User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Doctors Endorsing Products on X May Not Disclose Company Ties
Lead author Aaron Mitchell, MD, MPH, a medical oncologist at Memorial Sloan Kettering Cancer Center in New York City, told this news organization that he and his colleagues undertook the study in part to see whether physicians were adhering to professional and industry guidelines regarding marketing communications.
The team reviewed posts by physicians on X during 2022, looking for key words that might indicate that the posts were intended as endorsements of a product. The researchers then delved into the Centers for Medicare and Medicaid Services Open Payments database to see how many of those identified as having endorsed a product were paid by the manufacturers.
What Dr. Mitchell found concerned him, he said.
Overall, the researchers identified 28 physician endorsers who received a total of $1.4 million from sponsors in 2022. Among these, 26 physicians (93%) received payments from the product’s manufacturer, totaling $713,976, and 24 physicians (86%) accepted payments related to the endorsed drug or device, totaling $492,098.
While most did disclose that the posts were sponsored — by adding the word “sponsored” or using #sponsored — nine physicians did not.
Although 28 physician endorsers represent a “small fraction” of the overall number of physicians who use X, each endorsement was ultimately posted dozens, if not hundreds of times, said Dr. Mitchell. In fact, he said he saw the same particular endorsement post every time he opened his X app for months.
Overall, Dr. Mitchell noted that it’s less about the fact that the endorsements are occurring on social media and more that there are these paid endorsements taking place at all.
Among the physician specialties promoting a product, urologists and oncologists dominated. Almost one third were urologists, and 57% were oncologists — six medical oncologists, six radiation oncologists, and four gynecologic oncologists. Of the remaining three physicians, two were internists and one was a pulmonary and critical care medicine specialist.
The authors tracked posts from physicians and industry accounts. Many of the posts on industry accounts were physician testimonials, usually videos. Almost half — 8 of 17 — of those testimonials did not disclose that the doctor was being paid by the manufacturer. In another case, a physician did not disclose that they were paid to endorse a white paper.
Fifteen promotional posts were for a Boston Scientific product, followed by six for GlaxoSmithKline, two for Eisai, two for Exelixis, and one each for AstraZeneca, Novartis, and Pfizer.
In general, Dr. Mitchell said, industry guidelines suggest that manufacturer-paid speakers or consultants should have well-regarded expertise in the area they are being asked to weigh in on, but most physician endorsers in the study were not key opinion leaders or experts.
The authors examined the paid endorsers’ H-index — a measure of academic productivity provided by Scopus. Overall, 19 of the 28 physicians had an H-index below 20, which is considered less accomplished, and 14 had no published research related to the endorsed product.
Ten received payments from manufacturers for research purposes, and only one received research payments related to the endorsed product ($224,577).
“Physicians’ participation in industry marketing raises questions regarding professionalism and their responsibilities as patient advocates,” the JAMA authors wrote.
The study was supported by grants from the National Cancer Institute. Dr. Mitchell reported no relevant financial relationships. Coauthors Samer Al Hadidi, MD, reported receiving personal fees from Pfizer, Sanofi, and Janssen during the conduct of the study, and Timothy S. Anderson, MD, reported receiving grants from the National Institute on Aging, the American Heart Association, and the American College of Cardiology, and receiving consulting fees from the American Medical Student Association. Dr. Anderson is also an associate editor of JAMA Internal Medicine.
A version of this article appeared on Medscape.com.
Lead author Aaron Mitchell, MD, MPH, a medical oncologist at Memorial Sloan Kettering Cancer Center in New York City, told this news organization that he and his colleagues undertook the study in part to see whether physicians were adhering to professional and industry guidelines regarding marketing communications.
The team reviewed posts by physicians on X during 2022, looking for key words that might indicate that the posts were intended as endorsements of a product. The researchers then delved into the Centers for Medicare and Medicaid Services Open Payments database to see how many of those identified as having endorsed a product were paid by the manufacturers.
What Dr. Mitchell found concerned him, he said.
Overall, the researchers identified 28 physician endorsers who received a total of $1.4 million from sponsors in 2022. Among these, 26 physicians (93%) received payments from the product’s manufacturer, totaling $713,976, and 24 physicians (86%) accepted payments related to the endorsed drug or device, totaling $492,098.
While most did disclose that the posts were sponsored — by adding the word “sponsored” or using #sponsored — nine physicians did not.
Although 28 physician endorsers represent a “small fraction” of the overall number of physicians who use X, each endorsement was ultimately posted dozens, if not hundreds of times, said Dr. Mitchell. In fact, he said he saw the same particular endorsement post every time he opened his X app for months.
Overall, Dr. Mitchell noted that it’s less about the fact that the endorsements are occurring on social media and more that there are these paid endorsements taking place at all.
Among the physician specialties promoting a product, urologists and oncologists dominated. Almost one third were urologists, and 57% were oncologists — six medical oncologists, six radiation oncologists, and four gynecologic oncologists. Of the remaining three physicians, two were internists and one was a pulmonary and critical care medicine specialist.
The authors tracked posts from physicians and industry accounts. Many of the posts on industry accounts were physician testimonials, usually videos. Almost half — 8 of 17 — of those testimonials did not disclose that the doctor was being paid by the manufacturer. In another case, a physician did not disclose that they were paid to endorse a white paper.
Fifteen promotional posts were for a Boston Scientific product, followed by six for GlaxoSmithKline, two for Eisai, two for Exelixis, and one each for AstraZeneca, Novartis, and Pfizer.
In general, Dr. Mitchell said, industry guidelines suggest that manufacturer-paid speakers or consultants should have well-regarded expertise in the area they are being asked to weigh in on, but most physician endorsers in the study were not key opinion leaders or experts.
The authors examined the paid endorsers’ H-index — a measure of academic productivity provided by Scopus. Overall, 19 of the 28 physicians had an H-index below 20, which is considered less accomplished, and 14 had no published research related to the endorsed product.
Ten received payments from manufacturers for research purposes, and only one received research payments related to the endorsed product ($224,577).
“Physicians’ participation in industry marketing raises questions regarding professionalism and their responsibilities as patient advocates,” the JAMA authors wrote.
The study was supported by grants from the National Cancer Institute. Dr. Mitchell reported no relevant financial relationships. Coauthors Samer Al Hadidi, MD, reported receiving personal fees from Pfizer, Sanofi, and Janssen during the conduct of the study, and Timothy S. Anderson, MD, reported receiving grants from the National Institute on Aging, the American Heart Association, and the American College of Cardiology, and receiving consulting fees from the American Medical Student Association. Dr. Anderson is also an associate editor of JAMA Internal Medicine.
A version of this article appeared on Medscape.com.
Lead author Aaron Mitchell, MD, MPH, a medical oncologist at Memorial Sloan Kettering Cancer Center in New York City, told this news organization that he and his colleagues undertook the study in part to see whether physicians were adhering to professional and industry guidelines regarding marketing communications.
The team reviewed posts by physicians on X during 2022, looking for key words that might indicate that the posts were intended as endorsements of a product. The researchers then delved into the Centers for Medicare and Medicaid Services Open Payments database to see how many of those identified as having endorsed a product were paid by the manufacturers.
What Dr. Mitchell found concerned him, he said.
Overall, the researchers identified 28 physician endorsers who received a total of $1.4 million from sponsors in 2022. Among these, 26 physicians (93%) received payments from the product’s manufacturer, totaling $713,976, and 24 physicians (86%) accepted payments related to the endorsed drug or device, totaling $492,098.
While most did disclose that the posts were sponsored — by adding the word “sponsored” or using #sponsored — nine physicians did not.
Although 28 physician endorsers represent a “small fraction” of the overall number of physicians who use X, each endorsement was ultimately posted dozens, if not hundreds of times, said Dr. Mitchell. In fact, he said he saw the same particular endorsement post every time he opened his X app for months.
Overall, Dr. Mitchell noted that it’s less about the fact that the endorsements are occurring on social media and more that there are these paid endorsements taking place at all.
Among the physician specialties promoting a product, urologists and oncologists dominated. Almost one third were urologists, and 57% were oncologists — six medical oncologists, six radiation oncologists, and four gynecologic oncologists. Of the remaining three physicians, two were internists and one was a pulmonary and critical care medicine specialist.
The authors tracked posts from physicians and industry accounts. Many of the posts on industry accounts were physician testimonials, usually videos. Almost half — 8 of 17 — of those testimonials did not disclose that the doctor was being paid by the manufacturer. In another case, a physician did not disclose that they were paid to endorse a white paper.
Fifteen promotional posts were for a Boston Scientific product, followed by six for GlaxoSmithKline, two for Eisai, two for Exelixis, and one each for AstraZeneca, Novartis, and Pfizer.
In general, Dr. Mitchell said, industry guidelines suggest that manufacturer-paid speakers or consultants should have well-regarded expertise in the area they are being asked to weigh in on, but most physician endorsers in the study were not key opinion leaders or experts.
The authors examined the paid endorsers’ H-index — a measure of academic productivity provided by Scopus. Overall, 19 of the 28 physicians had an H-index below 20, which is considered less accomplished, and 14 had no published research related to the endorsed product.
Ten received payments from manufacturers for research purposes, and only one received research payments related to the endorsed product ($224,577).
“Physicians’ participation in industry marketing raises questions regarding professionalism and their responsibilities as patient advocates,” the JAMA authors wrote.
The study was supported by grants from the National Cancer Institute. Dr. Mitchell reported no relevant financial relationships. Coauthors Samer Al Hadidi, MD, reported receiving personal fees from Pfizer, Sanofi, and Janssen during the conduct of the study, and Timothy S. Anderson, MD, reported receiving grants from the National Institute on Aging, the American Heart Association, and the American College of Cardiology, and receiving consulting fees from the American Medical Student Association. Dr. Anderson is also an associate editor of JAMA Internal Medicine.
A version of this article appeared on Medscape.com.
One Patient Changed This Oncologist’s View of Hope. Here’s How.
CHICAGO — Carlos, a 21-year-old, lay in a hospital bed, barely clinging to life. Following a stem cell transplant for leukemia, Carlos had developed a life-threatening case of graft-vs-host disease.
But Carlos’ mother had faith.
“I have hope things will get better,” she said, via interpreter, to Richard Leiter, MD, a palliative care doctor in training at that time.
“I hope they will,” Dr. Leiter told her.
“I should have stopped there,” said Dr. Leiter, recounting an early-career lesson on hope during the ASCO Voices session at the American Society of Clinical Oncology annual meeting. “But in my eagerness to show my attending and myself that I could handle this conversation, I kept going, mistakenly.”
“But none of us think they will,” Dr. Leiter continued.
Carlos’ mother looked Dr. Leiter in the eye. “You want him to die,” she said.
“I knew, even then, that she was right,” recalled Dr. Leiter, now a palliative care physician at Dana-Farber Cancer Institute and Brigham and Women’s Hospital and an assistant professor of medicine at Harvard Medical School, Boston.
Although there was nothing he could do to save Carlos, Dr. Leiter also couldn’t sit with the extreme suffering. “The pain was too great,” Dr. Leiter said. “I needed her to adopt our narrative that we had done everything we could to help him live, and now, we would do everything we could to help his death be a comfortable one.”
But looking back, Dr. Leiter realized, “How could we have asked her to accept what was fundamentally unacceptable, to comprehend the incomprehensible?”
The Importance of Hope
Alan B. Astrow, MD, said during an ASCO symposium on “The Art and Science of Hope.”
“How we think about hope directly influences patient care,” said Dr. Astrow, chief of hematology and medical oncology at NewYork-Presbyterian Brooklyn Methodist Hospital and a professor of clinical medicine at Weill Cornell Medicine in New York City.
Hope, whatever it turns out to be neurobiologically, is “very much a gift” that underlies human existence, he said.
Physicians have the capacity to restore or shatter a patient’s hopes, and those who come to understand the importance of hope will wish to extend the gift to others, Dr. Astrow said.
Asking patients about their hopes is the “golden question,” Steven Z. Pantilat, MD, said at the symposium. “When you think about the future, what do you hope for?”
Often, the answers reveal not only “things beyond a cure that matter tremendously to the patient but things that we can help with,” said Dr. Pantilat, professor and chief of the Division of Palliative Medicine at the University of California San Francisco.
Dr. Pantilat recalled a patient with advanced pancreatic cancer who wished to see her daughter’s wedding in 10 months. He knew that was unlikely, but the discussion led to another solution.
Her daughter moved the wedding to the ICU.
Hope can persist and uplift even in the darkest of times, and “as clinicians, we need to be in the true hope business,” he said.
While some patients may wish for a cure, others may want more time with family or comfort in the face of suffering. People can “hope for all the things that can still be, despite the fact that there’s a lot of things that can’t,” he said.
However, fear that a patient will hope for a cure, and that the difficult discussions to follow might destroy hope or lead to false hope, sometimes means physicians won’t begin the conversation.
“We want to be honest with our patients — compassionate and kind, but honest — when we talk about their hopes,” Dr. Pantilat explained. Sometimes that means he needs to tell patients, “I wish that could happen. I wish I had a treatment that could make your cancer go away, but unfortunately, I don’t. So let’s think about what else we can do to help you.”
Having these difficult discussions matters. The evidence, although limited, indicates that feeling hopeful can improve patients’ well-being and may even boost their cancer outcomes.
One recent study found, for instance, that patients who reported feeling more hopeful also had lower levels of depression and anxiety. Early research also suggests that greater levels of hope may have a hand in reducing inflammation in patients with ovarian cancer and could even improve survival in some patients with advanced cancer.
For Dr. Leiter, while these lessons came early in his career as a palliative care physician, they persist and influence his practice today.
“I know that I could not have prevented Carlos’ death. None of us could have, and none of us could have protected his mother from the unimaginable grief that will stay with her for the rest of her life,” he said. “But I could have made things just a little bit less difficult for her.
“I could have acted as her guide rather than her cross-examiner,” he continued, explaining that he now sees hope as “a generous collaborator” that can coexist with rising creatinine levels, failing livers, and fears about intubation.
“As clinicians, we can always find space to hope with our patients and their families,” he said. “So now, years later when I sit with a terrified and grieving family and they tell me they hope their loved one gets better, I remember Carlos’ mother’s eyes piercing mine ... and I know how to respond: ‘I hope so, too.’ And I do.”
A version of this article appeared on Medscape.com.
CHICAGO — Carlos, a 21-year-old, lay in a hospital bed, barely clinging to life. Following a stem cell transplant for leukemia, Carlos had developed a life-threatening case of graft-vs-host disease.
But Carlos’ mother had faith.
“I have hope things will get better,” she said, via interpreter, to Richard Leiter, MD, a palliative care doctor in training at that time.
“I hope they will,” Dr. Leiter told her.
“I should have stopped there,” said Dr. Leiter, recounting an early-career lesson on hope during the ASCO Voices session at the American Society of Clinical Oncology annual meeting. “But in my eagerness to show my attending and myself that I could handle this conversation, I kept going, mistakenly.”
“But none of us think they will,” Dr. Leiter continued.
Carlos’ mother looked Dr. Leiter in the eye. “You want him to die,” she said.
“I knew, even then, that she was right,” recalled Dr. Leiter, now a palliative care physician at Dana-Farber Cancer Institute and Brigham and Women’s Hospital and an assistant professor of medicine at Harvard Medical School, Boston.
Although there was nothing he could do to save Carlos, Dr. Leiter also couldn’t sit with the extreme suffering. “The pain was too great,” Dr. Leiter said. “I needed her to adopt our narrative that we had done everything we could to help him live, and now, we would do everything we could to help his death be a comfortable one.”
But looking back, Dr. Leiter realized, “How could we have asked her to accept what was fundamentally unacceptable, to comprehend the incomprehensible?”
The Importance of Hope
Alan B. Astrow, MD, said during an ASCO symposium on “The Art and Science of Hope.”
“How we think about hope directly influences patient care,” said Dr. Astrow, chief of hematology and medical oncology at NewYork-Presbyterian Brooklyn Methodist Hospital and a professor of clinical medicine at Weill Cornell Medicine in New York City.
Hope, whatever it turns out to be neurobiologically, is “very much a gift” that underlies human existence, he said.
Physicians have the capacity to restore or shatter a patient’s hopes, and those who come to understand the importance of hope will wish to extend the gift to others, Dr. Astrow said.
Asking patients about their hopes is the “golden question,” Steven Z. Pantilat, MD, said at the symposium. “When you think about the future, what do you hope for?”
Often, the answers reveal not only “things beyond a cure that matter tremendously to the patient but things that we can help with,” said Dr. Pantilat, professor and chief of the Division of Palliative Medicine at the University of California San Francisco.
Dr. Pantilat recalled a patient with advanced pancreatic cancer who wished to see her daughter’s wedding in 10 months. He knew that was unlikely, but the discussion led to another solution.
Her daughter moved the wedding to the ICU.
Hope can persist and uplift even in the darkest of times, and “as clinicians, we need to be in the true hope business,” he said.
While some patients may wish for a cure, others may want more time with family or comfort in the face of suffering. People can “hope for all the things that can still be, despite the fact that there’s a lot of things that can’t,” he said.
However, fear that a patient will hope for a cure, and that the difficult discussions to follow might destroy hope or lead to false hope, sometimes means physicians won’t begin the conversation.
“We want to be honest with our patients — compassionate and kind, but honest — when we talk about their hopes,” Dr. Pantilat explained. Sometimes that means he needs to tell patients, “I wish that could happen. I wish I had a treatment that could make your cancer go away, but unfortunately, I don’t. So let’s think about what else we can do to help you.”
Having these difficult discussions matters. The evidence, although limited, indicates that feeling hopeful can improve patients’ well-being and may even boost their cancer outcomes.
One recent study found, for instance, that patients who reported feeling more hopeful also had lower levels of depression and anxiety. Early research also suggests that greater levels of hope may have a hand in reducing inflammation in patients with ovarian cancer and could even improve survival in some patients with advanced cancer.
For Dr. Leiter, while these lessons came early in his career as a palliative care physician, they persist and influence his practice today.
“I know that I could not have prevented Carlos’ death. None of us could have, and none of us could have protected his mother from the unimaginable grief that will stay with her for the rest of her life,” he said. “But I could have made things just a little bit less difficult for her.
“I could have acted as her guide rather than her cross-examiner,” he continued, explaining that he now sees hope as “a generous collaborator” that can coexist with rising creatinine levels, failing livers, and fears about intubation.
“As clinicians, we can always find space to hope with our patients and their families,” he said. “So now, years later when I sit with a terrified and grieving family and they tell me they hope their loved one gets better, I remember Carlos’ mother’s eyes piercing mine ... and I know how to respond: ‘I hope so, too.’ And I do.”
A version of this article appeared on Medscape.com.
CHICAGO — Carlos, a 21-year-old, lay in a hospital bed, barely clinging to life. Following a stem cell transplant for leukemia, Carlos had developed a life-threatening case of graft-vs-host disease.
But Carlos’ mother had faith.
“I have hope things will get better,” she said, via interpreter, to Richard Leiter, MD, a palliative care doctor in training at that time.
“I hope they will,” Dr. Leiter told her.
“I should have stopped there,” said Dr. Leiter, recounting an early-career lesson on hope during the ASCO Voices session at the American Society of Clinical Oncology annual meeting. “But in my eagerness to show my attending and myself that I could handle this conversation, I kept going, mistakenly.”
“But none of us think they will,” Dr. Leiter continued.
Carlos’ mother looked Dr. Leiter in the eye. “You want him to die,” she said.
“I knew, even then, that she was right,” recalled Dr. Leiter, now a palliative care physician at Dana-Farber Cancer Institute and Brigham and Women’s Hospital and an assistant professor of medicine at Harvard Medical School, Boston.
Although there was nothing he could do to save Carlos, Dr. Leiter also couldn’t sit with the extreme suffering. “The pain was too great,” Dr. Leiter said. “I needed her to adopt our narrative that we had done everything we could to help him live, and now, we would do everything we could to help his death be a comfortable one.”
But looking back, Dr. Leiter realized, “How could we have asked her to accept what was fundamentally unacceptable, to comprehend the incomprehensible?”
The Importance of Hope
Alan B. Astrow, MD, said during an ASCO symposium on “The Art and Science of Hope.”
“How we think about hope directly influences patient care,” said Dr. Astrow, chief of hematology and medical oncology at NewYork-Presbyterian Brooklyn Methodist Hospital and a professor of clinical medicine at Weill Cornell Medicine in New York City.
Hope, whatever it turns out to be neurobiologically, is “very much a gift” that underlies human existence, he said.
Physicians have the capacity to restore or shatter a patient’s hopes, and those who come to understand the importance of hope will wish to extend the gift to others, Dr. Astrow said.
Asking patients about their hopes is the “golden question,” Steven Z. Pantilat, MD, said at the symposium. “When you think about the future, what do you hope for?”
Often, the answers reveal not only “things beyond a cure that matter tremendously to the patient but things that we can help with,” said Dr. Pantilat, professor and chief of the Division of Palliative Medicine at the University of California San Francisco.
Dr. Pantilat recalled a patient with advanced pancreatic cancer who wished to see her daughter’s wedding in 10 months. He knew that was unlikely, but the discussion led to another solution.
Her daughter moved the wedding to the ICU.
Hope can persist and uplift even in the darkest of times, and “as clinicians, we need to be in the true hope business,” he said.
While some patients may wish for a cure, others may want more time with family or comfort in the face of suffering. People can “hope for all the things that can still be, despite the fact that there’s a lot of things that can’t,” he said.
However, fear that a patient will hope for a cure, and that the difficult discussions to follow might destroy hope or lead to false hope, sometimes means physicians won’t begin the conversation.
“We want to be honest with our patients — compassionate and kind, but honest — when we talk about their hopes,” Dr. Pantilat explained. Sometimes that means he needs to tell patients, “I wish that could happen. I wish I had a treatment that could make your cancer go away, but unfortunately, I don’t. So let’s think about what else we can do to help you.”
Having these difficult discussions matters. The evidence, although limited, indicates that feeling hopeful can improve patients’ well-being and may even boost their cancer outcomes.
One recent study found, for instance, that patients who reported feeling more hopeful also had lower levels of depression and anxiety. Early research also suggests that greater levels of hope may have a hand in reducing inflammation in patients with ovarian cancer and could even improve survival in some patients with advanced cancer.
For Dr. Leiter, while these lessons came early in his career as a palliative care physician, they persist and influence his practice today.
“I know that I could not have prevented Carlos’ death. None of us could have, and none of us could have protected his mother from the unimaginable grief that will stay with her for the rest of her life,” he said. “But I could have made things just a little bit less difficult for her.
“I could have acted as her guide rather than her cross-examiner,” he continued, explaining that he now sees hope as “a generous collaborator” that can coexist with rising creatinine levels, failing livers, and fears about intubation.
“As clinicians, we can always find space to hope with our patients and their families,” he said. “So now, years later when I sit with a terrified and grieving family and they tell me they hope their loved one gets better, I remember Carlos’ mother’s eyes piercing mine ... and I know how to respond: ‘I hope so, too.’ And I do.”
A version of this article appeared on Medscape.com.
FROM ASCO 2024
Photoprotection: Benefits of Sunscreens With Iron Oxide
CHICAGO — One of the more recent developments in sunscreen technology is the addition of iron oxide to mineral sunscreens.
Iron oxide is “an excellent pigment” that absorbs and blocks visible light, which is particularly important in individuals with Fitzpatrick skin types III-VI, Zoe D. Draelos, MD, consulting professor of dermatology at Duke University, Durham, North Carolina, said at the Pigmentation Disorders Exchange symposium.
Susan C. Taylor, MD, professor of dermatology at the University of Pennsylvania, Philadelphia, who spoke at the conference, also recommended tinted sunscreen with iron oxide for patients with skin of color. “It still needs to be broad spectrum,” she said, “and at least an SPF [Sun Protection Factor] 30.”
When blended with mineral sunscreens, iron oxide can reduce transmission of visible light by 90% and can protect patients from hyperpigmentation. Iron oxide comes in different colors blended together for various degrees of tinting.
Dr. Taylor noted that iron oxide is listed under the inactive ingredients. “The literature indicates a 3% concentration to aim for, but we don’t know the concentration in most of the products,” she added.
During her presentation, Dr. Draelos noted that inorganic sunscreens, such as zinc oxide and titanium oxides, are highly effective but make the skin white and pasty. To address this issue, many companies are now grinding these materials into such small particles that they are transparent.
“That’s great, except the smaller the particle is, the less UV [ultraviolet] radiation it reflects and that lowers the [SPF],” she said.
In addition to providing photoprotection, sunscreens in general provide protection from nanoparticles in tobacco and combustion, such as traffic exhaust, which can harm skin over time. “Moisturizers and sunscreens are the best way to protect against pollution and tobacco nanoparticle damage, which can contribute to inflammation,” she noted. They create a film over the skin and trap the nanoparticles.
Start the Patient Visit With a Photoprotection Talk
At the meeting, Dr. Taylor recommended that for all patients with hypopigmentation and hyperpigmentation disorders, “treatment really begins with photoprotection.”
She acknowledged that photoprotection discussions, including the basics of seeking shade, wearing protective clothing, and avoiding midday sun, often come at the end of the patient visit but she urged dermatologists to make that the first topic instead.
Dr. Taylor said a question often asked of patients of color about prolonged sun exposure — whether their skin turns bright red after too much sun — may get a negative reply. The better question is whether the patient has experienced tender skin after too much sun — which can signify a sunburn, she said.
Dr. Draelos reported no relevant financial relationships. Dr. Taylor reported financial relationships and grant support from multiple pharmaceutical companies.
A version of this article first appeared on Medscape.com.
CHICAGO — One of the more recent developments in sunscreen technology is the addition of iron oxide to mineral sunscreens.
Iron oxide is “an excellent pigment” that absorbs and blocks visible light, which is particularly important in individuals with Fitzpatrick skin types III-VI, Zoe D. Draelos, MD, consulting professor of dermatology at Duke University, Durham, North Carolina, said at the Pigmentation Disorders Exchange symposium.
Susan C. Taylor, MD, professor of dermatology at the University of Pennsylvania, Philadelphia, who spoke at the conference, also recommended tinted sunscreen with iron oxide for patients with skin of color. “It still needs to be broad spectrum,” she said, “and at least an SPF [Sun Protection Factor] 30.”
When blended with mineral sunscreens, iron oxide can reduce transmission of visible light by 90% and can protect patients from hyperpigmentation. Iron oxide comes in different colors blended together for various degrees of tinting.
Dr. Taylor noted that iron oxide is listed under the inactive ingredients. “The literature indicates a 3% concentration to aim for, but we don’t know the concentration in most of the products,” she added.
During her presentation, Dr. Draelos noted that inorganic sunscreens, such as zinc oxide and titanium oxides, are highly effective but make the skin white and pasty. To address this issue, many companies are now grinding these materials into such small particles that they are transparent.
“That’s great, except the smaller the particle is, the less UV [ultraviolet] radiation it reflects and that lowers the [SPF],” she said.
In addition to providing photoprotection, sunscreens in general provide protection from nanoparticles in tobacco and combustion, such as traffic exhaust, which can harm skin over time. “Moisturizers and sunscreens are the best way to protect against pollution and tobacco nanoparticle damage, which can contribute to inflammation,” she noted. They create a film over the skin and trap the nanoparticles.
Start the Patient Visit With a Photoprotection Talk
At the meeting, Dr. Taylor recommended that for all patients with hypopigmentation and hyperpigmentation disorders, “treatment really begins with photoprotection.”
She acknowledged that photoprotection discussions, including the basics of seeking shade, wearing protective clothing, and avoiding midday sun, often come at the end of the patient visit but she urged dermatologists to make that the first topic instead.
Dr. Taylor said a question often asked of patients of color about prolonged sun exposure — whether their skin turns bright red after too much sun — may get a negative reply. The better question is whether the patient has experienced tender skin after too much sun — which can signify a sunburn, she said.
Dr. Draelos reported no relevant financial relationships. Dr. Taylor reported financial relationships and grant support from multiple pharmaceutical companies.
A version of this article first appeared on Medscape.com.
CHICAGO — One of the more recent developments in sunscreen technology is the addition of iron oxide to mineral sunscreens.
Iron oxide is “an excellent pigment” that absorbs and blocks visible light, which is particularly important in individuals with Fitzpatrick skin types III-VI, Zoe D. Draelos, MD, consulting professor of dermatology at Duke University, Durham, North Carolina, said at the Pigmentation Disorders Exchange symposium.
Susan C. Taylor, MD, professor of dermatology at the University of Pennsylvania, Philadelphia, who spoke at the conference, also recommended tinted sunscreen with iron oxide for patients with skin of color. “It still needs to be broad spectrum,” she said, “and at least an SPF [Sun Protection Factor] 30.”
When blended with mineral sunscreens, iron oxide can reduce transmission of visible light by 90% and can protect patients from hyperpigmentation. Iron oxide comes in different colors blended together for various degrees of tinting.
Dr. Taylor noted that iron oxide is listed under the inactive ingredients. “The literature indicates a 3% concentration to aim for, but we don’t know the concentration in most of the products,” she added.
During her presentation, Dr. Draelos noted that inorganic sunscreens, such as zinc oxide and titanium oxides, are highly effective but make the skin white and pasty. To address this issue, many companies are now grinding these materials into such small particles that they are transparent.
“That’s great, except the smaller the particle is, the less UV [ultraviolet] radiation it reflects and that lowers the [SPF],” she said.
In addition to providing photoprotection, sunscreens in general provide protection from nanoparticles in tobacco and combustion, such as traffic exhaust, which can harm skin over time. “Moisturizers and sunscreens are the best way to protect against pollution and tobacco nanoparticle damage, which can contribute to inflammation,” she noted. They create a film over the skin and trap the nanoparticles.
Start the Patient Visit With a Photoprotection Talk
At the meeting, Dr. Taylor recommended that for all patients with hypopigmentation and hyperpigmentation disorders, “treatment really begins with photoprotection.”
She acknowledged that photoprotection discussions, including the basics of seeking shade, wearing protective clothing, and avoiding midday sun, often come at the end of the patient visit but she urged dermatologists to make that the first topic instead.
Dr. Taylor said a question often asked of patients of color about prolonged sun exposure — whether their skin turns bright red after too much sun — may get a negative reply. The better question is whether the patient has experienced tender skin after too much sun — which can signify a sunburn, she said.
Dr. Draelos reported no relevant financial relationships. Dr. Taylor reported financial relationships and grant support from multiple pharmaceutical companies.
A version of this article first appeared on Medscape.com.
Inpatient Management of Hidradenitis Suppurativa: A Delphi Consensus Study
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition that affects approximately 0.1% of the US population.1,2 Severe disease or HS flares can lead patients to seek care through the emergency department (ED), with some requiring inpatient admission. 3 Inpatient hospitalization of patients with HS has increased over the last 2 decades, and patients with HS utilize emergency and inpatient care more frequently than those with other dermatologic conditions.4,5 Minority patients and those of lower socioeconomic status are more likely to present to the ED for HS management due to limited access to care and other existing comorbid conditions. 4 In a 2022 study of the Nationwide Readmissions Database, the authors looked at hospital readmission rates of patients with HS compared with those with heart failure—both patient populations with chronic debilitating conditions. Results indicated that the hospital readmission rates for patients with HS surpassed those of patients with heart failure for that year, highlighting the need for improved inpatient management of HS.6
Patients with HS present to the ED with severe pain, fever, wound care, or the need for surgical intervention. The ED and inpatient hospital setting are locations in which physicians may not be as familiar with the diagnosis or treatment of HS, specifically flares or severe disease. 7 The inpatient care setting provides access to certain resources that can be challenging to obtain in the outpatient clinical setting, such as social workers and pain specialists, but also can prove challenging in obtaining other resources for HS management, such as advanced medical therapies. Given the increase in hospital- based care for HS and lack of widespread inpatient access to dermatology and HS experts, consensus recommendations for management of HS in the acute hospital setting would be beneficial. In our study, we sought to generate a collection of expert consensus statements providers can refer to when managing patients with HS in the inpatient setting.
Methods
The study team at the Wake Forest University School of Medicine (Winston-Salem, North Carolina)(M.N., R.P., L.C.S.) developed an initial set of consensus statements based on current published HS treatment guidelines,8,9 publications on management of inpatient HS,3 published supportive care guidelines for Stevens-Johnson syndrome, 10 and personal clinical experience in managing inpatient HS, which resulted in 50 statements organized into the following categories: overall care, wound care, genital care, pain management, infection control, medical management, surgical management, nutrition, and transitional care guidelines. This study was approved by the Wake Forest University institutional review board (IRB00084257).
Participant Recruitment—Dermatologists were identified for participation in the study based on membership in the Society of Dermatology Hospitalists and the Hidradenitis Suppurativa Foundation or authorship of publications relevant to HS or inpatient dermatology. Dermatologists from larger academic institutions with HS specialty clinics and inpatient dermatology services also were identified. Participants were invited via email and could suggest other experts for inclusion. A total of 31 dermatologists were invited to participate in the study, with 26 agreeing to participate. All participating dermatologists were practicing in the United States.
Delphi Study—In the first round of the Delphi study, the participants were sent an online survey via REDCap in which they were asked to rank the appropriateness of each of the proposed 50 guideline statements on a scale of 1 (very inappropriate) to 9 (very appropriate). Participants also were able to provide commentary and feedback on each of the statements. Survey results were analyzed using the RAND/ UCLA Appropriateness Method.11 For each statement, the median rating for appropriateness, interpercentile range (IPR), IPR adjusted for symmetry, and disagreement index (DI) were calculated (DI=IPR/IPR adjusted for symmetry). The 30th and 70th percentiles were used in the DI calculation as the upper and lower limits, respectively. A median rating for appropriateness of 1.0 to 3.9 was considered “inappropriate,” 4.0 to 6.9 was considered “uncertain appropriateness,” and 7.0 to 9.0 was “appropriate.” A DI value greater than or equal to 1 indicated a lack of consensus regarding the appropriateness of the statement. Following each round, participants received a copy of their responses along with the group median rank of each statement. Statements that did not reach consensus in the first Delphi round were revised based on feedback received by the participants, and a second survey with 14 statements was sent via REDCap 2 weeks later. The RAND/UCLA Appropriateness Method also was applied to this second Delphi round. After the second survey, participants received a copy of anonymized comments regarding the consensus statements and were allowed to provide additional final commentary to be included in the discussion of these recommendations.
Results
Twenty-six dermatologists completed the first-round survey, and 24 participants completed the second-round survey. All participants self-identified as having expertise in either HS (n=22 [85%]) or inpatient dermatology (n=17 [65%]), and 13 (50%) participants self-identified as experts in both HS and inpatient dermatology. All participants, except 1, were affiliated with an academic health system with inpatient dermatology services. The average length of time in practice as a dermatologist was 10 years (median, 9 years [range, 3–27 years]).
Of the 50 initial proposed consensus statements, 26 (52%) achieved consensus after the first round; 21 statements revealed DI calculations that did not achieve consensus. Two statements achieved consensus but received median ratings for appropriateness, indicating uncertain appropriateness; because of this, 1 statement was removed and 1 was revised based on participant feedback, resulting in 13 revised statements (eTable 1). Controversial topics in the consensus process included obtaining wound cultures and meaningful culture data interpretation, use of specific biologic medications in the inpatient setting, and use of intravenous ertapenem. Participant responses to these topics are discussed in detail below. Of these secondround statements, all achieved consensus. The final set of consensus statements can be found in eTable 2.
Comment
Our Delphi consensus study combined the expertise of both dermatologists who care for patients with HS and those with inpatient dermatology experience to produce a set of recommendations for the management of HS in the hospital care setting. A strength of this study is inclusion of many national leaders in both HS and inpatient dermatology, with some participants having developed the previously published HS treatment guidelines and others having participated in inpatient dermatology Delphi studies.8-10 The expertise is further strengthened by the geographically diverse institutional representation within the United States.
The final consensus recommendations included 40 statements covering a range of patient care issues, including use of appropriate inpatient subspecialists (care team), supportive care measures (wound care, pain control, genital care), disease-oriented treatment (medical management, surgical management), inpatient complications (infection control, nutrition), and successful transition back to outpatient management (transitional care). These recommendations are meant to serve as a resource for providers to consider when taking care of inpatient HS flares, recognizing that the complexity and individual circumstances of each patient are unique.
Delphi Consensus Recommendations Compared to Prior Guidelines—Several recommendations in the current study align with the previously published North American clinical management guidelines for HS.8,9 Our recommendations agree with prior guidelines on the importance of disease staging and pain assessment using validated assessment tools as well as screening for HS comorbidities. There also is agreement in the potential benefit of involving pain specialists in the development of a comprehensive pain management plan. The inpatient care setting provides a unique opportunity to engage multiple specialists and collaborate on patient care in a timely manner. Our recommendations regarding surgical care also align with established guidelines in recommending incision and drainage as an acute bedside procedure best utilized for symptom relief in inflamed abscesses and relegating most other surgical management to the outpatient setting. Wound care recommendations also are similar, with our expert participants agreeing on individualizing dressing choices based on wound characteristics. A benefit of inpatient wound care is access to skilled nursing for dressing changes and potentially improved access to more sophisticated dressing materials. Our recommendations differ from the prior guidelines in our focus on severe HS, HS flares, and HS complications, which constitute the majority of inpatient disease management. We provide additional guidance on management of secondary infections, perianal fistulous disease, and importantly transitional care to optimize discharge planning.
Differing Opinions in Our Analysis—Despite the success of our Delphi consensus process, there were some differing opinions regarding certain aspects of inpatient HS management, which is to be expected given the lack of strong evidence-based research to support some of the recommended practices. There were differing opinions on the utility of wound culture data, with some participants feeling culture data could help with antibiotic susceptibility and resistance patterns, while others felt wound cultures represent bacterial colonization or biofilm formation.
Initial consensus statements in the first Delphi round were created for individual biologic medications but did not achieve consensus, and feedback on the use of biologics in the inpatient environment was mixed, largely due to logistic and insurance issues. Many participants felt biologic medication cost, difficulty obtaining inpatient reimbursement, health care resource utilization, and availability of biologics in different hospital systems prevented recommending the use of specific biologics during hospitalization. The one exception was in the case of a hospitalized patient who was already receiving infliximab for HS: there was consensus on ensuring the patient dosing was maximized, if appropriate, to 10 mg/kg.12 Ertapenem use also was controversial, with some participants using it as a bridge therapy to either outpatient biologic use or surgery, while others felt it was onerous and difficult to establish reliable access to secure intravenous administration and regular dosing once the patient left the inpatient setting.13 Others said they have experienced objections from infectious disease colleagues on the use of intravenous antibiotics, citing antibiotic stewardship concerns.
Patient Care in the Inpatient Setting—Prior literature suggests patients admitted as inpatients for HS tend to be of lower socioeconomic status and are admitted to larger urban teaching hospitals.14,15 Patients with lower socioeconomic status have increased difficulty accessing health care resources; therefore, inpatient admission serves as an opportunity to provide a holistic HS assessment and coordinate resources for chronic outpatient management.
Study Limitations—This Delphi consensus study has some limitations. The existing literature on inpatient management of HS is limited, challenging our ability to assess the extent to which these published recommendations are already being implemented. Additionally, the study included HS and inpatient dermatology experts from the United States, which means the recommendations may not be generalizable to other countries. Most participants practiced dermatology at large tertiary care academic medical centers, which may limit the ability to implement recommendations in all US inpatient care settings such as small community-based hospitals; however, many of the supportive care guidelines such as pain control, wound care, nutritional support, and social work should be achievable in most inpatient care settings.
Conclusion
Given the increase in inpatient and ED health care utilization for HS, there is an urgent need for expert consensus recommendations on inpatient management of this unique patient population, which requires complex multidisciplinary care. Our recommendations are a resource for providers to utilize and potentially improve the standard of care we provide these patients.
Acknowledgment—We thank the Wake Forest University Clinical and Translational Science Institute (Winston- Salem, North Carolina) for providing statistical help.
- Garg A, Kirby JS, Lavian J, et al. Sex- and age-adjusted population analysis of prevalence estimates for hidradenitis suppurativa in the United States. JAMA Dermatol. 2017;153:760-764.
- Ingram JR. The epidemiology of hidradenitis suppurativa. Br J Dermatol. 2020;183:990-998. doi:10.1111/bjd.19435
- Charrow A, Savage KT, Flood K, et al. Hidradenitis suppurativa for the dermatologic hospitalist. Cutis. 2019;104:276-280.
- Anzaldi L, Perkins JA, Byrd AS, et al. Characterizing inpatient hospitalizations for hidradenitis suppurativa in the United States. J Am Acad Dermatol. 2020;82:510-513. doi:10.1016/j.jaad.2019.09.019
- Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614. doi:10.1016/j.jaad.2015.06.053
- Edigin E, Kaul S, Eseaton PO, et al. At 180 days hidradenitis suppurativa readmission rate is comparable to heart failure: analysis of the nationwide readmissions database. J Am Acad Dermatol. 2022;87:188-192. doi:10.1016/j.jaad.2021.06.894
- Kirby JS, Miller JJ, Adams DR, et al. Health care utilization patterns and costs for patients with hidradenitis suppurativa. JAMA Dermatol. 2014;150:937-944. doi:10.1001/jamadermatol.2014.691
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi:10.1016/j .jaad.2019.02.067
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101. doi:10.1016/j.jaad.2019.02.068
- Seminario-Vidal L, Kroshinsky D, Malachowski SJ, et al. Society of Dermatology Hospitalists supportive care guidelines for the management of Stevens-Johnson syndrome/toxic epidermal necrolysis in adults. J Am Acad Dermatol. 2020;82:1553-1567. doi:10.1016/j .jaad.2020.02.066
- Fitch K, Bernstein SJ, Burnand B, et al. The RAND/UCLA Appropriateness Method: User’s Manual. Rand; 2001.
- Oskardmay AN, Miles JA, Sayed CJ. Determining the optimal dose of infliximab for treatment of hidradenitis suppurativa. J Am Acad Dermatol. 2019;81:702-708. doi:10.1016/j.jaad.2019.05.022
- Join-Lambert O, Coignard-Biehler H, Jais JP, et al. Efficacy of ertapenem in severe hidradenitis suppurativa: a pilot study in a cohort of 30 consecutive patients. J Antimicrob Chemother. 2016;71:513-520. doi:10.1093/jac/dkv361
- Khanna R, Whang KA, Huang AH, et al. Inpatient burden of hidradenitis suppurativa in the United States: analysis of the 2016 National Inpatient Sample. J Dermatolog Treat. 2022;33:1150-1152. doi:10.1080/09 546634.2020.1773380
- Patel A, Patel A, Solanki D, et al. Hidradenitis suppurativa in the United States: insights from the national inpatient sample (2008-2017) on contemporary trends in demographics, hospitalization rates, chronic comorbid conditions, and mortality. Cureus. 2022;14:E24755. doi:10.7759/cureus.24755
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition that affects approximately 0.1% of the US population.1,2 Severe disease or HS flares can lead patients to seek care through the emergency department (ED), with some requiring inpatient admission. 3 Inpatient hospitalization of patients with HS has increased over the last 2 decades, and patients with HS utilize emergency and inpatient care more frequently than those with other dermatologic conditions.4,5 Minority patients and those of lower socioeconomic status are more likely to present to the ED for HS management due to limited access to care and other existing comorbid conditions. 4 In a 2022 study of the Nationwide Readmissions Database, the authors looked at hospital readmission rates of patients with HS compared with those with heart failure—both patient populations with chronic debilitating conditions. Results indicated that the hospital readmission rates for patients with HS surpassed those of patients with heart failure for that year, highlighting the need for improved inpatient management of HS.6
Patients with HS present to the ED with severe pain, fever, wound care, or the need for surgical intervention. The ED and inpatient hospital setting are locations in which physicians may not be as familiar with the diagnosis or treatment of HS, specifically flares or severe disease. 7 The inpatient care setting provides access to certain resources that can be challenging to obtain in the outpatient clinical setting, such as social workers and pain specialists, but also can prove challenging in obtaining other resources for HS management, such as advanced medical therapies. Given the increase in hospital- based care for HS and lack of widespread inpatient access to dermatology and HS experts, consensus recommendations for management of HS in the acute hospital setting would be beneficial. In our study, we sought to generate a collection of expert consensus statements providers can refer to when managing patients with HS in the inpatient setting.
Methods
The study team at the Wake Forest University School of Medicine (Winston-Salem, North Carolina)(M.N., R.P., L.C.S.) developed an initial set of consensus statements based on current published HS treatment guidelines,8,9 publications on management of inpatient HS,3 published supportive care guidelines for Stevens-Johnson syndrome, 10 and personal clinical experience in managing inpatient HS, which resulted in 50 statements organized into the following categories: overall care, wound care, genital care, pain management, infection control, medical management, surgical management, nutrition, and transitional care guidelines. This study was approved by the Wake Forest University institutional review board (IRB00084257).
Participant Recruitment—Dermatologists were identified for participation in the study based on membership in the Society of Dermatology Hospitalists and the Hidradenitis Suppurativa Foundation or authorship of publications relevant to HS or inpatient dermatology. Dermatologists from larger academic institutions with HS specialty clinics and inpatient dermatology services also were identified. Participants were invited via email and could suggest other experts for inclusion. A total of 31 dermatologists were invited to participate in the study, with 26 agreeing to participate. All participating dermatologists were practicing in the United States.
Delphi Study—In the first round of the Delphi study, the participants were sent an online survey via REDCap in which they were asked to rank the appropriateness of each of the proposed 50 guideline statements on a scale of 1 (very inappropriate) to 9 (very appropriate). Participants also were able to provide commentary and feedback on each of the statements. Survey results were analyzed using the RAND/ UCLA Appropriateness Method.11 For each statement, the median rating for appropriateness, interpercentile range (IPR), IPR adjusted for symmetry, and disagreement index (DI) were calculated (DI=IPR/IPR adjusted for symmetry). The 30th and 70th percentiles were used in the DI calculation as the upper and lower limits, respectively. A median rating for appropriateness of 1.0 to 3.9 was considered “inappropriate,” 4.0 to 6.9 was considered “uncertain appropriateness,” and 7.0 to 9.0 was “appropriate.” A DI value greater than or equal to 1 indicated a lack of consensus regarding the appropriateness of the statement. Following each round, participants received a copy of their responses along with the group median rank of each statement. Statements that did not reach consensus in the first Delphi round were revised based on feedback received by the participants, and a second survey with 14 statements was sent via REDCap 2 weeks later. The RAND/UCLA Appropriateness Method also was applied to this second Delphi round. After the second survey, participants received a copy of anonymized comments regarding the consensus statements and were allowed to provide additional final commentary to be included in the discussion of these recommendations.
Results
Twenty-six dermatologists completed the first-round survey, and 24 participants completed the second-round survey. All participants self-identified as having expertise in either HS (n=22 [85%]) or inpatient dermatology (n=17 [65%]), and 13 (50%) participants self-identified as experts in both HS and inpatient dermatology. All participants, except 1, were affiliated with an academic health system with inpatient dermatology services. The average length of time in practice as a dermatologist was 10 years (median, 9 years [range, 3–27 years]).
Of the 50 initial proposed consensus statements, 26 (52%) achieved consensus after the first round; 21 statements revealed DI calculations that did not achieve consensus. Two statements achieved consensus but received median ratings for appropriateness, indicating uncertain appropriateness; because of this, 1 statement was removed and 1 was revised based on participant feedback, resulting in 13 revised statements (eTable 1). Controversial topics in the consensus process included obtaining wound cultures and meaningful culture data interpretation, use of specific biologic medications in the inpatient setting, and use of intravenous ertapenem. Participant responses to these topics are discussed in detail below. Of these secondround statements, all achieved consensus. The final set of consensus statements can be found in eTable 2.
Comment
Our Delphi consensus study combined the expertise of both dermatologists who care for patients with HS and those with inpatient dermatology experience to produce a set of recommendations for the management of HS in the hospital care setting. A strength of this study is inclusion of many national leaders in both HS and inpatient dermatology, with some participants having developed the previously published HS treatment guidelines and others having participated in inpatient dermatology Delphi studies.8-10 The expertise is further strengthened by the geographically diverse institutional representation within the United States.
The final consensus recommendations included 40 statements covering a range of patient care issues, including use of appropriate inpatient subspecialists (care team), supportive care measures (wound care, pain control, genital care), disease-oriented treatment (medical management, surgical management), inpatient complications (infection control, nutrition), and successful transition back to outpatient management (transitional care). These recommendations are meant to serve as a resource for providers to consider when taking care of inpatient HS flares, recognizing that the complexity and individual circumstances of each patient are unique.
Delphi Consensus Recommendations Compared to Prior Guidelines—Several recommendations in the current study align with the previously published North American clinical management guidelines for HS.8,9 Our recommendations agree with prior guidelines on the importance of disease staging and pain assessment using validated assessment tools as well as screening for HS comorbidities. There also is agreement in the potential benefit of involving pain specialists in the development of a comprehensive pain management plan. The inpatient care setting provides a unique opportunity to engage multiple specialists and collaborate on patient care in a timely manner. Our recommendations regarding surgical care also align with established guidelines in recommending incision and drainage as an acute bedside procedure best utilized for symptom relief in inflamed abscesses and relegating most other surgical management to the outpatient setting. Wound care recommendations also are similar, with our expert participants agreeing on individualizing dressing choices based on wound characteristics. A benefit of inpatient wound care is access to skilled nursing for dressing changes and potentially improved access to more sophisticated dressing materials. Our recommendations differ from the prior guidelines in our focus on severe HS, HS flares, and HS complications, which constitute the majority of inpatient disease management. We provide additional guidance on management of secondary infections, perianal fistulous disease, and importantly transitional care to optimize discharge planning.
Differing Opinions in Our Analysis—Despite the success of our Delphi consensus process, there were some differing opinions regarding certain aspects of inpatient HS management, which is to be expected given the lack of strong evidence-based research to support some of the recommended practices. There were differing opinions on the utility of wound culture data, with some participants feeling culture data could help with antibiotic susceptibility and resistance patterns, while others felt wound cultures represent bacterial colonization or biofilm formation.
Initial consensus statements in the first Delphi round were created for individual biologic medications but did not achieve consensus, and feedback on the use of biologics in the inpatient environment was mixed, largely due to logistic and insurance issues. Many participants felt biologic medication cost, difficulty obtaining inpatient reimbursement, health care resource utilization, and availability of biologics in different hospital systems prevented recommending the use of specific biologics during hospitalization. The one exception was in the case of a hospitalized patient who was already receiving infliximab for HS: there was consensus on ensuring the patient dosing was maximized, if appropriate, to 10 mg/kg.12 Ertapenem use also was controversial, with some participants using it as a bridge therapy to either outpatient biologic use or surgery, while others felt it was onerous and difficult to establish reliable access to secure intravenous administration and regular dosing once the patient left the inpatient setting.13 Others said they have experienced objections from infectious disease colleagues on the use of intravenous antibiotics, citing antibiotic stewardship concerns.
Patient Care in the Inpatient Setting—Prior literature suggests patients admitted as inpatients for HS tend to be of lower socioeconomic status and are admitted to larger urban teaching hospitals.14,15 Patients with lower socioeconomic status have increased difficulty accessing health care resources; therefore, inpatient admission serves as an opportunity to provide a holistic HS assessment and coordinate resources for chronic outpatient management.
Study Limitations—This Delphi consensus study has some limitations. The existing literature on inpatient management of HS is limited, challenging our ability to assess the extent to which these published recommendations are already being implemented. Additionally, the study included HS and inpatient dermatology experts from the United States, which means the recommendations may not be generalizable to other countries. Most participants practiced dermatology at large tertiary care academic medical centers, which may limit the ability to implement recommendations in all US inpatient care settings such as small community-based hospitals; however, many of the supportive care guidelines such as pain control, wound care, nutritional support, and social work should be achievable in most inpatient care settings.
Conclusion
Given the increase in inpatient and ED health care utilization for HS, there is an urgent need for expert consensus recommendations on inpatient management of this unique patient population, which requires complex multidisciplinary care. Our recommendations are a resource for providers to utilize and potentially improve the standard of care we provide these patients.
Acknowledgment—We thank the Wake Forest University Clinical and Translational Science Institute (Winston- Salem, North Carolina) for providing statistical help.
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition that affects approximately 0.1% of the US population.1,2 Severe disease or HS flares can lead patients to seek care through the emergency department (ED), with some requiring inpatient admission. 3 Inpatient hospitalization of patients with HS has increased over the last 2 decades, and patients with HS utilize emergency and inpatient care more frequently than those with other dermatologic conditions.4,5 Minority patients and those of lower socioeconomic status are more likely to present to the ED for HS management due to limited access to care and other existing comorbid conditions. 4 In a 2022 study of the Nationwide Readmissions Database, the authors looked at hospital readmission rates of patients with HS compared with those with heart failure—both patient populations with chronic debilitating conditions. Results indicated that the hospital readmission rates for patients with HS surpassed those of patients with heart failure for that year, highlighting the need for improved inpatient management of HS.6
Patients with HS present to the ED with severe pain, fever, wound care, or the need for surgical intervention. The ED and inpatient hospital setting are locations in which physicians may not be as familiar with the diagnosis or treatment of HS, specifically flares or severe disease. 7 The inpatient care setting provides access to certain resources that can be challenging to obtain in the outpatient clinical setting, such as social workers and pain specialists, but also can prove challenging in obtaining other resources for HS management, such as advanced medical therapies. Given the increase in hospital- based care for HS and lack of widespread inpatient access to dermatology and HS experts, consensus recommendations for management of HS in the acute hospital setting would be beneficial. In our study, we sought to generate a collection of expert consensus statements providers can refer to when managing patients with HS in the inpatient setting.
Methods
The study team at the Wake Forest University School of Medicine (Winston-Salem, North Carolina)(M.N., R.P., L.C.S.) developed an initial set of consensus statements based on current published HS treatment guidelines,8,9 publications on management of inpatient HS,3 published supportive care guidelines for Stevens-Johnson syndrome, 10 and personal clinical experience in managing inpatient HS, which resulted in 50 statements organized into the following categories: overall care, wound care, genital care, pain management, infection control, medical management, surgical management, nutrition, and transitional care guidelines. This study was approved by the Wake Forest University institutional review board (IRB00084257).
Participant Recruitment—Dermatologists were identified for participation in the study based on membership in the Society of Dermatology Hospitalists and the Hidradenitis Suppurativa Foundation or authorship of publications relevant to HS or inpatient dermatology. Dermatologists from larger academic institutions with HS specialty clinics and inpatient dermatology services also were identified. Participants were invited via email and could suggest other experts for inclusion. A total of 31 dermatologists were invited to participate in the study, with 26 agreeing to participate. All participating dermatologists were practicing in the United States.
Delphi Study—In the first round of the Delphi study, the participants were sent an online survey via REDCap in which they were asked to rank the appropriateness of each of the proposed 50 guideline statements on a scale of 1 (very inappropriate) to 9 (very appropriate). Participants also were able to provide commentary and feedback on each of the statements. Survey results were analyzed using the RAND/ UCLA Appropriateness Method.11 For each statement, the median rating for appropriateness, interpercentile range (IPR), IPR adjusted for symmetry, and disagreement index (DI) were calculated (DI=IPR/IPR adjusted for symmetry). The 30th and 70th percentiles were used in the DI calculation as the upper and lower limits, respectively. A median rating for appropriateness of 1.0 to 3.9 was considered “inappropriate,” 4.0 to 6.9 was considered “uncertain appropriateness,” and 7.0 to 9.0 was “appropriate.” A DI value greater than or equal to 1 indicated a lack of consensus regarding the appropriateness of the statement. Following each round, participants received a copy of their responses along with the group median rank of each statement. Statements that did not reach consensus in the first Delphi round were revised based on feedback received by the participants, and a second survey with 14 statements was sent via REDCap 2 weeks later. The RAND/UCLA Appropriateness Method also was applied to this second Delphi round. After the second survey, participants received a copy of anonymized comments regarding the consensus statements and were allowed to provide additional final commentary to be included in the discussion of these recommendations.
Results
Twenty-six dermatologists completed the first-round survey, and 24 participants completed the second-round survey. All participants self-identified as having expertise in either HS (n=22 [85%]) or inpatient dermatology (n=17 [65%]), and 13 (50%) participants self-identified as experts in both HS and inpatient dermatology. All participants, except 1, were affiliated with an academic health system with inpatient dermatology services. The average length of time in practice as a dermatologist was 10 years (median, 9 years [range, 3–27 years]).
Of the 50 initial proposed consensus statements, 26 (52%) achieved consensus after the first round; 21 statements revealed DI calculations that did not achieve consensus. Two statements achieved consensus but received median ratings for appropriateness, indicating uncertain appropriateness; because of this, 1 statement was removed and 1 was revised based on participant feedback, resulting in 13 revised statements (eTable 1). Controversial topics in the consensus process included obtaining wound cultures and meaningful culture data interpretation, use of specific biologic medications in the inpatient setting, and use of intravenous ertapenem. Participant responses to these topics are discussed in detail below. Of these secondround statements, all achieved consensus. The final set of consensus statements can be found in eTable 2.
Comment
Our Delphi consensus study combined the expertise of both dermatologists who care for patients with HS and those with inpatient dermatology experience to produce a set of recommendations for the management of HS in the hospital care setting. A strength of this study is inclusion of many national leaders in both HS and inpatient dermatology, with some participants having developed the previously published HS treatment guidelines and others having participated in inpatient dermatology Delphi studies.8-10 The expertise is further strengthened by the geographically diverse institutional representation within the United States.
The final consensus recommendations included 40 statements covering a range of patient care issues, including use of appropriate inpatient subspecialists (care team), supportive care measures (wound care, pain control, genital care), disease-oriented treatment (medical management, surgical management), inpatient complications (infection control, nutrition), and successful transition back to outpatient management (transitional care). These recommendations are meant to serve as a resource for providers to consider when taking care of inpatient HS flares, recognizing that the complexity and individual circumstances of each patient are unique.
Delphi Consensus Recommendations Compared to Prior Guidelines—Several recommendations in the current study align with the previously published North American clinical management guidelines for HS.8,9 Our recommendations agree with prior guidelines on the importance of disease staging and pain assessment using validated assessment tools as well as screening for HS comorbidities. There also is agreement in the potential benefit of involving pain specialists in the development of a comprehensive pain management plan. The inpatient care setting provides a unique opportunity to engage multiple specialists and collaborate on patient care in a timely manner. Our recommendations regarding surgical care also align with established guidelines in recommending incision and drainage as an acute bedside procedure best utilized for symptom relief in inflamed abscesses and relegating most other surgical management to the outpatient setting. Wound care recommendations also are similar, with our expert participants agreeing on individualizing dressing choices based on wound characteristics. A benefit of inpatient wound care is access to skilled nursing for dressing changes and potentially improved access to more sophisticated dressing materials. Our recommendations differ from the prior guidelines in our focus on severe HS, HS flares, and HS complications, which constitute the majority of inpatient disease management. We provide additional guidance on management of secondary infections, perianal fistulous disease, and importantly transitional care to optimize discharge planning.
Differing Opinions in Our Analysis—Despite the success of our Delphi consensus process, there were some differing opinions regarding certain aspects of inpatient HS management, which is to be expected given the lack of strong evidence-based research to support some of the recommended practices. There were differing opinions on the utility of wound culture data, with some participants feeling culture data could help with antibiotic susceptibility and resistance patterns, while others felt wound cultures represent bacterial colonization or biofilm formation.
Initial consensus statements in the first Delphi round were created for individual biologic medications but did not achieve consensus, and feedback on the use of biologics in the inpatient environment was mixed, largely due to logistic and insurance issues. Many participants felt biologic medication cost, difficulty obtaining inpatient reimbursement, health care resource utilization, and availability of biologics in different hospital systems prevented recommending the use of specific biologics during hospitalization. The one exception was in the case of a hospitalized patient who was already receiving infliximab for HS: there was consensus on ensuring the patient dosing was maximized, if appropriate, to 10 mg/kg.12 Ertapenem use also was controversial, with some participants using it as a bridge therapy to either outpatient biologic use or surgery, while others felt it was onerous and difficult to establish reliable access to secure intravenous administration and regular dosing once the patient left the inpatient setting.13 Others said they have experienced objections from infectious disease colleagues on the use of intravenous antibiotics, citing antibiotic stewardship concerns.
Patient Care in the Inpatient Setting—Prior literature suggests patients admitted as inpatients for HS tend to be of lower socioeconomic status and are admitted to larger urban teaching hospitals.14,15 Patients with lower socioeconomic status have increased difficulty accessing health care resources; therefore, inpatient admission serves as an opportunity to provide a holistic HS assessment and coordinate resources for chronic outpatient management.
Study Limitations—This Delphi consensus study has some limitations. The existing literature on inpatient management of HS is limited, challenging our ability to assess the extent to which these published recommendations are already being implemented. Additionally, the study included HS and inpatient dermatology experts from the United States, which means the recommendations may not be generalizable to other countries. Most participants practiced dermatology at large tertiary care academic medical centers, which may limit the ability to implement recommendations in all US inpatient care settings such as small community-based hospitals; however, many of the supportive care guidelines such as pain control, wound care, nutritional support, and social work should be achievable in most inpatient care settings.
Conclusion
Given the increase in inpatient and ED health care utilization for HS, there is an urgent need for expert consensus recommendations on inpatient management of this unique patient population, which requires complex multidisciplinary care. Our recommendations are a resource for providers to utilize and potentially improve the standard of care we provide these patients.
Acknowledgment—We thank the Wake Forest University Clinical and Translational Science Institute (Winston- Salem, North Carolina) for providing statistical help.
- Garg A, Kirby JS, Lavian J, et al. Sex- and age-adjusted population analysis of prevalence estimates for hidradenitis suppurativa in the United States. JAMA Dermatol. 2017;153:760-764.
- Ingram JR. The epidemiology of hidradenitis suppurativa. Br J Dermatol. 2020;183:990-998. doi:10.1111/bjd.19435
- Charrow A, Savage KT, Flood K, et al. Hidradenitis suppurativa for the dermatologic hospitalist. Cutis. 2019;104:276-280.
- Anzaldi L, Perkins JA, Byrd AS, et al. Characterizing inpatient hospitalizations for hidradenitis suppurativa in the United States. J Am Acad Dermatol. 2020;82:510-513. doi:10.1016/j.jaad.2019.09.019
- Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614. doi:10.1016/j.jaad.2015.06.053
- Edigin E, Kaul S, Eseaton PO, et al. At 180 days hidradenitis suppurativa readmission rate is comparable to heart failure: analysis of the nationwide readmissions database. J Am Acad Dermatol. 2022;87:188-192. doi:10.1016/j.jaad.2021.06.894
- Kirby JS, Miller JJ, Adams DR, et al. Health care utilization patterns and costs for patients with hidradenitis suppurativa. JAMA Dermatol. 2014;150:937-944. doi:10.1001/jamadermatol.2014.691
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi:10.1016/j .jaad.2019.02.067
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101. doi:10.1016/j.jaad.2019.02.068
- Seminario-Vidal L, Kroshinsky D, Malachowski SJ, et al. Society of Dermatology Hospitalists supportive care guidelines for the management of Stevens-Johnson syndrome/toxic epidermal necrolysis in adults. J Am Acad Dermatol. 2020;82:1553-1567. doi:10.1016/j .jaad.2020.02.066
- Fitch K, Bernstein SJ, Burnand B, et al. The RAND/UCLA Appropriateness Method: User’s Manual. Rand; 2001.
- Oskardmay AN, Miles JA, Sayed CJ. Determining the optimal dose of infliximab for treatment of hidradenitis suppurativa. J Am Acad Dermatol. 2019;81:702-708. doi:10.1016/j.jaad.2019.05.022
- Join-Lambert O, Coignard-Biehler H, Jais JP, et al. Efficacy of ertapenem in severe hidradenitis suppurativa: a pilot study in a cohort of 30 consecutive patients. J Antimicrob Chemother. 2016;71:513-520. doi:10.1093/jac/dkv361
- Khanna R, Whang KA, Huang AH, et al. Inpatient burden of hidradenitis suppurativa in the United States: analysis of the 2016 National Inpatient Sample. J Dermatolog Treat. 2022;33:1150-1152. doi:10.1080/09 546634.2020.1773380
- Patel A, Patel A, Solanki D, et al. Hidradenitis suppurativa in the United States: insights from the national inpatient sample (2008-2017) on contemporary trends in demographics, hospitalization rates, chronic comorbid conditions, and mortality. Cureus. 2022;14:E24755. doi:10.7759/cureus.24755
- Garg A, Kirby JS, Lavian J, et al. Sex- and age-adjusted population analysis of prevalence estimates for hidradenitis suppurativa in the United States. JAMA Dermatol. 2017;153:760-764.
- Ingram JR. The epidemiology of hidradenitis suppurativa. Br J Dermatol. 2020;183:990-998. doi:10.1111/bjd.19435
- Charrow A, Savage KT, Flood K, et al. Hidradenitis suppurativa for the dermatologic hospitalist. Cutis. 2019;104:276-280.
- Anzaldi L, Perkins JA, Byrd AS, et al. Characterizing inpatient hospitalizations for hidradenitis suppurativa in the United States. J Am Acad Dermatol. 2020;82:510-513. doi:10.1016/j.jaad.2019.09.019
- Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614. doi:10.1016/j.jaad.2015.06.053
- Edigin E, Kaul S, Eseaton PO, et al. At 180 days hidradenitis suppurativa readmission rate is comparable to heart failure: analysis of the nationwide readmissions database. J Am Acad Dermatol. 2022;87:188-192. doi:10.1016/j.jaad.2021.06.894
- Kirby JS, Miller JJ, Adams DR, et al. Health care utilization patterns and costs for patients with hidradenitis suppurativa. JAMA Dermatol. 2014;150:937-944. doi:10.1001/jamadermatol.2014.691
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi:10.1016/j .jaad.2019.02.067
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101. doi:10.1016/j.jaad.2019.02.068
- Seminario-Vidal L, Kroshinsky D, Malachowski SJ, et al. Society of Dermatology Hospitalists supportive care guidelines for the management of Stevens-Johnson syndrome/toxic epidermal necrolysis in adults. J Am Acad Dermatol. 2020;82:1553-1567. doi:10.1016/j .jaad.2020.02.066
- Fitch K, Bernstein SJ, Burnand B, et al. The RAND/UCLA Appropriateness Method: User’s Manual. Rand; 2001.
- Oskardmay AN, Miles JA, Sayed CJ. Determining the optimal dose of infliximab for treatment of hidradenitis suppurativa. J Am Acad Dermatol. 2019;81:702-708. doi:10.1016/j.jaad.2019.05.022
- Join-Lambert O, Coignard-Biehler H, Jais JP, et al. Efficacy of ertapenem in severe hidradenitis suppurativa: a pilot study in a cohort of 30 consecutive patients. J Antimicrob Chemother. 2016;71:513-520. doi:10.1093/jac/dkv361
- Khanna R, Whang KA, Huang AH, et al. Inpatient burden of hidradenitis suppurativa in the United States: analysis of the 2016 National Inpatient Sample. J Dermatolog Treat. 2022;33:1150-1152. doi:10.1080/09 546634.2020.1773380
- Patel A, Patel A, Solanki D, et al. Hidradenitis suppurativa in the United States: insights from the national inpatient sample (2008-2017) on contemporary trends in demographics, hospitalization rates, chronic comorbid conditions, and mortality. Cureus. 2022;14:E24755. doi:10.7759/cureus.24755
Practice Points
- Given the increase in hospital-based care for hidradenitis suppurativa (HS) and the lack of widespread inpatient access to dermatology and HS experts, consensus recommendations for management of HS in the acute hospital setting would be beneficial.
- Our Delphi study yielded 40 statements that reached consensus covering a range of patient care issues (eg, appropriate inpatient subspecialists [care team]), supportive care measures (wound care, pain control, genital care), disease-oriented treatment (medical management, surgical management), inpatient complications (infection control, nutrition), and successful transition to outpatient management (transitional care).
- These recommendations serve as an important resource for providers caring for inpatients with HS and represent a successful collaboration between inpatient dermatology and HS experts.
Targeting JAK Inhibitors in Severe Alopecia Areata
Alopecia areata (AA) is an autoimmune disease that affects 2% of the population. Janus kinase (JAK) signaling has been shown to improve outcomes for many patients with severe AA, as outlined by Dr Brittany Craiglow, associate professor adjunct, Yale School of Medicine, New Haven, Connecticut.
Dr Craiglow reports that in AA, hair loss can be caused by key cytokines, including interleukin 15 and interferon gamma. These cytokines use the JAK-STAT pathway to transmit their signal. JAK inhibitors, which interfere with that pathway, are showing to be an effective treatment that can lead to hair growth.
Two JAK inhibitors have received US Food and Drug Administration approval for treatment of severe AA. Oral JAK1/2 inhibitor baricitinib has been approved for patients aged 18 years or older. The oral JAK3 inhibitor ritlecitinib has been approved for patients aged 12 years or older.
Dr Craiglow looks at clinical trials involving these JAK inhibitors. The results show that patients are more likely to respond to treatment earlier in the disease process. The study also found that patients with less severe hair loss (50%-94%) respond better than did those with severe hair loss.
Finally, Dr Craiglow explores the topic of JAK inhibitor selection. She notes that different medications will hit different JAK proteins, the failure of one JAK inhibitor does not always predict failure of another. Dr Craiglow points to current efficacy of these targeted therapies and expresses optimism about the future of personalized medicine in treating patients with severe AA.
--
Brittany Craiglow, MD, Associate Professor Adjunct, Department of Dermatology, Yale University School of Medicine, New Haven, Connecticut
Brittany Craiglow, MD, has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for:
AbbVie; BiologicsMD; Dermavant; Incyte; Eli Lilly; Pfizer; Regeneron; Sanofi-Genzyme; Sun Pharmaceuticals
Serve(d) as a speaker or a member of a speaker’s bureau for: AbbVie; Incyte; Eli Lilly; Pfizer; Regeneron; Sanofi-Genzyme
Alopecia areata (AA) is an autoimmune disease that affects 2% of the population. Janus kinase (JAK) signaling has been shown to improve outcomes for many patients with severe AA, as outlined by Dr Brittany Craiglow, associate professor adjunct, Yale School of Medicine, New Haven, Connecticut.
Dr Craiglow reports that in AA, hair loss can be caused by key cytokines, including interleukin 15 and interferon gamma. These cytokines use the JAK-STAT pathway to transmit their signal. JAK inhibitors, which interfere with that pathway, are showing to be an effective treatment that can lead to hair growth.
Two JAK inhibitors have received US Food and Drug Administration approval for treatment of severe AA. Oral JAK1/2 inhibitor baricitinib has been approved for patients aged 18 years or older. The oral JAK3 inhibitor ritlecitinib has been approved for patients aged 12 years or older.
Dr Craiglow looks at clinical trials involving these JAK inhibitors. The results show that patients are more likely to respond to treatment earlier in the disease process. The study also found that patients with less severe hair loss (50%-94%) respond better than did those with severe hair loss.
Finally, Dr Craiglow explores the topic of JAK inhibitor selection. She notes that different medications will hit different JAK proteins, the failure of one JAK inhibitor does not always predict failure of another. Dr Craiglow points to current efficacy of these targeted therapies and expresses optimism about the future of personalized medicine in treating patients with severe AA.
--
Brittany Craiglow, MD, Associate Professor Adjunct, Department of Dermatology, Yale University School of Medicine, New Haven, Connecticut
Brittany Craiglow, MD, has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for:
AbbVie; BiologicsMD; Dermavant; Incyte; Eli Lilly; Pfizer; Regeneron; Sanofi-Genzyme; Sun Pharmaceuticals
Serve(d) as a speaker or a member of a speaker’s bureau for: AbbVie; Incyte; Eli Lilly; Pfizer; Regeneron; Sanofi-Genzyme
Alopecia areata (AA) is an autoimmune disease that affects 2% of the population. Janus kinase (JAK) signaling has been shown to improve outcomes for many patients with severe AA, as outlined by Dr Brittany Craiglow, associate professor adjunct, Yale School of Medicine, New Haven, Connecticut.
Dr Craiglow reports that in AA, hair loss can be caused by key cytokines, including interleukin 15 and interferon gamma. These cytokines use the JAK-STAT pathway to transmit their signal. JAK inhibitors, which interfere with that pathway, are showing to be an effective treatment that can lead to hair growth.
Two JAK inhibitors have received US Food and Drug Administration approval for treatment of severe AA. Oral JAK1/2 inhibitor baricitinib has been approved for patients aged 18 years or older. The oral JAK3 inhibitor ritlecitinib has been approved for patients aged 12 years or older.
Dr Craiglow looks at clinical trials involving these JAK inhibitors. The results show that patients are more likely to respond to treatment earlier in the disease process. The study also found that patients with less severe hair loss (50%-94%) respond better than did those with severe hair loss.
Finally, Dr Craiglow explores the topic of JAK inhibitor selection. She notes that different medications will hit different JAK proteins, the failure of one JAK inhibitor does not always predict failure of another. Dr Craiglow points to current efficacy of these targeted therapies and expresses optimism about the future of personalized medicine in treating patients with severe AA.
--
Brittany Craiglow, MD, Associate Professor Adjunct, Department of Dermatology, Yale University School of Medicine, New Haven, Connecticut
Brittany Craiglow, MD, has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for:
AbbVie; BiologicsMD; Dermavant; Incyte; Eli Lilly; Pfizer; Regeneron; Sanofi-Genzyme; Sun Pharmaceuticals
Serve(d) as a speaker or a member of a speaker’s bureau for: AbbVie; Incyte; Eli Lilly; Pfizer; Regeneron; Sanofi-Genzyme

DEA Training Mandate: 8 Hours of My Life I’d Like Back
It’s time to renew two of my three narcotic prescribing licenses. For the first time in my career, I’ve waffled on whether the financial outlay to the US Drug Enforcement Agency (DEA) is worth it.
At $888 each, I’ve considered letting two licenses lapse because I only work part-time in Montana. But several friends advised me to keep a “spare” in case I transfer to a new location.
I thought about just paying the fees until I could do a little more research, but there is no mechanism for a refund unless I die within the first year of the 3-year cycle, provide incorrect credit card digits, or accidentally duplicate payments.
The renewal fee is just part of the issue.
Mandatory 8-Hour Training
I also received an alert about the requirement for more “narcotics prescribing education” thanks to the Medication Access and Training Expansion Act (MATE).
The requirement seems counterintuitive because opioid prescribing has decreased for the 10th consecutive year, according to the AMA Overdose Epidemic Report. The continuing rise in overdose deaths is largely due to illegitimate manufacturing of synthetic opioids.
I’ve written zero outpatient narcotics prescriptions in the past 6 years, and I’ve written very few in my 33 years of practice. My use is limited to intravenous morphine for flash pulmonary edema or refractory angina, but unless you graduated from a training program within 5 years of the June 2023 mandate or are boarded in addiction medicine, there is no way to escape the 8-hour education requirement.
The problem is that these courses are never just 8 hours in duration. After signing up for one such CME course that cost $150, I was still dying of boredom and at risk for DVT 4 days later. That’s how long it took to sit through.
Instead of the 30 seconds it should have taken to review the simple instructions to deliver Narcan, there were scores of screens followed by juvenile quizlets and cartoons. All but about 2 hours out of the 4 days is now relegated to that category of “hours of my life that I can never get back.” Additionally, none of that mandatory “education” will change my prescribing habits one whit.
And beware the penalty. 
Of course, I would always be truthful when asked to check the box on the DEA renewal application attesting to my having completed the required education. On the outside chance that you plan to check the yes box without completing the relevant courses, those found guilty of such false claims could be fined up to $250,000 and subject to “not more than four years in prison,” or both. Yikes! 
Larry Houck, a former DEA investigator, explained that “[t]here are lot of people who are coming up for renewal and log on but still don’t know this is a requirement.” Neither ignorance nor complacency is an acceptable defense.
Changes Needed
The only good thing that came of those 4 long days of opioid education was a motivation to drive change in our current licensing and educational experience. Why not use this opportunity to reform the DEA-physician/prescriber relationship?
The educational requirements should be curtailed for those of us who do not provide outpatient narcotic prescriptions even if we use inpatient opioids. Meds with low abuse potential should be rescheduled to minimize who gets caught in the broad net of the education requirement.
We should reduce overregulation of the legitimate prescribers by lowering, instead of increasing, licensing fees. We should change to a single license number that covers every state. In this digital age, there is no legitimate excuse to prevent this from happening.
After all, the settlements from opioid manufacturers and distributors will in time total $50 billion. It seems that at least some of the responsibilities of the DEA could shift to states, cities, and towns.
My friend Siamak Karimian, MD, who provides locum services in multiple states, pays for seven active DEA licenses every 3 years. He pointed out the hypocrisy in the current regulatory system: “It’s funny that you can have only one DEA or state license and work for the government in all other states or territories with no limits, including the VA, Indian healthcare systems, or prison systems.”
All other prescribers require a separate DEA number for every state. Ultimately, you’d think tracking prescriptions for a single DEA number should be far simpler than tracking someone with seven.
Competent physicians not guilty of criminal overprescribing seem to be the last to be considered in nearly every healthcare endeavor these days. It would be refreshing if they would reduce our fees and prevent this waste of our time.
And while we are at it, perhaps a more fitting punishment is due for Richard Sackler and all the Purdue Pharma–affiliated family members. The Sacklers will pay out $6 billion in exchange for immunity against civil litigation. That doesn’t seem like much when they are worth $11 billion.
Perhaps they should be made to take an 8-hour course on opioid prescribing, annually and in perpetuity. Let’s see them complete a few quizlets and sit through screens of instruction on how to administer Naloxone. Of course, that would be a mild punishment for those who manufactured a drug that killed hundreds of thousands. But it would be a start.
Dr. Walton-Shirley, a clinical cardiologist in Nashville, Tennessee, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
It’s time to renew two of my three narcotic prescribing licenses. For the first time in my career, I’ve waffled on whether the financial outlay to the US Drug Enforcement Agency (DEA) is worth it.
At $888 each, I’ve considered letting two licenses lapse because I only work part-time in Montana. But several friends advised me to keep a “spare” in case I transfer to a new location.
I thought about just paying the fees until I could do a little more research, but there is no mechanism for a refund unless I die within the first year of the 3-year cycle, provide incorrect credit card digits, or accidentally duplicate payments.
The renewal fee is just part of the issue.
Mandatory 8-Hour Training
I also received an alert about the requirement for more “narcotics prescribing education” thanks to the Medication Access and Training Expansion Act (MATE).
The requirement seems counterintuitive because opioid prescribing has decreased for the 10th consecutive year, according to the AMA Overdose Epidemic Report. The continuing rise in overdose deaths is largely due to illegitimate manufacturing of synthetic opioids.
I’ve written zero outpatient narcotics prescriptions in the past 6 years, and I’ve written very few in my 33 years of practice. My use is limited to intravenous morphine for flash pulmonary edema or refractory angina, but unless you graduated from a training program within 5 years of the June 2023 mandate or are boarded in addiction medicine, there is no way to escape the 8-hour education requirement.
The problem is that these courses are never just 8 hours in duration. After signing up for one such CME course that cost $150, I was still dying of boredom and at risk for DVT 4 days later. That’s how long it took to sit through.
Instead of the 30 seconds it should have taken to review the simple instructions to deliver Narcan, there were scores of screens followed by juvenile quizlets and cartoons. All but about 2 hours out of the 4 days is now relegated to that category of “hours of my life that I can never get back.” Additionally, none of that mandatory “education” will change my prescribing habits one whit.
And beware the penalty.
Of course, I would always be truthful when asked to check the box on the DEA renewal application attesting to my having completed the required education. On the outside chance that you plan to check the yes box without completing the relevant courses, those found guilty of such false claims could be fined up to $250,000 and subject to “not more than four years in prison,” or both. Yikes!
Larry Houck, a former DEA investigator, explained that “[t]here are lot of people who are coming up for renewal and log on but still don’t know this is a requirement.” Neither ignorance nor complacency is an acceptable defense.
Changes Needed
The only good thing that came of those 4 long days of opioid education was a motivation to drive change in our current licensing and educational experience. Why not use this opportunity to reform the DEA-physician/prescriber relationship?
The educational requirements should be curtailed for those of us who do not provide outpatient narcotic prescriptions even if we use inpatient opioids. Meds with low abuse potential should be rescheduled to minimize who gets caught in the broad net of the education requirement.
We should reduce overregulation of the legitimate prescribers by lowering, instead of increasing, licensing fees. We should change to a single license number that covers every state. In this digital age, there is no legitimate excuse to prevent this from happening.
After all, the settlements from opioid manufacturers and distributors will in time total $50 billion. It seems that at least some of the responsibilities of the DEA could shift to states, cities, and towns.
My friend Siamak Karimian, MD, who provides locum services in multiple states, pays for seven active DEA licenses every 3 years. He pointed out the hypocrisy in the current regulatory system: “It’s funny that you can have only one DEA or state license and work for the government in all other states or territories with no limits, including the VA, Indian healthcare systems, or prison systems.”
All other prescribers require a separate DEA number for every state. Ultimately, you’d think tracking prescriptions for a single DEA number should be far simpler than tracking someone with seven.
Competent physicians not guilty of criminal overprescribing seem to be the last to be considered in nearly every healthcare endeavor these days. It would be refreshing if they would reduce our fees and prevent this waste of our time.
And while we are at it, perhaps a more fitting punishment is due for Richard Sackler and all the Purdue Pharma–affiliated family members. The Sacklers will pay out $6 billion in exchange for immunity against civil litigation. That doesn’t seem like much when they are worth $11 billion.
Perhaps they should be made to take an 8-hour course on opioid prescribing, annually and in perpetuity. Let’s see them complete a few quizlets and sit through screens of instruction on how to administer Naloxone. Of course, that would be a mild punishment for those who manufactured a drug that killed hundreds of thousands. But it would be a start.
Dr. Walton-Shirley, a clinical cardiologist in Nashville, Tennessee, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
It’s time to renew two of my three narcotic prescribing licenses. For the first time in my career, I’ve waffled on whether the financial outlay to the US Drug Enforcement Agency (DEA) is worth it.
At $888 each, I’ve considered letting two licenses lapse because I only work part-time in Montana. But several friends advised me to keep a “spare” in case I transfer to a new location.
I thought about just paying the fees until I could do a little more research, but there is no mechanism for a refund unless I die within the first year of the 3-year cycle, provide incorrect credit card digits, or accidentally duplicate payments.
The renewal fee is just part of the issue.
Mandatory 8-Hour Training
I also received an alert about the requirement for more “narcotics prescribing education” thanks to the Medication Access and Training Expansion Act (MATE).
The requirement seems counterintuitive because opioid prescribing has decreased for the 10th consecutive year, according to the AMA Overdose Epidemic Report. The continuing rise in overdose deaths is largely due to illegitimate manufacturing of synthetic opioids.
I’ve written zero outpatient narcotics prescriptions in the past 6 years, and I’ve written very few in my 33 years of practice. My use is limited to intravenous morphine for flash pulmonary edema or refractory angina, but unless you graduated from a training program within 5 years of the June 2023 mandate or are boarded in addiction medicine, there is no way to escape the 8-hour education requirement.
The problem is that these courses are never just 8 hours in duration. After signing up for one such CME course that cost $150, I was still dying of boredom and at risk for DVT 4 days later. That’s how long it took to sit through.
Instead of the 30 seconds it should have taken to review the simple instructions to deliver Narcan, there were scores of screens followed by juvenile quizlets and cartoons. All but about 2 hours out of the 4 days is now relegated to that category of “hours of my life that I can never get back.” Additionally, none of that mandatory “education” will change my prescribing habits one whit.
And beware the penalty.
Of course, I would always be truthful when asked to check the box on the DEA renewal application attesting to my having completed the required education. On the outside chance that you plan to check the yes box without completing the relevant courses, those found guilty of such false claims could be fined up to $250,000 and subject to “not more than four years in prison,” or both. Yikes!
Larry Houck, a former DEA investigator, explained that “[t]here are lot of people who are coming up for renewal and log on but still don’t know this is a requirement.” Neither ignorance nor complacency is an acceptable defense.
Changes Needed
The only good thing that came of those 4 long days of opioid education was a motivation to drive change in our current licensing and educational experience. Why not use this opportunity to reform the DEA-physician/prescriber relationship?
The educational requirements should be curtailed for those of us who do not provide outpatient narcotic prescriptions even if we use inpatient opioids. Meds with low abuse potential should be rescheduled to minimize who gets caught in the broad net of the education requirement.
We should reduce overregulation of the legitimate prescribers by lowering, instead of increasing, licensing fees. We should change to a single license number that covers every state. In this digital age, there is no legitimate excuse to prevent this from happening.
After all, the settlements from opioid manufacturers and distributors will in time total $50 billion. It seems that at least some of the responsibilities of the DEA could shift to states, cities, and towns.
My friend Siamak Karimian, MD, who provides locum services in multiple states, pays for seven active DEA licenses every 3 years. He pointed out the hypocrisy in the current regulatory system: “It’s funny that you can have only one DEA or state license and work for the government in all other states or territories with no limits, including the VA, Indian healthcare systems, or prison systems.”
All other prescribers require a separate DEA number for every state. Ultimately, you’d think tracking prescriptions for a single DEA number should be far simpler than tracking someone with seven.
Competent physicians not guilty of criminal overprescribing seem to be the last to be considered in nearly every healthcare endeavor these days. It would be refreshing if they would reduce our fees and prevent this waste of our time.
And while we are at it, perhaps a more fitting punishment is due for Richard Sackler and all the Purdue Pharma–affiliated family members. The Sacklers will pay out $6 billion in exchange for immunity against civil litigation. That doesn’t seem like much when they are worth $11 billion.
Perhaps they should be made to take an 8-hour course on opioid prescribing, annually and in perpetuity. Let’s see them complete a few quizlets and sit through screens of instruction on how to administer Naloxone. Of course, that would be a mild punishment for those who manufactured a drug that killed hundreds of thousands. But it would be a start.
Dr. Walton-Shirley, a clinical cardiologist in Nashville, Tennessee, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
What’s in a Name: Defining Difficult-to-Treat axSpA and PsA
Despite an expanding arsenal of disease-modifying antirheumatic drugs (DMARDs), many patients with axial spondyloarthritis (axSpA) and psoriatic arthritis (PsA) still struggle to reach remission even after trying multiple advanced treatments.
Now, international groups of experts are working to better define these “difficult-to-treat” patients to both inform care and improve selection of participants for future clinical trials.
“The idea is rather simple, and the need is relatively ubiquitous,” Denis Poddubnyy, MD, of the Charité – Universitätsmedizin Berlin and the German Rheumatism Research Center Berlin, both in Berlin, Germany, said in an interview. He is the co-primary investigator for the ongoing Assessment of SpondyloArthritis International Society (ASAS) project to develop a consensus definition of difficult-to-treat axSpA.
According to ASAS, only 40%-50% of patients with axSpA achieve a 40% improvement in ASAS response criteria (ASAS40), and few (10%-20%) achieve remission in the first 4-6 months of treatment.

“If you look into current clinical guidelines, you will see that there is no clear guidance,” on how to manage these patients, Dr. Poddubnyy continued. “In other similar recommendations for the treatment of axSpA, the only point which is clearly made with regards to nonresponders to effective anti-inflammatory treatment is to ‘check the diagnosis.’”
Multiple Reasons for Nonresponse
“While the term difficult-to-treat can refer to refractory disease, that is not the only reason why a patient might not be responding to medication. In fact, it’s likely that truly biologically refractory disease makes up only a fraction of cases that respond inadequately to treatment,” said Shikha Singla, MD, who directs the psoriatic arthritis program at the Medical College of Wisconsin in Milwaukee. She is also involved with the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) initiative to define Difficult-to-Treat and Complex-to-Manage PsA.

“Apart from the persistent articular and periarticular inflammation, there could be multiple noninflammatory factors that may be contributing to this treatment-resistant disease, including comorbid conditions such as obesity, cardiovascular disease, fibromyalgia, and even social factors such as limited access to medications,” she told this news organization. “Given these complexities, it is a matter of supreme importance to recognize and carefully delineate the elements that contribute to treatment refractory disease: Is it truly the inflammation, or are there noninflammatory components that are causing the treatment failure, or a combination of the two?”
Other contributing factors could be depression, hypersensitization, and comorbidities that prevent certain treatment approaches, added Fabian Proft, MD, also of Charité – Universitätsmedizin Berlin. Dr. Proft discussed these difficult-to-treat definition efforts at the recent Spondyloarthritis Research and Treatment Network (SPARTAN) annual meeting held in Cleveland. Patients also might not be taking their medication regularly and may be seeking alternative medicine approaches, he said.

“There is a quite clear consensus within the community” that differentiation between these two groups is needed, Dr. Proft said.
The Definitions
Terminology for these two groups can vary by professional society. The European Alliance of Associations for Rheumatology (EULAR) published a definition for “difficult-to-treat” rheumatoid arthritis (RA) that includes cases with “both inflammatory activity and/or noninflammatory complaints.”
The definition includes three criteria:
1) Treatment according to EULAR recommendation and failure of at least two biologic DMARDs (bDMARDs) or targeted synthetic DMARDs (tsDMARDs) (with different mechanisms of action) after failing conventional synthetic DMARD therapy (unless contraindicated)
2) Signs suggestive of active/progressive disease, including at least one of the following:
- Moderate disease activity (according to validated composite measures including joint counts)
- Signs (including acute phase reactants and imaging) and/or symptoms suggestive of active disease, whether joint-related or other
- Inability to taper glucocorticoid treatment
- Rapid radiographic progression (with or without signs of active disease)
- RA symptoms that are causing a reduction in quality of life
3) Symptom/sign management perceived as problematic by the rheumatologist or the patient
All three criteria must be met.
Both GRAPPA and ASAS plan to use the term “difficult-to-treat” or “treatment refractory” to describe true biologically refractory inflammatory disease and are categorizing the larger, heterogeneous group of nonresponders as “difficult-to-manage” (ASAS) or “complex-to-manage” (GRAPPA).
According to Dr. Poddubnyy, the agreed ASAS definition of difficult-to-manage has several similarities with EULAR’s RA definition, including three pillars:
- Treatment according to existing recommendations and failure of at least two different bDMARDs or tsDMARDs with different mechanisms
- Having signs and symptoms of disease (measured by high disease activity by certain disease activity indexes, persistently elevated C-reactive protein, inflammation on MRI, or rapid radiographic spinal progression)
- Symptoms/signs of disease that are considered problematic by the provider or patient
The definition was approved in January, and the manuscript is in the works, Dr. Poddubnyy said.
The GRAPPA project on PsA is still in its early stages, which so far has included a comprehensive literature review as well as a survey of GRAPPA members across 47 countries. The group is generally in agreement that two separate definitions for nonresponse to treatment are necessary, and that the “difficult-to-treat” definition — which identifies true refractory disease — should include objective signs of inflammation, Dr. Singla said.
Looking Forward
The next step of the ASAS project is to “define the pathway” from difficult-to-manage axSpA to treatment refractory disease, Dr. Poddubnyy said.
“What should be ruled out in order to exclude so-called noninflammatory causes of pain?” he continued. “It will require some Delphi exercises and [a] consensus approach.”
Proft anticipates that this treatment refractory definition in both axSpA and PsA will be most useful in research, rather than clinical practice.
“It is really important to have unified definition criteria to shape as homogeneous a cohort as possible,” he said, for future clinical trials in this population.
On the other hand, the complex/difficult-to-manage definition may be more useful for clinical practice, Dr. Proft thought.
“If you see a patient not responding to treatment, the easiest thing you can do would be to change treatment,” like swapping one biologic for another, Dr. Poddubnyy added, “but this would not be the right approach in every patient.” One goal of these initiatives is to give guidance on “what things should be looked after or excluded before you conclude this is biological [nonresponse],” he said.
Dr. Singla consults for AbbVie, Janssen, and UCB and received research funding from Eli Lilly. Dr. Poddubnyy disclosed serving as a speaker, consultant, and/or research grant recipient for multiple companies including AbbVie, Lilly, Merck Sharp and Dohme, Novartis, Pfizer, GlaxoSmithKline, Novartis, and UCB. Dr. Proft reported receiving research grants, consultant fees, or support for attending meetings and/or travel from Amgen, AbbVie, Bristol Myers Squibb, Celgene, Eli Lilly, Janssen, Merck Sharp and Dohme, Novartis, Pfizer, Roche, UCB, Medscape Medical News, Galapagos, and Hexal. Dr. Proft also participants on a data safety monitoring board or advisory board for AbbVie, Celgene, Janssen, Novartis, and UCB.
A version of this article appeared on Medscape.com.
Despite an expanding arsenal of disease-modifying antirheumatic drugs (DMARDs), many patients with axial spondyloarthritis (axSpA) and psoriatic arthritis (PsA) still struggle to reach remission even after trying multiple advanced treatments.
Now, international groups of experts are working to better define these “difficult-to-treat” patients to both inform care and improve selection of participants for future clinical trials.
“The idea is rather simple, and the need is relatively ubiquitous,” Denis Poddubnyy, MD, of the Charité – Universitätsmedizin Berlin and the German Rheumatism Research Center Berlin, both in Berlin, Germany, said in an interview. He is the co-primary investigator for the ongoing Assessment of SpondyloArthritis International Society (ASAS) project to develop a consensus definition of difficult-to-treat axSpA.
According to ASAS, only 40%-50% of patients with axSpA achieve a 40% improvement in ASAS response criteria (ASAS40), and few (10%-20%) achieve remission in the first 4-6 months of treatment.
“If you look into current clinical guidelines, you will see that there is no clear guidance,” on how to manage these patients, Dr. Poddubnyy continued. “In other similar recommendations for the treatment of axSpA, the only point which is clearly made with regards to nonresponders to effective anti-inflammatory treatment is to ‘check the diagnosis.’”
Multiple Reasons for Nonresponse
“While the term difficult-to-treat can refer to refractory disease, that is not the only reason why a patient might not be responding to medication. In fact, it’s likely that truly biologically refractory disease makes up only a fraction of cases that respond inadequately to treatment,” said Shikha Singla, MD, who directs the psoriatic arthritis program at the Medical College of Wisconsin in Milwaukee. She is also involved with the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) initiative to define Difficult-to-Treat and Complex-to-Manage PsA.
“Apart from the persistent articular and periarticular inflammation, there could be multiple noninflammatory factors that may be contributing to this treatment-resistant disease, including comorbid conditions such as obesity, cardiovascular disease, fibromyalgia, and even social factors such as limited access to medications,” she told this news organization. “Given these complexities, it is a matter of supreme importance to recognize and carefully delineate the elements that contribute to treatment refractory disease: Is it truly the inflammation, or are there noninflammatory components that are causing the treatment failure, or a combination of the two?”
Other contributing factors could be depression, hypersensitization, and comorbidities that prevent certain treatment approaches, added Fabian Proft, MD, also of Charité – Universitätsmedizin Berlin. Dr. Proft discussed these difficult-to-treat definition efforts at the recent Spondyloarthritis Research and Treatment Network (SPARTAN) annual meeting held in Cleveland. Patients also might not be taking their medication regularly and may be seeking alternative medicine approaches, he said.
“There is a quite clear consensus within the community” that differentiation between these two groups is needed, Dr. Proft said.
The Definitions
Terminology for these two groups can vary by professional society. The European Alliance of Associations for Rheumatology (EULAR) published a definition for “difficult-to-treat” rheumatoid arthritis (RA) that includes cases with “both inflammatory activity and/or noninflammatory complaints.”
The definition includes three criteria:
1) Treatment according to EULAR recommendation and failure of at least two biologic DMARDs (bDMARDs) or targeted synthetic DMARDs (tsDMARDs) (with different mechanisms of action) after failing conventional synthetic DMARD therapy (unless contraindicated)
2) Signs suggestive of active/progressive disease, including at least one of the following:
- Moderate disease activity (according to validated composite measures including joint counts)
- Signs (including acute phase reactants and imaging) and/or symptoms suggestive of active disease, whether joint-related or other
- Inability to taper glucocorticoid treatment
- Rapid radiographic progression (with or without signs of active disease)
- RA symptoms that are causing a reduction in quality of life
3) Symptom/sign management perceived as problematic by the rheumatologist or the patient
All three criteria must be met.
Both GRAPPA and ASAS plan to use the term “difficult-to-treat” or “treatment refractory” to describe true biologically refractory inflammatory disease and are categorizing the larger, heterogeneous group of nonresponders as “difficult-to-manage” (ASAS) or “complex-to-manage” (GRAPPA).
According to Dr. Poddubnyy, the agreed ASAS definition of difficult-to-manage has several similarities with EULAR’s RA definition, including three pillars:
- Treatment according to existing recommendations and failure of at least two different bDMARDs or tsDMARDs with different mechanisms
- Having signs and symptoms of disease (measured by high disease activity by certain disease activity indexes, persistently elevated C-reactive protein, inflammation on MRI, or rapid radiographic spinal progression)
- Symptoms/signs of disease that are considered problematic by the provider or patient
The definition was approved in January, and the manuscript is in the works, Dr. Poddubnyy said.
The GRAPPA project on PsA is still in its early stages, which so far has included a comprehensive literature review as well as a survey of GRAPPA members across 47 countries. The group is generally in agreement that two separate definitions for nonresponse to treatment are necessary, and that the “difficult-to-treat” definition — which identifies true refractory disease — should include objective signs of inflammation, Dr. Singla said.
Looking Forward
The next step of the ASAS project is to “define the pathway” from difficult-to-manage axSpA to treatment refractory disease, Dr. Poddubnyy said.
“What should be ruled out in order to exclude so-called noninflammatory causes of pain?” he continued. “It will require some Delphi exercises and [a] consensus approach.”
Proft anticipates that this treatment refractory definition in both axSpA and PsA will be most useful in research, rather than clinical practice.
“It is really important to have unified definition criteria to shape as homogeneous a cohort as possible,” he said, for future clinical trials in this population.
On the other hand, the complex/difficult-to-manage definition may be more useful for clinical practice, Dr. Proft thought.
“If you see a patient not responding to treatment, the easiest thing you can do would be to change treatment,” like swapping one biologic for another, Dr. Poddubnyy added, “but this would not be the right approach in every patient.” One goal of these initiatives is to give guidance on “what things should be looked after or excluded before you conclude this is biological [nonresponse],” he said.
Dr. Singla consults for AbbVie, Janssen, and UCB and received research funding from Eli Lilly. Dr. Poddubnyy disclosed serving as a speaker, consultant, and/or research grant recipient for multiple companies including AbbVie, Lilly, Merck Sharp and Dohme, Novartis, Pfizer, GlaxoSmithKline, Novartis, and UCB. Dr. Proft reported receiving research grants, consultant fees, or support for attending meetings and/or travel from Amgen, AbbVie, Bristol Myers Squibb, Celgene, Eli Lilly, Janssen, Merck Sharp and Dohme, Novartis, Pfizer, Roche, UCB, Medscape Medical News, Galapagos, and Hexal. Dr. Proft also participants on a data safety monitoring board or advisory board for AbbVie, Celgene, Janssen, Novartis, and UCB.
A version of this article appeared on Medscape.com.
Despite an expanding arsenal of disease-modifying antirheumatic drugs (DMARDs), many patients with axial spondyloarthritis (axSpA) and psoriatic arthritis (PsA) still struggle to reach remission even after trying multiple advanced treatments.
Now, international groups of experts are working to better define these “difficult-to-treat” patients to both inform care and improve selection of participants for future clinical trials.
“The idea is rather simple, and the need is relatively ubiquitous,” Denis Poddubnyy, MD, of the Charité – Universitätsmedizin Berlin and the German Rheumatism Research Center Berlin, both in Berlin, Germany, said in an interview. He is the co-primary investigator for the ongoing Assessment of SpondyloArthritis International Society (ASAS) project to develop a consensus definition of difficult-to-treat axSpA.
According to ASAS, only 40%-50% of patients with axSpA achieve a 40% improvement in ASAS response criteria (ASAS40), and few (10%-20%) achieve remission in the first 4-6 months of treatment.
“If you look into current clinical guidelines, you will see that there is no clear guidance,” on how to manage these patients, Dr. Poddubnyy continued. “In other similar recommendations for the treatment of axSpA, the only point which is clearly made with regards to nonresponders to effective anti-inflammatory treatment is to ‘check the diagnosis.’”
Multiple Reasons for Nonresponse
“While the term difficult-to-treat can refer to refractory disease, that is not the only reason why a patient might not be responding to medication. In fact, it’s likely that truly biologically refractory disease makes up only a fraction of cases that respond inadequately to treatment,” said Shikha Singla, MD, who directs the psoriatic arthritis program at the Medical College of Wisconsin in Milwaukee. She is also involved with the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) initiative to define Difficult-to-Treat and Complex-to-Manage PsA.
“Apart from the persistent articular and periarticular inflammation, there could be multiple noninflammatory factors that may be contributing to this treatment-resistant disease, including comorbid conditions such as obesity, cardiovascular disease, fibromyalgia, and even social factors such as limited access to medications,” she told this news organization. “Given these complexities, it is a matter of supreme importance to recognize and carefully delineate the elements that contribute to treatment refractory disease: Is it truly the inflammation, or are there noninflammatory components that are causing the treatment failure, or a combination of the two?”
Other contributing factors could be depression, hypersensitization, and comorbidities that prevent certain treatment approaches, added Fabian Proft, MD, also of Charité – Universitätsmedizin Berlin. Dr. Proft discussed these difficult-to-treat definition efforts at the recent Spondyloarthritis Research and Treatment Network (SPARTAN) annual meeting held in Cleveland. Patients also might not be taking their medication regularly and may be seeking alternative medicine approaches, he said.
“There is a quite clear consensus within the community” that differentiation between these two groups is needed, Dr. Proft said.
The Definitions
Terminology for these two groups can vary by professional society. The European Alliance of Associations for Rheumatology (EULAR) published a definition for “difficult-to-treat” rheumatoid arthritis (RA) that includes cases with “both inflammatory activity and/or noninflammatory complaints.”
The definition includes three criteria:
1) Treatment according to EULAR recommendation and failure of at least two biologic DMARDs (bDMARDs) or targeted synthetic DMARDs (tsDMARDs) (with different mechanisms of action) after failing conventional synthetic DMARD therapy (unless contraindicated)
2) Signs suggestive of active/progressive disease, including at least one of the following:
- Moderate disease activity (according to validated composite measures including joint counts)
- Signs (including acute phase reactants and imaging) and/or symptoms suggestive of active disease, whether joint-related or other
- Inability to taper glucocorticoid treatment
- Rapid radiographic progression (with or without signs of active disease)
- RA symptoms that are causing a reduction in quality of life
3) Symptom/sign management perceived as problematic by the rheumatologist or the patient
All three criteria must be met.
Both GRAPPA and ASAS plan to use the term “difficult-to-treat” or “treatment refractory” to describe true biologically refractory inflammatory disease and are categorizing the larger, heterogeneous group of nonresponders as “difficult-to-manage” (ASAS) or “complex-to-manage” (GRAPPA).
According to Dr. Poddubnyy, the agreed ASAS definition of difficult-to-manage has several similarities with EULAR’s RA definition, including three pillars:
- Treatment according to existing recommendations and failure of at least two different bDMARDs or tsDMARDs with different mechanisms
- Having signs and symptoms of disease (measured by high disease activity by certain disease activity indexes, persistently elevated C-reactive protein, inflammation on MRI, or rapid radiographic spinal progression)
- Symptoms/signs of disease that are considered problematic by the provider or patient
The definition was approved in January, and the manuscript is in the works, Dr. Poddubnyy said.
The GRAPPA project on PsA is still in its early stages, which so far has included a comprehensive literature review as well as a survey of GRAPPA members across 47 countries. The group is generally in agreement that two separate definitions for nonresponse to treatment are necessary, and that the “difficult-to-treat” definition — which identifies true refractory disease — should include objective signs of inflammation, Dr. Singla said.
Looking Forward
The next step of the ASAS project is to “define the pathway” from difficult-to-manage axSpA to treatment refractory disease, Dr. Poddubnyy said.
“What should be ruled out in order to exclude so-called noninflammatory causes of pain?” he continued. “It will require some Delphi exercises and [a] consensus approach.”
Proft anticipates that this treatment refractory definition in both axSpA and PsA will be most useful in research, rather than clinical practice.
“It is really important to have unified definition criteria to shape as homogeneous a cohort as possible,” he said, for future clinical trials in this population.
On the other hand, the complex/difficult-to-manage definition may be more useful for clinical practice, Dr. Proft thought.
“If you see a patient not responding to treatment, the easiest thing you can do would be to change treatment,” like swapping one biologic for another, Dr. Poddubnyy added, “but this would not be the right approach in every patient.” One goal of these initiatives is to give guidance on “what things should be looked after or excluded before you conclude this is biological [nonresponse],” he said.
Dr. Singla consults for AbbVie, Janssen, and UCB and received research funding from Eli Lilly. Dr. Poddubnyy disclosed serving as a speaker, consultant, and/or research grant recipient for multiple companies including AbbVie, Lilly, Merck Sharp and Dohme, Novartis, Pfizer, GlaxoSmithKline, Novartis, and UCB. Dr. Proft reported receiving research grants, consultant fees, or support for attending meetings and/or travel from Amgen, AbbVie, Bristol Myers Squibb, Celgene, Eli Lilly, Janssen, Merck Sharp and Dohme, Novartis, Pfizer, Roche, UCB, Medscape Medical News, Galapagos, and Hexal. Dr. Proft also participants on a data safety monitoring board or advisory board for AbbVie, Celgene, Janssen, Novartis, and UCB.
A version of this article appeared on Medscape.com.
FROM SPARTAN 2024
Autoantibodies Nonspecific to Systemic Sclerosis May Play Role in ILD Prediction
VIENNA — Anti-Ro/SSA antibodies may help predict which patients with systemic sclerosis (SSc) are at a greater risk for interstitial lung disease (ILD) and may serve as a biomarker to guide screening, according to an analysis of data from a large European cohort.
The researchers were led by Blaž Burja, MD, PhD, a physician-scientist at the Center of Experimental Rheumatology, University Hospital Zürich, Switzerland, who reported that anti-Ro/SSA antibodies are a risk factor for ILD, with an odds ratio of 1.24, in patients with SSc.
At the annual European Congress of Rheumatology, he presented the findings of the study that aimed to find out if SSc-nonspecific antibodies might help better risk-stratify patients with SSc, focusing on lung involvement. “Among them, anti-Ro/SSA antibodies have been shown to be associated with interstitial lung disease in different connective tissue diseases,” Dr. Burja pointed out.
“A total of 15% of all patients in the SSc cohort presented with anti-Ro/SSA antibodies, and this subgroup presented with distinct clinical features: Importantly, higher prevalence of ILD and lower DLCO% [diffusing capacity of the lungs for carbon monoxide] in patients with established ILD,” reported Dr. Burja. “However, these anti-Ro/SSA antibodies do not predict ILD progression, death, or overall disease progression.”
Based on the findings, Dr. Burja suggested that these antibodies be incorporated into routine clinical practice to identify patients with SSc who have a high risk for ILD. He noted that “this has specific importance in clinical settings without availability of high-resolution computed tomography (HRCT), where anti-Ro/SSA antibodies could represent an additional biomarker to guide the screening process, in particular, in patients without SSc-specific antibodies.”
Caroline Ospelt, MD, PhD, co-moderator of the session and scientific program chair of EULAR 2024, told this news organization that the study was unique in its approach to studying ILD risk by “looking outside the box, so not just at specific antibodies but whether cross-disease antibodies may have value in stratifying patients and help predict risk of lung involvement and possibly monitor these patients.”
Dr. Ospelt, professor of experimental rheumatology at University Hospital Zürich, who was not involved in the study, noted: “It might also be the case that we could adapt this concept and use these antibodies in other rheumatic diseases, too, not just systemic sclerosis, to predict lung involvement.”
Risk-Stratifying With SSc-Nonspecific Antibodies
Dr. Burja explained that despite better stratification of patients with SSc with SSc-specific antibodies, “in clinical practice, we see large heterogeneity, and individual prognosis with regards to outcomes is still unpredictable, so we wanted to know whether by using nonspecific autoantibodies we might be better able to risk-stratify these patients.”
A study population of 4421 with at least one follow-up visit, including 3060 patients with available follow-up serologic data, was drawn from the European Scleroderma Trials and Research group database (n = 22,482). Of these 3060 patients, 461 were positive for anti-Ro/SSA antibodies and 2599 were negative. The researchers analyzed the relationships between baseline characteristics and the development or progression of ILD over 2.7 years of follow-up. Incident, de novo ILD was defined based on its presence on HRCT, and progression was defined by whether the percentage of predicted forced vital capacity (FVC%) dropped ≥ 10%, FVC% dropped 5%-9% in association with a DLCO% drop ≥ 15%, or FVC% dropped > 5%. Deaths from all causes and prognostic factors for the progression of lung fibrosis during follow-up were recorded.
High Prevalence of ILD With Anti-Ro/SSA Antibodies in SSc
At baseline, patients with anti-Ro/SSA antibodies were aged 55-56 years, 84%-87% were women, and muscular involvement was present in 18% of patients positive for anti-Ro/SSA antibodies and 12.5% of those who were negative (P < .001). According to HRCT, ILD was present in 56.2% of patients positive for anti-Ro/SSA antibodies and in 47.8% of those who were negative (P = .001). FVC% was 92.5% in patients positive for anti-Ro/SSA antibodies and 95.7% in those who were negative (P = .002). DLCO% was 66.9% in patients positive for anti-Ro/SSA antibodies and 71% in those who were negative (P < .001).
“A total of 15% of all SSc patients presented as positive for anti-Ro/SSA antibodies, and these patients all presented with higher prevalence of SSA-nonspecific antibodies, too: Of note, those with anti-La/SSB and anti-U1/RNP and rheumatoid factor,” Dr. Burja reported.
In patients with anti-U1/RNP autoantibodies, 1% were positive and 4% were negative for anti-Ro/SSA antibodies; in those with anti-La/SSB autoantibodies, 17% were positive and 1% were negative for anti-Ro/SSA antibodies; and in those with rheumatoid factor, 28% were positive and 14% were negative for anti-Ro/SSA antibodies.
Dr. Burja pointed out that the average disease duration in the study cohort at baseline was 7 years, “and at this timepoint, we expect to see some common disease manifestations. Specifically, higher muscular involvement and higher ILD based on HRCT.
“We decided to focus on patients with established ILD at baseline,” said Dr. Burja. “Anti-Ro/SSA-positive patients with established ILD at baseline presented with lower DLCO values at 59% in patients positive for anti-Ro/SSA antibodies and 61% for those who were negative.”
After conducting a multivariable analysis of 14,066 healthcare visits and adjusting for known risk factors for ILD, the researchers concluded that anti-Ro/SSA antibodies are an independent risk factor for ILD, with an odds ratio of 1.24 (95% CI, 1.07-1.44; P = .006). They also determined that anti-Ro/SSA antibodies are a risk factor for lower DLCO values in patients with ILD, with a regression coefficient of −1.93.
The researchers then explored the progression of ILD and overall disease progression and survival during the follow-up period in a longitudinal analysis. “However, anti-Ro/SSA antibodies were not found to predict the progression of ILD,” reported Dr. Burja, adding that this was true regardless of the definition of ILD progression used. “Nor did anti-Ro/SSA antibodies do not predict survival or overall disease progression.”
Dr. Burja pointed out the limitations in his study, including the lack of standardized criteria for all centers to assess anti-Ro/SSA positivity; there was a lack of discrimination between anti-Ro52 and anti-Ro60 subtypes, and there were no standardized applicable criteria to study lung progression in SSc.
Dr. Burja and Dr. Ospelt had no relevant financial disclosures.
A version of this article appeared on Medscape.com.
VIENNA — Anti-Ro/SSA antibodies may help predict which patients with systemic sclerosis (SSc) are at a greater risk for interstitial lung disease (ILD) and may serve as a biomarker to guide screening, according to an analysis of data from a large European cohort.
The researchers were led by Blaž Burja, MD, PhD, a physician-scientist at the Center of Experimental Rheumatology, University Hospital Zürich, Switzerland, who reported that anti-Ro/SSA antibodies are a risk factor for ILD, with an odds ratio of 1.24, in patients with SSc.
At the annual European Congress of Rheumatology, he presented the findings of the study that aimed to find out if SSc-nonspecific antibodies might help better risk-stratify patients with SSc, focusing on lung involvement. “Among them, anti-Ro/SSA antibodies have been shown to be associated with interstitial lung disease in different connective tissue diseases,” Dr. Burja pointed out.
“A total of 15% of all patients in the SSc cohort presented with anti-Ro/SSA antibodies, and this subgroup presented with distinct clinical features: Importantly, higher prevalence of ILD and lower DLCO% [diffusing capacity of the lungs for carbon monoxide] in patients with established ILD,” reported Dr. Burja. “However, these anti-Ro/SSA antibodies do not predict ILD progression, death, or overall disease progression.”
Based on the findings, Dr. Burja suggested that these antibodies be incorporated into routine clinical practice to identify patients with SSc who have a high risk for ILD. He noted that “this has specific importance in clinical settings without availability of high-resolution computed tomography (HRCT), where anti-Ro/SSA antibodies could represent an additional biomarker to guide the screening process, in particular, in patients without SSc-specific antibodies.”
Caroline Ospelt, MD, PhD, co-moderator of the session and scientific program chair of EULAR 2024, told this news organization that the study was unique in its approach to studying ILD risk by “looking outside the box, so not just at specific antibodies but whether cross-disease antibodies may have value in stratifying patients and help predict risk of lung involvement and possibly monitor these patients.”
Dr. Ospelt, professor of experimental rheumatology at University Hospital Zürich, who was not involved in the study, noted: “It might also be the case that we could adapt this concept and use these antibodies in other rheumatic diseases, too, not just systemic sclerosis, to predict lung involvement.”
Risk-Stratifying With SSc-Nonspecific Antibodies
Dr. Burja explained that despite better stratification of patients with SSc with SSc-specific antibodies, “in clinical practice, we see large heterogeneity, and individual prognosis with regards to outcomes is still unpredictable, so we wanted to know whether by using nonspecific autoantibodies we might be better able to risk-stratify these patients.”
A study population of 4421 with at least one follow-up visit, including 3060 patients with available follow-up serologic data, was drawn from the European Scleroderma Trials and Research group database (n = 22,482). Of these 3060 patients, 461 were positive for anti-Ro/SSA antibodies and 2599 were negative. The researchers analyzed the relationships between baseline characteristics and the development or progression of ILD over 2.7 years of follow-up. Incident, de novo ILD was defined based on its presence on HRCT, and progression was defined by whether the percentage of predicted forced vital capacity (FVC%) dropped ≥ 10%, FVC% dropped 5%-9% in association with a DLCO% drop ≥ 15%, or FVC% dropped > 5%. Deaths from all causes and prognostic factors for the progression of lung fibrosis during follow-up were recorded.
High Prevalence of ILD With Anti-Ro/SSA Antibodies in SSc
At baseline, patients with anti-Ro/SSA antibodies were aged 55-56 years, 84%-87% were women, and muscular involvement was present in 18% of patients positive for anti-Ro/SSA antibodies and 12.5% of those who were negative (P < .001). According to HRCT, ILD was present in 56.2% of patients positive for anti-Ro/SSA antibodies and in 47.8% of those who were negative (P = .001). FVC% was 92.5% in patients positive for anti-Ro/SSA antibodies and 95.7% in those who were negative (P = .002). DLCO% was 66.9% in patients positive for anti-Ro/SSA antibodies and 71% in those who were negative (P < .001).
“A total of 15% of all SSc patients presented as positive for anti-Ro/SSA antibodies, and these patients all presented with higher prevalence of SSA-nonspecific antibodies, too: Of note, those with anti-La/SSB and anti-U1/RNP and rheumatoid factor,” Dr. Burja reported.
In patients with anti-U1/RNP autoantibodies, 1% were positive and 4% were negative for anti-Ro/SSA antibodies; in those with anti-La/SSB autoantibodies, 17% were positive and 1% were negative for anti-Ro/SSA antibodies; and in those with rheumatoid factor, 28% were positive and 14% were negative for anti-Ro/SSA antibodies.
Dr. Burja pointed out that the average disease duration in the study cohort at baseline was 7 years, “and at this timepoint, we expect to see some common disease manifestations. Specifically, higher muscular involvement and higher ILD based on HRCT.
“We decided to focus on patients with established ILD at baseline,” said Dr. Burja. “Anti-Ro/SSA-positive patients with established ILD at baseline presented with lower DLCO values at 59% in patients positive for anti-Ro/SSA antibodies and 61% for those who were negative.”
After conducting a multivariable analysis of 14,066 healthcare visits and adjusting for known risk factors for ILD, the researchers concluded that anti-Ro/SSA antibodies are an independent risk factor for ILD, with an odds ratio of 1.24 (95% CI, 1.07-1.44; P = .006). They also determined that anti-Ro/SSA antibodies are a risk factor for lower DLCO values in patients with ILD, with a regression coefficient of −1.93.
The researchers then explored the progression of ILD and overall disease progression and survival during the follow-up period in a longitudinal analysis. “However, anti-Ro/SSA antibodies were not found to predict the progression of ILD,” reported Dr. Burja, adding that this was true regardless of the definition of ILD progression used. “Nor did anti-Ro/SSA antibodies do not predict survival or overall disease progression.”
Dr. Burja pointed out the limitations in his study, including the lack of standardized criteria for all centers to assess anti-Ro/SSA positivity; there was a lack of discrimination between anti-Ro52 and anti-Ro60 subtypes, and there were no standardized applicable criteria to study lung progression in SSc.
Dr. Burja and Dr. Ospelt had no relevant financial disclosures.
A version of this article appeared on Medscape.com.
VIENNA — Anti-Ro/SSA antibodies may help predict which patients with systemic sclerosis (SSc) are at a greater risk for interstitial lung disease (ILD) and may serve as a biomarker to guide screening, according to an analysis of data from a large European cohort.
The researchers were led by Blaž Burja, MD, PhD, a physician-scientist at the Center of Experimental Rheumatology, University Hospital Zürich, Switzerland, who reported that anti-Ro/SSA antibodies are a risk factor for ILD, with an odds ratio of 1.24, in patients with SSc.
At the annual European Congress of Rheumatology, he presented the findings of the study that aimed to find out if SSc-nonspecific antibodies might help better risk-stratify patients with SSc, focusing on lung involvement. “Among them, anti-Ro/SSA antibodies have been shown to be associated with interstitial lung disease in different connective tissue diseases,” Dr. Burja pointed out.
“A total of 15% of all patients in the SSc cohort presented with anti-Ro/SSA antibodies, and this subgroup presented with distinct clinical features: Importantly, higher prevalence of ILD and lower DLCO% [diffusing capacity of the lungs for carbon monoxide] in patients with established ILD,” reported Dr. Burja. “However, these anti-Ro/SSA antibodies do not predict ILD progression, death, or overall disease progression.”
Based on the findings, Dr. Burja suggested that these antibodies be incorporated into routine clinical practice to identify patients with SSc who have a high risk for ILD. He noted that “this has specific importance in clinical settings without availability of high-resolution computed tomography (HRCT), where anti-Ro/SSA antibodies could represent an additional biomarker to guide the screening process, in particular, in patients without SSc-specific antibodies.”
Caroline Ospelt, MD, PhD, co-moderator of the session and scientific program chair of EULAR 2024, told this news organization that the study was unique in its approach to studying ILD risk by “looking outside the box, so not just at specific antibodies but whether cross-disease antibodies may have value in stratifying patients and help predict risk of lung involvement and possibly monitor these patients.”
Dr. Ospelt, professor of experimental rheumatology at University Hospital Zürich, who was not involved in the study, noted: “It might also be the case that we could adapt this concept and use these antibodies in other rheumatic diseases, too, not just systemic sclerosis, to predict lung involvement.”
Risk-Stratifying With SSc-Nonspecific Antibodies
Dr. Burja explained that despite better stratification of patients with SSc with SSc-specific antibodies, “in clinical practice, we see large heterogeneity, and individual prognosis with regards to outcomes is still unpredictable, so we wanted to know whether by using nonspecific autoantibodies we might be better able to risk-stratify these patients.”
A study population of 4421 with at least one follow-up visit, including 3060 patients with available follow-up serologic data, was drawn from the European Scleroderma Trials and Research group database (n = 22,482). Of these 3060 patients, 461 were positive for anti-Ro/SSA antibodies and 2599 were negative. The researchers analyzed the relationships between baseline characteristics and the development or progression of ILD over 2.7 years of follow-up. Incident, de novo ILD was defined based on its presence on HRCT, and progression was defined by whether the percentage of predicted forced vital capacity (FVC%) dropped ≥ 10%, FVC% dropped 5%-9% in association with a DLCO% drop ≥ 15%, or FVC% dropped > 5%. Deaths from all causes and prognostic factors for the progression of lung fibrosis during follow-up were recorded.
High Prevalence of ILD With Anti-Ro/SSA Antibodies in SSc
At baseline, patients with anti-Ro/SSA antibodies were aged 55-56 years, 84%-87% were women, and muscular involvement was present in 18% of patients positive for anti-Ro/SSA antibodies and 12.5% of those who were negative (P < .001). According to HRCT, ILD was present in 56.2% of patients positive for anti-Ro/SSA antibodies and in 47.8% of those who were negative (P = .001). FVC% was 92.5% in patients positive for anti-Ro/SSA antibodies and 95.7% in those who were negative (P = .002). DLCO% was 66.9% in patients positive for anti-Ro/SSA antibodies and 71% in those who were negative (P < .001).
“A total of 15% of all SSc patients presented as positive for anti-Ro/SSA antibodies, and these patients all presented with higher prevalence of SSA-nonspecific antibodies, too: Of note, those with anti-La/SSB and anti-U1/RNP and rheumatoid factor,” Dr. Burja reported.
In patients with anti-U1/RNP autoantibodies, 1% were positive and 4% were negative for anti-Ro/SSA antibodies; in those with anti-La/SSB autoantibodies, 17% were positive and 1% were negative for anti-Ro/SSA antibodies; and in those with rheumatoid factor, 28% were positive and 14% were negative for anti-Ro/SSA antibodies.
Dr. Burja pointed out that the average disease duration in the study cohort at baseline was 7 years, “and at this timepoint, we expect to see some common disease manifestations. Specifically, higher muscular involvement and higher ILD based on HRCT.
“We decided to focus on patients with established ILD at baseline,” said Dr. Burja. “Anti-Ro/SSA-positive patients with established ILD at baseline presented with lower DLCO values at 59% in patients positive for anti-Ro/SSA antibodies and 61% for those who were negative.”
After conducting a multivariable analysis of 14,066 healthcare visits and adjusting for known risk factors for ILD, the researchers concluded that anti-Ro/SSA antibodies are an independent risk factor for ILD, with an odds ratio of 1.24 (95% CI, 1.07-1.44; P = .006). They also determined that anti-Ro/SSA antibodies are a risk factor for lower DLCO values in patients with ILD, with a regression coefficient of −1.93.
The researchers then explored the progression of ILD and overall disease progression and survival during the follow-up period in a longitudinal analysis. “However, anti-Ro/SSA antibodies were not found to predict the progression of ILD,” reported Dr. Burja, adding that this was true regardless of the definition of ILD progression used. “Nor did anti-Ro/SSA antibodies do not predict survival or overall disease progression.”
Dr. Burja pointed out the limitations in his study, including the lack of standardized criteria for all centers to assess anti-Ro/SSA positivity; there was a lack of discrimination between anti-Ro52 and anti-Ro60 subtypes, and there were no standardized applicable criteria to study lung progression in SSc.
Dr. Burja and Dr. Ospelt had no relevant financial disclosures.
A version of this article appeared on Medscape.com.
FROM EULAR 2024
Reticulated Brownish Erythema on the Lower Back
The Diagnosis: Erythema Ab Igne
Based on the patient's long-standing history of back pain treated with heating pads as well as the normal laboratory findings and skin examination, a diagnosis of erythema ab igne (EAI) was made.
Erythema ab igne presents as reticulated brownish erythema or hyperpigmentation on sites exposed to prolonged use of heat sources such as heating pads, laptops, and space heaters. Erythema ab igne most commonly affects the lower back, thighs, or legs1-6; however, EAI can appear on atypical sites such as the forehead and eyebrows due to newer technology (eg, virtual reality headsets).7 The level of heat required for EAI to occur is below the threshold for thermal burns (<45 °C [113 °F]).1 Erythema ab igne can occur at any age, and woman are more commonly affected than men.8 The pathophysiology currently is unknown; however, recurrent and prolonged heat exposure may damage superficial vessels. As a result, hemosiderin accumulates in the skin, and hyperpigmentation subsequently occurs.9
The diagnosis of EAI is clinical, and early stages of the rash present as blanching reticulated erythema in areas associated with heat exposure. If the offending source of heat is not removed, EAI can progress to nonblanching, fixed, hyperpigmented plaques with skin atrophy, bullae, or hyperkeratosis. Patients often are asymptomatic; however, mild burning may occur.2 Histopathology reveals cellular atypia, epidermal atrophy, dilation of dermal blood vessels, a minute inflammatory infiltrate, and keratinocyte apoptosis.10 Skin biopsy may be necessary in cases of suspected malignancy due to chronic heat exposure. Lesions that ulcerate or evolve should raise suspicion for malignancy.11 Squamous cell carcinoma is the most common malignancy associated with EAI; other malignancies that may manifest include basal cell carcinoma, Merkel cell carcinoma, or cutaneous marginal zone lymphoma.2,12-14
Erythema ab igne often is mistaken for livedo reticularis, which appears more erythematous without hyperpigmentation or epidermal changes and may be associated with a pathologic state.15 The differential diagnosis in our patient, who was in her 40s with a history of fatigue and joint pain, included livedo reticularis associated with lupus; however, the history of heating pad use, normal laboratory findings, and presence of epidermal changes suggested EAI. Lupus typically affects the hand and knee joints.16 Additionally, livedo reticularis more commonly appears on the legs.15
Other differentials for EAI include livedo racemosa, cutaneous T-cell lymphoma, and cutis marmorata. Livedo racemosa presents with broken rings of erythema in young to middle-aged women and primarily affects the trunk and proximal limbs. It is associated with an underlying condition such as polyarteritis nodosa and less commonly with lupus erythematosus with antiphospholipid or Sneddon syndrome.15,17 Cutaneous T-cell lymphoma typically manifests with poikilodermatous patches larger than the palm, especially in covered areas of skin.18 Cutis marmorata is transient and temperature dependent.9
The key intervention for EAI is removal of the offending heat source.2 Patients should be counseled that the erythema and hyperpigmentation may take months to years to resolve. Topical hydroquinone or tretinoin may be used in cases of persistent hyperpigmentation.19 Patients who continue to use heating pads for long-standing pain should be advised to limit their use to short intervals without occlusion. If malignancy is a concern, a biopsy should be performed.20
- Wipf AJ, Brown MR. Malignant transformation of erythema ab igne. JAAD Case Rep. 2022;26:85-87. doi:10.1016/j.jdcr.2022.06.018
- Sigmon JR, Cantrell J, Teague D, et al. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35:676-678. doi:10.1097/DAD.0b013e3182871648
- Patel DP. The evolving nomenclature of erythema ab igne-redness from fire. JAMA Dermatol. 2017;153:685. doi:10.1001/jamadermatol.2017.2021
- Arnold AW, Itin PH. Laptop computer-induced erythema ab igne in a child and review of the literature. Pediatrics. 2010;126:E1227-E1230. doi:10.1542/peds.2010-1390
- Riahi RR, Cohen PR. Laptop-induced erythema ab igne: report and review of literature. Dermatol Online J. 2012;18:5.
- Haleem Z, Philip J, Muhammad S. Erythema ab igne: a rare presentation of toasted skin syndrome with the use of a space heater. Cureus. 2021;13:e13401. doi:10.7759/cureus.13401
- Moreau T, Benzaquen M, Gueissaz F. Erythema ab igne after using a virtual reality headset: a new phenomenon to know. J Eur Acad Dermatol Venereol. 2022;36:E932-E933. doi:10.1111/jdv.18371
- Ozturk M, An I. Clinical features and etiology of patients with erythema ab igne: a retrospective multicenter study. J Cosmet Dermatol. 2020;19:1774-1779. doi:10.1111/jocd.13210
- Gmuca S, Yu J, Weiss PF, et al. Erythema ab igne in an adolescent with chronic pain: an alarming cutaneous eruption from heat exposure. Pediatr Emerg Care. 2020;36:E236-E238. doi:10.1097 /PEC.0000000000001460
- Wells A, Desai A, Rudnick EW, et al. Erythema ab igne with features resembling keratosis lichenoides chronica. J Cutan Pathol. 2021;48:151-153. doi:10.1111/cup.13885
- Milchak M, Smucker J, Chung CG, et al. Erythema ab igne due to heating pad use: a case report and review of clinical presentation, prevention, and complications. Case Rep Med. 2016;2016:1862480. doi:10.1155/2016/1862480
- Daneshvar E, Seraji S, Kamyab-Hesari K, et al. Basal cell carcinoma associated with erythema ab igne. Dermatol Online J. 2020;26:13030 /qt3kz985b4.
- Jones CS, Tyring SK, Lee PC, et al. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1988;124:110-113.
- Wharton J, Roffwarg D, Miller J, et al. Cutaneous marginal zone lymphoma arising in the setting of erythema ab igne. J Am Acad Dermatol. 2010;62:1080-1081. doi:10.1016/j.jaad.2009.08.005
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103 /2229-5178.164493
- Grossman JM. Lupus arthritis. Best Pract Res Clin Rheumatol. 2009;23:495-506. doi:10.1016/j.berh.2009.04.003
- Aria AB, Chen L, Silapunt S. Erythema ab igne from heating pad use: a report of three clinical cases and a differential diagnosis. Cureus. 2018;10:E2635. doi:10.7759/cureus.2635
- Wilcox RA. Cutaneous T-cell lymphoma: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2017;92:1085-1102. doi:10.1002/ajh.24876
- Pennitz A, Kinberger M, Avila Valle G, et al. Self-applied topical interventions for melasma: a systematic review and meta-analysis of data from randomized, investigator-blinded clinical trials. Br J Dermatol. 2022;187:309-317.
- Sahl WJ, Taira JW. Erythema ab igne: treatment with 5-fluorouracil cream. J Am Acad Dermatol. 1992;27:109-110.
The Diagnosis: Erythema Ab Igne
Based on the patient's long-standing history of back pain treated with heating pads as well as the normal laboratory findings and skin examination, a diagnosis of erythema ab igne (EAI) was made.
Erythema ab igne presents as reticulated brownish erythema or hyperpigmentation on sites exposed to prolonged use of heat sources such as heating pads, laptops, and space heaters. Erythema ab igne most commonly affects the lower back, thighs, or legs1-6; however, EAI can appear on atypical sites such as the forehead and eyebrows due to newer technology (eg, virtual reality headsets).7 The level of heat required for EAI to occur is below the threshold for thermal burns (<45 °C [113 °F]).1 Erythema ab igne can occur at any age, and woman are more commonly affected than men.8 The pathophysiology currently is unknown; however, recurrent and prolonged heat exposure may damage superficial vessels. As a result, hemosiderin accumulates in the skin, and hyperpigmentation subsequently occurs.9
The diagnosis of EAI is clinical, and early stages of the rash present as blanching reticulated erythema in areas associated with heat exposure. If the offending source of heat is not removed, EAI can progress to nonblanching, fixed, hyperpigmented plaques with skin atrophy, bullae, or hyperkeratosis. Patients often are asymptomatic; however, mild burning may occur.2 Histopathology reveals cellular atypia, epidermal atrophy, dilation of dermal blood vessels, a minute inflammatory infiltrate, and keratinocyte apoptosis.10 Skin biopsy may be necessary in cases of suspected malignancy due to chronic heat exposure. Lesions that ulcerate or evolve should raise suspicion for malignancy.11 Squamous cell carcinoma is the most common malignancy associated with EAI; other malignancies that may manifest include basal cell carcinoma, Merkel cell carcinoma, or cutaneous marginal zone lymphoma.2,12-14
Erythema ab igne often is mistaken for livedo reticularis, which appears more erythematous without hyperpigmentation or epidermal changes and may be associated with a pathologic state.15 The differential diagnosis in our patient, who was in her 40s with a history of fatigue and joint pain, included livedo reticularis associated with lupus; however, the history of heating pad use, normal laboratory findings, and presence of epidermal changes suggested EAI. Lupus typically affects the hand and knee joints.16 Additionally, livedo reticularis more commonly appears on the legs.15
Other differentials for EAI include livedo racemosa, cutaneous T-cell lymphoma, and cutis marmorata. Livedo racemosa presents with broken rings of erythema in young to middle-aged women and primarily affects the trunk and proximal limbs. It is associated with an underlying condition such as polyarteritis nodosa and less commonly with lupus erythematosus with antiphospholipid or Sneddon syndrome.15,17 Cutaneous T-cell lymphoma typically manifests with poikilodermatous patches larger than the palm, especially in covered areas of skin.18 Cutis marmorata is transient and temperature dependent.9
The key intervention for EAI is removal of the offending heat source.2 Patients should be counseled that the erythema and hyperpigmentation may take months to years to resolve. Topical hydroquinone or tretinoin may be used in cases of persistent hyperpigmentation.19 Patients who continue to use heating pads for long-standing pain should be advised to limit their use to short intervals without occlusion. If malignancy is a concern, a biopsy should be performed.20
The Diagnosis: Erythema Ab Igne
Based on the patient's long-standing history of back pain treated with heating pads as well as the normal laboratory findings and skin examination, a diagnosis of erythema ab igne (EAI) was made.
Erythema ab igne presents as reticulated brownish erythema or hyperpigmentation on sites exposed to prolonged use of heat sources such as heating pads, laptops, and space heaters. Erythema ab igne most commonly affects the lower back, thighs, or legs1-6; however, EAI can appear on atypical sites such as the forehead and eyebrows due to newer technology (eg, virtual reality headsets).7 The level of heat required for EAI to occur is below the threshold for thermal burns (<45 °C [113 °F]).1 Erythema ab igne can occur at any age, and woman are more commonly affected than men.8 The pathophysiology currently is unknown; however, recurrent and prolonged heat exposure may damage superficial vessels. As a result, hemosiderin accumulates in the skin, and hyperpigmentation subsequently occurs.9
The diagnosis of EAI is clinical, and early stages of the rash present as blanching reticulated erythema in areas associated with heat exposure. If the offending source of heat is not removed, EAI can progress to nonblanching, fixed, hyperpigmented plaques with skin atrophy, bullae, or hyperkeratosis. Patients often are asymptomatic; however, mild burning may occur.2 Histopathology reveals cellular atypia, epidermal atrophy, dilation of dermal blood vessels, a minute inflammatory infiltrate, and keratinocyte apoptosis.10 Skin biopsy may be necessary in cases of suspected malignancy due to chronic heat exposure. Lesions that ulcerate or evolve should raise suspicion for malignancy.11 Squamous cell carcinoma is the most common malignancy associated with EAI; other malignancies that may manifest include basal cell carcinoma, Merkel cell carcinoma, or cutaneous marginal zone lymphoma.2,12-14
Erythema ab igne often is mistaken for livedo reticularis, which appears more erythematous without hyperpigmentation or epidermal changes and may be associated with a pathologic state.15 The differential diagnosis in our patient, who was in her 40s with a history of fatigue and joint pain, included livedo reticularis associated with lupus; however, the history of heating pad use, normal laboratory findings, and presence of epidermal changes suggested EAI. Lupus typically affects the hand and knee joints.16 Additionally, livedo reticularis more commonly appears on the legs.15
Other differentials for EAI include livedo racemosa, cutaneous T-cell lymphoma, and cutis marmorata. Livedo racemosa presents with broken rings of erythema in young to middle-aged women and primarily affects the trunk and proximal limbs. It is associated with an underlying condition such as polyarteritis nodosa and less commonly with lupus erythematosus with antiphospholipid or Sneddon syndrome.15,17 Cutaneous T-cell lymphoma typically manifests with poikilodermatous patches larger than the palm, especially in covered areas of skin.18 Cutis marmorata is transient and temperature dependent.9
The key intervention for EAI is removal of the offending heat source.2 Patients should be counseled that the erythema and hyperpigmentation may take months to years to resolve. Topical hydroquinone or tretinoin may be used in cases of persistent hyperpigmentation.19 Patients who continue to use heating pads for long-standing pain should be advised to limit their use to short intervals without occlusion. If malignancy is a concern, a biopsy should be performed.20
- Wipf AJ, Brown MR. Malignant transformation of erythema ab igne. JAAD Case Rep. 2022;26:85-87. doi:10.1016/j.jdcr.2022.06.018
- Sigmon JR, Cantrell J, Teague D, et al. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35:676-678. doi:10.1097/DAD.0b013e3182871648
- Patel DP. The evolving nomenclature of erythema ab igne-redness from fire. JAMA Dermatol. 2017;153:685. doi:10.1001/jamadermatol.2017.2021
- Arnold AW, Itin PH. Laptop computer-induced erythema ab igne in a child and review of the literature. Pediatrics. 2010;126:E1227-E1230. doi:10.1542/peds.2010-1390
- Riahi RR, Cohen PR. Laptop-induced erythema ab igne: report and review of literature. Dermatol Online J. 2012;18:5.
- Haleem Z, Philip J, Muhammad S. Erythema ab igne: a rare presentation of toasted skin syndrome with the use of a space heater. Cureus. 2021;13:e13401. doi:10.7759/cureus.13401
- Moreau T, Benzaquen M, Gueissaz F. Erythema ab igne after using a virtual reality headset: a new phenomenon to know. J Eur Acad Dermatol Venereol. 2022;36:E932-E933. doi:10.1111/jdv.18371
- Ozturk M, An I. Clinical features and etiology of patients with erythema ab igne: a retrospective multicenter study. J Cosmet Dermatol. 2020;19:1774-1779. doi:10.1111/jocd.13210
- Gmuca S, Yu J, Weiss PF, et al. Erythema ab igne in an adolescent with chronic pain: an alarming cutaneous eruption from heat exposure. Pediatr Emerg Care. 2020;36:E236-E238. doi:10.1097 /PEC.0000000000001460
- Wells A, Desai A, Rudnick EW, et al. Erythema ab igne with features resembling keratosis lichenoides chronica. J Cutan Pathol. 2021;48:151-153. doi:10.1111/cup.13885
- Milchak M, Smucker J, Chung CG, et al. Erythema ab igne due to heating pad use: a case report and review of clinical presentation, prevention, and complications. Case Rep Med. 2016;2016:1862480. doi:10.1155/2016/1862480
- Daneshvar E, Seraji S, Kamyab-Hesari K, et al. Basal cell carcinoma associated with erythema ab igne. Dermatol Online J. 2020;26:13030 /qt3kz985b4.
- Jones CS, Tyring SK, Lee PC, et al. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1988;124:110-113.
- Wharton J, Roffwarg D, Miller J, et al. Cutaneous marginal zone lymphoma arising in the setting of erythema ab igne. J Am Acad Dermatol. 2010;62:1080-1081. doi:10.1016/j.jaad.2009.08.005
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103 /2229-5178.164493
- Grossman JM. Lupus arthritis. Best Pract Res Clin Rheumatol. 2009;23:495-506. doi:10.1016/j.berh.2009.04.003
- Aria AB, Chen L, Silapunt S. Erythema ab igne from heating pad use: a report of three clinical cases and a differential diagnosis. Cureus. 2018;10:E2635. doi:10.7759/cureus.2635
- Wilcox RA. Cutaneous T-cell lymphoma: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2017;92:1085-1102. doi:10.1002/ajh.24876
- Pennitz A, Kinberger M, Avila Valle G, et al. Self-applied topical interventions for melasma: a systematic review and meta-analysis of data from randomized, investigator-blinded clinical trials. Br J Dermatol. 2022;187:309-317.
- Sahl WJ, Taira JW. Erythema ab igne: treatment with 5-fluorouracil cream. J Am Acad Dermatol. 1992;27:109-110.
- Wipf AJ, Brown MR. Malignant transformation of erythema ab igne. JAAD Case Rep. 2022;26:85-87. doi:10.1016/j.jdcr.2022.06.018
- Sigmon JR, Cantrell J, Teague D, et al. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35:676-678. doi:10.1097/DAD.0b013e3182871648
- Patel DP. The evolving nomenclature of erythema ab igne-redness from fire. JAMA Dermatol. 2017;153:685. doi:10.1001/jamadermatol.2017.2021
- Arnold AW, Itin PH. Laptop computer-induced erythema ab igne in a child and review of the literature. Pediatrics. 2010;126:E1227-E1230. doi:10.1542/peds.2010-1390
- Riahi RR, Cohen PR. Laptop-induced erythema ab igne: report and review of literature. Dermatol Online J. 2012;18:5.
- Haleem Z, Philip J, Muhammad S. Erythema ab igne: a rare presentation of toasted skin syndrome with the use of a space heater. Cureus. 2021;13:e13401. doi:10.7759/cureus.13401
- Moreau T, Benzaquen M, Gueissaz F. Erythema ab igne after using a virtual reality headset: a new phenomenon to know. J Eur Acad Dermatol Venereol. 2022;36:E932-E933. doi:10.1111/jdv.18371
- Ozturk M, An I. Clinical features and etiology of patients with erythema ab igne: a retrospective multicenter study. J Cosmet Dermatol. 2020;19:1774-1779. doi:10.1111/jocd.13210
- Gmuca S, Yu J, Weiss PF, et al. Erythema ab igne in an adolescent with chronic pain: an alarming cutaneous eruption from heat exposure. Pediatr Emerg Care. 2020;36:E236-E238. doi:10.1097 /PEC.0000000000001460
- Wells A, Desai A, Rudnick EW, et al. Erythema ab igne with features resembling keratosis lichenoides chronica. J Cutan Pathol. 2021;48:151-153. doi:10.1111/cup.13885
- Milchak M, Smucker J, Chung CG, et al. Erythema ab igne due to heating pad use: a case report and review of clinical presentation, prevention, and complications. Case Rep Med. 2016;2016:1862480. doi:10.1155/2016/1862480
- Daneshvar E, Seraji S, Kamyab-Hesari K, et al. Basal cell carcinoma associated with erythema ab igne. Dermatol Online J. 2020;26:13030 /qt3kz985b4.
- Jones CS, Tyring SK, Lee PC, et al. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1988;124:110-113.
- Wharton J, Roffwarg D, Miller J, et al. Cutaneous marginal zone lymphoma arising in the setting of erythema ab igne. J Am Acad Dermatol. 2010;62:1080-1081. doi:10.1016/j.jaad.2009.08.005
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103 /2229-5178.164493
- Grossman JM. Lupus arthritis. Best Pract Res Clin Rheumatol. 2009;23:495-506. doi:10.1016/j.berh.2009.04.003
- Aria AB, Chen L, Silapunt S. Erythema ab igne from heating pad use: a report of three clinical cases and a differential diagnosis. Cureus. 2018;10:E2635. doi:10.7759/cureus.2635
- Wilcox RA. Cutaneous T-cell lymphoma: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2017;92:1085-1102. doi:10.1002/ajh.24876
- Pennitz A, Kinberger M, Avila Valle G, et al. Self-applied topical interventions for melasma: a systematic review and meta-analysis of data from randomized, investigator-blinded clinical trials. Br J Dermatol. 2022;187:309-317.
- Sahl WJ, Taira JW. Erythema ab igne: treatment with 5-fluorouracil cream. J Am Acad Dermatol. 1992;27:109-110.
A 42-year-old woman presented with an asymptomatic, erythematous, lacelike rash on the lower back of 8 months’ duration that was first noticed by her husband. The patient had a long-standing history of chronic fatigue and lower back pain treated with acetaminophen, diclofenac gel, and heating pads. Physical examination revealed reticulated brownish erythema confined to the lower back. Laboratory findings were unremarkable.
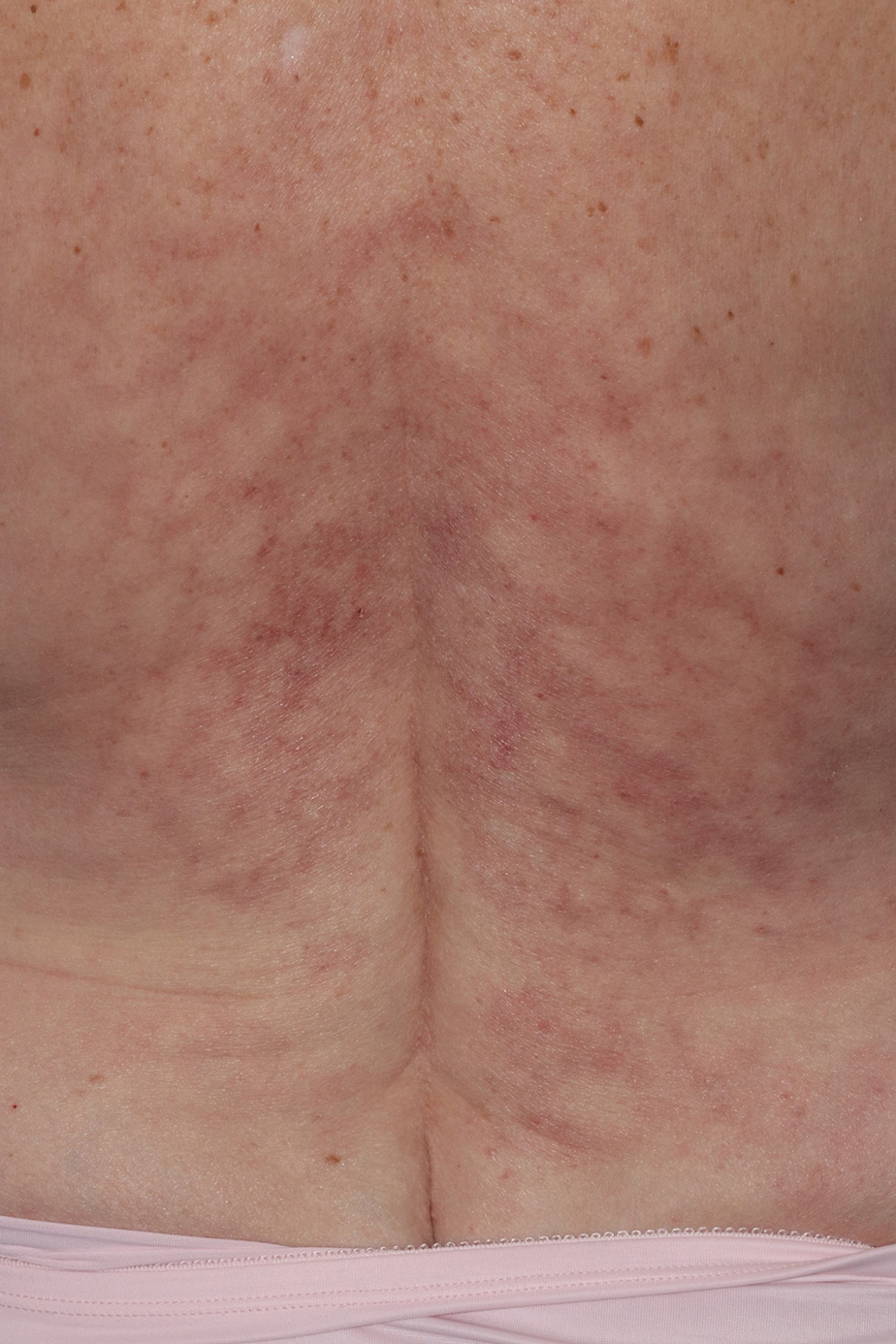
FDA Expands Repotrectinib Label to All NTRK Gene Fusion+ Solid Tumors
The approval is a label expansion for the tyrosine kinase inhibitor (TKI), which received initial clearance in November 2023 for locally advanced or metastatic ROS1-positive non–small cell lung cancer.
NTRK gene fusions are genetic abnormalities wherein part of the NTRK gene fuses with an unrelated gene. The abnormal gene can then produce an oncogenic protein. Although rare, these mutations are found in many cancer types.
The approval, for adult and pediatric patients aged 12 years or older, was based on the single-arm open-label TRIDENT-1 trial in 88 adults with locally advanced or metastatic NTRK gene fusion solid tumors.
In the 40 patients who were TKI-naive, the overall response rate was 58%, and the median duration of response was not estimable. In the 48 patients who had a TKI previously, the overall response rate was 50% and median duration of response was 9.9 months.
In 20% or more of participants, treatment caused dizziness, dysgeusia, peripheral neuropathy, constipation, dyspnea, fatigue, ataxia, cognitive impairment, muscular weakness, and nausea.
Labeling warns of central nervous system reactions, interstitial lung disease/pneumonitis, hepatotoxicity, myalgia with creatine phosphokinase elevation, hyperuricemia, bone fractures, and embryo-fetal toxicity.
The recommended dose is 160 mg orally once daily for 14 days then increased to 160 mg twice daily until disease progression or unacceptable toxicity.
Sixty 40-mg capsules cost around $7,644, according to drugs.com.
A version of this article appeared on Medscape.com.
The approval is a label expansion for the tyrosine kinase inhibitor (TKI), which received initial clearance in November 2023 for locally advanced or metastatic ROS1-positive non–small cell lung cancer.
NTRK gene fusions are genetic abnormalities wherein part of the NTRK gene fuses with an unrelated gene. The abnormal gene can then produce an oncogenic protein. Although rare, these mutations are found in many cancer types.
The approval, for adult and pediatric patients aged 12 years or older, was based on the single-arm open-label TRIDENT-1 trial in 88 adults with locally advanced or metastatic NTRK gene fusion solid tumors.
In the 40 patients who were TKI-naive, the overall response rate was 58%, and the median duration of response was not estimable. In the 48 patients who had a TKI previously, the overall response rate was 50% and median duration of response was 9.9 months.
In 20% or more of participants, treatment caused dizziness, dysgeusia, peripheral neuropathy, constipation, dyspnea, fatigue, ataxia, cognitive impairment, muscular weakness, and nausea.
Labeling warns of central nervous system reactions, interstitial lung disease/pneumonitis, hepatotoxicity, myalgia with creatine phosphokinase elevation, hyperuricemia, bone fractures, and embryo-fetal toxicity.
The recommended dose is 160 mg orally once daily for 14 days then increased to 160 mg twice daily until disease progression or unacceptable toxicity.
Sixty 40-mg capsules cost around $7,644, according to drugs.com.
A version of this article appeared on Medscape.com.
The approval is a label expansion for the tyrosine kinase inhibitor (TKI), which received initial clearance in November 2023 for locally advanced or metastatic ROS1-positive non–small cell lung cancer.
NTRK gene fusions are genetic abnormalities wherein part of the NTRK gene fuses with an unrelated gene. The abnormal gene can then produce an oncogenic protein. Although rare, these mutations are found in many cancer types.
The approval, for adult and pediatric patients aged 12 years or older, was based on the single-arm open-label TRIDENT-1 trial in 88 adults with locally advanced or metastatic NTRK gene fusion solid tumors.
In the 40 patients who were TKI-naive, the overall response rate was 58%, and the median duration of response was not estimable. In the 48 patients who had a TKI previously, the overall response rate was 50% and median duration of response was 9.9 months.
In 20% or more of participants, treatment caused dizziness, dysgeusia, peripheral neuropathy, constipation, dyspnea, fatigue, ataxia, cognitive impairment, muscular weakness, and nausea.
Labeling warns of central nervous system reactions, interstitial lung disease/pneumonitis, hepatotoxicity, myalgia with creatine phosphokinase elevation, hyperuricemia, bone fractures, and embryo-fetal toxicity.
The recommended dose is 160 mg orally once daily for 14 days then increased to 160 mg twice daily until disease progression or unacceptable toxicity.
Sixty 40-mg capsules cost around $7,644, according to drugs.com.
A version of this article appeared on Medscape.com.