User login
Hope, hepatology, and social determinants of health
Welcome to the April edition of GI & Hepatology News. April has always been a month where we have a sense of renewal and hope. For those of us living in northern climates, the distinct change in daylight and the melting of the snow (finally) both lifts us from the doldrums of winter darkness. In just over a month, we will gather in Washington for Digestive Disease Week® (DDW). I have seen a preview of AGA plenary sessions (basic science and clinical). They will be terrific. We will hear about advances in areas such as the microbiome, IBD-related inflammatory pathways, new insights into functional bowel disorders, and myriad new therapeutics (both medical and device) for us to share with our patients.
Substantial work is being done to better define an IBD severity index. These metrics are of critical importance for clinical researchers to use as we investigate the efficacy and effectiveness of new IBD drugs. You can also read about incorporating psychological care in the management of chronic diseases – a topic becoming more important as we expand our focus beyond just the biology of disease and into social determinants of health as we continue our transition to value-based reimbursement. Another topic included this month (and to which several DDW sessions are dedicated) is the devastating impact of opiates on our patients.

We have included a number of hepatology articles this month, such as the front-page story on NASH and its relationship with hepatocellular cancer. Pioglitazone benefits NASH patients with and without type 2 diabetes and biomarkers may predict liver transplant failures. There are selected articles about Barrett’s esophagus progression and risk stratification for colorectal cancer.
From Washington, we have received some good news. Please see the AGA commentary on the proposed budget. We were reminded last month about how Federal politics can impact U.S. medicine. With the (very late) reauthorization of the Children’s Health Insurance Plan (CHIP), we saw how political dysfunction can impact millions of American family’s lives. Changes in 340-B funding, continued transition from commercial to government payers, a tightening labor market, relentless increases in overhead expenses, all combine to reduce financial margins of both academic and nonacademic health systems. Economic pressures are leading to massive consolidations within the health care delivery system. Vertical integrations now have supplanted horizontal integrations as the industry trend. This situation that will impact many of our independent gastroenterology practices as demand-side management by large national corporations increases.
John I. Allen, MD, MBA, AGAF
Editor in Chief
Welcome to the April edition of GI & Hepatology News. April has always been a month where we have a sense of renewal and hope. For those of us living in northern climates, the distinct change in daylight and the melting of the snow (finally) both lifts us from the doldrums of winter darkness. In just over a month, we will gather in Washington for Digestive Disease Week® (DDW). I have seen a preview of AGA plenary sessions (basic science and clinical). They will be terrific. We will hear about advances in areas such as the microbiome, IBD-related inflammatory pathways, new insights into functional bowel disorders, and myriad new therapeutics (both medical and device) for us to share with our patients.
Substantial work is being done to better define an IBD severity index. These metrics are of critical importance for clinical researchers to use as we investigate the efficacy and effectiveness of new IBD drugs. You can also read about incorporating psychological care in the management of chronic diseases – a topic becoming more important as we expand our focus beyond just the biology of disease and into social determinants of health as we continue our transition to value-based reimbursement. Another topic included this month (and to which several DDW sessions are dedicated) is the devastating impact of opiates on our patients.
We have included a number of hepatology articles this month, such as the front-page story on NASH and its relationship with hepatocellular cancer. Pioglitazone benefits NASH patients with and without type 2 diabetes and biomarkers may predict liver transplant failures. There are selected articles about Barrett’s esophagus progression and risk stratification for colorectal cancer.
From Washington, we have received some good news. Please see the AGA commentary on the proposed budget. We were reminded last month about how Federal politics can impact U.S. medicine. With the (very late) reauthorization of the Children’s Health Insurance Plan (CHIP), we saw how political dysfunction can impact millions of American family’s lives. Changes in 340-B funding, continued transition from commercial to government payers, a tightening labor market, relentless increases in overhead expenses, all combine to reduce financial margins of both academic and nonacademic health systems. Economic pressures are leading to massive consolidations within the health care delivery system. Vertical integrations now have supplanted horizontal integrations as the industry trend. This situation that will impact many of our independent gastroenterology practices as demand-side management by large national corporations increases.
John I. Allen, MD, MBA, AGAF
Editor in Chief
Welcome to the April edition of GI & Hepatology News. April has always been a month where we have a sense of renewal and hope. For those of us living in northern climates, the distinct change in daylight and the melting of the snow (finally) both lifts us from the doldrums of winter darkness. In just over a month, we will gather in Washington for Digestive Disease Week® (DDW). I have seen a preview of AGA plenary sessions (basic science and clinical). They will be terrific. We will hear about advances in areas such as the microbiome, IBD-related inflammatory pathways, new insights into functional bowel disorders, and myriad new therapeutics (both medical and device) for us to share with our patients.
Substantial work is being done to better define an IBD severity index. These metrics are of critical importance for clinical researchers to use as we investigate the efficacy and effectiveness of new IBD drugs. You can also read about incorporating psychological care in the management of chronic diseases – a topic becoming more important as we expand our focus beyond just the biology of disease and into social determinants of health as we continue our transition to value-based reimbursement. Another topic included this month (and to which several DDW sessions are dedicated) is the devastating impact of opiates on our patients.
We have included a number of hepatology articles this month, such as the front-page story on NASH and its relationship with hepatocellular cancer. Pioglitazone benefits NASH patients with and without type 2 diabetes and biomarkers may predict liver transplant failures. There are selected articles about Barrett’s esophagus progression and risk stratification for colorectal cancer.
From Washington, we have received some good news. Please see the AGA commentary on the proposed budget. We were reminded last month about how Federal politics can impact U.S. medicine. With the (very late) reauthorization of the Children’s Health Insurance Plan (CHIP), we saw how political dysfunction can impact millions of American family’s lives. Changes in 340-B funding, continued transition from commercial to government payers, a tightening labor market, relentless increases in overhead expenses, all combine to reduce financial margins of both academic and nonacademic health systems. Economic pressures are leading to massive consolidations within the health care delivery system. Vertical integrations now have supplanted horizontal integrations as the industry trend. This situation that will impact many of our independent gastroenterology practices as demand-side management by large national corporations increases.
John I. Allen, MD, MBA, AGAF
Editor in Chief

Pain Management in an Opioid Epidemic What’s Appropriate, What’s Safe
CE/CME No: CR-1804
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Understand the basic pharmacology of opioid medications and how they affect pain.
• Apply a stepwise approach to pain management, based on the World Health Organization's "pain ladder."
• Communicate to patients the key educational points on the risks of opioid use.
• Identify strategies to deter or detect opioid misuse or abubse.
FACULTY
Deborah Salani is an Associate Professor of Clinical and Director of the Accelerated BSN Program, Nichole A. Crenshaw is an Assistant Professor of Clinical and Program Director for the Adult Gerontology Acute Care Nurse Practitioner Program, Brenda Owusu is an Assistant Professor of Clinical and Program Director for the Adult Gerontology Primary Care Nurse Practitioner Program, and Juan M. Gonzalez is an Assistant Professor of Clinical and Program Director for the Family Nurse Practitioner Program, at the University of Miami School of Nursing and Health Studies in Coral Gables, Florida.
The authors have no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through March 31, 2019.
Article begins on next page >>
Abuse of prescribed controlled substances—particularly opioid analgesics—and associated morbidity and mortality are a serious public health problem. The response to this crisis must include prevention, early identification, and appropriate treatment of addiction. Prescribing NPs and PAs must understand how to manage acute and chronic pain while also being attentive to signs of drug seeking and opioid misuse and abuse. The information and tools outlined in this article can equip providers to combat the opioid epidemic.
Controlled prescription drug abuse and its associated morbidity and mortality are a serious public health problem globally. In 2015, more than 29 million people worldwide misused and abused drugs, according to the United Nations Office on Drugs and Crime.1 Opioid use disorders account for approximately 70% of that estimate.
In the US, the mortality associated with this abuse has been devastating. Between 1999 and 2014, drug overdose deaths nearly tripled; in 2014 alone, there were 47,055 such fatalities, 61% of which involved opioids.2,3 Since 2000, unintentional overdose deaths from opioids have increased by 200%.3 Overdose deaths associated with natural and semisynthetic opioids (the most commonly prescribed pain relievers) increased 9% from 2013 to 2014, while those associated with synthetic opioids (fentanyl and tramadol) nearly doubled in the same period.3
Further contributing to the problem, a person addicted to prescription opioid drugs is 40 times more likely to be addicted to heroin, compared to someone who is not addicted to opioids.4 Deaths related to heroin overdose continue to dramatically increase.3
A call to action
In August 2016, former US Surgeon General Vivek Murthy, MD, sent a personal letter to more than 2.3 million health care providers, seeking their assistance in addressing the prescription opioid crisis.5 Murthy acknowledged the challenges providers face when attempting to strike a balance between treating a patient’s pain and reducing the risk for opioid addiction. He explained that clinicians are uniquely situated to end this crisis, and he asked providers to pledge to “turn the tide” by taking three actions
- Become more educated about treating pain safely and effectively.
- Screen patients for opioid use disorder and make the appropriate evidence-based treatment referrals.
- Discuss and treat addiction as a chronic disorder.5
To help stem the epidemic of controlled prescription drug abuse, NPs and PAs must be knowledgeable about patient safety issues, including how to identify patients at risk for opioid misuse and recognize signs of misuse or abuse. This article aims to educate providers who have prescriptive authority about the pharmacology of opioids; safe and effective prescribing of these drugs; and how to identify and manage misuse and abuse.
Continue to: OVERVIEW OF PAIN
OVERVIEW OF PAIN
Pain, considered the fifth vital sign, is one of the more common reasons that people seek treatment from a health care provider. Pain is a personal, individual, subjective experience: It is whatever the patient says it is and exists whenever the patient says it does. Pain is an unpleasant sensory and emotional experience associated with actual or potential tissue damage.
Pain is classified as acute or chronic. Acute pain is a sudden but temporary, self-limiting response to some type of bodily injury; it generally lasts less than six months. Chronic pain is often associated with prolonged diseases such as cancer, fibromyalgia, and osteoarthritis; it persists for six months or longer.
Pain can be separated into two categories: nociceptive and neuropathic. Nociceptive pain originates from peripheral or visceral nociceptors as a result of injury and comprises somatic and visceral pain. Somatic pain is caused by injury to soft tissue, connective tissue, and bone; the classic description is a sharp, well-localized discomfort. Visceral pain originates from an organ or deeper structure; it is commonly described as dull, poorly localized, and sensitive to stretch, ischemia, and inflammation.
Neuropathic pain is an abnormal processing of pain stimuli by the peripheral nervous system or central nervous system (CNS) and can result from injury or inflammation to a nerve. Neuropathic pain is usually described by patients as electric, burning, and/or shooting. Examples include pain associated with cancer, diabetic neuropathy, and phantom-limb sensation (following amputation).
The physiologic experience of pain follows a defined set of phases. First is transduction, which occurs at the moment of injury or trauma; sensory nerve endings convert the noxious stimulus into a nerve impulse. Second is transmission of the pain impulse to the spinal column by means of chemical messengers known as neurotransmitters.
After the pain impulse reaches the spinal tract, it continues to the brain, at which point there is perception, the third step in the process. This leads to modulation (also known as anti-nociception). In this fourth step, neurons that originate in the brainstem are activated, releasing neurotransmitters that inhibit transmission of pain. Modulation occurs in several areas of the CNS and involves the neurotransmitters serotonin, norepinephrine, and endogenous opioids (eg, ß-endorphin).6 During modulation, the limbic nervous system provokes a response to the painful stimulus, triggering endogenous opioids to bind to opioid receptors.7
Continue to: ROLE OF OPIOIDS IN PAIN MANAGEMENT
ROLE OF OPIOIDS IN PAIN MANAGEMENT
Opioids have been used to control pain for centuries. They are extracted from the opium poppy plant, Papaver somniferum. From the substance extracted, roughly 9% to 14% is morphine and 0.8% to 2.5% is codeine.7 Opioids are used to treat many symptoms and ailments, including diarrhea, moderate to severe pain, and persistent cough.
Opioids work through receptors in the CNS, including mu, kappa, and delta opioid receptors and the opioid-like receptor nociceptin.7 The principal receptors associated with pain physiology and inhibition are mu and kappa. (Morphine, the gold standard for treating severe pain, is an opioid agonist that binds to mu and kappa receptors.)
Most opioids that are used clinically bind to mu receptors; these drugs provide analgesia but also present the risk for adverse effects, such as decreased respiratory drive, miosis, and decreased motor function of the gastrointestinal (GI) tract, which can lead to constipation.8 Because mu receptors are located mainly in the brain and spinal cord (as well as the GI tract), opioids also produce a feeling of euphoria that can lead to dependence.
Kappa receptors, in contrast, are located mainly in the limbic system, diencephalic area, and spinal cord. When these receptors are activated, they can produce spinal analgesia, dyspnea, dependence, and dysphoria.
Delta receptors also play a role in pain management and are associated with emotional and affective components of the experience of pain.7 They are largely located in the brain; when activated, they can lead to spinal and supraspinal anesthesia, as well as decreased gastric motility.8 Delta receptors have not been studied as much as mu and kappa receptors, but it has been suggested that they play a role in psychologic dependency.
Depending on the effect that a drug has on these receptors, it can be considered a full (or pure) opioid agonist, a partial agonist, or a mixed agonist–antagonist. By binding to opioid receptors, opioid agonists provide pain relief. Health care providers often prescribe a full agonist, such as morphine, hydrocodone, codeine, or oxycodone, to treat pain. Partial agonists, such as buprenorphine and butorphanol, often decrease activity at mu receptor sites. Mixed agonist–antagonists either block or bind opioids at receptor sites.9 Medications that block mu and kappa receptors are considered opioid antagonists, which are used not to treat pain but rather to reverse the effect of opioids (eg, naloxone).6
Table 1 lists commonly used opioids, their analgesic duration, and the standard approved dosages.10
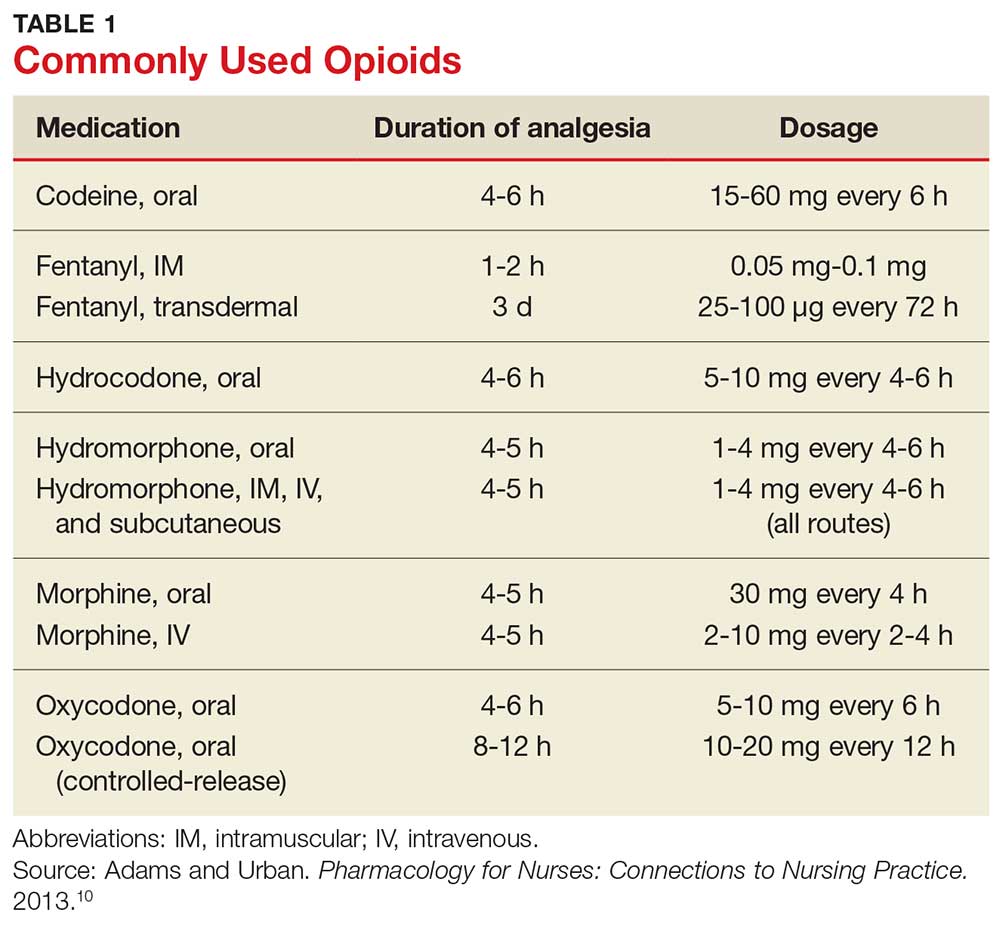
Continue to: A STEPWISE APPROACH TO PAIN MANAGEMENT
A STEPWISE APPROACH TO PAIN MANAGEMENT
On January 1, 2018, The Joint Commission (JNC) implemented new and revised standards to ensure that all patients receive appropriate assessment and management of their pain. While these standards apply to accredited hospitals, they provide a solid framework for assessing and treating pain in any patient. JNC now requires that patients be included in the development of treatment plans, which should encompass realistic expectations and reasonable goals, and that providers promote safe opioid use by identifying and monitoring high-risk patients.11
One valuable tool that can help clinicians fulfill the obligation to provide safe and effective pain management is the World Health Organization’s “pain ladder” (see Figure 1).12 Originally released in 1986 to address cancer pain in the pediatric population, this tool has proven validity. It has since been expanded to guide treatment of pain in other patient populations. In addition, the steps of the “pain ladder” provide useful information on the clinical examination and documentation of pain, principles of pharmacotherapeutic management, and considerations when using different analgesics.
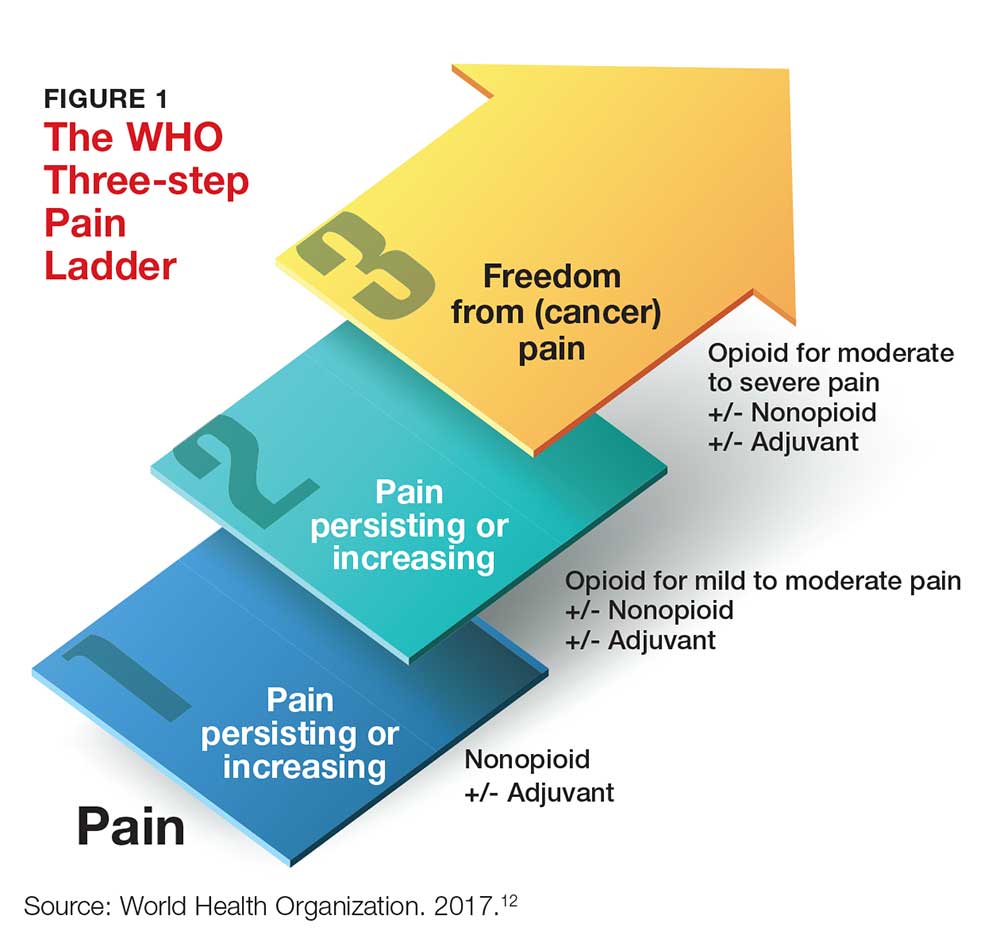
Pain is assessed on a scale of 1 to 10, with 1 representing the least pain. Medication recommendations are as follows
- For mild pain (ie, a score of 1-3): acetaminophen, NSAIDs, or other nonopioids.
- For moderate pain (pain score, 4-6): an opioid (eg, hydrocodone), with or without an adjunct medication.
- For severe pain (pain score, 7-10) or pain that has not responded to previous therapies: a stronger opioid (eg, morphine, hydromorphone, fentanyl), with or without an adjuvant drug.12
In all cases, patients should be informed about both pharmacotherapeutic and nonpharmacotherapeutic options. The latter include hypnosis, relaxation techniques, acupuncture, physical therapy, application of heat and cold, and electro-analgesia.
Pharmacologic options at any “step” of the ladder carry the risk for adverse effects. Thus, NPs and PAs who prescribe these medications need to apprise patients of the potential harms associated with their treatment.
Acetaminophen. Patients should be instructed on the safe use of acetaminophen, particularly with regard to dosing, since liver damage can occur. Patients should not take more than 4,000 mg in a 24-hour period, and each dose should not exceed 1,000 mg.
NSAIDs. These drugs are often used for short-term management of mild and moderate pain. Patients should be instructed to take these agents with food to decrease GI upset. Other common adverse effects include GI bleed or perforation and renal insufficiency or failure.
Opioids. Depending on which class of receptors an opioid medication targets, patients may develop any of the following: constipation, decreased GI motility, nausea, hypotension, urinary retention, euphoria, pruritus, miosis, dependence, respiratory depression, and sedation. It is important for NPs and PAs who prescribe these medications to remain vigilant for adverse effects and complications from opioid use and to educate the patient and his/her family about possible complications.6
Patients must be instructed not to drink alcohol or take other CNS depressants while taking an opioid. They should be advised about the dangers of operating heavy equipment or engaging in other activities that require mental and physical alertness, since opioids can cause drowsiness. Among the GI effects of some opioids (nausea, vomiting) is constipation—so patients should also be educated on the need to increase fluid intake and include high-fiber foods in their diet.9
But most important of all, patients taking an opioid should be informed that there is the potential for physical dependency and abuse with these agents, and these agents should be used only for acute, severe pain.
Continue to: DETECTING & MANAGING PRESCRIPTION DRUG MISUSE & ABUSE
DETECTING & MANAGING PRESCRIPTION DRUG MISUSE & ABUSE
Every patient has a right to adequate and safe pain control—but NPs and PAs must be aware of the potential for some patients to misuse opioids by taking them in a different way than intended, in a different quantity than prescribed, or without a prescription.13 Having prescriptive authority confers an obligation for NPs and PAs to recognize the prevalence of drug misuse and its impact on patients, families, and society.
Regrettably, there is lack of clarity in the literature about specific characteristics and demographic data that can help determine who is at risk for opioid misuse.14 For example, risk factors that have been associated with drug misuse include a personal or family history of substance abuse; younger age; and an ongoing psychiatric condition.
In contrast, Kennedy and colleagues determined that patients seeking prescription opioids for misuse or abuse tend to be older; be of Caucasian background; have a history of overdose; be receiving methadone maintenance therapy; and have been incarcerated.15 In addition, several characteristics—having moderate or extreme pain, disability, or a history of being refused pain medication—were also associated with a history of seeking prescription opioids to abuse.15
This diverse set of variables underscores the importance of obtaining and documenting a complete history from patients who are experiencing (and seeking relief of) pain; performing a thorough physical exam; and asking specific questions about the patient’s level of pain and the potential for misuse of pain medication.
Gathering this information may help identify patients at risk for opioid misuse or abuse. Furthermore, it ensures that a patient’s chronic pain is not being undertreated and that he/she is not being undeservedly labeled or judged as a drug seeker or abuser.
Continue to: Tools and strategies for appropriate use of opioids
Tools and strategies for appropriate use of opioids
There are tools and strategies available to ensure proper use of opioids for managing chronic noncancer pain. Urine drug testing, screening tools for opioid abuse, prescription drug monitoring programs, and opioid treatment agreements should be considered for patients who require prescription opioids to treat pain.15
Urine drug testing. The CDC recommends that prescribing clinicians perform urine drug testing before initiating opioid therapy and at least annually afterward. It can be used to assess for prescription medications generally, controlled prescription drugs specifically, and substances of abuse.16 Urine drug testing can mitigate the risk for misuse or overdose of opioids, as well as identify patients who were prescribed an opioid but are not taking it. The prescribing provider is responsible for explaining to the patient why urine testing is being done, performing confirmatory testing, and discussing results with the patient.
Risk-assessment tools. A number of web-based tools help the prescribing provider assess a patient’s risk for misuse or abuse of opioids and other substances. They fall into three general categories of use: assessing patients being considered for long-term opioid therapy; assessing for misuse once opioid treatment is initiated; and addressing the potential for substance abuse generally.17-24 Table 2 lists examples. Although screening tools are not 100% accurate at identifying who is a substance abuser, they do alert the provider that a potential problem exists and needs to be explored. As such, they should be considered one component of comprehensive risk assessment, monitoring, and mitigation.25
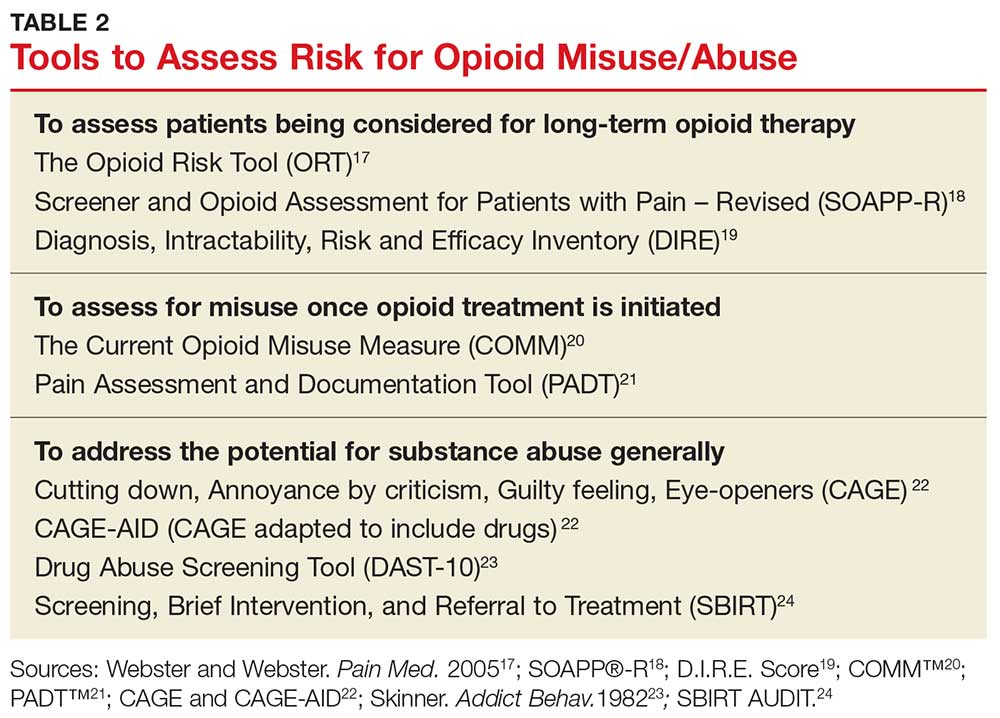
Prescription drug monitoring programs (PDMPs). NPs and PAs must also be aware of “doctor shopping,” in which a person seeks prescriptions from multiple providers (often under false pretenses) and has them filled at multiple pharmacies. PDMPs are designed to monitor for suspected abuse, diversion, or inappropriate prescribing. These state-run electronic databases track the amount of controlled substances prescribed, dispensed, and refilled for a given patient.26 This information can assist providers in identifying high-risk patients who may benefit from an early intervention program.27 Once a patient is identified as having an opioid use disorder, NPs and PAs must provide appropriate referral to an evidence-based practice for treatment of abuse. It is essential to recognize that an opioid use disorder is a chronic illness and that relapses occur.
Opioid treatment agreements. These have been presented as a strategy to prevent prescription drug abuse; however, there is little evidence to support their effectiveness in preventing medication misuse, abuse, or diversion of opioids. In fact, research has shown that such agreements can put the patient–provider therapeutic relationship at risk for disruption, since patients may feel mistrusted or stigmatized by the suggestion that they might behave inappropriately.28 The position of the American Pain Society and the American Academy of Pain Management is that patients and clinicians should have ongoing discussions about chronic opioid therapy that include goals, expectations, risks, and alternatives to opioids.29 If a written agreement is used, it needs to address the patient’s and the clinician’s responsibilities and expectations in managing chronic pain.28
Continue to: Additional resources for providers
Additional resources for providers
Many other resources are available for prescribers of controlled substances. For example, the CDC has published guidelines for prescribing opioids to patients with chronic pain, with a goal of increasing patient–provider communication.16 Additional goals include improving the safety of opioid use, maintaining the effectiveness of treatment, and reducing the necessity and practice of long-term therapy.
The FDA has also published a blueprint on how opioid analgesics can be formulated to deter abuse and, thus, be safer.30 Although directed at the pharmaceutical industry—the FDA encourages manufacturers to develop abuse-deterrent mechanisms, such as physical and chemical barriers, aversion technology, and new delivery systems—the guidance may enlighten providers on how abusers can alter or manipulate oral opioids to achieve the desired effects.30

CONCLUSION
Because NPs and PAs are authorized to prescribe Schedule II-V drugs in their scope of practice, they must have knowledge of drug-seeking behaviors and drug misuse before they prescribe opioids for pain relief. They must be attentive to patients’ pain-control needs and consider how to avoid or reduce the potential for misuse and abuse. Understanding the experience of pain and how opioids modulate it, as well as using available risk-assessment strategies, will help providers offer safe, effective treatment to their patients.
1. United Nations Office on Drugs and Crime. World drug report 2015. www.unodc.org/documents/wdr2015/World_Drug_Report_2015.pdf. Accessed March 21, 2018.
2. Rudd RA, Seth P, David F, Scholl L. Increases in drug and opioid-involved overdose deaths—United States, 2010-2015. MMWR Morb Mortal Wkly Rep. 2016;65(5051): 1445-1452.
3. Rudd RA, Aleshire N, Zibbell JE, Gladden RM. Increases in drug and opioid overdose deaths—United States, 2000-2014. MMWR Morb Mortal Wkly Rep. 2016;64(5051):1378-1382.
4. Jones CM, Logan J, Gladden RM, Bohm MK. Vital signs: demographic and substance use trends among heroin users—United States, 2002-2013. MMWR Morb Mortal Wkly Rep. 2015;64(26):719-725.
5. US Department of Health and Human Services. United States Surgeon General. Letter from the Surgeon General. 2016. https://turnthetiderx.org/#. Accessed March 21, 2018.
6. Arcangelo VP, Peterson AM, Wilbur V, Reinhold JA. Pharmacotherapeutics for Advanced Practice. 4th ed. Philadelphia, PA: Wolters Kluwer; 2017:1-23.
7. Adams MP, Holland N, Urban CQ. Pharmacology for Nurses: A Pathophysiologic Approach. 5th ed. Upper Saddle River, NJ: Pearson Education; 2016:239-252.
8. Grossman S, Porth CM. Somatosensory function, pain, and headache. In: Porth’s Pathophysiology: Concepts of Altered Health States. 9th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2014:441.
9. Woo TM, Robinson MV. Pain management: Acute and chronic pain. In: Pharmacotherapeutics for Advanced Practice Nurse Prescribers. 4th ed. Philadelphia, PA: FA Davis; 2015:1361.
10. Adams MP, Urban CQ. Pharmacology for Nurses: Connections to Nursing Practice. 2nd ed. Upper Saddle River, NJ: Pearson Education; 2013:437.
11. Joint Commission enhances pain assessment and management requirements for accredited hospitals. The Joint Commission Perspectives. 2017;37(7):1-4. www.jointcommission.org/assets/1/18/Joint_Commission_Enhances_Pain_Assessment_and_Management_Requirements_for_Accredited_Hospitals1.PDF. Accessed March 21, 2018.
12. World Health Organization. WHO’s cancer pain ladder for adults. 2017. www.who.int/cancer/palliative/painladder/en/. Accessed March 21, 2018.
13. National Institutes of Health. National Institute on Drug Abuse. Opioids: brief description. www.drugabuse.gov/drugs-abuse/opioids. Accessed March 21, 2018.
14. Hudspeth RS. Safe opioid prescribing for adults by nurse practitioners: Part 1. Patient history and assessment standards and techniques. J Nurse Pract. 2016;12(3):141-148.
15. Kennedy MC, Kerr T, DeBeck K, et al. Seeking prescription opioids from physicians for nonmedical use among people who inject drugs in a Canadian setting. Am J Addict. 2016;25(4):275-282.
16. US Department of Health and Human Services. CDC. CDC guideline for prescribing opioids for chronic pain—United States, 2016. www.cdc.gov/mmwr/volumes/65/rr/rr6501e1.htm. Accessed March 21, 2018.
17. Webster LR, Webster R. Predicting aberrant behaviors in opioid‐treated patients: preliminary validation of the Opioid Risk Tool. Pain Med. 2005;6(6):432. [Tool available at www.drugabuse.gov/sites/default/files/files/OpioidRiskTool.pdf.] Accessed March 21, 2018.
18. Screener and Opioid Assessment for Patients with Pain—Revised (SOAPP®-R). http://nationalpaincentre.mcmaster.ca/documents/soapp_r_sample_watermark.pdf. Accessed March 21, 2018.
19. D.I.R.E. Score: Patient Selection for Chronic Opioid Analgesia. www.ucdenver.edu/academics/colleges/PublicHealth/research/centers/CHWE/Documents/D.I.R.E.%20Score.pdf. Accessed March 21, 2018.
20. Current Opioid Misuse Measure (COMM)™. www.opioidprescribing.com/documents/09-comm-inflexxion.pdf. Accessed March 21, 2018.
21. Pain Assessment and Documentation Tool (PADT™). www.ucdenver.edu/academics/colleges/PublicHealth/research/centers/CHWE/Documents/Pain%20Assess ment%20Documentation%20Tool%20%28PADT%29.pdf. Accessed March 21, 2018.
22. The CAGE and CAGE-AID Questionnaires. www.ucdenver.edu/academics/colleges/PublicHealth/research/centers/CHWE/Documents/CAGE-AID.pdf. Accessed March 21, 2018.
23. Skinner HA. The drug abuse screening test. Addict Behav. 1982;7(4):363-371. [DAST-10 available at https://cde.drugabuse.gov/sites/nida_cde/files/DrugAbuseScreeningTest_2014Mar24.pdf.] Accessed March 21, 2018.
24. SBIRT AUDIT forms (English and Spanish). www.communitycarenc.org/media/tool-resource-files/sbirt-audit-forms.pdf. Accessed March 21, 2018.
25. Cheattle MD. Risk assessment: safe opioid prescribing tools. 2017. https://www.practicalpainmanagement.com/resource-centers/opioid-prescribing-monitoring/risk-assessment-safe-opioid-prescribing-tools. Accessed March 21, 2018.
26. Ali MM, Dowd WN, Classen T, et al. Prescription drug monitoring programs, nonmedical use of prescription drugs, and heroin use: evidence from the National Survey of Drug Use and Health. Addict Behav. 2017;69:65-77.
27. US Department of Health and Human Services. CDC. Drug overdose deaths hit record numbers in 2014. www.cdc.gov/media/releases/2015/p1218-drug-overdose.html. Accessed March 21, 2018.
28. McGee S, Silverman RD. Treatment agreements, informed consent, and the role of state medical boards in opioid prescribing. Pain Med. 2015;16(1):25-29.
29. Chou R, Fanciullo GJ, Fine PG, et al; for the American Pain Society–American Academy of Pain Medicine Opioids Guidelines Panel. Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J Pain. 2009;10(2):113-130.
30. US Department of Health and Human Services. FDA Center for Drug Evaluation and Research. Abuse-deterrent opioids—evaluation and labeling guidance for industry. 2015. www.fda.gov/downloads/Drugs/Guid ances/UCM334743.pdf. Accessed March 21, 2018.
CE/CME No: CR-1804
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Understand the basic pharmacology of opioid medications and how they affect pain.
• Apply a stepwise approach to pain management, based on the World Health Organization's "pain ladder."
• Communicate to patients the key educational points on the risks of opioid use.
• Identify strategies to deter or detect opioid misuse or abubse.
FACULTY
Deborah Salani is an Associate Professor of Clinical and Director of the Accelerated BSN Program, Nichole A. Crenshaw is an Assistant Professor of Clinical and Program Director for the Adult Gerontology Acute Care Nurse Practitioner Program, Brenda Owusu is an Assistant Professor of Clinical and Program Director for the Adult Gerontology Primary Care Nurse Practitioner Program, and Juan M. Gonzalez is an Assistant Professor of Clinical and Program Director for the Family Nurse Practitioner Program, at the University of Miami School of Nursing and Health Studies in Coral Gables, Florida.
The authors have no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through March 31, 2019.
Article begins on next page >>
Abuse of prescribed controlled substances—particularly opioid analgesics—and associated morbidity and mortality are a serious public health problem. The response to this crisis must include prevention, early identification, and appropriate treatment of addiction. Prescribing NPs and PAs must understand how to manage acute and chronic pain while also being attentive to signs of drug seeking and opioid misuse and abuse. The information and tools outlined in this article can equip providers to combat the opioid epidemic.
Controlled prescription drug abuse and its associated morbidity and mortality are a serious public health problem globally. In 2015, more than 29 million people worldwide misused and abused drugs, according to the United Nations Office on Drugs and Crime.1 Opioid use disorders account for approximately 70% of that estimate.
In the US, the mortality associated with this abuse has been devastating. Between 1999 and 2014, drug overdose deaths nearly tripled; in 2014 alone, there were 47,055 such fatalities, 61% of which involved opioids.2,3 Since 2000, unintentional overdose deaths from opioids have increased by 200%.3 Overdose deaths associated with natural and semisynthetic opioids (the most commonly prescribed pain relievers) increased 9% from 2013 to 2014, while those associated with synthetic opioids (fentanyl and tramadol) nearly doubled in the same period.3
Further contributing to the problem, a person addicted to prescription opioid drugs is 40 times more likely to be addicted to heroin, compared to someone who is not addicted to opioids.4 Deaths related to heroin overdose continue to dramatically increase.3
A call to action
In August 2016, former US Surgeon General Vivek Murthy, MD, sent a personal letter to more than 2.3 million health care providers, seeking their assistance in addressing the prescription opioid crisis.5 Murthy acknowledged the challenges providers face when attempting to strike a balance between treating a patient’s pain and reducing the risk for opioid addiction. He explained that clinicians are uniquely situated to end this crisis, and he asked providers to pledge to “turn the tide” by taking three actions
- Become more educated about treating pain safely and effectively.
- Screen patients for opioid use disorder and make the appropriate evidence-based treatment referrals.
- Discuss and treat addiction as a chronic disorder.5
To help stem the epidemic of controlled prescription drug abuse, NPs and PAs must be knowledgeable about patient safety issues, including how to identify patients at risk for opioid misuse and recognize signs of misuse or abuse. This article aims to educate providers who have prescriptive authority about the pharmacology of opioids; safe and effective prescribing of these drugs; and how to identify and manage misuse and abuse.
Continue to: OVERVIEW OF PAIN
OVERVIEW OF PAIN
Pain, considered the fifth vital sign, is one of the more common reasons that people seek treatment from a health care provider. Pain is a personal, individual, subjective experience: It is whatever the patient says it is and exists whenever the patient says it does. Pain is an unpleasant sensory and emotional experience associated with actual or potential tissue damage.
Pain is classified as acute or chronic. Acute pain is a sudden but temporary, self-limiting response to some type of bodily injury; it generally lasts less than six months. Chronic pain is often associated with prolonged diseases such as cancer, fibromyalgia, and osteoarthritis; it persists for six months or longer.
Pain can be separated into two categories: nociceptive and neuropathic. Nociceptive pain originates from peripheral or visceral nociceptors as a result of injury and comprises somatic and visceral pain. Somatic pain is caused by injury to soft tissue, connective tissue, and bone; the classic description is a sharp, well-localized discomfort. Visceral pain originates from an organ or deeper structure; it is commonly described as dull, poorly localized, and sensitive to stretch, ischemia, and inflammation.
Neuropathic pain is an abnormal processing of pain stimuli by the peripheral nervous system or central nervous system (CNS) and can result from injury or inflammation to a nerve. Neuropathic pain is usually described by patients as electric, burning, and/or shooting. Examples include pain associated with cancer, diabetic neuropathy, and phantom-limb sensation (following amputation).
The physiologic experience of pain follows a defined set of phases. First is transduction, which occurs at the moment of injury or trauma; sensory nerve endings convert the noxious stimulus into a nerve impulse. Second is transmission of the pain impulse to the spinal column by means of chemical messengers known as neurotransmitters.
After the pain impulse reaches the spinal tract, it continues to the brain, at which point there is perception, the third step in the process. This leads to modulation (also known as anti-nociception). In this fourth step, neurons that originate in the brainstem are activated, releasing neurotransmitters that inhibit transmission of pain. Modulation occurs in several areas of the CNS and involves the neurotransmitters serotonin, norepinephrine, and endogenous opioids (eg, ß-endorphin).6 During modulation, the limbic nervous system provokes a response to the painful stimulus, triggering endogenous opioids to bind to opioid receptors.7
Continue to: ROLE OF OPIOIDS IN PAIN MANAGEMENT
ROLE OF OPIOIDS IN PAIN MANAGEMENT
Opioids have been used to control pain for centuries. They are extracted from the opium poppy plant, Papaver somniferum. From the substance extracted, roughly 9% to 14% is morphine and 0.8% to 2.5% is codeine.7 Opioids are used to treat many symptoms and ailments, including diarrhea, moderate to severe pain, and persistent cough.
Opioids work through receptors in the CNS, including mu, kappa, and delta opioid receptors and the opioid-like receptor nociceptin.7 The principal receptors associated with pain physiology and inhibition are mu and kappa. (Morphine, the gold standard for treating severe pain, is an opioid agonist that binds to mu and kappa receptors.)
Most opioids that are used clinically bind to mu receptors; these drugs provide analgesia but also present the risk for adverse effects, such as decreased respiratory drive, miosis, and decreased motor function of the gastrointestinal (GI) tract, which can lead to constipation.8 Because mu receptors are located mainly in the brain and spinal cord (as well as the GI tract), opioids also produce a feeling of euphoria that can lead to dependence.
Kappa receptors, in contrast, are located mainly in the limbic system, diencephalic area, and spinal cord. When these receptors are activated, they can produce spinal analgesia, dyspnea, dependence, and dysphoria.
Delta receptors also play a role in pain management and are associated with emotional and affective components of the experience of pain.7 They are largely located in the brain; when activated, they can lead to spinal and supraspinal anesthesia, as well as decreased gastric motility.8 Delta receptors have not been studied as much as mu and kappa receptors, but it has been suggested that they play a role in psychologic dependency.
Depending on the effect that a drug has on these receptors, it can be considered a full (or pure) opioid agonist, a partial agonist, or a mixed agonist–antagonist. By binding to opioid receptors, opioid agonists provide pain relief. Health care providers often prescribe a full agonist, such as morphine, hydrocodone, codeine, or oxycodone, to treat pain. Partial agonists, such as buprenorphine and butorphanol, often decrease activity at mu receptor sites. Mixed agonist–antagonists either block or bind opioids at receptor sites.9 Medications that block mu and kappa receptors are considered opioid antagonists, which are used not to treat pain but rather to reverse the effect of opioids (eg, naloxone).6
Table 1 lists commonly used opioids, their analgesic duration, and the standard approved dosages.10
Continue to: A STEPWISE APPROACH TO PAIN MANAGEMENT
A STEPWISE APPROACH TO PAIN MANAGEMENT
On January 1, 2018, The Joint Commission (JNC) implemented new and revised standards to ensure that all patients receive appropriate assessment and management of their pain. While these standards apply to accredited hospitals, they provide a solid framework for assessing and treating pain in any patient. JNC now requires that patients be included in the development of treatment plans, which should encompass realistic expectations and reasonable goals, and that providers promote safe opioid use by identifying and monitoring high-risk patients.11
One valuable tool that can help clinicians fulfill the obligation to provide safe and effective pain management is the World Health Organization’s “pain ladder” (see Figure 1).12 Originally released in 1986 to address cancer pain in the pediatric population, this tool has proven validity. It has since been expanded to guide treatment of pain in other patient populations. In addition, the steps of the “pain ladder” provide useful information on the clinical examination and documentation of pain, principles of pharmacotherapeutic management, and considerations when using different analgesics.
Pain is assessed on a scale of 1 to 10, with 1 representing the least pain. Medication recommendations are as follows
- For mild pain (ie, a score of 1-3): acetaminophen, NSAIDs, or other nonopioids.
- For moderate pain (pain score, 4-6): an opioid (eg, hydrocodone), with or without an adjunct medication.
- For severe pain (pain score, 7-10) or pain that has not responded to previous therapies: a stronger opioid (eg, morphine, hydromorphone, fentanyl), with or without an adjuvant drug.12
In all cases, patients should be informed about both pharmacotherapeutic and nonpharmacotherapeutic options. The latter include hypnosis, relaxation techniques, acupuncture, physical therapy, application of heat and cold, and electro-analgesia.
Pharmacologic options at any “step” of the ladder carry the risk for adverse effects. Thus, NPs and PAs who prescribe these medications need to apprise patients of the potential harms associated with their treatment.
Acetaminophen. Patients should be instructed on the safe use of acetaminophen, particularly with regard to dosing, since liver damage can occur. Patients should not take more than 4,000 mg in a 24-hour period, and each dose should not exceed 1,000 mg.
NSAIDs. These drugs are often used for short-term management of mild and moderate pain. Patients should be instructed to take these agents with food to decrease GI upset. Other common adverse effects include GI bleed or perforation and renal insufficiency or failure.
Opioids. Depending on which class of receptors an opioid medication targets, patients may develop any of the following: constipation, decreased GI motility, nausea, hypotension, urinary retention, euphoria, pruritus, miosis, dependence, respiratory depression, and sedation. It is important for NPs and PAs who prescribe these medications to remain vigilant for adverse effects and complications from opioid use and to educate the patient and his/her family about possible complications.6
Patients must be instructed not to drink alcohol or take other CNS depressants while taking an opioid. They should be advised about the dangers of operating heavy equipment or engaging in other activities that require mental and physical alertness, since opioids can cause drowsiness. Among the GI effects of some opioids (nausea, vomiting) is constipation—so patients should also be educated on the need to increase fluid intake and include high-fiber foods in their diet.9
But most important of all, patients taking an opioid should be informed that there is the potential for physical dependency and abuse with these agents, and these agents should be used only for acute, severe pain.
Continue to: DETECTING & MANAGING PRESCRIPTION DRUG MISUSE & ABUSE
DETECTING & MANAGING PRESCRIPTION DRUG MISUSE & ABUSE
Every patient has a right to adequate and safe pain control—but NPs and PAs must be aware of the potential for some patients to misuse opioids by taking them in a different way than intended, in a different quantity than prescribed, or without a prescription.13 Having prescriptive authority confers an obligation for NPs and PAs to recognize the prevalence of drug misuse and its impact on patients, families, and society.
Regrettably, there is lack of clarity in the literature about specific characteristics and demographic data that can help determine who is at risk for opioid misuse.14 For example, risk factors that have been associated with drug misuse include a personal or family history of substance abuse; younger age; and an ongoing psychiatric condition.
In contrast, Kennedy and colleagues determined that patients seeking prescription opioids for misuse or abuse tend to be older; be of Caucasian background; have a history of overdose; be receiving methadone maintenance therapy; and have been incarcerated.15 In addition, several characteristics—having moderate or extreme pain, disability, or a history of being refused pain medication—were also associated with a history of seeking prescription opioids to abuse.15
This diverse set of variables underscores the importance of obtaining and documenting a complete history from patients who are experiencing (and seeking relief of) pain; performing a thorough physical exam; and asking specific questions about the patient’s level of pain and the potential for misuse of pain medication.
Gathering this information may help identify patients at risk for opioid misuse or abuse. Furthermore, it ensures that a patient’s chronic pain is not being undertreated and that he/she is not being undeservedly labeled or judged as a drug seeker or abuser.
Continue to: Tools and strategies for appropriate use of opioids
Tools and strategies for appropriate use of opioids
There are tools and strategies available to ensure proper use of opioids for managing chronic noncancer pain. Urine drug testing, screening tools for opioid abuse, prescription drug monitoring programs, and opioid treatment agreements should be considered for patients who require prescription opioids to treat pain.15
Urine drug testing. The CDC recommends that prescribing clinicians perform urine drug testing before initiating opioid therapy and at least annually afterward. It can be used to assess for prescription medications generally, controlled prescription drugs specifically, and substances of abuse.16 Urine drug testing can mitigate the risk for misuse or overdose of opioids, as well as identify patients who were prescribed an opioid but are not taking it. The prescribing provider is responsible for explaining to the patient why urine testing is being done, performing confirmatory testing, and discussing results with the patient.
Risk-assessment tools. A number of web-based tools help the prescribing provider assess a patient’s risk for misuse or abuse of opioids and other substances. They fall into three general categories of use: assessing patients being considered for long-term opioid therapy; assessing for misuse once opioid treatment is initiated; and addressing the potential for substance abuse generally.17-24 Table 2 lists examples. Although screening tools are not 100% accurate at identifying who is a substance abuser, they do alert the provider that a potential problem exists and needs to be explored. As such, they should be considered one component of comprehensive risk assessment, monitoring, and mitigation.25
Prescription drug monitoring programs (PDMPs). NPs and PAs must also be aware of “doctor shopping,” in which a person seeks prescriptions from multiple providers (often under false pretenses) and has them filled at multiple pharmacies. PDMPs are designed to monitor for suspected abuse, diversion, or inappropriate prescribing. These state-run electronic databases track the amount of controlled substances prescribed, dispensed, and refilled for a given patient.26 This information can assist providers in identifying high-risk patients who may benefit from an early intervention program.27 Once a patient is identified as having an opioid use disorder, NPs and PAs must provide appropriate referral to an evidence-based practice for treatment of abuse. It is essential to recognize that an opioid use disorder is a chronic illness and that relapses occur.
Opioid treatment agreements. These have been presented as a strategy to prevent prescription drug abuse; however, there is little evidence to support their effectiveness in preventing medication misuse, abuse, or diversion of opioids. In fact, research has shown that such agreements can put the patient–provider therapeutic relationship at risk for disruption, since patients may feel mistrusted or stigmatized by the suggestion that they might behave inappropriately.28 The position of the American Pain Society and the American Academy of Pain Management is that patients and clinicians should have ongoing discussions about chronic opioid therapy that include goals, expectations, risks, and alternatives to opioids.29 If a written agreement is used, it needs to address the patient’s and the clinician’s responsibilities and expectations in managing chronic pain.28
Continue to: Additional resources for providers
Additional resources for providers
Many other resources are available for prescribers of controlled substances. For example, the CDC has published guidelines for prescribing opioids to patients with chronic pain, with a goal of increasing patient–provider communication.16 Additional goals include improving the safety of opioid use, maintaining the effectiveness of treatment, and reducing the necessity and practice of long-term therapy.
The FDA has also published a blueprint on how opioid analgesics can be formulated to deter abuse and, thus, be safer.30 Although directed at the pharmaceutical industry—the FDA encourages manufacturers to develop abuse-deterrent mechanisms, such as physical and chemical barriers, aversion technology, and new delivery systems—the guidance may enlighten providers on how abusers can alter or manipulate oral opioids to achieve the desired effects.30
CONCLUSION
Because NPs and PAs are authorized to prescribe Schedule II-V drugs in their scope of practice, they must have knowledge of drug-seeking behaviors and drug misuse before they prescribe opioids for pain relief. They must be attentive to patients’ pain-control needs and consider how to avoid or reduce the potential for misuse and abuse. Understanding the experience of pain and how opioids modulate it, as well as using available risk-assessment strategies, will help providers offer safe, effective treatment to their patients.
CE/CME No: CR-1804
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Understand the basic pharmacology of opioid medications and how they affect pain.
• Apply a stepwise approach to pain management, based on the World Health Organization's "pain ladder."
• Communicate to patients the key educational points on the risks of opioid use.
• Identify strategies to deter or detect opioid misuse or abubse.
FACULTY
Deborah Salani is an Associate Professor of Clinical and Director of the Accelerated BSN Program, Nichole A. Crenshaw is an Assistant Professor of Clinical and Program Director for the Adult Gerontology Acute Care Nurse Practitioner Program, Brenda Owusu is an Assistant Professor of Clinical and Program Director for the Adult Gerontology Primary Care Nurse Practitioner Program, and Juan M. Gonzalez is an Assistant Professor of Clinical and Program Director for the Family Nurse Practitioner Program, at the University of Miami School of Nursing and Health Studies in Coral Gables, Florida.
The authors have no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through March 31, 2019.
Article begins on next page >>
Abuse of prescribed controlled substances—particularly opioid analgesics—and associated morbidity and mortality are a serious public health problem. The response to this crisis must include prevention, early identification, and appropriate treatment of addiction. Prescribing NPs and PAs must understand how to manage acute and chronic pain while also being attentive to signs of drug seeking and opioid misuse and abuse. The information and tools outlined in this article can equip providers to combat the opioid epidemic.
Controlled prescription drug abuse and its associated morbidity and mortality are a serious public health problem globally. In 2015, more than 29 million people worldwide misused and abused drugs, according to the United Nations Office on Drugs and Crime.1 Opioid use disorders account for approximately 70% of that estimate.
In the US, the mortality associated with this abuse has been devastating. Between 1999 and 2014, drug overdose deaths nearly tripled; in 2014 alone, there were 47,055 such fatalities, 61% of which involved opioids.2,3 Since 2000, unintentional overdose deaths from opioids have increased by 200%.3 Overdose deaths associated with natural and semisynthetic opioids (the most commonly prescribed pain relievers) increased 9% from 2013 to 2014, while those associated with synthetic opioids (fentanyl and tramadol) nearly doubled in the same period.3
Further contributing to the problem, a person addicted to prescription opioid drugs is 40 times more likely to be addicted to heroin, compared to someone who is not addicted to opioids.4 Deaths related to heroin overdose continue to dramatically increase.3
A call to action
In August 2016, former US Surgeon General Vivek Murthy, MD, sent a personal letter to more than 2.3 million health care providers, seeking their assistance in addressing the prescription opioid crisis.5 Murthy acknowledged the challenges providers face when attempting to strike a balance between treating a patient’s pain and reducing the risk for opioid addiction. He explained that clinicians are uniquely situated to end this crisis, and he asked providers to pledge to “turn the tide” by taking three actions
- Become more educated about treating pain safely and effectively.
- Screen patients for opioid use disorder and make the appropriate evidence-based treatment referrals.
- Discuss and treat addiction as a chronic disorder.5
To help stem the epidemic of controlled prescription drug abuse, NPs and PAs must be knowledgeable about patient safety issues, including how to identify patients at risk for opioid misuse and recognize signs of misuse or abuse. This article aims to educate providers who have prescriptive authority about the pharmacology of opioids; safe and effective prescribing of these drugs; and how to identify and manage misuse and abuse.
Continue to: OVERVIEW OF PAIN
OVERVIEW OF PAIN
Pain, considered the fifth vital sign, is one of the more common reasons that people seek treatment from a health care provider. Pain is a personal, individual, subjective experience: It is whatever the patient says it is and exists whenever the patient says it does. Pain is an unpleasant sensory and emotional experience associated with actual or potential tissue damage.
Pain is classified as acute or chronic. Acute pain is a sudden but temporary, self-limiting response to some type of bodily injury; it generally lasts less than six months. Chronic pain is often associated with prolonged diseases such as cancer, fibromyalgia, and osteoarthritis; it persists for six months or longer.
Pain can be separated into two categories: nociceptive and neuropathic. Nociceptive pain originates from peripheral or visceral nociceptors as a result of injury and comprises somatic and visceral pain. Somatic pain is caused by injury to soft tissue, connective tissue, and bone; the classic description is a sharp, well-localized discomfort. Visceral pain originates from an organ or deeper structure; it is commonly described as dull, poorly localized, and sensitive to stretch, ischemia, and inflammation.
Neuropathic pain is an abnormal processing of pain stimuli by the peripheral nervous system or central nervous system (CNS) and can result from injury or inflammation to a nerve. Neuropathic pain is usually described by patients as electric, burning, and/or shooting. Examples include pain associated with cancer, diabetic neuropathy, and phantom-limb sensation (following amputation).
The physiologic experience of pain follows a defined set of phases. First is transduction, which occurs at the moment of injury or trauma; sensory nerve endings convert the noxious stimulus into a nerve impulse. Second is transmission of the pain impulse to the spinal column by means of chemical messengers known as neurotransmitters.
After the pain impulse reaches the spinal tract, it continues to the brain, at which point there is perception, the third step in the process. This leads to modulation (also known as anti-nociception). In this fourth step, neurons that originate in the brainstem are activated, releasing neurotransmitters that inhibit transmission of pain. Modulation occurs in several areas of the CNS and involves the neurotransmitters serotonin, norepinephrine, and endogenous opioids (eg, ß-endorphin).6 During modulation, the limbic nervous system provokes a response to the painful stimulus, triggering endogenous opioids to bind to opioid receptors.7
Continue to: ROLE OF OPIOIDS IN PAIN MANAGEMENT
ROLE OF OPIOIDS IN PAIN MANAGEMENT
Opioids have been used to control pain for centuries. They are extracted from the opium poppy plant, Papaver somniferum. From the substance extracted, roughly 9% to 14% is morphine and 0.8% to 2.5% is codeine.7 Opioids are used to treat many symptoms and ailments, including diarrhea, moderate to severe pain, and persistent cough.
Opioids work through receptors in the CNS, including mu, kappa, and delta opioid receptors and the opioid-like receptor nociceptin.7 The principal receptors associated with pain physiology and inhibition are mu and kappa. (Morphine, the gold standard for treating severe pain, is an opioid agonist that binds to mu and kappa receptors.)
Most opioids that are used clinically bind to mu receptors; these drugs provide analgesia but also present the risk for adverse effects, such as decreased respiratory drive, miosis, and decreased motor function of the gastrointestinal (GI) tract, which can lead to constipation.8 Because mu receptors are located mainly in the brain and spinal cord (as well as the GI tract), opioids also produce a feeling of euphoria that can lead to dependence.
Kappa receptors, in contrast, are located mainly in the limbic system, diencephalic area, and spinal cord. When these receptors are activated, they can produce spinal analgesia, dyspnea, dependence, and dysphoria.
Delta receptors also play a role in pain management and are associated with emotional and affective components of the experience of pain.7 They are largely located in the brain; when activated, they can lead to spinal and supraspinal anesthesia, as well as decreased gastric motility.8 Delta receptors have not been studied as much as mu and kappa receptors, but it has been suggested that they play a role in psychologic dependency.
Depending on the effect that a drug has on these receptors, it can be considered a full (or pure) opioid agonist, a partial agonist, or a mixed agonist–antagonist. By binding to opioid receptors, opioid agonists provide pain relief. Health care providers often prescribe a full agonist, such as morphine, hydrocodone, codeine, or oxycodone, to treat pain. Partial agonists, such as buprenorphine and butorphanol, often decrease activity at mu receptor sites. Mixed agonist–antagonists either block or bind opioids at receptor sites.9 Medications that block mu and kappa receptors are considered opioid antagonists, which are used not to treat pain but rather to reverse the effect of opioids (eg, naloxone).6
Table 1 lists commonly used opioids, their analgesic duration, and the standard approved dosages.10
Continue to: A STEPWISE APPROACH TO PAIN MANAGEMENT
A STEPWISE APPROACH TO PAIN MANAGEMENT
On January 1, 2018, The Joint Commission (JNC) implemented new and revised standards to ensure that all patients receive appropriate assessment and management of their pain. While these standards apply to accredited hospitals, they provide a solid framework for assessing and treating pain in any patient. JNC now requires that patients be included in the development of treatment plans, which should encompass realistic expectations and reasonable goals, and that providers promote safe opioid use by identifying and monitoring high-risk patients.11
One valuable tool that can help clinicians fulfill the obligation to provide safe and effective pain management is the World Health Organization’s “pain ladder” (see Figure 1).12 Originally released in 1986 to address cancer pain in the pediatric population, this tool has proven validity. It has since been expanded to guide treatment of pain in other patient populations. In addition, the steps of the “pain ladder” provide useful information on the clinical examination and documentation of pain, principles of pharmacotherapeutic management, and considerations when using different analgesics.
Pain is assessed on a scale of 1 to 10, with 1 representing the least pain. Medication recommendations are as follows
- For mild pain (ie, a score of 1-3): acetaminophen, NSAIDs, or other nonopioids.
- For moderate pain (pain score, 4-6): an opioid (eg, hydrocodone), with or without an adjunct medication.
- For severe pain (pain score, 7-10) or pain that has not responded to previous therapies: a stronger opioid (eg, morphine, hydromorphone, fentanyl), with or without an adjuvant drug.12
In all cases, patients should be informed about both pharmacotherapeutic and nonpharmacotherapeutic options. The latter include hypnosis, relaxation techniques, acupuncture, physical therapy, application of heat and cold, and electro-analgesia.
Pharmacologic options at any “step” of the ladder carry the risk for adverse effects. Thus, NPs and PAs who prescribe these medications need to apprise patients of the potential harms associated with their treatment.
Acetaminophen. Patients should be instructed on the safe use of acetaminophen, particularly with regard to dosing, since liver damage can occur. Patients should not take more than 4,000 mg in a 24-hour period, and each dose should not exceed 1,000 mg.
NSAIDs. These drugs are often used for short-term management of mild and moderate pain. Patients should be instructed to take these agents with food to decrease GI upset. Other common adverse effects include GI bleed or perforation and renal insufficiency or failure.
Opioids. Depending on which class of receptors an opioid medication targets, patients may develop any of the following: constipation, decreased GI motility, nausea, hypotension, urinary retention, euphoria, pruritus, miosis, dependence, respiratory depression, and sedation. It is important for NPs and PAs who prescribe these medications to remain vigilant for adverse effects and complications from opioid use and to educate the patient and his/her family about possible complications.6
Patients must be instructed not to drink alcohol or take other CNS depressants while taking an opioid. They should be advised about the dangers of operating heavy equipment or engaging in other activities that require mental and physical alertness, since opioids can cause drowsiness. Among the GI effects of some opioids (nausea, vomiting) is constipation—so patients should also be educated on the need to increase fluid intake and include high-fiber foods in their diet.9
But most important of all, patients taking an opioid should be informed that there is the potential for physical dependency and abuse with these agents, and these agents should be used only for acute, severe pain.
Continue to: DETECTING & MANAGING PRESCRIPTION DRUG MISUSE & ABUSE
DETECTING & MANAGING PRESCRIPTION DRUG MISUSE & ABUSE
Every patient has a right to adequate and safe pain control—but NPs and PAs must be aware of the potential for some patients to misuse opioids by taking them in a different way than intended, in a different quantity than prescribed, or without a prescription.13 Having prescriptive authority confers an obligation for NPs and PAs to recognize the prevalence of drug misuse and its impact on patients, families, and society.
Regrettably, there is lack of clarity in the literature about specific characteristics and demographic data that can help determine who is at risk for opioid misuse.14 For example, risk factors that have been associated with drug misuse include a personal or family history of substance abuse; younger age; and an ongoing psychiatric condition.
In contrast, Kennedy and colleagues determined that patients seeking prescription opioids for misuse or abuse tend to be older; be of Caucasian background; have a history of overdose; be receiving methadone maintenance therapy; and have been incarcerated.15 In addition, several characteristics—having moderate or extreme pain, disability, or a history of being refused pain medication—were also associated with a history of seeking prescription opioids to abuse.15
This diverse set of variables underscores the importance of obtaining and documenting a complete history from patients who are experiencing (and seeking relief of) pain; performing a thorough physical exam; and asking specific questions about the patient’s level of pain and the potential for misuse of pain medication.
Gathering this information may help identify patients at risk for opioid misuse or abuse. Furthermore, it ensures that a patient’s chronic pain is not being undertreated and that he/she is not being undeservedly labeled or judged as a drug seeker or abuser.
Continue to: Tools and strategies for appropriate use of opioids
Tools and strategies for appropriate use of opioids
There are tools and strategies available to ensure proper use of opioids for managing chronic noncancer pain. Urine drug testing, screening tools for opioid abuse, prescription drug monitoring programs, and opioid treatment agreements should be considered for patients who require prescription opioids to treat pain.15
Urine drug testing. The CDC recommends that prescribing clinicians perform urine drug testing before initiating opioid therapy and at least annually afterward. It can be used to assess for prescription medications generally, controlled prescription drugs specifically, and substances of abuse.16 Urine drug testing can mitigate the risk for misuse or overdose of opioids, as well as identify patients who were prescribed an opioid but are not taking it. The prescribing provider is responsible for explaining to the patient why urine testing is being done, performing confirmatory testing, and discussing results with the patient.
Risk-assessment tools. A number of web-based tools help the prescribing provider assess a patient’s risk for misuse or abuse of opioids and other substances. They fall into three general categories of use: assessing patients being considered for long-term opioid therapy; assessing for misuse once opioid treatment is initiated; and addressing the potential for substance abuse generally.17-24 Table 2 lists examples. Although screening tools are not 100% accurate at identifying who is a substance abuser, they do alert the provider that a potential problem exists and needs to be explored. As such, they should be considered one component of comprehensive risk assessment, monitoring, and mitigation.25
Prescription drug monitoring programs (PDMPs). NPs and PAs must also be aware of “doctor shopping,” in which a person seeks prescriptions from multiple providers (often under false pretenses) and has them filled at multiple pharmacies. PDMPs are designed to monitor for suspected abuse, diversion, or inappropriate prescribing. These state-run electronic databases track the amount of controlled substances prescribed, dispensed, and refilled for a given patient.26 This information can assist providers in identifying high-risk patients who may benefit from an early intervention program.27 Once a patient is identified as having an opioid use disorder, NPs and PAs must provide appropriate referral to an evidence-based practice for treatment of abuse. It is essential to recognize that an opioid use disorder is a chronic illness and that relapses occur.
Opioid treatment agreements. These have been presented as a strategy to prevent prescription drug abuse; however, there is little evidence to support their effectiveness in preventing medication misuse, abuse, or diversion of opioids. In fact, research has shown that such agreements can put the patient–provider therapeutic relationship at risk for disruption, since patients may feel mistrusted or stigmatized by the suggestion that they might behave inappropriately.28 The position of the American Pain Society and the American Academy of Pain Management is that patients and clinicians should have ongoing discussions about chronic opioid therapy that include goals, expectations, risks, and alternatives to opioids.29 If a written agreement is used, it needs to address the patient’s and the clinician’s responsibilities and expectations in managing chronic pain.28
Continue to: Additional resources for providers
Additional resources for providers
Many other resources are available for prescribers of controlled substances. For example, the CDC has published guidelines for prescribing opioids to patients with chronic pain, with a goal of increasing patient–provider communication.16 Additional goals include improving the safety of opioid use, maintaining the effectiveness of treatment, and reducing the necessity and practice of long-term therapy.
The FDA has also published a blueprint on how opioid analgesics can be formulated to deter abuse and, thus, be safer.30 Although directed at the pharmaceutical industry—the FDA encourages manufacturers to develop abuse-deterrent mechanisms, such as physical and chemical barriers, aversion technology, and new delivery systems—the guidance may enlighten providers on how abusers can alter or manipulate oral opioids to achieve the desired effects.30
CONCLUSION
Because NPs and PAs are authorized to prescribe Schedule II-V drugs in their scope of practice, they must have knowledge of drug-seeking behaviors and drug misuse before they prescribe opioids for pain relief. They must be attentive to patients’ pain-control needs and consider how to avoid or reduce the potential for misuse and abuse. Understanding the experience of pain and how opioids modulate it, as well as using available risk-assessment strategies, will help providers offer safe, effective treatment to their patients.
1. United Nations Office on Drugs and Crime. World drug report 2015. www.unodc.org/documents/wdr2015/World_Drug_Report_2015.pdf. Accessed March 21, 2018.
2. Rudd RA, Seth P, David F, Scholl L. Increases in drug and opioid-involved overdose deaths—United States, 2010-2015. MMWR Morb Mortal Wkly Rep. 2016;65(5051): 1445-1452.
3. Rudd RA, Aleshire N, Zibbell JE, Gladden RM. Increases in drug and opioid overdose deaths—United States, 2000-2014. MMWR Morb Mortal Wkly Rep. 2016;64(5051):1378-1382.
4. Jones CM, Logan J, Gladden RM, Bohm MK. Vital signs: demographic and substance use trends among heroin users—United States, 2002-2013. MMWR Morb Mortal Wkly Rep. 2015;64(26):719-725.
5. US Department of Health and Human Services. United States Surgeon General. Letter from the Surgeon General. 2016. https://turnthetiderx.org/#. Accessed March 21, 2018.
6. Arcangelo VP, Peterson AM, Wilbur V, Reinhold JA. Pharmacotherapeutics for Advanced Practice. 4th ed. Philadelphia, PA: Wolters Kluwer; 2017:1-23.
7. Adams MP, Holland N, Urban CQ. Pharmacology for Nurses: A Pathophysiologic Approach. 5th ed. Upper Saddle River, NJ: Pearson Education; 2016:239-252.
8. Grossman S, Porth CM. Somatosensory function, pain, and headache. In: Porth’s Pathophysiology: Concepts of Altered Health States. 9th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2014:441.
9. Woo TM, Robinson MV. Pain management: Acute and chronic pain. In: Pharmacotherapeutics for Advanced Practice Nurse Prescribers. 4th ed. Philadelphia, PA: FA Davis; 2015:1361.
10. Adams MP, Urban CQ. Pharmacology for Nurses: Connections to Nursing Practice. 2nd ed. Upper Saddle River, NJ: Pearson Education; 2013:437.
11. Joint Commission enhances pain assessment and management requirements for accredited hospitals. The Joint Commission Perspectives. 2017;37(7):1-4. www.jointcommission.org/assets/1/18/Joint_Commission_Enhances_Pain_Assessment_and_Management_Requirements_for_Accredited_Hospitals1.PDF. Accessed March 21, 2018.
12. World Health Organization. WHO’s cancer pain ladder for adults. 2017. www.who.int/cancer/palliative/painladder/en/. Accessed March 21, 2018.
13. National Institutes of Health. National Institute on Drug Abuse. Opioids: brief description. www.drugabuse.gov/drugs-abuse/opioids. Accessed March 21, 2018.
14. Hudspeth RS. Safe opioid prescribing for adults by nurse practitioners: Part 1. Patient history and assessment standards and techniques. J Nurse Pract. 2016;12(3):141-148.
15. Kennedy MC, Kerr T, DeBeck K, et al. Seeking prescription opioids from physicians for nonmedical use among people who inject drugs in a Canadian setting. Am J Addict. 2016;25(4):275-282.
16. US Department of Health and Human Services. CDC. CDC guideline for prescribing opioids for chronic pain—United States, 2016. www.cdc.gov/mmwr/volumes/65/rr/rr6501e1.htm. Accessed March 21, 2018.
17. Webster LR, Webster R. Predicting aberrant behaviors in opioid‐treated patients: preliminary validation of the Opioid Risk Tool. Pain Med. 2005;6(6):432. [Tool available at www.drugabuse.gov/sites/default/files/files/OpioidRiskTool.pdf.] Accessed March 21, 2018.
18. Screener and Opioid Assessment for Patients with Pain—Revised (SOAPP®-R). http://nationalpaincentre.mcmaster.ca/documents/soapp_r_sample_watermark.pdf. Accessed March 21, 2018.
19. D.I.R.E. Score: Patient Selection for Chronic Opioid Analgesia. www.ucdenver.edu/academics/colleges/PublicHealth/research/centers/CHWE/Documents/D.I.R.E.%20Score.pdf. Accessed March 21, 2018.
20. Current Opioid Misuse Measure (COMM)™. www.opioidprescribing.com/documents/09-comm-inflexxion.pdf. Accessed March 21, 2018.
21. Pain Assessment and Documentation Tool (PADT™). www.ucdenver.edu/academics/colleges/PublicHealth/research/centers/CHWE/Documents/Pain%20Assess ment%20Documentation%20Tool%20%28PADT%29.pdf. Accessed March 21, 2018.
22. The CAGE and CAGE-AID Questionnaires. www.ucdenver.edu/academics/colleges/PublicHealth/research/centers/CHWE/Documents/CAGE-AID.pdf. Accessed March 21, 2018.
23. Skinner HA. The drug abuse screening test. Addict Behav. 1982;7(4):363-371. [DAST-10 available at https://cde.drugabuse.gov/sites/nida_cde/files/DrugAbuseScreeningTest_2014Mar24.pdf.] Accessed March 21, 2018.
24. SBIRT AUDIT forms (English and Spanish). www.communitycarenc.org/media/tool-resource-files/sbirt-audit-forms.pdf. Accessed March 21, 2018.
25. Cheattle MD. Risk assessment: safe opioid prescribing tools. 2017. https://www.practicalpainmanagement.com/resource-centers/opioid-prescribing-monitoring/risk-assessment-safe-opioid-prescribing-tools. Accessed March 21, 2018.
26. Ali MM, Dowd WN, Classen T, et al. Prescription drug monitoring programs, nonmedical use of prescription drugs, and heroin use: evidence from the National Survey of Drug Use and Health. Addict Behav. 2017;69:65-77.
27. US Department of Health and Human Services. CDC. Drug overdose deaths hit record numbers in 2014. www.cdc.gov/media/releases/2015/p1218-drug-overdose.html. Accessed March 21, 2018.
28. McGee S, Silverman RD. Treatment agreements, informed consent, and the role of state medical boards in opioid prescribing. Pain Med. 2015;16(1):25-29.
29. Chou R, Fanciullo GJ, Fine PG, et al; for the American Pain Society–American Academy of Pain Medicine Opioids Guidelines Panel. Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J Pain. 2009;10(2):113-130.
30. US Department of Health and Human Services. FDA Center for Drug Evaluation and Research. Abuse-deterrent opioids—evaluation and labeling guidance for industry. 2015. www.fda.gov/downloads/Drugs/Guid ances/UCM334743.pdf. Accessed March 21, 2018.
1. United Nations Office on Drugs and Crime. World drug report 2015. www.unodc.org/documents/wdr2015/World_Drug_Report_2015.pdf. Accessed March 21, 2018.
2. Rudd RA, Seth P, David F, Scholl L. Increases in drug and opioid-involved overdose deaths—United States, 2010-2015. MMWR Morb Mortal Wkly Rep. 2016;65(5051): 1445-1452.
3. Rudd RA, Aleshire N, Zibbell JE, Gladden RM. Increases in drug and opioid overdose deaths—United States, 2000-2014. MMWR Morb Mortal Wkly Rep. 2016;64(5051):1378-1382.
4. Jones CM, Logan J, Gladden RM, Bohm MK. Vital signs: demographic and substance use trends among heroin users—United States, 2002-2013. MMWR Morb Mortal Wkly Rep. 2015;64(26):719-725.
5. US Department of Health and Human Services. United States Surgeon General. Letter from the Surgeon General. 2016. https://turnthetiderx.org/#. Accessed March 21, 2018.
6. Arcangelo VP, Peterson AM, Wilbur V, Reinhold JA. Pharmacotherapeutics for Advanced Practice. 4th ed. Philadelphia, PA: Wolters Kluwer; 2017:1-23.
7. Adams MP, Holland N, Urban CQ. Pharmacology for Nurses: A Pathophysiologic Approach. 5th ed. Upper Saddle River, NJ: Pearson Education; 2016:239-252.
8. Grossman S, Porth CM. Somatosensory function, pain, and headache. In: Porth’s Pathophysiology: Concepts of Altered Health States. 9th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2014:441.
9. Woo TM, Robinson MV. Pain management: Acute and chronic pain. In: Pharmacotherapeutics for Advanced Practice Nurse Prescribers. 4th ed. Philadelphia, PA: FA Davis; 2015:1361.
10. Adams MP, Urban CQ. Pharmacology for Nurses: Connections to Nursing Practice. 2nd ed. Upper Saddle River, NJ: Pearson Education; 2013:437.
11. Joint Commission enhances pain assessment and management requirements for accredited hospitals. The Joint Commission Perspectives. 2017;37(7):1-4. www.jointcommission.org/assets/1/18/Joint_Commission_Enhances_Pain_Assessment_and_Management_Requirements_for_Accredited_Hospitals1.PDF. Accessed March 21, 2018.
12. World Health Organization. WHO’s cancer pain ladder for adults. 2017. www.who.int/cancer/palliative/painladder/en/. Accessed March 21, 2018.
13. National Institutes of Health. National Institute on Drug Abuse. Opioids: brief description. www.drugabuse.gov/drugs-abuse/opioids. Accessed March 21, 2018.
14. Hudspeth RS. Safe opioid prescribing for adults by nurse practitioners: Part 1. Patient history and assessment standards and techniques. J Nurse Pract. 2016;12(3):141-148.
15. Kennedy MC, Kerr T, DeBeck K, et al. Seeking prescription opioids from physicians for nonmedical use among people who inject drugs in a Canadian setting. Am J Addict. 2016;25(4):275-282.
16. US Department of Health and Human Services. CDC. CDC guideline for prescribing opioids for chronic pain—United States, 2016. www.cdc.gov/mmwr/volumes/65/rr/rr6501e1.htm. Accessed March 21, 2018.
17. Webster LR, Webster R. Predicting aberrant behaviors in opioid‐treated patients: preliminary validation of the Opioid Risk Tool. Pain Med. 2005;6(6):432. [Tool available at www.drugabuse.gov/sites/default/files/files/OpioidRiskTool.pdf.] Accessed March 21, 2018.
18. Screener and Opioid Assessment for Patients with Pain—Revised (SOAPP®-R). http://nationalpaincentre.mcmaster.ca/documents/soapp_r_sample_watermark.pdf. Accessed March 21, 2018.
19. D.I.R.E. Score: Patient Selection for Chronic Opioid Analgesia. www.ucdenver.edu/academics/colleges/PublicHealth/research/centers/CHWE/Documents/D.I.R.E.%20Score.pdf. Accessed March 21, 2018.
20. Current Opioid Misuse Measure (COMM)™. www.opioidprescribing.com/documents/09-comm-inflexxion.pdf. Accessed March 21, 2018.
21. Pain Assessment and Documentation Tool (PADT™). www.ucdenver.edu/academics/colleges/PublicHealth/research/centers/CHWE/Documents/Pain%20Assess ment%20Documentation%20Tool%20%28PADT%29.pdf. Accessed March 21, 2018.
22. The CAGE and CAGE-AID Questionnaires. www.ucdenver.edu/academics/colleges/PublicHealth/research/centers/CHWE/Documents/CAGE-AID.pdf. Accessed March 21, 2018.
23. Skinner HA. The drug abuse screening test. Addict Behav. 1982;7(4):363-371. [DAST-10 available at https://cde.drugabuse.gov/sites/nida_cde/files/DrugAbuseScreeningTest_2014Mar24.pdf.] Accessed March 21, 2018.
24. SBIRT AUDIT forms (English and Spanish). www.communitycarenc.org/media/tool-resource-files/sbirt-audit-forms.pdf. Accessed March 21, 2018.
25. Cheattle MD. Risk assessment: safe opioid prescribing tools. 2017. https://www.practicalpainmanagement.com/resource-centers/opioid-prescribing-monitoring/risk-assessment-safe-opioid-prescribing-tools. Accessed March 21, 2018.
26. Ali MM, Dowd WN, Classen T, et al. Prescription drug monitoring programs, nonmedical use of prescription drugs, and heroin use: evidence from the National Survey of Drug Use and Health. Addict Behav. 2017;69:65-77.
27. US Department of Health and Human Services. CDC. Drug overdose deaths hit record numbers in 2014. www.cdc.gov/media/releases/2015/p1218-drug-overdose.html. Accessed March 21, 2018.
28. McGee S, Silverman RD. Treatment agreements, informed consent, and the role of state medical boards in opioid prescribing. Pain Med. 2015;16(1):25-29.
29. Chou R, Fanciullo GJ, Fine PG, et al; for the American Pain Society–American Academy of Pain Medicine Opioids Guidelines Panel. Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J Pain. 2009;10(2):113-130.
30. US Department of Health and Human Services. FDA Center for Drug Evaluation and Research. Abuse-deterrent opioids—evaluation and labeling guidance for industry. 2015. www.fda.gov/downloads/Drugs/Guid ances/UCM334743.pdf. Accessed March 21, 2018.

SHM Research, Innovation, and Clinical Vignettes, 2018 Abstracts
SHM Research, Innovation, and Clinical Vignettes Abstracts, Hospital Medicine 2018, April 8-11
SHM Research, Innovation, and Clinical Vignettes Abstracts, Hospital Medicine 2018, April 8-11
SHM Research, Innovation, and Clinical Vignettes Abstracts, Hospital Medicine 2018, April 8-11
Cracking the clinician educator code in gastroenterology
For gastroenterologists who enter academic medicine, the most common career track is that of clinician educator (CE). Although most academic gastroenterologists are CEs, their career paths vary substantially, and expectations for promotion can be much less explicit compared with those of physician scientists. This delineation of different pathways in academic gastroenterology starts as early as the fellowship application process, before the implications are understood. Furthermore, many community gastroenterologists have appointments within academic medical centers, which typically fall into the realm of CEs.
A review of all gastroenterology and hepatology fellowship program websites listed on the American Gastroenterological Association website showed that 33 of 175 (18.8%) programs endorse distinctly different tracks, usually distinguishing traditional research (i.e., basic science, epidemiology, or outcomes) from clinical care of patients (i.e., clinician educator or clinical scholar). One of the most common words appearing in descriptions of both tracks was “clinical,” highlighting that a good clinician educator or researcher is, first and foremost, a good clinician.
With clinical duties requiring the majority of a CE’s time and efforts, a reasonable assumption is that CEs are clinicians who teach trainees via lectures, clinic, endoscopy, and/or inpatient rounds. Although such educational endeavors form the backbone of a CE’s scholarly activities, what constitutes scholarship for CEs is much more diverse than many people realize. The variability in what a career as a CE may look like can be both an obstacle and an opportunity. Included in the category of CE are community clinicians who have a stake in the education of residents and fellows and play an important role in trainee learning. Sherbino et al1 defined a CE as “a clinician active in health professional practice who applies theory to education practice, engages in education scholarship, and serves as a consultant to other health professionals on education issues.”
Because we recognize that many community and academic gastroenterologists spend the majority of their education efforts teaching trainees, we have made every effort to ensure that the five recommendations listed later are equally pertinent to all gastroenterologists who devote any portion of their careers to educating trainees, colleagues, allied health professionals, as well as patients. For example, a CE who primarily teaches trainees still can benefit from learning how to better document their efforts, receive mentorship as an educator, take everyday activities and convert them into scholarship, share teaching materials with broader audiences, and learn new teaching techniques without ever opening a book on education theory. For community-based physicians, this can assist in obtaining recognition from the academic centers for their teaching efforts. We hope that the five recommendations that follow will serve to guide those just setting out on the CE path, as well as for those who have trodden it for some time.
Number 1: Maintain a current curriculum vitae and teaching portfolio
All CEs must have two critical instruments to document their accomplishments to their institutions and to the field: a curriculum vitae (CV) and a teaching portfolio. These items also are very important when the time comes for promotion because they validate one’s accomplishments, both quantitatively and qualitatively. Knowing the criteria for promotion as a CE is critical for shaping one’s career, and we recommend checking with an individual’s institution for its specific requirements regarding formats for both the CV and the teaching portfolio, which typically are available from the academic promotion committee. Because most fellows and faculty are familiar with the format of a CV, we will focus on the teaching portfolio.
For most fellows and many faculty, the teaching portfolio is a new and/or less well understood entity. Unlike a CV, the teaching portfolio presents teaching activities not only as a collection or list, but also provides evidence of the influence the work has had on others, in a much more personal way. A few tips are listed on putting together a teaching portfolio. However, the most important advice we can offer is this: one should save all evidence of teaching including unsolicited letters and e-mails from learners and colleagues.
If your institution does not have a teaching portfolio template, we recommend using a pre-existing format. Several examples from academic medicine can be found on the Internet or on MedEdPORTAL, an open-access repository of educational content provided by the Association of American Medical Colleges. One such tool is the Educator Portfolio Template of the Academic Pediatric Association’s Educational Scholars Program (available: https://www.academicpeds.org/education/educator_portfolio_template.cfm). The Association of American Medical Colleges Group on Education Affairs held a consensus conference in 2006, from which five educational categories were defined: teaching, learner assessment, curriculum development, mentoring and advising, and educational leadership and administration.2 These categories can serve as an arrangement for a teaching portfolio. We also recommend that you include both educational research/scholarship and web-based educational materials such as online learning modules, YouTube videos, blogs, and wikis as a part of a teaching portfolio. For each project highlighted in the teaching portfolio, we recommend reflecting on and writing down how the project shows the quantity and quality of the work.
Quantity of work in the teaching portfolio refers to more than a mere cataloging of published peer-reviewed articles and book chapters, courses taught, presentations given, and so forth (which should be included in the CV). Instead, it documents time spent in teaching activities, how often teaching occurs, the number and types of learners involved, and how the activity fits into a training program.
Quality of work can include how innovative methods were crafted and implemented to customize teaching in creative ways to accomplish specific learning objectives. This description renders the contents of the teaching portfolio more than merely a sketch of work activities documented by numbers, and tells a story about what occurred. When documenting evidence of quality, provide comparative measures whenever possible. Quality of teaching also can be illustrated by evaluations, pretests and posttests, and as complimentary e-mails and letters from learners and other faculty members. The description of teaching activities also shows one’s flexibility as an educator, and the greater the breadth of experiences, the better. A CE also must document within the portfolio how the teaching activity drew from existing literature and best practices and/or contributed to the medical education field and its body of knowledge. Above all else, we recommend collecting evidence on teaching to both provide evidence of one’s teaching skills and to gather data on which to improve.

The teaching portfolio templates begin with a personal statement outlining why one teaches (i.e., teaching philosophy). In a teaching portfolio, it is important to include details of how impact was defined or determined with regard to teaching endeavors, how the feedback from formal evaluative processes was used to mold one’s future activities as an educator, and what strategies will be implemented to improve teaching to meet the needs of diverse and changing groups of learners.
Both the CV and teaching portfolio should be updated continually – we recommend at least quarterly (or as articles are published, courses are taught, abstracts are presented, and so forth) – to ensure that nothing is overlooked or forgotten.
Number 2: Mentors and mentees
Every CE needs to have a primary mentor, typically a more senior faculty member with an interest in and experience with mentoring, as well as a commitment to fostering the mentee’s professional growth. It may be difficult to find a mentor when starting out as a junior faculty member or when changing academic institutions. Once you have a mentor, take ownership for the success of the relationship by managing-up, by organizing all the meetings, exceeding (not just meeting) deadlines, and by communicating needs and information in a way the mentor prefers. Rustgi and Hecht3 wrap up their article on mentorship with a pathway that highlights the following components for a successful mentoring relationship: regular meetings, specific goals and measurable outcomes, manuscript and grant writing, presentation skills and efficiency, and navigating the complexities of regulatory affairs such as institutional review boards. Although many of these tenets hold true for both clinician researchers and clinician educators, Farrell et al4 offer four steps to finding a mentor for clinician educators, as follows. Step 1: self-reflection and assessment: critically assessing one’s competence as a teacher, educational administrator, or researcher; determining what prior education projects have been successful and why; and defining career goals and the current relationship to them. Step 2: identification of areas needing development: examples may include teaching skills, curriculum innovation, evaluation/assessment, educational research, time management, negotiation skills, grantsmanship, scholarly writing, and presentation skills; identify specific questions regarding the type of help needed. Step 3: matchmaking: determine qualities (personal and professional) desired in a mentor, and search for candidates with the help of colleagues. Step 4: engagement with a mentor: explain why you desire mentorship, career goals, current academic role(s), your perceived needs, and recognize and acknowledge appreciation for your mentor’s time and energy.
One caution is to avoid having too many primary mentors. A mentee may assume the perspective that it takes a village when it comes to seeking and providing mentoring. Although having clinical, research, and/or personal mentors can be helpful, having too many mentors can make it difficult to meet regularly enough to allow for the mentee–mentor relationship to grow. Instead of a network of mentors, build a web of minimentors, or coaches, to serve as consultants, coaches, and accountability partners, and tap into this network as needed. Mentors are involved longitudinally with mentees and tend to provide general career and project-specific guidance, whereas coaches tend to be involved in specific projects or areas of focus of a mentee.
In addition to having their own mentors, CEs quickly will find opportunities themselves to serve as mentors to more junior faculty, fellows, residents, and students. Indeed, one measure of a successful mentor–mentee relationship is the development of the mentee into a new mentor for future generations.
Number 3: Think broadly about scholarship
Traditionally, the definition of scholarship has been very narrow and usually is related to the number of publications and grants one receives. Beginning with Boyer’s work in 1990, the definition of scholarship has expanded at academic institutions beyond the concept of traditional research.5 Medical education scholarship most often is guided and judged by six core qualitative standards of excellence, known as “Glassick’s criteria”6: clear goals, adequate preparation, appropriate methods, significant results, effective presentation, and reflective critique. The key to scholarship is that it builds on or adds to the field, is made public, and thus available for peer-review.
CE projects can be categorized in many ways, but we recommend broadening the classic notions of research with which we have been indoctrinated. Golub’s7 2016 editorial in the Journal of the American Medical Association, “Looking Inward and Reflecting Back: Medical Education and Journal of the American Medical Association,” highlights the range of research questions and methodologies, which include ethics, behavioral psychology, diversity of patient care and the workforce, medical education research, quality and value of care, well-being of trainees and faculty, and health informatics. If one breaks down daily tasks, countless opportunities for scholarly projects will emerge. One need look no further for opportunities than the countless opportunities for quality improvement research that avail themselves daily, with examples ranging from reducing variation in cirrhosis care to improving adenoma detection rates. Quality improvement is an important method of scholarship for both academic and community-based physicians, which also can contribute toward Part IV of Maintenance of Certification requirements. CEs also can engage in educational scholarship other than research by using these same principles. To transform your teaching into scholarship you should examine the activities you perform or a problem that needs to be solved, apply information or a solution based on best practices or what is known from the literature, and then share the results/products with others (peer-review). Crites et al8 provide practical guidelines for developing education research questions, designing and implementing scholarly activities, and interpreting the scope and impact of education scholarship.
In addition, reaching beyond one’s department to other departments, as well as participating in educational scholarly activities on regional and national levels, is important as one’s career progresses. Well-connected and diverse networks are information highways by which one’s work can be amplified to achieve a greater impact, and from which many opportunities will be shared.
Number 4: Share broadly
Scholarship activities of both academic and community-based CEs can target many audiences, including medical students, residents, and fellows; faculty; other health professions; or even patients and the community. Knowing who will be the recipients or end-users can help to identify which types of projects may be most rewarding and make the greatest impact. Consider sharing curricula, evaluation tools, and other educational products with colleagues at other institutions who ask for them. Request acknowledgment for the development of the materials and ask for written feedback on how these products are being used and what impact they have had on learners.
One education model used to assess the impact and target of education interventions is known as Kirkpatrick’s9 hierarchy, which traditionally included the following four levels: reaction (level 1), learning (level 2), behavior (level 3), and results (level 4). The model has been adapted by the British Medical Journal’s Best Evidence in Medical Education collaboration to medical education with the following modifications in levels as follows.9,10 Level 1: participation: focused on learners’ views of the learning experience including content, presentation, and teaching methods. Level 2a: modification of attitudes/perceptions: focused on changes in attitudes or perceptions between participant groups toward the intervention. Level 2b: modification of knowledge/skills: for knowledge, focused on the acquisition of concepts, procedures, and principles; for skills, focused on the acquisition of problem solving, psychomotor, and social skills. Level 3: behavioral change: focused on the transfer of learning to the workplace or willingness of learners to apply new knowledge and skills. Level 4a: change in organizational practice: focused on wider changes in the organization or delivery of care attributable to an educational program. Level 4b: focused on improvements in the health and well-being of patients as a direct result of an education initiative.
Similar to more traditional clinical research, education research needs to be performed in a scholarly fashion and shared with a wider audience. In addition to submitting research to gastroenterology journals (e.g., Gastroenterology’s Mentoring, Education, and Training Corner), education research can be submitted to education journals such as the Association of American Medical Colleges’ Academic Medicine, the Association for the Study of Medical Education’s Medical Education, the Accreditation Council for Graduate Medical Education’s Journal of Graduate Medical Education, or the European Association for Medical Education in Europe’s Medical Teacher; online education warehouses such as MedEdPORTAL (www.mededportal.org) or MERLOT (www.merlot.org); and national conferences as workshops. Also, one should keep in mind that opportunities arise on a regular basis to share educational videos or images in forums such as the American Society for Gastrointestinal Endoscopy’s video journal VideoGIE, The American Journal of Gastroenterology’s video of the month, and Clinical Gastroenterology and Hepatology’s Images of the Month.
Number 5: Ongoing professional development
Continuing Medical Education is a standard requirement to maintain an active medical license because it shows ongoing efforts to remain up-to-date with changes in medicine. Similar opportunities exist with respect to further development as an educator. Given the multitude of manners in which these opportunities can be divided, we have compiled recommendations for resources on educational scholarship based on level of experience and desired level of engagement (Table 1).
Summary
The framework provided should help guide the gastroenterologist on the path of becoming an effective clinician educator in gastroenterology. The diversity of what a career as a clinician educator can entail is unlimited. The success of the future of medical education and our careers requires not only that every clinician educator be productive, but also that each one brings a unique passion to work each day to share. The authors would like to thank all those clinician educators who contributed to our education, and look forward to learning from you in the future.
Acknowledgments
The authors thank Dr Lee Ligon, Center for Research, Innovation, and Scholarship, Department of Pediatrics, Baylor College of Medicine, for providing editorial assistance.
References
1. Sherbino, J., Frank, J.R., Snell, L. Defining the key roles and competencies of the clinician-educator of the 21st century: a national mixed-methods study. Acad Med. 2014;89:783-9.
2. Simpson, D., Fincher, R.M., Hafler, J.P., et al. Advancing educators and education by defining the components and evidence associated with educational scholarship. Med Educ. 2007;41:1002-9.
3. Rustgi, A.K. Hecht, G.A. Mentorship in academic medicine. Gastroenterology. 2011;141:789-92.
4. Farrell, S.E., Digioia, N.M., Broderick, K.B., et al. Mentoring for clinician-educators. Acad Emerg Med. 2004;11:1346-50.
5. Boyer, E.L. Scholarship reconsidered: priorities of the professoriate. Carnegie Foundation for the Advancement of Teaching, Princeton, NJ; 1990.
6. Glassick, C.E., Taylor-Huber, M., Maeroff, G.I., et al. Scholarship assessed: evaluation of the professoriate. Jossey-Bass, San Francisco; 1997.
7. Golub, R.M. Looking inward and reflecting back medical education and JAMA. JAMA. 2016;316:2200-3.
8. Crites, G.E., Gaines, J.K., Cottrell, S., et al. Medical education scholarship: an introductory guide: AMEE guide no. 89. Med Teach. 2014;36:657-74.
9. Kirkpatrick, D.L. Evaluation of training. In: R. Craig, L. Bittel (Eds.) Training and development handbook. McGraw-Hill, New York; 1967: 87-112.
10. Littlewood, S., Ypinazar, V., Margolis, S.A., et al. Early practical experience and the social responsiveness of clinical education: systematic review. BMJ. 2005;331:387-91.
Dr. Shapiro is a gastroenterology fellow in the department of medicine, section of gastroenterology; Dr. Gould Suarez is an associate professor in the department of medicine, section of gastroenterology and associate program director of the gastroenterology fellowship; and Dr. Turner is an associate professor of pediatrics, vice chair of education, associate program director for house staff education, section of academic general pediatrics, and director for research, innovation, and scholarship, Baylor College of Medicine, Houston. The authors disclose no conflicts.
For gastroenterologists who enter academic medicine, the most common career track is that of clinician educator (CE). Although most academic gastroenterologists are CEs, their career paths vary substantially, and expectations for promotion can be much less explicit compared with those of physician scientists. This delineation of different pathways in academic gastroenterology starts as early as the fellowship application process, before the implications are understood. Furthermore, many community gastroenterologists have appointments within academic medical centers, which typically fall into the realm of CEs.
A review of all gastroenterology and hepatology fellowship program websites listed on the American Gastroenterological Association website showed that 33 of 175 (18.8%) programs endorse distinctly different tracks, usually distinguishing traditional research (i.e., basic science, epidemiology, or outcomes) from clinical care of patients (i.e., clinician educator or clinical scholar). One of the most common words appearing in descriptions of both tracks was “clinical,” highlighting that a good clinician educator or researcher is, first and foremost, a good clinician.
With clinical duties requiring the majority of a CE’s time and efforts, a reasonable assumption is that CEs are clinicians who teach trainees via lectures, clinic, endoscopy, and/or inpatient rounds. Although such educational endeavors form the backbone of a CE’s scholarly activities, what constitutes scholarship for CEs is much more diverse than many people realize. The variability in what a career as a CE may look like can be both an obstacle and an opportunity. Included in the category of CE are community clinicians who have a stake in the education of residents and fellows and play an important role in trainee learning. Sherbino et al1 defined a CE as “a clinician active in health professional practice who applies theory to education practice, engages in education scholarship, and serves as a consultant to other health professionals on education issues.”
Because we recognize that many community and academic gastroenterologists spend the majority of their education efforts teaching trainees, we have made every effort to ensure that the five recommendations listed later are equally pertinent to all gastroenterologists who devote any portion of their careers to educating trainees, colleagues, allied health professionals, as well as patients. For example, a CE who primarily teaches trainees still can benefit from learning how to better document their efforts, receive mentorship as an educator, take everyday activities and convert them into scholarship, share teaching materials with broader audiences, and learn new teaching techniques without ever opening a book on education theory. For community-based physicians, this can assist in obtaining recognition from the academic centers for their teaching efforts. We hope that the five recommendations that follow will serve to guide those just setting out on the CE path, as well as for those who have trodden it for some time.
Number 1: Maintain a current curriculum vitae and teaching portfolio
All CEs must have two critical instruments to document their accomplishments to their institutions and to the field: a curriculum vitae (CV) and a teaching portfolio. These items also are very important when the time comes for promotion because they validate one’s accomplishments, both quantitatively and qualitatively. Knowing the criteria for promotion as a CE is critical for shaping one’s career, and we recommend checking with an individual’s institution for its specific requirements regarding formats for both the CV and the teaching portfolio, which typically are available from the academic promotion committee. Because most fellows and faculty are familiar with the format of a CV, we will focus on the teaching portfolio.
For most fellows and many faculty, the teaching portfolio is a new and/or less well understood entity. Unlike a CV, the teaching portfolio presents teaching activities not only as a collection or list, but also provides evidence of the influence the work has had on others, in a much more personal way. A few tips are listed on putting together a teaching portfolio. However, the most important advice we can offer is this: one should save all evidence of teaching including unsolicited letters and e-mails from learners and colleagues.
If your institution does not have a teaching portfolio template, we recommend using a pre-existing format. Several examples from academic medicine can be found on the Internet or on MedEdPORTAL, an open-access repository of educational content provided by the Association of American Medical Colleges. One such tool is the Educator Portfolio Template of the Academic Pediatric Association’s Educational Scholars Program (available: https://www.academicpeds.org/education/educator_portfolio_template.cfm). The Association of American Medical Colleges Group on Education Affairs held a consensus conference in 2006, from which five educational categories were defined: teaching, learner assessment, curriculum development, mentoring and advising, and educational leadership and administration.2 These categories can serve as an arrangement for a teaching portfolio. We also recommend that you include both educational research/scholarship and web-based educational materials such as online learning modules, YouTube videos, blogs, and wikis as a part of a teaching portfolio. For each project highlighted in the teaching portfolio, we recommend reflecting on and writing down how the project shows the quantity and quality of the work.
Quantity of work in the teaching portfolio refers to more than a mere cataloging of published peer-reviewed articles and book chapters, courses taught, presentations given, and so forth (which should be included in the CV). Instead, it documents time spent in teaching activities, how often teaching occurs, the number and types of learners involved, and how the activity fits into a training program.
Quality of work can include how innovative methods were crafted and implemented to customize teaching in creative ways to accomplish specific learning objectives. This description renders the contents of the teaching portfolio more than merely a sketch of work activities documented by numbers, and tells a story about what occurred. When documenting evidence of quality, provide comparative measures whenever possible. Quality of teaching also can be illustrated by evaluations, pretests and posttests, and as complimentary e-mails and letters from learners and other faculty members. The description of teaching activities also shows one’s flexibility as an educator, and the greater the breadth of experiences, the better. A CE also must document within the portfolio how the teaching activity drew from existing literature and best practices and/or contributed to the medical education field and its body of knowledge. Above all else, we recommend collecting evidence on teaching to both provide evidence of one’s teaching skills and to gather data on which to improve.
The teaching portfolio templates begin with a personal statement outlining why one teaches (i.e., teaching philosophy). In a teaching portfolio, it is important to include details of how impact was defined or determined with regard to teaching endeavors, how the feedback from formal evaluative processes was used to mold one’s future activities as an educator, and what strategies will be implemented to improve teaching to meet the needs of diverse and changing groups of learners.
Both the CV and teaching portfolio should be updated continually – we recommend at least quarterly (or as articles are published, courses are taught, abstracts are presented, and so forth) – to ensure that nothing is overlooked or forgotten.
Number 2: Mentors and mentees
Every CE needs to have a primary mentor, typically a more senior faculty member with an interest in and experience with mentoring, as well as a commitment to fostering the mentee’s professional growth. It may be difficult to find a mentor when starting out as a junior faculty member or when changing academic institutions. Once you have a mentor, take ownership for the success of the relationship by managing-up, by organizing all the meetings, exceeding (not just meeting) deadlines, and by communicating needs and information in a way the mentor prefers. Rustgi and Hecht3 wrap up their article on mentorship with a pathway that highlights the following components for a successful mentoring relationship: regular meetings, specific goals and measurable outcomes, manuscript and grant writing, presentation skills and efficiency, and navigating the complexities of regulatory affairs such as institutional review boards. Although many of these tenets hold true for both clinician researchers and clinician educators, Farrell et al4 offer four steps to finding a mentor for clinician educators, as follows. Step 1: self-reflection and assessment: critically assessing one’s competence as a teacher, educational administrator, or researcher; determining what prior education projects have been successful and why; and defining career goals and the current relationship to them. Step 2: identification of areas needing development: examples may include teaching skills, curriculum innovation, evaluation/assessment, educational research, time management, negotiation skills, grantsmanship, scholarly writing, and presentation skills; identify specific questions regarding the type of help needed. Step 3: matchmaking: determine qualities (personal and professional) desired in a mentor, and search for candidates with the help of colleagues. Step 4: engagement with a mentor: explain why you desire mentorship, career goals, current academic role(s), your perceived needs, and recognize and acknowledge appreciation for your mentor’s time and energy.
One caution is to avoid having too many primary mentors. A mentee may assume the perspective that it takes a village when it comes to seeking and providing mentoring. Although having clinical, research, and/or personal mentors can be helpful, having too many mentors can make it difficult to meet regularly enough to allow for the mentee–mentor relationship to grow. Instead of a network of mentors, build a web of minimentors, or coaches, to serve as consultants, coaches, and accountability partners, and tap into this network as needed. Mentors are involved longitudinally with mentees and tend to provide general career and project-specific guidance, whereas coaches tend to be involved in specific projects or areas of focus of a mentee.
In addition to having their own mentors, CEs quickly will find opportunities themselves to serve as mentors to more junior faculty, fellows, residents, and students. Indeed, one measure of a successful mentor–mentee relationship is the development of the mentee into a new mentor for future generations.
Number 3: Think broadly about scholarship
Traditionally, the definition of scholarship has been very narrow and usually is related to the number of publications and grants one receives. Beginning with Boyer’s work in 1990, the definition of scholarship has expanded at academic institutions beyond the concept of traditional research.5 Medical education scholarship most often is guided and judged by six core qualitative standards of excellence, known as “Glassick’s criteria”6: clear goals, adequate preparation, appropriate methods, significant results, effective presentation, and reflective critique. The key to scholarship is that it builds on or adds to the field, is made public, and thus available for peer-review.
CE projects can be categorized in many ways, but we recommend broadening the classic notions of research with which we have been indoctrinated. Golub’s7 2016 editorial in the Journal of the American Medical Association, “Looking Inward and Reflecting Back: Medical Education and Journal of the American Medical Association,” highlights the range of research questions and methodologies, which include ethics, behavioral psychology, diversity of patient care and the workforce, medical education research, quality and value of care, well-being of trainees and faculty, and health informatics. If one breaks down daily tasks, countless opportunities for scholarly projects will emerge. One need look no further for opportunities than the countless opportunities for quality improvement research that avail themselves daily, with examples ranging from reducing variation in cirrhosis care to improving adenoma detection rates. Quality improvement is an important method of scholarship for both academic and community-based physicians, which also can contribute toward Part IV of Maintenance of Certification requirements. CEs also can engage in educational scholarship other than research by using these same principles. To transform your teaching into scholarship you should examine the activities you perform or a problem that needs to be solved, apply information or a solution based on best practices or what is known from the literature, and then share the results/products with others (peer-review). Crites et al8 provide practical guidelines for developing education research questions, designing and implementing scholarly activities, and interpreting the scope and impact of education scholarship.
In addition, reaching beyond one’s department to other departments, as well as participating in educational scholarly activities on regional and national levels, is important as one’s career progresses. Well-connected and diverse networks are information highways by which one’s work can be amplified to achieve a greater impact, and from which many opportunities will be shared.
Number 4: Share broadly
Scholarship activities of both academic and community-based CEs can target many audiences, including medical students, residents, and fellows; faculty; other health professions; or even patients and the community. Knowing who will be the recipients or end-users can help to identify which types of projects may be most rewarding and make the greatest impact. Consider sharing curricula, evaluation tools, and other educational products with colleagues at other institutions who ask for them. Request acknowledgment for the development of the materials and ask for written feedback on how these products are being used and what impact they have had on learners.
One education model used to assess the impact and target of education interventions is known as Kirkpatrick’s9 hierarchy, which traditionally included the following four levels: reaction (level 1), learning (level 2), behavior (level 3), and results (level 4). The model has been adapted by the British Medical Journal’s Best Evidence in Medical Education collaboration to medical education with the following modifications in levels as follows.9,10 Level 1: participation: focused on learners’ views of the learning experience including content, presentation, and teaching methods. Level 2a: modification of attitudes/perceptions: focused on changes in attitudes or perceptions between participant groups toward the intervention. Level 2b: modification of knowledge/skills: for knowledge, focused on the acquisition of concepts, procedures, and principles; for skills, focused on the acquisition of problem solving, psychomotor, and social skills. Level 3: behavioral change: focused on the transfer of learning to the workplace or willingness of learners to apply new knowledge and skills. Level 4a: change in organizational practice: focused on wider changes in the organization or delivery of care attributable to an educational program. Level 4b: focused on improvements in the health and well-being of patients as a direct result of an education initiative.
Similar to more traditional clinical research, education research needs to be performed in a scholarly fashion and shared with a wider audience. In addition to submitting research to gastroenterology journals (e.g., Gastroenterology’s Mentoring, Education, and Training Corner), education research can be submitted to education journals such as the Association of American Medical Colleges’ Academic Medicine, the Association for the Study of Medical Education’s Medical Education, the Accreditation Council for Graduate Medical Education’s Journal of Graduate Medical Education, or the European Association for Medical Education in Europe’s Medical Teacher; online education warehouses such as MedEdPORTAL (www.mededportal.org) or MERLOT (www.merlot.org); and national conferences as workshops. Also, one should keep in mind that opportunities arise on a regular basis to share educational videos or images in forums such as the American Society for Gastrointestinal Endoscopy’s video journal VideoGIE, The American Journal of Gastroenterology’s video of the month, and Clinical Gastroenterology and Hepatology’s Images of the Month.
Number 5: Ongoing professional development
Continuing Medical Education is a standard requirement to maintain an active medical license because it shows ongoing efforts to remain up-to-date with changes in medicine. Similar opportunities exist with respect to further development as an educator. Given the multitude of manners in which these opportunities can be divided, we have compiled recommendations for resources on educational scholarship based on level of experience and desired level of engagement (Table 1).
Summary
The framework provided should help guide the gastroenterologist on the path of becoming an effective clinician educator in gastroenterology. The diversity of what a career as a clinician educator can entail is unlimited. The success of the future of medical education and our careers requires not only that every clinician educator be productive, but also that each one brings a unique passion to work each day to share. The authors would like to thank all those clinician educators who contributed to our education, and look forward to learning from you in the future.
Acknowledgments
The authors thank Dr Lee Ligon, Center for Research, Innovation, and Scholarship, Department of Pediatrics, Baylor College of Medicine, for providing editorial assistance.
References
1. Sherbino, J., Frank, J.R., Snell, L. Defining the key roles and competencies of the clinician-educator of the 21st century: a national mixed-methods study. Acad Med. 2014;89:783-9.
2. Simpson, D., Fincher, R.M., Hafler, J.P., et al. Advancing educators and education by defining the components and evidence associated with educational scholarship. Med Educ. 2007;41:1002-9.
3. Rustgi, A.K. Hecht, G.A. Mentorship in academic medicine. Gastroenterology. 2011;141:789-92.
4. Farrell, S.E., Digioia, N.M., Broderick, K.B., et al. Mentoring for clinician-educators. Acad Emerg Med. 2004;11:1346-50.
5. Boyer, E.L. Scholarship reconsidered: priorities of the professoriate. Carnegie Foundation for the Advancement of Teaching, Princeton, NJ; 1990.
6. Glassick, C.E., Taylor-Huber, M., Maeroff, G.I., et al. Scholarship assessed: evaluation of the professoriate. Jossey-Bass, San Francisco; 1997.
7. Golub, R.M. Looking inward and reflecting back medical education and JAMA. JAMA. 2016;316:2200-3.
8. Crites, G.E., Gaines, J.K., Cottrell, S., et al. Medical education scholarship: an introductory guide: AMEE guide no. 89. Med Teach. 2014;36:657-74.
9. Kirkpatrick, D.L. Evaluation of training. In: R. Craig, L. Bittel (Eds.) Training and development handbook. McGraw-Hill, New York; 1967: 87-112.
10. Littlewood, S., Ypinazar, V., Margolis, S.A., et al. Early practical experience and the social responsiveness of clinical education: systematic review. BMJ. 2005;331:387-91.
Dr. Shapiro is a gastroenterology fellow in the department of medicine, section of gastroenterology; Dr. Gould Suarez is an associate professor in the department of medicine, section of gastroenterology and associate program director of the gastroenterology fellowship; and Dr. Turner is an associate professor of pediatrics, vice chair of education, associate program director for house staff education, section of academic general pediatrics, and director for research, innovation, and scholarship, Baylor College of Medicine, Houston. The authors disclose no conflicts.
For gastroenterologists who enter academic medicine, the most common career track is that of clinician educator (CE). Although most academic gastroenterologists are CEs, their career paths vary substantially, and expectations for promotion can be much less explicit compared with those of physician scientists. This delineation of different pathways in academic gastroenterology starts as early as the fellowship application process, before the implications are understood. Furthermore, many community gastroenterologists have appointments within academic medical centers, which typically fall into the realm of CEs.
A review of all gastroenterology and hepatology fellowship program websites listed on the American Gastroenterological Association website showed that 33 of 175 (18.8%) programs endorse distinctly different tracks, usually distinguishing traditional research (i.e., basic science, epidemiology, or outcomes) from clinical care of patients (i.e., clinician educator or clinical scholar). One of the most common words appearing in descriptions of both tracks was “clinical,” highlighting that a good clinician educator or researcher is, first and foremost, a good clinician.
With clinical duties requiring the majority of a CE’s time and efforts, a reasonable assumption is that CEs are clinicians who teach trainees via lectures, clinic, endoscopy, and/or inpatient rounds. Although such educational endeavors form the backbone of a CE’s scholarly activities, what constitutes scholarship for CEs is much more diverse than many people realize. The variability in what a career as a CE may look like can be both an obstacle and an opportunity. Included in the category of CE are community clinicians who have a stake in the education of residents and fellows and play an important role in trainee learning. Sherbino et al1 defined a CE as “a clinician active in health professional practice who applies theory to education practice, engages in education scholarship, and serves as a consultant to other health professionals on education issues.”
Because we recognize that many community and academic gastroenterologists spend the majority of their education efforts teaching trainees, we have made every effort to ensure that the five recommendations listed later are equally pertinent to all gastroenterologists who devote any portion of their careers to educating trainees, colleagues, allied health professionals, as well as patients. For example, a CE who primarily teaches trainees still can benefit from learning how to better document their efforts, receive mentorship as an educator, take everyday activities and convert them into scholarship, share teaching materials with broader audiences, and learn new teaching techniques without ever opening a book on education theory. For community-based physicians, this can assist in obtaining recognition from the academic centers for their teaching efforts. We hope that the five recommendations that follow will serve to guide those just setting out on the CE path, as well as for those who have trodden it for some time.
Number 1: Maintain a current curriculum vitae and teaching portfolio
All CEs must have two critical instruments to document their accomplishments to their institutions and to the field: a curriculum vitae (CV) and a teaching portfolio. These items also are very important when the time comes for promotion because they validate one’s accomplishments, both quantitatively and qualitatively. Knowing the criteria for promotion as a CE is critical for shaping one’s career, and we recommend checking with an individual’s institution for its specific requirements regarding formats for both the CV and the teaching portfolio, which typically are available from the academic promotion committee. Because most fellows and faculty are familiar with the format of a CV, we will focus on the teaching portfolio.
For most fellows and many faculty, the teaching portfolio is a new and/or less well understood entity. Unlike a CV, the teaching portfolio presents teaching activities not only as a collection or list, but also provides evidence of the influence the work has had on others, in a much more personal way. A few tips are listed on putting together a teaching portfolio. However, the most important advice we can offer is this: one should save all evidence of teaching including unsolicited letters and e-mails from learners and colleagues.
If your institution does not have a teaching portfolio template, we recommend using a pre-existing format. Several examples from academic medicine can be found on the Internet or on MedEdPORTAL, an open-access repository of educational content provided by the Association of American Medical Colleges. One such tool is the Educator Portfolio Template of the Academic Pediatric Association’s Educational Scholars Program (available: https://www.academicpeds.org/education/educator_portfolio_template.cfm). The Association of American Medical Colleges Group on Education Affairs held a consensus conference in 2006, from which five educational categories were defined: teaching, learner assessment, curriculum development, mentoring and advising, and educational leadership and administration.2 These categories can serve as an arrangement for a teaching portfolio. We also recommend that you include both educational research/scholarship and web-based educational materials such as online learning modules, YouTube videos, blogs, and wikis as a part of a teaching portfolio. For each project highlighted in the teaching portfolio, we recommend reflecting on and writing down how the project shows the quantity and quality of the work.
Quantity of work in the teaching portfolio refers to more than a mere cataloging of published peer-reviewed articles and book chapters, courses taught, presentations given, and so forth (which should be included in the CV). Instead, it documents time spent in teaching activities, how often teaching occurs, the number and types of learners involved, and how the activity fits into a training program.
Quality of work can include how innovative methods were crafted and implemented to customize teaching in creative ways to accomplish specific learning objectives. This description renders the contents of the teaching portfolio more than merely a sketch of work activities documented by numbers, and tells a story about what occurred. When documenting evidence of quality, provide comparative measures whenever possible. Quality of teaching also can be illustrated by evaluations, pretests and posttests, and as complimentary e-mails and letters from learners and other faculty members. The description of teaching activities also shows one’s flexibility as an educator, and the greater the breadth of experiences, the better. A CE also must document within the portfolio how the teaching activity drew from existing literature and best practices and/or contributed to the medical education field and its body of knowledge. Above all else, we recommend collecting evidence on teaching to both provide evidence of one’s teaching skills and to gather data on which to improve.
The teaching portfolio templates begin with a personal statement outlining why one teaches (i.e., teaching philosophy). In a teaching portfolio, it is important to include details of how impact was defined or determined with regard to teaching endeavors, how the feedback from formal evaluative processes was used to mold one’s future activities as an educator, and what strategies will be implemented to improve teaching to meet the needs of diverse and changing groups of learners.
Both the CV and teaching portfolio should be updated continually – we recommend at least quarterly (or as articles are published, courses are taught, abstracts are presented, and so forth) – to ensure that nothing is overlooked or forgotten.
Number 2: Mentors and mentees
Every CE needs to have a primary mentor, typically a more senior faculty member with an interest in and experience with mentoring, as well as a commitment to fostering the mentee’s professional growth. It may be difficult to find a mentor when starting out as a junior faculty member or when changing academic institutions. Once you have a mentor, take ownership for the success of the relationship by managing-up, by organizing all the meetings, exceeding (not just meeting) deadlines, and by communicating needs and information in a way the mentor prefers. Rustgi and Hecht3 wrap up their article on mentorship with a pathway that highlights the following components for a successful mentoring relationship: regular meetings, specific goals and measurable outcomes, manuscript and grant writing, presentation skills and efficiency, and navigating the complexities of regulatory affairs such as institutional review boards. Although many of these tenets hold true for both clinician researchers and clinician educators, Farrell et al4 offer four steps to finding a mentor for clinician educators, as follows. Step 1: self-reflection and assessment: critically assessing one’s competence as a teacher, educational administrator, or researcher; determining what prior education projects have been successful and why; and defining career goals and the current relationship to them. Step 2: identification of areas needing development: examples may include teaching skills, curriculum innovation, evaluation/assessment, educational research, time management, negotiation skills, grantsmanship, scholarly writing, and presentation skills; identify specific questions regarding the type of help needed. Step 3: matchmaking: determine qualities (personal and professional) desired in a mentor, and search for candidates with the help of colleagues. Step 4: engagement with a mentor: explain why you desire mentorship, career goals, current academic role(s), your perceived needs, and recognize and acknowledge appreciation for your mentor’s time and energy.
One caution is to avoid having too many primary mentors. A mentee may assume the perspective that it takes a village when it comes to seeking and providing mentoring. Although having clinical, research, and/or personal mentors can be helpful, having too many mentors can make it difficult to meet regularly enough to allow for the mentee–mentor relationship to grow. Instead of a network of mentors, build a web of minimentors, or coaches, to serve as consultants, coaches, and accountability partners, and tap into this network as needed. Mentors are involved longitudinally with mentees and tend to provide general career and project-specific guidance, whereas coaches tend to be involved in specific projects or areas of focus of a mentee.
In addition to having their own mentors, CEs quickly will find opportunities themselves to serve as mentors to more junior faculty, fellows, residents, and students. Indeed, one measure of a successful mentor–mentee relationship is the development of the mentee into a new mentor for future generations.
Number 3: Think broadly about scholarship
Traditionally, the definition of scholarship has been very narrow and usually is related to the number of publications and grants one receives. Beginning with Boyer’s work in 1990, the definition of scholarship has expanded at academic institutions beyond the concept of traditional research.5 Medical education scholarship most often is guided and judged by six core qualitative standards of excellence, known as “Glassick’s criteria”6: clear goals, adequate preparation, appropriate methods, significant results, effective presentation, and reflective critique. The key to scholarship is that it builds on or adds to the field, is made public, and thus available for peer-review.
CE projects can be categorized in many ways, but we recommend broadening the classic notions of research with which we have been indoctrinated. Golub’s7 2016 editorial in the Journal of the American Medical Association, “Looking Inward and Reflecting Back: Medical Education and Journal of the American Medical Association,” highlights the range of research questions and methodologies, which include ethics, behavioral psychology, diversity of patient care and the workforce, medical education research, quality and value of care, well-being of trainees and faculty, and health informatics. If one breaks down daily tasks, countless opportunities for scholarly projects will emerge. One need look no further for opportunities than the countless opportunities for quality improvement research that avail themselves daily, with examples ranging from reducing variation in cirrhosis care to improving adenoma detection rates. Quality improvement is an important method of scholarship for both academic and community-based physicians, which also can contribute toward Part IV of Maintenance of Certification requirements. CEs also can engage in educational scholarship other than research by using these same principles. To transform your teaching into scholarship you should examine the activities you perform or a problem that needs to be solved, apply information or a solution based on best practices or what is known from the literature, and then share the results/products with others (peer-review). Crites et al8 provide practical guidelines for developing education research questions, designing and implementing scholarly activities, and interpreting the scope and impact of education scholarship.
In addition, reaching beyond one’s department to other departments, as well as participating in educational scholarly activities on regional and national levels, is important as one’s career progresses. Well-connected and diverse networks are information highways by which one’s work can be amplified to achieve a greater impact, and from which many opportunities will be shared.
Number 4: Share broadly
Scholarship activities of both academic and community-based CEs can target many audiences, including medical students, residents, and fellows; faculty; other health professions; or even patients and the community. Knowing who will be the recipients or end-users can help to identify which types of projects may be most rewarding and make the greatest impact. Consider sharing curricula, evaluation tools, and other educational products with colleagues at other institutions who ask for them. Request acknowledgment for the development of the materials and ask for written feedback on how these products are being used and what impact they have had on learners.
One education model used to assess the impact and target of education interventions is known as Kirkpatrick’s9 hierarchy, which traditionally included the following four levels: reaction (level 1), learning (level 2), behavior (level 3), and results (level 4). The model has been adapted by the British Medical Journal’s Best Evidence in Medical Education collaboration to medical education with the following modifications in levels as follows.9,10 Level 1: participation: focused on learners’ views of the learning experience including content, presentation, and teaching methods. Level 2a: modification of attitudes/perceptions: focused on changes in attitudes or perceptions between participant groups toward the intervention. Level 2b: modification of knowledge/skills: for knowledge, focused on the acquisition of concepts, procedures, and principles; for skills, focused on the acquisition of problem solving, psychomotor, and social skills. Level 3: behavioral change: focused on the transfer of learning to the workplace or willingness of learners to apply new knowledge and skills. Level 4a: change in organizational practice: focused on wider changes in the organization or delivery of care attributable to an educational program. Level 4b: focused on improvements in the health and well-being of patients as a direct result of an education initiative.
Similar to more traditional clinical research, education research needs to be performed in a scholarly fashion and shared with a wider audience. In addition to submitting research to gastroenterology journals (e.g., Gastroenterology’s Mentoring, Education, and Training Corner), education research can be submitted to education journals such as the Association of American Medical Colleges’ Academic Medicine, the Association for the Study of Medical Education’s Medical Education, the Accreditation Council for Graduate Medical Education’s Journal of Graduate Medical Education, or the European Association for Medical Education in Europe’s Medical Teacher; online education warehouses such as MedEdPORTAL (www.mededportal.org) or MERLOT (www.merlot.org); and national conferences as workshops. Also, one should keep in mind that opportunities arise on a regular basis to share educational videos or images in forums such as the American Society for Gastrointestinal Endoscopy’s video journal VideoGIE, The American Journal of Gastroenterology’s video of the month, and Clinical Gastroenterology and Hepatology’s Images of the Month.
Number 5: Ongoing professional development
Continuing Medical Education is a standard requirement to maintain an active medical license because it shows ongoing efforts to remain up-to-date with changes in medicine. Similar opportunities exist with respect to further development as an educator. Given the multitude of manners in which these opportunities can be divided, we have compiled recommendations for resources on educational scholarship based on level of experience and desired level of engagement (Table 1).
Summary
The framework provided should help guide the gastroenterologist on the path of becoming an effective clinician educator in gastroenterology. The diversity of what a career as a clinician educator can entail is unlimited. The success of the future of medical education and our careers requires not only that every clinician educator be productive, but also that each one brings a unique passion to work each day to share. The authors would like to thank all those clinician educators who contributed to our education, and look forward to learning from you in the future.
Acknowledgments
The authors thank Dr Lee Ligon, Center for Research, Innovation, and Scholarship, Department of Pediatrics, Baylor College of Medicine, for providing editorial assistance.
References
1. Sherbino, J., Frank, J.R., Snell, L. Defining the key roles and competencies of the clinician-educator of the 21st century: a national mixed-methods study. Acad Med. 2014;89:783-9.
2. Simpson, D., Fincher, R.M., Hafler, J.P., et al. Advancing educators and education by defining the components and evidence associated with educational scholarship. Med Educ. 2007;41:1002-9.
3. Rustgi, A.K. Hecht, G.A. Mentorship in academic medicine. Gastroenterology. 2011;141:789-92.
4. Farrell, S.E., Digioia, N.M., Broderick, K.B., et al. Mentoring for clinician-educators. Acad Emerg Med. 2004;11:1346-50.
5. Boyer, E.L. Scholarship reconsidered: priorities of the professoriate. Carnegie Foundation for the Advancement of Teaching, Princeton, NJ; 1990.
6. Glassick, C.E., Taylor-Huber, M., Maeroff, G.I., et al. Scholarship assessed: evaluation of the professoriate. Jossey-Bass, San Francisco; 1997.
7. Golub, R.M. Looking inward and reflecting back medical education and JAMA. JAMA. 2016;316:2200-3.
8. Crites, G.E., Gaines, J.K., Cottrell, S., et al. Medical education scholarship: an introductory guide: AMEE guide no. 89. Med Teach. 2014;36:657-74.
9. Kirkpatrick, D.L. Evaluation of training. In: R. Craig, L. Bittel (Eds.) Training and development handbook. McGraw-Hill, New York; 1967: 87-112.
10. Littlewood, S., Ypinazar, V., Margolis, S.A., et al. Early practical experience and the social responsiveness of clinical education: systematic review. BMJ. 2005;331:387-91.
Dr. Shapiro is a gastroenterology fellow in the department of medicine, section of gastroenterology; Dr. Gould Suarez is an associate professor in the department of medicine, section of gastroenterology and associate program director of the gastroenterology fellowship; and Dr. Turner is an associate professor of pediatrics, vice chair of education, associate program director for house staff education, section of academic general pediatrics, and director for research, innovation, and scholarship, Baylor College of Medicine, Houston. The authors disclose no conflicts.
Smokers face higher infection risk after hernia operations
Jonah Stulberg, MD, FACS, is stickler about requiring patients to stop smoking at least 3 months before hernia surgery. He even uses urine tests to confirm whether they actually quit. A study by Dr. Stulberg and his colleagues supports this approach: .
The finding held up even after the researchers controlled for various factors. “Our findings are in agreement with other findings in higher risk surgeries, and they provide evidence that low-risk surgeries are not exempt from the risks associated with smoking,” said Dr. Stulberg in an interview. “Our data would suggest that there is significant clinical benefit to encouraging smoking cessation before elective hernia repair.”

The researchers launched the study to better understand how smoking affects complication rates in light of the fact that “surgeons in the U.S. tend to offer low-risk elective surgical procedures to patients who are actively smoking despite overwhelming evidence that smoking increases surgical risks,” Dr. Stulberg said.
The researchers tracked 220,629 patients in the American College of Surgeons National Surgical Quality Improvement Project (NSQIP) database who underwent several types of elective hernia repair from 2011 to 2014.
Just over 18% of the patients said they’d smoked over the past year; they were more likely to be younger (median age, 50 for smokers vs. 57 for nonsmokers). Smokers also were more likely to be black, to be underweight, and to consume two or more alcoholic beverages per day (P less than .05).
The researchers tracked serious complications in the 30 days after surgery such as death, sepsis, and readmission.
Complications developed in 6.34% of smokers and 4.72% of nonsmokers (P less than .001). Numerous kinds of complications were more common in the smokers prior to adjustment: death, return to the operating room, readmission, and transfusion plus wound, pulmonary, thromboembolic and cardiac complications.
The researchers adjusted their statistics to account for factors such as ethnicity, sex, body mass index, preexisting comorbidities, and type of hernia operation. They found that risk of all complications was higher in smokers, compared with nonsmokers (odds ratio, 1.30) as were several other complications: death (OR, 1.53), return to operating room (OR, 1.23), readmission (OR, 1.24), wound complication (OR, 1.36), sepsis/septic shock (OR, 1.31), pulmonary complication (OR 1.77-2.30) and cardiac complication (OR, 1.27-1.43).
Only transfusion (OR, 0.90) and thromboembolic (OR, 0.87) complications were less likely in smokers.
The researchers noted that the statistics don’t allow them to analyze whether it makes any difference if smokers quit shortly before their procedures. Still, Dr. Stulberg stands by his you-must-quit-smoking-before-surgery edict. “I believe that their active smoking habit is a bigger health threat than their asymptomatic hernia, and therefore feel the right thing to do as their physician is support them through their smoking cessation,” he said. “I offer counseling and nicotine replacement if needed. I have very good quit rates and would encourage other surgeons to do the same.”
Dr. Stulberg noted that he can’t point to evidence supporting his requirement that patients quit at least 3 months before surgery. “Most data out there says the farther away from your last cigarette, the better,” he said. “But there isn’t any magic to 3 months, other than I believe it will lead to a higher likelihood of permanent cessation.”
Northwestern Memorial Hospital and Northwestern University funded the study. Four of the nine authors, including Dr. Stulberg, report various disclosures that are not directly related to the study, including funding from government agencies, physician organizations, Health Care Services Corporation and Blue Cross Blue Shield of Illinois, Mallinckrodt, and Northwestern University. The other authors report no disclosures.
SOURCE: DeLancey JO et al. Am J Surg. 2018 Mar 6. doi: 10.1016/j.amjsurg.2018.03.004.
Jonah Stulberg, MD, FACS, is stickler about requiring patients to stop smoking at least 3 months before hernia surgery. He even uses urine tests to confirm whether they actually quit. A study by Dr. Stulberg and his colleagues supports this approach: .
The finding held up even after the researchers controlled for various factors. “Our findings are in agreement with other findings in higher risk surgeries, and they provide evidence that low-risk surgeries are not exempt from the risks associated with smoking,” said Dr. Stulberg in an interview. “Our data would suggest that there is significant clinical benefit to encouraging smoking cessation before elective hernia repair.”
The researchers launched the study to better understand how smoking affects complication rates in light of the fact that “surgeons in the U.S. tend to offer low-risk elective surgical procedures to patients who are actively smoking despite overwhelming evidence that smoking increases surgical risks,” Dr. Stulberg said.
The researchers tracked 220,629 patients in the American College of Surgeons National Surgical Quality Improvement Project (NSQIP) database who underwent several types of elective hernia repair from 2011 to 2014.
Just over 18% of the patients said they’d smoked over the past year; they were more likely to be younger (median age, 50 for smokers vs. 57 for nonsmokers). Smokers also were more likely to be black, to be underweight, and to consume two or more alcoholic beverages per day (P less than .05).
The researchers tracked serious complications in the 30 days after surgery such as death, sepsis, and readmission.
Complications developed in 6.34% of smokers and 4.72% of nonsmokers (P less than .001). Numerous kinds of complications were more common in the smokers prior to adjustment: death, return to the operating room, readmission, and transfusion plus wound, pulmonary, thromboembolic and cardiac complications.
The researchers adjusted their statistics to account for factors such as ethnicity, sex, body mass index, preexisting comorbidities, and type of hernia operation. They found that risk of all complications was higher in smokers, compared with nonsmokers (odds ratio, 1.30) as were several other complications: death (OR, 1.53), return to operating room (OR, 1.23), readmission (OR, 1.24), wound complication (OR, 1.36), sepsis/septic shock (OR, 1.31), pulmonary complication (OR 1.77-2.30) and cardiac complication (OR, 1.27-1.43).
Only transfusion (OR, 0.90) and thromboembolic (OR, 0.87) complications were less likely in smokers.
The researchers noted that the statistics don’t allow them to analyze whether it makes any difference if smokers quit shortly before their procedures. Still, Dr. Stulberg stands by his you-must-quit-smoking-before-surgery edict. “I believe that their active smoking habit is a bigger health threat than their asymptomatic hernia, and therefore feel the right thing to do as their physician is support them through their smoking cessation,” he said. “I offer counseling and nicotine replacement if needed. I have very good quit rates and would encourage other surgeons to do the same.”
Dr. Stulberg noted that he can’t point to evidence supporting his requirement that patients quit at least 3 months before surgery. “Most data out there says the farther away from your last cigarette, the better,” he said. “But there isn’t any magic to 3 months, other than I believe it will lead to a higher likelihood of permanent cessation.”
Northwestern Memorial Hospital and Northwestern University funded the study. Four of the nine authors, including Dr. Stulberg, report various disclosures that are not directly related to the study, including funding from government agencies, physician organizations, Health Care Services Corporation and Blue Cross Blue Shield of Illinois, Mallinckrodt, and Northwestern University. The other authors report no disclosures.
SOURCE: DeLancey JO et al. Am J Surg. 2018 Mar 6. doi: 10.1016/j.amjsurg.2018.03.004.
Jonah Stulberg, MD, FACS, is stickler about requiring patients to stop smoking at least 3 months before hernia surgery. He even uses urine tests to confirm whether they actually quit. A study by Dr. Stulberg and his colleagues supports this approach: .
The finding held up even after the researchers controlled for various factors. “Our findings are in agreement with other findings in higher risk surgeries, and they provide evidence that low-risk surgeries are not exempt from the risks associated with smoking,” said Dr. Stulberg in an interview. “Our data would suggest that there is significant clinical benefit to encouraging smoking cessation before elective hernia repair.”
The researchers launched the study to better understand how smoking affects complication rates in light of the fact that “surgeons in the U.S. tend to offer low-risk elective surgical procedures to patients who are actively smoking despite overwhelming evidence that smoking increases surgical risks,” Dr. Stulberg said.
The researchers tracked 220,629 patients in the American College of Surgeons National Surgical Quality Improvement Project (NSQIP) database who underwent several types of elective hernia repair from 2011 to 2014.
Just over 18% of the patients said they’d smoked over the past year; they were more likely to be younger (median age, 50 for smokers vs. 57 for nonsmokers). Smokers also were more likely to be black, to be underweight, and to consume two or more alcoholic beverages per day (P less than .05).
The researchers tracked serious complications in the 30 days after surgery such as death, sepsis, and readmission.
Complications developed in 6.34% of smokers and 4.72% of nonsmokers (P less than .001). Numerous kinds of complications were more common in the smokers prior to adjustment: death, return to the operating room, readmission, and transfusion plus wound, pulmonary, thromboembolic and cardiac complications.
The researchers adjusted their statistics to account for factors such as ethnicity, sex, body mass index, preexisting comorbidities, and type of hernia operation. They found that risk of all complications was higher in smokers, compared with nonsmokers (odds ratio, 1.30) as were several other complications: death (OR, 1.53), return to operating room (OR, 1.23), readmission (OR, 1.24), wound complication (OR, 1.36), sepsis/septic shock (OR, 1.31), pulmonary complication (OR 1.77-2.30) and cardiac complication (OR, 1.27-1.43).
Only transfusion (OR, 0.90) and thromboembolic (OR, 0.87) complications were less likely in smokers.
The researchers noted that the statistics don’t allow them to analyze whether it makes any difference if smokers quit shortly before their procedures. Still, Dr. Stulberg stands by his you-must-quit-smoking-before-surgery edict. “I believe that their active smoking habit is a bigger health threat than their asymptomatic hernia, and therefore feel the right thing to do as their physician is support them through their smoking cessation,” he said. “I offer counseling and nicotine replacement if needed. I have very good quit rates and would encourage other surgeons to do the same.”
Dr. Stulberg noted that he can’t point to evidence supporting his requirement that patients quit at least 3 months before surgery. “Most data out there says the farther away from your last cigarette, the better,” he said. “But there isn’t any magic to 3 months, other than I believe it will lead to a higher likelihood of permanent cessation.”
Northwestern Memorial Hospital and Northwestern University funded the study. Four of the nine authors, including Dr. Stulberg, report various disclosures that are not directly related to the study, including funding from government agencies, physician organizations, Health Care Services Corporation and Blue Cross Blue Shield of Illinois, Mallinckrodt, and Northwestern University. The other authors report no disclosures.
SOURCE: DeLancey JO et al. Am J Surg. 2018 Mar 6. doi: 10.1016/j.amjsurg.2018.03.004.
FROM AMERICAN JOURNAL OF SURGERY
Key clinical point: Smokers are more likely than are nonsmokers to develop serious complications after elective hernia surgery.
Major finding: The adjusted risk of serious complications after elective hernia surgery is higher (odds ratio, 1.30) in smokers than nonsmokers.
Study details: Retrospective study of ACS NSQIP data on 220,629 patients in the United States (18% smokers) who underwent elective hernia operations during 2011-2014.
Disclosures: Northwestern Memorial Hospital and Northwestern University funded the study. Four of the nine authors reported various disclosures. The other authors report no disclosures.
Source: DeLancey JO et al. Am J Surg. 2018 Mar 6. doi: 10.1016/j.amjsurg.2018.03.004.
Model predicted Barrett’s esophagus progression
A scoring model encompassing just four traits accurately predicted which patients with Barrett’s esophagus were most likely to develop high-grade dysplasia or esophageal adenocarcinoma, researchers reported in the April issue of Gastroenterology (2017 Dec 19. doi: 10.1053/j.gastro.2017.12.009).
Those risk factors included sex, smoking, length of Barrett’s esophagus, and the presence of baseline low-grade dysplasia, said Sravanthi Parasa, MD, of Swedish Medical Center, Seattle, and her associates. For example, a male with a history of smoking found to have a 5-cm, nondysplastic Barrett’s esophagus on histology during his index endoscopy would fall into the model’s intermediate risk category, with a 0.7% annual risk of progression to high-grade dysplasia or esophageal adenocarcinoma, they explained. “This model has the potential to complement molecular biomarker panels currently in development,” they wrote.
Barrett’s esophagus increases the risk of esophageal adenocarcinoma by anywhere from 30 to 125 times, a range that reflects the multifactorial nature of progression and the hypothesis that not all patients with Barrett’s esophagus should undergo the same frequency of endoscopic surveillance, said the researchers. To incorporate predictors of progression into a single model, they analyzed prospective data from nearly 3,000 patients with Barrett’s esophagus who were followed for a median of 6 years at five centers in the United States and one center in the Netherlands. At baseline, patients were an average of 55 years old (standard deviation, 20 years), 84% were men, 88% were white, and the average Barrett’s esophagus length was 3.7 cm (SD, 3.2 cm).
The researchers created the model by starting with many demographic and clinical candidate variables and then using backward selection to eliminate those that did not predict progression with a P value of .05 or less. This is the same method used in the Framingham Heart Study, they noted. In all, 154 (6%) patients with Barrett’s esophagus developed high-grade dysplasia or esophageal adenocarcinoma, with an annual progression rate of about 1%. The significant predictors of progression included male sex, smoking, length of Barrett’s esophagus, and low-grade dysplasia at baseline. A model that included only these four variables distinguished progressors from nonprogressors with a c statistic of 0.76 (95% confidence interval, 0.72 to 0.80; P less than .001). Using 30% of patients as an internal validation cohort, the model’s calibration slope was 0.99 and its calibration intercept was -0.09 cohort (perfectly calibrated models have a slope of 1.0 and an intercept of 0.0).
Therefore, the model was well calibrated and did an appropriate job of identifying risk groups, the investigators concluded. Considering that the overall risk of Barrett’s esophagus progression is low, using this model could help avoid excess costs and burdens of unnecessary surveillance, they added. “We recognize that there is a key interest in contemporary medical research whether a marker (e.g. molecular, genetic) could add to incremental value of a risk progression score,” they wrote. “This can be an area of future research.”
There were no funding sources. Dr. Parasa had no disclosures. One coinvestigator disclosed ties to Cook Medical, CDx Diagnostics, and Cosmo Pharmaceuticals.
SOURCE: Parasa S et al. Gastroenterology. 2017 Dec 19. doi: 10.1053/j.gastro.2017.12.009.
Barrett’s esophagus (BE) is the only known precursor lesion to esophageal adenocarcinoma (EAC), a rapidly rising cancer in the Western world, which has a poor 5-year survival rate of less than 20%. Management strategies to affect EAC incidence include screening and surveillance, with current guidelines recommending surveillance for all patients with a diagnosis of BE.
However, there are several challenges associated with adopting BE surveillance for all patients: It is estimated that anywhere from 2 million to 5 million U.S. adults may harbor BE, and the overall risk of BE progression to EAC is low (approximately 0.2%-0.4% annually). Both of these factors influence the cost-effectiveness of a global BE surveillance program.
Hence, a risk-stratification score that can distinguish BE patients who are at high risk for progression to high-grade dysplasia (HGD) and/or EAC from those whose disease will not progress will be extremely useful. This concept would be similar to other risk-scoring mechanisms, such as the MELD score for progression in liver disease.

With use of a large multicenter cohort of patients with BE (more than 4,500 patients), this is the first risk-prediction score developed and validated using baseline demographic and endoscopy information to determine risk of progression. Readily available factors such as patient sex, smoking status, BE length, and confirmed histology were identified as risk factors for progression, which could then generate a score determining the individual patient’s risk of progression. Such a simple scoring system has the potential of tailoring management based on the risk factors. In the future, inclusion of molecular biomarkers along with this score may further enhance its potential for personalized medicine in BE patients.
Prateek Sharma, MD, is a professor of medicine of University of Kansas, Kansas City. He has no conflicts of interest.
Barrett’s esophagus (BE) is the only known precursor lesion to esophageal adenocarcinoma (EAC), a rapidly rising cancer in the Western world, which has a poor 5-year survival rate of less than 20%. Management strategies to affect EAC incidence include screening and surveillance, with current guidelines recommending surveillance for all patients with a diagnosis of BE.
However, there are several challenges associated with adopting BE surveillance for all patients: It is estimated that anywhere from 2 million to 5 million U.S. adults may harbor BE, and the overall risk of BE progression to EAC is low (approximately 0.2%-0.4% annually). Both of these factors influence the cost-effectiveness of a global BE surveillance program.
Hence, a risk-stratification score that can distinguish BE patients who are at high risk for progression to high-grade dysplasia (HGD) and/or EAC from those whose disease will not progress will be extremely useful. This concept would be similar to other risk-scoring mechanisms, such as the MELD score for progression in liver disease.
With use of a large multicenter cohort of patients with BE (more than 4,500 patients), this is the first risk-prediction score developed and validated using baseline demographic and endoscopy information to determine risk of progression. Readily available factors such as patient sex, smoking status, BE length, and confirmed histology were identified as risk factors for progression, which could then generate a score determining the individual patient’s risk of progression. Such a simple scoring system has the potential of tailoring management based on the risk factors. In the future, inclusion of molecular biomarkers along with this score may further enhance its potential for personalized medicine in BE patients.
Prateek Sharma, MD, is a professor of medicine of University of Kansas, Kansas City. He has no conflicts of interest.
Barrett’s esophagus (BE) is the only known precursor lesion to esophageal adenocarcinoma (EAC), a rapidly rising cancer in the Western world, which has a poor 5-year survival rate of less than 20%. Management strategies to affect EAC incidence include screening and surveillance, with current guidelines recommending surveillance for all patients with a diagnosis of BE.
However, there are several challenges associated with adopting BE surveillance for all patients: It is estimated that anywhere from 2 million to 5 million U.S. adults may harbor BE, and the overall risk of BE progression to EAC is low (approximately 0.2%-0.4% annually). Both of these factors influence the cost-effectiveness of a global BE surveillance program.
Hence, a risk-stratification score that can distinguish BE patients who are at high risk for progression to high-grade dysplasia (HGD) and/or EAC from those whose disease will not progress will be extremely useful. This concept would be similar to other risk-scoring mechanisms, such as the MELD score for progression in liver disease.
With use of a large multicenter cohort of patients with BE (more than 4,500 patients), this is the first risk-prediction score developed and validated using baseline demographic and endoscopy information to determine risk of progression. Readily available factors such as patient sex, smoking status, BE length, and confirmed histology were identified as risk factors for progression, which could then generate a score determining the individual patient’s risk of progression. Such a simple scoring system has the potential of tailoring management based on the risk factors. In the future, inclusion of molecular biomarkers along with this score may further enhance its potential for personalized medicine in BE patients.
Prateek Sharma, MD, is a professor of medicine of University of Kansas, Kansas City. He has no conflicts of interest.
A scoring model encompassing just four traits accurately predicted which patients with Barrett’s esophagus were most likely to develop high-grade dysplasia or esophageal adenocarcinoma, researchers reported in the April issue of Gastroenterology (2017 Dec 19. doi: 10.1053/j.gastro.2017.12.009).
Those risk factors included sex, smoking, length of Barrett’s esophagus, and the presence of baseline low-grade dysplasia, said Sravanthi Parasa, MD, of Swedish Medical Center, Seattle, and her associates. For example, a male with a history of smoking found to have a 5-cm, nondysplastic Barrett’s esophagus on histology during his index endoscopy would fall into the model’s intermediate risk category, with a 0.7% annual risk of progression to high-grade dysplasia or esophageal adenocarcinoma, they explained. “This model has the potential to complement molecular biomarker panels currently in development,” they wrote.
Barrett’s esophagus increases the risk of esophageal adenocarcinoma by anywhere from 30 to 125 times, a range that reflects the multifactorial nature of progression and the hypothesis that not all patients with Barrett’s esophagus should undergo the same frequency of endoscopic surveillance, said the researchers. To incorporate predictors of progression into a single model, they analyzed prospective data from nearly 3,000 patients with Barrett’s esophagus who were followed for a median of 6 years at five centers in the United States and one center in the Netherlands. At baseline, patients were an average of 55 years old (standard deviation, 20 years), 84% were men, 88% were white, and the average Barrett’s esophagus length was 3.7 cm (SD, 3.2 cm).
The researchers created the model by starting with many demographic and clinical candidate variables and then using backward selection to eliminate those that did not predict progression with a P value of .05 or less. This is the same method used in the Framingham Heart Study, they noted. In all, 154 (6%) patients with Barrett’s esophagus developed high-grade dysplasia or esophageal adenocarcinoma, with an annual progression rate of about 1%. The significant predictors of progression included male sex, smoking, length of Barrett’s esophagus, and low-grade dysplasia at baseline. A model that included only these four variables distinguished progressors from nonprogressors with a c statistic of 0.76 (95% confidence interval, 0.72 to 0.80; P less than .001). Using 30% of patients as an internal validation cohort, the model’s calibration slope was 0.99 and its calibration intercept was -0.09 cohort (perfectly calibrated models have a slope of 1.0 and an intercept of 0.0).
Therefore, the model was well calibrated and did an appropriate job of identifying risk groups, the investigators concluded. Considering that the overall risk of Barrett’s esophagus progression is low, using this model could help avoid excess costs and burdens of unnecessary surveillance, they added. “We recognize that there is a key interest in contemporary medical research whether a marker (e.g. molecular, genetic) could add to incremental value of a risk progression score,” they wrote. “This can be an area of future research.”
There were no funding sources. Dr. Parasa had no disclosures. One coinvestigator disclosed ties to Cook Medical, CDx Diagnostics, and Cosmo Pharmaceuticals.
SOURCE: Parasa S et al. Gastroenterology. 2017 Dec 19. doi: 10.1053/j.gastro.2017.12.009.
A scoring model encompassing just four traits accurately predicted which patients with Barrett’s esophagus were most likely to develop high-grade dysplasia or esophageal adenocarcinoma, researchers reported in the April issue of Gastroenterology (2017 Dec 19. doi: 10.1053/j.gastro.2017.12.009).
Those risk factors included sex, smoking, length of Barrett’s esophagus, and the presence of baseline low-grade dysplasia, said Sravanthi Parasa, MD, of Swedish Medical Center, Seattle, and her associates. For example, a male with a history of smoking found to have a 5-cm, nondysplastic Barrett’s esophagus on histology during his index endoscopy would fall into the model’s intermediate risk category, with a 0.7% annual risk of progression to high-grade dysplasia or esophageal adenocarcinoma, they explained. “This model has the potential to complement molecular biomarker panels currently in development,” they wrote.
Barrett’s esophagus increases the risk of esophageal adenocarcinoma by anywhere from 30 to 125 times, a range that reflects the multifactorial nature of progression and the hypothesis that not all patients with Barrett’s esophagus should undergo the same frequency of endoscopic surveillance, said the researchers. To incorporate predictors of progression into a single model, they analyzed prospective data from nearly 3,000 patients with Barrett’s esophagus who were followed for a median of 6 years at five centers in the United States and one center in the Netherlands. At baseline, patients were an average of 55 years old (standard deviation, 20 years), 84% were men, 88% were white, and the average Barrett’s esophagus length was 3.7 cm (SD, 3.2 cm).
The researchers created the model by starting with many demographic and clinical candidate variables and then using backward selection to eliminate those that did not predict progression with a P value of .05 or less. This is the same method used in the Framingham Heart Study, they noted. In all, 154 (6%) patients with Barrett’s esophagus developed high-grade dysplasia or esophageal adenocarcinoma, with an annual progression rate of about 1%. The significant predictors of progression included male sex, smoking, length of Barrett’s esophagus, and low-grade dysplasia at baseline. A model that included only these four variables distinguished progressors from nonprogressors with a c statistic of 0.76 (95% confidence interval, 0.72 to 0.80; P less than .001). Using 30% of patients as an internal validation cohort, the model’s calibration slope was 0.99 and its calibration intercept was -0.09 cohort (perfectly calibrated models have a slope of 1.0 and an intercept of 0.0).
Therefore, the model was well calibrated and did an appropriate job of identifying risk groups, the investigators concluded. Considering that the overall risk of Barrett’s esophagus progression is low, using this model could help avoid excess costs and burdens of unnecessary surveillance, they added. “We recognize that there is a key interest in contemporary medical research whether a marker (e.g. molecular, genetic) could add to incremental value of a risk progression score,” they wrote. “This can be an area of future research.”
There were no funding sources. Dr. Parasa had no disclosures. One coinvestigator disclosed ties to Cook Medical, CDx Diagnostics, and Cosmo Pharmaceuticals.
SOURCE: Parasa S et al. Gastroenterology. 2017 Dec 19. doi: 10.1053/j.gastro.2017.12.009.
FROM GASTROENTEROLOGY
Key clinical point: A model containing four risk factors identified patients with Barrett’s esophagus at significantly increased risk of progression to high-grade dysplasia or esophageal adenocarcinoma.
Major finding: Scores assigned identified patients with BE that progressed to HGD or EAC with a c statistic of 0.76 (95% CI, 0.72 to 0.80; P less than .001).
Data source: A multicenter, longitudinal study of 2,697 patients with Barrett’s esophagus.
Disclosures: There were no funding sources. Dr. Parasa had no disclosures. One coinvestigator disclosed ties to Cook Medical, CDx Diagnostics, and Cosmo Pharmaceuticals.
Source: Parasa S et al. Gastroenterology. 2017 Dec 19. doi: 10.1053/j.gastro.2017.12.009.
Sustaining the evolution of PAs in hospital medicine
Editor’s note: Each month, SHM puts the spotlight on some of our most active members who are making substantial contributions to hospital medicine. Visit www.hospitalmedicine.org for more information on how you can lend your expertise to help SHM improve the care of hospitalized patients.
This month, The Hospitalist spotlights Meredith K. Wold, PA-C, APC supervisor, Hospital Medicine and Critical Care, at Regions Hospital in St. Paul, Minn., and adjunct faculty, Augsburg University Physician Assistant Program. Ms. Wold is a long-time member of SHM and the recipient of this year’s Clinical Excellence Award for Nurse Practitioners and Physician Assistants.
How did you first hear of SHM and why did you decide to become a member?
I’ve always recognized the importance of engaging in a community beyond my daily practice. Shortly after starting my career in hospital medicine, I quickly recognized this was a belief shared and cultivated by my hospital medicine group as well. Our HM group at HealthPartners has a long history of SHM participation. As our advanced practice clinician (APC) group grew, I knew engagement at the national level was critical to ensure that our ongoing evolution was supported, sustained, and shared.
What does it mean to you to receive SHM’s Clinical Excellence Award for nurse practitioners and physician assistants?
Being awarded the SHM Clinical Excellence Award is remarkable. I work alongside really, really amazing people, and every day I strive toward the exceptionally high bar they set. I’m passionate and committed to hospital medicine, and I’m so very grateful this is appreciated.
Which SHM conferences have you attended? Tell us about some of the highlights from these courses.
The first SHM annual conference I attended was in 2008 in sunny San Diego. I’d been a physician assistant (PA) for barely a year. I remember being so energized by the passion and commitment of the speakers and attendees. I harnessed that energy and spent the next several years being part of a growing APC group at Regions Hospital in St. Paul, Minn., where our HM group holds partnership and innovation at its core. You can imagine my excitement when I was asked to speak about APC practice models at HM16. Fellow APC Emily Thornhill Davis and I spoke to a standing-room only audience! Emily and I partnered again as faculty at HM17. I look forward to being part of a panel discussion at HM18 in Orlando (alongside some SHM trailblazers!).
Closer to home, I’ve taken advantage of phenomenal opportunities hosted by our local chapter of SHM. My colleagues Benji Mathews, MD, and Kreegan Reierson, MD, have led Point-of-Care Ultrasound (POCUS) training courses regionally and nationally. Their comprehensive, hands-on course ensured that I had the foundation to incorporate portable ultrasound into my practice. Thank goodness for their refresher course as well; my skills were rusty after a long maternity leave!
Given the tremendous clinical growth I have absorbed through local and national SHM offerings, I look forward to my leadership and operations skills being bolstered at SHM’s Leadership Academy this fall in Vancouver. As APCs hold more and more vital roles within HM groups, it’s integral that, along the way, our leadership skills are recognized and honed as well.
As an SHM member of over 10 years, what has been most valuable for you as a physician assistant?
The relationships. Networking, sharing ideas, pushing the status quo with other like-minded clinicians from around the country is invigorating. Because of SHM, I have an APC network from coast to coast – a lattice of clinicians that are linked by dedication and enthusiasm to hospital medicine.
What advice do you have for early-career physician assistants looking to work in hospital medicine?
Find a hospital medicine group whose culture allows and supports your growth as an advanced practice clinician. In an exemplary HM model, the delegated autonomy of an APC should widen and deepen over time. Seek out a team that appreciates the importance of this evolution.
Ms. Steele is marketing communications specialist at the Society of Hospital Medicine.
Editor’s note: Each month, SHM puts the spotlight on some of our most active members who are making substantial contributions to hospital medicine. Visit www.hospitalmedicine.org for more information on how you can lend your expertise to help SHM improve the care of hospitalized patients.
This month, The Hospitalist spotlights Meredith K. Wold, PA-C, APC supervisor, Hospital Medicine and Critical Care, at Regions Hospital in St. Paul, Minn., and adjunct faculty, Augsburg University Physician Assistant Program. Ms. Wold is a long-time member of SHM and the recipient of this year’s Clinical Excellence Award for Nurse Practitioners and Physician Assistants.
How did you first hear of SHM and why did you decide to become a member?
I’ve always recognized the importance of engaging in a community beyond my daily practice. Shortly after starting my career in hospital medicine, I quickly recognized this was a belief shared and cultivated by my hospital medicine group as well. Our HM group at HealthPartners has a long history of SHM participation. As our advanced practice clinician (APC) group grew, I knew engagement at the national level was critical to ensure that our ongoing evolution was supported, sustained, and shared.
What does it mean to you to receive SHM’s Clinical Excellence Award for nurse practitioners and physician assistants?
Being awarded the SHM Clinical Excellence Award is remarkable. I work alongside really, really amazing people, and every day I strive toward the exceptionally high bar they set. I’m passionate and committed to hospital medicine, and I’m so very grateful this is appreciated.
Which SHM conferences have you attended? Tell us about some of the highlights from these courses.
The first SHM annual conference I attended was in 2008 in sunny San Diego. I’d been a physician assistant (PA) for barely a year. I remember being so energized by the passion and commitment of the speakers and attendees. I harnessed that energy and spent the next several years being part of a growing APC group at Regions Hospital in St. Paul, Minn., where our HM group holds partnership and innovation at its core. You can imagine my excitement when I was asked to speak about APC practice models at HM16. Fellow APC Emily Thornhill Davis and I spoke to a standing-room only audience! Emily and I partnered again as faculty at HM17. I look forward to being part of a panel discussion at HM18 in Orlando (alongside some SHM trailblazers!).
Closer to home, I’ve taken advantage of phenomenal opportunities hosted by our local chapter of SHM. My colleagues Benji Mathews, MD, and Kreegan Reierson, MD, have led Point-of-Care Ultrasound (POCUS) training courses regionally and nationally. Their comprehensive, hands-on course ensured that I had the foundation to incorporate portable ultrasound into my practice. Thank goodness for their refresher course as well; my skills were rusty after a long maternity leave!
Given the tremendous clinical growth I have absorbed through local and national SHM offerings, I look forward to my leadership and operations skills being bolstered at SHM’s Leadership Academy this fall in Vancouver. As APCs hold more and more vital roles within HM groups, it’s integral that, along the way, our leadership skills are recognized and honed as well.
As an SHM member of over 10 years, what has been most valuable for you as a physician assistant?
The relationships. Networking, sharing ideas, pushing the status quo with other like-minded clinicians from around the country is invigorating. Because of SHM, I have an APC network from coast to coast – a lattice of clinicians that are linked by dedication and enthusiasm to hospital medicine.
What advice do you have for early-career physician assistants looking to work in hospital medicine?
Find a hospital medicine group whose culture allows and supports your growth as an advanced practice clinician. In an exemplary HM model, the delegated autonomy of an APC should widen and deepen over time. Seek out a team that appreciates the importance of this evolution.
Ms. Steele is marketing communications specialist at the Society of Hospital Medicine.
Editor’s note: Each month, SHM puts the spotlight on some of our most active members who are making substantial contributions to hospital medicine. Visit www.hospitalmedicine.org for more information on how you can lend your expertise to help SHM improve the care of hospitalized patients.
This month, The Hospitalist spotlights Meredith K. Wold, PA-C, APC supervisor, Hospital Medicine and Critical Care, at Regions Hospital in St. Paul, Minn., and adjunct faculty, Augsburg University Physician Assistant Program. Ms. Wold is a long-time member of SHM and the recipient of this year’s Clinical Excellence Award for Nurse Practitioners and Physician Assistants.
How did you first hear of SHM and why did you decide to become a member?
I’ve always recognized the importance of engaging in a community beyond my daily practice. Shortly after starting my career in hospital medicine, I quickly recognized this was a belief shared and cultivated by my hospital medicine group as well. Our HM group at HealthPartners has a long history of SHM participation. As our advanced practice clinician (APC) group grew, I knew engagement at the national level was critical to ensure that our ongoing evolution was supported, sustained, and shared.
What does it mean to you to receive SHM’s Clinical Excellence Award for nurse practitioners and physician assistants?
Being awarded the SHM Clinical Excellence Award is remarkable. I work alongside really, really amazing people, and every day I strive toward the exceptionally high bar they set. I’m passionate and committed to hospital medicine, and I’m so very grateful this is appreciated.
Which SHM conferences have you attended? Tell us about some of the highlights from these courses.
The first SHM annual conference I attended was in 2008 in sunny San Diego. I’d been a physician assistant (PA) for barely a year. I remember being so energized by the passion and commitment of the speakers and attendees. I harnessed that energy and spent the next several years being part of a growing APC group at Regions Hospital in St. Paul, Minn., where our HM group holds partnership and innovation at its core. You can imagine my excitement when I was asked to speak about APC practice models at HM16. Fellow APC Emily Thornhill Davis and I spoke to a standing-room only audience! Emily and I partnered again as faculty at HM17. I look forward to being part of a panel discussion at HM18 in Orlando (alongside some SHM trailblazers!).
Closer to home, I’ve taken advantage of phenomenal opportunities hosted by our local chapter of SHM. My colleagues Benji Mathews, MD, and Kreegan Reierson, MD, have led Point-of-Care Ultrasound (POCUS) training courses regionally and nationally. Their comprehensive, hands-on course ensured that I had the foundation to incorporate portable ultrasound into my practice. Thank goodness for their refresher course as well; my skills were rusty after a long maternity leave!
Given the tremendous clinical growth I have absorbed through local and national SHM offerings, I look forward to my leadership and operations skills being bolstered at SHM’s Leadership Academy this fall in Vancouver. As APCs hold more and more vital roles within HM groups, it’s integral that, along the way, our leadership skills are recognized and honed as well.
As an SHM member of over 10 years, what has been most valuable for you as a physician assistant?
The relationships. Networking, sharing ideas, pushing the status quo with other like-minded clinicians from around the country is invigorating. Because of SHM, I have an APC network from coast to coast – a lattice of clinicians that are linked by dedication and enthusiasm to hospital medicine.
What advice do you have for early-career physician assistants looking to work in hospital medicine?
Find a hospital medicine group whose culture allows and supports your growth as an advanced practice clinician. In an exemplary HM model, the delegated autonomy of an APC should widen and deepen over time. Seek out a team that appreciates the importance of this evolution.
Ms. Steele is marketing communications specialist at the Society of Hospital Medicine.
VIDEO: Intestinal remodeling contributes to HbA1c drop after Roux-en-Y gastric bypass
CHICAGO – Of medical and surgical tactics to tackle long-term weight loss, , and gene expression in the Roux limb may hold the key to the surgery’s efficacy, according to an ongoing study.
“We know that Roux-en-Y gastric bypass surgery is highly effective as not only a weight-loss therapy, but more and more we’re appreciating its role as a diabetes therapy as well,” said Margaret Stefater, MD, PhD, speaking in an interview at the annual meeting of the Endocrine Society.
The study, she said, was designed to learn more about the intestine’s contribution to the salubrious effect that Roux-en-Y surgery has on diabetes.
“We used microarray in order to characterize gene expression in the intestine” to gain a broad understanding of the processes that are altered after surgery, said Dr. Stefater, a pediatric endocrinology fellow at Boston Children’s Hospital. More specifically, though, the study looked at an individual’s changes in gene expression over time and correlated those changes with that patient’s clinical picture.
The data reported by Dr. Stefater and shared in a press conference, represent part of an ongoing longitudinal prospective study of 32 patients.
“The study aims to characterize gene expression for the first postoperative year,” and findings from the first 6 postoperative months of 19 patients were shared at the meeting, said Dr. Stefater. “This is the first look at our cohort.”
So far, she and her colleagues have compared gene expression using microarray at 1 month and 6 months post-surgery, comparing change across time and change from baseline data.
From hundreds of candidate genes, Dr. Stefater and her colleagues have developed a smaller gene list that, even in the first postoperative month, is predictive of changes in hemoglobin A1c levels over time. “Remarkably, the changes in a select list of genes out to 1 month is actually able to predict hemoglobin A1c levels out to 1 year,” she said. “This speaks to the fact that biological reprogramming in the intestine is somehow related to glycemic response in patients.
“We hope that by understanding these processes, we can home in on those processes that are most likely to be mechanistically responsible for these changes, and then to reverse-engineer this surgery to identify processes or targets which may be good places to start when we think about creating better, or nonsurgical, therapies for people who have obesity and diabetes,” said Dr. Stefater.
Dr. Stefater reported no relevant financial disclosures.
SOURCE: Stefater MA et al. ENDO 2018, Abstract OR 12-6.
CHICAGO – Of medical and surgical tactics to tackle long-term weight loss, , and gene expression in the Roux limb may hold the key to the surgery’s efficacy, according to an ongoing study.
“We know that Roux-en-Y gastric bypass surgery is highly effective as not only a weight-loss therapy, but more and more we’re appreciating its role as a diabetes therapy as well,” said Margaret Stefater, MD, PhD, speaking in an interview at the annual meeting of the Endocrine Society.
The study, she said, was designed to learn more about the intestine’s contribution to the salubrious effect that Roux-en-Y surgery has on diabetes.
“We used microarray in order to characterize gene expression in the intestine” to gain a broad understanding of the processes that are altered after surgery, said Dr. Stefater, a pediatric endocrinology fellow at Boston Children’s Hospital. More specifically, though, the study looked at an individual’s changes in gene expression over time and correlated those changes with that patient’s clinical picture.
The data reported by Dr. Stefater and shared in a press conference, represent part of an ongoing longitudinal prospective study of 32 patients.
“The study aims to characterize gene expression for the first postoperative year,” and findings from the first 6 postoperative months of 19 patients were shared at the meeting, said Dr. Stefater. “This is the first look at our cohort.”
So far, she and her colleagues have compared gene expression using microarray at 1 month and 6 months post-surgery, comparing change across time and change from baseline data.
From hundreds of candidate genes, Dr. Stefater and her colleagues have developed a smaller gene list that, even in the first postoperative month, is predictive of changes in hemoglobin A1c levels over time. “Remarkably, the changes in a select list of genes out to 1 month is actually able to predict hemoglobin A1c levels out to 1 year,” she said. “This speaks to the fact that biological reprogramming in the intestine is somehow related to glycemic response in patients.
“We hope that by understanding these processes, we can home in on those processes that are most likely to be mechanistically responsible for these changes, and then to reverse-engineer this surgery to identify processes or targets which may be good places to start when we think about creating better, or nonsurgical, therapies for people who have obesity and diabetes,” said Dr. Stefater.
Dr. Stefater reported no relevant financial disclosures.
SOURCE: Stefater MA et al. ENDO 2018, Abstract OR 12-6.
CHICAGO – Of medical and surgical tactics to tackle long-term weight loss, , and gene expression in the Roux limb may hold the key to the surgery’s efficacy, according to an ongoing study.
“We know that Roux-en-Y gastric bypass surgery is highly effective as not only a weight-loss therapy, but more and more we’re appreciating its role as a diabetes therapy as well,” said Margaret Stefater, MD, PhD, speaking in an interview at the annual meeting of the Endocrine Society.
The study, she said, was designed to learn more about the intestine’s contribution to the salubrious effect that Roux-en-Y surgery has on diabetes.
“We used microarray in order to characterize gene expression in the intestine” to gain a broad understanding of the processes that are altered after surgery, said Dr. Stefater, a pediatric endocrinology fellow at Boston Children’s Hospital. More specifically, though, the study looked at an individual’s changes in gene expression over time and correlated those changes with that patient’s clinical picture.
The data reported by Dr. Stefater and shared in a press conference, represent part of an ongoing longitudinal prospective study of 32 patients.
“The study aims to characterize gene expression for the first postoperative year,” and findings from the first 6 postoperative months of 19 patients were shared at the meeting, said Dr. Stefater. “This is the first look at our cohort.”
So far, she and her colleagues have compared gene expression using microarray at 1 month and 6 months post-surgery, comparing change across time and change from baseline data.
From hundreds of candidate genes, Dr. Stefater and her colleagues have developed a smaller gene list that, even in the first postoperative month, is predictive of changes in hemoglobin A1c levels over time. “Remarkably, the changes in a select list of genes out to 1 month is actually able to predict hemoglobin A1c levels out to 1 year,” she said. “This speaks to the fact that biological reprogramming in the intestine is somehow related to glycemic response in patients.
“We hope that by understanding these processes, we can home in on those processes that are most likely to be mechanistically responsible for these changes, and then to reverse-engineer this surgery to identify processes or targets which may be good places to start when we think about creating better, or nonsurgical, therapies for people who have obesity and diabetes,” said Dr. Stefater.
Dr. Stefater reported no relevant financial disclosures.
SOURCE: Stefater MA et al. ENDO 2018, Abstract OR 12-6.
REPORTING FROM ENDO 2018
Protocol helped identify hospitalized children at risk for VTE
SAN DIEGO – Following simple institutional care guidelines helped clinicians identify pediatric patients at moderate-to-severe risk of venous thromboembolism (VTE), results from a single-center study showed.
“Hospital-acquired VTE is on the rise in the pediatric population,” lead study author Emily Southard, MD, said at the biennial summit of the Thrombosis & Hemostasis Societies of North America. “This consists of a DVT or [pulmonary embolism] 48 hours or more after admission, or any time at the site of a central venous catheter.”
One published study found a 70% increased incidence in the pediatric population from 2001-2007 (Pediatrics 2009;124[4]:1001-8). More than half of the children in that study (63%) had at least one coexisting complex medical condition, with malignancy being the most common.

Hospital-acquired VTE cases tend to harbor a number of complications, said Dr. Southard, who is a pediatric hematology/oncology fellow at Children’s Hospital Colorado, Aurora. For example, 15%-20% of patients with a DVT will have a pulmonary embolism (PE) as well, 26% of patients with upper or lower extremity DVT develop post-thrombotic syndrome, and 3% of patients with PE develop chronic pulmonary hypertension.
“Medical costs are also impacted,” she said. “The cost for a hospital-acquired VTE in pediatrics increased the length of stay by about 8 days and increased the cost of hospital admission by more than $27,000.”
Known risk factors for VTE in this patient population include ICU admission (Odds Ratio, 2.14), presence of a central venous catheter (OR, 2.12), mechanical ventilation (OR, 1.56), and prolonged admission (OR, 1.03 for each day).
Risk factors in pediatric trauma patients include ICU admission (OR, 6.25), transfusion of blood products (OR, 2.1), lower extremity fracture (OR, 1.8), and neurosurgery (OR, 2.13). She and her associates hypothesized that understanding the relative contributions of clinical, biological, and genetic risk factors for pediatric VTE would help appropriately risk-stratify patients and allow better prophylactic approaches.
In 2012, Children’s Hospital Colorado implemented a VTE risk assessment tool as part of a hospital-wide patient safety initiative. The assessment is triggered via an Epic Best Practice Advisory to complete in certain higher-risk patients, including ICU patients, hematology/oncology floor patients, any patients with a central line catheter, and those who are over age 12 and obese.
Clinicians also assess for risk factors such as significant infection, recent surgery, and personal or family history of thrombophilia. Next, they classify each patient’s risk of hospital-acquired VTE as high, moderate, or low risk.
In a pilot study, Dr. Southard and her associates set out to validate the accuracy of the institution’s VTE risk assessment tool since it was implemented in 2012. She presented findings from 215 hospital-acquired VTE cases in patients younger than age 18, compared with age-matched inpatient controls. Data from patients under 6 months of age is available after October 2016, coinciding with a change in definition of pediatric hospital-acquired VTE.
Most hospital-acquired VTE patients (77.2%) ranged in age from 1-17 years. The number of patients admitted for a trauma diagnosis was similar between VTE cases and controls (7.4% vs. 7.9%, respectively). However, compared with controls, a significantly greater number of VTE cases were immobile (41.8% vs. 10.3%, respectively), required ICU admission (86.4% vs. 26.5%), had a central venous catheter (80.4% vs. 10.9%), had a positive blood culture (16.7% vs. 1.9%), required surgery or a medical procedure (57.7% vs. 36.7%), and had a longer procedure time (a mean of 151 vs. 133 minutes).
The researchers also found that upon initial admission, 7.9% of VTE cases were identified as high risk and another 21.9% were identified as moderate risk, compared with 1.2% and 3.7% in the controls, respectively.
“Patients identified as moderate or high risk for VTE were generally more medically complex patients,” Dr. Southard said.
Future directions of this project include expanding the patient population that has a risk assessment performed.
Dr. Southard reported having no financial disclosures.
SOURCE: Southard E et al. THSNA 2018.
SAN DIEGO – Following simple institutional care guidelines helped clinicians identify pediatric patients at moderate-to-severe risk of venous thromboembolism (VTE), results from a single-center study showed.
“Hospital-acquired VTE is on the rise in the pediatric population,” lead study author Emily Southard, MD, said at the biennial summit of the Thrombosis & Hemostasis Societies of North America. “This consists of a DVT or [pulmonary embolism] 48 hours or more after admission, or any time at the site of a central venous catheter.”
One published study found a 70% increased incidence in the pediatric population from 2001-2007 (Pediatrics 2009;124[4]:1001-8). More than half of the children in that study (63%) had at least one coexisting complex medical condition, with malignancy being the most common.
Hospital-acquired VTE cases tend to harbor a number of complications, said Dr. Southard, who is a pediatric hematology/oncology fellow at Children’s Hospital Colorado, Aurora. For example, 15%-20% of patients with a DVT will have a pulmonary embolism (PE) as well, 26% of patients with upper or lower extremity DVT develop post-thrombotic syndrome, and 3% of patients with PE develop chronic pulmonary hypertension.
“Medical costs are also impacted,” she said. “The cost for a hospital-acquired VTE in pediatrics increased the length of stay by about 8 days and increased the cost of hospital admission by more than $27,000.”
Known risk factors for VTE in this patient population include ICU admission (Odds Ratio, 2.14), presence of a central venous catheter (OR, 2.12), mechanical ventilation (OR, 1.56), and prolonged admission (OR, 1.03 for each day).
Risk factors in pediatric trauma patients include ICU admission (OR, 6.25), transfusion of blood products (OR, 2.1), lower extremity fracture (OR, 1.8), and neurosurgery (OR, 2.13). She and her associates hypothesized that understanding the relative contributions of clinical, biological, and genetic risk factors for pediatric VTE would help appropriately risk-stratify patients and allow better prophylactic approaches.
In 2012, Children’s Hospital Colorado implemented a VTE risk assessment tool as part of a hospital-wide patient safety initiative. The assessment is triggered via an Epic Best Practice Advisory to complete in certain higher-risk patients, including ICU patients, hematology/oncology floor patients, any patients with a central line catheter, and those who are over age 12 and obese.
Clinicians also assess for risk factors such as significant infection, recent surgery, and personal or family history of thrombophilia. Next, they classify each patient’s risk of hospital-acquired VTE as high, moderate, or low risk.
In a pilot study, Dr. Southard and her associates set out to validate the accuracy of the institution’s VTE risk assessment tool since it was implemented in 2012. She presented findings from 215 hospital-acquired VTE cases in patients younger than age 18, compared with age-matched inpatient controls. Data from patients under 6 months of age is available after October 2016, coinciding with a change in definition of pediatric hospital-acquired VTE.
Most hospital-acquired VTE patients (77.2%) ranged in age from 1-17 years. The number of patients admitted for a trauma diagnosis was similar between VTE cases and controls (7.4% vs. 7.9%, respectively). However, compared with controls, a significantly greater number of VTE cases were immobile (41.8% vs. 10.3%, respectively), required ICU admission (86.4% vs. 26.5%), had a central venous catheter (80.4% vs. 10.9%), had a positive blood culture (16.7% vs. 1.9%), required surgery or a medical procedure (57.7% vs. 36.7%), and had a longer procedure time (a mean of 151 vs. 133 minutes).
The researchers also found that upon initial admission, 7.9% of VTE cases were identified as high risk and another 21.9% were identified as moderate risk, compared with 1.2% and 3.7% in the controls, respectively.
“Patients identified as moderate or high risk for VTE were generally more medically complex patients,” Dr. Southard said.
Future directions of this project include expanding the patient population that has a risk assessment performed.
Dr. Southard reported having no financial disclosures.
SOURCE: Southard E et al. THSNA 2018.
SAN DIEGO – Following simple institutional care guidelines helped clinicians identify pediatric patients at moderate-to-severe risk of venous thromboembolism (VTE), results from a single-center study showed.
“Hospital-acquired VTE is on the rise in the pediatric population,” lead study author Emily Southard, MD, said at the biennial summit of the Thrombosis & Hemostasis Societies of North America. “This consists of a DVT or [pulmonary embolism] 48 hours or more after admission, or any time at the site of a central venous catheter.”
One published study found a 70% increased incidence in the pediatric population from 2001-2007 (Pediatrics 2009;124[4]:1001-8). More than half of the children in that study (63%) had at least one coexisting complex medical condition, with malignancy being the most common.
Hospital-acquired VTE cases tend to harbor a number of complications, said Dr. Southard, who is a pediatric hematology/oncology fellow at Children’s Hospital Colorado, Aurora. For example, 15%-20% of patients with a DVT will have a pulmonary embolism (PE) as well, 26% of patients with upper or lower extremity DVT develop post-thrombotic syndrome, and 3% of patients with PE develop chronic pulmonary hypertension.
“Medical costs are also impacted,” she said. “The cost for a hospital-acquired VTE in pediatrics increased the length of stay by about 8 days and increased the cost of hospital admission by more than $27,000.”
Known risk factors for VTE in this patient population include ICU admission (Odds Ratio, 2.14), presence of a central venous catheter (OR, 2.12), mechanical ventilation (OR, 1.56), and prolonged admission (OR, 1.03 for each day).
Risk factors in pediatric trauma patients include ICU admission (OR, 6.25), transfusion of blood products (OR, 2.1), lower extremity fracture (OR, 1.8), and neurosurgery (OR, 2.13). She and her associates hypothesized that understanding the relative contributions of clinical, biological, and genetic risk factors for pediatric VTE would help appropriately risk-stratify patients and allow better prophylactic approaches.
In 2012, Children’s Hospital Colorado implemented a VTE risk assessment tool as part of a hospital-wide patient safety initiative. The assessment is triggered via an Epic Best Practice Advisory to complete in certain higher-risk patients, including ICU patients, hematology/oncology floor patients, any patients with a central line catheter, and those who are over age 12 and obese.
Clinicians also assess for risk factors such as significant infection, recent surgery, and personal or family history of thrombophilia. Next, they classify each patient’s risk of hospital-acquired VTE as high, moderate, or low risk.
In a pilot study, Dr. Southard and her associates set out to validate the accuracy of the institution’s VTE risk assessment tool since it was implemented in 2012. She presented findings from 215 hospital-acquired VTE cases in patients younger than age 18, compared with age-matched inpatient controls. Data from patients under 6 months of age is available after October 2016, coinciding with a change in definition of pediatric hospital-acquired VTE.
Most hospital-acquired VTE patients (77.2%) ranged in age from 1-17 years. The number of patients admitted for a trauma diagnosis was similar between VTE cases and controls (7.4% vs. 7.9%, respectively). However, compared with controls, a significantly greater number of VTE cases were immobile (41.8% vs. 10.3%, respectively), required ICU admission (86.4% vs. 26.5%), had a central venous catheter (80.4% vs. 10.9%), had a positive blood culture (16.7% vs. 1.9%), required surgery or a medical procedure (57.7% vs. 36.7%), and had a longer procedure time (a mean of 151 vs. 133 minutes).
The researchers also found that upon initial admission, 7.9% of VTE cases were identified as high risk and another 21.9% were identified as moderate risk, compared with 1.2% and 3.7% in the controls, respectively.
“Patients identified as moderate or high risk for VTE were generally more medically complex patients,” Dr. Southard said.
Future directions of this project include expanding the patient population that has a risk assessment performed.
Dr. Southard reported having no financial disclosures.
SOURCE: Southard E et al. THSNA 2018.
REPORTING FROM THSNA 2018
Key clinical point:
Major finding: A significantly greater number of VTE patients were immobile (41.8% vs. 10.3%, respectively), required ICU admission (86.4% vs. 26.5%), and had a central venous catheter (80.4% vs. 10.9%), compared with controls.
Study details: A retrospective analysis of 215 hospital-acquired VTE cases in patients younger than age 18.
Disclosures: Dr. Southard reported having no financial disclosures.
Source: Southard E et al. THSNA 2018.
Certifications, training to increase addiction medicine specialists
Two new workforce developments aim to increase the number of addiction medicine specialists and provide new training opportunities in the subspecialty.
The American Board of Medical Specialties (ABMS) recently certified its first formal wave of addiction medicine physicians, adding 1,200 specialists to the field. Addiction medicine was first recognized as a subspecialty by ABMS in 2015, followed by the first certification exam in 2017.

The two developments “will change the landscape in substance use prevention, early intervention, and in addiction treatment and management,” said Lon R. Hays, MD, president of The Addiction Medicine Foundation, in Chevy Chase, Md., and director of the addiction medicine fellowship program at the University of Kentucky, Lexington.
“Many more trained physicians will be available to address the opioid crisis and other addictions,” Dr. Hays said in a statement. “They will also be able to help prevent and intervene early with unhealthy substance use in all its forms. For the first time, when aspiring physicians consider a career path, they will now have as an available choice an addiction medicine specialty that meets the highest standards of medicine.”

“When the American Board of Medical Specialties welcomed addiction medicine as its newest subspecialty, it in a lot of ways, legitimized our discipline,” Dr. Brennan said in an interview. “The American Board of Medical Specialties really represents the ‘House of Medicine.’ Being able to enter into that, it gives us a measure of credibility in the eyes of the public, and it basically codifies that these physicians who have passed this board exam have achieved a level of competency and knowledge that makes them trustworthy and safe to provide care to folks suffering from addiction.”
While the 1,200 additional addiction medicine specialists are an improvement, many more are needed, Dr. Brennan said, adding that he is optimistic that the new addiction medicine training opportunities provided by ACGME will help achieve higher numbers.
“For addiction medicine, we’ve had fellowships for about 10 years, but the funding for those fellowships was really challenging,” Dr. Brennan said. “Once you get ACGME-accredited, it gives you the ability to partake of [Centers for Medicare & Medicaid Services] funding that funds most of the graduate medical education residency fellowship spots in the United States. ACGME is the gold standard. I think that makes us much more potentially attractive for graduating physicians who are finishing their residencies.”
The certification of new addiction specialists is welcome news, particularly in the midst of the current epidemic, added Clif Knight, MD, senior vice president for education for the American Academy of Family Physicians.
“This is really good news [especially considering], the difficulty that the country is having with so much addiction – of course opioids are in the forefront – but there are so many different types of addiction,” he said in an interview. “This is good news that the certification is available and that physicians are pursuing obtaining additional expertise and recognition in their ability to treat addictions.”

Dr. Knight expressed appreciation that the ACGME training opportunities in addiction medicine are open to doctors of various specialties, he said.
“A lot of times subspecialty fellowships are only available to one specialty,” said Dr. Knight. “In this case, it looks like they’ve been very deliberate to make this available to multiple specialties. I think that really sends an important message that this is not just one specialty that is focused on [addiction medicine], but really should be something that all specialties are engaged and involved in.”
*This article was updated on 4/2/2018.
Two new workforce developments aim to increase the number of addiction medicine specialists and provide new training opportunities in the subspecialty.
The American Board of Medical Specialties (ABMS) recently certified its first formal wave of addiction medicine physicians, adding 1,200 specialists to the field. Addiction medicine was first recognized as a subspecialty by ABMS in 2015, followed by the first certification exam in 2017.
The two developments “will change the landscape in substance use prevention, early intervention, and in addiction treatment and management,” said Lon R. Hays, MD, president of The Addiction Medicine Foundation, in Chevy Chase, Md., and director of the addiction medicine fellowship program at the University of Kentucky, Lexington.
“Many more trained physicians will be available to address the opioid crisis and other addictions,” Dr. Hays said in a statement. “They will also be able to help prevent and intervene early with unhealthy substance use in all its forms. For the first time, when aspiring physicians consider a career path, they will now have as an available choice an addiction medicine specialty that meets the highest standards of medicine.”
“When the American Board of Medical Specialties welcomed addiction medicine as its newest subspecialty, it in a lot of ways, legitimized our discipline,” Dr. Brennan said in an interview. “The American Board of Medical Specialties really represents the ‘House of Medicine.’ Being able to enter into that, it gives us a measure of credibility in the eyes of the public, and it basically codifies that these physicians who have passed this board exam have achieved a level of competency and knowledge that makes them trustworthy and safe to provide care to folks suffering from addiction.”
While the 1,200 additional addiction medicine specialists are an improvement, many more are needed, Dr. Brennan said, adding that he is optimistic that the new addiction medicine training opportunities provided by ACGME will help achieve higher numbers.
“For addiction medicine, we’ve had fellowships for about 10 years, but the funding for those fellowships was really challenging,” Dr. Brennan said. “Once you get ACGME-accredited, it gives you the ability to partake of [Centers for Medicare & Medicaid Services] funding that funds most of the graduate medical education residency fellowship spots in the United States. ACGME is the gold standard. I think that makes us much more potentially attractive for graduating physicians who are finishing their residencies.”
The certification of new addiction specialists is welcome news, particularly in the midst of the current epidemic, added Clif Knight, MD, senior vice president for education for the American Academy of Family Physicians.
“This is really good news [especially considering], the difficulty that the country is having with so much addiction – of course opioids are in the forefront – but there are so many different types of addiction,” he said in an interview. “This is good news that the certification is available and that physicians are pursuing obtaining additional expertise and recognition in their ability to treat addictions.”
Dr. Knight expressed appreciation that the ACGME training opportunities in addiction medicine are open to doctors of various specialties, he said.
“A lot of times subspecialty fellowships are only available to one specialty,” said Dr. Knight. “In this case, it looks like they’ve been very deliberate to make this available to multiple specialties. I think that really sends an important message that this is not just one specialty that is focused on [addiction medicine], but really should be something that all specialties are engaged and involved in.”
*This article was updated on 4/2/2018.
Two new workforce developments aim to increase the number of addiction medicine specialists and provide new training opportunities in the subspecialty.
The American Board of Medical Specialties (ABMS) recently certified its first formal wave of addiction medicine physicians, adding 1,200 specialists to the field. Addiction medicine was first recognized as a subspecialty by ABMS in 2015, followed by the first certification exam in 2017.
The two developments “will change the landscape in substance use prevention, early intervention, and in addiction treatment and management,” said Lon R. Hays, MD, president of The Addiction Medicine Foundation, in Chevy Chase, Md., and director of the addiction medicine fellowship program at the University of Kentucky, Lexington.
“Many more trained physicians will be available to address the opioid crisis and other addictions,” Dr. Hays said in a statement. “They will also be able to help prevent and intervene early with unhealthy substance use in all its forms. For the first time, when aspiring physicians consider a career path, they will now have as an available choice an addiction medicine specialty that meets the highest standards of medicine.”
“When the American Board of Medical Specialties welcomed addiction medicine as its newest subspecialty, it in a lot of ways, legitimized our discipline,” Dr. Brennan said in an interview. “The American Board of Medical Specialties really represents the ‘House of Medicine.’ Being able to enter into that, it gives us a measure of credibility in the eyes of the public, and it basically codifies that these physicians who have passed this board exam have achieved a level of competency and knowledge that makes them trustworthy and safe to provide care to folks suffering from addiction.”
While the 1,200 additional addiction medicine specialists are an improvement, many more are needed, Dr. Brennan said, adding that he is optimistic that the new addiction medicine training opportunities provided by ACGME will help achieve higher numbers.
“For addiction medicine, we’ve had fellowships for about 10 years, but the funding for those fellowships was really challenging,” Dr. Brennan said. “Once you get ACGME-accredited, it gives you the ability to partake of [Centers for Medicare & Medicaid Services] funding that funds most of the graduate medical education residency fellowship spots in the United States. ACGME is the gold standard. I think that makes us much more potentially attractive for graduating physicians who are finishing their residencies.”
The certification of new addiction specialists is welcome news, particularly in the midst of the current epidemic, added Clif Knight, MD, senior vice president for education for the American Academy of Family Physicians.
“This is really good news [especially considering], the difficulty that the country is having with so much addiction – of course opioids are in the forefront – but there are so many different types of addiction,” he said in an interview. “This is good news that the certification is available and that physicians are pursuing obtaining additional expertise and recognition in their ability to treat addictions.”
Dr. Knight expressed appreciation that the ACGME training opportunities in addiction medicine are open to doctors of various specialties, he said.
“A lot of times subspecialty fellowships are only available to one specialty,” said Dr. Knight. “In this case, it looks like they’ve been very deliberate to make this available to multiple specialties. I think that really sends an important message that this is not just one specialty that is focused on [addiction medicine], but really should be something that all specialties are engaged and involved in.”
*This article was updated on 4/2/2018.