User login
Melancholic, psychotic depression may protect against ECT cognitive effects
Patients with severe melancholic or psychotic depression are more likely to respond to ECT, and preliminary evidence indicates they’re also protected against ECT-induced cognitive impairment, Linda van Diermen, MD, PhD, reported at the virtual congress of the European College of Neuropsychopharmacology.

Over the decades many small, underpowered studies have looked at possible predictors of ECT response and remission, with no consensus being reached. In an effort to bring a measure of clarity, Dr. van Diermen and her coinvestigators performed a meta-analysis of 34 published studies in accord with the PRISMA-P (Preferred Reporting Items for Systematic Review and Meta-analysis Protocols) guidelines and published their findings in the British Journal of Psychiatry. They scrutinized three potential predictors of response: the presence of psychotic features, melancholic depression with psychomotor symptoms, and older age.
Psychotic depression was associated with a 1.7-fold increased likelihood of response to ECT and a 1.5-fold increased odds of remission, compared with that of ECT-treated patients without psychotic depression. Older age was also a statistically significant predictor of response. However, the findings on melancholic depression were inconclusive, with only five studies with inconsistent results being available, said Dr. van Diermen, a psychiatrist at the University of Antwerp (Belgium).
She was quick to point out that, although psychotic depression and older age were statistically significant predictors of heightened likelihood of ECT response, they are of only limited clinical significance in treatment decision-making. The ECT response rate was 79% in patients with psychotic depression but still quite good at 71% in those without psychotic depression. Moreover, the average age of remitters was 59.7 years, compared with 55.4 years in nonresponders, a difference too small to be useful in guiding clinical treatment decisions.
“Although we did a meta-analysis in more than 3,200 patients that confirmed the superior effects of ECT in older patients and we recommended it at that time as one of the elements to guide decision-making when you consider ECT, our present, more detailed look at the interdependence of the predictors leads us to reconsider this statement. We now venture that age has been given too much weight in the past decades.”
A closer look at ECT response predictors
The studies included in the meta-analysis assessed psychotic depression and melancholic features as ECT response predictors in the typical binary way employed in clinical practice: yes/no, either present or absent. Dr. van Diermer hypothesized that a more in-depth assessment of the severity of those factors would boost their predictive power.
She found that this was indeed the case for melancholic depression as evaluated by three tools for measuring psychomotor symptoms, a core feature of this form of depression. She and her coinvestigators assessed psychomotor functioning in 65 adults with major depressive disorder before, during, and after ECT using the clinician-rated CORE scale, which measures psychomotor retardation, agitation, and noninteractiveness. In addition, the investigators had the subjects wear an accelerometer and complete a timed fine-motor drawing test.
The 41 patients with melancholic depression with psychomotor symptoms as defined by a CORE score of 8 or more were 4.9-fold more likely to reach an ECT response than were those with nonmelancholic depression. A lower baseline daytime activity level as assessed by accelerometer was also a significant predictor of increased likelihood of response, as were slower times on the drawing test.
In contrast, the investigators found that more detailed assessment of psychotic depression using the validated Psychotic Depression Assessment Scale (PDAS) was predictive of the likelihood of ECT response, but not any more so than the simple presence or absence of psychotic symptoms (J ECT. 2019 Dec;35[4]:238-44).
“In our sample, better measurement of psychotic symptoms did not improve prediction, but better measurement of psychomotor symptoms did seem to be valuable,” according to the psychiatrist.
Protection against ECT’s cognitive side effects?
Dr. van Diermen and colleagues assessed short- and long-term changes in global cognitive functioning in 65 consecutive patients treated with ECT for a major depressive episode by administering the Montreal Cognitive Assessment (MoCA) at baseline, before the third ECT session, and 1 week, 3 months, and 6 months after completing their treatment course.
During ECT, the investigators documented a limited decrease in cognitive functioning at the group level, which rebounded during the 6 months after ECT. But although there was no significant difference between MoCA scores at baseline and 6 months follow-up after ECT in the overall group of study participants, that doesn’t tell the full story. Six months after completing their course of ECT, 18% of patients demonstrated improved cognitive functioning, compared with baseline, but 8% had significantly worse cognitive functioning than pretreatment.
“Saying that ECT has no cognitive effects seems to be somewhat wrong to me. It has cognitive effects for certain people, and it will be interesting to know which people,” Dr. van Diermen said.
In what she termed “a very, very preliminary analysis,” she found that the patients with psychotic or melancholic depression were markedly less likely to have long-term cognitive impairment as defined by a worse MoCA score, compared with baseline, both at 6 months and one or more intermediate time points. Only 1 of 31 patients with psychotic depression fell into that poor cognitive outcome category, as did 4 patients with melancholic depression, compared with 12 patients without psychotic depression and 9 without melancholic depression. This, Dr. van Diermen believes, is the first report of an apparent protective effect of melancholic or psychotic depression against ECT-induced long-term cognitive worsening.
“Replication of our results is definitely necessary in larger patient samples,” she cautioned.
Dr. van Diermen reported having no financial conflicts regarding her presentation.
SOURCE: van Diermen L. ECNP 2020, Session EDU03.
Patients with severe melancholic or psychotic depression are more likely to respond to ECT, and preliminary evidence indicates they’re also protected against ECT-induced cognitive impairment, Linda van Diermen, MD, PhD, reported at the virtual congress of the European College of Neuropsychopharmacology.
Over the decades many small, underpowered studies have looked at possible predictors of ECT response and remission, with no consensus being reached. In an effort to bring a measure of clarity, Dr. van Diermen and her coinvestigators performed a meta-analysis of 34 published studies in accord with the PRISMA-P (Preferred Reporting Items for Systematic Review and Meta-analysis Protocols) guidelines and published their findings in the British Journal of Psychiatry. They scrutinized three potential predictors of response: the presence of psychotic features, melancholic depression with psychomotor symptoms, and older age.
Psychotic depression was associated with a 1.7-fold increased likelihood of response to ECT and a 1.5-fold increased odds of remission, compared with that of ECT-treated patients without psychotic depression. Older age was also a statistically significant predictor of response. However, the findings on melancholic depression were inconclusive, with only five studies with inconsistent results being available, said Dr. van Diermen, a psychiatrist at the University of Antwerp (Belgium).
She was quick to point out that, although psychotic depression and older age were statistically significant predictors of heightened likelihood of ECT response, they are of only limited clinical significance in treatment decision-making. The ECT response rate was 79% in patients with psychotic depression but still quite good at 71% in those without psychotic depression. Moreover, the average age of remitters was 59.7 years, compared with 55.4 years in nonresponders, a difference too small to be useful in guiding clinical treatment decisions.
“Although we did a meta-analysis in more than 3,200 patients that confirmed the superior effects of ECT in older patients and we recommended it at that time as one of the elements to guide decision-making when you consider ECT, our present, more detailed look at the interdependence of the predictors leads us to reconsider this statement. We now venture that age has been given too much weight in the past decades.”
A closer look at ECT response predictors
The studies included in the meta-analysis assessed psychotic depression and melancholic features as ECT response predictors in the typical binary way employed in clinical practice: yes/no, either present or absent. Dr. van Diermer hypothesized that a more in-depth assessment of the severity of those factors would boost their predictive power.
She found that this was indeed the case for melancholic depression as evaluated by three tools for measuring psychomotor symptoms, a core feature of this form of depression. She and her coinvestigators assessed psychomotor functioning in 65 adults with major depressive disorder before, during, and after ECT using the clinician-rated CORE scale, which measures psychomotor retardation, agitation, and noninteractiveness. In addition, the investigators had the subjects wear an accelerometer and complete a timed fine-motor drawing test.
The 41 patients with melancholic depression with psychomotor symptoms as defined by a CORE score of 8 or more were 4.9-fold more likely to reach an ECT response than were those with nonmelancholic depression. A lower baseline daytime activity level as assessed by accelerometer was also a significant predictor of increased likelihood of response, as were slower times on the drawing test.
In contrast, the investigators found that more detailed assessment of psychotic depression using the validated Psychotic Depression Assessment Scale (PDAS) was predictive of the likelihood of ECT response, but not any more so than the simple presence or absence of psychotic symptoms (J ECT. 2019 Dec;35[4]:238-44).
“In our sample, better measurement of psychotic symptoms did not improve prediction, but better measurement of psychomotor symptoms did seem to be valuable,” according to the psychiatrist.
Protection against ECT’s cognitive side effects?
Dr. van Diermen and colleagues assessed short- and long-term changes in global cognitive functioning in 65 consecutive patients treated with ECT for a major depressive episode by administering the Montreal Cognitive Assessment (MoCA) at baseline, before the third ECT session, and 1 week, 3 months, and 6 months after completing their treatment course.
During ECT, the investigators documented a limited decrease in cognitive functioning at the group level, which rebounded during the 6 months after ECT. But although there was no significant difference between MoCA scores at baseline and 6 months follow-up after ECT in the overall group of study participants, that doesn’t tell the full story. Six months after completing their course of ECT, 18% of patients demonstrated improved cognitive functioning, compared with baseline, but 8% had significantly worse cognitive functioning than pretreatment.
“Saying that ECT has no cognitive effects seems to be somewhat wrong to me. It has cognitive effects for certain people, and it will be interesting to know which people,” Dr. van Diermen said.
In what she termed “a very, very preliminary analysis,” she found that the patients with psychotic or melancholic depression were markedly less likely to have long-term cognitive impairment as defined by a worse MoCA score, compared with baseline, both at 6 months and one or more intermediate time points. Only 1 of 31 patients with psychotic depression fell into that poor cognitive outcome category, as did 4 patients with melancholic depression, compared with 12 patients without psychotic depression and 9 without melancholic depression. This, Dr. van Diermen believes, is the first report of an apparent protective effect of melancholic or psychotic depression against ECT-induced long-term cognitive worsening.
“Replication of our results is definitely necessary in larger patient samples,” she cautioned.
Dr. van Diermen reported having no financial conflicts regarding her presentation.
SOURCE: van Diermen L. ECNP 2020, Session EDU03.
Patients with severe melancholic or psychotic depression are more likely to respond to ECT, and preliminary evidence indicates they’re also protected against ECT-induced cognitive impairment, Linda van Diermen, MD, PhD, reported at the virtual congress of the European College of Neuropsychopharmacology.
Over the decades many small, underpowered studies have looked at possible predictors of ECT response and remission, with no consensus being reached. In an effort to bring a measure of clarity, Dr. van Diermen and her coinvestigators performed a meta-analysis of 34 published studies in accord with the PRISMA-P (Preferred Reporting Items for Systematic Review and Meta-analysis Protocols) guidelines and published their findings in the British Journal of Psychiatry. They scrutinized three potential predictors of response: the presence of psychotic features, melancholic depression with psychomotor symptoms, and older age.
Psychotic depression was associated with a 1.7-fold increased likelihood of response to ECT and a 1.5-fold increased odds of remission, compared with that of ECT-treated patients without psychotic depression. Older age was also a statistically significant predictor of response. However, the findings on melancholic depression were inconclusive, with only five studies with inconsistent results being available, said Dr. van Diermen, a psychiatrist at the University of Antwerp (Belgium).
She was quick to point out that, although psychotic depression and older age were statistically significant predictors of heightened likelihood of ECT response, they are of only limited clinical significance in treatment decision-making. The ECT response rate was 79% in patients with psychotic depression but still quite good at 71% in those without psychotic depression. Moreover, the average age of remitters was 59.7 years, compared with 55.4 years in nonresponders, a difference too small to be useful in guiding clinical treatment decisions.
“Although we did a meta-analysis in more than 3,200 patients that confirmed the superior effects of ECT in older patients and we recommended it at that time as one of the elements to guide decision-making when you consider ECT, our present, more detailed look at the interdependence of the predictors leads us to reconsider this statement. We now venture that age has been given too much weight in the past decades.”
A closer look at ECT response predictors
The studies included in the meta-analysis assessed psychotic depression and melancholic features as ECT response predictors in the typical binary way employed in clinical practice: yes/no, either present or absent. Dr. van Diermer hypothesized that a more in-depth assessment of the severity of those factors would boost their predictive power.
She found that this was indeed the case for melancholic depression as evaluated by three tools for measuring psychomotor symptoms, a core feature of this form of depression. She and her coinvestigators assessed psychomotor functioning in 65 adults with major depressive disorder before, during, and after ECT using the clinician-rated CORE scale, which measures psychomotor retardation, agitation, and noninteractiveness. In addition, the investigators had the subjects wear an accelerometer and complete a timed fine-motor drawing test.
The 41 patients with melancholic depression with psychomotor symptoms as defined by a CORE score of 8 or more were 4.9-fold more likely to reach an ECT response than were those with nonmelancholic depression. A lower baseline daytime activity level as assessed by accelerometer was also a significant predictor of increased likelihood of response, as were slower times on the drawing test.
In contrast, the investigators found that more detailed assessment of psychotic depression using the validated Psychotic Depression Assessment Scale (PDAS) was predictive of the likelihood of ECT response, but not any more so than the simple presence or absence of psychotic symptoms (J ECT. 2019 Dec;35[4]:238-44).
“In our sample, better measurement of psychotic symptoms did not improve prediction, but better measurement of psychomotor symptoms did seem to be valuable,” according to the psychiatrist.
Protection against ECT’s cognitive side effects?
Dr. van Diermen and colleagues assessed short- and long-term changes in global cognitive functioning in 65 consecutive patients treated with ECT for a major depressive episode by administering the Montreal Cognitive Assessment (MoCA) at baseline, before the third ECT session, and 1 week, 3 months, and 6 months after completing their treatment course.
During ECT, the investigators documented a limited decrease in cognitive functioning at the group level, which rebounded during the 6 months after ECT. But although there was no significant difference between MoCA scores at baseline and 6 months follow-up after ECT in the overall group of study participants, that doesn’t tell the full story. Six months after completing their course of ECT, 18% of patients demonstrated improved cognitive functioning, compared with baseline, but 8% had significantly worse cognitive functioning than pretreatment.
“Saying that ECT has no cognitive effects seems to be somewhat wrong to me. It has cognitive effects for certain people, and it will be interesting to know which people,” Dr. van Diermen said.
In what she termed “a very, very preliminary analysis,” she found that the patients with psychotic or melancholic depression were markedly less likely to have long-term cognitive impairment as defined by a worse MoCA score, compared with baseline, both at 6 months and one or more intermediate time points. Only 1 of 31 patients with psychotic depression fell into that poor cognitive outcome category, as did 4 patients with melancholic depression, compared with 12 patients without psychotic depression and 9 without melancholic depression. This, Dr. van Diermen believes, is the first report of an apparent protective effect of melancholic or psychotic depression against ECT-induced long-term cognitive worsening.
“Replication of our results is definitely necessary in larger patient samples,” she cautioned.
Dr. van Diermen reported having no financial conflicts regarding her presentation.
SOURCE: van Diermen L. ECNP 2020, Session EDU03.
FROM ECNP 2020

Non-Whites remain sorely underrepresented in phase 3 psoriasis trials
Non-White patient participation in phase 3 therapeutic trials for plaque psoriasis is less than 15%, according to a recently published analysis of data from the ClinicalTrials.gov database.
The exact figure drawn from the survey of 82 trials was 14.2%, but 20 (24%) of the trials did not include ethnoracial data at all, and only 65% of those with data had complete data, according to a report in the British Journal of Dermatology by a team of investigators from the department of dermatology at the University of California, San Francisco.
“The remaining studies reported the percentage of white participants only or white participants and one additional ethnoracial group,” reported the investigators, led by Vidhatha D. Reddy, a medical student at UCSF.
The investigators broke down participation by race in all phase 3 plaque psoriasis trials that enrolled adults and had posted results by May 2020. Data from trials of medications yet to be approved were excluded.
Most trials were multinational. The medications evaluated included 11 biologics, 10 topicals, 2 oral systemic agents, and a phosphodiesterase type-4 inhibitor. The 82 trials included in this analysis enrolled 48,846 collectively.
From trials that identified race, 85.8% of 39,161 participants were White, 3.09% of 25,565 patients were Black, 19.55% of 11,364 patients were Hispanic or Latino, and 9.21% of 30,009 patients were Asian. Of trials that included Native Americans or Pacific Islanders, fewer than 2% of participants represented this category.
Non-White patients remain underrepresented even when recognizing differences in the prevalence of psoriasis. For example, one recent survey found the U.S, prevalence of psoriasis to be about half as great in Blacks as it is in Whites (1.9% vs. 3.9%), but the representation of Blacks in the phase 3 trials evaluated by Mr. Reddy and colleagues was more than 20 times lower.
There are many reasons to suspect that lack of diversification in psoriasis trials is impeding optimal care in those underrepresented. Of several examples offered by the authors, one involved differential responses to adalimumab among patients with hidradenitis suppurativa with genetic variants in the BCL2 gene, but the authors reported racially associated genetic differences are not uncommon.
“Estimates have shown that approximately one-fifth of newly developed medications demonstrate interracial/ethnic variability in regard to various factors, such as pharmacokinetics, safety and efficacy profiles, dosing, and pharmacogenetics,” Mr. Reddy and his coinvestigators stated.
Although racial diversity in the design and recruitment for clinical trials has not been a priority in trials involving psoriasis, other skin diseases, or most diseases in general, the authors cited some evidence that this is changing.
“Since 2017, research funded by the National Institutes of Health has been required to report race and ethnicity of participants following an amendment to the Health Revitalization Act,” according to the authors, who suggested that other such initiatives are needed. They advocated “explicit goals to increase recruitment of people of color” as a standard step in clinical trial conduct.
Hypertension trials were cited as an example in which diversity has made a difference.
“Although Black patients are at an elevated risk of developing hypertension, it was not until the enrollment of a substantial proportion of black participants in ALLHAT (Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial) that enough data on Black patients were available to make specific treatment recommendations in this population,” they noted.
Impossible to know treatment benefits without ethnoracial data

Another investigator who has considered this issue, Junko Takeshita, MD, PhD, an assistant professor of dermatology at the University of Pennsylvania, Philadelphia, agreed.
“Lack of diversity among participants in phase 3 clinical trials for psoriasis is a problem,” said Dr. Takeshita, who led a study of racial differences in perceptions of psoriasis therapies that was published last year.
In that study, “my research group not only found differences in perceptions about biologics between Black and White patients with psoriasis, but we have also shown that Black patients with psoriasis are less likely to receive biologic treatment,” she reported. There are many explanations. For example, she found in another study that Black patients are underrepresented in direct-to-consumer advertisements for biologics.
This problem is not unique to psoriasis. Underrepresentation of Blacks and other ethnoracial groups is true of other skin diseases and many diseases in general, according to Dr. Takeshita. However, she cautioned that the 3% figure for Black participation in psoriasis trials reported by Mr. Reddy and colleagues is not necessarily reflective of trials in the United States.
“This study included international study sites that are recruiting patients from populations with different demographics than the U.S.,” she noted. By including sites with only Asian patients or countries with few Blacks in the population, it dilutes Black representation. She would expect the exact proportion of Black participants to be somewhat higher even if they are “still likely to be underrepresented” if the analysis has been limited to U.S. data.
The research had no funding source. Three of the nine authors reported financial relationships with pharmaceutical companies.
SOURCE: Reddy VD et al. Br J Dermatol. 2020 Sep 17. doi: 10.1111/bjd.19468.
Non-White patient participation in phase 3 therapeutic trials for plaque psoriasis is less than 15%, according to a recently published analysis of data from the ClinicalTrials.gov database.
The exact figure drawn from the survey of 82 trials was 14.2%, but 20 (24%) of the trials did not include ethnoracial data at all, and only 65% of those with data had complete data, according to a report in the British Journal of Dermatology by a team of investigators from the department of dermatology at the University of California, San Francisco.
“The remaining studies reported the percentage of white participants only or white participants and one additional ethnoracial group,” reported the investigators, led by Vidhatha D. Reddy, a medical student at UCSF.
The investigators broke down participation by race in all phase 3 plaque psoriasis trials that enrolled adults and had posted results by May 2020. Data from trials of medications yet to be approved were excluded.
Most trials were multinational. The medications evaluated included 11 biologics, 10 topicals, 2 oral systemic agents, and a phosphodiesterase type-4 inhibitor. The 82 trials included in this analysis enrolled 48,846 collectively.
From trials that identified race, 85.8% of 39,161 participants were White, 3.09% of 25,565 patients were Black, 19.55% of 11,364 patients were Hispanic or Latino, and 9.21% of 30,009 patients were Asian. Of trials that included Native Americans or Pacific Islanders, fewer than 2% of participants represented this category.
Non-White patients remain underrepresented even when recognizing differences in the prevalence of psoriasis. For example, one recent survey found the U.S, prevalence of psoriasis to be about half as great in Blacks as it is in Whites (1.9% vs. 3.9%), but the representation of Blacks in the phase 3 trials evaluated by Mr. Reddy and colleagues was more than 20 times lower.
There are many reasons to suspect that lack of diversification in psoriasis trials is impeding optimal care in those underrepresented. Of several examples offered by the authors, one involved differential responses to adalimumab among patients with hidradenitis suppurativa with genetic variants in the BCL2 gene, but the authors reported racially associated genetic differences are not uncommon.
“Estimates have shown that approximately one-fifth of newly developed medications demonstrate interracial/ethnic variability in regard to various factors, such as pharmacokinetics, safety and efficacy profiles, dosing, and pharmacogenetics,” Mr. Reddy and his coinvestigators stated.
Although racial diversity in the design and recruitment for clinical trials has not been a priority in trials involving psoriasis, other skin diseases, or most diseases in general, the authors cited some evidence that this is changing.
“Since 2017, research funded by the National Institutes of Health has been required to report race and ethnicity of participants following an amendment to the Health Revitalization Act,” according to the authors, who suggested that other such initiatives are needed. They advocated “explicit goals to increase recruitment of people of color” as a standard step in clinical trial conduct.
Hypertension trials were cited as an example in which diversity has made a difference.
“Although Black patients are at an elevated risk of developing hypertension, it was not until the enrollment of a substantial proportion of black participants in ALLHAT (Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial) that enough data on Black patients were available to make specific treatment recommendations in this population,” they noted.
Impossible to know treatment benefits without ethnoracial data
Another investigator who has considered this issue, Junko Takeshita, MD, PhD, an assistant professor of dermatology at the University of Pennsylvania, Philadelphia, agreed.
“Lack of diversity among participants in phase 3 clinical trials for psoriasis is a problem,” said Dr. Takeshita, who led a study of racial differences in perceptions of psoriasis therapies that was published last year.
In that study, “my research group not only found differences in perceptions about biologics between Black and White patients with psoriasis, but we have also shown that Black patients with psoriasis are less likely to receive biologic treatment,” she reported. There are many explanations. For example, she found in another study that Black patients are underrepresented in direct-to-consumer advertisements for biologics.
This problem is not unique to psoriasis. Underrepresentation of Blacks and other ethnoracial groups is true of other skin diseases and many diseases in general, according to Dr. Takeshita. However, she cautioned that the 3% figure for Black participation in psoriasis trials reported by Mr. Reddy and colleagues is not necessarily reflective of trials in the United States.
“This study included international study sites that are recruiting patients from populations with different demographics than the U.S.,” she noted. By including sites with only Asian patients or countries with few Blacks in the population, it dilutes Black representation. She would expect the exact proportion of Black participants to be somewhat higher even if they are “still likely to be underrepresented” if the analysis has been limited to U.S. data.
The research had no funding source. Three of the nine authors reported financial relationships with pharmaceutical companies.
SOURCE: Reddy VD et al. Br J Dermatol. 2020 Sep 17. doi: 10.1111/bjd.19468.
Non-White patient participation in phase 3 therapeutic trials for plaque psoriasis is less than 15%, according to a recently published analysis of data from the ClinicalTrials.gov database.
The exact figure drawn from the survey of 82 trials was 14.2%, but 20 (24%) of the trials did not include ethnoracial data at all, and only 65% of those with data had complete data, according to a report in the British Journal of Dermatology by a team of investigators from the department of dermatology at the University of California, San Francisco.
“The remaining studies reported the percentage of white participants only or white participants and one additional ethnoracial group,” reported the investigators, led by Vidhatha D. Reddy, a medical student at UCSF.
The investigators broke down participation by race in all phase 3 plaque psoriasis trials that enrolled adults and had posted results by May 2020. Data from trials of medications yet to be approved were excluded.
Most trials were multinational. The medications evaluated included 11 biologics, 10 topicals, 2 oral systemic agents, and a phosphodiesterase type-4 inhibitor. The 82 trials included in this analysis enrolled 48,846 collectively.
From trials that identified race, 85.8% of 39,161 participants were White, 3.09% of 25,565 patients were Black, 19.55% of 11,364 patients were Hispanic or Latino, and 9.21% of 30,009 patients were Asian. Of trials that included Native Americans or Pacific Islanders, fewer than 2% of participants represented this category.
Non-White patients remain underrepresented even when recognizing differences in the prevalence of psoriasis. For example, one recent survey found the U.S, prevalence of psoriasis to be about half as great in Blacks as it is in Whites (1.9% vs. 3.9%), but the representation of Blacks in the phase 3 trials evaluated by Mr. Reddy and colleagues was more than 20 times lower.
There are many reasons to suspect that lack of diversification in psoriasis trials is impeding optimal care in those underrepresented. Of several examples offered by the authors, one involved differential responses to adalimumab among patients with hidradenitis suppurativa with genetic variants in the BCL2 gene, but the authors reported racially associated genetic differences are not uncommon.
“Estimates have shown that approximately one-fifth of newly developed medications demonstrate interracial/ethnic variability in regard to various factors, such as pharmacokinetics, safety and efficacy profiles, dosing, and pharmacogenetics,” Mr. Reddy and his coinvestigators stated.
Although racial diversity in the design and recruitment for clinical trials has not been a priority in trials involving psoriasis, other skin diseases, or most diseases in general, the authors cited some evidence that this is changing.
“Since 2017, research funded by the National Institutes of Health has been required to report race and ethnicity of participants following an amendment to the Health Revitalization Act,” according to the authors, who suggested that other such initiatives are needed. They advocated “explicit goals to increase recruitment of people of color” as a standard step in clinical trial conduct.
Hypertension trials were cited as an example in which diversity has made a difference.
“Although Black patients are at an elevated risk of developing hypertension, it was not until the enrollment of a substantial proportion of black participants in ALLHAT (Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial) that enough data on Black patients were available to make specific treatment recommendations in this population,” they noted.
Impossible to know treatment benefits without ethnoracial data
Another investigator who has considered this issue, Junko Takeshita, MD, PhD, an assistant professor of dermatology at the University of Pennsylvania, Philadelphia, agreed.
“Lack of diversity among participants in phase 3 clinical trials for psoriasis is a problem,” said Dr. Takeshita, who led a study of racial differences in perceptions of psoriasis therapies that was published last year.
In that study, “my research group not only found differences in perceptions about biologics between Black and White patients with psoriasis, but we have also shown that Black patients with psoriasis are less likely to receive biologic treatment,” she reported. There are many explanations. For example, she found in another study that Black patients are underrepresented in direct-to-consumer advertisements for biologics.
This problem is not unique to psoriasis. Underrepresentation of Blacks and other ethnoracial groups is true of other skin diseases and many diseases in general, according to Dr. Takeshita. However, she cautioned that the 3% figure for Black participation in psoriasis trials reported by Mr. Reddy and colleagues is not necessarily reflective of trials in the United States.
“This study included international study sites that are recruiting patients from populations with different demographics than the U.S.,” she noted. By including sites with only Asian patients or countries with few Blacks in the population, it dilutes Black representation. She would expect the exact proportion of Black participants to be somewhat higher even if they are “still likely to be underrepresented” if the analysis has been limited to U.S. data.
The research had no funding source. Three of the nine authors reported financial relationships with pharmaceutical companies.
SOURCE: Reddy VD et al. Br J Dermatol. 2020 Sep 17. doi: 10.1111/bjd.19468.
FROM THE BRITISH JOURNAL OF DERMATOLOGY
Link between vitamin D and ICU outcomes unclear
We can “stop putting money on vitamin D” to help patients who require critical care, said Todd Rice, MD, FCCP.
“Results from vitamin D trials have not been uniformly one way, but they have been pretty uniformly disappointing,” Dr. Rice, from Vanderbilt University Medical Center, Nashville, Tenn., reported at the annual meeting of the American College of Chest Physicians.
Low levels of vitamin D in critically ill COVID-19 patients have been reported in numerous recent studies, and researchers are looking for ways to boost those levels and improve outcomes.
We are seeing “the exact same story” in the critically ill COVID-19 population as we see in the general ICU population, said Dr. Rice. “The whole scenario is repeating itself. I’m pessimistic.”
Still, vitamin D levels can be elevated so, in theory, “the concept makes sense,” he said. There is evidence that, “when given enterally, the levels rise nicely” and vitamin D is absorbed reasonably well.” But is that enough?
When patients are admitted to the ICU, some biomarkers in the body are too high and others are too low. Vitamin D is often too low. So far, though, “supplementing vitamin D in the ICU has not significantly improved outcomes,” said Dr. Rice.
In the Vitamin D to Improve Outcomes by Leveraging Early Treatment (VIOLET) trial, Dr. Rice and colleagues found no statistical benefit when a 540,000 IU boost of vitamin D was administered to 2,624 critically ill patients, as reported by Medscape Medical News.
“Early administration of high-dose enteral vitamin D3 did not provide an advantage over placebo with respect to 90-day mortality or other nonfatal outcomes among critically ill, vitamin D–deficient patients,” the researchers write in their recent report.
In fact, VIOLET ended before enrollment had reached the planned 3,000-patient cohort because the statistical analysis clearly did not show benefit. Those enrolled were in the ICU because of, among other things, pneumonia, sepsis, the need for mechanical ventilation or vasopressors, and risk for acute respiratory distress syndrome.
“It doesn’t look like vitamin D is going to be the answer to our critical care problems,” Dr. Rice said in an interview.
Maintenance dose needed?
One theory suggests that VIOLET might have failed because a maintenance dose is needed after the initial boost of vitamin D.
In the ongoing VITDALIZE trial, critically ill patients with severe vitamin D deficiency (12 ng/mL or less at admission) receive an initial 540,000-IU dose followed by 4,000 IU per day.
The highly anticipated VITDALIZE results are expected in the middle of next year, Dr. Rice reported, so “let’s wait to see.”
“Vitamin D may not have an acute effect,” he theorized. “We can raise your levels, but that doesn’t give you all the benefits of having a sufficient level for a long period of time.”
Another theory suggests that a low level of vitamin D is simply a signal of the severity of disease, not a direct influence on disease pathology.
Some observational data have shown an association between low levels of vitamin D and outcomes in COVID-19 patients (Nutrients. 2020 May 9;12[5]:1359; medRxiv 2020 Apr 24. doi: 10.1101/2020.04.24.20075838; JAMA Netw Open. 2020;3[9]:e2019722; FEBS J. 2020 Jul 23;10.1111/febs.15495; Clin Endocrinol [Oxf]. 2020 Jul 3;10.1111/cen.14276), but some have shown no association (medRxiv. 2020 Jun 26. doi: 10.1101/2020.06.26.20140921; J Public Health [Oxf]. 2020 Aug 18;42[3]:451-60).
Dr. Rice conducted a search of Clinicaltrials.gov immediately before his presentation on Sunday, and found 41 ongoing interventional studies – “not observational studies” – looking at COVID-19 and vitamin D.
“They’re recruiting, they’re enrolling; hopefully we’ll have data soon,” he said.
Researchers have checked a lot of boxes with a resounding yes on the vitamin D question, so there’s reason to think an association does exist for ICU patients, whether or not they have COVID-19.
“Is there a theoretical benefit of vitamin D in the ICU?” Dr. Rice asked. “Yes. Is vitamin D deficient in patients in the ICU? Yes. Is that deficiency associated with poor outcomes? Yes. Can it be replaced safely? Yes.”
However, “we’re not really sure that it improves outcomes,” he said.
A chronic issue?
“Do you think it’s really an issue of the patients being critically ill with vitamin D,” or is it “a chronic issue of having low vitamin D?” asked session moderator Antine Stenbit, MD, PhD, from the University of California, San Diego.
“We don’t know for sure,” Dr. Rice said. Vitamin D might not have a lot of acute effects; it might have effects that are chronic, that work with levels over a period of time, he explained.
“It’s not clear we can correct that with a single dose or with a few days of giving a level that is adequate,” he acknowledged.
Dr. Rice is an investigator in the PETAL network. Dr. Stenbit disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
We can “stop putting money on vitamin D” to help patients who require critical care, said Todd Rice, MD, FCCP.
“Results from vitamin D trials have not been uniformly one way, but they have been pretty uniformly disappointing,” Dr. Rice, from Vanderbilt University Medical Center, Nashville, Tenn., reported at the annual meeting of the American College of Chest Physicians.
Low levels of vitamin D in critically ill COVID-19 patients have been reported in numerous recent studies, and researchers are looking for ways to boost those levels and improve outcomes.
We are seeing “the exact same story” in the critically ill COVID-19 population as we see in the general ICU population, said Dr. Rice. “The whole scenario is repeating itself. I’m pessimistic.”
Still, vitamin D levels can be elevated so, in theory, “the concept makes sense,” he said. There is evidence that, “when given enterally, the levels rise nicely” and vitamin D is absorbed reasonably well.” But is that enough?
When patients are admitted to the ICU, some biomarkers in the body are too high and others are too low. Vitamin D is often too low. So far, though, “supplementing vitamin D in the ICU has not significantly improved outcomes,” said Dr. Rice.
In the Vitamin D to Improve Outcomes by Leveraging Early Treatment (VIOLET) trial, Dr. Rice and colleagues found no statistical benefit when a 540,000 IU boost of vitamin D was administered to 2,624 critically ill patients, as reported by Medscape Medical News.
“Early administration of high-dose enteral vitamin D3 did not provide an advantage over placebo with respect to 90-day mortality or other nonfatal outcomes among critically ill, vitamin D–deficient patients,” the researchers write in their recent report.
In fact, VIOLET ended before enrollment had reached the planned 3,000-patient cohort because the statistical analysis clearly did not show benefit. Those enrolled were in the ICU because of, among other things, pneumonia, sepsis, the need for mechanical ventilation or vasopressors, and risk for acute respiratory distress syndrome.
“It doesn’t look like vitamin D is going to be the answer to our critical care problems,” Dr. Rice said in an interview.
Maintenance dose needed?
One theory suggests that VIOLET might have failed because a maintenance dose is needed after the initial boost of vitamin D.
In the ongoing VITDALIZE trial, critically ill patients with severe vitamin D deficiency (12 ng/mL or less at admission) receive an initial 540,000-IU dose followed by 4,000 IU per day.
The highly anticipated VITDALIZE results are expected in the middle of next year, Dr. Rice reported, so “let’s wait to see.”
“Vitamin D may not have an acute effect,” he theorized. “We can raise your levels, but that doesn’t give you all the benefits of having a sufficient level for a long period of time.”
Another theory suggests that a low level of vitamin D is simply a signal of the severity of disease, not a direct influence on disease pathology.
Some observational data have shown an association between low levels of vitamin D and outcomes in COVID-19 patients (Nutrients. 2020 May 9;12[5]:1359; medRxiv 2020 Apr 24. doi: 10.1101/2020.04.24.20075838; JAMA Netw Open. 2020;3[9]:e2019722; FEBS J. 2020 Jul 23;10.1111/febs.15495; Clin Endocrinol [Oxf]. 2020 Jul 3;10.1111/cen.14276), but some have shown no association (medRxiv. 2020 Jun 26. doi: 10.1101/2020.06.26.20140921; J Public Health [Oxf]. 2020 Aug 18;42[3]:451-60).
Dr. Rice conducted a search of Clinicaltrials.gov immediately before his presentation on Sunday, and found 41 ongoing interventional studies – “not observational studies” – looking at COVID-19 and vitamin D.
“They’re recruiting, they’re enrolling; hopefully we’ll have data soon,” he said.
Researchers have checked a lot of boxes with a resounding yes on the vitamin D question, so there’s reason to think an association does exist for ICU patients, whether or not they have COVID-19.
“Is there a theoretical benefit of vitamin D in the ICU?” Dr. Rice asked. “Yes. Is vitamin D deficient in patients in the ICU? Yes. Is that deficiency associated with poor outcomes? Yes. Can it be replaced safely? Yes.”
However, “we’re not really sure that it improves outcomes,” he said.
A chronic issue?
“Do you think it’s really an issue of the patients being critically ill with vitamin D,” or is it “a chronic issue of having low vitamin D?” asked session moderator Antine Stenbit, MD, PhD, from the University of California, San Diego.
“We don’t know for sure,” Dr. Rice said. Vitamin D might not have a lot of acute effects; it might have effects that are chronic, that work with levels over a period of time, he explained.
“It’s not clear we can correct that with a single dose or with a few days of giving a level that is adequate,” he acknowledged.
Dr. Rice is an investigator in the PETAL network. Dr. Stenbit disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
We can “stop putting money on vitamin D” to help patients who require critical care, said Todd Rice, MD, FCCP.
“Results from vitamin D trials have not been uniformly one way, but they have been pretty uniformly disappointing,” Dr. Rice, from Vanderbilt University Medical Center, Nashville, Tenn., reported at the annual meeting of the American College of Chest Physicians.
Low levels of vitamin D in critically ill COVID-19 patients have been reported in numerous recent studies, and researchers are looking for ways to boost those levels and improve outcomes.
We are seeing “the exact same story” in the critically ill COVID-19 population as we see in the general ICU population, said Dr. Rice. “The whole scenario is repeating itself. I’m pessimistic.”
Still, vitamin D levels can be elevated so, in theory, “the concept makes sense,” he said. There is evidence that, “when given enterally, the levels rise nicely” and vitamin D is absorbed reasonably well.” But is that enough?
When patients are admitted to the ICU, some biomarkers in the body are too high and others are too low. Vitamin D is often too low. So far, though, “supplementing vitamin D in the ICU has not significantly improved outcomes,” said Dr. Rice.
In the Vitamin D to Improve Outcomes by Leveraging Early Treatment (VIOLET) trial, Dr. Rice and colleagues found no statistical benefit when a 540,000 IU boost of vitamin D was administered to 2,624 critically ill patients, as reported by Medscape Medical News.
“Early administration of high-dose enteral vitamin D3 did not provide an advantage over placebo with respect to 90-day mortality or other nonfatal outcomes among critically ill, vitamin D–deficient patients,” the researchers write in their recent report.
In fact, VIOLET ended before enrollment had reached the planned 3,000-patient cohort because the statistical analysis clearly did not show benefit. Those enrolled were in the ICU because of, among other things, pneumonia, sepsis, the need for mechanical ventilation or vasopressors, and risk for acute respiratory distress syndrome.
“It doesn’t look like vitamin D is going to be the answer to our critical care problems,” Dr. Rice said in an interview.
Maintenance dose needed?
One theory suggests that VIOLET might have failed because a maintenance dose is needed after the initial boost of vitamin D.
In the ongoing VITDALIZE trial, critically ill patients with severe vitamin D deficiency (12 ng/mL or less at admission) receive an initial 540,000-IU dose followed by 4,000 IU per day.
The highly anticipated VITDALIZE results are expected in the middle of next year, Dr. Rice reported, so “let’s wait to see.”
“Vitamin D may not have an acute effect,” he theorized. “We can raise your levels, but that doesn’t give you all the benefits of having a sufficient level for a long period of time.”
Another theory suggests that a low level of vitamin D is simply a signal of the severity of disease, not a direct influence on disease pathology.
Some observational data have shown an association between low levels of vitamin D and outcomes in COVID-19 patients (Nutrients. 2020 May 9;12[5]:1359; medRxiv 2020 Apr 24. doi: 10.1101/2020.04.24.20075838; JAMA Netw Open. 2020;3[9]:e2019722; FEBS J. 2020 Jul 23;10.1111/febs.15495; Clin Endocrinol [Oxf]. 2020 Jul 3;10.1111/cen.14276), but some have shown no association (medRxiv. 2020 Jun 26. doi: 10.1101/2020.06.26.20140921; J Public Health [Oxf]. 2020 Aug 18;42[3]:451-60).
Dr. Rice conducted a search of Clinicaltrials.gov immediately before his presentation on Sunday, and found 41 ongoing interventional studies – “not observational studies” – looking at COVID-19 and vitamin D.
“They’re recruiting, they’re enrolling; hopefully we’ll have data soon,” he said.
Researchers have checked a lot of boxes with a resounding yes on the vitamin D question, so there’s reason to think an association does exist for ICU patients, whether or not they have COVID-19.
“Is there a theoretical benefit of vitamin D in the ICU?” Dr. Rice asked. “Yes. Is vitamin D deficient in patients in the ICU? Yes. Is that deficiency associated with poor outcomes? Yes. Can it be replaced safely? Yes.”
However, “we’re not really sure that it improves outcomes,” he said.
A chronic issue?
“Do you think it’s really an issue of the patients being critically ill with vitamin D,” or is it “a chronic issue of having low vitamin D?” asked session moderator Antine Stenbit, MD, PhD, from the University of California, San Diego.
“We don’t know for sure,” Dr. Rice said. Vitamin D might not have a lot of acute effects; it might have effects that are chronic, that work with levels over a period of time, he explained.
“It’s not clear we can correct that with a single dose or with a few days of giving a level that is adequate,” he acknowledged.
Dr. Rice is an investigator in the PETAL network. Dr. Stenbit disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
FROM CHEST 2020
Include irritability in ADHD suicidality risk assessments
Irritability appears to be a potent independent predictor of increased risk for suicidality in children and adolescents with ADHD, Tomer Levy, MD, said at the virtual congress of the European College of Neuropsychopharmacology.
While there is ample evidence that ADHD is associated with increased suicidality, Dr. Levy’s recent study involving 1,516 youths aged 6-17 years attending an outpatient ADHD clinic demonstrated that this increased risk is mediated by depression and irritability in roughly equal measures. Moreover, upon controlling for those two factors in a multivariate analysis, ADHD symptoms, per se, had no direct effect on risk of suicidality as defined by suidical ideation, attempts, or self-harm.
The clinical take-home message is that assessing irritability, as well as depression, may bolster an estimate of suicidality and help in managing suicidal risk in ADHD, according to Dr. Levy, a child and adolescent psychiatrist at the Hospital for Sick Children, Toronto, and head of behavioral regulation services at the Geha Mental Health Center in Petah Tikva, Israel.
The study included separate parent- and teacher-structured reports of the youths’ ADHD symptoms, suicidality, depression, irritability, and anxiety.
In multivariate analyses, parent-reported depression accounted for 39.1% of the association between ADHD symptoms and suicidality, while irritability symptoms mediated 36.8% of the total effect. In the teachers’ reports, depression and irritability symptoms accounted for 45.3% and 38.4% of the association. Anxiety symptoms mediated 19% of the relationship between ADHD and suicidality by parental report but had no significant impact on the association according to teacher report in the recently published study.
Dr. Levy noted that, in the DSM-5, irritability cuts across diagnostic categories. It is not only a core dimension of ADHD, but of the other externalizing disorders – conduct disorder and oppositional defiant disorder – as well, and also of neurodevelopmental, internalizing, and stress-related disorders.
Interventional studies aimed at dampening irritability as a potential strategy to reduce suicidality haven’t yet been done, but they deserve research priority status, in Dr. Levy’s view. Numerous functional dimensions that influence irritability are potential targets, including aggression, negative affect, low tolerance of frustration, skewed threat perception, and impaired self-regulation, according to the psychiatrist.
Most suicidal youths are attempting to cope with mental disorders. The most prevalent of these are major depressive disorder and dysthymia, followed by externalizing disorders. And among the externalizing disorders, conduct disorder stands out in terms of the magnitude of associated suicidality risk. In a large Taiwanese national study including 3,711 adolescents with conduct disorder and 14,844 age- and sex-matched controls, conduct disorder was associated with an adjusted 5.17-fold increased risk of subsequent suicide attempts over the next 10 years in a multivariate regression analysis adjusted for other psychiatric comorbidities and demographics.
In addition to depression, irritability symptoms, and conduct problems, other risk factors that should be part of a suicidality assessment in children and adolescents with ADHD include substance use, anxiety, poor family support, and bullying and/or being bullied. But, perhaps surprisingly, not impulsivity, Dr. Levy said.
“There is a widely held perception that impulsivity imparts a risk for suicidality, and especially in the transition from ideation to attempt. However, more recent evidence fails to show a convincing association,” according to Dr. Levy.
He reported having no financial conflicts regarding his presentation.
SOURCE: Levy T. ECNP 2020, Session EDU.02.
Irritability appears to be a potent independent predictor of increased risk for suicidality in children and adolescents with ADHD, Tomer Levy, MD, said at the virtual congress of the European College of Neuropsychopharmacology.
While there is ample evidence that ADHD is associated with increased suicidality, Dr. Levy’s recent study involving 1,516 youths aged 6-17 years attending an outpatient ADHD clinic demonstrated that this increased risk is mediated by depression and irritability in roughly equal measures. Moreover, upon controlling for those two factors in a multivariate analysis, ADHD symptoms, per se, had no direct effect on risk of suicidality as defined by suidical ideation, attempts, or self-harm.
The clinical take-home message is that assessing irritability, as well as depression, may bolster an estimate of suicidality and help in managing suicidal risk in ADHD, according to Dr. Levy, a child and adolescent psychiatrist at the Hospital for Sick Children, Toronto, and head of behavioral regulation services at the Geha Mental Health Center in Petah Tikva, Israel.
The study included separate parent- and teacher-structured reports of the youths’ ADHD symptoms, suicidality, depression, irritability, and anxiety.
In multivariate analyses, parent-reported depression accounted for 39.1% of the association between ADHD symptoms and suicidality, while irritability symptoms mediated 36.8% of the total effect. In the teachers’ reports, depression and irritability symptoms accounted for 45.3% and 38.4% of the association. Anxiety symptoms mediated 19% of the relationship between ADHD and suicidality by parental report but had no significant impact on the association according to teacher report in the recently published study.
Dr. Levy noted that, in the DSM-5, irritability cuts across diagnostic categories. It is not only a core dimension of ADHD, but of the other externalizing disorders – conduct disorder and oppositional defiant disorder – as well, and also of neurodevelopmental, internalizing, and stress-related disorders.
Interventional studies aimed at dampening irritability as a potential strategy to reduce suicidality haven’t yet been done, but they deserve research priority status, in Dr. Levy’s view. Numerous functional dimensions that influence irritability are potential targets, including aggression, negative affect, low tolerance of frustration, skewed threat perception, and impaired self-regulation, according to the psychiatrist.
Most suicidal youths are attempting to cope with mental disorders. The most prevalent of these are major depressive disorder and dysthymia, followed by externalizing disorders. And among the externalizing disorders, conduct disorder stands out in terms of the magnitude of associated suicidality risk. In a large Taiwanese national study including 3,711 adolescents with conduct disorder and 14,844 age- and sex-matched controls, conduct disorder was associated with an adjusted 5.17-fold increased risk of subsequent suicide attempts over the next 10 years in a multivariate regression analysis adjusted for other psychiatric comorbidities and demographics.
In addition to depression, irritability symptoms, and conduct problems, other risk factors that should be part of a suicidality assessment in children and adolescents with ADHD include substance use, anxiety, poor family support, and bullying and/or being bullied. But, perhaps surprisingly, not impulsivity, Dr. Levy said.
“There is a widely held perception that impulsivity imparts a risk for suicidality, and especially in the transition from ideation to attempt. However, more recent evidence fails to show a convincing association,” according to Dr. Levy.
He reported having no financial conflicts regarding his presentation.
SOURCE: Levy T. ECNP 2020, Session EDU.02.
Irritability appears to be a potent independent predictor of increased risk for suicidality in children and adolescents with ADHD, Tomer Levy, MD, said at the virtual congress of the European College of Neuropsychopharmacology.
While there is ample evidence that ADHD is associated with increased suicidality, Dr. Levy’s recent study involving 1,516 youths aged 6-17 years attending an outpatient ADHD clinic demonstrated that this increased risk is mediated by depression and irritability in roughly equal measures. Moreover, upon controlling for those two factors in a multivariate analysis, ADHD symptoms, per se, had no direct effect on risk of suicidality as defined by suidical ideation, attempts, or self-harm.
The clinical take-home message is that assessing irritability, as well as depression, may bolster an estimate of suicidality and help in managing suicidal risk in ADHD, according to Dr. Levy, a child and adolescent psychiatrist at the Hospital for Sick Children, Toronto, and head of behavioral regulation services at the Geha Mental Health Center in Petah Tikva, Israel.
The study included separate parent- and teacher-structured reports of the youths’ ADHD symptoms, suicidality, depression, irritability, and anxiety.
In multivariate analyses, parent-reported depression accounted for 39.1% of the association between ADHD symptoms and suicidality, while irritability symptoms mediated 36.8% of the total effect. In the teachers’ reports, depression and irritability symptoms accounted for 45.3% and 38.4% of the association. Anxiety symptoms mediated 19% of the relationship between ADHD and suicidality by parental report but had no significant impact on the association according to teacher report in the recently published study.
Dr. Levy noted that, in the DSM-5, irritability cuts across diagnostic categories. It is not only a core dimension of ADHD, but of the other externalizing disorders – conduct disorder and oppositional defiant disorder – as well, and also of neurodevelopmental, internalizing, and stress-related disorders.
Interventional studies aimed at dampening irritability as a potential strategy to reduce suicidality haven’t yet been done, but they deserve research priority status, in Dr. Levy’s view. Numerous functional dimensions that influence irritability are potential targets, including aggression, negative affect, low tolerance of frustration, skewed threat perception, and impaired self-regulation, according to the psychiatrist.
Most suicidal youths are attempting to cope with mental disorders. The most prevalent of these are major depressive disorder and dysthymia, followed by externalizing disorders. And among the externalizing disorders, conduct disorder stands out in terms of the magnitude of associated suicidality risk. In a large Taiwanese national study including 3,711 adolescents with conduct disorder and 14,844 age- and sex-matched controls, conduct disorder was associated with an adjusted 5.17-fold increased risk of subsequent suicide attempts over the next 10 years in a multivariate regression analysis adjusted for other psychiatric comorbidities and demographics.
In addition to depression, irritability symptoms, and conduct problems, other risk factors that should be part of a suicidality assessment in children and adolescents with ADHD include substance use, anxiety, poor family support, and bullying and/or being bullied. But, perhaps surprisingly, not impulsivity, Dr. Levy said.
“There is a widely held perception that impulsivity imparts a risk for suicidality, and especially in the transition from ideation to attempt. However, more recent evidence fails to show a convincing association,” according to Dr. Levy.
He reported having no financial conflicts regarding his presentation.
SOURCE: Levy T. ECNP 2020, Session EDU.02.
FROM ECNP 2020
Key clinical point: Assessment of irritability symptoms and depression may be helpful in managing suicidality risk in ADHD.
Major finding: Parent- and teacher-reported depression and irritability symptoms mediated up to 84% of the association between pediatric ADHD and suicidality.
Study details: This cross-sectional study examined the role of irritability, depression, and anxiety in suicidality among 1,516 children and adolescents at an outpatient ADHD clinic.
Disclosures: The presenter reported having no financial conflicts regarding his study.
Source: Levy T. ECNP 2020, Session EDU.02.
COVID-19 transforms medical education: No ‘back to normal’
The COVID-19 pandemic has thrown a monkey wrench into the medical education landscape across the entire health care spectrum, disrupting the plans of medical students, residents, fellows, and program directors.
As cases of COVID-19 spread across the United States in early 2020, it became clear to training program directors that immediate action was required to meet the needs of medical learners. The challenges were unlike those surrounding the Ebola virus in 2014, “where we could more easily prevent students and trainees from exposure due to the fact that there were simply not significant numbers of cases in the United States,” Tiffany Murano, MD, said at a Society for Critical Care virtual meeting: COVID-19: What’s Next. Dr. Murano is professor of emergency medicine at Rutgers New Jersey Medical School, Newark, and president-elect of the Council of Residency Directors in Emergency Medicine. “COVID was a completely different scenario. We quickly realized that not only was personal protective equipment in short supply, but we also lacked the testing and tracking capabilities for potential exposures. Medical students and other supportive workers who were considered nonessential were removed from the clinical setting. This was after a trial of limiting who the students saw, essentially dampening the risk of exposure. But this proved to be flawed as COVID patients presented with symptoms that were unexpected.”
To complicate matters, she continued, many medical clinics either shut down, had limited access, or converted to telemedicine. Elective surgeries were canceled. This led to an overall pause in clinical medical student rotations and no direct patient care activities. As social distancing mandates were instituted, licensing examination testing centers were closed, and exams and on-campus activities were postponed.
Limiting trainee exposure
On the graduate medical education front, some training programs attempted to limit exposure of their trainees to persons under investigation for COVID-19. “As the number of COVID cases grew and encompassed most of what we were seeing in the hospital, it was obvious that residents had to play a vital part in the care of these patients,” said Dr. Murano, who is also a member of the American Council of Graduate Medical Education’s emergency review and recognition committee. “However, there was a consensus among all of the specialties that the procedures that posed the highest risk of exposure would be limited to the most senior or experienced trainees or professionals, and closely supervised by the faculty.”
ACGME activities such as accreditation site visits, clinical environment learning reviews, self-study, and resident and faculty surveys were suspended, postponed, or modified in some way, she said. The ACGME created stages of COVID status to guide sponsoring institutions to suspend learning curricula in order for patients to be cared for. Stage 1 was business as usual, “so there was no significant impact on patient care,” Dr. Murano said. “Stage 2 was increased but manageable clinical demand, while stage 3 was pandemic emergency status, where there were extraordinary circumstances where the clinical demand was so high and strenuous that the routine patient care and education really needed to be reconfigured in order to care for the patients.”
New requirements to manage training
The ACGME also implemented four requirements to manage training that were consistent among institutions, regardless of their COVID stage status. These included making sure that trainees continued to be held to work-hour limit requirements, ensuring adequate resources for training, ensuring that all residents had the appropriate level of supervision at all times, and allowing fellows to function in the core specialty in which they completed their residency training. “This was only possible if the fellows were ABMS [American Board of Medical Specialties] or AOA [American Osteopathic Association] board-eligible, or certified in their core specialty,” Dr. Murano said. “The fellows had to be appointed to the medical staff at the sponsoring institution, and their time spent on the core specialty service would be limited to 20% of their annual education time in any academic year.”
Mindful that there may have been trainees who required a 2-week quarantine period following exposure or potential exposure to COVID-19, some specialty boards showed leniency in residency time required to sit for the written exam. “Testing centers were being forced to close to observe social distancing requirements and heed sanitation recommendations, so exams were either canceled or postponed,” Dr. Murano said. “This posed a special concern for the board certification process, and those specialties with oral examinations had to make a heavy decision regarding whether or not they would allow these exams to take place. Naturally, travel among institutions was suspended or limited, or had quarantine requirements upon returning home from endemic areas. Conferences were either being canceled or converted to virtual formats.”
Subani Chandra, MD, FCCP, of the division of pulmonary, allergy, and critical care medicine at Columbia University, New York, is the internal medicine residency program director and the associate vice-chair of education for the department of medicine, and she recognized the problem created for medical trainees by the changes necessitated by the pandemic.
“The variability in caseloads and clinical exposure has given thrust to the move toward competency-based assessments rather than number- or time-based criteria for determining proficiency and graduation,” she wrote in an email interview. In addition, she noted the impact on medical meetings and the need to adapt. “Early on, before large regional and national conferences adapted to a virtual format, many were canceled altogether. Students, residents, and fellows expecting to have the opportunity to present their scholarly work were suddenly no longer able to do so. Understanding the importance of scholarly interaction, the virtual format of CHEST 2020 is designed with opportunities to present, interact with experts in the field, ask questions, network, and meet mentors.”
No return to ‘normal’
By April 2020, cases in the northeast continued to rise, particularly in the New York, New Jersey, and Connecticut region. “These states were essentially shut down in order to contain spread of the virus,” she said. “This was a real turning point because we realized that things were not going to return to ‘normal’ in the foreseeable future.” With the clinical experience essentially halted for medical students during this time, some medical schools allowed their senior students who met requirements to graduate early. “There were a lot of mixed feelings about this, recognizing that PPE [personal protective equipment] was still in short supply in many areas,” Dr. Murano said. “So, institutions took on these early graduates into roles in which they were not learners in particular, but rather medical workers. They were helping with informatics and technology, telehealth, virtual or telephone call follow-ups, and other tasks like this. There was a movement to virtual learning for the preclinical undergraduate learners, so classes were now online, recorded, or livestreamed.”
Early graduation, matching, and residencies
On April 3, the ACGME released a statement regarding graduating students early and appointing them early to the clinical learning environment. “They pointed out that institutions that were in emergency pandemic status lacked the ability to offer the comprehensive orientation and training in PPE and direct supervision required for new residents at the start of their residency,” Dr. Murano said. “Their opinion maintained that graduating medical students matriculate in their previously matched program, the National Resident Match Program start date, or other date that would be nationally determined to be the beginning of the 2020-2021 academic year.”
As May 2020 rolled around, the overriding feeling was uncertainty regarding when, if, and how medical schools were going to open in the early summer and fall. “There was also uncertainty about how graduating medical students were going to function in their new role as residents,” she said. “Same for the graduating residents. There were some who had signed contracts for jobs months before, and had them rescinded, and physicians were being furloughed due to financial hardships that institutions faced. There was also postponement of board certification exams, so people were uncertain about when they would become board certified.”
July 2020 ushered in what Dr. Murano characterized as “a whole new level of stress.” For medical students in particular, “we were entering the application season for residency positions,” she said. “Due to travel restrictions placed by various states and institutions, away rotations were limited or nonexistent. Application release dates through the Electronic Residency Application Service were moved to later in the year. The United States Medical Licensing Examination clinical skills exam was suspended, and there were modifications made for Education Commission for Foreign Medical Graduates requirements. Letters of recommendation were also going to be limited, so there had to be some degree of leniency within specialties to take a more holistic approach to review of applications for residencies.”
On the graduate medical education front, the ACGME sunsetted the initial stages and created two categories: nonemergency, which was formerly stages 1 and 2, and emergency, which was formerly stage 3. “All emergency stages are applied for and granted at 1-month intervals,” Dr. Murano said. Board certification exams were modified to accommodate either later exams or online formats, and specialties with oral examinations faced the task of potentially creating virtual oral exams.
Despite the challenges, Dr. Chandra has seen medical training programs respond with new ideas. “The flexibility and agile adaptability of the entire educational enterprise has been remarkable. The inherent uncertainty in a very dynamic and changing learning environment can be challenging. Recognizing this, many programs are creating additional ways to support the mental, emotional, physical, and financial health of students, residents, and fellows and all health care workers. The importance of this innovative response cannot be overstated.”
New learning formats
The pandemic forced Dr. Murano and other medical educators to consider unorthodox learning formats, and virtual learning took center stage. “Residency programs had shared national livestream conferences and grand rounds, and there were virtual curricula made for medical students as well as virtual simulation,” she said. “Telemedicine and telehealth really became important parts of education as well, as this may have been the only face-to-face contact that students and residents had with patients who had non–COVID-related complaints.”
To level the playing field for medical residents during this unprecedented time, a work group of the Coalition for Physician Accountability developed a set of recommendations that include limiting the number of letters of recommendation accepted, limiting the number of away rotations, and allowing alternative or less conventional letters of recommendation. “Keeping an open mind and taking a more holistic approach to applicants has really been needed during this time,” Dr. Murano said. “Virtual interview days have been agreed upon for all specialties. They’re safer, and they allow for students to virtually meet faculty and residents from distant programs that in the past would have been a deterrent due to distance and travel costs. This is not without its own downside, as it’s difficult to determine how well a student will fit into a program without [him or her] actually visiting the institution.”
Dr. Chandra agreed that virtual interviews are necessary but have inherent limitations. However, “we will all learn a lot, and very likely the future process will blend the benefits of both virtual and in-person interviews.”
‘We need to keep moving forward’
Dr. Murano concluded her presentation by noting that the COVID-19 pandemic has created opportunities for growth and innovation in medical education, “so we need to keep moving forward. I’ve heard many say that they can’t wait for things to go back to normal. But I think it’s important to go ahead to new and better ways of learning. We’re now thinking outside of the typical education model and are embracing technology and alternative means of education. We don’t know yet if this education is better, worse, or equivalent to traditional methods, but that will be determined and studied in months and years to come, so we’re certainly looking to the future.”
Dr. Murano and Dr. Chandra reported having no financial disclosures.
The COVID-19 pandemic has thrown a monkey wrench into the medical education landscape across the entire health care spectrum, disrupting the plans of medical students, residents, fellows, and program directors.
As cases of COVID-19 spread across the United States in early 2020, it became clear to training program directors that immediate action was required to meet the needs of medical learners. The challenges were unlike those surrounding the Ebola virus in 2014, “where we could more easily prevent students and trainees from exposure due to the fact that there were simply not significant numbers of cases in the United States,” Tiffany Murano, MD, said at a Society for Critical Care virtual meeting: COVID-19: What’s Next. Dr. Murano is professor of emergency medicine at Rutgers New Jersey Medical School, Newark, and president-elect of the Council of Residency Directors in Emergency Medicine. “COVID was a completely different scenario. We quickly realized that not only was personal protective equipment in short supply, but we also lacked the testing and tracking capabilities for potential exposures. Medical students and other supportive workers who were considered nonessential were removed from the clinical setting. This was after a trial of limiting who the students saw, essentially dampening the risk of exposure. But this proved to be flawed as COVID patients presented with symptoms that were unexpected.”
To complicate matters, she continued, many medical clinics either shut down, had limited access, or converted to telemedicine. Elective surgeries were canceled. This led to an overall pause in clinical medical student rotations and no direct patient care activities. As social distancing mandates were instituted, licensing examination testing centers were closed, and exams and on-campus activities were postponed.
Limiting trainee exposure
On the graduate medical education front, some training programs attempted to limit exposure of their trainees to persons under investigation for COVID-19. “As the number of COVID cases grew and encompassed most of what we were seeing in the hospital, it was obvious that residents had to play a vital part in the care of these patients,” said Dr. Murano, who is also a member of the American Council of Graduate Medical Education’s emergency review and recognition committee. “However, there was a consensus among all of the specialties that the procedures that posed the highest risk of exposure would be limited to the most senior or experienced trainees or professionals, and closely supervised by the faculty.”
ACGME activities such as accreditation site visits, clinical environment learning reviews, self-study, and resident and faculty surveys were suspended, postponed, or modified in some way, she said. The ACGME created stages of COVID status to guide sponsoring institutions to suspend learning curricula in order for patients to be cared for. Stage 1 was business as usual, “so there was no significant impact on patient care,” Dr. Murano said. “Stage 2 was increased but manageable clinical demand, while stage 3 was pandemic emergency status, where there were extraordinary circumstances where the clinical demand was so high and strenuous that the routine patient care and education really needed to be reconfigured in order to care for the patients.”
New requirements to manage training
The ACGME also implemented four requirements to manage training that were consistent among institutions, regardless of their COVID stage status. These included making sure that trainees continued to be held to work-hour limit requirements, ensuring adequate resources for training, ensuring that all residents had the appropriate level of supervision at all times, and allowing fellows to function in the core specialty in which they completed their residency training. “This was only possible if the fellows were ABMS [American Board of Medical Specialties] or AOA [American Osteopathic Association] board-eligible, or certified in their core specialty,” Dr. Murano said. “The fellows had to be appointed to the medical staff at the sponsoring institution, and their time spent on the core specialty service would be limited to 20% of their annual education time in any academic year.”
Mindful that there may have been trainees who required a 2-week quarantine period following exposure or potential exposure to COVID-19, some specialty boards showed leniency in residency time required to sit for the written exam. “Testing centers were being forced to close to observe social distancing requirements and heed sanitation recommendations, so exams were either canceled or postponed,” Dr. Murano said. “This posed a special concern for the board certification process, and those specialties with oral examinations had to make a heavy decision regarding whether or not they would allow these exams to take place. Naturally, travel among institutions was suspended or limited, or had quarantine requirements upon returning home from endemic areas. Conferences were either being canceled or converted to virtual formats.”
Subani Chandra, MD, FCCP, of the division of pulmonary, allergy, and critical care medicine at Columbia University, New York, is the internal medicine residency program director and the associate vice-chair of education for the department of medicine, and she recognized the problem created for medical trainees by the changes necessitated by the pandemic.
“The variability in caseloads and clinical exposure has given thrust to the move toward competency-based assessments rather than number- or time-based criteria for determining proficiency and graduation,” she wrote in an email interview. In addition, she noted the impact on medical meetings and the need to adapt. “Early on, before large regional and national conferences adapted to a virtual format, many were canceled altogether. Students, residents, and fellows expecting to have the opportunity to present their scholarly work were suddenly no longer able to do so. Understanding the importance of scholarly interaction, the virtual format of CHEST 2020 is designed with opportunities to present, interact with experts in the field, ask questions, network, and meet mentors.”
No return to ‘normal’
By April 2020, cases in the northeast continued to rise, particularly in the New York, New Jersey, and Connecticut region. “These states were essentially shut down in order to contain spread of the virus,” she said. “This was a real turning point because we realized that things were not going to return to ‘normal’ in the foreseeable future.” With the clinical experience essentially halted for medical students during this time, some medical schools allowed their senior students who met requirements to graduate early. “There were a lot of mixed feelings about this, recognizing that PPE [personal protective equipment] was still in short supply in many areas,” Dr. Murano said. “So, institutions took on these early graduates into roles in which they were not learners in particular, but rather medical workers. They were helping with informatics and technology, telehealth, virtual or telephone call follow-ups, and other tasks like this. There was a movement to virtual learning for the preclinical undergraduate learners, so classes were now online, recorded, or livestreamed.”
Early graduation, matching, and residencies
On April 3, the ACGME released a statement regarding graduating students early and appointing them early to the clinical learning environment. “They pointed out that institutions that were in emergency pandemic status lacked the ability to offer the comprehensive orientation and training in PPE and direct supervision required for new residents at the start of their residency,” Dr. Murano said. “Their opinion maintained that graduating medical students matriculate in their previously matched program, the National Resident Match Program start date, or other date that would be nationally determined to be the beginning of the 2020-2021 academic year.”
As May 2020 rolled around, the overriding feeling was uncertainty regarding when, if, and how medical schools were going to open in the early summer and fall. “There was also uncertainty about how graduating medical students were going to function in their new role as residents,” she said. “Same for the graduating residents. There were some who had signed contracts for jobs months before, and had them rescinded, and physicians were being furloughed due to financial hardships that institutions faced. There was also postponement of board certification exams, so people were uncertain about when they would become board certified.”
July 2020 ushered in what Dr. Murano characterized as “a whole new level of stress.” For medical students in particular, “we were entering the application season for residency positions,” she said. “Due to travel restrictions placed by various states and institutions, away rotations were limited or nonexistent. Application release dates through the Electronic Residency Application Service were moved to later in the year. The United States Medical Licensing Examination clinical skills exam was suspended, and there were modifications made for Education Commission for Foreign Medical Graduates requirements. Letters of recommendation were also going to be limited, so there had to be some degree of leniency within specialties to take a more holistic approach to review of applications for residencies.”
On the graduate medical education front, the ACGME sunsetted the initial stages and created two categories: nonemergency, which was formerly stages 1 and 2, and emergency, which was formerly stage 3. “All emergency stages are applied for and granted at 1-month intervals,” Dr. Murano said. Board certification exams were modified to accommodate either later exams or online formats, and specialties with oral examinations faced the task of potentially creating virtual oral exams.
Despite the challenges, Dr. Chandra has seen medical training programs respond with new ideas. “The flexibility and agile adaptability of the entire educational enterprise has been remarkable. The inherent uncertainty in a very dynamic and changing learning environment can be challenging. Recognizing this, many programs are creating additional ways to support the mental, emotional, physical, and financial health of students, residents, and fellows and all health care workers. The importance of this innovative response cannot be overstated.”
New learning formats
The pandemic forced Dr. Murano and other medical educators to consider unorthodox learning formats, and virtual learning took center stage. “Residency programs had shared national livestream conferences and grand rounds, and there were virtual curricula made for medical students as well as virtual simulation,” she said. “Telemedicine and telehealth really became important parts of education as well, as this may have been the only face-to-face contact that students and residents had with patients who had non–COVID-related complaints.”
To level the playing field for medical residents during this unprecedented time, a work group of the Coalition for Physician Accountability developed a set of recommendations that include limiting the number of letters of recommendation accepted, limiting the number of away rotations, and allowing alternative or less conventional letters of recommendation. “Keeping an open mind and taking a more holistic approach to applicants has really been needed during this time,” Dr. Murano said. “Virtual interview days have been agreed upon for all specialties. They’re safer, and they allow for students to virtually meet faculty and residents from distant programs that in the past would have been a deterrent due to distance and travel costs. This is not without its own downside, as it’s difficult to determine how well a student will fit into a program without [him or her] actually visiting the institution.”
Dr. Chandra agreed that virtual interviews are necessary but have inherent limitations. However, “we will all learn a lot, and very likely the future process will blend the benefits of both virtual and in-person interviews.”
‘We need to keep moving forward’
Dr. Murano concluded her presentation by noting that the COVID-19 pandemic has created opportunities for growth and innovation in medical education, “so we need to keep moving forward. I’ve heard many say that they can’t wait for things to go back to normal. But I think it’s important to go ahead to new and better ways of learning. We’re now thinking outside of the typical education model and are embracing technology and alternative means of education. We don’t know yet if this education is better, worse, or equivalent to traditional methods, but that will be determined and studied in months and years to come, so we’re certainly looking to the future.”
Dr. Murano and Dr. Chandra reported having no financial disclosures.
The COVID-19 pandemic has thrown a monkey wrench into the medical education landscape across the entire health care spectrum, disrupting the plans of medical students, residents, fellows, and program directors.
As cases of COVID-19 spread across the United States in early 2020, it became clear to training program directors that immediate action was required to meet the needs of medical learners. The challenges were unlike those surrounding the Ebola virus in 2014, “where we could more easily prevent students and trainees from exposure due to the fact that there were simply not significant numbers of cases in the United States,” Tiffany Murano, MD, said at a Society for Critical Care virtual meeting: COVID-19: What’s Next. Dr. Murano is professor of emergency medicine at Rutgers New Jersey Medical School, Newark, and president-elect of the Council of Residency Directors in Emergency Medicine. “COVID was a completely different scenario. We quickly realized that not only was personal protective equipment in short supply, but we also lacked the testing and tracking capabilities for potential exposures. Medical students and other supportive workers who were considered nonessential were removed from the clinical setting. This was after a trial of limiting who the students saw, essentially dampening the risk of exposure. But this proved to be flawed as COVID patients presented with symptoms that were unexpected.”
To complicate matters, she continued, many medical clinics either shut down, had limited access, or converted to telemedicine. Elective surgeries were canceled. This led to an overall pause in clinical medical student rotations and no direct patient care activities. As social distancing mandates were instituted, licensing examination testing centers were closed, and exams and on-campus activities were postponed.
Limiting trainee exposure
On the graduate medical education front, some training programs attempted to limit exposure of their trainees to persons under investigation for COVID-19. “As the number of COVID cases grew and encompassed most of what we were seeing in the hospital, it was obvious that residents had to play a vital part in the care of these patients,” said Dr. Murano, who is also a member of the American Council of Graduate Medical Education’s emergency review and recognition committee. “However, there was a consensus among all of the specialties that the procedures that posed the highest risk of exposure would be limited to the most senior or experienced trainees or professionals, and closely supervised by the faculty.”
ACGME activities such as accreditation site visits, clinical environment learning reviews, self-study, and resident and faculty surveys were suspended, postponed, or modified in some way, she said. The ACGME created stages of COVID status to guide sponsoring institutions to suspend learning curricula in order for patients to be cared for. Stage 1 was business as usual, “so there was no significant impact on patient care,” Dr. Murano said. “Stage 2 was increased but manageable clinical demand, while stage 3 was pandemic emergency status, where there were extraordinary circumstances where the clinical demand was so high and strenuous that the routine patient care and education really needed to be reconfigured in order to care for the patients.”
New requirements to manage training
The ACGME also implemented four requirements to manage training that were consistent among institutions, regardless of their COVID stage status. These included making sure that trainees continued to be held to work-hour limit requirements, ensuring adequate resources for training, ensuring that all residents had the appropriate level of supervision at all times, and allowing fellows to function in the core specialty in which they completed their residency training. “This was only possible if the fellows were ABMS [American Board of Medical Specialties] or AOA [American Osteopathic Association] board-eligible, or certified in their core specialty,” Dr. Murano said. “The fellows had to be appointed to the medical staff at the sponsoring institution, and their time spent on the core specialty service would be limited to 20% of their annual education time in any academic year.”
Mindful that there may have been trainees who required a 2-week quarantine period following exposure or potential exposure to COVID-19, some specialty boards showed leniency in residency time required to sit for the written exam. “Testing centers were being forced to close to observe social distancing requirements and heed sanitation recommendations, so exams were either canceled or postponed,” Dr. Murano said. “This posed a special concern for the board certification process, and those specialties with oral examinations had to make a heavy decision regarding whether or not they would allow these exams to take place. Naturally, travel among institutions was suspended or limited, or had quarantine requirements upon returning home from endemic areas. Conferences were either being canceled or converted to virtual formats.”
Subani Chandra, MD, FCCP, of the division of pulmonary, allergy, and critical care medicine at Columbia University, New York, is the internal medicine residency program director and the associate vice-chair of education for the department of medicine, and she recognized the problem created for medical trainees by the changes necessitated by the pandemic.
“The variability in caseloads and clinical exposure has given thrust to the move toward competency-based assessments rather than number- or time-based criteria for determining proficiency and graduation,” she wrote in an email interview. In addition, she noted the impact on medical meetings and the need to adapt. “Early on, before large regional and national conferences adapted to a virtual format, many were canceled altogether. Students, residents, and fellows expecting to have the opportunity to present their scholarly work were suddenly no longer able to do so. Understanding the importance of scholarly interaction, the virtual format of CHEST 2020 is designed with opportunities to present, interact with experts in the field, ask questions, network, and meet mentors.”
No return to ‘normal’
By April 2020, cases in the northeast continued to rise, particularly in the New York, New Jersey, and Connecticut region. “These states were essentially shut down in order to contain spread of the virus,” she said. “This was a real turning point because we realized that things were not going to return to ‘normal’ in the foreseeable future.” With the clinical experience essentially halted for medical students during this time, some medical schools allowed their senior students who met requirements to graduate early. “There were a lot of mixed feelings about this, recognizing that PPE [personal protective equipment] was still in short supply in many areas,” Dr. Murano said. “So, institutions took on these early graduates into roles in which they were not learners in particular, but rather medical workers. They were helping with informatics and technology, telehealth, virtual or telephone call follow-ups, and other tasks like this. There was a movement to virtual learning for the preclinical undergraduate learners, so classes were now online, recorded, or livestreamed.”
Early graduation, matching, and residencies
On April 3, the ACGME released a statement regarding graduating students early and appointing them early to the clinical learning environment. “They pointed out that institutions that were in emergency pandemic status lacked the ability to offer the comprehensive orientation and training in PPE and direct supervision required for new residents at the start of their residency,” Dr. Murano said. “Their opinion maintained that graduating medical students matriculate in their previously matched program, the National Resident Match Program start date, or other date that would be nationally determined to be the beginning of the 2020-2021 academic year.”
As May 2020 rolled around, the overriding feeling was uncertainty regarding when, if, and how medical schools were going to open in the early summer and fall. “There was also uncertainty about how graduating medical students were going to function in their new role as residents,” she said. “Same for the graduating residents. There were some who had signed contracts for jobs months before, and had them rescinded, and physicians were being furloughed due to financial hardships that institutions faced. There was also postponement of board certification exams, so people were uncertain about when they would become board certified.”
July 2020 ushered in what Dr. Murano characterized as “a whole new level of stress.” For medical students in particular, “we were entering the application season for residency positions,” she said. “Due to travel restrictions placed by various states and institutions, away rotations were limited or nonexistent. Application release dates through the Electronic Residency Application Service were moved to later in the year. The United States Medical Licensing Examination clinical skills exam was suspended, and there were modifications made for Education Commission for Foreign Medical Graduates requirements. Letters of recommendation were also going to be limited, so there had to be some degree of leniency within specialties to take a more holistic approach to review of applications for residencies.”
On the graduate medical education front, the ACGME sunsetted the initial stages and created two categories: nonemergency, which was formerly stages 1 and 2, and emergency, which was formerly stage 3. “All emergency stages are applied for and granted at 1-month intervals,” Dr. Murano said. Board certification exams were modified to accommodate either later exams or online formats, and specialties with oral examinations faced the task of potentially creating virtual oral exams.
Despite the challenges, Dr. Chandra has seen medical training programs respond with new ideas. “The flexibility and agile adaptability of the entire educational enterprise has been remarkable. The inherent uncertainty in a very dynamic and changing learning environment can be challenging. Recognizing this, many programs are creating additional ways to support the mental, emotional, physical, and financial health of students, residents, and fellows and all health care workers. The importance of this innovative response cannot be overstated.”
New learning formats
The pandemic forced Dr. Murano and other medical educators to consider unorthodox learning formats, and virtual learning took center stage. “Residency programs had shared national livestream conferences and grand rounds, and there were virtual curricula made for medical students as well as virtual simulation,” she said. “Telemedicine and telehealth really became important parts of education as well, as this may have been the only face-to-face contact that students and residents had with patients who had non–COVID-related complaints.”
To level the playing field for medical residents during this unprecedented time, a work group of the Coalition for Physician Accountability developed a set of recommendations that include limiting the number of letters of recommendation accepted, limiting the number of away rotations, and allowing alternative or less conventional letters of recommendation. “Keeping an open mind and taking a more holistic approach to applicants has really been needed during this time,” Dr. Murano said. “Virtual interview days have been agreed upon for all specialties. They’re safer, and they allow for students to virtually meet faculty and residents from distant programs that in the past would have been a deterrent due to distance and travel costs. This is not without its own downside, as it’s difficult to determine how well a student will fit into a program without [him or her] actually visiting the institution.”
Dr. Chandra agreed that virtual interviews are necessary but have inherent limitations. However, “we will all learn a lot, and very likely the future process will blend the benefits of both virtual and in-person interviews.”
‘We need to keep moving forward’
Dr. Murano concluded her presentation by noting that the COVID-19 pandemic has created opportunities for growth and innovation in medical education, “so we need to keep moving forward. I’ve heard many say that they can’t wait for things to go back to normal. But I think it’s important to go ahead to new and better ways of learning. We’re now thinking outside of the typical education model and are embracing technology and alternative means of education. We don’t know yet if this education is better, worse, or equivalent to traditional methods, but that will be determined and studied in months and years to come, so we’re certainly looking to the future.”
Dr. Murano and Dr. Chandra reported having no financial disclosures.
FROM AN SCCM VIRTUAL MEETING
Irritable Baby With Weight Loss and a Periorificial and Truncal Rash
The Diagnosis: Acrodermatitis Enteropathica
Acrodermatitis enteropathica (AE) was the presumptive diagnosis. Oral supplementation with zinc sulfate 3 mg/kg/d was started immediately after a zinc level was ordered. A low zinc level of 15 µg/dL (reference range, 56-134 µg/dL) eventually was obtained. The lesions began to fade in 2 days along with return of normal feeding and disposition, and the patient was discharged with continued zinc supplementation.
Acrodermatitis enteropathica is an autosomal-recessive condition resulting in severe zinc deficiency caused by a defect of dietary zinc absorption in the duodenum and jejunum.1 It occurs in 1 in 500,000 individuals with no gender or racial predilection. It can be acquired or inherited.2 Recognition of clinical symptoms is essential due to potential death if untreated. Zinc is an important trace element required for the proper functioning of all cells and plays a large role in the metabolism of protein, carbohydrates, and vitamin A. Zinc deficiency impairs immune function, leading to bacterial infections. It also is a cofactor of numerous metal enzymes such as alkaline phosphatase, RNA polymerase, and numerous digestive enzymes.3
Our laboratory analysis revealed low alkaline phosphatase and zinc levels, which led to the diagnosis of AE; unfortunately, these levels can be ambiguous.4 There are many causes of acquired zinc deficiency, including premature birth, low birth weight, zinc deficiency in maternal milk, exclusive parenteral nutrition, malabsorption syndromes such as Crohn disease and celiac disease, alcoholism, low calcium and phytate (cereal grain) diet, and kwashiorkor.5 The hereditary deficiency of zinc classically is known as AE and is caused by an autosomal-recessive mutation of the SLC39A4 gene on chromosome arm 8q24.3, which determines a congenital partial or total deficiency of the zinc transporter protein ZIP4.6
The clinical manifestations of acquired zinc deficiency and AE are similar and consist of 3 essential symptoms: periorificial dermatitis, alopecia, and diarrhea. Unfortunately, this clinical triad is complete in only 20% of patients with AE.3 For example, our patient was too young for an alopecia determination. The disease typically presents with eczematous papules and sometimes vesiculobullous or pustular lesions located around perioral and acral areas (Figure 1) as well as the anogenital region (Figures 2 and 3). The severity of the skin lesions is variable.7 Our patient also presented with eczematous truncal papules on the chest (Figure 4). Acrodermatitis enteropathica usually presents during childhood after weaning. Along with the aforementioned skin findings, other symptoms in infancy can include diarrhea, mood changes, and anorexia. In school-aged children and toddlers, zinc deficiency is characterized by growth retardation, alopecia, weight loss, and recurrent infections.
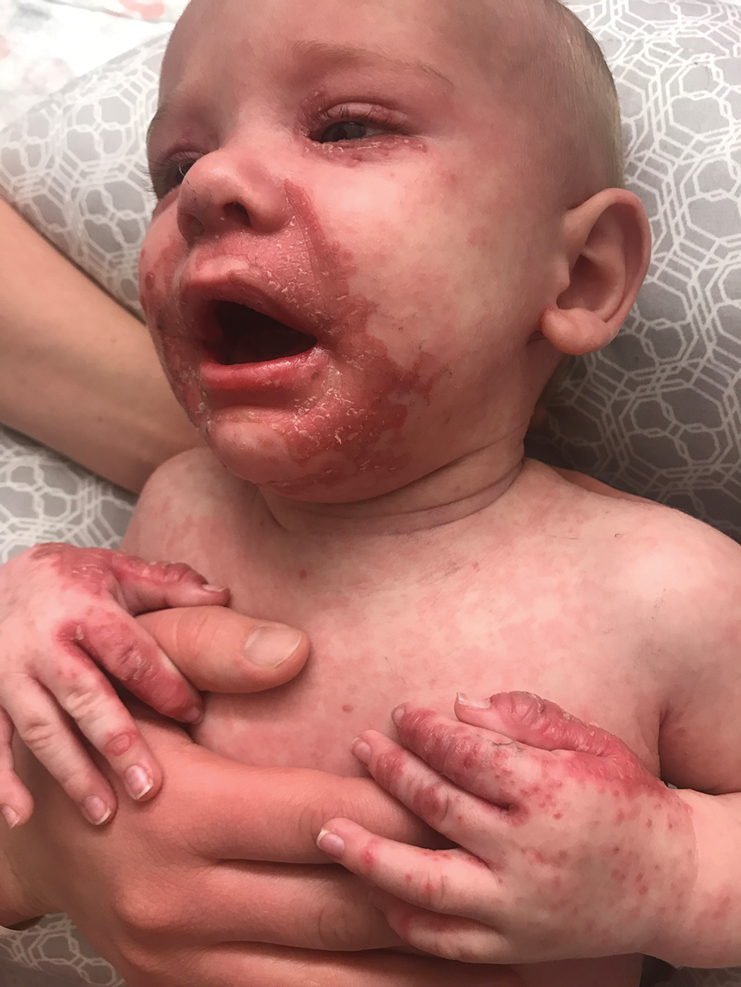

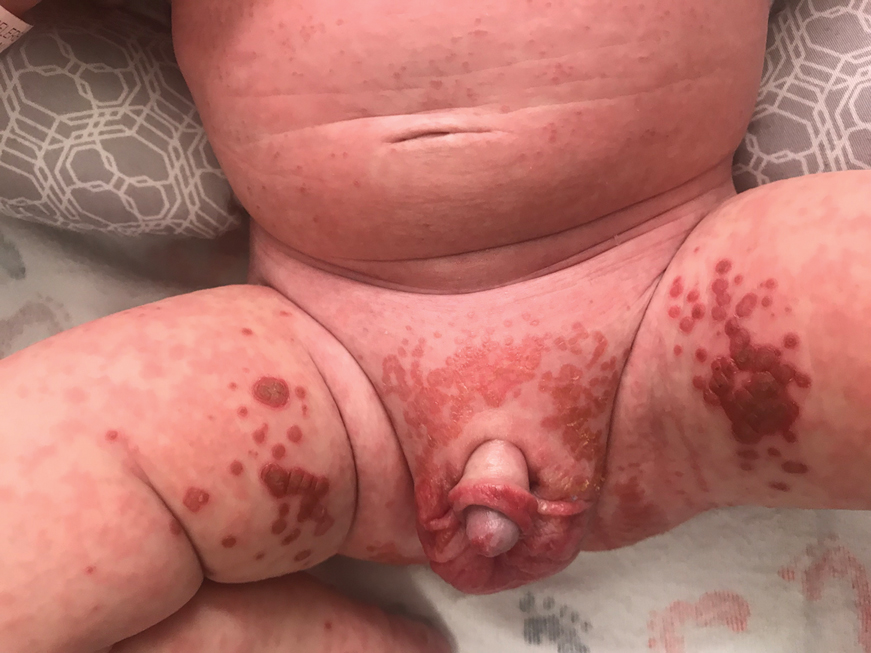
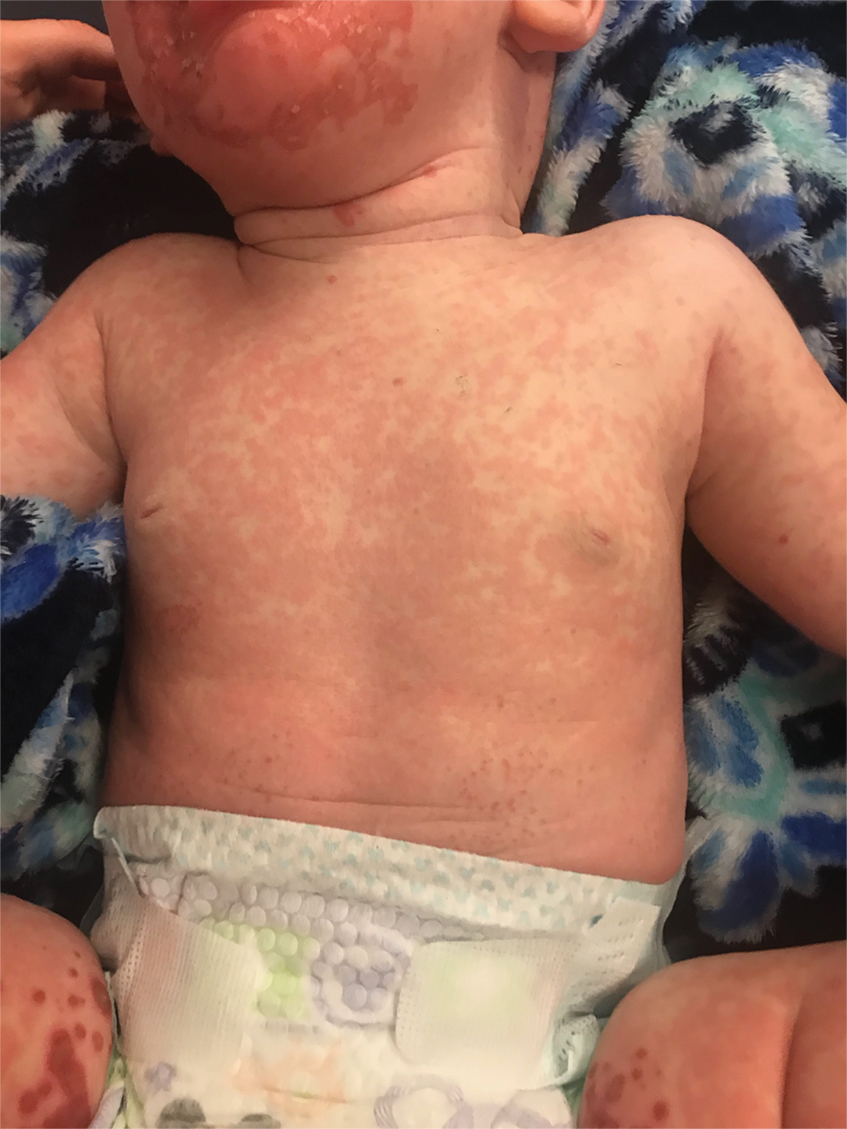
In the differential diagnosis, the clinical presentation of biotin deficiency involves abnormalities of the hair, skin, nails, and central nervous system (eg, seizures, ataxia, deafness).8 Cystic fibrosis presentation depends on the multiorgan involvement, but neonates often present with failure to thrive.9 Essential fatty acid deficiency presents clinically as dermatitis, alopecia, and thrombocytopenia, but a complete blood cell count with platelets was within reference range in our patient.10 Langerhans cell histiocytosis presents with perineal and postauricular lesions, but the skin biopsy did not confirm this diagnosis in our patient.11 Histopathologic examination of the buttock biopsy in our patient revealed nonspecific epidermal hyperplasia with acanthosis as well as clustered necrotic keratinocytes with vacuolization and parakeratosis.
Most clinicians who suspect AE treat with a therapeutic supplementation of zinc sulfate 3 mg/kg/d while awaiting laboratory results. Acrodermatitis enteropathica is a rare condition, and early recognition of skin findings is important because misdiagnosis can lead to infections, malnutrition, and possibly death.
- Sehgal VN, Jain S. Acrodermatitis enteropathica. Clin Dermatol. 2000;18:745-748.
- Van Wouwe JP. Clinical and laboratory assessment of zinc deficiency in Dutch children: a review. Biol Trace Elem Res. 1995;49:211-225.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Van Wouwe JP. Clinical and laboratory diagnosis of acrodermatitis enteropathica. Eur J Pediatr. 1989;149:2-8.
- Perafán-Riveros C, França LF, Alves AC, et al. Acrodermatitisenteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Kury S, Dréno B, Bézieau S, et al. Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet. 2002;31:239-240.
- Nistor N, Ciontu L, Frasinariu OE, et al. Acrodermatitis enteropathica: a case report. Medicine. 2016;95:E3553.
- Gratias T. Biotin deficiency. Medscape website. https://emedicine.medscape.com/article/984803-overview. Updated October 22, 2018. Accessed October 15, 2020.
- Sharma G. Cystic fibrosis. Medscape website. https://emedicine.medscape.com/article/1001602-overview. Updated September 28, 2018. Accessed October 15, 2020.
- Morley JE. Essential fatty acid deficiency. Merck Manual website. https://www.merckmanuals.com/professional/nutritional-disorders/undernutrition/essential-fatty-acid-deficiency. Updated January 2020. Accessed October 15, 2020.
- Shea CR. Langerhans cell histiocytosis. Medscape website. https://emedicine.medscape.com/article/1100579-overview. Updated June 12, 2020. Accessed October 15, 2020.
The Diagnosis: Acrodermatitis Enteropathica
Acrodermatitis enteropathica (AE) was the presumptive diagnosis. Oral supplementation with zinc sulfate 3 mg/kg/d was started immediately after a zinc level was ordered. A low zinc level of 15 µg/dL (reference range, 56-134 µg/dL) eventually was obtained. The lesions began to fade in 2 days along with return of normal feeding and disposition, and the patient was discharged with continued zinc supplementation.
Acrodermatitis enteropathica is an autosomal-recessive condition resulting in severe zinc deficiency caused by a defect of dietary zinc absorption in the duodenum and jejunum.1 It occurs in 1 in 500,000 individuals with no gender or racial predilection. It can be acquired or inherited.2 Recognition of clinical symptoms is essential due to potential death if untreated. Zinc is an important trace element required for the proper functioning of all cells and plays a large role in the metabolism of protein, carbohydrates, and vitamin A. Zinc deficiency impairs immune function, leading to bacterial infections. It also is a cofactor of numerous metal enzymes such as alkaline phosphatase, RNA polymerase, and numerous digestive enzymes.3
Our laboratory analysis revealed low alkaline phosphatase and zinc levels, which led to the diagnosis of AE; unfortunately, these levels can be ambiguous.4 There are many causes of acquired zinc deficiency, including premature birth, low birth weight, zinc deficiency in maternal milk, exclusive parenteral nutrition, malabsorption syndromes such as Crohn disease and celiac disease, alcoholism, low calcium and phytate (cereal grain) diet, and kwashiorkor.5 The hereditary deficiency of zinc classically is known as AE and is caused by an autosomal-recessive mutation of the SLC39A4 gene on chromosome arm 8q24.3, which determines a congenital partial or total deficiency of the zinc transporter protein ZIP4.6
The clinical manifestations of acquired zinc deficiency and AE are similar and consist of 3 essential symptoms: periorificial dermatitis, alopecia, and diarrhea. Unfortunately, this clinical triad is complete in only 20% of patients with AE.3 For example, our patient was too young for an alopecia determination. The disease typically presents with eczematous papules and sometimes vesiculobullous or pustular lesions located around perioral and acral areas (Figure 1) as well as the anogenital region (Figures 2 and 3). The severity of the skin lesions is variable.7 Our patient also presented with eczematous truncal papules on the chest (Figure 4). Acrodermatitis enteropathica usually presents during childhood after weaning. Along with the aforementioned skin findings, other symptoms in infancy can include diarrhea, mood changes, and anorexia. In school-aged children and toddlers, zinc deficiency is characterized by growth retardation, alopecia, weight loss, and recurrent infections.
In the differential diagnosis, the clinical presentation of biotin deficiency involves abnormalities of the hair, skin, nails, and central nervous system (eg, seizures, ataxia, deafness).8 Cystic fibrosis presentation depends on the multiorgan involvement, but neonates often present with failure to thrive.9 Essential fatty acid deficiency presents clinically as dermatitis, alopecia, and thrombocytopenia, but a complete blood cell count with platelets was within reference range in our patient.10 Langerhans cell histiocytosis presents with perineal and postauricular lesions, but the skin biopsy did not confirm this diagnosis in our patient.11 Histopathologic examination of the buttock biopsy in our patient revealed nonspecific epidermal hyperplasia with acanthosis as well as clustered necrotic keratinocytes with vacuolization and parakeratosis.
Most clinicians who suspect AE treat with a therapeutic supplementation of zinc sulfate 3 mg/kg/d while awaiting laboratory results. Acrodermatitis enteropathica is a rare condition, and early recognition of skin findings is important because misdiagnosis can lead to infections, malnutrition, and possibly death.
The Diagnosis: Acrodermatitis Enteropathica
Acrodermatitis enteropathica (AE) was the presumptive diagnosis. Oral supplementation with zinc sulfate 3 mg/kg/d was started immediately after a zinc level was ordered. A low zinc level of 15 µg/dL (reference range, 56-134 µg/dL) eventually was obtained. The lesions began to fade in 2 days along with return of normal feeding and disposition, and the patient was discharged with continued zinc supplementation.
Acrodermatitis enteropathica is an autosomal-recessive condition resulting in severe zinc deficiency caused by a defect of dietary zinc absorption in the duodenum and jejunum.1 It occurs in 1 in 500,000 individuals with no gender or racial predilection. It can be acquired or inherited.2 Recognition of clinical symptoms is essential due to potential death if untreated. Zinc is an important trace element required for the proper functioning of all cells and plays a large role in the metabolism of protein, carbohydrates, and vitamin A. Zinc deficiency impairs immune function, leading to bacterial infections. It also is a cofactor of numerous metal enzymes such as alkaline phosphatase, RNA polymerase, and numerous digestive enzymes.3
Our laboratory analysis revealed low alkaline phosphatase and zinc levels, which led to the diagnosis of AE; unfortunately, these levels can be ambiguous.4 There are many causes of acquired zinc deficiency, including premature birth, low birth weight, zinc deficiency in maternal milk, exclusive parenteral nutrition, malabsorption syndromes such as Crohn disease and celiac disease, alcoholism, low calcium and phytate (cereal grain) diet, and kwashiorkor.5 The hereditary deficiency of zinc classically is known as AE and is caused by an autosomal-recessive mutation of the SLC39A4 gene on chromosome arm 8q24.3, which determines a congenital partial or total deficiency of the zinc transporter protein ZIP4.6
The clinical manifestations of acquired zinc deficiency and AE are similar and consist of 3 essential symptoms: periorificial dermatitis, alopecia, and diarrhea. Unfortunately, this clinical triad is complete in only 20% of patients with AE.3 For example, our patient was too young for an alopecia determination. The disease typically presents with eczematous papules and sometimes vesiculobullous or pustular lesions located around perioral and acral areas (Figure 1) as well as the anogenital region (Figures 2 and 3). The severity of the skin lesions is variable.7 Our patient also presented with eczematous truncal papules on the chest (Figure 4). Acrodermatitis enteropathica usually presents during childhood after weaning. Along with the aforementioned skin findings, other symptoms in infancy can include diarrhea, mood changes, and anorexia. In school-aged children and toddlers, zinc deficiency is characterized by growth retardation, alopecia, weight loss, and recurrent infections.
In the differential diagnosis, the clinical presentation of biotin deficiency involves abnormalities of the hair, skin, nails, and central nervous system (eg, seizures, ataxia, deafness).8 Cystic fibrosis presentation depends on the multiorgan involvement, but neonates often present with failure to thrive.9 Essential fatty acid deficiency presents clinically as dermatitis, alopecia, and thrombocytopenia, but a complete blood cell count with platelets was within reference range in our patient.10 Langerhans cell histiocytosis presents with perineal and postauricular lesions, but the skin biopsy did not confirm this diagnosis in our patient.11 Histopathologic examination of the buttock biopsy in our patient revealed nonspecific epidermal hyperplasia with acanthosis as well as clustered necrotic keratinocytes with vacuolization and parakeratosis.
Most clinicians who suspect AE treat with a therapeutic supplementation of zinc sulfate 3 mg/kg/d while awaiting laboratory results. Acrodermatitis enteropathica is a rare condition, and early recognition of skin findings is important because misdiagnosis can lead to infections, malnutrition, and possibly death.
- Sehgal VN, Jain S. Acrodermatitis enteropathica. Clin Dermatol. 2000;18:745-748.
- Van Wouwe JP. Clinical and laboratory assessment of zinc deficiency in Dutch children: a review. Biol Trace Elem Res. 1995;49:211-225.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Van Wouwe JP. Clinical and laboratory diagnosis of acrodermatitis enteropathica. Eur J Pediatr. 1989;149:2-8.
- Perafán-Riveros C, França LF, Alves AC, et al. Acrodermatitisenteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Kury S, Dréno B, Bézieau S, et al. Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet. 2002;31:239-240.
- Nistor N, Ciontu L, Frasinariu OE, et al. Acrodermatitis enteropathica: a case report. Medicine. 2016;95:E3553.
- Gratias T. Biotin deficiency. Medscape website. https://emedicine.medscape.com/article/984803-overview. Updated October 22, 2018. Accessed October 15, 2020.
- Sharma G. Cystic fibrosis. Medscape website. https://emedicine.medscape.com/article/1001602-overview. Updated September 28, 2018. Accessed October 15, 2020.
- Morley JE. Essential fatty acid deficiency. Merck Manual website. https://www.merckmanuals.com/professional/nutritional-disorders/undernutrition/essential-fatty-acid-deficiency. Updated January 2020. Accessed October 15, 2020.
- Shea CR. Langerhans cell histiocytosis. Medscape website. https://emedicine.medscape.com/article/1100579-overview. Updated June 12, 2020. Accessed October 15, 2020.
- Sehgal VN, Jain S. Acrodermatitis enteropathica. Clin Dermatol. 2000;18:745-748.
- Van Wouwe JP. Clinical and laboratory assessment of zinc deficiency in Dutch children: a review. Biol Trace Elem Res. 1995;49:211-225.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Van Wouwe JP. Clinical and laboratory diagnosis of acrodermatitis enteropathica. Eur J Pediatr. 1989;149:2-8.
- Perafán-Riveros C, França LF, Alves AC, et al. Acrodermatitisenteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Kury S, Dréno B, Bézieau S, et al. Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet. 2002;31:239-240.
- Nistor N, Ciontu L, Frasinariu OE, et al. Acrodermatitis enteropathica: a case report. Medicine. 2016;95:E3553.
- Gratias T. Biotin deficiency. Medscape website. https://emedicine.medscape.com/article/984803-overview. Updated October 22, 2018. Accessed October 15, 2020.
- Sharma G. Cystic fibrosis. Medscape website. https://emedicine.medscape.com/article/1001602-overview. Updated September 28, 2018. Accessed October 15, 2020.
- Morley JE. Essential fatty acid deficiency. Merck Manual website. https://www.merckmanuals.com/professional/nutritional-disorders/undernutrition/essential-fatty-acid-deficiency. Updated January 2020. Accessed October 15, 2020.
- Shea CR. Langerhans cell histiocytosis. Medscape website. https://emedicine.medscape.com/article/1100579-overview. Updated June 12, 2020. Accessed October 15, 2020.
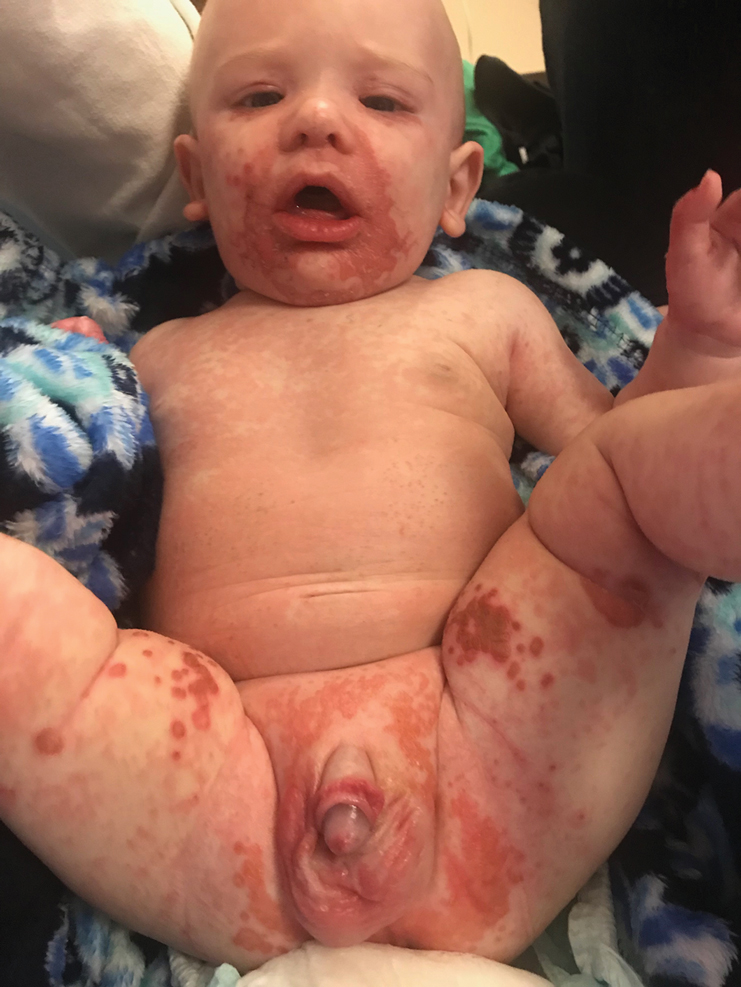
A 4-month-old infant boy presented to the pediatric hospital unit with a rash, fever, and failure to thrive. Prior to admission, the patient was treated for impetigo by a community dermatologist. After not responding to treatment, he was admitted and given intravenous acyclovir for 1 day by the pediatric hospitalist, and the dermatology service was consulted. The parents reported the patient had diarrhea for 1 month and a worsening rash over the last 2 weeks. The mother was breastfeeding. Physical examination revealed a fever (temperature, 38.9°C [102°F]) and an irritable infant whose growth curve had fallen from the 50th to 15th percentile since the 2-month well-baby examination. He had a fine, red, papular truncal rash with confluent plaques in a periorificial distribution that spared the inguinal skin folds, with some vesicles in a herpetiform presentation on the thighs as well as inflammation on the feet and hands. A complete blood cell count was within reference range, but the alkaline phosphatase level was low at 53 U/L (reference range, 72–307 U/L). A herpes simplex virus test was negative. A human immunodeficiency virus test and skin biopsy were performed.
Efforts to close the ‘AYA gap’ in lymphoma
In the 1970s, cancer survival was poor for young children and older adults in the United States, as shown by data published in the Journal of the National Cancer Institute.
Great progress has been made since the 1970s, but improvements in outcome have been less impressive for cancer patients aged 15-39 years, as shown by research published in Cancer.

Patients aged 15-39 years have been designated by the National Institutes of Health (NIH) as “adolescents and young adults (AYAs),” and the lag in survival benefit has been termed “the AYA gap.”
The AYA gap persists in lymphoma patients, and an expert panel recently outlined differences between lymphoma in AYAs and lymphoma in other age groups.
The experts spoke at a special session of the AACR Virtual Meeting: Advances in Malignant Lymphoma moderated by Somali M. Smith, MD, of the University of Chicago.
Factors that contribute to the AYA gap
About 89,000 AYAs are diagnosed with cancer each year in the United States, according to data from the National Cancer Institute (NCI). Lymphomas and thyroid cancer are the most common cancers among younger AYAs, aged 15-24 years.
In a report commissioned by the NIH in 2006, many factors contributing to the AYA gap were identified. Chief among them were:
- Limitations in access to care.
- Delayed diagnosis.
- Inconsistency in treatment and follow-up.
- Long-term toxicity (fertility, second malignancies, and cardiovascular disease).
These factors compromise health-related survival, even when cancer-specific survival is improved.
Panelist Kara Kelly, MD, of Roswell Park Comprehensive Cancer Center in Buffalo, N.Y., noted that there are additional unique challenges for AYAs with cancer. These include:
- Pubertal changes.
- Developmental transition to independence.
- Societal impediments such as insurance coverage and disparities in access to specialized centers.
- Psychosocial factors such as health literacy and adherence to treatment and follow-up.
Focusing on lymphoma specifically, Dr. Kelly noted that lymphoma biology differs across the age spectrum and by race and ethnicity. Both tumor and host factors require further study, she said.
Clinical trial access for AYAs
Dr. Kelly emphasized that, unfortunately, clinical research participation is low among AYAs. A major impediment is that adult clinical trials historically required participants to be at least 18 years old.
In addition, there has not been a focused effort to educate AYAs about regulatory safeguards to ensure safety and the promise of enhanced benefit to them in NCI Cancer Trials Network (NCTN) trials. As a result, the refusal rate is high.
A multi-stakeholder workshop, convened in May 2016 by the American Society of Clinical Oncology and Friends of Cancer Research, outlined opportunities for expanding trial eligibility to include children younger than 18 years in first-in-human and other adult cancer clinical trials, enhancing their access to new agents, without compromising safety.
Recently, collaborative efforts between the adult and children’s NCTN research groups have included AYAs in studies addressing cancers that span the age spectrum, including lymphoma.
However, as Dr. Kelly noted, there are differences in AYA lymphoid malignancy types with a transition from more pediatric to more adult types.
Hodgkin lymphoma and primary mediastinal B-cell lymphoma
Panelist Lisa G. Roth, MD, of Weill Cornell Medicine, New York, reviewed the genomic landscape of Hodgkin lymphoma (HL) and primary mediastinal B-cell lymphoma (PMBCL).
Dr. Roth explained that both HL and PMBCL are derived from thymic B cells, predominantly affect the mediastinum, and are CD30-positive lymphomas. Both are characterized by upregulation of JAK/STAT and NF-kappaB as well as overexpression of PD-L1.
Dr. Roth noted that HL is challenging to sequence by standard methods because Reed Sternberg (HRS) cells represent less than 1% of the cellular infiltrate. Recurrently mutated genes in HL cluster by histologic subtype.
Whole-exome sequencing of HRS cells show loss of beta-2 microglobulin and MHC-1 expression, HLA-B, NF-kappaB signaling, and JAK-STAT signaling, according to data published in Blood Advances in 2019.
Dr. Roth’s lab performed immunohistochemistry on tissue microarrays in 145 cases of HL (unpublished data). Results showed that loss of beta-2 microglobulin is more common in younger HL patients. For other alterations, there were too few cases to know.
Dr. Roth’s lab is a member of a pediatric/AYA HL sequencing multi-institutional consortium that has been able to extract DNA and RNA from samples submitted for whole-exome sequencing. The consortium’s goal is to shed light on implications of other genomic alterations that may differ by age in HL patients.
Dr. Roth cited research showing that PMBCL shares molecular alterations similar to those of HL. Alterations in PMBCL suggest dysregulated cellular signaling and immune evasion mechanisms (e.g., deletions in MHC type 1 and 2, beta-2 microglobulin, JAK-STAT, and NF-kappaB mutations) that provide opportunities to study novel agents, according to data published in Blood in 2019.
By early 2021, the S1826 and ANHL1931 studies, which have no age restriction, will be available to AYA lymphoma patients with HL and PMBCL, respectively, Dr. Roth said.
Follicular lymphoma: Clinical features by age
Panelist Abner Louissaint Jr, MD, PhD, of Massachusetts General Hospital in Boston, discussed age-related differences in follicular lymphoma (FL).
He noted that FL typically presents at an advanced stage, with low- or high-grade histology. It is increasingly common in adults in their 50s and 60s, representing 20% of all lymphomas. FL is rare in children and AYAs.
Dr. Louissaint explained that the typical flow cytometric findings in FL are BCL2 translocations, occurring in up to 85%-90% of low-grade and 50% of high-grade cases. The t(14;18)(q32;q21) translocation juxtaposes BCL2 on 18q21 to regulatory sequences and enhances the expression of elements of the Ig heavy chain.
Malignant cells in FL patients express CD20, CD10, CD21, and BCL2 (in contrast to normal germinal centers) and overexpress BCL6 (in contrast to normal follicles), Dr. Louissaint noted. He said the Ki-67 proliferative index of the malignant cells is typically low.
Pediatric-type FL is rare, but case series show clinical, pathologic, and molecular features that are distinctive from adult FL, Dr. Louissaint explained.
He then discussed the features of pediatric-type FL in multiple domains. In the clinical domain, there is a male predilection, and stage tends to be low. There is frequent involvement of nodes of the head and neck region and rare involvement of internal lymph node chains.
Pathologically, the malignant cells appear high grade, with architectural effacement, expansile follicular pattern, large lymphocyte size, and an elevated proliferation index. In contrast to adult FL, malignant cells in pediatric-type FL lack aberrant BCL2 expression.
Most importantly, for pediatric-type FL, the prognosis is excellent with durable remissions after surgical excision, Dr. Louissaint said.
Follicular lymphoma: Molecular features by age
Because of the excellent prognosis in pediatric-type FL, it is important to assess whether young adults with FL have adult-type or pediatric-type lesions, Dr. Louissaint said.
He cited many studies showing differences in adult and pediatric-type FL. In adult FL, the mutational landscape is characterized by frequent chromatin-modifying mutations in genes such as CREBBP, KM22D, and EP300.
In contrast, in pediatric-type FL, there are frequent activating MAPK pathway mutations, including mutations in the negative regulatory domain of MAP2K1. These mutations are not seen in adult FL.
Dr. Louissaint noted that there may be mutations in epigenetic modifiers (CREBBP, TNFRSF14) in both adult and pediatric-type FL. However, CREBBP is very unusual in pediatric-type FL and common in adult FL. This suggests the alterations in pediatric-type FL do not simply represent an early stage of the same disease as adult FL.
Despite a high proliferating fraction and absence of BCL2/BCL6/IRF4 rearrangements in pediatric-type FL, the presence of these features was associated with dramatic difference in progression-free survival, according to research published in Blood in 2012.
A distinct entity
In 2016, the World Health Organization recognized pediatric-type FL as a distinct entity, with the following diagnostic criteria (published in Blood):
- At least partial effacement of nodal architecture, expansile follicles, intermediate-size blastoid cells, and no component of diffuse large B-cell lymphoma.
- Immunohistochemistry showing BCL6 positivity, BCL2 negativity or weak positivity, and a high proliferative fraction.
- Genomic studies showing no BCL2 amplification.
- Clinical features of nodal disease in the head and neck region, early clinical stage, age younger than 40 years, typically in a male with no internal nodes involved.
When FL occurs in AYAs, the diagnostic findings of pediatric-type FL suggest the patient will do well with conservative management (e.g., excision alone), Dr. Louissaint noted.
Two sizes do not fit all
The strategies that have improved cancer outcomes since the 1970s for children and older adults have been much less successful for AYAs with cancer.
As an oncologic community, we should not allow the AYA gap to persist. As always, the solutions are likely to involve focused clinical research, education, and communication. Effort will need to be targeted specifically to the AYA population.
Since health-related mortality is high even when cancer-specific outcomes improve, adopting and maintaining a healthy lifestyle must be a key part of the discussion with these young patients.
The biologic differences associated with AYA lymphomas demand participation in clinical trials.
Oncologists should vigorously support removing impediments to the participation of AYAs in prospective clinical trials, stratified (but unrestricted) by age, with careful analysis of patient-reported outcomes, late adverse effects, and biospecimen collection.
As Dr. Kelly noted in the question-and-answer period, the Children’s Oncology Group has an existing biobank of paraffin-embedded tumor samples, DNA from lymphoma specimens, plasma, and sera with clinically annotated data that can be given to investigators upon request and justification.
Going beyond eligibility for clinical trials
Unfortunately, we will likely find that broadening eligibility criteria is the “low-hanging fruit.” There are protocol-, patient-, and physician-related obstacles, according to a review published in Cancer in 2019.
Patient-related obstacles include fear of toxicity, uncertainty about placebos, a steep learning curve for health literacy, insurance-related impediments, and other access-related issues.
Discussions will need to be tailored to the AYA population. Frank, early conversations about fertility, sexuality, financial hardship, career advancement, work-life balance, and cognitive risks may not only facilitate treatment planning but also encourage the trust that is essential for patients to enroll in trials.
The investment in time, multidisciplinary staff and physician involvement, and potential delays in treatment initiation may be painful and inconvenient, but the benefits for long-term health outcomes and personal-professional relationships will be gratifying beyond measure.
Dr. Smith disclosed relationships with Genentech/Roche, Celgene, TGTX, Karyopharm, Janssen, and Bantem. Dr. Roth disclosed relationships with Janssen, ADC Therapeutics, and Celgene. Dr. Kelly and Dr. Louissaint had no financial relationships to disclose.
Dr. Lyss was a community-based medical oncologist and clinical researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers, as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
In the 1970s, cancer survival was poor for young children and older adults in the United States, as shown by data published in the Journal of the National Cancer Institute.
Great progress has been made since the 1970s, but improvements in outcome have been less impressive for cancer patients aged 15-39 years, as shown by research published in Cancer.
Patients aged 15-39 years have been designated by the National Institutes of Health (NIH) as “adolescents and young adults (AYAs),” and the lag in survival benefit has been termed “the AYA gap.”
The AYA gap persists in lymphoma patients, and an expert panel recently outlined differences between lymphoma in AYAs and lymphoma in other age groups.
The experts spoke at a special session of the AACR Virtual Meeting: Advances in Malignant Lymphoma moderated by Somali M. Smith, MD, of the University of Chicago.
Factors that contribute to the AYA gap
About 89,000 AYAs are diagnosed with cancer each year in the United States, according to data from the National Cancer Institute (NCI). Lymphomas and thyroid cancer are the most common cancers among younger AYAs, aged 15-24 years.
In a report commissioned by the NIH in 2006, many factors contributing to the AYA gap were identified. Chief among them were:
- Limitations in access to care.
- Delayed diagnosis.
- Inconsistency in treatment and follow-up.
- Long-term toxicity (fertility, second malignancies, and cardiovascular disease).
These factors compromise health-related survival, even when cancer-specific survival is improved.
Panelist Kara Kelly, MD, of Roswell Park Comprehensive Cancer Center in Buffalo, N.Y., noted that there are additional unique challenges for AYAs with cancer. These include:
- Pubertal changes.
- Developmental transition to independence.
- Societal impediments such as insurance coverage and disparities in access to specialized centers.
- Psychosocial factors such as health literacy and adherence to treatment and follow-up.
Focusing on lymphoma specifically, Dr. Kelly noted that lymphoma biology differs across the age spectrum and by race and ethnicity. Both tumor and host factors require further study, she said.
Clinical trial access for AYAs
Dr. Kelly emphasized that, unfortunately, clinical research participation is low among AYAs. A major impediment is that adult clinical trials historically required participants to be at least 18 years old.
In addition, there has not been a focused effort to educate AYAs about regulatory safeguards to ensure safety and the promise of enhanced benefit to them in NCI Cancer Trials Network (NCTN) trials. As a result, the refusal rate is high.
A multi-stakeholder workshop, convened in May 2016 by the American Society of Clinical Oncology and Friends of Cancer Research, outlined opportunities for expanding trial eligibility to include children younger than 18 years in first-in-human and other adult cancer clinical trials, enhancing their access to new agents, without compromising safety.
Recently, collaborative efforts between the adult and children’s NCTN research groups have included AYAs in studies addressing cancers that span the age spectrum, including lymphoma.
However, as Dr. Kelly noted, there are differences in AYA lymphoid malignancy types with a transition from more pediatric to more adult types.
Hodgkin lymphoma and primary mediastinal B-cell lymphoma
Panelist Lisa G. Roth, MD, of Weill Cornell Medicine, New York, reviewed the genomic landscape of Hodgkin lymphoma (HL) and primary mediastinal B-cell lymphoma (PMBCL).
Dr. Roth explained that both HL and PMBCL are derived from thymic B cells, predominantly affect the mediastinum, and are CD30-positive lymphomas. Both are characterized by upregulation of JAK/STAT and NF-kappaB as well as overexpression of PD-L1.
Dr. Roth noted that HL is challenging to sequence by standard methods because Reed Sternberg (HRS) cells represent less than 1% of the cellular infiltrate. Recurrently mutated genes in HL cluster by histologic subtype.
Whole-exome sequencing of HRS cells show loss of beta-2 microglobulin and MHC-1 expression, HLA-B, NF-kappaB signaling, and JAK-STAT signaling, according to data published in Blood Advances in 2019.
Dr. Roth’s lab performed immunohistochemistry on tissue microarrays in 145 cases of HL (unpublished data). Results showed that loss of beta-2 microglobulin is more common in younger HL patients. For other alterations, there were too few cases to know.
Dr. Roth’s lab is a member of a pediatric/AYA HL sequencing multi-institutional consortium that has been able to extract DNA and RNA from samples submitted for whole-exome sequencing. The consortium’s goal is to shed light on implications of other genomic alterations that may differ by age in HL patients.
Dr. Roth cited research showing that PMBCL shares molecular alterations similar to those of HL. Alterations in PMBCL suggest dysregulated cellular signaling and immune evasion mechanisms (e.g., deletions in MHC type 1 and 2, beta-2 microglobulin, JAK-STAT, and NF-kappaB mutations) that provide opportunities to study novel agents, according to data published in Blood in 2019.
By early 2021, the S1826 and ANHL1931 studies, which have no age restriction, will be available to AYA lymphoma patients with HL and PMBCL, respectively, Dr. Roth said.
Follicular lymphoma: Clinical features by age
Panelist Abner Louissaint Jr, MD, PhD, of Massachusetts General Hospital in Boston, discussed age-related differences in follicular lymphoma (FL).
He noted that FL typically presents at an advanced stage, with low- or high-grade histology. It is increasingly common in adults in their 50s and 60s, representing 20% of all lymphomas. FL is rare in children and AYAs.
Dr. Louissaint explained that the typical flow cytometric findings in FL are BCL2 translocations, occurring in up to 85%-90% of low-grade and 50% of high-grade cases. The t(14;18)(q32;q21) translocation juxtaposes BCL2 on 18q21 to regulatory sequences and enhances the expression of elements of the Ig heavy chain.
Malignant cells in FL patients express CD20, CD10, CD21, and BCL2 (in contrast to normal germinal centers) and overexpress BCL6 (in contrast to normal follicles), Dr. Louissaint noted. He said the Ki-67 proliferative index of the malignant cells is typically low.
Pediatric-type FL is rare, but case series show clinical, pathologic, and molecular features that are distinctive from adult FL, Dr. Louissaint explained.
He then discussed the features of pediatric-type FL in multiple domains. In the clinical domain, there is a male predilection, and stage tends to be low. There is frequent involvement of nodes of the head and neck region and rare involvement of internal lymph node chains.
Pathologically, the malignant cells appear high grade, with architectural effacement, expansile follicular pattern, large lymphocyte size, and an elevated proliferation index. In contrast to adult FL, malignant cells in pediatric-type FL lack aberrant BCL2 expression.
Most importantly, for pediatric-type FL, the prognosis is excellent with durable remissions after surgical excision, Dr. Louissaint said.
Follicular lymphoma: Molecular features by age
Because of the excellent prognosis in pediatric-type FL, it is important to assess whether young adults with FL have adult-type or pediatric-type lesions, Dr. Louissaint said.
He cited many studies showing differences in adult and pediatric-type FL. In adult FL, the mutational landscape is characterized by frequent chromatin-modifying mutations in genes such as CREBBP, KM22D, and EP300.
In contrast, in pediatric-type FL, there are frequent activating MAPK pathway mutations, including mutations in the negative regulatory domain of MAP2K1. These mutations are not seen in adult FL.
Dr. Louissaint noted that there may be mutations in epigenetic modifiers (CREBBP, TNFRSF14) in both adult and pediatric-type FL. However, CREBBP is very unusual in pediatric-type FL and common in adult FL. This suggests the alterations in pediatric-type FL do not simply represent an early stage of the same disease as adult FL.
Despite a high proliferating fraction and absence of BCL2/BCL6/IRF4 rearrangements in pediatric-type FL, the presence of these features was associated with dramatic difference in progression-free survival, according to research published in Blood in 2012.
A distinct entity
In 2016, the World Health Organization recognized pediatric-type FL as a distinct entity, with the following diagnostic criteria (published in Blood):
- At least partial effacement of nodal architecture, expansile follicles, intermediate-size blastoid cells, and no component of diffuse large B-cell lymphoma.
- Immunohistochemistry showing BCL6 positivity, BCL2 negativity or weak positivity, and a high proliferative fraction.
- Genomic studies showing no BCL2 amplification.
- Clinical features of nodal disease in the head and neck region, early clinical stage, age younger than 40 years, typically in a male with no internal nodes involved.
When FL occurs in AYAs, the diagnostic findings of pediatric-type FL suggest the patient will do well with conservative management (e.g., excision alone), Dr. Louissaint noted.
Two sizes do not fit all
The strategies that have improved cancer outcomes since the 1970s for children and older adults have been much less successful for AYAs with cancer.
As an oncologic community, we should not allow the AYA gap to persist. As always, the solutions are likely to involve focused clinical research, education, and communication. Effort will need to be targeted specifically to the AYA population.
Since health-related mortality is high even when cancer-specific outcomes improve, adopting and maintaining a healthy lifestyle must be a key part of the discussion with these young patients.
The biologic differences associated with AYA lymphomas demand participation in clinical trials.
Oncologists should vigorously support removing impediments to the participation of AYAs in prospective clinical trials, stratified (but unrestricted) by age, with careful analysis of patient-reported outcomes, late adverse effects, and biospecimen collection.
As Dr. Kelly noted in the question-and-answer period, the Children’s Oncology Group has an existing biobank of paraffin-embedded tumor samples, DNA from lymphoma specimens, plasma, and sera with clinically annotated data that can be given to investigators upon request and justification.
Going beyond eligibility for clinical trials
Unfortunately, we will likely find that broadening eligibility criteria is the “low-hanging fruit.” There are protocol-, patient-, and physician-related obstacles, according to a review published in Cancer in 2019.
Patient-related obstacles include fear of toxicity, uncertainty about placebos, a steep learning curve for health literacy, insurance-related impediments, and other access-related issues.
Discussions will need to be tailored to the AYA population. Frank, early conversations about fertility, sexuality, financial hardship, career advancement, work-life balance, and cognitive risks may not only facilitate treatment planning but also encourage the trust that is essential for patients to enroll in trials.
The investment in time, multidisciplinary staff and physician involvement, and potential delays in treatment initiation may be painful and inconvenient, but the benefits for long-term health outcomes and personal-professional relationships will be gratifying beyond measure.
Dr. Smith disclosed relationships with Genentech/Roche, Celgene, TGTX, Karyopharm, Janssen, and Bantem. Dr. Roth disclosed relationships with Janssen, ADC Therapeutics, and Celgene. Dr. Kelly and Dr. Louissaint had no financial relationships to disclose.
Dr. Lyss was a community-based medical oncologist and clinical researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers, as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
In the 1970s, cancer survival was poor for young children and older adults in the United States, as shown by data published in the Journal of the National Cancer Institute.
Great progress has been made since the 1970s, but improvements in outcome have been less impressive for cancer patients aged 15-39 years, as shown by research published in Cancer.
Patients aged 15-39 years have been designated by the National Institutes of Health (NIH) as “adolescents and young adults (AYAs),” and the lag in survival benefit has been termed “the AYA gap.”
The AYA gap persists in lymphoma patients, and an expert panel recently outlined differences between lymphoma in AYAs and lymphoma in other age groups.
The experts spoke at a special session of the AACR Virtual Meeting: Advances in Malignant Lymphoma moderated by Somali M. Smith, MD, of the University of Chicago.
Factors that contribute to the AYA gap
About 89,000 AYAs are diagnosed with cancer each year in the United States, according to data from the National Cancer Institute (NCI). Lymphomas and thyroid cancer are the most common cancers among younger AYAs, aged 15-24 years.
In a report commissioned by the NIH in 2006, many factors contributing to the AYA gap were identified. Chief among them were:
- Limitations in access to care.
- Delayed diagnosis.
- Inconsistency in treatment and follow-up.
- Long-term toxicity (fertility, second malignancies, and cardiovascular disease).
These factors compromise health-related survival, even when cancer-specific survival is improved.
Panelist Kara Kelly, MD, of Roswell Park Comprehensive Cancer Center in Buffalo, N.Y., noted that there are additional unique challenges for AYAs with cancer. These include:
- Pubertal changes.
- Developmental transition to independence.
- Societal impediments such as insurance coverage and disparities in access to specialized centers.
- Psychosocial factors such as health literacy and adherence to treatment and follow-up.
Focusing on lymphoma specifically, Dr. Kelly noted that lymphoma biology differs across the age spectrum and by race and ethnicity. Both tumor and host factors require further study, she said.
Clinical trial access for AYAs
Dr. Kelly emphasized that, unfortunately, clinical research participation is low among AYAs. A major impediment is that adult clinical trials historically required participants to be at least 18 years old.
In addition, there has not been a focused effort to educate AYAs about regulatory safeguards to ensure safety and the promise of enhanced benefit to them in NCI Cancer Trials Network (NCTN) trials. As a result, the refusal rate is high.
A multi-stakeholder workshop, convened in May 2016 by the American Society of Clinical Oncology and Friends of Cancer Research, outlined opportunities for expanding trial eligibility to include children younger than 18 years in first-in-human and other adult cancer clinical trials, enhancing their access to new agents, without compromising safety.
Recently, collaborative efforts between the adult and children’s NCTN research groups have included AYAs in studies addressing cancers that span the age spectrum, including lymphoma.
However, as Dr. Kelly noted, there are differences in AYA lymphoid malignancy types with a transition from more pediatric to more adult types.
Hodgkin lymphoma and primary mediastinal B-cell lymphoma
Panelist Lisa G. Roth, MD, of Weill Cornell Medicine, New York, reviewed the genomic landscape of Hodgkin lymphoma (HL) and primary mediastinal B-cell lymphoma (PMBCL).
Dr. Roth explained that both HL and PMBCL are derived from thymic B cells, predominantly affect the mediastinum, and are CD30-positive lymphomas. Both are characterized by upregulation of JAK/STAT and NF-kappaB as well as overexpression of PD-L1.
Dr. Roth noted that HL is challenging to sequence by standard methods because Reed Sternberg (HRS) cells represent less than 1% of the cellular infiltrate. Recurrently mutated genes in HL cluster by histologic subtype.
Whole-exome sequencing of HRS cells show loss of beta-2 microglobulin and MHC-1 expression, HLA-B, NF-kappaB signaling, and JAK-STAT signaling, according to data published in Blood Advances in 2019.
Dr. Roth’s lab performed immunohistochemistry on tissue microarrays in 145 cases of HL (unpublished data). Results showed that loss of beta-2 microglobulin is more common in younger HL patients. For other alterations, there were too few cases to know.
Dr. Roth’s lab is a member of a pediatric/AYA HL sequencing multi-institutional consortium that has been able to extract DNA and RNA from samples submitted for whole-exome sequencing. The consortium’s goal is to shed light on implications of other genomic alterations that may differ by age in HL patients.
Dr. Roth cited research showing that PMBCL shares molecular alterations similar to those of HL. Alterations in PMBCL suggest dysregulated cellular signaling and immune evasion mechanisms (e.g., deletions in MHC type 1 and 2, beta-2 microglobulin, JAK-STAT, and NF-kappaB mutations) that provide opportunities to study novel agents, according to data published in Blood in 2019.
By early 2021, the S1826 and ANHL1931 studies, which have no age restriction, will be available to AYA lymphoma patients with HL and PMBCL, respectively, Dr. Roth said.
Follicular lymphoma: Clinical features by age
Panelist Abner Louissaint Jr, MD, PhD, of Massachusetts General Hospital in Boston, discussed age-related differences in follicular lymphoma (FL).
He noted that FL typically presents at an advanced stage, with low- or high-grade histology. It is increasingly common in adults in their 50s and 60s, representing 20% of all lymphomas. FL is rare in children and AYAs.
Dr. Louissaint explained that the typical flow cytometric findings in FL are BCL2 translocations, occurring in up to 85%-90% of low-grade and 50% of high-grade cases. The t(14;18)(q32;q21) translocation juxtaposes BCL2 on 18q21 to regulatory sequences and enhances the expression of elements of the Ig heavy chain.
Malignant cells in FL patients express CD20, CD10, CD21, and BCL2 (in contrast to normal germinal centers) and overexpress BCL6 (in contrast to normal follicles), Dr. Louissaint noted. He said the Ki-67 proliferative index of the malignant cells is typically low.
Pediatric-type FL is rare, but case series show clinical, pathologic, and molecular features that are distinctive from adult FL, Dr. Louissaint explained.
He then discussed the features of pediatric-type FL in multiple domains. In the clinical domain, there is a male predilection, and stage tends to be low. There is frequent involvement of nodes of the head and neck region and rare involvement of internal lymph node chains.
Pathologically, the malignant cells appear high grade, with architectural effacement, expansile follicular pattern, large lymphocyte size, and an elevated proliferation index. In contrast to adult FL, malignant cells in pediatric-type FL lack aberrant BCL2 expression.
Most importantly, for pediatric-type FL, the prognosis is excellent with durable remissions after surgical excision, Dr. Louissaint said.
Follicular lymphoma: Molecular features by age
Because of the excellent prognosis in pediatric-type FL, it is important to assess whether young adults with FL have adult-type or pediatric-type lesions, Dr. Louissaint said.
He cited many studies showing differences in adult and pediatric-type FL. In adult FL, the mutational landscape is characterized by frequent chromatin-modifying mutations in genes such as CREBBP, KM22D, and EP300.
In contrast, in pediatric-type FL, there are frequent activating MAPK pathway mutations, including mutations in the negative regulatory domain of MAP2K1. These mutations are not seen in adult FL.
Dr. Louissaint noted that there may be mutations in epigenetic modifiers (CREBBP, TNFRSF14) in both adult and pediatric-type FL. However, CREBBP is very unusual in pediatric-type FL and common in adult FL. This suggests the alterations in pediatric-type FL do not simply represent an early stage of the same disease as adult FL.
Despite a high proliferating fraction and absence of BCL2/BCL6/IRF4 rearrangements in pediatric-type FL, the presence of these features was associated with dramatic difference in progression-free survival, according to research published in Blood in 2012.
A distinct entity
In 2016, the World Health Organization recognized pediatric-type FL as a distinct entity, with the following diagnostic criteria (published in Blood):
- At least partial effacement of nodal architecture, expansile follicles, intermediate-size blastoid cells, and no component of diffuse large B-cell lymphoma.
- Immunohistochemistry showing BCL6 positivity, BCL2 negativity or weak positivity, and a high proliferative fraction.
- Genomic studies showing no BCL2 amplification.
- Clinical features of nodal disease in the head and neck region, early clinical stage, age younger than 40 years, typically in a male with no internal nodes involved.
When FL occurs in AYAs, the diagnostic findings of pediatric-type FL suggest the patient will do well with conservative management (e.g., excision alone), Dr. Louissaint noted.
Two sizes do not fit all
The strategies that have improved cancer outcomes since the 1970s for children and older adults have been much less successful for AYAs with cancer.
As an oncologic community, we should not allow the AYA gap to persist. As always, the solutions are likely to involve focused clinical research, education, and communication. Effort will need to be targeted specifically to the AYA population.
Since health-related mortality is high even when cancer-specific outcomes improve, adopting and maintaining a healthy lifestyle must be a key part of the discussion with these young patients.
The biologic differences associated with AYA lymphomas demand participation in clinical trials.
Oncologists should vigorously support removing impediments to the participation of AYAs in prospective clinical trials, stratified (but unrestricted) by age, with careful analysis of patient-reported outcomes, late adverse effects, and biospecimen collection.
As Dr. Kelly noted in the question-and-answer period, the Children’s Oncology Group has an existing biobank of paraffin-embedded tumor samples, DNA from lymphoma specimens, plasma, and sera with clinically annotated data that can be given to investigators upon request and justification.
Going beyond eligibility for clinical trials
Unfortunately, we will likely find that broadening eligibility criteria is the “low-hanging fruit.” There are protocol-, patient-, and physician-related obstacles, according to a review published in Cancer in 2019.
Patient-related obstacles include fear of toxicity, uncertainty about placebos, a steep learning curve for health literacy, insurance-related impediments, and other access-related issues.
Discussions will need to be tailored to the AYA population. Frank, early conversations about fertility, sexuality, financial hardship, career advancement, work-life balance, and cognitive risks may not only facilitate treatment planning but also encourage the trust that is essential for patients to enroll in trials.
The investment in time, multidisciplinary staff and physician involvement, and potential delays in treatment initiation may be painful and inconvenient, but the benefits for long-term health outcomes and personal-professional relationships will be gratifying beyond measure.
Dr. Smith disclosed relationships with Genentech/Roche, Celgene, TGTX, Karyopharm, Janssen, and Bantem. Dr. Roth disclosed relationships with Janssen, ADC Therapeutics, and Celgene. Dr. Kelly and Dr. Louissaint had no financial relationships to disclose.
Dr. Lyss was a community-based medical oncologist and clinical researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers, as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
FROM AACR ADVANCES IN MALIGNANT LYMPHOMA 2020
Is patient suicide in psychiatry a medical error?
When Rodney Vivian, MD, a psychiatrist in Cincinnati, was sued for medical malpractice after a psychiatric inpatient died by suicide, he recalls being naive about the process and how difficult it would be. “I was thinking that truth and common sense would prevail. How stupid I was,” he said.

Although Dr. Vivian, who was at the time the medical director of a hospital psychiatric unit in Ohio, was found not liable in two appeals, the legal process dragged on for 6 years, creating an emotional roller coaster of sadness, fear, vulnerability, and anxiety.
“The lawsuit took a big chunk out of me, and there was a sense of unfairness. It was incredibly humiliating and destructive; and it did not make me a better person or psychiatrist,” Dr. Vivian said.
Dr. Vivian is just one of the many psychiatrists who have had their world turned upside down after a patient suicide. When such events occur, grief-stricken families often point the finger at the treating psychiatrist. Although lawsuits are rare in psychiatry, patient suicide can lead to a myriad of emotional, legal, and career consequences.
Tyler Black, MD, child and adolescent psychiatrist and assistant clinical professor at the University of British Columbia, Vancouver, likens patient suicide to “a nuclear bomb” but emphasizes the importance of not classifying such events as a medical error or assigning blame.
“Starting with the assumption that suicide is always avoidable is not evidence based,” Dr. Black said.
Although patient suicide can occur across medicine, the odds are alarmingly high in psychiatry.

“There’s at least a 50-50 chance that a psychiatrist is going to face the suicide of a patient,” said Eric Plakun, MD, medical director/CEO at the Austen Riggs Center, Stockbridge, Mass. a hospital-based facility that offers a continuum of psychiatric treatment. Quoting forensic psychiatrist Robert Simon, Dr. Plakun said: “There are two kinds of psychiatrists – those who have had a patient die by suicide, and those who will.”
Research from 2015 shows that, among specialists, psychiatrists are among the least likely to be sued. A 2007/2008 Physician Survey from the American Medical Association showed that 22.2% of psychiatrists had been sued for malpractice; the probability that they would face a claim each year was only 2.6%. However, failure to prevent suicide is one of the top reasons for lawsuits.
One report from 2008 suggests that 20%-68% of psychiatrists will lose a patient to suicide. A report cowritten by Dr. Plakun in 2005 notes that about one in six psychiatric interns and one in three psychiatric residents will experience a patient suicide some time during their training. The authors added that 50% of all psychiatrists will have a patient die by suicide during their career. That risk stays at about 50% for future patients even after a clinician experiences the death of a previous patient.
Although mental health professionals prevail in up to 80% of suicide-related malpractice cases, such events are still emotionally devastating for everyone involved.
When a patient dies by suicide, it is a huge event, he noted. “It fuels a lot of fear and a lot of guilt, worry, and sadness.”

Paul S. Appelbaum, MD, past president of the American Psychiatric Association and Dollard Professor of Psychiatry, Medicine, and Law at Columbia University, New York, noted that patient suicide will happen.
The problem with many administrators who talk about a target of “zero suicide” is that when suicide does occur, it can lead to the erroneous conclusion that someone is to blame, said Dr. Appelbaum, who is also director of the Center for Law, Ethics, and Psychiatry at Columbia University. “That’s not necessarily true and contributes to finger-pointing.”
Stopping the blame game
Dr. Black’s first experience as a psychiatry resident was arriving at the hospital and finding the body of a patient who had died by suicide by hanging. Although he did not know the patient, Dr. Black said he had a strong emotional response that was coupled with an intense and sometimes confusing reaction by the hospital administration, including what he called “nonsensical banning” of pencils on the ward.

Dr. Black is now the medical director of emergency psychiatry at BC Children’s Hospital in Vancouver and specializes in suicidology and emergency/crisis youth mental health care. He said during a recent live chat on Twitter that he does not predict suicide but instead “assesses risk,” meaning he examines potential risk factors in his patients.
“If systems and administrators (and consulting doctors) could recognize this, the ‘blame game’ would severely decrease. From the advocacy end, we have to stop seeing suicide as a ‘medical error,’ ” Dr. Black tweeted.
“There’s a strong administrative push, especially in the face of suicide, to dive into the [occurrence] as if it must be that an error was made,” he said in an interview.
To help counteract any potential finger-pointing, Dr. Black created a free-to-download patient risk assessment document called the Assessment of Suicide and Risk Inventory (ASARI) for use at every patient visit.
“ASARI was designed to walk an assessor through their thinking process such that they can put all of their thoughts down on one piece of paper. It makes it a better communication document, and it’s definitely better medicolegal documentation,” he said.
Dr. Appelbaum noted that, although having documentation is beneficial, “I don’t think that you necessarily need to separate actions that are ‘protective’ from actions that are intended to help a patient.”
However, he pointed out that, if a psychiatrist conforms to or exceeds the standard of care, including conducting appropriate suicide risk assessments, developing an appropriate treatment plan, and keeping comprehensive documentation, these measures “should provide an effective defense to claims of malpractice or negligence.”
‘Horrendous event’
Dr. Vivian said that, during his 40-year career in psychiatry, there have been about 12 “office patients” who died by suicide. However, nothing prepared him for the fallout from a lawsuit.
In 2010, a patient who had overdosed was transferred to the psychiatric unit of Mercy Health–Clermont (Ohio) Hospital, where Dr. Vivian was the admitting physician. Although the hospital staff was ordered to check on her every 15 minutes, her husband found her unconscious from a hanging attempt when he came to visit the next evening. After she was transferred to the ICU, she was taken off life support and died a few days later.
“It was a horrendous event,” Dr. Vivian said.
The family sued the hospital, and the matter was settled out of court without Dr. Vivian’s knowledge. The family also filed a separate lawsuit against Dr. Vivian, which went to trial 3 years later.
“My insurance company’s claims person was very supportive and wanted me to not settle. She agreed that I didn’t do anything wrong and that I needed to face this,” he added.
In the first trial, a jury found Dr. Vivian not liable. Six months later, the plaintiff’s attorney filed an appeal. A year after the first trial, the court of appeals also came back with a new ruling in his favor and, in a subsequent appeal, the Ohio Supreme Court also ruled in his favor.
Dr. Vivian noted that there really are no winners in these situations. “Even though the jury ruled in my favor, there was never a sense of ‘success.’ I could never feel good about what happened.” He was told the insurance company spent more than $300,000 on his defense.
Although he no longer performs psychiatric inpatient admissions, Dr. Vivian continues to work in private practice and provides psychiatric consultation to patients at a local medical center.
“I consider my work as a blessing in my life, and I continue to learn from my patients,” he said.
‘Will I be sued?’
Dr. Appelbaum noted there is a difference between a malpractice claim that may be filed and a “payout” to plaintiffs because of a negotiated resolution of a case or an award that is made at trial.
Malpractice insurers may raise the rates of a physician who has been found at fault in one or more legal actions in which financial settlements have been paid out, he said.
The issue in any malpractice case is whether the psychiatrist met the standard of care, which is traditionally defined as “skill and learning that is ordinarily possessed and exercised by members of that profession in good standing.”
“No physician is expected to be the guarantor of a good outcome of a case. Sometimes things go wrong. Merely because there’s a bad outcome, merely because a suicide has occurred, doesn’t mean that the psychiatrist was negligent,” Dr. Appelbaum said.
He believes all large centers should have a “clear-cut plan” in place to assist clinicians in the event of a patient suicide. Such plans should help in dealing with stress from losing a patient and should provide guidance about how to handle any potential lawsuit.
For those worried that a patient’s suicide will shadow them through their career, Dr. Appelbaum said that it can happen, especially in cases involving a financial settlement against the clinician.
Such cases must be reported to the national practitioner data bank, where they can be accessed by any licensing body in any state when physicians apply for a medical license.
In addition, Dr. Appelbaum pointed out that licensure, medical staff, and malpractice applications typically require disclosure of a history of successful or unsuccessful claims filed against a physician. Although that may be limited to the past 10 years, the requirement can go on indefinitely.
Beware how you share
Dr. Plakun noted that there is a sense of isolation for a clinician in cases of patient suicide and that physicians often turn inward. He added that, although it is important to talk with others, in institutions, this is best done in a “peer-review, protected space” – and perhaps with a lawyer present.
However, Dr. Appelbaum warned that sharing information, even in this type of setting, may not offer legal protection. Talking to others in order to get some emotional support is permitted once the statute of limitations for filing a claim has lapsed or if a claim has been closed.
Discussing a case of patient suicide with peers prior to that can have serious legal implications, he added. Colleagues can be called to testify in any resulting legal case and disclose what was said during such conversations.
“The typical advice that a risk manager, a claims manager, or an attorney would give to a clinician is, don’t talk to other people about it other than the lawyer or claims manager who’s dealing with the case,” he noted.
That said, there are three general exceptions to this rule. These include attorney-client privilege, any matters discussed with the physician’s own therapist, and, “depending on the state, there are varying protections for what’s considered ‘peer review.’ ”
For instance, when hospitals implement a formal review process after an event, what is said during discovery may be protected. However, not all states have such protection. That’s why it is important to understand what the law is in your particular state, said Dr. Appelbaum.
Support for psychiatrists
Kaz J. Nelson, MD, psychiatrist and associate professor at the University of Minnesota, Minneapolis, also works with high-risk populations, including those with acute suicidality and self-injury.

During a recent chat on patient suicide, Dr. Nelson tweeted: “Sadly in our field, suicide is not an IF but a WHEN. Don’t keep the inevitable shame and sadness to yourself.”
Dr. Nelson agreed with Dr. Black that it’s important to look into these occurrences as a quality improvement measure, but not as a way to assign blame. Preparing for potential patient loss “and having very solid, very supportive, very inclusive ‘postvention’ procedures” is critical, she noted. “When you don’t have these policies and procedures in place and have them very transparent, it creates a culture of silence around the issue.”
Dr. Plakun reiterated the importance of not staying silent. “We can’t simply surrender to the idea of not talking about patient suicide. We have to find a way to speak.”
A version of this story originally appeared on Medscape.com.
When Rodney Vivian, MD, a psychiatrist in Cincinnati, was sued for medical malpractice after a psychiatric inpatient died by suicide, he recalls being naive about the process and how difficult it would be. “I was thinking that truth and common sense would prevail. How stupid I was,” he said.
Although Dr. Vivian, who was at the time the medical director of a hospital psychiatric unit in Ohio, was found not liable in two appeals, the legal process dragged on for 6 years, creating an emotional roller coaster of sadness, fear, vulnerability, and anxiety.
“The lawsuit took a big chunk out of me, and there was a sense of unfairness. It was incredibly humiliating and destructive; and it did not make me a better person or psychiatrist,” Dr. Vivian said.
Dr. Vivian is just one of the many psychiatrists who have had their world turned upside down after a patient suicide. When such events occur, grief-stricken families often point the finger at the treating psychiatrist. Although lawsuits are rare in psychiatry, patient suicide can lead to a myriad of emotional, legal, and career consequences.
Tyler Black, MD, child and adolescent psychiatrist and assistant clinical professor at the University of British Columbia, Vancouver, likens patient suicide to “a nuclear bomb” but emphasizes the importance of not classifying such events as a medical error or assigning blame.
“Starting with the assumption that suicide is always avoidable is not evidence based,” Dr. Black said.
Although patient suicide can occur across medicine, the odds are alarmingly high in psychiatry.
“There’s at least a 50-50 chance that a psychiatrist is going to face the suicide of a patient,” said Eric Plakun, MD, medical director/CEO at the Austen Riggs Center, Stockbridge, Mass. a hospital-based facility that offers a continuum of psychiatric treatment. Quoting forensic psychiatrist Robert Simon, Dr. Plakun said: “There are two kinds of psychiatrists – those who have had a patient die by suicide, and those who will.”
Research from 2015 shows that, among specialists, psychiatrists are among the least likely to be sued. A 2007/2008 Physician Survey from the American Medical Association showed that 22.2% of psychiatrists had been sued for malpractice; the probability that they would face a claim each year was only 2.6%. However, failure to prevent suicide is one of the top reasons for lawsuits.
One report from 2008 suggests that 20%-68% of psychiatrists will lose a patient to suicide. A report cowritten by Dr. Plakun in 2005 notes that about one in six psychiatric interns and one in three psychiatric residents will experience a patient suicide some time during their training. The authors added that 50% of all psychiatrists will have a patient die by suicide during their career. That risk stays at about 50% for future patients even after a clinician experiences the death of a previous patient.
Although mental health professionals prevail in up to 80% of suicide-related malpractice cases, such events are still emotionally devastating for everyone involved.
When a patient dies by suicide, it is a huge event, he noted. “It fuels a lot of fear and a lot of guilt, worry, and sadness.”
Paul S. Appelbaum, MD, past president of the American Psychiatric Association and Dollard Professor of Psychiatry, Medicine, and Law at Columbia University, New York, noted that patient suicide will happen.
The problem with many administrators who talk about a target of “zero suicide” is that when suicide does occur, it can lead to the erroneous conclusion that someone is to blame, said Dr. Appelbaum, who is also director of the Center for Law, Ethics, and Psychiatry at Columbia University. “That’s not necessarily true and contributes to finger-pointing.”
Stopping the blame game
Dr. Black’s first experience as a psychiatry resident was arriving at the hospital and finding the body of a patient who had died by suicide by hanging. Although he did not know the patient, Dr. Black said he had a strong emotional response that was coupled with an intense and sometimes confusing reaction by the hospital administration, including what he called “nonsensical banning” of pencils on the ward.
Dr. Black is now the medical director of emergency psychiatry at BC Children’s Hospital in Vancouver and specializes in suicidology and emergency/crisis youth mental health care. He said during a recent live chat on Twitter that he does not predict suicide but instead “assesses risk,” meaning he examines potential risk factors in his patients.
“If systems and administrators (and consulting doctors) could recognize this, the ‘blame game’ would severely decrease. From the advocacy end, we have to stop seeing suicide as a ‘medical error,’ ” Dr. Black tweeted.
“There’s a strong administrative push, especially in the face of suicide, to dive into the [occurrence] as if it must be that an error was made,” he said in an interview.
To help counteract any potential finger-pointing, Dr. Black created a free-to-download patient risk assessment document called the Assessment of Suicide and Risk Inventory (ASARI) for use at every patient visit.
“ASARI was designed to walk an assessor through their thinking process such that they can put all of their thoughts down on one piece of paper. It makes it a better communication document, and it’s definitely better medicolegal documentation,” he said.
Dr. Appelbaum noted that, although having documentation is beneficial, “I don’t think that you necessarily need to separate actions that are ‘protective’ from actions that are intended to help a patient.”
However, he pointed out that, if a psychiatrist conforms to or exceeds the standard of care, including conducting appropriate suicide risk assessments, developing an appropriate treatment plan, and keeping comprehensive documentation, these measures “should provide an effective defense to claims of malpractice or negligence.”
‘Horrendous event’
Dr. Vivian said that, during his 40-year career in psychiatry, there have been about 12 “office patients” who died by suicide. However, nothing prepared him for the fallout from a lawsuit.
In 2010, a patient who had overdosed was transferred to the psychiatric unit of Mercy Health–Clermont (Ohio) Hospital, where Dr. Vivian was the admitting physician. Although the hospital staff was ordered to check on her every 15 minutes, her husband found her unconscious from a hanging attempt when he came to visit the next evening. After she was transferred to the ICU, she was taken off life support and died a few days later.
“It was a horrendous event,” Dr. Vivian said.
The family sued the hospital, and the matter was settled out of court without Dr. Vivian’s knowledge. The family also filed a separate lawsuit against Dr. Vivian, which went to trial 3 years later.
“My insurance company’s claims person was very supportive and wanted me to not settle. She agreed that I didn’t do anything wrong and that I needed to face this,” he added.
In the first trial, a jury found Dr. Vivian not liable. Six months later, the plaintiff’s attorney filed an appeal. A year after the first trial, the court of appeals also came back with a new ruling in his favor and, in a subsequent appeal, the Ohio Supreme Court also ruled in his favor.
Dr. Vivian noted that there really are no winners in these situations. “Even though the jury ruled in my favor, there was never a sense of ‘success.’ I could never feel good about what happened.” He was told the insurance company spent more than $300,000 on his defense.
Although he no longer performs psychiatric inpatient admissions, Dr. Vivian continues to work in private practice and provides psychiatric consultation to patients at a local medical center.
“I consider my work as a blessing in my life, and I continue to learn from my patients,” he said.
‘Will I be sued?’
Dr. Appelbaum noted there is a difference between a malpractice claim that may be filed and a “payout” to plaintiffs because of a negotiated resolution of a case or an award that is made at trial.
Malpractice insurers may raise the rates of a physician who has been found at fault in one or more legal actions in which financial settlements have been paid out, he said.
The issue in any malpractice case is whether the psychiatrist met the standard of care, which is traditionally defined as “skill and learning that is ordinarily possessed and exercised by members of that profession in good standing.”
“No physician is expected to be the guarantor of a good outcome of a case. Sometimes things go wrong. Merely because there’s a bad outcome, merely because a suicide has occurred, doesn’t mean that the psychiatrist was negligent,” Dr. Appelbaum said.
He believes all large centers should have a “clear-cut plan” in place to assist clinicians in the event of a patient suicide. Such plans should help in dealing with stress from losing a patient and should provide guidance about how to handle any potential lawsuit.
For those worried that a patient’s suicide will shadow them through their career, Dr. Appelbaum said that it can happen, especially in cases involving a financial settlement against the clinician.
Such cases must be reported to the national practitioner data bank, where they can be accessed by any licensing body in any state when physicians apply for a medical license.
In addition, Dr. Appelbaum pointed out that licensure, medical staff, and malpractice applications typically require disclosure of a history of successful or unsuccessful claims filed against a physician. Although that may be limited to the past 10 years, the requirement can go on indefinitely.
Beware how you share
Dr. Plakun noted that there is a sense of isolation for a clinician in cases of patient suicide and that physicians often turn inward. He added that, although it is important to talk with others, in institutions, this is best done in a “peer-review, protected space” – and perhaps with a lawyer present.
However, Dr. Appelbaum warned that sharing information, even in this type of setting, may not offer legal protection. Talking to others in order to get some emotional support is permitted once the statute of limitations for filing a claim has lapsed or if a claim has been closed.
Discussing a case of patient suicide with peers prior to that can have serious legal implications, he added. Colleagues can be called to testify in any resulting legal case and disclose what was said during such conversations.
“The typical advice that a risk manager, a claims manager, or an attorney would give to a clinician is, don’t talk to other people about it other than the lawyer or claims manager who’s dealing with the case,” he noted.
That said, there are three general exceptions to this rule. These include attorney-client privilege, any matters discussed with the physician’s own therapist, and, “depending on the state, there are varying protections for what’s considered ‘peer review.’ ”
For instance, when hospitals implement a formal review process after an event, what is said during discovery may be protected. However, not all states have such protection. That’s why it is important to understand what the law is in your particular state, said Dr. Appelbaum.
Support for psychiatrists
Kaz J. Nelson, MD, psychiatrist and associate professor at the University of Minnesota, Minneapolis, also works with high-risk populations, including those with acute suicidality and self-injury.
During a recent chat on patient suicide, Dr. Nelson tweeted: “Sadly in our field, suicide is not an IF but a WHEN. Don’t keep the inevitable shame and sadness to yourself.”
Dr. Nelson agreed with Dr. Black that it’s important to look into these occurrences as a quality improvement measure, but not as a way to assign blame. Preparing for potential patient loss “and having very solid, very supportive, very inclusive ‘postvention’ procedures” is critical, she noted. “When you don’t have these policies and procedures in place and have them very transparent, it creates a culture of silence around the issue.”
Dr. Plakun reiterated the importance of not staying silent. “We can’t simply surrender to the idea of not talking about patient suicide. We have to find a way to speak.”
A version of this story originally appeared on Medscape.com.
When Rodney Vivian, MD, a psychiatrist in Cincinnati, was sued for medical malpractice after a psychiatric inpatient died by suicide, he recalls being naive about the process and how difficult it would be. “I was thinking that truth and common sense would prevail. How stupid I was,” he said.
Although Dr. Vivian, who was at the time the medical director of a hospital psychiatric unit in Ohio, was found not liable in two appeals, the legal process dragged on for 6 years, creating an emotional roller coaster of sadness, fear, vulnerability, and anxiety.
“The lawsuit took a big chunk out of me, and there was a sense of unfairness. It was incredibly humiliating and destructive; and it did not make me a better person or psychiatrist,” Dr. Vivian said.
Dr. Vivian is just one of the many psychiatrists who have had their world turned upside down after a patient suicide. When such events occur, grief-stricken families often point the finger at the treating psychiatrist. Although lawsuits are rare in psychiatry, patient suicide can lead to a myriad of emotional, legal, and career consequences.
Tyler Black, MD, child and adolescent psychiatrist and assistant clinical professor at the University of British Columbia, Vancouver, likens patient suicide to “a nuclear bomb” but emphasizes the importance of not classifying such events as a medical error or assigning blame.
“Starting with the assumption that suicide is always avoidable is not evidence based,” Dr. Black said.
Although patient suicide can occur across medicine, the odds are alarmingly high in psychiatry.
“There’s at least a 50-50 chance that a psychiatrist is going to face the suicide of a patient,” said Eric Plakun, MD, medical director/CEO at the Austen Riggs Center, Stockbridge, Mass. a hospital-based facility that offers a continuum of psychiatric treatment. Quoting forensic psychiatrist Robert Simon, Dr. Plakun said: “There are two kinds of psychiatrists – those who have had a patient die by suicide, and those who will.”
Research from 2015 shows that, among specialists, psychiatrists are among the least likely to be sued. A 2007/2008 Physician Survey from the American Medical Association showed that 22.2% of psychiatrists had been sued for malpractice; the probability that they would face a claim each year was only 2.6%. However, failure to prevent suicide is one of the top reasons for lawsuits.
One report from 2008 suggests that 20%-68% of psychiatrists will lose a patient to suicide. A report cowritten by Dr. Plakun in 2005 notes that about one in six psychiatric interns and one in three psychiatric residents will experience a patient suicide some time during their training. The authors added that 50% of all psychiatrists will have a patient die by suicide during their career. That risk stays at about 50% for future patients even after a clinician experiences the death of a previous patient.
Although mental health professionals prevail in up to 80% of suicide-related malpractice cases, such events are still emotionally devastating for everyone involved.
When a patient dies by suicide, it is a huge event, he noted. “It fuels a lot of fear and a lot of guilt, worry, and sadness.”
Paul S. Appelbaum, MD, past president of the American Psychiatric Association and Dollard Professor of Psychiatry, Medicine, and Law at Columbia University, New York, noted that patient suicide will happen.
The problem with many administrators who talk about a target of “zero suicide” is that when suicide does occur, it can lead to the erroneous conclusion that someone is to blame, said Dr. Appelbaum, who is also director of the Center for Law, Ethics, and Psychiatry at Columbia University. “That’s not necessarily true and contributes to finger-pointing.”
Stopping the blame game
Dr. Black’s first experience as a psychiatry resident was arriving at the hospital and finding the body of a patient who had died by suicide by hanging. Although he did not know the patient, Dr. Black said he had a strong emotional response that was coupled with an intense and sometimes confusing reaction by the hospital administration, including what he called “nonsensical banning” of pencils on the ward.
Dr. Black is now the medical director of emergency psychiatry at BC Children’s Hospital in Vancouver and specializes in suicidology and emergency/crisis youth mental health care. He said during a recent live chat on Twitter that he does not predict suicide but instead “assesses risk,” meaning he examines potential risk factors in his patients.
“If systems and administrators (and consulting doctors) could recognize this, the ‘blame game’ would severely decrease. From the advocacy end, we have to stop seeing suicide as a ‘medical error,’ ” Dr. Black tweeted.
“There’s a strong administrative push, especially in the face of suicide, to dive into the [occurrence] as if it must be that an error was made,” he said in an interview.
To help counteract any potential finger-pointing, Dr. Black created a free-to-download patient risk assessment document called the Assessment of Suicide and Risk Inventory (ASARI) for use at every patient visit.
“ASARI was designed to walk an assessor through their thinking process such that they can put all of their thoughts down on one piece of paper. It makes it a better communication document, and it’s definitely better medicolegal documentation,” he said.
Dr. Appelbaum noted that, although having documentation is beneficial, “I don’t think that you necessarily need to separate actions that are ‘protective’ from actions that are intended to help a patient.”
However, he pointed out that, if a psychiatrist conforms to or exceeds the standard of care, including conducting appropriate suicide risk assessments, developing an appropriate treatment plan, and keeping comprehensive documentation, these measures “should provide an effective defense to claims of malpractice or negligence.”
‘Horrendous event’
Dr. Vivian said that, during his 40-year career in psychiatry, there have been about 12 “office patients” who died by suicide. However, nothing prepared him for the fallout from a lawsuit.
In 2010, a patient who had overdosed was transferred to the psychiatric unit of Mercy Health–Clermont (Ohio) Hospital, where Dr. Vivian was the admitting physician. Although the hospital staff was ordered to check on her every 15 minutes, her husband found her unconscious from a hanging attempt when he came to visit the next evening. After she was transferred to the ICU, she was taken off life support and died a few days later.
“It was a horrendous event,” Dr. Vivian said.
The family sued the hospital, and the matter was settled out of court without Dr. Vivian’s knowledge. The family also filed a separate lawsuit against Dr. Vivian, which went to trial 3 years later.
“My insurance company’s claims person was very supportive and wanted me to not settle. She agreed that I didn’t do anything wrong and that I needed to face this,” he added.
In the first trial, a jury found Dr. Vivian not liable. Six months later, the plaintiff’s attorney filed an appeal. A year after the first trial, the court of appeals also came back with a new ruling in his favor and, in a subsequent appeal, the Ohio Supreme Court also ruled in his favor.
Dr. Vivian noted that there really are no winners in these situations. “Even though the jury ruled in my favor, there was never a sense of ‘success.’ I could never feel good about what happened.” He was told the insurance company spent more than $300,000 on his defense.
Although he no longer performs psychiatric inpatient admissions, Dr. Vivian continues to work in private practice and provides psychiatric consultation to patients at a local medical center.
“I consider my work as a blessing in my life, and I continue to learn from my patients,” he said.
‘Will I be sued?’
Dr. Appelbaum noted there is a difference between a malpractice claim that may be filed and a “payout” to plaintiffs because of a negotiated resolution of a case or an award that is made at trial.
Malpractice insurers may raise the rates of a physician who has been found at fault in one or more legal actions in which financial settlements have been paid out, he said.
The issue in any malpractice case is whether the psychiatrist met the standard of care, which is traditionally defined as “skill and learning that is ordinarily possessed and exercised by members of that profession in good standing.”
“No physician is expected to be the guarantor of a good outcome of a case. Sometimes things go wrong. Merely because there’s a bad outcome, merely because a suicide has occurred, doesn’t mean that the psychiatrist was negligent,” Dr. Appelbaum said.
He believes all large centers should have a “clear-cut plan” in place to assist clinicians in the event of a patient suicide. Such plans should help in dealing with stress from losing a patient and should provide guidance about how to handle any potential lawsuit.
For those worried that a patient’s suicide will shadow them through their career, Dr. Appelbaum said that it can happen, especially in cases involving a financial settlement against the clinician.
Such cases must be reported to the national practitioner data bank, where they can be accessed by any licensing body in any state when physicians apply for a medical license.
In addition, Dr. Appelbaum pointed out that licensure, medical staff, and malpractice applications typically require disclosure of a history of successful or unsuccessful claims filed against a physician. Although that may be limited to the past 10 years, the requirement can go on indefinitely.
Beware how you share
Dr. Plakun noted that there is a sense of isolation for a clinician in cases of patient suicide and that physicians often turn inward. He added that, although it is important to talk with others, in institutions, this is best done in a “peer-review, protected space” – and perhaps with a lawyer present.
However, Dr. Appelbaum warned that sharing information, even in this type of setting, may not offer legal protection. Talking to others in order to get some emotional support is permitted once the statute of limitations for filing a claim has lapsed or if a claim has been closed.
Discussing a case of patient suicide with peers prior to that can have serious legal implications, he added. Colleagues can be called to testify in any resulting legal case and disclose what was said during such conversations.
“The typical advice that a risk manager, a claims manager, or an attorney would give to a clinician is, don’t talk to other people about it other than the lawyer or claims manager who’s dealing with the case,” he noted.
That said, there are three general exceptions to this rule. These include attorney-client privilege, any matters discussed with the physician’s own therapist, and, “depending on the state, there are varying protections for what’s considered ‘peer review.’ ”
For instance, when hospitals implement a formal review process after an event, what is said during discovery may be protected. However, not all states have such protection. That’s why it is important to understand what the law is in your particular state, said Dr. Appelbaum.
Support for psychiatrists
Kaz J. Nelson, MD, psychiatrist and associate professor at the University of Minnesota, Minneapolis, also works with high-risk populations, including those with acute suicidality and self-injury.
During a recent chat on patient suicide, Dr. Nelson tweeted: “Sadly in our field, suicide is not an IF but a WHEN. Don’t keep the inevitable shame and sadness to yourself.”
Dr. Nelson agreed with Dr. Black that it’s important to look into these occurrences as a quality improvement measure, but not as a way to assign blame. Preparing for potential patient loss “and having very solid, very supportive, very inclusive ‘postvention’ procedures” is critical, she noted. “When you don’t have these policies and procedures in place and have them very transparent, it creates a culture of silence around the issue.”
Dr. Plakun reiterated the importance of not staying silent. “We can’t simply surrender to the idea of not talking about patient suicide. We have to find a way to speak.”
A version of this story originally appeared on Medscape.com.
How VA Nurses are Coping With the COVID-19 Pandemic
The tsunami we call COVID-19 has threatened to overwhelm everything in its path, with devastating effects that can be hard to quantify or qualify. But the Nurses Organization of Veterans Affairs (NOVA) has taken on the fight. Earlier this year, NOVA surveyed its members to learn how nurses felt the US Department of Veterans Affairs (VA) was managing personal protective equipment (PPE), testing, communications, staffing needs, and other issues. Following the survey NOVA conducted in-depth interviews with nurses at the VA Boston Healthcare System to better understand how the pandemic was affecting nurses across the Veterans Health Administration (VHA).
The first survey, which was conducted in March and April 2020, included the following questions:
- Do you feel prepared for COVID-19?
- Do you feel your facility’s supply of PPE is adequate?
- Are you aware of the protocol for the distribution of equipment/supplies at your facility?
- Are staff being tested for the virus, and if so, when, and are there enough test kits available for staff and patients?
- Do you believe your facility is properly handling staff who have been exposed to COVID-19 but are asymptomatic?
With the May survey, NOVA aimed to get an update on how things were progressing. This survey asked how the pandemic was affecting members personally, including questions such as: Do you feel you are being supported—mentally and physically? Has VA offered to provide staff mental health counselors or others to help mitigate stress during the crisis?
Results revealed inconsistencies and some confusion. For example, communication among leadership, staff, and veterans continued to change rapidly, causing some misunderstanding overall, respondents said. Some pointed to “e-mail overload” and weekly updates that didn’t work. Others felt communication was “reactive” and “bare minimum”—not “proactive and informative.”
Most (74%) respondents felt that access to PPE was inadequate, and many did not know the protocol for distribution or what supplies were on hand, while 47% felt ill prepared for any COVID-19 onslaught. Although things had improved somewhat by May, > 85% of the respondents said they were reusing what was provided daily and were still finding it difficult to get PPE when needed.
According to respondents, testing was pretty much nonexistent. When asked whether staff were being tested and whether tests for both staff and patients were available, the answer was a resounding No (80%). Nurses’ comments ranged from “staff are not being tested, even if they have been exposed,” to “there are not enough tests for patients, let alone for frontline staff.”
The lack of tests compounded stress. Helen Motroni, ADN, RN, spinal cord injury staff nurse, said, “I have been tested twice for direct exposure to the coronavirus in the past 3 weeks. Luckily, the results were negative, but waiting for the test results was extremely stressful because I have 2 little boys at home. While waiting for results, I self-quarantined and was terrified that I possibly brought the virus home to my children. I never left my room and would talk with my boys through the door and FaceTime. My 8-year-old asked me why I couldn’t stay home so I would not get sick. I explained to him that if every nurse did that, there would be nobody to help those that are sick and suffering.”
As summer approached, testing throughout most of the states remained scarce and contact tracing was a struggle at many facilities. It seemed that, even if testing was available, only those at high risk would be tested.
Moreover, quarantine and sick leave for those who were COVID positive remained a concern, as numbers of those exposed increased. It was widely known and reported that staffing levels within the VHA were inadequate prior to the months going into the pandemic; survey respondents wondered how they would handle vacancies and take care of outside patients as part of the Fourth Mission if staff got sick. One COVID-compelled solution (which NOVA has advocated for years) included expedited hiring practices. Timely application and quicker onboarding enabled VA to hire within weeks rather than months. Since March, VA has hired > 20,000 new employees.
Multiplied Multitasking
The crisis, along with potential and actual staff shortages, has meant that many nurses have been doing double and triple duty—at the least. Danielle Newman, MSN, RN, clinical resource nurse, specialty and outpatient clinics, said, “Throughout the COVID crisis, I have been a direct care provider, an educator, a member of the float pool, an infection preventionist, a colleague, a therapist, and part of a support system to many.… As a PPE nurse educator, I visit every floor in the hospital and help educate staff on how to properly don and doff PPE, as well as monitor the doffing area and assist staff through the doffing process. I have also been involved in obtaining nasal swabs on veterans as well as staff in order to isolate and slow the progression of the virus.”
Some nurses were deployed, some redeployed, and some volunteered for the COVID front. James Murphy, BSN, RN, an emergency department nurse, answered the call. “During the first few weeks of the outbreak and with all its uncertainty, I volunteered to be a COVID nurse.… Seeing how the pandemic has changed practice was an experience.”
Peter Russo, MSN, RN, an OR assistant nurse manager, also found new experience. When the pandemic began to take hold, he was reassigned to Urgent Care and then to the Incident Management Team (IMT). “I asked my associate chief nurse, ‘What is the IMT?’ After my first 2 days on this team, my head was spinning with loads of information, trying to understand and grasp my role as the IMT Nursing Clinical Lead.”
After the third day, his preceptor was redeployed back to the endoscopy department. “I quickly realized my main role as a clinical leader on this team,” Russo says, “was to support and troubleshoot any and all nursing problems, concerns and or questions related to the COVID-19 pandemic among the 3 campuses. One of the more complex challenges was to identify the nursing burn rate of our PPE, especially the N95 masks in all of our COVID units.”
The new experience benefited, him, though. “When changes in policy or procedure happen during the course of an hour, it was one of my responsibilities to notify the nursing staff across the board. I now feel much more confident and competent offering suggestions [about] the many issues that arise.”
Because of the pandemic, all elective surgeries were cancelled and the ambulatory surgery unit where perioperative nurse Valentina Ward, BSN, RN, MPS, CPPS, worked was closed. “As a result,” she says, “I received an accelerated course and was reassigned where I was needed the most. … Serving veterans during the global crisis means so much to me. Despite all that is going on, and the challenges that exist, I have never been prouder to be a frontline nurse.”
The Importance of Maintaining ‘Normal’
COVID-19 seems to subsume everything around it, but there are still health care needs that have nothing to do with the virus. What happens to those patients and their care?
Melissa Varela-Manso, MSN, RN, CRRN, is an inpatient nurse case manager who works with a dedicated team to coordinate admissions and discharges for veterans on the acute Spinal Cord Injury (SCI) unit. “During this pandemic,” she says, “our veterans continue to have non-COVID medical conditions that warrant attention and hospitalization. Therefore, we are following the SCI veterans with non-COVID health issues on the inpatient unit. We also follow our SCI veterans on other units who are being ruled out for COVID and those who have tested positive. We are working diligently to guarantee each veteran gets the quality care they need and deserve while following the evolving guidelines.”
One of the ways VA health care teams have kept things stable for patients, while also ensuring their safety, is through telehealth. VA introduced telehealth in 2003 and is now the nation’s largest telehealth provider. In 2019, > 900,000 veterans received care through telehealth. But it took something like COVID-19 to really show the strengths and utility of being able to care for patients electronically. At the beginning of March, VA conducted about 2,500 telehealth video sessions daily. By June, that had jumped to nearly 25,000—a 1,000% increase.
Sherry Clement, BSN, RN, CFCN, CWCN, CCCN, is the VA Coordinated Transitional Care (C-TraC) Program case manager assigned to the COVID Outpatient Call Team, managing COVID-positive outpatients via telephone and VA video connect. “Working with the call team has been a great collaboration between primary care, telehealth, palliative care, infection control, and occupational health,” she says. “It is amazing to see how fast and efficient our team came together to create contact protocols, consults, notes, and a working plan to organize and oversee the needs of our COVID positive veterans."
Increased usage of telehealth also has meant community-based outpatient clinics continue to be operational, says Karen Boenig, MSN, APRN, ANP-BC, a primary care nurse manager. “Nurses and providers are conducting routine and urgent visits via VA Video Connect and telephone.… At the Framingham clinic, I am working with the Tele-Flu Clinic, which assesses veterans virtually who have symptoms of COVID-19. By utilizing virtual care visits, we can continue providing health care services to our veterans, which minimizes patient and provider exposure and supports social distancing.”
Florence Would be Proud
It seems fitting that 2020 is also the International Year of the Nurse, in celebration of Florence Nightingale’s 200th birthday. That passionate reformer would appreciate NOVA’s mission: “Shaping and Influencing healthcare in the Department of Veterans Affairs.”
In a NOVA blog post (www.vanurse.org), Teresa Morris, director of advocacy and government relations, says, “Thanks to those who responded to our 2 COVID-19 surveys, we were able to use the information as a firsthand look into how nurses felt about VA’s work and response during the Pandemic. The testimony touched on PPE, communication, testing, and comments and concerns that you had during the height of the virus.” She added, “I think some areas that spiked early had less time to prepare while others (MidWest/West) spiked later and used lessons learned.”
“We discussed the findings from the survey and thanked Congress for providing the $19.6 billion in emergency supplemental funding for VA. The funds helped to hire new staff, provide overtime pay, and purchase supplies needed for those caring for veterans and other COVID patients,” She continued.
In May, VA Secretary Robert Wilkie announced his plan to phase in reopening various VA facilities. Respondents were concerned about how the opening up of VA facilities would go as cases continue to spike around the country. They remain committed to their patients, however.
“Never was there a time in the history of nursing that I can say we are needed more,” says Cecilia McVey, MHA, RN, FAAN, Associate Director for Nursing and Patient Care Services, “and nurses make a difference. As you do every day, you are sacrificing more than you may have anticipated when you signed on to be a nurse. Never has it been more important to believe in yourself, in our profession, in the scientists that are guiding us and in ourselves. What you are doing is critical and your sacrifices have not gone unnoticed. I am thankful for all you do—every day—now, and in the future.”
The tsunami we call COVID-19 has threatened to overwhelm everything in its path, with devastating effects that can be hard to quantify or qualify. But the Nurses Organization of Veterans Affairs (NOVA) has taken on the fight. Earlier this year, NOVA surveyed its members to learn how nurses felt the US Department of Veterans Affairs (VA) was managing personal protective equipment (PPE), testing, communications, staffing needs, and other issues. Following the survey NOVA conducted in-depth interviews with nurses at the VA Boston Healthcare System to better understand how the pandemic was affecting nurses across the Veterans Health Administration (VHA).
The first survey, which was conducted in March and April 2020, included the following questions:
- Do you feel prepared for COVID-19?
- Do you feel your facility’s supply of PPE is adequate?
- Are you aware of the protocol for the distribution of equipment/supplies at your facility?
- Are staff being tested for the virus, and if so, when, and are there enough test kits available for staff and patients?
- Do you believe your facility is properly handling staff who have been exposed to COVID-19 but are asymptomatic?
With the May survey, NOVA aimed to get an update on how things were progressing. This survey asked how the pandemic was affecting members personally, including questions such as: Do you feel you are being supported—mentally and physically? Has VA offered to provide staff mental health counselors or others to help mitigate stress during the crisis?
Results revealed inconsistencies and some confusion. For example, communication among leadership, staff, and veterans continued to change rapidly, causing some misunderstanding overall, respondents said. Some pointed to “e-mail overload” and weekly updates that didn’t work. Others felt communication was “reactive” and “bare minimum”—not “proactive and informative.”
Most (74%) respondents felt that access to PPE was inadequate, and many did not know the protocol for distribution or what supplies were on hand, while 47% felt ill prepared for any COVID-19 onslaught. Although things had improved somewhat by May, > 85% of the respondents said they were reusing what was provided daily and were still finding it difficult to get PPE when needed.
According to respondents, testing was pretty much nonexistent. When asked whether staff were being tested and whether tests for both staff and patients were available, the answer was a resounding No (80%). Nurses’ comments ranged from “staff are not being tested, even if they have been exposed,” to “there are not enough tests for patients, let alone for frontline staff.”
The lack of tests compounded stress. Helen Motroni, ADN, RN, spinal cord injury staff nurse, said, “I have been tested twice for direct exposure to the coronavirus in the past 3 weeks. Luckily, the results were negative, but waiting for the test results was extremely stressful because I have 2 little boys at home. While waiting for results, I self-quarantined and was terrified that I possibly brought the virus home to my children. I never left my room and would talk with my boys through the door and FaceTime. My 8-year-old asked me why I couldn’t stay home so I would not get sick. I explained to him that if every nurse did that, there would be nobody to help those that are sick and suffering.”
As summer approached, testing throughout most of the states remained scarce and contact tracing was a struggle at many facilities. It seemed that, even if testing was available, only those at high risk would be tested.
Moreover, quarantine and sick leave for those who were COVID positive remained a concern, as numbers of those exposed increased. It was widely known and reported that staffing levels within the VHA were inadequate prior to the months going into the pandemic; survey respondents wondered how they would handle vacancies and take care of outside patients as part of the Fourth Mission if staff got sick. One COVID-compelled solution (which NOVA has advocated for years) included expedited hiring practices. Timely application and quicker onboarding enabled VA to hire within weeks rather than months. Since March, VA has hired > 20,000 new employees.
Multiplied Multitasking
The crisis, along with potential and actual staff shortages, has meant that many nurses have been doing double and triple duty—at the least. Danielle Newman, MSN, RN, clinical resource nurse, specialty and outpatient clinics, said, “Throughout the COVID crisis, I have been a direct care provider, an educator, a member of the float pool, an infection preventionist, a colleague, a therapist, and part of a support system to many.… As a PPE nurse educator, I visit every floor in the hospital and help educate staff on how to properly don and doff PPE, as well as monitor the doffing area and assist staff through the doffing process. I have also been involved in obtaining nasal swabs on veterans as well as staff in order to isolate and slow the progression of the virus.”
Some nurses were deployed, some redeployed, and some volunteered for the COVID front. James Murphy, BSN, RN, an emergency department nurse, answered the call. “During the first few weeks of the outbreak and with all its uncertainty, I volunteered to be a COVID nurse.… Seeing how the pandemic has changed practice was an experience.”
Peter Russo, MSN, RN, an OR assistant nurse manager, also found new experience. When the pandemic began to take hold, he was reassigned to Urgent Care and then to the Incident Management Team (IMT). “I asked my associate chief nurse, ‘What is the IMT?’ After my first 2 days on this team, my head was spinning with loads of information, trying to understand and grasp my role as the IMT Nursing Clinical Lead.”
After the third day, his preceptor was redeployed back to the endoscopy department. “I quickly realized my main role as a clinical leader on this team,” Russo says, “was to support and troubleshoot any and all nursing problems, concerns and or questions related to the COVID-19 pandemic among the 3 campuses. One of the more complex challenges was to identify the nursing burn rate of our PPE, especially the N95 masks in all of our COVID units.”
The new experience benefited, him, though. “When changes in policy or procedure happen during the course of an hour, it was one of my responsibilities to notify the nursing staff across the board. I now feel much more confident and competent offering suggestions [about] the many issues that arise.”
Because of the pandemic, all elective surgeries were cancelled and the ambulatory surgery unit where perioperative nurse Valentina Ward, BSN, RN, MPS, CPPS, worked was closed. “As a result,” she says, “I received an accelerated course and was reassigned where I was needed the most. … Serving veterans during the global crisis means so much to me. Despite all that is going on, and the challenges that exist, I have never been prouder to be a frontline nurse.”
The Importance of Maintaining ‘Normal’
COVID-19 seems to subsume everything around it, but there are still health care needs that have nothing to do with the virus. What happens to those patients and their care?
Melissa Varela-Manso, MSN, RN, CRRN, is an inpatient nurse case manager who works with a dedicated team to coordinate admissions and discharges for veterans on the acute Spinal Cord Injury (SCI) unit. “During this pandemic,” she says, “our veterans continue to have non-COVID medical conditions that warrant attention and hospitalization. Therefore, we are following the SCI veterans with non-COVID health issues on the inpatient unit. We also follow our SCI veterans on other units who are being ruled out for COVID and those who have tested positive. We are working diligently to guarantee each veteran gets the quality care they need and deserve while following the evolving guidelines.”
One of the ways VA health care teams have kept things stable for patients, while also ensuring their safety, is through telehealth. VA introduced telehealth in 2003 and is now the nation’s largest telehealth provider. In 2019, > 900,000 veterans received care through telehealth. But it took something like COVID-19 to really show the strengths and utility of being able to care for patients electronically. At the beginning of March, VA conducted about 2,500 telehealth video sessions daily. By June, that had jumped to nearly 25,000—a 1,000% increase.
Sherry Clement, BSN, RN, CFCN, CWCN, CCCN, is the VA Coordinated Transitional Care (C-TraC) Program case manager assigned to the COVID Outpatient Call Team, managing COVID-positive outpatients via telephone and VA video connect. “Working with the call team has been a great collaboration between primary care, telehealth, palliative care, infection control, and occupational health,” she says. “It is amazing to see how fast and efficient our team came together to create contact protocols, consults, notes, and a working plan to organize and oversee the needs of our COVID positive veterans."
Increased usage of telehealth also has meant community-based outpatient clinics continue to be operational, says Karen Boenig, MSN, APRN, ANP-BC, a primary care nurse manager. “Nurses and providers are conducting routine and urgent visits via VA Video Connect and telephone.… At the Framingham clinic, I am working with the Tele-Flu Clinic, which assesses veterans virtually who have symptoms of COVID-19. By utilizing virtual care visits, we can continue providing health care services to our veterans, which minimizes patient and provider exposure and supports social distancing.”
Florence Would be Proud
It seems fitting that 2020 is also the International Year of the Nurse, in celebration of Florence Nightingale’s 200th birthday. That passionate reformer would appreciate NOVA’s mission: “Shaping and Influencing healthcare in the Department of Veterans Affairs.”
In a NOVA blog post (www.vanurse.org), Teresa Morris, director of advocacy and government relations, says, “Thanks to those who responded to our 2 COVID-19 surveys, we were able to use the information as a firsthand look into how nurses felt about VA’s work and response during the Pandemic. The testimony touched on PPE, communication, testing, and comments and concerns that you had during the height of the virus.” She added, “I think some areas that spiked early had less time to prepare while others (MidWest/West) spiked later and used lessons learned.”
“We discussed the findings from the survey and thanked Congress for providing the $19.6 billion in emergency supplemental funding for VA. The funds helped to hire new staff, provide overtime pay, and purchase supplies needed for those caring for veterans and other COVID patients,” She continued.
In May, VA Secretary Robert Wilkie announced his plan to phase in reopening various VA facilities. Respondents were concerned about how the opening up of VA facilities would go as cases continue to spike around the country. They remain committed to their patients, however.
“Never was there a time in the history of nursing that I can say we are needed more,” says Cecilia McVey, MHA, RN, FAAN, Associate Director for Nursing and Patient Care Services, “and nurses make a difference. As you do every day, you are sacrificing more than you may have anticipated when you signed on to be a nurse. Never has it been more important to believe in yourself, in our profession, in the scientists that are guiding us and in ourselves. What you are doing is critical and your sacrifices have not gone unnoticed. I am thankful for all you do—every day—now, and in the future.”
The tsunami we call COVID-19 has threatened to overwhelm everything in its path, with devastating effects that can be hard to quantify or qualify. But the Nurses Organization of Veterans Affairs (NOVA) has taken on the fight. Earlier this year, NOVA surveyed its members to learn how nurses felt the US Department of Veterans Affairs (VA) was managing personal protective equipment (PPE), testing, communications, staffing needs, and other issues. Following the survey NOVA conducted in-depth interviews with nurses at the VA Boston Healthcare System to better understand how the pandemic was affecting nurses across the Veterans Health Administration (VHA).
The first survey, which was conducted in March and April 2020, included the following questions:
- Do you feel prepared for COVID-19?
- Do you feel your facility’s supply of PPE is adequate?
- Are you aware of the protocol for the distribution of equipment/supplies at your facility?
- Are staff being tested for the virus, and if so, when, and are there enough test kits available for staff and patients?
- Do you believe your facility is properly handling staff who have been exposed to COVID-19 but are asymptomatic?
With the May survey, NOVA aimed to get an update on how things were progressing. This survey asked how the pandemic was affecting members personally, including questions such as: Do you feel you are being supported—mentally and physically? Has VA offered to provide staff mental health counselors or others to help mitigate stress during the crisis?
Results revealed inconsistencies and some confusion. For example, communication among leadership, staff, and veterans continued to change rapidly, causing some misunderstanding overall, respondents said. Some pointed to “e-mail overload” and weekly updates that didn’t work. Others felt communication was “reactive” and “bare minimum”—not “proactive and informative.”
Most (74%) respondents felt that access to PPE was inadequate, and many did not know the protocol for distribution or what supplies were on hand, while 47% felt ill prepared for any COVID-19 onslaught. Although things had improved somewhat by May, > 85% of the respondents said they were reusing what was provided daily and were still finding it difficult to get PPE when needed.
According to respondents, testing was pretty much nonexistent. When asked whether staff were being tested and whether tests for both staff and patients were available, the answer was a resounding No (80%). Nurses’ comments ranged from “staff are not being tested, even if they have been exposed,” to “there are not enough tests for patients, let alone for frontline staff.”
The lack of tests compounded stress. Helen Motroni, ADN, RN, spinal cord injury staff nurse, said, “I have been tested twice for direct exposure to the coronavirus in the past 3 weeks. Luckily, the results were negative, but waiting for the test results was extremely stressful because I have 2 little boys at home. While waiting for results, I self-quarantined and was terrified that I possibly brought the virus home to my children. I never left my room and would talk with my boys through the door and FaceTime. My 8-year-old asked me why I couldn’t stay home so I would not get sick. I explained to him that if every nurse did that, there would be nobody to help those that are sick and suffering.”
As summer approached, testing throughout most of the states remained scarce and contact tracing was a struggle at many facilities. It seemed that, even if testing was available, only those at high risk would be tested.
Moreover, quarantine and sick leave for those who were COVID positive remained a concern, as numbers of those exposed increased. It was widely known and reported that staffing levels within the VHA were inadequate prior to the months going into the pandemic; survey respondents wondered how they would handle vacancies and take care of outside patients as part of the Fourth Mission if staff got sick. One COVID-compelled solution (which NOVA has advocated for years) included expedited hiring practices. Timely application and quicker onboarding enabled VA to hire within weeks rather than months. Since March, VA has hired > 20,000 new employees.
Multiplied Multitasking
The crisis, along with potential and actual staff shortages, has meant that many nurses have been doing double and triple duty—at the least. Danielle Newman, MSN, RN, clinical resource nurse, specialty and outpatient clinics, said, “Throughout the COVID crisis, I have been a direct care provider, an educator, a member of the float pool, an infection preventionist, a colleague, a therapist, and part of a support system to many.… As a PPE nurse educator, I visit every floor in the hospital and help educate staff on how to properly don and doff PPE, as well as monitor the doffing area and assist staff through the doffing process. I have also been involved in obtaining nasal swabs on veterans as well as staff in order to isolate and slow the progression of the virus.”
Some nurses were deployed, some redeployed, and some volunteered for the COVID front. James Murphy, BSN, RN, an emergency department nurse, answered the call. “During the first few weeks of the outbreak and with all its uncertainty, I volunteered to be a COVID nurse.… Seeing how the pandemic has changed practice was an experience.”
Peter Russo, MSN, RN, an OR assistant nurse manager, also found new experience. When the pandemic began to take hold, he was reassigned to Urgent Care and then to the Incident Management Team (IMT). “I asked my associate chief nurse, ‘What is the IMT?’ After my first 2 days on this team, my head was spinning with loads of information, trying to understand and grasp my role as the IMT Nursing Clinical Lead.”
After the third day, his preceptor was redeployed back to the endoscopy department. “I quickly realized my main role as a clinical leader on this team,” Russo says, “was to support and troubleshoot any and all nursing problems, concerns and or questions related to the COVID-19 pandemic among the 3 campuses. One of the more complex challenges was to identify the nursing burn rate of our PPE, especially the N95 masks in all of our COVID units.”
The new experience benefited, him, though. “When changes in policy or procedure happen during the course of an hour, it was one of my responsibilities to notify the nursing staff across the board. I now feel much more confident and competent offering suggestions [about] the many issues that arise.”
Because of the pandemic, all elective surgeries were cancelled and the ambulatory surgery unit where perioperative nurse Valentina Ward, BSN, RN, MPS, CPPS, worked was closed. “As a result,” she says, “I received an accelerated course and was reassigned where I was needed the most. … Serving veterans during the global crisis means so much to me. Despite all that is going on, and the challenges that exist, I have never been prouder to be a frontline nurse.”
The Importance of Maintaining ‘Normal’
COVID-19 seems to subsume everything around it, but there are still health care needs that have nothing to do with the virus. What happens to those patients and their care?
Melissa Varela-Manso, MSN, RN, CRRN, is an inpatient nurse case manager who works with a dedicated team to coordinate admissions and discharges for veterans on the acute Spinal Cord Injury (SCI) unit. “During this pandemic,” she says, “our veterans continue to have non-COVID medical conditions that warrant attention and hospitalization. Therefore, we are following the SCI veterans with non-COVID health issues on the inpatient unit. We also follow our SCI veterans on other units who are being ruled out for COVID and those who have tested positive. We are working diligently to guarantee each veteran gets the quality care they need and deserve while following the evolving guidelines.”
One of the ways VA health care teams have kept things stable for patients, while also ensuring their safety, is through telehealth. VA introduced telehealth in 2003 and is now the nation’s largest telehealth provider. In 2019, > 900,000 veterans received care through telehealth. But it took something like COVID-19 to really show the strengths and utility of being able to care for patients electronically. At the beginning of March, VA conducted about 2,500 telehealth video sessions daily. By June, that had jumped to nearly 25,000—a 1,000% increase.
Sherry Clement, BSN, RN, CFCN, CWCN, CCCN, is the VA Coordinated Transitional Care (C-TraC) Program case manager assigned to the COVID Outpatient Call Team, managing COVID-positive outpatients via telephone and VA video connect. “Working with the call team has been a great collaboration between primary care, telehealth, palliative care, infection control, and occupational health,” she says. “It is amazing to see how fast and efficient our team came together to create contact protocols, consults, notes, and a working plan to organize and oversee the needs of our COVID positive veterans."
Increased usage of telehealth also has meant community-based outpatient clinics continue to be operational, says Karen Boenig, MSN, APRN, ANP-BC, a primary care nurse manager. “Nurses and providers are conducting routine and urgent visits via VA Video Connect and telephone.… At the Framingham clinic, I am working with the Tele-Flu Clinic, which assesses veterans virtually who have symptoms of COVID-19. By utilizing virtual care visits, we can continue providing health care services to our veterans, which minimizes patient and provider exposure and supports social distancing.”
Florence Would be Proud
It seems fitting that 2020 is also the International Year of the Nurse, in celebration of Florence Nightingale’s 200th birthday. That passionate reformer would appreciate NOVA’s mission: “Shaping and Influencing healthcare in the Department of Veterans Affairs.”
In a NOVA blog post (www.vanurse.org), Teresa Morris, director of advocacy and government relations, says, “Thanks to those who responded to our 2 COVID-19 surveys, we were able to use the information as a firsthand look into how nurses felt about VA’s work and response during the Pandemic. The testimony touched on PPE, communication, testing, and comments and concerns that you had during the height of the virus.” She added, “I think some areas that spiked early had less time to prepare while others (MidWest/West) spiked later and used lessons learned.”
“We discussed the findings from the survey and thanked Congress for providing the $19.6 billion in emergency supplemental funding for VA. The funds helped to hire new staff, provide overtime pay, and purchase supplies needed for those caring for veterans and other COVID patients,” She continued.
In May, VA Secretary Robert Wilkie announced his plan to phase in reopening various VA facilities. Respondents were concerned about how the opening up of VA facilities would go as cases continue to spike around the country. They remain committed to their patients, however.
“Never was there a time in the history of nursing that I can say we are needed more,” says Cecilia McVey, MHA, RN, FAAN, Associate Director for Nursing and Patient Care Services, “and nurses make a difference. As you do every day, you are sacrificing more than you may have anticipated when you signed on to be a nurse. Never has it been more important to believe in yourself, in our profession, in the scientists that are guiding us and in ourselves. What you are doing is critical and your sacrifices have not gone unnoticed. I am thankful for all you do—every day—now, and in the future.”
Cancer researchers cross over to COVID-19 clinical trials
When the first reports emerged of “cytokine storm” in patients with severe COVID-19, all eyes turned to cancer research. Oncologists have years of experience reigning in “cytokine release syndrome” (CRS) in patients treated with chimeric antigen receptor (CAR) therapies for advanced blood cancers.
There was hope that drugs used to quell CRS in patients with cancer would be effective in patients with severe COVID. But the promise of a quick fix with oncology medications has yet to be fully realized.
Part of the problem is that the two conditions, while analogous, are “not the same,” said Nirali Shah, MD, head of the hematologic malignancies section in the pediatric oncology branch at the National Cancer Institute.
“You have to understand the underlying pathophysiology, what triggers the inflammation,” Dr. Shah said.
CAR T–related CRS is caused by activated T cells in patients with cancer who often do not have an infection, she explained. In contrast, cytokine storm in COVID-19 is triggered by a viral pathogen that can drive “out of control” inflammation. These differences may explain why drugs work in the first instance, but not in the second, she added. Drugs that inhibit interleukin-6 (such as tocilizumab, sarilumab, and siltuximab) are used with great success to dampen down the CRS in patients receiving CAR therapy for blood cancers. And although trials of these agents in patients with COVID are still ongoing, initial results are disappointing.
The first global, phase 3 randomized controlled trial of tocilizumab in severe COVID-19 failed to meet its primary endpoint of improved clinical status, and it did not meet its secondary endpoint of improved mortality at week 4.
In its recent recommendations, the National Institutes of Health noted a lack of data to support the efficacy of IL-6 inhibitors in COVID-19, and recommended against their use, except as part of a clinical trial.
Trimming the tree vs. cutting it down
As researchers have begun to decode the immune process underlying severe COVID-19, they have turned to other cancer drugs to tame cytokine storm.
Louis Staudt, MD, PhD, and Wyndham Wilson, MD, PhD, both at the NCI, think that cytokine storm in COVID-19 is driven by macrophages, which trigger release of multiple cytokines.
For years, the pair have been studying lymphoid tumors. Dr. Staudt is chief of the lymphoid malignancies branch at the NCI, and Wilson is head of the lymphoma therapeutics section. In past work, Dr. Staudt discovered that inhibiting an enzyme called bruton tyrosine kinase (BTK) dampens macrophage function.
When the pandemic began, Dr. Staudt and Dr. Wilson realized that singling out just one cytokine like IL-6 may not be enough. They thought that a more effective approach may be to target macrophages with a BTK inhibitor called acalabrutinib (Calquence), which would inhibit multiple cytokines at the same time.
Dr. Staudt likens the immune response to a tree, with the macrophages composing the tree trunk and the limbs made up of individual cytokines.
“Targeting macrophages is getting at the trunk of the problem,” he said. “You’re only cutting off the limbs with tocilizumab.”
In just 3 days, Dr. Staudt and Dr. Wilson went from concept to approval to launching a prospective, observational study. The study took place at five centers in the US, and included 19 patients hospitalized with COVID-19; the results were published in Science Immunology. Over a treatment course of 14 days, the majority of patients treated off-label with acalabrutinib improved, some within 24 hours. Eight of 11 patients on supplemental oxygen were discharged on room air. Four of eight patients on ventilators were extubated, with two of these discharged on room air. Two patients on ventilators died. No discernible toxicity was noted.
Analyses also showed increased BTK activity and elevated IL-6 levels in monocytes – precursors of macrophages – in patients with severe COVID-19, compared with healthy volunteers.
“We showed that the target of acalabrutinib was active in the immune cells of patients with severe COVID-19,” Dr. Staudt said. “So we have the target. We have the drug to hit the target. And we have an apparent clinical benefit.”
Those three things were compelling enough to launch the CALAVI phase 2 trial, an open-label, randomized, controlled trial, sponsored by AstraZeneca and the NCI, that is being conducted in the United States and internationally. It is testing acalabrutinib with best supportive care versus BSC alone in people hospitalized with COVID-19. The trial is scheduled to be completed on Nov. 26.
Preliminary insights from this trial are expected soon. “These are not insights that we will likely publish, but they are important insights that will lead to the launch of a definitive double-blind, randomized, phase 3 trial, which we hope to launch in the next month or so,” Dr. Wilson said.
Targeting inflammation and infection simultaneously
Other scientists are investigating inhibitors of Janus kinase (JAK), a family of enzymes that play a key role in orchestrating immune responses, particularly cytokines. Interest in JAK inhibition to control hyperinflammation in cancer goes back at least 15 years, and drugs that act as JAK inhibitors are already approved for use in the treatment of myelofibrosis (ruxolitinib [Jakafi], fedratinib [Inrebic]) and also for rheumatoid arthritis (upadacitinib [Rinvoq], baricitinib [Olumiant]).
“It wasn’t a huge leap for those of us with a lot of understanding of JAK inhibitors to propose taking them into the clinic to treat patients with COVID-19,” commented John Mascarenhas, MD, the leader of clinical investigation in the myeloproliferative disorders program at the Icahn School of Medicine at Mount Sinai, New York.
Dr. Mascarenhas is also principal investigator of the PRE-VENT trial, which is comparing the investigational JAK2 inhibitor pacritinib plus standard of care to standard of care alone in patients hospitalized with severe COVID-19, with and without cancer. The trial is sponsored by CTI BioPharma (manufacturer of pacritinib), and is taking place at 10 sites in the United States.
In a move that may raise eyebrows, PRE-VENT skipped phase 1 and 2 and went straight to phase 3. Pacritinib has yet to receive FDA approval and has mostly been studied in myelofibrosis, an intensely inflammatory disease.
The decision was based on trials of pacritinib in hematologic malignancies and also on results from a phase 2 study in China that found possible clinical benefit for the JAK 1/2 inhibitor ruxolitinib in 43 patients hospitalized with severe COVID-19, although results were not statistically significant, Dr. Mascarenhas explained.
Recent results from Lilly’s ACTT-2 study have provided further support for the role of JAK inhibitors in treating cytokine storm. ACTT-2 is a phase 3, double-blind, placebo-controlled, randomized, controlled trial sponsored by the NIH and NIAID comparing the JAK 1/2 inhibitor baricitinib plus the antiviral remdesivir with remdesivir alone in patients hospitalized with COVID-19. In September, Lilly announced that the trial met its primary endpoint of decreased time to recovery in patients who received baricitinib in combination with remdesivir.
But pacritinib’s mechanism of action may take things a step further. The drug selectively inhibits JAK2 and spares JAK1, which is important for antiviral activity in the immune system. Also, in vitro data suggests pacritinib may simultaneously reduce inflammation and fight off the virus by selectively inhibiting two additional enzymes and two other receptors.
“The rationale to me is very strong for using pacritinib,” Dr. Mascarenhas said. “I think this approach was bold but appropriate.”
The main safety concern with pacritinib could be bleeding, especially among patients on anticoagulants, Dr. Mascarenhas said. Because some patients with severe COVID-19 tend to develop blood clots, anticoagulation has become the standard of care at many institutions.
Because the trial is just beginning – only a minority of the total planned population of 358 patients has been enrolled – no interim results are available.
Right drug, wrong time?
IL-6 inhibition could still have a role to play in COVID-19, but the trick could be in the timing. Most of the trials so far have studied tocilizumab in patients with severe COVID-19, many of whom were already on ventilators. At that point, it may be too late to reverse the damage that has already taken place.
One of the main reasons tocilizumab works so well in CRS after CAR T therapy is that oncologists have learned how to use it early, often within 24 hours of fever onset. Oncologists use the American Society for Transplantation and Cellular Therapy consensus grading system, which helps them identify CRS when it is easier to control.
But applying the ASTCT grading system to COVID-19 is problematic. “Almost by definition, patients hospitalized with COVID-19 have low oxygen levels, which throws off the scale,” said Joshua Hill, MD, an infectious disease specialist at Fred Hutchinson Cancer Research Center in Seattle, who has research expertise in infectious complications after CAR T therapy.
“The key is to intervene earlier to prevent damage to the lungs and other end organs. We don’t have anything magical that will reverse that damage,” Dr. Hill said.
Results from the phase 3 trial EMPACTA trial (sponsored by Genentech) seem to bear this out. EMPACTA is evaluating use of tocilizumab in hospitalized patients with less severe COVID-19 who do not yet require mechanical ventilation. The trial is notable for being the first global phase 3 trial to demonstrate efficacy for tocilizumab vs placebo in hospitalized patients with COVID-19 pneumonia, and for including a high percentage of racial/ethnic minorities (85% of 389 participants), who have been hard hit by the pandemic and have historically been underrepresented in drug trials.
Last month, Roche announced that EMPACTA met its primary endpoint. Results showed that patients hospitalized with COVID-19 pneumonia who received tocilizumab plus standard of care were 44% less likely to go on mechanical ventilation or die, compared with those who received placebo plus standard of care (P = .0348), although there were no statistically significant differences in death by day 28 between tocilizumab and placebo (10.4% vs. 8.6%, P = .5146).
However, earlier administration of tocilizumab raises another issue. IL-6 and its pathway are important for clearing viral infections. Using tocilizumab in the context of an ongoing infection could raise safety issues.
Also, tocilizumab sticks around in the body for a relatively long time. In the treatment of rheumatoid arthritis, it is dosed once a month, and it carries a black box warning for reactivation of tuberculosis.
Whereas results from EMPACTA showed similar rates of infection associated with tocilizumab and placebo (10% vs. 11%), at least one other study has found increased rates of superinfection in patients with severe COVID-19 who received tocilizumab. Overall, though, the drug was associated with decreased risk of death in the latter study.
A phase 2 trial called COVIDOSE is tackling the safety issue. COVIDOSE is evaluating whether low-dose tocilizumab is effective in noncritical COVID-19 patients, with the idea that lower doses could be safer. Early results published as a preprint before peer review indicated that low-dose tocilizumab (ranging from 40 mg to 200 mg) was associated with clinical improvement in 32 noncritical patients hospitalized with COVID-19.
Five patients (15.6%) developed bacterial superinfections, and five (15.6%) died by 28-day follow-up, although there wasn’t a perfect “overlap” between these groups of patients. Bacterial superinfection was not the cause of death in all five patients who died, and not all patients who died developed bacterial superinfections, according to senior author Pankti Reid, MD, MPH, an assistant professor of medicine at the University of Chicago.
Results from COVIDOSE also showed that treatment with tocilizumab did not seem to affect the ability of patients to develop antibodies against COVID-19. The results set the stage for a larger randomized, controlled trial (still ongoing) to determine the optimal dose of tocilizumab.
Still, Dr. Hill urges caution.
Many of these immunomodulators have been used only in the context of a clinical trial, or only for patients with terminal cancer and no other treatment options. In patients with cancer, these drugs have been studied and have shown an “acceptable safety profile,” according to Dr. Shah.
But this is a different situation, and when it comes to repurposing them to relatively healthy patients with COVID-19, Dr. Hill emphasized the need for careful research.
“We’re always very concerned about giving drugs that suppress the immune response if people have active infections,” Dr. Hill said. “Often times we think it makes things worse, and it typically does.”
Dr. Mascarenhas reported institutional research funding from CTI Biopharma. Dr. Hill, Dr. Staudt, Dr. Wilson, and Dr. Shah disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
When the first reports emerged of “cytokine storm” in patients with severe COVID-19, all eyes turned to cancer research. Oncologists have years of experience reigning in “cytokine release syndrome” (CRS) in patients treated with chimeric antigen receptor (CAR) therapies for advanced blood cancers.
There was hope that drugs used to quell CRS in patients with cancer would be effective in patients with severe COVID. But the promise of a quick fix with oncology medications has yet to be fully realized.
Part of the problem is that the two conditions, while analogous, are “not the same,” said Nirali Shah, MD, head of the hematologic malignancies section in the pediatric oncology branch at the National Cancer Institute.
“You have to understand the underlying pathophysiology, what triggers the inflammation,” Dr. Shah said.
CAR T–related CRS is caused by activated T cells in patients with cancer who often do not have an infection, she explained. In contrast, cytokine storm in COVID-19 is triggered by a viral pathogen that can drive “out of control” inflammation. These differences may explain why drugs work in the first instance, but not in the second, she added. Drugs that inhibit interleukin-6 (such as tocilizumab, sarilumab, and siltuximab) are used with great success to dampen down the CRS in patients receiving CAR therapy for blood cancers. And although trials of these agents in patients with COVID are still ongoing, initial results are disappointing.
The first global, phase 3 randomized controlled trial of tocilizumab in severe COVID-19 failed to meet its primary endpoint of improved clinical status, and it did not meet its secondary endpoint of improved mortality at week 4.
In its recent recommendations, the National Institutes of Health noted a lack of data to support the efficacy of IL-6 inhibitors in COVID-19, and recommended against their use, except as part of a clinical trial.
Trimming the tree vs. cutting it down
As researchers have begun to decode the immune process underlying severe COVID-19, they have turned to other cancer drugs to tame cytokine storm.
Louis Staudt, MD, PhD, and Wyndham Wilson, MD, PhD, both at the NCI, think that cytokine storm in COVID-19 is driven by macrophages, which trigger release of multiple cytokines.
For years, the pair have been studying lymphoid tumors. Dr. Staudt is chief of the lymphoid malignancies branch at the NCI, and Wilson is head of the lymphoma therapeutics section. In past work, Dr. Staudt discovered that inhibiting an enzyme called bruton tyrosine kinase (BTK) dampens macrophage function.
When the pandemic began, Dr. Staudt and Dr. Wilson realized that singling out just one cytokine like IL-6 may not be enough. They thought that a more effective approach may be to target macrophages with a BTK inhibitor called acalabrutinib (Calquence), which would inhibit multiple cytokines at the same time.
Dr. Staudt likens the immune response to a tree, with the macrophages composing the tree trunk and the limbs made up of individual cytokines.
“Targeting macrophages is getting at the trunk of the problem,” he said. “You’re only cutting off the limbs with tocilizumab.”
In just 3 days, Dr. Staudt and Dr. Wilson went from concept to approval to launching a prospective, observational study. The study took place at five centers in the US, and included 19 patients hospitalized with COVID-19; the results were published in Science Immunology. Over a treatment course of 14 days, the majority of patients treated off-label with acalabrutinib improved, some within 24 hours. Eight of 11 patients on supplemental oxygen were discharged on room air. Four of eight patients on ventilators were extubated, with two of these discharged on room air. Two patients on ventilators died. No discernible toxicity was noted.
Analyses also showed increased BTK activity and elevated IL-6 levels in monocytes – precursors of macrophages – in patients with severe COVID-19, compared with healthy volunteers.
“We showed that the target of acalabrutinib was active in the immune cells of patients with severe COVID-19,” Dr. Staudt said. “So we have the target. We have the drug to hit the target. And we have an apparent clinical benefit.”
Those three things were compelling enough to launch the CALAVI phase 2 trial, an open-label, randomized, controlled trial, sponsored by AstraZeneca and the NCI, that is being conducted in the United States and internationally. It is testing acalabrutinib with best supportive care versus BSC alone in people hospitalized with COVID-19. The trial is scheduled to be completed on Nov. 26.
Preliminary insights from this trial are expected soon. “These are not insights that we will likely publish, but they are important insights that will lead to the launch of a definitive double-blind, randomized, phase 3 trial, which we hope to launch in the next month or so,” Dr. Wilson said.
Targeting inflammation and infection simultaneously
Other scientists are investigating inhibitors of Janus kinase (JAK), a family of enzymes that play a key role in orchestrating immune responses, particularly cytokines. Interest in JAK inhibition to control hyperinflammation in cancer goes back at least 15 years, and drugs that act as JAK inhibitors are already approved for use in the treatment of myelofibrosis (ruxolitinib [Jakafi], fedratinib [Inrebic]) and also for rheumatoid arthritis (upadacitinib [Rinvoq], baricitinib [Olumiant]).
“It wasn’t a huge leap for those of us with a lot of understanding of JAK inhibitors to propose taking them into the clinic to treat patients with COVID-19,” commented John Mascarenhas, MD, the leader of clinical investigation in the myeloproliferative disorders program at the Icahn School of Medicine at Mount Sinai, New York.
Dr. Mascarenhas is also principal investigator of the PRE-VENT trial, which is comparing the investigational JAK2 inhibitor pacritinib plus standard of care to standard of care alone in patients hospitalized with severe COVID-19, with and without cancer. The trial is sponsored by CTI BioPharma (manufacturer of pacritinib), and is taking place at 10 sites in the United States.
In a move that may raise eyebrows, PRE-VENT skipped phase 1 and 2 and went straight to phase 3. Pacritinib has yet to receive FDA approval and has mostly been studied in myelofibrosis, an intensely inflammatory disease.
The decision was based on trials of pacritinib in hematologic malignancies and also on results from a phase 2 study in China that found possible clinical benefit for the JAK 1/2 inhibitor ruxolitinib in 43 patients hospitalized with severe COVID-19, although results were not statistically significant, Dr. Mascarenhas explained.
Recent results from Lilly’s ACTT-2 study have provided further support for the role of JAK inhibitors in treating cytokine storm. ACTT-2 is a phase 3, double-blind, placebo-controlled, randomized, controlled trial sponsored by the NIH and NIAID comparing the JAK 1/2 inhibitor baricitinib plus the antiviral remdesivir with remdesivir alone in patients hospitalized with COVID-19. In September, Lilly announced that the trial met its primary endpoint of decreased time to recovery in patients who received baricitinib in combination with remdesivir.
But pacritinib’s mechanism of action may take things a step further. The drug selectively inhibits JAK2 and spares JAK1, which is important for antiviral activity in the immune system. Also, in vitro data suggests pacritinib may simultaneously reduce inflammation and fight off the virus by selectively inhibiting two additional enzymes and two other receptors.
“The rationale to me is very strong for using pacritinib,” Dr. Mascarenhas said. “I think this approach was bold but appropriate.”
The main safety concern with pacritinib could be bleeding, especially among patients on anticoagulants, Dr. Mascarenhas said. Because some patients with severe COVID-19 tend to develop blood clots, anticoagulation has become the standard of care at many institutions.
Because the trial is just beginning – only a minority of the total planned population of 358 patients has been enrolled – no interim results are available.
Right drug, wrong time?
IL-6 inhibition could still have a role to play in COVID-19, but the trick could be in the timing. Most of the trials so far have studied tocilizumab in patients with severe COVID-19, many of whom were already on ventilators. At that point, it may be too late to reverse the damage that has already taken place.
One of the main reasons tocilizumab works so well in CRS after CAR T therapy is that oncologists have learned how to use it early, often within 24 hours of fever onset. Oncologists use the American Society for Transplantation and Cellular Therapy consensus grading system, which helps them identify CRS when it is easier to control.
But applying the ASTCT grading system to COVID-19 is problematic. “Almost by definition, patients hospitalized with COVID-19 have low oxygen levels, which throws off the scale,” said Joshua Hill, MD, an infectious disease specialist at Fred Hutchinson Cancer Research Center in Seattle, who has research expertise in infectious complications after CAR T therapy.
“The key is to intervene earlier to prevent damage to the lungs and other end organs. We don’t have anything magical that will reverse that damage,” Dr. Hill said.
Results from the phase 3 trial EMPACTA trial (sponsored by Genentech) seem to bear this out. EMPACTA is evaluating use of tocilizumab in hospitalized patients with less severe COVID-19 who do not yet require mechanical ventilation. The trial is notable for being the first global phase 3 trial to demonstrate efficacy for tocilizumab vs placebo in hospitalized patients with COVID-19 pneumonia, and for including a high percentage of racial/ethnic minorities (85% of 389 participants), who have been hard hit by the pandemic and have historically been underrepresented in drug trials.
Last month, Roche announced that EMPACTA met its primary endpoint. Results showed that patients hospitalized with COVID-19 pneumonia who received tocilizumab plus standard of care were 44% less likely to go on mechanical ventilation or die, compared with those who received placebo plus standard of care (P = .0348), although there were no statistically significant differences in death by day 28 between tocilizumab and placebo (10.4% vs. 8.6%, P = .5146).
However, earlier administration of tocilizumab raises another issue. IL-6 and its pathway are important for clearing viral infections. Using tocilizumab in the context of an ongoing infection could raise safety issues.
Also, tocilizumab sticks around in the body for a relatively long time. In the treatment of rheumatoid arthritis, it is dosed once a month, and it carries a black box warning for reactivation of tuberculosis.
Whereas results from EMPACTA showed similar rates of infection associated with tocilizumab and placebo (10% vs. 11%), at least one other study has found increased rates of superinfection in patients with severe COVID-19 who received tocilizumab. Overall, though, the drug was associated with decreased risk of death in the latter study.
A phase 2 trial called COVIDOSE is tackling the safety issue. COVIDOSE is evaluating whether low-dose tocilizumab is effective in noncritical COVID-19 patients, with the idea that lower doses could be safer. Early results published as a preprint before peer review indicated that low-dose tocilizumab (ranging from 40 mg to 200 mg) was associated with clinical improvement in 32 noncritical patients hospitalized with COVID-19.
Five patients (15.6%) developed bacterial superinfections, and five (15.6%) died by 28-day follow-up, although there wasn’t a perfect “overlap” between these groups of patients. Bacterial superinfection was not the cause of death in all five patients who died, and not all patients who died developed bacterial superinfections, according to senior author Pankti Reid, MD, MPH, an assistant professor of medicine at the University of Chicago.
Results from COVIDOSE also showed that treatment with tocilizumab did not seem to affect the ability of patients to develop antibodies against COVID-19. The results set the stage for a larger randomized, controlled trial (still ongoing) to determine the optimal dose of tocilizumab.
Still, Dr. Hill urges caution.
Many of these immunomodulators have been used only in the context of a clinical trial, or only for patients with terminal cancer and no other treatment options. In patients with cancer, these drugs have been studied and have shown an “acceptable safety profile,” according to Dr. Shah.
But this is a different situation, and when it comes to repurposing them to relatively healthy patients with COVID-19, Dr. Hill emphasized the need for careful research.
“We’re always very concerned about giving drugs that suppress the immune response if people have active infections,” Dr. Hill said. “Often times we think it makes things worse, and it typically does.”
Dr. Mascarenhas reported institutional research funding from CTI Biopharma. Dr. Hill, Dr. Staudt, Dr. Wilson, and Dr. Shah disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
When the first reports emerged of “cytokine storm” in patients with severe COVID-19, all eyes turned to cancer research. Oncologists have years of experience reigning in “cytokine release syndrome” (CRS) in patients treated with chimeric antigen receptor (CAR) therapies for advanced blood cancers.
There was hope that drugs used to quell CRS in patients with cancer would be effective in patients with severe COVID. But the promise of a quick fix with oncology medications has yet to be fully realized.
Part of the problem is that the two conditions, while analogous, are “not the same,” said Nirali Shah, MD, head of the hematologic malignancies section in the pediatric oncology branch at the National Cancer Institute.
“You have to understand the underlying pathophysiology, what triggers the inflammation,” Dr. Shah said.
CAR T–related CRS is caused by activated T cells in patients with cancer who often do not have an infection, she explained. In contrast, cytokine storm in COVID-19 is triggered by a viral pathogen that can drive “out of control” inflammation. These differences may explain why drugs work in the first instance, but not in the second, she added. Drugs that inhibit interleukin-6 (such as tocilizumab, sarilumab, and siltuximab) are used with great success to dampen down the CRS in patients receiving CAR therapy for blood cancers. And although trials of these agents in patients with COVID are still ongoing, initial results are disappointing.
The first global, phase 3 randomized controlled trial of tocilizumab in severe COVID-19 failed to meet its primary endpoint of improved clinical status, and it did not meet its secondary endpoint of improved mortality at week 4.
In its recent recommendations, the National Institutes of Health noted a lack of data to support the efficacy of IL-6 inhibitors in COVID-19, and recommended against their use, except as part of a clinical trial.
Trimming the tree vs. cutting it down
As researchers have begun to decode the immune process underlying severe COVID-19, they have turned to other cancer drugs to tame cytokine storm.
Louis Staudt, MD, PhD, and Wyndham Wilson, MD, PhD, both at the NCI, think that cytokine storm in COVID-19 is driven by macrophages, which trigger release of multiple cytokines.
For years, the pair have been studying lymphoid tumors. Dr. Staudt is chief of the lymphoid malignancies branch at the NCI, and Wilson is head of the lymphoma therapeutics section. In past work, Dr. Staudt discovered that inhibiting an enzyme called bruton tyrosine kinase (BTK) dampens macrophage function.
When the pandemic began, Dr. Staudt and Dr. Wilson realized that singling out just one cytokine like IL-6 may not be enough. They thought that a more effective approach may be to target macrophages with a BTK inhibitor called acalabrutinib (Calquence), which would inhibit multiple cytokines at the same time.
Dr. Staudt likens the immune response to a tree, with the macrophages composing the tree trunk and the limbs made up of individual cytokines.
“Targeting macrophages is getting at the trunk of the problem,” he said. “You’re only cutting off the limbs with tocilizumab.”
In just 3 days, Dr. Staudt and Dr. Wilson went from concept to approval to launching a prospective, observational study. The study took place at five centers in the US, and included 19 patients hospitalized with COVID-19; the results were published in Science Immunology. Over a treatment course of 14 days, the majority of patients treated off-label with acalabrutinib improved, some within 24 hours. Eight of 11 patients on supplemental oxygen were discharged on room air. Four of eight patients on ventilators were extubated, with two of these discharged on room air. Two patients on ventilators died. No discernible toxicity was noted.
Analyses also showed increased BTK activity and elevated IL-6 levels in monocytes – precursors of macrophages – in patients with severe COVID-19, compared with healthy volunteers.
“We showed that the target of acalabrutinib was active in the immune cells of patients with severe COVID-19,” Dr. Staudt said. “So we have the target. We have the drug to hit the target. And we have an apparent clinical benefit.”
Those three things were compelling enough to launch the CALAVI phase 2 trial, an open-label, randomized, controlled trial, sponsored by AstraZeneca and the NCI, that is being conducted in the United States and internationally. It is testing acalabrutinib with best supportive care versus BSC alone in people hospitalized with COVID-19. The trial is scheduled to be completed on Nov. 26.
Preliminary insights from this trial are expected soon. “These are not insights that we will likely publish, but they are important insights that will lead to the launch of a definitive double-blind, randomized, phase 3 trial, which we hope to launch in the next month or so,” Dr. Wilson said.
Targeting inflammation and infection simultaneously
Other scientists are investigating inhibitors of Janus kinase (JAK), a family of enzymes that play a key role in orchestrating immune responses, particularly cytokines. Interest in JAK inhibition to control hyperinflammation in cancer goes back at least 15 years, and drugs that act as JAK inhibitors are already approved for use in the treatment of myelofibrosis (ruxolitinib [Jakafi], fedratinib [Inrebic]) and also for rheumatoid arthritis (upadacitinib [Rinvoq], baricitinib [Olumiant]).
“It wasn’t a huge leap for those of us with a lot of understanding of JAK inhibitors to propose taking them into the clinic to treat patients with COVID-19,” commented John Mascarenhas, MD, the leader of clinical investigation in the myeloproliferative disorders program at the Icahn School of Medicine at Mount Sinai, New York.
Dr. Mascarenhas is also principal investigator of the PRE-VENT trial, which is comparing the investigational JAK2 inhibitor pacritinib plus standard of care to standard of care alone in patients hospitalized with severe COVID-19, with and without cancer. The trial is sponsored by CTI BioPharma (manufacturer of pacritinib), and is taking place at 10 sites in the United States.
In a move that may raise eyebrows, PRE-VENT skipped phase 1 and 2 and went straight to phase 3. Pacritinib has yet to receive FDA approval and has mostly been studied in myelofibrosis, an intensely inflammatory disease.
The decision was based on trials of pacritinib in hematologic malignancies and also on results from a phase 2 study in China that found possible clinical benefit for the JAK 1/2 inhibitor ruxolitinib in 43 patients hospitalized with severe COVID-19, although results were not statistically significant, Dr. Mascarenhas explained.
Recent results from Lilly’s ACTT-2 study have provided further support for the role of JAK inhibitors in treating cytokine storm. ACTT-2 is a phase 3, double-blind, placebo-controlled, randomized, controlled trial sponsored by the NIH and NIAID comparing the JAK 1/2 inhibitor baricitinib plus the antiviral remdesivir with remdesivir alone in patients hospitalized with COVID-19. In September, Lilly announced that the trial met its primary endpoint of decreased time to recovery in patients who received baricitinib in combination with remdesivir.
But pacritinib’s mechanism of action may take things a step further. The drug selectively inhibits JAK2 and spares JAK1, which is important for antiviral activity in the immune system. Also, in vitro data suggests pacritinib may simultaneously reduce inflammation and fight off the virus by selectively inhibiting two additional enzymes and two other receptors.
“The rationale to me is very strong for using pacritinib,” Dr. Mascarenhas said. “I think this approach was bold but appropriate.”
The main safety concern with pacritinib could be bleeding, especially among patients on anticoagulants, Dr. Mascarenhas said. Because some patients with severe COVID-19 tend to develop blood clots, anticoagulation has become the standard of care at many institutions.
Because the trial is just beginning – only a minority of the total planned population of 358 patients has been enrolled – no interim results are available.
Right drug, wrong time?
IL-6 inhibition could still have a role to play in COVID-19, but the trick could be in the timing. Most of the trials so far have studied tocilizumab in patients with severe COVID-19, many of whom were already on ventilators. At that point, it may be too late to reverse the damage that has already taken place.
One of the main reasons tocilizumab works so well in CRS after CAR T therapy is that oncologists have learned how to use it early, often within 24 hours of fever onset. Oncologists use the American Society for Transplantation and Cellular Therapy consensus grading system, which helps them identify CRS when it is easier to control.
But applying the ASTCT grading system to COVID-19 is problematic. “Almost by definition, patients hospitalized with COVID-19 have low oxygen levels, which throws off the scale,” said Joshua Hill, MD, an infectious disease specialist at Fred Hutchinson Cancer Research Center in Seattle, who has research expertise in infectious complications after CAR T therapy.
“The key is to intervene earlier to prevent damage to the lungs and other end organs. We don’t have anything magical that will reverse that damage,” Dr. Hill said.
Results from the phase 3 trial EMPACTA trial (sponsored by Genentech) seem to bear this out. EMPACTA is evaluating use of tocilizumab in hospitalized patients with less severe COVID-19 who do not yet require mechanical ventilation. The trial is notable for being the first global phase 3 trial to demonstrate efficacy for tocilizumab vs placebo in hospitalized patients with COVID-19 pneumonia, and for including a high percentage of racial/ethnic minorities (85% of 389 participants), who have been hard hit by the pandemic and have historically been underrepresented in drug trials.
Last month, Roche announced that EMPACTA met its primary endpoint. Results showed that patients hospitalized with COVID-19 pneumonia who received tocilizumab plus standard of care were 44% less likely to go on mechanical ventilation or die, compared with those who received placebo plus standard of care (P = .0348), although there were no statistically significant differences in death by day 28 between tocilizumab and placebo (10.4% vs. 8.6%, P = .5146).
However, earlier administration of tocilizumab raises another issue. IL-6 and its pathway are important for clearing viral infections. Using tocilizumab in the context of an ongoing infection could raise safety issues.
Also, tocilizumab sticks around in the body for a relatively long time. In the treatment of rheumatoid arthritis, it is dosed once a month, and it carries a black box warning for reactivation of tuberculosis.
Whereas results from EMPACTA showed similar rates of infection associated with tocilizumab and placebo (10% vs. 11%), at least one other study has found increased rates of superinfection in patients with severe COVID-19 who received tocilizumab. Overall, though, the drug was associated with decreased risk of death in the latter study.
A phase 2 trial called COVIDOSE is tackling the safety issue. COVIDOSE is evaluating whether low-dose tocilizumab is effective in noncritical COVID-19 patients, with the idea that lower doses could be safer. Early results published as a preprint before peer review indicated that low-dose tocilizumab (ranging from 40 mg to 200 mg) was associated with clinical improvement in 32 noncritical patients hospitalized with COVID-19.
Five patients (15.6%) developed bacterial superinfections, and five (15.6%) died by 28-day follow-up, although there wasn’t a perfect “overlap” between these groups of patients. Bacterial superinfection was not the cause of death in all five patients who died, and not all patients who died developed bacterial superinfections, according to senior author Pankti Reid, MD, MPH, an assistant professor of medicine at the University of Chicago.
Results from COVIDOSE also showed that treatment with tocilizumab did not seem to affect the ability of patients to develop antibodies against COVID-19. The results set the stage for a larger randomized, controlled trial (still ongoing) to determine the optimal dose of tocilizumab.
Still, Dr. Hill urges caution.
Many of these immunomodulators have been used only in the context of a clinical trial, or only for patients with terminal cancer and no other treatment options. In patients with cancer, these drugs have been studied and have shown an “acceptable safety profile,” according to Dr. Shah.
But this is a different situation, and when it comes to repurposing them to relatively healthy patients with COVID-19, Dr. Hill emphasized the need for careful research.
“We’re always very concerned about giving drugs that suppress the immune response if people have active infections,” Dr. Hill said. “Often times we think it makes things worse, and it typically does.”
Dr. Mascarenhas reported institutional research funding from CTI Biopharma. Dr. Hill, Dr. Staudt, Dr. Wilson, and Dr. Shah disclosed no relevant financial relationships.
This article first appeared on Medscape.com.