User login
New Weapon Against Malaria
Malaria is a major challenge in more than 100 countries. In 2015, nearly half the countries in the world had ongoing malaria transmission, according to Dr. Eileen Villasante, head of the Malaria Department at the Naval Medical Research Center (NMRC). But NMRC researchers may have found a new way to meet that challenge. They identified a novel highly protective malaria antigen, Plasmodium yoelii E140.
E140 is found in multiple stages of the life cycle of the malaria parasite, including sporozoites, liver stages, and blood stages, Villasante said. The researchers found that E140 induced up to 100% sterile protection, persisting for at least 3 months. They are now at work on a vaccine with the antigen.
Malaria is a major challenge in more than 100 countries. In 2015, nearly half the countries in the world had ongoing malaria transmission, according to Dr. Eileen Villasante, head of the Malaria Department at the Naval Medical Research Center (NMRC). But NMRC researchers may have found a new way to meet that challenge. They identified a novel highly protective malaria antigen, Plasmodium yoelii E140.
E140 is found in multiple stages of the life cycle of the malaria parasite, including sporozoites, liver stages, and blood stages, Villasante said. The researchers found that E140 induced up to 100% sterile protection, persisting for at least 3 months. They are now at work on a vaccine with the antigen.
Malaria is a major challenge in more than 100 countries. In 2015, nearly half the countries in the world had ongoing malaria transmission, according to Dr. Eileen Villasante, head of the Malaria Department at the Naval Medical Research Center (NMRC). But NMRC researchers may have found a new way to meet that challenge. They identified a novel highly protective malaria antigen, Plasmodium yoelii E140.
E140 is found in multiple stages of the life cycle of the malaria parasite, including sporozoites, liver stages, and blood stages, Villasante said. The researchers found that E140 induced up to 100% sterile protection, persisting for at least 3 months. They are now at work on a vaccine with the antigen.
Enzyme may be target for MM treatment
The enzyme ADAR1 may be a therapeutic target for multiple myeloma (MM), according to research published in Nature Communications.
Investigators found that high ADAR1 levels correlate with reduced survival rates in MM patients.
The team also discovered that blocking ADAR1 reduced regeneration of high-risk MM in serially transplantable patient-derived xenografts.
The investigators believe that JAK2 inhibitors could be used to dampen ADAR1 activity and ultimately prevent progression or relapse in patients with MM.
“Despite new therapies, it’s virtually inevitable that a patient with multiple myeloma will experience relapse of the disease at some point,” said study author Catriona Jamieson, MD, PhD, of University of California, San Diego, in La Jolla.
“That’s why it’s exciting that this discovery may allow us to detect the disease earlier and address the root cause.”
Dr Jamieson and her colleagues knew that ADAR1 is normally expressed during fetal development to help blood cells form. The enzyme edits the sequence of RNA and is known to promote cancer progression and resistance to therapy.
With previous work, Dr Jamieson’s team described ADAR1’s contributions to chronic myeloid leukemia (CML). The enzyme’s RNA-editing activity boosts leukemic stem cells, giving rise to CML, increasing disease recurrence, and allowing CML to resist treatment.
For their current study, Dr Jamieson and her colleagues investigated ADAR1’s role in MM, first analyzing a database of nearly 800 MM patient samples.
The investigators found that patients with low ADAR1 levels in their tumor cells survived longer than patients with high ADAR1 levels.
While more than 90% of patients with low ADAR1 levels survived longer than 2 years after their initial diagnosis, fewer than 70% of patients with high ADAR1 levels did the same.
The investigators also created a humanized mouse model of MM and found that silencing the ADAR1 gene reduced the engraftment of MM.
“This is a difficult disease to model in animals; there isn’t a single gene we can manipulate to mimic multiple myeloma,” said study author Leslie A. Crews, PhD, of University of California, San Diego.
“This study is important, in part because we now have a new xenograft model that will, for the first time, allow us to apply new biomarkers to better predict disease progression and test new therapeutics.”
To advance their findings from this study, the investigators are exploring ways to leverage ADAR1 to detect MM progression as early as possible.
They are also testing inhibitors of JAK2, a molecule that influences ADAR1 activity, for their ability to eliminate cancer stem cells in MM models.
“Several major advances in recent years have been good news for multiple myeloma patients, but those new drugs only target terminally differentiated cancer cells and, thus, can only reduce the bulk of the tumor,” Dr Jamieson said.
“They don’t get to the root cause of disease development, progression, and relapse—cancer stem cells—the way inhibiting ADAR1 does. I like to call our approach ‘precision regenerative medicine.’” ![]()
The enzyme ADAR1 may be a therapeutic target for multiple myeloma (MM), according to research published in Nature Communications.
Investigators found that high ADAR1 levels correlate with reduced survival rates in MM patients.
The team also discovered that blocking ADAR1 reduced regeneration of high-risk MM in serially transplantable patient-derived xenografts.
The investigators believe that JAK2 inhibitors could be used to dampen ADAR1 activity and ultimately prevent progression or relapse in patients with MM.
“Despite new therapies, it’s virtually inevitable that a patient with multiple myeloma will experience relapse of the disease at some point,” said study author Catriona Jamieson, MD, PhD, of University of California, San Diego, in La Jolla.
“That’s why it’s exciting that this discovery may allow us to detect the disease earlier and address the root cause.”
Dr Jamieson and her colleagues knew that ADAR1 is normally expressed during fetal development to help blood cells form. The enzyme edits the sequence of RNA and is known to promote cancer progression and resistance to therapy.
With previous work, Dr Jamieson’s team described ADAR1’s contributions to chronic myeloid leukemia (CML). The enzyme’s RNA-editing activity boosts leukemic stem cells, giving rise to CML, increasing disease recurrence, and allowing CML to resist treatment.
For their current study, Dr Jamieson and her colleagues investigated ADAR1’s role in MM, first analyzing a database of nearly 800 MM patient samples.
The investigators found that patients with low ADAR1 levels in their tumor cells survived longer than patients with high ADAR1 levels.
While more than 90% of patients with low ADAR1 levels survived longer than 2 years after their initial diagnosis, fewer than 70% of patients with high ADAR1 levels did the same.
The investigators also created a humanized mouse model of MM and found that silencing the ADAR1 gene reduced the engraftment of MM.
“This is a difficult disease to model in animals; there isn’t a single gene we can manipulate to mimic multiple myeloma,” said study author Leslie A. Crews, PhD, of University of California, San Diego.
“This study is important, in part because we now have a new xenograft model that will, for the first time, allow us to apply new biomarkers to better predict disease progression and test new therapeutics.”
To advance their findings from this study, the investigators are exploring ways to leverage ADAR1 to detect MM progression as early as possible.
They are also testing inhibitors of JAK2, a molecule that influences ADAR1 activity, for their ability to eliminate cancer stem cells in MM models.
“Several major advances in recent years have been good news for multiple myeloma patients, but those new drugs only target terminally differentiated cancer cells and, thus, can only reduce the bulk of the tumor,” Dr Jamieson said.
“They don’t get to the root cause of disease development, progression, and relapse—cancer stem cells—the way inhibiting ADAR1 does. I like to call our approach ‘precision regenerative medicine.’” ![]()
The enzyme ADAR1 may be a therapeutic target for multiple myeloma (MM), according to research published in Nature Communications.
Investigators found that high ADAR1 levels correlate with reduced survival rates in MM patients.
The team also discovered that blocking ADAR1 reduced regeneration of high-risk MM in serially transplantable patient-derived xenografts.
The investigators believe that JAK2 inhibitors could be used to dampen ADAR1 activity and ultimately prevent progression or relapse in patients with MM.
“Despite new therapies, it’s virtually inevitable that a patient with multiple myeloma will experience relapse of the disease at some point,” said study author Catriona Jamieson, MD, PhD, of University of California, San Diego, in La Jolla.
“That’s why it’s exciting that this discovery may allow us to detect the disease earlier and address the root cause.”
Dr Jamieson and her colleagues knew that ADAR1 is normally expressed during fetal development to help blood cells form. The enzyme edits the sequence of RNA and is known to promote cancer progression and resistance to therapy.
With previous work, Dr Jamieson’s team described ADAR1’s contributions to chronic myeloid leukemia (CML). The enzyme’s RNA-editing activity boosts leukemic stem cells, giving rise to CML, increasing disease recurrence, and allowing CML to resist treatment.
For their current study, Dr Jamieson and her colleagues investigated ADAR1’s role in MM, first analyzing a database of nearly 800 MM patient samples.
The investigators found that patients with low ADAR1 levels in their tumor cells survived longer than patients with high ADAR1 levels.
While more than 90% of patients with low ADAR1 levels survived longer than 2 years after their initial diagnosis, fewer than 70% of patients with high ADAR1 levels did the same.
The investigators also created a humanized mouse model of MM and found that silencing the ADAR1 gene reduced the engraftment of MM.
“This is a difficult disease to model in animals; there isn’t a single gene we can manipulate to mimic multiple myeloma,” said study author Leslie A. Crews, PhD, of University of California, San Diego.
“This study is important, in part because we now have a new xenograft model that will, for the first time, allow us to apply new biomarkers to better predict disease progression and test new therapeutics.”
To advance their findings from this study, the investigators are exploring ways to leverage ADAR1 to detect MM progression as early as possible.
They are also testing inhibitors of JAK2, a molecule that influences ADAR1 activity, for their ability to eliminate cancer stem cells in MM models.
“Several major advances in recent years have been good news for multiple myeloma patients, but those new drugs only target terminally differentiated cancer cells and, thus, can only reduce the bulk of the tumor,” Dr Jamieson said.
“They don’t get to the root cause of disease development, progression, and relapse—cancer stem cells—the way inhibiting ADAR1 does. I like to call our approach ‘precision regenerative medicine.’” ![]()
Health Canada approves midostaurin for AML

Health Canada has approved use of the multi-targeted kinase inhibitor midostaurin (Rydapt™).
This makes midostaurin the first targeted therapy approved to treat FLT3-mutated acute myeloid leukemia (AML) in Canada.
Midostaurin is approved for use with standard cytarabine and daunorubicin induction and cytarabine consolidation for the treatment of adults with newly diagnosed FLT3-mutated AML.
Health Canada’s approval of midostaurin is based on results from the phase 3 RATIFY trial, which were published in NEJM in August.
In RATIFY, researchers compared midostaurin plus standard chemotherapy to placebo plus standard chemotherapy in 717 adults younger than age 60 who had FLT3-mutated AML.
The median overall survival was significantly longer in the midostaurin arm than the placebo arm—74.7 months and 25.6 months, respectively (hazard ratio=0.77, P=0.016).
And the median event-free survival was significantly longer in the midostaurin arm than the placebo arm—8.2 months and 3.0 months, respectively (hazard ratio=0.78, P=0.004).
The most frequent adverse events (AEs) in the midostaurin arm (occurring in at least 20% of patients) were febrile neutropenia, nausea, vomiting, mucositis, headache, musculoskeletal pain, petechiae, device-related infection, epistaxis, hyperglycemia, and upper respiratory tract infection.
The most frequent grade 3/4 AEs (occurring in at least 10% of patients) were febrile neutropenia, device-related infection, and mucositis.
Nine percent of patients in the midostaurin arm stopped treatment due to AEs, as did 6% in the placebo arm. ![]()
Health Canada has approved use of the multi-targeted kinase inhibitor midostaurin (Rydapt™).
This makes midostaurin the first targeted therapy approved to treat FLT3-mutated acute myeloid leukemia (AML) in Canada.
Midostaurin is approved for use with standard cytarabine and daunorubicin induction and cytarabine consolidation for the treatment of adults with newly diagnosed FLT3-mutated AML.
Health Canada’s approval of midostaurin is based on results from the phase 3 RATIFY trial, which were published in NEJM in August.
In RATIFY, researchers compared midostaurin plus standard chemotherapy to placebo plus standard chemotherapy in 717 adults younger than age 60 who had FLT3-mutated AML.
The median overall survival was significantly longer in the midostaurin arm than the placebo arm—74.7 months and 25.6 months, respectively (hazard ratio=0.77, P=0.016).
And the median event-free survival was significantly longer in the midostaurin arm than the placebo arm—8.2 months and 3.0 months, respectively (hazard ratio=0.78, P=0.004).
The most frequent adverse events (AEs) in the midostaurin arm (occurring in at least 20% of patients) were febrile neutropenia, nausea, vomiting, mucositis, headache, musculoskeletal pain, petechiae, device-related infection, epistaxis, hyperglycemia, and upper respiratory tract infection.
The most frequent grade 3/4 AEs (occurring in at least 10% of patients) were febrile neutropenia, device-related infection, and mucositis.
Nine percent of patients in the midostaurin arm stopped treatment due to AEs, as did 6% in the placebo arm. ![]()
Health Canada has approved use of the multi-targeted kinase inhibitor midostaurin (Rydapt™).
This makes midostaurin the first targeted therapy approved to treat FLT3-mutated acute myeloid leukemia (AML) in Canada.
Midostaurin is approved for use with standard cytarabine and daunorubicin induction and cytarabine consolidation for the treatment of adults with newly diagnosed FLT3-mutated AML.
Health Canada’s approval of midostaurin is based on results from the phase 3 RATIFY trial, which were published in NEJM in August.
In RATIFY, researchers compared midostaurin plus standard chemotherapy to placebo plus standard chemotherapy in 717 adults younger than age 60 who had FLT3-mutated AML.
The median overall survival was significantly longer in the midostaurin arm than the placebo arm—74.7 months and 25.6 months, respectively (hazard ratio=0.77, P=0.016).
And the median event-free survival was significantly longer in the midostaurin arm than the placebo arm—8.2 months and 3.0 months, respectively (hazard ratio=0.78, P=0.004).
The most frequent adverse events (AEs) in the midostaurin arm (occurring in at least 20% of patients) were febrile neutropenia, nausea, vomiting, mucositis, headache, musculoskeletal pain, petechiae, device-related infection, epistaxis, hyperglycemia, and upper respiratory tract infection.
The most frequent grade 3/4 AEs (occurring in at least 10% of patients) were febrile neutropenia, device-related infection, and mucositis.
Nine percent of patients in the midostaurin arm stopped treatment due to AEs, as did 6% in the placebo arm. ![]()
Companies launch generic busulfan in US

Two companies have announced the US launch of a generic busulfan product, Myleran Injection.
Mylan NV and Aspen have partnered to develop Myleran (busulfan) Injection, 60 mg/10 mL (6 mg/mL) Single-dose Vial, a generic version of Otsuka Pharmaceutical’s Busulfex® Injection.
The US Food and Drug Administration approved Myleran Injection for use in combination with cyclophosphamide as a conditioning regimen prior to allogeneic hematopoietic stem cell transplant in patients with chronic myeloid leukemia. ![]()
Two companies have announced the US launch of a generic busulfan product, Myleran Injection.
Mylan NV and Aspen have partnered to develop Myleran (busulfan) Injection, 60 mg/10 mL (6 mg/mL) Single-dose Vial, a generic version of Otsuka Pharmaceutical’s Busulfex® Injection.
The US Food and Drug Administration approved Myleran Injection for use in combination with cyclophosphamide as a conditioning regimen prior to allogeneic hematopoietic stem cell transplant in patients with chronic myeloid leukemia. ![]()
Two companies have announced the US launch of a generic busulfan product, Myleran Injection.
Mylan NV and Aspen have partnered to develop Myleran (busulfan) Injection, 60 mg/10 mL (6 mg/mL) Single-dose Vial, a generic version of Otsuka Pharmaceutical’s Busulfex® Injection.
The US Food and Drug Administration approved Myleran Injection for use in combination with cyclophosphamide as a conditioning regimen prior to allogeneic hematopoietic stem cell transplant in patients with chronic myeloid leukemia. ![]()
A (Not So) Humerus Situation
ANSWER
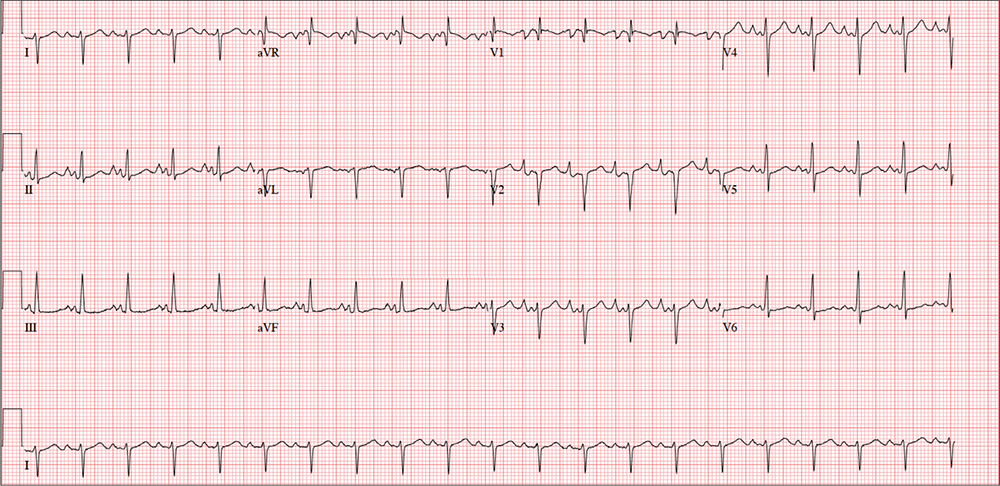
This ECG shows evidence of sinus tachycardia with biatrial enlargement, right-axis deviation, right ventricular hypertrophy, and poor R-wave progression consistent with a septal MI.
Sinus tachycardia is signified by an atrial rate > 100 beats/min. The markedly notched P waves in leads I and II and biphasic P waves in lead V1 suggest biatrial enlargement. Right-axis deviation is diagnosed based on the R axis of 119°, and right ventricular hypertrophy is indicated by the right-axis deviation, a QR pattern in lead V1, and an R wave ≥ 5 mm in lead aVR. All of the above are findings seen with a history of pulmonary hypertension.
Poor R-wave progression in leads V1 to V4 suggests a septal MI of indeterminate age; however, there is no history of previous infarction.
The patient was diagnosed with a complex fracture of the left humerus and referred to orthopedics for repair. At his request, a pulmonary medicine consultation was ordered so that he could establish care in this facility.
ANSWER
This ECG shows evidence of sinus tachycardia with biatrial enlargement, right-axis deviation, right ventricular hypertrophy, and poor R-wave progression consistent with a septal MI.
Sinus tachycardia is signified by an atrial rate > 100 beats/min. The markedly notched P waves in leads I and II and biphasic P waves in lead V1 suggest biatrial enlargement. Right-axis deviation is diagnosed based on the R axis of 119°, and right ventricular hypertrophy is indicated by the right-axis deviation, a QR pattern in lead V1, and an R wave ≥ 5 mm in lead aVR. All of the above are findings seen with a history of pulmonary hypertension.
Poor R-wave progression in leads V1 to V4 suggests a septal MI of indeterminate age; however, there is no history of previous infarction.
The patient was diagnosed with a complex fracture of the left humerus and referred to orthopedics for repair. At his request, a pulmonary medicine consultation was ordered so that he could establish care in this facility.
ANSWER
This ECG shows evidence of sinus tachycardia with biatrial enlargement, right-axis deviation, right ventricular hypertrophy, and poor R-wave progression consistent with a septal MI.
Sinus tachycardia is signified by an atrial rate > 100 beats/min. The markedly notched P waves in leads I and II and biphasic P waves in lead V1 suggest biatrial enlargement. Right-axis deviation is diagnosed based on the R axis of 119°, and right ventricular hypertrophy is indicated by the right-axis deviation, a QR pattern in lead V1, and an R wave ≥ 5 mm in lead aVR. All of the above are findings seen with a history of pulmonary hypertension.
Poor R-wave progression in leads V1 to V4 suggests a septal MI of indeterminate age; however, there is no history of previous infarction.
The patient was diagnosed with a complex fracture of the left humerus and referred to orthopedics for repair. At his request, a pulmonary medicine consultation was ordered so that he could establish care in this facility.
While clearing leaves from his gutters, a 44-year-old man falls approximately 12 feet, hitting his left upper arm on the concrete curb. His neighbor sees him fall and comes quickly to his aid; after stabilizing the arm, the neighbor drives him to your facility. The patient reports feeling and hearing a snap and says he suspects he has fractured his humerus.
Medical history is remarkable for idiopathic pulmonary arterial hypertension, diagnosed at age 30. Symptoms at that time included dyspnea with exercise (and later, at rest), fatigue, palpitations, near-syncope, and lower extremity swelling. He has a pulmonologist at another facility but admits he’s not compliant with medications or appointments because he doesn’t like his doctor. He hasn’t had an exacerbation in the past two months.
Surgical history includes a tonsillectomy as a child.
His current medications include multiple inhalers (he can’t remember any names) and tadalafil, which he takes daily. He’s supposed to take warfarin but says he hasn’t done so for at least six months. He has no known drug allergies.
Two of his four siblings have pulmonary hypertension; the family had genetic testing performed to rule out a gene mutation. His father had cardiac sarcoidosis and died of an arrhythmia. His mother is in good health.
The patient, a welder, is on medical disability. He initially denies smoking, then admits to having two or three cigarettes a day. He drinks three beers a day and “a few more” on the weekends. He denies current illicit drug use but details heavy methamphetamine use in his early 20s.
Review of systems is noncontributory; he says he’s in a lot of pain and “does not want to go into my life story.” He denies shortness of breath or chest pain.
Vital signs include a blood pressure of 162/98 mm Hg; pulse, 120 beats/min; respiratory rate, 18 breaths/min-1; and temperature, 96.4°F. His weight is 214 lb and his height, 74 in.
Physical exam reveals an anxious male in apparent pain, holding his left arm against his side with his right hand. A cursory HEENT exam reveals no obvious trauma. His lungs have scattered crackles in all lung fields. The cardiac exam reveals a regular rhythm at a rate of 120 beats/min. There are no murmurs or rubs. The abdomen is soft and nontender.
The left upper arm has multiple abrasions, and there is point tenderness and swelling in the mid-portion of the humerus. There is no visual evidence of a compound fracture, but the bone appears displaced. Aside from additional abrasions on the left hip and lower leg, the remainder of the physical exam is unremarkable.
Given the history of pulmonary hypertension and findings of tachycardia on physical exam, an ECG is obtained prior to sending the patient to radiology. The ECG reveals a ventricular rate of 122 beats/min; PR interval, 164 ms; QRS duration, 68 ms; QT/QTc interval, 310/441 ms; P axis, 15°; R axis, 119°; and T axis, 37°. What is your interpretation?
Mammography screening’s benefits for breast cancer mortality questioned
Twenty-four years’ worth of data from the Netherlands’ mammography screening program suggest that it has achieved only a marginal impact on breast cancer mortality, according to a paper published online Dec. 5 in the British Medical Journal.
Researchers used data on the stage-specific incidence of breast cancer in the Netherlands during 1989-2012 and estimated the mortality effect of a nationwide, population-based mammography breast cancer screening program, which was introduced in 1988 and targeted women aged 50-75 years.
While there were considerable increases in the incidence of in situ tumors and stage 1 cancers during 1989-2012, the incidence of stages 2-4 cancers was relatively stable over this period. In women aged more than 50 years, who would have been eligible to participate in the screening program, the incidence of stages 2-4 cancers decreased by a nonsignificant 0.16%; from 168/100,000 women in 1989 to 166/100,000 in 2012.
Even when researchers limited their analysis to the period from 1995 to 2012, when the screening program was fully operational and participation rates were around 80%, the incidence of stages 2-4 cancers remained steady. It was also stable in women younger than 50 years, who would not have been eligible for screening.
To estimate the effects of mammography screening on mortality, researchers assumed a scenario without efficient treatment of breast cancer, in which the mortality increase of 0.09% per year seen from 1967 to 1995 would persist until 2012. Under this scenario, they calculated that screening would have at least prevented the predicted 2% increase in breast cancer mortality over that period. Combined with the 0.16% nonsignificant reduction in stages 2-4 cancers over the 23 years of screening, this equated to a total mortality decrease of around 3%.
This reduction paled in comparison to the contributions made by improved treatment and patient management, which the authors suggested would be associated with a 28% reduction in mortality.
“The data on advanced breast cancer in the Netherlands indicate that the Dutch national mammography screening programme would have had little influence on the decreases in breast cancer mortality observed over the past 24 years,” wrote Philippe Autier, MD, and his colleagues from the International Prevention Research Institute in Lyon, France. “This conclusion accords with the mounting evidence that randomised trials have overestimated the ability of mammography screening to reduce the risk of deaths from breast cancer in the entire life period after first exposure to mammography screening.”
However, mammography screening also was associated with a sixfold increase in the incidence of in situ cancers among women aged 50-74 years, over the 23-year study period.
The incidence of stage 1 cancers tripled in woman aged 50-69 years, and increased 3.5-fold in those aged 70-74 years. In comparison, over the same period the rates of stage 1 cancers in women younger than 50 years or older than 75 years increased 1.3-fold.
This amounted to a 50% increase in in situ and stage 1 cancers diagnosed among women who were invited to screening, compared with those younger than 50 years. Even in a best-case scenario, the advent of digital mammography would mean 10,038 overdiagnosed cancers for 640 breast cancer deaths prevented by screening.
“Thus for 1 woman who would not die from breast cancer because of screening, about 16 women would be overdiagnosed with an in situ or a stage 1 cancer,” the authors wrote. “Hence, the advent of digital technologies has probably worsened the overdiagnosis problem without clear evidence for improvements in the ability of screening to curb the risk of breast cancer death.”
The study was partly supported by the International Prevention Research Institute. No conflicts of interest were declared.
SOURCE: Autier P et al. BMJ. 2017 Dec 5;359:j5224.
Twenty-four years’ worth of data from the Netherlands’ mammography screening program suggest that it has achieved only a marginal impact on breast cancer mortality, according to a paper published online Dec. 5 in the British Medical Journal.
Researchers used data on the stage-specific incidence of breast cancer in the Netherlands during 1989-2012 and estimated the mortality effect of a nationwide, population-based mammography breast cancer screening program, which was introduced in 1988 and targeted women aged 50-75 years.
While there were considerable increases in the incidence of in situ tumors and stage 1 cancers during 1989-2012, the incidence of stages 2-4 cancers was relatively stable over this period. In women aged more than 50 years, who would have been eligible to participate in the screening program, the incidence of stages 2-4 cancers decreased by a nonsignificant 0.16%; from 168/100,000 women in 1989 to 166/100,000 in 2012.
Even when researchers limited their analysis to the period from 1995 to 2012, when the screening program was fully operational and participation rates were around 80%, the incidence of stages 2-4 cancers remained steady. It was also stable in women younger than 50 years, who would not have been eligible for screening.
To estimate the effects of mammography screening on mortality, researchers assumed a scenario without efficient treatment of breast cancer, in which the mortality increase of 0.09% per year seen from 1967 to 1995 would persist until 2012. Under this scenario, they calculated that screening would have at least prevented the predicted 2% increase in breast cancer mortality over that period. Combined with the 0.16% nonsignificant reduction in stages 2-4 cancers over the 23 years of screening, this equated to a total mortality decrease of around 3%.
This reduction paled in comparison to the contributions made by improved treatment and patient management, which the authors suggested would be associated with a 28% reduction in mortality.
“The data on advanced breast cancer in the Netherlands indicate that the Dutch national mammography screening programme would have had little influence on the decreases in breast cancer mortality observed over the past 24 years,” wrote Philippe Autier, MD, and his colleagues from the International Prevention Research Institute in Lyon, France. “This conclusion accords with the mounting evidence that randomised trials have overestimated the ability of mammography screening to reduce the risk of deaths from breast cancer in the entire life period after first exposure to mammography screening.”
However, mammography screening also was associated with a sixfold increase in the incidence of in situ cancers among women aged 50-74 years, over the 23-year study period.
The incidence of stage 1 cancers tripled in woman aged 50-69 years, and increased 3.5-fold in those aged 70-74 years. In comparison, over the same period the rates of stage 1 cancers in women younger than 50 years or older than 75 years increased 1.3-fold.
This amounted to a 50% increase in in situ and stage 1 cancers diagnosed among women who were invited to screening, compared with those younger than 50 years. Even in a best-case scenario, the advent of digital mammography would mean 10,038 overdiagnosed cancers for 640 breast cancer deaths prevented by screening.
“Thus for 1 woman who would not die from breast cancer because of screening, about 16 women would be overdiagnosed with an in situ or a stage 1 cancer,” the authors wrote. “Hence, the advent of digital technologies has probably worsened the overdiagnosis problem without clear evidence for improvements in the ability of screening to curb the risk of breast cancer death.”
The study was partly supported by the International Prevention Research Institute. No conflicts of interest were declared.
SOURCE: Autier P et al. BMJ. 2017 Dec 5;359:j5224.
Twenty-four years’ worth of data from the Netherlands’ mammography screening program suggest that it has achieved only a marginal impact on breast cancer mortality, according to a paper published online Dec. 5 in the British Medical Journal.
Researchers used data on the stage-specific incidence of breast cancer in the Netherlands during 1989-2012 and estimated the mortality effect of a nationwide, population-based mammography breast cancer screening program, which was introduced in 1988 and targeted women aged 50-75 years.
While there were considerable increases in the incidence of in situ tumors and stage 1 cancers during 1989-2012, the incidence of stages 2-4 cancers was relatively stable over this period. In women aged more than 50 years, who would have been eligible to participate in the screening program, the incidence of stages 2-4 cancers decreased by a nonsignificant 0.16%; from 168/100,000 women in 1989 to 166/100,000 in 2012.
Even when researchers limited their analysis to the period from 1995 to 2012, when the screening program was fully operational and participation rates were around 80%, the incidence of stages 2-4 cancers remained steady. It was also stable in women younger than 50 years, who would not have been eligible for screening.
To estimate the effects of mammography screening on mortality, researchers assumed a scenario without efficient treatment of breast cancer, in which the mortality increase of 0.09% per year seen from 1967 to 1995 would persist until 2012. Under this scenario, they calculated that screening would have at least prevented the predicted 2% increase in breast cancer mortality over that period. Combined with the 0.16% nonsignificant reduction in stages 2-4 cancers over the 23 years of screening, this equated to a total mortality decrease of around 3%.
This reduction paled in comparison to the contributions made by improved treatment and patient management, which the authors suggested would be associated with a 28% reduction in mortality.
“The data on advanced breast cancer in the Netherlands indicate that the Dutch national mammography screening programme would have had little influence on the decreases in breast cancer mortality observed over the past 24 years,” wrote Philippe Autier, MD, and his colleagues from the International Prevention Research Institute in Lyon, France. “This conclusion accords with the mounting evidence that randomised trials have overestimated the ability of mammography screening to reduce the risk of deaths from breast cancer in the entire life period after first exposure to mammography screening.”
However, mammography screening also was associated with a sixfold increase in the incidence of in situ cancers among women aged 50-74 years, over the 23-year study period.
The incidence of stage 1 cancers tripled in woman aged 50-69 years, and increased 3.5-fold in those aged 70-74 years. In comparison, over the same period the rates of stage 1 cancers in women younger than 50 years or older than 75 years increased 1.3-fold.
This amounted to a 50% increase in in situ and stage 1 cancers diagnosed among women who were invited to screening, compared with those younger than 50 years. Even in a best-case scenario, the advent of digital mammography would mean 10,038 overdiagnosed cancers for 640 breast cancer deaths prevented by screening.
“Thus for 1 woman who would not die from breast cancer because of screening, about 16 women would be overdiagnosed with an in situ or a stage 1 cancer,” the authors wrote. “Hence, the advent of digital technologies has probably worsened the overdiagnosis problem without clear evidence for improvements in the ability of screening to curb the risk of breast cancer death.”
The study was partly supported by the International Prevention Research Institute. No conflicts of interest were declared.
SOURCE: Autier P et al. BMJ. 2017 Dec 5;359:j5224.
FROM BMJ
Key clinical point: Data from the Netherlands’ mammography screening program suggests it has only achieved a small, nonsignificant reduction in breast cancer mortality at a cost of a significant increase in overdiagnosis of stage 1 and in situ breast cancers.
Major finding: The incidence of stages 2-4 cancers decreased only by a nonsignificant 0.16% over the 23 years of mammography screening in the Netherlands.
Data source: Population-based study using 23 years worth of data from the Netherlands.
Disclosures: The study was partly supported by the International Prevention Research Institute. No conflicts of interest were declared.
Source: Autier P et al. BMJ. 2017 Dec 5;359:j5224.
Arthritis prevalence higher than previously thought, especially in adults under 65
The prevalence of arthritis in the United States is much higher than current estimates indicate, especially among adults under 65 years of age, a study showed.
The higher prevalence can be largely attributed to “the previous underestimate of arthritis in adults between 18-64 years of age,” according to S. Reza Jafarzadeh, PhD, and David T. Felson , MD, both of Boston University. Using a new surveillance model, they estimated that 91.2 million adults in the United States (36.8%) had arthritis in 2015, compared with a previously reported national estimate of 54.4 million adults (22.7%). Of these, 61.1 million were between 18 and 64 years of age (Arthritis Rheumatol. 2017 Nov 27. doi: 10.1002/art.40355).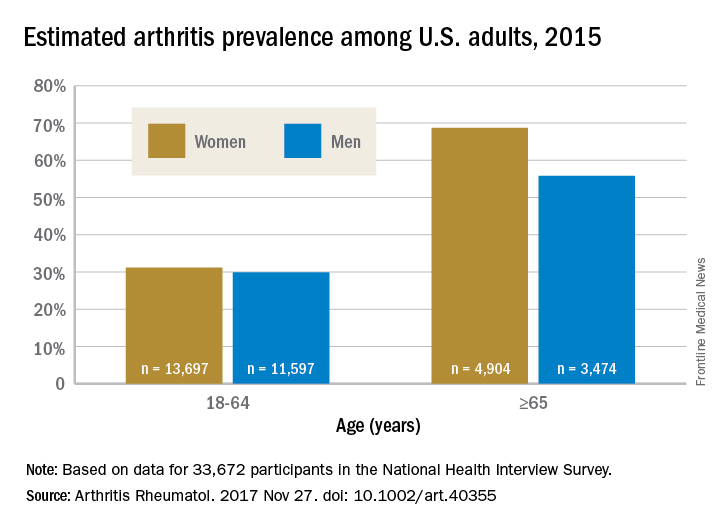
Arthritis prevalence was 29.9% in men aged 18-64 years (95% probability interval, 23.4-42.3), 31.2% in women aged 18-64 years (95% PI, 25.8-44.1), 55.8% in men aged 65 years and older (95% PI, 49.9-70.4), and 68.7% in women aged 65 years and older (95% PI, 62.1-79.9), the authors reported.
Among respondents aged 18-64 years, 19.3% of men and 16.7% of women reported that they had chronic joint symptoms but no physician-diagnosed arthritis. Among those 65 years of age or older, 15.7% of men and 13.5% of women responded that they had chronic joint symptoms without physician-diagnosed arthritis.
Current methodology for estimating arthritis prevalence is based on a single survey question asking whether a health care provider has ever told the patient that he or she has arthritis, a method that has previously been shown to have a sensitivity of 68.8% in adults 65 years of age and older and 52.5% for those aged 45-64 years, Dr. Jafarzadeh and Dr. Felson reported. “Such a low sensitivity, especially in a younger population, where almost half of true arthritis cases are missed, results in substantial misclassification and underestimation of prevalence and would have a detrimental effect for planning and needs assessment,” the authors wrote.
The two additional questions on joint pain, aching, and stiffness that the investigators included in the study captured “a substantial (i.e., 65%-80%) fraction of the population with arthritis, who are between 18-64 years of age, but are misclassified as healthy by the doctor-diagnosed arthritis criterion due to low sensitivity,” they said.
The study authors also speculated that younger patients might be more likely to ignore symptoms or visit a doctor less often.
“Further studies are needed to evaluate potential changes in the specific causes of arthritis, especially among adults below the age of 65,” they concluded.
The study was supported by a grant from the National Institutes of Health. The investigators did not disclose any other conflicts of interest.
By including two additional survey criteria in their study of arthritis prevalence, Dr. Jafarzadeh and Dr. Felson introduced a method that “may be considerably more accurate than prior estimates that use the single NHIS [National Health Interview Survey] item on doctor-diagnosed arthritis,” said Jeffrey N. Katz, MD, in an editorial accompanying the study.
The study raises important questions about how “arthritis” is defined, as well as how prevalence estimates could affect policy agendas, and could potentially have “far-reaching consequences” related to investment in research, prevention, and treatment, he added.
Dr. Katz is with the Orthopedic and Arthritis Center for Outcomes Research at Brigham and Women’s Hospital in Boston (Arthritis Rheumatol. 2017 Nov 27. doi: 10.1002/art.40357). No conflicts of interest were disclosed.
By including two additional survey criteria in their study of arthritis prevalence, Dr. Jafarzadeh and Dr. Felson introduced a method that “may be considerably more accurate than prior estimates that use the single NHIS [National Health Interview Survey] item on doctor-diagnosed arthritis,” said Jeffrey N. Katz, MD, in an editorial accompanying the study.
The study raises important questions about how “arthritis” is defined, as well as how prevalence estimates could affect policy agendas, and could potentially have “far-reaching consequences” related to investment in research, prevention, and treatment, he added.
Dr. Katz is with the Orthopedic and Arthritis Center for Outcomes Research at Brigham and Women’s Hospital in Boston (Arthritis Rheumatol. 2017 Nov 27. doi: 10.1002/art.40357). No conflicts of interest were disclosed.
By including two additional survey criteria in their study of arthritis prevalence, Dr. Jafarzadeh and Dr. Felson introduced a method that “may be considerably more accurate than prior estimates that use the single NHIS [National Health Interview Survey] item on doctor-diagnosed arthritis,” said Jeffrey N. Katz, MD, in an editorial accompanying the study.
The study raises important questions about how “arthritis” is defined, as well as how prevalence estimates could affect policy agendas, and could potentially have “far-reaching consequences” related to investment in research, prevention, and treatment, he added.
Dr. Katz is with the Orthopedic and Arthritis Center for Outcomes Research at Brigham and Women’s Hospital in Boston (Arthritis Rheumatol. 2017 Nov 27. doi: 10.1002/art.40357). No conflicts of interest were disclosed.
The prevalence of arthritis in the United States is much higher than current estimates indicate, especially among adults under 65 years of age, a study showed.
The higher prevalence can be largely attributed to “the previous underestimate of arthritis in adults between 18-64 years of age,” according to S. Reza Jafarzadeh, PhD, and David T. Felson , MD, both of Boston University. Using a new surveillance model, they estimated that 91.2 million adults in the United States (36.8%) had arthritis in 2015, compared with a previously reported national estimate of 54.4 million adults (22.7%). Of these, 61.1 million were between 18 and 64 years of age (Arthritis Rheumatol. 2017 Nov 27. doi: 10.1002/art.40355).
Arthritis prevalence was 29.9% in men aged 18-64 years (95% probability interval, 23.4-42.3), 31.2% in women aged 18-64 years (95% PI, 25.8-44.1), 55.8% in men aged 65 years and older (95% PI, 49.9-70.4), and 68.7% in women aged 65 years and older (95% PI, 62.1-79.9), the authors reported.
Among respondents aged 18-64 years, 19.3% of men and 16.7% of women reported that they had chronic joint symptoms but no physician-diagnosed arthritis. Among those 65 years of age or older, 15.7% of men and 13.5% of women responded that they had chronic joint symptoms without physician-diagnosed arthritis.
Current methodology for estimating arthritis prevalence is based on a single survey question asking whether a health care provider has ever told the patient that he or she has arthritis, a method that has previously been shown to have a sensitivity of 68.8% in adults 65 years of age and older and 52.5% for those aged 45-64 years, Dr. Jafarzadeh and Dr. Felson reported. “Such a low sensitivity, especially in a younger population, where almost half of true arthritis cases are missed, results in substantial misclassification and underestimation of prevalence and would have a detrimental effect for planning and needs assessment,” the authors wrote.
The two additional questions on joint pain, aching, and stiffness that the investigators included in the study captured “a substantial (i.e., 65%-80%) fraction of the population with arthritis, who are between 18-64 years of age, but are misclassified as healthy by the doctor-diagnosed arthritis criterion due to low sensitivity,” they said.
The study authors also speculated that younger patients might be more likely to ignore symptoms or visit a doctor less often.
“Further studies are needed to evaluate potential changes in the specific causes of arthritis, especially among adults below the age of 65,” they concluded.
The study was supported by a grant from the National Institutes of Health. The investigators did not disclose any other conflicts of interest.
The prevalence of arthritis in the United States is much higher than current estimates indicate, especially among adults under 65 years of age, a study showed.
The higher prevalence can be largely attributed to “the previous underestimate of arthritis in adults between 18-64 years of age,” according to S. Reza Jafarzadeh, PhD, and David T. Felson , MD, both of Boston University. Using a new surveillance model, they estimated that 91.2 million adults in the United States (36.8%) had arthritis in 2015, compared with a previously reported national estimate of 54.4 million adults (22.7%). Of these, 61.1 million were between 18 and 64 years of age (Arthritis Rheumatol. 2017 Nov 27. doi: 10.1002/art.40355).
Arthritis prevalence was 29.9% in men aged 18-64 years (95% probability interval, 23.4-42.3), 31.2% in women aged 18-64 years (95% PI, 25.8-44.1), 55.8% in men aged 65 years and older (95% PI, 49.9-70.4), and 68.7% in women aged 65 years and older (95% PI, 62.1-79.9), the authors reported.
Among respondents aged 18-64 years, 19.3% of men and 16.7% of women reported that they had chronic joint symptoms but no physician-diagnosed arthritis. Among those 65 years of age or older, 15.7% of men and 13.5% of women responded that they had chronic joint symptoms without physician-diagnosed arthritis.
Current methodology for estimating arthritis prevalence is based on a single survey question asking whether a health care provider has ever told the patient that he or she has arthritis, a method that has previously been shown to have a sensitivity of 68.8% in adults 65 years of age and older and 52.5% for those aged 45-64 years, Dr. Jafarzadeh and Dr. Felson reported. “Such a low sensitivity, especially in a younger population, where almost half of true arthritis cases are missed, results in substantial misclassification and underestimation of prevalence and would have a detrimental effect for planning and needs assessment,” the authors wrote.
The two additional questions on joint pain, aching, and stiffness that the investigators included in the study captured “a substantial (i.e., 65%-80%) fraction of the population with arthritis, who are between 18-64 years of age, but are misclassified as healthy by the doctor-diagnosed arthritis criterion due to low sensitivity,” they said.
The study authors also speculated that younger patients might be more likely to ignore symptoms or visit a doctor less often.
“Further studies are needed to evaluate potential changes in the specific causes of arthritis, especially among adults below the age of 65,” they concluded.
The study was supported by a grant from the National Institutes of Health. The investigators did not disclose any other conflicts of interest.
FROM ARTHRITIS & RHEUMATOLOGY
Key clinical point:
Major finding: An estimated 91.2 million adults in the United States (36.8%) had arthritis in 2015, compared with a previously reported national estimate of 54.4 million adults (22.7%).
Data source: Data from 33,672 respondents to the 2015 National Health Interview Survey.
Disclosures: The study was supported by a grant from the National Institutes of Health. The investigators did not disclose any other conflicts of interest.
Alarm reductions don’t improve ICU response times
TORONTO – It will take more than a reduction in alarms to address the issue of alarm fatigue in the ICU; a change in the ICU staff culture is needed, suggests new research.
“It may take years to recondition clinicians [to realize] that alarms are actionable and must get a response,” Afua Kunadu, MD, said during her presentation on the study at the CHEST annual meeting. Results from prior studies had suggested that as many as 99% of clinical alarms do not result in clinical intervention, noted Dr. Kunadu, an internal medicine physician at Harlem Hospital Center in New York.

She described the program, which started in the 20-bed adult ICU of Harlem Hospital Center, following a 2014 National Patient Safety Goal issued by The Joint Commission to improve the safety of clinical alarm systems by reducing unneeded alarms and alarm fatigue. The Harlem Hospital task force that ran the program began with an audit of alarms that went off in the ICU and used the results to identify the three most common alarms: bedside cardiac monitors, infusion pumps, and mechanical ventilators. The task force arranged to reset the default settings on these devices to decrease alarm frequency and boost the clinical importance of each alarm that still sounded. Concurrently, they ran educational sessions about the new alarm thresholds, the anticipated drop in alarm number, and the increased urgency to respond to the remaining alarms very quickly for the ICU staff.
The raised thresholds effectively cut the number of alarms. The average number of alarms per patient per hour fell from 4.5 at baseline during September 2016 to about 2 after 1 month, during December 2016. Then the rate further declined to reach a steady nadir that stayed at about 1.3 alarms per patient per hour 4 months into the program.
But timely responses, measured as the percentage of alarm responses occurring within 60 seconds after the alarm went off, fell from 60% at 1 month into the program down to 12% after 4 months, Dr. Kunadu reported.
She had no disclosures.
[email protected]
On Twitter @mitchelzoler

TORONTO – It will take more than a reduction in alarms to address the issue of alarm fatigue in the ICU; a change in the ICU staff culture is needed, suggests new research.
“It may take years to recondition clinicians [to realize] that alarms are actionable and must get a response,” Afua Kunadu, MD, said during her presentation on the study at the CHEST annual meeting. Results from prior studies had suggested that as many as 99% of clinical alarms do not result in clinical intervention, noted Dr. Kunadu, an internal medicine physician at Harlem Hospital Center in New York.
She described the program, which started in the 20-bed adult ICU of Harlem Hospital Center, following a 2014 National Patient Safety Goal issued by The Joint Commission to improve the safety of clinical alarm systems by reducing unneeded alarms and alarm fatigue. The Harlem Hospital task force that ran the program began with an audit of alarms that went off in the ICU and used the results to identify the three most common alarms: bedside cardiac monitors, infusion pumps, and mechanical ventilators. The task force arranged to reset the default settings on these devices to decrease alarm frequency and boost the clinical importance of each alarm that still sounded. Concurrently, they ran educational sessions about the new alarm thresholds, the anticipated drop in alarm number, and the increased urgency to respond to the remaining alarms very quickly for the ICU staff.
The raised thresholds effectively cut the number of alarms. The average number of alarms per patient per hour fell from 4.5 at baseline during September 2016 to about 2 after 1 month, during December 2016. Then the rate further declined to reach a steady nadir that stayed at about 1.3 alarms per patient per hour 4 months into the program.
But timely responses, measured as the percentage of alarm responses occurring within 60 seconds after the alarm went off, fell from 60% at 1 month into the program down to 12% after 4 months, Dr. Kunadu reported.
She had no disclosures.
[email protected]
On Twitter @mitchelzoler
TORONTO – It will take more than a reduction in alarms to address the issue of alarm fatigue in the ICU; a change in the ICU staff culture is needed, suggests new research.
“It may take years to recondition clinicians [to realize] that alarms are actionable and must get a response,” Afua Kunadu, MD, said during her presentation on the study at the CHEST annual meeting. Results from prior studies had suggested that as many as 99% of clinical alarms do not result in clinical intervention, noted Dr. Kunadu, an internal medicine physician at Harlem Hospital Center in New York.
She described the program, which started in the 20-bed adult ICU of Harlem Hospital Center, following a 2014 National Patient Safety Goal issued by The Joint Commission to improve the safety of clinical alarm systems by reducing unneeded alarms and alarm fatigue. The Harlem Hospital task force that ran the program began with an audit of alarms that went off in the ICU and used the results to identify the three most common alarms: bedside cardiac monitors, infusion pumps, and mechanical ventilators. The task force arranged to reset the default settings on these devices to decrease alarm frequency and boost the clinical importance of each alarm that still sounded. Concurrently, they ran educational sessions about the new alarm thresholds, the anticipated drop in alarm number, and the increased urgency to respond to the remaining alarms very quickly for the ICU staff.
The raised thresholds effectively cut the number of alarms. The average number of alarms per patient per hour fell from 4.5 at baseline during September 2016 to about 2 after 1 month, during December 2016. Then the rate further declined to reach a steady nadir that stayed at about 1.3 alarms per patient per hour 4 months into the program.
But timely responses, measured as the percentage of alarm responses occurring within 60 seconds after the alarm went off, fell from 60% at 1 month into the program down to 12% after 4 months, Dr. Kunadu reported.
She had no disclosures.
[email protected]
On Twitter @mitchelzoler
AT CHEST 2017
Key clinical point:
Major finding: Average alarms/patient/hour fell from 4.5 to 1.3, but the percentage of responses in less than 60 seconds fell from 60% to 12%.
Data source: An observational study at a single adult ICU in the United States.
Disclosures: Dr. Kunadu had no disclosures.
Major venous injury tied to adverse events in aortic reconstruction
Although uncommon, major venous injury during surgery for aortic reconstruction can result in massive blood loss resulting in increased morbidity and mortality, according to the results of a retrospective review conducted by Sachinder S. Hans, MD, and colleagues, and reported online in the Annals of Vascular Surgery.
Of 945 patients undergoing major aortic reconstruction, 723 (76.5%) underwent open abdominal aortic aneurysm (AAA) repair/iliac aneurysm repair; 222 patients (23.5%) underwent aortofemoral grafting (AFG). The number of units of packed red blood cells transfused, location of injured vessel, type of repair, postoperative morbidity, and mortality were collected in a vascular registry on a continuous basis. All patients identified with iliac vein/inferior vena cava/femoral vein injury had follow-up noninvasive venous examination of the lower extremities.
A total of 17 of 945 patients (1.9%) suffered 18 major venous injuries during aortic reconstruction according to Dr. Hans and his colleagues at St. John Macomb Hospital, Warren, Mich. These injuries comprised four inferior vena cava injuries, 10 iliac vein injuries, and four left renal vein injuries (Ann Vasc Surg. 2017. doi: 10.1016/j.avsg.2017.08.004).
Overall, 16 of the 18 injuries occurred during open AAA repair (7 for ruptured AAA, and 9 for intact). Two of the patients with venous injury died (11.8%), one from uncontrolled bleeding from a tear in the right iliac during repair of a ruptured AAA, and the second from disseminated intravascular complication following repair of ruptured AAA. The remaining two major venous injuries occurred during redo AFG (1 out of 6 total) and primary AFG (1 out of 216 total).
The following risk factors were also observed: The majority of the patients experiencing major venous injury were men (83%; P = .002), and the presence of periarterial inflammation (P = .006) and associated iliac aneurysm (P = .05) were significantly associated with major venous injury among the AAA patients.
The researchers suggested the following tips to lessen the likelihood of major venous injury: “Prevention of major venous injury is not always possible; however, keeping dissection plane close to arterial wall, avoiding passage of vessel loops or tapes around the neck of the aorta and iliac bifurcation, particularly in patients with surrounding inflammation and ligating venous tributaries crossing the aorta as they are joining the inferior vena cava may help reduce incidence of such injuries.”
They also suggested that surgeons should be cognizant of the serious complication that major venous injury was for patients undergoing aortic reconstruction, and to be aware that “the incidence of such injury is higher during the repair of ruptured AAA and redo aortofemoral grafting.”
The authors received no study funding and reported that they had no conflicts.
Although uncommon, major venous injury during surgery for aortic reconstruction can result in massive blood loss resulting in increased morbidity and mortality, according to the results of a retrospective review conducted by Sachinder S. Hans, MD, and colleagues, and reported online in the Annals of Vascular Surgery.
Of 945 patients undergoing major aortic reconstruction, 723 (76.5%) underwent open abdominal aortic aneurysm (AAA) repair/iliac aneurysm repair; 222 patients (23.5%) underwent aortofemoral grafting (AFG). The number of units of packed red blood cells transfused, location of injured vessel, type of repair, postoperative morbidity, and mortality were collected in a vascular registry on a continuous basis. All patients identified with iliac vein/inferior vena cava/femoral vein injury had follow-up noninvasive venous examination of the lower extremities.
A total of 17 of 945 patients (1.9%) suffered 18 major venous injuries during aortic reconstruction according to Dr. Hans and his colleagues at St. John Macomb Hospital, Warren, Mich. These injuries comprised four inferior vena cava injuries, 10 iliac vein injuries, and four left renal vein injuries (Ann Vasc Surg. 2017. doi: 10.1016/j.avsg.2017.08.004).
Overall, 16 of the 18 injuries occurred during open AAA repair (7 for ruptured AAA, and 9 for intact). Two of the patients with venous injury died (11.8%), one from uncontrolled bleeding from a tear in the right iliac during repair of a ruptured AAA, and the second from disseminated intravascular complication following repair of ruptured AAA. The remaining two major venous injuries occurred during redo AFG (1 out of 6 total) and primary AFG (1 out of 216 total).
The following risk factors were also observed: The majority of the patients experiencing major venous injury were men (83%; P = .002), and the presence of periarterial inflammation (P = .006) and associated iliac aneurysm (P = .05) were significantly associated with major venous injury among the AAA patients.
The researchers suggested the following tips to lessen the likelihood of major venous injury: “Prevention of major venous injury is not always possible; however, keeping dissection plane close to arterial wall, avoiding passage of vessel loops or tapes around the neck of the aorta and iliac bifurcation, particularly in patients with surrounding inflammation and ligating venous tributaries crossing the aorta as they are joining the inferior vena cava may help reduce incidence of such injuries.”
They also suggested that surgeons should be cognizant of the serious complication that major venous injury was for patients undergoing aortic reconstruction, and to be aware that “the incidence of such injury is higher during the repair of ruptured AAA and redo aortofemoral grafting.”
The authors received no study funding and reported that they had no conflicts.
Although uncommon, major venous injury during surgery for aortic reconstruction can result in massive blood loss resulting in increased morbidity and mortality, according to the results of a retrospective review conducted by Sachinder S. Hans, MD, and colleagues, and reported online in the Annals of Vascular Surgery.
Of 945 patients undergoing major aortic reconstruction, 723 (76.5%) underwent open abdominal aortic aneurysm (AAA) repair/iliac aneurysm repair; 222 patients (23.5%) underwent aortofemoral grafting (AFG). The number of units of packed red blood cells transfused, location of injured vessel, type of repair, postoperative morbidity, and mortality were collected in a vascular registry on a continuous basis. All patients identified with iliac vein/inferior vena cava/femoral vein injury had follow-up noninvasive venous examination of the lower extremities.
A total of 17 of 945 patients (1.9%) suffered 18 major venous injuries during aortic reconstruction according to Dr. Hans and his colleagues at St. John Macomb Hospital, Warren, Mich. These injuries comprised four inferior vena cava injuries, 10 iliac vein injuries, and four left renal vein injuries (Ann Vasc Surg. 2017. doi: 10.1016/j.avsg.2017.08.004).
Overall, 16 of the 18 injuries occurred during open AAA repair (7 for ruptured AAA, and 9 for intact). Two of the patients with venous injury died (11.8%), one from uncontrolled bleeding from a tear in the right iliac during repair of a ruptured AAA, and the second from disseminated intravascular complication following repair of ruptured AAA. The remaining two major venous injuries occurred during redo AFG (1 out of 6 total) and primary AFG (1 out of 216 total).
The following risk factors were also observed: The majority of the patients experiencing major venous injury were men (83%; P = .002), and the presence of periarterial inflammation (P = .006) and associated iliac aneurysm (P = .05) were significantly associated with major venous injury among the AAA patients.
The researchers suggested the following tips to lessen the likelihood of major venous injury: “Prevention of major venous injury is not always possible; however, keeping dissection plane close to arterial wall, avoiding passage of vessel loops or tapes around the neck of the aorta and iliac bifurcation, particularly in patients with surrounding inflammation and ligating venous tributaries crossing the aorta as they are joining the inferior vena cava may help reduce incidence of such injuries.”
They also suggested that surgeons should be cognizant of the serious complication that major venous injury was for patients undergoing aortic reconstruction, and to be aware that “the incidence of such injury is higher during the repair of ruptured AAA and redo aortofemoral grafting.”
The authors received no study funding and reported that they had no conflicts.
FROM THE ANNALS OF VASCULAR SURGERY
Key clinical point:
Major finding: A total of 17/945 patients suffered major venous injuries during aortic reconstruction.
Data source: A retrospective review of 945 patients undergoing aortic reconstruction at two sites.
Disclosures: The authors received no study funding and reported that they had no conflicts.
Physician health programs: ‘Diagnosing for dollars’?
As medicine struggles with rising rates of physician burnout, dissatisfaction, depression, and suicide, one solution comes in the form of Physician Health Programs, or PHPs. These organizations were originally started by volunteer physicians, often doctors in recovery, and funded by medical societies, as a way of providing help while maintaining confidentiality. Now, they are run by independent corporations, by medical societies in some states, and sometimes by hospitals or health systems. The services they offer vary by PHP, and they may have relationships with state licensing boards. While they can provide a gateway to help for a troubled doctor, there has also been concern about the services that are being provided.
Louise Andrew, MD, JD, served as the liaison from the American College of Emergency Physicians (ACEP) to the the Federation of State Medical Boards from 2006 to 2014. In an online forum called Collective Wisdom, Andrew talked about the benefits of Physician Health Programs as entities that are helpful to stuggling doctors and urged her colleagues to use them as a safe alternative to suffering in silence.
More recently, Dr. Andrew has become concerned that PHPs may have taken on the role of what is more akin to “diagnosing for dollars.” In her May, 2016 column in Emergency Physician’s Monthly, Andrew noted, “A decade later, and my convictions have changed dramatically. Horror stories that colleagues related to me while I chaired ACEP’s Personal and Professional WellBeing Committee cannot all be isolated events. For example, physicians who self-referred to the PHP for management of stress and depression were reportedly railroaded into incredibly expensive and inconvenient out-of-state drug and alcohol treatment programs, even when there was no coexisting drug or alcohol problem.”
Dr. Andrew is not the only one voicing concerns about PHPs. In “Physician Health Programs: More harm than good?” (Medscape, Aug. 19, 2015), Pauline Anderson wrote about a several problems that have surfaced. In North Carolina, the state audited the PHPs after complaints that they were mandating physicians to lengthy and expensive inpatient programs. The complaints asserted that the physicians had no recourse and were not able to see their records. “The state auditor’s report found no abuse by North Carolina’s PHP. However, there was a caveat – the report determined that abuse could occur and potentially go undetected.
“It also found that the North Carolina PHP created the appearance of conflicts of interest by allowing the centers to provide both patient evaluation and treatments and that procedures did not ensure that physicians receive quality evaluations and treatment because the PHP had no documented criteria for selecting treatment centers and did not adequately monitor them.”
Finally, in a Florida Fox4News story, “Are FL doctors and nurses being sent to rehab unnecessarily? Accusations: Overdiagnosing; overcharging” (Nov. 16, 2017), reporters Katie Lagrone and Matthew Apthorp wrote about financial incentives for evaluators to refer doctors to inpatient substance abuse facilities.

The American Psychiatric Association has made it a priority to address physician burnout and mental health. Richard F. Summers, MD, APA Trustee-at-Large noted: “State PHPs are an essential resource for physicians, but there is a tremendous diversity in quality and approach. It is critical that these programs include attention to mental health problems as well as addiction, and that they support individual physicians’ treatment and journey toward well-being. They need to be accessible, private, and high quality, and they should be staffed by excellent psychiatrists and other mental health professionals.”
PHPs provide a much-needed and wanted service. But if the goal is to provide mental health and substance abuse services to physicians who are struggling – to prevent physicians from burning out, leaving medicine, and dying of suicide – then any whiff of corruption and any fear of professional repercussions become a reason not to use these services. If they are to be helpful, physicians must feel safe using them.
Dr. Miller is coauthor with Annette Hanson, MD, of “Committed: The Battle Over Involuntary Psychiatric Care” (Baltimore: Johns Hopkins University Press, 2016).
As medicine struggles with rising rates of physician burnout, dissatisfaction, depression, and suicide, one solution comes in the form of Physician Health Programs, or PHPs. These organizations were originally started by volunteer physicians, often doctors in recovery, and funded by medical societies, as a way of providing help while maintaining confidentiality. Now, they are run by independent corporations, by medical societies in some states, and sometimes by hospitals or health systems. The services they offer vary by PHP, and they may have relationships with state licensing boards. While they can provide a gateway to help for a troubled doctor, there has also been concern about the services that are being provided.
Louise Andrew, MD, JD, served as the liaison from the American College of Emergency Physicians (ACEP) to the the Federation of State Medical Boards from 2006 to 2014. In an online forum called Collective Wisdom, Andrew talked about the benefits of Physician Health Programs as entities that are helpful to stuggling doctors and urged her colleagues to use them as a safe alternative to suffering in silence.
More recently, Dr. Andrew has become concerned that PHPs may have taken on the role of what is more akin to “diagnosing for dollars.” In her May, 2016 column in Emergency Physician’s Monthly, Andrew noted, “A decade later, and my convictions have changed dramatically. Horror stories that colleagues related to me while I chaired ACEP’s Personal and Professional WellBeing Committee cannot all be isolated events. For example, physicians who self-referred to the PHP for management of stress and depression were reportedly railroaded into incredibly expensive and inconvenient out-of-state drug and alcohol treatment programs, even when there was no coexisting drug or alcohol problem.”
Dr. Andrew is not the only one voicing concerns about PHPs. In “Physician Health Programs: More harm than good?” (Medscape, Aug. 19, 2015), Pauline Anderson wrote about a several problems that have surfaced. In North Carolina, the state audited the PHPs after complaints that they were mandating physicians to lengthy and expensive inpatient programs. The complaints asserted that the physicians had no recourse and were not able to see their records. “The state auditor’s report found no abuse by North Carolina’s PHP. However, there was a caveat – the report determined that abuse could occur and potentially go undetected.
“It also found that the North Carolina PHP created the appearance of conflicts of interest by allowing the centers to provide both patient evaluation and treatments and that procedures did not ensure that physicians receive quality evaluations and treatment because the PHP had no documented criteria for selecting treatment centers and did not adequately monitor them.”
Finally, in a Florida Fox4News story, “Are FL doctors and nurses being sent to rehab unnecessarily? Accusations: Overdiagnosing; overcharging” (Nov. 16, 2017), reporters Katie Lagrone and Matthew Apthorp wrote about financial incentives for evaluators to refer doctors to inpatient substance abuse facilities.
The American Psychiatric Association has made it a priority to address physician burnout and mental health. Richard F. Summers, MD, APA Trustee-at-Large noted: “State PHPs are an essential resource for physicians, but there is a tremendous diversity in quality and approach. It is critical that these programs include attention to mental health problems as well as addiction, and that they support individual physicians’ treatment and journey toward well-being. They need to be accessible, private, and high quality, and they should be staffed by excellent psychiatrists and other mental health professionals.”
PHPs provide a much-needed and wanted service. But if the goal is to provide mental health and substance abuse services to physicians who are struggling – to prevent physicians from burning out, leaving medicine, and dying of suicide – then any whiff of corruption and any fear of professional repercussions become a reason not to use these services. If they are to be helpful, physicians must feel safe using them.
Dr. Miller is coauthor with Annette Hanson, MD, of “Committed: The Battle Over Involuntary Psychiatric Care” (Baltimore: Johns Hopkins University Press, 2016).
As medicine struggles with rising rates of physician burnout, dissatisfaction, depression, and suicide, one solution comes in the form of Physician Health Programs, or PHPs. These organizations were originally started by volunteer physicians, often doctors in recovery, and funded by medical societies, as a way of providing help while maintaining confidentiality. Now, they are run by independent corporations, by medical societies in some states, and sometimes by hospitals or health systems. The services they offer vary by PHP, and they may have relationships with state licensing boards. While they can provide a gateway to help for a troubled doctor, there has also been concern about the services that are being provided.
Louise Andrew, MD, JD, served as the liaison from the American College of Emergency Physicians (ACEP) to the the Federation of State Medical Boards from 2006 to 2014. In an online forum called Collective Wisdom, Andrew talked about the benefits of Physician Health Programs as entities that are helpful to stuggling doctors and urged her colleagues to use them as a safe alternative to suffering in silence.
More recently, Dr. Andrew has become concerned that PHPs may have taken on the role of what is more akin to “diagnosing for dollars.” In her May, 2016 column in Emergency Physician’s Monthly, Andrew noted, “A decade later, and my convictions have changed dramatically. Horror stories that colleagues related to me while I chaired ACEP’s Personal and Professional WellBeing Committee cannot all be isolated events. For example, physicians who self-referred to the PHP for management of stress and depression were reportedly railroaded into incredibly expensive and inconvenient out-of-state drug and alcohol treatment programs, even when there was no coexisting drug or alcohol problem.”
Dr. Andrew is not the only one voicing concerns about PHPs. In “Physician Health Programs: More harm than good?” (Medscape, Aug. 19, 2015), Pauline Anderson wrote about a several problems that have surfaced. In North Carolina, the state audited the PHPs after complaints that they were mandating physicians to lengthy and expensive inpatient programs. The complaints asserted that the physicians had no recourse and were not able to see their records. “The state auditor’s report found no abuse by North Carolina’s PHP. However, there was a caveat – the report determined that abuse could occur and potentially go undetected.
“It also found that the North Carolina PHP created the appearance of conflicts of interest by allowing the centers to provide both patient evaluation and treatments and that procedures did not ensure that physicians receive quality evaluations and treatment because the PHP had no documented criteria for selecting treatment centers and did not adequately monitor them.”
Finally, in a Florida Fox4News story, “Are FL doctors and nurses being sent to rehab unnecessarily? Accusations: Overdiagnosing; overcharging” (Nov. 16, 2017), reporters Katie Lagrone and Matthew Apthorp wrote about financial incentives for evaluators to refer doctors to inpatient substance abuse facilities.
The American Psychiatric Association has made it a priority to address physician burnout and mental health. Richard F. Summers, MD, APA Trustee-at-Large noted: “State PHPs are an essential resource for physicians, but there is a tremendous diversity in quality and approach. It is critical that these programs include attention to mental health problems as well as addiction, and that they support individual physicians’ treatment and journey toward well-being. They need to be accessible, private, and high quality, and they should be staffed by excellent psychiatrists and other mental health professionals.”
PHPs provide a much-needed and wanted service. But if the goal is to provide mental health and substance abuse services to physicians who are struggling – to prevent physicians from burning out, leaving medicine, and dying of suicide – then any whiff of corruption and any fear of professional repercussions become a reason not to use these services. If they are to be helpful, physicians must feel safe using them.
Dr. Miller is coauthor with Annette Hanson, MD, of “Committed: The Battle Over Involuntary Psychiatric Care” (Baltimore: Johns Hopkins University Press, 2016).