User login
Antibiotic stewardship: Optimizing antibiotic use in an era of increasing resistance and rising costs
RELEASE DATE: January 15, 2011 EXPIRATION DATE: January 31, 2012
Estimated time to complete the activity: 1 hour 45 minutes
Jointly sponsored by Postgraduate Institute for Medicine and Global Education Exchange, Inc
This activity is supported by an educational grant from Merck & Co., Inc.
Program Description
With antimicrobial resistance on the rise and very few new pharmaceutical agents in development, a well‐managed antimicrobial stewardship program in the hospital becomes the first‐line defense against the emergence of resistance and provides not only a cost‐containment measure but also ensures the continued efficacy of available antimicrobials. A successful stewardship program knows and understands the local epidemiology and utilizes a multidisciplinary strategy to ensure the selection of an appropriate antibiotic at the right dose for the right duration. In addition, stewardship in the community‐based parenteral antiinfective therapy (CoPAT) program provides an opportunity for an integrated patient‐centric model of care as well as continunity of care when patients transition from inpatient to outpatient setting.
Learning Objectives
-
Explain the impact of multidisciplinary antimicrobial stewardship programs on the emergence and transmission of antimicrobial‐resistant microorganisms
-
Identify potential challenges and controversies related to the implementation of antimicrobial stewardship programs in health systems
-
Use pharmacokinetic and pharmacodynamic data to facilitate appropriate antimicrobial use
-
Describe the role of CoPAT in an integrated patient‐centric model of antimicrobial stewardship and pharmaco epidemiology
-
Illustrate how a CoPAT model of care can be used to mitigate complications and prevent emergency room visits and readmissions
Target Audience
This activity has been designed to meet the educational needs of hospitalists and other healthcare providers involved in the treatment of patients with infectious diseases.
Accreditation Statement
This activity has been planned and implemented in accordance with the Essential Areas and policies of the Accreditation Council for Continuing Medical Education (ACCME) through the joint sponsorship of Postgraduate Institute for Medicine (PIM) and Global Education Exchange, Inc. (GLOBEX). PIM is accredited by the ACCME to provide continuing medical education for physicians.
Credit Designation
Postgraduate Institute for Medicine designates this educational activity for a maximum of 1.75 AMA PRA Category 1 Credit(s). Physicians should only claim credit commensurate with the extent of their participation in the activity.
Faculty
James Pile, MD [Chairman] Director, Division of Hospital Medicine Case Western Reserve University at MetroHealth Medical Center Cleveland, Ohio Steven M. Gordon, MD Chairman, Department of Infectious Disease Cleveland Clinic, Cleveland, Ohio Nabin Shrestha, MD Department of Infectious Disease Cleveland Clinic, Cleveland, Ohio Susan J. Rehm, MD Vice Chair, Department of Infectious Disease Cleveland Clinic, Cleveland, Ohio Thomas Lodise, PharmD Associate Professor, Albany College of Pharmacy Albany, New York Jill Butterfield, PharmD Albany College of Pharmacy Albany, New York Arjun Srinivasan, MD Division of Healthcare Quality Promotion Centers for Disease Control and Prevention Atlanta, Georgia Christopher A. Ohl, MD Associate Professor Internal Medicine‐Infectious Diseases Wake Forest University, Baptist Medical Center, Winston‐Salem, North Carolina Vera P. Luther, MD, Assistant Professor of Medicine Section of Infectious Diseases and Department of Internal Medicine Wake Forest University School of Medicine Winston‐Salem, North Carolina
Disclosure of Conflicts of Interest
The Postgraduate Institute for Medicine (PIM) assesses conflict of interest with its instructors, planners, managers and other individuals who are in a position to control the content of CME activities. All relevant conflicts of interest that are identified are thoroughly vetted by PIM for fair balance, scientific objectivity of studies utilized in this activity, and patient care recommendations. PIM is committed to providing its learners with high quality CME activities and related materials that promote improvements or quality in healthcare and not a specific proprietary business interest of a commercial interest.
The faculty reported the following financial relationships or relationships to products or devices they or their spouse/life partner have with commercial interests related to the content of this CME activity:0
| Name of Faculty or Presenter | Reported Financial Relationship |
|---|---|
| James Pile, MD | Consulting Fees: Pfizer |
| CME Symposia: URL Pharma | |
| Honorarium: Merck & Co. | |
| Steven M. Gordon, MD | Honorarium: Merck & Co. |
| Thomas Lodise, PharmD | Consulting Fees: Astellas Pharma, Cubist Pharmaceuticals, Forest Laboratories, Merck & Co. Inc., Pfizer |
| Grants: Astellas Pharma, Cubist Pharmaceuticals, Merck & Co. Inc., Pfizer | |
| Speaker's Bureau: Astellas Pharma, Cubist | |
| Honorarium: Merck & Co. | |
| Jill Butterfield, PhamD | No real or apparent conflicts of interest to report |
| Nabin Shrestha, MD | No real or apparent conflicts of interest to report |
| Susan J. Rehm, MD | No real or apparent conflicts of interest to report |
| Arjun Srinivasan, MD | No real or apparent conflicts of interest to report |
| Christopher A. Ohl, MD | Consulting Fees: CDC, Cubist, FDA, Ortho‐McNeil, Pfizer, USDA |
| Speaker's Bureau: CDC, Cubist, FDA, Ortho‐McNeil, Pfizer, USDA | |
| Honorarium: Merck & Co. | |
| Vera P. Luther, MD | No real or apparent conflicts of interest to report |
The planners and managers reported the following financial relationships or relationships to products or devices they or their spouse/life partner have with commercial interests related to the content of this CME activity:0, 0
| Name of Planner or Manager | Reported Financial Relationship |
|---|---|
| Meri D. Pozo, PhD | No real or apparent conflicts of interest to report |
| Jan Hixon, RN, BSN, MSN | No real or apparent conflicts of interest to report |
| Trace Hutchison, PharmD | No real or apparent conflicts of interest to report |
| Julia Kimball, RN, BSN | No real or apparent conflicts of interest to report |
| Samantha Mattiucci, PharmD | No real or apparent conflicts of interest to report |
| Jan Schultz, RN, MSN, CCMEP | No real or apparent conflicts of interest to report |
| Patricia Staples, MSN, NP‐C, CCRN | No real or apparent conflicts of interest to report |
| Name of Editor | Reported Financial Relationship |
|---|---|
| Daniel Brotman, MD, FHM | Honorarium from Wiley‐Blackwell for service as the Supplement Editor |
| Thomas Baudendistel, MD | Honorarium from Wiley‐Blackwell for service as the CME Editor |
Disclosure of Unlabeled Use
This educational activity may contain discussion of published and/or investigational uses of agents that are not indicated by the FDA. Postgraduate Institute for Medicine (PIM), Global Education Exchange, Inc. (GLOBEX) and Merck & Co., Inc. do not recommend the use of any agent outside of the labeled indications.
The opinions expressed in the educational activity are those of the faculty and do not necessarily represent the views of PIM, GLOBEX and Merck & Co., Inc. Please refer to the official prescribing information for each product for discussion of approved indications, contraindications, and warnings.
Method of Participation:
There are no fees for participating and receiving CME credit for this activity. During the period January 15, 2011 through January 31, 2012, participants must read the learning objectives and faculty disclosures and study the educational activity.
PIM supports Green CME by offering your Request for Credit online. If you wish to receive acknowledgment for completing this activity, please complete the post‐test and evaluation on
Media:
Journal supplement
Disclaimer
Participants have an implied responsibility to use the newly acquired information to enhance patient outcomes and their own professional development. The information presented in this activity is not meant to serve as a guideline for patient management. Any procedures, medications, or other courses of diagnosis or treatment discussed or suggested in this activity should not be used by clinicians without evaluation of their patient's conditions and possible contraindications on dangers in use, review of any applicable manufacturer's product information, and comparison with recommendations of other authorities.
RELEASE DATE: January 15, 2011 EXPIRATION DATE: January 31, 2012
Estimated time to complete the activity: 1 hour 45 minutes
Jointly sponsored by Postgraduate Institute for Medicine and Global Education Exchange, Inc
This activity is supported by an educational grant from Merck & Co., Inc.
Program Description
With antimicrobial resistance on the rise and very few new pharmaceutical agents in development, a well‐managed antimicrobial stewardship program in the hospital becomes the first‐line defense against the emergence of resistance and provides not only a cost‐containment measure but also ensures the continued efficacy of available antimicrobials. A successful stewardship program knows and understands the local epidemiology and utilizes a multidisciplinary strategy to ensure the selection of an appropriate antibiotic at the right dose for the right duration. In addition, stewardship in the community‐based parenteral antiinfective therapy (CoPAT) program provides an opportunity for an integrated patient‐centric model of care as well as continunity of care when patients transition from inpatient to outpatient setting.
Learning Objectives
-
Explain the impact of multidisciplinary antimicrobial stewardship programs on the emergence and transmission of antimicrobial‐resistant microorganisms
-
Identify potential challenges and controversies related to the implementation of antimicrobial stewardship programs in health systems
-
Use pharmacokinetic and pharmacodynamic data to facilitate appropriate antimicrobial use
-
Describe the role of CoPAT in an integrated patient‐centric model of antimicrobial stewardship and pharmaco epidemiology
-
Illustrate how a CoPAT model of care can be used to mitigate complications and prevent emergency room visits and readmissions
Target Audience
This activity has been designed to meet the educational needs of hospitalists and other healthcare providers involved in the treatment of patients with infectious diseases.
Accreditation Statement
This activity has been planned and implemented in accordance with the Essential Areas and policies of the Accreditation Council for Continuing Medical Education (ACCME) through the joint sponsorship of Postgraduate Institute for Medicine (PIM) and Global Education Exchange, Inc. (GLOBEX). PIM is accredited by the ACCME to provide continuing medical education for physicians.
Credit Designation
Postgraduate Institute for Medicine designates this educational activity for a maximum of 1.75 AMA PRA Category 1 Credit(s). Physicians should only claim credit commensurate with the extent of their participation in the activity.
Faculty
James Pile, MD [Chairman] Director, Division of Hospital Medicine Case Western Reserve University at MetroHealth Medical Center Cleveland, Ohio Steven M. Gordon, MD Chairman, Department of Infectious Disease Cleveland Clinic, Cleveland, Ohio Nabin Shrestha, MD Department of Infectious Disease Cleveland Clinic, Cleveland, Ohio Susan J. Rehm, MD Vice Chair, Department of Infectious Disease Cleveland Clinic, Cleveland, Ohio Thomas Lodise, PharmD Associate Professor, Albany College of Pharmacy Albany, New York Jill Butterfield, PharmD Albany College of Pharmacy Albany, New York Arjun Srinivasan, MD Division of Healthcare Quality Promotion Centers for Disease Control and Prevention Atlanta, Georgia Christopher A. Ohl, MD Associate Professor Internal Medicine‐Infectious Diseases Wake Forest University, Baptist Medical Center, Winston‐Salem, North Carolina Vera P. Luther, MD, Assistant Professor of Medicine Section of Infectious Diseases and Department of Internal Medicine Wake Forest University School of Medicine Winston‐Salem, North Carolina
Disclosure of Conflicts of Interest
The Postgraduate Institute for Medicine (PIM) assesses conflict of interest with its instructors, planners, managers and other individuals who are in a position to control the content of CME activities. All relevant conflicts of interest that are identified are thoroughly vetted by PIM for fair balance, scientific objectivity of studies utilized in this activity, and patient care recommendations. PIM is committed to providing its learners with high quality CME activities and related materials that promote improvements or quality in healthcare and not a specific proprietary business interest of a commercial interest.
The faculty reported the following financial relationships or relationships to products or devices they or their spouse/life partner have with commercial interests related to the content of this CME activity:0
| Name of Faculty or Presenter | Reported Financial Relationship |
|---|---|
| James Pile, MD | Consulting Fees: Pfizer |
| CME Symposia: URL Pharma | |
| Honorarium: Merck & Co. | |
| Steven M. Gordon, MD | Honorarium: Merck & Co. |
| Thomas Lodise, PharmD | Consulting Fees: Astellas Pharma, Cubist Pharmaceuticals, Forest Laboratories, Merck & Co. Inc., Pfizer |
| Grants: Astellas Pharma, Cubist Pharmaceuticals, Merck & Co. Inc., Pfizer | |
| Speaker's Bureau: Astellas Pharma, Cubist | |
| Honorarium: Merck & Co. | |
| Jill Butterfield, PhamD | No real or apparent conflicts of interest to report |
| Nabin Shrestha, MD | No real or apparent conflicts of interest to report |
| Susan J. Rehm, MD | No real or apparent conflicts of interest to report |
| Arjun Srinivasan, MD | No real or apparent conflicts of interest to report |
| Christopher A. Ohl, MD | Consulting Fees: CDC, Cubist, FDA, Ortho‐McNeil, Pfizer, USDA |
| Speaker's Bureau: CDC, Cubist, FDA, Ortho‐McNeil, Pfizer, USDA | |
| Honorarium: Merck & Co. | |
| Vera P. Luther, MD | No real or apparent conflicts of interest to report |
The planners and managers reported the following financial relationships or relationships to products or devices they or their spouse/life partner have with commercial interests related to the content of this CME activity:0, 0
| Name of Planner or Manager | Reported Financial Relationship |
|---|---|
| Meri D. Pozo, PhD | No real or apparent conflicts of interest to report |
| Jan Hixon, RN, BSN, MSN | No real or apparent conflicts of interest to report |
| Trace Hutchison, PharmD | No real or apparent conflicts of interest to report |
| Julia Kimball, RN, BSN | No real or apparent conflicts of interest to report |
| Samantha Mattiucci, PharmD | No real or apparent conflicts of interest to report |
| Jan Schultz, RN, MSN, CCMEP | No real or apparent conflicts of interest to report |
| Patricia Staples, MSN, NP‐C, CCRN | No real or apparent conflicts of interest to report |
| Name of Editor | Reported Financial Relationship |
|---|---|
| Daniel Brotman, MD, FHM | Honorarium from Wiley‐Blackwell for service as the Supplement Editor |
| Thomas Baudendistel, MD | Honorarium from Wiley‐Blackwell for service as the CME Editor |
Disclosure of Unlabeled Use
This educational activity may contain discussion of published and/or investigational uses of agents that are not indicated by the FDA. Postgraduate Institute for Medicine (PIM), Global Education Exchange, Inc. (GLOBEX) and Merck & Co., Inc. do not recommend the use of any agent outside of the labeled indications.
The opinions expressed in the educational activity are those of the faculty and do not necessarily represent the views of PIM, GLOBEX and Merck & Co., Inc. Please refer to the official prescribing information for each product for discussion of approved indications, contraindications, and warnings.
Method of Participation:
There are no fees for participating and receiving CME credit for this activity. During the period January 15, 2011 through January 31, 2012, participants must read the learning objectives and faculty disclosures and study the educational activity.
PIM supports Green CME by offering your Request for Credit online. If you wish to receive acknowledgment for completing this activity, please complete the post‐test and evaluation on
Media:
Journal supplement
Disclaimer
Participants have an implied responsibility to use the newly acquired information to enhance patient outcomes and their own professional development. The information presented in this activity is not meant to serve as a guideline for patient management. Any procedures, medications, or other courses of diagnosis or treatment discussed or suggested in this activity should not be used by clinicians without evaluation of their patient's conditions and possible contraindications on dangers in use, review of any applicable manufacturer's product information, and comparison with recommendations of other authorities.
RELEASE DATE: January 15, 2011 EXPIRATION DATE: January 31, 2012
Estimated time to complete the activity: 1 hour 45 minutes
Jointly sponsored by Postgraduate Institute for Medicine and Global Education Exchange, Inc
This activity is supported by an educational grant from Merck & Co., Inc.
Program Description
With antimicrobial resistance on the rise and very few new pharmaceutical agents in development, a well‐managed antimicrobial stewardship program in the hospital becomes the first‐line defense against the emergence of resistance and provides not only a cost‐containment measure but also ensures the continued efficacy of available antimicrobials. A successful stewardship program knows and understands the local epidemiology and utilizes a multidisciplinary strategy to ensure the selection of an appropriate antibiotic at the right dose for the right duration. In addition, stewardship in the community‐based parenteral antiinfective therapy (CoPAT) program provides an opportunity for an integrated patient‐centric model of care as well as continunity of care when patients transition from inpatient to outpatient setting.
Learning Objectives
-
Explain the impact of multidisciplinary antimicrobial stewardship programs on the emergence and transmission of antimicrobial‐resistant microorganisms
-
Identify potential challenges and controversies related to the implementation of antimicrobial stewardship programs in health systems
-
Use pharmacokinetic and pharmacodynamic data to facilitate appropriate antimicrobial use
-
Describe the role of CoPAT in an integrated patient‐centric model of antimicrobial stewardship and pharmaco epidemiology
-
Illustrate how a CoPAT model of care can be used to mitigate complications and prevent emergency room visits and readmissions
Target Audience
This activity has been designed to meet the educational needs of hospitalists and other healthcare providers involved in the treatment of patients with infectious diseases.
Accreditation Statement
This activity has been planned and implemented in accordance with the Essential Areas and policies of the Accreditation Council for Continuing Medical Education (ACCME) through the joint sponsorship of Postgraduate Institute for Medicine (PIM) and Global Education Exchange, Inc. (GLOBEX). PIM is accredited by the ACCME to provide continuing medical education for physicians.
Credit Designation
Postgraduate Institute for Medicine designates this educational activity for a maximum of 1.75 AMA PRA Category 1 Credit(s). Physicians should only claim credit commensurate with the extent of their participation in the activity.
Faculty
James Pile, MD [Chairman] Director, Division of Hospital Medicine Case Western Reserve University at MetroHealth Medical Center Cleveland, Ohio Steven M. Gordon, MD Chairman, Department of Infectious Disease Cleveland Clinic, Cleveland, Ohio Nabin Shrestha, MD Department of Infectious Disease Cleveland Clinic, Cleveland, Ohio Susan J. Rehm, MD Vice Chair, Department of Infectious Disease Cleveland Clinic, Cleveland, Ohio Thomas Lodise, PharmD Associate Professor, Albany College of Pharmacy Albany, New York Jill Butterfield, PharmD Albany College of Pharmacy Albany, New York Arjun Srinivasan, MD Division of Healthcare Quality Promotion Centers for Disease Control and Prevention Atlanta, Georgia Christopher A. Ohl, MD Associate Professor Internal Medicine‐Infectious Diseases Wake Forest University, Baptist Medical Center, Winston‐Salem, North Carolina Vera P. Luther, MD, Assistant Professor of Medicine Section of Infectious Diseases and Department of Internal Medicine Wake Forest University School of Medicine Winston‐Salem, North Carolina
Disclosure of Conflicts of Interest
The Postgraduate Institute for Medicine (PIM) assesses conflict of interest with its instructors, planners, managers and other individuals who are in a position to control the content of CME activities. All relevant conflicts of interest that are identified are thoroughly vetted by PIM for fair balance, scientific objectivity of studies utilized in this activity, and patient care recommendations. PIM is committed to providing its learners with high quality CME activities and related materials that promote improvements or quality in healthcare and not a specific proprietary business interest of a commercial interest.
The faculty reported the following financial relationships or relationships to products or devices they or their spouse/life partner have with commercial interests related to the content of this CME activity:0
| Name of Faculty or Presenter | Reported Financial Relationship |
|---|---|
| James Pile, MD | Consulting Fees: Pfizer |
| CME Symposia: URL Pharma | |
| Honorarium: Merck & Co. | |
| Steven M. Gordon, MD | Honorarium: Merck & Co. |
| Thomas Lodise, PharmD | Consulting Fees: Astellas Pharma, Cubist Pharmaceuticals, Forest Laboratories, Merck & Co. Inc., Pfizer |
| Grants: Astellas Pharma, Cubist Pharmaceuticals, Merck & Co. Inc., Pfizer | |
| Speaker's Bureau: Astellas Pharma, Cubist | |
| Honorarium: Merck & Co. | |
| Jill Butterfield, PhamD | No real or apparent conflicts of interest to report |
| Nabin Shrestha, MD | No real or apparent conflicts of interest to report |
| Susan J. Rehm, MD | No real or apparent conflicts of interest to report |
| Arjun Srinivasan, MD | No real or apparent conflicts of interest to report |
| Christopher A. Ohl, MD | Consulting Fees: CDC, Cubist, FDA, Ortho‐McNeil, Pfizer, USDA |
| Speaker's Bureau: CDC, Cubist, FDA, Ortho‐McNeil, Pfizer, USDA | |
| Honorarium: Merck & Co. | |
| Vera P. Luther, MD | No real or apparent conflicts of interest to report |
The planners and managers reported the following financial relationships or relationships to products or devices they or their spouse/life partner have with commercial interests related to the content of this CME activity:0, 0
| Name of Planner or Manager | Reported Financial Relationship |
|---|---|
| Meri D. Pozo, PhD | No real or apparent conflicts of interest to report |
| Jan Hixon, RN, BSN, MSN | No real or apparent conflicts of interest to report |
| Trace Hutchison, PharmD | No real or apparent conflicts of interest to report |
| Julia Kimball, RN, BSN | No real or apparent conflicts of interest to report |
| Samantha Mattiucci, PharmD | No real or apparent conflicts of interest to report |
| Jan Schultz, RN, MSN, CCMEP | No real or apparent conflicts of interest to report |
| Patricia Staples, MSN, NP‐C, CCRN | No real or apparent conflicts of interest to report |
| Name of Editor | Reported Financial Relationship |
|---|---|
| Daniel Brotman, MD, FHM | Honorarium from Wiley‐Blackwell for service as the Supplement Editor |
| Thomas Baudendistel, MD | Honorarium from Wiley‐Blackwell for service as the CME Editor |
Disclosure of Unlabeled Use
This educational activity may contain discussion of published and/or investigational uses of agents that are not indicated by the FDA. Postgraduate Institute for Medicine (PIM), Global Education Exchange, Inc. (GLOBEX) and Merck & Co., Inc. do not recommend the use of any agent outside of the labeled indications.
The opinions expressed in the educational activity are those of the faculty and do not necessarily represent the views of PIM, GLOBEX and Merck & Co., Inc. Please refer to the official prescribing information for each product for discussion of approved indications, contraindications, and warnings.
Method of Participation:
There are no fees for participating and receiving CME credit for this activity. During the period January 15, 2011 through January 31, 2012, participants must read the learning objectives and faculty disclosures and study the educational activity.
PIM supports Green CME by offering your Request for Credit online. If you wish to receive acknowledgment for completing this activity, please complete the post‐test and evaluation on
Media:
Journal supplement
Disclaimer
Participants have an implied responsibility to use the newly acquired information to enhance patient outcomes and their own professional development. The information presented in this activity is not meant to serve as a guideline for patient management. Any procedures, medications, or other courses of diagnosis or treatment discussed or suggested in this activity should not be used by clinicians without evaluation of their patient's conditions and possible contraindications on dangers in use, review of any applicable manufacturer's product information, and comparison with recommendations of other authorities.
Antimicrobial Stewardship: Optimizing Antibiotic Use
Clinicians want to use antimicrobials as effectively as possible, but good intentions frequently fail to translate into adherence to best available evidence. This was recognized as early as 1956, when Earnest Jawetz noted that [The physician] is under great pressure to prescribe the newest, best, broadest antibiotic preparation, prescribe it for any complaint whatever, quickly, and preferably without worrying too much about specific etiologic diagnosis or proper indication of the drug.1 That was true in 1956, and is more so in 2010.
The term that has come to describe the collection of practices intended to optimize antimicrobial therapy is antimicrobial stewardship. The label is a relatively new one; it appears to have been coined in the mid‐1990s, and has slowly crept into the medical lexicon since then. However, many clinicians remain uncertain of exactly what antimicrobial stewardship means or what practices characterize it. The 2007 Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA) guidelines for developing institutional programs to enhance antimicrobial use define antimicrobial stewardship as an activity that includes appropriate selection, dosing, route, and duration of antimicrobial therapy, with a primary goal of optimizing clinical outcomes while minimizing unintended consequences of antimicrobial use.2
In 2004, IDSA published a document titled Bad Bugs, No Drugs that noted a marked decrease in research and development of new antimicrobials within the pharmaceutical industry, at the same time that the number of antibiotic‐resistant bacteria was dramatically increasing, particularly within healthcare settings.3, 4 Since that time, trends in antimicrobial drug development have not appreciably improved, and governmental agencies have been largely silent or ineffective in addressing the problem.58 The growth of multidrug‐resistant and sometimes pan‐resistant pathogens, at a time when newer agents with novel mechanisms of action are not available nor expected for years to come, is particularly troublesome. This scenario highlights the necessity of using currently available agents in a manner that prolongs their effectiveness and reduces further emergence and spread of resistant pathogensie, the crucial need for improved antimicrobial stewardship.
Antimicrobial stewardship recognizes that antimicrobials are a unique category of drugs, with potential to affect treatment outcomes in patients for whom the drug was not originally intended. For example, cardiac and oncologic medications can be costly, and may certainly be toxic, but their effects are limited to the patient that directly receives them. In contrast, the effects of antimicrobials prescribed to a patient in the hospital (or outside the hospital, for that matter), can ripple across the hospital system and beyond. This reflects the fact that suboptimal use of antimicrobials may induce or otherwise promote the development of antimicrobial‐resistant pathogens, with potential wide‐ranging impact.9, 10 Misuse or overuse of antimicrobials can also promote colonization and overgrowth of potentially toxic pathogens within hospitalized patients, such as Clostridium difficile, that may then be transferred widely within the hospital or other healthcare institutions, particularly when infection control measures are also less than optimal. This unique societal feature of antimicrobial drugs highlights the vital importance of antimicrobial stewardship within the hospital setting, especially at a time when few novel agents exist in the developmental pipeline, at least for gram‐negative pathogens.
Are hospitalists stakeholders in this? The answer is a resounding Yes, for several reasons. Hospitalists are the dominant prescribers of antibiotics and other antimicrobials in the United States, almost certainly prescribing more antibiotics in the hospital setting than infectious disease specialists, intensivists, or any other medical specialty or subspecialty. As such, hospitalists play a central role in the optimization of antimicrobial use in the hospital setting, including minimizing negative consequences arising from antimicrobial drug misuse or overuse. In addition, the role hospitalists play in this process is likely to increase in the future. The field of hospital medicine has grown from modest beginnings in the mid‐1990s to approximately 30,000 hospitalists in the United States today.11 Based on a sample of Medicare beneficiaries, 6% of general internists were identified as hospitalists in 1995 versus 19% in 2006, and the percentage of claims for inpatient services provided by general internists who were hospitalists increased from 9% to 37%.12 Current estimates are that >50% of medical inpatients in the United States are cared for by hospitalists (personal communication, J. Miller, Society of Hospital Medicine). Importantly, hospitalists manage patients not only on regular medical floors, but also in intensive care units (ICUs), where antimicrobial resistance is a particular problem.13, 14 Finally, hospitalists serve as gatekeepers for patients leaving the hospital on either oral or intravenous antibiotics or other antimicrobials. As such, via interactions with primary care physicians, hospitalists can play a key role in improving antimicrobial stewardship not only within, but also beyond, the hospital setting.
Hospital medicine is the first medical specialty to make patient safety and quality improvement central principles of its practice, and antimicrobial stewardship is an under‐recognized but central tenet of patient safety. As a consequence, hospitalists are natural allies of stewardship programs. Indeed, antimicrobial stewardship may be considered an example of the Holy Grail of quality improvement; ie, an intervention that improves outcomes while leading to cost savings, and should thus resonate with all hospitalists.
A number of clinical practice guidelines or campaigns have been developed by professional societies and governmental organizations in the United States2, 1526 and other countries2730 for the promotion of improved antimicrobial stewardship and/or infection control in hospitals and long‐term care facilities. The goals of these initiatives, campaigns, or guidelines are to provide information that can be utilized to prevent or slow the development and spread of hospital‐acquired infections, particularly those involving antimicrobial‐resistant pathogens. In addition, the United States Congress is currently considering the STAAR (Strategies to Address Antimicrobial Resistance) Act to encourage the use of innovative governmental and private approaches to combat this critical and expanding problem.31
This supplement to the Journal of Hospital Medicine contains several articles of interest to hospitalists seeking to positively impact patient care through improvements in antimicrobial stewardship. The first article by Christopher Ohl, MD, examines general principles of antimicrobial stewardship programs as they apply to inpatient facilities. The second article by Thomas Lodise, PharmD, explores the effective use of pharmacokinetic‐pharmacodynamic principles in antimicrobial stewardship. In the third article, Steven Gordon, MD, moves beyond the hospital setting to discuss antimicrobial stewardship at the time of hospital discharge and beyond. Finally, Arjun Srinivasan, MD, outlines several practical ways in which hospitalists may take leadership roles in stewardship efforts at their institutions. Our hope is that the supplement will be thought‐provoking, and ultimately lead to greater partnership between hospitalists and other stakeholders in antimicrobial stewardship.
- .Antimicrobial chemotherapy.Annu Rev Microbiol.1956;10:85–114.
- ,,, et al.Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America guidelines for developing an institutional program to enhance antimicrobial stewardship.Clin Infect Dis.2007;44:159–177.
- Bad bugs, no drugs: as antibiotic R2004.
- ,,,,.Trends in antimicrobial drug development: implications for the future.Clin Infect Dis.2004;38:1279–1286.
- ,,, et al.Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America.Clin Infect Dis.2009;48:1–12.
- .Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE.J Infect Dis.2008;197:1079–1081.
- ,,, et al.The epidemic of antibiotic‐resistant infections: a call to action for the medical community from the Infectious Diseases Society of America.Clin Infect Dis.2008;46:155–164.
- ,,,,,.Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America.Clin Infect Dis.2006;42:657–668.
- .“Collateral damage” from cephalosporin or quinolone antibiotic therapy.Clin Infect Dis.2004;38(Suppl 4):S341–345.
- .Collateral damage and what the future might hold. The need to balance prudent antibiotic utilization and stewardship with effective patient management.Int J Infect Dis.2006;10:S17–S24.
- .Hospitalists and intensivists: partners in caring for the critically ill‐−the time has come.J Hosp Med.2010;5:1–3.
- ,,,.Growth in the care of older patients by hospitalists in the United States.N Engl J Med.2009;360:1102–1112.
- ,,.Epidemiology and control of antibiotic resistance in the intensive care unit.Curr Opin Infect Dis.2004;17:309–316.
- .Bench‐to‐bedside review: antimicrobial utilization strategies aimed at preventing the emergence of bacterial resistance in the intensive care unit.Crit Care.2005;9:459–464.
- Centers for Disease Control and Prevention. Get smart: know when antibiotics work. Available at http://www.cdc.gov/getsmart/. Accessed June 20,2010.
- The Society for Healthcare Epidemiology of America. Compendium of strategies to prevent healthcare‐associated infections in acute care hospitals. Available at http://www.shea‐online.org/about/compendium.cfm. Accessed June 20,2010.
- ,,, et al.Therapeutic monitoring of vancomycin in adult patients: a consensus review of the American Society of Health‐System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists.Am J Health Syst Pharm.2009;66:82–98.
- Centers for Disease Control and Prevention. Department of Health and Human Services. Infection control in healthcare settings. Available at http://www.cdc.gov/nidod/dhqp/. Accessed February 17,2010.
- Centers for Disease Control and Prevention. Healthcare Infection Control Practices Advisory Committee (HICPAC). Available at http://www.cdc. gov/hicpac/. Accessed February 18,2010.
- Centers for Disease Control and Prevention. Campaign to prevent antimicrobial resistance in healthcare settings. Available at http://www.cdc. gov/drugresistance/healthcare/default.htm. Accessed February 18,2010.
- ,.Guideline for Hand Hygiene in Health‐Care Settings: recommendations of the Healthcare Infection Control Practices Advisory Committee and the HICPAC/SHEA/APIC/IDSA Hand Hygiene Task Force.Infect Control Hosp Epidemiol.2002;23:S3–40.
- ,,, et al.Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the society for healthcare epidemiology of America (SHEA) and the infectious diseases society of America (IDSA).Infect Control Hosp Epidemiol.2010;31:431–455.
- ,,, et al.SHEA guideline for preventing nosocomial transmission of multidrug‐resistant strains of Staphylococcus aureus and enterococcus.Infect Control Hosp Epidemiol.2003;24:362–386.
- ,,, et al.Society for Healthcare Epidemiology of America and Infectious Diseases Society of America Joint Committee on the Prevention of Antimicrobial Resistance: guidelines for the prevention of antimicrobial resistance in hospitals.Clin Infect Dis.1997;25:584–599.
- ,,, et al.SHEA/APIC guideline: infection prevention and control in the long‐term care facility, July 2008.Infect Control Hosp Epidemiol.2008;29:785–814.
- ,,, et al.A compendium of strategies to prevent healthcare‐associated infections in acute care hospitals.Infect Control Hosp Epidemiol.2008;29 Suppl 1:S12–21.
- Australian Commission on Safety and Quality in Health Care (2009). Windows into Safety and Quality in Health Care 2009, ASCQHC, Sydney, Australia. Available at http://www.safetyandquality.gov/au. Accessed June 20, 2010.
- ,,,.Antibiotic stewardship implementation in the EU: the way forward.Expert Rev Anti Infect Ther.2009;7:1175–1183.
- ,,,.Optimization of antibiotic use in hospitals−antimicrobial stewardship and the EU project ABS international.Chemotherapy.2008;54:260–267.
- ,,,,,.European Antibiotic Awareness Day, 2008−The first European‐wide public information campaign on prudent antibiotic use: methods and survey of participating countries.Euro Surveill.2009;14:19280.
- The Infectious Diseases Society of America. Strategies to Address Antimicrobial Resistance (STAAR) Act. Available at http://www.idsociety.org/STAARAct.htm. Accessed June 20,2010.
Clinicians want to use antimicrobials as effectively as possible, but good intentions frequently fail to translate into adherence to best available evidence. This was recognized as early as 1956, when Earnest Jawetz noted that [The physician] is under great pressure to prescribe the newest, best, broadest antibiotic preparation, prescribe it for any complaint whatever, quickly, and preferably without worrying too much about specific etiologic diagnosis or proper indication of the drug.1 That was true in 1956, and is more so in 2010.
The term that has come to describe the collection of practices intended to optimize antimicrobial therapy is antimicrobial stewardship. The label is a relatively new one; it appears to have been coined in the mid‐1990s, and has slowly crept into the medical lexicon since then. However, many clinicians remain uncertain of exactly what antimicrobial stewardship means or what practices characterize it. The 2007 Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA) guidelines for developing institutional programs to enhance antimicrobial use define antimicrobial stewardship as an activity that includes appropriate selection, dosing, route, and duration of antimicrobial therapy, with a primary goal of optimizing clinical outcomes while minimizing unintended consequences of antimicrobial use.2
In 2004, IDSA published a document titled Bad Bugs, No Drugs that noted a marked decrease in research and development of new antimicrobials within the pharmaceutical industry, at the same time that the number of antibiotic‐resistant bacteria was dramatically increasing, particularly within healthcare settings.3, 4 Since that time, trends in antimicrobial drug development have not appreciably improved, and governmental agencies have been largely silent or ineffective in addressing the problem.58 The growth of multidrug‐resistant and sometimes pan‐resistant pathogens, at a time when newer agents with novel mechanisms of action are not available nor expected for years to come, is particularly troublesome. This scenario highlights the necessity of using currently available agents in a manner that prolongs their effectiveness and reduces further emergence and spread of resistant pathogensie, the crucial need for improved antimicrobial stewardship.
Antimicrobial stewardship recognizes that antimicrobials are a unique category of drugs, with potential to affect treatment outcomes in patients for whom the drug was not originally intended. For example, cardiac and oncologic medications can be costly, and may certainly be toxic, but their effects are limited to the patient that directly receives them. In contrast, the effects of antimicrobials prescribed to a patient in the hospital (or outside the hospital, for that matter), can ripple across the hospital system and beyond. This reflects the fact that suboptimal use of antimicrobials may induce or otherwise promote the development of antimicrobial‐resistant pathogens, with potential wide‐ranging impact.9, 10 Misuse or overuse of antimicrobials can also promote colonization and overgrowth of potentially toxic pathogens within hospitalized patients, such as Clostridium difficile, that may then be transferred widely within the hospital or other healthcare institutions, particularly when infection control measures are also less than optimal. This unique societal feature of antimicrobial drugs highlights the vital importance of antimicrobial stewardship within the hospital setting, especially at a time when few novel agents exist in the developmental pipeline, at least for gram‐negative pathogens.
Are hospitalists stakeholders in this? The answer is a resounding Yes, for several reasons. Hospitalists are the dominant prescribers of antibiotics and other antimicrobials in the United States, almost certainly prescribing more antibiotics in the hospital setting than infectious disease specialists, intensivists, or any other medical specialty or subspecialty. As such, hospitalists play a central role in the optimization of antimicrobial use in the hospital setting, including minimizing negative consequences arising from antimicrobial drug misuse or overuse. In addition, the role hospitalists play in this process is likely to increase in the future. The field of hospital medicine has grown from modest beginnings in the mid‐1990s to approximately 30,000 hospitalists in the United States today.11 Based on a sample of Medicare beneficiaries, 6% of general internists were identified as hospitalists in 1995 versus 19% in 2006, and the percentage of claims for inpatient services provided by general internists who were hospitalists increased from 9% to 37%.12 Current estimates are that >50% of medical inpatients in the United States are cared for by hospitalists (personal communication, J. Miller, Society of Hospital Medicine). Importantly, hospitalists manage patients not only on regular medical floors, but also in intensive care units (ICUs), where antimicrobial resistance is a particular problem.13, 14 Finally, hospitalists serve as gatekeepers for patients leaving the hospital on either oral or intravenous antibiotics or other antimicrobials. As such, via interactions with primary care physicians, hospitalists can play a key role in improving antimicrobial stewardship not only within, but also beyond, the hospital setting.
Hospital medicine is the first medical specialty to make patient safety and quality improvement central principles of its practice, and antimicrobial stewardship is an under‐recognized but central tenet of patient safety. As a consequence, hospitalists are natural allies of stewardship programs. Indeed, antimicrobial stewardship may be considered an example of the Holy Grail of quality improvement; ie, an intervention that improves outcomes while leading to cost savings, and should thus resonate with all hospitalists.
A number of clinical practice guidelines or campaigns have been developed by professional societies and governmental organizations in the United States2, 1526 and other countries2730 for the promotion of improved antimicrobial stewardship and/or infection control in hospitals and long‐term care facilities. The goals of these initiatives, campaigns, or guidelines are to provide information that can be utilized to prevent or slow the development and spread of hospital‐acquired infections, particularly those involving antimicrobial‐resistant pathogens. In addition, the United States Congress is currently considering the STAAR (Strategies to Address Antimicrobial Resistance) Act to encourage the use of innovative governmental and private approaches to combat this critical and expanding problem.31
This supplement to the Journal of Hospital Medicine contains several articles of interest to hospitalists seeking to positively impact patient care through improvements in antimicrobial stewardship. The first article by Christopher Ohl, MD, examines general principles of antimicrobial stewardship programs as they apply to inpatient facilities. The second article by Thomas Lodise, PharmD, explores the effective use of pharmacokinetic‐pharmacodynamic principles in antimicrobial stewardship. In the third article, Steven Gordon, MD, moves beyond the hospital setting to discuss antimicrobial stewardship at the time of hospital discharge and beyond. Finally, Arjun Srinivasan, MD, outlines several practical ways in which hospitalists may take leadership roles in stewardship efforts at their institutions. Our hope is that the supplement will be thought‐provoking, and ultimately lead to greater partnership between hospitalists and other stakeholders in antimicrobial stewardship.
Clinicians want to use antimicrobials as effectively as possible, but good intentions frequently fail to translate into adherence to best available evidence. This was recognized as early as 1956, when Earnest Jawetz noted that [The physician] is under great pressure to prescribe the newest, best, broadest antibiotic preparation, prescribe it for any complaint whatever, quickly, and preferably without worrying too much about specific etiologic diagnosis or proper indication of the drug.1 That was true in 1956, and is more so in 2010.
The term that has come to describe the collection of practices intended to optimize antimicrobial therapy is antimicrobial stewardship. The label is a relatively new one; it appears to have been coined in the mid‐1990s, and has slowly crept into the medical lexicon since then. However, many clinicians remain uncertain of exactly what antimicrobial stewardship means or what practices characterize it. The 2007 Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA) guidelines for developing institutional programs to enhance antimicrobial use define antimicrobial stewardship as an activity that includes appropriate selection, dosing, route, and duration of antimicrobial therapy, with a primary goal of optimizing clinical outcomes while minimizing unintended consequences of antimicrobial use.2
In 2004, IDSA published a document titled Bad Bugs, No Drugs that noted a marked decrease in research and development of new antimicrobials within the pharmaceutical industry, at the same time that the number of antibiotic‐resistant bacteria was dramatically increasing, particularly within healthcare settings.3, 4 Since that time, trends in antimicrobial drug development have not appreciably improved, and governmental agencies have been largely silent or ineffective in addressing the problem.58 The growth of multidrug‐resistant and sometimes pan‐resistant pathogens, at a time when newer agents with novel mechanisms of action are not available nor expected for years to come, is particularly troublesome. This scenario highlights the necessity of using currently available agents in a manner that prolongs their effectiveness and reduces further emergence and spread of resistant pathogensie, the crucial need for improved antimicrobial stewardship.
Antimicrobial stewardship recognizes that antimicrobials are a unique category of drugs, with potential to affect treatment outcomes in patients for whom the drug was not originally intended. For example, cardiac and oncologic medications can be costly, and may certainly be toxic, but their effects are limited to the patient that directly receives them. In contrast, the effects of antimicrobials prescribed to a patient in the hospital (or outside the hospital, for that matter), can ripple across the hospital system and beyond. This reflects the fact that suboptimal use of antimicrobials may induce or otherwise promote the development of antimicrobial‐resistant pathogens, with potential wide‐ranging impact.9, 10 Misuse or overuse of antimicrobials can also promote colonization and overgrowth of potentially toxic pathogens within hospitalized patients, such as Clostridium difficile, that may then be transferred widely within the hospital or other healthcare institutions, particularly when infection control measures are also less than optimal. This unique societal feature of antimicrobial drugs highlights the vital importance of antimicrobial stewardship within the hospital setting, especially at a time when few novel agents exist in the developmental pipeline, at least for gram‐negative pathogens.
Are hospitalists stakeholders in this? The answer is a resounding Yes, for several reasons. Hospitalists are the dominant prescribers of antibiotics and other antimicrobials in the United States, almost certainly prescribing more antibiotics in the hospital setting than infectious disease specialists, intensivists, or any other medical specialty or subspecialty. As such, hospitalists play a central role in the optimization of antimicrobial use in the hospital setting, including minimizing negative consequences arising from antimicrobial drug misuse or overuse. In addition, the role hospitalists play in this process is likely to increase in the future. The field of hospital medicine has grown from modest beginnings in the mid‐1990s to approximately 30,000 hospitalists in the United States today.11 Based on a sample of Medicare beneficiaries, 6% of general internists were identified as hospitalists in 1995 versus 19% in 2006, and the percentage of claims for inpatient services provided by general internists who were hospitalists increased from 9% to 37%.12 Current estimates are that >50% of medical inpatients in the United States are cared for by hospitalists (personal communication, J. Miller, Society of Hospital Medicine). Importantly, hospitalists manage patients not only on regular medical floors, but also in intensive care units (ICUs), where antimicrobial resistance is a particular problem.13, 14 Finally, hospitalists serve as gatekeepers for patients leaving the hospital on either oral or intravenous antibiotics or other antimicrobials. As such, via interactions with primary care physicians, hospitalists can play a key role in improving antimicrobial stewardship not only within, but also beyond, the hospital setting.
Hospital medicine is the first medical specialty to make patient safety and quality improvement central principles of its practice, and antimicrobial stewardship is an under‐recognized but central tenet of patient safety. As a consequence, hospitalists are natural allies of stewardship programs. Indeed, antimicrobial stewardship may be considered an example of the Holy Grail of quality improvement; ie, an intervention that improves outcomes while leading to cost savings, and should thus resonate with all hospitalists.
A number of clinical practice guidelines or campaigns have been developed by professional societies and governmental organizations in the United States2, 1526 and other countries2730 for the promotion of improved antimicrobial stewardship and/or infection control in hospitals and long‐term care facilities. The goals of these initiatives, campaigns, or guidelines are to provide information that can be utilized to prevent or slow the development and spread of hospital‐acquired infections, particularly those involving antimicrobial‐resistant pathogens. In addition, the United States Congress is currently considering the STAAR (Strategies to Address Antimicrobial Resistance) Act to encourage the use of innovative governmental and private approaches to combat this critical and expanding problem.31
This supplement to the Journal of Hospital Medicine contains several articles of interest to hospitalists seeking to positively impact patient care through improvements in antimicrobial stewardship. The first article by Christopher Ohl, MD, examines general principles of antimicrobial stewardship programs as they apply to inpatient facilities. The second article by Thomas Lodise, PharmD, explores the effective use of pharmacokinetic‐pharmacodynamic principles in antimicrobial stewardship. In the third article, Steven Gordon, MD, moves beyond the hospital setting to discuss antimicrobial stewardship at the time of hospital discharge and beyond. Finally, Arjun Srinivasan, MD, outlines several practical ways in which hospitalists may take leadership roles in stewardship efforts at their institutions. Our hope is that the supplement will be thought‐provoking, and ultimately lead to greater partnership between hospitalists and other stakeholders in antimicrobial stewardship.
- .Antimicrobial chemotherapy.Annu Rev Microbiol.1956;10:85–114.
- ,,, et al.Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America guidelines for developing an institutional program to enhance antimicrobial stewardship.Clin Infect Dis.2007;44:159–177.
- Bad bugs, no drugs: as antibiotic R2004.
- ,,,,.Trends in antimicrobial drug development: implications for the future.Clin Infect Dis.2004;38:1279–1286.
- ,,, et al.Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America.Clin Infect Dis.2009;48:1–12.
- .Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE.J Infect Dis.2008;197:1079–1081.
- ,,, et al.The epidemic of antibiotic‐resistant infections: a call to action for the medical community from the Infectious Diseases Society of America.Clin Infect Dis.2008;46:155–164.
- ,,,,,.Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America.Clin Infect Dis.2006;42:657–668.
- .“Collateral damage” from cephalosporin or quinolone antibiotic therapy.Clin Infect Dis.2004;38(Suppl 4):S341–345.
- .Collateral damage and what the future might hold. The need to balance prudent antibiotic utilization and stewardship with effective patient management.Int J Infect Dis.2006;10:S17–S24.
- .Hospitalists and intensivists: partners in caring for the critically ill‐−the time has come.J Hosp Med.2010;5:1–3.
- ,,,.Growth in the care of older patients by hospitalists in the United States.N Engl J Med.2009;360:1102–1112.
- ,,.Epidemiology and control of antibiotic resistance in the intensive care unit.Curr Opin Infect Dis.2004;17:309–316.
- .Bench‐to‐bedside review: antimicrobial utilization strategies aimed at preventing the emergence of bacterial resistance in the intensive care unit.Crit Care.2005;9:459–464.
- Centers for Disease Control and Prevention. Get smart: know when antibiotics work. Available at http://www.cdc.gov/getsmart/. Accessed June 20,2010.
- The Society for Healthcare Epidemiology of America. Compendium of strategies to prevent healthcare‐associated infections in acute care hospitals. Available at http://www.shea‐online.org/about/compendium.cfm. Accessed June 20,2010.
- ,,, et al.Therapeutic monitoring of vancomycin in adult patients: a consensus review of the American Society of Health‐System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists.Am J Health Syst Pharm.2009;66:82–98.
- Centers for Disease Control and Prevention. Department of Health and Human Services. Infection control in healthcare settings. Available at http://www.cdc.gov/nidod/dhqp/. Accessed February 17,2010.
- Centers for Disease Control and Prevention. Healthcare Infection Control Practices Advisory Committee (HICPAC). Available at http://www.cdc. gov/hicpac/. Accessed February 18,2010.
- Centers for Disease Control and Prevention. Campaign to prevent antimicrobial resistance in healthcare settings. Available at http://www.cdc. gov/drugresistance/healthcare/default.htm. Accessed February 18,2010.
- ,.Guideline for Hand Hygiene in Health‐Care Settings: recommendations of the Healthcare Infection Control Practices Advisory Committee and the HICPAC/SHEA/APIC/IDSA Hand Hygiene Task Force.Infect Control Hosp Epidemiol.2002;23:S3–40.
- ,,, et al.Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the society for healthcare epidemiology of America (SHEA) and the infectious diseases society of America (IDSA).Infect Control Hosp Epidemiol.2010;31:431–455.
- ,,, et al.SHEA guideline for preventing nosocomial transmission of multidrug‐resistant strains of Staphylococcus aureus and enterococcus.Infect Control Hosp Epidemiol.2003;24:362–386.
- ,,, et al.Society for Healthcare Epidemiology of America and Infectious Diseases Society of America Joint Committee on the Prevention of Antimicrobial Resistance: guidelines for the prevention of antimicrobial resistance in hospitals.Clin Infect Dis.1997;25:584–599.
- ,,, et al.SHEA/APIC guideline: infection prevention and control in the long‐term care facility, July 2008.Infect Control Hosp Epidemiol.2008;29:785–814.
- ,,, et al.A compendium of strategies to prevent healthcare‐associated infections in acute care hospitals.Infect Control Hosp Epidemiol.2008;29 Suppl 1:S12–21.
- Australian Commission on Safety and Quality in Health Care (2009). Windows into Safety and Quality in Health Care 2009, ASCQHC, Sydney, Australia. Available at http://www.safetyandquality.gov/au. Accessed June 20, 2010.
- ,,,.Antibiotic stewardship implementation in the EU: the way forward.Expert Rev Anti Infect Ther.2009;7:1175–1183.
- ,,,.Optimization of antibiotic use in hospitals−antimicrobial stewardship and the EU project ABS international.Chemotherapy.2008;54:260–267.
- ,,,,,.European Antibiotic Awareness Day, 2008−The first European‐wide public information campaign on prudent antibiotic use: methods and survey of participating countries.Euro Surveill.2009;14:19280.
- The Infectious Diseases Society of America. Strategies to Address Antimicrobial Resistance (STAAR) Act. Available at http://www.idsociety.org/STAARAct.htm. Accessed June 20,2010.
- .Antimicrobial chemotherapy.Annu Rev Microbiol.1956;10:85–114.
- ,,, et al.Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America guidelines for developing an institutional program to enhance antimicrobial stewardship.Clin Infect Dis.2007;44:159–177.
- Bad bugs, no drugs: as antibiotic R2004.
- ,,,,.Trends in antimicrobial drug development: implications for the future.Clin Infect Dis.2004;38:1279–1286.
- ,,, et al.Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America.Clin Infect Dis.2009;48:1–12.
- .Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE.J Infect Dis.2008;197:1079–1081.
- ,,, et al.The epidemic of antibiotic‐resistant infections: a call to action for the medical community from the Infectious Diseases Society of America.Clin Infect Dis.2008;46:155–164.
- ,,,,,.Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America.Clin Infect Dis.2006;42:657–668.
- .“Collateral damage” from cephalosporin or quinolone antibiotic therapy.Clin Infect Dis.2004;38(Suppl 4):S341–345.
- .Collateral damage and what the future might hold. The need to balance prudent antibiotic utilization and stewardship with effective patient management.Int J Infect Dis.2006;10:S17–S24.
- .Hospitalists and intensivists: partners in caring for the critically ill‐−the time has come.J Hosp Med.2010;5:1–3.
- ,,,.Growth in the care of older patients by hospitalists in the United States.N Engl J Med.2009;360:1102–1112.
- ,,.Epidemiology and control of antibiotic resistance in the intensive care unit.Curr Opin Infect Dis.2004;17:309–316.
- .Bench‐to‐bedside review: antimicrobial utilization strategies aimed at preventing the emergence of bacterial resistance in the intensive care unit.Crit Care.2005;9:459–464.
- Centers for Disease Control and Prevention. Get smart: know when antibiotics work. Available at http://www.cdc.gov/getsmart/. Accessed June 20,2010.
- The Society for Healthcare Epidemiology of America. Compendium of strategies to prevent healthcare‐associated infections in acute care hospitals. Available at http://www.shea‐online.org/about/compendium.cfm. Accessed June 20,2010.
- ,,, et al.Therapeutic monitoring of vancomycin in adult patients: a consensus review of the American Society of Health‐System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists.Am J Health Syst Pharm.2009;66:82–98.
- Centers for Disease Control and Prevention. Department of Health and Human Services. Infection control in healthcare settings. Available at http://www.cdc.gov/nidod/dhqp/. Accessed February 17,2010.
- Centers for Disease Control and Prevention. Healthcare Infection Control Practices Advisory Committee (HICPAC). Available at http://www.cdc. gov/hicpac/. Accessed February 18,2010.
- Centers for Disease Control and Prevention. Campaign to prevent antimicrobial resistance in healthcare settings. Available at http://www.cdc. gov/drugresistance/healthcare/default.htm. Accessed February 18,2010.
- ,.Guideline for Hand Hygiene in Health‐Care Settings: recommendations of the Healthcare Infection Control Practices Advisory Committee and the HICPAC/SHEA/APIC/IDSA Hand Hygiene Task Force.Infect Control Hosp Epidemiol.2002;23:S3–40.
- ,,, et al.Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the society for healthcare epidemiology of America (SHEA) and the infectious diseases society of America (IDSA).Infect Control Hosp Epidemiol.2010;31:431–455.
- ,,, et al.SHEA guideline for preventing nosocomial transmission of multidrug‐resistant strains of Staphylococcus aureus and enterococcus.Infect Control Hosp Epidemiol.2003;24:362–386.
- ,,, et al.Society for Healthcare Epidemiology of America and Infectious Diseases Society of America Joint Committee on the Prevention of Antimicrobial Resistance: guidelines for the prevention of antimicrobial resistance in hospitals.Clin Infect Dis.1997;25:584–599.
- ,,, et al.SHEA/APIC guideline: infection prevention and control in the long‐term care facility, July 2008.Infect Control Hosp Epidemiol.2008;29:785–814.
- ,,, et al.A compendium of strategies to prevent healthcare‐associated infections in acute care hospitals.Infect Control Hosp Epidemiol.2008;29 Suppl 1:S12–21.
- Australian Commission on Safety and Quality in Health Care (2009). Windows into Safety and Quality in Health Care 2009, ASCQHC, Sydney, Australia. Available at http://www.safetyandquality.gov/au. Accessed June 20, 2010.
- ,,,.Antibiotic stewardship implementation in the EU: the way forward.Expert Rev Anti Infect Ther.2009;7:1175–1183.
- ,,,.Optimization of antibiotic use in hospitals−antimicrobial stewardship and the EU project ABS international.Chemotherapy.2008;54:260–267.
- ,,,,,.European Antibiotic Awareness Day, 2008−The first European‐wide public information campaign on prudent antibiotic use: methods and survey of participating countries.Euro Surveill.2009;14:19280.
- The Infectious Diseases Society of America. Strategies to Address Antimicrobial Resistance (STAAR) Act. Available at http://www.idsociety.org/STAARAct.htm. Accessed June 20,2010.
CDC on Antimicrobial Stewardship
What if there was a quality improvement initiative that had been proven in multiple, peer‐reviewed publications to improve individual patient outcomes, reduce the overall burden of antimicrobial resistance, and save healthcare dollars? Surely such an initiative would enjoy widespread, if not uniform, adoption by health care facilities. Antimicrobial stewardship is just such an intervention. Ensuring that hospitalized patients receive the right antimicrobial, at the right dose, at the right time, and for the right duration has been shown to reduce mortality,1 reduce the risks of Clostridium difficileassociated diarrhea,2 shorten length of stay,3 reduce overall antimicrobial resistance within the facility,4 and save money.5 Yet despite these benefits, antimicrobial stewardship programs and interventions are far from the norm in US hospitals.
There are 2 important myths about antimicrobial stewardship that likely contribute substantially to the gap between the recognized benefits and implementation of stewardship interventions. Dispelling these myths is a crucial step in promoting wider adoption efforts to improve antimicrobial use. The first myth stems from the very name antimicrobial stewardship program, which has created a misperception that optimal inpatient antimicrobial use is only possible in settings with formal stewardship programs that are staffed by infectious diseases (ID) physicians and pharmacist. The best guidelines on implementing stewardship programs, developed by the Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America,6 may have contributed to this misperception by suggesting that optimal programs require dedicated time from both an ID physician and an ID‐trained pharmacist. However, many hospitals do not have ID physicians on staff, and the vast majority do not have access to an ID pharmacist who is comfortable with antimicrobial stewardship. Although these traditionally staffed programs have well‐proven benefits and are an excellent goal, they are not feasible in many hospitals. However, different types of stewardship interventions, led by a variety of health care providers and specialists, also have well‐proven benefits. Although these latter experiences are much less likely to appear in peer‐reviewed medical journals, experts in antimicrobial stewardship indicate that they often hear about very successful stewardship interventions being led by groups like general clinical pharmacists, intensivists, and hospitalists. Workshops on antimicrobial stewardship are often full of attendees who are successfully improving antimicrobial use in facilities that represent the full spectrum of US hospitals: large and small, urban and rural, teaching and nonteaching. Indeed, I prefer the term antimicrobial stewardship programs and interventions to convey that improving antimicrobial use can be done, and done well, even without the ideally staffed program.
The second myth is that the only goal of stewardship programs and interventions is to stop clinicians from using antimicrobials. This misperception has led to counterproductive attitudes toward stewardship programs and interventions in some facilities. Without question, stopping unnecessary antimicrobial use is an important aspect of stewardship interventions that has well‐established benefits for patients and hospitals. That one third to one half of all inpatient antimicrobial use might be unnecessary, combined with the growing problem of C. difficile, certainly supports the goal of reducing inappropriate antimicrobial use. However, the primary goal of stewardship is to optimize antimicrobial therapy. In many instances, this does involve stopping unneeded antimicrobials, and because stopping antibiotics has the most readily demonstrable benefits on patient and financial outcomes, interventions with this aim are the subject of nearly all published studies. However, anyone who has worked on stewardship interventions can describe numerous instances when the recommendation provided was to broaden or lengthen antimicrobial therapy. Moreover, surveys indicate that, far from viewing stewardship as an intrusion or infringement on their autonomy, clinicians appreciate and even want the assistance that these efforts provide.7
If stewardship has substantial proven benefits, can be implemented in nearly any hospital setting, and is welcomed by providers, what can be done to move toward broader implementation? I believe that engaging hospitalists more fully in stewardship efforts will be a critical step in this direction. Hospitalists already provide a substantial portion of all inpatient care in the United States, and the numbers of hospitalists are growing rapidly. Moreover, they are increasingly taking the lead in a variety of quality improvement initiatives. Hence, hospitalists are ideally positioned and well suited to move stewardship efforts forward. Some, including hospitalists, have also suggested that developing a practical stewardship implementation framework would be helpful in promoting these interventions.
This suggestion has led the Centers for Disease Control and Prevention's (CDC) Get Smart for Healthcare campaign to partner with the Institute for Healthcare Improvement (IHI) and a variety of external experts (including a hospitalist) to develop such a framework using the IHI's Driver Diagram and Change Package methodology. The driver diagram seeks to identify a core set of highly influential practices that lead to a desired outcome. For optimizing antimicrobial use, the primary drivers that were identified by experts include: 1) timely and appropriate initiation of antibiotics; 2) appropriate administration and de‐escalation of therapy; 3) data monitoring and transparency (measuring and feeding back to clinicians data on antimicrobial use and resistance); and 4) improving stewardship infrastructure, knowledge, and engagement in antimicrobial stewardship efforts. Once these drivers were identified, the expert panel then identified a number of specific practices, or change concepts, that would support progress toward each driver. Now that the Driver Diagram and Change Package has been drafted, the CDC and IHI are collaborating on a pilot testing effort and are working to ensure that a substantial number of the pilot projects are led by hospitalists. Our goal is that the Driver Diagram and Change Package will be honed and refined with the help of hospitalists so that the end result will be a highly implementable set of antimicrobial stewardship interventions that can be widely applied by hospitalists around the country.
However, we need not wait for finalization of the Driver Diagram and Change Package to begin a productive collaboration on antimicrobial stewardship. In addition to the project with IHI, Get Smart for Healthcare is working to identify a variety of resources that would be useful in implementing and improving stewardship efforts. To that end, we would love to hear from any hospitalists who would like to share their experiences with stewardship interventions or who have tools (eg, order sets), ideas (eg, particularly successful intervention projects), or success stories. They can be e‐mailed to [email protected].
For now, I would like to suggest that there are 4 antibiotic quality improvement projects hospitalists would be ideally suited to lead. The first is ensuring that all antibiotic orders include a Dose, Duration, Indication. Efforts to improve antibiotic use are often hampered because the nonprescribing providers are not sure why the patient is on antibiotics. This problem is amplified when patients are transitioned from one provider to another or when multiple providers are involved. Specifying the duration and indication in all antibiotic orders will ensure that treatments continue for the right amount of time and would allow therapy to be stopped if the initially suspected infection is ruled out or altered if another infection is identified. The second improvement project is developing a process to ensure that any patient with a positive blood culture is on the appropriate therapy. This is a relatively straightforward intervention that is based on the patient's own microbiology results, and it ensures the optimal therapy of a serious infection. Third is the development of an intervention to encourage the reassessment of patients who are started on antibiotics for community‐acquired pneumonia (CAP). Several hospitalists have suggested that the pressure to initiate therapy quickly in cases of CAP often leads to overtreatment. Interventions that encourage a reexamination of the CAP diagnosis when the clinical situation has stabilized would likely reduce this overtreatment. And the fourth improvement project is ensuring that urinary tract infections (UTIs) in hospitalized patients are properly diagnosed and treated. Work done by hospitalists at the University of Michigan suggests that improving the diagnosis and treatment of UTIs would have a significant impact on improving antibiotic use.8 Currently, the CDC is collaborating with these investigators to develop protocols and tools to improve the treatment of inpatient UTIs.
The time to promote aggressive implementation of antimicrobial stewardship interventions has come. Clinicians are increasingly encountering infections for which there are very limited or, in some cases, no good treatment optionsand there are very few new antibiotics on the horizon. Many groups are advocating for expanded efforts to develop new antibiotics.9 Although this is crucial, it is just as important that we work now to aggressively improve the use of the agents we have. Not only might this extend the life of our current agents, but it will also help ensure that any new agents will enjoy longer periods of effectiveness. Indeed, failing to inextricably link the development of new antibiotics with efforts to improve antibiotic use is akin to buying a new car to drive on a road full of potholes. Fortunately, there are a number of interventions that have proven successful; we now need to determine how best to apply these interventions in more settings. We want and need the involvement of hospitalists in these efforts. Yes, improving antimicrobial stewardship will require investments, but past experience tells us that the alternative could prove far more costly.
- .Inadequate antimicrobial treatment: an important determinant of outcome for hospitalized patients.Clin Infect Dis.2000;31:S131–S138.
- ,,, et al.Impact of a reduction in the use of high‐risk antibiotics on the course of an epidemic of Clostridium difficile‐associated disease caused by the hypervirulent NAP1/027 strain.Clin Infect Dis.2007;45(Suppl 2):S112–S121.
- ,,, et al.Early transition to oral antibiotic therapy for community‐acquired pneumonia: duration of therapy, clinical outcomes, and cost analysis.Respir Med.1998;92:1032–1039.
- ,,, et al.A hospitalwide intervention program to optimize the quality of antibiotic use: impact on prescribing practice, antibiotic consumption, cost savings, and bacterial resistance.Clin Infect Dis.2003;37:180–186.
- ,,, et al.A World Wide Web‐based antimicrobial stewardship program improves efficiency, communication, and user satisfaction and reduces cost in a tertiary care pediatric medical center.Clin Infect Dis.2008;47:747–753.
- ,,, et al.Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America guidelines for developing an institutional program to enhance antimicrobial stewardship.Clin Infect Dis.2007;44:159–177.
- ,,, et al.A knowledge, attitudes and beliefs survey of housestaff physicians from various specialties concerning antimicrobial use and resistance.Arch Intern Med.2004;164:1451–1456.
- ,,, et al.Importance of urinary tract infection to antibiotic use among hospitalized patients.Infect Control Hosp Epidemiol.2009;30:193–195.
- Infectious Diseases Society of America.The 10 × '20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020.Clin Infect Dis.2010;50:1081–1083.
What if there was a quality improvement initiative that had been proven in multiple, peer‐reviewed publications to improve individual patient outcomes, reduce the overall burden of antimicrobial resistance, and save healthcare dollars? Surely such an initiative would enjoy widespread, if not uniform, adoption by health care facilities. Antimicrobial stewardship is just such an intervention. Ensuring that hospitalized patients receive the right antimicrobial, at the right dose, at the right time, and for the right duration has been shown to reduce mortality,1 reduce the risks of Clostridium difficileassociated diarrhea,2 shorten length of stay,3 reduce overall antimicrobial resistance within the facility,4 and save money.5 Yet despite these benefits, antimicrobial stewardship programs and interventions are far from the norm in US hospitals.
There are 2 important myths about antimicrobial stewardship that likely contribute substantially to the gap between the recognized benefits and implementation of stewardship interventions. Dispelling these myths is a crucial step in promoting wider adoption efforts to improve antimicrobial use. The first myth stems from the very name antimicrobial stewardship program, which has created a misperception that optimal inpatient antimicrobial use is only possible in settings with formal stewardship programs that are staffed by infectious diseases (ID) physicians and pharmacist. The best guidelines on implementing stewardship programs, developed by the Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America,6 may have contributed to this misperception by suggesting that optimal programs require dedicated time from both an ID physician and an ID‐trained pharmacist. However, many hospitals do not have ID physicians on staff, and the vast majority do not have access to an ID pharmacist who is comfortable with antimicrobial stewardship. Although these traditionally staffed programs have well‐proven benefits and are an excellent goal, they are not feasible in many hospitals. However, different types of stewardship interventions, led by a variety of health care providers and specialists, also have well‐proven benefits. Although these latter experiences are much less likely to appear in peer‐reviewed medical journals, experts in antimicrobial stewardship indicate that they often hear about very successful stewardship interventions being led by groups like general clinical pharmacists, intensivists, and hospitalists. Workshops on antimicrobial stewardship are often full of attendees who are successfully improving antimicrobial use in facilities that represent the full spectrum of US hospitals: large and small, urban and rural, teaching and nonteaching. Indeed, I prefer the term antimicrobial stewardship programs and interventions to convey that improving antimicrobial use can be done, and done well, even without the ideally staffed program.
The second myth is that the only goal of stewardship programs and interventions is to stop clinicians from using antimicrobials. This misperception has led to counterproductive attitudes toward stewardship programs and interventions in some facilities. Without question, stopping unnecessary antimicrobial use is an important aspect of stewardship interventions that has well‐established benefits for patients and hospitals. That one third to one half of all inpatient antimicrobial use might be unnecessary, combined with the growing problem of C. difficile, certainly supports the goal of reducing inappropriate antimicrobial use. However, the primary goal of stewardship is to optimize antimicrobial therapy. In many instances, this does involve stopping unneeded antimicrobials, and because stopping antibiotics has the most readily demonstrable benefits on patient and financial outcomes, interventions with this aim are the subject of nearly all published studies. However, anyone who has worked on stewardship interventions can describe numerous instances when the recommendation provided was to broaden or lengthen antimicrobial therapy. Moreover, surveys indicate that, far from viewing stewardship as an intrusion or infringement on their autonomy, clinicians appreciate and even want the assistance that these efforts provide.7
If stewardship has substantial proven benefits, can be implemented in nearly any hospital setting, and is welcomed by providers, what can be done to move toward broader implementation? I believe that engaging hospitalists more fully in stewardship efforts will be a critical step in this direction. Hospitalists already provide a substantial portion of all inpatient care in the United States, and the numbers of hospitalists are growing rapidly. Moreover, they are increasingly taking the lead in a variety of quality improvement initiatives. Hence, hospitalists are ideally positioned and well suited to move stewardship efforts forward. Some, including hospitalists, have also suggested that developing a practical stewardship implementation framework would be helpful in promoting these interventions.
This suggestion has led the Centers for Disease Control and Prevention's (CDC) Get Smart for Healthcare campaign to partner with the Institute for Healthcare Improvement (IHI) and a variety of external experts (including a hospitalist) to develop such a framework using the IHI's Driver Diagram and Change Package methodology. The driver diagram seeks to identify a core set of highly influential practices that lead to a desired outcome. For optimizing antimicrobial use, the primary drivers that were identified by experts include: 1) timely and appropriate initiation of antibiotics; 2) appropriate administration and de‐escalation of therapy; 3) data monitoring and transparency (measuring and feeding back to clinicians data on antimicrobial use and resistance); and 4) improving stewardship infrastructure, knowledge, and engagement in antimicrobial stewardship efforts. Once these drivers were identified, the expert panel then identified a number of specific practices, or change concepts, that would support progress toward each driver. Now that the Driver Diagram and Change Package has been drafted, the CDC and IHI are collaborating on a pilot testing effort and are working to ensure that a substantial number of the pilot projects are led by hospitalists. Our goal is that the Driver Diagram and Change Package will be honed and refined with the help of hospitalists so that the end result will be a highly implementable set of antimicrobial stewardship interventions that can be widely applied by hospitalists around the country.
However, we need not wait for finalization of the Driver Diagram and Change Package to begin a productive collaboration on antimicrobial stewardship. In addition to the project with IHI, Get Smart for Healthcare is working to identify a variety of resources that would be useful in implementing and improving stewardship efforts. To that end, we would love to hear from any hospitalists who would like to share their experiences with stewardship interventions or who have tools (eg, order sets), ideas (eg, particularly successful intervention projects), or success stories. They can be e‐mailed to [email protected].
For now, I would like to suggest that there are 4 antibiotic quality improvement projects hospitalists would be ideally suited to lead. The first is ensuring that all antibiotic orders include a Dose, Duration, Indication. Efforts to improve antibiotic use are often hampered because the nonprescribing providers are not sure why the patient is on antibiotics. This problem is amplified when patients are transitioned from one provider to another or when multiple providers are involved. Specifying the duration and indication in all antibiotic orders will ensure that treatments continue for the right amount of time and would allow therapy to be stopped if the initially suspected infection is ruled out or altered if another infection is identified. The second improvement project is developing a process to ensure that any patient with a positive blood culture is on the appropriate therapy. This is a relatively straightforward intervention that is based on the patient's own microbiology results, and it ensures the optimal therapy of a serious infection. Third is the development of an intervention to encourage the reassessment of patients who are started on antibiotics for community‐acquired pneumonia (CAP). Several hospitalists have suggested that the pressure to initiate therapy quickly in cases of CAP often leads to overtreatment. Interventions that encourage a reexamination of the CAP diagnosis when the clinical situation has stabilized would likely reduce this overtreatment. And the fourth improvement project is ensuring that urinary tract infections (UTIs) in hospitalized patients are properly diagnosed and treated. Work done by hospitalists at the University of Michigan suggests that improving the diagnosis and treatment of UTIs would have a significant impact on improving antibiotic use.8 Currently, the CDC is collaborating with these investigators to develop protocols and tools to improve the treatment of inpatient UTIs.
The time to promote aggressive implementation of antimicrobial stewardship interventions has come. Clinicians are increasingly encountering infections for which there are very limited or, in some cases, no good treatment optionsand there are very few new antibiotics on the horizon. Many groups are advocating for expanded efforts to develop new antibiotics.9 Although this is crucial, it is just as important that we work now to aggressively improve the use of the agents we have. Not only might this extend the life of our current agents, but it will also help ensure that any new agents will enjoy longer periods of effectiveness. Indeed, failing to inextricably link the development of new antibiotics with efforts to improve antibiotic use is akin to buying a new car to drive on a road full of potholes. Fortunately, there are a number of interventions that have proven successful; we now need to determine how best to apply these interventions in more settings. We want and need the involvement of hospitalists in these efforts. Yes, improving antimicrobial stewardship will require investments, but past experience tells us that the alternative could prove far more costly.
What if there was a quality improvement initiative that had been proven in multiple, peer‐reviewed publications to improve individual patient outcomes, reduce the overall burden of antimicrobial resistance, and save healthcare dollars? Surely such an initiative would enjoy widespread, if not uniform, adoption by health care facilities. Antimicrobial stewardship is just such an intervention. Ensuring that hospitalized patients receive the right antimicrobial, at the right dose, at the right time, and for the right duration has been shown to reduce mortality,1 reduce the risks of Clostridium difficileassociated diarrhea,2 shorten length of stay,3 reduce overall antimicrobial resistance within the facility,4 and save money.5 Yet despite these benefits, antimicrobial stewardship programs and interventions are far from the norm in US hospitals.
There are 2 important myths about antimicrobial stewardship that likely contribute substantially to the gap between the recognized benefits and implementation of stewardship interventions. Dispelling these myths is a crucial step in promoting wider adoption efforts to improve antimicrobial use. The first myth stems from the very name antimicrobial stewardship program, which has created a misperception that optimal inpatient antimicrobial use is only possible in settings with formal stewardship programs that are staffed by infectious diseases (ID) physicians and pharmacist. The best guidelines on implementing stewardship programs, developed by the Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America,6 may have contributed to this misperception by suggesting that optimal programs require dedicated time from both an ID physician and an ID‐trained pharmacist. However, many hospitals do not have ID physicians on staff, and the vast majority do not have access to an ID pharmacist who is comfortable with antimicrobial stewardship. Although these traditionally staffed programs have well‐proven benefits and are an excellent goal, they are not feasible in many hospitals. However, different types of stewardship interventions, led by a variety of health care providers and specialists, also have well‐proven benefits. Although these latter experiences are much less likely to appear in peer‐reviewed medical journals, experts in antimicrobial stewardship indicate that they often hear about very successful stewardship interventions being led by groups like general clinical pharmacists, intensivists, and hospitalists. Workshops on antimicrobial stewardship are often full of attendees who are successfully improving antimicrobial use in facilities that represent the full spectrum of US hospitals: large and small, urban and rural, teaching and nonteaching. Indeed, I prefer the term antimicrobial stewardship programs and interventions to convey that improving antimicrobial use can be done, and done well, even without the ideally staffed program.
The second myth is that the only goal of stewardship programs and interventions is to stop clinicians from using antimicrobials. This misperception has led to counterproductive attitudes toward stewardship programs and interventions in some facilities. Without question, stopping unnecessary antimicrobial use is an important aspect of stewardship interventions that has well‐established benefits for patients and hospitals. That one third to one half of all inpatient antimicrobial use might be unnecessary, combined with the growing problem of C. difficile, certainly supports the goal of reducing inappropriate antimicrobial use. However, the primary goal of stewardship is to optimize antimicrobial therapy. In many instances, this does involve stopping unneeded antimicrobials, and because stopping antibiotics has the most readily demonstrable benefits on patient and financial outcomes, interventions with this aim are the subject of nearly all published studies. However, anyone who has worked on stewardship interventions can describe numerous instances when the recommendation provided was to broaden or lengthen antimicrobial therapy. Moreover, surveys indicate that, far from viewing stewardship as an intrusion or infringement on their autonomy, clinicians appreciate and even want the assistance that these efforts provide.7
If stewardship has substantial proven benefits, can be implemented in nearly any hospital setting, and is welcomed by providers, what can be done to move toward broader implementation? I believe that engaging hospitalists more fully in stewardship efforts will be a critical step in this direction. Hospitalists already provide a substantial portion of all inpatient care in the United States, and the numbers of hospitalists are growing rapidly. Moreover, they are increasingly taking the lead in a variety of quality improvement initiatives. Hence, hospitalists are ideally positioned and well suited to move stewardship efforts forward. Some, including hospitalists, have also suggested that developing a practical stewardship implementation framework would be helpful in promoting these interventions.
This suggestion has led the Centers for Disease Control and Prevention's (CDC) Get Smart for Healthcare campaign to partner with the Institute for Healthcare Improvement (IHI) and a variety of external experts (including a hospitalist) to develop such a framework using the IHI's Driver Diagram and Change Package methodology. The driver diagram seeks to identify a core set of highly influential practices that lead to a desired outcome. For optimizing antimicrobial use, the primary drivers that were identified by experts include: 1) timely and appropriate initiation of antibiotics; 2) appropriate administration and de‐escalation of therapy; 3) data monitoring and transparency (measuring and feeding back to clinicians data on antimicrobial use and resistance); and 4) improving stewardship infrastructure, knowledge, and engagement in antimicrobial stewardship efforts. Once these drivers were identified, the expert panel then identified a number of specific practices, or change concepts, that would support progress toward each driver. Now that the Driver Diagram and Change Package has been drafted, the CDC and IHI are collaborating on a pilot testing effort and are working to ensure that a substantial number of the pilot projects are led by hospitalists. Our goal is that the Driver Diagram and Change Package will be honed and refined with the help of hospitalists so that the end result will be a highly implementable set of antimicrobial stewardship interventions that can be widely applied by hospitalists around the country.
However, we need not wait for finalization of the Driver Diagram and Change Package to begin a productive collaboration on antimicrobial stewardship. In addition to the project with IHI, Get Smart for Healthcare is working to identify a variety of resources that would be useful in implementing and improving stewardship efforts. To that end, we would love to hear from any hospitalists who would like to share their experiences with stewardship interventions or who have tools (eg, order sets), ideas (eg, particularly successful intervention projects), or success stories. They can be e‐mailed to [email protected].
For now, I would like to suggest that there are 4 antibiotic quality improvement projects hospitalists would be ideally suited to lead. The first is ensuring that all antibiotic orders include a Dose, Duration, Indication. Efforts to improve antibiotic use are often hampered because the nonprescribing providers are not sure why the patient is on antibiotics. This problem is amplified when patients are transitioned from one provider to another or when multiple providers are involved. Specifying the duration and indication in all antibiotic orders will ensure that treatments continue for the right amount of time and would allow therapy to be stopped if the initially suspected infection is ruled out or altered if another infection is identified. The second improvement project is developing a process to ensure that any patient with a positive blood culture is on the appropriate therapy. This is a relatively straightforward intervention that is based on the patient's own microbiology results, and it ensures the optimal therapy of a serious infection. Third is the development of an intervention to encourage the reassessment of patients who are started on antibiotics for community‐acquired pneumonia (CAP). Several hospitalists have suggested that the pressure to initiate therapy quickly in cases of CAP often leads to overtreatment. Interventions that encourage a reexamination of the CAP diagnosis when the clinical situation has stabilized would likely reduce this overtreatment. And the fourth improvement project is ensuring that urinary tract infections (UTIs) in hospitalized patients are properly diagnosed and treated. Work done by hospitalists at the University of Michigan suggests that improving the diagnosis and treatment of UTIs would have a significant impact on improving antibiotic use.8 Currently, the CDC is collaborating with these investigators to develop protocols and tools to improve the treatment of inpatient UTIs.
The time to promote aggressive implementation of antimicrobial stewardship interventions has come. Clinicians are increasingly encountering infections for which there are very limited or, in some cases, no good treatment optionsand there are very few new antibiotics on the horizon. Many groups are advocating for expanded efforts to develop new antibiotics.9 Although this is crucial, it is just as important that we work now to aggressively improve the use of the agents we have. Not only might this extend the life of our current agents, but it will also help ensure that any new agents will enjoy longer periods of effectiveness. Indeed, failing to inextricably link the development of new antibiotics with efforts to improve antibiotic use is akin to buying a new car to drive on a road full of potholes. Fortunately, there are a number of interventions that have proven successful; we now need to determine how best to apply these interventions in more settings. We want and need the involvement of hospitalists in these efforts. Yes, improving antimicrobial stewardship will require investments, but past experience tells us that the alternative could prove far more costly.
- .Inadequate antimicrobial treatment: an important determinant of outcome for hospitalized patients.Clin Infect Dis.2000;31:S131–S138.
- ,,, et al.Impact of a reduction in the use of high‐risk antibiotics on the course of an epidemic of Clostridium difficile‐associated disease caused by the hypervirulent NAP1/027 strain.Clin Infect Dis.2007;45(Suppl 2):S112–S121.
- ,,, et al.Early transition to oral antibiotic therapy for community‐acquired pneumonia: duration of therapy, clinical outcomes, and cost analysis.Respir Med.1998;92:1032–1039.
- ,,, et al.A hospitalwide intervention program to optimize the quality of antibiotic use: impact on prescribing practice, antibiotic consumption, cost savings, and bacterial resistance.Clin Infect Dis.2003;37:180–186.
- ,,, et al.A World Wide Web‐based antimicrobial stewardship program improves efficiency, communication, and user satisfaction and reduces cost in a tertiary care pediatric medical center.Clin Infect Dis.2008;47:747–753.
- ,,, et al.Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America guidelines for developing an institutional program to enhance antimicrobial stewardship.Clin Infect Dis.2007;44:159–177.
- ,,, et al.A knowledge, attitudes and beliefs survey of housestaff physicians from various specialties concerning antimicrobial use and resistance.Arch Intern Med.2004;164:1451–1456.
- ,,, et al.Importance of urinary tract infection to antibiotic use among hospitalized patients.Infect Control Hosp Epidemiol.2009;30:193–195.
- Infectious Diseases Society of America.The 10 × '20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020.Clin Infect Dis.2010;50:1081–1083.
- .Inadequate antimicrobial treatment: an important determinant of outcome for hospitalized patients.Clin Infect Dis.2000;31:S131–S138.
- ,,, et al.Impact of a reduction in the use of high‐risk antibiotics on the course of an epidemic of Clostridium difficile‐associated disease caused by the hypervirulent NAP1/027 strain.Clin Infect Dis.2007;45(Suppl 2):S112–S121.
- ,,, et al.Early transition to oral antibiotic therapy for community‐acquired pneumonia: duration of therapy, clinical outcomes, and cost analysis.Respir Med.1998;92:1032–1039.
- ,,, et al.A hospitalwide intervention program to optimize the quality of antibiotic use: impact on prescribing practice, antibiotic consumption, cost savings, and bacterial resistance.Clin Infect Dis.2003;37:180–186.
- ,,, et al.A World Wide Web‐based antimicrobial stewardship program improves efficiency, communication, and user satisfaction and reduces cost in a tertiary care pediatric medical center.Clin Infect Dis.2008;47:747–753.
- ,,, et al.Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America guidelines for developing an institutional program to enhance antimicrobial stewardship.Clin Infect Dis.2007;44:159–177.
- ,,, et al.A knowledge, attitudes and beliefs survey of housestaff physicians from various specialties concerning antimicrobial use and resistance.Arch Intern Med.2004;164:1451–1456.
- ,,, et al.Importance of urinary tract infection to antibiotic use among hospitalized patients.Infect Control Hosp Epidemiol.2009;30:193–195.
- Infectious Diseases Society of America.The 10 × '20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020.Clin Infect Dis.2010;50:1081–1083.
Use of Pharmacodynamic Principles to Inform β‐Lactam Dosing
Tremendous strides have been made over the last 25 years in understanding the relationship between antimicrobial exposure and response.14 Many clinicians consider antimicrobial drug pharmacokinetics (PK) and pharmacodynamics (PD) a rather esoteric or academic topic without practical applicability or clinical utility. However, it is becoming increasingly clear, particularly as less‐susceptible pathogens emerge, that consideration of PK/PD in dose selection is essential for optimizing antimicrobial therapy and, as such, is a core component of effective antimicrobial stewardship and patient care. Antimicrobial therapy can fail if an appropriate agent is selected but the dosing regimen does not provide adequate exposure against the infecting pathogens, especially at the site of infection.5, 6
The 2007 guidelines from the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA) for developing institutional antimicrobial stewardship programs highlight dose optimization as one of the key strategies for enhancing antimicrobial stewardship.7 More specifically, they recommend optimizing dosing by focusing on individual patient characteristics, causative organism and site of infection, and the PK/PD characteristics of the drug. With advances in mathematical modeling (Monte Carlo simulation), it is possible to apply our understanding of PK/PD to clinical practice and design empiric regimens that have a high probability of achieving the PD target linked to effect. These mathematical modeling techniques have an array of other utilities and have become the standard methodologies for assessing the clinical viability of both experimental and approved antimicrobials.8, 9 Furthermore, the Clinical and Laboratory Standards Institute (CLSI) has recently begun to incorporate results from PK/PD analyses in determining MIC breakpoints.10 This paper provides a general overview of antimicrobial PD before demonstrating how to apply PD principles to clinical practice through the use of Monte Carlo simulation (MCS). Piperacillin/tazobactam (TZP) is used as a motivating example for this latter purpose.
Pharmacokinetics and Pharmacodynamics: Parameters and Principles
Pharmacokinetics describes the actions of the body on an administered drug, whereas PD describes the actions of the administered drug on the body. In essence, PK refers to the movement of the drug within the body, including absorption, distribution, metabolism, and excretion. Conversely, PD refers to the effects of the drug on the body, or its physiologic actions. A drug's PD is defined by its mechanism of action, and includes both desired and undesired effects. Typically, PK and PD work together to best define or predict the full range of effects of an administered drug on an individual patient, as described in greater detail below.
The Minimum Inhibitory Concentration
The MIC is the PD parameter most often used to describe the relationship between antimicrobial drug and physiologic activity. The MIC is defined as the lowest or minimum antimicrobial concentration that inhibits visible microbial growth in artificial medium after a fixed incubation time.10, 11 This is typically determined by placing a known quantity of bacteria (or other microorganism) into multiple test tubes, and then adding increasing concentrations of a particular antibiotic, typically in log2 dilution, into consecutive tubes. The lowest antibiotic concentration that inhibits bacterial growth is then defined as the MIC for that drug‐pathogen pairing.
While useful as a quantitative measure of drug activity or potency, the MIC is not without limitations.12 The MIC does not mimic physiologic conditions. The MIC is a static measure (fixed concentration of drug in an artificial growth medium for a fixed period of time) and is not reflective of the concentration‐time profile one would typically observe in patients; drug concentrations change throughout the dosing interval. Because the MIC only measures growth inhibition, it does not reflect the rate at which bacteria are killed, nor can it identify if a dosekill response relationship exists for a particular antibiotic‐pathogen pairing. Furthermore, the MIC only quantifies net growth over an 1824‐hour observation period. Killing and regrowth may well occur during this period, as long as the net growth is zero. Finally, the MIC does not account for the post‐antibiotic effects of antibiotics. Most antibiotics, depending on the pathogen and drug class, exhibit some persistence of bacteriostatic or bactericidal activity after the drug concentration at the target site has dropped below the MIC. This activity has been described as the post‐antibiotic effect,1315 post‐antibiotic sub‐MIC effect,1317 or post‐antibiotic leukocyte enhancement effect.18, 19
Common Pharmacodynamic Measures
Examination of PK measures of drug exposure (eg, serum/tissue concentrations) in relation to the MIC surmounts many of the limitations of the MIC and provides much better prediction of antimicrobial effect than the MIC or exposure profile alone. The 3 most common PK/PD indices (sometimes abbreviated as PD measures) used to predict drug response are: 1) the ratio of the maximal free drug concentration to the MIC (fCmax:MIC), 2) the ratio of the free area under the concentration‐time curve to the MIC (fAUC:MIC), and 3) the duration of time free drug concentrations remain above the MIC (fT>MIC).24, 20, 21 The PD parameter most predictive of outcomes varies by drug class (Table 1).20
| Antibiotic | Optimal PD measure(s) |
|---|---|
| |
| Aminoglycosides | Cmax:MIC; AUC:MIC |
| ‐lactams | |
| Penicillins | T>MIC |
| Cephalosporins | T>MIC |
| Carbapenems | T>MIC |
| Monobactams | T>MIC |
| Clindamycin | AUC:MIC |
| Fluoroquinolones | AUC:MIC, Cmax:MIC |
| Glycopeptides/lipopeptides | |
| Daptomycin | AUC:MIC, Cmax:MIC |
| Oritavancin | T>MIC, Cmax:MIC |
| Vancomycin | AUC:MIC |
| Linezolid | AUC:MIC |
| Macrolides | |
| Azithromycin | AUC:MIC |
| Clarithromycin | AUC:MIC |
| Telithromycin | AUC:MIC |
| Metronidazole | AUC:MIC, Cmax:MIC |
| Tetracyclines | |
| Doxycycline | AUC:MIC |
| Tigecycline | AUC:MIC |
Certain antibiotics exhibit concentration‐dependent bactericidal activity, while others exhibit time‐dependent activity (Table 1).24, 20 For concentration‐dependent antibiotics, a doseresponse relationship exists and the therapeutic goal is to maximize exposure at the target site. Alternatively, the activity of time‐dependent antibiotics is not dependent on the intensity of exposure but is a function of the duration of time concentrations are above the MIC during the dosing interval. For the time‐dependent antibiotics like the ‐lactams, concentrations do not have to remain above the MIC for the entire dosing interval, and the fraction of the dosing interval required for maximal bacterial effect varies for the different types of ‐lactams. Although the precise fT > MIC varies for different drugbacteria combinations, bacteriostatic effects are typically observed when the free drug concentration exceeds the MIC for 3540%, 30%, and 20% of the dosing interval for the cephalosporins, penicillins, and carbapenems, respectively. Near‐maximal bactericidal effects require 6070%, 50%, and 40% fT > MIC, respectively, for these ‐lactam classes.3, 4
It is important to note that it is the free (or unbound) fraction of drug that determines its ability to penetrate tissues and exert its microbiological effect.3, 4, 22 This was demonstrated as early as the 1940s with penicillin. There are occasionally exceptions, mostly with the therapy of gram‐positive infections. Daptomycin is one such example; protein binding is approximately 9092% (free drug 810%), but the agent behaves as if the drug is approximately 75% bound (25% free).23 Nonetheless, the guiding principle is that protein binding can have an adverse impact on the PD and microbiological activity of an antibacterial agent.
Monte Carlo Simulation
With advances in mathematical modeling, it is possible to apply our understanding of antimicrobial PD to clinical practice.12 In particular, MCS can be used to integrate PK, PD, and local microbiologic surveillance data to design antibiotic regimens that have a high probability of achieving the PD target linked to effect against the range of pathogens encountered in clinical practice. In short, MCS is a technique that incorporates the variability in PK among potential patients (between‐patient variability) when predicting antibiotic exposures, and allows calculation of the probability for obtaining a critical target exposure for the range of possible MIC values.12 If a number of volunteers or patients are given an antibiotic, there will be true variability in the observed concentration time profiles between people. For example, the peak serum concentrations and AUC0‐24h will vary between individuals. In essence, MCS is a mathematical modeling technique that simulates the dispersion or full spread of concentration‐time exposure values (eg, peak concentration, area under the curve) that would be seen in a large population after administration of a specific drug dose or regimen. Once the distribution in concentration‐time profiles is determined, the probability of achieving the PD target at each MIC value for a given MIC range (ie, probability of target attainment [PTA] profile) is ascertained.
There are several steps in the MCS process. First, a PK model for the antibiotic under study is embedded into the MCS. The mean PK parameters (eg, volume, clearance, intercompartmental transfer constant) and associated variability (variance and covariance) from the selected PK model are used to create a multivariate distribution of PK parameters. From this multivariate distribution, the MCS randomly selects a set of PK parameters, and these randomly selected PK parameters are used to simulate a concentration‐time profile for a virtual subject based on the desired antibiotic dosing regimen. This process is repeated a specified number of times (eg, 5000, 1000) to simulate the distribution of concentration‐time profiles one would expect to see in the population. Once the specified number of virtual patients has been simulated (eg, 10,000 virtual patients), the proportion of the simulated population that achieves the critical exposure target (eg, 50% fT > MIC) at each MIC value for a given MIC range can be calculated. Because the relationship between drug exposure and effect is expressed as a ratio (eg, AUC:MIC, Cmax:MIC, T:MIC), a unique drug exposure:MIC ratio and PTA exists for each unique MIC value within the distribution.12
In clinical practice, a distribution of MIC values exists for a given organism or infection. Therefore, the final step is determining the overall PTA for the distribution of organisms encountered clinically. As previously mentioned, the PTA is determined at each MIC value within a given MIC range. Because the fraction of organisms collected at each MIC value is known, the overall or weighted PTA average can be calculated by multiplying the PTA for a specific MIC and the proportion of isolates with that MIC. This product is calculated for each MIC value within the MIC distribution. The overall PTA is then calculated by summing the products (PTA at a given MIC value x proportion of isolates with that MIC value) of the MIC values encountered within the distribution.12
A key element for these simulations is the estimation of the PK parameters and their associated dispersion (variance and covariance). Pharmacokinetic data, especially for new compounds, are usually limited to data from healthy volunteer studies. Caution should be exercised when generalizing the results of volunteer studies to the population of interest. Volunteer studies are often considered as the most conservative evaluation of a new drug; volunteers are young and healthy, likely to have the highest drug clearances and shortest half lives. However, when one performs MCS, the measure of central tendency (high drug clearance, short half‐lives) is only part of the story. Because MCS are explicitly creating a distribution, it is important to understand the measure of dispersion. Secondary to the limited variation surrounding PK parameters from healthy volunteer studies, it is possible that they overestimate the PTA. Applicability to the target population must always be considered.12
Motivating Example: Piperacillin‐tazobactam
Piperacillin‐tazobactam (TZP) is an acylureido‐penicillinbeta‐lactamase inhibitor combination and is frequently used as first‐line empirical therapy for healthcare‐associated infections. Like all ‐lactams, the PD parameter most predictive of its efficacy is fT > MIC, and its activity is optimized when free drug concentrations exceed the MIC for 50% of the dosing interval (50% fT > MIC). Because it is used empirically, it is critical that the TZP regimens used in practice have a high probability of achieving 50% fT > MIC against the range of MIC values likely to be encountered in a given institution.
Since the MIC of the infecting pathogen is often not available at the start of therapy, clinicians frequently rely on the hospital antibiogram to determine the utility of an antibiotic as an empiric agent. The range of MIC values reported as susceptible in clinical practice is based on the CLSI susceptibility interpretive criteria. For TZP, Enterobacteriaceae and Acinetobacter baumannii isolates with MIC values 16 mg/L are considered susceptible. The CLSI breakpoint for Pseudomonas aeruginosa is higher and isolates with MIC values 64/4 mg/L are considered susceptible.11
It is important to recognize that these CLSI TZP susceptibility breakpoints were established prior to our current understanding of ‐lactam PD and are higher relative to other ‐lactams. It was not until sometime after the establishment of the TZP susceptibility interpretive criteria were MCS studies performed to determine the ability of the US Food and Drug Administration (FDA)‐approved TZP dosing regimens in achieving 50% fT > MIC against the range of MICs deemed susceptible by CLSI.
The first study to characterize the ability of standard TZP dosing (0.5‐hour infusion of 3.375 g every 6 hours) in achieving 50% T > MIC in its targeted population for the range of MIC values deemed susceptible by CLSI was published in 2004 by our group. Employing a population PK model derived in hospitalized patients, TZP 3.375 grams administered every 6 h provided high PTA rates for MICs of 8/4 mg/L (ie, 8 mg/L for piperacillin and 4 mg/L for tazobactam) when hospitalized‐patient data were used (Figure 1).24 In clinical situations in which the MICs are expected to be 16/4 mg/L, the results of the MCS indicate that caution should be exercised when using standard TZP dosing. More recently, DeRyke et al evaluated the PD profile of the TZP nosocomial dosing scheme (0.5‐hour infusion of 4.5 g every 6 hours). Using the same population PK model employed as our study, DeRyke and colleagues noted a slightly improved PTA profile at a MIC value of 16/4 mg/L with the TZP nosocomial pneumonia dosing scheme relative to standard dosing. However, the PTA was still suboptimal for MIC values 32/4 mg/L (Figure 1).25
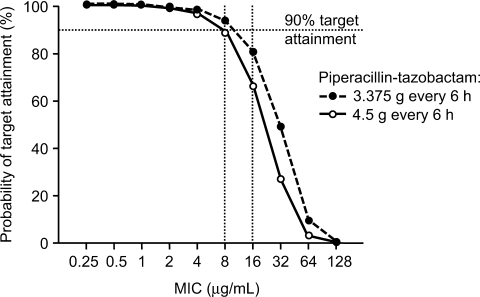
These findings are concerning because the TZP CLSI susceptibility breakpoint for non‐lactose fermenting Gram‐negative bacteria is 64/4 mg/L.11 In essence, the conventional and nosocomial pneumonia TZP dosing schemes provide a suboptimal PD profile for a substantial portion of the MIC distribution deemed susceptible by CLSI. The clinical relevance of this is highlighted by a study by Tam and coworkers examining the efficacy of TZP in hospitalized patients with bacteremia due to P. aeruginosa (Figure 2).26 This retrospective cohort study examined 30‐day mortality among patients who received appropriate empiric therapy between 2002 and 2006. Therapy was defined as appropriate if: 1) ‐lactam treatment (in doses appropriate for renal function as recommended by the manufacturer) was started within 24 hours of blood culture collection, and 2) the isolate was found to be susceptible to the ‐lactam agent selected. The cohort was stratified by the TZP piperacillin MIC (3264 mg/L vs. 16 mg/L) and 30‐day mortality rates were compared within MIC strata between patients who received TZP or an alternative ‐lactam with activity against Pseudomonas aeruginosa. A total of 34 episodes with MICs of 32 or 64 mg/L were identified. Seven of these cases were empirically treated with TZP, while the remaining 27 received other ‐lactam agents. Forty‐nine episodes of P. aeruginosa bacteremias had MIC values 16 mg/L. Of these 49, 10 were empirically treated with TZP and the remaining 39 were treated with other ‐lactams. The results showed that the 30‐day mortality rate was significantly higher among patients treated with TZP versus control‐treated patients with isolates possessing a MIC of either 32 or 64 mg/L (86% vs. 22%, P value = 0.004), while there was no significant difference between the two treatment groups for isolates with a MIC of up to 16 mg/L (30% vs. 21%, P = 0.673). Interestingly, patients treated with a non‐TZP ‐lactam antibiotic had 30‐day mortality rates of 21%, regardless of the TZP MIC value. Collectively, these findings and the results of the TZP MCS studies highlight the importance of considering PTA data when evaluating the utility of an antibiotic dosing scheme. These data also cast uncertainty on the appropriateness of the current TZP CLSI susceptibility breakpoint in connection with the conventional dosing TZP strategies. The current CLSI interpretation of TZP susceptibility for non‐lactose‐fermenting gram‐negatives may inadvertently provide misleading guidance to clinicians for optimal patient care.
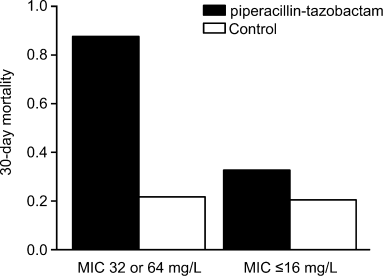
Dosing Strategies to Improve the Probability of Target Attainment Profile of ‐lactams
Three potential dosing strategies used to improve the PTA of a ‐lactam against the range of pathogens encountered in various clinical situations include: 1) increasing the dose, 2) increasing the dosing frequency, or 3) increasing the duration of infusion.12 Intuitively, it makes sense to simply increase the drug dose. However, as demonstrated in the aforementioned TZP MCS studies, increasing the TZP dose from 3.375 grams to 4.5 grams every 6 hours had a minimal impact on the PTA profile.24, 25 To increase fT >MIC by 1 half‐life, the dose would need to be doubled. Since most ‐lactams have a half‐life of 30 minutes to 1 hour, doubling the dose only provides an extra 30 minutes or hour above the MIC, which would not be expected to have much clinical impact. In addition, doubling the dose is not cost effective since it doubles drug acquisition costs.12, 27
Increasing the dosing frequency is a viable option and may be the optimal strategy in certain situations.12 However, it is often associated with increased drug acquisition costs (more doses per day) relative to the parent regimen and may not be a viable option from a nursing and pharmacy perspective due to increased administration and preparation time. In addition, there may be a higher potential for toxicity because a greater amount of drug is given per day.
Extending the infusion time is another ‐lactam dose optimization strategy that is becoming more commonly used in clinical practice. Administering a dose of a ‐lactam agent as an infusion longer than the conventional 0.51.0‐hour infusion duration has 2 main effects. First, it produces a lower peak concentration of the drug.24 Because the bacterial kill rate for these agents is not concentration‐dependent, this does not present a major disadvantage.3, 4, 2830 Second, the drug concentrations remain in excess of the MIC for a longer period of time. Because this is what drives antibacterial effect for ‐lactams, this will yield a more favorable PTA profile. It should also be noted that this can be done with less frequent drug dosing.27
Extending the infusion time can be accomplished by either prolonging the infusion time for a major portion of the dosing interval (prolonged infusion) or administering continuously throughout the day (continuous infusion). From a PD profiling viewpoint, the two infusion methodologies yield nearly identical PTA profiles. This was highlighted in the 2007 study by Kim et al, which compared PTA rates between intermittent (0.5 hour), prolonged (4 hours), and continuous infusions of TZP (Figure 3). In their study, the PTA curves for prolonged and continuous infusion TZP were superimposable and superior to the intermittent infusion regimen for MIC values in excess of 4 mg/L.31
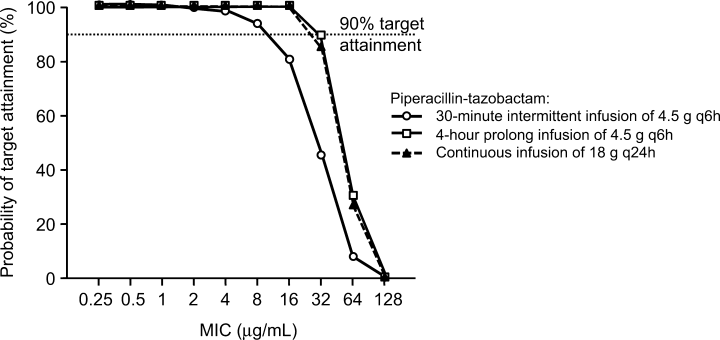
There are several practicalities to consider when differentiating prolonged and continuous infusion methods. The principle advantages of continuous infusion are once‐daily administration and reduced costs for labor, supplies, and administration.12, 27 The major disadvantages of continuous infusion are the need for a dedicated line for infusion (which often leads to drug compatibility issues), issues of drug stability and waste, and lack of ambulation for the patient. The need for a dedicated infusion line is particularly impractical for patients with limited intravenous access or those requiring multiple daily infusions. In addition, continuous infusion often requires insertion of a central line, which places patients at unnecessary risk of secondary catheter‐related infection.12 Continuous infusion solutions are typically prepared as 24‐hour infusions containing the total daily amount of drug. Considerable drug wastage can occur with early discontinuation of therapy; all drug within the solution needs to be wasted and cannot be reused if the order is discontinued prior to scheduled completion.
Prolonged infusion provides many of the benefits of intermittent dosing, but with the PD advantages of continuous infusion. Administration of the infusion for a prolonged time, but not continuously, obviates the need to have a dedicated intravenous line just for ‐lactam continuous infusion. It also achieves the targeted fT > MIC at a total daily dose less than standard ‐lactam dosing methods. Drug wastage is also minimized because the intermittent administration formulations are used; there is no need to prepare antibiotic solutions for 24‐hour periods. Prolonged infusion also allows the patient to be ambulatory for much of the day. The potential disadvantages of prolonged infusion relative to continuous infusion include the increased use of labor, supplies, and administration resources. Although minimized, there is still the need to schedule or time the administration of incompatible drugs.12, 27
Data Examining the Outcomes Associated With Prolonged and Continuous ‐lactam Infusions
Over the years, a number of randomized controlled trials (RCTs) and observational studies have compared outcomes between extended and intermittent ‐lactam infusions. These studies, mostly small scale in nature, involved a number of different ‐lactam antibiotics and various infectious etiologies. To ascertain if there are any clinical benefits in extending the infusion duration (prolonged and continuous), Roberts and colleagues performed a systematic review of available data on PubMed (January 1950 to November 2007), EMBASE (1966 to November 2007), and the Cochrane Controlled Trial Register (updated November 2007).32 Randomized controlled trials were meta‐analyzed, and observational studies were reviewed. Among a total of 59 potentially RCTs, 14 involving a total of 846 patients from nine countries were deemed appropriate for meta‐analysis. The use of continuous infusion of a ‐lactam antibiotic was not associated with an improvement in clinical cure (n=755 patients; odds ratio: 1.04, 95% confidence interval: 0.741.46, P = 0.83) or mortality (n=541 patients; odds ratio: 1.00, 95% confidence interval: 0.482.06, P = 1.00). In contrast, the observational studies showed that ‐lactam administration by extended or continuous infusion confers an improvement in clinical cure and this was most pronounced in critically ill patients being treated for gram‐negative bacterial infections.
There are several possible explanations for the discrepancy in results between the meta‐analysis and observational studies. First, disease severity in the studies included in the meta‐analysis was generally low, as evidenced by low mortality rates in the majority of studies. Second, a diverse group of patients and infection types were included in the RCTs, which increased the heterogeneity of the cohort analyzed. Third, a higher antibiotic dose was used in the intermittent administration group in all RCTs except one. Fourth, microbiologic and PK/PD data were not available for the majority of RCTs. Collectively, the null result from the meta‐analysis and positive data from the nonrandomized studies suggest that prolonged or continuous infusion ‐lactams is unlikely to be advantageous for all hospitalized patient populations, but may be beneficial for specific groups, such as critically ill patients with higher MIC pathogens.
The benefits of prolonged ‐lactam infusion among critically ill patients were highlighted by the study performed at Albany Medical Center Hospital.27 Based on a MCS, prolonged infusion TZP (3.375 grams administered over a 4‐hour period every 8 hours) was identified as an alternative means to the intermittent TZP dosing (3.375 grams administered over 30 minutes every 4 or 6 hours) and adopted as the standard TZP dosing scheme in February 2002. Prior to February 2002, all patients received traditional infusion TZP; after this time, all patients received prolonged infusion TZP. To evaluate the impact of the automatic dose substitution program, 14‐day mortality and hospital length of stay post‐culture collection were compared between patients who received either intermittent or prolonged TZP infusion for a TZP‐susceptible P. aeruginosa infection between 2000 and 2004.27 The study was restricted to P. aeruginosa infections for several reasons. First, patients with P. aeruginosa represented a relatively homogenous patient population; this attribute minimized confounding and increased the ability to detect differences between treatment groups according to intervention. Second, patients with P. aeruginosa infections are more dependent on antimicrobial therapy than other populations, since patients infected with P. aeruginosa are frequently critically ill and often have an impaired innate immune system.33, 34 Third, P. aeruginosa isolates typically have a higher range of MICs to TZP than other organisms, and the benefits of optimizing fT>MIC were thought to be better elucidated in this patient population.35, 36
In patients who were identified as having the greatest risk for 14‐day mortality (Acute Physiology and Chronic Health Evaluation [APACHE] II score 17), there was a significantly lower 14‐day mortality rate and a shorter median hospital LOS after culture sample collection for patients who received prolonged infusion, compared with patients who received intermittent infusion (Figure 4). No differences between prolonged infusion and intermittent infusion of TZP were observed with respect to outcome in patients at lowest risk for death (APACHE II score 17). These findings support the notion that critically ill patients who have P. aeruginosa infection are most dependent upon drug exposure for good clinical outcomes. The results also suggest that improved outcomes can be achieved by optimizing antibiotic PD in this population. Furthermore, the results highlight the importance of examining the influence of treatment within a population at greatest risk for the outcome of interest.27
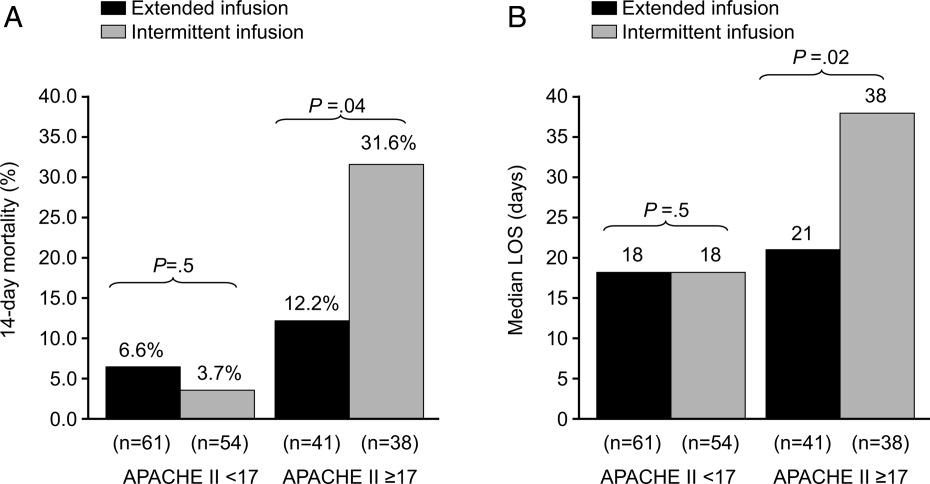
In addition to potential clinical benefits, prolonged infusions can provide cost savings by minimizing the amount of drug used per day. Prolonged infusion typically achieves the targeted fT > MIC at a total daily dose less than standard ‐lactam dosing methods. For example, TZP purchases totaled $275,000 the year before conversion at Albany Medical Center Hospital. Switching to the prolonged infusion strategy reduced the total daily dose by 25%50% (by 13 doses per day) representing a savings of $68,750$135,750 in annual direct drug acquisition costs.27
Additional Pharmacokinetic and Pharmacodynamic Considerations
When assessing the PK/PD of an antibiotic, it is also important to consider concentrations achieved at the site of infection. Most MCS studies have focused on free concentrations in plasma. Whereas free concentrations in plasma are often viewed as an acceptable approximation for free concentrations at the site of infection, this is not always the case. Of particular concern is in the treatment of lower respiratory tract infections. For ‐lactams, it was commonly believed that plasma and epithelial lining fluid (ELF) of the alveolar space concentrations were comparable; antibiotic concentrations in ELF are currently used to estimate the penetration of antibiotics into the respiratory tract. However, the median ELF/plasma penetration ratio for meropenem among patients with ventilator‐associated pneumonia (VAP) is only 25%.37 The only way to achieve a favorable fT > MIC PD profile at the site of infection with meropenem is to administer higher doses over prolonged periods of time (Figure 4). In light of the meropenem ELF data, data available on concentrations at the site of infection, particularly difficult‐to‐penetrate sites, such as ELF and cerebrospinal fluid, should be considered before designing dosing scheme for implementation into clinical practice.
Up to this point, this review has been focused on PD targets of clinical success. The next frontier in PK/PD is identifying antibiotic dosing schemes and drug combinations that minimize the emergence of resistance. Data available to date suggest that PD targets for resistance prevention are typically 24‐fold higher than PD targets for success. Tam et al showed that for meropenem, the PD target needed to suppress the emergence of resistance in P. aeruginosa was a Cmin:MIC ratio of 1.7.38 Further study is still needed in the area of resistance suppression but the current data suggest that obtaining the PK/PD target against the range of MIC encountered clinically is not likely with conventional ‐lactam dosing and will most likely require more intensive regimens administered over extended periods of time.38
Arguments Against Extended ‐lactam Infusions
Limited clinical trial data and lack of FDA approval are frequently cited as the major clinical barriers for implementing extended ‐lactam infusions into practice. Unfortunately, there is a relative dearth of large‐scale randomized clinical data supporting extending the infusion of ‐lactam therapy. In addition, the package inserts for the various ‐lactam antibiotics do not provide support for these prolonged infusion dosing.
While these are valid concerns, the clinical support for intermittent ‐lactam infusions is also limited. The clinical data are largely limited to complicated intra‐abdominal infections, complicated skin and soft tissue infections, complicated urinary tract infections, and community‐acquired pneumonia. None of the intermittently administered ‐lactams currently have an indication for bacteremia (except imipenem for bacterial septicemia), and there are only limited indications for hospital‐acquired pneumonia (HAP) or VAP (imipenem for lower respiratory tract infections and TZP in combination with an aminoglycoside for HAP). In addition, the clinical trials of intermittent ‐lactam infusion regimens have commonly assessed clinical response at the test‐of‐cure visit or after completion of therapy. Arguably, this is not a very clinically meaningful endpoint for the types of infections commonly encountered on a day‐to‐day basis in today's world, where mixed diagnoses and infecting pathogens are often seen. Most important, the bacteria have evolved since the early clinical trials used to obtain FDA approval, and those outdated studies do not address the resistance profiles currently observed in clinical practice.
Conclusions
Understanding exposure‐response relationships is critical when designing antibiotic dosing schemes. In the absence of therapeutic drug monitoring, MCS can be used to design antibiotic regimens that have a high probability of attaining the PD target linked to effect against the range of MICs likely to be encountered in clinical practice. When considering ‐lactam therapy for critically ill patients likely infected with high‐MIC or reduced‐susceptibility pathogens, a prolonged or continuous infusion regimen should be considered. Compared with intermittent dosing, prolonged infusion of ‐lactams is typically associated with improved PTA, as potential benefits of cost savings, and an enhanced PD profile at the site of infection.
- ,,,.Pharmacokinetic‐pharmacodynamic considerations in the design of hospital‐acquired or ventilator‐associated bacterial pneumonia studies: look before you leap!Clin Infect Dis.2010;51(Suppl 1):S103–S110.
- ,,, et al.Pharmacokinetics‐pharmacodynamics of antimicrobial therapy: it's not just for mice anymore.Clin Infect Dis.2007;44(1):79–86.
- .Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men.Clin Infect Dis.1998;26(1):1–10; quiz 11–12.
- .Antimicrobial pharmacodynamics: critical interactions of ‘bug and drug’.Nat Rev Microbiol.2004;2(4):289–300.
- ,,,,.Length of stay and hospital costs associated with a pharmacodynamic‐based clinical pathway for empiric antibiotic choice for ventilator‐associated pneumonia.Pharmacotherapy.2010;30(5):453–462.
- ,,, et al.Pharmacodynamic‐based clinical pathway for empiric antibiotic choice in patients with ventilator‐associated pneumonia.J Crit Care.2010;25(1):69–77.
- ,,, et al.Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America guidelines for developing an institutional program to enhance antimicrobial stewardship.Clin Infect Dis.2007;44(2):159–177.
- ,,, et al.How modeling and simulation have enhanced decision making in new drug development.J Pharmacokinet Pharmacodyn.2005;32(2):185–197.
- ,,,,.PK/PD: new insights for antibacterial and antiviral applications.Curr Opin Pharmacol. Oct2008;8(5):549–556.
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing; Twentieth Informational Supplement. CLSI document M100‐S20. Wayne, Pennsylvania: Clinical and Laboratory Standards Institute; 2010.
- Clinical and Laboratory Standards Institute/NCCLS. Performance standards for Antimicrobial disc diffusion tests; Approved standards. 9th ed. CLSI Document M2‐M9. Wayne, PA: Clinical and Laboratory Standards Institute; 2006.
- ,,.Application of antimicrobial pharmacodynamic concepts into clinical practice: focus on beta‐lactam antibiotics: insights from the Society of Infectious Diseases Pharmacists.Pharmacotherapy.2006;26(9):1320–1332.
- ,,,.Postantibiotic effect of imipenem on Pseudomonas aeruginosa.Antimicrob Agents Chemother.1984;26(5):678–682.
- ,,.Postantibiotic, postantibiotic sub‐MIC, and subinhibitory effects of PGE‐9509924, ciprofloxacin, and levofloxacin.Antimicrob Agents Chemother.2003;47(10):3352–3356.
- ,,, et al.In vitro evaluation of CBR‐2092, a novel rifamycin‐quinolone hybrid antibiotic: microbiology profiling studies with staphylococci and streptococci.Antimicrob Agents Chemother.2008;52(7):2324–2334.
- ,.The post‐antibiotic sub‐MIC effect in vitro and in vivo.J Antimicrob Chemother.1993;31(Suppl D):159–166.
- .Pharmacodynamic effects of subinhibitory antibiotic concentrations.Int J Antimicrob Agents.2001;17(1):1–8.
- ,,.Postantibiotic leukocyte enhancement: increased susceptibility of bacteria pretreated with antibiotics to activity of leukocytes.Rev Infect Dis.1981;3(1):38–44.
- ,.Enhancement of leukocyte activity against Escherichia coli after brief exposure to chloramphenicol.Antimicrob Agents Chemother.1979;16(6):695–700.
- ,.Pharmacokinetic and pharmacodynamic parameters of antimicrobials: potential for providing dosing regimens that are less vulnerable to resistance.Clin Pharmacokinet.2009;48(8):517–528.
- ,.Pharmacokinetics/pharmacodynamics of antibacterials in the intensive care unit: setting appropriate dosing regimens.Int J Antimicrob Agents.2008;32(4):294–301.
- ,,.Effect of protein binding on antibiotic activity in vivo.J Antimicrob Chemother.1983;11(3):233–238.
- ,,,,.Determining the active fraction of daptomycin against MRSA by evaluating bactericidal activity in the presence of protein and pharmacodynamic (PD) modeling. 49th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy.2009;A1‐1270.
- ,,,,.Pharmacodynamic profiling of piperacillin in the presence of tazobactam in patients through the use of population pharmacokinetic models and Monte Carlo simulation.Antimicrob Agents Chemother.2004;48(12):4718–4724.
- ,,.Reevaluation of current susceptibility breakpoints for Gram‐negative rods based on pharmacodynamic assessment.Diagn Microbiol Infect Dis.2007;58(3):337–344.
- ,,, et al.Outcomes of bacteremia due to Pseudomonas aeruginosa with reduced susceptibility to piperacillin‐tazobactam: implications on the appropriateness of the resistance breakpoint.Clin Infect Dis. 152008;46(6):862–867.
- ,,.Piperacillin‐tazobactam for Pseudomonas aeruginosa infection: clinical implications of an extended‐infusion dosing strategy.Clin Infect Dis. 12007;44(3):357–363.
- ,.Pharmacokinetics and pharmacodynamics of antibiotics in otitis media.Pediatr Infect Dis J.1996;15(3):255–259.
- .Interrelationship between pharmacokinetics and pharmacodynamics in determining dosage regimens for broad‐spectrum cephalosporins.Diagn Microbiol Infect Dis.1995;22(1–2):89–96.
- .How does a patient maximally benefit from anti‐infective chemotherapy?Clin Infect Dis.2004;39(8):1245–1246.
- ,,,.Optimal dosing of piperacillin‐tazobactam for the treatment of Pseudomonas aeruginosa infections: prolonged or continuous infusion?Pharmacotherapy.2007;27(11):1490–1497.
- ,,,,.A systematic review on clinical benefits of continuous administration of beta‐lactam antibiotics.Crit Care Med.2009;37(6):2071–2078.
- ,,.Pharmacokinetic/pharmacodynamic modeling can help guide targeted antimicrobial therapy for nosocomial gram‐negative infections in critically ill patients.Diagn Microbiol Infect Dis.2004;48(2):125–130.
- ,,, et al.Pseudomonas aeruginosa bloodstream infection: importance of appropriate initial antimicrobial treatment.Antimicrob Agents Chemother.2005;49(4):1306–1311.
- ,,,.Assessment of pathogen occurrences and resistance profiles among infected patients in the intensive care unit: report from the SENTRY Antimicrobial Surveillance Program (North America, 2001).Int J Antimicrob Agents.2004;24(2):111–118.
- ,,.Results from the Meropenem Yearly Susceptibility Test Information Collection (MYSTIC) Programme: report of the 2001 data from 15 United States medical centres.Int J Antimicrob Agents.2004;23(1):52–59.
- ,,, et al.Penetration of meropenem into epithelial lining fluid of patients with ventilator‐associated pneumonia. Presented at the 48th Interscience Conference on Antimicrobial Agents and Chemotherapy/46th Annual Meeting of the Infectious Diseases Society of America. Washington DC,2008. Abstr 1889.
- ,,, et al.Optimization of meropenem minimum concentration/MIC ratio to suppress in vitro resistance of Pseudomonas aeruginosa.Antimicrob Agents Chemother. In press.
Tremendous strides have been made over the last 25 years in understanding the relationship between antimicrobial exposure and response.14 Many clinicians consider antimicrobial drug pharmacokinetics (PK) and pharmacodynamics (PD) a rather esoteric or academic topic without practical applicability or clinical utility. However, it is becoming increasingly clear, particularly as less‐susceptible pathogens emerge, that consideration of PK/PD in dose selection is essential for optimizing antimicrobial therapy and, as such, is a core component of effective antimicrobial stewardship and patient care. Antimicrobial therapy can fail if an appropriate agent is selected but the dosing regimen does not provide adequate exposure against the infecting pathogens, especially at the site of infection.5, 6
The 2007 guidelines from the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA) for developing institutional antimicrobial stewardship programs highlight dose optimization as one of the key strategies for enhancing antimicrobial stewardship.7 More specifically, they recommend optimizing dosing by focusing on individual patient characteristics, causative organism and site of infection, and the PK/PD characteristics of the drug. With advances in mathematical modeling (Monte Carlo simulation), it is possible to apply our understanding of PK/PD to clinical practice and design empiric regimens that have a high probability of achieving the PD target linked to effect. These mathematical modeling techniques have an array of other utilities and have become the standard methodologies for assessing the clinical viability of both experimental and approved antimicrobials.8, 9 Furthermore, the Clinical and Laboratory Standards Institute (CLSI) has recently begun to incorporate results from PK/PD analyses in determining MIC breakpoints.10 This paper provides a general overview of antimicrobial PD before demonstrating how to apply PD principles to clinical practice through the use of Monte Carlo simulation (MCS). Piperacillin/tazobactam (TZP) is used as a motivating example for this latter purpose.
Pharmacokinetics and Pharmacodynamics: Parameters and Principles
Pharmacokinetics describes the actions of the body on an administered drug, whereas PD describes the actions of the administered drug on the body. In essence, PK refers to the movement of the drug within the body, including absorption, distribution, metabolism, and excretion. Conversely, PD refers to the effects of the drug on the body, or its physiologic actions. A drug's PD is defined by its mechanism of action, and includes both desired and undesired effects. Typically, PK and PD work together to best define or predict the full range of effects of an administered drug on an individual patient, as described in greater detail below.
The Minimum Inhibitory Concentration
The MIC is the PD parameter most often used to describe the relationship between antimicrobial drug and physiologic activity. The MIC is defined as the lowest or minimum antimicrobial concentration that inhibits visible microbial growth in artificial medium after a fixed incubation time.10, 11 This is typically determined by placing a known quantity of bacteria (or other microorganism) into multiple test tubes, and then adding increasing concentrations of a particular antibiotic, typically in log2 dilution, into consecutive tubes. The lowest antibiotic concentration that inhibits bacterial growth is then defined as the MIC for that drug‐pathogen pairing.
While useful as a quantitative measure of drug activity or potency, the MIC is not without limitations.12 The MIC does not mimic physiologic conditions. The MIC is a static measure (fixed concentration of drug in an artificial growth medium for a fixed period of time) and is not reflective of the concentration‐time profile one would typically observe in patients; drug concentrations change throughout the dosing interval. Because the MIC only measures growth inhibition, it does not reflect the rate at which bacteria are killed, nor can it identify if a dosekill response relationship exists for a particular antibiotic‐pathogen pairing. Furthermore, the MIC only quantifies net growth over an 1824‐hour observation period. Killing and regrowth may well occur during this period, as long as the net growth is zero. Finally, the MIC does not account for the post‐antibiotic effects of antibiotics. Most antibiotics, depending on the pathogen and drug class, exhibit some persistence of bacteriostatic or bactericidal activity after the drug concentration at the target site has dropped below the MIC. This activity has been described as the post‐antibiotic effect,1315 post‐antibiotic sub‐MIC effect,1317 or post‐antibiotic leukocyte enhancement effect.18, 19
Common Pharmacodynamic Measures
Examination of PK measures of drug exposure (eg, serum/tissue concentrations) in relation to the MIC surmounts many of the limitations of the MIC and provides much better prediction of antimicrobial effect than the MIC or exposure profile alone. The 3 most common PK/PD indices (sometimes abbreviated as PD measures) used to predict drug response are: 1) the ratio of the maximal free drug concentration to the MIC (fCmax:MIC), 2) the ratio of the free area under the concentration‐time curve to the MIC (fAUC:MIC), and 3) the duration of time free drug concentrations remain above the MIC (fT>MIC).24, 20, 21 The PD parameter most predictive of outcomes varies by drug class (Table 1).20
| Antibiotic | Optimal PD measure(s) |
|---|---|
| |
| Aminoglycosides | Cmax:MIC; AUC:MIC |
| ‐lactams | |
| Penicillins | T>MIC |
| Cephalosporins | T>MIC |
| Carbapenems | T>MIC |
| Monobactams | T>MIC |
| Clindamycin | AUC:MIC |
| Fluoroquinolones | AUC:MIC, Cmax:MIC |
| Glycopeptides/lipopeptides | |
| Daptomycin | AUC:MIC, Cmax:MIC |
| Oritavancin | T>MIC, Cmax:MIC |
| Vancomycin | AUC:MIC |
| Linezolid | AUC:MIC |
| Macrolides | |
| Azithromycin | AUC:MIC |
| Clarithromycin | AUC:MIC |
| Telithromycin | AUC:MIC |
| Metronidazole | AUC:MIC, Cmax:MIC |
| Tetracyclines | |
| Doxycycline | AUC:MIC |
| Tigecycline | AUC:MIC |
Certain antibiotics exhibit concentration‐dependent bactericidal activity, while others exhibit time‐dependent activity (Table 1).24, 20 For concentration‐dependent antibiotics, a doseresponse relationship exists and the therapeutic goal is to maximize exposure at the target site. Alternatively, the activity of time‐dependent antibiotics is not dependent on the intensity of exposure but is a function of the duration of time concentrations are above the MIC during the dosing interval. For the time‐dependent antibiotics like the ‐lactams, concentrations do not have to remain above the MIC for the entire dosing interval, and the fraction of the dosing interval required for maximal bacterial effect varies for the different types of ‐lactams. Although the precise fT > MIC varies for different drugbacteria combinations, bacteriostatic effects are typically observed when the free drug concentration exceeds the MIC for 3540%, 30%, and 20% of the dosing interval for the cephalosporins, penicillins, and carbapenems, respectively. Near‐maximal bactericidal effects require 6070%, 50%, and 40% fT > MIC, respectively, for these ‐lactam classes.3, 4
It is important to note that it is the free (or unbound) fraction of drug that determines its ability to penetrate tissues and exert its microbiological effect.3, 4, 22 This was demonstrated as early as the 1940s with penicillin. There are occasionally exceptions, mostly with the therapy of gram‐positive infections. Daptomycin is one such example; protein binding is approximately 9092% (free drug 810%), but the agent behaves as if the drug is approximately 75% bound (25% free).23 Nonetheless, the guiding principle is that protein binding can have an adverse impact on the PD and microbiological activity of an antibacterial agent.
Monte Carlo Simulation
With advances in mathematical modeling, it is possible to apply our understanding of antimicrobial PD to clinical practice.12 In particular, MCS can be used to integrate PK, PD, and local microbiologic surveillance data to design antibiotic regimens that have a high probability of achieving the PD target linked to effect against the range of pathogens encountered in clinical practice. In short, MCS is a technique that incorporates the variability in PK among potential patients (between‐patient variability) when predicting antibiotic exposures, and allows calculation of the probability for obtaining a critical target exposure for the range of possible MIC values.12 If a number of volunteers or patients are given an antibiotic, there will be true variability in the observed concentration time profiles between people. For example, the peak serum concentrations and AUC0‐24h will vary between individuals. In essence, MCS is a mathematical modeling technique that simulates the dispersion or full spread of concentration‐time exposure values (eg, peak concentration, area under the curve) that would be seen in a large population after administration of a specific drug dose or regimen. Once the distribution in concentration‐time profiles is determined, the probability of achieving the PD target at each MIC value for a given MIC range (ie, probability of target attainment [PTA] profile) is ascertained.
There are several steps in the MCS process. First, a PK model for the antibiotic under study is embedded into the MCS. The mean PK parameters (eg, volume, clearance, intercompartmental transfer constant) and associated variability (variance and covariance) from the selected PK model are used to create a multivariate distribution of PK parameters. From this multivariate distribution, the MCS randomly selects a set of PK parameters, and these randomly selected PK parameters are used to simulate a concentration‐time profile for a virtual subject based on the desired antibiotic dosing regimen. This process is repeated a specified number of times (eg, 5000, 1000) to simulate the distribution of concentration‐time profiles one would expect to see in the population. Once the specified number of virtual patients has been simulated (eg, 10,000 virtual patients), the proportion of the simulated population that achieves the critical exposure target (eg, 50% fT > MIC) at each MIC value for a given MIC range can be calculated. Because the relationship between drug exposure and effect is expressed as a ratio (eg, AUC:MIC, Cmax:MIC, T:MIC), a unique drug exposure:MIC ratio and PTA exists for each unique MIC value within the distribution.12
In clinical practice, a distribution of MIC values exists for a given organism or infection. Therefore, the final step is determining the overall PTA for the distribution of organisms encountered clinically. As previously mentioned, the PTA is determined at each MIC value within a given MIC range. Because the fraction of organisms collected at each MIC value is known, the overall or weighted PTA average can be calculated by multiplying the PTA for a specific MIC and the proportion of isolates with that MIC. This product is calculated for each MIC value within the MIC distribution. The overall PTA is then calculated by summing the products (PTA at a given MIC value x proportion of isolates with that MIC value) of the MIC values encountered within the distribution.12
A key element for these simulations is the estimation of the PK parameters and their associated dispersion (variance and covariance). Pharmacokinetic data, especially for new compounds, are usually limited to data from healthy volunteer studies. Caution should be exercised when generalizing the results of volunteer studies to the population of interest. Volunteer studies are often considered as the most conservative evaluation of a new drug; volunteers are young and healthy, likely to have the highest drug clearances and shortest half lives. However, when one performs MCS, the measure of central tendency (high drug clearance, short half‐lives) is only part of the story. Because MCS are explicitly creating a distribution, it is important to understand the measure of dispersion. Secondary to the limited variation surrounding PK parameters from healthy volunteer studies, it is possible that they overestimate the PTA. Applicability to the target population must always be considered.12
Motivating Example: Piperacillin‐tazobactam
Piperacillin‐tazobactam (TZP) is an acylureido‐penicillinbeta‐lactamase inhibitor combination and is frequently used as first‐line empirical therapy for healthcare‐associated infections. Like all ‐lactams, the PD parameter most predictive of its efficacy is fT > MIC, and its activity is optimized when free drug concentrations exceed the MIC for 50% of the dosing interval (50% fT > MIC). Because it is used empirically, it is critical that the TZP regimens used in practice have a high probability of achieving 50% fT > MIC against the range of MIC values likely to be encountered in a given institution.
Since the MIC of the infecting pathogen is often not available at the start of therapy, clinicians frequently rely on the hospital antibiogram to determine the utility of an antibiotic as an empiric agent. The range of MIC values reported as susceptible in clinical practice is based on the CLSI susceptibility interpretive criteria. For TZP, Enterobacteriaceae and Acinetobacter baumannii isolates with MIC values 16 mg/L are considered susceptible. The CLSI breakpoint for Pseudomonas aeruginosa is higher and isolates with MIC values 64/4 mg/L are considered susceptible.11
It is important to recognize that these CLSI TZP susceptibility breakpoints were established prior to our current understanding of ‐lactam PD and are higher relative to other ‐lactams. It was not until sometime after the establishment of the TZP susceptibility interpretive criteria were MCS studies performed to determine the ability of the US Food and Drug Administration (FDA)‐approved TZP dosing regimens in achieving 50% fT > MIC against the range of MICs deemed susceptible by CLSI.
The first study to characterize the ability of standard TZP dosing (0.5‐hour infusion of 3.375 g every 6 hours) in achieving 50% T > MIC in its targeted population for the range of MIC values deemed susceptible by CLSI was published in 2004 by our group. Employing a population PK model derived in hospitalized patients, TZP 3.375 grams administered every 6 h provided high PTA rates for MICs of 8/4 mg/L (ie, 8 mg/L for piperacillin and 4 mg/L for tazobactam) when hospitalized‐patient data were used (Figure 1).24 In clinical situations in which the MICs are expected to be 16/4 mg/L, the results of the MCS indicate that caution should be exercised when using standard TZP dosing. More recently, DeRyke et al evaluated the PD profile of the TZP nosocomial dosing scheme (0.5‐hour infusion of 4.5 g every 6 hours). Using the same population PK model employed as our study, DeRyke and colleagues noted a slightly improved PTA profile at a MIC value of 16/4 mg/L with the TZP nosocomial pneumonia dosing scheme relative to standard dosing. However, the PTA was still suboptimal for MIC values 32/4 mg/L (Figure 1).25
These findings are concerning because the TZP CLSI susceptibility breakpoint for non‐lactose fermenting Gram‐negative bacteria is 64/4 mg/L.11 In essence, the conventional and nosocomial pneumonia TZP dosing schemes provide a suboptimal PD profile for a substantial portion of the MIC distribution deemed susceptible by CLSI. The clinical relevance of this is highlighted by a study by Tam and coworkers examining the efficacy of TZP in hospitalized patients with bacteremia due to P. aeruginosa (Figure 2).26 This retrospective cohort study examined 30‐day mortality among patients who received appropriate empiric therapy between 2002 and 2006. Therapy was defined as appropriate if: 1) ‐lactam treatment (in doses appropriate for renal function as recommended by the manufacturer) was started within 24 hours of blood culture collection, and 2) the isolate was found to be susceptible to the ‐lactam agent selected. The cohort was stratified by the TZP piperacillin MIC (3264 mg/L vs. 16 mg/L) and 30‐day mortality rates were compared within MIC strata between patients who received TZP or an alternative ‐lactam with activity against Pseudomonas aeruginosa. A total of 34 episodes with MICs of 32 or 64 mg/L were identified. Seven of these cases were empirically treated with TZP, while the remaining 27 received other ‐lactam agents. Forty‐nine episodes of P. aeruginosa bacteremias had MIC values 16 mg/L. Of these 49, 10 were empirically treated with TZP and the remaining 39 were treated with other ‐lactams. The results showed that the 30‐day mortality rate was significantly higher among patients treated with TZP versus control‐treated patients with isolates possessing a MIC of either 32 or 64 mg/L (86% vs. 22%, P value = 0.004), while there was no significant difference between the two treatment groups for isolates with a MIC of up to 16 mg/L (30% vs. 21%, P = 0.673). Interestingly, patients treated with a non‐TZP ‐lactam antibiotic had 30‐day mortality rates of 21%, regardless of the TZP MIC value. Collectively, these findings and the results of the TZP MCS studies highlight the importance of considering PTA data when evaluating the utility of an antibiotic dosing scheme. These data also cast uncertainty on the appropriateness of the current TZP CLSI susceptibility breakpoint in connection with the conventional dosing TZP strategies. The current CLSI interpretation of TZP susceptibility for non‐lactose‐fermenting gram‐negatives may inadvertently provide misleading guidance to clinicians for optimal patient care.
Dosing Strategies to Improve the Probability of Target Attainment Profile of ‐lactams
Three potential dosing strategies used to improve the PTA of a ‐lactam against the range of pathogens encountered in various clinical situations include: 1) increasing the dose, 2) increasing the dosing frequency, or 3) increasing the duration of infusion.12 Intuitively, it makes sense to simply increase the drug dose. However, as demonstrated in the aforementioned TZP MCS studies, increasing the TZP dose from 3.375 grams to 4.5 grams every 6 hours had a minimal impact on the PTA profile.24, 25 To increase fT >MIC by 1 half‐life, the dose would need to be doubled. Since most ‐lactams have a half‐life of 30 minutes to 1 hour, doubling the dose only provides an extra 30 minutes or hour above the MIC, which would not be expected to have much clinical impact. In addition, doubling the dose is not cost effective since it doubles drug acquisition costs.12, 27
Increasing the dosing frequency is a viable option and may be the optimal strategy in certain situations.12 However, it is often associated with increased drug acquisition costs (more doses per day) relative to the parent regimen and may not be a viable option from a nursing and pharmacy perspective due to increased administration and preparation time. In addition, there may be a higher potential for toxicity because a greater amount of drug is given per day.
Extending the infusion time is another ‐lactam dose optimization strategy that is becoming more commonly used in clinical practice. Administering a dose of a ‐lactam agent as an infusion longer than the conventional 0.51.0‐hour infusion duration has 2 main effects. First, it produces a lower peak concentration of the drug.24 Because the bacterial kill rate for these agents is not concentration‐dependent, this does not present a major disadvantage.3, 4, 2830 Second, the drug concentrations remain in excess of the MIC for a longer period of time. Because this is what drives antibacterial effect for ‐lactams, this will yield a more favorable PTA profile. It should also be noted that this can be done with less frequent drug dosing.27
Extending the infusion time can be accomplished by either prolonging the infusion time for a major portion of the dosing interval (prolonged infusion) or administering continuously throughout the day (continuous infusion). From a PD profiling viewpoint, the two infusion methodologies yield nearly identical PTA profiles. This was highlighted in the 2007 study by Kim et al, which compared PTA rates between intermittent (0.5 hour), prolonged (4 hours), and continuous infusions of TZP (Figure 3). In their study, the PTA curves for prolonged and continuous infusion TZP were superimposable and superior to the intermittent infusion regimen for MIC values in excess of 4 mg/L.31
There are several practicalities to consider when differentiating prolonged and continuous infusion methods. The principle advantages of continuous infusion are once‐daily administration and reduced costs for labor, supplies, and administration.12, 27 The major disadvantages of continuous infusion are the need for a dedicated line for infusion (which often leads to drug compatibility issues), issues of drug stability and waste, and lack of ambulation for the patient. The need for a dedicated infusion line is particularly impractical for patients with limited intravenous access or those requiring multiple daily infusions. In addition, continuous infusion often requires insertion of a central line, which places patients at unnecessary risk of secondary catheter‐related infection.12 Continuous infusion solutions are typically prepared as 24‐hour infusions containing the total daily amount of drug. Considerable drug wastage can occur with early discontinuation of therapy; all drug within the solution needs to be wasted and cannot be reused if the order is discontinued prior to scheduled completion.
Prolonged infusion provides many of the benefits of intermittent dosing, but with the PD advantages of continuous infusion. Administration of the infusion for a prolonged time, but not continuously, obviates the need to have a dedicated intravenous line just for ‐lactam continuous infusion. It also achieves the targeted fT > MIC at a total daily dose less than standard ‐lactam dosing methods. Drug wastage is also minimized because the intermittent administration formulations are used; there is no need to prepare antibiotic solutions for 24‐hour periods. Prolonged infusion also allows the patient to be ambulatory for much of the day. The potential disadvantages of prolonged infusion relative to continuous infusion include the increased use of labor, supplies, and administration resources. Although minimized, there is still the need to schedule or time the administration of incompatible drugs.12, 27
Data Examining the Outcomes Associated With Prolonged and Continuous ‐lactam Infusions
Over the years, a number of randomized controlled trials (RCTs) and observational studies have compared outcomes between extended and intermittent ‐lactam infusions. These studies, mostly small scale in nature, involved a number of different ‐lactam antibiotics and various infectious etiologies. To ascertain if there are any clinical benefits in extending the infusion duration (prolonged and continuous), Roberts and colleagues performed a systematic review of available data on PubMed (January 1950 to November 2007), EMBASE (1966 to November 2007), and the Cochrane Controlled Trial Register (updated November 2007).32 Randomized controlled trials were meta‐analyzed, and observational studies were reviewed. Among a total of 59 potentially RCTs, 14 involving a total of 846 patients from nine countries were deemed appropriate for meta‐analysis. The use of continuous infusion of a ‐lactam antibiotic was not associated with an improvement in clinical cure (n=755 patients; odds ratio: 1.04, 95% confidence interval: 0.741.46, P = 0.83) or mortality (n=541 patients; odds ratio: 1.00, 95% confidence interval: 0.482.06, P = 1.00). In contrast, the observational studies showed that ‐lactam administration by extended or continuous infusion confers an improvement in clinical cure and this was most pronounced in critically ill patients being treated for gram‐negative bacterial infections.
There are several possible explanations for the discrepancy in results between the meta‐analysis and observational studies. First, disease severity in the studies included in the meta‐analysis was generally low, as evidenced by low mortality rates in the majority of studies. Second, a diverse group of patients and infection types were included in the RCTs, which increased the heterogeneity of the cohort analyzed. Third, a higher antibiotic dose was used in the intermittent administration group in all RCTs except one. Fourth, microbiologic and PK/PD data were not available for the majority of RCTs. Collectively, the null result from the meta‐analysis and positive data from the nonrandomized studies suggest that prolonged or continuous infusion ‐lactams is unlikely to be advantageous for all hospitalized patient populations, but may be beneficial for specific groups, such as critically ill patients with higher MIC pathogens.
The benefits of prolonged ‐lactam infusion among critically ill patients were highlighted by the study performed at Albany Medical Center Hospital.27 Based on a MCS, prolonged infusion TZP (3.375 grams administered over a 4‐hour period every 8 hours) was identified as an alternative means to the intermittent TZP dosing (3.375 grams administered over 30 minutes every 4 or 6 hours) and adopted as the standard TZP dosing scheme in February 2002. Prior to February 2002, all patients received traditional infusion TZP; after this time, all patients received prolonged infusion TZP. To evaluate the impact of the automatic dose substitution program, 14‐day mortality and hospital length of stay post‐culture collection were compared between patients who received either intermittent or prolonged TZP infusion for a TZP‐susceptible P. aeruginosa infection between 2000 and 2004.27 The study was restricted to P. aeruginosa infections for several reasons. First, patients with P. aeruginosa represented a relatively homogenous patient population; this attribute minimized confounding and increased the ability to detect differences between treatment groups according to intervention. Second, patients with P. aeruginosa infections are more dependent on antimicrobial therapy than other populations, since patients infected with P. aeruginosa are frequently critically ill and often have an impaired innate immune system.33, 34 Third, P. aeruginosa isolates typically have a higher range of MICs to TZP than other organisms, and the benefits of optimizing fT>MIC were thought to be better elucidated in this patient population.35, 36
In patients who were identified as having the greatest risk for 14‐day mortality (Acute Physiology and Chronic Health Evaluation [APACHE] II score 17), there was a significantly lower 14‐day mortality rate and a shorter median hospital LOS after culture sample collection for patients who received prolonged infusion, compared with patients who received intermittent infusion (Figure 4). No differences between prolonged infusion and intermittent infusion of TZP were observed with respect to outcome in patients at lowest risk for death (APACHE II score 17). These findings support the notion that critically ill patients who have P. aeruginosa infection are most dependent upon drug exposure for good clinical outcomes. The results also suggest that improved outcomes can be achieved by optimizing antibiotic PD in this population. Furthermore, the results highlight the importance of examining the influence of treatment within a population at greatest risk for the outcome of interest.27
In addition to potential clinical benefits, prolonged infusions can provide cost savings by minimizing the amount of drug used per day. Prolonged infusion typically achieves the targeted fT > MIC at a total daily dose less than standard ‐lactam dosing methods. For example, TZP purchases totaled $275,000 the year before conversion at Albany Medical Center Hospital. Switching to the prolonged infusion strategy reduced the total daily dose by 25%50% (by 13 doses per day) representing a savings of $68,750$135,750 in annual direct drug acquisition costs.27
Additional Pharmacokinetic and Pharmacodynamic Considerations
When assessing the PK/PD of an antibiotic, it is also important to consider concentrations achieved at the site of infection. Most MCS studies have focused on free concentrations in plasma. Whereas free concentrations in plasma are often viewed as an acceptable approximation for free concentrations at the site of infection, this is not always the case. Of particular concern is in the treatment of lower respiratory tract infections. For ‐lactams, it was commonly believed that plasma and epithelial lining fluid (ELF) of the alveolar space concentrations were comparable; antibiotic concentrations in ELF are currently used to estimate the penetration of antibiotics into the respiratory tract. However, the median ELF/plasma penetration ratio for meropenem among patients with ventilator‐associated pneumonia (VAP) is only 25%.37 The only way to achieve a favorable fT > MIC PD profile at the site of infection with meropenem is to administer higher doses over prolonged periods of time (Figure 4). In light of the meropenem ELF data, data available on concentrations at the site of infection, particularly difficult‐to‐penetrate sites, such as ELF and cerebrospinal fluid, should be considered before designing dosing scheme for implementation into clinical practice.
Up to this point, this review has been focused on PD targets of clinical success. The next frontier in PK/PD is identifying antibiotic dosing schemes and drug combinations that minimize the emergence of resistance. Data available to date suggest that PD targets for resistance prevention are typically 24‐fold higher than PD targets for success. Tam et al showed that for meropenem, the PD target needed to suppress the emergence of resistance in P. aeruginosa was a Cmin:MIC ratio of 1.7.38 Further study is still needed in the area of resistance suppression but the current data suggest that obtaining the PK/PD target against the range of MIC encountered clinically is not likely with conventional ‐lactam dosing and will most likely require more intensive regimens administered over extended periods of time.38
Arguments Against Extended ‐lactam Infusions
Limited clinical trial data and lack of FDA approval are frequently cited as the major clinical barriers for implementing extended ‐lactam infusions into practice. Unfortunately, there is a relative dearth of large‐scale randomized clinical data supporting extending the infusion of ‐lactam therapy. In addition, the package inserts for the various ‐lactam antibiotics do not provide support for these prolonged infusion dosing.
While these are valid concerns, the clinical support for intermittent ‐lactam infusions is also limited. The clinical data are largely limited to complicated intra‐abdominal infections, complicated skin and soft tissue infections, complicated urinary tract infections, and community‐acquired pneumonia. None of the intermittently administered ‐lactams currently have an indication for bacteremia (except imipenem for bacterial septicemia), and there are only limited indications for hospital‐acquired pneumonia (HAP) or VAP (imipenem for lower respiratory tract infections and TZP in combination with an aminoglycoside for HAP). In addition, the clinical trials of intermittent ‐lactam infusion regimens have commonly assessed clinical response at the test‐of‐cure visit or after completion of therapy. Arguably, this is not a very clinically meaningful endpoint for the types of infections commonly encountered on a day‐to‐day basis in today's world, where mixed diagnoses and infecting pathogens are often seen. Most important, the bacteria have evolved since the early clinical trials used to obtain FDA approval, and those outdated studies do not address the resistance profiles currently observed in clinical practice.
Conclusions
Understanding exposure‐response relationships is critical when designing antibiotic dosing schemes. In the absence of therapeutic drug monitoring, MCS can be used to design antibiotic regimens that have a high probability of attaining the PD target linked to effect against the range of MICs likely to be encountered in clinical practice. When considering ‐lactam therapy for critically ill patients likely infected with high‐MIC or reduced‐susceptibility pathogens, a prolonged or continuous infusion regimen should be considered. Compared with intermittent dosing, prolonged infusion of ‐lactams is typically associated with improved PTA, as potential benefits of cost savings, and an enhanced PD profile at the site of infection.
Tremendous strides have been made over the last 25 years in understanding the relationship between antimicrobial exposure and response.14 Many clinicians consider antimicrobial drug pharmacokinetics (PK) and pharmacodynamics (PD) a rather esoteric or academic topic without practical applicability or clinical utility. However, it is becoming increasingly clear, particularly as less‐susceptible pathogens emerge, that consideration of PK/PD in dose selection is essential for optimizing antimicrobial therapy and, as such, is a core component of effective antimicrobial stewardship and patient care. Antimicrobial therapy can fail if an appropriate agent is selected but the dosing regimen does not provide adequate exposure against the infecting pathogens, especially at the site of infection.5, 6
The 2007 guidelines from the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA) for developing institutional antimicrobial stewardship programs highlight dose optimization as one of the key strategies for enhancing antimicrobial stewardship.7 More specifically, they recommend optimizing dosing by focusing on individual patient characteristics, causative organism and site of infection, and the PK/PD characteristics of the drug. With advances in mathematical modeling (Monte Carlo simulation), it is possible to apply our understanding of PK/PD to clinical practice and design empiric regimens that have a high probability of achieving the PD target linked to effect. These mathematical modeling techniques have an array of other utilities and have become the standard methodologies for assessing the clinical viability of both experimental and approved antimicrobials.8, 9 Furthermore, the Clinical and Laboratory Standards Institute (CLSI) has recently begun to incorporate results from PK/PD analyses in determining MIC breakpoints.10 This paper provides a general overview of antimicrobial PD before demonstrating how to apply PD principles to clinical practice through the use of Monte Carlo simulation (MCS). Piperacillin/tazobactam (TZP) is used as a motivating example for this latter purpose.
Pharmacokinetics and Pharmacodynamics: Parameters and Principles
Pharmacokinetics describes the actions of the body on an administered drug, whereas PD describes the actions of the administered drug on the body. In essence, PK refers to the movement of the drug within the body, including absorption, distribution, metabolism, and excretion. Conversely, PD refers to the effects of the drug on the body, or its physiologic actions. A drug's PD is defined by its mechanism of action, and includes both desired and undesired effects. Typically, PK and PD work together to best define or predict the full range of effects of an administered drug on an individual patient, as described in greater detail below.
The Minimum Inhibitory Concentration
The MIC is the PD parameter most often used to describe the relationship between antimicrobial drug and physiologic activity. The MIC is defined as the lowest or minimum antimicrobial concentration that inhibits visible microbial growth in artificial medium after a fixed incubation time.10, 11 This is typically determined by placing a known quantity of bacteria (or other microorganism) into multiple test tubes, and then adding increasing concentrations of a particular antibiotic, typically in log2 dilution, into consecutive tubes. The lowest antibiotic concentration that inhibits bacterial growth is then defined as the MIC for that drug‐pathogen pairing.
While useful as a quantitative measure of drug activity or potency, the MIC is not without limitations.12 The MIC does not mimic physiologic conditions. The MIC is a static measure (fixed concentration of drug in an artificial growth medium for a fixed period of time) and is not reflective of the concentration‐time profile one would typically observe in patients; drug concentrations change throughout the dosing interval. Because the MIC only measures growth inhibition, it does not reflect the rate at which bacteria are killed, nor can it identify if a dosekill response relationship exists for a particular antibiotic‐pathogen pairing. Furthermore, the MIC only quantifies net growth over an 1824‐hour observation period. Killing and regrowth may well occur during this period, as long as the net growth is zero. Finally, the MIC does not account for the post‐antibiotic effects of antibiotics. Most antibiotics, depending on the pathogen and drug class, exhibit some persistence of bacteriostatic or bactericidal activity after the drug concentration at the target site has dropped below the MIC. This activity has been described as the post‐antibiotic effect,1315 post‐antibiotic sub‐MIC effect,1317 or post‐antibiotic leukocyte enhancement effect.18, 19
Common Pharmacodynamic Measures
Examination of PK measures of drug exposure (eg, serum/tissue concentrations) in relation to the MIC surmounts many of the limitations of the MIC and provides much better prediction of antimicrobial effect than the MIC or exposure profile alone. The 3 most common PK/PD indices (sometimes abbreviated as PD measures) used to predict drug response are: 1) the ratio of the maximal free drug concentration to the MIC (fCmax:MIC), 2) the ratio of the free area under the concentration‐time curve to the MIC (fAUC:MIC), and 3) the duration of time free drug concentrations remain above the MIC (fT>MIC).24, 20, 21 The PD parameter most predictive of outcomes varies by drug class (Table 1).20
| Antibiotic | Optimal PD measure(s) |
|---|---|
| |
| Aminoglycosides | Cmax:MIC; AUC:MIC |
| ‐lactams | |
| Penicillins | T>MIC |
| Cephalosporins | T>MIC |
| Carbapenems | T>MIC |
| Monobactams | T>MIC |
| Clindamycin | AUC:MIC |
| Fluoroquinolones | AUC:MIC, Cmax:MIC |
| Glycopeptides/lipopeptides | |
| Daptomycin | AUC:MIC, Cmax:MIC |
| Oritavancin | T>MIC, Cmax:MIC |
| Vancomycin | AUC:MIC |
| Linezolid | AUC:MIC |
| Macrolides | |
| Azithromycin | AUC:MIC |
| Clarithromycin | AUC:MIC |
| Telithromycin | AUC:MIC |
| Metronidazole | AUC:MIC, Cmax:MIC |
| Tetracyclines | |
| Doxycycline | AUC:MIC |
| Tigecycline | AUC:MIC |
Certain antibiotics exhibit concentration‐dependent bactericidal activity, while others exhibit time‐dependent activity (Table 1).24, 20 For concentration‐dependent antibiotics, a doseresponse relationship exists and the therapeutic goal is to maximize exposure at the target site. Alternatively, the activity of time‐dependent antibiotics is not dependent on the intensity of exposure but is a function of the duration of time concentrations are above the MIC during the dosing interval. For the time‐dependent antibiotics like the ‐lactams, concentrations do not have to remain above the MIC for the entire dosing interval, and the fraction of the dosing interval required for maximal bacterial effect varies for the different types of ‐lactams. Although the precise fT > MIC varies for different drugbacteria combinations, bacteriostatic effects are typically observed when the free drug concentration exceeds the MIC for 3540%, 30%, and 20% of the dosing interval for the cephalosporins, penicillins, and carbapenems, respectively. Near‐maximal bactericidal effects require 6070%, 50%, and 40% fT > MIC, respectively, for these ‐lactam classes.3, 4
It is important to note that it is the free (or unbound) fraction of drug that determines its ability to penetrate tissues and exert its microbiological effect.3, 4, 22 This was demonstrated as early as the 1940s with penicillin. There are occasionally exceptions, mostly with the therapy of gram‐positive infections. Daptomycin is one such example; protein binding is approximately 9092% (free drug 810%), but the agent behaves as if the drug is approximately 75% bound (25% free).23 Nonetheless, the guiding principle is that protein binding can have an adverse impact on the PD and microbiological activity of an antibacterial agent.
Monte Carlo Simulation
With advances in mathematical modeling, it is possible to apply our understanding of antimicrobial PD to clinical practice.12 In particular, MCS can be used to integrate PK, PD, and local microbiologic surveillance data to design antibiotic regimens that have a high probability of achieving the PD target linked to effect against the range of pathogens encountered in clinical practice. In short, MCS is a technique that incorporates the variability in PK among potential patients (between‐patient variability) when predicting antibiotic exposures, and allows calculation of the probability for obtaining a critical target exposure for the range of possible MIC values.12 If a number of volunteers or patients are given an antibiotic, there will be true variability in the observed concentration time profiles between people. For example, the peak serum concentrations and AUC0‐24h will vary between individuals. In essence, MCS is a mathematical modeling technique that simulates the dispersion or full spread of concentration‐time exposure values (eg, peak concentration, area under the curve) that would be seen in a large population after administration of a specific drug dose or regimen. Once the distribution in concentration‐time profiles is determined, the probability of achieving the PD target at each MIC value for a given MIC range (ie, probability of target attainment [PTA] profile) is ascertained.
There are several steps in the MCS process. First, a PK model for the antibiotic under study is embedded into the MCS. The mean PK parameters (eg, volume, clearance, intercompartmental transfer constant) and associated variability (variance and covariance) from the selected PK model are used to create a multivariate distribution of PK parameters. From this multivariate distribution, the MCS randomly selects a set of PK parameters, and these randomly selected PK parameters are used to simulate a concentration‐time profile for a virtual subject based on the desired antibiotic dosing regimen. This process is repeated a specified number of times (eg, 5000, 1000) to simulate the distribution of concentration‐time profiles one would expect to see in the population. Once the specified number of virtual patients has been simulated (eg, 10,000 virtual patients), the proportion of the simulated population that achieves the critical exposure target (eg, 50% fT > MIC) at each MIC value for a given MIC range can be calculated. Because the relationship between drug exposure and effect is expressed as a ratio (eg, AUC:MIC, Cmax:MIC, T:MIC), a unique drug exposure:MIC ratio and PTA exists for each unique MIC value within the distribution.12
In clinical practice, a distribution of MIC values exists for a given organism or infection. Therefore, the final step is determining the overall PTA for the distribution of organisms encountered clinically. As previously mentioned, the PTA is determined at each MIC value within a given MIC range. Because the fraction of organisms collected at each MIC value is known, the overall or weighted PTA average can be calculated by multiplying the PTA for a specific MIC and the proportion of isolates with that MIC. This product is calculated for each MIC value within the MIC distribution. The overall PTA is then calculated by summing the products (PTA at a given MIC value x proportion of isolates with that MIC value) of the MIC values encountered within the distribution.12
A key element for these simulations is the estimation of the PK parameters and their associated dispersion (variance and covariance). Pharmacokinetic data, especially for new compounds, are usually limited to data from healthy volunteer studies. Caution should be exercised when generalizing the results of volunteer studies to the population of interest. Volunteer studies are often considered as the most conservative evaluation of a new drug; volunteers are young and healthy, likely to have the highest drug clearances and shortest half lives. However, when one performs MCS, the measure of central tendency (high drug clearance, short half‐lives) is only part of the story. Because MCS are explicitly creating a distribution, it is important to understand the measure of dispersion. Secondary to the limited variation surrounding PK parameters from healthy volunteer studies, it is possible that they overestimate the PTA. Applicability to the target population must always be considered.12
Motivating Example: Piperacillin‐tazobactam
Piperacillin‐tazobactam (TZP) is an acylureido‐penicillinbeta‐lactamase inhibitor combination and is frequently used as first‐line empirical therapy for healthcare‐associated infections. Like all ‐lactams, the PD parameter most predictive of its efficacy is fT > MIC, and its activity is optimized when free drug concentrations exceed the MIC for 50% of the dosing interval (50% fT > MIC). Because it is used empirically, it is critical that the TZP regimens used in practice have a high probability of achieving 50% fT > MIC against the range of MIC values likely to be encountered in a given institution.
Since the MIC of the infecting pathogen is often not available at the start of therapy, clinicians frequently rely on the hospital antibiogram to determine the utility of an antibiotic as an empiric agent. The range of MIC values reported as susceptible in clinical practice is based on the CLSI susceptibility interpretive criteria. For TZP, Enterobacteriaceae and Acinetobacter baumannii isolates with MIC values 16 mg/L are considered susceptible. The CLSI breakpoint for Pseudomonas aeruginosa is higher and isolates with MIC values 64/4 mg/L are considered susceptible.11
It is important to recognize that these CLSI TZP susceptibility breakpoints were established prior to our current understanding of ‐lactam PD and are higher relative to other ‐lactams. It was not until sometime after the establishment of the TZP susceptibility interpretive criteria were MCS studies performed to determine the ability of the US Food and Drug Administration (FDA)‐approved TZP dosing regimens in achieving 50% fT > MIC against the range of MICs deemed susceptible by CLSI.
The first study to characterize the ability of standard TZP dosing (0.5‐hour infusion of 3.375 g every 6 hours) in achieving 50% T > MIC in its targeted population for the range of MIC values deemed susceptible by CLSI was published in 2004 by our group. Employing a population PK model derived in hospitalized patients, TZP 3.375 grams administered every 6 h provided high PTA rates for MICs of 8/4 mg/L (ie, 8 mg/L for piperacillin and 4 mg/L for tazobactam) when hospitalized‐patient data were used (Figure 1).24 In clinical situations in which the MICs are expected to be 16/4 mg/L, the results of the MCS indicate that caution should be exercised when using standard TZP dosing. More recently, DeRyke et al evaluated the PD profile of the TZP nosocomial dosing scheme (0.5‐hour infusion of 4.5 g every 6 hours). Using the same population PK model employed as our study, DeRyke and colleagues noted a slightly improved PTA profile at a MIC value of 16/4 mg/L with the TZP nosocomial pneumonia dosing scheme relative to standard dosing. However, the PTA was still suboptimal for MIC values 32/4 mg/L (Figure 1).25
These findings are concerning because the TZP CLSI susceptibility breakpoint for non‐lactose fermenting Gram‐negative bacteria is 64/4 mg/L.11 In essence, the conventional and nosocomial pneumonia TZP dosing schemes provide a suboptimal PD profile for a substantial portion of the MIC distribution deemed susceptible by CLSI. The clinical relevance of this is highlighted by a study by Tam and coworkers examining the efficacy of TZP in hospitalized patients with bacteremia due to P. aeruginosa (Figure 2).26 This retrospective cohort study examined 30‐day mortality among patients who received appropriate empiric therapy between 2002 and 2006. Therapy was defined as appropriate if: 1) ‐lactam treatment (in doses appropriate for renal function as recommended by the manufacturer) was started within 24 hours of blood culture collection, and 2) the isolate was found to be susceptible to the ‐lactam agent selected. The cohort was stratified by the TZP piperacillin MIC (3264 mg/L vs. 16 mg/L) and 30‐day mortality rates were compared within MIC strata between patients who received TZP or an alternative ‐lactam with activity against Pseudomonas aeruginosa. A total of 34 episodes with MICs of 32 or 64 mg/L were identified. Seven of these cases were empirically treated with TZP, while the remaining 27 received other ‐lactam agents. Forty‐nine episodes of P. aeruginosa bacteremias had MIC values 16 mg/L. Of these 49, 10 were empirically treated with TZP and the remaining 39 were treated with other ‐lactams. The results showed that the 30‐day mortality rate was significantly higher among patients treated with TZP versus control‐treated patients with isolates possessing a MIC of either 32 or 64 mg/L (86% vs. 22%, P value = 0.004), while there was no significant difference between the two treatment groups for isolates with a MIC of up to 16 mg/L (30% vs. 21%, P = 0.673). Interestingly, patients treated with a non‐TZP ‐lactam antibiotic had 30‐day mortality rates of 21%, regardless of the TZP MIC value. Collectively, these findings and the results of the TZP MCS studies highlight the importance of considering PTA data when evaluating the utility of an antibiotic dosing scheme. These data also cast uncertainty on the appropriateness of the current TZP CLSI susceptibility breakpoint in connection with the conventional dosing TZP strategies. The current CLSI interpretation of TZP susceptibility for non‐lactose‐fermenting gram‐negatives may inadvertently provide misleading guidance to clinicians for optimal patient care.
Dosing Strategies to Improve the Probability of Target Attainment Profile of ‐lactams
Three potential dosing strategies used to improve the PTA of a ‐lactam against the range of pathogens encountered in various clinical situations include: 1) increasing the dose, 2) increasing the dosing frequency, or 3) increasing the duration of infusion.12 Intuitively, it makes sense to simply increase the drug dose. However, as demonstrated in the aforementioned TZP MCS studies, increasing the TZP dose from 3.375 grams to 4.5 grams every 6 hours had a minimal impact on the PTA profile.24, 25 To increase fT >MIC by 1 half‐life, the dose would need to be doubled. Since most ‐lactams have a half‐life of 30 minutes to 1 hour, doubling the dose only provides an extra 30 minutes or hour above the MIC, which would not be expected to have much clinical impact. In addition, doubling the dose is not cost effective since it doubles drug acquisition costs.12, 27
Increasing the dosing frequency is a viable option and may be the optimal strategy in certain situations.12 However, it is often associated with increased drug acquisition costs (more doses per day) relative to the parent regimen and may not be a viable option from a nursing and pharmacy perspective due to increased administration and preparation time. In addition, there may be a higher potential for toxicity because a greater amount of drug is given per day.
Extending the infusion time is another ‐lactam dose optimization strategy that is becoming more commonly used in clinical practice. Administering a dose of a ‐lactam agent as an infusion longer than the conventional 0.51.0‐hour infusion duration has 2 main effects. First, it produces a lower peak concentration of the drug.24 Because the bacterial kill rate for these agents is not concentration‐dependent, this does not present a major disadvantage.3, 4, 2830 Second, the drug concentrations remain in excess of the MIC for a longer period of time. Because this is what drives antibacterial effect for ‐lactams, this will yield a more favorable PTA profile. It should also be noted that this can be done with less frequent drug dosing.27
Extending the infusion time can be accomplished by either prolonging the infusion time for a major portion of the dosing interval (prolonged infusion) or administering continuously throughout the day (continuous infusion). From a PD profiling viewpoint, the two infusion methodologies yield nearly identical PTA profiles. This was highlighted in the 2007 study by Kim et al, which compared PTA rates between intermittent (0.5 hour), prolonged (4 hours), and continuous infusions of TZP (Figure 3). In their study, the PTA curves for prolonged and continuous infusion TZP were superimposable and superior to the intermittent infusion regimen for MIC values in excess of 4 mg/L.31
There are several practicalities to consider when differentiating prolonged and continuous infusion methods. The principle advantages of continuous infusion are once‐daily administration and reduced costs for labor, supplies, and administration.12, 27 The major disadvantages of continuous infusion are the need for a dedicated line for infusion (which often leads to drug compatibility issues), issues of drug stability and waste, and lack of ambulation for the patient. The need for a dedicated infusion line is particularly impractical for patients with limited intravenous access or those requiring multiple daily infusions. In addition, continuous infusion often requires insertion of a central line, which places patients at unnecessary risk of secondary catheter‐related infection.12 Continuous infusion solutions are typically prepared as 24‐hour infusions containing the total daily amount of drug. Considerable drug wastage can occur with early discontinuation of therapy; all drug within the solution needs to be wasted and cannot be reused if the order is discontinued prior to scheduled completion.
Prolonged infusion provides many of the benefits of intermittent dosing, but with the PD advantages of continuous infusion. Administration of the infusion for a prolonged time, but not continuously, obviates the need to have a dedicated intravenous line just for ‐lactam continuous infusion. It also achieves the targeted fT > MIC at a total daily dose less than standard ‐lactam dosing methods. Drug wastage is also minimized because the intermittent administration formulations are used; there is no need to prepare antibiotic solutions for 24‐hour periods. Prolonged infusion also allows the patient to be ambulatory for much of the day. The potential disadvantages of prolonged infusion relative to continuous infusion include the increased use of labor, supplies, and administration resources. Although minimized, there is still the need to schedule or time the administration of incompatible drugs.12, 27
Data Examining the Outcomes Associated With Prolonged and Continuous ‐lactam Infusions
Over the years, a number of randomized controlled trials (RCTs) and observational studies have compared outcomes between extended and intermittent ‐lactam infusions. These studies, mostly small scale in nature, involved a number of different ‐lactam antibiotics and various infectious etiologies. To ascertain if there are any clinical benefits in extending the infusion duration (prolonged and continuous), Roberts and colleagues performed a systematic review of available data on PubMed (January 1950 to November 2007), EMBASE (1966 to November 2007), and the Cochrane Controlled Trial Register (updated November 2007).32 Randomized controlled trials were meta‐analyzed, and observational studies were reviewed. Among a total of 59 potentially RCTs, 14 involving a total of 846 patients from nine countries were deemed appropriate for meta‐analysis. The use of continuous infusion of a ‐lactam antibiotic was not associated with an improvement in clinical cure (n=755 patients; odds ratio: 1.04, 95% confidence interval: 0.741.46, P = 0.83) or mortality (n=541 patients; odds ratio: 1.00, 95% confidence interval: 0.482.06, P = 1.00). In contrast, the observational studies showed that ‐lactam administration by extended or continuous infusion confers an improvement in clinical cure and this was most pronounced in critically ill patients being treated for gram‐negative bacterial infections.
There are several possible explanations for the discrepancy in results between the meta‐analysis and observational studies. First, disease severity in the studies included in the meta‐analysis was generally low, as evidenced by low mortality rates in the majority of studies. Second, a diverse group of patients and infection types were included in the RCTs, which increased the heterogeneity of the cohort analyzed. Third, a higher antibiotic dose was used in the intermittent administration group in all RCTs except one. Fourth, microbiologic and PK/PD data were not available for the majority of RCTs. Collectively, the null result from the meta‐analysis and positive data from the nonrandomized studies suggest that prolonged or continuous infusion ‐lactams is unlikely to be advantageous for all hospitalized patient populations, but may be beneficial for specific groups, such as critically ill patients with higher MIC pathogens.
The benefits of prolonged ‐lactam infusion among critically ill patients were highlighted by the study performed at Albany Medical Center Hospital.27 Based on a MCS, prolonged infusion TZP (3.375 grams administered over a 4‐hour period every 8 hours) was identified as an alternative means to the intermittent TZP dosing (3.375 grams administered over 30 minutes every 4 or 6 hours) and adopted as the standard TZP dosing scheme in February 2002. Prior to February 2002, all patients received traditional infusion TZP; after this time, all patients received prolonged infusion TZP. To evaluate the impact of the automatic dose substitution program, 14‐day mortality and hospital length of stay post‐culture collection were compared between patients who received either intermittent or prolonged TZP infusion for a TZP‐susceptible P. aeruginosa infection between 2000 and 2004.27 The study was restricted to P. aeruginosa infections for several reasons. First, patients with P. aeruginosa represented a relatively homogenous patient population; this attribute minimized confounding and increased the ability to detect differences between treatment groups according to intervention. Second, patients with P. aeruginosa infections are more dependent on antimicrobial therapy than other populations, since patients infected with P. aeruginosa are frequently critically ill and often have an impaired innate immune system.33, 34 Third, P. aeruginosa isolates typically have a higher range of MICs to TZP than other organisms, and the benefits of optimizing fT>MIC were thought to be better elucidated in this patient population.35, 36
In patients who were identified as having the greatest risk for 14‐day mortality (Acute Physiology and Chronic Health Evaluation [APACHE] II score 17), there was a significantly lower 14‐day mortality rate and a shorter median hospital LOS after culture sample collection for patients who received prolonged infusion, compared with patients who received intermittent infusion (Figure 4). No differences between prolonged infusion and intermittent infusion of TZP were observed with respect to outcome in patients at lowest risk for death (APACHE II score 17). These findings support the notion that critically ill patients who have P. aeruginosa infection are most dependent upon drug exposure for good clinical outcomes. The results also suggest that improved outcomes can be achieved by optimizing antibiotic PD in this population. Furthermore, the results highlight the importance of examining the influence of treatment within a population at greatest risk for the outcome of interest.27
In addition to potential clinical benefits, prolonged infusions can provide cost savings by minimizing the amount of drug used per day. Prolonged infusion typically achieves the targeted fT > MIC at a total daily dose less than standard ‐lactam dosing methods. For example, TZP purchases totaled $275,000 the year before conversion at Albany Medical Center Hospital. Switching to the prolonged infusion strategy reduced the total daily dose by 25%50% (by 13 doses per day) representing a savings of $68,750$135,750 in annual direct drug acquisition costs.27
Additional Pharmacokinetic and Pharmacodynamic Considerations
When assessing the PK/PD of an antibiotic, it is also important to consider concentrations achieved at the site of infection. Most MCS studies have focused on free concentrations in plasma. Whereas free concentrations in plasma are often viewed as an acceptable approximation for free concentrations at the site of infection, this is not always the case. Of particular concern is in the treatment of lower respiratory tract infections. For ‐lactams, it was commonly believed that plasma and epithelial lining fluid (ELF) of the alveolar space concentrations were comparable; antibiotic concentrations in ELF are currently used to estimate the penetration of antibiotics into the respiratory tract. However, the median ELF/plasma penetration ratio for meropenem among patients with ventilator‐associated pneumonia (VAP) is only 25%.37 The only way to achieve a favorable fT > MIC PD profile at the site of infection with meropenem is to administer higher doses over prolonged periods of time (Figure 4). In light of the meropenem ELF data, data available on concentrations at the site of infection, particularly difficult‐to‐penetrate sites, such as ELF and cerebrospinal fluid, should be considered before designing dosing scheme for implementation into clinical practice.
Up to this point, this review has been focused on PD targets of clinical success. The next frontier in PK/PD is identifying antibiotic dosing schemes and drug combinations that minimize the emergence of resistance. Data available to date suggest that PD targets for resistance prevention are typically 24‐fold higher than PD targets for success. Tam et al showed that for meropenem, the PD target needed to suppress the emergence of resistance in P. aeruginosa was a Cmin:MIC ratio of 1.7.38 Further study is still needed in the area of resistance suppression but the current data suggest that obtaining the PK/PD target against the range of MIC encountered clinically is not likely with conventional ‐lactam dosing and will most likely require more intensive regimens administered over extended periods of time.38
Arguments Against Extended ‐lactam Infusions
Limited clinical trial data and lack of FDA approval are frequently cited as the major clinical barriers for implementing extended ‐lactam infusions into practice. Unfortunately, there is a relative dearth of large‐scale randomized clinical data supporting extending the infusion of ‐lactam therapy. In addition, the package inserts for the various ‐lactam antibiotics do not provide support for these prolonged infusion dosing.
While these are valid concerns, the clinical support for intermittent ‐lactam infusions is also limited. The clinical data are largely limited to complicated intra‐abdominal infections, complicated skin and soft tissue infections, complicated urinary tract infections, and community‐acquired pneumonia. None of the intermittently administered ‐lactams currently have an indication for bacteremia (except imipenem for bacterial septicemia), and there are only limited indications for hospital‐acquired pneumonia (HAP) or VAP (imipenem for lower respiratory tract infections and TZP in combination with an aminoglycoside for HAP). In addition, the clinical trials of intermittent ‐lactam infusion regimens have commonly assessed clinical response at the test‐of‐cure visit or after completion of therapy. Arguably, this is not a very clinically meaningful endpoint for the types of infections commonly encountered on a day‐to‐day basis in today's world, where mixed diagnoses and infecting pathogens are often seen. Most important, the bacteria have evolved since the early clinical trials used to obtain FDA approval, and those outdated studies do not address the resistance profiles currently observed in clinical practice.
Conclusions
Understanding exposure‐response relationships is critical when designing antibiotic dosing schemes. In the absence of therapeutic drug monitoring, MCS can be used to design antibiotic regimens that have a high probability of attaining the PD target linked to effect against the range of MICs likely to be encountered in clinical practice. When considering ‐lactam therapy for critically ill patients likely infected with high‐MIC or reduced‐susceptibility pathogens, a prolonged or continuous infusion regimen should be considered. Compared with intermittent dosing, prolonged infusion of ‐lactams is typically associated with improved PTA, as potential benefits of cost savings, and an enhanced PD profile at the site of infection.
- ,,,.Pharmacokinetic‐pharmacodynamic considerations in the design of hospital‐acquired or ventilator‐associated bacterial pneumonia studies: look before you leap!Clin Infect Dis.2010;51(Suppl 1):S103–S110.
- ,,, et al.Pharmacokinetics‐pharmacodynamics of antimicrobial therapy: it's not just for mice anymore.Clin Infect Dis.2007;44(1):79–86.
- .Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men.Clin Infect Dis.1998;26(1):1–10; quiz 11–12.
- .Antimicrobial pharmacodynamics: critical interactions of ‘bug and drug’.Nat Rev Microbiol.2004;2(4):289–300.
- ,,,,.Length of stay and hospital costs associated with a pharmacodynamic‐based clinical pathway for empiric antibiotic choice for ventilator‐associated pneumonia.Pharmacotherapy.2010;30(5):453–462.
- ,,, et al.Pharmacodynamic‐based clinical pathway for empiric antibiotic choice in patients with ventilator‐associated pneumonia.J Crit Care.2010;25(1):69–77.
- ,,, et al.Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America guidelines for developing an institutional program to enhance antimicrobial stewardship.Clin Infect Dis.2007;44(2):159–177.
- ,,, et al.How modeling and simulation have enhanced decision making in new drug development.J Pharmacokinet Pharmacodyn.2005;32(2):185–197.
- ,,,,.PK/PD: new insights for antibacterial and antiviral applications.Curr Opin Pharmacol. Oct2008;8(5):549–556.
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing; Twentieth Informational Supplement. CLSI document M100‐S20. Wayne, Pennsylvania: Clinical and Laboratory Standards Institute; 2010.
- Clinical and Laboratory Standards Institute/NCCLS. Performance standards for Antimicrobial disc diffusion tests; Approved standards. 9th ed. CLSI Document M2‐M9. Wayne, PA: Clinical and Laboratory Standards Institute; 2006.
- ,,.Application of antimicrobial pharmacodynamic concepts into clinical practice: focus on beta‐lactam antibiotics: insights from the Society of Infectious Diseases Pharmacists.Pharmacotherapy.2006;26(9):1320–1332.
- ,,,.Postantibiotic effect of imipenem on Pseudomonas aeruginosa.Antimicrob Agents Chemother.1984;26(5):678–682.
- ,,.Postantibiotic, postantibiotic sub‐MIC, and subinhibitory effects of PGE‐9509924, ciprofloxacin, and levofloxacin.Antimicrob Agents Chemother.2003;47(10):3352–3356.
- ,,, et al.In vitro evaluation of CBR‐2092, a novel rifamycin‐quinolone hybrid antibiotic: microbiology profiling studies with staphylococci and streptococci.Antimicrob Agents Chemother.2008;52(7):2324–2334.
- ,.The post‐antibiotic sub‐MIC effect in vitro and in vivo.J Antimicrob Chemother.1993;31(Suppl D):159–166.
- .Pharmacodynamic effects of subinhibitory antibiotic concentrations.Int J Antimicrob Agents.2001;17(1):1–8.
- ,,.Postantibiotic leukocyte enhancement: increased susceptibility of bacteria pretreated with antibiotics to activity of leukocytes.Rev Infect Dis.1981;3(1):38–44.
- ,.Enhancement of leukocyte activity against Escherichia coli after brief exposure to chloramphenicol.Antimicrob Agents Chemother.1979;16(6):695–700.
- ,.Pharmacokinetic and pharmacodynamic parameters of antimicrobials: potential for providing dosing regimens that are less vulnerable to resistance.Clin Pharmacokinet.2009;48(8):517–528.
- ,.Pharmacokinetics/pharmacodynamics of antibacterials in the intensive care unit: setting appropriate dosing regimens.Int J Antimicrob Agents.2008;32(4):294–301.
- ,,.Effect of protein binding on antibiotic activity in vivo.J Antimicrob Chemother.1983;11(3):233–238.
- ,,,,.Determining the active fraction of daptomycin against MRSA by evaluating bactericidal activity in the presence of protein and pharmacodynamic (PD) modeling. 49th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy.2009;A1‐1270.
- ,,,,.Pharmacodynamic profiling of piperacillin in the presence of tazobactam in patients through the use of population pharmacokinetic models and Monte Carlo simulation.Antimicrob Agents Chemother.2004;48(12):4718–4724.
- ,,.Reevaluation of current susceptibility breakpoints for Gram‐negative rods based on pharmacodynamic assessment.Diagn Microbiol Infect Dis.2007;58(3):337–344.
- ,,, et al.Outcomes of bacteremia due to Pseudomonas aeruginosa with reduced susceptibility to piperacillin‐tazobactam: implications on the appropriateness of the resistance breakpoint.Clin Infect Dis. 152008;46(6):862–867.
- ,,.Piperacillin‐tazobactam for Pseudomonas aeruginosa infection: clinical implications of an extended‐infusion dosing strategy.Clin Infect Dis. 12007;44(3):357–363.
- ,.Pharmacokinetics and pharmacodynamics of antibiotics in otitis media.Pediatr Infect Dis J.1996;15(3):255–259.
- .Interrelationship between pharmacokinetics and pharmacodynamics in determining dosage regimens for broad‐spectrum cephalosporins.Diagn Microbiol Infect Dis.1995;22(1–2):89–96.
- .How does a patient maximally benefit from anti‐infective chemotherapy?Clin Infect Dis.2004;39(8):1245–1246.
- ,,,.Optimal dosing of piperacillin‐tazobactam for the treatment of Pseudomonas aeruginosa infections: prolonged or continuous infusion?Pharmacotherapy.2007;27(11):1490–1497.
- ,,,,.A systematic review on clinical benefits of continuous administration of beta‐lactam antibiotics.Crit Care Med.2009;37(6):2071–2078.
- ,,.Pharmacokinetic/pharmacodynamic modeling can help guide targeted antimicrobial therapy for nosocomial gram‐negative infections in critically ill patients.Diagn Microbiol Infect Dis.2004;48(2):125–130.
- ,,, et al.Pseudomonas aeruginosa bloodstream infection: importance of appropriate initial antimicrobial treatment.Antimicrob Agents Chemother.2005;49(4):1306–1311.
- ,,,.Assessment of pathogen occurrences and resistance profiles among infected patients in the intensive care unit: report from the SENTRY Antimicrobial Surveillance Program (North America, 2001).Int J Antimicrob Agents.2004;24(2):111–118.
- ,,.Results from the Meropenem Yearly Susceptibility Test Information Collection (MYSTIC) Programme: report of the 2001 data from 15 United States medical centres.Int J Antimicrob Agents.2004;23(1):52–59.
- ,,, et al.Penetration of meropenem into epithelial lining fluid of patients with ventilator‐associated pneumonia. Presented at the 48th Interscience Conference on Antimicrobial Agents and Chemotherapy/46th Annual Meeting of the Infectious Diseases Society of America. Washington DC,2008. Abstr 1889.
- ,,, et al.Optimization of meropenem minimum concentration/MIC ratio to suppress in vitro resistance of Pseudomonas aeruginosa.Antimicrob Agents Chemother. In press.
- ,,,.Pharmacokinetic‐pharmacodynamic considerations in the design of hospital‐acquired or ventilator‐associated bacterial pneumonia studies: look before you leap!Clin Infect Dis.2010;51(Suppl 1):S103–S110.
- ,,, et al.Pharmacokinetics‐pharmacodynamics of antimicrobial therapy: it's not just for mice anymore.Clin Infect Dis.2007;44(1):79–86.
- .Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men.Clin Infect Dis.1998;26(1):1–10; quiz 11–12.
- .Antimicrobial pharmacodynamics: critical interactions of ‘bug and drug’.Nat Rev Microbiol.2004;2(4):289–300.
- ,,,,.Length of stay and hospital costs associated with a pharmacodynamic‐based clinical pathway for empiric antibiotic choice for ventilator‐associated pneumonia.Pharmacotherapy.2010;30(5):453–462.
- ,,, et al.Pharmacodynamic‐based clinical pathway for empiric antibiotic choice in patients with ventilator‐associated pneumonia.J Crit Care.2010;25(1):69–77.
- ,,, et al.Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America guidelines for developing an institutional program to enhance antimicrobial stewardship.Clin Infect Dis.2007;44(2):159–177.
- ,,, et al.How modeling and simulation have enhanced decision making in new drug development.J Pharmacokinet Pharmacodyn.2005;32(2):185–197.
- ,,,,.PK/PD: new insights for antibacterial and antiviral applications.Curr Opin Pharmacol. Oct2008;8(5):549–556.
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing; Twentieth Informational Supplement. CLSI document M100‐S20. Wayne, Pennsylvania: Clinical and Laboratory Standards Institute; 2010.
- Clinical and Laboratory Standards Institute/NCCLS. Performance standards for Antimicrobial disc diffusion tests; Approved standards. 9th ed. CLSI Document M2‐M9. Wayne, PA: Clinical and Laboratory Standards Institute; 2006.
- ,,.Application of antimicrobial pharmacodynamic concepts into clinical practice: focus on beta‐lactam antibiotics: insights from the Society of Infectious Diseases Pharmacists.Pharmacotherapy.2006;26(9):1320–1332.
- ,,,.Postantibiotic effect of imipenem on Pseudomonas aeruginosa.Antimicrob Agents Chemother.1984;26(5):678–682.
- ,,.Postantibiotic, postantibiotic sub‐MIC, and subinhibitory effects of PGE‐9509924, ciprofloxacin, and levofloxacin.Antimicrob Agents Chemother.2003;47(10):3352–3356.
- ,,, et al.In vitro evaluation of CBR‐2092, a novel rifamycin‐quinolone hybrid antibiotic: microbiology profiling studies with staphylococci and streptococci.Antimicrob Agents Chemother.2008;52(7):2324–2334.
- ,.The post‐antibiotic sub‐MIC effect in vitro and in vivo.J Antimicrob Chemother.1993;31(Suppl D):159–166.
- .Pharmacodynamic effects of subinhibitory antibiotic concentrations.Int J Antimicrob Agents.2001;17(1):1–8.
- ,,.Postantibiotic leukocyte enhancement: increased susceptibility of bacteria pretreated with antibiotics to activity of leukocytes.Rev Infect Dis.1981;3(1):38–44.
- ,.Enhancement of leukocyte activity against Escherichia coli after brief exposure to chloramphenicol.Antimicrob Agents Chemother.1979;16(6):695–700.
- ,.Pharmacokinetic and pharmacodynamic parameters of antimicrobials: potential for providing dosing regimens that are less vulnerable to resistance.Clin Pharmacokinet.2009;48(8):517–528.
- ,.Pharmacokinetics/pharmacodynamics of antibacterials in the intensive care unit: setting appropriate dosing regimens.Int J Antimicrob Agents.2008;32(4):294–301.
- ,,.Effect of protein binding on antibiotic activity in vivo.J Antimicrob Chemother.1983;11(3):233–238.
- ,,,,.Determining the active fraction of daptomycin against MRSA by evaluating bactericidal activity in the presence of protein and pharmacodynamic (PD) modeling. 49th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy.2009;A1‐1270.
- ,,,,.Pharmacodynamic profiling of piperacillin in the presence of tazobactam in patients through the use of population pharmacokinetic models and Monte Carlo simulation.Antimicrob Agents Chemother.2004;48(12):4718–4724.
- ,,.Reevaluation of current susceptibility breakpoints for Gram‐negative rods based on pharmacodynamic assessment.Diagn Microbiol Infect Dis.2007;58(3):337–344.
- ,,, et al.Outcomes of bacteremia due to Pseudomonas aeruginosa with reduced susceptibility to piperacillin‐tazobactam: implications on the appropriateness of the resistance breakpoint.Clin Infect Dis. 152008;46(6):862–867.
- ,,.Piperacillin‐tazobactam for Pseudomonas aeruginosa infection: clinical implications of an extended‐infusion dosing strategy.Clin Infect Dis. 12007;44(3):357–363.
- ,.Pharmacokinetics and pharmacodynamics of antibiotics in otitis media.Pediatr Infect Dis J.1996;15(3):255–259.
- .Interrelationship between pharmacokinetics and pharmacodynamics in determining dosage regimens for broad‐spectrum cephalosporins.Diagn Microbiol Infect Dis.1995;22(1–2):89–96.
- .How does a patient maximally benefit from anti‐infective chemotherapy?Clin Infect Dis.2004;39(8):1245–1246.
- ,,,.Optimal dosing of piperacillin‐tazobactam for the treatment of Pseudomonas aeruginosa infections: prolonged or continuous infusion?Pharmacotherapy.2007;27(11):1490–1497.
- ,,,,.A systematic review on clinical benefits of continuous administration of beta‐lactam antibiotics.Crit Care Med.2009;37(6):2071–2078.
- ,,.Pharmacokinetic/pharmacodynamic modeling can help guide targeted antimicrobial therapy for nosocomial gram‐negative infections in critically ill patients.Diagn Microbiol Infect Dis.2004;48(2):125–130.
- ,,, et al.Pseudomonas aeruginosa bloodstream infection: importance of appropriate initial antimicrobial treatment.Antimicrob Agents Chemother.2005;49(4):1306–1311.
- ,,,.Assessment of pathogen occurrences and resistance profiles among infected patients in the intensive care unit: report from the SENTRY Antimicrobial Surveillance Program (North America, 2001).Int J Antimicrob Agents.2004;24(2):111–118.
- ,,.Results from the Meropenem Yearly Susceptibility Test Information Collection (MYSTIC) Programme: report of the 2001 data from 15 United States medical centres.Int J Antimicrob Agents.2004;23(1):52–59.
- ,,, et al.Penetration of meropenem into epithelial lining fluid of patients with ventilator‐associated pneumonia. Presented at the 48th Interscience Conference on Antimicrobial Agents and Chemotherapy/46th Annual Meeting of the Infectious Diseases Society of America. Washington DC,2008. Abstr 1889.
- ,,, et al.Optimization of meropenem minimum concentration/MIC ratio to suppress in vitro resistance of Pseudomonas aeruginosa.Antimicrob Agents Chemother. In press.
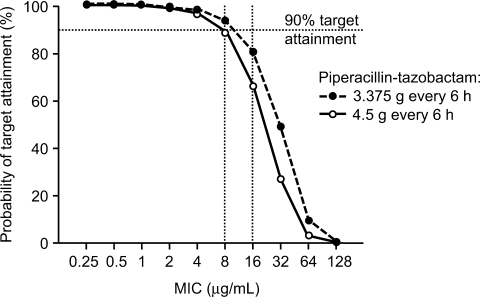
How to Get Ahead in Community-Based Hospital Medicine
What does it take to advance your career in the community hospital setting? Good communication and relationship skills top the list, according to Joe Metcalf, MD, director of the Hospital Medicine Service at Faith Regional Health Services in Norfolk, Neb. When Dr. Metcalf started the hospitalist service at the 227-bed regional referral center three and a half years ago, he was not looking for "brilliant" physicians. Instead, he says, "I was looking for excellent communicators."
"Patients are not interested in exceedingly smart clinicians," Dr. Metcalf says. "They are interested in clinicians who are willing to sit at the bedside, hold the elderly patient’s hand, listen to their concerns, and communicate the plans of care in ways they can understand."
Listening is a part of communication that is often neglected, he says.
Versatile and Teachable
Family-practice-trained, ED-hospitalist Joseph Babbitt, MD, works at 22-bed Blue Hill Memorial Hospital in Blue Hill, Maine. The key to success for hospitalists in the small-community-hospital setting is versatility, he says. It’s not unusual for hospitalists at Blue Hill to perform cardiac stress tests for a cardiology colleague or provide perioperative comanagement for surgical colleagues, he adds.
Dr. Babbitt underscores the willingness to acquire a broad range of skills. Dr. Metcalf agrees, and explains it’s one of the reasons he looks for "teachability" in hospitalist job candidates.
"It goes without saying that physicians must be proficient clinicians," he says, "but what I’m also listening for is whether they truly have a passion for patient-centered care."
Dr. Metcalf values honesty in his team members, and respects hospitalists who ask for help with their difficult cases.
Wired for Small
When it comes to community hospital settings, size matters. The hospital setting and job description should fit your preferences. Dr. Babbitt advises candidates to "listen to your gut" when interviewing for community hospital jobs. "If your reaction after checking out a job prospect is, ‘Well, I think I can make this work,’ then don’t try to make it work, because it probably won’t," he says. "You’re either ‘wired for small’ or you’re not."
Residents can prepare for a new job at a small community hospital. Dr. Babbitt recalls one young physician who, after a medical school rotation in a small hospital, realized that he was interested in becoming a "small-hospital hospitalist." He acquired additional training in emergency medicine because he identified it as a skill set that he needed to strengthen.
One thing is for sure: Variety is the order of the day in community settings. "You can bet," Dr. Babbitt says, "that the job you’re hired for is not going to be the job you’re doing in a year. Something about it will have changed."
Gretchen Henkel is a frequent contributor to The Hospitalist.
SOUND CAREER ADVICE
For more tips and strategies for advancing or establishing a career in hospital medicine, visit the SHM Career Center.
What does it take to advance your career in the community hospital setting? Good communication and relationship skills top the list, according to Joe Metcalf, MD, director of the Hospital Medicine Service at Faith Regional Health Services in Norfolk, Neb. When Dr. Metcalf started the hospitalist service at the 227-bed regional referral center three and a half years ago, he was not looking for "brilliant" physicians. Instead, he says, "I was looking for excellent communicators."
"Patients are not interested in exceedingly smart clinicians," Dr. Metcalf says. "They are interested in clinicians who are willing to sit at the bedside, hold the elderly patient’s hand, listen to their concerns, and communicate the plans of care in ways they can understand."
Listening is a part of communication that is often neglected, he says.
Versatile and Teachable
Family-practice-trained, ED-hospitalist Joseph Babbitt, MD, works at 22-bed Blue Hill Memorial Hospital in Blue Hill, Maine. The key to success for hospitalists in the small-community-hospital setting is versatility, he says. It’s not unusual for hospitalists at Blue Hill to perform cardiac stress tests for a cardiology colleague or provide perioperative comanagement for surgical colleagues, he adds.
Dr. Babbitt underscores the willingness to acquire a broad range of skills. Dr. Metcalf agrees, and explains it’s one of the reasons he looks for "teachability" in hospitalist job candidates.
"It goes without saying that physicians must be proficient clinicians," he says, "but what I’m also listening for is whether they truly have a passion for patient-centered care."
Dr. Metcalf values honesty in his team members, and respects hospitalists who ask for help with their difficult cases.
Wired for Small
When it comes to community hospital settings, size matters. The hospital setting and job description should fit your preferences. Dr. Babbitt advises candidates to "listen to your gut" when interviewing for community hospital jobs. "If your reaction after checking out a job prospect is, ‘Well, I think I can make this work,’ then don’t try to make it work, because it probably won’t," he says. "You’re either ‘wired for small’ or you’re not."
Residents can prepare for a new job at a small community hospital. Dr. Babbitt recalls one young physician who, after a medical school rotation in a small hospital, realized that he was interested in becoming a "small-hospital hospitalist." He acquired additional training in emergency medicine because he identified it as a skill set that he needed to strengthen.
One thing is for sure: Variety is the order of the day in community settings. "You can bet," Dr. Babbitt says, "that the job you’re hired for is not going to be the job you’re doing in a year. Something about it will have changed."
Gretchen Henkel is a frequent contributor to The Hospitalist.
SOUND CAREER ADVICE
For more tips and strategies for advancing or establishing a career in hospital medicine, visit the SHM Career Center.
What does it take to advance your career in the community hospital setting? Good communication and relationship skills top the list, according to Joe Metcalf, MD, director of the Hospital Medicine Service at Faith Regional Health Services in Norfolk, Neb. When Dr. Metcalf started the hospitalist service at the 227-bed regional referral center three and a half years ago, he was not looking for "brilliant" physicians. Instead, he says, "I was looking for excellent communicators."
"Patients are not interested in exceedingly smart clinicians," Dr. Metcalf says. "They are interested in clinicians who are willing to sit at the bedside, hold the elderly patient’s hand, listen to their concerns, and communicate the plans of care in ways they can understand."
Listening is a part of communication that is often neglected, he says.
Versatile and Teachable
Family-practice-trained, ED-hospitalist Joseph Babbitt, MD, works at 22-bed Blue Hill Memorial Hospital in Blue Hill, Maine. The key to success for hospitalists in the small-community-hospital setting is versatility, he says. It’s not unusual for hospitalists at Blue Hill to perform cardiac stress tests for a cardiology colleague or provide perioperative comanagement for surgical colleagues, he adds.
Dr. Babbitt underscores the willingness to acquire a broad range of skills. Dr. Metcalf agrees, and explains it’s one of the reasons he looks for "teachability" in hospitalist job candidates.
"It goes without saying that physicians must be proficient clinicians," he says, "but what I’m also listening for is whether they truly have a passion for patient-centered care."
Dr. Metcalf values honesty in his team members, and respects hospitalists who ask for help with their difficult cases.
Wired for Small
When it comes to community hospital settings, size matters. The hospital setting and job description should fit your preferences. Dr. Babbitt advises candidates to "listen to your gut" when interviewing for community hospital jobs. "If your reaction after checking out a job prospect is, ‘Well, I think I can make this work,’ then don’t try to make it work, because it probably won’t," he says. "You’re either ‘wired for small’ or you’re not."
Residents can prepare for a new job at a small community hospital. Dr. Babbitt recalls one young physician who, after a medical school rotation in a small hospital, realized that he was interested in becoming a "small-hospital hospitalist." He acquired additional training in emergency medicine because he identified it as a skill set that he needed to strengthen.
One thing is for sure: Variety is the order of the day in community settings. "You can bet," Dr. Babbitt says, "that the job you’re hired for is not going to be the job you’re doing in a year. Something about it will have changed."
Gretchen Henkel is a frequent contributor to The Hospitalist.
SOUND CAREER ADVICE
For more tips and strategies for advancing or establishing a career in hospital medicine, visit the SHM Career Center.
The Next Generation of Anticoagulants?
The popularity of the next generation of anticoagulation therapies could be dependent on whether reversing agents for the newest drugs can be developed, says a hospitalist who heads an antithrombotic clinic.
In October, the FDA approved dabigatran etexilate (Pradaxa) for atrial fibrillation (AF) patients. In a noninferiority study published last month, investigators found that treatment with oral rivaroxaban alone (15mg twice daily for three weeks, followed by 20mg once daily) showed effectiveness versus subcutaneous enoxaparin followed by a vitamin K antagonist. In relation to the primary outcome of recurrent DVT, rivaroxaban had noninferior efficacy (36 events [2.1%], vs. 51 events, 0.44 to 1.04; P<0.001) (N Engl J Med. 2010;363:2499-2510).
Another study, dubbed ROCKET-AF (PDF) and unveiled at an American Heart Association meeting in November, reported that rivaroxaban was noninferior to warfarin in the treatment of stroke and non-CNS embolism. Study patients treated with rivaroxaban exhibited significantly less events (1.71) per 100 patient-years (188 patients) compared with those on warfarin (2.16; 241 patients; P<0.001 for noninferiority, P=0.018 for superiority).
A third medication, apixaban, which also acts as a direct
fact Xa inhibitor, is currently being tested in clinical trials.
Geno Merli, MD, senior vice president and chief medical officer at Thomas Jefferson University Hospital and head of the Jefferson Antithrombotic Therapy Service, both in Philadelphia, says one of the most pressing issues with the Xa inhibitors is that there is not yet a reversing agent for the drugs should complications arise. “I can reverse Coumadin,” Dr. Merli says. “I can give vitamin K or fresh frozen plasma. You’re giving back the factors that were affected.”
Dr. Merli adds that pharmaceutical companies already are working on development of reversing agents and antibodies, but until those are approved, some physicians might shy away from new anticoagulant therapies. Still, he encourages physicians to get the medications added to their respective hospitals’ medicine cabinets as quickly as feasible.
“You’ve got to have it on your formulary because you have to know the drug,” Dr. Merli says. “You have to have it for the doctor who will choose to use it or the patient who comes in already on it.”
The popularity of the next generation of anticoagulation therapies could be dependent on whether reversing agents for the newest drugs can be developed, says a hospitalist who heads an antithrombotic clinic.
In October, the FDA approved dabigatran etexilate (Pradaxa) for atrial fibrillation (AF) patients. In a noninferiority study published last month, investigators found that treatment with oral rivaroxaban alone (15mg twice daily for three weeks, followed by 20mg once daily) showed effectiveness versus subcutaneous enoxaparin followed by a vitamin K antagonist. In relation to the primary outcome of recurrent DVT, rivaroxaban had noninferior efficacy (36 events [2.1%], vs. 51 events, 0.44 to 1.04; P<0.001) (N Engl J Med. 2010;363:2499-2510).
Another study, dubbed ROCKET-AF (PDF) and unveiled at an American Heart Association meeting in November, reported that rivaroxaban was noninferior to warfarin in the treatment of stroke and non-CNS embolism. Study patients treated with rivaroxaban exhibited significantly less events (1.71) per 100 patient-years (188 patients) compared with those on warfarin (2.16; 241 patients; P<0.001 for noninferiority, P=0.018 for superiority).
A third medication, apixaban, which also acts as a direct
fact Xa inhibitor, is currently being tested in clinical trials.
Geno Merli, MD, senior vice president and chief medical officer at Thomas Jefferson University Hospital and head of the Jefferson Antithrombotic Therapy Service, both in Philadelphia, says one of the most pressing issues with the Xa inhibitors is that there is not yet a reversing agent for the drugs should complications arise. “I can reverse Coumadin,” Dr. Merli says. “I can give vitamin K or fresh frozen plasma. You’re giving back the factors that were affected.”
Dr. Merli adds that pharmaceutical companies already are working on development of reversing agents and antibodies, but until those are approved, some physicians might shy away from new anticoagulant therapies. Still, he encourages physicians to get the medications added to their respective hospitals’ medicine cabinets as quickly as feasible.
“You’ve got to have it on your formulary because you have to know the drug,” Dr. Merli says. “You have to have it for the doctor who will choose to use it or the patient who comes in already on it.”
The popularity of the next generation of anticoagulation therapies could be dependent on whether reversing agents for the newest drugs can be developed, says a hospitalist who heads an antithrombotic clinic.
In October, the FDA approved dabigatran etexilate (Pradaxa) for atrial fibrillation (AF) patients. In a noninferiority study published last month, investigators found that treatment with oral rivaroxaban alone (15mg twice daily for three weeks, followed by 20mg once daily) showed effectiveness versus subcutaneous enoxaparin followed by a vitamin K antagonist. In relation to the primary outcome of recurrent DVT, rivaroxaban had noninferior efficacy (36 events [2.1%], vs. 51 events, 0.44 to 1.04; P<0.001) (N Engl J Med. 2010;363:2499-2510).
Another study, dubbed ROCKET-AF (PDF) and unveiled at an American Heart Association meeting in November, reported that rivaroxaban was noninferior to warfarin in the treatment of stroke and non-CNS embolism. Study patients treated with rivaroxaban exhibited significantly less events (1.71) per 100 patient-years (188 patients) compared with those on warfarin (2.16; 241 patients; P<0.001 for noninferiority, P=0.018 for superiority).
A third medication, apixaban, which also acts as a direct
fact Xa inhibitor, is currently being tested in clinical trials.
Geno Merli, MD, senior vice president and chief medical officer at Thomas Jefferson University Hospital and head of the Jefferson Antithrombotic Therapy Service, both in Philadelphia, says one of the most pressing issues with the Xa inhibitors is that there is not yet a reversing agent for the drugs should complications arise. “I can reverse Coumadin,” Dr. Merli says. “I can give vitamin K or fresh frozen plasma. You’re giving back the factors that were affected.”
Dr. Merli adds that pharmaceutical companies already are working on development of reversing agents and antibodies, but until those are approved, some physicians might shy away from new anticoagulant therapies. Still, he encourages physicians to get the medications added to their respective hospitals’ medicine cabinets as quickly as feasible.
“You’ve got to have it on your formulary because you have to know the drug,” Dr. Merli says. “You have to have it for the doctor who will choose to use it or the patient who comes in already on it.”
In the Literature: Research You Need to Know
Clinical question: What is the relative efficacy of trimethoprim/sulfamethoxazole (TMP/sulfa) versus ciprofloxacin for the treatment of severe exacerbations of COPD?
Background: The use of antimicrobials in the treatment of COPD exacerbations is well accepted, with the original studies using amoxicillin, TMP/sulfa, and tetracyclines. Whether newer antimicrobial agents offer greater efficacy versus these standard agents remains uncertain.
Study design: Randomized, double-blind, placebo-controlled (double-dummy), noninferiority trial.
Setting: Two academic medical ICUs in Tunisia.
Synopsis: Consecutive patients (n=170) with severe exacerbations of COPD requiring mechanical ventilation were randomized to standard medical therapy plus either TMP/sulfa or ciprofloxacin. Patients had a prior diagnosis of COPD and the clinical presence of purulent sputum and respiratory failure. The study excluded those who were immunosuppressed, had significant hepatic or renal disease, pneumonia, recent antibiotic use, active cancer, or inability to take oral medications.
The primary endpoint of hospital death and the need for an additional course of antibiotics was no different between the groups (16.4% with TMP/sulfa versus 15.3% with ciprofloxacin). The mean exacerbation-free interval, days of mechanical ventilation, and length of stay were no different. Noninferiority was defined as <10% relative difference.
Bottom line: TMP/sulfa was noninferior to ciprofloxacin in the treatment of severe exacerbations of COPD requiring mechanical ventilation.
Citation: Nouira S, Marghli S, Besbes L, et al. Standard versus newer antibacterial agents in the treatment of severe acute exacerbations of chronic obstructive pulmonary disease: a randomized trial of trimethoprim-sulfamethoxazole versus ciprofloxacin. Clin Inf Dis. 2010;51:143-149.
For more physician reviews of HM-related research, visit our website.
Clinical question: What is the relative efficacy of trimethoprim/sulfamethoxazole (TMP/sulfa) versus ciprofloxacin for the treatment of severe exacerbations of COPD?
Background: The use of antimicrobials in the treatment of COPD exacerbations is well accepted, with the original studies using amoxicillin, TMP/sulfa, and tetracyclines. Whether newer antimicrobial agents offer greater efficacy versus these standard agents remains uncertain.
Study design: Randomized, double-blind, placebo-controlled (double-dummy), noninferiority trial.
Setting: Two academic medical ICUs in Tunisia.
Synopsis: Consecutive patients (n=170) with severe exacerbations of COPD requiring mechanical ventilation were randomized to standard medical therapy plus either TMP/sulfa or ciprofloxacin. Patients had a prior diagnosis of COPD and the clinical presence of purulent sputum and respiratory failure. The study excluded those who were immunosuppressed, had significant hepatic or renal disease, pneumonia, recent antibiotic use, active cancer, or inability to take oral medications.
The primary endpoint of hospital death and the need for an additional course of antibiotics was no different between the groups (16.4% with TMP/sulfa versus 15.3% with ciprofloxacin). The mean exacerbation-free interval, days of mechanical ventilation, and length of stay were no different. Noninferiority was defined as <10% relative difference.
Bottom line: TMP/sulfa was noninferior to ciprofloxacin in the treatment of severe exacerbations of COPD requiring mechanical ventilation.
Citation: Nouira S, Marghli S, Besbes L, et al. Standard versus newer antibacterial agents in the treatment of severe acute exacerbations of chronic obstructive pulmonary disease: a randomized trial of trimethoprim-sulfamethoxazole versus ciprofloxacin. Clin Inf Dis. 2010;51:143-149.
For more physician reviews of HM-related research, visit our website.
Clinical question: What is the relative efficacy of trimethoprim/sulfamethoxazole (TMP/sulfa) versus ciprofloxacin for the treatment of severe exacerbations of COPD?
Background: The use of antimicrobials in the treatment of COPD exacerbations is well accepted, with the original studies using amoxicillin, TMP/sulfa, and tetracyclines. Whether newer antimicrobial agents offer greater efficacy versus these standard agents remains uncertain.
Study design: Randomized, double-blind, placebo-controlled (double-dummy), noninferiority trial.
Setting: Two academic medical ICUs in Tunisia.
Synopsis: Consecutive patients (n=170) with severe exacerbations of COPD requiring mechanical ventilation were randomized to standard medical therapy plus either TMP/sulfa or ciprofloxacin. Patients had a prior diagnosis of COPD and the clinical presence of purulent sputum and respiratory failure. The study excluded those who were immunosuppressed, had significant hepatic or renal disease, pneumonia, recent antibiotic use, active cancer, or inability to take oral medications.
The primary endpoint of hospital death and the need for an additional course of antibiotics was no different between the groups (16.4% with TMP/sulfa versus 15.3% with ciprofloxacin). The mean exacerbation-free interval, days of mechanical ventilation, and length of stay were no different. Noninferiority was defined as <10% relative difference.
Bottom line: TMP/sulfa was noninferior to ciprofloxacin in the treatment of severe exacerbations of COPD requiring mechanical ventilation.
Citation: Nouira S, Marghli S, Besbes L, et al. Standard versus newer antibacterial agents in the treatment of severe acute exacerbations of chronic obstructive pulmonary disease: a randomized trial of trimethoprim-sulfamethoxazole versus ciprofloxacin. Clin Inf Dis. 2010;51:143-149.
For more physician reviews of HM-related research, visit our website.
Pneumonia Readmission Validation
Hospital readmissions are emblematic of the numerous challenges facing the US health care system. Despite high levels of spending, nearly 20% of Medicare beneficiaries are readmitted within 30 days of hospital discharge, many readmissions are considered preventable, and rates vary widely by hospital and region.1 Further, while readmissions have been estimated to cost taxpayers as much as $17 billion annually, the current fee‐for‐service method of paying for the acute care needs of seniors rewards hospitals financially for readmission, not their prevention.2
Pneumonia is the second most common reason for hospitalization among Medicare beneficiaries, accounting for approximately 650,000 admissions annually,3 and has been a focus of national quality‐improvement efforts for more than a decade.4, 5 Despite improvements in key processes of care, rates of readmission within 30 days of discharge following a hospitalization for pneumonia have been reported to vary from 10% to 24%.68 Among several factors, readmissions are believed to be influenced by the quality of both inpatient and outpatient care, and by care‐coordination activities occurring in the transition from inpatient to outpatient status.912
Public reporting of hospital performance is considered a key strategy for improving quality, reducing costs, and increasing the value of hospital care, both in the US and worldwide.13 In 2009, the Centers for Medicare & Medicaid Services (CMS) expanded its reporting initiatives by adding risk‐adjusted hospital readmission rates for acute myocardial infarction, heart failure, and pneumonia to the Hospital Compare website.14, 15 Readmission rates are an attractive focus for public reporting for several reasons. First, in contrast to most process‐based measures of quality (eg, whether a patient with pneumonia received a particular antibiotic), a readmission is an adverse outcome that matters to patients and families.16 Second, unlike process measures whose assessment requires detailed review of medical records, readmissions can be easily determined from standard hospital claims. Finally, readmissions are costly, and their prevention could yield substantial savings to society.
A necessary prerequisite for public reporting of readmission is a validated, risk‐adjusted measure that can be used to track performance over time and can facilitate comparisons across institutions. Toward this end, we describe the development, validation, and results of a National Quality Forum‐approved and CMS‐adopted model to estimate hospital‐specific, risk‐standardized, 30‐day readmission rates for Medicare patients hospitalized with pneumonia.17
METHODS
Data Sources
We used 20052006 claims data from Medicare inpatient, outpatient, and carrier (physician) Standard Analytic Files to develop and validate the administrative model. The Medicare Enrollment Database was used to determine Medicare fee‐for‐service enrollment and mortality statuses. A medical record model, used for additional validation of the administrative model, was developed using information abstracted from the charts of 75,616 pneumonia cases from 19982001 as part of the National Pneumonia Project, a CMS quality improvement initiative.18
Study Cohort
We identified hospitalizations of patients 65 years of age and older with a principal diagnosis of pneumonia (International Classification of Diseases, 9th Revision, Clinical Modification codes 480.XX, 481, 482.XX, 483.X, 485, 486, 487.0) as potential index pneumonia admissions. Because our focus was readmission for patients discharged from acute care settings, we excluded admissions in which patients died or were transferred to another acute care facility. Additionally, we restricted analysis to patients who had been enrolled in fee‐for‐service Medicare Parts A and B, for at least 12 months prior to their pneumonia hospitalization, so that we could use diagnostic codes from all inpatient and outpatient encounters during that period to enhance identification of comorbidities.
Outcome
The outcome was 30‐day readmission, defined as occurrence of at least one hospitalization for any cause within 30 days of discharge after an index admission. Readmissions were identified from hospital claims data, and were attributed to the hospital that had discharged the patient. A 30‐day time frame was selected because it is a clinically meaningful period during which hospitals can be expected to collaborate with other organizations and providers to implement measures to reduce the risk of rehospitalization.
Candidate and Final Model Variables
Candidate variables for the administrative claims model were selected by a clinician team from 189 diagnostic groups included in the Hierarchical Condition Category (HCC) clinical classification system.19 The HCC clinical classification system was developed for CMS in preparation for all‐encounter risk adjustment for Medicare Advantage (managed care). Under the HCC algorithm, the 15,000+ ICD‐9‐CM diagnosis codes are assigned to one of 189 clinically‐coherent condition categories (CCs). We used the April 2008 version of the ICD‐9‐CM to CC assignment map, which is maintained by CMS and posted at
The final risk‐adjustment model included 39 variables selected by the team of clinicians and analysts, primarily based on their clinical relevance but also with knowledge of the strength of their statistical association with readmission outcome (Table 1). For each patient, the presence or absence of these conditions was assessed from multiple sources, including secondary diagnoses during the index admission, principal and secondary diagnoses from hospital admissions in the 12 months prior to the index admission, and diagnoses from hospital outpatient and physician encounters 12 months before the index admission. A small number of CCs were considered to represent potential complications of care (eg, bleeding). Because we did not want to adjust for complications of care occurring during the index admission, a patient was not considered to have one of these conditions unless it was also present in at least one encounter prior to the index admission.
| Variable | Frequencies | Estimate | Standard Error | Odds Ratio | 95% CI | |
|---|---|---|---|---|---|---|
| ||||||
| Intercept | 2.395 | 0.021 | ||||
| Age 65 (years above 65, continuous) | 0.0001 | 0.001 | 1.000 | 0.998 | 1.001 | |
| Male | 45 | 0.071 | 0.012 | 1.073 | 1.048 | 1.099 |
| History of CABG | 5.2 | 0.179 | 0.027 | 0.836 | 0.793 | 0.881 |
| Metastatic cancer and acute leukemia (CC 7) | 4.3 | 0.177 | 0.029 | 1.194 | 1.128 | 1.263 |
| Lung, upper digestive tract, and other severe cancers (CC 8) | 6.0 | 0.256 | 0.024 | 1.292 | 1.232 | 1.354 |
| Diabetes and DM complications (CC 15‐20, 119, 120) | 36 | 0.059 | 0.012 | 1.061 | 1.036 | 1.087 |
| Disorders of fluid/electrolyte/acid‐base (CC 22, 23) | 34 | 0.149 | 0.013 | 1.160 | 1.131 | 1.191 |
| Iron deficiency and other/unspecified anemias and blood disease (CC 47) | 46 | 0.118 | 0.012 | 1.126 | 1.099 | 1.153 |
| Other psychiatric disorders (CC 60) | 12 | 0.108 | 0.017 | 1.114 | 1.077 | 1.151 |
| Cardio‐respiratory failure and shock (CC 79) | 16 | 0.114 | 0.016 | 1.121 | 1.087 | 1.156 |
| Congestive heart failure (CC 80) | 39 | 0.151 | 0.014 | 1.163 | 1.133 | 1.194 |
| Chronic atherosclerosis (CC 83, 84) | 47 | 0.051 | 0.013 | 1.053 | 1.027 | 1.079 |
| Valvular and rheumatic heart disease (CC 86) | 23 | 0.062 | 0.014 | 1.064 | 1.036 | 1.093 |
| Arrhythmias (CC 92, 93) | 38 | 0.126 | 0.013 | 1.134 | 1.107 | 1.163 |
| Vascular or circulatory disease (CC 104‐106) | 38 | 0.088 | 0.012 | 1.092 | 1.066 | 1.119 |
| COPD (CC 108) | 58 | 0.186 | 0.013 | 1.205 | 1.175 | 1.235 |
| Fibrosis of lung and other chronic lung disorders (CC 109) | 17 | 0.086 | 0.015 | 1.090 | 1.059 | 1.122 |
| Renal failure (CC 131) | 17 | 0.147 | 0.016 | 1.158 | 1.122 | 1.196 |
| Protein‐calorie malnutrition (CC 21) | 7.9 | 0.121 | 0.020 | 1.129 | 1.086 | 1.173 |
| History of infection (CC 1, 3‐6) | 35 | 0.068 | 0.012 | 1.071 | 1.045 | 1.097 |
| Severe hematological disorders (CC 44) | 3.6 | 0.117 | 0.028 | 1.125 | 1.064 | 1.188 |
| Decubitus ulcer or chronic skin ulcer (CC 148, 149) | 10 | 0.101 | 0.018 | 1.106 | 1.067 | 1.146 |
| History of pneumonia (CC 111‐113) | 44 | 0.065 | 0.013 | 1.067 | 1.041 | 1.094 |
| Vertebral fractures (CC 157) | 5.1 | 0.113 | 0.024 | 1.120 | 1.068 | 1.174 |
| Other injuries (CC 162) | 32 | 0.061 | 0.012 | 1.063 | 1.038 | 1.089 |
| Urinary tract infection (CC 135) | 26 | 0.064 | 0.014 | 1.066 | 1.038 | 1.095 |
| Lymphatic, head and neck, brain, and other major cancers; breast, prostate, colorectal, and other cancers and tumors (CC 9‐10) | 16 | 0.050 | 0.016 | 1.051 | 1.018 | 1.084 |
| End‐stage renal disease or dialysis (CC 129, 130) | 1.9 | 0.131 | 0.037 | 1.140 | 1.060 | 1.226 |
| Drug/alcohol abuse/dependence/psychosis (CC 51‐53) | 12 | 0.081 | 0.017 | 1.084 | 1.048 | 1.121 |
| Septicemia/shock (CC 2) | 6.3 | 0.094 | 0.022 | 1.098 | 1.052 | 1.146 |
| Other gastrointestinal disorders (CC 36) | 56 | 0.073 | 0.012 | 1.076 | 1.051 | 1.102 |
| Acute coronary syndrome (CC 81, 82) | 8.3 | 0.126 | 0.019 | 1.134 | 1.092 | 1.178 |
| Pleural effusion/pneumothorax (CC 114) | 12 | 0.083 | 0.017 | 1.086 | 1.051 | 1.123 |
| Other urinary tract disorders (CC 136) | 24 | 0.059 | 0.014 | 1.061 | 1.033 | 1.090 |
| Stroke (CC 95, 96) | 10 | 0.047 | 0.019 | 1.049 | 1.011 | 1.088 |
| Dementia and senility (CC 49, 50) | 27 | 0.031 | 0.014 | 1.031 | 1.004 | 1.059 |
| Hemiplegia, paraplegia, paralysis, functional disability (CC 67‐69, 100‐102, 177, 178) | 7.4 | 0.068 | 0.021 | 1.070 | 1.026 | 1.116 |
| Other lung disorders (CC 115) | 45 | 0.005 | 0.012 | 1.005 | 0.982 | 1.030 |
| Major psychiatric disorders (CC 54‐56) | 11 | 0.038 | 0.018 | 1.038 | 1.003 | 1.075 |
| Asthma (CC 110) | 12 | 0.006 | 0.018 | 1.006 | 0.972 | 1.041 |
Model Derivation
For the development of the administrative claims model, we randomly sampled half of 2006 hospitalizations that met inclusion criteria. To assess model performance at the patient level, we calculated the area under the receiver operating curve (AUC), and calculated observed readmission rates in the lowest and highest deciles on the basis of predicted readmission probabilities. We also compared performance with a null model, a model that adjusted for age and sex, and a model that included all candidate variables.20
Risk‐Standardized Readmission Rates
Using hierarchical logistic regression, we modeled the log‐odds of readmission within 30 days of discharge from an index pneumonia admission as a function of patient demographic and clinical characteristics, and a random hospital‐specific intercept. This strategy accounts for within‐hospital correlation, or clustering, of observed outcomes, and models the assumption that underlying differences in quality among hospitals being evaluated lead to systematic differences in outcomes. We then calculated hospital‐specific readmission rates as the ratio of predicted‐to‐expected readmissions (similar to observed/expected ratio), multiplied by the national unadjusted ratea form of indirect standardization. Predicted number of readmissions in each hospital is estimated given the same patient mix and its estimated hospital‐specific intercept. Expected number of readmissions in each hospital is estimated using its patient mix and the average hospital‐specific intercept. To assess hospital performance in any given year, we re‐estimate model coefficients using that year's data.
Model Validation: Administrative Claims
We compared the model performance in the development sample with its performance in the sample from the 2006 data that was not selected for the development set, and separately among pneumonia admissions in 2005. The model was recalibrated in each validation set.
Model Validation: Medical Record Abstraction
We developed a separate medical record‐based model of readmission risk using information from charts that had previously been abstracted as part of CMS's National Pneumonia Project. To select variables for this model, the clinician team: 1) reviewed the list of variables that were included in a medical record model that was previously developed for validating the National Quality Forum‐approved pneumonia mortality measure; 2) reviewed a list of other potential candidate variables available in the National Pneumonia Project dataset; and 3) reviewed variables that emerged as potentially important predictors of readmission, based on a systematic review of the literature that was conducted as part of measure development. This selection process resulted in a final medical record model that included 35 variables.
We linked patients in the National Pneumonia Project cohort to their Medicare claims data, including claims from one year before the index hospitalization, so that we could calculate risk‐standardized readmission rates in this cohort separately using medical record and claims‐based models. This analysis was conducted at the state level, for the 50 states plus the District of Columbia and Puerto Rico, because medical record data were unavailable in sufficient numbers to permit hospital‐level comparisons. To examine the relationship between risk‐standardized rates obtained from medical record and administrative data models, we estimated a linear regression model describing the association between the two rates, weighting each state by number of index hospitalizations, and calculated the correlation coefficient and the intercept and slope of this equation. A slope close to 1 and an intercept close to 0 would provide evidence that risk‐standardized state readmission rates from the medical record and claims models were similar. We also calculated the difference between state risk‐standardized readmission rates from the two models.
Analyses were conducted with the use of SAS version 9.1.3 (SAS Institute Inc, Cary, NC). Models were fitted separately for the National Pneumonia Project and 2006 cohort. We estimated the hierarchical models using the GLIMMIX procedure in SAS. The Human Investigation Committee at the Yale School of Medicine approved an exemption for the authors to use CMS claims and enrollment data for research analyses and publication.
RESULTS
Model Derivation and Performance
After exclusions were applied, the 2006 sample included 453,251 pneumonia hospitalizations (Figure 1). The development sample consisted of 226,545 hospitalizations at 4675 hospitals, with an overall unadjusted 30‐day readmission rate of 17.4%. In 11,694 index cases (5.2%), the patient died within 30 days without being readmitted. Median readmission rate was 16.3%, 25th and 75th percentile rates were 11.1% and 21.3%, and at the 10th and 90th percentile, hospital readmission rates ranged from 4.6% to 26.7% (Figure 2).
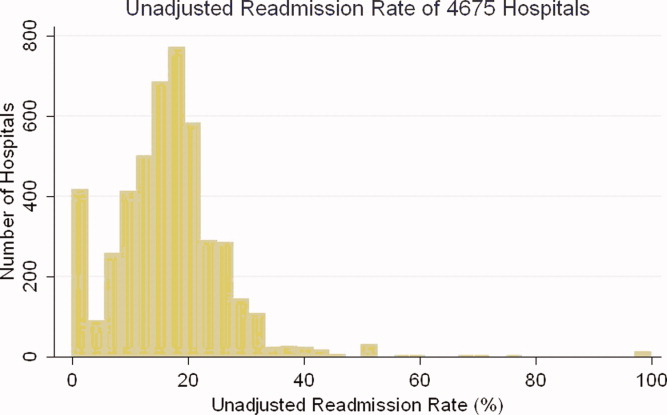
The claims model included 39 variables (age, sex, and 37 clinical variables) (Table 1). The mean age of the cohort was 80.0 years, with 55.5% women and 11.1% nonwhite patients. Mean observed readmission rate in the development sample ranged from 9% in the lowest decile of predicted pneumonia readmission rates to 32% in the highest predicted decile, a range of 23%. The AUC was 0.63. For comparison, a model with only age and sex had an AUC of 0.51, and a model with all candidate variables had an AUC equal to 0.63 (Table 2).
| Calibration (0, 1)* | Discrimination | Residuals Lack of Fit (Pearson Residual Fall %) | Model 2 (No. of Covariates) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Predictive Ability (Lowest Decile, Highest Decile) | AUC | (<2) | (2, 0) | (0, 2) | (2+) | ||||
| |||||||||
| Development sample | |||||||||
| 2006 | (1st half) N = 226,545 | (0, 1) | (0.09, 0.32) | 0.63 | 0 | 82.62 | 7.39 | 9.99 | 6,843 (40) |
| Validation sample | |||||||||
| 2006 | (2nd half) N = 226,706 | (0.002, 0.997) | (0.09, 0.31) | 0.63 | 0 | 82.55 | 7.45 | 9.99 | 6,870 (40) |
| 2005 | N = 536,015 | (0.035, 1.008) | (0.08, 0.31) | 0.63 | 0 | 82.67 | 7.31 | 10.03 | 16,241 (40) |
Hospital Risk‐Standardized Readmission Rates
Risk‐standardized readmission rates varied across hospitals (Figure 3). Median risk‐standardized readmission rate was 17.3%, and the 25th and 75th percentiles were 16.9% and 17.9%, respectively. The 5th percentile was 16.0% and the 95th percentile was 19.1%. Odds of readmission for a hospital one standard deviation above average was 1.4 times that of a hospital one standard deviation below average.
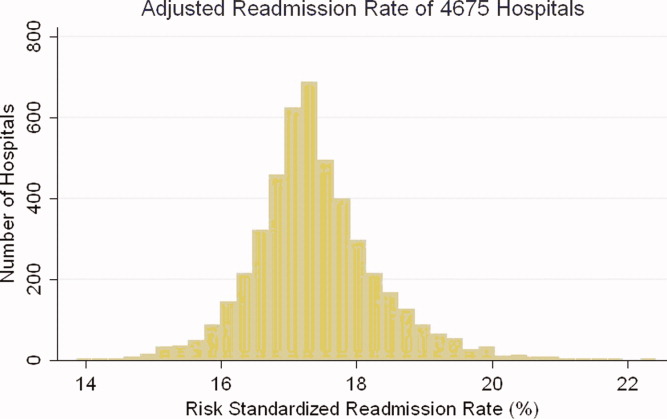
Administrative Model Validation
In the remaining 50% of pneumonia index hospitalizations from 2006, and the entire 2005 cohort, regression coefficients and standard errors of model variables were similar to those in the development data set. Model performance using 2005 data was consistent with model performance using the 2006 development and validation half‐samples (Table 2).
Medical Record Validation
After exclusions, the medical record sample taken from the National Pneumonia Project included 47,429 cases, with an unadjusted 30‐day readmission rate of 17.0%. The final medical record risk‐adjustment model included a total of 35 variables, whose prevalence and association with readmission risk varied modestly (Table 3). Performance of the medical record and administrative models was similar (areas under the ROC curve 0.59 and 0.63, respectively) (Table 4). Additionally, in the administrative model, predicted readmission rates ranged from 8% in the lowest predicted decile to 30% in the highest predicted decile, while in the medical record model, the corresponding rates varied from 10% to 26%.
| Variable | Percent | Estimate | Standard Error | Odds Ratio | 95% CI | |
|---|---|---|---|---|---|---|
| ||||||
| Age 65, mean (SD) | 15.24 (7.87) | 0.003 | 0.002 | 0.997 | 0.993 | 1.000 |
| Male | 46.18 | 0.122 | 0.025 | 1.130 | 1.075 | 1.188 |
| Nursing home resident | 17.71 | 0.035 | 0.037 | 1.036 | 0.963 | 1.114 |
| Neoplastic disease | 6.80 | 0.130 | 0.049 | 1.139 | 1.034 | 1.254 |
| Liver disease | 1.04 | 0.089 | 0.123 | 0.915 | 0.719 | 1.164 |
| History of heart failure | 28.98 | 0.234 | 0.029 | 1.264 | 1.194 | 1.339 |
| History of renal disease | 8.51 | 0.188 | 0.047 | 1.206 | 1.100 | 1.323 |
| Altered mental status | 17.95 | 0.009 | 0.034 | 1.009 | 0.944 | 1.080 |
| Pleural effusion | 21.20 | 0.165 | 0.030 | 1.179 | 1.111 | 1.251 |
| BUN 30 mg/dl | 23.28 | 0.160 | 0.033 | 1.174 | 1.100 | 1.252 |
| BUN missing | 14.56 | 0.101 | 0.185 | 0.904 | 0.630 | 1.298 |
| Systolic BP <90 mmHg | 2.95 | 0.068 | 0.070 | 1.070 | 0.932 | 1.228 |
| Systolic BP missing | 11.21 | 0.149 | 0.425 | 1.160 | 0.504 | 2.669 |
| Pulse 125/min | 7.73 | 0.036 | 0.047 | 1.036 | 0.945 | 1.137 |
| Pulse missing | 11.22 | 0.210 | 0.405 | 1.234 | 0.558 | 2.729 |
| Respiratory rate 30/min | 16.38 | 0.079 | 0.034 | 1.082 | 1.012 | 1.157 |
| Respiratory rate missing | 11.39 | 0.204 | 0.240 | 1.226 | 0.765 | 1.964 |
| Sodium <130 mmol/L | 4.82 | 0.136 | 0.057 | 1.145 | 1.025 | 1.280 |
| Sodium missing | 14.39 | 0.049 | 0.143 | 1.050 | 0.793 | 1.391 |
| Glucose 250 mg/dl | 5.19 | 0.005 | 0.057 | 0.995 | 0.889 | 1.114 |
| Glucose missing | 15.44 | 0.156 | 0.105 | 0.855 | 0.696 | 1.051 |
| Hematocrit <30% | 7.77 | 0.270 | 0.044 | 1.310 | 1.202 | 1.428 |
| Hematocrit missing | 13.62 | 0.071 | 0.135 | 0.932 | 0.715 | 1.215 |
| Creatinine 2.5 mg/dL | 4.68 | 0.109 | 0.062 | 1.115 | 0.989 | 1.258 |
| Creatinine missing | 14.63 | 0.200 | 0.167 | 1.221 | 0.880 | 1.695 |
| WBC 6‐12 b/L | 38.04 | 0.021 | 0.049 | 0.979 | 0.889 | 1.079 |
| WBC >12 b/L | 41.45 | 0.068 | 0.049 | 0.934 | 0.848 | 1.029 |
| WBC missing | 12.85 | 0.167 | 0.162 | 1.181 | 0.860 | 1.623 |
| Immunosuppressive therapy | 15.01 | 0.347 | 0.035 | 1.415 | 1.321 | 1.516 |
| Chronic lung disease | 42.16 | 0.137 | 0.028 | 1.147 | 1.086 | 1.211 |
| Coronary artery disease | 39.57 | 0.150 | 0.028 | 1.162 | 1.100 | 1.227 |
| Diabetes mellitus | 20.90 | 0.137 | 0.033 | 1.147 | 1.076 | 1.223 |
| Alcohol/drug abuse | 3.40 | 0.099 | 0.071 | 0.906 | 0.788 | 1.041 |
| Dementia/Alzheimer's disease | 16.38 | 0.125 | 0.038 | 1.133 | 1.052 | 1.222 |
| Splenectomy | 0.44 | 0.016 | 0.186 | 1.016 | 0.706 | 1.463 |
| Model | Calibration (0, 1)* | Discrimination | Residuals Lack of Fit (Pearson Residual Fall %) | Model 2 (No. of Covariates) | ||||
|---|---|---|---|---|---|---|---|---|
| Predictive Ability (Lowest Decile, Highest Decile) | AUC | (<2) | (2, 0) | (0, 2) | (2+) | |||
| ||||||||
| Medical Record Model Development Sample (NP) | ||||||||
| N = 47,429 No. of 30‐day readmissions = 8,042 | (1, 0) | (0.10, 0.26) | 0.59 | 0 | 83.04 | 5.28 | 11.68 | 710 (35) |
| Linked Administrative Model Validation Sample | ||||||||
| N = 47,429 No. of 30‐day readmissions = 8,042 | (1, 0) | (0.08, 0.30) | 0.63 | 0 | 83.04 | 6.94 | 10.01 | 1,414 (40) |
The correlation coefficient of the estimated state‐specific standardized readmission rates from the administrative and medical record models was 0.96, and the proportion of the variance explained by the model was 0.92 (Figure 4).
DISCUSSION
We have described the development, validation, and results of a hospital, 30‐day, risk‐standardized readmission model for pneumonia that was created to support current federal transparency initiatives. The model uses administrative claims data from Medicare fee‐for‐service patients and produces results that are comparable to a model based on information obtained through manual abstraction of medical records. We observed an overall 30‐day readmission rate of 17%, and our analyses revealed substantial variation across US hospitals, suggesting that improvement by lower performing institutions is an achievable goal.
Because more than one in six pneumonia patients are rehospitalized shortly after discharge, and because pneumonia hospitalizations represent an enormous expense to the Medicare program, prevention of readmissions is now widely recognized to offer a substantial opportunity to improve patient outcomes while simultaneously lowering health care costs. Accordingly, promotion of strategies to reduce readmission rates has become a key priority for payers and quality‐improvement organizations. These range from policy‐level attempts to stimulate change, such as publicly reporting hospital readmission rates on government websites, to establishing accreditation standardssuch as the Joint Commission's requirement to accurately reconcile medications, to the creation of quality improvement collaboratives focused on sharing best practices across institutions. Regardless of the approach taken, a valid, risk‐adjusted measure of performance is required to evaluate and track performance over time. The measure we have described meets the National Quality Forum's measure evaluation criteria in that it addresses an important clinical topic for which there appears to be significant opportunities for improvement, the measure is precisely defined and has been subjected to validity and reliability testing, it is risk‐adjusted based on patient clinical factors present at the start of care, is feasible to produce, and is understandable by a broad range of potential users.21 Because hospitalists are the physicians primarily responsible for the care of patients with pneumonia at US hospitals, and because they frequently serve as the physician champions for quality improvement activities related to pneumonia, it is especially important that they maintain a thorough understanding of the measures and methodologies underlying current efforts to measure hospital performance.
Several features of our approach warrant additional comment. First, we deliberately chose to measure all readmission events rather than attempt to discriminate between potentially preventable and nonpreventable readmissions. From the patient perspective, readmission for any reason is a concern, and limiting the measure to pneumonia‐related readmissions could make it susceptible to gaming by hospitals. Moreover, determining whether a readmission is related to a potential quality problem is not straightforward. For example, a patient with pneumonia whose discharge medications were prescribed incorrectly may be readmitted with a hip fracture following an episode of syncope. It would be inappropriate to treat this readmission as unrelated to the care the patient received for pneumonia. Additionally, while our approach does not presume that every readmission is preventable, the goal is to reduce the risk of readmissions generally (not just in narrowly defined subpopulations), and successful interventions to reduce rehospitalization have typically demonstrated reductions in all‐cause readmission.9, 22 Second, deaths that occurred within 30 days of discharge, yet that were not accompanied by a hospital readmission, were not counted as a readmission outcome. While it may seem inappropriate to treat a postdischarge death as a nonevent (rather than censoring or excluding such cases), alternative analytic approaches, such as using a hierarchical survival model, are not currently computationally feasible with large national data sets. Fortunately, only a relatively small proportion of discharges fell into this category (5.2% of index cases in the 2006 development sample died within 30 days of discharge without being readmitted). An alternative approach to handling the competing outcome of death would have been to use a composite outcome of readmission or death. However, we believe that it is important to report the outcomes separately because factors that predict readmission and mortality may differ, and when making comparisons across hospitals it would not be possible to determine whether differences in rate were due to readmission or mortality. Third, while the patient‐level readmission model showed only modest discrimination, we intentionally excluded covariates such as race and socioeconomic status, as well as in‐hospital events and potential complications of care, and whether patients were discharged home or to a skilled nursing facility. While these variables could have improved predictive ability, they may be directly or indirectly related to quality or supply factors that should not be included in a model that seeks to control for patient clinical characteristics. For example, if hospitals with a large share of poor patients have higher readmission rates, then including income in the model will obscure differences that are important to identify. While we believe that the decision to exclude such factors in the model is in the best interest of patients, and supports efforts to reduce health inequality in society more generally, we also recognize that hospitals that care for a disproportionate share of poor patients are likely to require additional resources to overcome these social factors. Fourth, we limited the analysis to patients with a principal diagnosis of pneumonia, and chose not to also include those with a principal diagnosis of sepsis or respiratory failure coupled with a secondary diagnosis of pneumonia. While the broader definition is used by CMS in the National Pneumonia Project, that initiative relied on chart abstraction to differentiate pneumonia present at the time of admission from cases developing as a complication of hospitalization. Additionally, we did not attempt to differentiate between community‐acquired and healthcare‐associated pneumonia, however our approach is consistent with the National Pneumonia Project and Pneumonia Patient Outcomes Research Team.18 Fifth, while our model estimates readmission rates at the hospital level, we recognize that readmissions are influenced by a complex and extensive range of factors. In this context, greater cooperation between hospitals and other care providers will almost certainly be required in order to achieve dramatic improvement in readmission rates, which in turn will depend upon changes to the way serious illness is paid for. Some options that have recently been described include imposing financial penalties for early readmission, extending the boundaries of case‐based payment beyond hospital discharge, and bundling payments between hospitals and physicians.2325
Our measure has several limitations. First, our models were developed and validated using Medicare data, and the results may not apply to pneumonia patients less than 65 years of age. However, most patients hospitalized with pneumonia in the US are 65 or older. In addition, we were unable to test the model with a Medicare managed care population, because data are not currently available on such patients. Finally, the medical record‐based validation was conducted by state‐level analysis because the sample size was insufficient to carry this out at the hospital level.
In conclusion, more than 17% of Medicare beneficiaries are readmitted within 30 days following discharge after a hospitalization for pneumonia, and rates vary substantially across institutions. The development of a valid measure of hospital performance and public reporting are important first steps towards focusing attention on this problem. Actual improvement will now depend on whether hospitals and partner organizations are successful at identifying and implementing effective methods to prevent readmission.
- ,,.Rehospitalizations among patients in the Medicare Fee‐for‐Service Program.N Engl J Med.2009;360(14):1418–1428.
- Medicare Payment Advisory Commission.Report to the Congress: Promoting Greater Efficiency in Medicare.2007.
- ,,,,. HCUP Facts and Figures: Statistics on Hospital‐based Care in the United States, 2007.2009. Available at: http://www.hcup‐us.ahrq.gov/reports.jsp. Accessed November 7, 2009.
- Centers for Medicare 353(3):255–264.
- ,,,.Trends in postdischarge mortality and readmissions: has length of stay declined too far?Arch Intern Med.2004;164(5):538–544.
- ,,,.Short‐term outcomes and their predictors for patients hospitalized with community‐acquired pneumonia.Heart Lung.2004;33(5):301–307.
- ,,, et al.Improved clinical outcomes with utilization of a community‐acquired pneumonia guideline.Chest.2006;130(3):794–799.
- ,,,,.Associations between initial antimicrobial therapy and medical outcomes for hospitalized elderly patients with pneumonia.Arch Intern Med.1999;159(21):2562–2572.
- ,.Hospital readmissions as a measure of quality of health care: advantages and limitations.Arch Intern Med.2000;160(8):1074–1081.
- ,,,.The care transitions intervention: results of a randomized controlled trial.Arch Intern Med.2006;166(17):1822–1828.
- Corrigan JM, Eden J, Smith BM, eds.Leadership by Example: Coordinating Government Roles in Improving Health Care Quality. Committee on Enhancing Federal Healthcare Quality Programs.Washington, DC:National Academies Press,2003.
- Medicare.gov—Hospital Compare. Available at: http://www.hospitalcompare.hhs.gov/Hospital/Search/Welcome.asp?version=default1(1):29–37.
- ,,,,.Measuring performance for treating heart attacks and heart failure: the case for outcomes measurement.Health Aff.2007;26(1):75–85.
- NQF‐Endorsed® Standards. Available at: http://www.qualityforum.org/Measures_List.aspx. Accessed November 6,2009.
- ,,,,.Timing of antibiotic administration and outcomes for Medicare patients hospitalized with community‐acquired pneumonia.Arch Intern Med.2004;164(6):637–644.
- ,,. Diagnostic Cost Group Hierarchical Condition Category Models for Medicare Risk Adjustment. Report prepared for the Health Care Financing Administration. Health Economics Research, Inc;2000. Available at: http://www.cms.hhs.gov/Reports/Reports/ItemDetail.asp?ItemID=CMS023176. Accessed November 7, 2009.
- .Regression Modeling Strategies: With Applications to Linear Models, Logistic Regression, and Survival Analysis.1st ed.New York:Springer;2006.
- National Quality Forum—Measure Evaluation Criteria.2008. Available at: http://www.qualityforum.org/uploadedFiles/Quality_Forum/Measuring_Performance/Consensus_Development_Process%E2%80%99s_Principle/EvalCriteria2008–08‐28Final.pdf?n=4701.
- ,,, et al.Comprehensive discharge planning and home follow‐up of hospitalized elders: a randomized clinical trial.JAMA.1999;281(7):613–620.
- .Paying for care episodes and care coordination.N Engl J Med.2007;356(11):1166–1168.
- .Health care reform—toward more freedom, and responsibility, for physicians.N Engl J Med.2009;361(6):623–628.
- .Beyond pay for performance—emerging models of provider‐payment reform.N Engl J Med.2008;359(12):1197–1200.
Hospital readmissions are emblematic of the numerous challenges facing the US health care system. Despite high levels of spending, nearly 20% of Medicare beneficiaries are readmitted within 30 days of hospital discharge, many readmissions are considered preventable, and rates vary widely by hospital and region.1 Further, while readmissions have been estimated to cost taxpayers as much as $17 billion annually, the current fee‐for‐service method of paying for the acute care needs of seniors rewards hospitals financially for readmission, not their prevention.2
Pneumonia is the second most common reason for hospitalization among Medicare beneficiaries, accounting for approximately 650,000 admissions annually,3 and has been a focus of national quality‐improvement efforts for more than a decade.4, 5 Despite improvements in key processes of care, rates of readmission within 30 days of discharge following a hospitalization for pneumonia have been reported to vary from 10% to 24%.68 Among several factors, readmissions are believed to be influenced by the quality of both inpatient and outpatient care, and by care‐coordination activities occurring in the transition from inpatient to outpatient status.912
Public reporting of hospital performance is considered a key strategy for improving quality, reducing costs, and increasing the value of hospital care, both in the US and worldwide.13 In 2009, the Centers for Medicare & Medicaid Services (CMS) expanded its reporting initiatives by adding risk‐adjusted hospital readmission rates for acute myocardial infarction, heart failure, and pneumonia to the Hospital Compare website.14, 15 Readmission rates are an attractive focus for public reporting for several reasons. First, in contrast to most process‐based measures of quality (eg, whether a patient with pneumonia received a particular antibiotic), a readmission is an adverse outcome that matters to patients and families.16 Second, unlike process measures whose assessment requires detailed review of medical records, readmissions can be easily determined from standard hospital claims. Finally, readmissions are costly, and their prevention could yield substantial savings to society.
A necessary prerequisite for public reporting of readmission is a validated, risk‐adjusted measure that can be used to track performance over time and can facilitate comparisons across institutions. Toward this end, we describe the development, validation, and results of a National Quality Forum‐approved and CMS‐adopted model to estimate hospital‐specific, risk‐standardized, 30‐day readmission rates for Medicare patients hospitalized with pneumonia.17
METHODS
Data Sources
We used 20052006 claims data from Medicare inpatient, outpatient, and carrier (physician) Standard Analytic Files to develop and validate the administrative model. The Medicare Enrollment Database was used to determine Medicare fee‐for‐service enrollment and mortality statuses. A medical record model, used for additional validation of the administrative model, was developed using information abstracted from the charts of 75,616 pneumonia cases from 19982001 as part of the National Pneumonia Project, a CMS quality improvement initiative.18
Study Cohort
We identified hospitalizations of patients 65 years of age and older with a principal diagnosis of pneumonia (International Classification of Diseases, 9th Revision, Clinical Modification codes 480.XX, 481, 482.XX, 483.X, 485, 486, 487.0) as potential index pneumonia admissions. Because our focus was readmission for patients discharged from acute care settings, we excluded admissions in which patients died or were transferred to another acute care facility. Additionally, we restricted analysis to patients who had been enrolled in fee‐for‐service Medicare Parts A and B, for at least 12 months prior to their pneumonia hospitalization, so that we could use diagnostic codes from all inpatient and outpatient encounters during that period to enhance identification of comorbidities.
Outcome
The outcome was 30‐day readmission, defined as occurrence of at least one hospitalization for any cause within 30 days of discharge after an index admission. Readmissions were identified from hospital claims data, and were attributed to the hospital that had discharged the patient. A 30‐day time frame was selected because it is a clinically meaningful period during which hospitals can be expected to collaborate with other organizations and providers to implement measures to reduce the risk of rehospitalization.
Candidate and Final Model Variables
Candidate variables for the administrative claims model were selected by a clinician team from 189 diagnostic groups included in the Hierarchical Condition Category (HCC) clinical classification system.19 The HCC clinical classification system was developed for CMS in preparation for all‐encounter risk adjustment for Medicare Advantage (managed care). Under the HCC algorithm, the 15,000+ ICD‐9‐CM diagnosis codes are assigned to one of 189 clinically‐coherent condition categories (CCs). We used the April 2008 version of the ICD‐9‐CM to CC assignment map, which is maintained by CMS and posted at
The final risk‐adjustment model included 39 variables selected by the team of clinicians and analysts, primarily based on their clinical relevance but also with knowledge of the strength of their statistical association with readmission outcome (Table 1). For each patient, the presence or absence of these conditions was assessed from multiple sources, including secondary diagnoses during the index admission, principal and secondary diagnoses from hospital admissions in the 12 months prior to the index admission, and diagnoses from hospital outpatient and physician encounters 12 months before the index admission. A small number of CCs were considered to represent potential complications of care (eg, bleeding). Because we did not want to adjust for complications of care occurring during the index admission, a patient was not considered to have one of these conditions unless it was also present in at least one encounter prior to the index admission.
| Variable | Frequencies | Estimate | Standard Error | Odds Ratio | 95% CI | |
|---|---|---|---|---|---|---|
| ||||||
| Intercept | 2.395 | 0.021 | ||||
| Age 65 (years above 65, continuous) | 0.0001 | 0.001 | 1.000 | 0.998 | 1.001 | |
| Male | 45 | 0.071 | 0.012 | 1.073 | 1.048 | 1.099 |
| History of CABG | 5.2 | 0.179 | 0.027 | 0.836 | 0.793 | 0.881 |
| Metastatic cancer and acute leukemia (CC 7) | 4.3 | 0.177 | 0.029 | 1.194 | 1.128 | 1.263 |
| Lung, upper digestive tract, and other severe cancers (CC 8) | 6.0 | 0.256 | 0.024 | 1.292 | 1.232 | 1.354 |
| Diabetes and DM complications (CC 15‐20, 119, 120) | 36 | 0.059 | 0.012 | 1.061 | 1.036 | 1.087 |
| Disorders of fluid/electrolyte/acid‐base (CC 22, 23) | 34 | 0.149 | 0.013 | 1.160 | 1.131 | 1.191 |
| Iron deficiency and other/unspecified anemias and blood disease (CC 47) | 46 | 0.118 | 0.012 | 1.126 | 1.099 | 1.153 |
| Other psychiatric disorders (CC 60) | 12 | 0.108 | 0.017 | 1.114 | 1.077 | 1.151 |
| Cardio‐respiratory failure and shock (CC 79) | 16 | 0.114 | 0.016 | 1.121 | 1.087 | 1.156 |
| Congestive heart failure (CC 80) | 39 | 0.151 | 0.014 | 1.163 | 1.133 | 1.194 |
| Chronic atherosclerosis (CC 83, 84) | 47 | 0.051 | 0.013 | 1.053 | 1.027 | 1.079 |
| Valvular and rheumatic heart disease (CC 86) | 23 | 0.062 | 0.014 | 1.064 | 1.036 | 1.093 |
| Arrhythmias (CC 92, 93) | 38 | 0.126 | 0.013 | 1.134 | 1.107 | 1.163 |
| Vascular or circulatory disease (CC 104‐106) | 38 | 0.088 | 0.012 | 1.092 | 1.066 | 1.119 |
| COPD (CC 108) | 58 | 0.186 | 0.013 | 1.205 | 1.175 | 1.235 |
| Fibrosis of lung and other chronic lung disorders (CC 109) | 17 | 0.086 | 0.015 | 1.090 | 1.059 | 1.122 |
| Renal failure (CC 131) | 17 | 0.147 | 0.016 | 1.158 | 1.122 | 1.196 |
| Protein‐calorie malnutrition (CC 21) | 7.9 | 0.121 | 0.020 | 1.129 | 1.086 | 1.173 |
| History of infection (CC 1, 3‐6) | 35 | 0.068 | 0.012 | 1.071 | 1.045 | 1.097 |
| Severe hematological disorders (CC 44) | 3.6 | 0.117 | 0.028 | 1.125 | 1.064 | 1.188 |
| Decubitus ulcer or chronic skin ulcer (CC 148, 149) | 10 | 0.101 | 0.018 | 1.106 | 1.067 | 1.146 |
| History of pneumonia (CC 111‐113) | 44 | 0.065 | 0.013 | 1.067 | 1.041 | 1.094 |
| Vertebral fractures (CC 157) | 5.1 | 0.113 | 0.024 | 1.120 | 1.068 | 1.174 |
| Other injuries (CC 162) | 32 | 0.061 | 0.012 | 1.063 | 1.038 | 1.089 |
| Urinary tract infection (CC 135) | 26 | 0.064 | 0.014 | 1.066 | 1.038 | 1.095 |
| Lymphatic, head and neck, brain, and other major cancers; breast, prostate, colorectal, and other cancers and tumors (CC 9‐10) | 16 | 0.050 | 0.016 | 1.051 | 1.018 | 1.084 |
| End‐stage renal disease or dialysis (CC 129, 130) | 1.9 | 0.131 | 0.037 | 1.140 | 1.060 | 1.226 |
| Drug/alcohol abuse/dependence/psychosis (CC 51‐53) | 12 | 0.081 | 0.017 | 1.084 | 1.048 | 1.121 |
| Septicemia/shock (CC 2) | 6.3 | 0.094 | 0.022 | 1.098 | 1.052 | 1.146 |
| Other gastrointestinal disorders (CC 36) | 56 | 0.073 | 0.012 | 1.076 | 1.051 | 1.102 |
| Acute coronary syndrome (CC 81, 82) | 8.3 | 0.126 | 0.019 | 1.134 | 1.092 | 1.178 |
| Pleural effusion/pneumothorax (CC 114) | 12 | 0.083 | 0.017 | 1.086 | 1.051 | 1.123 |
| Other urinary tract disorders (CC 136) | 24 | 0.059 | 0.014 | 1.061 | 1.033 | 1.090 |
| Stroke (CC 95, 96) | 10 | 0.047 | 0.019 | 1.049 | 1.011 | 1.088 |
| Dementia and senility (CC 49, 50) | 27 | 0.031 | 0.014 | 1.031 | 1.004 | 1.059 |
| Hemiplegia, paraplegia, paralysis, functional disability (CC 67‐69, 100‐102, 177, 178) | 7.4 | 0.068 | 0.021 | 1.070 | 1.026 | 1.116 |
| Other lung disorders (CC 115) | 45 | 0.005 | 0.012 | 1.005 | 0.982 | 1.030 |
| Major psychiatric disorders (CC 54‐56) | 11 | 0.038 | 0.018 | 1.038 | 1.003 | 1.075 |
| Asthma (CC 110) | 12 | 0.006 | 0.018 | 1.006 | 0.972 | 1.041 |
Model Derivation
For the development of the administrative claims model, we randomly sampled half of 2006 hospitalizations that met inclusion criteria. To assess model performance at the patient level, we calculated the area under the receiver operating curve (AUC), and calculated observed readmission rates in the lowest and highest deciles on the basis of predicted readmission probabilities. We also compared performance with a null model, a model that adjusted for age and sex, and a model that included all candidate variables.20
Risk‐Standardized Readmission Rates
Using hierarchical logistic regression, we modeled the log‐odds of readmission within 30 days of discharge from an index pneumonia admission as a function of patient demographic and clinical characteristics, and a random hospital‐specific intercept. This strategy accounts for within‐hospital correlation, or clustering, of observed outcomes, and models the assumption that underlying differences in quality among hospitals being evaluated lead to systematic differences in outcomes. We then calculated hospital‐specific readmission rates as the ratio of predicted‐to‐expected readmissions (similar to observed/expected ratio), multiplied by the national unadjusted ratea form of indirect standardization. Predicted number of readmissions in each hospital is estimated given the same patient mix and its estimated hospital‐specific intercept. Expected number of readmissions in each hospital is estimated using its patient mix and the average hospital‐specific intercept. To assess hospital performance in any given year, we re‐estimate model coefficients using that year's data.
Model Validation: Administrative Claims
We compared the model performance in the development sample with its performance in the sample from the 2006 data that was not selected for the development set, and separately among pneumonia admissions in 2005. The model was recalibrated in each validation set.
Model Validation: Medical Record Abstraction
We developed a separate medical record‐based model of readmission risk using information from charts that had previously been abstracted as part of CMS's National Pneumonia Project. To select variables for this model, the clinician team: 1) reviewed the list of variables that were included in a medical record model that was previously developed for validating the National Quality Forum‐approved pneumonia mortality measure; 2) reviewed a list of other potential candidate variables available in the National Pneumonia Project dataset; and 3) reviewed variables that emerged as potentially important predictors of readmission, based on a systematic review of the literature that was conducted as part of measure development. This selection process resulted in a final medical record model that included 35 variables.
We linked patients in the National Pneumonia Project cohort to their Medicare claims data, including claims from one year before the index hospitalization, so that we could calculate risk‐standardized readmission rates in this cohort separately using medical record and claims‐based models. This analysis was conducted at the state level, for the 50 states plus the District of Columbia and Puerto Rico, because medical record data were unavailable in sufficient numbers to permit hospital‐level comparisons. To examine the relationship between risk‐standardized rates obtained from medical record and administrative data models, we estimated a linear regression model describing the association between the two rates, weighting each state by number of index hospitalizations, and calculated the correlation coefficient and the intercept and slope of this equation. A slope close to 1 and an intercept close to 0 would provide evidence that risk‐standardized state readmission rates from the medical record and claims models were similar. We also calculated the difference between state risk‐standardized readmission rates from the two models.
Analyses were conducted with the use of SAS version 9.1.3 (SAS Institute Inc, Cary, NC). Models were fitted separately for the National Pneumonia Project and 2006 cohort. We estimated the hierarchical models using the GLIMMIX procedure in SAS. The Human Investigation Committee at the Yale School of Medicine approved an exemption for the authors to use CMS claims and enrollment data for research analyses and publication.
RESULTS
Model Derivation and Performance
After exclusions were applied, the 2006 sample included 453,251 pneumonia hospitalizations (Figure 1). The development sample consisted of 226,545 hospitalizations at 4675 hospitals, with an overall unadjusted 30‐day readmission rate of 17.4%. In 11,694 index cases (5.2%), the patient died within 30 days without being readmitted. Median readmission rate was 16.3%, 25th and 75th percentile rates were 11.1% and 21.3%, and at the 10th and 90th percentile, hospital readmission rates ranged from 4.6% to 26.7% (Figure 2).
The claims model included 39 variables (age, sex, and 37 clinical variables) (Table 1). The mean age of the cohort was 80.0 years, with 55.5% women and 11.1% nonwhite patients. Mean observed readmission rate in the development sample ranged from 9% in the lowest decile of predicted pneumonia readmission rates to 32% in the highest predicted decile, a range of 23%. The AUC was 0.63. For comparison, a model with only age and sex had an AUC of 0.51, and a model with all candidate variables had an AUC equal to 0.63 (Table 2).
| Calibration (0, 1)* | Discrimination | Residuals Lack of Fit (Pearson Residual Fall %) | Model 2 (No. of Covariates) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Predictive Ability (Lowest Decile, Highest Decile) | AUC | (<2) | (2, 0) | (0, 2) | (2+) | ||||
| |||||||||
| Development sample | |||||||||
| 2006 | (1st half) N = 226,545 | (0, 1) | (0.09, 0.32) | 0.63 | 0 | 82.62 | 7.39 | 9.99 | 6,843 (40) |
| Validation sample | |||||||||
| 2006 | (2nd half) N = 226,706 | (0.002, 0.997) | (0.09, 0.31) | 0.63 | 0 | 82.55 | 7.45 | 9.99 | 6,870 (40) |
| 2005 | N = 536,015 | (0.035, 1.008) | (0.08, 0.31) | 0.63 | 0 | 82.67 | 7.31 | 10.03 | 16,241 (40) |
Hospital Risk‐Standardized Readmission Rates
Risk‐standardized readmission rates varied across hospitals (Figure 3). Median risk‐standardized readmission rate was 17.3%, and the 25th and 75th percentiles were 16.9% and 17.9%, respectively. The 5th percentile was 16.0% and the 95th percentile was 19.1%. Odds of readmission for a hospital one standard deviation above average was 1.4 times that of a hospital one standard deviation below average.
Administrative Model Validation
In the remaining 50% of pneumonia index hospitalizations from 2006, and the entire 2005 cohort, regression coefficients and standard errors of model variables were similar to those in the development data set. Model performance using 2005 data was consistent with model performance using the 2006 development and validation half‐samples (Table 2).
Medical Record Validation
After exclusions, the medical record sample taken from the National Pneumonia Project included 47,429 cases, with an unadjusted 30‐day readmission rate of 17.0%. The final medical record risk‐adjustment model included a total of 35 variables, whose prevalence and association with readmission risk varied modestly (Table 3). Performance of the medical record and administrative models was similar (areas under the ROC curve 0.59 and 0.63, respectively) (Table 4). Additionally, in the administrative model, predicted readmission rates ranged from 8% in the lowest predicted decile to 30% in the highest predicted decile, while in the medical record model, the corresponding rates varied from 10% to 26%.
| Variable | Percent | Estimate | Standard Error | Odds Ratio | 95% CI | |
|---|---|---|---|---|---|---|
| ||||||
| Age 65, mean (SD) | 15.24 (7.87) | 0.003 | 0.002 | 0.997 | 0.993 | 1.000 |
| Male | 46.18 | 0.122 | 0.025 | 1.130 | 1.075 | 1.188 |
| Nursing home resident | 17.71 | 0.035 | 0.037 | 1.036 | 0.963 | 1.114 |
| Neoplastic disease | 6.80 | 0.130 | 0.049 | 1.139 | 1.034 | 1.254 |
| Liver disease | 1.04 | 0.089 | 0.123 | 0.915 | 0.719 | 1.164 |
| History of heart failure | 28.98 | 0.234 | 0.029 | 1.264 | 1.194 | 1.339 |
| History of renal disease | 8.51 | 0.188 | 0.047 | 1.206 | 1.100 | 1.323 |
| Altered mental status | 17.95 | 0.009 | 0.034 | 1.009 | 0.944 | 1.080 |
| Pleural effusion | 21.20 | 0.165 | 0.030 | 1.179 | 1.111 | 1.251 |
| BUN 30 mg/dl | 23.28 | 0.160 | 0.033 | 1.174 | 1.100 | 1.252 |
| BUN missing | 14.56 | 0.101 | 0.185 | 0.904 | 0.630 | 1.298 |
| Systolic BP <90 mmHg | 2.95 | 0.068 | 0.070 | 1.070 | 0.932 | 1.228 |
| Systolic BP missing | 11.21 | 0.149 | 0.425 | 1.160 | 0.504 | 2.669 |
| Pulse 125/min | 7.73 | 0.036 | 0.047 | 1.036 | 0.945 | 1.137 |
| Pulse missing | 11.22 | 0.210 | 0.405 | 1.234 | 0.558 | 2.729 |
| Respiratory rate 30/min | 16.38 | 0.079 | 0.034 | 1.082 | 1.012 | 1.157 |
| Respiratory rate missing | 11.39 | 0.204 | 0.240 | 1.226 | 0.765 | 1.964 |
| Sodium <130 mmol/L | 4.82 | 0.136 | 0.057 | 1.145 | 1.025 | 1.280 |
| Sodium missing | 14.39 | 0.049 | 0.143 | 1.050 | 0.793 | 1.391 |
| Glucose 250 mg/dl | 5.19 | 0.005 | 0.057 | 0.995 | 0.889 | 1.114 |
| Glucose missing | 15.44 | 0.156 | 0.105 | 0.855 | 0.696 | 1.051 |
| Hematocrit <30% | 7.77 | 0.270 | 0.044 | 1.310 | 1.202 | 1.428 |
| Hematocrit missing | 13.62 | 0.071 | 0.135 | 0.932 | 0.715 | 1.215 |
| Creatinine 2.5 mg/dL | 4.68 | 0.109 | 0.062 | 1.115 | 0.989 | 1.258 |
| Creatinine missing | 14.63 | 0.200 | 0.167 | 1.221 | 0.880 | 1.695 |
| WBC 6‐12 b/L | 38.04 | 0.021 | 0.049 | 0.979 | 0.889 | 1.079 |
| WBC >12 b/L | 41.45 | 0.068 | 0.049 | 0.934 | 0.848 | 1.029 |
| WBC missing | 12.85 | 0.167 | 0.162 | 1.181 | 0.860 | 1.623 |
| Immunosuppressive therapy | 15.01 | 0.347 | 0.035 | 1.415 | 1.321 | 1.516 |
| Chronic lung disease | 42.16 | 0.137 | 0.028 | 1.147 | 1.086 | 1.211 |
| Coronary artery disease | 39.57 | 0.150 | 0.028 | 1.162 | 1.100 | 1.227 |
| Diabetes mellitus | 20.90 | 0.137 | 0.033 | 1.147 | 1.076 | 1.223 |
| Alcohol/drug abuse | 3.40 | 0.099 | 0.071 | 0.906 | 0.788 | 1.041 |
| Dementia/Alzheimer's disease | 16.38 | 0.125 | 0.038 | 1.133 | 1.052 | 1.222 |
| Splenectomy | 0.44 | 0.016 | 0.186 | 1.016 | 0.706 | 1.463 |
| Model | Calibration (0, 1)* | Discrimination | Residuals Lack of Fit (Pearson Residual Fall %) | Model 2 (No. of Covariates) | ||||
|---|---|---|---|---|---|---|---|---|
| Predictive Ability (Lowest Decile, Highest Decile) | AUC | (<2) | (2, 0) | (0, 2) | (2+) | |||
| ||||||||
| Medical Record Model Development Sample (NP) | ||||||||
| N = 47,429 No. of 30‐day readmissions = 8,042 | (1, 0) | (0.10, 0.26) | 0.59 | 0 | 83.04 | 5.28 | 11.68 | 710 (35) |
| Linked Administrative Model Validation Sample | ||||||||
| N = 47,429 No. of 30‐day readmissions = 8,042 | (1, 0) | (0.08, 0.30) | 0.63 | 0 | 83.04 | 6.94 | 10.01 | 1,414 (40) |
The correlation coefficient of the estimated state‐specific standardized readmission rates from the administrative and medical record models was 0.96, and the proportion of the variance explained by the model was 0.92 (Figure 4).
DISCUSSION
We have described the development, validation, and results of a hospital, 30‐day, risk‐standardized readmission model for pneumonia that was created to support current federal transparency initiatives. The model uses administrative claims data from Medicare fee‐for‐service patients and produces results that are comparable to a model based on information obtained through manual abstraction of medical records. We observed an overall 30‐day readmission rate of 17%, and our analyses revealed substantial variation across US hospitals, suggesting that improvement by lower performing institutions is an achievable goal.
Because more than one in six pneumonia patients are rehospitalized shortly after discharge, and because pneumonia hospitalizations represent an enormous expense to the Medicare program, prevention of readmissions is now widely recognized to offer a substantial opportunity to improve patient outcomes while simultaneously lowering health care costs. Accordingly, promotion of strategies to reduce readmission rates has become a key priority for payers and quality‐improvement organizations. These range from policy‐level attempts to stimulate change, such as publicly reporting hospital readmission rates on government websites, to establishing accreditation standardssuch as the Joint Commission's requirement to accurately reconcile medications, to the creation of quality improvement collaboratives focused on sharing best practices across institutions. Regardless of the approach taken, a valid, risk‐adjusted measure of performance is required to evaluate and track performance over time. The measure we have described meets the National Quality Forum's measure evaluation criteria in that it addresses an important clinical topic for which there appears to be significant opportunities for improvement, the measure is precisely defined and has been subjected to validity and reliability testing, it is risk‐adjusted based on patient clinical factors present at the start of care, is feasible to produce, and is understandable by a broad range of potential users.21 Because hospitalists are the physicians primarily responsible for the care of patients with pneumonia at US hospitals, and because they frequently serve as the physician champions for quality improvement activities related to pneumonia, it is especially important that they maintain a thorough understanding of the measures and methodologies underlying current efforts to measure hospital performance.
Several features of our approach warrant additional comment. First, we deliberately chose to measure all readmission events rather than attempt to discriminate between potentially preventable and nonpreventable readmissions. From the patient perspective, readmission for any reason is a concern, and limiting the measure to pneumonia‐related readmissions could make it susceptible to gaming by hospitals. Moreover, determining whether a readmission is related to a potential quality problem is not straightforward. For example, a patient with pneumonia whose discharge medications were prescribed incorrectly may be readmitted with a hip fracture following an episode of syncope. It would be inappropriate to treat this readmission as unrelated to the care the patient received for pneumonia. Additionally, while our approach does not presume that every readmission is preventable, the goal is to reduce the risk of readmissions generally (not just in narrowly defined subpopulations), and successful interventions to reduce rehospitalization have typically demonstrated reductions in all‐cause readmission.9, 22 Second, deaths that occurred within 30 days of discharge, yet that were not accompanied by a hospital readmission, were not counted as a readmission outcome. While it may seem inappropriate to treat a postdischarge death as a nonevent (rather than censoring or excluding such cases), alternative analytic approaches, such as using a hierarchical survival model, are not currently computationally feasible with large national data sets. Fortunately, only a relatively small proportion of discharges fell into this category (5.2% of index cases in the 2006 development sample died within 30 days of discharge without being readmitted). An alternative approach to handling the competing outcome of death would have been to use a composite outcome of readmission or death. However, we believe that it is important to report the outcomes separately because factors that predict readmission and mortality may differ, and when making comparisons across hospitals it would not be possible to determine whether differences in rate were due to readmission or mortality. Third, while the patient‐level readmission model showed only modest discrimination, we intentionally excluded covariates such as race and socioeconomic status, as well as in‐hospital events and potential complications of care, and whether patients were discharged home or to a skilled nursing facility. While these variables could have improved predictive ability, they may be directly or indirectly related to quality or supply factors that should not be included in a model that seeks to control for patient clinical characteristics. For example, if hospitals with a large share of poor patients have higher readmission rates, then including income in the model will obscure differences that are important to identify. While we believe that the decision to exclude such factors in the model is in the best interest of patients, and supports efforts to reduce health inequality in society more generally, we also recognize that hospitals that care for a disproportionate share of poor patients are likely to require additional resources to overcome these social factors. Fourth, we limited the analysis to patients with a principal diagnosis of pneumonia, and chose not to also include those with a principal diagnosis of sepsis or respiratory failure coupled with a secondary diagnosis of pneumonia. While the broader definition is used by CMS in the National Pneumonia Project, that initiative relied on chart abstraction to differentiate pneumonia present at the time of admission from cases developing as a complication of hospitalization. Additionally, we did not attempt to differentiate between community‐acquired and healthcare‐associated pneumonia, however our approach is consistent with the National Pneumonia Project and Pneumonia Patient Outcomes Research Team.18 Fifth, while our model estimates readmission rates at the hospital level, we recognize that readmissions are influenced by a complex and extensive range of factors. In this context, greater cooperation between hospitals and other care providers will almost certainly be required in order to achieve dramatic improvement in readmission rates, which in turn will depend upon changes to the way serious illness is paid for. Some options that have recently been described include imposing financial penalties for early readmission, extending the boundaries of case‐based payment beyond hospital discharge, and bundling payments between hospitals and physicians.2325
Our measure has several limitations. First, our models were developed and validated using Medicare data, and the results may not apply to pneumonia patients less than 65 years of age. However, most patients hospitalized with pneumonia in the US are 65 or older. In addition, we were unable to test the model with a Medicare managed care population, because data are not currently available on such patients. Finally, the medical record‐based validation was conducted by state‐level analysis because the sample size was insufficient to carry this out at the hospital level.
In conclusion, more than 17% of Medicare beneficiaries are readmitted within 30 days following discharge after a hospitalization for pneumonia, and rates vary substantially across institutions. The development of a valid measure of hospital performance and public reporting are important first steps towards focusing attention on this problem. Actual improvement will now depend on whether hospitals and partner organizations are successful at identifying and implementing effective methods to prevent readmission.
Hospital readmissions are emblematic of the numerous challenges facing the US health care system. Despite high levels of spending, nearly 20% of Medicare beneficiaries are readmitted within 30 days of hospital discharge, many readmissions are considered preventable, and rates vary widely by hospital and region.1 Further, while readmissions have been estimated to cost taxpayers as much as $17 billion annually, the current fee‐for‐service method of paying for the acute care needs of seniors rewards hospitals financially for readmission, not their prevention.2
Pneumonia is the second most common reason for hospitalization among Medicare beneficiaries, accounting for approximately 650,000 admissions annually,3 and has been a focus of national quality‐improvement efforts for more than a decade.4, 5 Despite improvements in key processes of care, rates of readmission within 30 days of discharge following a hospitalization for pneumonia have been reported to vary from 10% to 24%.68 Among several factors, readmissions are believed to be influenced by the quality of both inpatient and outpatient care, and by care‐coordination activities occurring in the transition from inpatient to outpatient status.912
Public reporting of hospital performance is considered a key strategy for improving quality, reducing costs, and increasing the value of hospital care, both in the US and worldwide.13 In 2009, the Centers for Medicare & Medicaid Services (CMS) expanded its reporting initiatives by adding risk‐adjusted hospital readmission rates for acute myocardial infarction, heart failure, and pneumonia to the Hospital Compare website.14, 15 Readmission rates are an attractive focus for public reporting for several reasons. First, in contrast to most process‐based measures of quality (eg, whether a patient with pneumonia received a particular antibiotic), a readmission is an adverse outcome that matters to patients and families.16 Second, unlike process measures whose assessment requires detailed review of medical records, readmissions can be easily determined from standard hospital claims. Finally, readmissions are costly, and their prevention could yield substantial savings to society.
A necessary prerequisite for public reporting of readmission is a validated, risk‐adjusted measure that can be used to track performance over time and can facilitate comparisons across institutions. Toward this end, we describe the development, validation, and results of a National Quality Forum‐approved and CMS‐adopted model to estimate hospital‐specific, risk‐standardized, 30‐day readmission rates for Medicare patients hospitalized with pneumonia.17
METHODS
Data Sources
We used 20052006 claims data from Medicare inpatient, outpatient, and carrier (physician) Standard Analytic Files to develop and validate the administrative model. The Medicare Enrollment Database was used to determine Medicare fee‐for‐service enrollment and mortality statuses. A medical record model, used for additional validation of the administrative model, was developed using information abstracted from the charts of 75,616 pneumonia cases from 19982001 as part of the National Pneumonia Project, a CMS quality improvement initiative.18
Study Cohort
We identified hospitalizations of patients 65 years of age and older with a principal diagnosis of pneumonia (International Classification of Diseases, 9th Revision, Clinical Modification codes 480.XX, 481, 482.XX, 483.X, 485, 486, 487.0) as potential index pneumonia admissions. Because our focus was readmission for patients discharged from acute care settings, we excluded admissions in which patients died or were transferred to another acute care facility. Additionally, we restricted analysis to patients who had been enrolled in fee‐for‐service Medicare Parts A and B, for at least 12 months prior to their pneumonia hospitalization, so that we could use diagnostic codes from all inpatient and outpatient encounters during that period to enhance identification of comorbidities.
Outcome
The outcome was 30‐day readmission, defined as occurrence of at least one hospitalization for any cause within 30 days of discharge after an index admission. Readmissions were identified from hospital claims data, and were attributed to the hospital that had discharged the patient. A 30‐day time frame was selected because it is a clinically meaningful period during which hospitals can be expected to collaborate with other organizations and providers to implement measures to reduce the risk of rehospitalization.
Candidate and Final Model Variables
Candidate variables for the administrative claims model were selected by a clinician team from 189 diagnostic groups included in the Hierarchical Condition Category (HCC) clinical classification system.19 The HCC clinical classification system was developed for CMS in preparation for all‐encounter risk adjustment for Medicare Advantage (managed care). Under the HCC algorithm, the 15,000+ ICD‐9‐CM diagnosis codes are assigned to one of 189 clinically‐coherent condition categories (CCs). We used the April 2008 version of the ICD‐9‐CM to CC assignment map, which is maintained by CMS and posted at
The final risk‐adjustment model included 39 variables selected by the team of clinicians and analysts, primarily based on their clinical relevance but also with knowledge of the strength of their statistical association with readmission outcome (Table 1). For each patient, the presence or absence of these conditions was assessed from multiple sources, including secondary diagnoses during the index admission, principal and secondary diagnoses from hospital admissions in the 12 months prior to the index admission, and diagnoses from hospital outpatient and physician encounters 12 months before the index admission. A small number of CCs were considered to represent potential complications of care (eg, bleeding). Because we did not want to adjust for complications of care occurring during the index admission, a patient was not considered to have one of these conditions unless it was also present in at least one encounter prior to the index admission.
| Variable | Frequencies | Estimate | Standard Error | Odds Ratio | 95% CI | |
|---|---|---|---|---|---|---|
| ||||||
| Intercept | 2.395 | 0.021 | ||||
| Age 65 (years above 65, continuous) | 0.0001 | 0.001 | 1.000 | 0.998 | 1.001 | |
| Male | 45 | 0.071 | 0.012 | 1.073 | 1.048 | 1.099 |
| History of CABG | 5.2 | 0.179 | 0.027 | 0.836 | 0.793 | 0.881 |
| Metastatic cancer and acute leukemia (CC 7) | 4.3 | 0.177 | 0.029 | 1.194 | 1.128 | 1.263 |
| Lung, upper digestive tract, and other severe cancers (CC 8) | 6.0 | 0.256 | 0.024 | 1.292 | 1.232 | 1.354 |
| Diabetes and DM complications (CC 15‐20, 119, 120) | 36 | 0.059 | 0.012 | 1.061 | 1.036 | 1.087 |
| Disorders of fluid/electrolyte/acid‐base (CC 22, 23) | 34 | 0.149 | 0.013 | 1.160 | 1.131 | 1.191 |
| Iron deficiency and other/unspecified anemias and blood disease (CC 47) | 46 | 0.118 | 0.012 | 1.126 | 1.099 | 1.153 |
| Other psychiatric disorders (CC 60) | 12 | 0.108 | 0.017 | 1.114 | 1.077 | 1.151 |
| Cardio‐respiratory failure and shock (CC 79) | 16 | 0.114 | 0.016 | 1.121 | 1.087 | 1.156 |
| Congestive heart failure (CC 80) | 39 | 0.151 | 0.014 | 1.163 | 1.133 | 1.194 |
| Chronic atherosclerosis (CC 83, 84) | 47 | 0.051 | 0.013 | 1.053 | 1.027 | 1.079 |
| Valvular and rheumatic heart disease (CC 86) | 23 | 0.062 | 0.014 | 1.064 | 1.036 | 1.093 |
| Arrhythmias (CC 92, 93) | 38 | 0.126 | 0.013 | 1.134 | 1.107 | 1.163 |
| Vascular or circulatory disease (CC 104‐106) | 38 | 0.088 | 0.012 | 1.092 | 1.066 | 1.119 |
| COPD (CC 108) | 58 | 0.186 | 0.013 | 1.205 | 1.175 | 1.235 |
| Fibrosis of lung and other chronic lung disorders (CC 109) | 17 | 0.086 | 0.015 | 1.090 | 1.059 | 1.122 |
| Renal failure (CC 131) | 17 | 0.147 | 0.016 | 1.158 | 1.122 | 1.196 |
| Protein‐calorie malnutrition (CC 21) | 7.9 | 0.121 | 0.020 | 1.129 | 1.086 | 1.173 |
| History of infection (CC 1, 3‐6) | 35 | 0.068 | 0.012 | 1.071 | 1.045 | 1.097 |
| Severe hematological disorders (CC 44) | 3.6 | 0.117 | 0.028 | 1.125 | 1.064 | 1.188 |
| Decubitus ulcer or chronic skin ulcer (CC 148, 149) | 10 | 0.101 | 0.018 | 1.106 | 1.067 | 1.146 |
| History of pneumonia (CC 111‐113) | 44 | 0.065 | 0.013 | 1.067 | 1.041 | 1.094 |
| Vertebral fractures (CC 157) | 5.1 | 0.113 | 0.024 | 1.120 | 1.068 | 1.174 |
| Other injuries (CC 162) | 32 | 0.061 | 0.012 | 1.063 | 1.038 | 1.089 |
| Urinary tract infection (CC 135) | 26 | 0.064 | 0.014 | 1.066 | 1.038 | 1.095 |
| Lymphatic, head and neck, brain, and other major cancers; breast, prostate, colorectal, and other cancers and tumors (CC 9‐10) | 16 | 0.050 | 0.016 | 1.051 | 1.018 | 1.084 |
| End‐stage renal disease or dialysis (CC 129, 130) | 1.9 | 0.131 | 0.037 | 1.140 | 1.060 | 1.226 |
| Drug/alcohol abuse/dependence/psychosis (CC 51‐53) | 12 | 0.081 | 0.017 | 1.084 | 1.048 | 1.121 |
| Septicemia/shock (CC 2) | 6.3 | 0.094 | 0.022 | 1.098 | 1.052 | 1.146 |
| Other gastrointestinal disorders (CC 36) | 56 | 0.073 | 0.012 | 1.076 | 1.051 | 1.102 |
| Acute coronary syndrome (CC 81, 82) | 8.3 | 0.126 | 0.019 | 1.134 | 1.092 | 1.178 |
| Pleural effusion/pneumothorax (CC 114) | 12 | 0.083 | 0.017 | 1.086 | 1.051 | 1.123 |
| Other urinary tract disorders (CC 136) | 24 | 0.059 | 0.014 | 1.061 | 1.033 | 1.090 |
| Stroke (CC 95, 96) | 10 | 0.047 | 0.019 | 1.049 | 1.011 | 1.088 |
| Dementia and senility (CC 49, 50) | 27 | 0.031 | 0.014 | 1.031 | 1.004 | 1.059 |
| Hemiplegia, paraplegia, paralysis, functional disability (CC 67‐69, 100‐102, 177, 178) | 7.4 | 0.068 | 0.021 | 1.070 | 1.026 | 1.116 |
| Other lung disorders (CC 115) | 45 | 0.005 | 0.012 | 1.005 | 0.982 | 1.030 |
| Major psychiatric disorders (CC 54‐56) | 11 | 0.038 | 0.018 | 1.038 | 1.003 | 1.075 |
| Asthma (CC 110) | 12 | 0.006 | 0.018 | 1.006 | 0.972 | 1.041 |
Model Derivation
For the development of the administrative claims model, we randomly sampled half of 2006 hospitalizations that met inclusion criteria. To assess model performance at the patient level, we calculated the area under the receiver operating curve (AUC), and calculated observed readmission rates in the lowest and highest deciles on the basis of predicted readmission probabilities. We also compared performance with a null model, a model that adjusted for age and sex, and a model that included all candidate variables.20
Risk‐Standardized Readmission Rates
Using hierarchical logistic regression, we modeled the log‐odds of readmission within 30 days of discharge from an index pneumonia admission as a function of patient demographic and clinical characteristics, and a random hospital‐specific intercept. This strategy accounts for within‐hospital correlation, or clustering, of observed outcomes, and models the assumption that underlying differences in quality among hospitals being evaluated lead to systematic differences in outcomes. We then calculated hospital‐specific readmission rates as the ratio of predicted‐to‐expected readmissions (similar to observed/expected ratio), multiplied by the national unadjusted ratea form of indirect standardization. Predicted number of readmissions in each hospital is estimated given the same patient mix and its estimated hospital‐specific intercept. Expected number of readmissions in each hospital is estimated using its patient mix and the average hospital‐specific intercept. To assess hospital performance in any given year, we re‐estimate model coefficients using that year's data.
Model Validation: Administrative Claims
We compared the model performance in the development sample with its performance in the sample from the 2006 data that was not selected for the development set, and separately among pneumonia admissions in 2005. The model was recalibrated in each validation set.
Model Validation: Medical Record Abstraction
We developed a separate medical record‐based model of readmission risk using information from charts that had previously been abstracted as part of CMS's National Pneumonia Project. To select variables for this model, the clinician team: 1) reviewed the list of variables that were included in a medical record model that was previously developed for validating the National Quality Forum‐approved pneumonia mortality measure; 2) reviewed a list of other potential candidate variables available in the National Pneumonia Project dataset; and 3) reviewed variables that emerged as potentially important predictors of readmission, based on a systematic review of the literature that was conducted as part of measure development. This selection process resulted in a final medical record model that included 35 variables.
We linked patients in the National Pneumonia Project cohort to their Medicare claims data, including claims from one year before the index hospitalization, so that we could calculate risk‐standardized readmission rates in this cohort separately using medical record and claims‐based models. This analysis was conducted at the state level, for the 50 states plus the District of Columbia and Puerto Rico, because medical record data were unavailable in sufficient numbers to permit hospital‐level comparisons. To examine the relationship between risk‐standardized rates obtained from medical record and administrative data models, we estimated a linear regression model describing the association between the two rates, weighting each state by number of index hospitalizations, and calculated the correlation coefficient and the intercept and slope of this equation. A slope close to 1 and an intercept close to 0 would provide evidence that risk‐standardized state readmission rates from the medical record and claims models were similar. We also calculated the difference between state risk‐standardized readmission rates from the two models.
Analyses were conducted with the use of SAS version 9.1.3 (SAS Institute Inc, Cary, NC). Models were fitted separately for the National Pneumonia Project and 2006 cohort. We estimated the hierarchical models using the GLIMMIX procedure in SAS. The Human Investigation Committee at the Yale School of Medicine approved an exemption for the authors to use CMS claims and enrollment data for research analyses and publication.
RESULTS
Model Derivation and Performance
After exclusions were applied, the 2006 sample included 453,251 pneumonia hospitalizations (Figure 1). The development sample consisted of 226,545 hospitalizations at 4675 hospitals, with an overall unadjusted 30‐day readmission rate of 17.4%. In 11,694 index cases (5.2%), the patient died within 30 days without being readmitted. Median readmission rate was 16.3%, 25th and 75th percentile rates were 11.1% and 21.3%, and at the 10th and 90th percentile, hospital readmission rates ranged from 4.6% to 26.7% (Figure 2).
The claims model included 39 variables (age, sex, and 37 clinical variables) (Table 1). The mean age of the cohort was 80.0 years, with 55.5% women and 11.1% nonwhite patients. Mean observed readmission rate in the development sample ranged from 9% in the lowest decile of predicted pneumonia readmission rates to 32% in the highest predicted decile, a range of 23%. The AUC was 0.63. For comparison, a model with only age and sex had an AUC of 0.51, and a model with all candidate variables had an AUC equal to 0.63 (Table 2).
| Calibration (0, 1)* | Discrimination | Residuals Lack of Fit (Pearson Residual Fall %) | Model 2 (No. of Covariates) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Predictive Ability (Lowest Decile, Highest Decile) | AUC | (<2) | (2, 0) | (0, 2) | (2+) | ||||
| |||||||||
| Development sample | |||||||||
| 2006 | (1st half) N = 226,545 | (0, 1) | (0.09, 0.32) | 0.63 | 0 | 82.62 | 7.39 | 9.99 | 6,843 (40) |
| Validation sample | |||||||||
| 2006 | (2nd half) N = 226,706 | (0.002, 0.997) | (0.09, 0.31) | 0.63 | 0 | 82.55 | 7.45 | 9.99 | 6,870 (40) |
| 2005 | N = 536,015 | (0.035, 1.008) | (0.08, 0.31) | 0.63 | 0 | 82.67 | 7.31 | 10.03 | 16,241 (40) |
Hospital Risk‐Standardized Readmission Rates
Risk‐standardized readmission rates varied across hospitals (Figure 3). Median risk‐standardized readmission rate was 17.3%, and the 25th and 75th percentiles were 16.9% and 17.9%, respectively. The 5th percentile was 16.0% and the 95th percentile was 19.1%. Odds of readmission for a hospital one standard deviation above average was 1.4 times that of a hospital one standard deviation below average.
Administrative Model Validation
In the remaining 50% of pneumonia index hospitalizations from 2006, and the entire 2005 cohort, regression coefficients and standard errors of model variables were similar to those in the development data set. Model performance using 2005 data was consistent with model performance using the 2006 development and validation half‐samples (Table 2).
Medical Record Validation
After exclusions, the medical record sample taken from the National Pneumonia Project included 47,429 cases, with an unadjusted 30‐day readmission rate of 17.0%. The final medical record risk‐adjustment model included a total of 35 variables, whose prevalence and association with readmission risk varied modestly (Table 3). Performance of the medical record and administrative models was similar (areas under the ROC curve 0.59 and 0.63, respectively) (Table 4). Additionally, in the administrative model, predicted readmission rates ranged from 8% in the lowest predicted decile to 30% in the highest predicted decile, while in the medical record model, the corresponding rates varied from 10% to 26%.
| Variable | Percent | Estimate | Standard Error | Odds Ratio | 95% CI | |
|---|---|---|---|---|---|---|
| ||||||
| Age 65, mean (SD) | 15.24 (7.87) | 0.003 | 0.002 | 0.997 | 0.993 | 1.000 |
| Male | 46.18 | 0.122 | 0.025 | 1.130 | 1.075 | 1.188 |
| Nursing home resident | 17.71 | 0.035 | 0.037 | 1.036 | 0.963 | 1.114 |
| Neoplastic disease | 6.80 | 0.130 | 0.049 | 1.139 | 1.034 | 1.254 |
| Liver disease | 1.04 | 0.089 | 0.123 | 0.915 | 0.719 | 1.164 |
| History of heart failure | 28.98 | 0.234 | 0.029 | 1.264 | 1.194 | 1.339 |
| History of renal disease | 8.51 | 0.188 | 0.047 | 1.206 | 1.100 | 1.323 |
| Altered mental status | 17.95 | 0.009 | 0.034 | 1.009 | 0.944 | 1.080 |
| Pleural effusion | 21.20 | 0.165 | 0.030 | 1.179 | 1.111 | 1.251 |
| BUN 30 mg/dl | 23.28 | 0.160 | 0.033 | 1.174 | 1.100 | 1.252 |
| BUN missing | 14.56 | 0.101 | 0.185 | 0.904 | 0.630 | 1.298 |
| Systolic BP <90 mmHg | 2.95 | 0.068 | 0.070 | 1.070 | 0.932 | 1.228 |
| Systolic BP missing | 11.21 | 0.149 | 0.425 | 1.160 | 0.504 | 2.669 |
| Pulse 125/min | 7.73 | 0.036 | 0.047 | 1.036 | 0.945 | 1.137 |
| Pulse missing | 11.22 | 0.210 | 0.405 | 1.234 | 0.558 | 2.729 |
| Respiratory rate 30/min | 16.38 | 0.079 | 0.034 | 1.082 | 1.012 | 1.157 |
| Respiratory rate missing | 11.39 | 0.204 | 0.240 | 1.226 | 0.765 | 1.964 |
| Sodium <130 mmol/L | 4.82 | 0.136 | 0.057 | 1.145 | 1.025 | 1.280 |
| Sodium missing | 14.39 | 0.049 | 0.143 | 1.050 | 0.793 | 1.391 |
| Glucose 250 mg/dl | 5.19 | 0.005 | 0.057 | 0.995 | 0.889 | 1.114 |
| Glucose missing | 15.44 | 0.156 | 0.105 | 0.855 | 0.696 | 1.051 |
| Hematocrit <30% | 7.77 | 0.270 | 0.044 | 1.310 | 1.202 | 1.428 |
| Hematocrit missing | 13.62 | 0.071 | 0.135 | 0.932 | 0.715 | 1.215 |
| Creatinine 2.5 mg/dL | 4.68 | 0.109 | 0.062 | 1.115 | 0.989 | 1.258 |
| Creatinine missing | 14.63 | 0.200 | 0.167 | 1.221 | 0.880 | 1.695 |
| WBC 6‐12 b/L | 38.04 | 0.021 | 0.049 | 0.979 | 0.889 | 1.079 |
| WBC >12 b/L | 41.45 | 0.068 | 0.049 | 0.934 | 0.848 | 1.029 |
| WBC missing | 12.85 | 0.167 | 0.162 | 1.181 | 0.860 | 1.623 |
| Immunosuppressive therapy | 15.01 | 0.347 | 0.035 | 1.415 | 1.321 | 1.516 |
| Chronic lung disease | 42.16 | 0.137 | 0.028 | 1.147 | 1.086 | 1.211 |
| Coronary artery disease | 39.57 | 0.150 | 0.028 | 1.162 | 1.100 | 1.227 |
| Diabetes mellitus | 20.90 | 0.137 | 0.033 | 1.147 | 1.076 | 1.223 |
| Alcohol/drug abuse | 3.40 | 0.099 | 0.071 | 0.906 | 0.788 | 1.041 |
| Dementia/Alzheimer's disease | 16.38 | 0.125 | 0.038 | 1.133 | 1.052 | 1.222 |
| Splenectomy | 0.44 | 0.016 | 0.186 | 1.016 | 0.706 | 1.463 |
| Model | Calibration (0, 1)* | Discrimination | Residuals Lack of Fit (Pearson Residual Fall %) | Model 2 (No. of Covariates) | ||||
|---|---|---|---|---|---|---|---|---|
| Predictive Ability (Lowest Decile, Highest Decile) | AUC | (<2) | (2, 0) | (0, 2) | (2+) | |||
| ||||||||
| Medical Record Model Development Sample (NP) | ||||||||
| N = 47,429 No. of 30‐day readmissions = 8,042 | (1, 0) | (0.10, 0.26) | 0.59 | 0 | 83.04 | 5.28 | 11.68 | 710 (35) |
| Linked Administrative Model Validation Sample | ||||||||
| N = 47,429 No. of 30‐day readmissions = 8,042 | (1, 0) | (0.08, 0.30) | 0.63 | 0 | 83.04 | 6.94 | 10.01 | 1,414 (40) |
The correlation coefficient of the estimated state‐specific standardized readmission rates from the administrative and medical record models was 0.96, and the proportion of the variance explained by the model was 0.92 (Figure 4).
DISCUSSION
We have described the development, validation, and results of a hospital, 30‐day, risk‐standardized readmission model for pneumonia that was created to support current federal transparency initiatives. The model uses administrative claims data from Medicare fee‐for‐service patients and produces results that are comparable to a model based on information obtained through manual abstraction of medical records. We observed an overall 30‐day readmission rate of 17%, and our analyses revealed substantial variation across US hospitals, suggesting that improvement by lower performing institutions is an achievable goal.
Because more than one in six pneumonia patients are rehospitalized shortly after discharge, and because pneumonia hospitalizations represent an enormous expense to the Medicare program, prevention of readmissions is now widely recognized to offer a substantial opportunity to improve patient outcomes while simultaneously lowering health care costs. Accordingly, promotion of strategies to reduce readmission rates has become a key priority for payers and quality‐improvement organizations. These range from policy‐level attempts to stimulate change, such as publicly reporting hospital readmission rates on government websites, to establishing accreditation standardssuch as the Joint Commission's requirement to accurately reconcile medications, to the creation of quality improvement collaboratives focused on sharing best practices across institutions. Regardless of the approach taken, a valid, risk‐adjusted measure of performance is required to evaluate and track performance over time. The measure we have described meets the National Quality Forum's measure evaluation criteria in that it addresses an important clinical topic for which there appears to be significant opportunities for improvement, the measure is precisely defined and has been subjected to validity and reliability testing, it is risk‐adjusted based on patient clinical factors present at the start of care, is feasible to produce, and is understandable by a broad range of potential users.21 Because hospitalists are the physicians primarily responsible for the care of patients with pneumonia at US hospitals, and because they frequently serve as the physician champions for quality improvement activities related to pneumonia, it is especially important that they maintain a thorough understanding of the measures and methodologies underlying current efforts to measure hospital performance.
Several features of our approach warrant additional comment. First, we deliberately chose to measure all readmission events rather than attempt to discriminate between potentially preventable and nonpreventable readmissions. From the patient perspective, readmission for any reason is a concern, and limiting the measure to pneumonia‐related readmissions could make it susceptible to gaming by hospitals. Moreover, determining whether a readmission is related to a potential quality problem is not straightforward. For example, a patient with pneumonia whose discharge medications were prescribed incorrectly may be readmitted with a hip fracture following an episode of syncope. It would be inappropriate to treat this readmission as unrelated to the care the patient received for pneumonia. Additionally, while our approach does not presume that every readmission is preventable, the goal is to reduce the risk of readmissions generally (not just in narrowly defined subpopulations), and successful interventions to reduce rehospitalization have typically demonstrated reductions in all‐cause readmission.9, 22 Second, deaths that occurred within 30 days of discharge, yet that were not accompanied by a hospital readmission, were not counted as a readmission outcome. While it may seem inappropriate to treat a postdischarge death as a nonevent (rather than censoring or excluding such cases), alternative analytic approaches, such as using a hierarchical survival model, are not currently computationally feasible with large national data sets. Fortunately, only a relatively small proportion of discharges fell into this category (5.2% of index cases in the 2006 development sample died within 30 days of discharge without being readmitted). An alternative approach to handling the competing outcome of death would have been to use a composite outcome of readmission or death. However, we believe that it is important to report the outcomes separately because factors that predict readmission and mortality may differ, and when making comparisons across hospitals it would not be possible to determine whether differences in rate were due to readmission or mortality. Third, while the patient‐level readmission model showed only modest discrimination, we intentionally excluded covariates such as race and socioeconomic status, as well as in‐hospital events and potential complications of care, and whether patients were discharged home or to a skilled nursing facility. While these variables could have improved predictive ability, they may be directly or indirectly related to quality or supply factors that should not be included in a model that seeks to control for patient clinical characteristics. For example, if hospitals with a large share of poor patients have higher readmission rates, then including income in the model will obscure differences that are important to identify. While we believe that the decision to exclude such factors in the model is in the best interest of patients, and supports efforts to reduce health inequality in society more generally, we also recognize that hospitals that care for a disproportionate share of poor patients are likely to require additional resources to overcome these social factors. Fourth, we limited the analysis to patients with a principal diagnosis of pneumonia, and chose not to also include those with a principal diagnosis of sepsis or respiratory failure coupled with a secondary diagnosis of pneumonia. While the broader definition is used by CMS in the National Pneumonia Project, that initiative relied on chart abstraction to differentiate pneumonia present at the time of admission from cases developing as a complication of hospitalization. Additionally, we did not attempt to differentiate between community‐acquired and healthcare‐associated pneumonia, however our approach is consistent with the National Pneumonia Project and Pneumonia Patient Outcomes Research Team.18 Fifth, while our model estimates readmission rates at the hospital level, we recognize that readmissions are influenced by a complex and extensive range of factors. In this context, greater cooperation between hospitals and other care providers will almost certainly be required in order to achieve dramatic improvement in readmission rates, which in turn will depend upon changes to the way serious illness is paid for. Some options that have recently been described include imposing financial penalties for early readmission, extending the boundaries of case‐based payment beyond hospital discharge, and bundling payments between hospitals and physicians.2325
Our measure has several limitations. First, our models were developed and validated using Medicare data, and the results may not apply to pneumonia patients less than 65 years of age. However, most patients hospitalized with pneumonia in the US are 65 or older. In addition, we were unable to test the model with a Medicare managed care population, because data are not currently available on such patients. Finally, the medical record‐based validation was conducted by state‐level analysis because the sample size was insufficient to carry this out at the hospital level.
In conclusion, more than 17% of Medicare beneficiaries are readmitted within 30 days following discharge after a hospitalization for pneumonia, and rates vary substantially across institutions. The development of a valid measure of hospital performance and public reporting are important first steps towards focusing attention on this problem. Actual improvement will now depend on whether hospitals and partner organizations are successful at identifying and implementing effective methods to prevent readmission.
- ,,.Rehospitalizations among patients in the Medicare Fee‐for‐Service Program.N Engl J Med.2009;360(14):1418–1428.
- Medicare Payment Advisory Commission.Report to the Congress: Promoting Greater Efficiency in Medicare.2007.
- ,,,,. HCUP Facts and Figures: Statistics on Hospital‐based Care in the United States, 2007.2009. Available at: http://www.hcup‐us.ahrq.gov/reports.jsp. Accessed November 7, 2009.
- Centers for Medicare 353(3):255–264.
- ,,,.Trends in postdischarge mortality and readmissions: has length of stay declined too far?Arch Intern Med.2004;164(5):538–544.
- ,,,.Short‐term outcomes and their predictors for patients hospitalized with community‐acquired pneumonia.Heart Lung.2004;33(5):301–307.
- ,,, et al.Improved clinical outcomes with utilization of a community‐acquired pneumonia guideline.Chest.2006;130(3):794–799.
- ,,,,.Associations between initial antimicrobial therapy and medical outcomes for hospitalized elderly patients with pneumonia.Arch Intern Med.1999;159(21):2562–2572.
- ,.Hospital readmissions as a measure of quality of health care: advantages and limitations.Arch Intern Med.2000;160(8):1074–1081.
- ,,,.The care transitions intervention: results of a randomized controlled trial.Arch Intern Med.2006;166(17):1822–1828.
- Corrigan JM, Eden J, Smith BM, eds.Leadership by Example: Coordinating Government Roles in Improving Health Care Quality. Committee on Enhancing Federal Healthcare Quality Programs.Washington, DC:National Academies Press,2003.
- Medicare.gov—Hospital Compare. Available at: http://www.hospitalcompare.hhs.gov/Hospital/Search/Welcome.asp?version=default1(1):29–37.
- ,,,,.Measuring performance for treating heart attacks and heart failure: the case for outcomes measurement.Health Aff.2007;26(1):75–85.
- NQF‐Endorsed® Standards. Available at: http://www.qualityforum.org/Measures_List.aspx. Accessed November 6,2009.
- ,,,,.Timing of antibiotic administration and outcomes for Medicare patients hospitalized with community‐acquired pneumonia.Arch Intern Med.2004;164(6):637–644.
- ,,. Diagnostic Cost Group Hierarchical Condition Category Models for Medicare Risk Adjustment. Report prepared for the Health Care Financing Administration. Health Economics Research, Inc;2000. Available at: http://www.cms.hhs.gov/Reports/Reports/ItemDetail.asp?ItemID=CMS023176. Accessed November 7, 2009.
- .Regression Modeling Strategies: With Applications to Linear Models, Logistic Regression, and Survival Analysis.1st ed.New York:Springer;2006.
- National Quality Forum—Measure Evaluation Criteria.2008. Available at: http://www.qualityforum.org/uploadedFiles/Quality_Forum/Measuring_Performance/Consensus_Development_Process%E2%80%99s_Principle/EvalCriteria2008–08‐28Final.pdf?n=4701.
- ,,, et al.Comprehensive discharge planning and home follow‐up of hospitalized elders: a randomized clinical trial.JAMA.1999;281(7):613–620.
- .Paying for care episodes and care coordination.N Engl J Med.2007;356(11):1166–1168.
- .Health care reform—toward more freedom, and responsibility, for physicians.N Engl J Med.2009;361(6):623–628.
- .Beyond pay for performance—emerging models of provider‐payment reform.N Engl J Med.2008;359(12):1197–1200.
- ,,.Rehospitalizations among patients in the Medicare Fee‐for‐Service Program.N Engl J Med.2009;360(14):1418–1428.
- Medicare Payment Advisory Commission.Report to the Congress: Promoting Greater Efficiency in Medicare.2007.
- ,,,,. HCUP Facts and Figures: Statistics on Hospital‐based Care in the United States, 2007.2009. Available at: http://www.hcup‐us.ahrq.gov/reports.jsp. Accessed November 7, 2009.
- Centers for Medicare 353(3):255–264.
- ,,,.Trends in postdischarge mortality and readmissions: has length of stay declined too far?Arch Intern Med.2004;164(5):538–544.
- ,,,.Short‐term outcomes and their predictors for patients hospitalized with community‐acquired pneumonia.Heart Lung.2004;33(5):301–307.
- ,,, et al.Improved clinical outcomes with utilization of a community‐acquired pneumonia guideline.Chest.2006;130(3):794–799.
- ,,,,.Associations between initial antimicrobial therapy and medical outcomes for hospitalized elderly patients with pneumonia.Arch Intern Med.1999;159(21):2562–2572.
- ,.Hospital readmissions as a measure of quality of health care: advantages and limitations.Arch Intern Med.2000;160(8):1074–1081.
- ,,,.The care transitions intervention: results of a randomized controlled trial.Arch Intern Med.2006;166(17):1822–1828.
- Corrigan JM, Eden J, Smith BM, eds.Leadership by Example: Coordinating Government Roles in Improving Health Care Quality. Committee on Enhancing Federal Healthcare Quality Programs.Washington, DC:National Academies Press,2003.
- Medicare.gov—Hospital Compare. Available at: http://www.hospitalcompare.hhs.gov/Hospital/Search/Welcome.asp?version=default1(1):29–37.
- ,,,,.Measuring performance for treating heart attacks and heart failure: the case for outcomes measurement.Health Aff.2007;26(1):75–85.
- NQF‐Endorsed® Standards. Available at: http://www.qualityforum.org/Measures_List.aspx. Accessed November 6,2009.
- ,,,,.Timing of antibiotic administration and outcomes for Medicare patients hospitalized with community‐acquired pneumonia.Arch Intern Med.2004;164(6):637–644.
- ,,. Diagnostic Cost Group Hierarchical Condition Category Models for Medicare Risk Adjustment. Report prepared for the Health Care Financing Administration. Health Economics Research, Inc;2000. Available at: http://www.cms.hhs.gov/Reports/Reports/ItemDetail.asp?ItemID=CMS023176. Accessed November 7, 2009.
- .Regression Modeling Strategies: With Applications to Linear Models, Logistic Regression, and Survival Analysis.1st ed.New York:Springer;2006.
- National Quality Forum—Measure Evaluation Criteria.2008. Available at: http://www.qualityforum.org/uploadedFiles/Quality_Forum/Measuring_Performance/Consensus_Development_Process%E2%80%99s_Principle/EvalCriteria2008–08‐28Final.pdf?n=4701.
- ,,, et al.Comprehensive discharge planning and home follow‐up of hospitalized elders: a randomized clinical trial.JAMA.1999;281(7):613–620.
- .Paying for care episodes and care coordination.N Engl J Med.2007;356(11):1166–1168.
- .Health care reform—toward more freedom, and responsibility, for physicians.N Engl J Med.2009;361(6):623–628.
- .Beyond pay for performance—emerging models of provider‐payment reform.N Engl J Med.2008;359(12):1197–1200.
Copyright © 2010 Society of Hospital Medicine
Turn to ACGME for Transfer, Resident Supervision Rules

I have doubts: Are there any guidelines about “bouncing” patients between teaching and nonteaching services in a teaching hospital?
Srikanth Seethala, MD
Pittsburgh
Dr. Hospitalist responds: Several thoughts came to my mind when I read your question. What did you mean by the term “bouncing”? When you refer to “nonteaching service,” are you referring to the cohort of inpatients in your teaching hospital cared for by attending physicians without the involvement of trainees? Of course, the most obvious question is what is causing your “doubt”?
As you may know, all U.S. postgraduate physician training programs are governed by the rules and standards set forth by the Accreditation Council for Graduate Medical Education (ACGME). You can find all of ACGME’s rules online at www.acgme.org. Regardless of whether you are a trainee or an attending physician, the ACGME expects the same interpretation and enforcement of their standards.
Our general medical service is divided into the resident-covered service and a separate, nonresident-covered service. Resident-covered service means IM residents are involved in the care of the patient under the supervision of an attending physician. No residents are involved in patient care on the nonresident-covered service. The development of our nonresident-covered service was clearly a product of ACGME duty-hour standards, which were originally enacted in 2003 and recently revised.
Our IM program has the same number of residents that we did before the new rules were put in place. Before 2003, we did not have a nonresident-covered medical service because we had a sufficient number of residents to care for all patients on our medical service. We found that the 2003 standards restricted the number of hours our residents could work in our hospital, so despite no change in the size of our medical service or the number of residents, we found ourselves without sufficient numbers of residents to meet the clinical demand. To meet this demand, we developed a hospitalist-run, nonresident-covered medical service.
We discussed a number of issues during the planning stages of our new service:
- How many hospitalist full-time equivalents (FTEs) would we need to staff this service?
- Would we have hospitalists physically in the hospital 24/7 or take call from outside the hospital?
- How much would it cost?
- Do we have two groups of hospitalist staff, one for the resident-covered service and a separate one for the nonresident-covered service? Or do we maintain one cohort of hospitalists and ask the staff to work on both the resident- and nonresident-covered services?
- Do we ask our hospitalists to rotate month by month or week by week, separately on the resident- and then the nonresident-covered service? Or do we ask hospitalists to see both patients on any given day?
- Do we geographically cohort our resident-covered patients on floors separate from our nonresident-covered patients?
The new rules fueled a lot of discussion between educators and trainees. Your question about the transfer of patients between resident- and nonresident-covered services does not surprise me. Some training programs tried to minimize the necessary number of attending level staff in the hospital by allowing trainees to “cross-cover,” or essentially care for patients on the nonresident-covered service, when the attending staff was not present in the hospital. It is my understanding that trainees are never allowed to cross-cover patients on the nonresident-covered service.
To my knowledge, however, there are no rules against transferring patients from the nonresident-covered service to the resident-covered service, or vice versa. TH
I have doubts: Are there any guidelines about “bouncing” patients between teaching and nonteaching services in a teaching hospital?
Srikanth Seethala, MD
Pittsburgh
Dr. Hospitalist responds: Several thoughts came to my mind when I read your question. What did you mean by the term “bouncing”? When you refer to “nonteaching service,” are you referring to the cohort of inpatients in your teaching hospital cared for by attending physicians without the involvement of trainees? Of course, the most obvious question is what is causing your “doubt”?
As you may know, all U.S. postgraduate physician training programs are governed by the rules and standards set forth by the Accreditation Council for Graduate Medical Education (ACGME). You can find all of ACGME’s rules online at www.acgme.org. Regardless of whether you are a trainee or an attending physician, the ACGME expects the same interpretation and enforcement of their standards.
Our general medical service is divided into the resident-covered service and a separate, nonresident-covered service. Resident-covered service means IM residents are involved in the care of the patient under the supervision of an attending physician. No residents are involved in patient care on the nonresident-covered service. The development of our nonresident-covered service was clearly a product of ACGME duty-hour standards, which were originally enacted in 2003 and recently revised.
Our IM program has the same number of residents that we did before the new rules were put in place. Before 2003, we did not have a nonresident-covered medical service because we had a sufficient number of residents to care for all patients on our medical service. We found that the 2003 standards restricted the number of hours our residents could work in our hospital, so despite no change in the size of our medical service or the number of residents, we found ourselves without sufficient numbers of residents to meet the clinical demand. To meet this demand, we developed a hospitalist-run, nonresident-covered medical service.
We discussed a number of issues during the planning stages of our new service:
- How many hospitalist full-time equivalents (FTEs) would we need to staff this service?
- Would we have hospitalists physically in the hospital 24/7 or take call from outside the hospital?
- How much would it cost?
- Do we have two groups of hospitalist staff, one for the resident-covered service and a separate one for the nonresident-covered service? Or do we maintain one cohort of hospitalists and ask the staff to work on both the resident- and nonresident-covered services?
- Do we ask our hospitalists to rotate month by month or week by week, separately on the resident- and then the nonresident-covered service? Or do we ask hospitalists to see both patients on any given day?
- Do we geographically cohort our resident-covered patients on floors separate from our nonresident-covered patients?
The new rules fueled a lot of discussion between educators and trainees. Your question about the transfer of patients between resident- and nonresident-covered services does not surprise me. Some training programs tried to minimize the necessary number of attending level staff in the hospital by allowing trainees to “cross-cover,” or essentially care for patients on the nonresident-covered service, when the attending staff was not present in the hospital. It is my understanding that trainees are never allowed to cross-cover patients on the nonresident-covered service.
To my knowledge, however, there are no rules against transferring patients from the nonresident-covered service to the resident-covered service, or vice versa. TH
I have doubts: Are there any guidelines about “bouncing” patients between teaching and nonteaching services in a teaching hospital?
Srikanth Seethala, MD
Pittsburgh
Dr. Hospitalist responds: Several thoughts came to my mind when I read your question. What did you mean by the term “bouncing”? When you refer to “nonteaching service,” are you referring to the cohort of inpatients in your teaching hospital cared for by attending physicians without the involvement of trainees? Of course, the most obvious question is what is causing your “doubt”?
As you may know, all U.S. postgraduate physician training programs are governed by the rules and standards set forth by the Accreditation Council for Graduate Medical Education (ACGME). You can find all of ACGME’s rules online at www.acgme.org. Regardless of whether you are a trainee or an attending physician, the ACGME expects the same interpretation and enforcement of their standards.
Our general medical service is divided into the resident-covered service and a separate, nonresident-covered service. Resident-covered service means IM residents are involved in the care of the patient under the supervision of an attending physician. No residents are involved in patient care on the nonresident-covered service. The development of our nonresident-covered service was clearly a product of ACGME duty-hour standards, which were originally enacted in 2003 and recently revised.
Our IM program has the same number of residents that we did before the new rules were put in place. Before 2003, we did not have a nonresident-covered medical service because we had a sufficient number of residents to care for all patients on our medical service. We found that the 2003 standards restricted the number of hours our residents could work in our hospital, so despite no change in the size of our medical service or the number of residents, we found ourselves without sufficient numbers of residents to meet the clinical demand. To meet this demand, we developed a hospitalist-run, nonresident-covered medical service.
We discussed a number of issues during the planning stages of our new service:
- How many hospitalist full-time equivalents (FTEs) would we need to staff this service?
- Would we have hospitalists physically in the hospital 24/7 or take call from outside the hospital?
- How much would it cost?
- Do we have two groups of hospitalist staff, one for the resident-covered service and a separate one for the nonresident-covered service? Or do we maintain one cohort of hospitalists and ask the staff to work on both the resident- and nonresident-covered services?
- Do we ask our hospitalists to rotate month by month or week by week, separately on the resident- and then the nonresident-covered service? Or do we ask hospitalists to see both patients on any given day?
- Do we geographically cohort our resident-covered patients on floors separate from our nonresident-covered patients?
The new rules fueled a lot of discussion between educators and trainees. Your question about the transfer of patients between resident- and nonresident-covered services does not surprise me. Some training programs tried to minimize the necessary number of attending level staff in the hospital by allowing trainees to “cross-cover,” or essentially care for patients on the nonresident-covered service, when the attending staff was not present in the hospital. It is my understanding that trainees are never allowed to cross-cover patients on the nonresident-covered service.
To my knowledge, however, there are no rules against transferring patients from the nonresident-covered service to the resident-covered service, or vice versa. TH
FPHM: A License to Drive Change

I was musing one morning about my day ahead. I was doing one of those subcortical activities of daily living in which the mind can wander freely. Have you ever jumped in the car with the intention of stopping at the store on the way home, only to find yourself pulling into your driveway after spending the drive contemplating those issues on your plate that day? You drive home on autopilot. It occurred to me that it can happen in much the same way in our daily practice of medicine—how easy it is to slip into autopilot when admitting patients and doing our daily rounds.
During this particular morning, I mulled over many things: the translocation between chromosomes 9 and 22 in CML, the obstructive PFTs one generally sees in cadmium exposure, debating whether to give corticosteroids or to induce delivery in a 33-week pregnant woman with HELLP syndrome. Maybe you’re wondering: Am I a physician practicing in a remote rural area that has no access to oncologists? Do I practice in an underserved industrial town next to an old battery factory? Or am I an old-fashioned GP who still delivers babies?
No, no, and no. I am a board-certified internist who was preparing for my Maintenance of Certification (MOC) examination.
A New Way of Thinking
I am 42 years old, I have a busy medical practice, I am the medical director of the 14-person HM group at Wentworth-Douglass Hospital in Dover, N.H., and I am the mother of two children, ages 9 and 11. And I found myself, on top of all these things, a student, too.
I’ve been practicing medicine for 11 years. I’ve gone from practicing primary care in a small community in Maine to working at a larger community medical center in New Hampshire, becoming a hospitalist in 2005, then taking on the job of director of my hospitalist group in 2006. With more than a decade of experience under my belt, I felt I had the depth of knowledge experience brings.
However, as I traveled through the process of preparing for the American Board of Internal Medicine’s (ABIM’s) new Focused Practice in Hospital Medicine (FPHM) secure exam, it began to dawn on me: Medicine is a complicated profession that not only requires careful attention to the details of every case, it also demands it. In order to avoid the pitfall of practicing distracted medicine, we must carefully foster our own continuing education.
Going through the studying process has enabled me to think about the medicine I practice in a much more academic way. True, I don’t necessarily need to know some of the things I’ve encountered in my study sessions for my everyday practice, but I find myself spouting off random facts to anyone who will listen—colleagues, nurses, even patients. “Did you know that only about one-fourth of crystalloid remains in the intravascular space, where the rest goes into the tissues?” “If the triglycerides in this fluid are greater than 115, this is a chylothorax!” I’m paying attention again to the theory and pathophysiology behind medical illnesses, not just to the drudgery of writing routine orders or checking off boxes on a protocol.
It has not been easy. Although I’ve known I needed to recertify in internal medicine since I took the exam the first time, I did not actively start looking into the exam and preparing until about a year and a half before my exam, when I talked to a colleague who had already started preparing. That’s when I learned that this was not only an exam, but also a process. This process is intended by the board to be an active part of maintaining certification during the 10 years before it is due again, not just to be crammed into the last year or two before certification expires. I recommend to anyone going through this process to familiarize yourself with the ABIM website (www.abim.org). Initially, it was a little unwieldy to maneuver around the site, and it wasn’t entirely clear to me what exactly was needed to recertify until I spent some time maneuvering through the site.
HM-Focused Pathway
To add to this, at around the time I was getting ready to register for the exam, it was announced that this would be the first year ABIM would be offering the FPHM pathway, which is designed to recognize those of us who concentrate our practices on hospital medicine. This to me was an excellent opportunity to recertify in a field in which I actively practice, hopefully making the exam more applicable to what I do, but the flip side was that no one would have taken this particular exam before. Admittedly, when I first signed up, I felt like I was either a guinea pig or a pioneer.
To obtain the FPHM, one must do not one but two projects requiring turning in data on process-improvement projects. Hospitalists who intend to certify with the FPHM will be well served by participating in safety, quality, and process-improvement projects, as we often already do. These projects can be used to complete the required Practice Improvement Modules.
Furthermore, I found that doing such projects is the best way to prepare for the new content, which deals specifically with HM on the actual exam. The internal-medicine topics were covered, just as they are in the nonfocused exam, and anyone who reviews for the exam with available study aids (e.g. review books, courses, or practice questions) will have adequate exposure to these topics.
However, a colleague in my HM group chose not to take the focused practice exam, largely because there was no previously established review material to use as study aids. I anticipate that future study aids will contain references to these questions, but for now I felt that material was adequately covered just by completing the Practice Improvement Modules and by being involved in process improvement projects at my hospital. In fact, attendance at one Institute for Healthcare Improvement (www.ihi.org/IHI/Programs) conference would probably cover the topics nicely.
To all of my colleages considering the MOC in FPHM exam, I wish you luck. I feel that any practicing hospitalist is likely to be able to satisfy the requirements of the FPHM pathway without doing too much more than they would in their daily practice or their usual exam preparation. I also found the ABIM staff useful and helpful, and recommend you use the “contact ABIM” link on their website with any questions.
Focused practice is exactly what we should be driving for. TH
Dr. Ammann is medical director of the hospital medicine division at Wentworth-Douglass Hospital in Dover, N.H.
I was musing one morning about my day ahead. I was doing one of those subcortical activities of daily living in which the mind can wander freely. Have you ever jumped in the car with the intention of stopping at the store on the way home, only to find yourself pulling into your driveway after spending the drive contemplating those issues on your plate that day? You drive home on autopilot. It occurred to me that it can happen in much the same way in our daily practice of medicine—how easy it is to slip into autopilot when admitting patients and doing our daily rounds.
During this particular morning, I mulled over many things: the translocation between chromosomes 9 and 22 in CML, the obstructive PFTs one generally sees in cadmium exposure, debating whether to give corticosteroids or to induce delivery in a 33-week pregnant woman with HELLP syndrome. Maybe you’re wondering: Am I a physician practicing in a remote rural area that has no access to oncologists? Do I practice in an underserved industrial town next to an old battery factory? Or am I an old-fashioned GP who still delivers babies?
No, no, and no. I am a board-certified internist who was preparing for my Maintenance of Certification (MOC) examination.
A New Way of Thinking
I am 42 years old, I have a busy medical practice, I am the medical director of the 14-person HM group at Wentworth-Douglass Hospital in Dover, N.H., and I am the mother of two children, ages 9 and 11. And I found myself, on top of all these things, a student, too.
I’ve been practicing medicine for 11 years. I’ve gone from practicing primary care in a small community in Maine to working at a larger community medical center in New Hampshire, becoming a hospitalist in 2005, then taking on the job of director of my hospitalist group in 2006. With more than a decade of experience under my belt, I felt I had the depth of knowledge experience brings.
However, as I traveled through the process of preparing for the American Board of Internal Medicine’s (ABIM’s) new Focused Practice in Hospital Medicine (FPHM) secure exam, it began to dawn on me: Medicine is a complicated profession that not only requires careful attention to the details of every case, it also demands it. In order to avoid the pitfall of practicing distracted medicine, we must carefully foster our own continuing education.
Going through the studying process has enabled me to think about the medicine I practice in a much more academic way. True, I don’t necessarily need to know some of the things I’ve encountered in my study sessions for my everyday practice, but I find myself spouting off random facts to anyone who will listen—colleagues, nurses, even patients. “Did you know that only about one-fourth of crystalloid remains in the intravascular space, where the rest goes into the tissues?” “If the triglycerides in this fluid are greater than 115, this is a chylothorax!” I’m paying attention again to the theory and pathophysiology behind medical illnesses, not just to the drudgery of writing routine orders or checking off boxes on a protocol.
It has not been easy. Although I’ve known I needed to recertify in internal medicine since I took the exam the first time, I did not actively start looking into the exam and preparing until about a year and a half before my exam, when I talked to a colleague who had already started preparing. That’s when I learned that this was not only an exam, but also a process. This process is intended by the board to be an active part of maintaining certification during the 10 years before it is due again, not just to be crammed into the last year or two before certification expires. I recommend to anyone going through this process to familiarize yourself with the ABIM website (www.abim.org). Initially, it was a little unwieldy to maneuver around the site, and it wasn’t entirely clear to me what exactly was needed to recertify until I spent some time maneuvering through the site.
HM-Focused Pathway
To add to this, at around the time I was getting ready to register for the exam, it was announced that this would be the first year ABIM would be offering the FPHM pathway, which is designed to recognize those of us who concentrate our practices on hospital medicine. This to me was an excellent opportunity to recertify in a field in which I actively practice, hopefully making the exam more applicable to what I do, but the flip side was that no one would have taken this particular exam before. Admittedly, when I first signed up, I felt like I was either a guinea pig or a pioneer.
To obtain the FPHM, one must do not one but two projects requiring turning in data on process-improvement projects. Hospitalists who intend to certify with the FPHM will be well served by participating in safety, quality, and process-improvement projects, as we often already do. These projects can be used to complete the required Practice Improvement Modules.
Furthermore, I found that doing such projects is the best way to prepare for the new content, which deals specifically with HM on the actual exam. The internal-medicine topics were covered, just as they are in the nonfocused exam, and anyone who reviews for the exam with available study aids (e.g. review books, courses, or practice questions) will have adequate exposure to these topics.
However, a colleague in my HM group chose not to take the focused practice exam, largely because there was no previously established review material to use as study aids. I anticipate that future study aids will contain references to these questions, but for now I felt that material was adequately covered just by completing the Practice Improvement Modules and by being involved in process improvement projects at my hospital. In fact, attendance at one Institute for Healthcare Improvement (www.ihi.org/IHI/Programs) conference would probably cover the topics nicely.
To all of my colleages considering the MOC in FPHM exam, I wish you luck. I feel that any practicing hospitalist is likely to be able to satisfy the requirements of the FPHM pathway without doing too much more than they would in their daily practice or their usual exam preparation. I also found the ABIM staff useful and helpful, and recommend you use the “contact ABIM” link on their website with any questions.
Focused practice is exactly what we should be driving for. TH
Dr. Ammann is medical director of the hospital medicine division at Wentworth-Douglass Hospital in Dover, N.H.
I was musing one morning about my day ahead. I was doing one of those subcortical activities of daily living in which the mind can wander freely. Have you ever jumped in the car with the intention of stopping at the store on the way home, only to find yourself pulling into your driveway after spending the drive contemplating those issues on your plate that day? You drive home on autopilot. It occurred to me that it can happen in much the same way in our daily practice of medicine—how easy it is to slip into autopilot when admitting patients and doing our daily rounds.
During this particular morning, I mulled over many things: the translocation between chromosomes 9 and 22 in CML, the obstructive PFTs one generally sees in cadmium exposure, debating whether to give corticosteroids or to induce delivery in a 33-week pregnant woman with HELLP syndrome. Maybe you’re wondering: Am I a physician practicing in a remote rural area that has no access to oncologists? Do I practice in an underserved industrial town next to an old battery factory? Or am I an old-fashioned GP who still delivers babies?
No, no, and no. I am a board-certified internist who was preparing for my Maintenance of Certification (MOC) examination.
A New Way of Thinking
I am 42 years old, I have a busy medical practice, I am the medical director of the 14-person HM group at Wentworth-Douglass Hospital in Dover, N.H., and I am the mother of two children, ages 9 and 11. And I found myself, on top of all these things, a student, too.
I’ve been practicing medicine for 11 years. I’ve gone from practicing primary care in a small community in Maine to working at a larger community medical center in New Hampshire, becoming a hospitalist in 2005, then taking on the job of director of my hospitalist group in 2006. With more than a decade of experience under my belt, I felt I had the depth of knowledge experience brings.
However, as I traveled through the process of preparing for the American Board of Internal Medicine’s (ABIM’s) new Focused Practice in Hospital Medicine (FPHM) secure exam, it began to dawn on me: Medicine is a complicated profession that not only requires careful attention to the details of every case, it also demands it. In order to avoid the pitfall of practicing distracted medicine, we must carefully foster our own continuing education.
Going through the studying process has enabled me to think about the medicine I practice in a much more academic way. True, I don’t necessarily need to know some of the things I’ve encountered in my study sessions for my everyday practice, but I find myself spouting off random facts to anyone who will listen—colleagues, nurses, even patients. “Did you know that only about one-fourth of crystalloid remains in the intravascular space, where the rest goes into the tissues?” “If the triglycerides in this fluid are greater than 115, this is a chylothorax!” I’m paying attention again to the theory and pathophysiology behind medical illnesses, not just to the drudgery of writing routine orders or checking off boxes on a protocol.
It has not been easy. Although I’ve known I needed to recertify in internal medicine since I took the exam the first time, I did not actively start looking into the exam and preparing until about a year and a half before my exam, when I talked to a colleague who had already started preparing. That’s when I learned that this was not only an exam, but also a process. This process is intended by the board to be an active part of maintaining certification during the 10 years before it is due again, not just to be crammed into the last year or two before certification expires. I recommend to anyone going through this process to familiarize yourself with the ABIM website (www.abim.org). Initially, it was a little unwieldy to maneuver around the site, and it wasn’t entirely clear to me what exactly was needed to recertify until I spent some time maneuvering through the site.
HM-Focused Pathway
To add to this, at around the time I was getting ready to register for the exam, it was announced that this would be the first year ABIM would be offering the FPHM pathway, which is designed to recognize those of us who concentrate our practices on hospital medicine. This to me was an excellent opportunity to recertify in a field in which I actively practice, hopefully making the exam more applicable to what I do, but the flip side was that no one would have taken this particular exam before. Admittedly, when I first signed up, I felt like I was either a guinea pig or a pioneer.
To obtain the FPHM, one must do not one but two projects requiring turning in data on process-improvement projects. Hospitalists who intend to certify with the FPHM will be well served by participating in safety, quality, and process-improvement projects, as we often already do. These projects can be used to complete the required Practice Improvement Modules.
Furthermore, I found that doing such projects is the best way to prepare for the new content, which deals specifically with HM on the actual exam. The internal-medicine topics were covered, just as they are in the nonfocused exam, and anyone who reviews for the exam with available study aids (e.g. review books, courses, or practice questions) will have adequate exposure to these topics.
However, a colleague in my HM group chose not to take the focused practice exam, largely because there was no previously established review material to use as study aids. I anticipate that future study aids will contain references to these questions, but for now I felt that material was adequately covered just by completing the Practice Improvement Modules and by being involved in process improvement projects at my hospital. In fact, attendance at one Institute for Healthcare Improvement (www.ihi.org/IHI/Programs) conference would probably cover the topics nicely.
To all of my colleages considering the MOC in FPHM exam, I wish you luck. I feel that any practicing hospitalist is likely to be able to satisfy the requirements of the FPHM pathway without doing too much more than they would in their daily practice or their usual exam preparation. I also found the ABIM staff useful and helpful, and recommend you use the “contact ABIM” link on their website with any questions.
Focused practice is exactly what we should be driving for. TH
Dr. Ammann is medical director of the hospital medicine division at Wentworth-Douglass Hospital in Dover, N.H.