User login
Doctor who lied about his age sentenced to 3 years for killing woman with botched marrow procedure
A mother-of-three was killed at her hospital appointment in the United Kingdom by a doctor who botched a routine procedure, a court has heard. On July 5, Dr. Isyaka Mamman was sentenced at Manchester Crown Court to 3 years imprisonment after pleading guilty to the manslaughter by gross negligence of his patient.
Dr. Mamman, 85, had already been suspended once by medical watchdogs for lying about his age and was sacked but then re-employed by the Royal Oldham Hospital, where he was responsible for a series of critical incidents before the fatal appointment, Manchester Crown Court heard.
The Nigerian-born doctor had also used various dates of birth and left his previous job through “poor performance.”
‘Highly dangerous’ procedure
Shahida Parveen, 48, had gone to the hospital with her husband, Khizar Mahmood, for investigations into possible myeloproliferative disorder.
A bone marrow biopsy had been advised and the routine procedure was allocated to Dr. Mamman, who was working as a specialty doctor in hematology, Andrew Thomas QC, prosecuting, told the hearing.
Normally, bone marrow samples are taken from the hip bone but Dr. Mamman failed to obtain a sample at the first attempt.
Instead, he attempted a rare and “highly dangerous” procedure of getting a sample from Ms. Parveen’s sternum – despite objections from the patient and her husband.
Dr. Mamman, using the wrong biopsy needle, missed the bone and pierced her pericardium, the sac containing the heart, causing massive internal bleeding.
Ms. Parveen lost consciousness as soon as the needle was inserted, with her husband running from the room shouting: “He killed her. I told him to stop three times and he did not listen. He killed her.”
A crash team arrived but Ms. Parveen was confirmed dead later the same day, September 3, 2018.
Controversy over his ‘true age’
Dr. Mamman qualified as a doctor in Nigeria in 1965 and had worked in the United Kingdom since 1991. From 2004 until the time of the fatal incident he was employed by the Pennine Acute Hospitals NHS Trust.
But his “true age” is a matter of “controversy,” the court heard, as his birthplace in rural Nigeria had no system of birth registration.
During his medical training he gave a date of birth of September 16, 1936, which meant that he was 21 years old when he began his medical training and 81 at the time of the fatal hospital incident.
But he knocked years off his age by adopting a birth date in 1941, provided to the NHS, suggesting he began his medical degree at the age of 16.
However, in about 2001 and approaching what was then the compulsory retirement age of 65, Dr. Mamman adopted an even later birth date – October 1947 – which he relied upon in an application for naturalisation as a British citizen – suggesting he started his degree course at the age of 10.
In 2004 he was found guilty of serious professional misconduct by the General Medical Council and suspended for 12 months for lying about his age.
The Pennine Trust sacked him but then re-employed him in 2006, after he had been restored to the register by the GMC, who accepted his date of birth to be 1943 – which meant he was 14 or 15 when he began his medical degree.
Dr. Mamman had left his previous employment with the Medway Trust because of “poor performance,” and in 2015 a formal complaint was made to the Oldham hospital when a patient complained he used “excessive force” during a bone marrow biopsy.
The patient was told that Dr. Mamman was in his 70s and his colleagues thought he should retire but they could not dismiss him purely because of his age. She was assured he would be put on light duties in future.
However, the same year there was another clinical incident, which resulted in serious injury to another patient, again during a bone marrow biopsy, and again involving a needle being inserted in the wrong place. The patient survived but has been left permanently disabled.
Michael Hayton, mitigating, said it was clear Dr. Mamman was a “failing” doctor and he should not have been allowed to continue treating patients.
He added: “He is not the only person at fault. He should not have been allowed to be in the position he was.
“There’s a grotesque catalogue of failings by the trust from 2015.”
This article contains information from PA Media.
A version of this article first appeared on Medscape.co.uk.
A mother-of-three was killed at her hospital appointment in the United Kingdom by a doctor who botched a routine procedure, a court has heard. On July 5, Dr. Isyaka Mamman was sentenced at Manchester Crown Court to 3 years imprisonment after pleading guilty to the manslaughter by gross negligence of his patient.
Dr. Mamman, 85, had already been suspended once by medical watchdogs for lying about his age and was sacked but then re-employed by the Royal Oldham Hospital, where he was responsible for a series of critical incidents before the fatal appointment, Manchester Crown Court heard.
The Nigerian-born doctor had also used various dates of birth and left his previous job through “poor performance.”
‘Highly dangerous’ procedure
Shahida Parveen, 48, had gone to the hospital with her husband, Khizar Mahmood, for investigations into possible myeloproliferative disorder.
A bone marrow biopsy had been advised and the routine procedure was allocated to Dr. Mamman, who was working as a specialty doctor in hematology, Andrew Thomas QC, prosecuting, told the hearing.
Normally, bone marrow samples are taken from the hip bone but Dr. Mamman failed to obtain a sample at the first attempt.
Instead, he attempted a rare and “highly dangerous” procedure of getting a sample from Ms. Parveen’s sternum – despite objections from the patient and her husband.
Dr. Mamman, using the wrong biopsy needle, missed the bone and pierced her pericardium, the sac containing the heart, causing massive internal bleeding.
Ms. Parveen lost consciousness as soon as the needle was inserted, with her husband running from the room shouting: “He killed her. I told him to stop three times and he did not listen. He killed her.”
A crash team arrived but Ms. Parveen was confirmed dead later the same day, September 3, 2018.
Controversy over his ‘true age’
Dr. Mamman qualified as a doctor in Nigeria in 1965 and had worked in the United Kingdom since 1991. From 2004 until the time of the fatal incident he was employed by the Pennine Acute Hospitals NHS Trust.
But his “true age” is a matter of “controversy,” the court heard, as his birthplace in rural Nigeria had no system of birth registration.
During his medical training he gave a date of birth of September 16, 1936, which meant that he was 21 years old when he began his medical training and 81 at the time of the fatal hospital incident.
But he knocked years off his age by adopting a birth date in 1941, provided to the NHS, suggesting he began his medical degree at the age of 16.
However, in about 2001 and approaching what was then the compulsory retirement age of 65, Dr. Mamman adopted an even later birth date – October 1947 – which he relied upon in an application for naturalisation as a British citizen – suggesting he started his degree course at the age of 10.
In 2004 he was found guilty of serious professional misconduct by the General Medical Council and suspended for 12 months for lying about his age.
The Pennine Trust sacked him but then re-employed him in 2006, after he had been restored to the register by the GMC, who accepted his date of birth to be 1943 – which meant he was 14 or 15 when he began his medical degree.
Dr. Mamman had left his previous employment with the Medway Trust because of “poor performance,” and in 2015 a formal complaint was made to the Oldham hospital when a patient complained he used “excessive force” during a bone marrow biopsy.
The patient was told that Dr. Mamman was in his 70s and his colleagues thought he should retire but they could not dismiss him purely because of his age. She was assured he would be put on light duties in future.
However, the same year there was another clinical incident, which resulted in serious injury to another patient, again during a bone marrow biopsy, and again involving a needle being inserted in the wrong place. The patient survived but has been left permanently disabled.
Michael Hayton, mitigating, said it was clear Dr. Mamman was a “failing” doctor and he should not have been allowed to continue treating patients.
He added: “He is not the only person at fault. He should not have been allowed to be in the position he was.
“There’s a grotesque catalogue of failings by the trust from 2015.”
This article contains information from PA Media.
A version of this article first appeared on Medscape.co.uk.
A mother-of-three was killed at her hospital appointment in the United Kingdom by a doctor who botched a routine procedure, a court has heard. On July 5, Dr. Isyaka Mamman was sentenced at Manchester Crown Court to 3 years imprisonment after pleading guilty to the manslaughter by gross negligence of his patient.
Dr. Mamman, 85, had already been suspended once by medical watchdogs for lying about his age and was sacked but then re-employed by the Royal Oldham Hospital, where he was responsible for a series of critical incidents before the fatal appointment, Manchester Crown Court heard.
The Nigerian-born doctor had also used various dates of birth and left his previous job through “poor performance.”
‘Highly dangerous’ procedure
Shahida Parveen, 48, had gone to the hospital with her husband, Khizar Mahmood, for investigations into possible myeloproliferative disorder.
A bone marrow biopsy had been advised and the routine procedure was allocated to Dr. Mamman, who was working as a specialty doctor in hematology, Andrew Thomas QC, prosecuting, told the hearing.
Normally, bone marrow samples are taken from the hip bone but Dr. Mamman failed to obtain a sample at the first attempt.
Instead, he attempted a rare and “highly dangerous” procedure of getting a sample from Ms. Parveen’s sternum – despite objections from the patient and her husband.
Dr. Mamman, using the wrong biopsy needle, missed the bone and pierced her pericardium, the sac containing the heart, causing massive internal bleeding.
Ms. Parveen lost consciousness as soon as the needle was inserted, with her husband running from the room shouting: “He killed her. I told him to stop three times and he did not listen. He killed her.”
A crash team arrived but Ms. Parveen was confirmed dead later the same day, September 3, 2018.
Controversy over his ‘true age’
Dr. Mamman qualified as a doctor in Nigeria in 1965 and had worked in the United Kingdom since 1991. From 2004 until the time of the fatal incident he was employed by the Pennine Acute Hospitals NHS Trust.
But his “true age” is a matter of “controversy,” the court heard, as his birthplace in rural Nigeria had no system of birth registration.
During his medical training he gave a date of birth of September 16, 1936, which meant that he was 21 years old when he began his medical training and 81 at the time of the fatal hospital incident.
But he knocked years off his age by adopting a birth date in 1941, provided to the NHS, suggesting he began his medical degree at the age of 16.
However, in about 2001 and approaching what was then the compulsory retirement age of 65, Dr. Mamman adopted an even later birth date – October 1947 – which he relied upon in an application for naturalisation as a British citizen – suggesting he started his degree course at the age of 10.
In 2004 he was found guilty of serious professional misconduct by the General Medical Council and suspended for 12 months for lying about his age.
The Pennine Trust sacked him but then re-employed him in 2006, after he had been restored to the register by the GMC, who accepted his date of birth to be 1943 – which meant he was 14 or 15 when he began his medical degree.
Dr. Mamman had left his previous employment with the Medway Trust because of “poor performance,” and in 2015 a formal complaint was made to the Oldham hospital when a patient complained he used “excessive force” during a bone marrow biopsy.
The patient was told that Dr. Mamman was in his 70s and his colleagues thought he should retire but they could not dismiss him purely because of his age. She was assured he would be put on light duties in future.
However, the same year there was another clinical incident, which resulted in serious injury to another patient, again during a bone marrow biopsy, and again involving a needle being inserted in the wrong place. The patient survived but has been left permanently disabled.
Michael Hayton, mitigating, said it was clear Dr. Mamman was a “failing” doctor and he should not have been allowed to continue treating patients.
He added: “He is not the only person at fault. He should not have been allowed to be in the position he was.
“There’s a grotesque catalogue of failings by the trust from 2015.”
This article contains information from PA Media.
A version of this article first appeared on Medscape.co.uk.
U.S. allows pharmacists to prescribe Paxlovid directly
The Food and Drug Administration revised the drug’s emergency use authorization on July 6, letting state-licensed pharmacists screen patients and determine if they are eligible for Paxlovid, according to The Associated Press.
Previously, only doctors could prescribe the antiviral drug, the AP reported. With some limits, pharmacists can now prescribe the medication for patients who face high risks for severe COVID-19.
“The FDA recognizes the important role pharmacists have played and continue to play in combating this pandemic,” Patrizia Cavazzoni, MD, director of the FDA’s Center for Drug Evaluation and Research, said in a statement.
“Since Paxlovid must be taken within 5 days after symptoms begin, authorizing state-licensed pharmacists to prescribe Paxlovid could expand access to timely treatment for some patients who are eligible to receive this drug for the treatment of COVID-19,” she said.
Tom Kraus, the vice president of government relations at the American Society of Health-System Pharmacists, said in a statement that the organization was “pleased to see the FDA remove this barrier to patients’ access to this critical treatment.”
“Pharmacists have played a vital role in our pandemic response efforts and are well-positioned to help patients, particularly those in rural and underserved communities, benefit from this medication,” he said.
But some doctor’s groups questioned the FDA’s move. Jack Resneck Jr., MD, the president of the American Medical Association, said in a statement that prescribing Paxlovid “requires knowledge of a patient’s medical history, as well as clinical monitoring for side effects and follow-up care to determine whether a patient is improving” – requirements that are “far beyond a pharmacist’s scope and training.”
“In the fight against a virus that has killed more than a million people in the United States and is still extremely present and transmissible, patients will get the best, most comprehensive care from physician-led teams – teams that include pharmacists. But, whenever possible, prescribing decisions should be made by a physician with knowledge of a patient’s medical history and the ability to follow up. To ensure the best possible care for COVID-19 patients, we urge people who test positive to discuss treatment options with their physician, if they have one,” he said.
After testing positive for COVID-19, patients should first consider seeking care from their regular health care provider or locating a Test-to-Treat site in their area, the FDA said. Although the latest update allows pharmacists to prescribe Paxlovid, community pharmacies that don’t yet take part in the Test-to-Treat program can decide if they will offer the prescription service to patients.
Paxlovid is authorized to treat mild to moderate COVID-19 in adults and in kids ages 12 and older who weigh at least 88 pounds. Patients who report a positive at-home test are eligible for Paxlovid under the FDA authorization.
If patients want to seek a prescription directly from a pharmacist, they should bring electronic or printed health records from the past year, including their most recent reports of blood work, so the pharmacist can review for kidney or liver problems. Pharmacists can also get this information from the patient’s health care provider.
In addition, patients should bring a list of all medications they are taking, including over-the-counter medications, so the pharmacist can screen for drugs that can have serious interactions with Paxlovid.
Under the limits in the updated FDA authorization, pharmacists should refer patients for more screening if Paxlovid isn’t a good option or if there’s not enough information to find out how well their kidneys or liver works, as well as potential drug interactions.
Paxlovid is intended for people with COVID-19 who face the highest risks for serious disease, the AP reported, including older adults and those with health conditions such as heart disease, obesity, cancer, or diabetes. It isn’t recommended for people with severe kidney or liver problems. A course of treatment requires three pills twice a day for 5 days.
A version of this article first appeared on WebMD.com.
The Food and Drug Administration revised the drug’s emergency use authorization on July 6, letting state-licensed pharmacists screen patients and determine if they are eligible for Paxlovid, according to The Associated Press.
Previously, only doctors could prescribe the antiviral drug, the AP reported. With some limits, pharmacists can now prescribe the medication for patients who face high risks for severe COVID-19.
“The FDA recognizes the important role pharmacists have played and continue to play in combating this pandemic,” Patrizia Cavazzoni, MD, director of the FDA’s Center for Drug Evaluation and Research, said in a statement.
“Since Paxlovid must be taken within 5 days after symptoms begin, authorizing state-licensed pharmacists to prescribe Paxlovid could expand access to timely treatment for some patients who are eligible to receive this drug for the treatment of COVID-19,” she said.
Tom Kraus, the vice president of government relations at the American Society of Health-System Pharmacists, said in a statement that the organization was “pleased to see the FDA remove this barrier to patients’ access to this critical treatment.”
“Pharmacists have played a vital role in our pandemic response efforts and are well-positioned to help patients, particularly those in rural and underserved communities, benefit from this medication,” he said.
But some doctor’s groups questioned the FDA’s move. Jack Resneck Jr., MD, the president of the American Medical Association, said in a statement that prescribing Paxlovid “requires knowledge of a patient’s medical history, as well as clinical monitoring for side effects and follow-up care to determine whether a patient is improving” – requirements that are “far beyond a pharmacist’s scope and training.”
“In the fight against a virus that has killed more than a million people in the United States and is still extremely present and transmissible, patients will get the best, most comprehensive care from physician-led teams – teams that include pharmacists. But, whenever possible, prescribing decisions should be made by a physician with knowledge of a patient’s medical history and the ability to follow up. To ensure the best possible care for COVID-19 patients, we urge people who test positive to discuss treatment options with their physician, if they have one,” he said.
After testing positive for COVID-19, patients should first consider seeking care from their regular health care provider or locating a Test-to-Treat site in their area, the FDA said. Although the latest update allows pharmacists to prescribe Paxlovid, community pharmacies that don’t yet take part in the Test-to-Treat program can decide if they will offer the prescription service to patients.
Paxlovid is authorized to treat mild to moderate COVID-19 in adults and in kids ages 12 and older who weigh at least 88 pounds. Patients who report a positive at-home test are eligible for Paxlovid under the FDA authorization.
If patients want to seek a prescription directly from a pharmacist, they should bring electronic or printed health records from the past year, including their most recent reports of blood work, so the pharmacist can review for kidney or liver problems. Pharmacists can also get this information from the patient’s health care provider.
In addition, patients should bring a list of all medications they are taking, including over-the-counter medications, so the pharmacist can screen for drugs that can have serious interactions with Paxlovid.
Under the limits in the updated FDA authorization, pharmacists should refer patients for more screening if Paxlovid isn’t a good option or if there’s not enough information to find out how well their kidneys or liver works, as well as potential drug interactions.
Paxlovid is intended for people with COVID-19 who face the highest risks for serious disease, the AP reported, including older adults and those with health conditions such as heart disease, obesity, cancer, or diabetes. It isn’t recommended for people with severe kidney or liver problems. A course of treatment requires three pills twice a day for 5 days.
A version of this article first appeared on WebMD.com.
The Food and Drug Administration revised the drug’s emergency use authorization on July 6, letting state-licensed pharmacists screen patients and determine if they are eligible for Paxlovid, according to The Associated Press.
Previously, only doctors could prescribe the antiviral drug, the AP reported. With some limits, pharmacists can now prescribe the medication for patients who face high risks for severe COVID-19.
“The FDA recognizes the important role pharmacists have played and continue to play in combating this pandemic,” Patrizia Cavazzoni, MD, director of the FDA’s Center for Drug Evaluation and Research, said in a statement.
“Since Paxlovid must be taken within 5 days after symptoms begin, authorizing state-licensed pharmacists to prescribe Paxlovid could expand access to timely treatment for some patients who are eligible to receive this drug for the treatment of COVID-19,” she said.
Tom Kraus, the vice president of government relations at the American Society of Health-System Pharmacists, said in a statement that the organization was “pleased to see the FDA remove this barrier to patients’ access to this critical treatment.”
“Pharmacists have played a vital role in our pandemic response efforts and are well-positioned to help patients, particularly those in rural and underserved communities, benefit from this medication,” he said.
But some doctor’s groups questioned the FDA’s move. Jack Resneck Jr., MD, the president of the American Medical Association, said in a statement that prescribing Paxlovid “requires knowledge of a patient’s medical history, as well as clinical monitoring for side effects and follow-up care to determine whether a patient is improving” – requirements that are “far beyond a pharmacist’s scope and training.”
“In the fight against a virus that has killed more than a million people in the United States and is still extremely present and transmissible, patients will get the best, most comprehensive care from physician-led teams – teams that include pharmacists. But, whenever possible, prescribing decisions should be made by a physician with knowledge of a patient’s medical history and the ability to follow up. To ensure the best possible care for COVID-19 patients, we urge people who test positive to discuss treatment options with their physician, if they have one,” he said.
After testing positive for COVID-19, patients should first consider seeking care from their regular health care provider or locating a Test-to-Treat site in their area, the FDA said. Although the latest update allows pharmacists to prescribe Paxlovid, community pharmacies that don’t yet take part in the Test-to-Treat program can decide if they will offer the prescription service to patients.
Paxlovid is authorized to treat mild to moderate COVID-19 in adults and in kids ages 12 and older who weigh at least 88 pounds. Patients who report a positive at-home test are eligible for Paxlovid under the FDA authorization.
If patients want to seek a prescription directly from a pharmacist, they should bring electronic or printed health records from the past year, including their most recent reports of blood work, so the pharmacist can review for kidney or liver problems. Pharmacists can also get this information from the patient’s health care provider.
In addition, patients should bring a list of all medications they are taking, including over-the-counter medications, so the pharmacist can screen for drugs that can have serious interactions with Paxlovid.
Under the limits in the updated FDA authorization, pharmacists should refer patients for more screening if Paxlovid isn’t a good option or if there’s not enough information to find out how well their kidneys or liver works, as well as potential drug interactions.
Paxlovid is intended for people with COVID-19 who face the highest risks for serious disease, the AP reported, including older adults and those with health conditions such as heart disease, obesity, cancer, or diabetes. It isn’t recommended for people with severe kidney or liver problems. A course of treatment requires three pills twice a day for 5 days.
A version of this article first appeared on WebMD.com.
WHO tracking new Omicron subvariant in India
The subvariant, a sublineage of BA.2 being called BA.2.75, has been reported in eight countries and hasn’t yet been declared a variant of concern.
“There’s been an emergence of a ‘could be’ subvariant. It’s been not yet officially called, but some people are referring to it as BA.2.75,” Soumya Swaminathan, MD, the WHO’s chief scientist, said in a video posted on Twitter.
The subvariant appears to have mutations similar to other contagious strains, she said, though there are a limited number of sequences available to analyze. How transmissible and severe it is, and how well it can evade our immunity, aren’t yet known.
“We have to wait and see, and of course, we are tracking it,” Dr. Swaminathan said.
The WHO committee responsible for analyzing global coronavirus data will label the subvariant officially and release more information as the situation warrants it, she said.
Public health experts around the world are also talking about the subvariant, which has been nicknamed Centaurus. BA.2.75 was first found in India in May and is now competing with BA.5, which has become dominant in the United States.
BA.2.75 has eight mutations beyond those seen in BA.5, which “could make immune escape worse than what we’re seeing now,” Eric Topol, MD, founder and director of the Scripps Research Translational Institute and editor-in-chief at Medscape, wrote in a Twitter post.
Individually, the extra mutations aren’t too concerning, “but all appearing together at once is another matter,” Tom Peacock, PhD, a virologist at Imperial College London, wrote in a Twitter post.
The “apparent rapid growth and wide geographical spread” are “worth keeping a close eye on,” he said.
BA.2.75 has been found in a handful of cases in the United States, Australia, Canada, Germany, Japan, New Zealand, and the United Kingdom. In India, the sequence accounts for about 23% of recent samples.
“It is really too early to know if BA.2.75 will take over relative to BA.2 or even relative to BA.5,” Ulrich Elling, PhD, a researcher at Australia’s Institute of Molecular Biotechnology, wrote in a Twitter post.
“Just to emphasize it again: While the distribution across Indian regions as well as internationally and the very rapid appearance makes it likely we are dealing with a variant spreading fast and spread widely already, the absolute data points are few,” he said.
Globally, coronavirus cases have increased nearly 30% during the past 2 weeks, the WHO said July 6. Four out of six of the WHO subregions reported an increase in the last week, with BA.4 and BA.5 driving waves in the United States and Europe.
A version of this article first appeared on WebMD.com.
The subvariant, a sublineage of BA.2 being called BA.2.75, has been reported in eight countries and hasn’t yet been declared a variant of concern.
“There’s been an emergence of a ‘could be’ subvariant. It’s been not yet officially called, but some people are referring to it as BA.2.75,” Soumya Swaminathan, MD, the WHO’s chief scientist, said in a video posted on Twitter.
The subvariant appears to have mutations similar to other contagious strains, she said, though there are a limited number of sequences available to analyze. How transmissible and severe it is, and how well it can evade our immunity, aren’t yet known.
“We have to wait and see, and of course, we are tracking it,” Dr. Swaminathan said.
The WHO committee responsible for analyzing global coronavirus data will label the subvariant officially and release more information as the situation warrants it, she said.
Public health experts around the world are also talking about the subvariant, which has been nicknamed Centaurus. BA.2.75 was first found in India in May and is now competing with BA.5, which has become dominant in the United States.
BA.2.75 has eight mutations beyond those seen in BA.5, which “could make immune escape worse than what we’re seeing now,” Eric Topol, MD, founder and director of the Scripps Research Translational Institute and editor-in-chief at Medscape, wrote in a Twitter post.
Individually, the extra mutations aren’t too concerning, “but all appearing together at once is another matter,” Tom Peacock, PhD, a virologist at Imperial College London, wrote in a Twitter post.
The “apparent rapid growth and wide geographical spread” are “worth keeping a close eye on,” he said.
BA.2.75 has been found in a handful of cases in the United States, Australia, Canada, Germany, Japan, New Zealand, and the United Kingdom. In India, the sequence accounts for about 23% of recent samples.
“It is really too early to know if BA.2.75 will take over relative to BA.2 or even relative to BA.5,” Ulrich Elling, PhD, a researcher at Australia’s Institute of Molecular Biotechnology, wrote in a Twitter post.
“Just to emphasize it again: While the distribution across Indian regions as well as internationally and the very rapid appearance makes it likely we are dealing with a variant spreading fast and spread widely already, the absolute data points are few,” he said.
Globally, coronavirus cases have increased nearly 30% during the past 2 weeks, the WHO said July 6. Four out of six of the WHO subregions reported an increase in the last week, with BA.4 and BA.5 driving waves in the United States and Europe.
A version of this article first appeared on WebMD.com.
The subvariant, a sublineage of BA.2 being called BA.2.75, has been reported in eight countries and hasn’t yet been declared a variant of concern.
“There’s been an emergence of a ‘could be’ subvariant. It’s been not yet officially called, but some people are referring to it as BA.2.75,” Soumya Swaminathan, MD, the WHO’s chief scientist, said in a video posted on Twitter.
The subvariant appears to have mutations similar to other contagious strains, she said, though there are a limited number of sequences available to analyze. How transmissible and severe it is, and how well it can evade our immunity, aren’t yet known.
“We have to wait and see, and of course, we are tracking it,” Dr. Swaminathan said.
The WHO committee responsible for analyzing global coronavirus data will label the subvariant officially and release more information as the situation warrants it, she said.
Public health experts around the world are also talking about the subvariant, which has been nicknamed Centaurus. BA.2.75 was first found in India in May and is now competing with BA.5, which has become dominant in the United States.
BA.2.75 has eight mutations beyond those seen in BA.5, which “could make immune escape worse than what we’re seeing now,” Eric Topol, MD, founder and director of the Scripps Research Translational Institute and editor-in-chief at Medscape, wrote in a Twitter post.
Individually, the extra mutations aren’t too concerning, “but all appearing together at once is another matter,” Tom Peacock, PhD, a virologist at Imperial College London, wrote in a Twitter post.
The “apparent rapid growth and wide geographical spread” are “worth keeping a close eye on,” he said.
BA.2.75 has been found in a handful of cases in the United States, Australia, Canada, Germany, Japan, New Zealand, and the United Kingdom. In India, the sequence accounts for about 23% of recent samples.
“It is really too early to know if BA.2.75 will take over relative to BA.2 or even relative to BA.5,” Ulrich Elling, PhD, a researcher at Australia’s Institute of Molecular Biotechnology, wrote in a Twitter post.
“Just to emphasize it again: While the distribution across Indian regions as well as internationally and the very rapid appearance makes it likely we are dealing with a variant spreading fast and spread widely already, the absolute data points are few,” he said.
Globally, coronavirus cases have increased nearly 30% during the past 2 weeks, the WHO said July 6. Four out of six of the WHO subregions reported an increase in the last week, with BA.4 and BA.5 driving waves in the United States and Europe.
A version of this article first appeared on WebMD.com.
Eczema severity, time spent on management strongly associated with overall disease burden
. However, AD severity and spending 11 hours or more per week managing the condition did correlate with higher overall disease burden.
“Research has documented the disease burden of AD, including its visible nature and the effect on itch and sleep, but knowledge gaps remain,” Aaron M. Drucker, MD, of the division of dermatology at the University of Toronto, and colleagues wrote in the study published online in JAMA Dermatology. “Gaps include a poor understanding of symptoms other than itch, patients’ treatment experience, and how different elements of burden of disease interact.”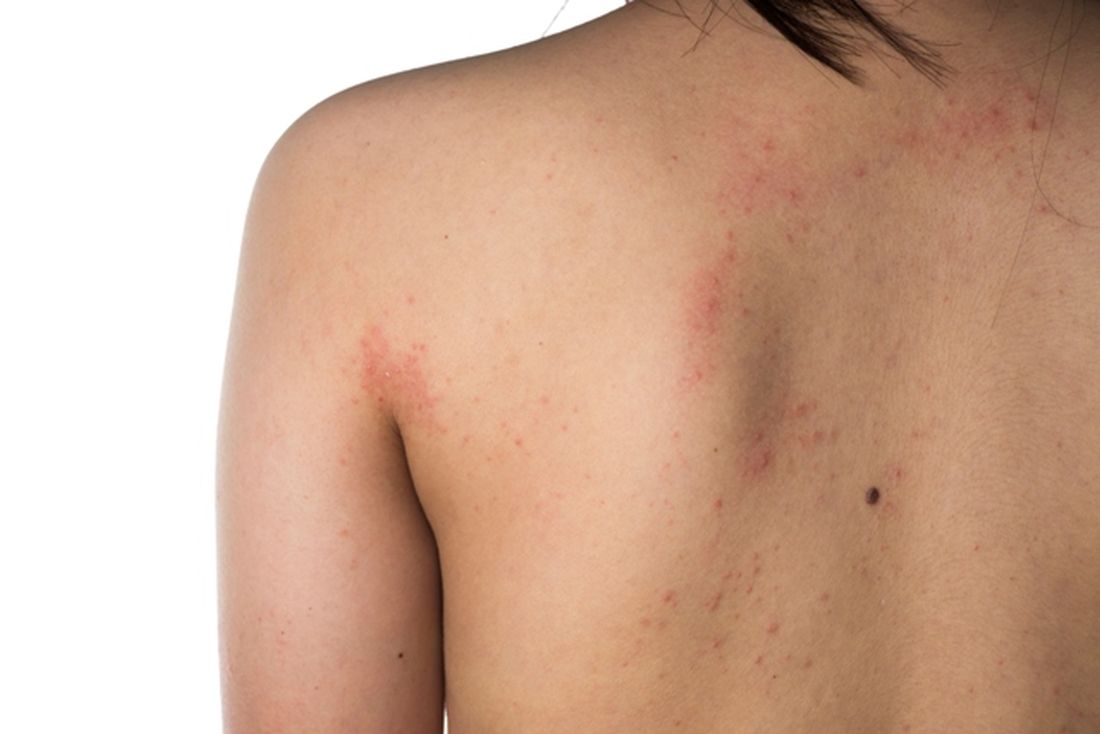
Dr. Drucker and colleagues collected data from an externally led patient-focused drug development survey on AD, a 32-item questionnaire that was administered electronically between Aug. 1, 2019, and Oct. 11, 2019. Respondents were asked to rate the overall impact of their AD in the past months and the specific elements of disease burden on a 1-5 scale, with 1 meaning no impact, and 5 meaning a significant impact. They were also asked to rate current mood changes and mood changes at the worst point of AD on a 4-point scale that ranged from “not present” to “severe.” The researchers used multivariable ordinal regression to examine associations between demographic and clinical variables and patient-reported overall AD impact scores.
Survey results
Of the 1,065 respondents, 33% were aged 18-34 years, 50% were aged 35-50 years, 17% were aged 65 years or older, and 83% were female. Nearly half (45%) reported having moderate AD, while 28% had severe AD. When asked about the overall disease burden of AD symptoms in the past month, 30% reported a significant impact on life, 28% reported a moderate impact score, 21% reported a high impact score, 18% reported a low impact score, and 3% of respondents reported no impact.
In the multivariable proportional odds analysis, moderate AD (odds ratio [OR], 4.13) and severe AD (OR, 13.63) were both associated with greater disease burden compared with mild AD. Also, spending 11 or more hours per week managing AD symptoms was associated with greater disease burden compared with 0 to 4 hours (an OR of 2.67 for 11-20 hours per week spent managing AD and OR of 5.34 for 21 or more hours per week spent managing AD).
Correlations between specific impact domains such as sleep, cognitive thinking, and physical activity and overall AD impact scores ranged from weak to moderate, and no individual aspect of disease burden correlated strongly with overall impact scores. The researchers observed similar results after they stratified the analysis by age, current severity, and time spent managing AD.
In other findings, 40% of study participants reported mild changes in mood related to their AD, 30% reported moderate changes, 9% reported severe changes, while the remainder reported no changes in mood. The variable most strongly associated with current mood changes was having severe AD at the time of the survey (OR 5.29).
Understanding of disease burden ‘limited’
“Atopic dermatitis is associated with an immense clinical burden,” said Raj Chovatiya, MD, PhD, assistant professor in the department of dermatology at Northwestern University, Chicago, who was asked to comment on the study. “However, our understanding of disease burden from the patient perspective is limited,” he added.

“Interestingly, no single specific element of disease burden was strongly correlated with overall burden, further supporting the complex, multidimensional nature” of the impact of AD, he said, noting that the study “highlights the need for clinicians to look beyond the skin when it comes to AD and underscores the need for additional research to better understand the patient and caregiver perspective.”
Zelma Chiesa Fuxench, MD, MSCE, assistant professor of dermatology at the University of Pennsylvania, Philadelphia, who was also asked to comment on the study, noted that aside from the well discussed impact and burden of itch and its impact on sleep loss, much remains to be learned about the full impact of AD, particularly among adults.

“For example, it is commonly accepted and expected that patients with more severe AD likely experience higher disease burden, but are there other factors that can influence this risk?” she asked. “Can we explain the high impact of AD disease aside from the level of disease severity, particularly among adults with AD?”
The study, she added, “is important because it provides additional insights into those possible factors, including ‘time spent managing their disease’ and ‘associated depression.’ In particular, understanding the association between ‘time spent managing their disease’ and higher disease burden is critical because, in my opinion, it emphasizes the need to develop better strategies for improving the care of patients with AD including the development of more efficacious and safer treatment strategies.”
Dr. Drucker and colleagues acknowledged certain limitations of the analysis, including its cross-sectional design, the potential for selection bias, and the fact that it did not use the patient-oriented outcome measure or the dermatology life quality index. “Further work to address the complex burden of AD, including strategies to reduce time spent managing AD, and understanding the fullness of the patient experience is needed,” they concluded.
The work was supported in part by a grant from the National Eczema Association (NEA). Dr. Drucker reported that he receives compensation from the British Journal of Dermatology (as reviewer and section editor), American Academy of Dermatology (guidelines writer), and NEA (grant reviewer). Coauthors representing the NEA and other patient organizations including the Allergy & Asthma Network, Asthma and Allergy Foundation of America, Global Parents for Eczema Research, and International Topical Steroid Awareness Network received organizational grants (Pfizer) and sponsorship funding for these analyses from AbbVie, Eli Lilly, Incyte, LEO Pharma, Regeneron Pharmaceuticals, and Sanofi Genzyme.
Dr. Chovatiya disclosed that he has served as an advisory board member, consultant, speaker, and/or investigator for AbbVie, Arcutis, Arena, Beiersdorf, Bristol Myers Squibb, Dermavant, Eli Lilly and Company, EPI Health, Incyte, L’Oréal, the NEA, Pfizer, Regeneron, Sanofi, and UCB.
Dr. Chiesa Fuxench disclosed that she has received research grants from Lilly, LEO Pharma, Regeneron, Sanofi, Tioga, and Vanda for work related to AD She has served as consultant for the Asthma and Allergy Foundation of America, NEA, AbbVie, Incyte Corporation, and Pfizer; and received honoraria for CME work in AD sponsored by education grants from Regeneron/Sanofi and Pfizer.
. However, AD severity and spending 11 hours or more per week managing the condition did correlate with higher overall disease burden.
“Research has documented the disease burden of AD, including its visible nature and the effect on itch and sleep, but knowledge gaps remain,” Aaron M. Drucker, MD, of the division of dermatology at the University of Toronto, and colleagues wrote in the study published online in JAMA Dermatology. “Gaps include a poor understanding of symptoms other than itch, patients’ treatment experience, and how different elements of burden of disease interact.”
Dr. Drucker and colleagues collected data from an externally led patient-focused drug development survey on AD, a 32-item questionnaire that was administered electronically between Aug. 1, 2019, and Oct. 11, 2019. Respondents were asked to rate the overall impact of their AD in the past months and the specific elements of disease burden on a 1-5 scale, with 1 meaning no impact, and 5 meaning a significant impact. They were also asked to rate current mood changes and mood changes at the worst point of AD on a 4-point scale that ranged from “not present” to “severe.” The researchers used multivariable ordinal regression to examine associations between demographic and clinical variables and patient-reported overall AD impact scores.
Survey results
Of the 1,065 respondents, 33% were aged 18-34 years, 50% were aged 35-50 years, 17% were aged 65 years or older, and 83% were female. Nearly half (45%) reported having moderate AD, while 28% had severe AD. When asked about the overall disease burden of AD symptoms in the past month, 30% reported a significant impact on life, 28% reported a moderate impact score, 21% reported a high impact score, 18% reported a low impact score, and 3% of respondents reported no impact.
In the multivariable proportional odds analysis, moderate AD (odds ratio [OR], 4.13) and severe AD (OR, 13.63) were both associated with greater disease burden compared with mild AD. Also, spending 11 or more hours per week managing AD symptoms was associated with greater disease burden compared with 0 to 4 hours (an OR of 2.67 for 11-20 hours per week spent managing AD and OR of 5.34 for 21 or more hours per week spent managing AD).
Correlations between specific impact domains such as sleep, cognitive thinking, and physical activity and overall AD impact scores ranged from weak to moderate, and no individual aspect of disease burden correlated strongly with overall impact scores. The researchers observed similar results after they stratified the analysis by age, current severity, and time spent managing AD.
In other findings, 40% of study participants reported mild changes in mood related to their AD, 30% reported moderate changes, 9% reported severe changes, while the remainder reported no changes in mood. The variable most strongly associated with current mood changes was having severe AD at the time of the survey (OR 5.29).
Understanding of disease burden ‘limited’
“Atopic dermatitis is associated with an immense clinical burden,” said Raj Chovatiya, MD, PhD, assistant professor in the department of dermatology at Northwestern University, Chicago, who was asked to comment on the study. “However, our understanding of disease burden from the patient perspective is limited,” he added.
“Interestingly, no single specific element of disease burden was strongly correlated with overall burden, further supporting the complex, multidimensional nature” of the impact of AD, he said, noting that the study “highlights the need for clinicians to look beyond the skin when it comes to AD and underscores the need for additional research to better understand the patient and caregiver perspective.”
Zelma Chiesa Fuxench, MD, MSCE, assistant professor of dermatology at the University of Pennsylvania, Philadelphia, who was also asked to comment on the study, noted that aside from the well discussed impact and burden of itch and its impact on sleep loss, much remains to be learned about the full impact of AD, particularly among adults.
“For example, it is commonly accepted and expected that patients with more severe AD likely experience higher disease burden, but are there other factors that can influence this risk?” she asked. “Can we explain the high impact of AD disease aside from the level of disease severity, particularly among adults with AD?”
The study, she added, “is important because it provides additional insights into those possible factors, including ‘time spent managing their disease’ and ‘associated depression.’ In particular, understanding the association between ‘time spent managing their disease’ and higher disease burden is critical because, in my opinion, it emphasizes the need to develop better strategies for improving the care of patients with AD including the development of more efficacious and safer treatment strategies.”
Dr. Drucker and colleagues acknowledged certain limitations of the analysis, including its cross-sectional design, the potential for selection bias, and the fact that it did not use the patient-oriented outcome measure or the dermatology life quality index. “Further work to address the complex burden of AD, including strategies to reduce time spent managing AD, and understanding the fullness of the patient experience is needed,” they concluded.
The work was supported in part by a grant from the National Eczema Association (NEA). Dr. Drucker reported that he receives compensation from the British Journal of Dermatology (as reviewer and section editor), American Academy of Dermatology (guidelines writer), and NEA (grant reviewer). Coauthors representing the NEA and other patient organizations including the Allergy & Asthma Network, Asthma and Allergy Foundation of America, Global Parents for Eczema Research, and International Topical Steroid Awareness Network received organizational grants (Pfizer) and sponsorship funding for these analyses from AbbVie, Eli Lilly, Incyte, LEO Pharma, Regeneron Pharmaceuticals, and Sanofi Genzyme.
Dr. Chovatiya disclosed that he has served as an advisory board member, consultant, speaker, and/or investigator for AbbVie, Arcutis, Arena, Beiersdorf, Bristol Myers Squibb, Dermavant, Eli Lilly and Company, EPI Health, Incyte, L’Oréal, the NEA, Pfizer, Regeneron, Sanofi, and UCB.
Dr. Chiesa Fuxench disclosed that she has received research grants from Lilly, LEO Pharma, Regeneron, Sanofi, Tioga, and Vanda for work related to AD She has served as consultant for the Asthma and Allergy Foundation of America, NEA, AbbVie, Incyte Corporation, and Pfizer; and received honoraria for CME work in AD sponsored by education grants from Regeneron/Sanofi and Pfizer.
. However, AD severity and spending 11 hours or more per week managing the condition did correlate with higher overall disease burden.
“Research has documented the disease burden of AD, including its visible nature and the effect on itch and sleep, but knowledge gaps remain,” Aaron M. Drucker, MD, of the division of dermatology at the University of Toronto, and colleagues wrote in the study published online in JAMA Dermatology. “Gaps include a poor understanding of symptoms other than itch, patients’ treatment experience, and how different elements of burden of disease interact.”
Dr. Drucker and colleagues collected data from an externally led patient-focused drug development survey on AD, a 32-item questionnaire that was administered electronically between Aug. 1, 2019, and Oct. 11, 2019. Respondents were asked to rate the overall impact of their AD in the past months and the specific elements of disease burden on a 1-5 scale, with 1 meaning no impact, and 5 meaning a significant impact. They were also asked to rate current mood changes and mood changes at the worst point of AD on a 4-point scale that ranged from “not present” to “severe.” The researchers used multivariable ordinal regression to examine associations between demographic and clinical variables and patient-reported overall AD impact scores.
Survey results
Of the 1,065 respondents, 33% were aged 18-34 years, 50% were aged 35-50 years, 17% were aged 65 years or older, and 83% were female. Nearly half (45%) reported having moderate AD, while 28% had severe AD. When asked about the overall disease burden of AD symptoms in the past month, 30% reported a significant impact on life, 28% reported a moderate impact score, 21% reported a high impact score, 18% reported a low impact score, and 3% of respondents reported no impact.
In the multivariable proportional odds analysis, moderate AD (odds ratio [OR], 4.13) and severe AD (OR, 13.63) were both associated with greater disease burden compared with mild AD. Also, spending 11 or more hours per week managing AD symptoms was associated with greater disease burden compared with 0 to 4 hours (an OR of 2.67 for 11-20 hours per week spent managing AD and OR of 5.34 for 21 or more hours per week spent managing AD).
Correlations between specific impact domains such as sleep, cognitive thinking, and physical activity and overall AD impact scores ranged from weak to moderate, and no individual aspect of disease burden correlated strongly with overall impact scores. The researchers observed similar results after they stratified the analysis by age, current severity, and time spent managing AD.
In other findings, 40% of study participants reported mild changes in mood related to their AD, 30% reported moderate changes, 9% reported severe changes, while the remainder reported no changes in mood. The variable most strongly associated with current mood changes was having severe AD at the time of the survey (OR 5.29).
Understanding of disease burden ‘limited’
“Atopic dermatitis is associated with an immense clinical burden,” said Raj Chovatiya, MD, PhD, assistant professor in the department of dermatology at Northwestern University, Chicago, who was asked to comment on the study. “However, our understanding of disease burden from the patient perspective is limited,” he added.
“Interestingly, no single specific element of disease burden was strongly correlated with overall burden, further supporting the complex, multidimensional nature” of the impact of AD, he said, noting that the study “highlights the need for clinicians to look beyond the skin when it comes to AD and underscores the need for additional research to better understand the patient and caregiver perspective.”
Zelma Chiesa Fuxench, MD, MSCE, assistant professor of dermatology at the University of Pennsylvania, Philadelphia, who was also asked to comment on the study, noted that aside from the well discussed impact and burden of itch and its impact on sleep loss, much remains to be learned about the full impact of AD, particularly among adults.
“For example, it is commonly accepted and expected that patients with more severe AD likely experience higher disease burden, but are there other factors that can influence this risk?” she asked. “Can we explain the high impact of AD disease aside from the level of disease severity, particularly among adults with AD?”
The study, she added, “is important because it provides additional insights into those possible factors, including ‘time spent managing their disease’ and ‘associated depression.’ In particular, understanding the association between ‘time spent managing their disease’ and higher disease burden is critical because, in my opinion, it emphasizes the need to develop better strategies for improving the care of patients with AD including the development of more efficacious and safer treatment strategies.”
Dr. Drucker and colleagues acknowledged certain limitations of the analysis, including its cross-sectional design, the potential for selection bias, and the fact that it did not use the patient-oriented outcome measure or the dermatology life quality index. “Further work to address the complex burden of AD, including strategies to reduce time spent managing AD, and understanding the fullness of the patient experience is needed,” they concluded.
The work was supported in part by a grant from the National Eczema Association (NEA). Dr. Drucker reported that he receives compensation from the British Journal of Dermatology (as reviewer and section editor), American Academy of Dermatology (guidelines writer), and NEA (grant reviewer). Coauthors representing the NEA and other patient organizations including the Allergy & Asthma Network, Asthma and Allergy Foundation of America, Global Parents for Eczema Research, and International Topical Steroid Awareness Network received organizational grants (Pfizer) and sponsorship funding for these analyses from AbbVie, Eli Lilly, Incyte, LEO Pharma, Regeneron Pharmaceuticals, and Sanofi Genzyme.
Dr. Chovatiya disclosed that he has served as an advisory board member, consultant, speaker, and/or investigator for AbbVie, Arcutis, Arena, Beiersdorf, Bristol Myers Squibb, Dermavant, Eli Lilly and Company, EPI Health, Incyte, L’Oréal, the NEA, Pfizer, Regeneron, Sanofi, and UCB.
Dr. Chiesa Fuxench disclosed that she has received research grants from Lilly, LEO Pharma, Regeneron, Sanofi, Tioga, and Vanda for work related to AD She has served as consultant for the Asthma and Allergy Foundation of America, NEA, AbbVie, Incyte Corporation, and Pfizer; and received honoraria for CME work in AD sponsored by education grants from Regeneron/Sanofi and Pfizer.
FROM JAMA DERMATOLOGY
Study explores gender differences in pediatric melanoma
INDIANAPOLIS – .
In addition, male gender was independently associated with increased mortality, but age was not.
Those are key findings from a retrospective cohort analysis of nearly 5,000 records from the National Cancer Database.

“There are multiple studies from primarily adult populations showing females with melanoma have a different presentation and better outcomes than males,” co-first author Rebecca M. Thiede, MD, a dermatologist at the University of Arizona, Tucson, said in an interview with this news organization in advance of the annual meeting of the Society for Pediatric Dermatology, where the abstract was presented during a poster session. “However, because melanoma is so rare in younger patients, little is known about gender differences in presentation and survival in pediatric and adolescent patients. To our knowledge, this is one of the largest studies to date in this population, and the first to explore gender differences in detail in pediatric and adolescent patients with melanoma.”
Working with co-first author Sabrina Dahak, a fourth-year medical student at the University of Arizona, Phoenix, Dr. Thiede and colleagues retrospectively analyzed the National Cancer Database to identify biopsy-confirmed invasive primary cutaneous melanoma cases diagnosed in patients 0-21 years of age between 2004 and 2018. The search yielded 4,645 cases, and the researchers used American Academy of Pediatrics definitions to categorize the patients by age, from infancy (birth to 2 years), to childhood (3-10 years), early adolescence (11-14 years), middle adolescence (15-17 years), and late adolescence (18-21 years). They used the Kaplan Meier analysis to determine overall survival and multivariate Cox regression to determine independent survival predictors.
Of the 4,645 pediatric melanoma cases, 63.4% were in females and 36.6% were in males, a difference that was significant (P < .001). Dr. Thiede and colleagues also observed a significant relationship between primary site and gender (P < .001). Primary sites included the trunk (34.3% of females vs. 32.9% of males, respectively), head and neck (16.4% vs. 30.9%), upper extremities (19.5% vs. 16%), lower extremities (27.9% vs. 16.5%), and “unspecified” (1.9% vs. 3.7%).
Females had higher rates of superficial spreading melanoma while males were affected by nodular melanoma more often. For example, the median Breslow depth was higher for males (1.05 mm; interquartile range [IQR] 0.50-2.31) than for females (0.80 mm; IQR, 0.40-1.67; P < .001).
Although females accounted for a higher percentage of cases than males overall, from birth to 17 years, a higher percentage of males than females were found to have later stage of melanoma at time of diagnosis: Females were more likely to be diagnosed with stage I disease (67.8%) than were males (53.6%), and males were more likely than were females to be diagnosed with stages II (15.9% vs. 12.3%), III (27.1% vs. 18.3%), and IV disease (3.3% vs. 1.6%; P < .001 for all).
In other findings, the 5- and 10-year overall survival rates were higher for females (95.9% and 93.9%, respectively) than for males (92.0% vs. 86.7%, respectively; P < .001). However, by age group, overall survival rates were similar between females and males among infants, children, and those in early adolescence – but not for those in middle adolescence (96.7% vs. 91.9%; P < .001) or late adolescence (95.7% vs. 90.4%; P < .001).
When the researchers adjusted for confounding variables, male gender was independently associated with an increased risk of death (adjusted hazard ratio 1.37; P < .001), but age was not.
“It was particularly surprising to see that even at such a young age, there is a significant difference in overall survival between males and females, where females have better outcomes than males,” Dr. Thiede said. “When examining pediatric and adolescent patients, it is essential to maintain cutaneous melanoma on the differential,” she advised. “It is important for clinicians to perform a thorough exam at annual visits particularly for those at high risk for melanoma to catch this rare but potentially devastating diagnosis.”
She acknowledged certain limitations of the study, including its reliance on one database, “as comparing multiple databases would strengthen the conclusions,” she said. “There was some missing data present in our dataset, and a large percentage of the histologic subtypes were unspecified, both of which are common issues with cancer registries. An additional limitation is related to the low death rates in adolescent and pediatric patients, which may impact the analysis related to survival and independent predictors of survival.”
Asked to comment on the study results, Carrie C. Coughlin, MD, who directs the section of pediatric dermatology Washington University/St. Louis Children’s Hospital, said that the finding that males were more likely to present with stage II or higher disease compared with females “could be related to their finding that females had more superficial spreading melanomas, whereas males had more nodular melanoma.” Those differences “could influence how providers evaluate melanocytic lesions in children,” she added.

Dr. Coughlin, who directs the pediatric dermatology fellowship at Washington University/St. Louis Children’s Hospital, said it was “interesting” that the authors found no association between older age and an increased risk of death. “It would be helpful to have more data about melanoma subtype, including information about Spitz or Spitzoid melanomas,” she said. “Also, knowing the distribution of melanoma across the age categories could provide more insight into their data.”
Ms. Dahak received an award from the National Cancer Institute to fund travel for presentation of this study at the SPD meeting. No other financial conflicts were reported by the researchers. Dr. Coughlin is on the board of the Pediatric Dermatology Research Alliance (PeDRA) and the International Immunosuppression and Transplant Skin Cancer Collaborative.
INDIANAPOLIS – .
In addition, male gender was independently associated with increased mortality, but age was not.
Those are key findings from a retrospective cohort analysis of nearly 5,000 records from the National Cancer Database.
“There are multiple studies from primarily adult populations showing females with melanoma have a different presentation and better outcomes than males,” co-first author Rebecca M. Thiede, MD, a dermatologist at the University of Arizona, Tucson, said in an interview with this news organization in advance of the annual meeting of the Society for Pediatric Dermatology, where the abstract was presented during a poster session. “However, because melanoma is so rare in younger patients, little is known about gender differences in presentation and survival in pediatric and adolescent patients. To our knowledge, this is one of the largest studies to date in this population, and the first to explore gender differences in detail in pediatric and adolescent patients with melanoma.”
Working with co-first author Sabrina Dahak, a fourth-year medical student at the University of Arizona, Phoenix, Dr. Thiede and colleagues retrospectively analyzed the National Cancer Database to identify biopsy-confirmed invasive primary cutaneous melanoma cases diagnosed in patients 0-21 years of age between 2004 and 2018. The search yielded 4,645 cases, and the researchers used American Academy of Pediatrics definitions to categorize the patients by age, from infancy (birth to 2 years), to childhood (3-10 years), early adolescence (11-14 years), middle adolescence (15-17 years), and late adolescence (18-21 years). They used the Kaplan Meier analysis to determine overall survival and multivariate Cox regression to determine independent survival predictors.
Of the 4,645 pediatric melanoma cases, 63.4% were in females and 36.6% were in males, a difference that was significant (P < .001). Dr. Thiede and colleagues also observed a significant relationship between primary site and gender (P < .001). Primary sites included the trunk (34.3% of females vs. 32.9% of males, respectively), head and neck (16.4% vs. 30.9%), upper extremities (19.5% vs. 16%), lower extremities (27.9% vs. 16.5%), and “unspecified” (1.9% vs. 3.7%).
Females had higher rates of superficial spreading melanoma while males were affected by nodular melanoma more often. For example, the median Breslow depth was higher for males (1.05 mm; interquartile range [IQR] 0.50-2.31) than for females (0.80 mm; IQR, 0.40-1.67; P < .001).
Although females accounted for a higher percentage of cases than males overall, from birth to 17 years, a higher percentage of males than females were found to have later stage of melanoma at time of diagnosis: Females were more likely to be diagnosed with stage I disease (67.8%) than were males (53.6%), and males were more likely than were females to be diagnosed with stages II (15.9% vs. 12.3%), III (27.1% vs. 18.3%), and IV disease (3.3% vs. 1.6%; P < .001 for all).
In other findings, the 5- and 10-year overall survival rates were higher for females (95.9% and 93.9%, respectively) than for males (92.0% vs. 86.7%, respectively; P < .001). However, by age group, overall survival rates were similar between females and males among infants, children, and those in early adolescence – but not for those in middle adolescence (96.7% vs. 91.9%; P < .001) or late adolescence (95.7% vs. 90.4%; P < .001).
When the researchers adjusted for confounding variables, male gender was independently associated with an increased risk of death (adjusted hazard ratio 1.37; P < .001), but age was not.
“It was particularly surprising to see that even at such a young age, there is a significant difference in overall survival between males and females, where females have better outcomes than males,” Dr. Thiede said. “When examining pediatric and adolescent patients, it is essential to maintain cutaneous melanoma on the differential,” she advised. “It is important for clinicians to perform a thorough exam at annual visits particularly for those at high risk for melanoma to catch this rare but potentially devastating diagnosis.”
She acknowledged certain limitations of the study, including its reliance on one database, “as comparing multiple databases would strengthen the conclusions,” she said. “There was some missing data present in our dataset, and a large percentage of the histologic subtypes were unspecified, both of which are common issues with cancer registries. An additional limitation is related to the low death rates in adolescent and pediatric patients, which may impact the analysis related to survival and independent predictors of survival.”
Asked to comment on the study results, Carrie C. Coughlin, MD, who directs the section of pediatric dermatology Washington University/St. Louis Children’s Hospital, said that the finding that males were more likely to present with stage II or higher disease compared with females “could be related to their finding that females had more superficial spreading melanomas, whereas males had more nodular melanoma.” Those differences “could influence how providers evaluate melanocytic lesions in children,” she added.
Dr. Coughlin, who directs the pediatric dermatology fellowship at Washington University/St. Louis Children’s Hospital, said it was “interesting” that the authors found no association between older age and an increased risk of death. “It would be helpful to have more data about melanoma subtype, including information about Spitz or Spitzoid melanomas,” she said. “Also, knowing the distribution of melanoma across the age categories could provide more insight into their data.”
Ms. Dahak received an award from the National Cancer Institute to fund travel for presentation of this study at the SPD meeting. No other financial conflicts were reported by the researchers. Dr. Coughlin is on the board of the Pediatric Dermatology Research Alliance (PeDRA) and the International Immunosuppression and Transplant Skin Cancer Collaborative.
INDIANAPOLIS – .
In addition, male gender was independently associated with increased mortality, but age was not.
Those are key findings from a retrospective cohort analysis of nearly 5,000 records from the National Cancer Database.
“There are multiple studies from primarily adult populations showing females with melanoma have a different presentation and better outcomes than males,” co-first author Rebecca M. Thiede, MD, a dermatologist at the University of Arizona, Tucson, said in an interview with this news organization in advance of the annual meeting of the Society for Pediatric Dermatology, where the abstract was presented during a poster session. “However, because melanoma is so rare in younger patients, little is known about gender differences in presentation and survival in pediatric and adolescent patients. To our knowledge, this is one of the largest studies to date in this population, and the first to explore gender differences in detail in pediatric and adolescent patients with melanoma.”
Working with co-first author Sabrina Dahak, a fourth-year medical student at the University of Arizona, Phoenix, Dr. Thiede and colleagues retrospectively analyzed the National Cancer Database to identify biopsy-confirmed invasive primary cutaneous melanoma cases diagnosed in patients 0-21 years of age between 2004 and 2018. The search yielded 4,645 cases, and the researchers used American Academy of Pediatrics definitions to categorize the patients by age, from infancy (birth to 2 years), to childhood (3-10 years), early adolescence (11-14 years), middle adolescence (15-17 years), and late adolescence (18-21 years). They used the Kaplan Meier analysis to determine overall survival and multivariate Cox regression to determine independent survival predictors.
Of the 4,645 pediatric melanoma cases, 63.4% were in females and 36.6% were in males, a difference that was significant (P < .001). Dr. Thiede and colleagues also observed a significant relationship between primary site and gender (P < .001). Primary sites included the trunk (34.3% of females vs. 32.9% of males, respectively), head and neck (16.4% vs. 30.9%), upper extremities (19.5% vs. 16%), lower extremities (27.9% vs. 16.5%), and “unspecified” (1.9% vs. 3.7%).
Females had higher rates of superficial spreading melanoma while males were affected by nodular melanoma more often. For example, the median Breslow depth was higher for males (1.05 mm; interquartile range [IQR] 0.50-2.31) than for females (0.80 mm; IQR, 0.40-1.67; P < .001).
Although females accounted for a higher percentage of cases than males overall, from birth to 17 years, a higher percentage of males than females were found to have later stage of melanoma at time of diagnosis: Females were more likely to be diagnosed with stage I disease (67.8%) than were males (53.6%), and males were more likely than were females to be diagnosed with stages II (15.9% vs. 12.3%), III (27.1% vs. 18.3%), and IV disease (3.3% vs. 1.6%; P < .001 for all).
In other findings, the 5- and 10-year overall survival rates were higher for females (95.9% and 93.9%, respectively) than for males (92.0% vs. 86.7%, respectively; P < .001). However, by age group, overall survival rates were similar between females and males among infants, children, and those in early adolescence – but not for those in middle adolescence (96.7% vs. 91.9%; P < .001) or late adolescence (95.7% vs. 90.4%; P < .001).
When the researchers adjusted for confounding variables, male gender was independently associated with an increased risk of death (adjusted hazard ratio 1.37; P < .001), but age was not.
“It was particularly surprising to see that even at such a young age, there is a significant difference in overall survival between males and females, where females have better outcomes than males,” Dr. Thiede said. “When examining pediatric and adolescent patients, it is essential to maintain cutaneous melanoma on the differential,” she advised. “It is important for clinicians to perform a thorough exam at annual visits particularly for those at high risk for melanoma to catch this rare but potentially devastating diagnosis.”
She acknowledged certain limitations of the study, including its reliance on one database, “as comparing multiple databases would strengthen the conclusions,” she said. “There was some missing data present in our dataset, and a large percentage of the histologic subtypes were unspecified, both of which are common issues with cancer registries. An additional limitation is related to the low death rates in adolescent and pediatric patients, which may impact the analysis related to survival and independent predictors of survival.”
Asked to comment on the study results, Carrie C. Coughlin, MD, who directs the section of pediatric dermatology Washington University/St. Louis Children’s Hospital, said that the finding that males were more likely to present with stage II or higher disease compared with females “could be related to their finding that females had more superficial spreading melanomas, whereas males had more nodular melanoma.” Those differences “could influence how providers evaluate melanocytic lesions in children,” she added.
Dr. Coughlin, who directs the pediatric dermatology fellowship at Washington University/St. Louis Children’s Hospital, said it was “interesting” that the authors found no association between older age and an increased risk of death. “It would be helpful to have more data about melanoma subtype, including information about Spitz or Spitzoid melanomas,” she said. “Also, knowing the distribution of melanoma across the age categories could provide more insight into their data.”
Ms. Dahak received an award from the National Cancer Institute to fund travel for presentation of this study at the SPD meeting. No other financial conflicts were reported by the researchers. Dr. Coughlin is on the board of the Pediatric Dermatology Research Alliance (PeDRA) and the International Immunosuppression and Transplant Skin Cancer Collaborative.
AT SPD 2022
Ruxolitinib found to benefit adolescents with vitiligo up to one year
INDIANAPOLIS – and a higher proportion responded at week 52, results from a pooled analysis of phase 3 data showed.
Currently, there is no treatment approved by the Food and Drug Administration to repigment patients with vitiligo, but the cream formulation of the Janus kinase inhibitor ruxolitinib was shown to be effective and have a favorable safety profile in patients aged 12 years and up in the phase 3 clinical trials, TRuE-V1 and TruE-V2. “We know that about half of patients will develop vitiligo by the age of 20, so there is a significant need to have treatments available for the pediatric population,” lead study author David Rosmarin, MD, told this news organization in advance of the annual meeting of the Society for Pediatric Dermatology.

In September 2021, topical ruxolitinib (Opzelura) was approved by the FDA for treating atopic dermatitis in nonimmunocompromised patients aged 12 years and older. The manufacturer, Incyte, has submitted an application for approval to the agency for treating vitiligo in patients ages 12 years and older based on 24-week results; the FDA is expected to make a decision by July 18.
For the current study, presented during a poster session at the meeting, Dr. Rosmarin, of the department of dermatology at Tufts Medical Center, Boston, and colleagues pooled efficacy and safety data for adolescent patients aged 12-17 years from the TRuE-V studies, which enrolled patients 12 years of age and older diagnosed with nonsegmental vitiligo with depigmentation covering up to 10% of total body surface area (BSA), including facial and total Vitiligo Area Scoring Index (F-VASI/T-VASI) scores of ≥ 0.5/≥ 3. Investigators randomized patients 2:1 to twice-daily 1.5% ruxolitinib cream or vehicle for 24 weeks, after which all patients could apply 1.5% ruxolitinib cream through week 52. Efficacy endpoints included the proportions of patients who achieved at least 75%, 50%, and 90% improvement from baseline in F-VASI scores (F-VASI75, F-VASI50, F-VASI90); the proportion of patients who achieved at least a 50% improvement from baseline in T-VASI (T-VASI50); the proportion of patients who achieved a Vitiligo Noticeability Scale (VNS) rating of 4 or 5; and percentage change from baseline in facial BSA (F-BSA). Safety and tolerability were also assessed.
For the pooled analysis, Dr. Rosmarin and colleagues reported results on 72 adolescents: 55 who received ruxolitinib cream and 17 who received vehicle. At week 24, 32.1% of adolescents treated with ruxolitinib cream achieved F-VASI75, compared with none of those in the vehicle group. Further, response rates at week 52 for patients who applied ruxolitinib cream from day 1 were as follows: F-VASI75, 48.0%; F-VASI50, 70.0%; F-VASI90, 24.0%; T-VASI50, 60.0%; VNS score of 4/5, 56.0%; and F-BSA mean percentage change from baseline, –41.9%.
Efficacy at week 52 among crossover patients (after 28 weeks of ruxolitinib cream) was consistent with week 24 data in patients who applied ruxolitinib cream from day 1.
“As we know that repigmentation takes time, about half of the patients achieved the F-VASI75 at the 52-week endpoint,” said Dr. Rosmarin, who is also vice-chair for research and education at Tufts Medical Center, Boston. “Particularly remarkable is that 60% of adolescents achieved a T-VASI50 [50% or more repigmentation of the whole body at the year mark] and over half the patients described their vitiligo as a lot less noticeable or no longer noticeable at the year mark.”
In terms of safety, treatment-related adverse events occurred in 12.9% of patients treated with ruxolitinib (no information was available on the specific events). Serious adverse events occurred in 1.4% of patients; none were considered related to treatment.
“Overall, these results are quite impressive,” Dr. Rosmarin said. “While it can be very challenging to repigment patients with vitiligo, ruxolitinib cream provides an effective option which can help many of my patients.” He acknowledged certain limitations of the analysis, including the fact that the TRuE-V studies were conducted during the COVID-19 pandemic, “which may have contributed to patients being lost to follow-up. Also, the majority of the patients had skin phototypes 1-3.”
Carrie C. Coughlin, MD, who was asked to comment on the study, said that patients with vitiligo need treatment options that are well-studied and covered by insurance. “This study is a great step forward in developing medications for this underserved patient population,” said Dr. Coughlin, who directs the section of pediatric dermatology at Washington University/St. Louis Children’s Hospital.
However, she continued, “the authors mention approximately 13% of patients had a treatment-related adverse reaction, but the abstract does not delineate these reactions.” In addition, the study was limited to children who had less than or equal to 10% body surface area involvement of vitiligo, she noted, adding that “more work is needed to learn about safety of application to larger surface areas.”
Going forward, “it will be important to learn the durability of response,” said Dr. Coughlin, who is also assistant professor of dermatology at Washington University in St. Louis. “Does the vitiligo return if patients stop applying the ruxolitinib cream?”
Dr. Rosmarin disclosed that he has received honoraria as a consultant for Incyte, AbbVie, Abcuro, AltruBio, Arena, Boehringer Ingelheim, Bristol Meyers Squibb, Celgene, Concert, CSL Behring, Dermavant, Dermira, Janssen, Kyowa Kirin, Lilly, Novartis, Pfizer, Regeneron, Revolo Biotherapeutics, Sanofi, Sun Pharmaceuticals, UCB, and VielaBio. He has also received research support from Incyte, AbbVie, Amgen, Bristol-Myers Squibb, Celgene, Dermira, Galderma, Janssen, Lilly, Merck, Novartis, Pfizer, and Regeneron; and has served as a paid speaker for Incyte, AbbVie, Amgen, Bristol-Myers Squibb, Celgene, Incyte, Janssen, Lilly, Novartis, Pfizer, Regeneron, and Sanofi. Dr. Coughlin is on the board of the Pediatric Dermatology Research Alliance and the International Immunosuppression and Transplant Skin Cancer Collaborative.
INDIANAPOLIS – and a higher proportion responded at week 52, results from a pooled analysis of phase 3 data showed.
Currently, there is no treatment approved by the Food and Drug Administration to repigment patients with vitiligo, but the cream formulation of the Janus kinase inhibitor ruxolitinib was shown to be effective and have a favorable safety profile in patients aged 12 years and up in the phase 3 clinical trials, TRuE-V1 and TruE-V2. “We know that about half of patients will develop vitiligo by the age of 20, so there is a significant need to have treatments available for the pediatric population,” lead study author David Rosmarin, MD, told this news organization in advance of the annual meeting of the Society for Pediatric Dermatology.
In September 2021, topical ruxolitinib (Opzelura) was approved by the FDA for treating atopic dermatitis in nonimmunocompromised patients aged 12 years and older. The manufacturer, Incyte, has submitted an application for approval to the agency for treating vitiligo in patients ages 12 years and older based on 24-week results; the FDA is expected to make a decision by July 18.
For the current study, presented during a poster session at the meeting, Dr. Rosmarin, of the department of dermatology at Tufts Medical Center, Boston, and colleagues pooled efficacy and safety data for adolescent patients aged 12-17 years from the TRuE-V studies, which enrolled patients 12 years of age and older diagnosed with nonsegmental vitiligo with depigmentation covering up to 10% of total body surface area (BSA), including facial and total Vitiligo Area Scoring Index (F-VASI/T-VASI) scores of ≥ 0.5/≥ 3. Investigators randomized patients 2:1 to twice-daily 1.5% ruxolitinib cream or vehicle for 24 weeks, after which all patients could apply 1.5% ruxolitinib cream through week 52. Efficacy endpoints included the proportions of patients who achieved at least 75%, 50%, and 90% improvement from baseline in F-VASI scores (F-VASI75, F-VASI50, F-VASI90); the proportion of patients who achieved at least a 50% improvement from baseline in T-VASI (T-VASI50); the proportion of patients who achieved a Vitiligo Noticeability Scale (VNS) rating of 4 or 5; and percentage change from baseline in facial BSA (F-BSA). Safety and tolerability were also assessed.
For the pooled analysis, Dr. Rosmarin and colleagues reported results on 72 adolescents: 55 who received ruxolitinib cream and 17 who received vehicle. At week 24, 32.1% of adolescents treated with ruxolitinib cream achieved F-VASI75, compared with none of those in the vehicle group. Further, response rates at week 52 for patients who applied ruxolitinib cream from day 1 were as follows: F-VASI75, 48.0%; F-VASI50, 70.0%; F-VASI90, 24.0%; T-VASI50, 60.0%; VNS score of 4/5, 56.0%; and F-BSA mean percentage change from baseline, –41.9%.
Efficacy at week 52 among crossover patients (after 28 weeks of ruxolitinib cream) was consistent with week 24 data in patients who applied ruxolitinib cream from day 1.
“As we know that repigmentation takes time, about half of the patients achieved the F-VASI75 at the 52-week endpoint,” said Dr. Rosmarin, who is also vice-chair for research and education at Tufts Medical Center, Boston. “Particularly remarkable is that 60% of adolescents achieved a T-VASI50 [50% or more repigmentation of the whole body at the year mark] and over half the patients described their vitiligo as a lot less noticeable or no longer noticeable at the year mark.”
In terms of safety, treatment-related adverse events occurred in 12.9% of patients treated with ruxolitinib (no information was available on the specific events). Serious adverse events occurred in 1.4% of patients; none were considered related to treatment.
“Overall, these results are quite impressive,” Dr. Rosmarin said. “While it can be very challenging to repigment patients with vitiligo, ruxolitinib cream provides an effective option which can help many of my patients.” He acknowledged certain limitations of the analysis, including the fact that the TRuE-V studies were conducted during the COVID-19 pandemic, “which may have contributed to patients being lost to follow-up. Also, the majority of the patients had skin phototypes 1-3.”
Carrie C. Coughlin, MD, who was asked to comment on the study, said that patients with vitiligo need treatment options that are well-studied and covered by insurance. “This study is a great step forward in developing medications for this underserved patient population,” said Dr. Coughlin, who directs the section of pediatric dermatology at Washington University/St. Louis Children’s Hospital.
However, she continued, “the authors mention approximately 13% of patients had a treatment-related adverse reaction, but the abstract does not delineate these reactions.” In addition, the study was limited to children who had less than or equal to 10% body surface area involvement of vitiligo, she noted, adding that “more work is needed to learn about safety of application to larger surface areas.”
Going forward, “it will be important to learn the durability of response,” said Dr. Coughlin, who is also assistant professor of dermatology at Washington University in St. Louis. “Does the vitiligo return if patients stop applying the ruxolitinib cream?”
Dr. Rosmarin disclosed that he has received honoraria as a consultant for Incyte, AbbVie, Abcuro, AltruBio, Arena, Boehringer Ingelheim, Bristol Meyers Squibb, Celgene, Concert, CSL Behring, Dermavant, Dermira, Janssen, Kyowa Kirin, Lilly, Novartis, Pfizer, Regeneron, Revolo Biotherapeutics, Sanofi, Sun Pharmaceuticals, UCB, and VielaBio. He has also received research support from Incyte, AbbVie, Amgen, Bristol-Myers Squibb, Celgene, Dermira, Galderma, Janssen, Lilly, Merck, Novartis, Pfizer, and Regeneron; and has served as a paid speaker for Incyte, AbbVie, Amgen, Bristol-Myers Squibb, Celgene, Incyte, Janssen, Lilly, Novartis, Pfizer, Regeneron, and Sanofi. Dr. Coughlin is on the board of the Pediatric Dermatology Research Alliance and the International Immunosuppression and Transplant Skin Cancer Collaborative.
INDIANAPOLIS – and a higher proportion responded at week 52, results from a pooled analysis of phase 3 data showed.
Currently, there is no treatment approved by the Food and Drug Administration to repigment patients with vitiligo, but the cream formulation of the Janus kinase inhibitor ruxolitinib was shown to be effective and have a favorable safety profile in patients aged 12 years and up in the phase 3 clinical trials, TRuE-V1 and TruE-V2. “We know that about half of patients will develop vitiligo by the age of 20, so there is a significant need to have treatments available for the pediatric population,” lead study author David Rosmarin, MD, told this news organization in advance of the annual meeting of the Society for Pediatric Dermatology.
In September 2021, topical ruxolitinib (Opzelura) was approved by the FDA for treating atopic dermatitis in nonimmunocompromised patients aged 12 years and older. The manufacturer, Incyte, has submitted an application for approval to the agency for treating vitiligo in patients ages 12 years and older based on 24-week results; the FDA is expected to make a decision by July 18.
For the current study, presented during a poster session at the meeting, Dr. Rosmarin, of the department of dermatology at Tufts Medical Center, Boston, and colleagues pooled efficacy and safety data for adolescent patients aged 12-17 years from the TRuE-V studies, which enrolled patients 12 years of age and older diagnosed with nonsegmental vitiligo with depigmentation covering up to 10% of total body surface area (BSA), including facial and total Vitiligo Area Scoring Index (F-VASI/T-VASI) scores of ≥ 0.5/≥ 3. Investigators randomized patients 2:1 to twice-daily 1.5% ruxolitinib cream or vehicle for 24 weeks, after which all patients could apply 1.5% ruxolitinib cream through week 52. Efficacy endpoints included the proportions of patients who achieved at least 75%, 50%, and 90% improvement from baseline in F-VASI scores (F-VASI75, F-VASI50, F-VASI90); the proportion of patients who achieved at least a 50% improvement from baseline in T-VASI (T-VASI50); the proportion of patients who achieved a Vitiligo Noticeability Scale (VNS) rating of 4 or 5; and percentage change from baseline in facial BSA (F-BSA). Safety and tolerability were also assessed.
For the pooled analysis, Dr. Rosmarin and colleagues reported results on 72 adolescents: 55 who received ruxolitinib cream and 17 who received vehicle. At week 24, 32.1% of adolescents treated with ruxolitinib cream achieved F-VASI75, compared with none of those in the vehicle group. Further, response rates at week 52 for patients who applied ruxolitinib cream from day 1 were as follows: F-VASI75, 48.0%; F-VASI50, 70.0%; F-VASI90, 24.0%; T-VASI50, 60.0%; VNS score of 4/5, 56.0%; and F-BSA mean percentage change from baseline, –41.9%.
Efficacy at week 52 among crossover patients (after 28 weeks of ruxolitinib cream) was consistent with week 24 data in patients who applied ruxolitinib cream from day 1.
“As we know that repigmentation takes time, about half of the patients achieved the F-VASI75 at the 52-week endpoint,” said Dr. Rosmarin, who is also vice-chair for research and education at Tufts Medical Center, Boston. “Particularly remarkable is that 60% of adolescents achieved a T-VASI50 [50% or more repigmentation of the whole body at the year mark] and over half the patients described their vitiligo as a lot less noticeable or no longer noticeable at the year mark.”
In terms of safety, treatment-related adverse events occurred in 12.9% of patients treated with ruxolitinib (no information was available on the specific events). Serious adverse events occurred in 1.4% of patients; none were considered related to treatment.
“Overall, these results are quite impressive,” Dr. Rosmarin said. “While it can be very challenging to repigment patients with vitiligo, ruxolitinib cream provides an effective option which can help many of my patients.” He acknowledged certain limitations of the analysis, including the fact that the TRuE-V studies were conducted during the COVID-19 pandemic, “which may have contributed to patients being lost to follow-up. Also, the majority of the patients had skin phototypes 1-3.”
Carrie C. Coughlin, MD, who was asked to comment on the study, said that patients with vitiligo need treatment options that are well-studied and covered by insurance. “This study is a great step forward in developing medications for this underserved patient population,” said Dr. Coughlin, who directs the section of pediatric dermatology at Washington University/St. Louis Children’s Hospital.
However, she continued, “the authors mention approximately 13% of patients had a treatment-related adverse reaction, but the abstract does not delineate these reactions.” In addition, the study was limited to children who had less than or equal to 10% body surface area involvement of vitiligo, she noted, adding that “more work is needed to learn about safety of application to larger surface areas.”
Going forward, “it will be important to learn the durability of response,” said Dr. Coughlin, who is also assistant professor of dermatology at Washington University in St. Louis. “Does the vitiligo return if patients stop applying the ruxolitinib cream?”
Dr. Rosmarin disclosed that he has received honoraria as a consultant for Incyte, AbbVie, Abcuro, AltruBio, Arena, Boehringer Ingelheim, Bristol Meyers Squibb, Celgene, Concert, CSL Behring, Dermavant, Dermira, Janssen, Kyowa Kirin, Lilly, Novartis, Pfizer, Regeneron, Revolo Biotherapeutics, Sanofi, Sun Pharmaceuticals, UCB, and VielaBio. He has also received research support from Incyte, AbbVie, Amgen, Bristol-Myers Squibb, Celgene, Dermira, Galderma, Janssen, Lilly, Merck, Novartis, Pfizer, and Regeneron; and has served as a paid speaker for Incyte, AbbVie, Amgen, Bristol-Myers Squibb, Celgene, Incyte, Janssen, Lilly, Novartis, Pfizer, Regeneron, and Sanofi. Dr. Coughlin is on the board of the Pediatric Dermatology Research Alliance and the International Immunosuppression and Transplant Skin Cancer Collaborative.
AT SPD 2022
Quality of life benefit exaggerated in some cancer studies
, according to a study published in JAMA Oncology.
The study found trials that failed to show improved quality of life often reported their quality of life outcomes more favorably. Non–immunotherapy-targeted drugs were found to lead to worse quality of life outcomes more often than did cytotoxic agents. And, while there is an association between quality of life benefit and overall survival, no such association was found with progression-free survival.
“In this study, we evaluated the outcomes of cancer drug trials with regard to patients’ quality of life and found that only a quarter of phase 3 cancer drug trials in the advanced-disease setting demonstrated improved quality of life,” wrote authors who were led by Bishal Gyawali, MD, PhD, of the Cancer Research Institute, Queen’s University, Kingston, Ont.
“Improved quality of life outcomes were associated with improved overall survival but not with improved progression-free survival. Importantly, almost half of the cancer drugs drug trials that showed improved progression-free survival showed no improved overall survival or quality of life (i.e., PFS-only benefit). Some reports included conclusions regarding quality of life (QOL) findings that were not directly supported by the trial data, particularly for inferior or non–statistically significant QOL outcomes, thereby framing the findings in a favorable light or downplaying detrimental effects of the study intervention on QOL. Furthermore, contrary to common perception, inferior QOL outcomes were more common with targeted drugs than cytotoxic drugs. Taken together, these findings have important policy implications,” the authors wrote.
These findings are based on the results of a cohort study of 45 phase 3 research clinical trials of 24,806 patients. Only a small percentage of patients showed QOL benefits. The study found that industry-funded clinical trial reports often framed QOL findings more favorably than was warranted by the data.
The study found improved QOL with experimental agents in 11 of 45 randomized controlled trials (24.4%). Studies that reported improved QOL were more likely to also show improved overall survival as compared with trials in which quality of life was not improved (7 of 11 [64%] versus 10 of 34 [29%] trials). For improved progression-free survival, however, there was no positive association (6 of 11 [55%] trials versus 17 of 34 [50%] trials without improved QOL). Among six trials reporting worsening QOL, three (50%) were trials of targeted drugs. Among 11 trials reporting improved QOL, 6 (55%) were trials of immunotherapy drugs. Among the 34 trials in which QOL was not improved compared with controls, the findings were framed favorably (versus neutrally or negatively) in the abstract or conclusions in 16 (47%), an observation that was statistically significantly associated with industry funding (chi-squared = 6.35; P = .01).
“It is important to clearly understand and communicate the effects of cancer drugs”
To fulfill the obligation to inform patients about proposed treatments, the authors wrote that it is important to clearly understand and communicate the effects of cancer drugs on patient quality of life alongside their effects on overall survival and intermediate end points such as progression-free survival. “Patients with advanced cancer expect treatment to help them live longer or have better lives,” the authors wrote. In that respect, in clinical trials of cancer medicines, overall survival and quality of life are the most important measures. Toxicity profiles and disease progression delays do not reliably predict quality of life, and studies have shown poor correlations between quality of life, overall survival, and progression-free survival. This raises the question of validity of progression-free survival as a surrogate endpoint. “Progression-free survival is meaningless without overall survival or quality of life gains,” Dr. Gyawali said in an interview.
Writing in The Lancet Oncology in March, Dr. Gyawali stated that, because progression free survival “does not directly measure how a patient feels or functions, or how long a patient lives, progression-free survival was not intended to inform clinical practice or establish whether a new therapy provides clinically meaningful benefits for patients. However, over the past 2 decades, it has become the most common primary endpoint in oncology clinical trials. We are deeply worried about how the term survival in this phrase can influence clinical practice and patient choices. We propose replacing the phrase progression-free survival with a less ambiguous term: progression-free interval.”
In JAMA Oncology, Dr. Gyawali aimed to elucidate relationships between QOL, overall survival, and progression-free survival, and to assess, as well, how QOL results are framed, especially in industry-sponsored research. When drug trials they analyzed showed no change in QOL but reported that QOL did not worsen or QOL was maintained rather than stating that QOL did not improve, or if there was downplaying of worse QOL outcomes, the study had favorable interpretation, Dr. Gyawali and associates wrote. The expectation of patients receiving cancer drugs would be improved QOL rather than “not worse” QOL, Dr. Gyawali said.
Regarding the finding that QOL outcomes were described as favorable in 47% of trials with unimproved QOL outcomes, Dr. Gyawali said, “the bias in reporting should be corrected by the reviewers and editors of journals. Also, quality of life reporting should be made mandatory. Without unbiased quality of life information, informed decision making on whether or not to use a certain drug is impossible. Patients and physicians need to know that information. Regulators can demand that this should be mandatory in all trials in noncurative settings.”
He remarked further on the worsening QOL in some targeted drug trials, “People tout chemo-free regimens as automatically having better quality of life, but that doesn’t seem to be the case. Targeted drugs can have a severe impact on quality of life, probably due to prolonged duration of side effects. Quality of life should be measured and reported for all drugs.”
Dr. Gyawali and associates noted the limitation in that several studies with negative QOL results are not published at all or are published after a considerable delay, so the present observations may understate the issues that have been raised.
Dr. Gyawali declared that he received no funding and disclosed no conflicts of interest for this study.
, according to a study published in JAMA Oncology.
The study found trials that failed to show improved quality of life often reported their quality of life outcomes more favorably. Non–immunotherapy-targeted drugs were found to lead to worse quality of life outcomes more often than did cytotoxic agents. And, while there is an association between quality of life benefit and overall survival, no such association was found with progression-free survival.
“In this study, we evaluated the outcomes of cancer drug trials with regard to patients’ quality of life and found that only a quarter of phase 3 cancer drug trials in the advanced-disease setting demonstrated improved quality of life,” wrote authors who were led by Bishal Gyawali, MD, PhD, of the Cancer Research Institute, Queen’s University, Kingston, Ont.
“Improved quality of life outcomes were associated with improved overall survival but not with improved progression-free survival. Importantly, almost half of the cancer drugs drug trials that showed improved progression-free survival showed no improved overall survival or quality of life (i.e., PFS-only benefit). Some reports included conclusions regarding quality of life (QOL) findings that were not directly supported by the trial data, particularly for inferior or non–statistically significant QOL outcomes, thereby framing the findings in a favorable light or downplaying detrimental effects of the study intervention on QOL. Furthermore, contrary to common perception, inferior QOL outcomes were more common with targeted drugs than cytotoxic drugs. Taken together, these findings have important policy implications,” the authors wrote.
These findings are based on the results of a cohort study of 45 phase 3 research clinical trials of 24,806 patients. Only a small percentage of patients showed QOL benefits. The study found that industry-funded clinical trial reports often framed QOL findings more favorably than was warranted by the data.
The study found improved QOL with experimental agents in 11 of 45 randomized controlled trials (24.4%). Studies that reported improved QOL were more likely to also show improved overall survival as compared with trials in which quality of life was not improved (7 of 11 [64%] versus 10 of 34 [29%] trials). For improved progression-free survival, however, there was no positive association (6 of 11 [55%] trials versus 17 of 34 [50%] trials without improved QOL). Among six trials reporting worsening QOL, three (50%) were trials of targeted drugs. Among 11 trials reporting improved QOL, 6 (55%) were trials of immunotherapy drugs. Among the 34 trials in which QOL was not improved compared with controls, the findings were framed favorably (versus neutrally or negatively) in the abstract or conclusions in 16 (47%), an observation that was statistically significantly associated with industry funding (chi-squared = 6.35; P = .01).
“It is important to clearly understand and communicate the effects of cancer drugs”
To fulfill the obligation to inform patients about proposed treatments, the authors wrote that it is important to clearly understand and communicate the effects of cancer drugs on patient quality of life alongside their effects on overall survival and intermediate end points such as progression-free survival. “Patients with advanced cancer expect treatment to help them live longer or have better lives,” the authors wrote. In that respect, in clinical trials of cancer medicines, overall survival and quality of life are the most important measures. Toxicity profiles and disease progression delays do not reliably predict quality of life, and studies have shown poor correlations between quality of life, overall survival, and progression-free survival. This raises the question of validity of progression-free survival as a surrogate endpoint. “Progression-free survival is meaningless without overall survival or quality of life gains,” Dr. Gyawali said in an interview.
Writing in The Lancet Oncology in March, Dr. Gyawali stated that, because progression free survival “does not directly measure how a patient feels or functions, or how long a patient lives, progression-free survival was not intended to inform clinical practice or establish whether a new therapy provides clinically meaningful benefits for patients. However, over the past 2 decades, it has become the most common primary endpoint in oncology clinical trials. We are deeply worried about how the term survival in this phrase can influence clinical practice and patient choices. We propose replacing the phrase progression-free survival with a less ambiguous term: progression-free interval.”
In JAMA Oncology, Dr. Gyawali aimed to elucidate relationships between QOL, overall survival, and progression-free survival, and to assess, as well, how QOL results are framed, especially in industry-sponsored research. When drug trials they analyzed showed no change in QOL but reported that QOL did not worsen or QOL was maintained rather than stating that QOL did not improve, or if there was downplaying of worse QOL outcomes, the study had favorable interpretation, Dr. Gyawali and associates wrote. The expectation of patients receiving cancer drugs would be improved QOL rather than “not worse” QOL, Dr. Gyawali said.
Regarding the finding that QOL outcomes were described as favorable in 47% of trials with unimproved QOL outcomes, Dr. Gyawali said, “the bias in reporting should be corrected by the reviewers and editors of journals. Also, quality of life reporting should be made mandatory. Without unbiased quality of life information, informed decision making on whether or not to use a certain drug is impossible. Patients and physicians need to know that information. Regulators can demand that this should be mandatory in all trials in noncurative settings.”
He remarked further on the worsening QOL in some targeted drug trials, “People tout chemo-free regimens as automatically having better quality of life, but that doesn’t seem to be the case. Targeted drugs can have a severe impact on quality of life, probably due to prolonged duration of side effects. Quality of life should be measured and reported for all drugs.”
Dr. Gyawali and associates noted the limitation in that several studies with negative QOL results are not published at all or are published after a considerable delay, so the present observations may understate the issues that have been raised.
Dr. Gyawali declared that he received no funding and disclosed no conflicts of interest for this study.
, according to a study published in JAMA Oncology.
The study found trials that failed to show improved quality of life often reported their quality of life outcomes more favorably. Non–immunotherapy-targeted drugs were found to lead to worse quality of life outcomes more often than did cytotoxic agents. And, while there is an association between quality of life benefit and overall survival, no such association was found with progression-free survival.
“In this study, we evaluated the outcomes of cancer drug trials with regard to patients’ quality of life and found that only a quarter of phase 3 cancer drug trials in the advanced-disease setting demonstrated improved quality of life,” wrote authors who were led by Bishal Gyawali, MD, PhD, of the Cancer Research Institute, Queen’s University, Kingston, Ont.
“Improved quality of life outcomes were associated with improved overall survival but not with improved progression-free survival. Importantly, almost half of the cancer drugs drug trials that showed improved progression-free survival showed no improved overall survival or quality of life (i.e., PFS-only benefit). Some reports included conclusions regarding quality of life (QOL) findings that were not directly supported by the trial data, particularly for inferior or non–statistically significant QOL outcomes, thereby framing the findings in a favorable light or downplaying detrimental effects of the study intervention on QOL. Furthermore, contrary to common perception, inferior QOL outcomes were more common with targeted drugs than cytotoxic drugs. Taken together, these findings have important policy implications,” the authors wrote.
These findings are based on the results of a cohort study of 45 phase 3 research clinical trials of 24,806 patients. Only a small percentage of patients showed QOL benefits. The study found that industry-funded clinical trial reports often framed QOL findings more favorably than was warranted by the data.
The study found improved QOL with experimental agents in 11 of 45 randomized controlled trials (24.4%). Studies that reported improved QOL were more likely to also show improved overall survival as compared with trials in which quality of life was not improved (7 of 11 [64%] versus 10 of 34 [29%] trials). For improved progression-free survival, however, there was no positive association (6 of 11 [55%] trials versus 17 of 34 [50%] trials without improved QOL). Among six trials reporting worsening QOL, three (50%) were trials of targeted drugs. Among 11 trials reporting improved QOL, 6 (55%) were trials of immunotherapy drugs. Among the 34 trials in which QOL was not improved compared with controls, the findings were framed favorably (versus neutrally or negatively) in the abstract or conclusions in 16 (47%), an observation that was statistically significantly associated with industry funding (chi-squared = 6.35; P = .01).
“It is important to clearly understand and communicate the effects of cancer drugs”
To fulfill the obligation to inform patients about proposed treatments, the authors wrote that it is important to clearly understand and communicate the effects of cancer drugs on patient quality of life alongside their effects on overall survival and intermediate end points such as progression-free survival. “Patients with advanced cancer expect treatment to help them live longer or have better lives,” the authors wrote. In that respect, in clinical trials of cancer medicines, overall survival and quality of life are the most important measures. Toxicity profiles and disease progression delays do not reliably predict quality of life, and studies have shown poor correlations between quality of life, overall survival, and progression-free survival. This raises the question of validity of progression-free survival as a surrogate endpoint. “Progression-free survival is meaningless without overall survival or quality of life gains,” Dr. Gyawali said in an interview.
Writing in The Lancet Oncology in March, Dr. Gyawali stated that, because progression free survival “does not directly measure how a patient feels or functions, or how long a patient lives, progression-free survival was not intended to inform clinical practice or establish whether a new therapy provides clinically meaningful benefits for patients. However, over the past 2 decades, it has become the most common primary endpoint in oncology clinical trials. We are deeply worried about how the term survival in this phrase can influence clinical practice and patient choices. We propose replacing the phrase progression-free survival with a less ambiguous term: progression-free interval.”
In JAMA Oncology, Dr. Gyawali aimed to elucidate relationships between QOL, overall survival, and progression-free survival, and to assess, as well, how QOL results are framed, especially in industry-sponsored research. When drug trials they analyzed showed no change in QOL but reported that QOL did not worsen or QOL was maintained rather than stating that QOL did not improve, or if there was downplaying of worse QOL outcomes, the study had favorable interpretation, Dr. Gyawali and associates wrote. The expectation of patients receiving cancer drugs would be improved QOL rather than “not worse” QOL, Dr. Gyawali said.
Regarding the finding that QOL outcomes were described as favorable in 47% of trials with unimproved QOL outcomes, Dr. Gyawali said, “the bias in reporting should be corrected by the reviewers and editors of journals. Also, quality of life reporting should be made mandatory. Without unbiased quality of life information, informed decision making on whether or not to use a certain drug is impossible. Patients and physicians need to know that information. Regulators can demand that this should be mandatory in all trials in noncurative settings.”
He remarked further on the worsening QOL in some targeted drug trials, “People tout chemo-free regimens as automatically having better quality of life, but that doesn’t seem to be the case. Targeted drugs can have a severe impact on quality of life, probably due to prolonged duration of side effects. Quality of life should be measured and reported for all drugs.”
Dr. Gyawali and associates noted the limitation in that several studies with negative QOL results are not published at all or are published after a considerable delay, so the present observations may understate the issues that have been raised.
Dr. Gyawali declared that he received no funding and disclosed no conflicts of interest for this study.
FROM JAMA ONCOLOGY
To vaccinate 6-month- to 5-year-olds against SARS-CoV-2 or not to vaccinate
A family’s decision to vaccinate their child is best made jointly with a trusted medical provider who knows the child and family. The American Academy of Pediatrics created a toolkit with resources for answering questions about the recently authorized SARS-CoV-2 mRNA vaccines (Pfizer and Moderna) for 6-month- to 5-year-olds with science-backed vaccine facts, including links to other useful AAP information websites, talking points, graphics, and videos.1

SARS-CoV-2 seasonality
SARS-CoV-2 is now endemic, not a once-a-year seasonal virus. Seasons (aka surges) will occur whenever a new variant arises (twice yearly since 2020, Omicron BA.4/BA.5 currently), or when enough vaccine holdouts, newborns, and/or those with waning of prior immunity (vaccine or infection induced) accrue.
Emergency use authorization submission data for mRNA vaccine responses in young children2,3
Moderna in 6-month- through 5-year-olds. Two 25-mcg doses given 4-8 weeks apart produced 37.8% (95% confidence interval, 20.9%-51.1%) protection against symptomatic Omicron SARS-CoV-2 infections through 3 months of follow-up. Immunobridging analysis of antibody responses compared to 18- to 25-year-olds (100-mcg doses) showed the children’s responses were noninferior. Thus, the committee inferred that vaccine effectiveness in children should be similar to that in 18- to 25-year-olds. Fever, irritability, or local reaction/pain occurred in two-thirds after the second dose. Grade 3 reactions were noted in less than 5%.
Pfizer in 6-month- through 4-year-olds. Three 3-mcg doses, two doses 3-8 weeks apart and the third dose at least 8 weeks later (median 16 weeks), produced 80.3% (95% CI, 13.9%-96.7%) protection against symptomatic COVID-19 during the 6 weeks after the third dose. Local and systemic reactions occurred in 63.8%; less than 5% had grade 3 reactions (fever in about 3%, irritability in 1.3%, fatigue in 0.8%) mostly after second dose.
Neither duration of follow-up is very long. The Moderna data tell me that a third primary dose would have been better but restarting the trial to evaluate third doses would have delayed Moderna’s EUA another 4-6 months. The three-dose Pfizer data look better but may not have been as good with another 6 weeks of follow-up.
Additional post-EUA data will be collected. Boosters will be needed when immunity from both vaccines wanes (one estimate is about 6 months after the primary series). The Advisory Committee on Immunization Practices noted in their deliberations that vaccine-induced antibody responses are higher and cross-neutralize variants (even Omicron) better than infection-induced immunity.4
Are there downsides to the vaccines? Naysayers question vaccinating children less than 5 years old with reasons containing enough “truth” that they catch people’s attention, for example, “young children don’t get very sick with COVID-19,” “most have been infected already,” “RNA for the spike protein stays in the body for months,” or “myocarditis.” Naysayers can quote references in reputable journals but seem to spin selected data out of context or quote unconfirmed data from the Vaccine Adverse Event Reporting System.
Reasons to vaccinate
- While children have milder disease than adults, mid-June 2022 surveillance indicated 50 hospitalizations and 1 pediatric death each day from SARS-CoV-2.5
- Vaccinating young children endows a foundation of vaccine-induced SARS-CoV-2 immunity that is superior to infection-induced immunity.4
- Long-term effects of large numbers of SARS-CoV-2 particles that enter every organ of a developing child have not been determined.
- Viral loads are lowered by prior vaccine; fewer viral replications lessen chances for newer variants to arise.
- Transmission is less in breakthrough infections than infections in the unvaccinated.
- Thirty percent of 5- to 11-year-olds hospitalized for SARS-CoV-2 had no underlying conditions;6 hospitalization rates in newborn to 4-year-olds have been the highest in the Omicron surge.7
- No myocarditis or pericarditis episodes have been detected in 6-month- to 11-year-old trials.
- The AAP and ACIP recommend the mRNA vaccines.
My thoughts are that SARS-CoV-2 vaccine is just another “routine” childhood vaccine that prepares children for healthier futures, pandemic or not, and the vaccines are as safe as other routine vaccines.
And like other pediatric vaccines, it should be no surprise that boosters will be needed, even if no newer variants than Omicron BA.4/BA.5 arise. But we know newer variants will arise and, similar to influenza vaccine, new formulations, perhaps with multiple SARS-CoV-2 strain antigens, will be needed every year or so. Everyone will get SARS-CoV-2 multiple times in their lives no matter how careful they are. So isn’t it good medical practice to establish early the best available foundation for maintaining lifelong SARS-CoV-2 immunity?
To me it is like pertussis. Most pertussis-infected children are sick enough to be hospitalized; very few die. They are miserable with illnesses that take weeks to months to subside. The worst disease usually occurs in unvaccinated young children or those with underlying conditions. Reactogenicity was reduced with acellular vaccine but resulted in less immunogenicity, so we give boosters at intervals that best match waning immunity. Circulating strains can be different than the vaccine strain, so protection against infection is 80%. Finally, even the safest vaccine may very rarely have sequelae. That is why The National Vaccine Injury Compensation Program was created. Yet the benefit-to-harm ratio for children and society favors universal pertussis vaccine use. And we vaccinate even those who have had pertussis because even infection-based immunity is incomplete and protection wanes. If arguments similar to those by SARS-CoV-2 vaccine naysayers were applied to acellular pertussis vaccine, it seems they would argue against pertussis vaccine for young children.
Another major issue has been “safety concerns” about the vaccines’ small amount of mRNA for the spike protein encased in microscopic lipid bubbles injected in the arm or leg. This mRNA is picked up by human cells, and in the cytoplasm (not the nucleus where our DNA resides) produces a limited supply of spike protein that is then picked up by antigen-presenting cells for short-lived distribution (days to 2 weeks at most) to regional lymph nodes where immune-memory processes are jump-started. Contrast that to even asymptomatic SARS-CoV-2 infection where multibillions of virus particles are produced for up to 14 days with access to every bodily organ that contains ACE-2 receptors (they all do). Each virus particle hijacks a human cell producing thousands of mRNA for spike protein (and multiple other SARS-CoV-2 proteins), eventually releasing multibillions of lipid fragments from the ruptured cell. Comparing the amount of these components in the mRNA vaccines to those from infection is like comparing a campfire to the many-thousand-acre wildfire. So, if one is worried about the effects of spike protein and lipid fragments, the limited localized amounts in mRNA vaccines should make one much less concerned than the enormous amounts circulating throughout the body as a result of a SARS-CoV-2 infection.
My take is that children 6-months to 5-years-old deserve SARS-CoV-2–induced vaccine protection and we can and should strongly recommend it as medical providers and child advocates.
*Dr. Harrison is professor, University of Missouri Kansas City School of Medicine, department of medicine, infectious diseases section, Kansas City. Email him at [email protected].
References
1. AAP. 2022 Jun 21. As COVID-19 vaccines become available for children ages 6 months to 4 years, AAP urges families to reach out to pediatricians to ask questions and access vaccine. www.aap.org.
2. CDC. Grading of recommendations, assessment, development, and evaluation (GRADE): Moderna COVID-19 vaccine for children aged 6 months–5 years. www.cdc.gov.
3. CDC. ACIP evidence to recommendations for use of Moderna COVID-19 vaccine in children ages 6 months–5 years and Pfizer-BioNTech COVID-19 vaccine in children ages 6 months–4 years under an emergency use authorization. www.cdc.gov.
4. Tang J et al. Nat Commun. 2022;13:2979.
5. Children and COVID-19: State Data Report. 2022 Jun 30. www.aap.org.
6. Shi DS et al. MMWR Morb Mortal Wkly Rep. 2022;71:574-81.
7. Marks KJ et al. MMWR Morb Mortal Wkly Rep. 2022;71:429-36.
Other good resources for families are https://getvaccineanswers.org/ or www.mayoclinic.org/diseases-conditions/coronavirus/in-depth/coronavirus-in-babies-and-children/art-20484405.
*This story was updated on July 19, 2022.
A family’s decision to vaccinate their child is best made jointly with a trusted medical provider who knows the child and family. The American Academy of Pediatrics created a toolkit with resources for answering questions about the recently authorized SARS-CoV-2 mRNA vaccines (Pfizer and Moderna) for 6-month- to 5-year-olds with science-backed vaccine facts, including links to other useful AAP information websites, talking points, graphics, and videos.1
SARS-CoV-2 seasonality
SARS-CoV-2 is now endemic, not a once-a-year seasonal virus. Seasons (aka surges) will occur whenever a new variant arises (twice yearly since 2020, Omicron BA.4/BA.5 currently), or when enough vaccine holdouts, newborns, and/or those with waning of prior immunity (vaccine or infection induced) accrue.
Emergency use authorization submission data for mRNA vaccine responses in young children2,3
Moderna in 6-month- through 5-year-olds. Two 25-mcg doses given 4-8 weeks apart produced 37.8% (95% confidence interval, 20.9%-51.1%) protection against symptomatic Omicron SARS-CoV-2 infections through 3 months of follow-up. Immunobridging analysis of antibody responses compared to 18- to 25-year-olds (100-mcg doses) showed the children’s responses were noninferior. Thus, the committee inferred that vaccine effectiveness in children should be similar to that in 18- to 25-year-olds. Fever, irritability, or local reaction/pain occurred in two-thirds after the second dose. Grade 3 reactions were noted in less than 5%.
Pfizer in 6-month- through 4-year-olds. Three 3-mcg doses, two doses 3-8 weeks apart and the third dose at least 8 weeks later (median 16 weeks), produced 80.3% (95% CI, 13.9%-96.7%) protection against symptomatic COVID-19 during the 6 weeks after the third dose. Local and systemic reactions occurred in 63.8%; less than 5% had grade 3 reactions (fever in about 3%, irritability in 1.3%, fatigue in 0.8%) mostly after second dose.
Neither duration of follow-up is very long. The Moderna data tell me that a third primary dose would have been better but restarting the trial to evaluate third doses would have delayed Moderna’s EUA another 4-6 months. The three-dose Pfizer data look better but may not have been as good with another 6 weeks of follow-up.
Additional post-EUA data will be collected. Boosters will be needed when immunity from both vaccines wanes (one estimate is about 6 months after the primary series). The Advisory Committee on Immunization Practices noted in their deliberations that vaccine-induced antibody responses are higher and cross-neutralize variants (even Omicron) better than infection-induced immunity.4
Are there downsides to the vaccines? Naysayers question vaccinating children less than 5 years old with reasons containing enough “truth” that they catch people’s attention, for example, “young children don’t get very sick with COVID-19,” “most have been infected already,” “RNA for the spike protein stays in the body for months,” or “myocarditis.” Naysayers can quote references in reputable journals but seem to spin selected data out of context or quote unconfirmed data from the Vaccine Adverse Event Reporting System.
Reasons to vaccinate
- While children have milder disease than adults, mid-June 2022 surveillance indicated 50 hospitalizations and 1 pediatric death each day from SARS-CoV-2.5
- Vaccinating young children endows a foundation of vaccine-induced SARS-CoV-2 immunity that is superior to infection-induced immunity.4
- Long-term effects of large numbers of SARS-CoV-2 particles that enter every organ of a developing child have not been determined.
- Viral loads are lowered by prior vaccine; fewer viral replications lessen chances for newer variants to arise.
- Transmission is less in breakthrough infections than infections in the unvaccinated.
- Thirty percent of 5- to 11-year-olds hospitalized for SARS-CoV-2 had no underlying conditions;6 hospitalization rates in newborn to 4-year-olds have been the highest in the Omicron surge.7
- No myocarditis or pericarditis episodes have been detected in 6-month- to 11-year-old trials.
- The AAP and ACIP recommend the mRNA vaccines.
My thoughts are that SARS-CoV-2 vaccine is just another “routine” childhood vaccine that prepares children for healthier futures, pandemic or not, and the vaccines are as safe as other routine vaccines.
And like other pediatric vaccines, it should be no surprise that boosters will be needed, even if no newer variants than Omicron BA.4/BA.5 arise. But we know newer variants will arise and, similar to influenza vaccine, new formulations, perhaps with multiple SARS-CoV-2 strain antigens, will be needed every year or so. Everyone will get SARS-CoV-2 multiple times in their lives no matter how careful they are. So isn’t it good medical practice to establish early the best available foundation for maintaining lifelong SARS-CoV-2 immunity?
To me it is like pertussis. Most pertussis-infected children are sick enough to be hospitalized; very few die. They are miserable with illnesses that take weeks to months to subside. The worst disease usually occurs in unvaccinated young children or those with underlying conditions. Reactogenicity was reduced with acellular vaccine but resulted in less immunogenicity, so we give boosters at intervals that best match waning immunity. Circulating strains can be different than the vaccine strain, so protection against infection is 80%. Finally, even the safest vaccine may very rarely have sequelae. That is why The National Vaccine Injury Compensation Program was created. Yet the benefit-to-harm ratio for children and society favors universal pertussis vaccine use. And we vaccinate even those who have had pertussis because even infection-based immunity is incomplete and protection wanes. If arguments similar to those by SARS-CoV-2 vaccine naysayers were applied to acellular pertussis vaccine, it seems they would argue against pertussis vaccine for young children.
Another major issue has been “safety concerns” about the vaccines’ small amount of mRNA for the spike protein encased in microscopic lipid bubbles injected in the arm or leg. This mRNA is picked up by human cells, and in the cytoplasm (not the nucleus where our DNA resides) produces a limited supply of spike protein that is then picked up by antigen-presenting cells for short-lived distribution (days to 2 weeks at most) to regional lymph nodes where immune-memory processes are jump-started. Contrast that to even asymptomatic SARS-CoV-2 infection where multibillions of virus particles are produced for up to 14 days with access to every bodily organ that contains ACE-2 receptors (they all do). Each virus particle hijacks a human cell producing thousands of mRNA for spike protein (and multiple other SARS-CoV-2 proteins), eventually releasing multibillions of lipid fragments from the ruptured cell. Comparing the amount of these components in the mRNA vaccines to those from infection is like comparing a campfire to the many-thousand-acre wildfire. So, if one is worried about the effects of spike protein and lipid fragments, the limited localized amounts in mRNA vaccines should make one much less concerned than the enormous amounts circulating throughout the body as a result of a SARS-CoV-2 infection.
My take is that children 6-months to 5-years-old deserve SARS-CoV-2–induced vaccine protection and we can and should strongly recommend it as medical providers and child advocates.
*Dr. Harrison is professor, University of Missouri Kansas City School of Medicine, department of medicine, infectious diseases section, Kansas City. Email him at [email protected].
References
1. AAP. 2022 Jun 21. As COVID-19 vaccines become available for children ages 6 months to 4 years, AAP urges families to reach out to pediatricians to ask questions and access vaccine. www.aap.org.
2. CDC. Grading of recommendations, assessment, development, and evaluation (GRADE): Moderna COVID-19 vaccine for children aged 6 months–5 years. www.cdc.gov.
3. CDC. ACIP evidence to recommendations for use of Moderna COVID-19 vaccine in children ages 6 months–5 years and Pfizer-BioNTech COVID-19 vaccine in children ages 6 months–4 years under an emergency use authorization. www.cdc.gov.
4. Tang J et al. Nat Commun. 2022;13:2979.
5. Children and COVID-19: State Data Report. 2022 Jun 30. www.aap.org.
6. Shi DS et al. MMWR Morb Mortal Wkly Rep. 2022;71:574-81.
7. Marks KJ et al. MMWR Morb Mortal Wkly Rep. 2022;71:429-36.
Other good resources for families are https://getvaccineanswers.org/ or www.mayoclinic.org/diseases-conditions/coronavirus/in-depth/coronavirus-in-babies-and-children/art-20484405.
*This story was updated on July 19, 2022.
A family’s decision to vaccinate their child is best made jointly with a trusted medical provider who knows the child and family. The American Academy of Pediatrics created a toolkit with resources for answering questions about the recently authorized SARS-CoV-2 mRNA vaccines (Pfizer and Moderna) for 6-month- to 5-year-olds with science-backed vaccine facts, including links to other useful AAP information websites, talking points, graphics, and videos.1
SARS-CoV-2 seasonality
SARS-CoV-2 is now endemic, not a once-a-year seasonal virus. Seasons (aka surges) will occur whenever a new variant arises (twice yearly since 2020, Omicron BA.4/BA.5 currently), or when enough vaccine holdouts, newborns, and/or those with waning of prior immunity (vaccine or infection induced) accrue.
Emergency use authorization submission data for mRNA vaccine responses in young children2,3
Moderna in 6-month- through 5-year-olds. Two 25-mcg doses given 4-8 weeks apart produced 37.8% (95% confidence interval, 20.9%-51.1%) protection against symptomatic Omicron SARS-CoV-2 infections through 3 months of follow-up. Immunobridging analysis of antibody responses compared to 18- to 25-year-olds (100-mcg doses) showed the children’s responses were noninferior. Thus, the committee inferred that vaccine effectiveness in children should be similar to that in 18- to 25-year-olds. Fever, irritability, or local reaction/pain occurred in two-thirds after the second dose. Grade 3 reactions were noted in less than 5%.
Pfizer in 6-month- through 4-year-olds. Three 3-mcg doses, two doses 3-8 weeks apart and the third dose at least 8 weeks later (median 16 weeks), produced 80.3% (95% CI, 13.9%-96.7%) protection against symptomatic COVID-19 during the 6 weeks after the third dose. Local and systemic reactions occurred in 63.8%; less than 5% had grade 3 reactions (fever in about 3%, irritability in 1.3%, fatigue in 0.8%) mostly after second dose.
Neither duration of follow-up is very long. The Moderna data tell me that a third primary dose would have been better but restarting the trial to evaluate third doses would have delayed Moderna’s EUA another 4-6 months. The three-dose Pfizer data look better but may not have been as good with another 6 weeks of follow-up.
Additional post-EUA data will be collected. Boosters will be needed when immunity from both vaccines wanes (one estimate is about 6 months after the primary series). The Advisory Committee on Immunization Practices noted in their deliberations that vaccine-induced antibody responses are higher and cross-neutralize variants (even Omicron) better than infection-induced immunity.4
Are there downsides to the vaccines? Naysayers question vaccinating children less than 5 years old with reasons containing enough “truth” that they catch people’s attention, for example, “young children don’t get very sick with COVID-19,” “most have been infected already,” “RNA for the spike protein stays in the body for months,” or “myocarditis.” Naysayers can quote references in reputable journals but seem to spin selected data out of context or quote unconfirmed data from the Vaccine Adverse Event Reporting System.
Reasons to vaccinate
- While children have milder disease than adults, mid-June 2022 surveillance indicated 50 hospitalizations and 1 pediatric death each day from SARS-CoV-2.5
- Vaccinating young children endows a foundation of vaccine-induced SARS-CoV-2 immunity that is superior to infection-induced immunity.4
- Long-term effects of large numbers of SARS-CoV-2 particles that enter every organ of a developing child have not been determined.
- Viral loads are lowered by prior vaccine; fewer viral replications lessen chances for newer variants to arise.
- Transmission is less in breakthrough infections than infections in the unvaccinated.
- Thirty percent of 5- to 11-year-olds hospitalized for SARS-CoV-2 had no underlying conditions;6 hospitalization rates in newborn to 4-year-olds have been the highest in the Omicron surge.7
- No myocarditis or pericarditis episodes have been detected in 6-month- to 11-year-old trials.
- The AAP and ACIP recommend the mRNA vaccines.
My thoughts are that SARS-CoV-2 vaccine is just another “routine” childhood vaccine that prepares children for healthier futures, pandemic or not, and the vaccines are as safe as other routine vaccines.
And like other pediatric vaccines, it should be no surprise that boosters will be needed, even if no newer variants than Omicron BA.4/BA.5 arise. But we know newer variants will arise and, similar to influenza vaccine, new formulations, perhaps with multiple SARS-CoV-2 strain antigens, will be needed every year or so. Everyone will get SARS-CoV-2 multiple times in their lives no matter how careful they are. So isn’t it good medical practice to establish early the best available foundation for maintaining lifelong SARS-CoV-2 immunity?
To me it is like pertussis. Most pertussis-infected children are sick enough to be hospitalized; very few die. They are miserable with illnesses that take weeks to months to subside. The worst disease usually occurs in unvaccinated young children or those with underlying conditions. Reactogenicity was reduced with acellular vaccine but resulted in less immunogenicity, so we give boosters at intervals that best match waning immunity. Circulating strains can be different than the vaccine strain, so protection against infection is 80%. Finally, even the safest vaccine may very rarely have sequelae. That is why The National Vaccine Injury Compensation Program was created. Yet the benefit-to-harm ratio for children and society favors universal pertussis vaccine use. And we vaccinate even those who have had pertussis because even infection-based immunity is incomplete and protection wanes. If arguments similar to those by SARS-CoV-2 vaccine naysayers were applied to acellular pertussis vaccine, it seems they would argue against pertussis vaccine for young children.
Another major issue has been “safety concerns” about the vaccines’ small amount of mRNA for the spike protein encased in microscopic lipid bubbles injected in the arm or leg. This mRNA is picked up by human cells, and in the cytoplasm (not the nucleus where our DNA resides) produces a limited supply of spike protein that is then picked up by antigen-presenting cells for short-lived distribution (days to 2 weeks at most) to regional lymph nodes where immune-memory processes are jump-started. Contrast that to even asymptomatic SARS-CoV-2 infection where multibillions of virus particles are produced for up to 14 days with access to every bodily organ that contains ACE-2 receptors (they all do). Each virus particle hijacks a human cell producing thousands of mRNA for spike protein (and multiple other SARS-CoV-2 proteins), eventually releasing multibillions of lipid fragments from the ruptured cell. Comparing the amount of these components in the mRNA vaccines to those from infection is like comparing a campfire to the many-thousand-acre wildfire. So, if one is worried about the effects of spike protein and lipid fragments, the limited localized amounts in mRNA vaccines should make one much less concerned than the enormous amounts circulating throughout the body as a result of a SARS-CoV-2 infection.
My take is that children 6-months to 5-years-old deserve SARS-CoV-2–induced vaccine protection and we can and should strongly recommend it as medical providers and child advocates.
*Dr. Harrison is professor, University of Missouri Kansas City School of Medicine, department of medicine, infectious diseases section, Kansas City. Email him at [email protected].
References
1. AAP. 2022 Jun 21. As COVID-19 vaccines become available for children ages 6 months to 4 years, AAP urges families to reach out to pediatricians to ask questions and access vaccine. www.aap.org.
2. CDC. Grading of recommendations, assessment, development, and evaluation (GRADE): Moderna COVID-19 vaccine for children aged 6 months–5 years. www.cdc.gov.
3. CDC. ACIP evidence to recommendations for use of Moderna COVID-19 vaccine in children ages 6 months–5 years and Pfizer-BioNTech COVID-19 vaccine in children ages 6 months–4 years under an emergency use authorization. www.cdc.gov.
4. Tang J et al. Nat Commun. 2022;13:2979.
5. Children and COVID-19: State Data Report. 2022 Jun 30. www.aap.org.
6. Shi DS et al. MMWR Morb Mortal Wkly Rep. 2022;71:574-81.
7. Marks KJ et al. MMWR Morb Mortal Wkly Rep. 2022;71:429-36.
Other good resources for families are https://getvaccineanswers.org/ or www.mayoclinic.org/diseases-conditions/coronavirus/in-depth/coronavirus-in-babies-and-children/art-20484405.
*This story was updated on July 19, 2022.
Bulevirtide reduces hepatitis D viral load in difficult-to-treat patients
Bulevirtide (Hepcludex) monotherapy significantly reduces the load of hepatitis delta virus (HDV) and is safe in difficult-to-treat patients with compensated cirrhosis and clinically significant portal hypertension, according to the results of an ongoing 1-year study.
In presenting a poster with these findings at the annual International Liver Congress, sponsored by the European Association for the Study of the Liver, lead author Elisabetta Degasperi, MD, from the Grand Hospital Maggiore Policlinico in Milan, said that they were important “because they confirm the safety of this drug in real life.”
Dr. Degasperi and colleagues showed that bulevirtide leads to a significant viral response in 78% of patients by week 48, which was measured using the outcome of greater than 2 log decline in HDV RNA from baseline.
Dr. Degasperi added that the research still needed to assess the longer-term benefits, but
Addressing an immense, unmet therapeutic need
HDV requires the presence of hepatitis B virus to replicate. Bulevirtide blocks the entry of HDV and hepatitis B virus into hepatocytes.
In July 2020, it was conditionally approved in the European Economic Area for use to treat chronic HDV infection in adults with compensated liver disease upon confirmation of HDV RNA in the blood. It currently remains an investigational agent in the United States, as well as outside of the EEA.
The ongoing trial led by Dr. Degasperi is specifically conducted in patients with compensated cirrhosis who also have clinically significant portal hypertension, where safety and efficacy are unknown.
Dr. Degasperi said in an interview that, although HDV was rare, there is nonetheless an “immense” need for effective therapies against it, especially in young patients with advanced liver disease.
“We have a lot of patients with hepatitis D who have not responded to other antiviral treatment. Right now, the only other available treatment is pegylated interferon,” she said. “Unfortunately, rates of sustained viral response to pegylated interferon are extremely low at around 30% of patients.”
Chronic HDV is the most severe form of viral hepatitis and can have mortality rates as high as 50% within 5 years in patients with cirrhosis.
The management of hepatitis D is also complicated by the fact that patients with advanced cirrhosis and clinically significant portal hypertension cannot be treated with pegylated interferon owing to lack of efficacy and safety reasons, including a high risk for decompensation and liver-related complications. Pegylated interferon is contraindicated in these patients.
Bulevirtide at 48 weeks: A closer look at the findings
Eighteen patients with HDV, compensated cirrhosis, and clinically significant portal hypertension were consecutively enrolled in this single-center, longitudinal study.
All received bulevirtide monotherapy at 2 mg/day and underwent monitoring every 2 months. They were also treated with nucleotide analogs for their hepatitis B virus, which was suppressed when they began bulevirtide.
Clinical and virologic characteristics were collected at baseline, at weeks 4 and 8, and then every 8 weeks thereafter.
Bulevirtide led to a significant viral response such that by week 48, HDV RNA declined by 3.1 log IU/mL (range, 0.2-4.6 log IU/mL), was undetectable in six patients (33%), and was less than 100 IU/L in 50% of patients. Two patients were nonresponders. In addition, 78% of patients achieved at least an HDV RNA 2 log decline from baseline.
There was also a normalization of biochemical response in the majority of patients.
Alanine aminotransferase normalization was seen in 89% of patients and declined by a median of 34 U/L (range, 15-76 U/L) over 48 weeks. Aspartate aminotransferase declined to 39 U/L (range, 21-92 U/L). A combined response was seen in 72% of patients, reported Dr. Degasperi.
“Previously, we only had results from a phase 2 study, so we had no idea of the results over such a long treatment period,” said Dr. Degasperi. “It is also the first time we have been able to treat these patients with such advanced disease that is so difficult to manage.”
“Real-world results are typically inferior to those from clinical trials, but the viral decline is comparable to phase 2 trials, and the first report of the phase 3 trial,” said Dr. Degasperi.
Gamma-glutamyltransferase, alpha-fetoprotein, immunoglobulin G, and gamma-globulin levels also improved, whereas hepatitis B surface antigen, hepatitis B virus RNA, hepatitis B core-related antigen, platelet, and bilirubin values did not significantly change.
“All patients were Child-Pugh score A, so well-compensated [disease]. However, they increased a little bit in liver function by week 48,” Dr. Degasperi said. “This was important for this very advanced disease population.”
She added that the safety profile was very favorable, with no adverse events, including no injection-site reactions.
There was an asymptomatic increase in serum bile acids. “No patients complained about itching or pruritus,” Dr. Degasperi said.
What’s ahead for bulevirtide?
In a comment, Marc Bourlière, MD, from Saint Joseph Hospital in Marseilles, France, welcomed the decrease in viral load.
“This is known to be beneficial in terms of reducing morbidity and mortality in hepatitis D,” he said. “Remember that this disease is very difficult to treat, and until now, we have had no drug available. Pegylated interferon achieves cure in only 30% of patients, and half of these relapse, so actually only 15% have a meaningful response from pegylated interferon.”
“The main issue is its use as a daily subcutaneous injection. In clinical practice, it is a little bit complicated to set up, but once done, it is quite well accepted,” he said.
“I’m impressed with these results to date because there are no other compounds that have, as yet, achieved such results. This is impressive,” he added. “But whether it translates into a long-term response we don’t yet know.”
Dr. Bourlière also noted the meaningful 2-point log decline, noting that “HDV RNA negativity where treatment can be stopped would be really meaningful, but this endpoint is hard to obtain.”
Dr. Bourlière is awaiting results of the current ongoing phase 2/3 study, which would help determine a possible final treatment duration. He is also curious to settle the ongoing debate about whether bulevirtide should be used alone or in combination.
“We need to combine bulevirtide with pegylated interferon in less-advanced patients, because we know it is more potent and active against the HDV RNA,” he said.
Dr. Degasperi has previously declared she was on the advisory board for AbbVie and has spoken and taught for Gilead, MSD, and AbbVie. Dr. Bourlière declared interests with all companies involved in the R&D of liver therapies.
A version of this article first appeared on Medscape.com.
Bulevirtide (Hepcludex) monotherapy significantly reduces the load of hepatitis delta virus (HDV) and is safe in difficult-to-treat patients with compensated cirrhosis and clinically significant portal hypertension, according to the results of an ongoing 1-year study.
In presenting a poster with these findings at the annual International Liver Congress, sponsored by the European Association for the Study of the Liver, lead author Elisabetta Degasperi, MD, from the Grand Hospital Maggiore Policlinico in Milan, said that they were important “because they confirm the safety of this drug in real life.”
Dr. Degasperi and colleagues showed that bulevirtide leads to a significant viral response in 78% of patients by week 48, which was measured using the outcome of greater than 2 log decline in HDV RNA from baseline.
Dr. Degasperi added that the research still needed to assess the longer-term benefits, but
Addressing an immense, unmet therapeutic need
HDV requires the presence of hepatitis B virus to replicate. Bulevirtide blocks the entry of HDV and hepatitis B virus into hepatocytes.
In July 2020, it was conditionally approved in the European Economic Area for use to treat chronic HDV infection in adults with compensated liver disease upon confirmation of HDV RNA in the blood. It currently remains an investigational agent in the United States, as well as outside of the EEA.
The ongoing trial led by Dr. Degasperi is specifically conducted in patients with compensated cirrhosis who also have clinically significant portal hypertension, where safety and efficacy are unknown.
Dr. Degasperi said in an interview that, although HDV was rare, there is nonetheless an “immense” need for effective therapies against it, especially in young patients with advanced liver disease.
“We have a lot of patients with hepatitis D who have not responded to other antiviral treatment. Right now, the only other available treatment is pegylated interferon,” she said. “Unfortunately, rates of sustained viral response to pegylated interferon are extremely low at around 30% of patients.”
Chronic HDV is the most severe form of viral hepatitis and can have mortality rates as high as 50% within 5 years in patients with cirrhosis.
The management of hepatitis D is also complicated by the fact that patients with advanced cirrhosis and clinically significant portal hypertension cannot be treated with pegylated interferon owing to lack of efficacy and safety reasons, including a high risk for decompensation and liver-related complications. Pegylated interferon is contraindicated in these patients.
Bulevirtide at 48 weeks: A closer look at the findings
Eighteen patients with HDV, compensated cirrhosis, and clinically significant portal hypertension were consecutively enrolled in this single-center, longitudinal study.
All received bulevirtide monotherapy at 2 mg/day and underwent monitoring every 2 months. They were also treated with nucleotide analogs for their hepatitis B virus, which was suppressed when they began bulevirtide.
Clinical and virologic characteristics were collected at baseline, at weeks 4 and 8, and then every 8 weeks thereafter.
Bulevirtide led to a significant viral response such that by week 48, HDV RNA declined by 3.1 log IU/mL (range, 0.2-4.6 log IU/mL), was undetectable in six patients (33%), and was less than 100 IU/L in 50% of patients. Two patients were nonresponders. In addition, 78% of patients achieved at least an HDV RNA 2 log decline from baseline.
There was also a normalization of biochemical response in the majority of patients.
Alanine aminotransferase normalization was seen in 89% of patients and declined by a median of 34 U/L (range, 15-76 U/L) over 48 weeks. Aspartate aminotransferase declined to 39 U/L (range, 21-92 U/L). A combined response was seen in 72% of patients, reported Dr. Degasperi.
“Previously, we only had results from a phase 2 study, so we had no idea of the results over such a long treatment period,” said Dr. Degasperi. “It is also the first time we have been able to treat these patients with such advanced disease that is so difficult to manage.”
“Real-world results are typically inferior to those from clinical trials, but the viral decline is comparable to phase 2 trials, and the first report of the phase 3 trial,” said Dr. Degasperi.
Gamma-glutamyltransferase, alpha-fetoprotein, immunoglobulin G, and gamma-globulin levels also improved, whereas hepatitis B surface antigen, hepatitis B virus RNA, hepatitis B core-related antigen, platelet, and bilirubin values did not significantly change.
“All patients were Child-Pugh score A, so well-compensated [disease]. However, they increased a little bit in liver function by week 48,” Dr. Degasperi said. “This was important for this very advanced disease population.”
She added that the safety profile was very favorable, with no adverse events, including no injection-site reactions.
There was an asymptomatic increase in serum bile acids. “No patients complained about itching or pruritus,” Dr. Degasperi said.
What’s ahead for bulevirtide?
In a comment, Marc Bourlière, MD, from Saint Joseph Hospital in Marseilles, France, welcomed the decrease in viral load.
“This is known to be beneficial in terms of reducing morbidity and mortality in hepatitis D,” he said. “Remember that this disease is very difficult to treat, and until now, we have had no drug available. Pegylated interferon achieves cure in only 30% of patients, and half of these relapse, so actually only 15% have a meaningful response from pegylated interferon.”
“The main issue is its use as a daily subcutaneous injection. In clinical practice, it is a little bit complicated to set up, but once done, it is quite well accepted,” he said.
“I’m impressed with these results to date because there are no other compounds that have, as yet, achieved such results. This is impressive,” he added. “But whether it translates into a long-term response we don’t yet know.”
Dr. Bourlière also noted the meaningful 2-point log decline, noting that “HDV RNA negativity where treatment can be stopped would be really meaningful, but this endpoint is hard to obtain.”
Dr. Bourlière is awaiting results of the current ongoing phase 2/3 study, which would help determine a possible final treatment duration. He is also curious to settle the ongoing debate about whether bulevirtide should be used alone or in combination.
“We need to combine bulevirtide with pegylated interferon in less-advanced patients, because we know it is more potent and active against the HDV RNA,” he said.
Dr. Degasperi has previously declared she was on the advisory board for AbbVie and has spoken and taught for Gilead, MSD, and AbbVie. Dr. Bourlière declared interests with all companies involved in the R&D of liver therapies.
A version of this article first appeared on Medscape.com.
Bulevirtide (Hepcludex) monotherapy significantly reduces the load of hepatitis delta virus (HDV) and is safe in difficult-to-treat patients with compensated cirrhosis and clinically significant portal hypertension, according to the results of an ongoing 1-year study.
In presenting a poster with these findings at the annual International Liver Congress, sponsored by the European Association for the Study of the Liver, lead author Elisabetta Degasperi, MD, from the Grand Hospital Maggiore Policlinico in Milan, said that they were important “because they confirm the safety of this drug in real life.”
Dr. Degasperi and colleagues showed that bulevirtide leads to a significant viral response in 78% of patients by week 48, which was measured using the outcome of greater than 2 log decline in HDV RNA from baseline.
Dr. Degasperi added that the research still needed to assess the longer-term benefits, but
Addressing an immense, unmet therapeutic need
HDV requires the presence of hepatitis B virus to replicate. Bulevirtide blocks the entry of HDV and hepatitis B virus into hepatocytes.
In July 2020, it was conditionally approved in the European Economic Area for use to treat chronic HDV infection in adults with compensated liver disease upon confirmation of HDV RNA in the blood. It currently remains an investigational agent in the United States, as well as outside of the EEA.
The ongoing trial led by Dr. Degasperi is specifically conducted in patients with compensated cirrhosis who also have clinically significant portal hypertension, where safety and efficacy are unknown.
Dr. Degasperi said in an interview that, although HDV was rare, there is nonetheless an “immense” need for effective therapies against it, especially in young patients with advanced liver disease.
“We have a lot of patients with hepatitis D who have not responded to other antiviral treatment. Right now, the only other available treatment is pegylated interferon,” she said. “Unfortunately, rates of sustained viral response to pegylated interferon are extremely low at around 30% of patients.”
Chronic HDV is the most severe form of viral hepatitis and can have mortality rates as high as 50% within 5 years in patients with cirrhosis.
The management of hepatitis D is also complicated by the fact that patients with advanced cirrhosis and clinically significant portal hypertension cannot be treated with pegylated interferon owing to lack of efficacy and safety reasons, including a high risk for decompensation and liver-related complications. Pegylated interferon is contraindicated in these patients.
Bulevirtide at 48 weeks: A closer look at the findings
Eighteen patients with HDV, compensated cirrhosis, and clinically significant portal hypertension were consecutively enrolled in this single-center, longitudinal study.
All received bulevirtide monotherapy at 2 mg/day and underwent monitoring every 2 months. They were also treated with nucleotide analogs for their hepatitis B virus, which was suppressed when they began bulevirtide.
Clinical and virologic characteristics were collected at baseline, at weeks 4 and 8, and then every 8 weeks thereafter.
Bulevirtide led to a significant viral response such that by week 48, HDV RNA declined by 3.1 log IU/mL (range, 0.2-4.6 log IU/mL), was undetectable in six patients (33%), and was less than 100 IU/L in 50% of patients. Two patients were nonresponders. In addition, 78% of patients achieved at least an HDV RNA 2 log decline from baseline.
There was also a normalization of biochemical response in the majority of patients.
Alanine aminotransferase normalization was seen in 89% of patients and declined by a median of 34 U/L (range, 15-76 U/L) over 48 weeks. Aspartate aminotransferase declined to 39 U/L (range, 21-92 U/L). A combined response was seen in 72% of patients, reported Dr. Degasperi.
“Previously, we only had results from a phase 2 study, so we had no idea of the results over such a long treatment period,” said Dr. Degasperi. “It is also the first time we have been able to treat these patients with such advanced disease that is so difficult to manage.”
“Real-world results are typically inferior to those from clinical trials, but the viral decline is comparable to phase 2 trials, and the first report of the phase 3 trial,” said Dr. Degasperi.
Gamma-glutamyltransferase, alpha-fetoprotein, immunoglobulin G, and gamma-globulin levels also improved, whereas hepatitis B surface antigen, hepatitis B virus RNA, hepatitis B core-related antigen, platelet, and bilirubin values did not significantly change.
“All patients were Child-Pugh score A, so well-compensated [disease]. However, they increased a little bit in liver function by week 48,” Dr. Degasperi said. “This was important for this very advanced disease population.”
She added that the safety profile was very favorable, with no adverse events, including no injection-site reactions.
There was an asymptomatic increase in serum bile acids. “No patients complained about itching or pruritus,” Dr. Degasperi said.
What’s ahead for bulevirtide?
In a comment, Marc Bourlière, MD, from Saint Joseph Hospital in Marseilles, France, welcomed the decrease in viral load.
“This is known to be beneficial in terms of reducing morbidity and mortality in hepatitis D,” he said. “Remember that this disease is very difficult to treat, and until now, we have had no drug available. Pegylated interferon achieves cure in only 30% of patients, and half of these relapse, so actually only 15% have a meaningful response from pegylated interferon.”
“The main issue is its use as a daily subcutaneous injection. In clinical practice, it is a little bit complicated to set up, but once done, it is quite well accepted,” he said.
“I’m impressed with these results to date because there are no other compounds that have, as yet, achieved such results. This is impressive,” he added. “But whether it translates into a long-term response we don’t yet know.”
Dr. Bourlière also noted the meaningful 2-point log decline, noting that “HDV RNA negativity where treatment can be stopped would be really meaningful, but this endpoint is hard to obtain.”
Dr. Bourlière is awaiting results of the current ongoing phase 2/3 study, which would help determine a possible final treatment duration. He is also curious to settle the ongoing debate about whether bulevirtide should be used alone or in combination.
“We need to combine bulevirtide with pegylated interferon in less-advanced patients, because we know it is more potent and active against the HDV RNA,” he said.
Dr. Degasperi has previously declared she was on the advisory board for AbbVie and has spoken and taught for Gilead, MSD, and AbbVie. Dr. Bourlière declared interests with all companies involved in the R&D of liver therapies.
A version of this article first appeared on Medscape.com.
FROM ILC 2022
When too much treatment creates more harm than good
Ann Marco, 73, who was diagnosed with ovarian cancer in late 2018, credits her oncology team for saving her life. They treated her with chemotherapy, debulking surgery, and more chemotherapy. But it is her second and current care team that helped restore Ms. Marco’s quality of life, directing her toward such resources as palliative care, physical therapy and counseling for her and her husband.
“I can’t say enough about my palliative care doctor. She helped me manage pain, and the fatigue associated with chemotherapy. When she noticed that my leg was swollen she suspected a blood clot and sent me for an ultrasound,” Ms. Marco said.
The ultrasound revealed that she did indeed have a blood clot, for which she received, and continues to receive, medication. “Because with ovarian cancer, you always have blood clots. So little things like that, though they’re not that little, have really helped me in my journey with this cancer,” Ms. Marco said.
That journey has had its ups and downs. One chemotherapy regimen was so intolerable she decided to discontinue it, with full support of her oncologist. I told her, I just want to live my life, whether that’s only 6 more months or 3 years, but I don’t want to live it like this. And she said, ‘Ann, we’re going to do what you want to do.’”
Nine months later, when her cancer started growing again, Ms. Marco returned to chemotherapy. But this regimen has been much more tolerable, and it also appears to be doing its job. A recent CT scan showed that the tumors are shrinking.
“They’ll never go away. I have metastatic cancer. But they’re smaller, and I was really thrilled about that. It’s the best news I’ve had in more than 3 years,” Ms. Marco said.
End-of-life aggressive care still common
, shows a study published in JCO Oncology Practice.
“We have good evidence that the types of aggressive end-of-life care we looked at in this paper are generally related to a lower quality of life for patients, poorer bereavement outcomes for their families, and even shorter duration survivals,” said lead author Megan A. Mullins, PhD, MPH, a postdoctoral research fellow at the University of Michigan in Ann Arbor. “This suggests there’s a disconnect between what people think aggressive care might do and what it’s doing.”
In their evaluation of variation in end-of-life care, Dr. Mullins and her colleagues analyzed SEER-Medicare data on 6,288 women with ovarian cancer who died between 2016 and 2020. They found that 51% of those women received some form of aggressive cancer care. The most common forms were not being admitted to hospice (28.9%), receiving an invasive procedure (20.7%) and being admitted to an intensive care unit (18.6%).
Dr. Mullins noted that since palliative care was officially recognized as a specialty in 2006, there has been increasing guidance for earlier integration of palliative care and reducing the aggressiveness of end-of-life care; both ASCO and the National Quality Form have standards advising against aggressive end-of-life care.
“But there are a lot of complicated factors that I think make it hard to move the needle in this area,” she said. “For one thing, particularly with ovarian cancer, women tend to have recurrences. I’ve spoken with physicians who got their patients through a difficult patch; they rebounded and they did fine. You don’t know for sure if that’s going to happen again if you try something else. Prognostication is not an exact science.”
Also, end-of-life discussions can be challenging conversations. “Nobody wants to take hope away from their patients. But there’s evidence to show that these conversations don’t actually reduce patients’ hopes – that’s a misconception,” Dr. Mullins said.
“It’s challenging. In the United States, we don’t like to talk about death and dying. But I think having these conversations earlier and more often can help make them a more regular part of care,” she said.
Brittany A. Davidson, MD, a gynecologic oncologist with Duke Health in Durham, N.C., who wrote an accompanying editorial, acknowledges that end-of-life can be fraught with fear, anxiety, and a lot of emotion. But she finds helping patients and their families navigate the ups and downs of their cancer one of the most rewarding aspects of her career as a physician.
“We want to help patients and their family members make these transitions as smoothly as possible,” she said.
A proponent of communications skills training for physicians in general, Dr. Brittany said doctors can learn to identify cues that patients are ready to have conversations about their end-of-life care.
“Those cues will help us facilitate conversations sooner rather than later so we’re not waiting until the very end,” she said.
What these conversations consist of varies depending on where the patient is in her cancer trajectory. In a patient with recurrent ovarian or recurrent uterine cancer, this might start with making sure the patient understands that while their cancer is treatable, it is very unlikely to be curable.
“I have often had patients who have been treated for cancer for several years and didn’t know their cancer wasn’t curable. How many missed opportunities have we overlooked?” Dr. Davidson said.
Then the conversation can turn to the goals of treatment. What’s important to the patient? “Are there events they want to be around for? Symptoms they want to avoid? Some patients really want to know what it’s going to be like to die. I try to take the lead from the patient. Ask what kind of information is helpful to them. Is it numbers? Is it symptoms? It’s really different for everybody,” Dr. Davidson said.
Although Dr. Mullins’s research and Dr. Davidson’s editorial suggest there’s room for improvement toward achieving goal-concordant care in gynecological cancers, Dr. Davidson suspects these patients might be faring a bit better than patients with other types of cancer based on her own anecdotal observations.
“One of the unique things about gynecologic oncology is that we have an amazing longitudinal relationship with our patients – we are not only their surgeons, we’re their oncologists. In other solid tumors, care is fractionated.
“That’s one of the reasons I love gynecologic oncology. I have the opportunity to know my patients through all the stages they experience as part of their cancer. I’d like to think that allows me a better opportunity to get to know them and help them recognize the value of palliative care,” Dr. Mullins said.
Ann Marco, 73, who was diagnosed with ovarian cancer in late 2018, credits her oncology team for saving her life. They treated her with chemotherapy, debulking surgery, and more chemotherapy. But it is her second and current care team that helped restore Ms. Marco’s quality of life, directing her toward such resources as palliative care, physical therapy and counseling for her and her husband.
“I can’t say enough about my palliative care doctor. She helped me manage pain, and the fatigue associated with chemotherapy. When she noticed that my leg was swollen she suspected a blood clot and sent me for an ultrasound,” Ms. Marco said.
The ultrasound revealed that she did indeed have a blood clot, for which she received, and continues to receive, medication. “Because with ovarian cancer, you always have blood clots. So little things like that, though they’re not that little, have really helped me in my journey with this cancer,” Ms. Marco said.
That journey has had its ups and downs. One chemotherapy regimen was so intolerable she decided to discontinue it, with full support of her oncologist. I told her, I just want to live my life, whether that’s only 6 more months or 3 years, but I don’t want to live it like this. And she said, ‘Ann, we’re going to do what you want to do.’”
Nine months later, when her cancer started growing again, Ms. Marco returned to chemotherapy. But this regimen has been much more tolerable, and it also appears to be doing its job. A recent CT scan showed that the tumors are shrinking.
“They’ll never go away. I have metastatic cancer. But they’re smaller, and I was really thrilled about that. It’s the best news I’ve had in more than 3 years,” Ms. Marco said.
End-of-life aggressive care still common
, shows a study published in JCO Oncology Practice.
“We have good evidence that the types of aggressive end-of-life care we looked at in this paper are generally related to a lower quality of life for patients, poorer bereavement outcomes for their families, and even shorter duration survivals,” said lead author Megan A. Mullins, PhD, MPH, a postdoctoral research fellow at the University of Michigan in Ann Arbor. “This suggests there’s a disconnect between what people think aggressive care might do and what it’s doing.”
In their evaluation of variation in end-of-life care, Dr. Mullins and her colleagues analyzed SEER-Medicare data on 6,288 women with ovarian cancer who died between 2016 and 2020. They found that 51% of those women received some form of aggressive cancer care. The most common forms were not being admitted to hospice (28.9%), receiving an invasive procedure (20.7%) and being admitted to an intensive care unit (18.6%).
Dr. Mullins noted that since palliative care was officially recognized as a specialty in 2006, there has been increasing guidance for earlier integration of palliative care and reducing the aggressiveness of end-of-life care; both ASCO and the National Quality Form have standards advising against aggressive end-of-life care.
“But there are a lot of complicated factors that I think make it hard to move the needle in this area,” she said. “For one thing, particularly with ovarian cancer, women tend to have recurrences. I’ve spoken with physicians who got their patients through a difficult patch; they rebounded and they did fine. You don’t know for sure if that’s going to happen again if you try something else. Prognostication is not an exact science.”
Also, end-of-life discussions can be challenging conversations. “Nobody wants to take hope away from their patients. But there’s evidence to show that these conversations don’t actually reduce patients’ hopes – that’s a misconception,” Dr. Mullins said.
“It’s challenging. In the United States, we don’t like to talk about death and dying. But I think having these conversations earlier and more often can help make them a more regular part of care,” she said.
Brittany A. Davidson, MD, a gynecologic oncologist with Duke Health in Durham, N.C., who wrote an accompanying editorial, acknowledges that end-of-life can be fraught with fear, anxiety, and a lot of emotion. But she finds helping patients and their families navigate the ups and downs of their cancer one of the most rewarding aspects of her career as a physician.
“We want to help patients and their family members make these transitions as smoothly as possible,” she said.
A proponent of communications skills training for physicians in general, Dr. Brittany said doctors can learn to identify cues that patients are ready to have conversations about their end-of-life care.
“Those cues will help us facilitate conversations sooner rather than later so we’re not waiting until the very end,” she said.
What these conversations consist of varies depending on where the patient is in her cancer trajectory. In a patient with recurrent ovarian or recurrent uterine cancer, this might start with making sure the patient understands that while their cancer is treatable, it is very unlikely to be curable.
“I have often had patients who have been treated for cancer for several years and didn’t know their cancer wasn’t curable. How many missed opportunities have we overlooked?” Dr. Davidson said.
Then the conversation can turn to the goals of treatment. What’s important to the patient? “Are there events they want to be around for? Symptoms they want to avoid? Some patients really want to know what it’s going to be like to die. I try to take the lead from the patient. Ask what kind of information is helpful to them. Is it numbers? Is it symptoms? It’s really different for everybody,” Dr. Davidson said.
Although Dr. Mullins’s research and Dr. Davidson’s editorial suggest there’s room for improvement toward achieving goal-concordant care in gynecological cancers, Dr. Davidson suspects these patients might be faring a bit better than patients with other types of cancer based on her own anecdotal observations.
“One of the unique things about gynecologic oncology is that we have an amazing longitudinal relationship with our patients – we are not only their surgeons, we’re their oncologists. In other solid tumors, care is fractionated.
“That’s one of the reasons I love gynecologic oncology. I have the opportunity to know my patients through all the stages they experience as part of their cancer. I’d like to think that allows me a better opportunity to get to know them and help them recognize the value of palliative care,” Dr. Mullins said.
Ann Marco, 73, who was diagnosed with ovarian cancer in late 2018, credits her oncology team for saving her life. They treated her with chemotherapy, debulking surgery, and more chemotherapy. But it is her second and current care team that helped restore Ms. Marco’s quality of life, directing her toward such resources as palliative care, physical therapy and counseling for her and her husband.
“I can’t say enough about my palliative care doctor. She helped me manage pain, and the fatigue associated with chemotherapy. When she noticed that my leg was swollen she suspected a blood clot and sent me for an ultrasound,” Ms. Marco said.
The ultrasound revealed that she did indeed have a blood clot, for which she received, and continues to receive, medication. “Because with ovarian cancer, you always have blood clots. So little things like that, though they’re not that little, have really helped me in my journey with this cancer,” Ms. Marco said.
That journey has had its ups and downs. One chemotherapy regimen was so intolerable she decided to discontinue it, with full support of her oncologist. I told her, I just want to live my life, whether that’s only 6 more months or 3 years, but I don’t want to live it like this. And she said, ‘Ann, we’re going to do what you want to do.’”
Nine months later, when her cancer started growing again, Ms. Marco returned to chemotherapy. But this regimen has been much more tolerable, and it also appears to be doing its job. A recent CT scan showed that the tumors are shrinking.
“They’ll never go away. I have metastatic cancer. But they’re smaller, and I was really thrilled about that. It’s the best news I’ve had in more than 3 years,” Ms. Marco said.
End-of-life aggressive care still common
, shows a study published in JCO Oncology Practice.
“We have good evidence that the types of aggressive end-of-life care we looked at in this paper are generally related to a lower quality of life for patients, poorer bereavement outcomes for their families, and even shorter duration survivals,” said lead author Megan A. Mullins, PhD, MPH, a postdoctoral research fellow at the University of Michigan in Ann Arbor. “This suggests there’s a disconnect between what people think aggressive care might do and what it’s doing.”
In their evaluation of variation in end-of-life care, Dr. Mullins and her colleagues analyzed SEER-Medicare data on 6,288 women with ovarian cancer who died between 2016 and 2020. They found that 51% of those women received some form of aggressive cancer care. The most common forms were not being admitted to hospice (28.9%), receiving an invasive procedure (20.7%) and being admitted to an intensive care unit (18.6%).
Dr. Mullins noted that since palliative care was officially recognized as a specialty in 2006, there has been increasing guidance for earlier integration of palliative care and reducing the aggressiveness of end-of-life care; both ASCO and the National Quality Form have standards advising against aggressive end-of-life care.
“But there are a lot of complicated factors that I think make it hard to move the needle in this area,” she said. “For one thing, particularly with ovarian cancer, women tend to have recurrences. I’ve spoken with physicians who got their patients through a difficult patch; they rebounded and they did fine. You don’t know for sure if that’s going to happen again if you try something else. Prognostication is not an exact science.”
Also, end-of-life discussions can be challenging conversations. “Nobody wants to take hope away from their patients. But there’s evidence to show that these conversations don’t actually reduce patients’ hopes – that’s a misconception,” Dr. Mullins said.
“It’s challenging. In the United States, we don’t like to talk about death and dying. But I think having these conversations earlier and more often can help make them a more regular part of care,” she said.
Brittany A. Davidson, MD, a gynecologic oncologist with Duke Health in Durham, N.C., who wrote an accompanying editorial, acknowledges that end-of-life can be fraught with fear, anxiety, and a lot of emotion. But she finds helping patients and their families navigate the ups and downs of their cancer one of the most rewarding aspects of her career as a physician.
“We want to help patients and their family members make these transitions as smoothly as possible,” she said.
A proponent of communications skills training for physicians in general, Dr. Brittany said doctors can learn to identify cues that patients are ready to have conversations about their end-of-life care.
“Those cues will help us facilitate conversations sooner rather than later so we’re not waiting until the very end,” she said.
What these conversations consist of varies depending on where the patient is in her cancer trajectory. In a patient with recurrent ovarian or recurrent uterine cancer, this might start with making sure the patient understands that while their cancer is treatable, it is very unlikely to be curable.
“I have often had patients who have been treated for cancer for several years and didn’t know their cancer wasn’t curable. How many missed opportunities have we overlooked?” Dr. Davidson said.
Then the conversation can turn to the goals of treatment. What’s important to the patient? “Are there events they want to be around for? Symptoms they want to avoid? Some patients really want to know what it’s going to be like to die. I try to take the lead from the patient. Ask what kind of information is helpful to them. Is it numbers? Is it symptoms? It’s really different for everybody,” Dr. Davidson said.
Although Dr. Mullins’s research and Dr. Davidson’s editorial suggest there’s room for improvement toward achieving goal-concordant care in gynecological cancers, Dr. Davidson suspects these patients might be faring a bit better than patients with other types of cancer based on her own anecdotal observations.
“One of the unique things about gynecologic oncology is that we have an amazing longitudinal relationship with our patients – we are not only their surgeons, we’re their oncologists. In other solid tumors, care is fractionated.
“That’s one of the reasons I love gynecologic oncology. I have the opportunity to know my patients through all the stages they experience as part of their cancer. I’d like to think that allows me a better opportunity to get to know them and help them recognize the value of palliative care,” Dr. Mullins said.