your first job, and you never close the gap, even as you gain experience and efficiency. Also, the more highly remunerated your specialty, the larger the gap. The gender pay gap joins a growing list of inequities within health care. Although physician compensation is not the most important, given that nearly all physicians are well-paid, and we have much more significant inequities that lead to direct patient harm, the reasons for this discrepancy warrant further consideration.
When I was first being educated about social inequity as part of work in social determinants of health, I made the error of using “inequality” and “inequity” interchangeably. The subtle yet important difference between the two terms was quickly described to me. Inequality is a gastroenterologist getting paid more money to do a colonoscopy than a family physician. Inequity is a female gastroenterologist getting paid less than a male gastroenterologist. Global Health Europe boldly identifies that “inequity is the result of failure.” In looking at the inequity inherent in the gender pay gap, I consider what failed and why.
I’m currently making a major career change, leaving an executive leadership position to return to full-time clinical practice. There is a significant pay decrease that will accompany this change because I am in a primary care specialty. Beyond that, I am considering two employment contracts from different systems to do a similar clinical role.
One of the questions my husband asked was which will pay more over the long run. This is difficult to discern because the compensation formula each health system uses is different, even though they are based on standard national benchmarking data. It is possible that women, in general, are like I am and look for factors other than compensation to make a job decision – assuming, like I do, that it will be close enough to not matter or is generally fair. In fact, while compensation is most certainly a consideration for me, once I determined that it was likely to be in the same ballpark, I stopped comparing. Even as the sole breadwinner in our family, I take this (probably faulty) approach.
It’s time to reconsider how we pay physicians
Women may be more likely to gloss over compensation details that men evaluate and negotiate carefully. To change this, women must first take responsibility for being an active, informed, and engaged part of compensation negotiations. In addition, employers who value gender pay equity must negotiate in good faith, keeping in mind the well-described vulnerabilities in discussions about pay. Finally, male and female mentors and leaders should actively coach female physicians on how to approach these conversations with confidence and skill.
In primary care, female physicians spend, on average, about 15% more time with their patients during a visit. Despite spending as much time in clinic seeing patients per week, they see fewer patients, thereby generating less revenue. For compensation plans that are based on productivity, the extra time spent costs money. In this case, it costs the female physicians lost compensation.
The way in which women are more likely to practice medicine, which includes the amount of time they spend with patients, may affect clinical outcomes without directly increasing productivity. A 2017 study demonstrated that elderly patients had lower rates of mortality and readmission when cared for by a female rather than a male physician. These findings require health systems to critically evaluate what compensation plans value and to promote an appropriate balance between quality of care, quantity of care, and style of care.
Although I’ve seen gender pay inequity as blatant as two different salaries for physicians doing the same work – one male and one female – I think this is uncommon. Like many forms of inequity, the outputs are often related to a failed system rather than solely a series of individual failures. Making compensation formulas gender-blind is an important step – but it is only the first step, not the last. Recognizing that the structure of a compensation formula may be biased toward a style of medical practice more likely to be espoused by one gender is necessary as well.
The data, including the findings of this recent study, clearly identify the gender pay gap that exists in medicine, as it does in many other fields, and that it is not explainable solely by differences in specialties, work hours, family status, or title.
To address the inequity, it is imperative that women engage with employers and leaders to both understand and develop skills around effective and appropriate compensation negotiation. Recognizing that compensation plans, especially those built on productivity models, may fail to place adequate value on gender-specific practice styles.
Jennifer Frank is a family physician, physician leader, wife, and mother in Northeast Wisconsin.
A version of this article first appeared on Medscape.com.
Vegetative Plaques on the Face
THE DIAGNOSIS: Vegetative Majocchi Granuloma
A biopsy and tissue culture showed acute dermal inflammation with granulomatous features and numerous fungal hyphae within the stratum corneum (Figure 1A), which were confirmed on GrocottGomori methenamine-silver staining (Figure 1B). Gram and Fite stains were negative for bacteria. A tissue culture speciated Trichophyton rubrum, which led to a diagnosis of deep dermatophyte infection (Majocchi granuloma) with a highly unusual clinical presentation of vegetative plaques. Predisposing factors included treatment with topical corticosteroids and possibly poor health and nutritional status at baseline. Our patient was treated with fluconazole 200 mg daily for 6 weeks, with near resolution of lesions at 3-week follow-up (Figure 2).
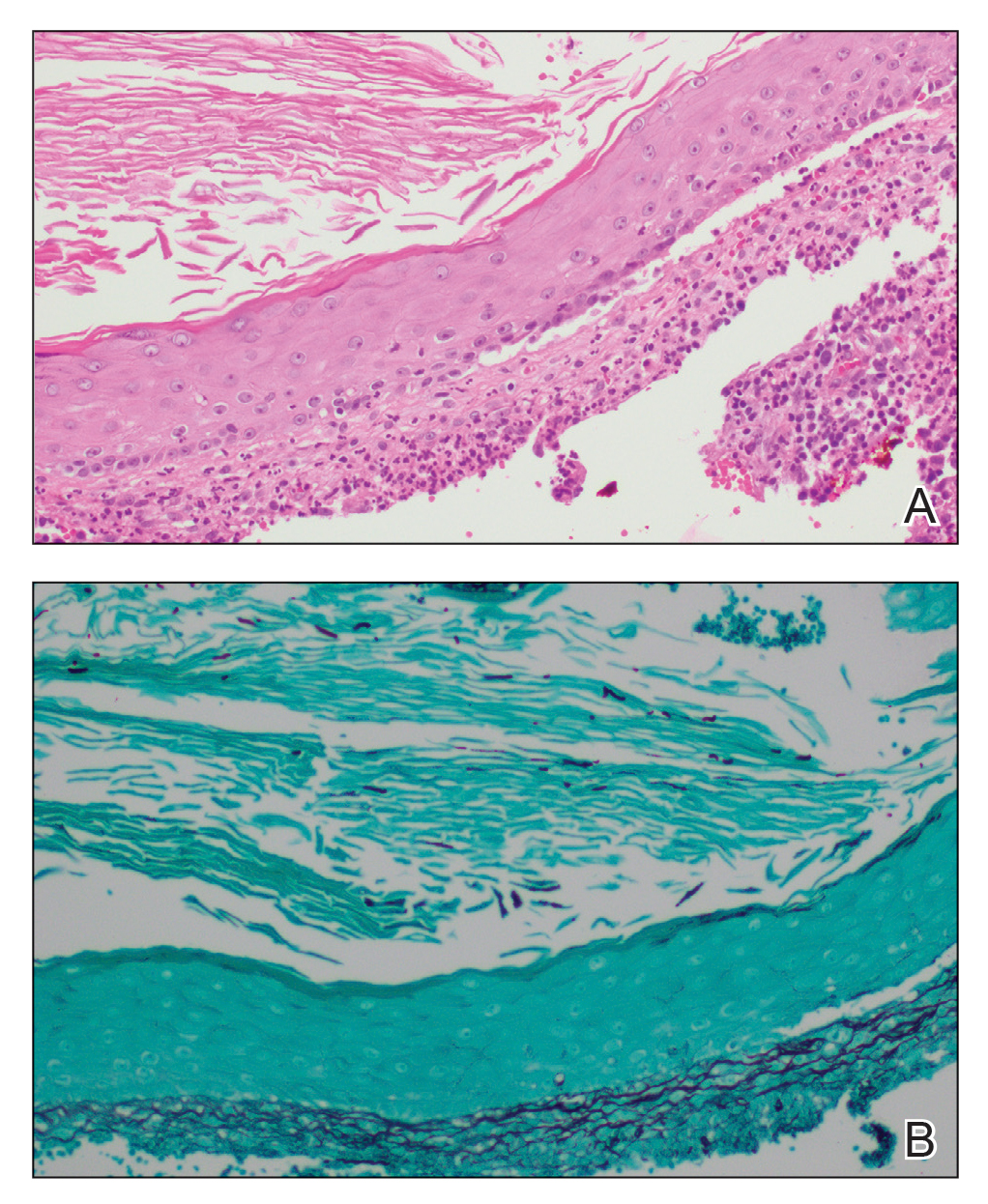
Dermatophytes are a common cause of superficial skin infections. The classic morphology consists of an annular scaly plaque; however, a wide variety of presentations have been observed (eg, verrucous, vesicular, pustular, granulomatous). Therefore, dermatophyte infections often mimic other dermatologic conditions, including atopic dermatitis, rosacea, psoriasis, bacterial abscess, erythema gyratum repens, lupus, granuloma annulare, cutaneous lymphoma, Hailey-Hailey disease, scarring alopecia, and syphilis.1
Notably, when dermatophytes grow downward along hair follicles causing deeper infection, disruption of the follicular wall can lead to an excessive inflammatory response with granulomatous features.2 Risk factors include cutaneous trauma, long-standing infection, immunocompromise, and treatment with topical corticosteroids.3 This disease evolution clinically appears as a nodule or infiltrated plaque, often without scale. The most well-known example is a kerion on the scalp. Elsewhere on the body, lesions often are termed Majocchi granulomas.2
Vegetative plaques, as seen in our patient, are a highly unusual morphology for deep tinea infection. Guanziroli et al4 reported a case of vegetative lesions on the forearm of a 67-year-old immunocompromised man that were successfully treated with a 3-month course of oral terbinafine after Trichophyton verrucosum was isolated. Skorepova et al5 reported a case of pyoderma vegetans triggered by recurrent Trichophyton mentagrophytes on the dorsal hands of a 64-year-old man with immunoglobulin deficiency of unknown etiology. The lesions were successfully treated with a prolonged course of doxycycline, topical triamcinolone, and intravenous immunoglobulin following 2 initial courses of terbinafine.
The differential diagnosis for vegetative lesions includes pemphigus vegetans, a vegetative variant of pyoderma gangrenosum; halogenoderma; and a variety of infections, including dimorphic fungi (histoplasmosis, blastomycosis), blastomycosislike pyoderma (bacterial), and candidiasis.6 These conditions usually can be distinguished based on histopathology. Clinically, pemphigus vegetans presents with pustules and vegetative lesions, as in our patient, but usually is more diffuse and favors the intertriginous areas. Histology likely would reveal foci of acantholysis and eosinophils. Vegetative pyoderma gangrenosum favors the trunk, particularly in sites of surgical trauma. In our patient, no lesions were present near the abdominal surgical sites, and there was no antecedent cribriform ulceration. Halogenoderma was a strong initial consideration given the localization, presence of large pustules, and history of numerous contrast computed tomography studies; however, our patient’s iodine levels were normal. Infectious etiologies including dimorphic fungi and blastomycosislike pyoderma generally are not restricted to the head and neck, and tissue culture helps exclude them. Vegetative lesions may occur in the setting of other infections, and tissue culture may be necessary to differentiate them if histopathology is not suggestive.
Deep dermatophyte infections require treatment with oral antifungals, as topicals do not penetrate adequately into the hair follicles. Exact regimens vary, but generally oral terbinafine or an oral azole (except ketoconazole) is administered for 2 to 6 weeks, with immunocompromise necessitating longer courses.
We present a rare case of vegetative Majocchi granuloma secondary to T rubrum infection. A dermatophyte infection should be included in the differential for vegetative lesions, especially in dense hair-bearing areas such as the beard. Treatment generally is straightforward with oral antifungals.
- Atzori L, Pau M, Aste N, et al. Dermatophyte infections mimicking other skin diseases: a 154-person case survey of tinea atypica in the district of Cagliari (Italy). Int J Dermatol. 2012;51:410-415.
- Ilkit M, Durdu M, Karakas M. Majocchi’s granuloma: a symptom complex caused by fungal pathogens. Med Mycol. 2012;50:449-457.
- Jevremovic L, Ilijin I, Kostic K, et al. Pyoderma vegetans—a case report. Serbian J Dermatol Venereol. 2017;9:22-28.
- Guanziroli E, Pavia G, Guttadauro A, et al. Deep dermatophytosis caused by Trichophyton verrucosum in an immunosuppressed patient: successful outcome with terbinafine. Mycopathologia. 2019;184:543-545.
- Skorepová M, Stuchlík D. Chronic pyoderma vegetans triggered by Trichophyton mentagrophytes. Mycoses. 2006;49:143-144.
- Reinholz M, Hermans C, Dietrich A, et al. A case of cutaneous vegetating candidiasis in a patient with keratitis-ichthyosis-deafness syndrome. J Eur Acad Dermatol Venereol. 2016;30:537-539.
THE DIAGNOSIS: Vegetative Majocchi Granuloma
A biopsy and tissue culture showed acute dermal inflammation with granulomatous features and numerous fungal hyphae within the stratum corneum (Figure 1A), which were confirmed on GrocottGomori methenamine-silver staining (Figure 1B). Gram and Fite stains were negative for bacteria. A tissue culture speciated Trichophyton rubrum, which led to a diagnosis of deep dermatophyte infection (Majocchi granuloma) with a highly unusual clinical presentation of vegetative plaques. Predisposing factors included treatment with topical corticosteroids and possibly poor health and nutritional status at baseline. Our patient was treated with fluconazole 200 mg daily for 6 weeks, with near resolution of lesions at 3-week follow-up (Figure 2).
Dermatophytes are a common cause of superficial skin infections. The classic morphology consists of an annular scaly plaque; however, a wide variety of presentations have been observed (eg, verrucous, vesicular, pustular, granulomatous). Therefore, dermatophyte infections often mimic other dermatologic conditions, including atopic dermatitis, rosacea, psoriasis, bacterial abscess, erythema gyratum repens, lupus, granuloma annulare, cutaneous lymphoma, Hailey-Hailey disease, scarring alopecia, and syphilis.1
Notably, when dermatophytes grow downward along hair follicles causing deeper infection, disruption of the follicular wall can lead to an excessive inflammatory response with granulomatous features.2 Risk factors include cutaneous trauma, long-standing infection, immunocompromise, and treatment with topical corticosteroids.3 This disease evolution clinically appears as a nodule or infiltrated plaque, often without scale. The most well-known example is a kerion on the scalp. Elsewhere on the body, lesions often are termed Majocchi granulomas.2
Vegetative plaques, as seen in our patient, are a highly unusual morphology for deep tinea infection. Guanziroli et al4 reported a case of vegetative lesions on the forearm of a 67-year-old immunocompromised man that were successfully treated with a 3-month course of oral terbinafine after Trichophyton verrucosum was isolated. Skorepova et al5 reported a case of pyoderma vegetans triggered by recurrent Trichophyton mentagrophytes on the dorsal hands of a 64-year-old man with immunoglobulin deficiency of unknown etiology. The lesions were successfully treated with a prolonged course of doxycycline, topical triamcinolone, and intravenous immunoglobulin following 2 initial courses of terbinafine.
The differential diagnosis for vegetative lesions includes pemphigus vegetans, a vegetative variant of pyoderma gangrenosum; halogenoderma; and a variety of infections, including dimorphic fungi (histoplasmosis, blastomycosis), blastomycosislike pyoderma (bacterial), and candidiasis.6 These conditions usually can be distinguished based on histopathology. Clinically, pemphigus vegetans presents with pustules and vegetative lesions, as in our patient, but usually is more diffuse and favors the intertriginous areas. Histology likely would reveal foci of acantholysis and eosinophils. Vegetative pyoderma gangrenosum favors the trunk, particularly in sites of surgical trauma. In our patient, no lesions were present near the abdominal surgical sites, and there was no antecedent cribriform ulceration. Halogenoderma was a strong initial consideration given the localization, presence of large pustules, and history of numerous contrast computed tomography studies; however, our patient’s iodine levels were normal. Infectious etiologies including dimorphic fungi and blastomycosislike pyoderma generally are not restricted to the head and neck, and tissue culture helps exclude them. Vegetative lesions may occur in the setting of other infections, and tissue culture may be necessary to differentiate them if histopathology is not suggestive.
Deep dermatophyte infections require treatment with oral antifungals, as topicals do not penetrate adequately into the hair follicles. Exact regimens vary, but generally oral terbinafine or an oral azole (except ketoconazole) is administered for 2 to 6 weeks, with immunocompromise necessitating longer courses.
We present a rare case of vegetative Majocchi granuloma secondary to T rubrum infection. A dermatophyte infection should be included in the differential for vegetative lesions, especially in dense hair-bearing areas such as the beard. Treatment generally is straightforward with oral antifungals.
THE DIAGNOSIS: Vegetative Majocchi Granuloma
A biopsy and tissue culture showed acute dermal inflammation with granulomatous features and numerous fungal hyphae within the stratum corneum (Figure 1A), which were confirmed on GrocottGomori methenamine-silver staining (Figure 1B). Gram and Fite stains were negative for bacteria. A tissue culture speciated Trichophyton rubrum, which led to a diagnosis of deep dermatophyte infection (Majocchi granuloma) with a highly unusual clinical presentation of vegetative plaques. Predisposing factors included treatment with topical corticosteroids and possibly poor health and nutritional status at baseline. Our patient was treated with fluconazole 200 mg daily for 6 weeks, with near resolution of lesions at 3-week follow-up (Figure 2).
Dermatophytes are a common cause of superficial skin infections. The classic morphology consists of an annular scaly plaque; however, a wide variety of presentations have been observed (eg, verrucous, vesicular, pustular, granulomatous). Therefore, dermatophyte infections often mimic other dermatologic conditions, including atopic dermatitis, rosacea, psoriasis, bacterial abscess, erythema gyratum repens, lupus, granuloma annulare, cutaneous lymphoma, Hailey-Hailey disease, scarring alopecia, and syphilis.1
Notably, when dermatophytes grow downward along hair follicles causing deeper infection, disruption of the follicular wall can lead to an excessive inflammatory response with granulomatous features.2 Risk factors include cutaneous trauma, long-standing infection, immunocompromise, and treatment with topical corticosteroids.3 This disease evolution clinically appears as a nodule or infiltrated plaque, often without scale. The most well-known example is a kerion on the scalp. Elsewhere on the body, lesions often are termed Majocchi granulomas.2
Vegetative plaques, as seen in our patient, are a highly unusual morphology for deep tinea infection. Guanziroli et al4 reported a case of vegetative lesions on the forearm of a 67-year-old immunocompromised man that were successfully treated with a 3-month course of oral terbinafine after Trichophyton verrucosum was isolated. Skorepova et al5 reported a case of pyoderma vegetans triggered by recurrent Trichophyton mentagrophytes on the dorsal hands of a 64-year-old man with immunoglobulin deficiency of unknown etiology. The lesions were successfully treated with a prolonged course of doxycycline, topical triamcinolone, and intravenous immunoglobulin following 2 initial courses of terbinafine.
The differential diagnosis for vegetative lesions includes pemphigus vegetans, a vegetative variant of pyoderma gangrenosum; halogenoderma; and a variety of infections, including dimorphic fungi (histoplasmosis, blastomycosis), blastomycosislike pyoderma (bacterial), and candidiasis.6 These conditions usually can be distinguished based on histopathology. Clinically, pemphigus vegetans presents with pustules and vegetative lesions, as in our patient, but usually is more diffuse and favors the intertriginous areas. Histology likely would reveal foci of acantholysis and eosinophils. Vegetative pyoderma gangrenosum favors the trunk, particularly in sites of surgical trauma. In our patient, no lesions were present near the abdominal surgical sites, and there was no antecedent cribriform ulceration. Halogenoderma was a strong initial consideration given the localization, presence of large pustules, and history of numerous contrast computed tomography studies; however, our patient’s iodine levels were normal. Infectious etiologies including dimorphic fungi and blastomycosislike pyoderma generally are not restricted to the head and neck, and tissue culture helps exclude them. Vegetative lesions may occur in the setting of other infections, and tissue culture may be necessary to differentiate them if histopathology is not suggestive.
Deep dermatophyte infections require treatment with oral antifungals, as topicals do not penetrate adequately into the hair follicles. Exact regimens vary, but generally oral terbinafine or an oral azole (except ketoconazole) is administered for 2 to 6 weeks, with immunocompromise necessitating longer courses.
We present a rare case of vegetative Majocchi granuloma secondary to T rubrum infection. A dermatophyte infection should be included in the differential for vegetative lesions, especially in dense hair-bearing areas such as the beard. Treatment generally is straightforward with oral antifungals.
- Atzori L, Pau M, Aste N, et al. Dermatophyte infections mimicking other skin diseases: a 154-person case survey of tinea atypica in the district of Cagliari (Italy). Int J Dermatol. 2012;51:410-415.
- Ilkit M, Durdu M, Karakas M. Majocchi’s granuloma: a symptom complex caused by fungal pathogens. Med Mycol. 2012;50:449-457.
- Jevremovic L, Ilijin I, Kostic K, et al. Pyoderma vegetans—a case report. Serbian J Dermatol Venereol. 2017;9:22-28.
- Guanziroli E, Pavia G, Guttadauro A, et al. Deep dermatophytosis caused by Trichophyton verrucosum in an immunosuppressed patient: successful outcome with terbinafine. Mycopathologia. 2019;184:543-545.
- Skorepová M, Stuchlík D. Chronic pyoderma vegetans triggered by Trichophyton mentagrophytes. Mycoses. 2006;49:143-144.
- Reinholz M, Hermans C, Dietrich A, et al. A case of cutaneous vegetating candidiasis in a patient with keratitis-ichthyosis-deafness syndrome. J Eur Acad Dermatol Venereol. 2016;30:537-539.
- Atzori L, Pau M, Aste N, et al. Dermatophyte infections mimicking other skin diseases: a 154-person case survey of tinea atypica in the district of Cagliari (Italy). Int J Dermatol. 2012;51:410-415.
- Ilkit M, Durdu M, Karakas M. Majocchi’s granuloma: a symptom complex caused by fungal pathogens. Med Mycol. 2012;50:449-457.
- Jevremovic L, Ilijin I, Kostic K, et al. Pyoderma vegetans—a case report. Serbian J Dermatol Venereol. 2017;9:22-28.
- Guanziroli E, Pavia G, Guttadauro A, et al. Deep dermatophytosis caused by Trichophyton verrucosum in an immunosuppressed patient: successful outcome with terbinafine. Mycopathologia. 2019;184:543-545.
- Skorepová M, Stuchlík D. Chronic pyoderma vegetans triggered by Trichophyton mentagrophytes. Mycoses. 2006;49:143-144.
- Reinholz M, Hermans C, Dietrich A, et al. A case of cutaneous vegetating candidiasis in a patient with keratitis-ichthyosis-deafness syndrome. J Eur Acad Dermatol Venereol. 2016;30:537-539.
An 86-year-old man was admitted to the hospital for sigmoid colon perforation secondary to ischemic colitis. His medical history consisted of sequelae from atherosclerotic vascular disease. He had no known personal or family history of skin disease. His bowel perforation was surgically repaired, and his clinical status was stabilized, enabling transfer to a transitional care hospital. His course was complicated by delayed healing of the midline abdominal surgical wounds, leading to multiple computed tomography studies with iodinated contrast. One week following arrival at the transitional care hospital, he was noted to have a pustular rash on the face. He was empirically treated with topical steroids, mupirocin, and sulfacetamide. The rash did not improve, and the appearance changed, at which point dermatology was consulted. On evaluation, the patient was afebrile with a normal white blood cell count. Physical examination revealed gray-brown, moist, vegetative plaques on the cheeks with a few large pustules as well as similar-appearing lesions on the neck and upper chest. Attempted removal of a portion of the plaque left an erosion.
Widespread Necrotizing Purpura and Lucio Phenomenon as the First Diagnostic Presentation of Diffuse Nonnodular Lepromatous Leprosy
Case Report
A 70-year-old man living in Esna, Luxor, Egypt presented to the Department of Rheumatology and Rehabilitation with widespread gangrenous skin lesions associated with ulcers of 2 weeks’ duration. One year prior, the patient had an insidious onset of nocturnal fever, bilateral leg edema, and numbness and a tingling sensation in both hands. He presented some laboratory and radiologic investigations that were performed at another hospital prior to the current presentation, which revealed thrombocytopenia, mild splenomegaly, and generalized lymphadenopathy. An excisional left axillary lymph node biopsy was performed at another hospital prior to the current presentation, and the pathology report provided by the patient described a reactive, foamy, histiocyte-rich lesion, suggesting a diagnosis of hemophagocytic lymphohistiocytosis. The patient had no diabetes or hypertension and no history of deep vein thrombosis, stroke, or unintentional weight loss. No medications were taken prior to the onset of the skin lesions, and his family history was irrelevant.
General examination at the current presentation revealed a fever (temperature, 101.3 °F [38.5 °C]), a normal heart rate (90 beats per minute), normal blood pressure (120/80 mmHg), normal respiratory rate (14 breaths per minute), accentuated heart sounds, and normal vesicular breathing without adventitious sounds. He had saddle nose, loss of the outer third of the eyebrows, and marked reduction in the density of the eyelashes (madarosis). Bilateral pitting edema of the legs also was present. Neurologic examination revealed hypoesthesia in a glove-and-stocking pattern, thickened peripheral nerves, and trophic changes over both hands; however, he had normal muscle power and deep reflexes. Joint examination revealed no abnormalities. Skin examination revealed widespread, reticulated, necrotizing, purpuric lesions on the arms, legs, abdomen, and ears, some associated with gangrenous ulcerations and hemorrhagic blisters. Scattered vasculitic ulcers and gangrenous patches were seen on the fingers. A gangrenous ulcer mimicking Fournier gangrene was seen involving the scrotal skin in addition to a gangrenous lesion on the glans penis (Figure 1–3). Unaffected skin appeared smooth, shiny, and edematous and showed no nodular lesions. Peripheral pulsations were intact.
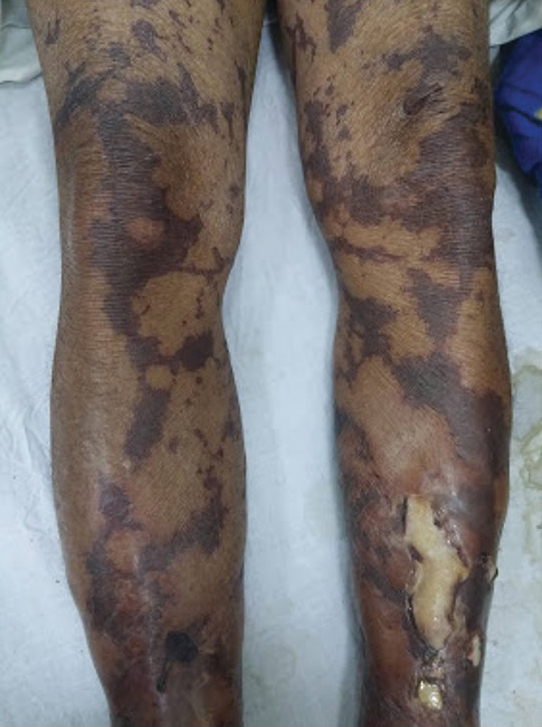
Positive findings from a wide panel of laboratory investigations included an elevated erythrocyte sedimentation rate (103 mm for the first hour [reference range, 0–22 mm]), high C-reactive protein (50.7 mg/L [reference range, up to 6 mg/L]), anemia (hemoglobin count, 7.3 g/dL [reference range, 13.5–17.5 g/dL]), thrombocytopenia (45×103/mm3 [reference range, 150×103/mm3), low serum albumin (2.3 g/dL [reference range, 3.4–5.4 g/dL]), elevated IgG and IgM anticardiolipin antibodies (IgG, 21.4 IgG phospholipid [GPL] units [reference range, <10 IgG phospholipid (GPL) units]; IgM, 59.4 IgM phospholipid (MPL) units [reference range, <7 IgM phospholipid (MPL) units]), positive lupus anticoagulant panel test, elevated anti-β2 glycoprotein antibodies (IgG, 17.5
Nerve conduction velocity showed axonal sensory polyneuropathy. Motor nerve conduction studies for median and ulnar nerves were within normal range. Lower-limb nerves assessment was limited by the ulcerated areas and marked edema. Echocardiography was unremarkable. Arterial Doppler studies were only available for the upper limbs and were unremarkable.
A punch biopsy was taken from one of the necrotizing purpuric lesions on the legs, and histopathologic examination revealed foci of epidermal necrosis and subepidermal separation and superficial and deep perivascular and periadnexal infiltrates extending into the fat lobules. The infiltrates were mainly made up of foamy macrophages, and some contained globi (lepra cells), in addition to lymphocytes and many neutrophils with nuclear dust. Blood vessels in the superficial and deep dermis and in the subcutaneous fat showed fibrinoid necrosis in their walls with neutrophils infiltrating the walls and thrombi in the lumens (Figure 4). Modified Ziehl-Neelsen staining revealed clumps of acid-fast lepra bacilli inside vascular lumina and endothelial cell lining and within the foamy macrophages (Figure 5). Slit-skin smear examination was performed twice and yielded negative results. The slide and paraffin block of the already performed lymph node biopsy were retrieved. Examination revealed aggregates of foamy histiocytes surrounded by lymphocytes and plasma cells replacing normal lymphoid follicles. Modified Ziehl-Neelsen stain was performed, and clusters of acid-fast bacilli were detected within the foamy histiocytic infiltrate (Figure 6).
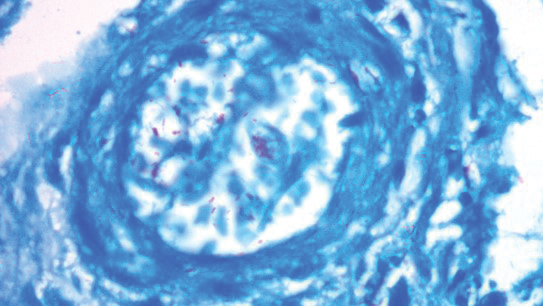
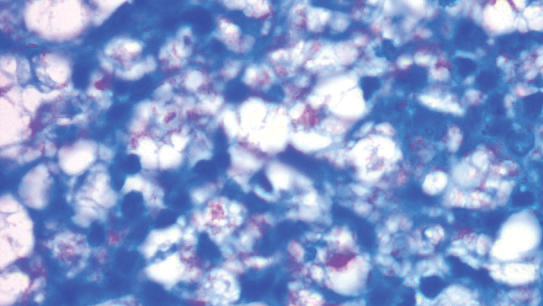
According to the results of the skin biopsy, the revised result of the lymph node biopsy, and the pattern of neurologic deficit together with clinical and laboratory correlation, the patient was diagnosed with diffuse nonnodular lepromatous leprosy presenting with Lucio phenomenon (Lucio leprosy) and associated with lepromatous lymphadenitis.
The patient received the following treatment: methylprednisolone 500 mg (intravenous pulse therapy) followed by daily oral administration of prednisolone 10 mg, rifampicin 300 mg, dapsone 100 mg, clofazimine 100 mg, acetylsalicylic acid 150 mg, and enoxaparin sodium 80 mg. In addition, the scrotal Fournier gangrene–like lesion was treated by surgical debridement followed by vacuum therapy. By the second week after treatment, the gangrenous lesions of the fingers developed a line of demarcation, and the skin infarctions started to recede.
Comment
Despite a decrease in its prevalence through a World Health Organization (WHO)–empowered eradication program, leprosy still represents a health problem in endemic areas.1,2 It is characterized by a wide range of immune responses to Mycobacterium leprae, displaying a spectrum of clinical and histopathologic manifestations that vary from the tuberculoid or paucibacillary pole with a strong cell-mediated immune response and fewer organisms to the lepromatous or multibacillary pole with weaker cell-mediated immune response and higher loads of organisms.3 In addition to its well-known cutaneous and neurologic manifestations, leprosy can present with a variety of manifestations, including constitutional symptoms, musculoskeletal manifestations, and serologic abnormalities; thus, leprosy can mimic rheumatoid arthritis, spondyloarthritis, and vasculitis—a pitfall that may result in misdiagnosis as a rheumatologic disorder.3-7
The chronic course of leprosy can be disrupted by acute, immunologically mediated reactions known as lepra reactions, of which there are 3 types.8 Type I lepra reactions are cell mediated and occur mainly in patients with borderline disease, often representing an upgrade toward the tuberculoid pole; less often they represent a downgrade reaction. Nerves become painful and swollen with possible loss of function, and skin lesions become edematous and tender; sometimes arthritis develops.9 Type II lepra reactions, also known as erythema nodosum leprosum (ENL), occur in borderline lepromatous and lepromatous patients with a high bacillary load. They are characterized by fever, body aches, tender cutaneous/subcutaneous nodules that may ulcerate, possible bullous lesions, painful nerve swellings, swollen joints, iritis, lymphadenitis, glomerulonephritis, epididymo-orchitis, and hepatic affection. Both immune-complex and delayed hypersensitivity reactions play a role in ENL.8,10 The third reaction is a rare aggressive type known as Lucio phenomenon or Lucio leprosy, which presents with irregular-shaped, angulated, or stellar necrotizing purpuric lesions (hemorrhagic infacrtions) developing mainly on the extremities. The lesions evolve into ulcers that heal with atrophic scarring.2,11 Lucio phenomenon develops as a result of thrombotic vascular occlusion secondary to massive invasion of vascular endothelial cells by lepra bacilli.2,11-14 Involvement of the scrotal skin, such as in our patient, is rare.
Lucio phenomenon mainly is seen in Mexico and Central America, and few cases have been documented in Cuba, South America, the United States, India, Polynesia, South Africa, and Southeast Asia.15-17 It specifically occurs in patients with untreated, diffuse, nonnodular lepromatous leprosy (pure and primitive diffuse lepromatous leprosy (DLL)/diffuse leprosy of Lucio and Latapí). This type of leprosy was first described by Lucio and Alvarado18 in 1852 as a distinct form of lepromatous leprosy characterized by widespread and dense infiltration of the whole skin by lepra bacilli without the typical nodular lesions of leprosy, rendering its diagnosis challenging, especially in sporadic cases. Other manifestations of DLL include complete alopecia of the eyebrows and eyelashes, destructive rhinitis, and areas of anhidrosis and dyesthesia.2
Latapí and Chévez-Zomora19 defined Lucio phenomenon in 1948 as a form of histopathologic vasculitis restricted to patients with DLL. Histopathologically, in addition to the infiltration of the skin with acid-fast bacilli–laden foamy histiocytes, lesions of Lucio phenomenon show features of necrotizing (leukocytoclastic) vasculitis with fibrinoid necrosis20 or vascular thrombi with minimal perivascular lymphocytic infiltrate and no evidence of vasculitis.11 Medium to large vessels in the deep dermis and subcutaneous tissue show infiltration of their walls with a large number of macrophages laden with acid-fast bacilli.11 Cases with histopathologic features mimicking antiphospholipid syndrome with endothelial cell proliferation, thrombosis, and mild mononuclear cell infiltrate also may be seen.20 In all cases, ischemic epidermal necrosis is seen, as well as acid-fast bacilli, both singly and in clusters (globi) within endothelial cells and inside blood vessel lumina.
Although Lucio phenomenon initially was thought to be immune-complex mediated like ENL, it has been suggested that the main trigger is thrombotic vascular occlusion secondary to massive invasion of the vascular endothelial cells by the lepra bacilli, resulting in necrosis.14 Bacterial lipopolysaccharides promote the release of IL-1 and tumor necrosis factor α, which in turn stimulate the production of prostaglandins, IL-6, and coagulation factor III, leading to vascular thrombosis and tissue necrosis.21,22 Moreover, antiphospholipid antibodies, which have been found to be induced in response to certain infectious agents in genetically predisposed individuals,23 have been reported in patients with leprosy, mainly in association with lepromatous leprosy. The reported prevalence of anticardiolipin antibodies ranged from 37% to 98%, whereas anti-β2-glycoprotein I antibodies ranged from 3% to 19%, and antiprothrombin antibodies ranged from 6% to 45%.24,25 Antiphospholipid antibodies have been reported to play a role in the pathogenesis of Lucio phenomenon.11,13,15,26 Our case supports this hypothesis with positive anticardiolipin antibodies, anti-β2 glycoprotein antibodies, and positive lupus anticoagulant.
In accordance with Curi et al,2 who reported 5 cases of DLL with Lucio phenomenon, our patient showed a similar presentation with positive inflammatory markers in association with a negative autoimmune profile (ANA, ANCA-C&P) and negative venereal disease research laboratory test. It is important to mention that a positive autoimmune profile (ANA, ANCA-C&P) can be present in leprotic patients, causing possible diagnostic confusion with collagen diseases.27,28
An interesting finding in our case was the negative slit-skin smear results. Although the specificity of slit-skin smear is 100%, as it directly demonstrates the presence of acid-fast bacilli,29 its sensitivity is low and varies from 10% to 50%.30 The detection of acid-fast bacilli in tissue sections is reported to be a better method for confirming the diagnosis of leprosy.31
The provisional impression of hemophagocytic lymphohistiocytosis in the lymph node biopsy in our patient was excluded upon detection of acid-fast bacilli in the foamy histiocytes infiltrating the lymph node; moreover, the normal serum lipids and serum ferritin argued against this diagnosis.32 Leprosy tends to involve the lymph nodes, particularly in borderline, borderline lepromatous, and lepromatous forms.33 The incidence of lymph node involvement accompanied by skin lesions with the presence of acid-fast bacilli in the lymph nodes is 92.2%.34
Our patient showed an excellent response to antileprotic treatment, which was administered according to the WHO multidrug therapy guidelines for multibacillary leprosy,35 combined with low-dose prednisolone, acetylsalicylic acid, and anticoagulant treatment. Thalidomide and high-dose prednisolone (60 mg/d) combined with antileprotic treatment also have been reported to be successful in managing recurrent infarctions in leprosy.36 The Fournier-like gangrenous ulcer of the scrotum was managed by surgical debridement and vacuum therapy.
It is noteworthy that the WHO elimination goal for leprosy was to reduce the prevalence to less than 1 case per 10,000 population. Egypt is among the first countries in North Africa and the Middle East regions to achieve this target supervised by the National Leprosy Control Program as early as 1994; this was further reduced to 0.33 cases per 10,000 population in 2004, and reduced again in 2009; however, certain foci showed a prevalence rate more than the elimination target, particularly in the cities of Qena (1.12) and Sohag (2.47).37 Esna, where our patient is from, is an endemic area in Egypt.38
Conclusion
1. World Health Organization. World Health Statistics: 2011. World Health Organization; 2011. https://www.who.int/gho/publications/world_health_statistics/EN_WHS2011_Full.pdf
2. Curi PF, Villaroel JS, Migliore N, et al. Lucio’s phenomenon: report of five cases. Clin Rheumatol. 2016;35:1397-1401.
3. Shrestha B, Li YQ, Fu P. Leprosy mimics adult onset Still’s disease in a Chinese patient. Egypt Rheumatol. 2018;40:217-220.
4. Prasad S, Misra R, Aggarwal A, et al. Leprosy revealed in a rheumatology clinic: a case series. Int J Rheum Dis. 2013;16:129-133.
5. Chao G, Fang L, Lu C. Leprosy with ANA positive mistaken for connective tissue disease. Clin Rheumatol. 2013;32:645-648.
6. Chauhan S, Wakhlu A, Agarwal V. Arthritis in leprosy. Rheumatology. 2010;49:2237-2242.
7. Rath D, Bhargava S, Kundu BK. Leprosy mimicking common rheumatologic entities: a trial for the clinician in the era of biologics. Case Rep Rheumatol. 2014;2014:429698.
8. Cuevas J, Rodríguez-Peralto JL, Carrillo R, et al. Erythema nodosum leprosum: reactional leprosy. Semin Cutan Med Surg. 2007;26:126-130.
9. Henriques CC, Lopéz B, Mestre T, et al. Leprosy and rheumatoid arthritis: consequence or association? BMJ Case Rep. 2012;13:1-4.
10. Vázquez-Botet M, Sánchez JL. Erythema nodosum leprosum. Int J Dermatol. 1987;26:436-437.
11. Nunzie E, Ortega Cabrera LV, Macanchi Moncayo FM, et al. Lucio leprosy with Lucio’s phenomenon, digital gangrene and anticardiolipin antibodies. Lepr Rev. 2014;85:194-200.
12. Salvi S, Chopra A. Leprosy in a rheumatology setting: a challenging mimic to expose. Clin Rheumatol. 2013;32:1557-1563.
13. Azulay-Abulafia L, Pereira SL, Hardmann D, et al. Lucio phenomenon. vasculitis or occlusive vasculopathy? Hautarzt. 2006;57:1101-1105.
14. Benard G, Sakai-Valente NY, Bianconcini Trindade MA. Concomittant Lucio phenomenon and erythema nodosum in a leprosy patient: clues for their distinct pathogenesis. Am J Dermatopathol. 2009;31:288-292.
15. Rocha RH, Emerich PS, Diniz LM, et al. Lucio’s phenomenon: exuberant case report and review of Brazilian cases. An Bras Dermatol. 2016;91(suppl 5):S60-S63.
16. Costa IM, Kawano LB, Pereira CP, et al. Lucio’s phenomenon: a case report and review of the literature. Int J Dermatol. 2005;44:566-571.
17. Kumari R, Thappa DM, Basu D. A fatal case of Lucio phenomenon from India. Dermatol Online J. 2008;14:10.
18. Lucio R, Alvarado I. Opúsculo Sobre el Mal de San Lázaro o Elefantiasis de los Griegos. M. Murguía; 1852.
19. Latapí F, Chévez-Zamora A. The “spotted” leprosy of Lucio: an introduction to its clinical and histological study. Int J Lepr. 1948;16:421-437.
20. Vargas OF. Diffuse leprosy of Lucio and Latapí: a histologic study. Lepr Rev. 2007;78:248-260.
21. Latapí FR, Chevez-Zamora A. La lepra manchada de Lucio. Rev Dermatol Mex. 1978;22:102-107.
22. Monteiro R, Abreu MA, Tiezzi MG, et al. Fenômeno de Lúcio: mais um caso relatado no Brasil. An Bras Dermatol. 2012;87:296-300.
23. Gharavi EE, Chaimovich H, Cucucrull E, et al. Induction of antiphospholipid antibodies by immunization with synthetic bacterial & viral peptides. Lupus. 1999;8:449-455.
24. de Larrañaga GF, Forastiero RR, Martinuzzo ME, et al. High prevalence of antiphospholipid antibodies in leprosy: evaluation of antigen reactivity. Lupus. 2000;9:594-600.
25. Loizou S, Singh S, Wypkema E, et al. Anticardiolipin, anti-beta(2)-glycoprotein I and antiprothrombin antibodies in black South African patients with infectious disease. Ann Rheum Dis. 2003;62:1106-1111.
26. Akerkar SM, Bichile LS. Leprosy & gangrene: a rare association; role of antiphospholipid antibodies. BMC Infect Dis. 2005,5:74.
27. Horta-Baas G, Hernández-Cabrera MF, Barile-Fabris LA, et al. Multibacillary leprosy mimicking systemic lupus erythematosus: case report and literature review. Lupus. 2015;24:1095-1102.
28. Pradhan V, Badakere SS, Shankar KU. Increased incidence of cytoplasmic ANCA (cANCA) and other auto antibodies in leprosy patients from western India. Lepr Rev. 2004;75:50-56.
29. Oskam L. Diagnosis and classification of leprosy. Lepr Rev. 2002;73:17-26.
30. Rao PN. Recent advances in the control programs and therapy of leprosy. Indian J Dermatol Venereol Leprol. 2004;70:269-276.
31. Rao PN, Pratap D, Ramana Reddy AV, et al. Evaluation of leprosy patients with 1 to 5 skin lesions with relevance to their grouping into paucibacillary or multibacillary disease. Indian J Dermatol Venereol Leprol. 2006;72:207-210.
32. Rosado FGN, Kim AS. Hemophagocytic lymphohistiocytosis. an update on diagnosis and pathogenesis. Am J Clin Pathol. 2013;139:713-727.
33. Kar HK, Mohanty HC, Mohanty GN, et al. Clinicopathological study of lymph node involvement in leprosy. Lepr India. 1983;55:725-738.
34. Gupta JC, Panda PK, Shrivastava KK, et al. A histopathologic study of lymph nodes in 43 cases of leprosy. Lepr India. 1978;50:196-203.
35. WHO Expert Committee on Leprosy. Seventh Report. World Health Organization; 1998. https://apps.who.int/iris/bitstream/handle/10665/42060/WHO_TRS_874.pdf?sequence=1&isAllowed=y
36. Misra DP, Parida JR, Chowdhury AC, et al. Lepra reaction with Lucio phenomenon mimicking cutaneous vasculitis. Case Rep Immunol. 2014;2014:641989.
37. Amer A, Mansour A. Epidemiological study of leprosy in Egypt: 2005-2009. Egypt J Dermatol Venereol. 2014;34:70-73.
38. World Health Organization. Screening campaign aims to eliminate leprosy in Egypt. Published May 9, 2018. Accessed September 8, 2021. http://www.emro.who.int/egy/egypt-events/last-miless-activities-on-eliminating-leprosy-from-egypt.html
Case Report
A 70-year-old man living in Esna, Luxor, Egypt presented to the Department of Rheumatology and Rehabilitation with widespread gangrenous skin lesions associated with ulcers of 2 weeks’ duration. One year prior, the patient had an insidious onset of nocturnal fever, bilateral leg edema, and numbness and a tingling sensation in both hands. He presented some laboratory and radiologic investigations that were performed at another hospital prior to the current presentation, which revealed thrombocytopenia, mild splenomegaly, and generalized lymphadenopathy. An excisional left axillary lymph node biopsy was performed at another hospital prior to the current presentation, and the pathology report provided by the patient described a reactive, foamy, histiocyte-rich lesion, suggesting a diagnosis of hemophagocytic lymphohistiocytosis. The patient had no diabetes or hypertension and no history of deep vein thrombosis, stroke, or unintentional weight loss. No medications were taken prior to the onset of the skin lesions, and his family history was irrelevant.
General examination at the current presentation revealed a fever (temperature, 101.3 °F [38.5 °C]), a normal heart rate (90 beats per minute), normal blood pressure (120/80 mmHg), normal respiratory rate (14 breaths per minute), accentuated heart sounds, and normal vesicular breathing without adventitious sounds. He had saddle nose, loss of the outer third of the eyebrows, and marked reduction in the density of the eyelashes (madarosis). Bilateral pitting edema of the legs also was present. Neurologic examination revealed hypoesthesia in a glove-and-stocking pattern, thickened peripheral nerves, and trophic changes over both hands; however, he had normal muscle power and deep reflexes. Joint examination revealed no abnormalities. Skin examination revealed widespread, reticulated, necrotizing, purpuric lesions on the arms, legs, abdomen, and ears, some associated with gangrenous ulcerations and hemorrhagic blisters. Scattered vasculitic ulcers and gangrenous patches were seen on the fingers. A gangrenous ulcer mimicking Fournier gangrene was seen involving the scrotal skin in addition to a gangrenous lesion on the glans penis (Figure 1–3). Unaffected skin appeared smooth, shiny, and edematous and showed no nodular lesions. Peripheral pulsations were intact.
Positive findings from a wide panel of laboratory investigations included an elevated erythrocyte sedimentation rate (103 mm for the first hour [reference range, 0–22 mm]), high C-reactive protein (50.7 mg/L [reference range, up to 6 mg/L]), anemia (hemoglobin count, 7.3 g/dL [reference range, 13.5–17.5 g/dL]), thrombocytopenia (45×103/mm3 [reference range, 150×103/mm3), low serum albumin (2.3 g/dL [reference range, 3.4–5.4 g/dL]), elevated IgG and IgM anticardiolipin antibodies (IgG, 21.4 IgG phospholipid [GPL] units [reference range, <10 IgG phospholipid (GPL) units]; IgM, 59.4 IgM phospholipid (MPL) units [reference range, <7 IgM phospholipid (MPL) units]), positive lupus anticoagulant panel test, elevated anti-β2 glycoprotein antibodies (IgG, 17.5
Nerve conduction velocity showed axonal sensory polyneuropathy. Motor nerve conduction studies for median and ulnar nerves were within normal range. Lower-limb nerves assessment was limited by the ulcerated areas and marked edema. Echocardiography was unremarkable. Arterial Doppler studies were only available for the upper limbs and were unremarkable.
A punch biopsy was taken from one of the necrotizing purpuric lesions on the legs, and histopathologic examination revealed foci of epidermal necrosis and subepidermal separation and superficial and deep perivascular and periadnexal infiltrates extending into the fat lobules. The infiltrates were mainly made up of foamy macrophages, and some contained globi (lepra cells), in addition to lymphocytes and many neutrophils with nuclear dust. Blood vessels in the superficial and deep dermis and in the subcutaneous fat showed fibrinoid necrosis in their walls with neutrophils infiltrating the walls and thrombi in the lumens (Figure 4). Modified Ziehl-Neelsen staining revealed clumps of acid-fast lepra bacilli inside vascular lumina and endothelial cell lining and within the foamy macrophages (Figure 5). Slit-skin smear examination was performed twice and yielded negative results. The slide and paraffin block of the already performed lymph node biopsy were retrieved. Examination revealed aggregates of foamy histiocytes surrounded by lymphocytes and plasma cells replacing normal lymphoid follicles. Modified Ziehl-Neelsen stain was performed, and clusters of acid-fast bacilli were detected within the foamy histiocytic infiltrate (Figure 6).
According to the results of the skin biopsy, the revised result of the lymph node biopsy, and the pattern of neurologic deficit together with clinical and laboratory correlation, the patient was diagnosed with diffuse nonnodular lepromatous leprosy presenting with Lucio phenomenon (Lucio leprosy) and associated with lepromatous lymphadenitis.
The patient received the following treatment: methylprednisolone 500 mg (intravenous pulse therapy) followed by daily oral administration of prednisolone 10 mg, rifampicin 300 mg, dapsone 100 mg, clofazimine 100 mg, acetylsalicylic acid 150 mg, and enoxaparin sodium 80 mg. In addition, the scrotal Fournier gangrene–like lesion was treated by surgical debridement followed by vacuum therapy. By the second week after treatment, the gangrenous lesions of the fingers developed a line of demarcation, and the skin infarctions started to recede.
Comment
Despite a decrease in its prevalence through a World Health Organization (WHO)–empowered eradication program, leprosy still represents a health problem in endemic areas.1,2 It is characterized by a wide range of immune responses to Mycobacterium leprae, displaying a spectrum of clinical and histopathologic manifestations that vary from the tuberculoid or paucibacillary pole with a strong cell-mediated immune response and fewer organisms to the lepromatous or multibacillary pole with weaker cell-mediated immune response and higher loads of organisms.3 In addition to its well-known cutaneous and neurologic manifestations, leprosy can present with a variety of manifestations, including constitutional symptoms, musculoskeletal manifestations, and serologic abnormalities; thus, leprosy can mimic rheumatoid arthritis, spondyloarthritis, and vasculitis—a pitfall that may result in misdiagnosis as a rheumatologic disorder.3-7
The chronic course of leprosy can be disrupted by acute, immunologically mediated reactions known as lepra reactions, of which there are 3 types.8 Type I lepra reactions are cell mediated and occur mainly in patients with borderline disease, often representing an upgrade toward the tuberculoid pole; less often they represent a downgrade reaction. Nerves become painful and swollen with possible loss of function, and skin lesions become edematous and tender; sometimes arthritis develops.9 Type II lepra reactions, also known as erythema nodosum leprosum (ENL), occur in borderline lepromatous and lepromatous patients with a high bacillary load. They are characterized by fever, body aches, tender cutaneous/subcutaneous nodules that may ulcerate, possible bullous lesions, painful nerve swellings, swollen joints, iritis, lymphadenitis, glomerulonephritis, epididymo-orchitis, and hepatic affection. Both immune-complex and delayed hypersensitivity reactions play a role in ENL.8,10 The third reaction is a rare aggressive type known as Lucio phenomenon or Lucio leprosy, which presents with irregular-shaped, angulated, or stellar necrotizing purpuric lesions (hemorrhagic infacrtions) developing mainly on the extremities. The lesions evolve into ulcers that heal with atrophic scarring.2,11 Lucio phenomenon develops as a result of thrombotic vascular occlusion secondary to massive invasion of vascular endothelial cells by lepra bacilli.2,11-14 Involvement of the scrotal skin, such as in our patient, is rare.
Lucio phenomenon mainly is seen in Mexico and Central America, and few cases have been documented in Cuba, South America, the United States, India, Polynesia, South Africa, and Southeast Asia.15-17 It specifically occurs in patients with untreated, diffuse, nonnodular lepromatous leprosy (pure and primitive diffuse lepromatous leprosy (DLL)/diffuse leprosy of Lucio and Latapí). This type of leprosy was first described by Lucio and Alvarado18 in 1852 as a distinct form of lepromatous leprosy characterized by widespread and dense infiltration of the whole skin by lepra bacilli without the typical nodular lesions of leprosy, rendering its diagnosis challenging, especially in sporadic cases. Other manifestations of DLL include complete alopecia of the eyebrows and eyelashes, destructive rhinitis, and areas of anhidrosis and dyesthesia.2
Latapí and Chévez-Zomora19 defined Lucio phenomenon in 1948 as a form of histopathologic vasculitis restricted to patients with DLL. Histopathologically, in addition to the infiltration of the skin with acid-fast bacilli–laden foamy histiocytes, lesions of Lucio phenomenon show features of necrotizing (leukocytoclastic) vasculitis with fibrinoid necrosis20 or vascular thrombi with minimal perivascular lymphocytic infiltrate and no evidence of vasculitis.11 Medium to large vessels in the deep dermis and subcutaneous tissue show infiltration of their walls with a large number of macrophages laden with acid-fast bacilli.11 Cases with histopathologic features mimicking antiphospholipid syndrome with endothelial cell proliferation, thrombosis, and mild mononuclear cell infiltrate also may be seen.20 In all cases, ischemic epidermal necrosis is seen, as well as acid-fast bacilli, both singly and in clusters (globi) within endothelial cells and inside blood vessel lumina.
Although Lucio phenomenon initially was thought to be immune-complex mediated like ENL, it has been suggested that the main trigger is thrombotic vascular occlusion secondary to massive invasion of the vascular endothelial cells by the lepra bacilli, resulting in necrosis.14 Bacterial lipopolysaccharides promote the release of IL-1 and tumor necrosis factor α, which in turn stimulate the production of prostaglandins, IL-6, and coagulation factor III, leading to vascular thrombosis and tissue necrosis.21,22 Moreover, antiphospholipid antibodies, which have been found to be induced in response to certain infectious agents in genetically predisposed individuals,23 have been reported in patients with leprosy, mainly in association with lepromatous leprosy. The reported prevalence of anticardiolipin antibodies ranged from 37% to 98%, whereas anti-β2-glycoprotein I antibodies ranged from 3% to 19%, and antiprothrombin antibodies ranged from 6% to 45%.24,25 Antiphospholipid antibodies have been reported to play a role in the pathogenesis of Lucio phenomenon.11,13,15,26 Our case supports this hypothesis with positive anticardiolipin antibodies, anti-β2 glycoprotein antibodies, and positive lupus anticoagulant.
In accordance with Curi et al,2 who reported 5 cases of DLL with Lucio phenomenon, our patient showed a similar presentation with positive inflammatory markers in association with a negative autoimmune profile (ANA, ANCA-C&P) and negative venereal disease research laboratory test. It is important to mention that a positive autoimmune profile (ANA, ANCA-C&P) can be present in leprotic patients, causing possible diagnostic confusion with collagen diseases.27,28
An interesting finding in our case was the negative slit-skin smear results. Although the specificity of slit-skin smear is 100%, as it directly demonstrates the presence of acid-fast bacilli,29 its sensitivity is low and varies from 10% to 50%.30 The detection of acid-fast bacilli in tissue sections is reported to be a better method for confirming the diagnosis of leprosy.31
The provisional impression of hemophagocytic lymphohistiocytosis in the lymph node biopsy in our patient was excluded upon detection of acid-fast bacilli in the foamy histiocytes infiltrating the lymph node; moreover, the normal serum lipids and serum ferritin argued against this diagnosis.32 Leprosy tends to involve the lymph nodes, particularly in borderline, borderline lepromatous, and lepromatous forms.33 The incidence of lymph node involvement accompanied by skin lesions with the presence of acid-fast bacilli in the lymph nodes is 92.2%.34
Our patient showed an excellent response to antileprotic treatment, which was administered according to the WHO multidrug therapy guidelines for multibacillary leprosy,35 combined with low-dose prednisolone, acetylsalicylic acid, and anticoagulant treatment. Thalidomide and high-dose prednisolone (60 mg/d) combined with antileprotic treatment also have been reported to be successful in managing recurrent infarctions in leprosy.36 The Fournier-like gangrenous ulcer of the scrotum was managed by surgical debridement and vacuum therapy.
It is noteworthy that the WHO elimination goal for leprosy was to reduce the prevalence to less than 1 case per 10,000 population. Egypt is among the first countries in North Africa and the Middle East regions to achieve this target supervised by the National Leprosy Control Program as early as 1994; this was further reduced to 0.33 cases per 10,000 population in 2004, and reduced again in 2009; however, certain foci showed a prevalence rate more than the elimination target, particularly in the cities of Qena (1.12) and Sohag (2.47).37 Esna, where our patient is from, is an endemic area in Egypt.38
Conclusion
Case Report
A 70-year-old man living in Esna, Luxor, Egypt presented to the Department of Rheumatology and Rehabilitation with widespread gangrenous skin lesions associated with ulcers of 2 weeks’ duration. One year prior, the patient had an insidious onset of nocturnal fever, bilateral leg edema, and numbness and a tingling sensation in both hands. He presented some laboratory and radiologic investigations that were performed at another hospital prior to the current presentation, which revealed thrombocytopenia, mild splenomegaly, and generalized lymphadenopathy. An excisional left axillary lymph node biopsy was performed at another hospital prior to the current presentation, and the pathology report provided by the patient described a reactive, foamy, histiocyte-rich lesion, suggesting a diagnosis of hemophagocytic lymphohistiocytosis. The patient had no diabetes or hypertension and no history of deep vein thrombosis, stroke, or unintentional weight loss. No medications were taken prior to the onset of the skin lesions, and his family history was irrelevant.
General examination at the current presentation revealed a fever (temperature, 101.3 °F [38.5 °C]), a normal heart rate (90 beats per minute), normal blood pressure (120/80 mmHg), normal respiratory rate (14 breaths per minute), accentuated heart sounds, and normal vesicular breathing without adventitious sounds. He had saddle nose, loss of the outer third of the eyebrows, and marked reduction in the density of the eyelashes (madarosis). Bilateral pitting edema of the legs also was present. Neurologic examination revealed hypoesthesia in a glove-and-stocking pattern, thickened peripheral nerves, and trophic changes over both hands; however, he had normal muscle power and deep reflexes. Joint examination revealed no abnormalities. Skin examination revealed widespread, reticulated, necrotizing, purpuric lesions on the arms, legs, abdomen, and ears, some associated with gangrenous ulcerations and hemorrhagic blisters. Scattered vasculitic ulcers and gangrenous patches were seen on the fingers. A gangrenous ulcer mimicking Fournier gangrene was seen involving the scrotal skin in addition to a gangrenous lesion on the glans penis (Figure 1–3). Unaffected skin appeared smooth, shiny, and edematous and showed no nodular lesions. Peripheral pulsations were intact.
Positive findings from a wide panel of laboratory investigations included an elevated erythrocyte sedimentation rate (103 mm for the first hour [reference range, 0–22 mm]), high C-reactive protein (50.7 mg/L [reference range, up to 6 mg/L]), anemia (hemoglobin count, 7.3 g/dL [reference range, 13.5–17.5 g/dL]), thrombocytopenia (45×103/mm3 [reference range, 150×103/mm3), low serum albumin (2.3 g/dL [reference range, 3.4–5.4 g/dL]), elevated IgG and IgM anticardiolipin antibodies (IgG, 21.4 IgG phospholipid [GPL] units [reference range, <10 IgG phospholipid (GPL) units]; IgM, 59.4 IgM phospholipid (MPL) units [reference range, <7 IgM phospholipid (MPL) units]), positive lupus anticoagulant panel test, elevated anti-β2 glycoprotein antibodies (IgG, 17.5
Nerve conduction velocity showed axonal sensory polyneuropathy. Motor nerve conduction studies for median and ulnar nerves were within normal range. Lower-limb nerves assessment was limited by the ulcerated areas and marked edema. Echocardiography was unremarkable. Arterial Doppler studies were only available for the upper limbs and were unremarkable.
A punch biopsy was taken from one of the necrotizing purpuric lesions on the legs, and histopathologic examination revealed foci of epidermal necrosis and subepidermal separation and superficial and deep perivascular and periadnexal infiltrates extending into the fat lobules. The infiltrates were mainly made up of foamy macrophages, and some contained globi (lepra cells), in addition to lymphocytes and many neutrophils with nuclear dust. Blood vessels in the superficial and deep dermis and in the subcutaneous fat showed fibrinoid necrosis in their walls with neutrophils infiltrating the walls and thrombi in the lumens (Figure 4). Modified Ziehl-Neelsen staining revealed clumps of acid-fast lepra bacilli inside vascular lumina and endothelial cell lining and within the foamy macrophages (Figure 5). Slit-skin smear examination was performed twice and yielded negative results. The slide and paraffin block of the already performed lymph node biopsy were retrieved. Examination revealed aggregates of foamy histiocytes surrounded by lymphocytes and plasma cells replacing normal lymphoid follicles. Modified Ziehl-Neelsen stain was performed, and clusters of acid-fast bacilli were detected within the foamy histiocytic infiltrate (Figure 6).
According to the results of the skin biopsy, the revised result of the lymph node biopsy, and the pattern of neurologic deficit together with clinical and laboratory correlation, the patient was diagnosed with diffuse nonnodular lepromatous leprosy presenting with Lucio phenomenon (Lucio leprosy) and associated with lepromatous lymphadenitis.
The patient received the following treatment: methylprednisolone 500 mg (intravenous pulse therapy) followed by daily oral administration of prednisolone 10 mg, rifampicin 300 mg, dapsone 100 mg, clofazimine 100 mg, acetylsalicylic acid 150 mg, and enoxaparin sodium 80 mg. In addition, the scrotal Fournier gangrene–like lesion was treated by surgical debridement followed by vacuum therapy. By the second week after treatment, the gangrenous lesions of the fingers developed a line of demarcation, and the skin infarctions started to recede.
Comment
Despite a decrease in its prevalence through a World Health Organization (WHO)–empowered eradication program, leprosy still represents a health problem in endemic areas.1,2 It is characterized by a wide range of immune responses to Mycobacterium leprae, displaying a spectrum of clinical and histopathologic manifestations that vary from the tuberculoid or paucibacillary pole with a strong cell-mediated immune response and fewer organisms to the lepromatous or multibacillary pole with weaker cell-mediated immune response and higher loads of organisms.3 In addition to its well-known cutaneous and neurologic manifestations, leprosy can present with a variety of manifestations, including constitutional symptoms, musculoskeletal manifestations, and serologic abnormalities; thus, leprosy can mimic rheumatoid arthritis, spondyloarthritis, and vasculitis—a pitfall that may result in misdiagnosis as a rheumatologic disorder.3-7
The chronic course of leprosy can be disrupted by acute, immunologically mediated reactions known as lepra reactions, of which there are 3 types.8 Type I lepra reactions are cell mediated and occur mainly in patients with borderline disease, often representing an upgrade toward the tuberculoid pole; less often they represent a downgrade reaction. Nerves become painful and swollen with possible loss of function, and skin lesions become edematous and tender; sometimes arthritis develops.9 Type II lepra reactions, also known as erythema nodosum leprosum (ENL), occur in borderline lepromatous and lepromatous patients with a high bacillary load. They are characterized by fever, body aches, tender cutaneous/subcutaneous nodules that may ulcerate, possible bullous lesions, painful nerve swellings, swollen joints, iritis, lymphadenitis, glomerulonephritis, epididymo-orchitis, and hepatic affection. Both immune-complex and delayed hypersensitivity reactions play a role in ENL.8,10 The third reaction is a rare aggressive type known as Lucio phenomenon or Lucio leprosy, which presents with irregular-shaped, angulated, or stellar necrotizing purpuric lesions (hemorrhagic infacrtions) developing mainly on the extremities. The lesions evolve into ulcers that heal with atrophic scarring.2,11 Lucio phenomenon develops as a result of thrombotic vascular occlusion secondary to massive invasion of vascular endothelial cells by lepra bacilli.2,11-14 Involvement of the scrotal skin, such as in our patient, is rare.
Lucio phenomenon mainly is seen in Mexico and Central America, and few cases have been documented in Cuba, South America, the United States, India, Polynesia, South Africa, and Southeast Asia.15-17 It specifically occurs in patients with untreated, diffuse, nonnodular lepromatous leprosy (pure and primitive diffuse lepromatous leprosy (DLL)/diffuse leprosy of Lucio and Latapí). This type of leprosy was first described by Lucio and Alvarado18 in 1852 as a distinct form of lepromatous leprosy characterized by widespread and dense infiltration of the whole skin by lepra bacilli without the typical nodular lesions of leprosy, rendering its diagnosis challenging, especially in sporadic cases. Other manifestations of DLL include complete alopecia of the eyebrows and eyelashes, destructive rhinitis, and areas of anhidrosis and dyesthesia.2
Latapí and Chévez-Zomora19 defined Lucio phenomenon in 1948 as a form of histopathologic vasculitis restricted to patients with DLL. Histopathologically, in addition to the infiltration of the skin with acid-fast bacilli–laden foamy histiocytes, lesions of Lucio phenomenon show features of necrotizing (leukocytoclastic) vasculitis with fibrinoid necrosis20 or vascular thrombi with minimal perivascular lymphocytic infiltrate and no evidence of vasculitis.11 Medium to large vessels in the deep dermis and subcutaneous tissue show infiltration of their walls with a large number of macrophages laden with acid-fast bacilli.11 Cases with histopathologic features mimicking antiphospholipid syndrome with endothelial cell proliferation, thrombosis, and mild mononuclear cell infiltrate also may be seen.20 In all cases, ischemic epidermal necrosis is seen, as well as acid-fast bacilli, both singly and in clusters (globi) within endothelial cells and inside blood vessel lumina.
Although Lucio phenomenon initially was thought to be immune-complex mediated like ENL, it has been suggested that the main trigger is thrombotic vascular occlusion secondary to massive invasion of the vascular endothelial cells by the lepra bacilli, resulting in necrosis.14 Bacterial lipopolysaccharides promote the release of IL-1 and tumor necrosis factor α, which in turn stimulate the production of prostaglandins, IL-6, and coagulation factor III, leading to vascular thrombosis and tissue necrosis.21,22 Moreover, antiphospholipid antibodies, which have been found to be induced in response to certain infectious agents in genetically predisposed individuals,23 have been reported in patients with leprosy, mainly in association with lepromatous leprosy. The reported prevalence of anticardiolipin antibodies ranged from 37% to 98%, whereas anti-β2-glycoprotein I antibodies ranged from 3% to 19%, and antiprothrombin antibodies ranged from 6% to 45%.24,25 Antiphospholipid antibodies have been reported to play a role in the pathogenesis of Lucio phenomenon.11,13,15,26 Our case supports this hypothesis with positive anticardiolipin antibodies, anti-β2 glycoprotein antibodies, and positive lupus anticoagulant.
In accordance with Curi et al,2 who reported 5 cases of DLL with Lucio phenomenon, our patient showed a similar presentation with positive inflammatory markers in association with a negative autoimmune profile (ANA, ANCA-C&P) and negative venereal disease research laboratory test. It is important to mention that a positive autoimmune profile (ANA, ANCA-C&P) can be present in leprotic patients, causing possible diagnostic confusion with collagen diseases.27,28
An interesting finding in our case was the negative slit-skin smear results. Although the specificity of slit-skin smear is 100%, as it directly demonstrates the presence of acid-fast bacilli,29 its sensitivity is low and varies from 10% to 50%.30 The detection of acid-fast bacilli in tissue sections is reported to be a better method for confirming the diagnosis of leprosy.31
The provisional impression of hemophagocytic lymphohistiocytosis in the lymph node biopsy in our patient was excluded upon detection of acid-fast bacilli in the foamy histiocytes infiltrating the lymph node; moreover, the normal serum lipids and serum ferritin argued against this diagnosis.32 Leprosy tends to involve the lymph nodes, particularly in borderline, borderline lepromatous, and lepromatous forms.33 The incidence of lymph node involvement accompanied by skin lesions with the presence of acid-fast bacilli in the lymph nodes is 92.2%.34
Our patient showed an excellent response to antileprotic treatment, which was administered according to the WHO multidrug therapy guidelines for multibacillary leprosy,35 combined with low-dose prednisolone, acetylsalicylic acid, and anticoagulant treatment. Thalidomide and high-dose prednisolone (60 mg/d) combined with antileprotic treatment also have been reported to be successful in managing recurrent infarctions in leprosy.36 The Fournier-like gangrenous ulcer of the scrotum was managed by surgical debridement and vacuum therapy.
It is noteworthy that the WHO elimination goal for leprosy was to reduce the prevalence to less than 1 case per 10,000 population. Egypt is among the first countries in North Africa and the Middle East regions to achieve this target supervised by the National Leprosy Control Program as early as 1994; this was further reduced to 0.33 cases per 10,000 population in 2004, and reduced again in 2009; however, certain foci showed a prevalence rate more than the elimination target, particularly in the cities of Qena (1.12) and Sohag (2.47).37 Esna, where our patient is from, is an endemic area in Egypt.38
Conclusion
1. World Health Organization. World Health Statistics: 2011. World Health Organization; 2011. https://www.who.int/gho/publications/world_health_statistics/EN_WHS2011_Full.pdf
2. Curi PF, Villaroel JS, Migliore N, et al. Lucio’s phenomenon: report of five cases. Clin Rheumatol. 2016;35:1397-1401.
3. Shrestha B, Li YQ, Fu P. Leprosy mimics adult onset Still’s disease in a Chinese patient. Egypt Rheumatol. 2018;40:217-220.
4. Prasad S, Misra R, Aggarwal A, et al. Leprosy revealed in a rheumatology clinic: a case series. Int J Rheum Dis. 2013;16:129-133.
5. Chao G, Fang L, Lu C. Leprosy with ANA positive mistaken for connective tissue disease. Clin Rheumatol. 2013;32:645-648.
6. Chauhan S, Wakhlu A, Agarwal V. Arthritis in leprosy. Rheumatology. 2010;49:2237-2242.
7. Rath D, Bhargava S, Kundu BK. Leprosy mimicking common rheumatologic entities: a trial for the clinician in the era of biologics. Case Rep Rheumatol. 2014;2014:429698.
8. Cuevas J, Rodríguez-Peralto JL, Carrillo R, et al. Erythema nodosum leprosum: reactional leprosy. Semin Cutan Med Surg. 2007;26:126-130.
9. Henriques CC, Lopéz B, Mestre T, et al. Leprosy and rheumatoid arthritis: consequence or association? BMJ Case Rep. 2012;13:1-4.
10. Vázquez-Botet M, Sánchez JL. Erythema nodosum leprosum. Int J Dermatol. 1987;26:436-437.
11. Nunzie E, Ortega Cabrera LV, Macanchi Moncayo FM, et al. Lucio leprosy with Lucio’s phenomenon, digital gangrene and anticardiolipin antibodies. Lepr Rev. 2014;85:194-200.
12. Salvi S, Chopra A. Leprosy in a rheumatology setting: a challenging mimic to expose. Clin Rheumatol. 2013;32:1557-1563.
13. Azulay-Abulafia L, Pereira SL, Hardmann D, et al. Lucio phenomenon. vasculitis or occlusive vasculopathy? Hautarzt. 2006;57:1101-1105.
14. Benard G, Sakai-Valente NY, Bianconcini Trindade MA. Concomittant Lucio phenomenon and erythema nodosum in a leprosy patient: clues for their distinct pathogenesis. Am J Dermatopathol. 2009;31:288-292.
15. Rocha RH, Emerich PS, Diniz LM, et al. Lucio’s phenomenon: exuberant case report and review of Brazilian cases. An Bras Dermatol. 2016;91(suppl 5):S60-S63.
16. Costa IM, Kawano LB, Pereira CP, et al. Lucio’s phenomenon: a case report and review of the literature. Int J Dermatol. 2005;44:566-571.
17. Kumari R, Thappa DM, Basu D. A fatal case of Lucio phenomenon from India. Dermatol Online J. 2008;14:10.
18. Lucio R, Alvarado I. Opúsculo Sobre el Mal de San Lázaro o Elefantiasis de los Griegos. M. Murguía; 1852.
19. Latapí F, Chévez-Zamora A. The “spotted” leprosy of Lucio: an introduction to its clinical and histological study. Int J Lepr. 1948;16:421-437.
20. Vargas OF. Diffuse leprosy of Lucio and Latapí: a histologic study. Lepr Rev. 2007;78:248-260.
21. Latapí FR, Chevez-Zamora A. La lepra manchada de Lucio. Rev Dermatol Mex. 1978;22:102-107.
22. Monteiro R, Abreu MA, Tiezzi MG, et al. Fenômeno de Lúcio: mais um caso relatado no Brasil. An Bras Dermatol. 2012;87:296-300.
23. Gharavi EE, Chaimovich H, Cucucrull E, et al. Induction of antiphospholipid antibodies by immunization with synthetic bacterial & viral peptides. Lupus. 1999;8:449-455.
24. de Larrañaga GF, Forastiero RR, Martinuzzo ME, et al. High prevalence of antiphospholipid antibodies in leprosy: evaluation of antigen reactivity. Lupus. 2000;9:594-600.
25. Loizou S, Singh S, Wypkema E, et al. Anticardiolipin, anti-beta(2)-glycoprotein I and antiprothrombin antibodies in black South African patients with infectious disease. Ann Rheum Dis. 2003;62:1106-1111.
26. Akerkar SM, Bichile LS. Leprosy & gangrene: a rare association; role of antiphospholipid antibodies. BMC Infect Dis. 2005,5:74.
27. Horta-Baas G, Hernández-Cabrera MF, Barile-Fabris LA, et al. Multibacillary leprosy mimicking systemic lupus erythematosus: case report and literature review. Lupus. 2015;24:1095-1102.
28. Pradhan V, Badakere SS, Shankar KU. Increased incidence of cytoplasmic ANCA (cANCA) and other auto antibodies in leprosy patients from western India. Lepr Rev. 2004;75:50-56.
29. Oskam L. Diagnosis and classification of leprosy. Lepr Rev. 2002;73:17-26.
30. Rao PN. Recent advances in the control programs and therapy of leprosy. Indian J Dermatol Venereol Leprol. 2004;70:269-276.
31. Rao PN, Pratap D, Ramana Reddy AV, et al. Evaluation of leprosy patients with 1 to 5 skin lesions with relevance to their grouping into paucibacillary or multibacillary disease. Indian J Dermatol Venereol Leprol. 2006;72:207-210.
32. Rosado FGN, Kim AS. Hemophagocytic lymphohistiocytosis. an update on diagnosis and pathogenesis. Am J Clin Pathol. 2013;139:713-727.
33. Kar HK, Mohanty HC, Mohanty GN, et al. Clinicopathological study of lymph node involvement in leprosy. Lepr India. 1983;55:725-738.
34. Gupta JC, Panda PK, Shrivastava KK, et al. A histopathologic study of lymph nodes in 43 cases of leprosy. Lepr India. 1978;50:196-203.
35. WHO Expert Committee on Leprosy. Seventh Report. World Health Organization; 1998. https://apps.who.int/iris/bitstream/handle/10665/42060/WHO_TRS_874.pdf?sequence=1&isAllowed=y
36. Misra DP, Parida JR, Chowdhury AC, et al. Lepra reaction with Lucio phenomenon mimicking cutaneous vasculitis. Case Rep Immunol. 2014;2014:641989.
37. Amer A, Mansour A. Epidemiological study of leprosy in Egypt: 2005-2009. Egypt J Dermatol Venereol. 2014;34:70-73.
38. World Health Organization. Screening campaign aims to eliminate leprosy in Egypt. Published May 9, 2018. Accessed September 8, 2021. http://www.emro.who.int/egy/egypt-events/last-miless-activities-on-eliminating-leprosy-from-egypt.html
1. World Health Organization. World Health Statistics: 2011. World Health Organization; 2011. https://www.who.int/gho/publications/world_health_statistics/EN_WHS2011_Full.pdf
2. Curi PF, Villaroel JS, Migliore N, et al. Lucio’s phenomenon: report of five cases. Clin Rheumatol. 2016;35:1397-1401.
3. Shrestha B, Li YQ, Fu P. Leprosy mimics adult onset Still’s disease in a Chinese patient. Egypt Rheumatol. 2018;40:217-220.
4. Prasad S, Misra R, Aggarwal A, et al. Leprosy revealed in a rheumatology clinic: a case series. Int J Rheum Dis. 2013;16:129-133.
5. Chao G, Fang L, Lu C. Leprosy with ANA positive mistaken for connective tissue disease. Clin Rheumatol. 2013;32:645-648.
6. Chauhan S, Wakhlu A, Agarwal V. Arthritis in leprosy. Rheumatology. 2010;49:2237-2242.
7. Rath D, Bhargava S, Kundu BK. Leprosy mimicking common rheumatologic entities: a trial for the clinician in the era of biologics. Case Rep Rheumatol. 2014;2014:429698.
8. Cuevas J, Rodríguez-Peralto JL, Carrillo R, et al. Erythema nodosum leprosum: reactional leprosy. Semin Cutan Med Surg. 2007;26:126-130.
9. Henriques CC, Lopéz B, Mestre T, et al. Leprosy and rheumatoid arthritis: consequence or association? BMJ Case Rep. 2012;13:1-4.
10. Vázquez-Botet M, Sánchez JL. Erythema nodosum leprosum. Int J Dermatol. 1987;26:436-437.
11. Nunzie E, Ortega Cabrera LV, Macanchi Moncayo FM, et al. Lucio leprosy with Lucio’s phenomenon, digital gangrene and anticardiolipin antibodies. Lepr Rev. 2014;85:194-200.
12. Salvi S, Chopra A. Leprosy in a rheumatology setting: a challenging mimic to expose. Clin Rheumatol. 2013;32:1557-1563.
13. Azulay-Abulafia L, Pereira SL, Hardmann D, et al. Lucio phenomenon. vasculitis or occlusive vasculopathy? Hautarzt. 2006;57:1101-1105.
14. Benard G, Sakai-Valente NY, Bianconcini Trindade MA. Concomittant Lucio phenomenon and erythema nodosum in a leprosy patient: clues for their distinct pathogenesis. Am J Dermatopathol. 2009;31:288-292.
15. Rocha RH, Emerich PS, Diniz LM, et al. Lucio’s phenomenon: exuberant case report and review of Brazilian cases. An Bras Dermatol. 2016;91(suppl 5):S60-S63.
16. Costa IM, Kawano LB, Pereira CP, et al. Lucio’s phenomenon: a case report and review of the literature. Int J Dermatol. 2005;44:566-571.
17. Kumari R, Thappa DM, Basu D. A fatal case of Lucio phenomenon from India. Dermatol Online J. 2008;14:10.
18. Lucio R, Alvarado I. Opúsculo Sobre el Mal de San Lázaro o Elefantiasis de los Griegos. M. Murguía; 1852.
19. Latapí F, Chévez-Zamora A. The “spotted” leprosy of Lucio: an introduction to its clinical and histological study. Int J Lepr. 1948;16:421-437.
20. Vargas OF. Diffuse leprosy of Lucio and Latapí: a histologic study. Lepr Rev. 2007;78:248-260.
21. Latapí FR, Chevez-Zamora A. La lepra manchada de Lucio. Rev Dermatol Mex. 1978;22:102-107.
22. Monteiro R, Abreu MA, Tiezzi MG, et al. Fenômeno de Lúcio: mais um caso relatado no Brasil. An Bras Dermatol. 2012;87:296-300.
23. Gharavi EE, Chaimovich H, Cucucrull E, et al. Induction of antiphospholipid antibodies by immunization with synthetic bacterial & viral peptides. Lupus. 1999;8:449-455.
24. de Larrañaga GF, Forastiero RR, Martinuzzo ME, et al. High prevalence of antiphospholipid antibodies in leprosy: evaluation of antigen reactivity. Lupus. 2000;9:594-600.
25. Loizou S, Singh S, Wypkema E, et al. Anticardiolipin, anti-beta(2)-glycoprotein I and antiprothrombin antibodies in black South African patients with infectious disease. Ann Rheum Dis. 2003;62:1106-1111.
26. Akerkar SM, Bichile LS. Leprosy & gangrene: a rare association; role of antiphospholipid antibodies. BMC Infect Dis. 2005,5:74.
27. Horta-Baas G, Hernández-Cabrera MF, Barile-Fabris LA, et al. Multibacillary leprosy mimicking systemic lupus erythematosus: case report and literature review. Lupus. 2015;24:1095-1102.
28. Pradhan V, Badakere SS, Shankar KU. Increased incidence of cytoplasmic ANCA (cANCA) and other auto antibodies in leprosy patients from western India. Lepr Rev. 2004;75:50-56.
29. Oskam L. Diagnosis and classification of leprosy. Lepr Rev. 2002;73:17-26.
30. Rao PN. Recent advances in the control programs and therapy of leprosy. Indian J Dermatol Venereol Leprol. 2004;70:269-276.
31. Rao PN, Pratap D, Ramana Reddy AV, et al. Evaluation of leprosy patients with 1 to 5 skin lesions with relevance to their grouping into paucibacillary or multibacillary disease. Indian J Dermatol Venereol Leprol. 2006;72:207-210.
32. Rosado FGN, Kim AS. Hemophagocytic lymphohistiocytosis. an update on diagnosis and pathogenesis. Am J Clin Pathol. 2013;139:713-727.
33. Kar HK, Mohanty HC, Mohanty GN, et al. Clinicopathological study of lymph node involvement in leprosy. Lepr India. 1983;55:725-738.
34. Gupta JC, Panda PK, Shrivastava KK, et al. A histopathologic study of lymph nodes in 43 cases of leprosy. Lepr India. 1978;50:196-203.
35. WHO Expert Committee on Leprosy. Seventh Report. World Health Organization; 1998. https://apps.who.int/iris/bitstream/handle/10665/42060/WHO_TRS_874.pdf?sequence=1&isAllowed=y
36. Misra DP, Parida JR, Chowdhury AC, et al. Lepra reaction with Lucio phenomenon mimicking cutaneous vasculitis. Case Rep Immunol. 2014;2014:641989.
37. Amer A, Mansour A. Epidemiological study of leprosy in Egypt: 2005-2009. Egypt J Dermatol Venereol. 2014;34:70-73.
38. World Health Organization. Screening campaign aims to eliminate leprosy in Egypt. Published May 9, 2018. Accessed September 8, 2021. http://www.emro.who.int/egy/egypt-events/last-miless-activities-on-eliminating-leprosy-from-egypt.html
Practice Points
- Leprosy is a great mimicker of many connective tissue diseases, including vasculitis.
- Antiphospholipid antibodies are involved in Lucio phenomenon.
- Prompt treatment is important in Lucio phenomenon to avoid morbidity and mortality.
A Fatal Case of Hemophagocytic Lymphohistiocytosis Secondary to Anti-MDA5–Positive Dermatomyositis
To the Editor:
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterized by bilateral, symmetrical, proximal muscle weakness and classic cutaneous manifestations.1 Patients with antibodies directed against melanoma differentiation–associated gene 5, MDA5, have a distinct presentation due to vasculopathy with more severe cutaneous ulcerations, palmar papules, alopecia, and an elevated risk of rapidly progressive interstitial lung disease.2 A ferritin level greater than 1600 ng/mL portends an increased risk for pulmonary disease and therefore can be of prognostic value.3 Further, patients with anti-MDA5 DM are at a lower risk of malignancy and are more likely to test negative for antinuclear antibodies in comparison to other patients with DM.2,4
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a potentially lethal condition whereby uncontrolled activation of histiocytes in the reticuloendothelial system causes hemophagocytosis and a hyperinflammatory state. Patients present with fever, splenomegaly, cytopenia, and hyperferritinemia.5 Autoimmune‐associated hemophagocytic syndrome (AAHS) describes HLH that develops in association with autoimmune conditions, most commonly systemic lupus erythematosus and adult-onset Still disease. Cases reported in association with DM exist but are few in number, and there is no standard-of-care treatment.6 We report a case of a woman with anti-MDA5 DM complicated by HLH and DM-associated liver injury.
A 50-year-old woman presented as a direct admit from the rheumatology clinic for diffuse muscle weakness of 8 months’ duration, 40-pound unintentional weight loss, pruritic rash, bilateral joint pains, dry eyes, dry mouth, and altered mental status. Four months prior, she presented to an outside hospital and was given a diagnosis of probable Sjögren syndrome and autoimmune hepatitis vs drug-induced liver injury. At that time, a workup was notable for antibodies against Sjögren syndrome–related antigen A, anti–smooth muscle antibodies, and transaminitis. Ultrasonography of the right upper quadrant revealed hepatic steatosis. The patient was started on oral prednisone and pilocarpine but had been off all medications for 1 month when she presented to our hospital.
On hospital admission, physical examination revealed a violaceous heliotrope rash; a v-sign on the chest; shawl sign; palmar papules with pits at the fingertips; and periungual erythema and ulcerations along the metacarpophalangeal joints, elbows, lateral feet, and upper eyelids (Figure 1). Laboratory workup showed the following results: white blood cell count, 4100/μL (reference range, 4000–11,000/μL); hemoglobin, 11.6 g/dL (reference range, 12–16 g/dL); platelet count, 100,000/μL (reference range, 150,000–450,000/μL); lactate dehydrogenase, 510 U/L (reference range, 80–225 U/L); alkaline phosphatase (ALP), 766 U/L (reference range, 30–120 U/L); alanine aminotransferase (ALT), 88 U/L (reference range, 10–40 U/L); aspartate aminotransferase (AST), 544 U/L (reference range, 10–40 U/L); total bilirubin, 4.2 mg/dL (reference range, 0.3–1.0 mg/dL); direct bilirubin, 3.7 mg/dL (reference range, 0.1–0.3 mg/dL); aldolase, 20.2 U/L (reference range, 1–7.5 U/L), creatine kinase, 180 U/L (reference range, 30–135 U/L); γ-glutamyltransferase (GGT), 2743 U/L (reference range, 8–40 U/L); high sensitivity C-reactive protein, 122.9 mg/L (low-risk reference range, <1.0 mg/L); triglycerides, 534 mg/dL (reference range, <150 mg/dL); ferritin, 3784 ng/mL (reference range, 24–307 ng/mL); antinuclear antibody, negative titer; antimitochondrial antibody, negative titer; soluble IL-2 receptor (CD25), 7000 U/mL (reference range, 189–846 U/mL); anti-Sjögren syndrome–related antigen A antibody, positive.
Magnetic resonance imaging of the shoulders showed diffuse soft-tissue edema. Computed tomography (CT) of the chest demonstrated parabronchial thickening and parenchymal bands suggestive of DM. An age-appropriate malignancy workup was negative, and results from a liver biopsy showed diffuse steatosis with no histologic evidence of autoimmune hepatitis. Punch biopsy results from a plaque on the left knee revealed vacuolar interface dermatitis with increased dermal mucin on colloidal iron staining, indicative of connective tissue disease (Figure 2). The patient was treated with intravenous (IV) methylprednisolone 250 mg twice daily for 2 days followed by oral prednisone 50 mg daily with IV immunoglobulin (IVIG) 0.4 mg/kg daily for 5 days. The patient’s symptoms improved, and she was discharged on oral prednisone 50 mg and mycophenolate mofetil 1000 mg twice daily with a plan for outpatient IVIG.
Two days after discharge, the patient was re-admitted for worsening muscle weakness; recalcitrant rash; new-onset hypophonia, dysphagia, and odynophagia; and intermittent fevers. Myositis panel results were positive for MDA5. Additionally, workup for HLH, which was initiated during the first hospital admission, revealed that she met 6 of 8 diagnostic criteria: intermittent fevers (maximum temperature, 38.2 °C), splenomegaly (12.6 cm on CT scan of abdomen), cytopenia in 2 cell lines (anemia, thrombocytopenia), hypertriglyceridemia, hyperferritinemia, and elevated IL-2 receptor (CD25). Based on these findings, the patient was diagnosed with anti-MDA5 DM associated with HLH.
The patient was started on IV methylprednisolone 1000 mg daily and received 1 rituximab infusion. Two days later, she experienced worsening fever with tachycardia, and a chest radiograph showed bibasilar infiltrates concerning for aspiration pneumonia, with sputum cultures growing Staphylococcus aureus. Due to the infection, the dosage of methylprednisolone was decreased to 16 mg 3 times daily and rituximab was stopped. The hematology department was consulted for the patient’s HLH, and due to her profound weakness and sepsis, the decision was made to hold initiation of etoposide, which, in addition to glucocorticoids, is considered first-line therapy for HLH. She subsequently experienced worsening hypoxia requiring intubation and received a second course of IVIG. Two days later, CT of the chest revealed progressive ground-glass opacities in the lower lobes of the lungs. The patient was then started on plasmapheresis every other day, hydroxychloroquine 200 mg daily, and IV methylprednisolone 1000 mg daily. Over the subsequent 6 days, she developed worsening renal failure, liver dysfunction, profound thrombocytopenia (13/μL), and acidemia. After extensive discussion with her family, the patient was transitioned to comfort care, and she died 33 days after the initial admission to our hospital.
Our case is a collection of several rare presentations: anti-MDA5 DM, with HLH and AAHS as complications of anti-MDA5 DM, and DM-associated liver injury. Anti-MDA5 DM is frequently refractory to conventional therapy, including high-dose glucocorticoids, cyclophosphamide, oral tacrolimus, and cyclosporine, and there currently is no single treatment algorithm.2 Lake and colleagues7 highlighted the importance of personalizing treatment of anti-MDA5 DM, as it can be one of the most aggressive rheumatologic diseases. We initially chose to treat our patient with high-dose methylprednisolone, IVIG, and rituximab. Kampylafka et al8 performed a retrospective analysis of the use of IVIG for DM as compared to standard therapy and demonstrated improved muscle and cutaneous involvement from a collection of 50 patients. Case reports have specifically revealed efficacy for the use of IVIG in patients with anti-MDA5 DM.9,10 Additionally, rituximab—an anti–B lymphocyte therapy—has been shown to be an effective supplemental therapy for cases of aggressive anti-MDA5 DM with associated interstitial lung disease, especially when conventional therapy has failed.11,12 Our patient’s sepsis secondary to S aureus pneumonia limited her to only receiving 1 dose of rituximab.
One promising treatment approach for anti-MDA5 DM recently published by Tsuji et al13 involves the use of combination therapy. In this prospective multicenter trial, patients were initially treated with a combination of high-dose glucocorticoids, oral tacrolimus, and IV cyclophosphamide. Plasmapheresis was then started for patients without symptomatic improvement. This method was compared to the more traditional step-up approach of high-dose steroids followed by another immunosuppressant. At 1-year follow-up, the combination therapy group demonstrated an 85% survival rate compared to 33% of historical controls.13
We suspect that our patient developed HLH and AAHS secondary to her underlying anti-MDA5 DM. Kumakura and Murakawa6 reported that among 116 cases of AAHS, 6.9% of cases were associated with DM, most commonly anti-Jo-1 DM. Hemophagocytic lymphohistiocytosis associated with anti-MDA5 DM has been described in only a few cases.14-16 The diagnosis of HLH is critical, as the treatments for HLH and DM differ. Both diseases manifest with hyperferritinemia—greater than 500 ng/mL in the case of HLH and 3784 ng/mL in our patient. Therefore, HLH can be easily overlooked. It is possible the rates of HLH associated with anti-MDA5 DM are higher than reported given their similar presentations.
Analogous to our case, Fujita et al15 reported a case of HLH associated with anti-MDA5 DM successfully treated with IV cyclophosphamide pulse therapy and plasmapheresis. The rationale for using plasmapheresis in anti-MDA5 DM is based on its success in patients with other antibody-mediated conditions such as Goodpasture syndrome and granulomatosis with polyangiitis.7 It is thought to expedite response to traditional treatment, and in the case described by Fujita et al,15 the patient received plasmapheresis 6 times total over the course of 9 days. The patient’s clinical symptoms, as well as platelet levels, liver enzymes, and ferritin value, improved.15 Our patient received 3 days of plasmapheresis with no improvement when the decision was made to discontinue plasmapheresis given her worsening clinical state.
Additionally, our patient had elevated hepatic enzymes (ALT, AST, ALP, GGT), and results of a liver biopsy demonstrated diffuse steatosis. We speculate her transaminitis was a complication of anti-MDA5 DM. Hepatocellular damage accompanying DM has been investigated in multiple studies and is most often defined as an elevated ALT.17-20 Improvement in ALT levels has been seen with DM treatment. However, investigators note that creatine kinase (CK) values often do not correlate with the resolution of the transaminitis, suggesting that CK denotes muscle damage whereas ALT represents separate liver damage.18-21
Nagashima et al22 highlighted that among 50 patients with DM without malignancy, only 20% presented with a transaminitis or elevated bilirubin. However, among those with liver injury, all were positive for antibodies against MDA5.22 The patients with anti-MDA5 DM liver dysfunction had higher ALT, ALP, and GGT levels compared to those without liver dysfunction. Similarly, in a retrospective review of 14 patients with anti-MDA5 DM, Gono and colleagues3 found elevated GGT levels and lower CK levels in comparison to patients with anti-aminoacyl-transfer RNA synthetase DM. Although liver enzymes can be elevated in patients with DM secondary to muscle damage, the authors argue that the specificity of GGT to the liver suggests intrinsic liver damage.3
The mechanism behind liver disease in anti-MDA5 DM is unclear, but it is hypothesized to be similar to nonalcoholic steatohepatitis.22 Other studies have revealed drug-induced hepatitis, hepatic congestion, nonspecific reactive hepatitis, metastatic liver tumor, primary biliary cholangitis, and autoimmune hepatitis as the etiology behind liver disease in their patients with DM.17-19 Liver biopsy results from patients with anti-MDA5 DM most commonly reveal hepatic steatosis, as seen in our patient, as well as hepatocyte ballooning and increased pigmented macrophages.22
We presented a case of anti-MDA5 DM complicated by HLH. Our patient had a fatal outcome despite aggressive treatment with high-dose methylprednisolone, IVIG, rituximab, and plasmapheresis. It is accepted that anti-MDA5 DM affects the lungs and skin, and our patient’s presentation also suggests liver involvement. In our case, onset of symptoms to fatality was approximately 1 year. It is essential to consider the diagnosis of HLH in all cases of anti-MDA5 DM given clinical disease overlap. Our patient could have benefited from earlier disease recognition and thus earlier aggressive therapy.
1. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344-347.
2. Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785.
3. Gono T, Kawaguchi Y, Satoh T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49:1713-1719.
4. Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34.
5. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. 2017;49:20-26.
6. Kumakura S, Murakawa Y. Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheum. 2014;66:2297-2307.
7. Lake M, George G, Summer R. Time to personalize the treatment of anti-MDA-5 associated lung disease. Ann Rheum Dis. 2019;78:E52.
8. Kampylafka EI, Kosmidis ML, Panagiotakos DB, et al. The effect of intravenous immunoglobulin (IVIG) treatment on patients with dermatomyositis: a 4-year follow-up study. Clin Exp Rheumatol. 2012;30:397-401.
9. Koguchi-Yoshioka H, Okiyama N, Iwamoto K, et al. Intravenous immunoglobulin contributes to the control of antimelanoma differentiation-associated protein 5 antibody-associated dermatomyositis with palmar violaceous macules/papules. Br J Dermatol. 2017;177:1442-1446.
10. Hamada-Ode K, Taniguchi Y, Kimata T, et al. High-dose intravenous immunoglobulin therapy for rapidly progressive interstitial pneumonitis accompanied by anti-melanoma differentiation-associated gene 5 antibody-positive amyopathic dermatomyositis. Eur J Rheumatol. 2015;2:83-85.
11. So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989.
12. Koichi Y, Aya Y, Megumi U, et al. A case of anti-MDA5-positive rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis ameliorated by rituximab, in addition to standard immunosuppressive treatment. Mod Rheumatol. 2017;27:536-540.
13. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol. 2020;72:488-498.
14. Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune-associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246.
15. Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478.
16. Gono T, Miyake K, Kawaguchi Y, et al. Hyperferritinaemia and macrophage activation in a patient with interstitial lung disease with clinically amyopathic DM. Rheumatology (Oxford). 2012;51:1336-1338.
17. Wada T, Abe G, Kudou, T, et al. Liver damage in patients with polymyositis and dermatomyositis. Kitasato Med Journal. 2016;46:40-46.
18. Takahashi A, Abe K, Yokokawa J, et al. Clinical features of liver dysfunction in collagen diseases. Hepatol Res. 2010;40:1092-1097.
19. Matsumoto T, Kobayashi S, Shimizu H, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver. 2000;20:366-373.
20. Shi Q, Niu J, Huang X, et al. Do muscle enzyme changes forecast liver injury in polymyositis/dermatomyositis patients treated with methylprednisolone and methotrexate? Ann Clin Lab Sci. 2016;46:266-269.
21. Noda S, Asano Y, Tamaki Z, et al. A case of dermatomyositis with “liver disease associated with rheumatoid diseases” positive for anti-liver-kidney microsome-1 antibody. Clin Rheumatol. 2010;29:941-943.
22. Nagashima T, Kamata Y, Iwamoto M, et al. Liver dysfunction in anti-melanoma differentiation-associated gene 5 antibody-positive patients with dermatomyositis. Rheumatol Int. 2019;39:901-909.
To the Editor:
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterized by bilateral, symmetrical, proximal muscle weakness and classic cutaneous manifestations.1 Patients with antibodies directed against melanoma differentiation–associated gene 5, MDA5, have a distinct presentation due to vasculopathy with more severe cutaneous ulcerations, palmar papules, alopecia, and an elevated risk of rapidly progressive interstitial lung disease.2 A ferritin level greater than 1600 ng/mL portends an increased risk for pulmonary disease and therefore can be of prognostic value.3 Further, patients with anti-MDA5 DM are at a lower risk of malignancy and are more likely to test negative for antinuclear antibodies in comparison to other patients with DM.2,4
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a potentially lethal condition whereby uncontrolled activation of histiocytes in the reticuloendothelial system causes hemophagocytosis and a hyperinflammatory state. Patients present with fever, splenomegaly, cytopenia, and hyperferritinemia.5 Autoimmune‐associated hemophagocytic syndrome (AAHS) describes HLH that develops in association with autoimmune conditions, most commonly systemic lupus erythematosus and adult-onset Still disease. Cases reported in association with DM exist but are few in number, and there is no standard-of-care treatment.6 We report a case of a woman with anti-MDA5 DM complicated by HLH and DM-associated liver injury.
A 50-year-old woman presented as a direct admit from the rheumatology clinic for diffuse muscle weakness of 8 months’ duration, 40-pound unintentional weight loss, pruritic rash, bilateral joint pains, dry eyes, dry mouth, and altered mental status. Four months prior, she presented to an outside hospital and was given a diagnosis of probable Sjögren syndrome and autoimmune hepatitis vs drug-induced liver injury. At that time, a workup was notable for antibodies against Sjögren syndrome–related antigen A, anti–smooth muscle antibodies, and transaminitis. Ultrasonography of the right upper quadrant revealed hepatic steatosis. The patient was started on oral prednisone and pilocarpine but had been off all medications for 1 month when she presented to our hospital.
On hospital admission, physical examination revealed a violaceous heliotrope rash; a v-sign on the chest; shawl sign; palmar papules with pits at the fingertips; and periungual erythema and ulcerations along the metacarpophalangeal joints, elbows, lateral feet, and upper eyelids (Figure 1). Laboratory workup showed the following results: white blood cell count, 4100/μL (reference range, 4000–11,000/μL); hemoglobin, 11.6 g/dL (reference range, 12–16 g/dL); platelet count, 100,000/μL (reference range, 150,000–450,000/μL); lactate dehydrogenase, 510 U/L (reference range, 80–225 U/L); alkaline phosphatase (ALP), 766 U/L (reference range, 30–120 U/L); alanine aminotransferase (ALT), 88 U/L (reference range, 10–40 U/L); aspartate aminotransferase (AST), 544 U/L (reference range, 10–40 U/L); total bilirubin, 4.2 mg/dL (reference range, 0.3–1.0 mg/dL); direct bilirubin, 3.7 mg/dL (reference range, 0.1–0.3 mg/dL); aldolase, 20.2 U/L (reference range, 1–7.5 U/L), creatine kinase, 180 U/L (reference range, 30–135 U/L); γ-glutamyltransferase (GGT), 2743 U/L (reference range, 8–40 U/L); high sensitivity C-reactive protein, 122.9 mg/L (low-risk reference range, <1.0 mg/L); triglycerides, 534 mg/dL (reference range, <150 mg/dL); ferritin, 3784 ng/mL (reference range, 24–307 ng/mL); antinuclear antibody, negative titer; antimitochondrial antibody, negative titer; soluble IL-2 receptor (CD25), 7000 U/mL (reference range, 189–846 U/mL); anti-Sjögren syndrome–related antigen A antibody, positive.
Magnetic resonance imaging of the shoulders showed diffuse soft-tissue edema. Computed tomography (CT) of the chest demonstrated parabronchial thickening and parenchymal bands suggestive of DM. An age-appropriate malignancy workup was negative, and results from a liver biopsy showed diffuse steatosis with no histologic evidence of autoimmune hepatitis. Punch biopsy results from a plaque on the left knee revealed vacuolar interface dermatitis with increased dermal mucin on colloidal iron staining, indicative of connective tissue disease (Figure 2). The patient was treated with intravenous (IV) methylprednisolone 250 mg twice daily for 2 days followed by oral prednisone 50 mg daily with IV immunoglobulin (IVIG) 0.4 mg/kg daily for 5 days. The patient’s symptoms improved, and she was discharged on oral prednisone 50 mg and mycophenolate mofetil 1000 mg twice daily with a plan for outpatient IVIG.
Two days after discharge, the patient was re-admitted for worsening muscle weakness; recalcitrant rash; new-onset hypophonia, dysphagia, and odynophagia; and intermittent fevers. Myositis panel results were positive for MDA5. Additionally, workup for HLH, which was initiated during the first hospital admission, revealed that she met 6 of 8 diagnostic criteria: intermittent fevers (maximum temperature, 38.2 °C), splenomegaly (12.6 cm on CT scan of abdomen), cytopenia in 2 cell lines (anemia, thrombocytopenia), hypertriglyceridemia, hyperferritinemia, and elevated IL-2 receptor (CD25). Based on these findings, the patient was diagnosed with anti-MDA5 DM associated with HLH.
The patient was started on IV methylprednisolone 1000 mg daily and received 1 rituximab infusion. Two days later, she experienced worsening fever with tachycardia, and a chest radiograph showed bibasilar infiltrates concerning for aspiration pneumonia, with sputum cultures growing Staphylococcus aureus. Due to the infection, the dosage of methylprednisolone was decreased to 16 mg 3 times daily and rituximab was stopped. The hematology department was consulted for the patient’s HLH, and due to her profound weakness and sepsis, the decision was made to hold initiation of etoposide, which, in addition to glucocorticoids, is considered first-line therapy for HLH. She subsequently experienced worsening hypoxia requiring intubation and received a second course of IVIG. Two days later, CT of the chest revealed progressive ground-glass opacities in the lower lobes of the lungs. The patient was then started on plasmapheresis every other day, hydroxychloroquine 200 mg daily, and IV methylprednisolone 1000 mg daily. Over the subsequent 6 days, she developed worsening renal failure, liver dysfunction, profound thrombocytopenia (13/μL), and acidemia. After extensive discussion with her family, the patient was transitioned to comfort care, and she died 33 days after the initial admission to our hospital.
Our case is a collection of several rare presentations: anti-MDA5 DM, with HLH and AAHS as complications of anti-MDA5 DM, and DM-associated liver injury. Anti-MDA5 DM is frequently refractory to conventional therapy, including high-dose glucocorticoids, cyclophosphamide, oral tacrolimus, and cyclosporine, and there currently is no single treatment algorithm.2 Lake and colleagues7 highlighted the importance of personalizing treatment of anti-MDA5 DM, as it can be one of the most aggressive rheumatologic diseases. We initially chose to treat our patient with high-dose methylprednisolone, IVIG, and rituximab. Kampylafka et al8 performed a retrospective analysis of the use of IVIG for DM as compared to standard therapy and demonstrated improved muscle and cutaneous involvement from a collection of 50 patients. Case reports have specifically revealed efficacy for the use of IVIG in patients with anti-MDA5 DM.9,10 Additionally, rituximab—an anti–B lymphocyte therapy—has been shown to be an effective supplemental therapy for cases of aggressive anti-MDA5 DM with associated interstitial lung disease, especially when conventional therapy has failed.11,12 Our patient’s sepsis secondary to S aureus pneumonia limited her to only receiving 1 dose of rituximab.
One promising treatment approach for anti-MDA5 DM recently published by Tsuji et al13 involves the use of combination therapy. In this prospective multicenter trial, patients were initially treated with a combination of high-dose glucocorticoids, oral tacrolimus, and IV cyclophosphamide. Plasmapheresis was then started for patients without symptomatic improvement. This method was compared to the more traditional step-up approach of high-dose steroids followed by another immunosuppressant. At 1-year follow-up, the combination therapy group demonstrated an 85% survival rate compared to 33% of historical controls.13
We suspect that our patient developed HLH and AAHS secondary to her underlying anti-MDA5 DM. Kumakura and Murakawa6 reported that among 116 cases of AAHS, 6.9% of cases were associated with DM, most commonly anti-Jo-1 DM. Hemophagocytic lymphohistiocytosis associated with anti-MDA5 DM has been described in only a few cases.14-16 The diagnosis of HLH is critical, as the treatments for HLH and DM differ. Both diseases manifest with hyperferritinemia—greater than 500 ng/mL in the case of HLH and 3784 ng/mL in our patient. Therefore, HLH can be easily overlooked. It is possible the rates of HLH associated with anti-MDA5 DM are higher than reported given their similar presentations.
Analogous to our case, Fujita et al15 reported a case of HLH associated with anti-MDA5 DM successfully treated with IV cyclophosphamide pulse therapy and plasmapheresis. The rationale for using plasmapheresis in anti-MDA5 DM is based on its success in patients with other antibody-mediated conditions such as Goodpasture syndrome and granulomatosis with polyangiitis.7 It is thought to expedite response to traditional treatment, and in the case described by Fujita et al,15 the patient received plasmapheresis 6 times total over the course of 9 days. The patient’s clinical symptoms, as well as platelet levels, liver enzymes, and ferritin value, improved.15 Our patient received 3 days of plasmapheresis with no improvement when the decision was made to discontinue plasmapheresis given her worsening clinical state.
Additionally, our patient had elevated hepatic enzymes (ALT, AST, ALP, GGT), and results of a liver biopsy demonstrated diffuse steatosis. We speculate her transaminitis was a complication of anti-MDA5 DM. Hepatocellular damage accompanying DM has been investigated in multiple studies and is most often defined as an elevated ALT.17-20 Improvement in ALT levels has been seen with DM treatment. However, investigators note that creatine kinase (CK) values often do not correlate with the resolution of the transaminitis, suggesting that CK denotes muscle damage whereas ALT represents separate liver damage.18-21
Nagashima et al22 highlighted that among 50 patients with DM without malignancy, only 20% presented with a transaminitis or elevated bilirubin. However, among those with liver injury, all were positive for antibodies against MDA5.22 The patients with anti-MDA5 DM liver dysfunction had higher ALT, ALP, and GGT levels compared to those without liver dysfunction. Similarly, in a retrospective review of 14 patients with anti-MDA5 DM, Gono and colleagues3 found elevated GGT levels and lower CK levels in comparison to patients with anti-aminoacyl-transfer RNA synthetase DM. Although liver enzymes can be elevated in patients with DM secondary to muscle damage, the authors argue that the specificity of GGT to the liver suggests intrinsic liver damage.3
The mechanism behind liver disease in anti-MDA5 DM is unclear, but it is hypothesized to be similar to nonalcoholic steatohepatitis.22 Other studies have revealed drug-induced hepatitis, hepatic congestion, nonspecific reactive hepatitis, metastatic liver tumor, primary biliary cholangitis, and autoimmune hepatitis as the etiology behind liver disease in their patients with DM.17-19 Liver biopsy results from patients with anti-MDA5 DM most commonly reveal hepatic steatosis, as seen in our patient, as well as hepatocyte ballooning and increased pigmented macrophages.22
We presented a case of anti-MDA5 DM complicated by HLH. Our patient had a fatal outcome despite aggressive treatment with high-dose methylprednisolone, IVIG, rituximab, and plasmapheresis. It is accepted that anti-MDA5 DM affects the lungs and skin, and our patient’s presentation also suggests liver involvement. In our case, onset of symptoms to fatality was approximately 1 year. It is essential to consider the diagnosis of HLH in all cases of anti-MDA5 DM given clinical disease overlap. Our patient could have benefited from earlier disease recognition and thus earlier aggressive therapy.
To the Editor:
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterized by bilateral, symmetrical, proximal muscle weakness and classic cutaneous manifestations.1 Patients with antibodies directed against melanoma differentiation–associated gene 5, MDA5, have a distinct presentation due to vasculopathy with more severe cutaneous ulcerations, palmar papules, alopecia, and an elevated risk of rapidly progressive interstitial lung disease.2 A ferritin level greater than 1600 ng/mL portends an increased risk for pulmonary disease and therefore can be of prognostic value.3 Further, patients with anti-MDA5 DM are at a lower risk of malignancy and are more likely to test negative for antinuclear antibodies in comparison to other patients with DM.2,4
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a potentially lethal condition whereby uncontrolled activation of histiocytes in the reticuloendothelial system causes hemophagocytosis and a hyperinflammatory state. Patients present with fever, splenomegaly, cytopenia, and hyperferritinemia.5 Autoimmune‐associated hemophagocytic syndrome (AAHS) describes HLH that develops in association with autoimmune conditions, most commonly systemic lupus erythematosus and adult-onset Still disease. Cases reported in association with DM exist but are few in number, and there is no standard-of-care treatment.6 We report a case of a woman with anti-MDA5 DM complicated by HLH and DM-associated liver injury.
A 50-year-old woman presented as a direct admit from the rheumatology clinic for diffuse muscle weakness of 8 months’ duration, 40-pound unintentional weight loss, pruritic rash, bilateral joint pains, dry eyes, dry mouth, and altered mental status. Four months prior, she presented to an outside hospital and was given a diagnosis of probable Sjögren syndrome and autoimmune hepatitis vs drug-induced liver injury. At that time, a workup was notable for antibodies against Sjögren syndrome–related antigen A, anti–smooth muscle antibodies, and transaminitis. Ultrasonography of the right upper quadrant revealed hepatic steatosis. The patient was started on oral prednisone and pilocarpine but had been off all medications for 1 month when she presented to our hospital.
On hospital admission, physical examination revealed a violaceous heliotrope rash; a v-sign on the chest; shawl sign; palmar papules with pits at the fingertips; and periungual erythema and ulcerations along the metacarpophalangeal joints, elbows, lateral feet, and upper eyelids (Figure 1). Laboratory workup showed the following results: white blood cell count, 4100/μL (reference range, 4000–11,000/μL); hemoglobin, 11.6 g/dL (reference range, 12–16 g/dL); platelet count, 100,000/μL (reference range, 150,000–450,000/μL); lactate dehydrogenase, 510 U/L (reference range, 80–225 U/L); alkaline phosphatase (ALP), 766 U/L (reference range, 30–120 U/L); alanine aminotransferase (ALT), 88 U/L (reference range, 10–40 U/L); aspartate aminotransferase (AST), 544 U/L (reference range, 10–40 U/L); total bilirubin, 4.2 mg/dL (reference range, 0.3–1.0 mg/dL); direct bilirubin, 3.7 mg/dL (reference range, 0.1–0.3 mg/dL); aldolase, 20.2 U/L (reference range, 1–7.5 U/L), creatine kinase, 180 U/L (reference range, 30–135 U/L); γ-glutamyltransferase (GGT), 2743 U/L (reference range, 8–40 U/L); high sensitivity C-reactive protein, 122.9 mg/L (low-risk reference range, <1.0 mg/L); triglycerides, 534 mg/dL (reference range, <150 mg/dL); ferritin, 3784 ng/mL (reference range, 24–307 ng/mL); antinuclear antibody, negative titer; antimitochondrial antibody, negative titer; soluble IL-2 receptor (CD25), 7000 U/mL (reference range, 189–846 U/mL); anti-Sjögren syndrome–related antigen A antibody, positive.
Magnetic resonance imaging of the shoulders showed diffuse soft-tissue edema. Computed tomography (CT) of the chest demonstrated parabronchial thickening and parenchymal bands suggestive of DM. An age-appropriate malignancy workup was negative, and results from a liver biopsy showed diffuse steatosis with no histologic evidence of autoimmune hepatitis. Punch biopsy results from a plaque on the left knee revealed vacuolar interface dermatitis with increased dermal mucin on colloidal iron staining, indicative of connective tissue disease (Figure 2). The patient was treated with intravenous (IV) methylprednisolone 250 mg twice daily for 2 days followed by oral prednisone 50 mg daily with IV immunoglobulin (IVIG) 0.4 mg/kg daily for 5 days. The patient’s symptoms improved, and she was discharged on oral prednisone 50 mg and mycophenolate mofetil 1000 mg twice daily with a plan for outpatient IVIG.
Two days after discharge, the patient was re-admitted for worsening muscle weakness; recalcitrant rash; new-onset hypophonia, dysphagia, and odynophagia; and intermittent fevers. Myositis panel results were positive for MDA5. Additionally, workup for HLH, which was initiated during the first hospital admission, revealed that she met 6 of 8 diagnostic criteria: intermittent fevers (maximum temperature, 38.2 °C), splenomegaly (12.6 cm on CT scan of abdomen), cytopenia in 2 cell lines (anemia, thrombocytopenia), hypertriglyceridemia, hyperferritinemia, and elevated IL-2 receptor (CD25). Based on these findings, the patient was diagnosed with anti-MDA5 DM associated with HLH.
The patient was started on IV methylprednisolone 1000 mg daily and received 1 rituximab infusion. Two days later, she experienced worsening fever with tachycardia, and a chest radiograph showed bibasilar infiltrates concerning for aspiration pneumonia, with sputum cultures growing Staphylococcus aureus. Due to the infection, the dosage of methylprednisolone was decreased to 16 mg 3 times daily and rituximab was stopped. The hematology department was consulted for the patient’s HLH, and due to her profound weakness and sepsis, the decision was made to hold initiation of etoposide, which, in addition to glucocorticoids, is considered first-line therapy for HLH. She subsequently experienced worsening hypoxia requiring intubation and received a second course of IVIG. Two days later, CT of the chest revealed progressive ground-glass opacities in the lower lobes of the lungs. The patient was then started on plasmapheresis every other day, hydroxychloroquine 200 mg daily, and IV methylprednisolone 1000 mg daily. Over the subsequent 6 days, she developed worsening renal failure, liver dysfunction, profound thrombocytopenia (13/μL), and acidemia. After extensive discussion with her family, the patient was transitioned to comfort care, and she died 33 days after the initial admission to our hospital.
Our case is a collection of several rare presentations: anti-MDA5 DM, with HLH and AAHS as complications of anti-MDA5 DM, and DM-associated liver injury. Anti-MDA5 DM is frequently refractory to conventional therapy, including high-dose glucocorticoids, cyclophosphamide, oral tacrolimus, and cyclosporine, and there currently is no single treatment algorithm.2 Lake and colleagues7 highlighted the importance of personalizing treatment of anti-MDA5 DM, as it can be one of the most aggressive rheumatologic diseases. We initially chose to treat our patient with high-dose methylprednisolone, IVIG, and rituximab. Kampylafka et al8 performed a retrospective analysis of the use of IVIG for DM as compared to standard therapy and demonstrated improved muscle and cutaneous involvement from a collection of 50 patients. Case reports have specifically revealed efficacy for the use of IVIG in patients with anti-MDA5 DM.9,10 Additionally, rituximab—an anti–B lymphocyte therapy—has been shown to be an effective supplemental therapy for cases of aggressive anti-MDA5 DM with associated interstitial lung disease, especially when conventional therapy has failed.11,12 Our patient’s sepsis secondary to S aureus pneumonia limited her to only receiving 1 dose of rituximab.
One promising treatment approach for anti-MDA5 DM recently published by Tsuji et al13 involves the use of combination therapy. In this prospective multicenter trial, patients were initially treated with a combination of high-dose glucocorticoids, oral tacrolimus, and IV cyclophosphamide. Plasmapheresis was then started for patients without symptomatic improvement. This method was compared to the more traditional step-up approach of high-dose steroids followed by another immunosuppressant. At 1-year follow-up, the combination therapy group demonstrated an 85% survival rate compared to 33% of historical controls.13
We suspect that our patient developed HLH and AAHS secondary to her underlying anti-MDA5 DM. Kumakura and Murakawa6 reported that among 116 cases of AAHS, 6.9% of cases were associated with DM, most commonly anti-Jo-1 DM. Hemophagocytic lymphohistiocytosis associated with anti-MDA5 DM has been described in only a few cases.14-16 The diagnosis of HLH is critical, as the treatments for HLH and DM differ. Both diseases manifest with hyperferritinemia—greater than 500 ng/mL in the case of HLH and 3784 ng/mL in our patient. Therefore, HLH can be easily overlooked. It is possible the rates of HLH associated with anti-MDA5 DM are higher than reported given their similar presentations.
Analogous to our case, Fujita et al15 reported a case of HLH associated with anti-MDA5 DM successfully treated with IV cyclophosphamide pulse therapy and plasmapheresis. The rationale for using plasmapheresis in anti-MDA5 DM is based on its success in patients with other antibody-mediated conditions such as Goodpasture syndrome and granulomatosis with polyangiitis.7 It is thought to expedite response to traditional treatment, and in the case described by Fujita et al,15 the patient received plasmapheresis 6 times total over the course of 9 days. The patient’s clinical symptoms, as well as platelet levels, liver enzymes, and ferritin value, improved.15 Our patient received 3 days of plasmapheresis with no improvement when the decision was made to discontinue plasmapheresis given her worsening clinical state.
Additionally, our patient had elevated hepatic enzymes (ALT, AST, ALP, GGT), and results of a liver biopsy demonstrated diffuse steatosis. We speculate her transaminitis was a complication of anti-MDA5 DM. Hepatocellular damage accompanying DM has been investigated in multiple studies and is most often defined as an elevated ALT.17-20 Improvement in ALT levels has been seen with DM treatment. However, investigators note that creatine kinase (CK) values often do not correlate with the resolution of the transaminitis, suggesting that CK denotes muscle damage whereas ALT represents separate liver damage.18-21
Nagashima et al22 highlighted that among 50 patients with DM without malignancy, only 20% presented with a transaminitis or elevated bilirubin. However, among those with liver injury, all were positive for antibodies against MDA5.22 The patients with anti-MDA5 DM liver dysfunction had higher ALT, ALP, and GGT levels compared to those without liver dysfunction. Similarly, in a retrospective review of 14 patients with anti-MDA5 DM, Gono and colleagues3 found elevated GGT levels and lower CK levels in comparison to patients with anti-aminoacyl-transfer RNA synthetase DM. Although liver enzymes can be elevated in patients with DM secondary to muscle damage, the authors argue that the specificity of GGT to the liver suggests intrinsic liver damage.3
The mechanism behind liver disease in anti-MDA5 DM is unclear, but it is hypothesized to be similar to nonalcoholic steatohepatitis.22 Other studies have revealed drug-induced hepatitis, hepatic congestion, nonspecific reactive hepatitis, metastatic liver tumor, primary biliary cholangitis, and autoimmune hepatitis as the etiology behind liver disease in their patients with DM.17-19 Liver biopsy results from patients with anti-MDA5 DM most commonly reveal hepatic steatosis, as seen in our patient, as well as hepatocyte ballooning and increased pigmented macrophages.22
We presented a case of anti-MDA5 DM complicated by HLH. Our patient had a fatal outcome despite aggressive treatment with high-dose methylprednisolone, IVIG, rituximab, and plasmapheresis. It is accepted that anti-MDA5 DM affects the lungs and skin, and our patient’s presentation also suggests liver involvement. In our case, onset of symptoms to fatality was approximately 1 year. It is essential to consider the diagnosis of HLH in all cases of anti-MDA5 DM given clinical disease overlap. Our patient could have benefited from earlier disease recognition and thus earlier aggressive therapy.
1. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344-347.
2. Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785.
3. Gono T, Kawaguchi Y, Satoh T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49:1713-1719.
4. Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34.
5. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. 2017;49:20-26.
6. Kumakura S, Murakawa Y. Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheum. 2014;66:2297-2307.
7. Lake M, George G, Summer R. Time to personalize the treatment of anti-MDA-5 associated lung disease. Ann Rheum Dis. 2019;78:E52.
8. Kampylafka EI, Kosmidis ML, Panagiotakos DB, et al. The effect of intravenous immunoglobulin (IVIG) treatment on patients with dermatomyositis: a 4-year follow-up study. Clin Exp Rheumatol. 2012;30:397-401.
9. Koguchi-Yoshioka H, Okiyama N, Iwamoto K, et al. Intravenous immunoglobulin contributes to the control of antimelanoma differentiation-associated protein 5 antibody-associated dermatomyositis with palmar violaceous macules/papules. Br J Dermatol. 2017;177:1442-1446.
10. Hamada-Ode K, Taniguchi Y, Kimata T, et al. High-dose intravenous immunoglobulin therapy for rapidly progressive interstitial pneumonitis accompanied by anti-melanoma differentiation-associated gene 5 antibody-positive amyopathic dermatomyositis. Eur J Rheumatol. 2015;2:83-85.
11. So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989.
12. Koichi Y, Aya Y, Megumi U, et al. A case of anti-MDA5-positive rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis ameliorated by rituximab, in addition to standard immunosuppressive treatment. Mod Rheumatol. 2017;27:536-540.
13. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol. 2020;72:488-498.
14. Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune-associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246.
15. Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478.
16. Gono T, Miyake K, Kawaguchi Y, et al. Hyperferritinaemia and macrophage activation in a patient with interstitial lung disease with clinically amyopathic DM. Rheumatology (Oxford). 2012;51:1336-1338.
17. Wada T, Abe G, Kudou, T, et al. Liver damage in patients with polymyositis and dermatomyositis. Kitasato Med Journal. 2016;46:40-46.
18. Takahashi A, Abe K, Yokokawa J, et al. Clinical features of liver dysfunction in collagen diseases. Hepatol Res. 2010;40:1092-1097.
19. Matsumoto T, Kobayashi S, Shimizu H, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver. 2000;20:366-373.
20. Shi Q, Niu J, Huang X, et al. Do muscle enzyme changes forecast liver injury in polymyositis/dermatomyositis patients treated with methylprednisolone and methotrexate? Ann Clin Lab Sci. 2016;46:266-269.
21. Noda S, Asano Y, Tamaki Z, et al. A case of dermatomyositis with “liver disease associated with rheumatoid diseases” positive for anti-liver-kidney microsome-1 antibody. Clin Rheumatol. 2010;29:941-943.
22. Nagashima T, Kamata Y, Iwamoto M, et al. Liver dysfunction in anti-melanoma differentiation-associated gene 5 antibody-positive patients with dermatomyositis. Rheumatol Int. 2019;39:901-909.
1. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344-347.
2. Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785.
3. Gono T, Kawaguchi Y, Satoh T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49:1713-1719.
4. Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34.
5. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. 2017;49:20-26.
6. Kumakura S, Murakawa Y. Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheum. 2014;66:2297-2307.
7. Lake M, George G, Summer R. Time to personalize the treatment of anti-MDA-5 associated lung disease. Ann Rheum Dis. 2019;78:E52.
8. Kampylafka EI, Kosmidis ML, Panagiotakos DB, et al. The effect of intravenous immunoglobulin (IVIG) treatment on patients with dermatomyositis: a 4-year follow-up study. Clin Exp Rheumatol. 2012;30:397-401.
9. Koguchi-Yoshioka H, Okiyama N, Iwamoto K, et al. Intravenous immunoglobulin contributes to the control of antimelanoma differentiation-associated protein 5 antibody-associated dermatomyositis with palmar violaceous macules/papules. Br J Dermatol. 2017;177:1442-1446.
10. Hamada-Ode K, Taniguchi Y, Kimata T, et al. High-dose intravenous immunoglobulin therapy for rapidly progressive interstitial pneumonitis accompanied by anti-melanoma differentiation-associated gene 5 antibody-positive amyopathic dermatomyositis. Eur J Rheumatol. 2015;2:83-85.
11. So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989.
12. Koichi Y, Aya Y, Megumi U, et al. A case of anti-MDA5-positive rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis ameliorated by rituximab, in addition to standard immunosuppressive treatment. Mod Rheumatol. 2017;27:536-540.
13. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol. 2020;72:488-498.
14. Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune-associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246.
15. Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478.
16. Gono T, Miyake K, Kawaguchi Y, et al. Hyperferritinaemia and macrophage activation in a patient with interstitial lung disease with clinically amyopathic DM. Rheumatology (Oxford). 2012;51:1336-1338.
17. Wada T, Abe G, Kudou, T, et al. Liver damage in patients with polymyositis and dermatomyositis. Kitasato Med Journal. 2016;46:40-46.
18. Takahashi A, Abe K, Yokokawa J, et al. Clinical features of liver dysfunction in collagen diseases. Hepatol Res. 2010;40:1092-1097.
19. Matsumoto T, Kobayashi S, Shimizu H, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver. 2000;20:366-373.
20. Shi Q, Niu J, Huang X, et al. Do muscle enzyme changes forecast liver injury in polymyositis/dermatomyositis patients treated with methylprednisolone and methotrexate? Ann Clin Lab Sci. 2016;46:266-269.
21. Noda S, Asano Y, Tamaki Z, et al. A case of dermatomyositis with “liver disease associated with rheumatoid diseases” positive for anti-liver-kidney microsome-1 antibody. Clin Rheumatol. 2010;29:941-943.
22. Nagashima T, Kamata Y, Iwamoto M, et al. Liver dysfunction in anti-melanoma differentiation-associated gene 5 antibody-positive patients with dermatomyositis. Rheumatol Int. 2019;39:901-909.
PRACTICE POINTS
- Anti-MDA5 (melanoma differentiation–associated gene 5 antibody)–positive dermatomyositis associated with hemophagocytic lymphohistiocytosis is a rare and aggressive condition associated with a poor prognosis, and there is no standard treatment.
- Dermatomyositis-associated liver injury is not well defined.
Acid series: Azelaic acid
However, it has many positive qualities, including being gentle enough to use daily and is safe to use in pregnancy. It is antibacterial, comedolytic, keratolytic, and has antioxidant activity. Unfortunately, in the last decade the formulations of azelaic acid have not been changed considerably. The 20% cream, 15% gel, and 15% foam vehicles are often too irritating and drying to be used in the population it is intended for: those with rosacea, or with inflamed or sensitive skin.

Azelaic acid is a dicarboxylic acid produced by Pityrosporum ovale. It inhibits the synthesis of cellular proteins and is bactericidal against Propionibacterium acnes and Staphylococcus epidermidis. Azelaic acid is both keratolytic and comedolytic by decreasing keratohyalin granules and reducing filaggrin in the epidermis. It not only scavenges free oxygen radicals, thereby reducing inflammation, but is also a tyrosinase inhibitor – making it a safe, non–hydroquinone-based alternative to skin lightening.
Azelaic acid has little toxicity, it is ingested regularly as it is found in wheat, barley, and rye. Topical side effects are usually mild and can subside with increased use. The most common side effects include erythema, local stinging, pruritus, scaling, and a burning sensation. It is considered safe in pregnancy and a great alternative to medications for acne in pregnant or nursing patients.
The largest constraint with azelaic acid preparations on the market – and most likely the reason it has not been more widely used for acne, rosacea, antiaging, and hyperpigmentation – is the formulation. The foam and gel preparations are irritating and difficult to use on dry or sensitive skin. The 20% cream preparations are slightly better tolerated; however, in vitro skin-penetration studies have shown that cutaneous penetration of azelaic acid is greater after application of a 15% gel (aqueous-based vehicle) and 15% foam (hydrophilic oil-in-water emulsion) as compared with the 20% cream formulations.

In my clinical experience, azelaic acid can only be used in rosacea patients with oily or nonsensitive skin. The majority of my rosacea patients cannot tolerate the burning sensation, albeit transient and mild. Acne patients who do not have dry skin and pregnant patients with mild acne are a great population for integrating azelaic acid into an acne regimen. I also use azelaic acid as an alternative for mild melasma and lentigines in patients who are tapering off hydroquinone or cannot use hydroquinone. In the future, we need better, creamier, nonirritating formulations to be developed and more studies of higher concentrations of this acid for both prescription/patient at-home use, as well as more elegant in-office localized peel systems using azelaic acid.
Dr. Talakoub and Dr. Wesley are cocontributors to this column. Dr. Talakoub is in private practice in McLean, Va. Dr. Wesley practices dermatology in Beverly Hills, Calif. This month’s column is by Dr. Talakoub. Write to them at [email protected]. They had no relevant disclosures.
References
Fitton A and Goa KL. Drugs. 1991 May;41(5):780-98.
Del Rosso JQ. J Clin Aesthet Dermatol. 2017 Mar;10(3):37-40.
Breathnach AC et al. Clin Dermatol. Apr-Jun 1989;7(2):106-19.
However, it has many positive qualities, including being gentle enough to use daily and is safe to use in pregnancy. It is antibacterial, comedolytic, keratolytic, and has antioxidant activity. Unfortunately, in the last decade the formulations of azelaic acid have not been changed considerably. The 20% cream, 15% gel, and 15% foam vehicles are often too irritating and drying to be used in the population it is intended for: those with rosacea, or with inflamed or sensitive skin.
Azelaic acid is a dicarboxylic acid produced by Pityrosporum ovale. It inhibits the synthesis of cellular proteins and is bactericidal against Propionibacterium acnes and Staphylococcus epidermidis. Azelaic acid is both keratolytic and comedolytic by decreasing keratohyalin granules and reducing filaggrin in the epidermis. It not only scavenges free oxygen radicals, thereby reducing inflammation, but is also a tyrosinase inhibitor – making it a safe, non–hydroquinone-based alternative to skin lightening.
Azelaic acid has little toxicity, it is ingested regularly as it is found in wheat, barley, and rye. Topical side effects are usually mild and can subside with increased use. The most common side effects include erythema, local stinging, pruritus, scaling, and a burning sensation. It is considered safe in pregnancy and a great alternative to medications for acne in pregnant or nursing patients.
The largest constraint with azelaic acid preparations on the market – and most likely the reason it has not been more widely used for acne, rosacea, antiaging, and hyperpigmentation – is the formulation. The foam and gel preparations are irritating and difficult to use on dry or sensitive skin. The 20% cream preparations are slightly better tolerated; however, in vitro skin-penetration studies have shown that cutaneous penetration of azelaic acid is greater after application of a 15% gel (aqueous-based vehicle) and 15% foam (hydrophilic oil-in-water emulsion) as compared with the 20% cream formulations.
In my clinical experience, azelaic acid can only be used in rosacea patients with oily or nonsensitive skin. The majority of my rosacea patients cannot tolerate the burning sensation, albeit transient and mild. Acne patients who do not have dry skin and pregnant patients with mild acne are a great population for integrating azelaic acid into an acne regimen. I also use azelaic acid as an alternative for mild melasma and lentigines in patients who are tapering off hydroquinone or cannot use hydroquinone. In the future, we need better, creamier, nonirritating formulations to be developed and more studies of higher concentrations of this acid for both prescription/patient at-home use, as well as more elegant in-office localized peel systems using azelaic acid.
Dr. Talakoub and Dr. Wesley are cocontributors to this column. Dr. Talakoub is in private practice in McLean, Va. Dr. Wesley practices dermatology in Beverly Hills, Calif. This month’s column is by Dr. Talakoub. Write to them at [email protected]. They had no relevant disclosures.
References
Fitton A and Goa KL. Drugs. 1991 May;41(5):780-98.
Del Rosso JQ. J Clin Aesthet Dermatol. 2017 Mar;10(3):37-40.
Breathnach AC et al. Clin Dermatol. Apr-Jun 1989;7(2):106-19.
However, it has many positive qualities, including being gentle enough to use daily and is safe to use in pregnancy. It is antibacterial, comedolytic, keratolytic, and has antioxidant activity. Unfortunately, in the last decade the formulations of azelaic acid have not been changed considerably. The 20% cream, 15% gel, and 15% foam vehicles are often too irritating and drying to be used in the population it is intended for: those with rosacea, or with inflamed or sensitive skin.
Azelaic acid is a dicarboxylic acid produced by Pityrosporum ovale. It inhibits the synthesis of cellular proteins and is bactericidal against Propionibacterium acnes and Staphylococcus epidermidis. Azelaic acid is both keratolytic and comedolytic by decreasing keratohyalin granules and reducing filaggrin in the epidermis. It not only scavenges free oxygen radicals, thereby reducing inflammation, but is also a tyrosinase inhibitor – making it a safe, non–hydroquinone-based alternative to skin lightening.
Azelaic acid has little toxicity, it is ingested regularly as it is found in wheat, barley, and rye. Topical side effects are usually mild and can subside with increased use. The most common side effects include erythema, local stinging, pruritus, scaling, and a burning sensation. It is considered safe in pregnancy and a great alternative to medications for acne in pregnant or nursing patients.
The largest constraint with azelaic acid preparations on the market – and most likely the reason it has not been more widely used for acne, rosacea, antiaging, and hyperpigmentation – is the formulation. The foam and gel preparations are irritating and difficult to use on dry or sensitive skin. The 20% cream preparations are slightly better tolerated; however, in vitro skin-penetration studies have shown that cutaneous penetration of azelaic acid is greater after application of a 15% gel (aqueous-based vehicle) and 15% foam (hydrophilic oil-in-water emulsion) as compared with the 20% cream formulations.
In my clinical experience, azelaic acid can only be used in rosacea patients with oily or nonsensitive skin. The majority of my rosacea patients cannot tolerate the burning sensation, albeit transient and mild. Acne patients who do not have dry skin and pregnant patients with mild acne are a great population for integrating azelaic acid into an acne regimen. I also use azelaic acid as an alternative for mild melasma and lentigines in patients who are tapering off hydroquinone or cannot use hydroquinone. In the future, we need better, creamier, nonirritating formulations to be developed and more studies of higher concentrations of this acid for both prescription/patient at-home use, as well as more elegant in-office localized peel systems using azelaic acid.
Dr. Talakoub and Dr. Wesley are cocontributors to this column. Dr. Talakoub is in private practice in McLean, Va. Dr. Wesley practices dermatology in Beverly Hills, Calif. This month’s column is by Dr. Talakoub. Write to them at [email protected]. They had no relevant disclosures.
References
Fitton A and Goa KL. Drugs. 1991 May;41(5):780-98.
Del Rosso JQ. J Clin Aesthet Dermatol. 2017 Mar;10(3):37-40.
Breathnach AC et al. Clin Dermatol. Apr-Jun 1989;7(2):106-19.