User login
Register for VRIC
"Hard Science: Calcification and Vascular Solutions” is the theme of this year’s Vascular Research Initiatives Conference (VRIC). The meeting will be held in Boston on May 13 and will focus on emerging vascular science and biology. Abstracts will cover topic areas including, but not limited to, vascular remodeling, stem cells and wound healing, arterial injury, and diabetes. Don’t miss out on the essential meeting for translational vascular science and interdisciplinary research – make your travel plans now.
"Hard Science: Calcification and Vascular Solutions” is the theme of this year’s Vascular Research Initiatives Conference (VRIC). The meeting will be held in Boston on May 13 and will focus on emerging vascular science and biology. Abstracts will cover topic areas including, but not limited to, vascular remodeling, stem cells and wound healing, arterial injury, and diabetes. Don’t miss out on the essential meeting for translational vascular science and interdisciplinary research – make your travel plans now.
"Hard Science: Calcification and Vascular Solutions” is the theme of this year’s Vascular Research Initiatives Conference (VRIC). The meeting will be held in Boston on May 13 and will focus on emerging vascular science and biology. Abstracts will cover topic areas including, but not limited to, vascular remodeling, stem cells and wound healing, arterial injury, and diabetes. Don’t miss out on the essential meeting for translational vascular science and interdisciplinary research – make your travel plans now.
What does a Google search for cosmetic and laser procedures reveal?
DENVER – of professional societies only 8% of the time, and even less frequently to websites of academic centers and peer-reviewed medical journals, results from a novel study showed.

“An increasing number of patients are seeking information about cosmetic and laser dermatology from online sources,” Jennifer L. Sawaya, MD, said in an interview in advance of the annual conference of the American Society for Laser Medicine and Surgery. “There are several studies that have discussed the role of the Internet and social media in dermatology. To our knowledge, this is the first study to specifically look at the results of Google search terms within our field to investigate which sources are providing this information.”
Dr. Sawaya, a fellow at Massachusetts General Hospital and the Wellman Center for Photomedicine, Boston, and her colleagues cross-measured keyword analytics provided by Zalea, an online resource on cosmetic treatments for consumers, with the most used Instagram hashtags to obtain 10 online keywords: body contouring, Botox, fillers, CoolSculpting, laser hair removal, tattoo removal, skin tightening, skin rejuvenation, cosmetic surgery, and liposuction. Next, they used an advanced Google search to obtain the top 25 search results for each of those 10 keywords and categorized information sources as professional societies, peer-reviewed journals, non–peer-reviewed online health information, news/media, device/cosmeceutical companies, clinical practices, academic centers, or medical spas.
Overall, the top search results came from clinical practices 23% of the time, followed by online health information sites (19%), medical spas (16%), and news/media (15%). A much smaller percentage of the search results came from professional societies (8%), academic centers (6%), and peer-reviewed medical journals (5%). Within the clinical practices and medical spas, nearly half of these sources were plastic surgeons, while board-certified dermatologists comprised only 21% of the clinical information sources.
When Dr. Sawaya and her associates evaluated the source of search results for each keyword, results varied. For example, search results for “body contouring” came most frequently from professional societies and clinical practices (20% each), “Botox” from news/media (36%), “fillers” from online health information (28%), “CoolSculpting” from clinical practices (40%), “laser hair removal” from news/media (32%), “tattoo removal” from medical spas (28%), “skin tightening” from news/media (24%), “skin rejuvenation” from medical spas (28%), “cosmetic surgery” from clinical practices (52%), and “liposuction” from online health information (36%).
“Our clinical take-home message is essentially a call for an increasing amount of evidence-based, academic content to be made available for online consumption,” Dr. Sawaya said. “In an era when patients seek a lot of medical information online and make important decisions through this manner, we have an obligation to understand what is out there and do our best to improve the quality of available information.”

She acknowledged certain limitations of the study, including the fact that results of a Google search may vary depending on the type of device used (mobile, desktop) as well as the location of the device. “An additional limitation is how the search history on the device may impact results,” she said. “To control for this, the device history, cache, and cookies were cleared prior to the search. Despite these controls, it is unclear how and to what extent prior searches affect the Google ranking algorithm. We acknowledge that the findings in this study reflect a single point in time and that the results of a Google search will change dynamically based on many factors. Finally, we acknowledge that our study is based on a single search engine site and that the trends we observe with Google may not be extrapolated to other online channels.”
Dr. Sawaya reported having no financial disclosures.
DENVER – of professional societies only 8% of the time, and even less frequently to websites of academic centers and peer-reviewed medical journals, results from a novel study showed.
“An increasing number of patients are seeking information about cosmetic and laser dermatology from online sources,” Jennifer L. Sawaya, MD, said in an interview in advance of the annual conference of the American Society for Laser Medicine and Surgery. “There are several studies that have discussed the role of the Internet and social media in dermatology. To our knowledge, this is the first study to specifically look at the results of Google search terms within our field to investigate which sources are providing this information.”
Dr. Sawaya, a fellow at Massachusetts General Hospital and the Wellman Center for Photomedicine, Boston, and her colleagues cross-measured keyword analytics provided by Zalea, an online resource on cosmetic treatments for consumers, with the most used Instagram hashtags to obtain 10 online keywords: body contouring, Botox, fillers, CoolSculpting, laser hair removal, tattoo removal, skin tightening, skin rejuvenation, cosmetic surgery, and liposuction. Next, they used an advanced Google search to obtain the top 25 search results for each of those 10 keywords and categorized information sources as professional societies, peer-reviewed journals, non–peer-reviewed online health information, news/media, device/cosmeceutical companies, clinical practices, academic centers, or medical spas.
Overall, the top search results came from clinical practices 23% of the time, followed by online health information sites (19%), medical spas (16%), and news/media (15%). A much smaller percentage of the search results came from professional societies (8%), academic centers (6%), and peer-reviewed medical journals (5%). Within the clinical practices and medical spas, nearly half of these sources were plastic surgeons, while board-certified dermatologists comprised only 21% of the clinical information sources.
When Dr. Sawaya and her associates evaluated the source of search results for each keyword, results varied. For example, search results for “body contouring” came most frequently from professional societies and clinical practices (20% each), “Botox” from news/media (36%), “fillers” from online health information (28%), “CoolSculpting” from clinical practices (40%), “laser hair removal” from news/media (32%), “tattoo removal” from medical spas (28%), “skin tightening” from news/media (24%), “skin rejuvenation” from medical spas (28%), “cosmetic surgery” from clinical practices (52%), and “liposuction” from online health information (36%).
“Our clinical take-home message is essentially a call for an increasing amount of evidence-based, academic content to be made available for online consumption,” Dr. Sawaya said. “In an era when patients seek a lot of medical information online and make important decisions through this manner, we have an obligation to understand what is out there and do our best to improve the quality of available information.”
She acknowledged certain limitations of the study, including the fact that results of a Google search may vary depending on the type of device used (mobile, desktop) as well as the location of the device. “An additional limitation is how the search history on the device may impact results,” she said. “To control for this, the device history, cache, and cookies were cleared prior to the search. Despite these controls, it is unclear how and to what extent prior searches affect the Google ranking algorithm. We acknowledge that the findings in this study reflect a single point in time and that the results of a Google search will change dynamically based on many factors. Finally, we acknowledge that our study is based on a single search engine site and that the trends we observe with Google may not be extrapolated to other online channels.”
Dr. Sawaya reported having no financial disclosures.
DENVER – of professional societies only 8% of the time, and even less frequently to websites of academic centers and peer-reviewed medical journals, results from a novel study showed.
“An increasing number of patients are seeking information about cosmetic and laser dermatology from online sources,” Jennifer L. Sawaya, MD, said in an interview in advance of the annual conference of the American Society for Laser Medicine and Surgery. “There are several studies that have discussed the role of the Internet and social media in dermatology. To our knowledge, this is the first study to specifically look at the results of Google search terms within our field to investigate which sources are providing this information.”
Dr. Sawaya, a fellow at Massachusetts General Hospital and the Wellman Center for Photomedicine, Boston, and her colleagues cross-measured keyword analytics provided by Zalea, an online resource on cosmetic treatments for consumers, with the most used Instagram hashtags to obtain 10 online keywords: body contouring, Botox, fillers, CoolSculpting, laser hair removal, tattoo removal, skin tightening, skin rejuvenation, cosmetic surgery, and liposuction. Next, they used an advanced Google search to obtain the top 25 search results for each of those 10 keywords and categorized information sources as professional societies, peer-reviewed journals, non–peer-reviewed online health information, news/media, device/cosmeceutical companies, clinical practices, academic centers, or medical spas.
Overall, the top search results came from clinical practices 23% of the time, followed by online health information sites (19%), medical spas (16%), and news/media (15%). A much smaller percentage of the search results came from professional societies (8%), academic centers (6%), and peer-reviewed medical journals (5%). Within the clinical practices and medical spas, nearly half of these sources were plastic surgeons, while board-certified dermatologists comprised only 21% of the clinical information sources.
When Dr. Sawaya and her associates evaluated the source of search results for each keyword, results varied. For example, search results for “body contouring” came most frequently from professional societies and clinical practices (20% each), “Botox” from news/media (36%), “fillers” from online health information (28%), “CoolSculpting” from clinical practices (40%), “laser hair removal” from news/media (32%), “tattoo removal” from medical spas (28%), “skin tightening” from news/media (24%), “skin rejuvenation” from medical spas (28%), “cosmetic surgery” from clinical practices (52%), and “liposuction” from online health information (36%).
“Our clinical take-home message is essentially a call for an increasing amount of evidence-based, academic content to be made available for online consumption,” Dr. Sawaya said. “In an era when patients seek a lot of medical information online and make important decisions through this manner, we have an obligation to understand what is out there and do our best to improve the quality of available information.”
She acknowledged certain limitations of the study, including the fact that results of a Google search may vary depending on the type of device used (mobile, desktop) as well as the location of the device. “An additional limitation is how the search history on the device may impact results,” she said. “To control for this, the device history, cache, and cookies were cleared prior to the search. Despite these controls, it is unclear how and to what extent prior searches affect the Google ranking algorithm. We acknowledge that the findings in this study reflect a single point in time and that the results of a Google search will change dynamically based on many factors. Finally, we acknowledge that our study is based on a single search engine site and that the trends we observe with Google may not be extrapolated to other online channels.”
Dr. Sawaya reported having no financial disclosures.
REPORTING FROM ASLMS 2019
Key clinical point: There is a paucity of online information regarding cosmetic and laser dermatology from professional societies and academic, peer-reviewed sources.
Major finding: Top Google search results came from clinical practices 23% of the time and from professional societies only 8% of the time.
Study details: An online review of 25 Google search results for 10 keywords associated with cosmetic and laser dermatology.
Disclosures: Dr. Sawaya reported having no financial disclosures.
Study finds pain perception disconnect during vascular laser procedures
DENVER – There is an apparent disconnect between the level of periprocedural pain experienced by patients during vascular laser procedures and what device manufacturers say that level of pain should be, results from a retrospective study showed.

“Although there is an abundance of research on how pain signals are transmitted in the nervous system and how pain is perceived among certain patient demographics, there is not much known about how pain perception differs from that put forth by industry,” Lauren Bonati, MD, said in an interview in advance of the annual conference of the American Society for Laser Medicine and Surgery. “This study is unique because we are questioning not whether pain perception is reproducible between patients, but rather if it reflects what industry and device manufacturers are telling us.”
Dr. Bonati, a dermatologist at Edwards, Colo.–based Mountain Dermatology Specialists, and her colleagues collected median and mode pain scores from a past clinical trial that investigated a dual wavelength laser used for different types of treatments. “The treatment type (laser wavelength and treatment area) was largely based on the severity of facial redness for each individual patient,” she explained. “The options were spot treatment, nose and cheeks, or a global facial treatment with either wavelength.” The researchers reviewed industry-provided materials to determine language regarding procedural pain, and they interviewed the clinical trial’s principal investigator about how pain expectations were set during the trial. Next, they transferred subject-reported pain scores and verbal pain descriptors to the validated Numerical Rating Scale and the Verbal Rating Scale, for comparison.
In all, 85 procedural pain scores were collected from 22 subject charts. The researchers found that the average procedural pain scores for treatment types reported by subjects were translated to entirely different verbal and numerical categories of pain from those described by industry materials. “It was surprising to see how vague pain descriptions can be in device manuals and industry materials, if even addressed at all,” Dr. Bonati said.
She advised clinicians to be wary of whom they rely on for information related to pain expectations. “Also, remember that wrongly set pain expectations can have physiologic and emotional effects that may positively or negatively impact patient experience,” Dr. Bonati said.
She acknowledged certain limitations of the study, including the fact that it was a review of a previously conducted clinical trial, “which is not a perfect representation of real-life clinic.”
She reported having no conflicts of interest.
DENVER – There is an apparent disconnect between the level of periprocedural pain experienced by patients during vascular laser procedures and what device manufacturers say that level of pain should be, results from a retrospective study showed.
“Although there is an abundance of research on how pain signals are transmitted in the nervous system and how pain is perceived among certain patient demographics, there is not much known about how pain perception differs from that put forth by industry,” Lauren Bonati, MD, said in an interview in advance of the annual conference of the American Society for Laser Medicine and Surgery. “This study is unique because we are questioning not whether pain perception is reproducible between patients, but rather if it reflects what industry and device manufacturers are telling us.”
Dr. Bonati, a dermatologist at Edwards, Colo.–based Mountain Dermatology Specialists, and her colleagues collected median and mode pain scores from a past clinical trial that investigated a dual wavelength laser used for different types of treatments. “The treatment type (laser wavelength and treatment area) was largely based on the severity of facial redness for each individual patient,” she explained. “The options were spot treatment, nose and cheeks, or a global facial treatment with either wavelength.” The researchers reviewed industry-provided materials to determine language regarding procedural pain, and they interviewed the clinical trial’s principal investigator about how pain expectations were set during the trial. Next, they transferred subject-reported pain scores and verbal pain descriptors to the validated Numerical Rating Scale and the Verbal Rating Scale, for comparison.
In all, 85 procedural pain scores were collected from 22 subject charts. The researchers found that the average procedural pain scores for treatment types reported by subjects were translated to entirely different verbal and numerical categories of pain from those described by industry materials. “It was surprising to see how vague pain descriptions can be in device manuals and industry materials, if even addressed at all,” Dr. Bonati said.
She advised clinicians to be wary of whom they rely on for information related to pain expectations. “Also, remember that wrongly set pain expectations can have physiologic and emotional effects that may positively or negatively impact patient experience,” Dr. Bonati said.
She acknowledged certain limitations of the study, including the fact that it was a review of a previously conducted clinical trial, “which is not a perfect representation of real-life clinic.”
She reported having no conflicts of interest.
DENVER – There is an apparent disconnect between the level of periprocedural pain experienced by patients during vascular laser procedures and what device manufacturers say that level of pain should be, results from a retrospective study showed.
“Although there is an abundance of research on how pain signals are transmitted in the nervous system and how pain is perceived among certain patient demographics, there is not much known about how pain perception differs from that put forth by industry,” Lauren Bonati, MD, said in an interview in advance of the annual conference of the American Society for Laser Medicine and Surgery. “This study is unique because we are questioning not whether pain perception is reproducible between patients, but rather if it reflects what industry and device manufacturers are telling us.”
Dr. Bonati, a dermatologist at Edwards, Colo.–based Mountain Dermatology Specialists, and her colleagues collected median and mode pain scores from a past clinical trial that investigated a dual wavelength laser used for different types of treatments. “The treatment type (laser wavelength and treatment area) was largely based on the severity of facial redness for each individual patient,” she explained. “The options were spot treatment, nose and cheeks, or a global facial treatment with either wavelength.” The researchers reviewed industry-provided materials to determine language regarding procedural pain, and they interviewed the clinical trial’s principal investigator about how pain expectations were set during the trial. Next, they transferred subject-reported pain scores and verbal pain descriptors to the validated Numerical Rating Scale and the Verbal Rating Scale, for comparison.
In all, 85 procedural pain scores were collected from 22 subject charts. The researchers found that the average procedural pain scores for treatment types reported by subjects were translated to entirely different verbal and numerical categories of pain from those described by industry materials. “It was surprising to see how vague pain descriptions can be in device manuals and industry materials, if even addressed at all,” Dr. Bonati said.
She advised clinicians to be wary of whom they rely on for information related to pain expectations. “Also, remember that wrongly set pain expectations can have physiologic and emotional effects that may positively or negatively impact patient experience,” Dr. Bonati said.
She acknowledged certain limitations of the study, including the fact that it was a review of a previously conducted clinical trial, “which is not a perfect representation of real-life clinic.”
She reported having no conflicts of interest.
REPORTING FROM ASLMS 2019
Key clinical point: Industry-provided materials failed to capture the range of procedural pain scores reported by patients undergoing a variety of vascular laser procedures.
Major finding: The average procedural pain scores for treatment types reported by subjects were translated to entirely different verbal and numerical categories of pain from those described by industry materials.
Study details: A retrospective evaluation of 85 procedural pain scores collected from 22 subject charts.
Disclosures: Dr. Bonati reported having no financial disclosures.
Trofinetide may benefit patients with Rett syndrome
Furthermore, the treatment is safe and well tolerated, according to results published online ahead of print March 27 in Neurology.

“These are very promising data for the Rett community that is currently without any [Food and Drug Administration]–approved treatment option,” Daniel Glaze, MD, said in a press release from Acadia Pharmaceuticals, which is developing trofinetide.
In 2017, a phase 2 study indicated that the drug was safe and tolerable when administered in doses of 70 mg/kg b.i.d. to adolescent and adult females with Rett syndrome. The study also provided initial evidence of the drug’s efficacy. Dr. Glaze, professor of pediatrics and neurology at Baylor College of Medicine in Houston, and his colleagues decided to conduct a larger phase 2 study that examined higher doses and a longer treatment duration.
The researchers first enrolled 62 participants in the study, all of whom received placebo b.i.d. for 14 days. They next randomized participants in equal groups to placebo or one of three twice-daily doses of trofinetide (i.e., 50 mg/kg, 100 mg/kg, and 200 mg/kg). After a blinded review of safety and tolerability data, Dr. Glaze and his colleagues enrolled 20 more participants and randomized them in equal groups to placebo or 200 mg/kg b.i.d. of trofinetide. This modification in study design was intended to increase the likelihood of detecting a clinical benefit. Randomized, double-blind treatment lasted for 42 days. Participants presented for a final visit at approximately 10 days after the treatment period ended.
A total of 82 girls aged 5-15 years participated in the study. They all met the 2010 diagnostic criteria for classic Rett syndrome, had a documented pathogenic MECP2 variant, were in the postregression stage of the syndrome, and had been stable on pharmacologic and behavioral treatments for at least 4 weeks. The sample’s mean age was 9.7 years, 94% were white, and mean weight was 26.1 kg. The treatment groups’ demographic characteristics were balanced.
All three doses of trofinetide were safe and tolerable. One participant in the 200-mg/kg b.i.d. group was withdrawn from the study at her parents’ request. Serious adverse events occurred in one control participant, one receiving the 100-mg/kg b.i.d. dose, and one receiving the 200-mg/kg b.i.d. dose. These serious adverse events were considered unrelated to study medication and resolved by the study’s end. The most common adverse events were diarrhea, vomiting, upper respiratory tract infection, and pyrexia. Most were mild or moderate and considered unrelated to trofinetide.
The 200-mg/kg b.i.d. dose of trofinetide significantly improved outcomes on the Rett Syndrome Behavior Questionnaire (RSBQ), Clinical Global Impression Scale–Improvement (CGI-I), and Rett Syndrome Domain Specific Concerns (RTT-DSC), compared with placebo. The change of the median RTT-DSC score was 15%, and the change of the mean RSBQ score was 16%. More than 20% of participants receiving 200 mg/kg b.i.d. of trofinetide were rated much improved on the CGI-I, compared with less than 5% of controls.
“Overall, the observed clinical improvement in the present pediatric trial was more manifest than in the previous trial, with younger age (i.e., greater neuroplasticity), higher doses (i.e., higher drug exposure), and longer drug treatment duration (i.e., 28 days in Rett-001 vs. 42 days in Rett-002) as potential contributors,” said Dr. Glaze and his colleagues. “Future studies aiming at replicating the results of this pediatric trial would benefit from including the RSBQ as a primary endpoint and should also consider evaluating RSBQ subscales such as the General Mood.”
Neuren Pharmaceuticals and Rettsyndrome.org funded the study. Dr. Glaze is a consultant to Neuren Pharmaceuticals.
SOURCE: Glaze DG et al. Neurology. 2019 Mar 27. doi: 10.1212/WNL.0000000000007316.
Furthermore, the treatment is safe and well tolerated, according to results published online ahead of print March 27 in Neurology.
“These are very promising data for the Rett community that is currently without any [Food and Drug Administration]–approved treatment option,” Daniel Glaze, MD, said in a press release from Acadia Pharmaceuticals, which is developing trofinetide.
In 2017, a phase 2 study indicated that the drug was safe and tolerable when administered in doses of 70 mg/kg b.i.d. to adolescent and adult females with Rett syndrome. The study also provided initial evidence of the drug’s efficacy. Dr. Glaze, professor of pediatrics and neurology at Baylor College of Medicine in Houston, and his colleagues decided to conduct a larger phase 2 study that examined higher doses and a longer treatment duration.
The researchers first enrolled 62 participants in the study, all of whom received placebo b.i.d. for 14 days. They next randomized participants in equal groups to placebo or one of three twice-daily doses of trofinetide (i.e., 50 mg/kg, 100 mg/kg, and 200 mg/kg). After a blinded review of safety and tolerability data, Dr. Glaze and his colleagues enrolled 20 more participants and randomized them in equal groups to placebo or 200 mg/kg b.i.d. of trofinetide. This modification in study design was intended to increase the likelihood of detecting a clinical benefit. Randomized, double-blind treatment lasted for 42 days. Participants presented for a final visit at approximately 10 days after the treatment period ended.
A total of 82 girls aged 5-15 years participated in the study. They all met the 2010 diagnostic criteria for classic Rett syndrome, had a documented pathogenic MECP2 variant, were in the postregression stage of the syndrome, and had been stable on pharmacologic and behavioral treatments for at least 4 weeks. The sample’s mean age was 9.7 years, 94% were white, and mean weight was 26.1 kg. The treatment groups’ demographic characteristics were balanced.
All three doses of trofinetide were safe and tolerable. One participant in the 200-mg/kg b.i.d. group was withdrawn from the study at her parents’ request. Serious adverse events occurred in one control participant, one receiving the 100-mg/kg b.i.d. dose, and one receiving the 200-mg/kg b.i.d. dose. These serious adverse events were considered unrelated to study medication and resolved by the study’s end. The most common adverse events were diarrhea, vomiting, upper respiratory tract infection, and pyrexia. Most were mild or moderate and considered unrelated to trofinetide.
The 200-mg/kg b.i.d. dose of trofinetide significantly improved outcomes on the Rett Syndrome Behavior Questionnaire (RSBQ), Clinical Global Impression Scale–Improvement (CGI-I), and Rett Syndrome Domain Specific Concerns (RTT-DSC), compared with placebo. The change of the median RTT-DSC score was 15%, and the change of the mean RSBQ score was 16%. More than 20% of participants receiving 200 mg/kg b.i.d. of trofinetide were rated much improved on the CGI-I, compared with less than 5% of controls.
“Overall, the observed clinical improvement in the present pediatric trial was more manifest than in the previous trial, with younger age (i.e., greater neuroplasticity), higher doses (i.e., higher drug exposure), and longer drug treatment duration (i.e., 28 days in Rett-001 vs. 42 days in Rett-002) as potential contributors,” said Dr. Glaze and his colleagues. “Future studies aiming at replicating the results of this pediatric trial would benefit from including the RSBQ as a primary endpoint and should also consider evaluating RSBQ subscales such as the General Mood.”
Neuren Pharmaceuticals and Rettsyndrome.org funded the study. Dr. Glaze is a consultant to Neuren Pharmaceuticals.
SOURCE: Glaze DG et al. Neurology. 2019 Mar 27. doi: 10.1212/WNL.0000000000007316.
Furthermore, the treatment is safe and well tolerated, according to results published online ahead of print March 27 in Neurology.
“These are very promising data for the Rett community that is currently without any [Food and Drug Administration]–approved treatment option,” Daniel Glaze, MD, said in a press release from Acadia Pharmaceuticals, which is developing trofinetide.
In 2017, a phase 2 study indicated that the drug was safe and tolerable when administered in doses of 70 mg/kg b.i.d. to adolescent and adult females with Rett syndrome. The study also provided initial evidence of the drug’s efficacy. Dr. Glaze, professor of pediatrics and neurology at Baylor College of Medicine in Houston, and his colleagues decided to conduct a larger phase 2 study that examined higher doses and a longer treatment duration.
The researchers first enrolled 62 participants in the study, all of whom received placebo b.i.d. for 14 days. They next randomized participants in equal groups to placebo or one of three twice-daily doses of trofinetide (i.e., 50 mg/kg, 100 mg/kg, and 200 mg/kg). After a blinded review of safety and tolerability data, Dr. Glaze and his colleagues enrolled 20 more participants and randomized them in equal groups to placebo or 200 mg/kg b.i.d. of trofinetide. This modification in study design was intended to increase the likelihood of detecting a clinical benefit. Randomized, double-blind treatment lasted for 42 days. Participants presented for a final visit at approximately 10 days after the treatment period ended.
A total of 82 girls aged 5-15 years participated in the study. They all met the 2010 diagnostic criteria for classic Rett syndrome, had a documented pathogenic MECP2 variant, were in the postregression stage of the syndrome, and had been stable on pharmacologic and behavioral treatments for at least 4 weeks. The sample’s mean age was 9.7 years, 94% were white, and mean weight was 26.1 kg. The treatment groups’ demographic characteristics were balanced.
All three doses of trofinetide were safe and tolerable. One participant in the 200-mg/kg b.i.d. group was withdrawn from the study at her parents’ request. Serious adverse events occurred in one control participant, one receiving the 100-mg/kg b.i.d. dose, and one receiving the 200-mg/kg b.i.d. dose. These serious adverse events were considered unrelated to study medication and resolved by the study’s end. The most common adverse events were diarrhea, vomiting, upper respiratory tract infection, and pyrexia. Most were mild or moderate and considered unrelated to trofinetide.
The 200-mg/kg b.i.d. dose of trofinetide significantly improved outcomes on the Rett Syndrome Behavior Questionnaire (RSBQ), Clinical Global Impression Scale–Improvement (CGI-I), and Rett Syndrome Domain Specific Concerns (RTT-DSC), compared with placebo. The change of the median RTT-DSC score was 15%, and the change of the mean RSBQ score was 16%. More than 20% of participants receiving 200 mg/kg b.i.d. of trofinetide were rated much improved on the CGI-I, compared with less than 5% of controls.
“Overall, the observed clinical improvement in the present pediatric trial was more manifest than in the previous trial, with younger age (i.e., greater neuroplasticity), higher doses (i.e., higher drug exposure), and longer drug treatment duration (i.e., 28 days in Rett-001 vs. 42 days in Rett-002) as potential contributors,” said Dr. Glaze and his colleagues. “Future studies aiming at replicating the results of this pediatric trial would benefit from including the RSBQ as a primary endpoint and should also consider evaluating RSBQ subscales such as the General Mood.”
Neuren Pharmaceuticals and Rettsyndrome.org funded the study. Dr. Glaze is a consultant to Neuren Pharmaceuticals.
SOURCE: Glaze DG et al. Neurology. 2019 Mar 27. doi: 10.1212/WNL.0000000000007316.
FROM NEUROLOGY
Key clinical point: Trofinetide provides clinically meaningful improvements in core symptoms of Rett syndrome.
Major finding: The 200 mg/kg b.i.d. dose of trofinetide improved outcomes on the Rett Syndrome Behavior Questionnaire by 16%.
Study details: A phase 2, double-blind, placebo-controlled study of 82 children and adolescents with Rett syndrome.
Disclosures: Neuren Pharmaceuticals and Rettsyndrome.org funded the study.
Source: Glaze DG et al. Neurology. 2019 Mar 27. doi: 10.1212/WNL.0000000000007316.
Proinflammatory diet may not trigger adult psoriasis, PsA, or AD
reported Alanna C. Bridgman of Queen’s University, Kingston, Ont., and her associates.

In a large, retrospective cohort study among women from the Nurses’ Health Study II (NHS-II), including 85,185 psoriasis participants and 63,443 atopic dermatitis participants, Ms. Bridgman and her associates sought to determine whether proinflammatory diet increased the risk of incident psoriasis, psoriatic arthritis, or atopic dermatitis. Clinicians administered food frequency questionnaires every 4 years beginning in 1991 among female nurses aged 25-42 years.
Food groups included in the evaluation were those most predictive of three plasma markers of inflammation: interleukin-6 (IL-6), C-reactive protein (CRP), and tumor necrosis factor–alpha R2 (TNF-R2). Proinflammatory foods included processed meat, red meat, organ meat, white fish, vegetables other than leafy green and dark yellow, refined grains, low- and high-energy drinks, and tomatoes. Anti-inflammatory foods included beer, wine, tea, coffee, dark yellow and green leafy vegetables, snacks such as popcorn and crackers, fruit juice, and pizza.
No association was found between proinflammatory diet and increased likelihood for incident psoriasis, psoriatic arthritis, or atopic dermatitis. Although proinflammatory dietary patterns were associated with psoriatic arthritis in the age-adjusted model, the hazard ratio was attenuated and found to be no longer statistically significant after adjustment for important confounders such as body mass index. In addition, no significant relationship between atopic dermatitis and proinflammatory diet was observed, they reported. The study was published in the Journal of the American Academy of Dermatology.
Ms. Bridgman and her associates measured dietary patterns using the Empirical Dietary Inflammatory Pattern (EDIP); dietary patterns measuring high on the EDIP scale were associated with higher levels of TNF-alpha, TNF-alpha R1, TNF-alpha R2, CRP, IL-6, and adiponectin. Psoriasis and psoriatic arthritis are Th1- and Th17-mediated diseases that exhibit higher serum levels of IL-6, CRP, and TNF-alpha, unlike atopic dermatitis, which is primarily a Th2-mediated condition featuring reduced involvement of the Th1/Th17 inflammatory cytokines.
Because a goal of the EDIP score was to “account for the overall effect of dietary patterns,” the researchers included in their analysis only those food groups that “explain the maximal variation in the three noted inflammatory biomarkers.”
All patients included in the study were questioned at baseline regarding their height and race/ethnicity. Weight, smoking status, and physical activity, and diagnoses of hypercholesterolemia, type 2 diabetes, cardiovascular disease, and asthma were monitored biennially.
Overall, patients with higher EDIP scores were found to have higher BMI, lower physical activity, and alcohol use, as well as increased rates of hypercholesterolemia and hypertension.
“Though we found no convincing evidence for an association with EDIP score for any of the investigated diseases, the results followed an internal pattern consistent with our hypotheses that higher EDIP scores would have more of an association with psoriatic disease than with atopic dermatitis,” the researchers wrote.
Citing recent evidence gathered in studies, such as the French NutriNet-Santé study, which demonstrated proinflammatory effects similar to those measured with the EDIP in cases where there was low adherence to the Mediterranean diet, the authors attributed their contradictory findings to “important methodological differences.” Unlike the NutriNet-Santé study, which classified psoriasis by severity, Ms. Bridgman and her colleagues examined the overall risk of incident psoriasis. “It is possible that a dietary index associated with more Th-2 inflammation would yield different results,” they noted.
The large sample size, prospectively collected dietary, and psoriatic disease data, as well as the ability to adjust for important confounding factors, were included among the strengths of the study.
That the participants were limited to U.S. women could be considered a limitation because the results may not be generalizable to other populations. The results also may not be relevant to child-onset disease because the patient population included only cases of adult-onset atopic dermatitis. Questionnaire-based diagnoses increase the likelihood of misclassification, so “dilution of the case pool with false-positive cases would bias our results towards the null,” they added.
Ultimately, the authors noted that proinflammatory diet may be associated with other health risks, but these do not warrant counseling patients concerning their possible impact in cases of psoriatic disease or atopic dermatitis.
The study was funded by Brown University department of dermatology and from Regeneron, Sanofi, the National Institutes of Health, and the National Cancer Institute. Two coauthors, one of whom has a patent pending for the nix-tix tick remover, disclosed ties with various companies.
SOURCE: Bridgman AC et al. J Am Acad Dermatol. 2019 Feb 21. pii: S0190-9622(19)30329-9.
reported Alanna C. Bridgman of Queen’s University, Kingston, Ont., and her associates.
In a large, retrospective cohort study among women from the Nurses’ Health Study II (NHS-II), including 85,185 psoriasis participants and 63,443 atopic dermatitis participants, Ms. Bridgman and her associates sought to determine whether proinflammatory diet increased the risk of incident psoriasis, psoriatic arthritis, or atopic dermatitis. Clinicians administered food frequency questionnaires every 4 years beginning in 1991 among female nurses aged 25-42 years.
Food groups included in the evaluation were those most predictive of three plasma markers of inflammation: interleukin-6 (IL-6), C-reactive protein (CRP), and tumor necrosis factor–alpha R2 (TNF-R2). Proinflammatory foods included processed meat, red meat, organ meat, white fish, vegetables other than leafy green and dark yellow, refined grains, low- and high-energy drinks, and tomatoes. Anti-inflammatory foods included beer, wine, tea, coffee, dark yellow and green leafy vegetables, snacks such as popcorn and crackers, fruit juice, and pizza.
No association was found between proinflammatory diet and increased likelihood for incident psoriasis, psoriatic arthritis, or atopic dermatitis. Although proinflammatory dietary patterns were associated with psoriatic arthritis in the age-adjusted model, the hazard ratio was attenuated and found to be no longer statistically significant after adjustment for important confounders such as body mass index. In addition, no significant relationship between atopic dermatitis and proinflammatory diet was observed, they reported. The study was published in the Journal of the American Academy of Dermatology.
Ms. Bridgman and her associates measured dietary patterns using the Empirical Dietary Inflammatory Pattern (EDIP); dietary patterns measuring high on the EDIP scale were associated with higher levels of TNF-alpha, TNF-alpha R1, TNF-alpha R2, CRP, IL-6, and adiponectin. Psoriasis and psoriatic arthritis are Th1- and Th17-mediated diseases that exhibit higher serum levels of IL-6, CRP, and TNF-alpha, unlike atopic dermatitis, which is primarily a Th2-mediated condition featuring reduced involvement of the Th1/Th17 inflammatory cytokines.
Because a goal of the EDIP score was to “account for the overall effect of dietary patterns,” the researchers included in their analysis only those food groups that “explain the maximal variation in the three noted inflammatory biomarkers.”
All patients included in the study were questioned at baseline regarding their height and race/ethnicity. Weight, smoking status, and physical activity, and diagnoses of hypercholesterolemia, type 2 diabetes, cardiovascular disease, and asthma were monitored biennially.
Overall, patients with higher EDIP scores were found to have higher BMI, lower physical activity, and alcohol use, as well as increased rates of hypercholesterolemia and hypertension.
“Though we found no convincing evidence for an association with EDIP score for any of the investigated diseases, the results followed an internal pattern consistent with our hypotheses that higher EDIP scores would have more of an association with psoriatic disease than with atopic dermatitis,” the researchers wrote.
Citing recent evidence gathered in studies, such as the French NutriNet-Santé study, which demonstrated proinflammatory effects similar to those measured with the EDIP in cases where there was low adherence to the Mediterranean diet, the authors attributed their contradictory findings to “important methodological differences.” Unlike the NutriNet-Santé study, which classified psoriasis by severity, Ms. Bridgman and her colleagues examined the overall risk of incident psoriasis. “It is possible that a dietary index associated with more Th-2 inflammation would yield different results,” they noted.
The large sample size, prospectively collected dietary, and psoriatic disease data, as well as the ability to adjust for important confounding factors, were included among the strengths of the study.
That the participants were limited to U.S. women could be considered a limitation because the results may not be generalizable to other populations. The results also may not be relevant to child-onset disease because the patient population included only cases of adult-onset atopic dermatitis. Questionnaire-based diagnoses increase the likelihood of misclassification, so “dilution of the case pool with false-positive cases would bias our results towards the null,” they added.
Ultimately, the authors noted that proinflammatory diet may be associated with other health risks, but these do not warrant counseling patients concerning their possible impact in cases of psoriatic disease or atopic dermatitis.
The study was funded by Brown University department of dermatology and from Regeneron, Sanofi, the National Institutes of Health, and the National Cancer Institute. Two coauthors, one of whom has a patent pending for the nix-tix tick remover, disclosed ties with various companies.
SOURCE: Bridgman AC et al. J Am Acad Dermatol. 2019 Feb 21. pii: S0190-9622(19)30329-9.
reported Alanna C. Bridgman of Queen’s University, Kingston, Ont., and her associates.
In a large, retrospective cohort study among women from the Nurses’ Health Study II (NHS-II), including 85,185 psoriasis participants and 63,443 atopic dermatitis participants, Ms. Bridgman and her associates sought to determine whether proinflammatory diet increased the risk of incident psoriasis, psoriatic arthritis, or atopic dermatitis. Clinicians administered food frequency questionnaires every 4 years beginning in 1991 among female nurses aged 25-42 years.
Food groups included in the evaluation were those most predictive of three plasma markers of inflammation: interleukin-6 (IL-6), C-reactive protein (CRP), and tumor necrosis factor–alpha R2 (TNF-R2). Proinflammatory foods included processed meat, red meat, organ meat, white fish, vegetables other than leafy green and dark yellow, refined grains, low- and high-energy drinks, and tomatoes. Anti-inflammatory foods included beer, wine, tea, coffee, dark yellow and green leafy vegetables, snacks such as popcorn and crackers, fruit juice, and pizza.
No association was found between proinflammatory diet and increased likelihood for incident psoriasis, psoriatic arthritis, or atopic dermatitis. Although proinflammatory dietary patterns were associated with psoriatic arthritis in the age-adjusted model, the hazard ratio was attenuated and found to be no longer statistically significant after adjustment for important confounders such as body mass index. In addition, no significant relationship between atopic dermatitis and proinflammatory diet was observed, they reported. The study was published in the Journal of the American Academy of Dermatology.
Ms. Bridgman and her associates measured dietary patterns using the Empirical Dietary Inflammatory Pattern (EDIP); dietary patterns measuring high on the EDIP scale were associated with higher levels of TNF-alpha, TNF-alpha R1, TNF-alpha R2, CRP, IL-6, and adiponectin. Psoriasis and psoriatic arthritis are Th1- and Th17-mediated diseases that exhibit higher serum levels of IL-6, CRP, and TNF-alpha, unlike atopic dermatitis, which is primarily a Th2-mediated condition featuring reduced involvement of the Th1/Th17 inflammatory cytokines.
Because a goal of the EDIP score was to “account for the overall effect of dietary patterns,” the researchers included in their analysis only those food groups that “explain the maximal variation in the three noted inflammatory biomarkers.”
All patients included in the study were questioned at baseline regarding their height and race/ethnicity. Weight, smoking status, and physical activity, and diagnoses of hypercholesterolemia, type 2 diabetes, cardiovascular disease, and asthma were monitored biennially.
Overall, patients with higher EDIP scores were found to have higher BMI, lower physical activity, and alcohol use, as well as increased rates of hypercholesterolemia and hypertension.
“Though we found no convincing evidence for an association with EDIP score for any of the investigated diseases, the results followed an internal pattern consistent with our hypotheses that higher EDIP scores would have more of an association with psoriatic disease than with atopic dermatitis,” the researchers wrote.
Citing recent evidence gathered in studies, such as the French NutriNet-Santé study, which demonstrated proinflammatory effects similar to those measured with the EDIP in cases where there was low adherence to the Mediterranean diet, the authors attributed their contradictory findings to “important methodological differences.” Unlike the NutriNet-Santé study, which classified psoriasis by severity, Ms. Bridgman and her colleagues examined the overall risk of incident psoriasis. “It is possible that a dietary index associated with more Th-2 inflammation would yield different results,” they noted.
The large sample size, prospectively collected dietary, and psoriatic disease data, as well as the ability to adjust for important confounding factors, were included among the strengths of the study.
That the participants were limited to U.S. women could be considered a limitation because the results may not be generalizable to other populations. The results also may not be relevant to child-onset disease because the patient population included only cases of adult-onset atopic dermatitis. Questionnaire-based diagnoses increase the likelihood of misclassification, so “dilution of the case pool with false-positive cases would bias our results towards the null,” they added.
Ultimately, the authors noted that proinflammatory diet may be associated with other health risks, but these do not warrant counseling patients concerning their possible impact in cases of psoriatic disease or atopic dermatitis.
The study was funded by Brown University department of dermatology and from Regeneron, Sanofi, the National Institutes of Health, and the National Cancer Institute. Two coauthors, one of whom has a patent pending for the nix-tix tick remover, disclosed ties with various companies.
SOURCE: Bridgman AC et al. J Am Acad Dermatol. 2019 Feb 21. pii: S0190-9622(19)30329-9.
FROM JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Key clinical point: Study results may not be generalizable to other study populations.
Major finding: No association was found between proinflammatory diet and increased likelihood for incident psoriasis, psoriatic arthritis, or atopic dermatitis in adult women.
Study details: Large retrospective cohort study of 85,185 psoriasis subjects and 63,443 atopic dermatitis subjects.
Disclosures: The study was funded by Brown University department of dermatology and from Regeneron, Sanofi, the National Institutes of Health, and the National Cancer Institute. Two coauthors, one of whom has a patent pending for the nix-tix tick remover, disclosed ties with various companies. Source: Bridgman AC et al. J Am Acad Dermatol. 2019 Feb 21. pii: S0190-9622(19)30329-9.
Flu shot can be given irrespective of the time of last methotrexate dose
Immune response to influenza vaccination in rheumatoid arthritis patients taking methotrexate appears to depend most on stopping the next two weekly doses of the drug rather than any effect from the timing of the last dose, new research concludes.
The new finding, reported in Annals of the Rheumatic Diseases, stems from a post hoc analysis of a randomized, controlled trial that Jin Kyun Park, MD, of Seoul (Korea) National University, and his colleagues had conducted earlier on immune response when patients stopped methotrexate for either 2 or 4 weeks after vaccination. While the main endpoint of that study showed no difference in the improvement in vaccine response with either stopping methotrexate for 2 or 4 weeks and no increase in disease activity with stopping for 2 weeks, it was unclear whether the timing of the last dose mattered when stopping for 2 weeks.
In a bid to identify the optimal time between the last dose of methotrexate and administration of a flu vaccine, Dr. Park and his colleagues conducted a post hoc analysis of the trial, which involved 316 patients with RA receiving methotrexate for 6 weeks or longer to continue (n = 156) or to hold methotrexate (n = 160) for 2 weeks after receiving a quadrivalent influenza vaccine containing H1N1, H3N2, B-Yamagata, and B-Victoria.
The study authors defined a positive vaccine response as a fourfold or greater increase in hemagglutination inhibition (HI) antibody titer. A satisfactory vaccine response was a positive response to two or more of four vaccine antigens.
Patients who stopped taking methotrexate were divided into eight subgroups according to the number of days between their last dose and their vaccination.
The research team reported that response to vaccine, fold increase in HI antibody titers, and postvaccination seroprotection rates were not associated with the time between the last methotrexate dose and the time of vaccination.
However, they conceded that “the absence of impact of the number of days between the last methotrexate dose and vaccination could be due to the small patient numbers in eight subgroups.”
Vaccine response also did not differ between patients who received the influenza vaccination within 3 days of the last methotrexate dose (n = 65) and those who received it between 4-7 days of the last methotrexate dose (n = 95).
Furthermore, RA disease activity, seropositivity, or use of conventional or biologic disease-modifying antirheumatic drugs did not have an impact on methotrexate discontinuation.
The authors concluded that vaccinations could be given irrespective of the time of the last methotrexate dose, and patients should be advised to skip two weekly doses following vaccination.
“This supports the notion that the effects of methotrexate on humeral immunity occur rapidly, despite the delayed effects on arthritis; therefore, the absence of methotrexate during the first 2 weeks postvaccination is critical for humoral immunity,” they wrote.
The study was sponsored by GC Pharma. One author disclosed serving as a consultant to Pfizer and receiving research grants from GC Pharma and Hanmi Pharma.
SOURCE: Park JK et al. Ann Rheum Dis. 2019 Mar 23. doi: 10.1136/annrheumdis-2019-215187.
Immune response to influenza vaccination in rheumatoid arthritis patients taking methotrexate appears to depend most on stopping the next two weekly doses of the drug rather than any effect from the timing of the last dose, new research concludes.
The new finding, reported in Annals of the Rheumatic Diseases, stems from a post hoc analysis of a randomized, controlled trial that Jin Kyun Park, MD, of Seoul (Korea) National University, and his colleagues had conducted earlier on immune response when patients stopped methotrexate for either 2 or 4 weeks after vaccination. While the main endpoint of that study showed no difference in the improvement in vaccine response with either stopping methotrexate for 2 or 4 weeks and no increase in disease activity with stopping for 2 weeks, it was unclear whether the timing of the last dose mattered when stopping for 2 weeks.
In a bid to identify the optimal time between the last dose of methotrexate and administration of a flu vaccine, Dr. Park and his colleagues conducted a post hoc analysis of the trial, which involved 316 patients with RA receiving methotrexate for 6 weeks or longer to continue (n = 156) or to hold methotrexate (n = 160) for 2 weeks after receiving a quadrivalent influenza vaccine containing H1N1, H3N2, B-Yamagata, and B-Victoria.
The study authors defined a positive vaccine response as a fourfold or greater increase in hemagglutination inhibition (HI) antibody titer. A satisfactory vaccine response was a positive response to two or more of four vaccine antigens.
Patients who stopped taking methotrexate were divided into eight subgroups according to the number of days between their last dose and their vaccination.
The research team reported that response to vaccine, fold increase in HI antibody titers, and postvaccination seroprotection rates were not associated with the time between the last methotrexate dose and the time of vaccination.
However, they conceded that “the absence of impact of the number of days between the last methotrexate dose and vaccination could be due to the small patient numbers in eight subgroups.”
Vaccine response also did not differ between patients who received the influenza vaccination within 3 days of the last methotrexate dose (n = 65) and those who received it between 4-7 days of the last methotrexate dose (n = 95).
Furthermore, RA disease activity, seropositivity, or use of conventional or biologic disease-modifying antirheumatic drugs did not have an impact on methotrexate discontinuation.
The authors concluded that vaccinations could be given irrespective of the time of the last methotrexate dose, and patients should be advised to skip two weekly doses following vaccination.
“This supports the notion that the effects of methotrexate on humeral immunity occur rapidly, despite the delayed effects on arthritis; therefore, the absence of methotrexate during the first 2 weeks postvaccination is critical for humoral immunity,” they wrote.
The study was sponsored by GC Pharma. One author disclosed serving as a consultant to Pfizer and receiving research grants from GC Pharma and Hanmi Pharma.
SOURCE: Park JK et al. Ann Rheum Dis. 2019 Mar 23. doi: 10.1136/annrheumdis-2019-215187.
Immune response to influenza vaccination in rheumatoid arthritis patients taking methotrexate appears to depend most on stopping the next two weekly doses of the drug rather than any effect from the timing of the last dose, new research concludes.
The new finding, reported in Annals of the Rheumatic Diseases, stems from a post hoc analysis of a randomized, controlled trial that Jin Kyun Park, MD, of Seoul (Korea) National University, and his colleagues had conducted earlier on immune response when patients stopped methotrexate for either 2 or 4 weeks after vaccination. While the main endpoint of that study showed no difference in the improvement in vaccine response with either stopping methotrexate for 2 or 4 weeks and no increase in disease activity with stopping for 2 weeks, it was unclear whether the timing of the last dose mattered when stopping for 2 weeks.
In a bid to identify the optimal time between the last dose of methotrexate and administration of a flu vaccine, Dr. Park and his colleagues conducted a post hoc analysis of the trial, which involved 316 patients with RA receiving methotrexate for 6 weeks or longer to continue (n = 156) or to hold methotrexate (n = 160) for 2 weeks after receiving a quadrivalent influenza vaccine containing H1N1, H3N2, B-Yamagata, and B-Victoria.
The study authors defined a positive vaccine response as a fourfold or greater increase in hemagglutination inhibition (HI) antibody titer. A satisfactory vaccine response was a positive response to two or more of four vaccine antigens.
Patients who stopped taking methotrexate were divided into eight subgroups according to the number of days between their last dose and their vaccination.
The research team reported that response to vaccine, fold increase in HI antibody titers, and postvaccination seroprotection rates were not associated with the time between the last methotrexate dose and the time of vaccination.
However, they conceded that “the absence of impact of the number of days between the last methotrexate dose and vaccination could be due to the small patient numbers in eight subgroups.”
Vaccine response also did not differ between patients who received the influenza vaccination within 3 days of the last methotrexate dose (n = 65) and those who received it between 4-7 days of the last methotrexate dose (n = 95).
Furthermore, RA disease activity, seropositivity, or use of conventional or biologic disease-modifying antirheumatic drugs did not have an impact on methotrexate discontinuation.
The authors concluded that vaccinations could be given irrespective of the time of the last methotrexate dose, and patients should be advised to skip two weekly doses following vaccination.
“This supports the notion that the effects of methotrexate on humeral immunity occur rapidly, despite the delayed effects on arthritis; therefore, the absence of methotrexate during the first 2 weeks postvaccination is critical for humoral immunity,” they wrote.
The study was sponsored by GC Pharma. One author disclosed serving as a consultant to Pfizer and receiving research grants from GC Pharma and Hanmi Pharma.
SOURCE: Park JK et al. Ann Rheum Dis. 2019 Mar 23. doi: 10.1136/annrheumdis-2019-215187.
FROM ANNALS OF THE RHEUMATIC DISEASES
Key clinical point:
Major finding: Response to vaccine, fold increase in HI antibody titers, and postvaccination seroprotection rates were not associated with the time between the last methotrexate dose and the time of vaccination.
Study details: A post hoc analysis of a randomized, controlled trial involving 316 patients with rheumatoid arthritis who continued or stopped methotrexate for 2 weeks following influenza vaccination.
Disclosures: The study was sponsored by GC Pharma. One author disclosed serving as a consultant to Pfizer and receiving research grants from GC Pharma and Hanmi Pharma.
Source: Park JK et al. Ann Rheum Dis. 2019 Mar 23. doi: 10.1136/annrheumdis-2019-215187
Investigative magnetic device found effective for skin tightening in a small study
DENVER – Patients treated with results from a small trial showed.

“There are many different modalities for tissue tightening, including lights, radiofrequency, ultrasound and thermal energy,” Jerome M. Garden, MD, said in an interview in advance of the annual conference of the American Society for Laser Medicine and Surgery. “The idea behind all of these technologies is to heat up the skin’s collagen and to stimulate further collagen production, which can then result in improved skin tightening and textural improvement.”
In a trial conducted at the Chicago-based Physicians Laser and Dermatology Institute, Dr. Garden and his colleagues evaluated a new technology for tissue tightening that involves magnetic energy. Developed by Rocky Mountain Biosystems and BioFusionary Corp., the investigative device produces a magnetic field in the targeted tissue, which then results in the heating and eventual tightening of the tissue. “By using magnetic energy, which relies on the polarity of the molecules, it allows for a safe way to target specifically the polar dermis, without heating the relatively dry epidermis or nonpolar adipose layer, resulting in a more tolerable and potentially safer alternative to tissue tightening,” said Dr. Garden, a dermatologist who is the director of the Physicians Laser and Dermatology Institute.
For the trial, 20 patients with facial and upper skin laxity underwent a mean of 4.3 treatment sessions with the 27MHz magnetic device that used a 3-cm spot size, with a minimum of 4 weeks between each session. No anesthetics or analgesics were used. “No gels or skin prep was performed before the treatment, other than a gentle soap beforehand,” Dr. Garden said. “A bland moisturizer was applied to the treated skin after treatments.” The majority of patients (85%) had paid for their procedures (a price comparable to other skin-tightening procedures), and two board-certified dermatologists evaluated both before and after photographs for overall improvement of skin laxity and texture. Follow-ups were done 2-4 months after the last treatment. The observers were not informed which photographs were before or after.
Dr. Garden reported that the observers correctly chose 19 out of 20 patients’ before and after photographs, and they rated the mean grade level of improvement as 43%. Nearly half of the patients (48%) were graded at 50% or greater improvement. At the same time, patients rated their own improvement as a mean 6.5 out of 10. Nearly half of patients graded their outcome at 7 or better, which was designated as “very satisfied.” The procedures were well tolerated, Dr. Garden said, and the most common side effects were minor transient erythema and edema. The erythema generally faded after 2-4 hours, and the mild edema lasted up to 24 hours.
“Magnetic energy is a new technology that can be used to treat lower face and neck laxity,” said Dr. Garden, who is also a professor of clinical dermatology at Northwestern University, Chicago. “We only treated patients with skin types I-IV, but we feel that this technology is likely safe for higher skin types as well.”
Rocky Mountain Biosystems and BioFusionary Corp. provided the device used for the study. Dr. Garden and his colleagues are currently extending the ongoing trial. He reported having no financial disclosures.
DENVER – Patients treated with results from a small trial showed.
“There are many different modalities for tissue tightening, including lights, radiofrequency, ultrasound and thermal energy,” Jerome M. Garden, MD, said in an interview in advance of the annual conference of the American Society for Laser Medicine and Surgery. “The idea behind all of these technologies is to heat up the skin’s collagen and to stimulate further collagen production, which can then result in improved skin tightening and textural improvement.”
In a trial conducted at the Chicago-based Physicians Laser and Dermatology Institute, Dr. Garden and his colleagues evaluated a new technology for tissue tightening that involves magnetic energy. Developed by Rocky Mountain Biosystems and BioFusionary Corp., the investigative device produces a magnetic field in the targeted tissue, which then results in the heating and eventual tightening of the tissue. “By using magnetic energy, which relies on the polarity of the molecules, it allows for a safe way to target specifically the polar dermis, without heating the relatively dry epidermis or nonpolar adipose layer, resulting in a more tolerable and potentially safer alternative to tissue tightening,” said Dr. Garden, a dermatologist who is the director of the Physicians Laser and Dermatology Institute.
For the trial, 20 patients with facial and upper skin laxity underwent a mean of 4.3 treatment sessions with the 27MHz magnetic device that used a 3-cm spot size, with a minimum of 4 weeks between each session. No anesthetics or analgesics were used. “No gels or skin prep was performed before the treatment, other than a gentle soap beforehand,” Dr. Garden said. “A bland moisturizer was applied to the treated skin after treatments.” The majority of patients (85%) had paid for their procedures (a price comparable to other skin-tightening procedures), and two board-certified dermatologists evaluated both before and after photographs for overall improvement of skin laxity and texture. Follow-ups were done 2-4 months after the last treatment. The observers were not informed which photographs were before or after.
Dr. Garden reported that the observers correctly chose 19 out of 20 patients’ before and after photographs, and they rated the mean grade level of improvement as 43%. Nearly half of the patients (48%) were graded at 50% or greater improvement. At the same time, patients rated their own improvement as a mean 6.5 out of 10. Nearly half of patients graded their outcome at 7 or better, which was designated as “very satisfied.” The procedures were well tolerated, Dr. Garden said, and the most common side effects were minor transient erythema and edema. The erythema generally faded after 2-4 hours, and the mild edema lasted up to 24 hours.
“Magnetic energy is a new technology that can be used to treat lower face and neck laxity,” said Dr. Garden, who is also a professor of clinical dermatology at Northwestern University, Chicago. “We only treated patients with skin types I-IV, but we feel that this technology is likely safe for higher skin types as well.”
Rocky Mountain Biosystems and BioFusionary Corp. provided the device used for the study. Dr. Garden and his colleagues are currently extending the ongoing trial. He reported having no financial disclosures.
DENVER – Patients treated with results from a small trial showed.
“There are many different modalities for tissue tightening, including lights, radiofrequency, ultrasound and thermal energy,” Jerome M. Garden, MD, said in an interview in advance of the annual conference of the American Society for Laser Medicine and Surgery. “The idea behind all of these technologies is to heat up the skin’s collagen and to stimulate further collagen production, which can then result in improved skin tightening and textural improvement.”
In a trial conducted at the Chicago-based Physicians Laser and Dermatology Institute, Dr. Garden and his colleagues evaluated a new technology for tissue tightening that involves magnetic energy. Developed by Rocky Mountain Biosystems and BioFusionary Corp., the investigative device produces a magnetic field in the targeted tissue, which then results in the heating and eventual tightening of the tissue. “By using magnetic energy, which relies on the polarity of the molecules, it allows for a safe way to target specifically the polar dermis, without heating the relatively dry epidermis or nonpolar adipose layer, resulting in a more tolerable and potentially safer alternative to tissue tightening,” said Dr. Garden, a dermatologist who is the director of the Physicians Laser and Dermatology Institute.
For the trial, 20 patients with facial and upper skin laxity underwent a mean of 4.3 treatment sessions with the 27MHz magnetic device that used a 3-cm spot size, with a minimum of 4 weeks between each session. No anesthetics or analgesics were used. “No gels or skin prep was performed before the treatment, other than a gentle soap beforehand,” Dr. Garden said. “A bland moisturizer was applied to the treated skin after treatments.” The majority of patients (85%) had paid for their procedures (a price comparable to other skin-tightening procedures), and two board-certified dermatologists evaluated both before and after photographs for overall improvement of skin laxity and texture. Follow-ups were done 2-4 months after the last treatment. The observers were not informed which photographs were before or after.
Dr. Garden reported that the observers correctly chose 19 out of 20 patients’ before and after photographs, and they rated the mean grade level of improvement as 43%. Nearly half of the patients (48%) were graded at 50% or greater improvement. At the same time, patients rated their own improvement as a mean 6.5 out of 10. Nearly half of patients graded their outcome at 7 or better, which was designated as “very satisfied.” The procedures were well tolerated, Dr. Garden said, and the most common side effects were minor transient erythema and edema. The erythema generally faded after 2-4 hours, and the mild edema lasted up to 24 hours.
“Magnetic energy is a new technology that can be used to treat lower face and neck laxity,” said Dr. Garden, who is also a professor of clinical dermatology at Northwestern University, Chicago. “We only treated patients with skin types I-IV, but we feel that this technology is likely safe for higher skin types as well.”
Rocky Mountain Biosystems and BioFusionary Corp. provided the device used for the study. Dr. Garden and his colleagues are currently extending the ongoing trial. He reported having no financial disclosures.
REPORTING FROM ASLMS 2019
Key clinical point: A device that delivers high magnetic energy was found safe and effective for treatment of skin laxity.
Major finding: Following treatment, dermatologists graded nearly half of the patients (48%) at 50% or greater improvement.
Study details: A single-center trial of 20 patients with facial and upper skin laxity who underwent a mean of 4.3 treatment sessions.
Disclosures: Rocky Mountain Biosystems and BioFusionary Corp. provided the device used for the study. Dr. Garden reported having no financial disclosures.
2018-2019 flu season: Going but not gone yet
The 2018-2019 flu season again showed real signs of ending as influenza activity levels dropped during the week ending March 23, according to the Centers for Disease Control and Prevention.
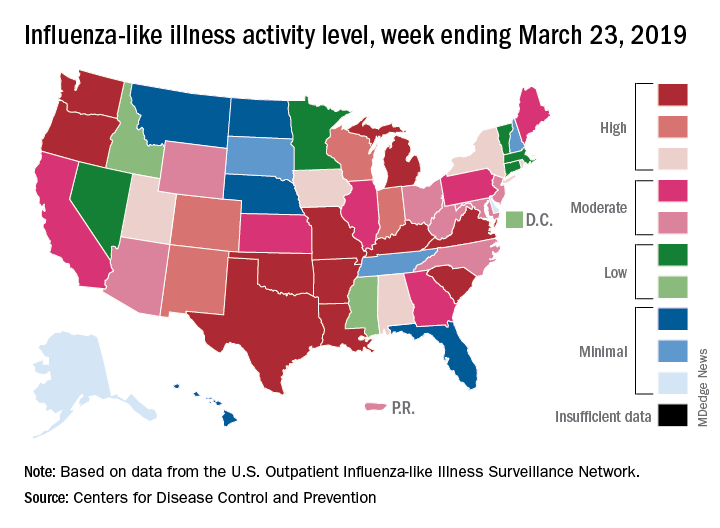
Despite those declines, however, current levels of influenza-like illness (ILI) activity are still elevated enough that the CDC issued a health advisory on March 28 to inform clinicians about the “increasing proportion of activity due to influenza A(H3N2) viruses, continued circulation of influenza A(H1N1) viruses, and low levels of influenza B viruses.”
The CDC’s weekly flu report, released March 29, does show that the overall burden is improving. The national proportion of outpatient visits for ILI dropped from 4.3% for the week ending March 16 to 3.8% for the latest reporting week, the CDC’s influenza division reported. The figure for March 16 was originally reported to be 4.4% but was revised in the new report.
The length of this years’ flu season, when measured as the number of weeks at or above the baseline level of 2.2%, is now 18 weeks. By this measure, the last five seasons have averaged 16 weeks, the CDC noted.
Influenza was considered widespread in 34 states and Puerto Rico for the week ending March 23, down from 44 states the previous week. The number of states at the highest level of ILI activity on the CDC’s 1-10 scale dropped from 20 to 11, and those in the high range (8-10) dropped from 26 to 20, data from the CDC’s Outpatient ILI Surveillance Network show.
There was one flu-related pediatric death during the week of March 23 but none reported from earlier weeks, which brings the total to 77 for the 2018-2019 season, the CDC said.
The 2018-2019 flu season again showed real signs of ending as influenza activity levels dropped during the week ending March 23, according to the Centers for Disease Control and Prevention.
Despite those declines, however, current levels of influenza-like illness (ILI) activity are still elevated enough that the CDC issued a health advisory on March 28 to inform clinicians about the “increasing proportion of activity due to influenza A(H3N2) viruses, continued circulation of influenza A(H1N1) viruses, and low levels of influenza B viruses.”
The CDC’s weekly flu report, released March 29, does show that the overall burden is improving. The national proportion of outpatient visits for ILI dropped from 4.3% for the week ending March 16 to 3.8% for the latest reporting week, the CDC’s influenza division reported. The figure for March 16 was originally reported to be 4.4% but was revised in the new report.
The length of this years’ flu season, when measured as the number of weeks at or above the baseline level of 2.2%, is now 18 weeks. By this measure, the last five seasons have averaged 16 weeks, the CDC noted.
Influenza was considered widespread in 34 states and Puerto Rico for the week ending March 23, down from 44 states the previous week. The number of states at the highest level of ILI activity on the CDC’s 1-10 scale dropped from 20 to 11, and those in the high range (8-10) dropped from 26 to 20, data from the CDC’s Outpatient ILI Surveillance Network show.
There was one flu-related pediatric death during the week of March 23 but none reported from earlier weeks, which brings the total to 77 for the 2018-2019 season, the CDC said.
The 2018-2019 flu season again showed real signs of ending as influenza activity levels dropped during the week ending March 23, according to the Centers for Disease Control and Prevention.
Despite those declines, however, current levels of influenza-like illness (ILI) activity are still elevated enough that the CDC issued a health advisory on March 28 to inform clinicians about the “increasing proportion of activity due to influenza A(H3N2) viruses, continued circulation of influenza A(H1N1) viruses, and low levels of influenza B viruses.”
The CDC’s weekly flu report, released March 29, does show that the overall burden is improving. The national proportion of outpatient visits for ILI dropped from 4.3% for the week ending March 16 to 3.8% for the latest reporting week, the CDC’s influenza division reported. The figure for March 16 was originally reported to be 4.4% but was revised in the new report.
The length of this years’ flu season, when measured as the number of weeks at or above the baseline level of 2.2%, is now 18 weeks. By this measure, the last five seasons have averaged 16 weeks, the CDC noted.
Influenza was considered widespread in 34 states and Puerto Rico for the week ending March 23, down from 44 states the previous week. The number of states at the highest level of ILI activity on the CDC’s 1-10 scale dropped from 20 to 11, and those in the high range (8-10) dropped from 26 to 20, data from the CDC’s Outpatient ILI Surveillance Network show.
There was one flu-related pediatric death during the week of March 23 but none reported from earlier weeks, which brings the total to 77 for the 2018-2019 season, the CDC said.
New renal, CV disease indication sought for canagliflozin
Janssen has announced that it has submitted a supplemental new drug application to the Food and Drug Administration to add an indication for canagliflozin (Invokana). The sodium-glucose cotransporter 2 is currently indicated, in addition to diet and exercise, for glycemic control in type 2 diabetes. However, in hopes of reducing the risks of end-stage kidney disease and of renal or cardiovascular death, according to a press release from the manufacturer.

If approved, canagliflozin will be the first diabetes medicine for the treatment of people living with type 2 diabetes and chronic kidney disease, according to the press release.
The application was based on the results of the phase 3 CREDENCE trial, a randomized, double-blind, placebo-controlled, multicenter study of 4,401 patients with type 2 diabetes, stage 2 or 3 chronic kidney disease, and macroalbuminuria. The patients received standard of care as well. The trial was stopped early, in July 2018, because it had met the prespecified criteria for efficacy. Data from the trial will be presented in mid-April at the annual meeting of the International Society of Nephrology World Congress of Nephrology in Melbourne.
Canagliflozin is contraindicated in patients with severe renal impairment (an estimated glomerular filtration rate of less than 30 mL/min per 1.73 m2), patients with end-stage renal disease, or patients on dialysis. Serious side effects associated with canagliflozin include ketoacidosis, kidney problems, hyperkalemia, serious urinary tract infections, and hypoglycemia. The most common side effects are yeast infections of the vagina or penis, and changes in urination.
The full prescribing information for canagliflozin is available on the FDA website.
Janssen has announced that it has submitted a supplemental new drug application to the Food and Drug Administration to add an indication for canagliflozin (Invokana). The sodium-glucose cotransporter 2 is currently indicated, in addition to diet and exercise, for glycemic control in type 2 diabetes. However, in hopes of reducing the risks of end-stage kidney disease and of renal or cardiovascular death, according to a press release from the manufacturer.
If approved, canagliflozin will be the first diabetes medicine for the treatment of people living with type 2 diabetes and chronic kidney disease, according to the press release.
The application was based on the results of the phase 3 CREDENCE trial, a randomized, double-blind, placebo-controlled, multicenter study of 4,401 patients with type 2 diabetes, stage 2 or 3 chronic kidney disease, and macroalbuminuria. The patients received standard of care as well. The trial was stopped early, in July 2018, because it had met the prespecified criteria for efficacy. Data from the trial will be presented in mid-April at the annual meeting of the International Society of Nephrology World Congress of Nephrology in Melbourne.
Canagliflozin is contraindicated in patients with severe renal impairment (an estimated glomerular filtration rate of less than 30 mL/min per 1.73 m2), patients with end-stage renal disease, or patients on dialysis. Serious side effects associated with canagliflozin include ketoacidosis, kidney problems, hyperkalemia, serious urinary tract infections, and hypoglycemia. The most common side effects are yeast infections of the vagina or penis, and changes in urination.
The full prescribing information for canagliflozin is available on the FDA website.
Janssen has announced that it has submitted a supplemental new drug application to the Food and Drug Administration to add an indication for canagliflozin (Invokana). The sodium-glucose cotransporter 2 is currently indicated, in addition to diet and exercise, for glycemic control in type 2 diabetes. However, in hopes of reducing the risks of end-stage kidney disease and of renal or cardiovascular death, according to a press release from the manufacturer.
If approved, canagliflozin will be the first diabetes medicine for the treatment of people living with type 2 diabetes and chronic kidney disease, according to the press release.
The application was based on the results of the phase 3 CREDENCE trial, a randomized, double-blind, placebo-controlled, multicenter study of 4,401 patients with type 2 diabetes, stage 2 or 3 chronic kidney disease, and macroalbuminuria. The patients received standard of care as well. The trial was stopped early, in July 2018, because it had met the prespecified criteria for efficacy. Data from the trial will be presented in mid-April at the annual meeting of the International Society of Nephrology World Congress of Nephrology in Melbourne.
Canagliflozin is contraindicated in patients with severe renal impairment (an estimated glomerular filtration rate of less than 30 mL/min per 1.73 m2), patients with end-stage renal disease, or patients on dialysis. Serious side effects associated with canagliflozin include ketoacidosis, kidney problems, hyperkalemia, serious urinary tract infections, and hypoglycemia. The most common side effects are yeast infections of the vagina or penis, and changes in urination.
The full prescribing information for canagliflozin is available on the FDA website.
HM19 Day One highlights: Pulmonary, critical care, and perioperative care updates (VIDEO)
Marina Farah, MD, MHA, and Kranthi Sitammagari, MD, editorial board members for The Hospitalist, discuss Day One highlights from HM19.

Marina Farah, MD, MHA, and Kranthi Sitammagari, MD, editorial board members for The Hospitalist, discuss Day One highlights from HM19.
Marina Farah, MD, MHA, and Kranthi Sitammagari, MD, editorial board members for The Hospitalist, discuss Day One highlights from HM19.
