User login
Rapid Bedside Diagnosis of Shock
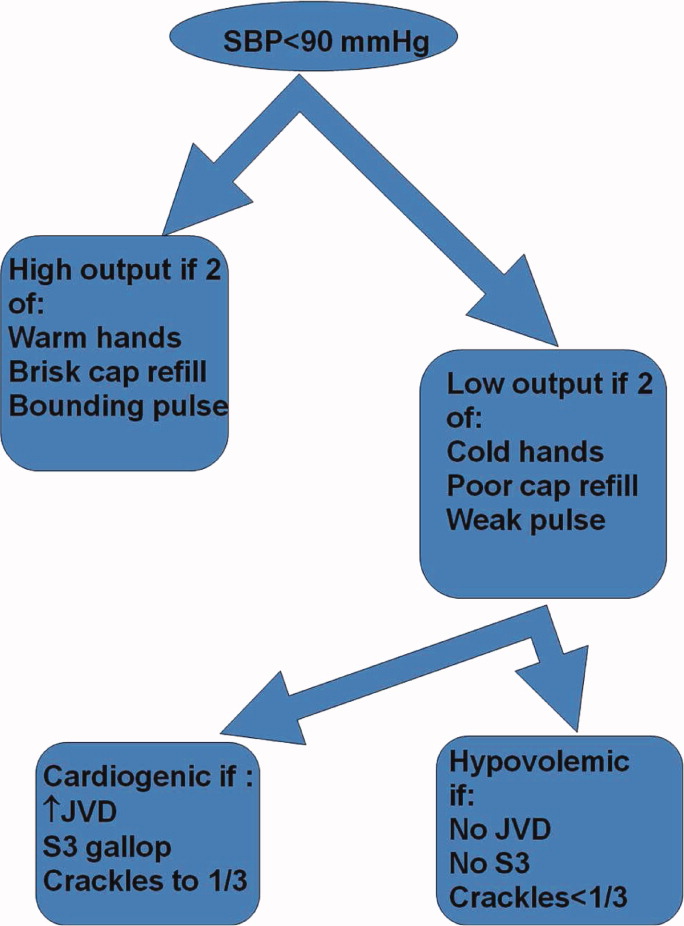
Shock has been defined as failure to deliver and/or utilize adequate amounts of oxygen1 and is a common cause of critical illness. Few studies have examined the predictive utility of bedside clinical examination in predicting the category of shock. Scholars have suggested a bedside approach that uses simple examination techniques and applied physiology to rapidly identify a patients' circulation as high vs. low cardiac output. Those with a high‐output examination are designated as high‐output, most often septic shock. Low‐output patients are further categorized as heart full or heart empty to distinguish cardiogenic from hypovolemic categories of shock, respectively.2 The predictive characteristics of this simple algorithm have not been studied. In this study, we examine the operating characteristics of selected elements of this algorithm when administered at the bedside by trainees in Internal Medicine.
Methods
This study was performed after approval of the Institutional Review Board; informed consent was waived. Patients with nonsurgical problems who present to the hospital or who develop sustained hypotension are managed by medical house officers on the intensive care and/or rapid response team with the supervision of patients' attending physicians. All house officers were asked to document explicitly in their assessment notes the following examination findings: finger capillary refill (same/quicker vs. slower than examiner's), hand skin temperature (same/warmer vs. cooler than examiner's) and pulse pressure (ie, same/wider vs. thinner than examiner's), presence or absence of crackles >1/3 from base on bilateral lung examination and jugular venous pressure (JVP) vs. 8 cmH2O. The documented examinations of either the rapid response team (PGY2; n = 14) or intensive care unit (ICU) resident (PGY3; n = 14) for patients evaluated between September 2008 and February 2009 were used for this study. Resuscitation was administered entirely by house officers, occasionally guided in person, but always supervised by attending physicians.
In May 2009, clinical data, including electrocardiograms/echocardiograms and laboratory (eg, cardiac enzymes, culture) results were abstracted from medical records of subjects. These were presented to a blinded senior clinician (DK) to review and apply evidence‐based or consensus criteria,36 whenever possible, to categorize the type of shock (septic vs. cardiogenic vs. hypovolemic) based on data acquired after the onset of shock. For example, patients with microbiologic and/or radiologic evidence of infection were classified as septic shock,1, 3, 4 those with acute left or right ventricular dysfunction on echocardiogram were classified as cardiogenic shock,1, 6 and those with clinical evidence of acute hemorrhage with hypovolemic shock.1, 5 While some of the patients were examined by DK as part of clinical care, he was blinded to the identity of patients and their algorithm‐related physical examination findings when he reviewed the abstracted data (>2 months after study closure) to adjudicate the final diagnosis of shock. These diagnoses were considered the reference standard for this study. The operating characteristics (sensitivity = true positive/true positive + false negative; specificity = true negative/true negative + false positive; negative predictive value (NPV) = true negative/all negatives; positive predictive value (PPV) = true positive/all positives; accuracy = true results/all results) were calculated for combinations of physical examination findings and correct final diagnosis (Figure 1).
Results
A total of 68 patients, averaging 71 16 years, were studied; 57% were male, and 66% were White, and 20% were Black. Table 1 lists characteristics of patients. A total of 37 patients were diagnosed as having septic shock, 11 had cardiogenic shock and 10 hypovolemic shock. Operating characteristics of the bedside examination techniques for predicting mechanism of shock are listed in Table 2. Capillary refill and skin temperature were 100% concordant yielding sensitivity of 89% (95% confidence interval [CI], 75‐97%), specificity of 68% (95% CI, 46‐83%), PPV of 77% (95% CI, 61‐88%), NPV of 84% (95% CI, 64‐96%) and overall accuracy of 79% (95% CI, 68‐88%) for diagnosis of high output (ie, septic shock). JVP 8 cmH2O was more accurate than crackles for predicting cardiogenic shock in low‐output patients with sensitivity of 82% (95% CI, 48‐98%), specificity of 79% (95% CI, 41‐95%), PPV of 75% (95% CI, 43‐95%), NPV of 85% (95% CI, 55‐98%), and overall accuracy of 80% (95%CI, 59‐93%). Using just skin temperature and JVP, the bedside approach misdiagnosed 19 of 75 cases (overall accuracy, 75%; 95% CI, 16‐37%).
| n Total | |
|---|---|
| |
| Gender, n (%) | n = 68 |
| Male | 39 (57) |
| Age, years | 71 16 |
| Race, n (%) | |
| White | 45 (66) |
| Black | 15 (22) |
| Hispanic | 7 (10) |
| Other | 1 (2) |
| High output, n (%) | n = 37 |
| Sepsis | |
| Pneumonia | 10 (27) |
| Urinary tract | 17 (46) |
| Skin | 3 (8) |
| Gastrointestinal | 5 (14) |
| Non‐infectious SIRS | 2 (5) |
| Low output heart full, n (%) | n = 18 |
| Pulmonary embolism | 3 (16) |
| AMI | 7 (40) |
| Cardiomyopathy | 5 (28) |
| Rhythm disturbance | 3 (16) |
| Low output heart empty, n (%) | n = 13 |
| Hemorrhagic | 9 (70) |
| NPO | 1 (7) |
| Diarrhea | 2 (14) |
| Adrenal crisis | 1 (7) |
| Prediction of SIRS | Capillary Refill Same/Faster (%) | Skin Same/ Warm (%) | Bounding Pulses (%) |
|---|---|---|---|
| |||
| Sensitivity | 89 | 89 | 65 |
| Specificity | 68 | 68 | 74 |
| Accuracy | 79 | 79 | 69 |
| Prediction of SIRS | Capillary Refill Same/Faster + Warm Skin + Bounding Pulse (%) | Capillary Refill Same/Faster + Warm Skin (%) | Any Other Combination of 2 (%) |
| Sensitivity | 62 | 89 | 62 |
| Specificity | 74 | 68 | 74 |
| Accuracy | 67 | 79 | 67 |
| Prediction of Cardiogenic | JVP (%) | Crackles (%) | JVP + Crackles (%) |
| Sensitivity | 82 | 55 | 55 |
| Specificity | 79 | 71 | 100 |
| Accuracy | 80 | 64 | 80 |
Discussion
This is the first study to examine the predictive characteristics of simple bedside physical examination techniques in correctly predicting the category/mechanism of shock. Overall, the algorithm performed well, and accurately predicted the category of shock in three‐quarters of patients. It also has the benefit of being very rapid, taking only seconds to complete, using bedside techniques that even inexperienced clinicians can apply.
Very few studies have examined the accuracy of examination techniques specifically for diagnosis of shock. In humans injected with endotoxin, body temperature and cardiac output increased, but skin temperature and capillary refill times are not well described.79 Schriger and Baraff10 reported that capillary refill >2 seconds was only 59% sensitive for diagnosing hypovolemia in patients with hypovolemic shock or orthostatic changes in blood pressure. Sensitivity was 77% in 13 patients with hypovolemic shock.10 However, some studies have demonstrated that age, sex, external temperature11 and fever12 can affect capillary refill times. Otieno et al.13 demonstrated a kappa statistic value of 0.49 for capillary refill 4 seconds, suggesting that reproducibility of this technique could be a major drawback. McGee et al.14 reviewed examination techniques for diagnosing hypovolemic states and concluded that postural changes in heart rate and blood pressure were the most accurate; capillary refill was not recommended. Stevenson and Perloff15 demonstrated that crackles and elevated JVP were absent in 18 of 43 patients with pulmonary capillary wedge pressures >22 mmHg. Butman et al.16 showed that elevated JVP was 82% accurate for predicting a wedge pressure >18 mmHg. Connors et al.17 demonstrated that clinicians' predictions of heart filling pressures and cardiac output were accurate (relative to pulmonary artery catheter measurements) in less than 50% of cases, though the examination techniques used were not qualified or quantified. No previous study has combined simple, semiobjective physical examination techniques for the purpose of distinguishing categories of shock.
Since identification of the pathogenesis of shock has important treatment/prognostic implications (eg, fluid and vasopressor therapies, early search for drainable focus of infection in sepsis, reestablishing vessel patency in myocardial infarction and pulmonary embolus), we believe that this simple, rapidly administered algorithm will prove useful in clinical medicine. In some clinical situations, the approach can lead to timely identification of the causative mechanism, allowing prompt definitive treatment. For example, a patient presenting with high‐output hypotension is so often sepsis/septic shock that treatment with antibiotics is justified (since success is time‐sensitive) even when the exact site/microbe has not yet been identified. Acute right heart overfilled low‐output hypotension should be considered pulmonary embolism (which also requires time‐sensitive therapies) until proven otherwise. Yet, a sizeable number of cases do not fit neatly into a single category. For example, 11% of patients with septic shock presented with cool extremities in the early phases of illness. In clinical decision‐making, 2 diagnostic‐therapeutic paradigms are common. In the first, the diagnosis is relatively certain and narrowly‐directed, mechanism‐specific treatment is appropriate. The second paradigm is 1 of significant uncertainty, when clinicians must treat empirically the most likely causes until more data become available to permit safe narrowing of therapies. For example, a patient presenting with hypotension, cool extremities, leukocytosis and apparent pneumonia should be treated empirically for septic shock while exploring explanations for the incongruous low‐output state (eg, profound hypovolemia, adrenal insufficiency, concurrent or precedent myocardial dysfunction). Patients often have several mechanisms contributing to hypotension. Since patients are not ideal forms, there can be no perfect decision‐tool; clinicians would be fool‐hardy to prematurely close decision‐making prior to definitive diagnosis. In the case of shock, such diagnostic arrogance would delay time‐sensitive therapies and thus contribute to morbidity and mortality. Nonetheless, this physical examination algorithmunderstanding its operating characteristics and limitationsmay add to the bedside clinician's diagnostic armamentarium.
Our study has several notable limitations. First, bedside examinations were performed by multiple observers who had limited (1 electronic mail) instruction on how to perform and document the data gathered for this study. So these results should be generalized cautiously until reproduced at other centers with greater numbers of observers (than the 28 of this study). The central supposition, that skin cooler, capillary refill longer, and pulse pressure more narrow than theirs, presupposes reasonable homogeneity of the normal state which is not necessarily true.11 Interobserver variability of physical examination further compromises the fidelity of findings recorded for this study.13 Since we conducted a retrospective review, and because of the emergency nature of the clinical problem, it would be difficult to conduct a study in which multiple examiners performed the same physical examinations to quantify interobserver variability. Irrespective, we would expect interobserver variability to systematically reduce accuracy; it is all‐the‐more impressive that trainees' examination results correctly diagnosed mechanism of shock in three‐quarters of cases. Also, examiners were not blinded to clinical history, so results of their examination could have been biased by their pre‐examination hypotheses of pathogenesis. Of course, they were not aware of the expert's final categorization of mechanism performed much later in time. Since there is no absolute reference standard for classification of the pathogenesis of shock, we depended upon careful review of selected data (same parameters for each patient) by a single senior investigatoralbeit armed with evidence‐based or consensus‐based standards of diagnosing shock. Finally, it can be argued that all forms of shock are mixed (with hypovolemia) early in the course; sepsis requires refilling of a leaky and dilated vasculature and the noncompliant ischemic ventricle often requires a higher filling pressure than normal to empty. To complicate even more, patients may have preexistent conditions (eg, chronic congestive heart failure, cirrhosis) that limit cardiovascular responses to acute shock. Our diagnostic approach was to identify the principal cause of the acute decompensation, assuming that many patients will have more than 1 single mechanism accounting for hypotension.
In conclusion, this is the first study to examine the utility of this simple physical examination algorithm to diagnose the mechanism of shock. Some have discounted or underemphasized examination techniques in favor of more time‐intensive and labor‐intensive diagnostic modalities, such as bedside echocardiography, which may waste precious time and resources. The simple physical examination algorithm assessed in this study has favorable operating characteristics and can be performed readily by even novice clinicians. If replicated at other centers and by greater numbers of observers, this approach could assist clinicians and teachers who train clinicians to rapidly diagnose and manage patients with shock.
- ,,, et al.Hemodynamic monitoring in shock and implications for management. International consensus conference. Paris, France, 27–28 April 2006.Intensive Care Med.2007;33:575–590.
- .The pathophysiology of the circulation in critical illness. In:Principles of Critical Care.New York:McGraw Hill;2005.
- ,,, et al.2001 SCCM/ESICM/ACCP/ATS/SIS. International Sepsis Definitions Conference.Crit Care Med.2003;31:1250–1256.
- ,,, et al.Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis.Chest.1992;101:1644–1655;
- ,,, et al.Resuscitation from severe hemorrhage.Crit Care Med.1996;24(2 Suppl):S12–S23.
- ,.Cardiogenic shock: current concepts and improving outcomes circulation.Circulation.2008;117:686–697.
- ,,, et al.The cardiovascular response of normal humans to the administration of endotoxin.N Engl J Med.1989;321:280–287.
- ,,,,,.Experimental endotoxemia in humans: análysis of cytokine release and coagulation, fibrinolytic, an complement pathways.Blood.1990;76:2520–2526.
- ,,,,.Peripheral resistance changes during shock in man.Angiology.1968;19:268–276.
- ,.Capillary refill—is it a useful predictor of hypovolemic states?Ann Emerg Med.1991;20:601–605.
- ,.Defining normal capillary refill: variation with age, sex, and temperature.Ann Emerg Med.1988;17:113–116.
- ,,,.Effect of fever on capillary refill time.Pediatr Emerg Care.1997;13:305–307.
- ,,,,,.Are bedside features of shock reproducible between different observers?Arch Dis Child.2004;89:977–979.
- ,,.Is this patient hypovolemic?JAMA.1999;281:1022–1029.
- ,.The limited reliability of physical signs for estimating hemodynamics in chronic heart failure.JAMA.1989;261:884–888.
- ,,,,.Bedside cardiovascular examination in patients with severe chronic heart failure: importance of rest or inducible jugular venous distension.J Am Coll Cardiol.1993;22:968–974.
- ,,.Evaluation of right‐heart catheterization in the critically ill patients without acute myocardial infarction.N Engl J Med.1983;308(5):263–267.
Shock has been defined as failure to deliver and/or utilize adequate amounts of oxygen1 and is a common cause of critical illness. Few studies have examined the predictive utility of bedside clinical examination in predicting the category of shock. Scholars have suggested a bedside approach that uses simple examination techniques and applied physiology to rapidly identify a patients' circulation as high vs. low cardiac output. Those with a high‐output examination are designated as high‐output, most often septic shock. Low‐output patients are further categorized as heart full or heart empty to distinguish cardiogenic from hypovolemic categories of shock, respectively.2 The predictive characteristics of this simple algorithm have not been studied. In this study, we examine the operating characteristics of selected elements of this algorithm when administered at the bedside by trainees in Internal Medicine.
Methods
This study was performed after approval of the Institutional Review Board; informed consent was waived. Patients with nonsurgical problems who present to the hospital or who develop sustained hypotension are managed by medical house officers on the intensive care and/or rapid response team with the supervision of patients' attending physicians. All house officers were asked to document explicitly in their assessment notes the following examination findings: finger capillary refill (same/quicker vs. slower than examiner's), hand skin temperature (same/warmer vs. cooler than examiner's) and pulse pressure (ie, same/wider vs. thinner than examiner's), presence or absence of crackles >1/3 from base on bilateral lung examination and jugular venous pressure (JVP) vs. 8 cmH2O. The documented examinations of either the rapid response team (PGY2; n = 14) or intensive care unit (ICU) resident (PGY3; n = 14) for patients evaluated between September 2008 and February 2009 were used for this study. Resuscitation was administered entirely by house officers, occasionally guided in person, but always supervised by attending physicians.
In May 2009, clinical data, including electrocardiograms/echocardiograms and laboratory (eg, cardiac enzymes, culture) results were abstracted from medical records of subjects. These were presented to a blinded senior clinician (DK) to review and apply evidence‐based or consensus criteria,36 whenever possible, to categorize the type of shock (septic vs. cardiogenic vs. hypovolemic) based on data acquired after the onset of shock. For example, patients with microbiologic and/or radiologic evidence of infection were classified as septic shock,1, 3, 4 those with acute left or right ventricular dysfunction on echocardiogram were classified as cardiogenic shock,1, 6 and those with clinical evidence of acute hemorrhage with hypovolemic shock.1, 5 While some of the patients were examined by DK as part of clinical care, he was blinded to the identity of patients and their algorithm‐related physical examination findings when he reviewed the abstracted data (>2 months after study closure) to adjudicate the final diagnosis of shock. These diagnoses were considered the reference standard for this study. The operating characteristics (sensitivity = true positive/true positive + false negative; specificity = true negative/true negative + false positive; negative predictive value (NPV) = true negative/all negatives; positive predictive value (PPV) = true positive/all positives; accuracy = true results/all results) were calculated for combinations of physical examination findings and correct final diagnosis (Figure 1).
Results
A total of 68 patients, averaging 71 16 years, were studied; 57% were male, and 66% were White, and 20% were Black. Table 1 lists characteristics of patients. A total of 37 patients were diagnosed as having septic shock, 11 had cardiogenic shock and 10 hypovolemic shock. Operating characteristics of the bedside examination techniques for predicting mechanism of shock are listed in Table 2. Capillary refill and skin temperature were 100% concordant yielding sensitivity of 89% (95% confidence interval [CI], 75‐97%), specificity of 68% (95% CI, 46‐83%), PPV of 77% (95% CI, 61‐88%), NPV of 84% (95% CI, 64‐96%) and overall accuracy of 79% (95% CI, 68‐88%) for diagnosis of high output (ie, septic shock). JVP 8 cmH2O was more accurate than crackles for predicting cardiogenic shock in low‐output patients with sensitivity of 82% (95% CI, 48‐98%), specificity of 79% (95% CI, 41‐95%), PPV of 75% (95% CI, 43‐95%), NPV of 85% (95% CI, 55‐98%), and overall accuracy of 80% (95%CI, 59‐93%). Using just skin temperature and JVP, the bedside approach misdiagnosed 19 of 75 cases (overall accuracy, 75%; 95% CI, 16‐37%).
| n Total | |
|---|---|
| |
| Gender, n (%) | n = 68 |
| Male | 39 (57) |
| Age, years | 71 16 |
| Race, n (%) | |
| White | 45 (66) |
| Black | 15 (22) |
| Hispanic | 7 (10) |
| Other | 1 (2) |
| High output, n (%) | n = 37 |
| Sepsis | |
| Pneumonia | 10 (27) |
| Urinary tract | 17 (46) |
| Skin | 3 (8) |
| Gastrointestinal | 5 (14) |
| Non‐infectious SIRS | 2 (5) |
| Low output heart full, n (%) | n = 18 |
| Pulmonary embolism | 3 (16) |
| AMI | 7 (40) |
| Cardiomyopathy | 5 (28) |
| Rhythm disturbance | 3 (16) |
| Low output heart empty, n (%) | n = 13 |
| Hemorrhagic | 9 (70) |
| NPO | 1 (7) |
| Diarrhea | 2 (14) |
| Adrenal crisis | 1 (7) |
| Prediction of SIRS | Capillary Refill Same/Faster (%) | Skin Same/ Warm (%) | Bounding Pulses (%) |
|---|---|---|---|
| |||
| Sensitivity | 89 | 89 | 65 |
| Specificity | 68 | 68 | 74 |
| Accuracy | 79 | 79 | 69 |
| Prediction of SIRS | Capillary Refill Same/Faster + Warm Skin + Bounding Pulse (%) | Capillary Refill Same/Faster + Warm Skin (%) | Any Other Combination of 2 (%) |
| Sensitivity | 62 | 89 | 62 |
| Specificity | 74 | 68 | 74 |
| Accuracy | 67 | 79 | 67 |
| Prediction of Cardiogenic | JVP (%) | Crackles (%) | JVP + Crackles (%) |
| Sensitivity | 82 | 55 | 55 |
| Specificity | 79 | 71 | 100 |
| Accuracy | 80 | 64 | 80 |
Discussion
This is the first study to examine the predictive characteristics of simple bedside physical examination techniques in correctly predicting the category/mechanism of shock. Overall, the algorithm performed well, and accurately predicted the category of shock in three‐quarters of patients. It also has the benefit of being very rapid, taking only seconds to complete, using bedside techniques that even inexperienced clinicians can apply.
Very few studies have examined the accuracy of examination techniques specifically for diagnosis of shock. In humans injected with endotoxin, body temperature and cardiac output increased, but skin temperature and capillary refill times are not well described.79 Schriger and Baraff10 reported that capillary refill >2 seconds was only 59% sensitive for diagnosing hypovolemia in patients with hypovolemic shock or orthostatic changes in blood pressure. Sensitivity was 77% in 13 patients with hypovolemic shock.10 However, some studies have demonstrated that age, sex, external temperature11 and fever12 can affect capillary refill times. Otieno et al.13 demonstrated a kappa statistic value of 0.49 for capillary refill 4 seconds, suggesting that reproducibility of this technique could be a major drawback. McGee et al.14 reviewed examination techniques for diagnosing hypovolemic states and concluded that postural changes in heart rate and blood pressure were the most accurate; capillary refill was not recommended. Stevenson and Perloff15 demonstrated that crackles and elevated JVP were absent in 18 of 43 patients with pulmonary capillary wedge pressures >22 mmHg. Butman et al.16 showed that elevated JVP was 82% accurate for predicting a wedge pressure >18 mmHg. Connors et al.17 demonstrated that clinicians' predictions of heart filling pressures and cardiac output were accurate (relative to pulmonary artery catheter measurements) in less than 50% of cases, though the examination techniques used were not qualified or quantified. No previous study has combined simple, semiobjective physical examination techniques for the purpose of distinguishing categories of shock.
Since identification of the pathogenesis of shock has important treatment/prognostic implications (eg, fluid and vasopressor therapies, early search for drainable focus of infection in sepsis, reestablishing vessel patency in myocardial infarction and pulmonary embolus), we believe that this simple, rapidly administered algorithm will prove useful in clinical medicine. In some clinical situations, the approach can lead to timely identification of the causative mechanism, allowing prompt definitive treatment. For example, a patient presenting with high‐output hypotension is so often sepsis/septic shock that treatment with antibiotics is justified (since success is time‐sensitive) even when the exact site/microbe has not yet been identified. Acute right heart overfilled low‐output hypotension should be considered pulmonary embolism (which also requires time‐sensitive therapies) until proven otherwise. Yet, a sizeable number of cases do not fit neatly into a single category. For example, 11% of patients with septic shock presented with cool extremities in the early phases of illness. In clinical decision‐making, 2 diagnostic‐therapeutic paradigms are common. In the first, the diagnosis is relatively certain and narrowly‐directed, mechanism‐specific treatment is appropriate. The second paradigm is 1 of significant uncertainty, when clinicians must treat empirically the most likely causes until more data become available to permit safe narrowing of therapies. For example, a patient presenting with hypotension, cool extremities, leukocytosis and apparent pneumonia should be treated empirically for septic shock while exploring explanations for the incongruous low‐output state (eg, profound hypovolemia, adrenal insufficiency, concurrent or precedent myocardial dysfunction). Patients often have several mechanisms contributing to hypotension. Since patients are not ideal forms, there can be no perfect decision‐tool; clinicians would be fool‐hardy to prematurely close decision‐making prior to definitive diagnosis. In the case of shock, such diagnostic arrogance would delay time‐sensitive therapies and thus contribute to morbidity and mortality. Nonetheless, this physical examination algorithmunderstanding its operating characteristics and limitationsmay add to the bedside clinician's diagnostic armamentarium.
Our study has several notable limitations. First, bedside examinations were performed by multiple observers who had limited (1 electronic mail) instruction on how to perform and document the data gathered for this study. So these results should be generalized cautiously until reproduced at other centers with greater numbers of observers (than the 28 of this study). The central supposition, that skin cooler, capillary refill longer, and pulse pressure more narrow than theirs, presupposes reasonable homogeneity of the normal state which is not necessarily true.11 Interobserver variability of physical examination further compromises the fidelity of findings recorded for this study.13 Since we conducted a retrospective review, and because of the emergency nature of the clinical problem, it would be difficult to conduct a study in which multiple examiners performed the same physical examinations to quantify interobserver variability. Irrespective, we would expect interobserver variability to systematically reduce accuracy; it is all‐the‐more impressive that trainees' examination results correctly diagnosed mechanism of shock in three‐quarters of cases. Also, examiners were not blinded to clinical history, so results of their examination could have been biased by their pre‐examination hypotheses of pathogenesis. Of course, they were not aware of the expert's final categorization of mechanism performed much later in time. Since there is no absolute reference standard for classification of the pathogenesis of shock, we depended upon careful review of selected data (same parameters for each patient) by a single senior investigatoralbeit armed with evidence‐based or consensus‐based standards of diagnosing shock. Finally, it can be argued that all forms of shock are mixed (with hypovolemia) early in the course; sepsis requires refilling of a leaky and dilated vasculature and the noncompliant ischemic ventricle often requires a higher filling pressure than normal to empty. To complicate even more, patients may have preexistent conditions (eg, chronic congestive heart failure, cirrhosis) that limit cardiovascular responses to acute shock. Our diagnostic approach was to identify the principal cause of the acute decompensation, assuming that many patients will have more than 1 single mechanism accounting for hypotension.
In conclusion, this is the first study to examine the utility of this simple physical examination algorithm to diagnose the mechanism of shock. Some have discounted or underemphasized examination techniques in favor of more time‐intensive and labor‐intensive diagnostic modalities, such as bedside echocardiography, which may waste precious time and resources. The simple physical examination algorithm assessed in this study has favorable operating characteristics and can be performed readily by even novice clinicians. If replicated at other centers and by greater numbers of observers, this approach could assist clinicians and teachers who train clinicians to rapidly diagnose and manage patients with shock.
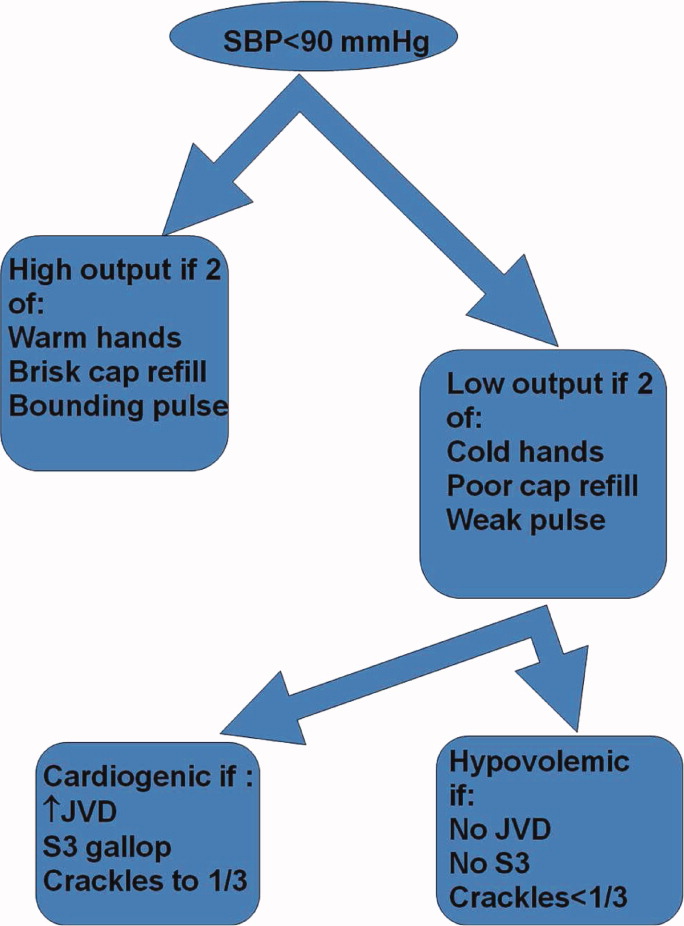
Shock has been defined as failure to deliver and/or utilize adequate amounts of oxygen1 and is a common cause of critical illness. Few studies have examined the predictive utility of bedside clinical examination in predicting the category of shock. Scholars have suggested a bedside approach that uses simple examination techniques and applied physiology to rapidly identify a patients' circulation as high vs. low cardiac output. Those with a high‐output examination are designated as high‐output, most often septic shock. Low‐output patients are further categorized as heart full or heart empty to distinguish cardiogenic from hypovolemic categories of shock, respectively.2 The predictive characteristics of this simple algorithm have not been studied. In this study, we examine the operating characteristics of selected elements of this algorithm when administered at the bedside by trainees in Internal Medicine.
Methods
This study was performed after approval of the Institutional Review Board; informed consent was waived. Patients with nonsurgical problems who present to the hospital or who develop sustained hypotension are managed by medical house officers on the intensive care and/or rapid response team with the supervision of patients' attending physicians. All house officers were asked to document explicitly in their assessment notes the following examination findings: finger capillary refill (same/quicker vs. slower than examiner's), hand skin temperature (same/warmer vs. cooler than examiner's) and pulse pressure (ie, same/wider vs. thinner than examiner's), presence or absence of crackles >1/3 from base on bilateral lung examination and jugular venous pressure (JVP) vs. 8 cmH2O. The documented examinations of either the rapid response team (PGY2; n = 14) or intensive care unit (ICU) resident (PGY3; n = 14) for patients evaluated between September 2008 and February 2009 were used for this study. Resuscitation was administered entirely by house officers, occasionally guided in person, but always supervised by attending physicians.
In May 2009, clinical data, including electrocardiograms/echocardiograms and laboratory (eg, cardiac enzymes, culture) results were abstracted from medical records of subjects. These were presented to a blinded senior clinician (DK) to review and apply evidence‐based or consensus criteria,36 whenever possible, to categorize the type of shock (septic vs. cardiogenic vs. hypovolemic) based on data acquired after the onset of shock. For example, patients with microbiologic and/or radiologic evidence of infection were classified as septic shock,1, 3, 4 those with acute left or right ventricular dysfunction on echocardiogram were classified as cardiogenic shock,1, 6 and those with clinical evidence of acute hemorrhage with hypovolemic shock.1, 5 While some of the patients were examined by DK as part of clinical care, he was blinded to the identity of patients and their algorithm‐related physical examination findings when he reviewed the abstracted data (>2 months after study closure) to adjudicate the final diagnosis of shock. These diagnoses were considered the reference standard for this study. The operating characteristics (sensitivity = true positive/true positive + false negative; specificity = true negative/true negative + false positive; negative predictive value (NPV) = true negative/all negatives; positive predictive value (PPV) = true positive/all positives; accuracy = true results/all results) were calculated for combinations of physical examination findings and correct final diagnosis (Figure 1).
Results
A total of 68 patients, averaging 71 16 years, were studied; 57% were male, and 66% were White, and 20% were Black. Table 1 lists characteristics of patients. A total of 37 patients were diagnosed as having septic shock, 11 had cardiogenic shock and 10 hypovolemic shock. Operating characteristics of the bedside examination techniques for predicting mechanism of shock are listed in Table 2. Capillary refill and skin temperature were 100% concordant yielding sensitivity of 89% (95% confidence interval [CI], 75‐97%), specificity of 68% (95% CI, 46‐83%), PPV of 77% (95% CI, 61‐88%), NPV of 84% (95% CI, 64‐96%) and overall accuracy of 79% (95% CI, 68‐88%) for diagnosis of high output (ie, septic shock). JVP 8 cmH2O was more accurate than crackles for predicting cardiogenic shock in low‐output patients with sensitivity of 82% (95% CI, 48‐98%), specificity of 79% (95% CI, 41‐95%), PPV of 75% (95% CI, 43‐95%), NPV of 85% (95% CI, 55‐98%), and overall accuracy of 80% (95%CI, 59‐93%). Using just skin temperature and JVP, the bedside approach misdiagnosed 19 of 75 cases (overall accuracy, 75%; 95% CI, 16‐37%).
| n Total | |
|---|---|
| |
| Gender, n (%) | n = 68 |
| Male | 39 (57) |
| Age, years | 71 16 |
| Race, n (%) | |
| White | 45 (66) |
| Black | 15 (22) |
| Hispanic | 7 (10) |
| Other | 1 (2) |
| High output, n (%) | n = 37 |
| Sepsis | |
| Pneumonia | 10 (27) |
| Urinary tract | 17 (46) |
| Skin | 3 (8) |
| Gastrointestinal | 5 (14) |
| Non‐infectious SIRS | 2 (5) |
| Low output heart full, n (%) | n = 18 |
| Pulmonary embolism | 3 (16) |
| AMI | 7 (40) |
| Cardiomyopathy | 5 (28) |
| Rhythm disturbance | 3 (16) |
| Low output heart empty, n (%) | n = 13 |
| Hemorrhagic | 9 (70) |
| NPO | 1 (7) |
| Diarrhea | 2 (14) |
| Adrenal crisis | 1 (7) |
| Prediction of SIRS | Capillary Refill Same/Faster (%) | Skin Same/ Warm (%) | Bounding Pulses (%) |
|---|---|---|---|
| |||
| Sensitivity | 89 | 89 | 65 |
| Specificity | 68 | 68 | 74 |
| Accuracy | 79 | 79 | 69 |
| Prediction of SIRS | Capillary Refill Same/Faster + Warm Skin + Bounding Pulse (%) | Capillary Refill Same/Faster + Warm Skin (%) | Any Other Combination of 2 (%) |
| Sensitivity | 62 | 89 | 62 |
| Specificity | 74 | 68 | 74 |
| Accuracy | 67 | 79 | 67 |
| Prediction of Cardiogenic | JVP (%) | Crackles (%) | JVP + Crackles (%) |
| Sensitivity | 82 | 55 | 55 |
| Specificity | 79 | 71 | 100 |
| Accuracy | 80 | 64 | 80 |
Discussion
This is the first study to examine the predictive characteristics of simple bedside physical examination techniques in correctly predicting the category/mechanism of shock. Overall, the algorithm performed well, and accurately predicted the category of shock in three‐quarters of patients. It also has the benefit of being very rapid, taking only seconds to complete, using bedside techniques that even inexperienced clinicians can apply.
Very few studies have examined the accuracy of examination techniques specifically for diagnosis of shock. In humans injected with endotoxin, body temperature and cardiac output increased, but skin temperature and capillary refill times are not well described.79 Schriger and Baraff10 reported that capillary refill >2 seconds was only 59% sensitive for diagnosing hypovolemia in patients with hypovolemic shock or orthostatic changes in blood pressure. Sensitivity was 77% in 13 patients with hypovolemic shock.10 However, some studies have demonstrated that age, sex, external temperature11 and fever12 can affect capillary refill times. Otieno et al.13 demonstrated a kappa statistic value of 0.49 for capillary refill 4 seconds, suggesting that reproducibility of this technique could be a major drawback. McGee et al.14 reviewed examination techniques for diagnosing hypovolemic states and concluded that postural changes in heart rate and blood pressure were the most accurate; capillary refill was not recommended. Stevenson and Perloff15 demonstrated that crackles and elevated JVP were absent in 18 of 43 patients with pulmonary capillary wedge pressures >22 mmHg. Butman et al.16 showed that elevated JVP was 82% accurate for predicting a wedge pressure >18 mmHg. Connors et al.17 demonstrated that clinicians' predictions of heart filling pressures and cardiac output were accurate (relative to pulmonary artery catheter measurements) in less than 50% of cases, though the examination techniques used were not qualified or quantified. No previous study has combined simple, semiobjective physical examination techniques for the purpose of distinguishing categories of shock.
Since identification of the pathogenesis of shock has important treatment/prognostic implications (eg, fluid and vasopressor therapies, early search for drainable focus of infection in sepsis, reestablishing vessel patency in myocardial infarction and pulmonary embolus), we believe that this simple, rapidly administered algorithm will prove useful in clinical medicine. In some clinical situations, the approach can lead to timely identification of the causative mechanism, allowing prompt definitive treatment. For example, a patient presenting with high‐output hypotension is so often sepsis/septic shock that treatment with antibiotics is justified (since success is time‐sensitive) even when the exact site/microbe has not yet been identified. Acute right heart overfilled low‐output hypotension should be considered pulmonary embolism (which also requires time‐sensitive therapies) until proven otherwise. Yet, a sizeable number of cases do not fit neatly into a single category. For example, 11% of patients with septic shock presented with cool extremities in the early phases of illness. In clinical decision‐making, 2 diagnostic‐therapeutic paradigms are common. In the first, the diagnosis is relatively certain and narrowly‐directed, mechanism‐specific treatment is appropriate. The second paradigm is 1 of significant uncertainty, when clinicians must treat empirically the most likely causes until more data become available to permit safe narrowing of therapies. For example, a patient presenting with hypotension, cool extremities, leukocytosis and apparent pneumonia should be treated empirically for septic shock while exploring explanations for the incongruous low‐output state (eg, profound hypovolemia, adrenal insufficiency, concurrent or precedent myocardial dysfunction). Patients often have several mechanisms contributing to hypotension. Since patients are not ideal forms, there can be no perfect decision‐tool; clinicians would be fool‐hardy to prematurely close decision‐making prior to definitive diagnosis. In the case of shock, such diagnostic arrogance would delay time‐sensitive therapies and thus contribute to morbidity and mortality. Nonetheless, this physical examination algorithmunderstanding its operating characteristics and limitationsmay add to the bedside clinician's diagnostic armamentarium.
Our study has several notable limitations. First, bedside examinations were performed by multiple observers who had limited (1 electronic mail) instruction on how to perform and document the data gathered for this study. So these results should be generalized cautiously until reproduced at other centers with greater numbers of observers (than the 28 of this study). The central supposition, that skin cooler, capillary refill longer, and pulse pressure more narrow than theirs, presupposes reasonable homogeneity of the normal state which is not necessarily true.11 Interobserver variability of physical examination further compromises the fidelity of findings recorded for this study.13 Since we conducted a retrospective review, and because of the emergency nature of the clinical problem, it would be difficult to conduct a study in which multiple examiners performed the same physical examinations to quantify interobserver variability. Irrespective, we would expect interobserver variability to systematically reduce accuracy; it is all‐the‐more impressive that trainees' examination results correctly diagnosed mechanism of shock in three‐quarters of cases. Also, examiners were not blinded to clinical history, so results of their examination could have been biased by their pre‐examination hypotheses of pathogenesis. Of course, they were not aware of the expert's final categorization of mechanism performed much later in time. Since there is no absolute reference standard for classification of the pathogenesis of shock, we depended upon careful review of selected data (same parameters for each patient) by a single senior investigatoralbeit armed with evidence‐based or consensus‐based standards of diagnosing shock. Finally, it can be argued that all forms of shock are mixed (with hypovolemia) early in the course; sepsis requires refilling of a leaky and dilated vasculature and the noncompliant ischemic ventricle often requires a higher filling pressure than normal to empty. To complicate even more, patients may have preexistent conditions (eg, chronic congestive heart failure, cirrhosis) that limit cardiovascular responses to acute shock. Our diagnostic approach was to identify the principal cause of the acute decompensation, assuming that many patients will have more than 1 single mechanism accounting for hypotension.
In conclusion, this is the first study to examine the utility of this simple physical examination algorithm to diagnose the mechanism of shock. Some have discounted or underemphasized examination techniques in favor of more time‐intensive and labor‐intensive diagnostic modalities, such as bedside echocardiography, which may waste precious time and resources. The simple physical examination algorithm assessed in this study has favorable operating characteristics and can be performed readily by even novice clinicians. If replicated at other centers and by greater numbers of observers, this approach could assist clinicians and teachers who train clinicians to rapidly diagnose and manage patients with shock.
- ,,, et al.Hemodynamic monitoring in shock and implications for management. International consensus conference. Paris, France, 27–28 April 2006.Intensive Care Med.2007;33:575–590.
- .The pathophysiology of the circulation in critical illness. In:Principles of Critical Care.New York:McGraw Hill;2005.
- ,,, et al.2001 SCCM/ESICM/ACCP/ATS/SIS. International Sepsis Definitions Conference.Crit Care Med.2003;31:1250–1256.
- ,,, et al.Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis.Chest.1992;101:1644–1655;
- ,,, et al.Resuscitation from severe hemorrhage.Crit Care Med.1996;24(2 Suppl):S12–S23.
- ,.Cardiogenic shock: current concepts and improving outcomes circulation.Circulation.2008;117:686–697.
- ,,, et al.The cardiovascular response of normal humans to the administration of endotoxin.N Engl J Med.1989;321:280–287.
- ,,,,,.Experimental endotoxemia in humans: análysis of cytokine release and coagulation, fibrinolytic, an complement pathways.Blood.1990;76:2520–2526.
- ,,,,.Peripheral resistance changes during shock in man.Angiology.1968;19:268–276.
- ,.Capillary refill—is it a useful predictor of hypovolemic states?Ann Emerg Med.1991;20:601–605.
- ,.Defining normal capillary refill: variation with age, sex, and temperature.Ann Emerg Med.1988;17:113–116.
- ,,,.Effect of fever on capillary refill time.Pediatr Emerg Care.1997;13:305–307.
- ,,,,,.Are bedside features of shock reproducible between different observers?Arch Dis Child.2004;89:977–979.
- ,,.Is this patient hypovolemic?JAMA.1999;281:1022–1029.
- ,.The limited reliability of physical signs for estimating hemodynamics in chronic heart failure.JAMA.1989;261:884–888.
- ,,,,.Bedside cardiovascular examination in patients with severe chronic heart failure: importance of rest or inducible jugular venous distension.J Am Coll Cardiol.1993;22:968–974.
- ,,.Evaluation of right‐heart catheterization in the critically ill patients without acute myocardial infarction.N Engl J Med.1983;308(5):263–267.
- ,,, et al.Hemodynamic monitoring in shock and implications for management. International consensus conference. Paris, France, 27–28 April 2006.Intensive Care Med.2007;33:575–590.
- .The pathophysiology of the circulation in critical illness. In:Principles of Critical Care.New York:McGraw Hill;2005.
- ,,, et al.2001 SCCM/ESICM/ACCP/ATS/SIS. International Sepsis Definitions Conference.Crit Care Med.2003;31:1250–1256.
- ,,, et al.Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis.Chest.1992;101:1644–1655;
- ,,, et al.Resuscitation from severe hemorrhage.Crit Care Med.1996;24(2 Suppl):S12–S23.
- ,.Cardiogenic shock: current concepts and improving outcomes circulation.Circulation.2008;117:686–697.
- ,,, et al.The cardiovascular response of normal humans to the administration of endotoxin.N Engl J Med.1989;321:280–287.
- ,,,,,.Experimental endotoxemia in humans: análysis of cytokine release and coagulation, fibrinolytic, an complement pathways.Blood.1990;76:2520–2526.
- ,,,,.Peripheral resistance changes during shock in man.Angiology.1968;19:268–276.
- ,.Capillary refill—is it a useful predictor of hypovolemic states?Ann Emerg Med.1991;20:601–605.
- ,.Defining normal capillary refill: variation with age, sex, and temperature.Ann Emerg Med.1988;17:113–116.
- ,,,.Effect of fever on capillary refill time.Pediatr Emerg Care.1997;13:305–307.
- ,,,,,.Are bedside features of shock reproducible between different observers?Arch Dis Child.2004;89:977–979.
- ,,.Is this patient hypovolemic?JAMA.1999;281:1022–1029.
- ,.The limited reliability of physical signs for estimating hemodynamics in chronic heart failure.JAMA.1989;261:884–888.
- ,,,,.Bedside cardiovascular examination in patients with severe chronic heart failure: importance of rest or inducible jugular venous distension.J Am Coll Cardiol.1993;22:968–974.
- ,,.Evaluation of right‐heart catheterization in the critically ill patients without acute myocardial infarction.N Engl J Med.1983;308(5):263–267.
Medication Reconciliation: A Consensus Statement From Stakeholders
Medication reconciliation is integral to reducing medication errors surrounding hospitalizations.1, 2 The practice of medication reconciliation requires a systematic and comprehensive review of all the medications a patient is currently taking to ensure that medications being added, changed, or discontinued are carefully evaluated with the goal of maintaining an accurate list; that this process is undertaken at every transition along the continuum of care; and that an accurate list of medications is available to the patient or family/caregiver and all providers involved in the patient's care, especially when a care handoff takes place. With regulators, payers and the public increasingly demanding action to reduce medication errors in hospitals, all health care providers must support efforts to achieve accurate medication reconciliation.1, 3
The Joint Commission's Definition of Medication
Any prescription medications, sample medications, herbal remedies, vitamins, nutraceuticals, vaccines, or over‐the‐counter drugs; diagnostic and contrast agents used on or administered to persons to diagnose, treat, or prevent disease or other abnormal conditions; radioactive medications, respiratory therapy treatments, parenteral nutrition, blood derivatives, and intravenous solutions (plain, with electrolytes and/or drugs); and any product designated by the Food and Drug Administration (FDA) as a drug. This definition of medication does not include enteral nutrition solutions (which are considered food products), oxygen, and other medical gases.
2010 Hospital Accreditation Standards,
The Joint Commission, 2010, p. GL19.
While conceptually straightforward, implementing medication reconciliation has proved to be very difficult in the myriad healthcare settings that exist. The disjointed nature of the American health care system and a conglomeration of paper and electronic systems for tracking medications synergize to thwart efforts to maintain an accurate, up‐to‐date medication list at every step along the care continuum. Although The Joint Commission defines medication for the purpose of its accreditation standards (see box), the healthcare community lacks a common understanding or agreement regarding what constitutes a medication. There is also confusion about who should ultimately be responsible for obtaining the patient's medication information, for performing the various steps in the reconciliation process, and for managing the multiple providers who alter the medication list but may not feel competent to perform reconciliation of medications outside their area of expertise safely. Importantly, there is also a lack of clarity around how patients and family/caregivers should be involved in the process.
Despite these challenges, medication reconciliation remains a critical patient safety activity that is supported by the organizations signing this consensus statement, (Table 1). Although medication reconciliation has an impact on medication safety in all care settings, this paper focuses on issues most germane to the continuum of care involving the hospital setting. The themes and issues discussed will likely apply to other care settings as well. In this paper, we also recommend several concrete steps that we believe should be initiated immediately to begin to reach the goal of optimizing the medication safety achievable through effective medication reconciliation.
Background
Medication reconciliation is intended to be a systematic extension of the medication history‐taking process that has been used by health care providers for decades. Its recent iteration was developed to ensure that medications were not added, omitted, or changed inadvertently during care transitions. It became codified, refined, and tested over the past decade through the efforts of a number of groups focused on medication safety including the Institute for Healthcare Improvement (IHI) and the Institute for Safe Medication Practices (ISMP). With the reinforcing adoption of medication reconciliation as National Patient Safety Goal (NPSG) No. 8 in 2005 by The Joint Commission, efforts to implement it became widespread in both hospital‐based and ambulatory settings.
Medication reconciliation has three steps, as described by IHI4:
-
Verification (collection of the patient's medication history);
-
Clarification (ensuring that the medications and doses are appropriate); and
-
Reconciliation (documentation of changes in the orders).
The details of the process vary by setting and by the availability of paper or electronic medical records. However, the essential steps remain the same, as does the need to perform reconciliation each time the patient transfers to a new setting or level of care. Table 2 lists the most common points at which medication reconciliation occurs in hospitalized patients.
|
| American Academy of Pediatrics |
| American Association of Critical‐Care Nurses |
| Consumers Advancing Patient Safety |
| Institute for Healthcare Improvement |
| Institute for Safe Medication Practices |
| The Joint Commission |
| Massachusetts Coalition for Prevention of Medical Errors |
| Microsoft Corporation |
| Northwestern Memorial Hospital and Northwestern University School of Medicine |
| Society of General Internal Medicine |
| Society of Hospital Medicine |
| University of California San Diego Medical Center |
Because of their complexity, organizations must take care to design their medication reconciliation processes systematically. IHI lists elements of a well‐designed medication reconciliation process as part of its 5 Million Lives Campaign How‐to Guide.4 Such a process:
-
Uses a patient centered approach.
-
Makes it easy to complete the process for all involved. Staff members recognize the what's‐in‐it‐for‐me aspect of the change.
-
Minimizes the opportunity for drug interactions and therapeutic duplications by making the patient's list of current medications available when clinicians prescribe new medications.
-
Provides the patient with an up‐to‐date list of medications.
-
Ensures that other providers who need to know have information about changes in a patient's medication plan.
Research on how adverse drug events (ADE) occur supports the need for tight control of medication orders at transitions in care. For instance:
-
In a study conducted at Mayo Health System in Wisconsin, poor communication of medical information at transition points was responsible for as many as 50% of all medication errors in the hospital and up to 20% of ADEs.5
-
Variances between the medications patients were taking prior to admission and their admission orders ranged from 30% to 70% in 2 literature reviews.1, 6
-
The largest study of medication reconciliation errors and risk factors at hospital admission documented that 36% of patients had errors in their admission orders.7
When The Joint Commission adopted medication reconciliation as NPSG No. 8 in 2005 it had 2 parts: Requirement 8Aa process must exist for comparing the patient's current medications with those ordered for the patient while under the care of the organization; and requirement 8Ba complete list of the patient's medications must be communicated to the next provider of service on transfer within or outside the organization and a complete list of medications must be provided to the patient on discharge.8
However, many hospitals found it difficult to implement medication reconciliation in a systematic way. There was also confusion among hospital staff and administration about the exact definition of medication reconciliation in terms of what it should entail.9 Given these difficulties, The Joint Commission announced that effective January 1, 2009, medication reconciliation would no longer be factored into an organization's accreditation decision or be considered for Requirements for Improvement. Additionally, The Joint Commission stated it is reviewing and revising the NPSG so that it will be ready to be released in January 2011 for implementation later that year.10
Recognizing the difficulty hospitals were having with meaningfully implementing medication reconciliation, the Society of Hospital Medicine convened a 1‐day conference on March 6, 2009, to obtain input from key stakeholders and focus on several critical domains relevant to the success of hospital‐based medication reconciliation. The Agency for Healthcare Research and Quality provided funding support for this conference through grant 1R13HS017520‐01.
An overarching theme emerged from the meeting: the need to reorient the focus of medication reconciliation away from that of an accreditation mandate and toward a broader view of patient safety. Forcing medication reconciliation via a requirement for accreditation tended to limit an organization's efforts to specific process measures. Addressing it as a more global patient safety issue takes into account the entire patient care experience and then opens the door to leverage nonclinical venues (e.g., medical home, family home, community, religious, and other social organizations, as well as social networking platforms) and engage the patient and family/caregivers to reinforce the importance of medication safety.
This white paper evolved from discussions at the March 2009 conference,11 and subsequent structured communication among attendees. Formal endorsement of this document was obtained from the organizations listed in Table 1. In this document, we explore several key issues in implementing clinically meaningful and patient‐centered medication reconciliation. We focus on building common language and understanding of the processes of and participants in medication reconciliation; consider issues of implementation and risk stratification; emphasize the need for research to identify best practices and discusses how to disseminate the findings; promote health information technology platforms that will support interoperable medication information exchange; support the formation of partnerships between patient care sites and nonclinical sites as well as utilizing social marketing opportunities to enhance opportunities for transmitting messages about medication safety; and reinforce the ongoing healthcare reform discussion which aims to align financial incentives with patient safety efforts. After each section, we offer concrete first steps to address the issues discussed.
| Admission: When clinicians reconcile the patient's medications taken at home or at a prior care setting with any new prescription orders to be prescribed by an admitting clinician. |
| Transfer (intra‐ or inter‐facility; with change of clinician or site of care): When clinicians review previous medication orders in light of the patient's clinical status, along with new orders or plans of care. |
| Discharge: When clinicians review all medications the patient was taking prior to being hospitalized, incorporating new prescriptions from the hospitalization and determining whether any medication should be added, discontinued, or modified while being mindful of therapeutic interchanges needed for formulary purposes. |
Methods
The invitation‐only meeting held on the Northwestern Medical Campus in Chicago, IL, brought together stakeholders representing professional, clinical, health care quality, consumer, and regulatory organizations (Table 3). The conference convened these participants with the goals of identifying barriers to meaningful implementation of medication reconciliation and developing a feasible plan toward its effective implementation in the hospital setting. At the meeting, all participants were divided into 1 of 4 groups, which held a facilitated discussion around 1 of 4 key relevant domains: (1) how to measure success in medication reconciliation; (2) key elements of successful strategies; (3) leveraging partnerships outside the hospital setting to support medication reconciliation; and (4) the roles of the patient and family/caregivers and health literacy. Individual group discussions were cofacilitated by experts in the content area. After each discussion, the small group then rotated to a different discussion. Ultimately, each group participated in all four discussions, which built iteratively on the content derived from the prior groups' insights. Key comments were then shared with the large group for further discussion. To help build consensus, these large group discussions were directed by professional facilitators.
| AACN American Association of Critical Care Nurses |
| AAFP American Academy of Family Physicians |
| AAP American Academy of Pediatrics |
| ACEP American College of Emergency Physicians |
| ACP American College of Physicians |
| AMA American Medical Association |
| AMSN Academy of Medical Surgical Nurses |
| ASHP American Society of Health‐System Pharmacists |
| ASHP Foundation American Society of Health‐System Pharmacists Foundation |
| CAPS Consumers Advancing Patient Safety |
| CMS Centers for Medicare and Medicaid Services |
| CMSA Case Management Society of America |
| HCI Hospitalist Consultants, Inc |
| IHI Institute for Healthcare Improvement |
| InCompass Health |
| ISMP Institute For Safe Medication Practice |
| JCR Joint Commission Resources |
| Massachusetts Coalition for Prevention of Medical Errors |
| Microsoft Corporation |
| Northwestern Memorial Hospital MATCH Program |
| NQF National Quality Forum |
| SGIM Society of General Internal Medicine |
| SHM Society of Hospital Medicine |
| The Joint Commission |
| UCSD Hospital Medicine |
| University of Oklahoma College of Pharmacy Tulsa |
After the meeting, attendees participated in 2 follow‐up conference calls to discuss issues raised at the conference and responses obtained from host organizations. They also subsequently participated in two focus groups with The Joint Commission, giving input on the revision of the medication reconciliation NPSG.
Results
Addressing Barriers to Medication Reconciliation
In order to implement successful medication reconciliation processes, one must build the steps with the patient and family/caregiver as the focus and demonstrate an understanding of the intent of these processes. At its roots, medication reconciliation was developed to ensure that clinicians do not inadvertently add, change, or omit medications and that changes made are communicated to all relevant caregivers.
A number of key issues with respect to successful medication reconciliation processes surfaced in discussions with stakeholders. We believe addressing these issues is necessary before meaningful and standardized implementation can be achieved. After each discussion below, we provide suggested first steps to address these issues.
1. Achieve Consensus on the Definition of Medication and Reconciliation
Despite proposed definitions of these terms by various organizations, there was little agreement about them in the healthcare community. This ambiguity contributed to general confusion about what actually constitutes medication reconciliation. There needs to be a single, clear, and broadly accepted definition of what constitutes a medication. For the purposes of medication reconciliation, the term medication should be broadly inclusive of substances that may have an impact on the patient's care and treatments as well as those substances that may interact with other therapies potentially used during the medical care episode. Illicit or recreational substances may also have impact on therapies considered and therefore may influence this definition.12 Concretely, this definition should encompass prescription and over‐the‐counter medications as well as herbal and dietary supplements.
The term reconciliation in its simplest form implies the process of verifying that a patient's current list of medications (including dose, route, and frequency) are correct and that the medications are currently medically necessary and safe. Reconciliation suggests a process which, by necessity, will vary based on clinical context and setting. Further defining this termand the process of reconciliation itselfshould be carried out using patient safety principles with a focus on patient‐ and family‐centeredness.
Designing hospital‐based medication reconciliation processes should:
-
Employ a multidisciplinary approach that involves nurses, pharmacists, and other appropriate personnel from the inpatient setting as well as ambulatory and community/retail areas, both ambulatory and inpatient physicians, and a patient/family representative;
-
Involve hospital leaders who support, provide guidance, and remove barriers for the multidisciplinary team working to implement the processes;
-
Clearly define the roles of each participant in the processes developed;
-
Include methods to assess and address any special needs due to the developmental stage, age, dependency, language or literacy levels of patients and their family/caregiver;
-
Use clinically relevant process measures (e.g., adherence to procedural steps) and outcome measures (e.g., change in the number of ADEs, unnecessary hospitalizations, or emergency department visits) where appropriate to assess the impact of the process;
-
Include feedback systems to allow for clinically significant process improvement.
Once a common understanding of the terms and intent of medication reconciliation is achieved, it will be important for accrediting organizations, medical societies, quality improvement organizations, and other interested parties to adopt the same language.
First Step
A consortium of clinical, quality, and regulatory stakeholders should work to achieve consensus on the definition for medication and the intent and expectations for the reconciliation process.
2. Clarify Roles and Responsibilities
Given the differences in organizational and practice structures in hospitals and the varying numbers of health professionals involved in a patient's care, no one process design will meet the needs of all sites. As it is clear that interdisciplinary teams are best suited to develop, implement, and carry out complex patient‐centered processes like medication reconciliation, it is crucial that all involved parties have clearly defined roles and responsibilities, including patients and their families/caregivers. It is also important to recognize that these responsibilities may change depending on the dependency or vulnerability of the patient (e.g., children or geriatric patients) or the transition of care being undertaken by the patient (i.e., admission, transfer, or discharge), thus requiring sites to develop clear policies about these roles and responsibilities and how they may change in various situations.
First Step
Individual sites must clearly define the roles and responsibilities of all parties directly involved in medication reconciliation as a part of designing local medication reconciliation processes.
3. Develop Measurement Tools
Ensuring that medication reconciliation processes result in clinically meaningful outcomes requires the development and standardization of a limited number of metrics that may be used by organizations and reported centrally for benchmarking. This core set of measures should be developed by clinical, quality, accreditation, and regulatory organizations (see #10 below) through a consensus building process utilizing multi‐stakeholder input. The set should be supplemented by additional site‐specific measures determined locally that focus on steps in the process itself and allow sites to perform continuous quality improvement. Sites should be encouraged to develop tools locally to support and facilitate organizational and professional adherence to medication reconciliation processes.
First Steps
Clinical, quality, accreditation, and regulatory organizations should develop reliable metrics to be assessed and reported.
The principles of patient‐centeredness and family/caregiver‐centeredness, the medical home, and clinical relevance must be central to the metrics chosen for quality and regulatory purposes.
4. Phased Implementation
Ultimately, comprehensive medication reconciliation processes need to be implemented in hospitals. However, to succeed in integrating complex processes like medication reconciliation into routine hospital practices, implementation may be facilitated by using a phased approach to allow for participants to adapt new processes and procedures to the local environment iteratively. While the most appropriate phased approach to implementation will vary by site and setting, options for phasing might include:
-
Starting with one clinical area or service.
-
Starting with either the admission or discharge reconciliation process.
-
Starting with a patient population at high risk for adverse events.
Irrespective of the phasing strategy employed, development of a clear and pragmatic schedule for the entire implementation process should be established. Phasing decisions should be made based on organizational resources and the clinical needs of the patient population within each clinical setting. As noted, the ultimate goal is to develop comprehensive reconciliation processes occurring during all significant care transitions (i.e., admission, service or site‐of‐care transfers, and discharge) for all hospitalized patients and involving all of their medications. Flexibility in design should be encouraged to ensure the processes can work within local workflow as long as progress toward this primary goal is made.
First Steps
Clinical sites should establish local, pragmatic priorities for a phased approach to implementation.
Tie the phased approach to a timeline or blueprint for programmatic expansion with ultimate plans for comprehensive implementation.
5. Develop Risk Stratification Systems
Medication‐related adverse events related to inadequate reconciliation are more likely to occur in hospitalized patients with certain identifiable risk factors. For example, the MATCH study documented that polypharmacy and age over 65 years were independently associated with increased risk for errors at the time of hospital admission.7 Other factors that may increase the likelihood of medication‐related adverse events at care transitions in the hospital might include: patients with multiple providers, developmental/cognitive impairment, dependency/vulnerability, multiple or high‐risk medications, or poor health literacy or limited English proficiency. Research is needed to elucidate these risk factors further.
An alert system for key risk factors for complications related to incompletely, inappropriately, or inaccurately completed medication reconciliation due to patient, clinician, or system factors should be developed, tested, and broadly implemented. Additionally, an alert system would help maintain vigilance toward this patient safety issue and, potentially, help focus additional resources on high‐risk patients. Such a tool has been tested in ambulatory settings.15
First Step
Additional research on inpatient predictors of failed medication reconciliation and ADE should be prioritized (see #6 below).
6. Study Interventions and Processes
Despite having been an NPSG since 2005, there is still a relative paucity of literature about broadly applicable and effective implementation strategies and demonstrated interventions that improve medication safety related to medication reconciliation. Some strategies that have shown to reduce medication errors at transitions include the involvement of pharmacist medication review on discharge16, 17 and the usefulness of planning by multidisciplinary groups.18 Other studies have outlined the continuing barriers to successful implementation of reconciliation, including the difficulty patients have in accurately recalling their current medications19 and the high cost in nurse and pharmacist time of tracking down a patient's ongoing prescriptions.20, 21 Studies evaluating potential solutions to overcome these and other common barriers are still needed.
Future research should focus on a comprehensive review of implementation strategies, (specifically including the role of health information technology‐based innovations) clinically relevant outcomes, and best practices, while being sensitive to the different needs of varying care settings (e.g., pediatric vs. adult centers, emergency departments vs. inpatient units, community hospital vs. academic medical center, etc.) as well as the resource requirements engendered in the interventions.
First Step
Funding agencies should explicitly prioritize outcomes‐focused medication reconciliation‐related projects (e.g., those which demonstrate a reduction in postdischarge ADE or reduced medication‐related emergency department visits). Previously identified successful strategies should be further investigated. Funded projects should explicitly partner with patients and family/caregivers and also include pediatric and adult patients, rural and urban locations of care, as well as academic and nonacademic hospital settings, to promote more broadly applicable results.
7. Disseminate Success
Best practices and lessons learned, especially those rigorously tested and driven by data, stratified by patient type, care setting (emergency department, intensive care, surgical ward, etc.) and institutional type (community, teaching, safety net, critical access, etc.) need to be disseminated so others can adopt and adapt them effectively. High‐quality case studies with clear explanations of successes, failures, and lessons learned may prove valuable sources of information. This knowledge should foster a learning community approach and accelerate implementation at new sites.
First Step
Hospitals, healthcare systems, as well as quality and regulatory agencies should develop mechanisms within reporting systems to track performance, identify notably successful sites, and publicly report and share methods and lessons learned from them.
8. Promote the Personal Health Record
A fully integrated and transferable personal health record should be accepted as the standard for health information storage and interoperability, giving both the patient (or family/caregiver) and clinical providers access and ownership. Both the HL7 Continuity of Care Document (CCD) and the Continuity of Care Record (CCR) meet these criteria. The CCR was endorsed by the American Society for Testing and Materials22 and a coalition of other medical societies.23 Notably, CCR and CCD were recently adopted as standards for structured electronic health record (EHR) exchange through the July 2010 publication of the Final Rule of the Health Information Technology for Economic and Clinical Health Act provision of the American Recovery and Reinvestment Act of 2009 (ARRA/HITECH) and is now part of the formal US Department of Health and Human Services certification criteria for EHR technologies.24
Mandating a content exchange standard such as the CCR or the CCD should also have the desired effect of ensuring that patients (and their caregivers) become increasingly involved in maintaining an accurate list of the medications they take. Additionally, systems must be sufficiently flexible to address the unique medication management needs of children and geriatric patients. An electronic version of a personal health record is a promising method for improving consistency across care platforms, but to be implemented effectively the record must be compatible across all settings, including, where possible, the patient's home. All health care organizations, pharmacy systems, and insurers, must make medication reconciliation‐related interoperability and accessibility a priority as they pursue information technology strategies.
First Step
Stakeholder organizations must send a clear and convincing message to legislators under the current atmosphere of health care reform, urging them to mandate that health information technology standards include interoperability and support platforms that are consistent with standards put forth in the 2009 HITECH Act Interim Final Rule for EHR certification.
9. Promote Partnerships
At a broader health care system level, leveraging existing partnerships and creating new ones among health care, public/private sector‐affiliated organizations (e.g., community and mail order pharmacies, pharmaceutical organizations and manufacturers, and insurers), and public health organizations are extremely important mechanisms for broader scale impact. This view recognizes the numerous opportunities to educate and influence patients about medication safety outside the dyadic relationship of the clinician and patient in traditional clinical settings. Partnerships between health care and public entities may capitalize on these opportunities to foster adoption of healthy medication practices (e.g., maintaining an accurate and updated medication list), thereby supporting medication reconciliation efforts when individuals encounter health care settings. Partnership and information sharing could be enhanced through the use of a central coordinating body or coalition. This body could generate a shared common vision and contribute expertise to the myriad issues in medication reconciliation.
Partnerships should utilize the following:
-
Social marketing techniques to engage the community. Included within this strategy must be a clear and compelling message that transmits the importance of safe medication practices. Current messages such as keep a list while important, do not offer enough of a sense of urgency or importance. A more powerful message could involve highly publicized medication errors or close calls that would resonate with a broad audience.
-
Local and national champions. Such individuals should be trusted for their health knowledge (e.g., television health care reporters) or be prominent, influential, and trusted figures in other circles (e.g., clergy, politicians, movie celebrities). Indeed, taking advantage of popular media by weaving a theme into a movie or television program about medication safety may prove effective.
Relevant partnerships would include:
-
Quality organizations partnering with other stakeholders to establish unambiguous and unified medication reconciliation standards across the care continuum.
-
Health systems partnering with community pharmacy providers to ensure an uninterrupted communication link in both the inpatient and outpatient settings.
-
Manufacturers and distributors of medications partnering with health care and public health organizations, the media, insurers and other constituents to promote the importance of maintaining and sharing an accurate list of medications.
-
Public health systems partnering with community‐based organizations to encourage and promote the established standards for medication safety through messaging and educational campaigns.
All partnerships must consider issues of patient language and literacy as well as the needs of vulnerable populations in the scope of their activities.
First Step
Public health agencies should partner with health care quality organizations and others to begin a national public campaign to increase the awareness of medication safety (the broader public health concept under which medication reconciliation would fall) and support the importance of the patient's role in maintaining an updated medication list at all times.
10. Align Financial Incentives With Newly Developed Regulatory and Accreditation Requirements
Implementing and performing medication reconciliation takes time, particularly at the outset of a new program. Time requirements and associated costs are major barriers to undertaking comprehensive medication reconciliation, despite its recognized importance for reducing avoidable injury to patients. At present, systems that impede efficiency and slow hospital throughput may be discouraged due to their potential for having an adverse impact on access, finances, and other aspects of care delivery. Moreover, the changed economic climate with reduced hospital fiscal margins limits resources for new initiatives. Currently, failed medication reconciliationand the related avoidable adverse events, culminating in readmission to the hospital or emergency departmentyields additional revenue for hospitals and other providers in some reimbursement models.
Alignment of financial incentives that ensured adequate time and resources for appropriate medication reconciliation processes would facilitate implementation. Additionally, start‐up funding to create and implement these processes needs to be made available.
One example illustrating efforts to align payment policy with medication safety efforts occurred when the Office of the National Coordinator (ONC), in publishing its Final Rule under the 2009 HITECH Act,24 endorsed the importance of financially supporting proper medication reconciliation, particularly at first encounter and transitions in care, by requiring EHR systems seeking certification under the rule to support the care team in the task of reconciliation. For example, vendors will have to support the ability to compare 2 or more medication lists electronically, create medication lists, drug allergy lists, perform drug formulary look‐ups, drug‐drug and drug‐allergy checks, and support creating patient summaries after each visit or post discharge that include medication lists. The ONC, in defining Meaningful Use for eligible health care organizations, included in that definition the goal of exchanging meaningful clinical information among the professional health care teams. This goal is demonstrated through organizations reporting that they performed medication reconciliation for at least 50% of transitions of care in which the patient is transitioned into the care of the eligible professional or admitted to the eligible hospital's or Critical Access Hospital's inpatient or emergency department. Organizations able to demonstrate this level of compliance, along with other Meaningful Use requirements, will be eligible to receive stimulus funds through 2015 and avoid financial penalties that begin after that period.
First Step
Future health care reform must address the misalignment of financial policies and structures, and provide financial incentives to support the development and implementation of better medication management systems and prevent avoidable rehospitalizations and emergency department visits resulting from medication‐related adverse events.
Conclusion
Medication reconciliation involves highly complex processes and is hampered by the disjointed nature of the American health care system. It is, however, a vital part of reducing ADE. If employed more broadly, it has the added benefits of enhancing communication among all providers of care and engaging patients and families/caregivers more consistently and meaningfully in their overall care.
Despite the difficulty of maintaining an accurate medication record in real time across disparate settings, reconciliation is a goal to which our organizations are committed. Given the wide range of healthcare organizations involved in providing medications to patients and the many agencies evaluating those efforts, we believed it would be helpful to provide an overarching set of goals to move medication reconciliation forward.
Our main message is this: Patient safety and patient/family‐centered care must be the principal drivers in the development and implementation of medication reconciliation systems. Ultimately this process is about ensuring that patients are receiving the most appropriate medications no matter where they are treated. With this document, we hope to bring to light the importance of creating and implementing a medication reconciliation program, addressing some barriers to success, and identifying potential solutions that will ensure utility and sustainability of this critical patient safety issue.
- ,,, et al.Unintended medication discrepancies at the time of hospital admission.Arch Intern Med.2005;165(4):424–429.
- .Prevention of medication errors in the pediatric inpatient setting. The American Academy of Pediatrics Policy Statement.Pediatrics.2003;112(2):431–436.
- ,,, et al.Medication reconciliation: a practical tool to reduce the risk of medication errors.J Crit Care.2003;18(4):201–205.
- Institute for Healthcare Improvement. 5 million lives getting started kit: preventing adverse drug events (medication reconciliation), how‐to guide. Available at: http://www.ihi.org/IHI/Programs/Campaign/ADEsMedReconciliation.htm. Published Oct. 1, 2008. Accessed September2010.
- ,.Medication safety: one organization's approach to the challenge.J Clin Outcomes Mana.2001;8(10):27–34.
- ,,,,,.Reconciliation of discrepancies in medication histories and admission orders of newly hospitalized patients.Am J Health Syst Pharm.2004;61(16):1689–1695.
- ,,, et al.Results of the Medications At Transitions and Clinical Handoffs (MATCH) Study: an analysis of medication reconciliation errors and risk factors at hospital admission.J Gen Intern Med.2010;25(5):441–447.
- Joint Commission on Accreditation of Healthcare Organizations.2005 Hospital Accreditation Standards, p.NPSG‐4.
- ,,,,.Brief communication: Results of a medication reconciliation survey from the 2006 Society of Hospital Medicine national meeting.J Hosp Med.2008;3(6):465–472.
- The Joint Commission.Approved: will not score medication reconciliation in 2009.Jt Comm Perspect.2009;29(3):1,3.
- Society of Hospital Medicine. Medication reconciliation: a team approach, conference summary. December 2009. Available at: http://www.hospitalmedicine.org/Content/NavigationMenu/QualityImprovement/QICurrentInitiativesandTrainingOpportunities/QI_Current_Initiativ.htm. Accessed September2010.
- The American Medical Association. The physician's role in medication reconciliation: issues, strategies and safety principles. 2007. Available at: http://www.ama‐assn.org/ama1/pub/upload/mm/370/med‐rec‐monograph.pdf. Accessed September2010.
- Institute of Safe Medication Practices. ISMP's list of high alert medications. 2008. Available at: http://www.ismp.org/Tools/highalertmedications.pdf. Accessed September2010.
- ,,,.Medication use leading to emergency department visits for adverse drug events in older adults.Ann Intern Med.2007;147(11):755–765
- ,,, et al.Experience with a trigger tool for identifying adverse drug events among older adults in ambulatory primary care.Qual Saf Health Care.2009;18(3):199–204.
- ,,, et al.Role of pharmacist counseling in preventing adverse drug events after hospitalization.Arch Intern Med.2006;166(5):565–571.
- ,,,,.Medication reconciliation at an academic medical center: implementation of a comprehensive program from admission to discharge.Am J Health Syst Pharm.2009;66(23):2126–2131.
- ,,,,,.Multidisciplinary approach to inpatient medication reconciliation in an academic setting.Am J Health Syst Pharm.2007;64(8):850–854.
- ,,.Lack of patient knowledge regarding hospital medications.J Hosp Med.2010;5(2):83–86.
- .The unexpected challenges of accurate medication reconciliation.Ann Emerg Med.2008;52(5):493–495.
- ,,,.Medication reconciliation in a rural trauma population.Ann Emerg Med.2008;52(5):483–491.
- ASTM International. ASTM E2369 ‐ 05e1 standard specification for continuity of care record (CCR). Available at: http://www.astm.org/Standards/E2369.htm. Accessed September2010.
- ,,.The continuity of care record.Am Fam Physician.2004;70(7):1220,1222–1223.
- Department of Health and Human Services. Health information technology: initial set of standards, implementation specifications, and certification criteria for electronic health record technology; final rule. Available at: http://edocket.access.gpo.gov/2010/pdf/2010–17210.pdf. Accessed September2010.
Medication reconciliation is integral to reducing medication errors surrounding hospitalizations.1, 2 The practice of medication reconciliation requires a systematic and comprehensive review of all the medications a patient is currently taking to ensure that medications being added, changed, or discontinued are carefully evaluated with the goal of maintaining an accurate list; that this process is undertaken at every transition along the continuum of care; and that an accurate list of medications is available to the patient or family/caregiver and all providers involved in the patient's care, especially when a care handoff takes place. With regulators, payers and the public increasingly demanding action to reduce medication errors in hospitals, all health care providers must support efforts to achieve accurate medication reconciliation.1, 3
The Joint Commission's Definition of Medication
Any prescription medications, sample medications, herbal remedies, vitamins, nutraceuticals, vaccines, or over‐the‐counter drugs; diagnostic and contrast agents used on or administered to persons to diagnose, treat, or prevent disease or other abnormal conditions; radioactive medications, respiratory therapy treatments, parenteral nutrition, blood derivatives, and intravenous solutions (plain, with electrolytes and/or drugs); and any product designated by the Food and Drug Administration (FDA) as a drug. This definition of medication does not include enteral nutrition solutions (which are considered food products), oxygen, and other medical gases.
2010 Hospital Accreditation Standards,
The Joint Commission, 2010, p. GL19.
While conceptually straightforward, implementing medication reconciliation has proved to be very difficult in the myriad healthcare settings that exist. The disjointed nature of the American health care system and a conglomeration of paper and electronic systems for tracking medications synergize to thwart efforts to maintain an accurate, up‐to‐date medication list at every step along the care continuum. Although The Joint Commission defines medication for the purpose of its accreditation standards (see box), the healthcare community lacks a common understanding or agreement regarding what constitutes a medication. There is also confusion about who should ultimately be responsible for obtaining the patient's medication information, for performing the various steps in the reconciliation process, and for managing the multiple providers who alter the medication list but may not feel competent to perform reconciliation of medications outside their area of expertise safely. Importantly, there is also a lack of clarity around how patients and family/caregivers should be involved in the process.
Despite these challenges, medication reconciliation remains a critical patient safety activity that is supported by the organizations signing this consensus statement, (Table 1). Although medication reconciliation has an impact on medication safety in all care settings, this paper focuses on issues most germane to the continuum of care involving the hospital setting. The themes and issues discussed will likely apply to other care settings as well. In this paper, we also recommend several concrete steps that we believe should be initiated immediately to begin to reach the goal of optimizing the medication safety achievable through effective medication reconciliation.
Background
Medication reconciliation is intended to be a systematic extension of the medication history‐taking process that has been used by health care providers for decades. Its recent iteration was developed to ensure that medications were not added, omitted, or changed inadvertently during care transitions. It became codified, refined, and tested over the past decade through the efforts of a number of groups focused on medication safety including the Institute for Healthcare Improvement (IHI) and the Institute for Safe Medication Practices (ISMP). With the reinforcing adoption of medication reconciliation as National Patient Safety Goal (NPSG) No. 8 in 2005 by The Joint Commission, efforts to implement it became widespread in both hospital‐based and ambulatory settings.
Medication reconciliation has three steps, as described by IHI4:
-
Verification (collection of the patient's medication history);
-
Clarification (ensuring that the medications and doses are appropriate); and
-
Reconciliation (documentation of changes in the orders).
The details of the process vary by setting and by the availability of paper or electronic medical records. However, the essential steps remain the same, as does the need to perform reconciliation each time the patient transfers to a new setting or level of care. Table 2 lists the most common points at which medication reconciliation occurs in hospitalized patients.
|
| American Academy of Pediatrics |
| American Association of Critical‐Care Nurses |
| Consumers Advancing Patient Safety |
| Institute for Healthcare Improvement |
| Institute for Safe Medication Practices |
| The Joint Commission |
| Massachusetts Coalition for Prevention of Medical Errors |
| Microsoft Corporation |
| Northwestern Memorial Hospital and Northwestern University School of Medicine |
| Society of General Internal Medicine |
| Society of Hospital Medicine |
| University of California San Diego Medical Center |
Because of their complexity, organizations must take care to design their medication reconciliation processes systematically. IHI lists elements of a well‐designed medication reconciliation process as part of its 5 Million Lives Campaign How‐to Guide.4 Such a process:
-
Uses a patient centered approach.
-
Makes it easy to complete the process for all involved. Staff members recognize the what's‐in‐it‐for‐me aspect of the change.
-
Minimizes the opportunity for drug interactions and therapeutic duplications by making the patient's list of current medications available when clinicians prescribe new medications.
-
Provides the patient with an up‐to‐date list of medications.
-
Ensures that other providers who need to know have information about changes in a patient's medication plan.
Research on how adverse drug events (ADE) occur supports the need for tight control of medication orders at transitions in care. For instance:
-
In a study conducted at Mayo Health System in Wisconsin, poor communication of medical information at transition points was responsible for as many as 50% of all medication errors in the hospital and up to 20% of ADEs.5
-
Variances between the medications patients were taking prior to admission and their admission orders ranged from 30% to 70% in 2 literature reviews.1, 6
-
The largest study of medication reconciliation errors and risk factors at hospital admission documented that 36% of patients had errors in their admission orders.7
When The Joint Commission adopted medication reconciliation as NPSG No. 8 in 2005 it had 2 parts: Requirement 8Aa process must exist for comparing the patient's current medications with those ordered for the patient while under the care of the organization; and requirement 8Ba complete list of the patient's medications must be communicated to the next provider of service on transfer within or outside the organization and a complete list of medications must be provided to the patient on discharge.8
However, many hospitals found it difficult to implement medication reconciliation in a systematic way. There was also confusion among hospital staff and administration about the exact definition of medication reconciliation in terms of what it should entail.9 Given these difficulties, The Joint Commission announced that effective January 1, 2009, medication reconciliation would no longer be factored into an organization's accreditation decision or be considered for Requirements for Improvement. Additionally, The Joint Commission stated it is reviewing and revising the NPSG so that it will be ready to be released in January 2011 for implementation later that year.10
Recognizing the difficulty hospitals were having with meaningfully implementing medication reconciliation, the Society of Hospital Medicine convened a 1‐day conference on March 6, 2009, to obtain input from key stakeholders and focus on several critical domains relevant to the success of hospital‐based medication reconciliation. The Agency for Healthcare Research and Quality provided funding support for this conference through grant 1R13HS017520‐01.
An overarching theme emerged from the meeting: the need to reorient the focus of medication reconciliation away from that of an accreditation mandate and toward a broader view of patient safety. Forcing medication reconciliation via a requirement for accreditation tended to limit an organization's efforts to specific process measures. Addressing it as a more global patient safety issue takes into account the entire patient care experience and then opens the door to leverage nonclinical venues (e.g., medical home, family home, community, religious, and other social organizations, as well as social networking platforms) and engage the patient and family/caregivers to reinforce the importance of medication safety.
This white paper evolved from discussions at the March 2009 conference,11 and subsequent structured communication among attendees. Formal endorsement of this document was obtained from the organizations listed in Table 1. In this document, we explore several key issues in implementing clinically meaningful and patient‐centered medication reconciliation. We focus on building common language and understanding of the processes of and participants in medication reconciliation; consider issues of implementation and risk stratification; emphasize the need for research to identify best practices and discusses how to disseminate the findings; promote health information technology platforms that will support interoperable medication information exchange; support the formation of partnerships between patient care sites and nonclinical sites as well as utilizing social marketing opportunities to enhance opportunities for transmitting messages about medication safety; and reinforce the ongoing healthcare reform discussion which aims to align financial incentives with patient safety efforts. After each section, we offer concrete first steps to address the issues discussed.
| Admission: When clinicians reconcile the patient's medications taken at home or at a prior care setting with any new prescription orders to be prescribed by an admitting clinician. |
| Transfer (intra‐ or inter‐facility; with change of clinician or site of care): When clinicians review previous medication orders in light of the patient's clinical status, along with new orders or plans of care. |
| Discharge: When clinicians review all medications the patient was taking prior to being hospitalized, incorporating new prescriptions from the hospitalization and determining whether any medication should be added, discontinued, or modified while being mindful of therapeutic interchanges needed for formulary purposes. |
Methods
The invitation‐only meeting held on the Northwestern Medical Campus in Chicago, IL, brought together stakeholders representing professional, clinical, health care quality, consumer, and regulatory organizations (Table 3). The conference convened these participants with the goals of identifying barriers to meaningful implementation of medication reconciliation and developing a feasible plan toward its effective implementation in the hospital setting. At the meeting, all participants were divided into 1 of 4 groups, which held a facilitated discussion around 1 of 4 key relevant domains: (1) how to measure success in medication reconciliation; (2) key elements of successful strategies; (3) leveraging partnerships outside the hospital setting to support medication reconciliation; and (4) the roles of the patient and family/caregivers and health literacy. Individual group discussions were cofacilitated by experts in the content area. After each discussion, the small group then rotated to a different discussion. Ultimately, each group participated in all four discussions, which built iteratively on the content derived from the prior groups' insights. Key comments were then shared with the large group for further discussion. To help build consensus, these large group discussions were directed by professional facilitators.
| AACN American Association of Critical Care Nurses |
| AAFP American Academy of Family Physicians |
| AAP American Academy of Pediatrics |
| ACEP American College of Emergency Physicians |
| ACP American College of Physicians |
| AMA American Medical Association |
| AMSN Academy of Medical Surgical Nurses |
| ASHP American Society of Health‐System Pharmacists |
| ASHP Foundation American Society of Health‐System Pharmacists Foundation |
| CAPS Consumers Advancing Patient Safety |
| CMS Centers for Medicare and Medicaid Services |
| CMSA Case Management Society of America |
| HCI Hospitalist Consultants, Inc |
| IHI Institute for Healthcare Improvement |
| InCompass Health |
| ISMP Institute For Safe Medication Practice |
| JCR Joint Commission Resources |
| Massachusetts Coalition for Prevention of Medical Errors |
| Microsoft Corporation |
| Northwestern Memorial Hospital MATCH Program |
| NQF National Quality Forum |
| SGIM Society of General Internal Medicine |
| SHM Society of Hospital Medicine |
| The Joint Commission |
| UCSD Hospital Medicine |
| University of Oklahoma College of Pharmacy Tulsa |
After the meeting, attendees participated in 2 follow‐up conference calls to discuss issues raised at the conference and responses obtained from host organizations. They also subsequently participated in two focus groups with The Joint Commission, giving input on the revision of the medication reconciliation NPSG.
Results
Addressing Barriers to Medication Reconciliation
In order to implement successful medication reconciliation processes, one must build the steps with the patient and family/caregiver as the focus and demonstrate an understanding of the intent of these processes. At its roots, medication reconciliation was developed to ensure that clinicians do not inadvertently add, change, or omit medications and that changes made are communicated to all relevant caregivers.
A number of key issues with respect to successful medication reconciliation processes surfaced in discussions with stakeholders. We believe addressing these issues is necessary before meaningful and standardized implementation can be achieved. After each discussion below, we provide suggested first steps to address these issues.
1. Achieve Consensus on the Definition of Medication and Reconciliation
Despite proposed definitions of these terms by various organizations, there was little agreement about them in the healthcare community. This ambiguity contributed to general confusion about what actually constitutes medication reconciliation. There needs to be a single, clear, and broadly accepted definition of what constitutes a medication. For the purposes of medication reconciliation, the term medication should be broadly inclusive of substances that may have an impact on the patient's care and treatments as well as those substances that may interact with other therapies potentially used during the medical care episode. Illicit or recreational substances may also have impact on therapies considered and therefore may influence this definition.12 Concretely, this definition should encompass prescription and over‐the‐counter medications as well as herbal and dietary supplements.
The term reconciliation in its simplest form implies the process of verifying that a patient's current list of medications (including dose, route, and frequency) are correct and that the medications are currently medically necessary and safe. Reconciliation suggests a process which, by necessity, will vary based on clinical context and setting. Further defining this termand the process of reconciliation itselfshould be carried out using patient safety principles with a focus on patient‐ and family‐centeredness.
Designing hospital‐based medication reconciliation processes should:
-
Employ a multidisciplinary approach that involves nurses, pharmacists, and other appropriate personnel from the inpatient setting as well as ambulatory and community/retail areas, both ambulatory and inpatient physicians, and a patient/family representative;
-
Involve hospital leaders who support, provide guidance, and remove barriers for the multidisciplinary team working to implement the processes;
-
Clearly define the roles of each participant in the processes developed;
-
Include methods to assess and address any special needs due to the developmental stage, age, dependency, language or literacy levels of patients and their family/caregiver;
-
Use clinically relevant process measures (e.g., adherence to procedural steps) and outcome measures (e.g., change in the number of ADEs, unnecessary hospitalizations, or emergency department visits) where appropriate to assess the impact of the process;
-
Include feedback systems to allow for clinically significant process improvement.
Once a common understanding of the terms and intent of medication reconciliation is achieved, it will be important for accrediting organizations, medical societies, quality improvement organizations, and other interested parties to adopt the same language.
First Step
A consortium of clinical, quality, and regulatory stakeholders should work to achieve consensus on the definition for medication and the intent and expectations for the reconciliation process.
2. Clarify Roles and Responsibilities
Given the differences in organizational and practice structures in hospitals and the varying numbers of health professionals involved in a patient's care, no one process design will meet the needs of all sites. As it is clear that interdisciplinary teams are best suited to develop, implement, and carry out complex patient‐centered processes like medication reconciliation, it is crucial that all involved parties have clearly defined roles and responsibilities, including patients and their families/caregivers. It is also important to recognize that these responsibilities may change depending on the dependency or vulnerability of the patient (e.g., children or geriatric patients) or the transition of care being undertaken by the patient (i.e., admission, transfer, or discharge), thus requiring sites to develop clear policies about these roles and responsibilities and how they may change in various situations.
First Step
Individual sites must clearly define the roles and responsibilities of all parties directly involved in medication reconciliation as a part of designing local medication reconciliation processes.
3. Develop Measurement Tools
Ensuring that medication reconciliation processes result in clinically meaningful outcomes requires the development and standardization of a limited number of metrics that may be used by organizations and reported centrally for benchmarking. This core set of measures should be developed by clinical, quality, accreditation, and regulatory organizations (see #10 below) through a consensus building process utilizing multi‐stakeholder input. The set should be supplemented by additional site‐specific measures determined locally that focus on steps in the process itself and allow sites to perform continuous quality improvement. Sites should be encouraged to develop tools locally to support and facilitate organizational and professional adherence to medication reconciliation processes.
First Steps
Clinical, quality, accreditation, and regulatory organizations should develop reliable metrics to be assessed and reported.
The principles of patient‐centeredness and family/caregiver‐centeredness, the medical home, and clinical relevance must be central to the metrics chosen for quality and regulatory purposes.
4. Phased Implementation
Ultimately, comprehensive medication reconciliation processes need to be implemented in hospitals. However, to succeed in integrating complex processes like medication reconciliation into routine hospital practices, implementation may be facilitated by using a phased approach to allow for participants to adapt new processes and procedures to the local environment iteratively. While the most appropriate phased approach to implementation will vary by site and setting, options for phasing might include:
-
Starting with one clinical area or service.
-
Starting with either the admission or discharge reconciliation process.
-
Starting with a patient population at high risk for adverse events.
Irrespective of the phasing strategy employed, development of a clear and pragmatic schedule for the entire implementation process should be established. Phasing decisions should be made based on organizational resources and the clinical needs of the patient population within each clinical setting. As noted, the ultimate goal is to develop comprehensive reconciliation processes occurring during all significant care transitions (i.e., admission, service or site‐of‐care transfers, and discharge) for all hospitalized patients and involving all of their medications. Flexibility in design should be encouraged to ensure the processes can work within local workflow as long as progress toward this primary goal is made.
First Steps
Clinical sites should establish local, pragmatic priorities for a phased approach to implementation.
Tie the phased approach to a timeline or blueprint for programmatic expansion with ultimate plans for comprehensive implementation.
5. Develop Risk Stratification Systems
Medication‐related adverse events related to inadequate reconciliation are more likely to occur in hospitalized patients with certain identifiable risk factors. For example, the MATCH study documented that polypharmacy and age over 65 years were independently associated with increased risk for errors at the time of hospital admission.7 Other factors that may increase the likelihood of medication‐related adverse events at care transitions in the hospital might include: patients with multiple providers, developmental/cognitive impairment, dependency/vulnerability, multiple or high‐risk medications, or poor health literacy or limited English proficiency. Research is needed to elucidate these risk factors further.
An alert system for key risk factors for complications related to incompletely, inappropriately, or inaccurately completed medication reconciliation due to patient, clinician, or system factors should be developed, tested, and broadly implemented. Additionally, an alert system would help maintain vigilance toward this patient safety issue and, potentially, help focus additional resources on high‐risk patients. Such a tool has been tested in ambulatory settings.15
First Step
Additional research on inpatient predictors of failed medication reconciliation and ADE should be prioritized (see #6 below).
6. Study Interventions and Processes
Despite having been an NPSG since 2005, there is still a relative paucity of literature about broadly applicable and effective implementation strategies and demonstrated interventions that improve medication safety related to medication reconciliation. Some strategies that have shown to reduce medication errors at transitions include the involvement of pharmacist medication review on discharge16, 17 and the usefulness of planning by multidisciplinary groups.18 Other studies have outlined the continuing barriers to successful implementation of reconciliation, including the difficulty patients have in accurately recalling their current medications19 and the high cost in nurse and pharmacist time of tracking down a patient's ongoing prescriptions.20, 21 Studies evaluating potential solutions to overcome these and other common barriers are still needed.
Future research should focus on a comprehensive review of implementation strategies, (specifically including the role of health information technology‐based innovations) clinically relevant outcomes, and best practices, while being sensitive to the different needs of varying care settings (e.g., pediatric vs. adult centers, emergency departments vs. inpatient units, community hospital vs. academic medical center, etc.) as well as the resource requirements engendered in the interventions.
First Step
Funding agencies should explicitly prioritize outcomes‐focused medication reconciliation‐related projects (e.g., those which demonstrate a reduction in postdischarge ADE or reduced medication‐related emergency department visits). Previously identified successful strategies should be further investigated. Funded projects should explicitly partner with patients and family/caregivers and also include pediatric and adult patients, rural and urban locations of care, as well as academic and nonacademic hospital settings, to promote more broadly applicable results.
7. Disseminate Success
Best practices and lessons learned, especially those rigorously tested and driven by data, stratified by patient type, care setting (emergency department, intensive care, surgical ward, etc.) and institutional type (community, teaching, safety net, critical access, etc.) need to be disseminated so others can adopt and adapt them effectively. High‐quality case studies with clear explanations of successes, failures, and lessons learned may prove valuable sources of information. This knowledge should foster a learning community approach and accelerate implementation at new sites.
First Step
Hospitals, healthcare systems, as well as quality and regulatory agencies should develop mechanisms within reporting systems to track performance, identify notably successful sites, and publicly report and share methods and lessons learned from them.
8. Promote the Personal Health Record
A fully integrated and transferable personal health record should be accepted as the standard for health information storage and interoperability, giving both the patient (or family/caregiver) and clinical providers access and ownership. Both the HL7 Continuity of Care Document (CCD) and the Continuity of Care Record (CCR) meet these criteria. The CCR was endorsed by the American Society for Testing and Materials22 and a coalition of other medical societies.23 Notably, CCR and CCD were recently adopted as standards for structured electronic health record (EHR) exchange through the July 2010 publication of the Final Rule of the Health Information Technology for Economic and Clinical Health Act provision of the American Recovery and Reinvestment Act of 2009 (ARRA/HITECH) and is now part of the formal US Department of Health and Human Services certification criteria for EHR technologies.24
Mandating a content exchange standard such as the CCR or the CCD should also have the desired effect of ensuring that patients (and their caregivers) become increasingly involved in maintaining an accurate list of the medications they take. Additionally, systems must be sufficiently flexible to address the unique medication management needs of children and geriatric patients. An electronic version of a personal health record is a promising method for improving consistency across care platforms, but to be implemented effectively the record must be compatible across all settings, including, where possible, the patient's home. All health care organizations, pharmacy systems, and insurers, must make medication reconciliation‐related interoperability and accessibility a priority as they pursue information technology strategies.
First Step
Stakeholder organizations must send a clear and convincing message to legislators under the current atmosphere of health care reform, urging them to mandate that health information technology standards include interoperability and support platforms that are consistent with standards put forth in the 2009 HITECH Act Interim Final Rule for EHR certification.
9. Promote Partnerships
At a broader health care system level, leveraging existing partnerships and creating new ones among health care, public/private sector‐affiliated organizations (e.g., community and mail order pharmacies, pharmaceutical organizations and manufacturers, and insurers), and public health organizations are extremely important mechanisms for broader scale impact. This view recognizes the numerous opportunities to educate and influence patients about medication safety outside the dyadic relationship of the clinician and patient in traditional clinical settings. Partnerships between health care and public entities may capitalize on these opportunities to foster adoption of healthy medication practices (e.g., maintaining an accurate and updated medication list), thereby supporting medication reconciliation efforts when individuals encounter health care settings. Partnership and information sharing could be enhanced through the use of a central coordinating body or coalition. This body could generate a shared common vision and contribute expertise to the myriad issues in medication reconciliation.
Partnerships should utilize the following:
-
Social marketing techniques to engage the community. Included within this strategy must be a clear and compelling message that transmits the importance of safe medication practices. Current messages such as keep a list while important, do not offer enough of a sense of urgency or importance. A more powerful message could involve highly publicized medication errors or close calls that would resonate with a broad audience.
-
Local and national champions. Such individuals should be trusted for their health knowledge (e.g., television health care reporters) or be prominent, influential, and trusted figures in other circles (e.g., clergy, politicians, movie celebrities). Indeed, taking advantage of popular media by weaving a theme into a movie or television program about medication safety may prove effective.
Relevant partnerships would include:
-
Quality organizations partnering with other stakeholders to establish unambiguous and unified medication reconciliation standards across the care continuum.
-
Health systems partnering with community pharmacy providers to ensure an uninterrupted communication link in both the inpatient and outpatient settings.
-
Manufacturers and distributors of medications partnering with health care and public health organizations, the media, insurers and other constituents to promote the importance of maintaining and sharing an accurate list of medications.
-
Public health systems partnering with community‐based organizations to encourage and promote the established standards for medication safety through messaging and educational campaigns.
All partnerships must consider issues of patient language and literacy as well as the needs of vulnerable populations in the scope of their activities.
First Step
Public health agencies should partner with health care quality organizations and others to begin a national public campaign to increase the awareness of medication safety (the broader public health concept under which medication reconciliation would fall) and support the importance of the patient's role in maintaining an updated medication list at all times.
10. Align Financial Incentives With Newly Developed Regulatory and Accreditation Requirements
Implementing and performing medication reconciliation takes time, particularly at the outset of a new program. Time requirements and associated costs are major barriers to undertaking comprehensive medication reconciliation, despite its recognized importance for reducing avoidable injury to patients. At present, systems that impede efficiency and slow hospital throughput may be discouraged due to their potential for having an adverse impact on access, finances, and other aspects of care delivery. Moreover, the changed economic climate with reduced hospital fiscal margins limits resources for new initiatives. Currently, failed medication reconciliationand the related avoidable adverse events, culminating in readmission to the hospital or emergency departmentyields additional revenue for hospitals and other providers in some reimbursement models.
Alignment of financial incentives that ensured adequate time and resources for appropriate medication reconciliation processes would facilitate implementation. Additionally, start‐up funding to create and implement these processes needs to be made available.
One example illustrating efforts to align payment policy with medication safety efforts occurred when the Office of the National Coordinator (ONC), in publishing its Final Rule under the 2009 HITECH Act,24 endorsed the importance of financially supporting proper medication reconciliation, particularly at first encounter and transitions in care, by requiring EHR systems seeking certification under the rule to support the care team in the task of reconciliation. For example, vendors will have to support the ability to compare 2 or more medication lists electronically, create medication lists, drug allergy lists, perform drug formulary look‐ups, drug‐drug and drug‐allergy checks, and support creating patient summaries after each visit or post discharge that include medication lists. The ONC, in defining Meaningful Use for eligible health care organizations, included in that definition the goal of exchanging meaningful clinical information among the professional health care teams. This goal is demonstrated through organizations reporting that they performed medication reconciliation for at least 50% of transitions of care in which the patient is transitioned into the care of the eligible professional or admitted to the eligible hospital's or Critical Access Hospital's inpatient or emergency department. Organizations able to demonstrate this level of compliance, along with other Meaningful Use requirements, will be eligible to receive stimulus funds through 2015 and avoid financial penalties that begin after that period.
First Step
Future health care reform must address the misalignment of financial policies and structures, and provide financial incentives to support the development and implementation of better medication management systems and prevent avoidable rehospitalizations and emergency department visits resulting from medication‐related adverse events.
Conclusion
Medication reconciliation involves highly complex processes and is hampered by the disjointed nature of the American health care system. It is, however, a vital part of reducing ADE. If employed more broadly, it has the added benefits of enhancing communication among all providers of care and engaging patients and families/caregivers more consistently and meaningfully in their overall care.
Despite the difficulty of maintaining an accurate medication record in real time across disparate settings, reconciliation is a goal to which our organizations are committed. Given the wide range of healthcare organizations involved in providing medications to patients and the many agencies evaluating those efforts, we believed it would be helpful to provide an overarching set of goals to move medication reconciliation forward.
Our main message is this: Patient safety and patient/family‐centered care must be the principal drivers in the development and implementation of medication reconciliation systems. Ultimately this process is about ensuring that patients are receiving the most appropriate medications no matter where they are treated. With this document, we hope to bring to light the importance of creating and implementing a medication reconciliation program, addressing some barriers to success, and identifying potential solutions that will ensure utility and sustainability of this critical patient safety issue.
Medication reconciliation is integral to reducing medication errors surrounding hospitalizations.1, 2 The practice of medication reconciliation requires a systematic and comprehensive review of all the medications a patient is currently taking to ensure that medications being added, changed, or discontinued are carefully evaluated with the goal of maintaining an accurate list; that this process is undertaken at every transition along the continuum of care; and that an accurate list of medications is available to the patient or family/caregiver and all providers involved in the patient's care, especially when a care handoff takes place. With regulators, payers and the public increasingly demanding action to reduce medication errors in hospitals, all health care providers must support efforts to achieve accurate medication reconciliation.1, 3
The Joint Commission's Definition of Medication
Any prescription medications, sample medications, herbal remedies, vitamins, nutraceuticals, vaccines, or over‐the‐counter drugs; diagnostic and contrast agents used on or administered to persons to diagnose, treat, or prevent disease or other abnormal conditions; radioactive medications, respiratory therapy treatments, parenteral nutrition, blood derivatives, and intravenous solutions (plain, with electrolytes and/or drugs); and any product designated by the Food and Drug Administration (FDA) as a drug. This definition of medication does not include enteral nutrition solutions (which are considered food products), oxygen, and other medical gases.
2010 Hospital Accreditation Standards,
The Joint Commission, 2010, p. GL19.
While conceptually straightforward, implementing medication reconciliation has proved to be very difficult in the myriad healthcare settings that exist. The disjointed nature of the American health care system and a conglomeration of paper and electronic systems for tracking medications synergize to thwart efforts to maintain an accurate, up‐to‐date medication list at every step along the care continuum. Although The Joint Commission defines medication for the purpose of its accreditation standards (see box), the healthcare community lacks a common understanding or agreement regarding what constitutes a medication. There is also confusion about who should ultimately be responsible for obtaining the patient's medication information, for performing the various steps in the reconciliation process, and for managing the multiple providers who alter the medication list but may not feel competent to perform reconciliation of medications outside their area of expertise safely. Importantly, there is also a lack of clarity around how patients and family/caregivers should be involved in the process.
Despite these challenges, medication reconciliation remains a critical patient safety activity that is supported by the organizations signing this consensus statement, (Table 1). Although medication reconciliation has an impact on medication safety in all care settings, this paper focuses on issues most germane to the continuum of care involving the hospital setting. The themes and issues discussed will likely apply to other care settings as well. In this paper, we also recommend several concrete steps that we believe should be initiated immediately to begin to reach the goal of optimizing the medication safety achievable through effective medication reconciliation.
Background
Medication reconciliation is intended to be a systematic extension of the medication history‐taking process that has been used by health care providers for decades. Its recent iteration was developed to ensure that medications were not added, omitted, or changed inadvertently during care transitions. It became codified, refined, and tested over the past decade through the efforts of a number of groups focused on medication safety including the Institute for Healthcare Improvement (IHI) and the Institute for Safe Medication Practices (ISMP). With the reinforcing adoption of medication reconciliation as National Patient Safety Goal (NPSG) No. 8 in 2005 by The Joint Commission, efforts to implement it became widespread in both hospital‐based and ambulatory settings.
Medication reconciliation has three steps, as described by IHI4:
-
Verification (collection of the patient's medication history);
-
Clarification (ensuring that the medications and doses are appropriate); and
-
Reconciliation (documentation of changes in the orders).
The details of the process vary by setting and by the availability of paper or electronic medical records. However, the essential steps remain the same, as does the need to perform reconciliation each time the patient transfers to a new setting or level of care. Table 2 lists the most common points at which medication reconciliation occurs in hospitalized patients.
|
| American Academy of Pediatrics |
| American Association of Critical‐Care Nurses |
| Consumers Advancing Patient Safety |
| Institute for Healthcare Improvement |
| Institute for Safe Medication Practices |
| The Joint Commission |
| Massachusetts Coalition for Prevention of Medical Errors |
| Microsoft Corporation |
| Northwestern Memorial Hospital and Northwestern University School of Medicine |
| Society of General Internal Medicine |
| Society of Hospital Medicine |
| University of California San Diego Medical Center |
Because of their complexity, organizations must take care to design their medication reconciliation processes systematically. IHI lists elements of a well‐designed medication reconciliation process as part of its 5 Million Lives Campaign How‐to Guide.4 Such a process:
-
Uses a patient centered approach.
-
Makes it easy to complete the process for all involved. Staff members recognize the what's‐in‐it‐for‐me aspect of the change.
-
Minimizes the opportunity for drug interactions and therapeutic duplications by making the patient's list of current medications available when clinicians prescribe new medications.
-
Provides the patient with an up‐to‐date list of medications.
-
Ensures that other providers who need to know have information about changes in a patient's medication plan.
Research on how adverse drug events (ADE) occur supports the need for tight control of medication orders at transitions in care. For instance:
-
In a study conducted at Mayo Health System in Wisconsin, poor communication of medical information at transition points was responsible for as many as 50% of all medication errors in the hospital and up to 20% of ADEs.5
-
Variances between the medications patients were taking prior to admission and their admission orders ranged from 30% to 70% in 2 literature reviews.1, 6
-
The largest study of medication reconciliation errors and risk factors at hospital admission documented that 36% of patients had errors in their admission orders.7
When The Joint Commission adopted medication reconciliation as NPSG No. 8 in 2005 it had 2 parts: Requirement 8Aa process must exist for comparing the patient's current medications with those ordered for the patient while under the care of the organization; and requirement 8Ba complete list of the patient's medications must be communicated to the next provider of service on transfer within or outside the organization and a complete list of medications must be provided to the patient on discharge.8
However, many hospitals found it difficult to implement medication reconciliation in a systematic way. There was also confusion among hospital staff and administration about the exact definition of medication reconciliation in terms of what it should entail.9 Given these difficulties, The Joint Commission announced that effective January 1, 2009, medication reconciliation would no longer be factored into an organization's accreditation decision or be considered for Requirements for Improvement. Additionally, The Joint Commission stated it is reviewing and revising the NPSG so that it will be ready to be released in January 2011 for implementation later that year.10
Recognizing the difficulty hospitals were having with meaningfully implementing medication reconciliation, the Society of Hospital Medicine convened a 1‐day conference on March 6, 2009, to obtain input from key stakeholders and focus on several critical domains relevant to the success of hospital‐based medication reconciliation. The Agency for Healthcare Research and Quality provided funding support for this conference through grant 1R13HS017520‐01.
An overarching theme emerged from the meeting: the need to reorient the focus of medication reconciliation away from that of an accreditation mandate and toward a broader view of patient safety. Forcing medication reconciliation via a requirement for accreditation tended to limit an organization's efforts to specific process measures. Addressing it as a more global patient safety issue takes into account the entire patient care experience and then opens the door to leverage nonclinical venues (e.g., medical home, family home, community, religious, and other social organizations, as well as social networking platforms) and engage the patient and family/caregivers to reinforce the importance of medication safety.
This white paper evolved from discussions at the March 2009 conference,11 and subsequent structured communication among attendees. Formal endorsement of this document was obtained from the organizations listed in Table 1. In this document, we explore several key issues in implementing clinically meaningful and patient‐centered medication reconciliation. We focus on building common language and understanding of the processes of and participants in medication reconciliation; consider issues of implementation and risk stratification; emphasize the need for research to identify best practices and discusses how to disseminate the findings; promote health information technology platforms that will support interoperable medication information exchange; support the formation of partnerships between patient care sites and nonclinical sites as well as utilizing social marketing opportunities to enhance opportunities for transmitting messages about medication safety; and reinforce the ongoing healthcare reform discussion which aims to align financial incentives with patient safety efforts. After each section, we offer concrete first steps to address the issues discussed.
| Admission: When clinicians reconcile the patient's medications taken at home or at a prior care setting with any new prescription orders to be prescribed by an admitting clinician. |
| Transfer (intra‐ or inter‐facility; with change of clinician or site of care): When clinicians review previous medication orders in light of the patient's clinical status, along with new orders or plans of care. |
| Discharge: When clinicians review all medications the patient was taking prior to being hospitalized, incorporating new prescriptions from the hospitalization and determining whether any medication should be added, discontinued, or modified while being mindful of therapeutic interchanges needed for formulary purposes. |
Methods
The invitation‐only meeting held on the Northwestern Medical Campus in Chicago, IL, brought together stakeholders representing professional, clinical, health care quality, consumer, and regulatory organizations (Table 3). The conference convened these participants with the goals of identifying barriers to meaningful implementation of medication reconciliation and developing a feasible plan toward its effective implementation in the hospital setting. At the meeting, all participants were divided into 1 of 4 groups, which held a facilitated discussion around 1 of 4 key relevant domains: (1) how to measure success in medication reconciliation; (2) key elements of successful strategies; (3) leveraging partnerships outside the hospital setting to support medication reconciliation; and (4) the roles of the patient and family/caregivers and health literacy. Individual group discussions were cofacilitated by experts in the content area. After each discussion, the small group then rotated to a different discussion. Ultimately, each group participated in all four discussions, which built iteratively on the content derived from the prior groups' insights. Key comments were then shared with the large group for further discussion. To help build consensus, these large group discussions were directed by professional facilitators.
| AACN American Association of Critical Care Nurses |
| AAFP American Academy of Family Physicians |
| AAP American Academy of Pediatrics |
| ACEP American College of Emergency Physicians |
| ACP American College of Physicians |
| AMA American Medical Association |
| AMSN Academy of Medical Surgical Nurses |
| ASHP American Society of Health‐System Pharmacists |
| ASHP Foundation American Society of Health‐System Pharmacists Foundation |
| CAPS Consumers Advancing Patient Safety |
| CMS Centers for Medicare and Medicaid Services |
| CMSA Case Management Society of America |
| HCI Hospitalist Consultants, Inc |
| IHI Institute for Healthcare Improvement |
| InCompass Health |
| ISMP Institute For Safe Medication Practice |
| JCR Joint Commission Resources |
| Massachusetts Coalition for Prevention of Medical Errors |
| Microsoft Corporation |
| Northwestern Memorial Hospital MATCH Program |
| NQF National Quality Forum |
| SGIM Society of General Internal Medicine |
| SHM Society of Hospital Medicine |
| The Joint Commission |
| UCSD Hospital Medicine |
| University of Oklahoma College of Pharmacy Tulsa |
After the meeting, attendees participated in 2 follow‐up conference calls to discuss issues raised at the conference and responses obtained from host organizations. They also subsequently participated in two focus groups with The Joint Commission, giving input on the revision of the medication reconciliation NPSG.
Results
Addressing Barriers to Medication Reconciliation
In order to implement successful medication reconciliation processes, one must build the steps with the patient and family/caregiver as the focus and demonstrate an understanding of the intent of these processes. At its roots, medication reconciliation was developed to ensure that clinicians do not inadvertently add, change, or omit medications and that changes made are communicated to all relevant caregivers.
A number of key issues with respect to successful medication reconciliation processes surfaced in discussions with stakeholders. We believe addressing these issues is necessary before meaningful and standardized implementation can be achieved. After each discussion below, we provide suggested first steps to address these issues.
1. Achieve Consensus on the Definition of Medication and Reconciliation
Despite proposed definitions of these terms by various organizations, there was little agreement about them in the healthcare community. This ambiguity contributed to general confusion about what actually constitutes medication reconciliation. There needs to be a single, clear, and broadly accepted definition of what constitutes a medication. For the purposes of medication reconciliation, the term medication should be broadly inclusive of substances that may have an impact on the patient's care and treatments as well as those substances that may interact with other therapies potentially used during the medical care episode. Illicit or recreational substances may also have impact on therapies considered and therefore may influence this definition.12 Concretely, this definition should encompass prescription and over‐the‐counter medications as well as herbal and dietary supplements.
The term reconciliation in its simplest form implies the process of verifying that a patient's current list of medications (including dose, route, and frequency) are correct and that the medications are currently medically necessary and safe. Reconciliation suggests a process which, by necessity, will vary based on clinical context and setting. Further defining this termand the process of reconciliation itselfshould be carried out using patient safety principles with a focus on patient‐ and family‐centeredness.
Designing hospital‐based medication reconciliation processes should:
-
Employ a multidisciplinary approach that involves nurses, pharmacists, and other appropriate personnel from the inpatient setting as well as ambulatory and community/retail areas, both ambulatory and inpatient physicians, and a patient/family representative;
-
Involve hospital leaders who support, provide guidance, and remove barriers for the multidisciplinary team working to implement the processes;
-
Clearly define the roles of each participant in the processes developed;
-
Include methods to assess and address any special needs due to the developmental stage, age, dependency, language or literacy levels of patients and their family/caregiver;
-
Use clinically relevant process measures (e.g., adherence to procedural steps) and outcome measures (e.g., change in the number of ADEs, unnecessary hospitalizations, or emergency department visits) where appropriate to assess the impact of the process;
-
Include feedback systems to allow for clinically significant process improvement.
Once a common understanding of the terms and intent of medication reconciliation is achieved, it will be important for accrediting organizations, medical societies, quality improvement organizations, and other interested parties to adopt the same language.
First Step
A consortium of clinical, quality, and regulatory stakeholders should work to achieve consensus on the definition for medication and the intent and expectations for the reconciliation process.
2. Clarify Roles and Responsibilities
Given the differences in organizational and practice structures in hospitals and the varying numbers of health professionals involved in a patient's care, no one process design will meet the needs of all sites. As it is clear that interdisciplinary teams are best suited to develop, implement, and carry out complex patient‐centered processes like medication reconciliation, it is crucial that all involved parties have clearly defined roles and responsibilities, including patients and their families/caregivers. It is also important to recognize that these responsibilities may change depending on the dependency or vulnerability of the patient (e.g., children or geriatric patients) or the transition of care being undertaken by the patient (i.e., admission, transfer, or discharge), thus requiring sites to develop clear policies about these roles and responsibilities and how they may change in various situations.
First Step
Individual sites must clearly define the roles and responsibilities of all parties directly involved in medication reconciliation as a part of designing local medication reconciliation processes.
3. Develop Measurement Tools
Ensuring that medication reconciliation processes result in clinically meaningful outcomes requires the development and standardization of a limited number of metrics that may be used by organizations and reported centrally for benchmarking. This core set of measures should be developed by clinical, quality, accreditation, and regulatory organizations (see #10 below) through a consensus building process utilizing multi‐stakeholder input. The set should be supplemented by additional site‐specific measures determined locally that focus on steps in the process itself and allow sites to perform continuous quality improvement. Sites should be encouraged to develop tools locally to support and facilitate organizational and professional adherence to medication reconciliation processes.
First Steps
Clinical, quality, accreditation, and regulatory organizations should develop reliable metrics to be assessed and reported.
The principles of patient‐centeredness and family/caregiver‐centeredness, the medical home, and clinical relevance must be central to the metrics chosen for quality and regulatory purposes.
4. Phased Implementation
Ultimately, comprehensive medication reconciliation processes need to be implemented in hospitals. However, to succeed in integrating complex processes like medication reconciliation into routine hospital practices, implementation may be facilitated by using a phased approach to allow for participants to adapt new processes and procedures to the local environment iteratively. While the most appropriate phased approach to implementation will vary by site and setting, options for phasing might include:
-
Starting with one clinical area or service.
-
Starting with either the admission or discharge reconciliation process.
-
Starting with a patient population at high risk for adverse events.
Irrespective of the phasing strategy employed, development of a clear and pragmatic schedule for the entire implementation process should be established. Phasing decisions should be made based on organizational resources and the clinical needs of the patient population within each clinical setting. As noted, the ultimate goal is to develop comprehensive reconciliation processes occurring during all significant care transitions (i.e., admission, service or site‐of‐care transfers, and discharge) for all hospitalized patients and involving all of their medications. Flexibility in design should be encouraged to ensure the processes can work within local workflow as long as progress toward this primary goal is made.
First Steps
Clinical sites should establish local, pragmatic priorities for a phased approach to implementation.
Tie the phased approach to a timeline or blueprint for programmatic expansion with ultimate plans for comprehensive implementation.
5. Develop Risk Stratification Systems
Medication‐related adverse events related to inadequate reconciliation are more likely to occur in hospitalized patients with certain identifiable risk factors. For example, the MATCH study documented that polypharmacy and age over 65 years were independently associated with increased risk for errors at the time of hospital admission.7 Other factors that may increase the likelihood of medication‐related adverse events at care transitions in the hospital might include: patients with multiple providers, developmental/cognitive impairment, dependency/vulnerability, multiple or high‐risk medications, or poor health literacy or limited English proficiency. Research is needed to elucidate these risk factors further.
An alert system for key risk factors for complications related to incompletely, inappropriately, or inaccurately completed medication reconciliation due to patient, clinician, or system factors should be developed, tested, and broadly implemented. Additionally, an alert system would help maintain vigilance toward this patient safety issue and, potentially, help focus additional resources on high‐risk patients. Such a tool has been tested in ambulatory settings.15
First Step
Additional research on inpatient predictors of failed medication reconciliation and ADE should be prioritized (see #6 below).
6. Study Interventions and Processes
Despite having been an NPSG since 2005, there is still a relative paucity of literature about broadly applicable and effective implementation strategies and demonstrated interventions that improve medication safety related to medication reconciliation. Some strategies that have shown to reduce medication errors at transitions include the involvement of pharmacist medication review on discharge16, 17 and the usefulness of planning by multidisciplinary groups.18 Other studies have outlined the continuing barriers to successful implementation of reconciliation, including the difficulty patients have in accurately recalling their current medications19 and the high cost in nurse and pharmacist time of tracking down a patient's ongoing prescriptions.20, 21 Studies evaluating potential solutions to overcome these and other common barriers are still needed.
Future research should focus on a comprehensive review of implementation strategies, (specifically including the role of health information technology‐based innovations) clinically relevant outcomes, and best practices, while being sensitive to the different needs of varying care settings (e.g., pediatric vs. adult centers, emergency departments vs. inpatient units, community hospital vs. academic medical center, etc.) as well as the resource requirements engendered in the interventions.
First Step
Funding agencies should explicitly prioritize outcomes‐focused medication reconciliation‐related projects (e.g., those which demonstrate a reduction in postdischarge ADE or reduced medication‐related emergency department visits). Previously identified successful strategies should be further investigated. Funded projects should explicitly partner with patients and family/caregivers and also include pediatric and adult patients, rural and urban locations of care, as well as academic and nonacademic hospital settings, to promote more broadly applicable results.
7. Disseminate Success
Best practices and lessons learned, especially those rigorously tested and driven by data, stratified by patient type, care setting (emergency department, intensive care, surgical ward, etc.) and institutional type (community, teaching, safety net, critical access, etc.) need to be disseminated so others can adopt and adapt them effectively. High‐quality case studies with clear explanations of successes, failures, and lessons learned may prove valuable sources of information. This knowledge should foster a learning community approach and accelerate implementation at new sites.
First Step
Hospitals, healthcare systems, as well as quality and regulatory agencies should develop mechanisms within reporting systems to track performance, identify notably successful sites, and publicly report and share methods and lessons learned from them.
8. Promote the Personal Health Record
A fully integrated and transferable personal health record should be accepted as the standard for health information storage and interoperability, giving both the patient (or family/caregiver) and clinical providers access and ownership. Both the HL7 Continuity of Care Document (CCD) and the Continuity of Care Record (CCR) meet these criteria. The CCR was endorsed by the American Society for Testing and Materials22 and a coalition of other medical societies.23 Notably, CCR and CCD were recently adopted as standards for structured electronic health record (EHR) exchange through the July 2010 publication of the Final Rule of the Health Information Technology for Economic and Clinical Health Act provision of the American Recovery and Reinvestment Act of 2009 (ARRA/HITECH) and is now part of the formal US Department of Health and Human Services certification criteria for EHR technologies.24
Mandating a content exchange standard such as the CCR or the CCD should also have the desired effect of ensuring that patients (and their caregivers) become increasingly involved in maintaining an accurate list of the medications they take. Additionally, systems must be sufficiently flexible to address the unique medication management needs of children and geriatric patients. An electronic version of a personal health record is a promising method for improving consistency across care platforms, but to be implemented effectively the record must be compatible across all settings, including, where possible, the patient's home. All health care organizations, pharmacy systems, and insurers, must make medication reconciliation‐related interoperability and accessibility a priority as they pursue information technology strategies.
First Step
Stakeholder organizations must send a clear and convincing message to legislators under the current atmosphere of health care reform, urging them to mandate that health information technology standards include interoperability and support platforms that are consistent with standards put forth in the 2009 HITECH Act Interim Final Rule for EHR certification.
9. Promote Partnerships
At a broader health care system level, leveraging existing partnerships and creating new ones among health care, public/private sector‐affiliated organizations (e.g., community and mail order pharmacies, pharmaceutical organizations and manufacturers, and insurers), and public health organizations are extremely important mechanisms for broader scale impact. This view recognizes the numerous opportunities to educate and influence patients about medication safety outside the dyadic relationship of the clinician and patient in traditional clinical settings. Partnerships between health care and public entities may capitalize on these opportunities to foster adoption of healthy medication practices (e.g., maintaining an accurate and updated medication list), thereby supporting medication reconciliation efforts when individuals encounter health care settings. Partnership and information sharing could be enhanced through the use of a central coordinating body or coalition. This body could generate a shared common vision and contribute expertise to the myriad issues in medication reconciliation.
Partnerships should utilize the following:
-
Social marketing techniques to engage the community. Included within this strategy must be a clear and compelling message that transmits the importance of safe medication practices. Current messages such as keep a list while important, do not offer enough of a sense of urgency or importance. A more powerful message could involve highly publicized medication errors or close calls that would resonate with a broad audience.
-
Local and national champions. Such individuals should be trusted for their health knowledge (e.g., television health care reporters) or be prominent, influential, and trusted figures in other circles (e.g., clergy, politicians, movie celebrities). Indeed, taking advantage of popular media by weaving a theme into a movie or television program about medication safety may prove effective.
Relevant partnerships would include:
-
Quality organizations partnering with other stakeholders to establish unambiguous and unified medication reconciliation standards across the care continuum.
-
Health systems partnering with community pharmacy providers to ensure an uninterrupted communication link in both the inpatient and outpatient settings.
-
Manufacturers and distributors of medications partnering with health care and public health organizations, the media, insurers and other constituents to promote the importance of maintaining and sharing an accurate list of medications.
-
Public health systems partnering with community‐based organizations to encourage and promote the established standards for medication safety through messaging and educational campaigns.
All partnerships must consider issues of patient language and literacy as well as the needs of vulnerable populations in the scope of their activities.
First Step
Public health agencies should partner with health care quality organizations and others to begin a national public campaign to increase the awareness of medication safety (the broader public health concept under which medication reconciliation would fall) and support the importance of the patient's role in maintaining an updated medication list at all times.
10. Align Financial Incentives With Newly Developed Regulatory and Accreditation Requirements
Implementing and performing medication reconciliation takes time, particularly at the outset of a new program. Time requirements and associated costs are major barriers to undertaking comprehensive medication reconciliation, despite its recognized importance for reducing avoidable injury to patients. At present, systems that impede efficiency and slow hospital throughput may be discouraged due to their potential for having an adverse impact on access, finances, and other aspects of care delivery. Moreover, the changed economic climate with reduced hospital fiscal margins limits resources for new initiatives. Currently, failed medication reconciliationand the related avoidable adverse events, culminating in readmission to the hospital or emergency departmentyields additional revenue for hospitals and other providers in some reimbursement models.
Alignment of financial incentives that ensured adequate time and resources for appropriate medication reconciliation processes would facilitate implementation. Additionally, start‐up funding to create and implement these processes needs to be made available.
One example illustrating efforts to align payment policy with medication safety efforts occurred when the Office of the National Coordinator (ONC), in publishing its Final Rule under the 2009 HITECH Act,24 endorsed the importance of financially supporting proper medication reconciliation, particularly at first encounter and transitions in care, by requiring EHR systems seeking certification under the rule to support the care team in the task of reconciliation. For example, vendors will have to support the ability to compare 2 or more medication lists electronically, create medication lists, drug allergy lists, perform drug formulary look‐ups, drug‐drug and drug‐allergy checks, and support creating patient summaries after each visit or post discharge that include medication lists. The ONC, in defining Meaningful Use for eligible health care organizations, included in that definition the goal of exchanging meaningful clinical information among the professional health care teams. This goal is demonstrated through organizations reporting that they performed medication reconciliation for at least 50% of transitions of care in which the patient is transitioned into the care of the eligible professional or admitted to the eligible hospital's or Critical Access Hospital's inpatient or emergency department. Organizations able to demonstrate this level of compliance, along with other Meaningful Use requirements, will be eligible to receive stimulus funds through 2015 and avoid financial penalties that begin after that period.
First Step
Future health care reform must address the misalignment of financial policies and structures, and provide financial incentives to support the development and implementation of better medication management systems and prevent avoidable rehospitalizations and emergency department visits resulting from medication‐related adverse events.
Conclusion
Medication reconciliation involves highly complex processes and is hampered by the disjointed nature of the American health care system. It is, however, a vital part of reducing ADE. If employed more broadly, it has the added benefits of enhancing communication among all providers of care and engaging patients and families/caregivers more consistently and meaningfully in their overall care.
Despite the difficulty of maintaining an accurate medication record in real time across disparate settings, reconciliation is a goal to which our organizations are committed. Given the wide range of healthcare organizations involved in providing medications to patients and the many agencies evaluating those efforts, we believed it would be helpful to provide an overarching set of goals to move medication reconciliation forward.
Our main message is this: Patient safety and patient/family‐centered care must be the principal drivers in the development and implementation of medication reconciliation systems. Ultimately this process is about ensuring that patients are receiving the most appropriate medications no matter where they are treated. With this document, we hope to bring to light the importance of creating and implementing a medication reconciliation program, addressing some barriers to success, and identifying potential solutions that will ensure utility and sustainability of this critical patient safety issue.
- ,,, et al.Unintended medication discrepancies at the time of hospital admission.Arch Intern Med.2005;165(4):424–429.
- .Prevention of medication errors in the pediatric inpatient setting. The American Academy of Pediatrics Policy Statement.Pediatrics.2003;112(2):431–436.
- ,,, et al.Medication reconciliation: a practical tool to reduce the risk of medication errors.J Crit Care.2003;18(4):201–205.
- Institute for Healthcare Improvement. 5 million lives getting started kit: preventing adverse drug events (medication reconciliation), how‐to guide. Available at: http://www.ihi.org/IHI/Programs/Campaign/ADEsMedReconciliation.htm. Published Oct. 1, 2008. Accessed September2010.
- ,.Medication safety: one organization's approach to the challenge.J Clin Outcomes Mana.2001;8(10):27–34.
- ,,,,,.Reconciliation of discrepancies in medication histories and admission orders of newly hospitalized patients.Am J Health Syst Pharm.2004;61(16):1689–1695.
- ,,, et al.Results of the Medications At Transitions and Clinical Handoffs (MATCH) Study: an analysis of medication reconciliation errors and risk factors at hospital admission.J Gen Intern Med.2010;25(5):441–447.
- Joint Commission on Accreditation of Healthcare Organizations.2005 Hospital Accreditation Standards, p.NPSG‐4.
- ,,,,.Brief communication: Results of a medication reconciliation survey from the 2006 Society of Hospital Medicine national meeting.J Hosp Med.2008;3(6):465–472.
- The Joint Commission.Approved: will not score medication reconciliation in 2009.Jt Comm Perspect.2009;29(3):1,3.
- Society of Hospital Medicine. Medication reconciliation: a team approach, conference summary. December 2009. Available at: http://www.hospitalmedicine.org/Content/NavigationMenu/QualityImprovement/QICurrentInitiativesandTrainingOpportunities/QI_Current_Initiativ.htm. Accessed September2010.
- The American Medical Association. The physician's role in medication reconciliation: issues, strategies and safety principles. 2007. Available at: http://www.ama‐assn.org/ama1/pub/upload/mm/370/med‐rec‐monograph.pdf. Accessed September2010.
- Institute of Safe Medication Practices. ISMP's list of high alert medications. 2008. Available at: http://www.ismp.org/Tools/highalertmedications.pdf. Accessed September2010.
- ,,,.Medication use leading to emergency department visits for adverse drug events in older adults.Ann Intern Med.2007;147(11):755–765
- ,,, et al.Experience with a trigger tool for identifying adverse drug events among older adults in ambulatory primary care.Qual Saf Health Care.2009;18(3):199–204.
- ,,, et al.Role of pharmacist counseling in preventing adverse drug events after hospitalization.Arch Intern Med.2006;166(5):565–571.
- ,,,,.Medication reconciliation at an academic medical center: implementation of a comprehensive program from admission to discharge.Am J Health Syst Pharm.2009;66(23):2126–2131.
- ,,,,,.Multidisciplinary approach to inpatient medication reconciliation in an academic setting.Am J Health Syst Pharm.2007;64(8):850–854.
- ,,.Lack of patient knowledge regarding hospital medications.J Hosp Med.2010;5(2):83–86.
- .The unexpected challenges of accurate medication reconciliation.Ann Emerg Med.2008;52(5):493–495.
- ,,,.Medication reconciliation in a rural trauma population.Ann Emerg Med.2008;52(5):483–491.
- ASTM International. ASTM E2369 ‐ 05e1 standard specification for continuity of care record (CCR). Available at: http://www.astm.org/Standards/E2369.htm. Accessed September2010.
- ,,.The continuity of care record.Am Fam Physician.2004;70(7):1220,1222–1223.
- Department of Health and Human Services. Health information technology: initial set of standards, implementation specifications, and certification criteria for electronic health record technology; final rule. Available at: http://edocket.access.gpo.gov/2010/pdf/2010–17210.pdf. Accessed September2010.
- ,,, et al.Unintended medication discrepancies at the time of hospital admission.Arch Intern Med.2005;165(4):424–429.
- .Prevention of medication errors in the pediatric inpatient setting. The American Academy of Pediatrics Policy Statement.Pediatrics.2003;112(2):431–436.
- ,,, et al.Medication reconciliation: a practical tool to reduce the risk of medication errors.J Crit Care.2003;18(4):201–205.
- Institute for Healthcare Improvement. 5 million lives getting started kit: preventing adverse drug events (medication reconciliation), how‐to guide. Available at: http://www.ihi.org/IHI/Programs/Campaign/ADEsMedReconciliation.htm. Published Oct. 1, 2008. Accessed September2010.
- ,.Medication safety: one organization's approach to the challenge.J Clin Outcomes Mana.2001;8(10):27–34.
- ,,,,,.Reconciliation of discrepancies in medication histories and admission orders of newly hospitalized patients.Am J Health Syst Pharm.2004;61(16):1689–1695.
- ,,, et al.Results of the Medications At Transitions and Clinical Handoffs (MATCH) Study: an analysis of medication reconciliation errors and risk factors at hospital admission.J Gen Intern Med.2010;25(5):441–447.
- Joint Commission on Accreditation of Healthcare Organizations.2005 Hospital Accreditation Standards, p.NPSG‐4.
- ,,,,.Brief communication: Results of a medication reconciliation survey from the 2006 Society of Hospital Medicine national meeting.J Hosp Med.2008;3(6):465–472.
- The Joint Commission.Approved: will not score medication reconciliation in 2009.Jt Comm Perspect.2009;29(3):1,3.
- Society of Hospital Medicine. Medication reconciliation: a team approach, conference summary. December 2009. Available at: http://www.hospitalmedicine.org/Content/NavigationMenu/QualityImprovement/QICurrentInitiativesandTrainingOpportunities/QI_Current_Initiativ.htm. Accessed September2010.
- The American Medical Association. The physician's role in medication reconciliation: issues, strategies and safety principles. 2007. Available at: http://www.ama‐assn.org/ama1/pub/upload/mm/370/med‐rec‐monograph.pdf. Accessed September2010.
- Institute of Safe Medication Practices. ISMP's list of high alert medications. 2008. Available at: http://www.ismp.org/Tools/highalertmedications.pdf. Accessed September2010.
- ,,,.Medication use leading to emergency department visits for adverse drug events in older adults.Ann Intern Med.2007;147(11):755–765
- ,,, et al.Experience with a trigger tool for identifying adverse drug events among older adults in ambulatory primary care.Qual Saf Health Care.2009;18(3):199–204.
- ,,, et al.Role of pharmacist counseling in preventing adverse drug events after hospitalization.Arch Intern Med.2006;166(5):565–571.
- ,,,,.Medication reconciliation at an academic medical center: implementation of a comprehensive program from admission to discharge.Am J Health Syst Pharm.2009;66(23):2126–2131.
- ,,,,,.Multidisciplinary approach to inpatient medication reconciliation in an academic setting.Am J Health Syst Pharm.2007;64(8):850–854.
- ,,.Lack of patient knowledge regarding hospital medications.J Hosp Med.2010;5(2):83–86.
- .The unexpected challenges of accurate medication reconciliation.Ann Emerg Med.2008;52(5):493–495.
- ,,,.Medication reconciliation in a rural trauma population.Ann Emerg Med.2008;52(5):483–491.
- ASTM International. ASTM E2369 ‐ 05e1 standard specification for continuity of care record (CCR). Available at: http://www.astm.org/Standards/E2369.htm. Accessed September2010.
- ,,.The continuity of care record.Am Fam Physician.2004;70(7):1220,1222–1223.
- Department of Health and Human Services. Health information technology: initial set of standards, implementation specifications, and certification criteria for electronic health record technology; final rule. Available at: http://edocket.access.gpo.gov/2010/pdf/2010–17210.pdf. Accessed September2010.
Role of Shock Index in ICU Transfers
The decision to transfer a patient to the intensive care unit (ICU) from a general care setting is complex and based not only on clinical findings and patient wishes but also on the understanding that ICU resources are limited and costly.1 Adding to the decision‐making complexity is the knowledge that patients who transfer to an ICU from a general medical unit comprise the highest mortality group of ICU patients, with the mortality rate directly proportional to both the time spent on the general medical unit1, 2 and the number of physiologic abnormalities before ICU admission.3, 4
Prior studies have shown that cardiac arrest and unplanned (unexpected) transfers to the ICU are preceded by a period of physiologic instability reflected in the vital signs.510 However, vital signs alone may not accurately indicate clinical condition. For example, a person may be able to maintain normal blood pressure and heart rate despite severe illness or may have abnormal vital signs at baseline, which may be the case for an otherwise healthy young woman who has baseline low systolic blood pressure. Also, noncritical conditions commonly seen in hospitalized medical or surgical patients, such as anxiety or pain, may increase the respiratory rate or heart rate. Conversely, certain common medications, such as ‐blockers, may mask or blunt the normal physiologic response to illness. Overall, the prevalence of abnormal physiologic variables is high among hospitalized adult patients irrespective of the presence of serious adverse events.11 This prevalence may be a reason why 2 recent studies of inpatient medical emergency teams (METs) or rapid response teams (RRTs), which generally rely on vital signs for activation, failed to show a decrease in adult mortality rates.12, 13
Given the complexity of interpreting single vital sign readings, we evaluated a simple and clinically intuitive variable, the shock index (SI) (heart rate/systolic blood pressure, a noninvasive indication of left ventricular function),14 as a potential marker of the need for intensive care. Allgwer and Burri14 first developed the SI in studies of patients with acute blood loss, intraabdominal bleeding, fat emboli, and severe infections. They observed that a healthy adult had a mean SI of 0.54 (standard deviation [SD], 0.021), while an index of 1.0 indicated threatened shock and indices greater than 1.5 were seen in volume‐deficient shock.
We hypothesized that an elevated SI is a differentiating factor between a patient who had an unplanned ICU transfer and a general medical patient who did not require this higher level of care. To our knowledge, the SI has not been studied for this application previously.
Patients and Methods
Study Design
We conducted a retrospective case‐control study of 50 consecutive general medical patients who had unplanned transfers to the ICU and 50 matched control patients, with the approval of the Mayo Clinic Institutional Review Board. All patients were admitted to a general medical unit, and only patients who previously provided permission for their records to be used in research were included in the study.
Patients
This study enrolled patients who were at least 18 years old and who were admitted to a single general medical unit for 24 hours or longer. Patients were excluded if they required a surgical intervention, were transferred from another hospital, received care on a different general medical unit at any time during the hospitalization, or were pregnant. Our data collection began at the patients' (cases and controls) arrival on the general medical unit; we did not include data from any evaluation (outpatient or emergency department) before hospital admission.
Case Definition
An unplanned transfer was defined as an episode of unexpected clinical deterioration in a general medical patient that necessitated transfer to the ICU, as opposed to a preemptive or elective transfer following a procedure. Patients with unplanned transfers from December 9, 2003, to December 29, 2004, were eligible for the study. Only the first transfer to the ICU was considered for patients who had multiple ICU transfers during a single hospitalization. Because these data were collected before METs or RRTs were introduced at our institution, the recommendation for ICU transfer was a joint decision by the primary care team and the ICU team.
Control Definition
The matched controls were identified from among patients admitted to the general medical unit from January 16, 2002, to December 13, 2004. To reduce the effect of the heterogeneity inherent in general medical patients, we matched controls for age (within 5 years of age of the corresponding case), admission diagnosis code, and patient care unit of admission and required that they were admitted for at least 24 hours before dismissal. Patients who had an ICU stay during the same admission were excluded. The median difference in admission dates between the cases and the controls was 327 days, and 26 of the 50 matched pairs had admission dates within 1 year of each other. This lengthy interval between cases and controls was a consequence of the low incidence of patients who met the matching criteria.
Setting
This study involved the general medical units and ICUs of the 1157‐bed Saint Mary's Hospital, an academic tertiary care facility at Mayo Clinic in Rochester, Minnesota.
Vital Sign Determination
Vital signs abstracted for this study included blood pressure, heart rate, respiratory rate, oxygen saturation, and temperature. The SI was calculated for each set of abstracted vital signs. Staff nurses were responsible for the routine measurement and recording of vital signs at least once every 8 hours, although in several instances not all parameters were checked. In accordance with nursing policy, values outside the defined parameters were rechecked by the nursing supervisor of each care unit and, if found to be abnormal, were conveyed to the patient's physician. This system meant that abnormal results were checked by numerous observers, with differences in the frequency of recordings for individual patients.
Data Collection
Demographic data and information on the vital signs were abstracted through a comprehensive chart review. Demographic data included age, sex, ethnicity, comorbid conditions, hospital care unit, date and time of admission, admission diagnosis, date and time of transfer to the ICU, length of stay, dismissal date, and disposition at discharge. Comorbid conditions were scored using the Charlson Comorbidity Index.15
Statistical Analysis
A sample size of 50 matched pairs provided 81% power to detect an odds ratio of 3.0 or greater between cases and controls, with a 0.05, 2‐tailed level of significance with McNemar test. Patient demographic characteristics were summarized by the frequencies for categorical data and by mean and SD for continuous data. Consistent with the study design, the McNemar test and conditional logistic model analyses were used to determine the association between the SI and the risk of unplanned ICU transfer. Shock indices for the cases and controls were compared with use of t test. A P value <0.05 was considered statistically significant. For the SI, we calculated the odds ratio and its 95% confidence interval (CI) and P value using different cut points. We did not perform a receiver operating characteristics analysis because matching of cases and controls greatly complicates estimation of the sensitivity and specificity of the SI;16 a cohort study is suggested to investigate this analysis further. All statistical analyses were performed by SAS version 9.1.3 software (SAS Institute Inc, Cary, NC).
Results
A total of 50 pairs of matching cases and controls was included in this study. Table 1 lists the source of admission, demographic characteristics, and numbers of deaths for cases and controls. There were no statistically significant differences in admission source, age, sex, ethnicity, admission care unit, or Charlson Comorbidity Index. Mean length of stay was 14.8 days (SD, 9.7 days) for the cases and 5.7 days (SD, 6.3 days; P < 0.001) for the controls. Admission diagnoses were classified on the basis of the organ system of involvement (Table 2). In 30 of 50 cases, the admission diagnosis and the reason for ICU transfer were related.
| Value | Cases (n = 50) | Controls (n = 50) | P Value* |
|---|---|---|---|
| |||
| Emergency department admission, No. (%) | 33 (66) | 28 (56) | 0.41 |
| Direct admission, No. (%) | 14 (28) | 15 (30) | 1.00 |
| Other admission, No. (%) | 3 (6) | 7 (14) | 0.32 |
| Age, mean (SD), years | 69.8 (15.7) | 70.3 (15.8) | 0.38 |
| Male sex, No. (%) | 26 (52) | 18 (36) | 0.12 |
| Ethnicity, No. (%) | 1.00 | ||
| White | 46 (92) | 46 (92) | |
| Other | 4 (8) | 4 (8) | |
| Charlson Comorbidity index, mean (SD) | 3.06 (2.31) | 2.66 (2.02) | 0.22 |
| Hospital stay, mean (SD), day | 14.8 (9.7) | 5.7 (6.3) | 0.0007 |
| Hospital deaths, No. | 9 | 1 | 0.008 |
| Deaths within 30 days, No. | 5 | 2 | 0.24 |
| Deaths within 6 months, No. | 9 | 6 | 0.40 |
| System | Primary Admission Diagnosis | No. of Cases |
|---|---|---|
| Constitutional | Fever, malaise, general symptoms | 7 |
| Cardiovascular | Hypertension, congestive heart failure, chest pain, peripheral vascular disease, edema | 5 |
| Dermatologic | Cellulitis, foot ulcer, skin rash | 3 |
| Gastrointestinal | Pancreatitis, gastrointestinal hemorrhage, nausea and vomiting, diarrhea, abdominal pain | 6 |
| Hematologic | Thrombocytopenia, abnormal coagulation | 2 |
| Musculoskeletal | Lymphedema, shoulder pain, lumbago, back ache, closed dorsal vertebral fracture | 7 |
| Neurologic | Delirium tremens, psychosis, convulsions | 3 |
| Pulmonary | Pneumonia, food or vomit aspiration pneumonitis, shortness of breath, respiratory abnormality | 13 |
| Renal | Hyperkalemia, acute renal failure, renal artery atherosclerosis | 4 |
We reviewed the vital signs and shock indices for the 24 hours before ICU transfer for each case and over the entire hospitalization for each control, to determine the worst set (the lowest systolic blood pressure and the highest heart rate, respiratory rate, and SI). The cases had 1 to 22 complete sets of vitals for the 24 hours before ICU transfer; the median number of sets was 3 and the mean was 4. The controls had 1 to 12 complete sets for the 24 hours before the worst SI: the median was 3 sets and the mean was 3. In 26 of 50 controls, the worst SI occurred within the first 24 hours after admission. There was a significant difference between the median values of the worst shock indices of the cases and the controls (0.87 vs. 0.72; P < 0.005).
Table 3 shows the different values of the SI and the corresponding odds ratio of unplanned ICU transfer for cases compared with controls. The difference was significant at an SI of 0.85 and greater, indicating a strong association with unplanned ICU transfer.
| Shock Index | P Value | Odds Ratio | 95% CI |
|---|---|---|---|
| |||
| 0.8 | 0.05 | 2.43 | 1.015.86 |
| 0.85 | 0.02 | 3.00 | 1.917.56 |
| 0.9 | 0.007 | 7.50 | 1.7232.78 |
| 0.95 | <0.03 | 5.50 | 1.2224.81 |
We also found that the patients who transferred to the ICU had a greater number of inpatient deaths (9 cases vs. 1 control; P = 0.008), which would be expected, but there was no difference in 30‐day or 6‐month mortality rate (Table 1). One patient died after 30 days and while still hospitalized.
Comparison between the temporal trend of vital signs and the SI of the cases for the 24 hours before ICU transfer is shown in Figure 1. This graph shows the median of all the worst values (minimum systolic blood pressure and maximum SI, heart rate, and respiratory rate) over the four 6‐hour time periods (24 hours) preceding ICU transfer. Of note, the change in vital signs is subtle even while the SI increased to more than 0.8 as the patients clinically worsened before transfer.
Discussion
In our comparison of the SI of 50 patients who required unplanned (unexpected) transfer to the ICU with the SI of 50 matched controls who did not require this higher level of care, we found that a SI of 0.85 or greater was significantly associated with unplanned transfer to the ICU. The cases had a significantly higher worst SI than the controls, and they also had a significantly longer hospital stay and higher inpatient mortality rate, as would be expected for a sicker patient population. These findings are important given that the SI may be useful for assessing illness severity, for helping determine the need for transfer to the ICU, or for activating METs or RRTs.
A major problem with providing optimal care for hospitalized general medical patients is the inherent difficulty in determining illness severity and clinical decline, especially when the decline occurs gradually. Existing consensus recommendations for ICU admission include both specific diagnoses and arbitrary objective criteria based on abnormal vital signs and laboratory values.27 Also, individual institutions may have their own ICU admission requirements, which may differ from these or RRT criteria. Although vital signs are important as a snapshot of basic physiologic function, a number of noncritical conditions may lead to abnormal vital signs, and not all abnormal vital signs are associated with an adverse clinical event. By relying solely on vital signs, clinicians may not recognize critical illness and therefore not transfer a patient to the ICU or may inappropriately transfer a patient who does not need ICU‐level care.
Markers of illness severity other than vital signs, such as the Acute Physiology and Chronic Health Evaluation (APACHE) score, have been shown to predict the death of ICU patients17, 18 but have been rarely studied outside the ICU setting.19 Also, calculating the APACHE score is cumbersome, and there is no cutoff score that defines when a patient should be transferred to the ICU. Subbe et al.,20 in their study to identify critically ill patients, found that introduction of a physiological scoring system (including MET or RRT activation scores) would have identified only a small number of additional patients as critically ill. Another common marker of illness severity, the 4 criteria of the systemic inflammatory response syndrome (temperature <36C or >38C; heart rate >90 beats per minute; respiratory rate >20 breaths per minute or PCO2 <32 mm Hg; and white blood cell count >12,000/L or <4000/L or with more than 10% band cells)21 may be too sensitive to use as a decision aid, since even a healthy person running after a bus could have 2 of the 4 criteria.22 Likewise, surgical patients may have transient leukocytosis due to a stress response independent of an infection.23
The SI may be more accurate than vital signs alone to determine illness severity and who is at risk for an unplanned transfer to the ICU. Birkhahn et al.24 concluded that the SI may be more useful in early hemorrhage than either heart rate or systolic blood pressure alone. Rady et al.25 showed that the SI used in the emergency department can identify critical illness with apparently stable vital signs, where an elevation of the SI above 0.9 was associated with an illness that was treated immediately with admission to the hospital and intensive therapy on admission. However, it is unclear whether the SI can be used to monitor ongoing treatment, because a previous study showed that the SI may be of limited value in the assessment of systemic oxygen transport and response to therapy in clinical septic shock.26 Of note, the SI is mostly independent of the effects of pain or anxiety, which cause a concurrent rise in heart rate and systolic blood pressure. Because the heart's left ventricular work is unchanged or may increase from the underlying catecholamine surge, the SI will be unchanged or may actually decrease.
Our study adds to the medical literature the findings that: (1) the SI may be useful as an indicator of illness severity and a triage tool in patients with no trauma but with various medical conditions, and (2) the SI showed a strong association with unplanned ICU transfer.
The main strength of our study is its case‐control design with matched controls. Also, by comparing groups from the same patient care unit, we sought to minimize the selection bias that can be inherent in case‐control studies. Limitations include the retrospective, nonrandomized study design and the fact that there may have been variations in vital sign measurements by the multiple caregivers. However, the vital signs were taken according to standard hospital practice and reflect real‐world conditions. Although generalizability may be somewhat limited because of our homogeneous patient population, our patients had a wide range of various medical illnesses, so our study should be applicable to other hospital settings, both academic and community‐based.
One of the main weaknesses of our study is that the results were not adjusted for the burden of comorbid conditions, although there were no statistically significant differences in the number of comorbid conditions among the cases and the controls (P = 0.96). Also, we did not directly compare the SI with vital signs alone to determine superiority.
The SI may be an important objective measure to help clinicians decide when patients need treatment that is more aggressive, assistance from a MET or an RRT, or a preemptive, rather than unplanned, transfer to an ICU. Although it is unlikely that a single measure will allow accurate triage of all medical or surgical patients, the SI may be a useful adjunct to clinical judgment and other objective measures in determining illness severity and clinical decline. Further prospective studies are needed to compare the role of the SI specifically with MET or RRT activation criteria, to clarify the role of comorbid conditions in unplanned transfers to the ICU, to validate the cut point for the SI in various disease states, and to assess its utility in patients with septic shock. Depending on these results, it may be beneficial to incorporate the SI into the electronic medical record as an automatic alert to identify patients at risk for ICU transfer.
Conclusions
The SI is an easily calculated composite index of heart rate and systolic blood pressure. An elevated SI of 0.85 can identify patients who are at risk for unplanned transfer to the ICU from general patient care units. Future studies will determine whether the SI is more accurate than simple vital signs as an indicator of clinical decline. If so, it may be useful as a trigger to activate METs or RRTs for treatment.
- ,.Outcome of intensive care patients in a group of British intensive care units.Crit Care Med.1998;26(8):1337–1345.
- ,,,.The longer patients are in hospital before Intensive Care admission the higher their mortality.Intensive Care Med.2004;30(10):1908–1913.
- ,.Physiological abnormalities in early warning scores are related to mortality in adult inpatients.Br J Anaesth.2004;92(6):882–884.
- ,,,,.Association between clinically abnormal observations and subsequent in‐hospital mortality: a prospective study.Resuscitation.2004;62(2):137–141.
- ,,,,,.Recognising clinical instability in hospital patients before cardiac arrest or unplanned admission to intensive care: a pilot study in a tertiary‐care hospital.Med J Aust.1999;171(1):22–25.
- ,,.Physiological values and procedures in the 24 h before ICU admission from the ward.Anaesthesia.1999;54(6):529–534.
- ,,, et al.Antecedents to hospital deaths.Intern Med J.2001;31(6):343–348.
- ,,, et al.A comparison of antecedents to cardiac arrests, deaths and emergency intensive care admissions in Australia and New Zealand, and the United Kingdom: the ACADEMIA study.Resuscitation.2004;62(3):275–282.
- ,,,,.Anticipating events of in‐hospital cardiac arrest.Eur J Emerg Med.2004;11(1):24–28.
- ,,, et al.Duration of life‐threatening antecedents prior to intensive care admission.Intensive Care Med.2002;28(11):1629–1634.
- ,,,.The prevalence of recordings of the signs of critical conditions and emergency responses in hospital wards: the SOCCER study.Resuscitation.2005;65(2):149–157.
- ,,,,,.Hospital‐wide code rates and mortality before and after implementation of a rapid response team.JAMA.2008;300(21):2506–2513.
- ,,,,.Rapid response teams: a systematic review and meta‐analysis.Arch Intern Med.2010;170(1):18–26.
- ,. [Shock index.]Dtsch Med Wochenschr.1967;92(43):1947–50. [German]
- ,,,.A new method of classifying prognostic comorbidity in longitudinal studies: development and validation.J Chronic Dis.1987;40(5):373–83.
- ,.Matching in studies of classification accuracy: implications for analysis, efficiency, and assessment of incremental value.Biometrics.2008;64(1):1–9.
- ,,,.APACHE II: a severity of disease classification system.Crit Care Med.1985;13(10):818–829.
- ,.Outcome prediction in critical care: the Acute Physiology and Chronic Health Evaluation models.Curr Opin Crit Care.2008;14(5):491–497.
- ,,.APACHE II predicts long‐term survival in COPD patients admitted to a general medical ward.J Gen Intern Med.2003;18(10):824–830.
- ,,,.Validation of physiological scoring systems in the accident and emergency department.Emerg Med J.2006;23(11):841–845.
- ,,, et al;2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference.Crit Care Med.2003;31(4):1250–1256.
- .Dear SIRS, I'm sorry to say that I don't like you...Crit Care Med.1997;25(2):372–374.
- ,.The inflammatory response to surgery and trauma.Curr Opin Crit Care.2006;12(4):325–332.
- ,,,,.Shock index in diagnosing early acute hypovolemia.Am J Emerg Med.2005;23(3):323–326.
- ,,,,.A comparison of the shock index and conventional vital signs to identify acute, critical illness in the emergency department.Ann Emerg Med.1994;24(4):685–690. Erratum in:Ann Emerg Med.year="1994"1994;24(6):1208.
- ,,,.Shock index: a re‐evaluation in acute circulatory failure.Resuscitation.1992;23(3):227–234.
- ,,,,, et al.Guidelines for intensive care unit admission, discharge, and triage. Task Force of the American College of Critical Care Medicine, Society of Critical Care Medicine.Crit Care Med.1999;27(3):633–638.
The decision to transfer a patient to the intensive care unit (ICU) from a general care setting is complex and based not only on clinical findings and patient wishes but also on the understanding that ICU resources are limited and costly.1 Adding to the decision‐making complexity is the knowledge that patients who transfer to an ICU from a general medical unit comprise the highest mortality group of ICU patients, with the mortality rate directly proportional to both the time spent on the general medical unit1, 2 and the number of physiologic abnormalities before ICU admission.3, 4
Prior studies have shown that cardiac arrest and unplanned (unexpected) transfers to the ICU are preceded by a period of physiologic instability reflected in the vital signs.510 However, vital signs alone may not accurately indicate clinical condition. For example, a person may be able to maintain normal blood pressure and heart rate despite severe illness or may have abnormal vital signs at baseline, which may be the case for an otherwise healthy young woman who has baseline low systolic blood pressure. Also, noncritical conditions commonly seen in hospitalized medical or surgical patients, such as anxiety or pain, may increase the respiratory rate or heart rate. Conversely, certain common medications, such as ‐blockers, may mask or blunt the normal physiologic response to illness. Overall, the prevalence of abnormal physiologic variables is high among hospitalized adult patients irrespective of the presence of serious adverse events.11 This prevalence may be a reason why 2 recent studies of inpatient medical emergency teams (METs) or rapid response teams (RRTs), which generally rely on vital signs for activation, failed to show a decrease in adult mortality rates.12, 13
Given the complexity of interpreting single vital sign readings, we evaluated a simple and clinically intuitive variable, the shock index (SI) (heart rate/systolic blood pressure, a noninvasive indication of left ventricular function),14 as a potential marker of the need for intensive care. Allgwer and Burri14 first developed the SI in studies of patients with acute blood loss, intraabdominal bleeding, fat emboli, and severe infections. They observed that a healthy adult had a mean SI of 0.54 (standard deviation [SD], 0.021), while an index of 1.0 indicated threatened shock and indices greater than 1.5 were seen in volume‐deficient shock.
We hypothesized that an elevated SI is a differentiating factor between a patient who had an unplanned ICU transfer and a general medical patient who did not require this higher level of care. To our knowledge, the SI has not been studied for this application previously.
Patients and Methods
Study Design
We conducted a retrospective case‐control study of 50 consecutive general medical patients who had unplanned transfers to the ICU and 50 matched control patients, with the approval of the Mayo Clinic Institutional Review Board. All patients were admitted to a general medical unit, and only patients who previously provided permission for their records to be used in research were included in the study.
Patients
This study enrolled patients who were at least 18 years old and who were admitted to a single general medical unit for 24 hours or longer. Patients were excluded if they required a surgical intervention, were transferred from another hospital, received care on a different general medical unit at any time during the hospitalization, or were pregnant. Our data collection began at the patients' (cases and controls) arrival on the general medical unit; we did not include data from any evaluation (outpatient or emergency department) before hospital admission.
Case Definition
An unplanned transfer was defined as an episode of unexpected clinical deterioration in a general medical patient that necessitated transfer to the ICU, as opposed to a preemptive or elective transfer following a procedure. Patients with unplanned transfers from December 9, 2003, to December 29, 2004, were eligible for the study. Only the first transfer to the ICU was considered for patients who had multiple ICU transfers during a single hospitalization. Because these data were collected before METs or RRTs were introduced at our institution, the recommendation for ICU transfer was a joint decision by the primary care team and the ICU team.
Control Definition
The matched controls were identified from among patients admitted to the general medical unit from January 16, 2002, to December 13, 2004. To reduce the effect of the heterogeneity inherent in general medical patients, we matched controls for age (within 5 years of age of the corresponding case), admission diagnosis code, and patient care unit of admission and required that they were admitted for at least 24 hours before dismissal. Patients who had an ICU stay during the same admission were excluded. The median difference in admission dates between the cases and the controls was 327 days, and 26 of the 50 matched pairs had admission dates within 1 year of each other. This lengthy interval between cases and controls was a consequence of the low incidence of patients who met the matching criteria.
Setting
This study involved the general medical units and ICUs of the 1157‐bed Saint Mary's Hospital, an academic tertiary care facility at Mayo Clinic in Rochester, Minnesota.
Vital Sign Determination
Vital signs abstracted for this study included blood pressure, heart rate, respiratory rate, oxygen saturation, and temperature. The SI was calculated for each set of abstracted vital signs. Staff nurses were responsible for the routine measurement and recording of vital signs at least once every 8 hours, although in several instances not all parameters were checked. In accordance with nursing policy, values outside the defined parameters were rechecked by the nursing supervisor of each care unit and, if found to be abnormal, were conveyed to the patient's physician. This system meant that abnormal results were checked by numerous observers, with differences in the frequency of recordings for individual patients.
Data Collection
Demographic data and information on the vital signs were abstracted through a comprehensive chart review. Demographic data included age, sex, ethnicity, comorbid conditions, hospital care unit, date and time of admission, admission diagnosis, date and time of transfer to the ICU, length of stay, dismissal date, and disposition at discharge. Comorbid conditions were scored using the Charlson Comorbidity Index.15
Statistical Analysis
A sample size of 50 matched pairs provided 81% power to detect an odds ratio of 3.0 or greater between cases and controls, with a 0.05, 2‐tailed level of significance with McNemar test. Patient demographic characteristics were summarized by the frequencies for categorical data and by mean and SD for continuous data. Consistent with the study design, the McNemar test and conditional logistic model analyses were used to determine the association between the SI and the risk of unplanned ICU transfer. Shock indices for the cases and controls were compared with use of t test. A P value <0.05 was considered statistically significant. For the SI, we calculated the odds ratio and its 95% confidence interval (CI) and P value using different cut points. We did not perform a receiver operating characteristics analysis because matching of cases and controls greatly complicates estimation of the sensitivity and specificity of the SI;16 a cohort study is suggested to investigate this analysis further. All statistical analyses were performed by SAS version 9.1.3 software (SAS Institute Inc, Cary, NC).
Results
A total of 50 pairs of matching cases and controls was included in this study. Table 1 lists the source of admission, demographic characteristics, and numbers of deaths for cases and controls. There were no statistically significant differences in admission source, age, sex, ethnicity, admission care unit, or Charlson Comorbidity Index. Mean length of stay was 14.8 days (SD, 9.7 days) for the cases and 5.7 days (SD, 6.3 days; P < 0.001) for the controls. Admission diagnoses were classified on the basis of the organ system of involvement (Table 2). In 30 of 50 cases, the admission diagnosis and the reason for ICU transfer were related.
| Value | Cases (n = 50) | Controls (n = 50) | P Value* |
|---|---|---|---|
| |||
| Emergency department admission, No. (%) | 33 (66) | 28 (56) | 0.41 |
| Direct admission, No. (%) | 14 (28) | 15 (30) | 1.00 |
| Other admission, No. (%) | 3 (6) | 7 (14) | 0.32 |
| Age, mean (SD), years | 69.8 (15.7) | 70.3 (15.8) | 0.38 |
| Male sex, No. (%) | 26 (52) | 18 (36) | 0.12 |
| Ethnicity, No. (%) | 1.00 | ||
| White | 46 (92) | 46 (92) | |
| Other | 4 (8) | 4 (8) | |
| Charlson Comorbidity index, mean (SD) | 3.06 (2.31) | 2.66 (2.02) | 0.22 |
| Hospital stay, mean (SD), day | 14.8 (9.7) | 5.7 (6.3) | 0.0007 |
| Hospital deaths, No. | 9 | 1 | 0.008 |
| Deaths within 30 days, No. | 5 | 2 | 0.24 |
| Deaths within 6 months, No. | 9 | 6 | 0.40 |
| System | Primary Admission Diagnosis | No. of Cases |
|---|---|---|
| Constitutional | Fever, malaise, general symptoms | 7 |
| Cardiovascular | Hypertension, congestive heart failure, chest pain, peripheral vascular disease, edema | 5 |
| Dermatologic | Cellulitis, foot ulcer, skin rash | 3 |
| Gastrointestinal | Pancreatitis, gastrointestinal hemorrhage, nausea and vomiting, diarrhea, abdominal pain | 6 |
| Hematologic | Thrombocytopenia, abnormal coagulation | 2 |
| Musculoskeletal | Lymphedema, shoulder pain, lumbago, back ache, closed dorsal vertebral fracture | 7 |
| Neurologic | Delirium tremens, psychosis, convulsions | 3 |
| Pulmonary | Pneumonia, food or vomit aspiration pneumonitis, shortness of breath, respiratory abnormality | 13 |
| Renal | Hyperkalemia, acute renal failure, renal artery atherosclerosis | 4 |
We reviewed the vital signs and shock indices for the 24 hours before ICU transfer for each case and over the entire hospitalization for each control, to determine the worst set (the lowest systolic blood pressure and the highest heart rate, respiratory rate, and SI). The cases had 1 to 22 complete sets of vitals for the 24 hours before ICU transfer; the median number of sets was 3 and the mean was 4. The controls had 1 to 12 complete sets for the 24 hours before the worst SI: the median was 3 sets and the mean was 3. In 26 of 50 controls, the worst SI occurred within the first 24 hours after admission. There was a significant difference between the median values of the worst shock indices of the cases and the controls (0.87 vs. 0.72; P < 0.005).
Table 3 shows the different values of the SI and the corresponding odds ratio of unplanned ICU transfer for cases compared with controls. The difference was significant at an SI of 0.85 and greater, indicating a strong association with unplanned ICU transfer.
| Shock Index | P Value | Odds Ratio | 95% CI |
|---|---|---|---|
| |||
| 0.8 | 0.05 | 2.43 | 1.015.86 |
| 0.85 | 0.02 | 3.00 | 1.917.56 |
| 0.9 | 0.007 | 7.50 | 1.7232.78 |
| 0.95 | <0.03 | 5.50 | 1.2224.81 |
We also found that the patients who transferred to the ICU had a greater number of inpatient deaths (9 cases vs. 1 control; P = 0.008), which would be expected, but there was no difference in 30‐day or 6‐month mortality rate (Table 1). One patient died after 30 days and while still hospitalized.
Comparison between the temporal trend of vital signs and the SI of the cases for the 24 hours before ICU transfer is shown in Figure 1. This graph shows the median of all the worst values (minimum systolic blood pressure and maximum SI, heart rate, and respiratory rate) over the four 6‐hour time periods (24 hours) preceding ICU transfer. Of note, the change in vital signs is subtle even while the SI increased to more than 0.8 as the patients clinically worsened before transfer.
Discussion
In our comparison of the SI of 50 patients who required unplanned (unexpected) transfer to the ICU with the SI of 50 matched controls who did not require this higher level of care, we found that a SI of 0.85 or greater was significantly associated with unplanned transfer to the ICU. The cases had a significantly higher worst SI than the controls, and they also had a significantly longer hospital stay and higher inpatient mortality rate, as would be expected for a sicker patient population. These findings are important given that the SI may be useful for assessing illness severity, for helping determine the need for transfer to the ICU, or for activating METs or RRTs.
A major problem with providing optimal care for hospitalized general medical patients is the inherent difficulty in determining illness severity and clinical decline, especially when the decline occurs gradually. Existing consensus recommendations for ICU admission include both specific diagnoses and arbitrary objective criteria based on abnormal vital signs and laboratory values.27 Also, individual institutions may have their own ICU admission requirements, which may differ from these or RRT criteria. Although vital signs are important as a snapshot of basic physiologic function, a number of noncritical conditions may lead to abnormal vital signs, and not all abnormal vital signs are associated with an adverse clinical event. By relying solely on vital signs, clinicians may not recognize critical illness and therefore not transfer a patient to the ICU or may inappropriately transfer a patient who does not need ICU‐level care.
Markers of illness severity other than vital signs, such as the Acute Physiology and Chronic Health Evaluation (APACHE) score, have been shown to predict the death of ICU patients17, 18 but have been rarely studied outside the ICU setting.19 Also, calculating the APACHE score is cumbersome, and there is no cutoff score that defines when a patient should be transferred to the ICU. Subbe et al.,20 in their study to identify critically ill patients, found that introduction of a physiological scoring system (including MET or RRT activation scores) would have identified only a small number of additional patients as critically ill. Another common marker of illness severity, the 4 criteria of the systemic inflammatory response syndrome (temperature <36C or >38C; heart rate >90 beats per minute; respiratory rate >20 breaths per minute or PCO2 <32 mm Hg; and white blood cell count >12,000/L or <4000/L or with more than 10% band cells)21 may be too sensitive to use as a decision aid, since even a healthy person running after a bus could have 2 of the 4 criteria.22 Likewise, surgical patients may have transient leukocytosis due to a stress response independent of an infection.23
The SI may be more accurate than vital signs alone to determine illness severity and who is at risk for an unplanned transfer to the ICU. Birkhahn et al.24 concluded that the SI may be more useful in early hemorrhage than either heart rate or systolic blood pressure alone. Rady et al.25 showed that the SI used in the emergency department can identify critical illness with apparently stable vital signs, where an elevation of the SI above 0.9 was associated with an illness that was treated immediately with admission to the hospital and intensive therapy on admission. However, it is unclear whether the SI can be used to monitor ongoing treatment, because a previous study showed that the SI may be of limited value in the assessment of systemic oxygen transport and response to therapy in clinical septic shock.26 Of note, the SI is mostly independent of the effects of pain or anxiety, which cause a concurrent rise in heart rate and systolic blood pressure. Because the heart's left ventricular work is unchanged or may increase from the underlying catecholamine surge, the SI will be unchanged or may actually decrease.
Our study adds to the medical literature the findings that: (1) the SI may be useful as an indicator of illness severity and a triage tool in patients with no trauma but with various medical conditions, and (2) the SI showed a strong association with unplanned ICU transfer.
The main strength of our study is its case‐control design with matched controls. Also, by comparing groups from the same patient care unit, we sought to minimize the selection bias that can be inherent in case‐control studies. Limitations include the retrospective, nonrandomized study design and the fact that there may have been variations in vital sign measurements by the multiple caregivers. However, the vital signs were taken according to standard hospital practice and reflect real‐world conditions. Although generalizability may be somewhat limited because of our homogeneous patient population, our patients had a wide range of various medical illnesses, so our study should be applicable to other hospital settings, both academic and community‐based.
One of the main weaknesses of our study is that the results were not adjusted for the burden of comorbid conditions, although there were no statistically significant differences in the number of comorbid conditions among the cases and the controls (P = 0.96). Also, we did not directly compare the SI with vital signs alone to determine superiority.
The SI may be an important objective measure to help clinicians decide when patients need treatment that is more aggressive, assistance from a MET or an RRT, or a preemptive, rather than unplanned, transfer to an ICU. Although it is unlikely that a single measure will allow accurate triage of all medical or surgical patients, the SI may be a useful adjunct to clinical judgment and other objective measures in determining illness severity and clinical decline. Further prospective studies are needed to compare the role of the SI specifically with MET or RRT activation criteria, to clarify the role of comorbid conditions in unplanned transfers to the ICU, to validate the cut point for the SI in various disease states, and to assess its utility in patients with septic shock. Depending on these results, it may be beneficial to incorporate the SI into the electronic medical record as an automatic alert to identify patients at risk for ICU transfer.
Conclusions
The SI is an easily calculated composite index of heart rate and systolic blood pressure. An elevated SI of 0.85 can identify patients who are at risk for unplanned transfer to the ICU from general patient care units. Future studies will determine whether the SI is more accurate than simple vital signs as an indicator of clinical decline. If so, it may be useful as a trigger to activate METs or RRTs for treatment.
The decision to transfer a patient to the intensive care unit (ICU) from a general care setting is complex and based not only on clinical findings and patient wishes but also on the understanding that ICU resources are limited and costly.1 Adding to the decision‐making complexity is the knowledge that patients who transfer to an ICU from a general medical unit comprise the highest mortality group of ICU patients, with the mortality rate directly proportional to both the time spent on the general medical unit1, 2 and the number of physiologic abnormalities before ICU admission.3, 4
Prior studies have shown that cardiac arrest and unplanned (unexpected) transfers to the ICU are preceded by a period of physiologic instability reflected in the vital signs.510 However, vital signs alone may not accurately indicate clinical condition. For example, a person may be able to maintain normal blood pressure and heart rate despite severe illness or may have abnormal vital signs at baseline, which may be the case for an otherwise healthy young woman who has baseline low systolic blood pressure. Also, noncritical conditions commonly seen in hospitalized medical or surgical patients, such as anxiety or pain, may increase the respiratory rate or heart rate. Conversely, certain common medications, such as ‐blockers, may mask or blunt the normal physiologic response to illness. Overall, the prevalence of abnormal physiologic variables is high among hospitalized adult patients irrespective of the presence of serious adverse events.11 This prevalence may be a reason why 2 recent studies of inpatient medical emergency teams (METs) or rapid response teams (RRTs), which generally rely on vital signs for activation, failed to show a decrease in adult mortality rates.12, 13
Given the complexity of interpreting single vital sign readings, we evaluated a simple and clinically intuitive variable, the shock index (SI) (heart rate/systolic blood pressure, a noninvasive indication of left ventricular function),14 as a potential marker of the need for intensive care. Allgwer and Burri14 first developed the SI in studies of patients with acute blood loss, intraabdominal bleeding, fat emboli, and severe infections. They observed that a healthy adult had a mean SI of 0.54 (standard deviation [SD], 0.021), while an index of 1.0 indicated threatened shock and indices greater than 1.5 were seen in volume‐deficient shock.
We hypothesized that an elevated SI is a differentiating factor between a patient who had an unplanned ICU transfer and a general medical patient who did not require this higher level of care. To our knowledge, the SI has not been studied for this application previously.
Patients and Methods
Study Design
We conducted a retrospective case‐control study of 50 consecutive general medical patients who had unplanned transfers to the ICU and 50 matched control patients, with the approval of the Mayo Clinic Institutional Review Board. All patients were admitted to a general medical unit, and only patients who previously provided permission for their records to be used in research were included in the study.
Patients
This study enrolled patients who were at least 18 years old and who were admitted to a single general medical unit for 24 hours or longer. Patients were excluded if they required a surgical intervention, were transferred from another hospital, received care on a different general medical unit at any time during the hospitalization, or were pregnant. Our data collection began at the patients' (cases and controls) arrival on the general medical unit; we did not include data from any evaluation (outpatient or emergency department) before hospital admission.
Case Definition
An unplanned transfer was defined as an episode of unexpected clinical deterioration in a general medical patient that necessitated transfer to the ICU, as opposed to a preemptive or elective transfer following a procedure. Patients with unplanned transfers from December 9, 2003, to December 29, 2004, were eligible for the study. Only the first transfer to the ICU was considered for patients who had multiple ICU transfers during a single hospitalization. Because these data were collected before METs or RRTs were introduced at our institution, the recommendation for ICU transfer was a joint decision by the primary care team and the ICU team.
Control Definition
The matched controls were identified from among patients admitted to the general medical unit from January 16, 2002, to December 13, 2004. To reduce the effect of the heterogeneity inherent in general medical patients, we matched controls for age (within 5 years of age of the corresponding case), admission diagnosis code, and patient care unit of admission and required that they were admitted for at least 24 hours before dismissal. Patients who had an ICU stay during the same admission were excluded. The median difference in admission dates between the cases and the controls was 327 days, and 26 of the 50 matched pairs had admission dates within 1 year of each other. This lengthy interval between cases and controls was a consequence of the low incidence of patients who met the matching criteria.
Setting
This study involved the general medical units and ICUs of the 1157‐bed Saint Mary's Hospital, an academic tertiary care facility at Mayo Clinic in Rochester, Minnesota.
Vital Sign Determination
Vital signs abstracted for this study included blood pressure, heart rate, respiratory rate, oxygen saturation, and temperature. The SI was calculated for each set of abstracted vital signs. Staff nurses were responsible for the routine measurement and recording of vital signs at least once every 8 hours, although in several instances not all parameters were checked. In accordance with nursing policy, values outside the defined parameters were rechecked by the nursing supervisor of each care unit and, if found to be abnormal, were conveyed to the patient's physician. This system meant that abnormal results were checked by numerous observers, with differences in the frequency of recordings for individual patients.
Data Collection
Demographic data and information on the vital signs were abstracted through a comprehensive chart review. Demographic data included age, sex, ethnicity, comorbid conditions, hospital care unit, date and time of admission, admission diagnosis, date and time of transfer to the ICU, length of stay, dismissal date, and disposition at discharge. Comorbid conditions were scored using the Charlson Comorbidity Index.15
Statistical Analysis
A sample size of 50 matched pairs provided 81% power to detect an odds ratio of 3.0 or greater between cases and controls, with a 0.05, 2‐tailed level of significance with McNemar test. Patient demographic characteristics were summarized by the frequencies for categorical data and by mean and SD for continuous data. Consistent with the study design, the McNemar test and conditional logistic model analyses were used to determine the association between the SI and the risk of unplanned ICU transfer. Shock indices for the cases and controls were compared with use of t test. A P value <0.05 was considered statistically significant. For the SI, we calculated the odds ratio and its 95% confidence interval (CI) and P value using different cut points. We did not perform a receiver operating characteristics analysis because matching of cases and controls greatly complicates estimation of the sensitivity and specificity of the SI;16 a cohort study is suggested to investigate this analysis further. All statistical analyses were performed by SAS version 9.1.3 software (SAS Institute Inc, Cary, NC).
Results
A total of 50 pairs of matching cases and controls was included in this study. Table 1 lists the source of admission, demographic characteristics, and numbers of deaths for cases and controls. There were no statistically significant differences in admission source, age, sex, ethnicity, admission care unit, or Charlson Comorbidity Index. Mean length of stay was 14.8 days (SD, 9.7 days) for the cases and 5.7 days (SD, 6.3 days; P < 0.001) for the controls. Admission diagnoses were classified on the basis of the organ system of involvement (Table 2). In 30 of 50 cases, the admission diagnosis and the reason for ICU transfer were related.
| Value | Cases (n = 50) | Controls (n = 50) | P Value* |
|---|---|---|---|
| |||
| Emergency department admission, No. (%) | 33 (66) | 28 (56) | 0.41 |
| Direct admission, No. (%) | 14 (28) | 15 (30) | 1.00 |
| Other admission, No. (%) | 3 (6) | 7 (14) | 0.32 |
| Age, mean (SD), years | 69.8 (15.7) | 70.3 (15.8) | 0.38 |
| Male sex, No. (%) | 26 (52) | 18 (36) | 0.12 |
| Ethnicity, No. (%) | 1.00 | ||
| White | 46 (92) | 46 (92) | |
| Other | 4 (8) | 4 (8) | |
| Charlson Comorbidity index, mean (SD) | 3.06 (2.31) | 2.66 (2.02) | 0.22 |
| Hospital stay, mean (SD), day | 14.8 (9.7) | 5.7 (6.3) | 0.0007 |
| Hospital deaths, No. | 9 | 1 | 0.008 |
| Deaths within 30 days, No. | 5 | 2 | 0.24 |
| Deaths within 6 months, No. | 9 | 6 | 0.40 |
| System | Primary Admission Diagnosis | No. of Cases |
|---|---|---|
| Constitutional | Fever, malaise, general symptoms | 7 |
| Cardiovascular | Hypertension, congestive heart failure, chest pain, peripheral vascular disease, edema | 5 |
| Dermatologic | Cellulitis, foot ulcer, skin rash | 3 |
| Gastrointestinal | Pancreatitis, gastrointestinal hemorrhage, nausea and vomiting, diarrhea, abdominal pain | 6 |
| Hematologic | Thrombocytopenia, abnormal coagulation | 2 |
| Musculoskeletal | Lymphedema, shoulder pain, lumbago, back ache, closed dorsal vertebral fracture | 7 |
| Neurologic | Delirium tremens, psychosis, convulsions | 3 |
| Pulmonary | Pneumonia, food or vomit aspiration pneumonitis, shortness of breath, respiratory abnormality | 13 |
| Renal | Hyperkalemia, acute renal failure, renal artery atherosclerosis | 4 |
We reviewed the vital signs and shock indices for the 24 hours before ICU transfer for each case and over the entire hospitalization for each control, to determine the worst set (the lowest systolic blood pressure and the highest heart rate, respiratory rate, and SI). The cases had 1 to 22 complete sets of vitals for the 24 hours before ICU transfer; the median number of sets was 3 and the mean was 4. The controls had 1 to 12 complete sets for the 24 hours before the worst SI: the median was 3 sets and the mean was 3. In 26 of 50 controls, the worst SI occurred within the first 24 hours after admission. There was a significant difference between the median values of the worst shock indices of the cases and the controls (0.87 vs. 0.72; P < 0.005).
Table 3 shows the different values of the SI and the corresponding odds ratio of unplanned ICU transfer for cases compared with controls. The difference was significant at an SI of 0.85 and greater, indicating a strong association with unplanned ICU transfer.
| Shock Index | P Value | Odds Ratio | 95% CI |
|---|---|---|---|
| |||
| 0.8 | 0.05 | 2.43 | 1.015.86 |
| 0.85 | 0.02 | 3.00 | 1.917.56 |
| 0.9 | 0.007 | 7.50 | 1.7232.78 |
| 0.95 | <0.03 | 5.50 | 1.2224.81 |
We also found that the patients who transferred to the ICU had a greater number of inpatient deaths (9 cases vs. 1 control; P = 0.008), which would be expected, but there was no difference in 30‐day or 6‐month mortality rate (Table 1). One patient died after 30 days and while still hospitalized.
Comparison between the temporal trend of vital signs and the SI of the cases for the 24 hours before ICU transfer is shown in Figure 1. This graph shows the median of all the worst values (minimum systolic blood pressure and maximum SI, heart rate, and respiratory rate) over the four 6‐hour time periods (24 hours) preceding ICU transfer. Of note, the change in vital signs is subtle even while the SI increased to more than 0.8 as the patients clinically worsened before transfer.
Discussion
In our comparison of the SI of 50 patients who required unplanned (unexpected) transfer to the ICU with the SI of 50 matched controls who did not require this higher level of care, we found that a SI of 0.85 or greater was significantly associated with unplanned transfer to the ICU. The cases had a significantly higher worst SI than the controls, and they also had a significantly longer hospital stay and higher inpatient mortality rate, as would be expected for a sicker patient population. These findings are important given that the SI may be useful for assessing illness severity, for helping determine the need for transfer to the ICU, or for activating METs or RRTs.
A major problem with providing optimal care for hospitalized general medical patients is the inherent difficulty in determining illness severity and clinical decline, especially when the decline occurs gradually. Existing consensus recommendations for ICU admission include both specific diagnoses and arbitrary objective criteria based on abnormal vital signs and laboratory values.27 Also, individual institutions may have their own ICU admission requirements, which may differ from these or RRT criteria. Although vital signs are important as a snapshot of basic physiologic function, a number of noncritical conditions may lead to abnormal vital signs, and not all abnormal vital signs are associated with an adverse clinical event. By relying solely on vital signs, clinicians may not recognize critical illness and therefore not transfer a patient to the ICU or may inappropriately transfer a patient who does not need ICU‐level care.
Markers of illness severity other than vital signs, such as the Acute Physiology and Chronic Health Evaluation (APACHE) score, have been shown to predict the death of ICU patients17, 18 but have been rarely studied outside the ICU setting.19 Also, calculating the APACHE score is cumbersome, and there is no cutoff score that defines when a patient should be transferred to the ICU. Subbe et al.,20 in their study to identify critically ill patients, found that introduction of a physiological scoring system (including MET or RRT activation scores) would have identified only a small number of additional patients as critically ill. Another common marker of illness severity, the 4 criteria of the systemic inflammatory response syndrome (temperature <36C or >38C; heart rate >90 beats per minute; respiratory rate >20 breaths per minute or PCO2 <32 mm Hg; and white blood cell count >12,000/L or <4000/L or with more than 10% band cells)21 may be too sensitive to use as a decision aid, since even a healthy person running after a bus could have 2 of the 4 criteria.22 Likewise, surgical patients may have transient leukocytosis due to a stress response independent of an infection.23
The SI may be more accurate than vital signs alone to determine illness severity and who is at risk for an unplanned transfer to the ICU. Birkhahn et al.24 concluded that the SI may be more useful in early hemorrhage than either heart rate or systolic blood pressure alone. Rady et al.25 showed that the SI used in the emergency department can identify critical illness with apparently stable vital signs, where an elevation of the SI above 0.9 was associated with an illness that was treated immediately with admission to the hospital and intensive therapy on admission. However, it is unclear whether the SI can be used to monitor ongoing treatment, because a previous study showed that the SI may be of limited value in the assessment of systemic oxygen transport and response to therapy in clinical septic shock.26 Of note, the SI is mostly independent of the effects of pain or anxiety, which cause a concurrent rise in heart rate and systolic blood pressure. Because the heart's left ventricular work is unchanged or may increase from the underlying catecholamine surge, the SI will be unchanged or may actually decrease.
Our study adds to the medical literature the findings that: (1) the SI may be useful as an indicator of illness severity and a triage tool in patients with no trauma but with various medical conditions, and (2) the SI showed a strong association with unplanned ICU transfer.
The main strength of our study is its case‐control design with matched controls. Also, by comparing groups from the same patient care unit, we sought to minimize the selection bias that can be inherent in case‐control studies. Limitations include the retrospective, nonrandomized study design and the fact that there may have been variations in vital sign measurements by the multiple caregivers. However, the vital signs were taken according to standard hospital practice and reflect real‐world conditions. Although generalizability may be somewhat limited because of our homogeneous patient population, our patients had a wide range of various medical illnesses, so our study should be applicable to other hospital settings, both academic and community‐based.
One of the main weaknesses of our study is that the results were not adjusted for the burden of comorbid conditions, although there were no statistically significant differences in the number of comorbid conditions among the cases and the controls (P = 0.96). Also, we did not directly compare the SI with vital signs alone to determine superiority.
The SI may be an important objective measure to help clinicians decide when patients need treatment that is more aggressive, assistance from a MET or an RRT, or a preemptive, rather than unplanned, transfer to an ICU. Although it is unlikely that a single measure will allow accurate triage of all medical or surgical patients, the SI may be a useful adjunct to clinical judgment and other objective measures in determining illness severity and clinical decline. Further prospective studies are needed to compare the role of the SI specifically with MET or RRT activation criteria, to clarify the role of comorbid conditions in unplanned transfers to the ICU, to validate the cut point for the SI in various disease states, and to assess its utility in patients with septic shock. Depending on these results, it may be beneficial to incorporate the SI into the electronic medical record as an automatic alert to identify patients at risk for ICU transfer.
Conclusions
The SI is an easily calculated composite index of heart rate and systolic blood pressure. An elevated SI of 0.85 can identify patients who are at risk for unplanned transfer to the ICU from general patient care units. Future studies will determine whether the SI is more accurate than simple vital signs as an indicator of clinical decline. If so, it may be useful as a trigger to activate METs or RRTs for treatment.
- ,.Outcome of intensive care patients in a group of British intensive care units.Crit Care Med.1998;26(8):1337–1345.
- ,,,.The longer patients are in hospital before Intensive Care admission the higher their mortality.Intensive Care Med.2004;30(10):1908–1913.
- ,.Physiological abnormalities in early warning scores are related to mortality in adult inpatients.Br J Anaesth.2004;92(6):882–884.
- ,,,,.Association between clinically abnormal observations and subsequent in‐hospital mortality: a prospective study.Resuscitation.2004;62(2):137–141.
- ,,,,,.Recognising clinical instability in hospital patients before cardiac arrest or unplanned admission to intensive care: a pilot study in a tertiary‐care hospital.Med J Aust.1999;171(1):22–25.
- ,,.Physiological values and procedures in the 24 h before ICU admission from the ward.Anaesthesia.1999;54(6):529–534.
- ,,, et al.Antecedents to hospital deaths.Intern Med J.2001;31(6):343–348.
- ,,, et al.A comparison of antecedents to cardiac arrests, deaths and emergency intensive care admissions in Australia and New Zealand, and the United Kingdom: the ACADEMIA study.Resuscitation.2004;62(3):275–282.
- ,,,,.Anticipating events of in‐hospital cardiac arrest.Eur J Emerg Med.2004;11(1):24–28.
- ,,, et al.Duration of life‐threatening antecedents prior to intensive care admission.Intensive Care Med.2002;28(11):1629–1634.
- ,,,.The prevalence of recordings of the signs of critical conditions and emergency responses in hospital wards: the SOCCER study.Resuscitation.2005;65(2):149–157.
- ,,,,,.Hospital‐wide code rates and mortality before and after implementation of a rapid response team.JAMA.2008;300(21):2506–2513.
- ,,,,.Rapid response teams: a systematic review and meta‐analysis.Arch Intern Med.2010;170(1):18–26.
- ,. [Shock index.]Dtsch Med Wochenschr.1967;92(43):1947–50. [German]
- ,,,.A new method of classifying prognostic comorbidity in longitudinal studies: development and validation.J Chronic Dis.1987;40(5):373–83.
- ,.Matching in studies of classification accuracy: implications for analysis, efficiency, and assessment of incremental value.Biometrics.2008;64(1):1–9.
- ,,,.APACHE II: a severity of disease classification system.Crit Care Med.1985;13(10):818–829.
- ,.Outcome prediction in critical care: the Acute Physiology and Chronic Health Evaluation models.Curr Opin Crit Care.2008;14(5):491–497.
- ,,.APACHE II predicts long‐term survival in COPD patients admitted to a general medical ward.J Gen Intern Med.2003;18(10):824–830.
- ,,,.Validation of physiological scoring systems in the accident and emergency department.Emerg Med J.2006;23(11):841–845.
- ,,, et al;2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference.Crit Care Med.2003;31(4):1250–1256.
- .Dear SIRS, I'm sorry to say that I don't like you...Crit Care Med.1997;25(2):372–374.
- ,.The inflammatory response to surgery and trauma.Curr Opin Crit Care.2006;12(4):325–332.
- ,,,,.Shock index in diagnosing early acute hypovolemia.Am J Emerg Med.2005;23(3):323–326.
- ,,,,.A comparison of the shock index and conventional vital signs to identify acute, critical illness in the emergency department.Ann Emerg Med.1994;24(4):685–690. Erratum in:Ann Emerg Med.year="1994"1994;24(6):1208.
- ,,,.Shock index: a re‐evaluation in acute circulatory failure.Resuscitation.1992;23(3):227–234.
- ,,,,, et al.Guidelines for intensive care unit admission, discharge, and triage. Task Force of the American College of Critical Care Medicine, Society of Critical Care Medicine.Crit Care Med.1999;27(3):633–638.
- ,.Outcome of intensive care patients in a group of British intensive care units.Crit Care Med.1998;26(8):1337–1345.
- ,,,.The longer patients are in hospital before Intensive Care admission the higher their mortality.Intensive Care Med.2004;30(10):1908–1913.
- ,.Physiological abnormalities in early warning scores are related to mortality in adult inpatients.Br J Anaesth.2004;92(6):882–884.
- ,,,,.Association between clinically abnormal observations and subsequent in‐hospital mortality: a prospective study.Resuscitation.2004;62(2):137–141.
- ,,,,,.Recognising clinical instability in hospital patients before cardiac arrest or unplanned admission to intensive care: a pilot study in a tertiary‐care hospital.Med J Aust.1999;171(1):22–25.
- ,,.Physiological values and procedures in the 24 h before ICU admission from the ward.Anaesthesia.1999;54(6):529–534.
- ,,, et al.Antecedents to hospital deaths.Intern Med J.2001;31(6):343–348.
- ,,, et al.A comparison of antecedents to cardiac arrests, deaths and emergency intensive care admissions in Australia and New Zealand, and the United Kingdom: the ACADEMIA study.Resuscitation.2004;62(3):275–282.
- ,,,,.Anticipating events of in‐hospital cardiac arrest.Eur J Emerg Med.2004;11(1):24–28.
- ,,, et al.Duration of life‐threatening antecedents prior to intensive care admission.Intensive Care Med.2002;28(11):1629–1634.
- ,,,.The prevalence of recordings of the signs of critical conditions and emergency responses in hospital wards: the SOCCER study.Resuscitation.2005;65(2):149–157.
- ,,,,,.Hospital‐wide code rates and mortality before and after implementation of a rapid response team.JAMA.2008;300(21):2506–2513.
- ,,,,.Rapid response teams: a systematic review and meta‐analysis.Arch Intern Med.2010;170(1):18–26.
- ,. [Shock index.]Dtsch Med Wochenschr.1967;92(43):1947–50. [German]
- ,,,.A new method of classifying prognostic comorbidity in longitudinal studies: development and validation.J Chronic Dis.1987;40(5):373–83.
- ,.Matching in studies of classification accuracy: implications for analysis, efficiency, and assessment of incremental value.Biometrics.2008;64(1):1–9.
- ,,,.APACHE II: a severity of disease classification system.Crit Care Med.1985;13(10):818–829.
- ,.Outcome prediction in critical care: the Acute Physiology and Chronic Health Evaluation models.Curr Opin Crit Care.2008;14(5):491–497.
- ,,.APACHE II predicts long‐term survival in COPD patients admitted to a general medical ward.J Gen Intern Med.2003;18(10):824–830.
- ,,,.Validation of physiological scoring systems in the accident and emergency department.Emerg Med J.2006;23(11):841–845.
- ,,, et al;2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference.Crit Care Med.2003;31(4):1250–1256.
- .Dear SIRS, I'm sorry to say that I don't like you...Crit Care Med.1997;25(2):372–374.
- ,.The inflammatory response to surgery and trauma.Curr Opin Crit Care.2006;12(4):325–332.
- ,,,,.Shock index in diagnosing early acute hypovolemia.Am J Emerg Med.2005;23(3):323–326.
- ,,,,.A comparison of the shock index and conventional vital signs to identify acute, critical illness in the emergency department.Ann Emerg Med.1994;24(4):685–690. Erratum in:Ann Emerg Med.year="1994"1994;24(6):1208.
- ,,,.Shock index: a re‐evaluation in acute circulatory failure.Resuscitation.1992;23(3):227–234.
- ,,,,, et al.Guidelines for intensive care unit admission, discharge, and triage. Task Force of the American College of Critical Care Medicine, Society of Critical Care Medicine.Crit Care Med.1999;27(3):633–638.
Copyright © 2010 Society of Hospital Medicine
Postcards from Our Students
During their junior medicine rotation, our students are asked to post to Blackboard (an online student forum) an anonymous essay about an issue of professionalism or ethics, either inspiring or troubling. In many ways, these vignettes are like postcards, written by visitors describing foreign cultures and norms. They represent a way for the students to debrief, but also provide an opportunity for us, as faculty, to reflect upon the way we practice and teach medicine. Many postingslike postcards from exotic or historic placesare inspiring stories of residents and faculty extending themselves for their patients. Unfortunately, unlike typical postcards, there are also essays that are troubling or provoking and challenge us to consider how we could improve the professional and ethical environment on our teams.
In order to begin a learning process with our faculty and housestaff, we have presented a number of these anonymous essays at both faculty and housestaff Department of Medicine conferences as well as our monthly hospital Ethics conference. The goal of these conferences was to gather as a moral community to reflect on our students' experience and consider ways in which our day to day practice as attendings could be informed by what they tell us. In addition, the junior medicine site directors have a session each quarter with their junior students to review some of the most significant issues brought up by their essays.
Practically, these vignettes and conferences serve three main purposes:
-
Raising Awareness: Many professional issues noted by our students occur under the radar. Attendings are often unaware of the issues of professionalism and/or ethics confronting our students and housestaff.
-
Exploring Attitudes: Some attending may underemphasize the importance of specific issues of professionalism and/or ethics. Open discussions at faculty or resident conferences create opportunities for individuals to reflect upon their own reactions and for the group to create a norm.
-
Sharing Skills: It is difficult to learn the practice of professionalism and ethics from a book. Skill in this area is gained primarily by experience. Conferences provide an excellent forum for seasoned physicians to share wisdom with less experienced physicians. In addition, important teaching points can be made: Students should not deliver bad news alone. Errors should be disclosed.
Following are 3 of the essays we presented, along with brief commentaries. At the end, we provide practical suggestions for individual attendings to improve the professional climate on their teams.
The Hospital Didn't Wait
Code. On 12, the surgical wards floor. Elise sprinted to the stairwell, dashed up to 12, and ran to the corner room as fast as she could. She could see the room before she got there. Instinctively, she started reviewing the steps she had memorized so many months ago. But when she finally arrived at the patient's bathroom, her thought process came to a jolting halt as she came upon the gruesome scene.
The 76‐year‐old patient had hanged himself with the cinching rope from his garment bag, and now dangled suspended from a high towel rack against the wall. Nurses from the floor started to file in, and without losing a beat Elise barked commands. Together they brought the man's body down to the floor, laid him on his back, and stripped off his hospital gown. Elise was in charge; deliberately but forcefully, she ordered a nurse to retrieve a defibrillator, and had another resident check for a pulse. There was none. Anesthesiology was here. Quickly and expertly, they shoved a plastic tube down his throat and began ventilation. The nurse placed on the electrodes between chest compressions then called to clear the body. Airway stepped back. The chest pumper stepped back. The body lurched forward as the defibrillator issued a long beep and discharged. Still no pulse. The cycle repeated.
Finally, Elise called a stop. Time of death, 19:37. By now there were about 20 people crammed in the patient room, all of whom had a separate role during the code. Some stayed behind, while the rest left to return to their interrupted work. The medical student didn't know what to think as he returned to the team room. His jaw was sorehad he been clenching it the whole time?and as he brought his hand up to rub his face, he saw that his knuckles were bloody. Somehow he had scraped them during the code. As he logged back into the computer to finish off his evening notes, he knew that he wouldn't have time to reflect until hours later when he returned home. Codes happened all the time. There was still work to be done in the hospital, and the hospital didn't wait.
The room had already been assigned to a patient waiting in the Emergency Department downstairs. That patient would be here in a few minutes. The hospital didn't wait.
When we presented this case in our conferences, there was universal agreement that such a traumatic event merits, even demands, team debriefing and processing. But in the real life aftermath of this traumatic event, the take‐home message for the medical student was that the hospital didn't wait for such discussions. We know this is not unique to our institution. In a study of 32 medical students who were asked to reflect on their most memorable patient death,1 debriefing sessions were rare and many students felt inadequately supported. While experienced clinicians may be accustomed to seeing patients die, students are new to the culture of the hospital, and have not had the chance to develop the defense mechanisms necessary to cope with this sort of experience. Angoff2 writes, As medical educators, we ought to ask our students how they are coping with long hours, fatigue, illness, suffering, and death. We ought to model and commend compassion and react to the deep feelings of our students in the same way we would teach them to react to the deep feelings of their patients.
I Told a Man Today That He Had Brain Cancer
The resident, intern, and I were huddled together in our team room when the report came back on the computer. New 3.5 2.3 1.7 cm contrast‐enhancing lesion seen anterior to genu of corpus callosum. Concerning for metastatic focus vs. lymphoma. Advise follow‐up. It wasn't unexpected but we had nevertheless been hoping for better.
The three of us went into his room and I was waiting to see how my resident would deliver the bad news, but she didn't. She simply said that we were continuing to do imaging studies and that a neurology team would be in touch. There were probably several reasons why she didn't tell him: not enough time, not her responsibility, or maybe she was just uncomfortable with it. Whatever the case, we left the room with my patient still oblivious to the awful mass now tangled in his head.
If my resident was taking a pass on this conversation, I knew it fell to me he needed to hear it from his primary team. I came back after rounds alone, sat down next to his bed, and told him that his MRI results had come back, and that I had unfortunate news.
I told him that the images showed that his lung cancer had spread to his brain.
I paused to give him a chance to let it sink in. He turned away and looked up at the ceiling.
Where is it? How big is it?
What now?
Reflecting on this case, our audiences were disturbed that a student would attempt this difficult conversation alone, while recognizing that the student clearly felt a sense of responsibility and desire to help his patient by sharing important information. We talked about how students may erroneously pick up a message that the team member who has spent the most time with a patient is the most obvious choice to have difficult conversations. We also noted that, unfortunately, sometimes students are directly asked by their team to shoulder this responsibility on their own. In this painful account, there is no mention of preparation, supervision, or support for the student before or after the encounter. The student perceived (rightly or wrongly) that the team leaders lacked comfort or skill to deliver the bad news, and stepped in. It is possible that the attending lacked the skill and ability to model an interaction, but more likely the deficit was in awareness and attitude. It is unlikely the attending knew that the student had this conversation alone. One of the major reasons we present these vignettes is to make attendings and housestaff more aware of issues that occur under their radar so that they can take preventative action. However, once the resident or attending found out that the student had this conversation alone, the student should be pulled aside for a 1:1 discussion. At the end of the day, the student should know that it was inappropriate to attempt this conversation alone
Rosenbaum3 reviewed a number of strategies to teach the skill of delivering bad news, from lecture and small group discussions to role play and standardized patients. When asked, students cited role‐modeling as the best way to learn how to deliver bad news.4 Observation of a veteran clinician provides a firm foundation for learning; but that is not enough. Unfortunately, we know from the literature (and our student vignettes suggest) that students and residents are unprepared to carry out these conversations properly, either because of misguided attitudes, lack of experience, or inadequate training.57 We conceptualize engaging in difficult conversations as a procedure, demanding a skill set. Mere observation of an expert executing this procedure is only a beginning. With any other skill, from successful completion of a lumbar puncture to initiating cardiopulmonary resuscitation (CPR), a student would never conclude that knowing the patient the best sufficiently credentials the student to undertake these procedures. We maintain that a difficult conversationbe it breaking bad news, discussing end‐of‐life care preferences, code status discussions, or prognosisis a clinical intervention, like any other procedure in medicine. If performed with skill and caution, it can bring about a stronger therapeutic relationship and increased support for the patient; if performed clumsily, it can lead to unintended adverse outcomes, including misunderstanding, mistrust, anxiety, and anger.
A Decimal Point Got Misplaced
On palliative care, I had a 90 year‐old man with end stage lung CA that presented to the ED with increasing SOB. The resident decided that giving him some morphine would be a good solution but was worried that too much would push him over the edge. He was thin; his O2 sats weren't that good After some discussion it was decided that 2.5 mg should be the starting amount. Unfortunately, when the note was written a decimal point got misplaced and he got 25 mg as a first dose. He ended up very sedated for most of the day but his breathing was ok.
The mistake was not discussed with the patient or the patient's family. While it did not cause any lasting harm, I wondered if telling the patient/patient's family that an error had been made would have been more ethically sound.
When we presented this case in our conferences, there was little controversy about whether the error should have been disclosed. The discussion did provide reinforcement for doing a simple but difficult task. Our analysis is that the nondiscussion of this error reflects a deficit in attitude and possibly skill. The team was aware of the error, but the resident and attending did not take the opportunity to disclose an error. They should have. We do not know whether the attending or resident felt unprepared to discuss this or were simply unimpressed with the adverse event. We do get the sense that the student did not feel comfortable raising the issue with the team. As such, it was a missed opportunity to seek help from any number of hospital resources and find encouragement to take on difficult encounters.
Much has been written about apologies.810 Disclosing errors and apologizing is the ethical standard, and many of our institutions have made it policy. Yet in the moment, it is embarrassing, anxiety provoking, and our concern about litigation looms large. Learning to do the right thing begins, perhaps with lectures and standardized patients, but only when students see it modeled by our housestaff and faculty, does it take root for good.
Our housestaff are quite good at managing medical issues, but they may still need help in creating the appropriate environment for professional learning and growth. This is 1 of the most important contributions an attending can make. We have emphasized that faculty have an important role to play in the area of professional development, reinforcing the rudimentary information preclinical students are presented with in the classroom and processing experiences residents are exposed to on a regular basis. If the hospital doesn't wait, then it is the attending physician's job to create the space and time for trainees to think about what is happening and ask if it could have been done better.
A number of seasoned clinical teachers have written about ways to improve teaching on the wards.11 Below, we will add to that discussion by considering practical ways to enhance learning about professionalism and ethics (see Table 1). Note should be made that while we focus on specific behaviors and activities, underlying all is the importance of availability, presence, and intention. Like all good teaching, these activities require planning and effort.
| Attending Activity | Examples |
|---|---|
| Creating an Open Climate | |
| Breaking Communication Barriers | Setting aside time for introductions and team building exercises at the beginning of a rotation, with attending participating equally with residents and students |
| Emphasizing attending availability to discuss or review problems of any kind | |
| Setting Clear Expectations | Emphasizing the importance of patient‐clinician or family‐clinician communication from the outset |
| Devoting some attending rounds to Difficult Conversations (e.g., breaking bad news or code status discussions) | |
| Explicitly stating that no ethical question is a stupid question and providing positive feedback for raising such questions for the team | |
| Regular Check‐ins | Establishing team communication rounds: 10 minutes every day to review a good, bad, or awkward interaction from the past day (e.g., family meeting, DNR discussion) |
| Setting aside time on rounds or during attending teaching sessions to explore the team's or an individual's emotional responses to a patient's death or deterioration | |
| Writing exercises that focus on our reactions to challenging situations that are shared with the group | |
| Supervision and Modeling | |
| Planning | Clarifying an agenda and practicing key phrases for a family meeting with the resident prior to meeting the family |
| Anticipating which patients may require a code status discussion and discussing a game plan on rounds | |
| Modeling | Students observe the attending facilitate a family meeting |
| Residents observe the attending apologizing for an error, no matter how small | |
| Attending thinks about an interpersonal conflict out loud and models asking patient‐relations for help | |
| Debriefing | Reviewing a family meeting with and giving feedback to the resident who facilitated |
| Reviewing a challenging code status discussion as a team |
Creating an Open Climate
The medical team, of which the attending, residents, and students are all a part, should not only be a unit that provides excellent medical care to its patients, but should also create a culture of continuous learning and improvement. As such, it is important to create a safe atmosphere where teachers are invested in the growth of their learners and learners feel free to question the prevailing logic and practice, including issues of professionalism and ethics. As Malcolm Gladwell12 describes in Outliers, Korean Air jets were crashing because subordinates were afraid to question their superiors. Once that culture changed, Korean Air safety improved dramatically. Similarly, breaking down some of the hierarchical barriers should improve the culture of a medical team. We typically make an effort to get to know our students and residents on a more personal basis: where they are from, who is in their family, what was their major, what are their interests outside of medicine, and what has been surprising to them in their training so far. Whether we set aside time when we first meet or e‐mail our questions before the first day, we aim for this to be 1 of the first team activities. We also share our own stories, making clear that the attending is part of the team, and not just an evaluating supervisor.
Vignette 1 describes the student's trauma of witnessing a code and the inability to process the event with anyone afterward. Failed resuscitation attempts are the most dramatic examples, but even expected deaths, nonfatal adverse events, and conflict between patients and providers may be traumatic for new trainees inexperienced with the reality of medicine. Attendings should be aware of these potentially traumatic events and make time to check in with the team members about how they are dealing with their emotions. Taking time on attending rounds, for example, allows the attending to not only model reflective practice and self‐care, but also elevates team support to a place traditionally reserved for discussions about diagnosis and treatment.
Supervision and Modeling
Vignettes 2 and 3 center around challenging communication tasks that require special training, including instruction, modeling, feedback, and practice. Unfortunately, as some of our student accounts document, many teaching opportunities are missed. As attendings, our duties include being aware of these opportunities, and being prepared to model competent patientor familydoctor interactions. Emphasizing the importance of the doctor‐patient relationship is in fact one of the key skills of an effective attending role model.13
When opportunities arise for any potentially difficult conversation, we make every effort to identify the issue, prebrief with the team about how to conduct the discussion, and either offer to model the conversation or be present to observe and provide feedback and debriefing afterwards. For example, by asking about all DNR discussions had with our patients, we gain insight into the skill level of our housestaff. As important, the housestaff understand that we believe that these conversations are vital to review during formal rounds, with the same attention we give to chest pain and electrocardiograms (ECGs).
Two key skills that develop with experience are the ability to know the limits of one's knowledge and to know when to ask for help. We try to be open about naming those limits and thinking about the other members of the larger healthcare team that may provide insight, skill, and expertise. We are used to doing this with medical questions (eg, asking the gastroenterology consult team to locate a source of bleeding). Asking our risk management, patient‐relations, or ethics services to assist with a difficult communication task or conflict with a family is no different, and often something the housestaff may not readily do.
We are grateful to our students and their postcards for the snapshots of our local medical culture. While we are gratified to read of excellent role modeling, we are also disappointed to read of situations which have left our students confused, demoralized and cynical. But if these exercises are to reach their full potential, they should tell us about where we would like to go, in addition to where we have been. We believe that our conferences have stimulated our faculty and housestaff to reflect on the professionalism lessons they are teaching. Reading the student postings has definitely affected our approach to teaching professionalism. They reinforce what every parent and educator knows: when it comes to teaching professionalism, communication and ethics, what matters most is the behavior of the teacher. Our words mean little if our actions do not live out what we espouse.
Acknowledgements
We are grateful for Michael Chan and his classmates from the NUFSM class of 2010 for their thoughtful essays. David Neely, Director of Undergraduate Education, Department of Medicine, Eytan Szmuilowicz, Palliative Medicine. Kathy Neely, Chairman of NMH Ethics Committee. Co‐director of Patient, Physician and Society. This article was previously published in this journal in Vol 5, Issue 5:E10E13 (2010) as online‐only.
- .This is just too awful; I just can't believe I experienced that.Acad Med.2005;80(7):634–640.
- .A piece of my mind.JAMA.2001;286(9):1017–1018.
- .Teaching medical students and residents skills for delivering bad news: a review of strategies.Acad Med.2004;79(2):107–117.
- .Third‐year medical students' experiences with dying patients during the internal medicine clerkship: a qualitative study of the informal curriculum.Acad Med.2005;80(7):641–647.
- ,,.How do medical residents discuss resuscitation with patients? Official journal of the Society for Research and Education in Primary Care Internal Medicine.J Gen Intern Med.1995;10(8):436–442.
- ,,.See one, do one, teach one? House staff experience discussing do‐not‐resuscitate orders.Arch Intern Med.1996;156(12):1285–1289.
- ,,,.Residents' end‐of‐life decision making with adult hospitalized patients: a review of the literature.Acad Med.2005:80(7)622–633.
- .Physician error and disclosure.Clin Obstet Gynecol.2008;51(4):700–708.
- .Revealing medical errors to your patients.Chest.2009;133:1064–1065.
- .Apology in medical practice.JAMA.2006;296:1401–1404.
- .What if Osler were one of us? Inpatient teaching today.J Gen Intern Med.1997;12(Suppl 2):S41–S48.
- Malcolm Gladwell.Outliers.New York:Little, Brown, Co.,2008: p.177–223.
- ,,,,.Attributes of excellent attending‐physician role models.N Engl J Med.1998;339(27):1986–1993.
During their junior medicine rotation, our students are asked to post to Blackboard (an online student forum) an anonymous essay about an issue of professionalism or ethics, either inspiring or troubling. In many ways, these vignettes are like postcards, written by visitors describing foreign cultures and norms. They represent a way for the students to debrief, but also provide an opportunity for us, as faculty, to reflect upon the way we practice and teach medicine. Many postingslike postcards from exotic or historic placesare inspiring stories of residents and faculty extending themselves for their patients. Unfortunately, unlike typical postcards, there are also essays that are troubling or provoking and challenge us to consider how we could improve the professional and ethical environment on our teams.
In order to begin a learning process with our faculty and housestaff, we have presented a number of these anonymous essays at both faculty and housestaff Department of Medicine conferences as well as our monthly hospital Ethics conference. The goal of these conferences was to gather as a moral community to reflect on our students' experience and consider ways in which our day to day practice as attendings could be informed by what they tell us. In addition, the junior medicine site directors have a session each quarter with their junior students to review some of the most significant issues brought up by their essays.
Practically, these vignettes and conferences serve three main purposes:
-
Raising Awareness: Many professional issues noted by our students occur under the radar. Attendings are often unaware of the issues of professionalism and/or ethics confronting our students and housestaff.
-
Exploring Attitudes: Some attending may underemphasize the importance of specific issues of professionalism and/or ethics. Open discussions at faculty or resident conferences create opportunities for individuals to reflect upon their own reactions and for the group to create a norm.
-
Sharing Skills: It is difficult to learn the practice of professionalism and ethics from a book. Skill in this area is gained primarily by experience. Conferences provide an excellent forum for seasoned physicians to share wisdom with less experienced physicians. In addition, important teaching points can be made: Students should not deliver bad news alone. Errors should be disclosed.
Following are 3 of the essays we presented, along with brief commentaries. At the end, we provide practical suggestions for individual attendings to improve the professional climate on their teams.
The Hospital Didn't Wait
Code. On 12, the surgical wards floor. Elise sprinted to the stairwell, dashed up to 12, and ran to the corner room as fast as she could. She could see the room before she got there. Instinctively, she started reviewing the steps she had memorized so many months ago. But when she finally arrived at the patient's bathroom, her thought process came to a jolting halt as she came upon the gruesome scene.
The 76‐year‐old patient had hanged himself with the cinching rope from his garment bag, and now dangled suspended from a high towel rack against the wall. Nurses from the floor started to file in, and without losing a beat Elise barked commands. Together they brought the man's body down to the floor, laid him on his back, and stripped off his hospital gown. Elise was in charge; deliberately but forcefully, she ordered a nurse to retrieve a defibrillator, and had another resident check for a pulse. There was none. Anesthesiology was here. Quickly and expertly, they shoved a plastic tube down his throat and began ventilation. The nurse placed on the electrodes between chest compressions then called to clear the body. Airway stepped back. The chest pumper stepped back. The body lurched forward as the defibrillator issued a long beep and discharged. Still no pulse. The cycle repeated.
Finally, Elise called a stop. Time of death, 19:37. By now there were about 20 people crammed in the patient room, all of whom had a separate role during the code. Some stayed behind, while the rest left to return to their interrupted work. The medical student didn't know what to think as he returned to the team room. His jaw was sorehad he been clenching it the whole time?and as he brought his hand up to rub his face, he saw that his knuckles were bloody. Somehow he had scraped them during the code. As he logged back into the computer to finish off his evening notes, he knew that he wouldn't have time to reflect until hours later when he returned home. Codes happened all the time. There was still work to be done in the hospital, and the hospital didn't wait.
The room had already been assigned to a patient waiting in the Emergency Department downstairs. That patient would be here in a few minutes. The hospital didn't wait.
When we presented this case in our conferences, there was universal agreement that such a traumatic event merits, even demands, team debriefing and processing. But in the real life aftermath of this traumatic event, the take‐home message for the medical student was that the hospital didn't wait for such discussions. We know this is not unique to our institution. In a study of 32 medical students who were asked to reflect on their most memorable patient death,1 debriefing sessions were rare and many students felt inadequately supported. While experienced clinicians may be accustomed to seeing patients die, students are new to the culture of the hospital, and have not had the chance to develop the defense mechanisms necessary to cope with this sort of experience. Angoff2 writes, As medical educators, we ought to ask our students how they are coping with long hours, fatigue, illness, suffering, and death. We ought to model and commend compassion and react to the deep feelings of our students in the same way we would teach them to react to the deep feelings of their patients.
I Told a Man Today That He Had Brain Cancer
The resident, intern, and I were huddled together in our team room when the report came back on the computer. New 3.5 2.3 1.7 cm contrast‐enhancing lesion seen anterior to genu of corpus callosum. Concerning for metastatic focus vs. lymphoma. Advise follow‐up. It wasn't unexpected but we had nevertheless been hoping for better.
The three of us went into his room and I was waiting to see how my resident would deliver the bad news, but she didn't. She simply said that we were continuing to do imaging studies and that a neurology team would be in touch. There were probably several reasons why she didn't tell him: not enough time, not her responsibility, or maybe she was just uncomfortable with it. Whatever the case, we left the room with my patient still oblivious to the awful mass now tangled in his head.
If my resident was taking a pass on this conversation, I knew it fell to me he needed to hear it from his primary team. I came back after rounds alone, sat down next to his bed, and told him that his MRI results had come back, and that I had unfortunate news.
I told him that the images showed that his lung cancer had spread to his brain.
I paused to give him a chance to let it sink in. He turned away and looked up at the ceiling.
Where is it? How big is it?
What now?
Reflecting on this case, our audiences were disturbed that a student would attempt this difficult conversation alone, while recognizing that the student clearly felt a sense of responsibility and desire to help his patient by sharing important information. We talked about how students may erroneously pick up a message that the team member who has spent the most time with a patient is the most obvious choice to have difficult conversations. We also noted that, unfortunately, sometimes students are directly asked by their team to shoulder this responsibility on their own. In this painful account, there is no mention of preparation, supervision, or support for the student before or after the encounter. The student perceived (rightly or wrongly) that the team leaders lacked comfort or skill to deliver the bad news, and stepped in. It is possible that the attending lacked the skill and ability to model an interaction, but more likely the deficit was in awareness and attitude. It is unlikely the attending knew that the student had this conversation alone. One of the major reasons we present these vignettes is to make attendings and housestaff more aware of issues that occur under their radar so that they can take preventative action. However, once the resident or attending found out that the student had this conversation alone, the student should be pulled aside for a 1:1 discussion. At the end of the day, the student should know that it was inappropriate to attempt this conversation alone
Rosenbaum3 reviewed a number of strategies to teach the skill of delivering bad news, from lecture and small group discussions to role play and standardized patients. When asked, students cited role‐modeling as the best way to learn how to deliver bad news.4 Observation of a veteran clinician provides a firm foundation for learning; but that is not enough. Unfortunately, we know from the literature (and our student vignettes suggest) that students and residents are unprepared to carry out these conversations properly, either because of misguided attitudes, lack of experience, or inadequate training.57 We conceptualize engaging in difficult conversations as a procedure, demanding a skill set. Mere observation of an expert executing this procedure is only a beginning. With any other skill, from successful completion of a lumbar puncture to initiating cardiopulmonary resuscitation (CPR), a student would never conclude that knowing the patient the best sufficiently credentials the student to undertake these procedures. We maintain that a difficult conversationbe it breaking bad news, discussing end‐of‐life care preferences, code status discussions, or prognosisis a clinical intervention, like any other procedure in medicine. If performed with skill and caution, it can bring about a stronger therapeutic relationship and increased support for the patient; if performed clumsily, it can lead to unintended adverse outcomes, including misunderstanding, mistrust, anxiety, and anger.
A Decimal Point Got Misplaced
On palliative care, I had a 90 year‐old man with end stage lung CA that presented to the ED with increasing SOB. The resident decided that giving him some morphine would be a good solution but was worried that too much would push him over the edge. He was thin; his O2 sats weren't that good After some discussion it was decided that 2.5 mg should be the starting amount. Unfortunately, when the note was written a decimal point got misplaced and he got 25 mg as a first dose. He ended up very sedated for most of the day but his breathing was ok.
The mistake was not discussed with the patient or the patient's family. While it did not cause any lasting harm, I wondered if telling the patient/patient's family that an error had been made would have been more ethically sound.
When we presented this case in our conferences, there was little controversy about whether the error should have been disclosed. The discussion did provide reinforcement for doing a simple but difficult task. Our analysis is that the nondiscussion of this error reflects a deficit in attitude and possibly skill. The team was aware of the error, but the resident and attending did not take the opportunity to disclose an error. They should have. We do not know whether the attending or resident felt unprepared to discuss this or were simply unimpressed with the adverse event. We do get the sense that the student did not feel comfortable raising the issue with the team. As such, it was a missed opportunity to seek help from any number of hospital resources and find encouragement to take on difficult encounters.
Much has been written about apologies.810 Disclosing errors and apologizing is the ethical standard, and many of our institutions have made it policy. Yet in the moment, it is embarrassing, anxiety provoking, and our concern about litigation looms large. Learning to do the right thing begins, perhaps with lectures and standardized patients, but only when students see it modeled by our housestaff and faculty, does it take root for good.
Our housestaff are quite good at managing medical issues, but they may still need help in creating the appropriate environment for professional learning and growth. This is 1 of the most important contributions an attending can make. We have emphasized that faculty have an important role to play in the area of professional development, reinforcing the rudimentary information preclinical students are presented with in the classroom and processing experiences residents are exposed to on a regular basis. If the hospital doesn't wait, then it is the attending physician's job to create the space and time for trainees to think about what is happening and ask if it could have been done better.
A number of seasoned clinical teachers have written about ways to improve teaching on the wards.11 Below, we will add to that discussion by considering practical ways to enhance learning about professionalism and ethics (see Table 1). Note should be made that while we focus on specific behaviors and activities, underlying all is the importance of availability, presence, and intention. Like all good teaching, these activities require planning and effort.
| Attending Activity | Examples |
|---|---|
| Creating an Open Climate | |
| Breaking Communication Barriers | Setting aside time for introductions and team building exercises at the beginning of a rotation, with attending participating equally with residents and students |
| Emphasizing attending availability to discuss or review problems of any kind | |
| Setting Clear Expectations | Emphasizing the importance of patient‐clinician or family‐clinician communication from the outset |
| Devoting some attending rounds to Difficult Conversations (e.g., breaking bad news or code status discussions) | |
| Explicitly stating that no ethical question is a stupid question and providing positive feedback for raising such questions for the team | |
| Regular Check‐ins | Establishing team communication rounds: 10 minutes every day to review a good, bad, or awkward interaction from the past day (e.g., family meeting, DNR discussion) |
| Setting aside time on rounds or during attending teaching sessions to explore the team's or an individual's emotional responses to a patient's death or deterioration | |
| Writing exercises that focus on our reactions to challenging situations that are shared with the group | |
| Supervision and Modeling | |
| Planning | Clarifying an agenda and practicing key phrases for a family meeting with the resident prior to meeting the family |
| Anticipating which patients may require a code status discussion and discussing a game plan on rounds | |
| Modeling | Students observe the attending facilitate a family meeting |
| Residents observe the attending apologizing for an error, no matter how small | |
| Attending thinks about an interpersonal conflict out loud and models asking patient‐relations for help | |
| Debriefing | Reviewing a family meeting with and giving feedback to the resident who facilitated |
| Reviewing a challenging code status discussion as a team |
Creating an Open Climate
The medical team, of which the attending, residents, and students are all a part, should not only be a unit that provides excellent medical care to its patients, but should also create a culture of continuous learning and improvement. As such, it is important to create a safe atmosphere where teachers are invested in the growth of their learners and learners feel free to question the prevailing logic and practice, including issues of professionalism and ethics. As Malcolm Gladwell12 describes in Outliers, Korean Air jets were crashing because subordinates were afraid to question their superiors. Once that culture changed, Korean Air safety improved dramatically. Similarly, breaking down some of the hierarchical barriers should improve the culture of a medical team. We typically make an effort to get to know our students and residents on a more personal basis: where they are from, who is in their family, what was their major, what are their interests outside of medicine, and what has been surprising to them in their training so far. Whether we set aside time when we first meet or e‐mail our questions before the first day, we aim for this to be 1 of the first team activities. We also share our own stories, making clear that the attending is part of the team, and not just an evaluating supervisor.
Vignette 1 describes the student's trauma of witnessing a code and the inability to process the event with anyone afterward. Failed resuscitation attempts are the most dramatic examples, but even expected deaths, nonfatal adverse events, and conflict between patients and providers may be traumatic for new trainees inexperienced with the reality of medicine. Attendings should be aware of these potentially traumatic events and make time to check in with the team members about how they are dealing with their emotions. Taking time on attending rounds, for example, allows the attending to not only model reflective practice and self‐care, but also elevates team support to a place traditionally reserved for discussions about diagnosis and treatment.
Supervision and Modeling
Vignettes 2 and 3 center around challenging communication tasks that require special training, including instruction, modeling, feedback, and practice. Unfortunately, as some of our student accounts document, many teaching opportunities are missed. As attendings, our duties include being aware of these opportunities, and being prepared to model competent patientor familydoctor interactions. Emphasizing the importance of the doctor‐patient relationship is in fact one of the key skills of an effective attending role model.13
When opportunities arise for any potentially difficult conversation, we make every effort to identify the issue, prebrief with the team about how to conduct the discussion, and either offer to model the conversation or be present to observe and provide feedback and debriefing afterwards. For example, by asking about all DNR discussions had with our patients, we gain insight into the skill level of our housestaff. As important, the housestaff understand that we believe that these conversations are vital to review during formal rounds, with the same attention we give to chest pain and electrocardiograms (ECGs).
Two key skills that develop with experience are the ability to know the limits of one's knowledge and to know when to ask for help. We try to be open about naming those limits and thinking about the other members of the larger healthcare team that may provide insight, skill, and expertise. We are used to doing this with medical questions (eg, asking the gastroenterology consult team to locate a source of bleeding). Asking our risk management, patient‐relations, or ethics services to assist with a difficult communication task or conflict with a family is no different, and often something the housestaff may not readily do.
We are grateful to our students and their postcards for the snapshots of our local medical culture. While we are gratified to read of excellent role modeling, we are also disappointed to read of situations which have left our students confused, demoralized and cynical. But if these exercises are to reach their full potential, they should tell us about where we would like to go, in addition to where we have been. We believe that our conferences have stimulated our faculty and housestaff to reflect on the professionalism lessons they are teaching. Reading the student postings has definitely affected our approach to teaching professionalism. They reinforce what every parent and educator knows: when it comes to teaching professionalism, communication and ethics, what matters most is the behavior of the teacher. Our words mean little if our actions do not live out what we espouse.
Acknowledgements
We are grateful for Michael Chan and his classmates from the NUFSM class of 2010 for their thoughtful essays. David Neely, Director of Undergraduate Education, Department of Medicine, Eytan Szmuilowicz, Palliative Medicine. Kathy Neely, Chairman of NMH Ethics Committee. Co‐director of Patient, Physician and Society. This article was previously published in this journal in Vol 5, Issue 5:E10E13 (2010) as online‐only.
During their junior medicine rotation, our students are asked to post to Blackboard (an online student forum) an anonymous essay about an issue of professionalism or ethics, either inspiring or troubling. In many ways, these vignettes are like postcards, written by visitors describing foreign cultures and norms. They represent a way for the students to debrief, but also provide an opportunity for us, as faculty, to reflect upon the way we practice and teach medicine. Many postingslike postcards from exotic or historic placesare inspiring stories of residents and faculty extending themselves for their patients. Unfortunately, unlike typical postcards, there are also essays that are troubling or provoking and challenge us to consider how we could improve the professional and ethical environment on our teams.
In order to begin a learning process with our faculty and housestaff, we have presented a number of these anonymous essays at both faculty and housestaff Department of Medicine conferences as well as our monthly hospital Ethics conference. The goal of these conferences was to gather as a moral community to reflect on our students' experience and consider ways in which our day to day practice as attendings could be informed by what they tell us. In addition, the junior medicine site directors have a session each quarter with their junior students to review some of the most significant issues brought up by their essays.
Practically, these vignettes and conferences serve three main purposes:
-
Raising Awareness: Many professional issues noted by our students occur under the radar. Attendings are often unaware of the issues of professionalism and/or ethics confronting our students and housestaff.
-
Exploring Attitudes: Some attending may underemphasize the importance of specific issues of professionalism and/or ethics. Open discussions at faculty or resident conferences create opportunities for individuals to reflect upon their own reactions and for the group to create a norm.
-
Sharing Skills: It is difficult to learn the practice of professionalism and ethics from a book. Skill in this area is gained primarily by experience. Conferences provide an excellent forum for seasoned physicians to share wisdom with less experienced physicians. In addition, important teaching points can be made: Students should not deliver bad news alone. Errors should be disclosed.
Following are 3 of the essays we presented, along with brief commentaries. At the end, we provide practical suggestions for individual attendings to improve the professional climate on their teams.
The Hospital Didn't Wait
Code. On 12, the surgical wards floor. Elise sprinted to the stairwell, dashed up to 12, and ran to the corner room as fast as she could. She could see the room before she got there. Instinctively, she started reviewing the steps she had memorized so many months ago. But when she finally arrived at the patient's bathroom, her thought process came to a jolting halt as she came upon the gruesome scene.
The 76‐year‐old patient had hanged himself with the cinching rope from his garment bag, and now dangled suspended from a high towel rack against the wall. Nurses from the floor started to file in, and without losing a beat Elise barked commands. Together they brought the man's body down to the floor, laid him on his back, and stripped off his hospital gown. Elise was in charge; deliberately but forcefully, she ordered a nurse to retrieve a defibrillator, and had another resident check for a pulse. There was none. Anesthesiology was here. Quickly and expertly, they shoved a plastic tube down his throat and began ventilation. The nurse placed on the electrodes between chest compressions then called to clear the body. Airway stepped back. The chest pumper stepped back. The body lurched forward as the defibrillator issued a long beep and discharged. Still no pulse. The cycle repeated.
Finally, Elise called a stop. Time of death, 19:37. By now there were about 20 people crammed in the patient room, all of whom had a separate role during the code. Some stayed behind, while the rest left to return to their interrupted work. The medical student didn't know what to think as he returned to the team room. His jaw was sorehad he been clenching it the whole time?and as he brought his hand up to rub his face, he saw that his knuckles were bloody. Somehow he had scraped them during the code. As he logged back into the computer to finish off his evening notes, he knew that he wouldn't have time to reflect until hours later when he returned home. Codes happened all the time. There was still work to be done in the hospital, and the hospital didn't wait.
The room had already been assigned to a patient waiting in the Emergency Department downstairs. That patient would be here in a few minutes. The hospital didn't wait.
When we presented this case in our conferences, there was universal agreement that such a traumatic event merits, even demands, team debriefing and processing. But in the real life aftermath of this traumatic event, the take‐home message for the medical student was that the hospital didn't wait for such discussions. We know this is not unique to our institution. In a study of 32 medical students who were asked to reflect on their most memorable patient death,1 debriefing sessions were rare and many students felt inadequately supported. While experienced clinicians may be accustomed to seeing patients die, students are new to the culture of the hospital, and have not had the chance to develop the defense mechanisms necessary to cope with this sort of experience. Angoff2 writes, As medical educators, we ought to ask our students how they are coping with long hours, fatigue, illness, suffering, and death. We ought to model and commend compassion and react to the deep feelings of our students in the same way we would teach them to react to the deep feelings of their patients.
I Told a Man Today That He Had Brain Cancer
The resident, intern, and I were huddled together in our team room when the report came back on the computer. New 3.5 2.3 1.7 cm contrast‐enhancing lesion seen anterior to genu of corpus callosum. Concerning for metastatic focus vs. lymphoma. Advise follow‐up. It wasn't unexpected but we had nevertheless been hoping for better.
The three of us went into his room and I was waiting to see how my resident would deliver the bad news, but she didn't. She simply said that we were continuing to do imaging studies and that a neurology team would be in touch. There were probably several reasons why she didn't tell him: not enough time, not her responsibility, or maybe she was just uncomfortable with it. Whatever the case, we left the room with my patient still oblivious to the awful mass now tangled in his head.
If my resident was taking a pass on this conversation, I knew it fell to me he needed to hear it from his primary team. I came back after rounds alone, sat down next to his bed, and told him that his MRI results had come back, and that I had unfortunate news.
I told him that the images showed that his lung cancer had spread to his brain.
I paused to give him a chance to let it sink in. He turned away and looked up at the ceiling.
Where is it? How big is it?
What now?
Reflecting on this case, our audiences were disturbed that a student would attempt this difficult conversation alone, while recognizing that the student clearly felt a sense of responsibility and desire to help his patient by sharing important information. We talked about how students may erroneously pick up a message that the team member who has spent the most time with a patient is the most obvious choice to have difficult conversations. We also noted that, unfortunately, sometimes students are directly asked by their team to shoulder this responsibility on their own. In this painful account, there is no mention of preparation, supervision, or support for the student before or after the encounter. The student perceived (rightly or wrongly) that the team leaders lacked comfort or skill to deliver the bad news, and stepped in. It is possible that the attending lacked the skill and ability to model an interaction, but more likely the deficit was in awareness and attitude. It is unlikely the attending knew that the student had this conversation alone. One of the major reasons we present these vignettes is to make attendings and housestaff more aware of issues that occur under their radar so that they can take preventative action. However, once the resident or attending found out that the student had this conversation alone, the student should be pulled aside for a 1:1 discussion. At the end of the day, the student should know that it was inappropriate to attempt this conversation alone
Rosenbaum3 reviewed a number of strategies to teach the skill of delivering bad news, from lecture and small group discussions to role play and standardized patients. When asked, students cited role‐modeling as the best way to learn how to deliver bad news.4 Observation of a veteran clinician provides a firm foundation for learning; but that is not enough. Unfortunately, we know from the literature (and our student vignettes suggest) that students and residents are unprepared to carry out these conversations properly, either because of misguided attitudes, lack of experience, or inadequate training.57 We conceptualize engaging in difficult conversations as a procedure, demanding a skill set. Mere observation of an expert executing this procedure is only a beginning. With any other skill, from successful completion of a lumbar puncture to initiating cardiopulmonary resuscitation (CPR), a student would never conclude that knowing the patient the best sufficiently credentials the student to undertake these procedures. We maintain that a difficult conversationbe it breaking bad news, discussing end‐of‐life care preferences, code status discussions, or prognosisis a clinical intervention, like any other procedure in medicine. If performed with skill and caution, it can bring about a stronger therapeutic relationship and increased support for the patient; if performed clumsily, it can lead to unintended adverse outcomes, including misunderstanding, mistrust, anxiety, and anger.
A Decimal Point Got Misplaced
On palliative care, I had a 90 year‐old man with end stage lung CA that presented to the ED with increasing SOB. The resident decided that giving him some morphine would be a good solution but was worried that too much would push him over the edge. He was thin; his O2 sats weren't that good After some discussion it was decided that 2.5 mg should be the starting amount. Unfortunately, when the note was written a decimal point got misplaced and he got 25 mg as a first dose. He ended up very sedated for most of the day but his breathing was ok.
The mistake was not discussed with the patient or the patient's family. While it did not cause any lasting harm, I wondered if telling the patient/patient's family that an error had been made would have been more ethically sound.
When we presented this case in our conferences, there was little controversy about whether the error should have been disclosed. The discussion did provide reinforcement for doing a simple but difficult task. Our analysis is that the nondiscussion of this error reflects a deficit in attitude and possibly skill. The team was aware of the error, but the resident and attending did not take the opportunity to disclose an error. They should have. We do not know whether the attending or resident felt unprepared to discuss this or were simply unimpressed with the adverse event. We do get the sense that the student did not feel comfortable raising the issue with the team. As such, it was a missed opportunity to seek help from any number of hospital resources and find encouragement to take on difficult encounters.
Much has been written about apologies.810 Disclosing errors and apologizing is the ethical standard, and many of our institutions have made it policy. Yet in the moment, it is embarrassing, anxiety provoking, and our concern about litigation looms large. Learning to do the right thing begins, perhaps with lectures and standardized patients, but only when students see it modeled by our housestaff and faculty, does it take root for good.
Our housestaff are quite good at managing medical issues, but they may still need help in creating the appropriate environment for professional learning and growth. This is 1 of the most important contributions an attending can make. We have emphasized that faculty have an important role to play in the area of professional development, reinforcing the rudimentary information preclinical students are presented with in the classroom and processing experiences residents are exposed to on a regular basis. If the hospital doesn't wait, then it is the attending physician's job to create the space and time for trainees to think about what is happening and ask if it could have been done better.
A number of seasoned clinical teachers have written about ways to improve teaching on the wards.11 Below, we will add to that discussion by considering practical ways to enhance learning about professionalism and ethics (see Table 1). Note should be made that while we focus on specific behaviors and activities, underlying all is the importance of availability, presence, and intention. Like all good teaching, these activities require planning and effort.
| Attending Activity | Examples |
|---|---|
| Creating an Open Climate | |
| Breaking Communication Barriers | Setting aside time for introductions and team building exercises at the beginning of a rotation, with attending participating equally with residents and students |
| Emphasizing attending availability to discuss or review problems of any kind | |
| Setting Clear Expectations | Emphasizing the importance of patient‐clinician or family‐clinician communication from the outset |
| Devoting some attending rounds to Difficult Conversations (e.g., breaking bad news or code status discussions) | |
| Explicitly stating that no ethical question is a stupid question and providing positive feedback for raising such questions for the team | |
| Regular Check‐ins | Establishing team communication rounds: 10 minutes every day to review a good, bad, or awkward interaction from the past day (e.g., family meeting, DNR discussion) |
| Setting aside time on rounds or during attending teaching sessions to explore the team's or an individual's emotional responses to a patient's death or deterioration | |
| Writing exercises that focus on our reactions to challenging situations that are shared with the group | |
| Supervision and Modeling | |
| Planning | Clarifying an agenda and practicing key phrases for a family meeting with the resident prior to meeting the family |
| Anticipating which patients may require a code status discussion and discussing a game plan on rounds | |
| Modeling | Students observe the attending facilitate a family meeting |
| Residents observe the attending apologizing for an error, no matter how small | |
| Attending thinks about an interpersonal conflict out loud and models asking patient‐relations for help | |
| Debriefing | Reviewing a family meeting with and giving feedback to the resident who facilitated |
| Reviewing a challenging code status discussion as a team |
Creating an Open Climate
The medical team, of which the attending, residents, and students are all a part, should not only be a unit that provides excellent medical care to its patients, but should also create a culture of continuous learning and improvement. As such, it is important to create a safe atmosphere where teachers are invested in the growth of their learners and learners feel free to question the prevailing logic and practice, including issues of professionalism and ethics. As Malcolm Gladwell12 describes in Outliers, Korean Air jets were crashing because subordinates were afraid to question their superiors. Once that culture changed, Korean Air safety improved dramatically. Similarly, breaking down some of the hierarchical barriers should improve the culture of a medical team. We typically make an effort to get to know our students and residents on a more personal basis: where they are from, who is in their family, what was their major, what are their interests outside of medicine, and what has been surprising to them in their training so far. Whether we set aside time when we first meet or e‐mail our questions before the first day, we aim for this to be 1 of the first team activities. We also share our own stories, making clear that the attending is part of the team, and not just an evaluating supervisor.
Vignette 1 describes the student's trauma of witnessing a code and the inability to process the event with anyone afterward. Failed resuscitation attempts are the most dramatic examples, but even expected deaths, nonfatal adverse events, and conflict between patients and providers may be traumatic for new trainees inexperienced with the reality of medicine. Attendings should be aware of these potentially traumatic events and make time to check in with the team members about how they are dealing with their emotions. Taking time on attending rounds, for example, allows the attending to not only model reflective practice and self‐care, but also elevates team support to a place traditionally reserved for discussions about diagnosis and treatment.
Supervision and Modeling
Vignettes 2 and 3 center around challenging communication tasks that require special training, including instruction, modeling, feedback, and practice. Unfortunately, as some of our student accounts document, many teaching opportunities are missed. As attendings, our duties include being aware of these opportunities, and being prepared to model competent patientor familydoctor interactions. Emphasizing the importance of the doctor‐patient relationship is in fact one of the key skills of an effective attending role model.13
When opportunities arise for any potentially difficult conversation, we make every effort to identify the issue, prebrief with the team about how to conduct the discussion, and either offer to model the conversation or be present to observe and provide feedback and debriefing afterwards. For example, by asking about all DNR discussions had with our patients, we gain insight into the skill level of our housestaff. As important, the housestaff understand that we believe that these conversations are vital to review during formal rounds, with the same attention we give to chest pain and electrocardiograms (ECGs).
Two key skills that develop with experience are the ability to know the limits of one's knowledge and to know when to ask for help. We try to be open about naming those limits and thinking about the other members of the larger healthcare team that may provide insight, skill, and expertise. We are used to doing this with medical questions (eg, asking the gastroenterology consult team to locate a source of bleeding). Asking our risk management, patient‐relations, or ethics services to assist with a difficult communication task or conflict with a family is no different, and often something the housestaff may not readily do.
We are grateful to our students and their postcards for the snapshots of our local medical culture. While we are gratified to read of excellent role modeling, we are also disappointed to read of situations which have left our students confused, demoralized and cynical. But if these exercises are to reach their full potential, they should tell us about where we would like to go, in addition to where we have been. We believe that our conferences have stimulated our faculty and housestaff to reflect on the professionalism lessons they are teaching. Reading the student postings has definitely affected our approach to teaching professionalism. They reinforce what every parent and educator knows: when it comes to teaching professionalism, communication and ethics, what matters most is the behavior of the teacher. Our words mean little if our actions do not live out what we espouse.
Acknowledgements
We are grateful for Michael Chan and his classmates from the NUFSM class of 2010 for their thoughtful essays. David Neely, Director of Undergraduate Education, Department of Medicine, Eytan Szmuilowicz, Palliative Medicine. Kathy Neely, Chairman of NMH Ethics Committee. Co‐director of Patient, Physician and Society. This article was previously published in this journal in Vol 5, Issue 5:E10E13 (2010) as online‐only.
- .This is just too awful; I just can't believe I experienced that.Acad Med.2005;80(7):634–640.
- .A piece of my mind.JAMA.2001;286(9):1017–1018.
- .Teaching medical students and residents skills for delivering bad news: a review of strategies.Acad Med.2004;79(2):107–117.
- .Third‐year medical students' experiences with dying patients during the internal medicine clerkship: a qualitative study of the informal curriculum.Acad Med.2005;80(7):641–647.
- ,,.How do medical residents discuss resuscitation with patients? Official journal of the Society for Research and Education in Primary Care Internal Medicine.J Gen Intern Med.1995;10(8):436–442.
- ,,.See one, do one, teach one? House staff experience discussing do‐not‐resuscitate orders.Arch Intern Med.1996;156(12):1285–1289.
- ,,,.Residents' end‐of‐life decision making with adult hospitalized patients: a review of the literature.Acad Med.2005:80(7)622–633.
- .Physician error and disclosure.Clin Obstet Gynecol.2008;51(4):700–708.
- .Revealing medical errors to your patients.Chest.2009;133:1064–1065.
- .Apology in medical practice.JAMA.2006;296:1401–1404.
- .What if Osler were one of us? Inpatient teaching today.J Gen Intern Med.1997;12(Suppl 2):S41–S48.
- Malcolm Gladwell.Outliers.New York:Little, Brown, Co.,2008: p.177–223.
- ,,,,.Attributes of excellent attending‐physician role models.N Engl J Med.1998;339(27):1986–1993.
- .This is just too awful; I just can't believe I experienced that.Acad Med.2005;80(7):634–640.
- .A piece of my mind.JAMA.2001;286(9):1017–1018.
- .Teaching medical students and residents skills for delivering bad news: a review of strategies.Acad Med.2004;79(2):107–117.
- .Third‐year medical students' experiences with dying patients during the internal medicine clerkship: a qualitative study of the informal curriculum.Acad Med.2005;80(7):641–647.
- ,,.How do medical residents discuss resuscitation with patients? Official journal of the Society for Research and Education in Primary Care Internal Medicine.J Gen Intern Med.1995;10(8):436–442.
- ,,.See one, do one, teach one? House staff experience discussing do‐not‐resuscitate orders.Arch Intern Med.1996;156(12):1285–1289.
- ,,,.Residents' end‐of‐life decision making with adult hospitalized patients: a review of the literature.Acad Med.2005:80(7)622–633.
- .Physician error and disclosure.Clin Obstet Gynecol.2008;51(4):700–708.
- .Revealing medical errors to your patients.Chest.2009;133:1064–1065.
- .Apology in medical practice.JAMA.2006;296:1401–1404.
- .What if Osler were one of us? Inpatient teaching today.J Gen Intern Med.1997;12(Suppl 2):S41–S48.
- Malcolm Gladwell.Outliers.New York:Little, Brown, Co.,2008: p.177–223.
- ,,,,.Attributes of excellent attending‐physician role models.N Engl J Med.1998;339(27):1986–1993.
Management of Inpatient Hyperglycemia
Hyperglycemia that develops acutely due to illness is associated with poor patient outcomes in hospitalized inpatients, especially those critically ill in the intensive care unit (ICU).18 In fact, those without a prior diagnosis of diabetes and therefore newly found to have hyperglycemia have worse outcomes than those who have a prior diagnosis of diabetes.1, 2, 4, 68 Many mechanisms have been put forward to explain the adverse outcomes related to hyperglycemia, including the release of counter‐regulatory hormones, increased lipolysis with free fatty acid release, the release of inflammatory cytokines and growth factors, increased reactive oxygen species with oxidative stress and altered immunoglobulin, and neutrophil phagocytic function.911
The practical importance of this was brought home by Furnary et al.12, 13 who showed that glycemic control using 3 days of intensive intravenous (IV) insulin therapy of diabetic patients undergoing cardiac surgical procedures was able to reduce significantly the risk of deep sternal wound infections and mortality and to bring these adverse outcomes to the same levels as those of nondiabetic patients. However, the benefits of intensive insulin therapy are not limited to those with diabetes and extend to those with critical illness‐induced hyperglycemia. In a landmark, randomized, prospective study from Belgium, van den Berghe et al.14 showed that the use of an intensive IV insulin protocol designed to maintain serum blood glucose 80 mg/dL to 110 mg/dL significantly decreased morbidity and mortality following admission to the surgical ICU (SICU). Of note, only 13% of the individuals in the study had a previously known diagnosis of diabetes, showing that hyperglycemia was common following SICU admission and glycemic control was beneficial regardless of diabetes status.14
These impressive benefits1214 led to the call for improved glycemic control in the hospital with glucose targets similar to those used in the Belgian study.15 The development of protocols for such treatment proceeded rapidly.16, 17 A meta‐analysis that reviewed 14 trials through May 1, 2006 of patients in SICUs showed a 31% reduction in mortality with intensive therapy, albeit at the expense of a substantially increased risk of hypoglycemia.18 Our own studies19 using 1 day of continuous insulin infusion followed by subcutaneous basal/bolus insulin for all hyperglycemic patients following coronary artery bypass surgery showed results similar to those of Furnary et al.12, 13
Subsequently, 3 large, multicenter studies of patients in medical ICU (MICU) and SICUs, the VISEP, NICE‐SUGAR, and GLUCONTROL studies, failed to show the benefit of intensive insulin therapy on mortality and all had very high rates of hypoglycemia.2022 The VISEP20 study was stopped prematurely because of excessive hypoglycemia in the intensive treatment arm and the GLUCONTROL study was stopped prematurely because of multiple protocol violations.22 The NICE‐SUGAR study actually showed an increased mortality in the intensively treated group21 but the target range for their control group was 140 mg/dL to 180 mg/dL rather than 180 mg/dL to 215 mg/dL and this likely accounted for the better mortality in their control group compared to other studies. Van den Berghe et al.,23 in a design similar to their earlier one in the SICU, found that intensive insulin therapy in the MICU resulted in significant reductions in new onset renal injury, MICU and hospital length of stay, and an improved ability to wean off mechanical ventilation; however, no improvement in mortality was found except for those whose MICU stay was >3 days duration. In a post hoc analysis of their combined SICU and MICU studies, van den Berghe et al.23 found that a glucose target of 110 mg/dL to 150 mg/dL accounted for about 75% of the mortality benefit with a low risk of hypoglycemia.24 A recent meta‐analysis that included data on 13,567 patients from 26 trials, including the NICE‐SUGAR study, concluded that although overall there was no mortality benefit from intensive insulin therapy there was benefit in the SICU but not in the MICU or mixed ICU units.25
As a result of these later studies, new recommendations from the American Association of Clinical Endocrinologists and the American Diabetes Association state that for optimal risk/benefit, the overall goal of inpatient treatment for most patients should be 140 mg/dL to 180 mg/dL, although a range of 110 mg/dL to 140 mg/dL may be appropriate for some patients.26 Stressed in this Consensus Statement is the need for experienced practitioners and systems to provide optimal implementation of protocols so as to provide adequate glycemic control without an undue amount of hypoglycemia. We have found that active individual patient management by experienced nurse practitioners who can modify existing protocols as needed provides better glycemic control with less hypoglycemia than nursing personnel adhering to a protocol without taking into account the myriad of factors affecting patients daily.16
Although hypoglycemia is certainly to be avoided and has been associated with increased mortality,6, 27 Kosiborod et al.7 showed that mortality in hyperglycemic patients following an acute myocardial infarction was not related to insulin‐induced hypoglycemia but to hypoglycemia unassociated with insulin use. In the latter case, the hypoglycemia is generally attributable to shock, sepsis, malnutrition, liver failure, renal failure, or multiorgan failure.
One of the potential hurdles to achievement of glycemic control in the critically ill is the labor‐intensive changes in patient care policies necessary to attain these goals. Particular concern lies in the ability of inpatient care providers to develop and implement successful insulin protocols. Intravenous insulin administration is effective and appropriate in the ICU and some non‐ICU settings, but administration of insulin subcutaneously is less nursing intensive and a more familiar hyperglycemia treatment option. However, glycemic control with subcutaneous insulin is only achieved using basal/bolus regimens and not with simple sliding‐scale regimens that omit basal insulin and attempt to treat rather than prevent hyperglycemia.28
In this issue of the Journal of Hospital Medicine, 3 articles deal with some of the practical aspects of inpatient hyperglycemia management. In ICU patients on continuous IV insulin infusions, Newton et al.29 demonstrated improved glycemic control without an increase in hypoglycemia when using a computer‐guided insulin algorithm using a hand‐held device (Glucommander) compared to a paper algorithm. A previous publication in JHM showed that when continuous insulin infusions were used on the regular hospital floors outside of the ICU, Smiley et al.30 found that 67% of patients achieved the targeted goal of 150 mg/dL by day 2. Wesorick et al.31 found that simply educating floor nurses as well as physicians and using standardized insulin protocols resulted in improved glycemic control and less hypoglycemia on inpatient services outside of the ICU. In the third paper, Ramos et al. found that those with glycosylated hemoglobin (HbA1c) levels >6.0%, a prior history of diabetes, or chronic steroid use did better with basal insulin than with no basal insulin when converting from insulin infusions.32 In contrast to their using only 40% of the stable insulin infusion rate for their basal dose, we found that 80% worked better.33 We also learned the hard way that overlap of the infusion by 2 hours to 4 hours after giving the basal insulin subcutaneous dose is just not carried out by the treating team because of timing and practical considerations. We now just give a dose of rapid acting insulin equal to 10% of the basal insulin dose at the time of the injection of the basal dose; this allows for the immediate cessation of the infusion without loss of glycemic control (Table 1).
|
| Measure HbA1c on admission to aid in discharge planning |
| Start insulin infusions in postoperative and other unstable patients if blood glucose >180 mg/dL on 2 or more occasions |
| Begin IV continuous insulin infusion using validated protocols |
| Glucose target: 140‐180 mg/dL* |
| When converting from IV to subcutaneous insulin |
| Give 80% of stable IV dose as glargine insulin |
| Give 10% of glargine dose as rapid acting insulin |
| Then stop insulin infusion |
| If starting with subcutaneous insulin without prior insulin infusion |
| Give 50% as long‐acting insulin (glargine or detemir) |
| Give 50% as rapid‐acting insulin, divided into 3 for the 3 meals |
Intensive insulin treatment in the ICU clearly results in better outcomes when compared to letting glucose levels remain greater than 200 mg/dL. A glucose target range of 140 mg/dL to 180 mg/dL provides improved mortality and morbidity with a low risk of hypoglycemia and is suitable for most hospitals. A more aggressive target range of 110 mg/dL to 140 mg/dL provides further improvement but increases the risk of hypoglycemia and would only be appropriate for those institutions with considerable experience with such therapy and demonstrated low rates of hypoglycemia. Work is still needed on devising the ideal treatment algorithm, regimens for conversion from IV to subcutaneous insulin, and discharge planning. However, the most important part of patient care we have found is the insertion of an intelligent and experienced brain between the patient and the insulin protocol.
- ,,, et al.Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: A systematic overview.Stroke.2001;32;2426–2432.
- ,,, et al.Hyperglycemia: an independent marker of in‐hospital mortality in patients with undiagnosed diabetes.J Clin Endocrinol Metab.2002;87;978–982.
- .Association between hyperglycemia and increased hospital mortality in a heterogeneous population of critically ill patients.Mayo Clin Proc.2003;78:1471–1478.
- ,,, et al.Influence of individual characteristics on outcome of glycemic control in intensive care unit patients with or without diabetes mellitus.Mayo Clin Proc.2005;80:1558–1567.
- ,,, et al.The relation between hyperglycemia and outcomes in 2,471 patients admitted to the hospital with community acquired pneumonia.Diabetes Care.2005;28:810–815.
- ,,, et al.Admission glucose and mortality in elderly patients hospitalized with acute myocardial infarction. Implications for patients with and without recognized diabetes.Circulation.2005;111:3078–3086.
- ,,, et al.Glucometrics in patients hospitalized with acute myocardial infarction. Defining the optimal outcomes‐based measure of risk.Circulation.2008;117:1018–1027.
- ,,, et al.Hyperglycemia‐related mortality in critically ill patients varies with admission diagnosis.Crit Care Med.2009;37:3001–3009.
- .Effect of insulin therapy on nonglycemic variables during acute illness.Endocr Pract.2004;10(Suppl 2):63–70.
- ,, et al.Therapy insight: the effect of tight glycemic control in acute illness.Nature Clin Pract Endocrinol Metab.2007;3:270–278.
- ,,.Stress hyperglycemia.Lancet.2009;373:1798–1807.
- ,,, et al.Continuous intravenous insulin infusion reduces the incidence of deep sternal wound infection in diabetic patients after cardiac surgical procedures.Ann Thorac Surg.1999;67:352–362.
- ,,, et al.Insulin infusion reduces mortality in patients undergoing coronary artery bypass grafting.J Thorac Cardiovasc Surg.2003;125:1007–1021.
- ,,, et al.Intensive insulin therapy in critically ill patients.N Engl J Med.2001;345:1359–1367.
- ,,, et al.American College of Endocrinology Position Statement on inpatient diabetes and metabolic control.Endocr Pract.2004;10:77–82.
- ,,, et al.Inpatient management of hyperglycemia: The Northwestern Experience.Endocr Pract.2006:12;491–505.
- ,,.Insulin infusion protocols for critically ill patients: a highlight of differences and similarities.Endocr Pract.2007;13:137–146.
- ,,, et al.Effect of perioperative insulin infusion on surgical morbidity and mortality: systematic review and meta‐analysis of randomized trials.Mayo Clin Proc.2008;83:418–430.
- ,,, et al.Reduction in surgical mortality and morbidity in diabetic patients undergoing cardiac surgery with a combined intravenous and subcutaneous insulin glucose management.Diabetes Care.2007;30:823–828.
- ,,, et al.Intensive insulin therapy and pentastarch resuscitation in severe sepsis.N Engl J Med.2008;358:125–139.
- ,,Su SY for the NICE‐SUGAR Study Investigators. Intensive versus conventional glucose control in critically ill patients.N Engl J Med.2009;360:1283–1297.
- ,,, et al.A prospective randomised multi‐centre controlled trial on tight glucose control by intensive insulin therapy in adult intensive care units: the Glucontrol Study.Intensive Care Med.2009;35:1738–1748.
- ,,, et al.Intensive insulin therapy in the medical ICU.N Engl J Med.2006;354:449–461.
- ,,, et al.Intensive insulin therapy in mixed medical/surgical intensive care units: benefit versus harm.Diabetes.2006;55:3151–3159.
- ,,, et al.Intensive insulin therapy and mortality among critically ill patients: a meta‐analysis including NIC‐SUGAR study data.CMAJ.2009;180:821–827.
- ,,, et al.American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control.Diabetes Care.2009;32:1119–1131.
- ,.Severe hypoglycemia in critically ill patients: risk factors and outcomes.Crit Care Med.2007;35:2262–2267.
- ,,, et al.Randomized study of basal‐bolus insulin therapy in the inpatient management of patients with type 2 diabetes (RABBIT 2 trial).Diabetes Care.2007;30:2181–2186.
- ,,, et al.A comparison study of continuous insulin infusion protocols in the Medical Intensive Care Unit: computer‐guided versus standard column‐based algorithms.J Hosp Med. 2010;5:432–437.
- ,,, et al.Safety and efficacy of continuous insulin infusion in noncritical care settings.J Hosp Med.2010;5(4):212–217.
- ,,, et al.Effects of an educational program and a standardized insulin order form on glycemic outcomes in non‐critically ill hospitalized patients.J Hosp Med. 2010;5:438–445.
- ,,, et al.Maintaining glycemic control when transitioning from infusion insulin: a protocol driven, multidisciplinary approach.J Hosp Med. 2010;5:446–451.
- ,,, et al.Conversion of intravenous insulin infusions to subcutaneously administered insulin glargine in patients with hyperglycemia.Endocr Pract.2006;12:641–650.
Hyperglycemia that develops acutely due to illness is associated with poor patient outcomes in hospitalized inpatients, especially those critically ill in the intensive care unit (ICU).18 In fact, those without a prior diagnosis of diabetes and therefore newly found to have hyperglycemia have worse outcomes than those who have a prior diagnosis of diabetes.1, 2, 4, 68 Many mechanisms have been put forward to explain the adverse outcomes related to hyperglycemia, including the release of counter‐regulatory hormones, increased lipolysis with free fatty acid release, the release of inflammatory cytokines and growth factors, increased reactive oxygen species with oxidative stress and altered immunoglobulin, and neutrophil phagocytic function.911
The practical importance of this was brought home by Furnary et al.12, 13 who showed that glycemic control using 3 days of intensive intravenous (IV) insulin therapy of diabetic patients undergoing cardiac surgical procedures was able to reduce significantly the risk of deep sternal wound infections and mortality and to bring these adverse outcomes to the same levels as those of nondiabetic patients. However, the benefits of intensive insulin therapy are not limited to those with diabetes and extend to those with critical illness‐induced hyperglycemia. In a landmark, randomized, prospective study from Belgium, van den Berghe et al.14 showed that the use of an intensive IV insulin protocol designed to maintain serum blood glucose 80 mg/dL to 110 mg/dL significantly decreased morbidity and mortality following admission to the surgical ICU (SICU). Of note, only 13% of the individuals in the study had a previously known diagnosis of diabetes, showing that hyperglycemia was common following SICU admission and glycemic control was beneficial regardless of diabetes status.14
These impressive benefits1214 led to the call for improved glycemic control in the hospital with glucose targets similar to those used in the Belgian study.15 The development of protocols for such treatment proceeded rapidly.16, 17 A meta‐analysis that reviewed 14 trials through May 1, 2006 of patients in SICUs showed a 31% reduction in mortality with intensive therapy, albeit at the expense of a substantially increased risk of hypoglycemia.18 Our own studies19 using 1 day of continuous insulin infusion followed by subcutaneous basal/bolus insulin for all hyperglycemic patients following coronary artery bypass surgery showed results similar to those of Furnary et al.12, 13
Subsequently, 3 large, multicenter studies of patients in medical ICU (MICU) and SICUs, the VISEP, NICE‐SUGAR, and GLUCONTROL studies, failed to show the benefit of intensive insulin therapy on mortality and all had very high rates of hypoglycemia.2022 The VISEP20 study was stopped prematurely because of excessive hypoglycemia in the intensive treatment arm and the GLUCONTROL study was stopped prematurely because of multiple protocol violations.22 The NICE‐SUGAR study actually showed an increased mortality in the intensively treated group21 but the target range for their control group was 140 mg/dL to 180 mg/dL rather than 180 mg/dL to 215 mg/dL and this likely accounted for the better mortality in their control group compared to other studies. Van den Berghe et al.,23 in a design similar to their earlier one in the SICU, found that intensive insulin therapy in the MICU resulted in significant reductions in new onset renal injury, MICU and hospital length of stay, and an improved ability to wean off mechanical ventilation; however, no improvement in mortality was found except for those whose MICU stay was >3 days duration. In a post hoc analysis of their combined SICU and MICU studies, van den Berghe et al.23 found that a glucose target of 110 mg/dL to 150 mg/dL accounted for about 75% of the mortality benefit with a low risk of hypoglycemia.24 A recent meta‐analysis that included data on 13,567 patients from 26 trials, including the NICE‐SUGAR study, concluded that although overall there was no mortality benefit from intensive insulin therapy there was benefit in the SICU but not in the MICU or mixed ICU units.25
As a result of these later studies, new recommendations from the American Association of Clinical Endocrinologists and the American Diabetes Association state that for optimal risk/benefit, the overall goal of inpatient treatment for most patients should be 140 mg/dL to 180 mg/dL, although a range of 110 mg/dL to 140 mg/dL may be appropriate for some patients.26 Stressed in this Consensus Statement is the need for experienced practitioners and systems to provide optimal implementation of protocols so as to provide adequate glycemic control without an undue amount of hypoglycemia. We have found that active individual patient management by experienced nurse practitioners who can modify existing protocols as needed provides better glycemic control with less hypoglycemia than nursing personnel adhering to a protocol without taking into account the myriad of factors affecting patients daily.16
Although hypoglycemia is certainly to be avoided and has been associated with increased mortality,6, 27 Kosiborod et al.7 showed that mortality in hyperglycemic patients following an acute myocardial infarction was not related to insulin‐induced hypoglycemia but to hypoglycemia unassociated with insulin use. In the latter case, the hypoglycemia is generally attributable to shock, sepsis, malnutrition, liver failure, renal failure, or multiorgan failure.
One of the potential hurdles to achievement of glycemic control in the critically ill is the labor‐intensive changes in patient care policies necessary to attain these goals. Particular concern lies in the ability of inpatient care providers to develop and implement successful insulin protocols. Intravenous insulin administration is effective and appropriate in the ICU and some non‐ICU settings, but administration of insulin subcutaneously is less nursing intensive and a more familiar hyperglycemia treatment option. However, glycemic control with subcutaneous insulin is only achieved using basal/bolus regimens and not with simple sliding‐scale regimens that omit basal insulin and attempt to treat rather than prevent hyperglycemia.28
In this issue of the Journal of Hospital Medicine, 3 articles deal with some of the practical aspects of inpatient hyperglycemia management. In ICU patients on continuous IV insulin infusions, Newton et al.29 demonstrated improved glycemic control without an increase in hypoglycemia when using a computer‐guided insulin algorithm using a hand‐held device (Glucommander) compared to a paper algorithm. A previous publication in JHM showed that when continuous insulin infusions were used on the regular hospital floors outside of the ICU, Smiley et al.30 found that 67% of patients achieved the targeted goal of 150 mg/dL by day 2. Wesorick et al.31 found that simply educating floor nurses as well as physicians and using standardized insulin protocols resulted in improved glycemic control and less hypoglycemia on inpatient services outside of the ICU. In the third paper, Ramos et al. found that those with glycosylated hemoglobin (HbA1c) levels >6.0%, a prior history of diabetes, or chronic steroid use did better with basal insulin than with no basal insulin when converting from insulin infusions.32 In contrast to their using only 40% of the stable insulin infusion rate for their basal dose, we found that 80% worked better.33 We also learned the hard way that overlap of the infusion by 2 hours to 4 hours after giving the basal insulin subcutaneous dose is just not carried out by the treating team because of timing and practical considerations. We now just give a dose of rapid acting insulin equal to 10% of the basal insulin dose at the time of the injection of the basal dose; this allows for the immediate cessation of the infusion without loss of glycemic control (Table 1).
|
| Measure HbA1c on admission to aid in discharge planning |
| Start insulin infusions in postoperative and other unstable patients if blood glucose >180 mg/dL on 2 or more occasions |
| Begin IV continuous insulin infusion using validated protocols |
| Glucose target: 140‐180 mg/dL* |
| When converting from IV to subcutaneous insulin |
| Give 80% of stable IV dose as glargine insulin |
| Give 10% of glargine dose as rapid acting insulin |
| Then stop insulin infusion |
| If starting with subcutaneous insulin without prior insulin infusion |
| Give 50% as long‐acting insulin (glargine or detemir) |
| Give 50% as rapid‐acting insulin, divided into 3 for the 3 meals |
Intensive insulin treatment in the ICU clearly results in better outcomes when compared to letting glucose levels remain greater than 200 mg/dL. A glucose target range of 140 mg/dL to 180 mg/dL provides improved mortality and morbidity with a low risk of hypoglycemia and is suitable for most hospitals. A more aggressive target range of 110 mg/dL to 140 mg/dL provides further improvement but increases the risk of hypoglycemia and would only be appropriate for those institutions with considerable experience with such therapy and demonstrated low rates of hypoglycemia. Work is still needed on devising the ideal treatment algorithm, regimens for conversion from IV to subcutaneous insulin, and discharge planning. However, the most important part of patient care we have found is the insertion of an intelligent and experienced brain between the patient and the insulin protocol.
Hyperglycemia that develops acutely due to illness is associated with poor patient outcomes in hospitalized inpatients, especially those critically ill in the intensive care unit (ICU).18 In fact, those without a prior diagnosis of diabetes and therefore newly found to have hyperglycemia have worse outcomes than those who have a prior diagnosis of diabetes.1, 2, 4, 68 Many mechanisms have been put forward to explain the adverse outcomes related to hyperglycemia, including the release of counter‐regulatory hormones, increased lipolysis with free fatty acid release, the release of inflammatory cytokines and growth factors, increased reactive oxygen species with oxidative stress and altered immunoglobulin, and neutrophil phagocytic function.911
The practical importance of this was brought home by Furnary et al.12, 13 who showed that glycemic control using 3 days of intensive intravenous (IV) insulin therapy of diabetic patients undergoing cardiac surgical procedures was able to reduce significantly the risk of deep sternal wound infections and mortality and to bring these adverse outcomes to the same levels as those of nondiabetic patients. However, the benefits of intensive insulin therapy are not limited to those with diabetes and extend to those with critical illness‐induced hyperglycemia. In a landmark, randomized, prospective study from Belgium, van den Berghe et al.14 showed that the use of an intensive IV insulin protocol designed to maintain serum blood glucose 80 mg/dL to 110 mg/dL significantly decreased morbidity and mortality following admission to the surgical ICU (SICU). Of note, only 13% of the individuals in the study had a previously known diagnosis of diabetes, showing that hyperglycemia was common following SICU admission and glycemic control was beneficial regardless of diabetes status.14
These impressive benefits1214 led to the call for improved glycemic control in the hospital with glucose targets similar to those used in the Belgian study.15 The development of protocols for such treatment proceeded rapidly.16, 17 A meta‐analysis that reviewed 14 trials through May 1, 2006 of patients in SICUs showed a 31% reduction in mortality with intensive therapy, albeit at the expense of a substantially increased risk of hypoglycemia.18 Our own studies19 using 1 day of continuous insulin infusion followed by subcutaneous basal/bolus insulin for all hyperglycemic patients following coronary artery bypass surgery showed results similar to those of Furnary et al.12, 13
Subsequently, 3 large, multicenter studies of patients in medical ICU (MICU) and SICUs, the VISEP, NICE‐SUGAR, and GLUCONTROL studies, failed to show the benefit of intensive insulin therapy on mortality and all had very high rates of hypoglycemia.2022 The VISEP20 study was stopped prematurely because of excessive hypoglycemia in the intensive treatment arm and the GLUCONTROL study was stopped prematurely because of multiple protocol violations.22 The NICE‐SUGAR study actually showed an increased mortality in the intensively treated group21 but the target range for their control group was 140 mg/dL to 180 mg/dL rather than 180 mg/dL to 215 mg/dL and this likely accounted for the better mortality in their control group compared to other studies. Van den Berghe et al.,23 in a design similar to their earlier one in the SICU, found that intensive insulin therapy in the MICU resulted in significant reductions in new onset renal injury, MICU and hospital length of stay, and an improved ability to wean off mechanical ventilation; however, no improvement in mortality was found except for those whose MICU stay was >3 days duration. In a post hoc analysis of their combined SICU and MICU studies, van den Berghe et al.23 found that a glucose target of 110 mg/dL to 150 mg/dL accounted for about 75% of the mortality benefit with a low risk of hypoglycemia.24 A recent meta‐analysis that included data on 13,567 patients from 26 trials, including the NICE‐SUGAR study, concluded that although overall there was no mortality benefit from intensive insulin therapy there was benefit in the SICU but not in the MICU or mixed ICU units.25
As a result of these later studies, new recommendations from the American Association of Clinical Endocrinologists and the American Diabetes Association state that for optimal risk/benefit, the overall goal of inpatient treatment for most patients should be 140 mg/dL to 180 mg/dL, although a range of 110 mg/dL to 140 mg/dL may be appropriate for some patients.26 Stressed in this Consensus Statement is the need for experienced practitioners and systems to provide optimal implementation of protocols so as to provide adequate glycemic control without an undue amount of hypoglycemia. We have found that active individual patient management by experienced nurse practitioners who can modify existing protocols as needed provides better glycemic control with less hypoglycemia than nursing personnel adhering to a protocol without taking into account the myriad of factors affecting patients daily.16
Although hypoglycemia is certainly to be avoided and has been associated with increased mortality,6, 27 Kosiborod et al.7 showed that mortality in hyperglycemic patients following an acute myocardial infarction was not related to insulin‐induced hypoglycemia but to hypoglycemia unassociated with insulin use. In the latter case, the hypoglycemia is generally attributable to shock, sepsis, malnutrition, liver failure, renal failure, or multiorgan failure.
One of the potential hurdles to achievement of glycemic control in the critically ill is the labor‐intensive changes in patient care policies necessary to attain these goals. Particular concern lies in the ability of inpatient care providers to develop and implement successful insulin protocols. Intravenous insulin administration is effective and appropriate in the ICU and some non‐ICU settings, but administration of insulin subcutaneously is less nursing intensive and a more familiar hyperglycemia treatment option. However, glycemic control with subcutaneous insulin is only achieved using basal/bolus regimens and not with simple sliding‐scale regimens that omit basal insulin and attempt to treat rather than prevent hyperglycemia.28
In this issue of the Journal of Hospital Medicine, 3 articles deal with some of the practical aspects of inpatient hyperglycemia management. In ICU patients on continuous IV insulin infusions, Newton et al.29 demonstrated improved glycemic control without an increase in hypoglycemia when using a computer‐guided insulin algorithm using a hand‐held device (Glucommander) compared to a paper algorithm. A previous publication in JHM showed that when continuous insulin infusions were used on the regular hospital floors outside of the ICU, Smiley et al.30 found that 67% of patients achieved the targeted goal of 150 mg/dL by day 2. Wesorick et al.31 found that simply educating floor nurses as well as physicians and using standardized insulin protocols resulted in improved glycemic control and less hypoglycemia on inpatient services outside of the ICU. In the third paper, Ramos et al. found that those with glycosylated hemoglobin (HbA1c) levels >6.0%, a prior history of diabetes, or chronic steroid use did better with basal insulin than with no basal insulin when converting from insulin infusions.32 In contrast to their using only 40% of the stable insulin infusion rate for their basal dose, we found that 80% worked better.33 We also learned the hard way that overlap of the infusion by 2 hours to 4 hours after giving the basal insulin subcutaneous dose is just not carried out by the treating team because of timing and practical considerations. We now just give a dose of rapid acting insulin equal to 10% of the basal insulin dose at the time of the injection of the basal dose; this allows for the immediate cessation of the infusion without loss of glycemic control (Table 1).
|
| Measure HbA1c on admission to aid in discharge planning |
| Start insulin infusions in postoperative and other unstable patients if blood glucose >180 mg/dL on 2 or more occasions |
| Begin IV continuous insulin infusion using validated protocols |
| Glucose target: 140‐180 mg/dL* |
| When converting from IV to subcutaneous insulin |
| Give 80% of stable IV dose as glargine insulin |
| Give 10% of glargine dose as rapid acting insulin |
| Then stop insulin infusion |
| If starting with subcutaneous insulin without prior insulin infusion |
| Give 50% as long‐acting insulin (glargine or detemir) |
| Give 50% as rapid‐acting insulin, divided into 3 for the 3 meals |
Intensive insulin treatment in the ICU clearly results in better outcomes when compared to letting glucose levels remain greater than 200 mg/dL. A glucose target range of 140 mg/dL to 180 mg/dL provides improved mortality and morbidity with a low risk of hypoglycemia and is suitable for most hospitals. A more aggressive target range of 110 mg/dL to 140 mg/dL provides further improvement but increases the risk of hypoglycemia and would only be appropriate for those institutions with considerable experience with such therapy and demonstrated low rates of hypoglycemia. Work is still needed on devising the ideal treatment algorithm, regimens for conversion from IV to subcutaneous insulin, and discharge planning. However, the most important part of patient care we have found is the insertion of an intelligent and experienced brain between the patient and the insulin protocol.
- ,,, et al.Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: A systematic overview.Stroke.2001;32;2426–2432.
- ,,, et al.Hyperglycemia: an independent marker of in‐hospital mortality in patients with undiagnosed diabetes.J Clin Endocrinol Metab.2002;87;978–982.
- .Association between hyperglycemia and increased hospital mortality in a heterogeneous population of critically ill patients.Mayo Clin Proc.2003;78:1471–1478.
- ,,, et al.Influence of individual characteristics on outcome of glycemic control in intensive care unit patients with or without diabetes mellitus.Mayo Clin Proc.2005;80:1558–1567.
- ,,, et al.The relation between hyperglycemia and outcomes in 2,471 patients admitted to the hospital with community acquired pneumonia.Diabetes Care.2005;28:810–815.
- ,,, et al.Admission glucose and mortality in elderly patients hospitalized with acute myocardial infarction. Implications for patients with and without recognized diabetes.Circulation.2005;111:3078–3086.
- ,,, et al.Glucometrics in patients hospitalized with acute myocardial infarction. Defining the optimal outcomes‐based measure of risk.Circulation.2008;117:1018–1027.
- ,,, et al.Hyperglycemia‐related mortality in critically ill patients varies with admission diagnosis.Crit Care Med.2009;37:3001–3009.
- .Effect of insulin therapy on nonglycemic variables during acute illness.Endocr Pract.2004;10(Suppl 2):63–70.
- ,, et al.Therapy insight: the effect of tight glycemic control in acute illness.Nature Clin Pract Endocrinol Metab.2007;3:270–278.
- ,,.Stress hyperglycemia.Lancet.2009;373:1798–1807.
- ,,, et al.Continuous intravenous insulin infusion reduces the incidence of deep sternal wound infection in diabetic patients after cardiac surgical procedures.Ann Thorac Surg.1999;67:352–362.
- ,,, et al.Insulin infusion reduces mortality in patients undergoing coronary artery bypass grafting.J Thorac Cardiovasc Surg.2003;125:1007–1021.
- ,,, et al.Intensive insulin therapy in critically ill patients.N Engl J Med.2001;345:1359–1367.
- ,,, et al.American College of Endocrinology Position Statement on inpatient diabetes and metabolic control.Endocr Pract.2004;10:77–82.
- ,,, et al.Inpatient management of hyperglycemia: The Northwestern Experience.Endocr Pract.2006:12;491–505.
- ,,.Insulin infusion protocols for critically ill patients: a highlight of differences and similarities.Endocr Pract.2007;13:137–146.
- ,,, et al.Effect of perioperative insulin infusion on surgical morbidity and mortality: systematic review and meta‐analysis of randomized trials.Mayo Clin Proc.2008;83:418–430.
- ,,, et al.Reduction in surgical mortality and morbidity in diabetic patients undergoing cardiac surgery with a combined intravenous and subcutaneous insulin glucose management.Diabetes Care.2007;30:823–828.
- ,,, et al.Intensive insulin therapy and pentastarch resuscitation in severe sepsis.N Engl J Med.2008;358:125–139.
- ,,Su SY for the NICE‐SUGAR Study Investigators. Intensive versus conventional glucose control in critically ill patients.N Engl J Med.2009;360:1283–1297.
- ,,, et al.A prospective randomised multi‐centre controlled trial on tight glucose control by intensive insulin therapy in adult intensive care units: the Glucontrol Study.Intensive Care Med.2009;35:1738–1748.
- ,,, et al.Intensive insulin therapy in the medical ICU.N Engl J Med.2006;354:449–461.
- ,,, et al.Intensive insulin therapy in mixed medical/surgical intensive care units: benefit versus harm.Diabetes.2006;55:3151–3159.
- ,,, et al.Intensive insulin therapy and mortality among critically ill patients: a meta‐analysis including NIC‐SUGAR study data.CMAJ.2009;180:821–827.
- ,,, et al.American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control.Diabetes Care.2009;32:1119–1131.
- ,.Severe hypoglycemia in critically ill patients: risk factors and outcomes.Crit Care Med.2007;35:2262–2267.
- ,,, et al.Randomized study of basal‐bolus insulin therapy in the inpatient management of patients with type 2 diabetes (RABBIT 2 trial).Diabetes Care.2007;30:2181–2186.
- ,,, et al.A comparison study of continuous insulin infusion protocols in the Medical Intensive Care Unit: computer‐guided versus standard column‐based algorithms.J Hosp Med. 2010;5:432–437.
- ,,, et al.Safety and efficacy of continuous insulin infusion in noncritical care settings.J Hosp Med.2010;5(4):212–217.
- ,,, et al.Effects of an educational program and a standardized insulin order form on glycemic outcomes in non‐critically ill hospitalized patients.J Hosp Med. 2010;5:438–445.
- ,,, et al.Maintaining glycemic control when transitioning from infusion insulin: a protocol driven, multidisciplinary approach.J Hosp Med. 2010;5:446–451.
- ,,, et al.Conversion of intravenous insulin infusions to subcutaneously administered insulin glargine in patients with hyperglycemia.Endocr Pract.2006;12:641–650.
- ,,, et al.Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: A systematic overview.Stroke.2001;32;2426–2432.
- ,,, et al.Hyperglycemia: an independent marker of in‐hospital mortality in patients with undiagnosed diabetes.J Clin Endocrinol Metab.2002;87;978–982.
- .Association between hyperglycemia and increased hospital mortality in a heterogeneous population of critically ill patients.Mayo Clin Proc.2003;78:1471–1478.
- ,,, et al.Influence of individual characteristics on outcome of glycemic control in intensive care unit patients with or without diabetes mellitus.Mayo Clin Proc.2005;80:1558–1567.
- ,,, et al.The relation between hyperglycemia and outcomes in 2,471 patients admitted to the hospital with community acquired pneumonia.Diabetes Care.2005;28:810–815.
- ,,, et al.Admission glucose and mortality in elderly patients hospitalized with acute myocardial infarction. Implications for patients with and without recognized diabetes.Circulation.2005;111:3078–3086.
- ,,, et al.Glucometrics in patients hospitalized with acute myocardial infarction. Defining the optimal outcomes‐based measure of risk.Circulation.2008;117:1018–1027.
- ,,, et al.Hyperglycemia‐related mortality in critically ill patients varies with admission diagnosis.Crit Care Med.2009;37:3001–3009.
- .Effect of insulin therapy on nonglycemic variables during acute illness.Endocr Pract.2004;10(Suppl 2):63–70.
- ,, et al.Therapy insight: the effect of tight glycemic control in acute illness.Nature Clin Pract Endocrinol Metab.2007;3:270–278.
- ,,.Stress hyperglycemia.Lancet.2009;373:1798–1807.
- ,,, et al.Continuous intravenous insulin infusion reduces the incidence of deep sternal wound infection in diabetic patients after cardiac surgical procedures.Ann Thorac Surg.1999;67:352–362.
- ,,, et al.Insulin infusion reduces mortality in patients undergoing coronary artery bypass grafting.J Thorac Cardiovasc Surg.2003;125:1007–1021.
- ,,, et al.Intensive insulin therapy in critically ill patients.N Engl J Med.2001;345:1359–1367.
- ,,, et al.American College of Endocrinology Position Statement on inpatient diabetes and metabolic control.Endocr Pract.2004;10:77–82.
- ,,, et al.Inpatient management of hyperglycemia: The Northwestern Experience.Endocr Pract.2006:12;491–505.
- ,,.Insulin infusion protocols for critically ill patients: a highlight of differences and similarities.Endocr Pract.2007;13:137–146.
- ,,, et al.Effect of perioperative insulin infusion on surgical morbidity and mortality: systematic review and meta‐analysis of randomized trials.Mayo Clin Proc.2008;83:418–430.
- ,,, et al.Reduction in surgical mortality and morbidity in diabetic patients undergoing cardiac surgery with a combined intravenous and subcutaneous insulin glucose management.Diabetes Care.2007;30:823–828.
- ,,, et al.Intensive insulin therapy and pentastarch resuscitation in severe sepsis.N Engl J Med.2008;358:125–139.
- ,,Su SY for the NICE‐SUGAR Study Investigators. Intensive versus conventional glucose control in critically ill patients.N Engl J Med.2009;360:1283–1297.
- ,,, et al.A prospective randomised multi‐centre controlled trial on tight glucose control by intensive insulin therapy in adult intensive care units: the Glucontrol Study.Intensive Care Med.2009;35:1738–1748.
- ,,, et al.Intensive insulin therapy in the medical ICU.N Engl J Med.2006;354:449–461.
- ,,, et al.Intensive insulin therapy in mixed medical/surgical intensive care units: benefit versus harm.Diabetes.2006;55:3151–3159.
- ,,, et al.Intensive insulin therapy and mortality among critically ill patients: a meta‐analysis including NIC‐SUGAR study data.CMAJ.2009;180:821–827.
- ,,, et al.American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control.Diabetes Care.2009;32:1119–1131.
- ,.Severe hypoglycemia in critically ill patients: risk factors and outcomes.Crit Care Med.2007;35:2262–2267.
- ,,, et al.Randomized study of basal‐bolus insulin therapy in the inpatient management of patients with type 2 diabetes (RABBIT 2 trial).Diabetes Care.2007;30:2181–2186.
- ,,, et al.A comparison study of continuous insulin infusion protocols in the Medical Intensive Care Unit: computer‐guided versus standard column‐based algorithms.J Hosp Med. 2010;5:432–437.
- ,,, et al.Safety and efficacy of continuous insulin infusion in noncritical care settings.J Hosp Med.2010;5(4):212–217.
- ,,, et al.Effects of an educational program and a standardized insulin order form on glycemic outcomes in non‐critically ill hospitalized patients.J Hosp Med. 2010;5:438–445.
- ,,, et al.Maintaining glycemic control when transitioning from infusion insulin: a protocol driven, multidisciplinary approach.J Hosp Med. 2010;5:446–451.
- ,,, et al.Conversion of intravenous insulin infusions to subcutaneously administered insulin glargine in patients with hyperglycemia.Endocr Pract.2006;12:641–650.
Continuing Medical Education Program in
If you wish to receive credit for this activity, which begins on the next page, please refer to the website:
Accreditation and Designation Statement
Blackwell Futura Media Services designates this educational activity for a 1 AMA PRA Category 1 Credit. Physicians should only claim credit commensurate with the extent of their participation in the activity.
Blackwell Futura Media Services is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
Educational Objectives
Continuous participation in the Journal of Hospital Medicine CME program will enable learners to be better able to:
-
Interpret clinical guidelines and their applications for higher quality and more efficient care for all hospitalized patients.
-
Describe the standard of care for common illnesses and conditions treated in the hospital; such as pneumonia, COPD exacerbation, acute coronary syndrome, HF exacerbation, glycemic control, venous thromboembolic disease, stroke, etc.
-
Discuss evidence‐based recommendations involving transitions of care, including the hospital discharge process.
-
Gain insights into the roles of hospitalists as medical educators, researchers, medical ethicists, palliative care providers, and hospital‐based geriatricians.
-
Incorporate best practices for hospitalist administration, including quality improvement, patient safety, practice management, leadership, and demonstrating hospitalist value.
-
Identify evidence‐based best practices and trends for both adult and pediatric hospital medicine.
Instructions on Receiving Credit
For information on applicability and acceptance of continuing medical education credit for this activity, please consult your professional licensing board.
This activity is designed to be completed within the time designated on the title page; physicians should claim only those credits that reflect the time actually spent in the activity. To successfully earn credit, participants must complete the activity during the valid credit period that is noted on the title page.
Follow these steps to earn credit:
-
Log on to
www.blackwellpublishing.com/cme . -
Read the target audience, learning objectives, and author disclosures.
-
Read the article in print or online format.
-
Reflect on the article.
-
Access the CME Exam, and choose the best answer to each question.
-
Complete the required evaluation component of the activity.
If you wish to receive credit for this activity, which begins on the next page, please refer to the website:
Accreditation and Designation Statement
Blackwell Futura Media Services designates this educational activity for a 1 AMA PRA Category 1 Credit. Physicians should only claim credit commensurate with the extent of their participation in the activity.
Blackwell Futura Media Services is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
Educational Objectives
Continuous participation in the Journal of Hospital Medicine CME program will enable learners to be better able to:
-
Interpret clinical guidelines and their applications for higher quality and more efficient care for all hospitalized patients.
-
Describe the standard of care for common illnesses and conditions treated in the hospital; such as pneumonia, COPD exacerbation, acute coronary syndrome, HF exacerbation, glycemic control, venous thromboembolic disease, stroke, etc.
-
Discuss evidence‐based recommendations involving transitions of care, including the hospital discharge process.
-
Gain insights into the roles of hospitalists as medical educators, researchers, medical ethicists, palliative care providers, and hospital‐based geriatricians.
-
Incorporate best practices for hospitalist administration, including quality improvement, patient safety, practice management, leadership, and demonstrating hospitalist value.
-
Identify evidence‐based best practices and trends for both adult and pediatric hospital medicine.
Instructions on Receiving Credit
For information on applicability and acceptance of continuing medical education credit for this activity, please consult your professional licensing board.
This activity is designed to be completed within the time designated on the title page; physicians should claim only those credits that reflect the time actually spent in the activity. To successfully earn credit, participants must complete the activity during the valid credit period that is noted on the title page.
Follow these steps to earn credit:
-
Log on to
www.blackwellpublishing.com/cme . -
Read the target audience, learning objectives, and author disclosures.
-
Read the article in print or online format.
-
Reflect on the article.
-
Access the CME Exam, and choose the best answer to each question.
-
Complete the required evaluation component of the activity.
If you wish to receive credit for this activity, which begins on the next page, please refer to the website:
Accreditation and Designation Statement
Blackwell Futura Media Services designates this educational activity for a 1 AMA PRA Category 1 Credit. Physicians should only claim credit commensurate with the extent of their participation in the activity.
Blackwell Futura Media Services is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
Educational Objectives
Continuous participation in the Journal of Hospital Medicine CME program will enable learners to be better able to:
-
Interpret clinical guidelines and their applications for higher quality and more efficient care for all hospitalized patients.
-
Describe the standard of care for common illnesses and conditions treated in the hospital; such as pneumonia, COPD exacerbation, acute coronary syndrome, HF exacerbation, glycemic control, venous thromboembolic disease, stroke, etc.
-
Discuss evidence‐based recommendations involving transitions of care, including the hospital discharge process.
-
Gain insights into the roles of hospitalists as medical educators, researchers, medical ethicists, palliative care providers, and hospital‐based geriatricians.
-
Incorporate best practices for hospitalist administration, including quality improvement, patient safety, practice management, leadership, and demonstrating hospitalist value.
-
Identify evidence‐based best practices and trends for both adult and pediatric hospital medicine.
Instructions on Receiving Credit
For information on applicability and acceptance of continuing medical education credit for this activity, please consult your professional licensing board.
This activity is designed to be completed within the time designated on the title page; physicians should claim only those credits that reflect the time actually spent in the activity. To successfully earn credit, participants must complete the activity during the valid credit period that is noted on the title page.
Follow these steps to earn credit:
-
Log on to
www.blackwellpublishing.com/cme . -
Read the target audience, learning objectives, and author disclosures.
-
Read the article in print or online format.
-
Reflect on the article.
-
Access the CME Exam, and choose the best answer to each question.
-
Complete the required evaluation component of the activity.
Transitioning From Infusion Insulin
Hyperglycemia due to diabetes or stress is prevalent in the intensive care unit (ICU) and general ward setting. Umpierrez et al.1 reported hyperglycemia in 38% of hospitalized ward patients with 26% having a known history of diabetes. While patients with hyperglycemia admitted to the ICU are primarily treated with infusion insulin, those on the general wards usually receive a subcutaneous regimen of insulin. How best to transition patients from infusion insulin to a subcutaneous regimen remains elusive and under evaluated.
A recent observational pilot study of 24 surgical and 17 cardiac/medical intensive care patients at our university‐based hospital found that glycemic control significantly deteriorated when patients with diabetes transitioned from infusion insulin to subcutaneous insulin. A total of 21 critical care patients with a history of diabetes failed to receive basal insulin prior to discontinuation of the drip and developed uncontrolled hyperglycemia (mean glucose Day 1 of 216 mg/dL and Day 2 of 197 mg/dL). Patients without a history of diabetes did well post transition with a mean glucose of 142 mg/dL Day 1 and 133 mg/dL Day 2. A similar study by Czosnowski et al.2 demonstrated a significant increase in blood glucose from 123 26 mg/dL to 168 50 mg/dL upon discontinuation of infusion insulin.
This failed transition is disappointing, especially in view of the existence of a reliable subcutaneous (SC) insulin order set at our institution, but not surprising, as this is an inherently complex process. The severity of illness, the amount and mode of nutritional intake, geographic location, and provider team may all be in flux at the time of this transition. A few centers have demonstrated that a much improved transition is possible,36 however many of these solutions involve technology or incremental personnel that may not be available or the descriptions may lack sufficient detail to implement theses strategies with confidence elsewhere.
Therefore, we designed and piloted a protocol, coordinated by a multidisciplinary team, to transition patients from infusion insulin to SC insulin. The successful implementation of this protocol could serve as a blueprint to other institutions without the need for additional technology or personnel.
Methods
Patient Population/Setting
This was a prospective study of patients admitted to either the medical/cardiac intensive care unit (MICU/CCU) or surgical intensive care unit (SICU) at an academic medical facility and placed on infusion insulin for >24 hours. The Institutional Review Board (IRB) approved the study for prospective chart review and anonymous results reporting without individual consent.
Patients in the SICU were initiated on infusion insulin after 2 blood glucose readings were above 150 mg/dL, whereas initiation was left to the discretion of the attending physician in the MICU/CCU. A computerized system created in‐house recommends insulin infusion rates based on point‐of‐care (POC) glucose measurements with a target range of 91 mg/dL to 150 mg/dL.
Inclusion/Exclusion Criteria
All patients on continuous insulin infusion admitted to the SICU or the MICU/CCU between May 2008 and September 2008 were evaluated for the study (Figure 1). Patients were excluded from analysis if they were on the infusion for less than 24 hours, had a liver transplant, were discharged within 48 hours of transition, were made comfort care or transitioned to an insulin pump. All other patients were included in the final analysis.
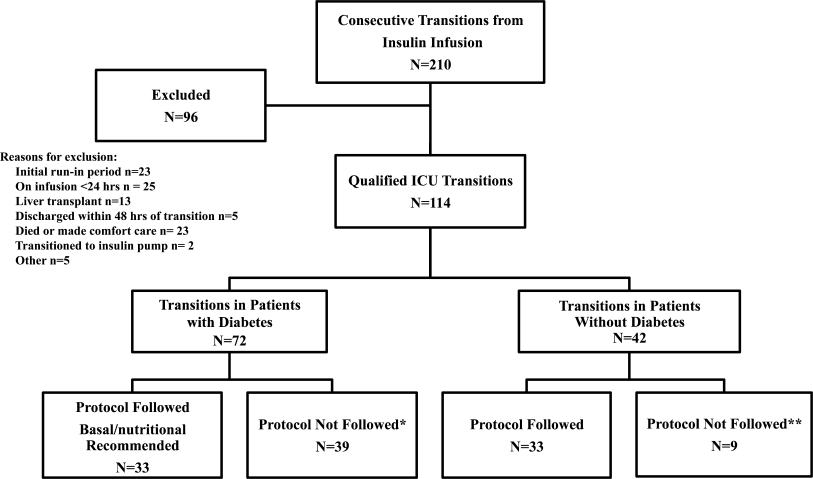
Transition Protocol
Step 1: Does the Patient Need Basal SC Insulin?
Patients were recommended to receive basal SC insulin if they either: (1) were on medications for diabetes; (2) had an A1c 6%; or (3) received the equivalent of 60 mg of prednisone; AND had an infusion rate 1 unit/hour (Supporting Information Appendix 1). Patients on infusion insulin due to stress hyperglycemia, regardless of the infusion rate, were not placed on basal SC insulin. Patients on high dose steroids due to spinal injuries were excluded because their duration of steroid use was typically less than 48 hours and usually ended prior to the time of transition. The protocol recommends premeal correctional insulin for those not qualifying for basal insulin.
In order to establish patients in need of basal/nutritional insulin we opted to use A1c as well as past medical history to identify patients with diabetes. The American Diabetes Association (ADA) has recently accepted using an A1c 6.5% to make a new diagnosis of diabetes.7 In a 2‐week trial prior to initiating the protocol we used a cut off A1c of 6.5%. However, we found that patients with an A1c of 6% to 6.5% had poor glucose control post transition; therefore we chose 6% as our identifier. In addition, using a cut off A1c of 6% was reported by Rohlfing et al.8 and Greci et al.9 to be more than 97% sensitive at identifying a new diagnosis of diabetes.
To ensure an A1c was ordered and available at the time of transition, critical care pharmacists were given Pharmacy and Therapeutics Committee authorization to order an A1c at the start of the infusion. Pharmacists would also guide the primary team through the protocol's recommendations as well as alert the project team when a patient was expected to transition.
Step 2: Evaluate the Patient's Nutritional Intake to Calculate the Total Daily Dose (TDD) of Insulin
TDD is the total amount of insulin needed to cover both the nutritional and basal requirements of a patient over the course of 24 hours. TDD was calculated by averaging the hourly drip rate over the prior 6 hours and multiplying by 20 if taking in full nutrition or 40 if taking minimal nutrition while on the drip. A higher multiplier was used for those on minimal nutrition with the expectation that their insulin requirements would double once tolerating a full diet. Full nutrition was defined as eating >50% of meals, on goal tube feeds, or receiving total parenteral nutrition (TPN). Minimal nutrition was defined as taking nothing by mouth (or NPO), tolerating <50% of meals, or on a clear liquid diet.
Step 3: Divide the TDD Into the Appropriate Components of Insulin Treatment (Basal, Nutritional and Correction), Depending on the Nutritional Status
In Step 3, the TDD was evenly divided into basal and nutritional insulin. A total of 50% of the TDD was given as glargine (Lantus) 2 hours prior to stopping the infusion. The remaining 50% was divided into nutritional components as either Regular insulin every 6 hours for patients on tube feeds or lispro (Humalog) before meals if tolerating an oral diet. For patients on minimal nutrition, the 50% nutritional insulin dose was not initiated until the patient was tolerating full nutrition.
The protocol recommended basal insulin administration 2 hours prior to infusion discontinuation as recommended by the American Association of Clinical Endocrinologists (AACE) and ADA consensus statement on inpatient glycemic control as well as pharmacokinetics.10, 11 For these reasons, failure to receive basal insulin prior to transition was viewed as failure to follow the protocol.
Safety features of the protocol included a maximum TDD of 100 units unless the patient was on >100 units/day of insulin prior to admission. A pager was carried by rotating hospitalists or pharmacist study investigators at all hours during the protocol implementation phase to answer any questions regarding a patient's transition.
Data Collection/Monitoring
A multidisciplinary team consisting of hospitalists, ICU pharmacists, critical care physicians and nursing representatives was assembled during the study period. This team was responsible for protocol implementation, data collection, and surveillance of patient response to the protocol. Educational sessions with house staff and nurses in each unit were held prior to the beginning of the study as well as continued monthly educational efforts during the study. In addition, biweekly huddles to review ongoing patient transitions as well as more formal monthly reviews were held.
The primary objective was to improve glycemic control, defined as the mean daily glucose, during the first 48 hours post transition without a significant increase in the percentage of patients with hypoglycemia (41‐70 mg/dL) or severe hypoglycemia (40 mg/dL). Secondary endpoints included the percent of patients with severe hyperglycemia (300 mg/dL), length of stay (LOS) calculated from the day of transition, number of restarts back onto infusion insulin within 72 hours of transition, and day‐weighted glucose mean up to 12 days following transition for patients with diabetes.
Glucose values were collected and averaged over 6‐hour periods for 48 hours post transition. For patients with diabetes, POC glucose values were collected up to 12 days of hospitalization. Day‐weighted means were obtained by calculating the mean glucose for each hospital day, averaged across all hospital days.12
Analysis
Subjects were divided by the presence or absence of diabetes. Those with diabetes were recommended to receive basal SC insulin during the transition period. Within each group, subjects were further divided by adherence to the protocol. Failure to transition per protocol was defined as: not receiving at least 80% of the recommended basal insulin dose, receiving the initial dose of insulin after the drip was discontinued, or receiving basal insulin when none was recommended.
Descriptive statistics within subgroups comparing age, gender, LOS by analysis of variance for continuous data and by chi‐square for nominal data, were compared. Twenty‐four and 48‐hour post transition mean glucose values and the 12 day weighted‐mean glucose were compared using analysis of variance (Stata ver. 10). All data are expressed as mean standard deviation with a significance value established at P < 0.05.
Results
A total of 210 episodes of infusion insulin in ICU patients were evaluated for the study from May of 2008 to September 2008 (Figure 1). Ninety‐six of these episodes were excluded, most commonly due to time on infusion insulin <24 hours or transition to comfort care. The remaining 114 infusions were eligible to use the protocol. Because the protocol recommends insulin therapy based on a diagnosis of diabetes, patients were further divided into these subcategories. Of these 114 transitions, the protocol was followed 66 times (58%).
Patients With Diabetes
(Table 1: Patient Demographics; Table 2: Insulin Use and Glycemic Control; Figure 2: Transition Graph).
| Patients With Diabetes | P Value | Patients Without Diabetes | P Value | |||
|---|---|---|---|---|---|---|
| Protocol Followed, n = 29 Patients* | Protocol NOT Followed, n = 33 Patients | Protocol Followed, n = 30 Patients | Protocol NOT Followed, n = 9 Patients | |||
| ||||||
| Average age, years, mean SD | 57.7 12.1 | 57.8 12.3 | 0.681 | 56.5 18.1 | 62.4 15.5 | 0.532 |
| Male patients | 21 (72%) | 21 (63%) | 0.58 | 20 (66%) | 7 (77%) | 0.691 |
| BMI | 30.7 7.2 | 28.6 6.8 | 0.180 | 27 5.4 | 25.2 3 | 0.081 |
| History of diabetes* | 18 (64%) | 25 (86%) | 0.07 | 0 | 0 | |
| Mean Hgb A1c (%) | 6.61.2 | 7.3 1.8 | 0.136 | 5.6 0.3 | 5.4 0.4 | 0.095 |
| Full nutrition | 26 (79%) | 24 (61%) | 0.131 | 23 (70%) | 9 (100%) | |
| On hemodialysis | 5 (17%) | 9 (27%) | 0.380 | 3 (10%) | 0 | |
| On >60 mg prednisone or equivalent per day | 7 (24%) | 10 (30%) | 0.632 | 0 | 0 | |
| Patients With Diabetes | P Value | Patients Without Diabetes | P Value | |||
|---|---|---|---|---|---|---|
| Protocol Followed, n = 33 transitions | Protocol NOT followed, n = 39 transitions | Protocol Followed, n = 33 transitions | Protocol NOT Followed, n = 9 transitions | |||
| ||||||
| Average infusion rate, hours | 3.96 3.15 | 3.74 3.64 | 0.1597 | 2.34 1.5 | 4.78 1.6 | <0.001 |
| Average BG on infusion insulin (mg/dL) | 122.5 27.5 | 122.5 31.8 | 0.844 | 115.1 22.7 | 127.5 27.2 | 0.006 |
| Average basal dose (units) given | 34.5 14.4 | 14.4 15.3 | <0.001 | 0 | 32.7 | <0.001 |
| Hours before () or after (+) infusion stopped basal insulin given | 1.13 0.9 | 11.6 9.3 | <0.001 | n/a | 0.33 | * |
| Average BG 6 hours post transition (mg/dL) | 143.7 39.4 | 182 62.5 | 0.019 | 150.2 54.9 | 142.1 34.1 | 0.624 |
| Average BG 0 to 24 hours post transition (mg/dL) | 167.98 50.24 | 211.02 81.01 | <0.001 | 150.24 54.9 | 150.12 32.4 | 0.600 |
| Total insulin used from 0 to 24 hours (units) | 65 32.2 | 26.7 25.4 | <0.001 | 3.2 4.1 | 51.3 30.3 | <0.001 |
| Average BG 25 to 48 hours post transition (mg/dL) | 176.1 55.25 | 218.2 88.54 | <0.001 | 153 35.3 | 154.4 46.7 | 0.711 |
| Total insulin used from 25 to 48 hours (units) | 60.5 35.4 | 28.1 24.4 | <0.001 | 2.8 3.8 | 44.9 34 | <0.001 |
| # of patients with severe hypoglycemia (<40 mg/dL) | 1 (3%) | 1 (2.6%) | * | 0 | 1 | * |
| # of patients with hypoglycemia (4170 mg/dL) | 3 (9%) | 2 (5.1%) | * | 1 | 0 | * |
| % of BG values in goal range (80180 mg/dL) (# in range/total #) | 60.2% (153/254) | 38.2% (104/272) | 0.004 | 80.1% (173/216) | 75.4% (49/65) | 0.83 |
| # of patients with severe hyperglycemia (>300 mg/dL) | 5 (15.2%) | 19 (48.7%) | 0.002 | 1 (3%) | 1 (11.1%) | * |
| LOS from transition (days) | 14.6 11.3 | 14 11.4 | 0.836 | 25.3 24.4 | 13.6 7.5 | 0.168 |
A total of 62 individual patients accounted for 72 separate transitions in patients with diabetes based on past medical history or an A1c 6% (n = 14). Of these 72 transitions, 33 (46%) adhered to the protocol while the remaining 39 (54%) transitions varied from the protocol at the treatment team's discretion. Despite similar insulin infusion rates and mean glucose values pretransition, patients with diabetes following the protocol had better glycemic control at both 24 hours and 48 hours after transition than those patients transitioned without the protocol. Day 1 mean blood glucose was 168 mg/dL vs. 211 mg/dL (P = <0.001) and day 2 mean blood glucose was 176 mg/dL vs. 218 mg/dL (P = <0.001) in protocol vs. nonprotocol patients with diabetes respectively (Figure 2).
There was a severe hypoglycemic event (40 mg/dL) in 1 patient with diabetes following the protocol and 1 patient not following the protocol within 48 hours of transition. Both events were secondary to nutritional‐insulin mismatch with emesis after insulin in one case and tube feeds being held in the second case. These findings were consistent with our prior examination of hypoglycemia cases.13 Severe hyperglycemia (glucose 300mg/dL) occurred in 5 (15 %) patients following the protocol vs. 19 (49%) patients not following protocol (P = 0.002.) Patients with diabetes following the protocol received significantly more insulin in the first 24 hours (mean of 65 units vs. 27 units, P 0.001) and 24 to 48 hours after transition (mean of 61 units vs. 28 units, p0.001) than those not following protocol.
An alternate method used at our institution and others14, 15 to calculate TDD is based on the patient's weight and body habitus. When we compared the projected TDD based on weight with the TDD using the transition protocol, we found that the weight based method was much less aggressive. For patients following the protocol, the weight based method projected a mean TDD of 46.3 16.9 units whereas the protocol projected a mean TDD of 65 33.2 units (P = 0.001).
Patients with diabetes following protocol received basal insulin an average of 1.13 hours prior to discontinuing the insulin infusion versus 11.6 hours after for those not following protocol.
Three patients with diabetes following the protocol and 3 patients with diabetes not following the protocol were restarted on infusion insulin within 72 hours of transition.
LOS from final transition to discharge was similar between protocol vs. nonprotocol patients (14.6 vs. 14 days, P = 0.836).
Figure 3 demonstrates that when used correctly, the protocol provides an extended period of glycemic control up to 12 days post transition. Patients transitioned per protocol had a day‐weighted mean glucose of 155 mg/dL vs. 184 mg/dL (P = 0.043) in patients not following protocol. There was only 1 glucose value less than 40 mg/dL between days 2 to 12 in the protocol group.
Patients Without Diabetes
Of the 39 individual patients without diabetes there were 42 transition events, 33 transitions (78.6%) were per protocol and placed on correctional insulin only. The remaining 9 transitions failed to follow protocol in that basal insulin was prescribed, but these patients maintained comparable glycemic control without an increase in hypoglycemic events. Following transition, patients without diabetes on protocol maintained a mean glucose of 150 mg/dL in the first 24 hours and 153 mg/dL in 24 to 48 hours post transition. They required a mean daily correctional insulin dose of 3.2 units on Day 1 and 2.8 units on Day 2 despite having an average drip rate of 2.3 units/hour at the time of transition (Table 2). There were no severe hypoglycemic events and 80% of blood sugars were within the goal range of 80 mg/dL to 180 mg/dL. Only 1 patient had a single blood glucose of >300mg/dL. No patient was restarted on infusion insulin once transitioned.
Patients without diabetes had a longer LOS after transition off of infusion insulin when compared to their diabetic counterparts (22 vs. 14 days).
Discussion
This study demonstrates the utility of hospitalist‐pharmacist collaboration in the creation and implementation of a safe and effective transition protocol for patients on infusion insulin. The protocol identifies patients appropriate for transition to a basal/nutritional insulin regimen versus those who will do well with premeal correctional insulin alone. Daily mean glucose was improved post transition for diabetic patients following the protocol compared to those not following the protocol without an increase in hypoglycemic events.
We found an equal number of insulin infusion restarts within 72 hours of transition and a similar LOS in protocol vs. nonprotocol patients with diabetes. The LOS was increased for patients without diabetes. This may be due to worse outcomes noted in patients with stress hyperglycemia in other studies.1, 16
The use of the higher multiplier for patients on minimal nutrition led to confusion among many protocol users. The protocol has since been modified to start by averaging the infusion rate over the prior 6 hours and then multiplying by 20 for all patients. This essentially calculates 80% of projected insulin requirements for the next 24 hours based on the patient's current needs. This calculation is then given as 50% basal and 50% nutritional for those on full nutrition vs. 100% basal for those on minimal nutrition. This protocol change has no impact on the amount of insulin received by the patient, but is more intuitive to providers. Instead of calculating the TDD as the projected requirement when full nutrition is obtained, the TDD is now calculated based on current insulin needs, and then doubled when patients who are receiving minimal nutrition advance to full nutrition.
Our study is limited by the lack of a true randomized control group. In lieu of this, we used our patients who did not follow protocol as our control. While not truly randomized, this group is comparable based on their age, gender mix, infusion rate, mean A1c, and projected TDD. This group was also similar to our preprotocol group mentioned in the Introduction.
Additional study limitations include the small number of nondiabetic patients not following the protocol (n = 9). We noted higher infusion rates in nondiabetics not following protocol versus those following protocol, which may have driven the primary team to give basal insulin. It is possible that these 9 patients were not yet ready to transition from infusion insulin or had other stressors not measured in our study. Unfortunately their small population size limits more extensive analysis.
The protocol was followed only 50% of the time for a variety of reasons. Patients who transitioned at night or on weekends were monitored by covering pharmacists and physicians who may not have been familiar with the protocol. Many physicians and nurses remain fearful of hypoglycemia and the outcomes of our study were not yet available for education. Some reported difficulty fully understanding how to use the protocol and why a higher multiplier was used for patients who were on minimal nutrition.
Efforts to improve adherence to the protocol are ongoing with some success, aided by the data demonstrating the safety and efficacy of the transition protocol.
Conclusion
By collaborating with ICU pharmacists we were able to design and implement a protocol that successfully and safely transitioned patients from infusion insulin to subcutaneous insulin. Patients following the protocol had a higher percentage of glucose values within the goal glucose range of 80 mg/dL to 180 mg/dL. In the future, we hope to automate the calculation of TDD and directly recommend a basal/bolus regimen for the clinical provider.
- ,,,,,.Hyperglycemia: an independent marker of in‐hospital mortality in patients with undiagnosed diabetes.J Clin Endocrinol Metab.2002;87:978–982.
- ,,,,,.Evaluation of glycemic control following discontinuation of an intensive insulin protocol.J Hosp Med.2009;4:28–34.
- ,,, et al.Inpatient management of hyperglycemia: the Northwestern experience.Endocr Pract.2006;12:491–505.
- ,,,.Intravenous insulin infusion therapy: indications, methods, and transition to subcutaneous insulin therapy.Endocr Pract.2004;10Suppl 2:71–80.
- ,,, et al.Conversion of intravenous insulin infusions to subcutaneously administered insulin glargine in patients with hyperglycemia.Endocr Pract.2006;12:641–650.
- ,.Effects of outcome on in‐hospital transition from intravenous insulin infusion to subcutaneous therapy.Am J Cardiol.2006;98:557–564.
- International Expert Committee report on the role of the A1C assay in the diagnosis of diabetes.Diabetes Care.2009;32:1327–1334.
- ,,, et al.Use of GHb (HbA1c) in screening for undiagnosed diabetes in the U.S. population.Diabetes Care.2000;23:187–191.
- ,,, et al.Utility of HbA(1c) levels for diabetes case finding in hospitalized patients with hyperglycemia.Diabetes Care.2003;26:1064–1068.
- ,,, et al.American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control.Endocr Pract.2009;15(4):353–369.
- ,,, et al.Pharmacokinetics and pharmacodynamics of subcutaneous injection of long‐acting human insulin analog glargine, NPH insulin, and ultralente human insulin and continuous subcutaneous infusion of insulin lispro.Diabetes.2000;49:2142–2148.
- ,,, et al.“Glucometrics”‐‐assessing the quality of inpatient glucose management.Diabetes Technol Ther.2006;8:560–569.
- ,,.Iatrogenic Inpatient Hypoglycemia: Risk Factors, Treatment, and Prevention: Analysis of Current Practice at an Academic Medical Center With Implications for Improvement Efforts.Diabetes Spectr.2008;21:241–247.
- ,,, et al.Management of diabetes and hyperglycemia in hospitals.Diabetes Care.2004;27:553–591.
- .Insulin management of diabetic patients on general medical and surgical floors.Endocr Pract.2006;12Suppl 3:86–90.
- ,,,.Inadequate blood glucose control is associated with in‐hospital mortality and morbidity in diabetic and nondiabetic patients undergoing cardiac surgery.Circulation.2008;118:113–123.
Hyperglycemia due to diabetes or stress is prevalent in the intensive care unit (ICU) and general ward setting. Umpierrez et al.1 reported hyperglycemia in 38% of hospitalized ward patients with 26% having a known history of diabetes. While patients with hyperglycemia admitted to the ICU are primarily treated with infusion insulin, those on the general wards usually receive a subcutaneous regimen of insulin. How best to transition patients from infusion insulin to a subcutaneous regimen remains elusive and under evaluated.
A recent observational pilot study of 24 surgical and 17 cardiac/medical intensive care patients at our university‐based hospital found that glycemic control significantly deteriorated when patients with diabetes transitioned from infusion insulin to subcutaneous insulin. A total of 21 critical care patients with a history of diabetes failed to receive basal insulin prior to discontinuation of the drip and developed uncontrolled hyperglycemia (mean glucose Day 1 of 216 mg/dL and Day 2 of 197 mg/dL). Patients without a history of diabetes did well post transition with a mean glucose of 142 mg/dL Day 1 and 133 mg/dL Day 2. A similar study by Czosnowski et al.2 demonstrated a significant increase in blood glucose from 123 26 mg/dL to 168 50 mg/dL upon discontinuation of infusion insulin.
This failed transition is disappointing, especially in view of the existence of a reliable subcutaneous (SC) insulin order set at our institution, but not surprising, as this is an inherently complex process. The severity of illness, the amount and mode of nutritional intake, geographic location, and provider team may all be in flux at the time of this transition. A few centers have demonstrated that a much improved transition is possible,36 however many of these solutions involve technology or incremental personnel that may not be available or the descriptions may lack sufficient detail to implement theses strategies with confidence elsewhere.
Therefore, we designed and piloted a protocol, coordinated by a multidisciplinary team, to transition patients from infusion insulin to SC insulin. The successful implementation of this protocol could serve as a blueprint to other institutions without the need for additional technology or personnel.
Methods
Patient Population/Setting
This was a prospective study of patients admitted to either the medical/cardiac intensive care unit (MICU/CCU) or surgical intensive care unit (SICU) at an academic medical facility and placed on infusion insulin for >24 hours. The Institutional Review Board (IRB) approved the study for prospective chart review and anonymous results reporting without individual consent.
Patients in the SICU were initiated on infusion insulin after 2 blood glucose readings were above 150 mg/dL, whereas initiation was left to the discretion of the attending physician in the MICU/CCU. A computerized system created in‐house recommends insulin infusion rates based on point‐of‐care (POC) glucose measurements with a target range of 91 mg/dL to 150 mg/dL.
Inclusion/Exclusion Criteria
All patients on continuous insulin infusion admitted to the SICU or the MICU/CCU between May 2008 and September 2008 were evaluated for the study (Figure 1). Patients were excluded from analysis if they were on the infusion for less than 24 hours, had a liver transplant, were discharged within 48 hours of transition, were made comfort care or transitioned to an insulin pump. All other patients were included in the final analysis.
Transition Protocol
Step 1: Does the Patient Need Basal SC Insulin?
Patients were recommended to receive basal SC insulin if they either: (1) were on medications for diabetes; (2) had an A1c 6%; or (3) received the equivalent of 60 mg of prednisone; AND had an infusion rate 1 unit/hour (Supporting Information Appendix 1). Patients on infusion insulin due to stress hyperglycemia, regardless of the infusion rate, were not placed on basal SC insulin. Patients on high dose steroids due to spinal injuries were excluded because their duration of steroid use was typically less than 48 hours and usually ended prior to the time of transition. The protocol recommends premeal correctional insulin for those not qualifying for basal insulin.
In order to establish patients in need of basal/nutritional insulin we opted to use A1c as well as past medical history to identify patients with diabetes. The American Diabetes Association (ADA) has recently accepted using an A1c 6.5% to make a new diagnosis of diabetes.7 In a 2‐week trial prior to initiating the protocol we used a cut off A1c of 6.5%. However, we found that patients with an A1c of 6% to 6.5% had poor glucose control post transition; therefore we chose 6% as our identifier. In addition, using a cut off A1c of 6% was reported by Rohlfing et al.8 and Greci et al.9 to be more than 97% sensitive at identifying a new diagnosis of diabetes.
To ensure an A1c was ordered and available at the time of transition, critical care pharmacists were given Pharmacy and Therapeutics Committee authorization to order an A1c at the start of the infusion. Pharmacists would also guide the primary team through the protocol's recommendations as well as alert the project team when a patient was expected to transition.
Step 2: Evaluate the Patient's Nutritional Intake to Calculate the Total Daily Dose (TDD) of Insulin
TDD is the total amount of insulin needed to cover both the nutritional and basal requirements of a patient over the course of 24 hours. TDD was calculated by averaging the hourly drip rate over the prior 6 hours and multiplying by 20 if taking in full nutrition or 40 if taking minimal nutrition while on the drip. A higher multiplier was used for those on minimal nutrition with the expectation that their insulin requirements would double once tolerating a full diet. Full nutrition was defined as eating >50% of meals, on goal tube feeds, or receiving total parenteral nutrition (TPN). Minimal nutrition was defined as taking nothing by mouth (or NPO), tolerating <50% of meals, or on a clear liquid diet.
Step 3: Divide the TDD Into the Appropriate Components of Insulin Treatment (Basal, Nutritional and Correction), Depending on the Nutritional Status
In Step 3, the TDD was evenly divided into basal and nutritional insulin. A total of 50% of the TDD was given as glargine (Lantus) 2 hours prior to stopping the infusion. The remaining 50% was divided into nutritional components as either Regular insulin every 6 hours for patients on tube feeds or lispro (Humalog) before meals if tolerating an oral diet. For patients on minimal nutrition, the 50% nutritional insulin dose was not initiated until the patient was tolerating full nutrition.
The protocol recommended basal insulin administration 2 hours prior to infusion discontinuation as recommended by the American Association of Clinical Endocrinologists (AACE) and ADA consensus statement on inpatient glycemic control as well as pharmacokinetics.10, 11 For these reasons, failure to receive basal insulin prior to transition was viewed as failure to follow the protocol.
Safety features of the protocol included a maximum TDD of 100 units unless the patient was on >100 units/day of insulin prior to admission. A pager was carried by rotating hospitalists or pharmacist study investigators at all hours during the protocol implementation phase to answer any questions regarding a patient's transition.
Data Collection/Monitoring
A multidisciplinary team consisting of hospitalists, ICU pharmacists, critical care physicians and nursing representatives was assembled during the study period. This team was responsible for protocol implementation, data collection, and surveillance of patient response to the protocol. Educational sessions with house staff and nurses in each unit were held prior to the beginning of the study as well as continued monthly educational efforts during the study. In addition, biweekly huddles to review ongoing patient transitions as well as more formal monthly reviews were held.
The primary objective was to improve glycemic control, defined as the mean daily glucose, during the first 48 hours post transition without a significant increase in the percentage of patients with hypoglycemia (41‐70 mg/dL) or severe hypoglycemia (40 mg/dL). Secondary endpoints included the percent of patients with severe hyperglycemia (300 mg/dL), length of stay (LOS) calculated from the day of transition, number of restarts back onto infusion insulin within 72 hours of transition, and day‐weighted glucose mean up to 12 days following transition for patients with diabetes.
Glucose values were collected and averaged over 6‐hour periods for 48 hours post transition. For patients with diabetes, POC glucose values were collected up to 12 days of hospitalization. Day‐weighted means were obtained by calculating the mean glucose for each hospital day, averaged across all hospital days.12
Analysis
Subjects were divided by the presence or absence of diabetes. Those with diabetes were recommended to receive basal SC insulin during the transition period. Within each group, subjects were further divided by adherence to the protocol. Failure to transition per protocol was defined as: not receiving at least 80% of the recommended basal insulin dose, receiving the initial dose of insulin after the drip was discontinued, or receiving basal insulin when none was recommended.
Descriptive statistics within subgroups comparing age, gender, LOS by analysis of variance for continuous data and by chi‐square for nominal data, were compared. Twenty‐four and 48‐hour post transition mean glucose values and the 12 day weighted‐mean glucose were compared using analysis of variance (Stata ver. 10). All data are expressed as mean standard deviation with a significance value established at P < 0.05.
Results
A total of 210 episodes of infusion insulin in ICU patients were evaluated for the study from May of 2008 to September 2008 (Figure 1). Ninety‐six of these episodes were excluded, most commonly due to time on infusion insulin <24 hours or transition to comfort care. The remaining 114 infusions were eligible to use the protocol. Because the protocol recommends insulin therapy based on a diagnosis of diabetes, patients were further divided into these subcategories. Of these 114 transitions, the protocol was followed 66 times (58%).
Patients With Diabetes
(Table 1: Patient Demographics; Table 2: Insulin Use and Glycemic Control; Figure 2: Transition Graph).
| Patients With Diabetes | P Value | Patients Without Diabetes | P Value | |||
|---|---|---|---|---|---|---|
| Protocol Followed, n = 29 Patients* | Protocol NOT Followed, n = 33 Patients | Protocol Followed, n = 30 Patients | Protocol NOT Followed, n = 9 Patients | |||
| ||||||
| Average age, years, mean SD | 57.7 12.1 | 57.8 12.3 | 0.681 | 56.5 18.1 | 62.4 15.5 | 0.532 |
| Male patients | 21 (72%) | 21 (63%) | 0.58 | 20 (66%) | 7 (77%) | 0.691 |
| BMI | 30.7 7.2 | 28.6 6.8 | 0.180 | 27 5.4 | 25.2 3 | 0.081 |
| History of diabetes* | 18 (64%) | 25 (86%) | 0.07 | 0 | 0 | |
| Mean Hgb A1c (%) | 6.61.2 | 7.3 1.8 | 0.136 | 5.6 0.3 | 5.4 0.4 | 0.095 |
| Full nutrition | 26 (79%) | 24 (61%) | 0.131 | 23 (70%) | 9 (100%) | |
| On hemodialysis | 5 (17%) | 9 (27%) | 0.380 | 3 (10%) | 0 | |
| On >60 mg prednisone or equivalent per day | 7 (24%) | 10 (30%) | 0.632 | 0 | 0 | |
| Patients With Diabetes | P Value | Patients Without Diabetes | P Value | |||
|---|---|---|---|---|---|---|
| Protocol Followed, n = 33 transitions | Protocol NOT followed, n = 39 transitions | Protocol Followed, n = 33 transitions | Protocol NOT Followed, n = 9 transitions | |||
| ||||||
| Average infusion rate, hours | 3.96 3.15 | 3.74 3.64 | 0.1597 | 2.34 1.5 | 4.78 1.6 | <0.001 |
| Average BG on infusion insulin (mg/dL) | 122.5 27.5 | 122.5 31.8 | 0.844 | 115.1 22.7 | 127.5 27.2 | 0.006 |
| Average basal dose (units) given | 34.5 14.4 | 14.4 15.3 | <0.001 | 0 | 32.7 | <0.001 |
| Hours before () or after (+) infusion stopped basal insulin given | 1.13 0.9 | 11.6 9.3 | <0.001 | n/a | 0.33 | * |
| Average BG 6 hours post transition (mg/dL) | 143.7 39.4 | 182 62.5 | 0.019 | 150.2 54.9 | 142.1 34.1 | 0.624 |
| Average BG 0 to 24 hours post transition (mg/dL) | 167.98 50.24 | 211.02 81.01 | <0.001 | 150.24 54.9 | 150.12 32.4 | 0.600 |
| Total insulin used from 0 to 24 hours (units) | 65 32.2 | 26.7 25.4 | <0.001 | 3.2 4.1 | 51.3 30.3 | <0.001 |
| Average BG 25 to 48 hours post transition (mg/dL) | 176.1 55.25 | 218.2 88.54 | <0.001 | 153 35.3 | 154.4 46.7 | 0.711 |
| Total insulin used from 25 to 48 hours (units) | 60.5 35.4 | 28.1 24.4 | <0.001 | 2.8 3.8 | 44.9 34 | <0.001 |
| # of patients with severe hypoglycemia (<40 mg/dL) | 1 (3%) | 1 (2.6%) | * | 0 | 1 | * |
| # of patients with hypoglycemia (4170 mg/dL) | 3 (9%) | 2 (5.1%) | * | 1 | 0 | * |
| % of BG values in goal range (80180 mg/dL) (# in range/total #) | 60.2% (153/254) | 38.2% (104/272) | 0.004 | 80.1% (173/216) | 75.4% (49/65) | 0.83 |
| # of patients with severe hyperglycemia (>300 mg/dL) | 5 (15.2%) | 19 (48.7%) | 0.002 | 1 (3%) | 1 (11.1%) | * |
| LOS from transition (days) | 14.6 11.3 | 14 11.4 | 0.836 | 25.3 24.4 | 13.6 7.5 | 0.168 |
A total of 62 individual patients accounted for 72 separate transitions in patients with diabetes based on past medical history or an A1c 6% (n = 14). Of these 72 transitions, 33 (46%) adhered to the protocol while the remaining 39 (54%) transitions varied from the protocol at the treatment team's discretion. Despite similar insulin infusion rates and mean glucose values pretransition, patients with diabetes following the protocol had better glycemic control at both 24 hours and 48 hours after transition than those patients transitioned without the protocol. Day 1 mean blood glucose was 168 mg/dL vs. 211 mg/dL (P = <0.001) and day 2 mean blood glucose was 176 mg/dL vs. 218 mg/dL (P = <0.001) in protocol vs. nonprotocol patients with diabetes respectively (Figure 2).
There was a severe hypoglycemic event (40 mg/dL) in 1 patient with diabetes following the protocol and 1 patient not following the protocol within 48 hours of transition. Both events were secondary to nutritional‐insulin mismatch with emesis after insulin in one case and tube feeds being held in the second case. These findings were consistent with our prior examination of hypoglycemia cases.13 Severe hyperglycemia (glucose 300mg/dL) occurred in 5 (15 %) patients following the protocol vs. 19 (49%) patients not following protocol (P = 0.002.) Patients with diabetes following the protocol received significantly more insulin in the first 24 hours (mean of 65 units vs. 27 units, P 0.001) and 24 to 48 hours after transition (mean of 61 units vs. 28 units, p0.001) than those not following protocol.
An alternate method used at our institution and others14, 15 to calculate TDD is based on the patient's weight and body habitus. When we compared the projected TDD based on weight with the TDD using the transition protocol, we found that the weight based method was much less aggressive. For patients following the protocol, the weight based method projected a mean TDD of 46.3 16.9 units whereas the protocol projected a mean TDD of 65 33.2 units (P = 0.001).
Patients with diabetes following protocol received basal insulin an average of 1.13 hours prior to discontinuing the insulin infusion versus 11.6 hours after for those not following protocol.
Three patients with diabetes following the protocol and 3 patients with diabetes not following the protocol were restarted on infusion insulin within 72 hours of transition.
LOS from final transition to discharge was similar between protocol vs. nonprotocol patients (14.6 vs. 14 days, P = 0.836).
Figure 3 demonstrates that when used correctly, the protocol provides an extended period of glycemic control up to 12 days post transition. Patients transitioned per protocol had a day‐weighted mean glucose of 155 mg/dL vs. 184 mg/dL (P = 0.043) in patients not following protocol. There was only 1 glucose value less than 40 mg/dL between days 2 to 12 in the protocol group.
Patients Without Diabetes
Of the 39 individual patients without diabetes there were 42 transition events, 33 transitions (78.6%) were per protocol and placed on correctional insulin only. The remaining 9 transitions failed to follow protocol in that basal insulin was prescribed, but these patients maintained comparable glycemic control without an increase in hypoglycemic events. Following transition, patients without diabetes on protocol maintained a mean glucose of 150 mg/dL in the first 24 hours and 153 mg/dL in 24 to 48 hours post transition. They required a mean daily correctional insulin dose of 3.2 units on Day 1 and 2.8 units on Day 2 despite having an average drip rate of 2.3 units/hour at the time of transition (Table 2). There were no severe hypoglycemic events and 80% of blood sugars were within the goal range of 80 mg/dL to 180 mg/dL. Only 1 patient had a single blood glucose of >300mg/dL. No patient was restarted on infusion insulin once transitioned.
Patients without diabetes had a longer LOS after transition off of infusion insulin when compared to their diabetic counterparts (22 vs. 14 days).
Discussion
This study demonstrates the utility of hospitalist‐pharmacist collaboration in the creation and implementation of a safe and effective transition protocol for patients on infusion insulin. The protocol identifies patients appropriate for transition to a basal/nutritional insulin regimen versus those who will do well with premeal correctional insulin alone. Daily mean glucose was improved post transition for diabetic patients following the protocol compared to those not following the protocol without an increase in hypoglycemic events.
We found an equal number of insulin infusion restarts within 72 hours of transition and a similar LOS in protocol vs. nonprotocol patients with diabetes. The LOS was increased for patients without diabetes. This may be due to worse outcomes noted in patients with stress hyperglycemia in other studies.1, 16
The use of the higher multiplier for patients on minimal nutrition led to confusion among many protocol users. The protocol has since been modified to start by averaging the infusion rate over the prior 6 hours and then multiplying by 20 for all patients. This essentially calculates 80% of projected insulin requirements for the next 24 hours based on the patient's current needs. This calculation is then given as 50% basal and 50% nutritional for those on full nutrition vs. 100% basal for those on minimal nutrition. This protocol change has no impact on the amount of insulin received by the patient, but is more intuitive to providers. Instead of calculating the TDD as the projected requirement when full nutrition is obtained, the TDD is now calculated based on current insulin needs, and then doubled when patients who are receiving minimal nutrition advance to full nutrition.
Our study is limited by the lack of a true randomized control group. In lieu of this, we used our patients who did not follow protocol as our control. While not truly randomized, this group is comparable based on their age, gender mix, infusion rate, mean A1c, and projected TDD. This group was also similar to our preprotocol group mentioned in the Introduction.
Additional study limitations include the small number of nondiabetic patients not following the protocol (n = 9). We noted higher infusion rates in nondiabetics not following protocol versus those following protocol, which may have driven the primary team to give basal insulin. It is possible that these 9 patients were not yet ready to transition from infusion insulin or had other stressors not measured in our study. Unfortunately their small population size limits more extensive analysis.
The protocol was followed only 50% of the time for a variety of reasons. Patients who transitioned at night or on weekends were monitored by covering pharmacists and physicians who may not have been familiar with the protocol. Many physicians and nurses remain fearful of hypoglycemia and the outcomes of our study were not yet available for education. Some reported difficulty fully understanding how to use the protocol and why a higher multiplier was used for patients who were on minimal nutrition.
Efforts to improve adherence to the protocol are ongoing with some success, aided by the data demonstrating the safety and efficacy of the transition protocol.
Conclusion
By collaborating with ICU pharmacists we were able to design and implement a protocol that successfully and safely transitioned patients from infusion insulin to subcutaneous insulin. Patients following the protocol had a higher percentage of glucose values within the goal glucose range of 80 mg/dL to 180 mg/dL. In the future, we hope to automate the calculation of TDD and directly recommend a basal/bolus regimen for the clinical provider.
Hyperglycemia due to diabetes or stress is prevalent in the intensive care unit (ICU) and general ward setting. Umpierrez et al.1 reported hyperglycemia in 38% of hospitalized ward patients with 26% having a known history of diabetes. While patients with hyperglycemia admitted to the ICU are primarily treated with infusion insulin, those on the general wards usually receive a subcutaneous regimen of insulin. How best to transition patients from infusion insulin to a subcutaneous regimen remains elusive and under evaluated.
A recent observational pilot study of 24 surgical and 17 cardiac/medical intensive care patients at our university‐based hospital found that glycemic control significantly deteriorated when patients with diabetes transitioned from infusion insulin to subcutaneous insulin. A total of 21 critical care patients with a history of diabetes failed to receive basal insulin prior to discontinuation of the drip and developed uncontrolled hyperglycemia (mean glucose Day 1 of 216 mg/dL and Day 2 of 197 mg/dL). Patients without a history of diabetes did well post transition with a mean glucose of 142 mg/dL Day 1 and 133 mg/dL Day 2. A similar study by Czosnowski et al.2 demonstrated a significant increase in blood glucose from 123 26 mg/dL to 168 50 mg/dL upon discontinuation of infusion insulin.
This failed transition is disappointing, especially in view of the existence of a reliable subcutaneous (SC) insulin order set at our institution, but not surprising, as this is an inherently complex process. The severity of illness, the amount and mode of nutritional intake, geographic location, and provider team may all be in flux at the time of this transition. A few centers have demonstrated that a much improved transition is possible,36 however many of these solutions involve technology or incremental personnel that may not be available or the descriptions may lack sufficient detail to implement theses strategies with confidence elsewhere.
Therefore, we designed and piloted a protocol, coordinated by a multidisciplinary team, to transition patients from infusion insulin to SC insulin. The successful implementation of this protocol could serve as a blueprint to other institutions without the need for additional technology or personnel.
Methods
Patient Population/Setting
This was a prospective study of patients admitted to either the medical/cardiac intensive care unit (MICU/CCU) or surgical intensive care unit (SICU) at an academic medical facility and placed on infusion insulin for >24 hours. The Institutional Review Board (IRB) approved the study for prospective chart review and anonymous results reporting without individual consent.
Patients in the SICU were initiated on infusion insulin after 2 blood glucose readings were above 150 mg/dL, whereas initiation was left to the discretion of the attending physician in the MICU/CCU. A computerized system created in‐house recommends insulin infusion rates based on point‐of‐care (POC) glucose measurements with a target range of 91 mg/dL to 150 mg/dL.
Inclusion/Exclusion Criteria
All patients on continuous insulin infusion admitted to the SICU or the MICU/CCU between May 2008 and September 2008 were evaluated for the study (Figure 1). Patients were excluded from analysis if they were on the infusion for less than 24 hours, had a liver transplant, were discharged within 48 hours of transition, were made comfort care or transitioned to an insulin pump. All other patients were included in the final analysis.
Transition Protocol
Step 1: Does the Patient Need Basal SC Insulin?
Patients were recommended to receive basal SC insulin if they either: (1) were on medications for diabetes; (2) had an A1c 6%; or (3) received the equivalent of 60 mg of prednisone; AND had an infusion rate 1 unit/hour (Supporting Information Appendix 1). Patients on infusion insulin due to stress hyperglycemia, regardless of the infusion rate, were not placed on basal SC insulin. Patients on high dose steroids due to spinal injuries were excluded because their duration of steroid use was typically less than 48 hours and usually ended prior to the time of transition. The protocol recommends premeal correctional insulin for those not qualifying for basal insulin.
In order to establish patients in need of basal/nutritional insulin we opted to use A1c as well as past medical history to identify patients with diabetes. The American Diabetes Association (ADA) has recently accepted using an A1c 6.5% to make a new diagnosis of diabetes.7 In a 2‐week trial prior to initiating the protocol we used a cut off A1c of 6.5%. However, we found that patients with an A1c of 6% to 6.5% had poor glucose control post transition; therefore we chose 6% as our identifier. In addition, using a cut off A1c of 6% was reported by Rohlfing et al.8 and Greci et al.9 to be more than 97% sensitive at identifying a new diagnosis of diabetes.
To ensure an A1c was ordered and available at the time of transition, critical care pharmacists were given Pharmacy and Therapeutics Committee authorization to order an A1c at the start of the infusion. Pharmacists would also guide the primary team through the protocol's recommendations as well as alert the project team when a patient was expected to transition.
Step 2: Evaluate the Patient's Nutritional Intake to Calculate the Total Daily Dose (TDD) of Insulin
TDD is the total amount of insulin needed to cover both the nutritional and basal requirements of a patient over the course of 24 hours. TDD was calculated by averaging the hourly drip rate over the prior 6 hours and multiplying by 20 if taking in full nutrition or 40 if taking minimal nutrition while on the drip. A higher multiplier was used for those on minimal nutrition with the expectation that their insulin requirements would double once tolerating a full diet. Full nutrition was defined as eating >50% of meals, on goal tube feeds, or receiving total parenteral nutrition (TPN). Minimal nutrition was defined as taking nothing by mouth (or NPO), tolerating <50% of meals, or on a clear liquid diet.
Step 3: Divide the TDD Into the Appropriate Components of Insulin Treatment (Basal, Nutritional and Correction), Depending on the Nutritional Status
In Step 3, the TDD was evenly divided into basal and nutritional insulin. A total of 50% of the TDD was given as glargine (Lantus) 2 hours prior to stopping the infusion. The remaining 50% was divided into nutritional components as either Regular insulin every 6 hours for patients on tube feeds or lispro (Humalog) before meals if tolerating an oral diet. For patients on minimal nutrition, the 50% nutritional insulin dose was not initiated until the patient was tolerating full nutrition.
The protocol recommended basal insulin administration 2 hours prior to infusion discontinuation as recommended by the American Association of Clinical Endocrinologists (AACE) and ADA consensus statement on inpatient glycemic control as well as pharmacokinetics.10, 11 For these reasons, failure to receive basal insulin prior to transition was viewed as failure to follow the protocol.
Safety features of the protocol included a maximum TDD of 100 units unless the patient was on >100 units/day of insulin prior to admission. A pager was carried by rotating hospitalists or pharmacist study investigators at all hours during the protocol implementation phase to answer any questions regarding a patient's transition.
Data Collection/Monitoring
A multidisciplinary team consisting of hospitalists, ICU pharmacists, critical care physicians and nursing representatives was assembled during the study period. This team was responsible for protocol implementation, data collection, and surveillance of patient response to the protocol. Educational sessions with house staff and nurses in each unit were held prior to the beginning of the study as well as continued monthly educational efforts during the study. In addition, biweekly huddles to review ongoing patient transitions as well as more formal monthly reviews were held.
The primary objective was to improve glycemic control, defined as the mean daily glucose, during the first 48 hours post transition without a significant increase in the percentage of patients with hypoglycemia (41‐70 mg/dL) or severe hypoglycemia (40 mg/dL). Secondary endpoints included the percent of patients with severe hyperglycemia (300 mg/dL), length of stay (LOS) calculated from the day of transition, number of restarts back onto infusion insulin within 72 hours of transition, and day‐weighted glucose mean up to 12 days following transition for patients with diabetes.
Glucose values were collected and averaged over 6‐hour periods for 48 hours post transition. For patients with diabetes, POC glucose values were collected up to 12 days of hospitalization. Day‐weighted means were obtained by calculating the mean glucose for each hospital day, averaged across all hospital days.12
Analysis
Subjects were divided by the presence or absence of diabetes. Those with diabetes were recommended to receive basal SC insulin during the transition period. Within each group, subjects were further divided by adherence to the protocol. Failure to transition per protocol was defined as: not receiving at least 80% of the recommended basal insulin dose, receiving the initial dose of insulin after the drip was discontinued, or receiving basal insulin when none was recommended.
Descriptive statistics within subgroups comparing age, gender, LOS by analysis of variance for continuous data and by chi‐square for nominal data, were compared. Twenty‐four and 48‐hour post transition mean glucose values and the 12 day weighted‐mean glucose were compared using analysis of variance (Stata ver. 10). All data are expressed as mean standard deviation with a significance value established at P < 0.05.
Results
A total of 210 episodes of infusion insulin in ICU patients were evaluated for the study from May of 2008 to September 2008 (Figure 1). Ninety‐six of these episodes were excluded, most commonly due to time on infusion insulin <24 hours or transition to comfort care. The remaining 114 infusions were eligible to use the protocol. Because the protocol recommends insulin therapy based on a diagnosis of diabetes, patients were further divided into these subcategories. Of these 114 transitions, the protocol was followed 66 times (58%).
Patients With Diabetes
(Table 1: Patient Demographics; Table 2: Insulin Use and Glycemic Control; Figure 2: Transition Graph).
| Patients With Diabetes | P Value | Patients Without Diabetes | P Value | |||
|---|---|---|---|---|---|---|
| Protocol Followed, n = 29 Patients* | Protocol NOT Followed, n = 33 Patients | Protocol Followed, n = 30 Patients | Protocol NOT Followed, n = 9 Patients | |||
| ||||||
| Average age, years, mean SD | 57.7 12.1 | 57.8 12.3 | 0.681 | 56.5 18.1 | 62.4 15.5 | 0.532 |
| Male patients | 21 (72%) | 21 (63%) | 0.58 | 20 (66%) | 7 (77%) | 0.691 |
| BMI | 30.7 7.2 | 28.6 6.8 | 0.180 | 27 5.4 | 25.2 3 | 0.081 |
| History of diabetes* | 18 (64%) | 25 (86%) | 0.07 | 0 | 0 | |
| Mean Hgb A1c (%) | 6.61.2 | 7.3 1.8 | 0.136 | 5.6 0.3 | 5.4 0.4 | 0.095 |
| Full nutrition | 26 (79%) | 24 (61%) | 0.131 | 23 (70%) | 9 (100%) | |
| On hemodialysis | 5 (17%) | 9 (27%) | 0.380 | 3 (10%) | 0 | |
| On >60 mg prednisone or equivalent per day | 7 (24%) | 10 (30%) | 0.632 | 0 | 0 | |
| Patients With Diabetes | P Value | Patients Without Diabetes | P Value | |||
|---|---|---|---|---|---|---|
| Protocol Followed, n = 33 transitions | Protocol NOT followed, n = 39 transitions | Protocol Followed, n = 33 transitions | Protocol NOT Followed, n = 9 transitions | |||
| ||||||
| Average infusion rate, hours | 3.96 3.15 | 3.74 3.64 | 0.1597 | 2.34 1.5 | 4.78 1.6 | <0.001 |
| Average BG on infusion insulin (mg/dL) | 122.5 27.5 | 122.5 31.8 | 0.844 | 115.1 22.7 | 127.5 27.2 | 0.006 |
| Average basal dose (units) given | 34.5 14.4 | 14.4 15.3 | <0.001 | 0 | 32.7 | <0.001 |
| Hours before () or after (+) infusion stopped basal insulin given | 1.13 0.9 | 11.6 9.3 | <0.001 | n/a | 0.33 | * |
| Average BG 6 hours post transition (mg/dL) | 143.7 39.4 | 182 62.5 | 0.019 | 150.2 54.9 | 142.1 34.1 | 0.624 |
| Average BG 0 to 24 hours post transition (mg/dL) | 167.98 50.24 | 211.02 81.01 | <0.001 | 150.24 54.9 | 150.12 32.4 | 0.600 |
| Total insulin used from 0 to 24 hours (units) | 65 32.2 | 26.7 25.4 | <0.001 | 3.2 4.1 | 51.3 30.3 | <0.001 |
| Average BG 25 to 48 hours post transition (mg/dL) | 176.1 55.25 | 218.2 88.54 | <0.001 | 153 35.3 | 154.4 46.7 | 0.711 |
| Total insulin used from 25 to 48 hours (units) | 60.5 35.4 | 28.1 24.4 | <0.001 | 2.8 3.8 | 44.9 34 | <0.001 |
| # of patients with severe hypoglycemia (<40 mg/dL) | 1 (3%) | 1 (2.6%) | * | 0 | 1 | * |
| # of patients with hypoglycemia (4170 mg/dL) | 3 (9%) | 2 (5.1%) | * | 1 | 0 | * |
| % of BG values in goal range (80180 mg/dL) (# in range/total #) | 60.2% (153/254) | 38.2% (104/272) | 0.004 | 80.1% (173/216) | 75.4% (49/65) | 0.83 |
| # of patients with severe hyperglycemia (>300 mg/dL) | 5 (15.2%) | 19 (48.7%) | 0.002 | 1 (3%) | 1 (11.1%) | * |
| LOS from transition (days) | 14.6 11.3 | 14 11.4 | 0.836 | 25.3 24.4 | 13.6 7.5 | 0.168 |
A total of 62 individual patients accounted for 72 separate transitions in patients with diabetes based on past medical history or an A1c 6% (n = 14). Of these 72 transitions, 33 (46%) adhered to the protocol while the remaining 39 (54%) transitions varied from the protocol at the treatment team's discretion. Despite similar insulin infusion rates and mean glucose values pretransition, patients with diabetes following the protocol had better glycemic control at both 24 hours and 48 hours after transition than those patients transitioned without the protocol. Day 1 mean blood glucose was 168 mg/dL vs. 211 mg/dL (P = <0.001) and day 2 mean blood glucose was 176 mg/dL vs. 218 mg/dL (P = <0.001) in protocol vs. nonprotocol patients with diabetes respectively (Figure 2).
There was a severe hypoglycemic event (40 mg/dL) in 1 patient with diabetes following the protocol and 1 patient not following the protocol within 48 hours of transition. Both events were secondary to nutritional‐insulin mismatch with emesis after insulin in one case and tube feeds being held in the second case. These findings were consistent with our prior examination of hypoglycemia cases.13 Severe hyperglycemia (glucose 300mg/dL) occurred in 5 (15 %) patients following the protocol vs. 19 (49%) patients not following protocol (P = 0.002.) Patients with diabetes following the protocol received significantly more insulin in the first 24 hours (mean of 65 units vs. 27 units, P 0.001) and 24 to 48 hours after transition (mean of 61 units vs. 28 units, p0.001) than those not following protocol.
An alternate method used at our institution and others14, 15 to calculate TDD is based on the patient's weight and body habitus. When we compared the projected TDD based on weight with the TDD using the transition protocol, we found that the weight based method was much less aggressive. For patients following the protocol, the weight based method projected a mean TDD of 46.3 16.9 units whereas the protocol projected a mean TDD of 65 33.2 units (P = 0.001).
Patients with diabetes following protocol received basal insulin an average of 1.13 hours prior to discontinuing the insulin infusion versus 11.6 hours after for those not following protocol.
Three patients with diabetes following the protocol and 3 patients with diabetes not following the protocol were restarted on infusion insulin within 72 hours of transition.
LOS from final transition to discharge was similar between protocol vs. nonprotocol patients (14.6 vs. 14 days, P = 0.836).
Figure 3 demonstrates that when used correctly, the protocol provides an extended period of glycemic control up to 12 days post transition. Patients transitioned per protocol had a day‐weighted mean glucose of 155 mg/dL vs. 184 mg/dL (P = 0.043) in patients not following protocol. There was only 1 glucose value less than 40 mg/dL between days 2 to 12 in the protocol group.
Patients Without Diabetes
Of the 39 individual patients without diabetes there were 42 transition events, 33 transitions (78.6%) were per protocol and placed on correctional insulin only. The remaining 9 transitions failed to follow protocol in that basal insulin was prescribed, but these patients maintained comparable glycemic control without an increase in hypoglycemic events. Following transition, patients without diabetes on protocol maintained a mean glucose of 150 mg/dL in the first 24 hours and 153 mg/dL in 24 to 48 hours post transition. They required a mean daily correctional insulin dose of 3.2 units on Day 1 and 2.8 units on Day 2 despite having an average drip rate of 2.3 units/hour at the time of transition (Table 2). There were no severe hypoglycemic events and 80% of blood sugars were within the goal range of 80 mg/dL to 180 mg/dL. Only 1 patient had a single blood glucose of >300mg/dL. No patient was restarted on infusion insulin once transitioned.
Patients without diabetes had a longer LOS after transition off of infusion insulin when compared to their diabetic counterparts (22 vs. 14 days).
Discussion
This study demonstrates the utility of hospitalist‐pharmacist collaboration in the creation and implementation of a safe and effective transition protocol for patients on infusion insulin. The protocol identifies patients appropriate for transition to a basal/nutritional insulin regimen versus those who will do well with premeal correctional insulin alone. Daily mean glucose was improved post transition for diabetic patients following the protocol compared to those not following the protocol without an increase in hypoglycemic events.
We found an equal number of insulin infusion restarts within 72 hours of transition and a similar LOS in protocol vs. nonprotocol patients with diabetes. The LOS was increased for patients without diabetes. This may be due to worse outcomes noted in patients with stress hyperglycemia in other studies.1, 16
The use of the higher multiplier for patients on minimal nutrition led to confusion among many protocol users. The protocol has since been modified to start by averaging the infusion rate over the prior 6 hours and then multiplying by 20 for all patients. This essentially calculates 80% of projected insulin requirements for the next 24 hours based on the patient's current needs. This calculation is then given as 50% basal and 50% nutritional for those on full nutrition vs. 100% basal for those on minimal nutrition. This protocol change has no impact on the amount of insulin received by the patient, but is more intuitive to providers. Instead of calculating the TDD as the projected requirement when full nutrition is obtained, the TDD is now calculated based on current insulin needs, and then doubled when patients who are receiving minimal nutrition advance to full nutrition.
Our study is limited by the lack of a true randomized control group. In lieu of this, we used our patients who did not follow protocol as our control. While not truly randomized, this group is comparable based on their age, gender mix, infusion rate, mean A1c, and projected TDD. This group was also similar to our preprotocol group mentioned in the Introduction.
Additional study limitations include the small number of nondiabetic patients not following the protocol (n = 9). We noted higher infusion rates in nondiabetics not following protocol versus those following protocol, which may have driven the primary team to give basal insulin. It is possible that these 9 patients were not yet ready to transition from infusion insulin or had other stressors not measured in our study. Unfortunately their small population size limits more extensive analysis.
The protocol was followed only 50% of the time for a variety of reasons. Patients who transitioned at night or on weekends were monitored by covering pharmacists and physicians who may not have been familiar with the protocol. Many physicians and nurses remain fearful of hypoglycemia and the outcomes of our study were not yet available for education. Some reported difficulty fully understanding how to use the protocol and why a higher multiplier was used for patients who were on minimal nutrition.
Efforts to improve adherence to the protocol are ongoing with some success, aided by the data demonstrating the safety and efficacy of the transition protocol.
Conclusion
By collaborating with ICU pharmacists we were able to design and implement a protocol that successfully and safely transitioned patients from infusion insulin to subcutaneous insulin. Patients following the protocol had a higher percentage of glucose values within the goal glucose range of 80 mg/dL to 180 mg/dL. In the future, we hope to automate the calculation of TDD and directly recommend a basal/bolus regimen for the clinical provider.
- ,,,,,.Hyperglycemia: an independent marker of in‐hospital mortality in patients with undiagnosed diabetes.J Clin Endocrinol Metab.2002;87:978–982.
- ,,,,,.Evaluation of glycemic control following discontinuation of an intensive insulin protocol.J Hosp Med.2009;4:28–34.
- ,,, et al.Inpatient management of hyperglycemia: the Northwestern experience.Endocr Pract.2006;12:491–505.
- ,,,.Intravenous insulin infusion therapy: indications, methods, and transition to subcutaneous insulin therapy.Endocr Pract.2004;10Suppl 2:71–80.
- ,,, et al.Conversion of intravenous insulin infusions to subcutaneously administered insulin glargine in patients with hyperglycemia.Endocr Pract.2006;12:641–650.
- ,.Effects of outcome on in‐hospital transition from intravenous insulin infusion to subcutaneous therapy.Am J Cardiol.2006;98:557–564.
- International Expert Committee report on the role of the A1C assay in the diagnosis of diabetes.Diabetes Care.2009;32:1327–1334.
- ,,, et al.Use of GHb (HbA1c) in screening for undiagnosed diabetes in the U.S. population.Diabetes Care.2000;23:187–191.
- ,,, et al.Utility of HbA(1c) levels for diabetes case finding in hospitalized patients with hyperglycemia.Diabetes Care.2003;26:1064–1068.
- ,,, et al.American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control.Endocr Pract.2009;15(4):353–369.
- ,,, et al.Pharmacokinetics and pharmacodynamics of subcutaneous injection of long‐acting human insulin analog glargine, NPH insulin, and ultralente human insulin and continuous subcutaneous infusion of insulin lispro.Diabetes.2000;49:2142–2148.
- ,,, et al.“Glucometrics”‐‐assessing the quality of inpatient glucose management.Diabetes Technol Ther.2006;8:560–569.
- ,,.Iatrogenic Inpatient Hypoglycemia: Risk Factors, Treatment, and Prevention: Analysis of Current Practice at an Academic Medical Center With Implications for Improvement Efforts.Diabetes Spectr.2008;21:241–247.
- ,,, et al.Management of diabetes and hyperglycemia in hospitals.Diabetes Care.2004;27:553–591.
- .Insulin management of diabetic patients on general medical and surgical floors.Endocr Pract.2006;12Suppl 3:86–90.
- ,,,.Inadequate blood glucose control is associated with in‐hospital mortality and morbidity in diabetic and nondiabetic patients undergoing cardiac surgery.Circulation.2008;118:113–123.
- ,,,,,.Hyperglycemia: an independent marker of in‐hospital mortality in patients with undiagnosed diabetes.J Clin Endocrinol Metab.2002;87:978–982.
- ,,,,,.Evaluation of glycemic control following discontinuation of an intensive insulin protocol.J Hosp Med.2009;4:28–34.
- ,,, et al.Inpatient management of hyperglycemia: the Northwestern experience.Endocr Pract.2006;12:491–505.
- ,,,.Intravenous insulin infusion therapy: indications, methods, and transition to subcutaneous insulin therapy.Endocr Pract.2004;10Suppl 2:71–80.
- ,,, et al.Conversion of intravenous insulin infusions to subcutaneously administered insulin glargine in patients with hyperglycemia.Endocr Pract.2006;12:641–650.
- ,.Effects of outcome on in‐hospital transition from intravenous insulin infusion to subcutaneous therapy.Am J Cardiol.2006;98:557–564.
- International Expert Committee report on the role of the A1C assay in the diagnosis of diabetes.Diabetes Care.2009;32:1327–1334.
- ,,, et al.Use of GHb (HbA1c) in screening for undiagnosed diabetes in the U.S. population.Diabetes Care.2000;23:187–191.
- ,,, et al.Utility of HbA(1c) levels for diabetes case finding in hospitalized patients with hyperglycemia.Diabetes Care.2003;26:1064–1068.
- ,,, et al.American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control.Endocr Pract.2009;15(4):353–369.
- ,,, et al.Pharmacokinetics and pharmacodynamics of subcutaneous injection of long‐acting human insulin analog glargine, NPH insulin, and ultralente human insulin and continuous subcutaneous infusion of insulin lispro.Diabetes.2000;49:2142–2148.
- ,,, et al.“Glucometrics”‐‐assessing the quality of inpatient glucose management.Diabetes Technol Ther.2006;8:560–569.
- ,,.Iatrogenic Inpatient Hypoglycemia: Risk Factors, Treatment, and Prevention: Analysis of Current Practice at an Academic Medical Center With Implications for Improvement Efforts.Diabetes Spectr.2008;21:241–247.
- ,,, et al.Management of diabetes and hyperglycemia in hospitals.Diabetes Care.2004;27:553–591.
- .Insulin management of diabetic patients on general medical and surgical floors.Endocr Pract.2006;12Suppl 3:86–90.
- ,,,.Inadequate blood glucose control is associated with in‐hospital mortality and morbidity in diabetic and nondiabetic patients undergoing cardiac surgery.Circulation.2008;118:113–123.
Copyright © 2010 Society of Hospital Medicine
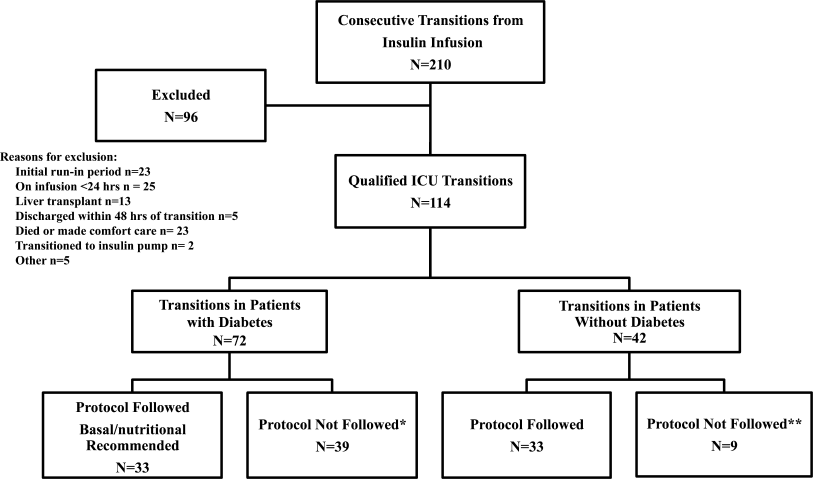
Researchers reveal structure of CXCR4
Credit: Raymond Stevens
Scripps Research Institute
A team of researchers has uncovered the structure of a cell surface receptor, CXCR4, which guides blood and immune cell movement throughout the body.
CXCR4 is also found on the surface of the human immunodeficiency virus (HIV), and helps the virus to enter blood cells.
The receptor is part of a group of about 700 proteins known as G protein-coupled receptors (GPCRs).
The team, led by Raymond C. Stevens, PhD, of Scripps Research Institute in La Jolla, California, and senior author of the study, already found the structures of two other GPCRs: the adrenergic receptor and A2A adenosine receptor.
CXCR4 belongs to a different group of GPCRs, one that binds to protein molecules called chemokines, responsible for steering blood and immune cells where they are needed.
The team used GPCR biochemistry, receptor stabilization, and X-ray crystallography to capture the first visual of a chemokine receptor bound to a ligand.
Unlike the adrenergic receptor and the A2A adenosine receptor, CXCR4 likes to form dimers.
“The dimerization observation was very intriguing,” said Dr Stevens. “We solved 5 different crystal structures in multiple crystal forms, and each one had the same dimer interface. It has long been debated how GPCRs might dimerize, if they did at all. This is the first solid observation about a consistent structural GPCR dimer.”
The team believes preventing dimerization might provide a new way to block CXCR4, which results in the release of hematopoietic stem cells from bone marrow into the bloodstream.
Currently, plerixafor injection is the only drug on the market that blocks CXCR4. Therapy that assists in the release of hematopoietic stem cells to the bloodstream is very useful following stem cell transplant.
Drugs that block CXCR4 are also useful in treating HIV infection.
Their findings were published in the October 7 issue of Science. ![]()
Credit: Raymond Stevens
Scripps Research Institute
A team of researchers has uncovered the structure of a cell surface receptor, CXCR4, which guides blood and immune cell movement throughout the body.
CXCR4 is also found on the surface of the human immunodeficiency virus (HIV), and helps the virus to enter blood cells.
The receptor is part of a group of about 700 proteins known as G protein-coupled receptors (GPCRs).
The team, led by Raymond C. Stevens, PhD, of Scripps Research Institute in La Jolla, California, and senior author of the study, already found the structures of two other GPCRs: the adrenergic receptor and A2A adenosine receptor.
CXCR4 belongs to a different group of GPCRs, one that binds to protein molecules called chemokines, responsible for steering blood and immune cells where they are needed.
The team used GPCR biochemistry, receptor stabilization, and X-ray crystallography to capture the first visual of a chemokine receptor bound to a ligand.
Unlike the adrenergic receptor and the A2A adenosine receptor, CXCR4 likes to form dimers.
“The dimerization observation was very intriguing,” said Dr Stevens. “We solved 5 different crystal structures in multiple crystal forms, and each one had the same dimer interface. It has long been debated how GPCRs might dimerize, if they did at all. This is the first solid observation about a consistent structural GPCR dimer.”
The team believes preventing dimerization might provide a new way to block CXCR4, which results in the release of hematopoietic stem cells from bone marrow into the bloodstream.
Currently, plerixafor injection is the only drug on the market that blocks CXCR4. Therapy that assists in the release of hematopoietic stem cells to the bloodstream is very useful following stem cell transplant.
Drugs that block CXCR4 are also useful in treating HIV infection.
Their findings were published in the October 7 issue of Science. ![]()
Credit: Raymond Stevens
Scripps Research Institute
A team of researchers has uncovered the structure of a cell surface receptor, CXCR4, which guides blood and immune cell movement throughout the body.
CXCR4 is also found on the surface of the human immunodeficiency virus (HIV), and helps the virus to enter blood cells.
The receptor is part of a group of about 700 proteins known as G protein-coupled receptors (GPCRs).
The team, led by Raymond C. Stevens, PhD, of Scripps Research Institute in La Jolla, California, and senior author of the study, already found the structures of two other GPCRs: the adrenergic receptor and A2A adenosine receptor.
CXCR4 belongs to a different group of GPCRs, one that binds to protein molecules called chemokines, responsible for steering blood and immune cells where they are needed.
The team used GPCR biochemistry, receptor stabilization, and X-ray crystallography to capture the first visual of a chemokine receptor bound to a ligand.
Unlike the adrenergic receptor and the A2A adenosine receptor, CXCR4 likes to form dimers.
“The dimerization observation was very intriguing,” said Dr Stevens. “We solved 5 different crystal structures in multiple crystal forms, and each one had the same dimer interface. It has long been debated how GPCRs might dimerize, if they did at all. This is the first solid observation about a consistent structural GPCR dimer.”
The team believes preventing dimerization might provide a new way to block CXCR4, which results in the release of hematopoietic stem cells from bone marrow into the bloodstream.
Currently, plerixafor injection is the only drug on the market that blocks CXCR4. Therapy that assists in the release of hematopoietic stem cells to the bloodstream is very useful following stem cell transplant.
Drugs that block CXCR4 are also useful in treating HIV infection.
Their findings were published in the October 7 issue of Science. ![]()
Anti-thrombotics, Bleeding Connection No Cause for Alarm
A new Archives of Internal Medicine study that shows the use of multiple anti-thrombotics increases the risk of bleeding in atrial fibrillation (AF) patients compared with warfarin monotherapy shouldn't change hospitalists' prescribing patterns, according to one physician.
Kurt Pfeifer, MD, FACP, program director of the Internal Medicine Residency program at Medical College in Milwaukee, says the research is important, as it adds to the knowledge base on the potential dangers of combining aspirin, warfarin, and clopidogrel. But even in cases where bleeding risks can be tripled, Dr. Pfeifer suggests hospitalists keep an eye on the risk-reward curve.
"It comes back to: Ask yourself, Do [patients] have a reason to be on it?" says Dr. Pfeifer, an associate professor who stays current on bleeding-risk research. "If they do, you better have a real reason for not putting them on it."
The study concluded that all combinations of the three medications in AF patients are associated with increased risk of both fatal and nonfatal bleeding (Arch Intern Med. 2010;170(16):1433-1441). The cohort study noted that "dual warfarin and clopidogrel therapy and triple therapy carried a more than 3-fold higher risk than did warfarin monotherapy."
Dr. Pfeifer doesn't discount the information, but notes that he would likely only discourage the use in patients with a particularly high risk for bleeding. However, he thinks the study is also a good reminder that HM practitioners should communicate the risks and therapies to both patients and their PCPs.
"It's important to know what the risks are, but it doesn't take away from what the indicators are," he says. "In most of these situations, I think you can feel better that you’ve academically addressed it."
A new Archives of Internal Medicine study that shows the use of multiple anti-thrombotics increases the risk of bleeding in atrial fibrillation (AF) patients compared with warfarin monotherapy shouldn't change hospitalists' prescribing patterns, according to one physician.
Kurt Pfeifer, MD, FACP, program director of the Internal Medicine Residency program at Medical College in Milwaukee, says the research is important, as it adds to the knowledge base on the potential dangers of combining aspirin, warfarin, and clopidogrel. But even in cases where bleeding risks can be tripled, Dr. Pfeifer suggests hospitalists keep an eye on the risk-reward curve.
"It comes back to: Ask yourself, Do [patients] have a reason to be on it?" says Dr. Pfeifer, an associate professor who stays current on bleeding-risk research. "If they do, you better have a real reason for not putting them on it."
The study concluded that all combinations of the three medications in AF patients are associated with increased risk of both fatal and nonfatal bleeding (Arch Intern Med. 2010;170(16):1433-1441). The cohort study noted that "dual warfarin and clopidogrel therapy and triple therapy carried a more than 3-fold higher risk than did warfarin monotherapy."
Dr. Pfeifer doesn't discount the information, but notes that he would likely only discourage the use in patients with a particularly high risk for bleeding. However, he thinks the study is also a good reminder that HM practitioners should communicate the risks and therapies to both patients and their PCPs.
"It's important to know what the risks are, but it doesn't take away from what the indicators are," he says. "In most of these situations, I think you can feel better that you’ve academically addressed it."
A new Archives of Internal Medicine study that shows the use of multiple anti-thrombotics increases the risk of bleeding in atrial fibrillation (AF) patients compared with warfarin monotherapy shouldn't change hospitalists' prescribing patterns, according to one physician.
Kurt Pfeifer, MD, FACP, program director of the Internal Medicine Residency program at Medical College in Milwaukee, says the research is important, as it adds to the knowledge base on the potential dangers of combining aspirin, warfarin, and clopidogrel. But even in cases where bleeding risks can be tripled, Dr. Pfeifer suggests hospitalists keep an eye on the risk-reward curve.
"It comes back to: Ask yourself, Do [patients] have a reason to be on it?" says Dr. Pfeifer, an associate professor who stays current on bleeding-risk research. "If they do, you better have a real reason for not putting them on it."
The study concluded that all combinations of the three medications in AF patients are associated with increased risk of both fatal and nonfatal bleeding (Arch Intern Med. 2010;170(16):1433-1441). The cohort study noted that "dual warfarin and clopidogrel therapy and triple therapy carried a more than 3-fold higher risk than did warfarin monotherapy."
Dr. Pfeifer doesn't discount the information, but notes that he would likely only discourage the use in patients with a particularly high risk for bleeding. However, he thinks the study is also a good reminder that HM practitioners should communicate the risks and therapies to both patients and their PCPs.
"It's important to know what the risks are, but it doesn't take away from what the indicators are," he says. "In most of these situations, I think you can feel better that you’ve academically addressed it."
MGMA Names Hospitalist CEO Physician Executive of the Year
Hospitalist leader Adam Singer, MD, is to be formally named physician executive of the year on Oct. 26 by the Medical Group Management Association (MGMA), a national association of 21,500 administrators and leaders of physician group practices. Dr. Singer, who learned of the award last month, is the first hospitalist honoree.
The award recognizes physician executives who have exhibited outstanding leadership and achieved exceptional performance in healthcare delivery.
Dr. Singer, 50, who is founder, CEO, and chief medical officer of IPC: The Hospitalist Company, took the North Hollywood, Calif.-based company public in 2008. A founding member of SHM, Dr. Singer's award represents a milestone in the growing acceptance of HM as a medical business and of its business model, says Dan Fuller, president of IN Compass Health of Alpharetta, Ga.
“We’re seeing more attention to hospitalist compensation models, payment incentives, and recognition of the need to align these with both productivity and quality,” says Dr. Fuller, a member of SHM's Practice Management Committee. The award “is great for Adam, but really exciting for our movement.”
Steven Deitelzweig, MD, chair of hospital medicine for Ochsner Health System in New Orleans and chair of SHM’s Practice Management Committee, says the MGMA honor reflects growing recognition of the role hospitalists will play in the changing business of hospital care under Affordable Care Act reforms. “Hospitalists will be the ones senior hospital leaders come to for help in figuring out healthcare reform,” he says.
Dr. Singer says the award recognizes his success, as a physician, and as an entrepreneur, and is a "signpost for hospital medicine as a whole. They're recognizing HM not just as a medical specialty that has emerged in recent years, but our success as a business," he says. "HM is a business, at least as we practice it at IPC. We've shown that it can be profitable as a standalone medical service, without requiring subsidization."
Hospitalist leader Adam Singer, MD, is to be formally named physician executive of the year on Oct. 26 by the Medical Group Management Association (MGMA), a national association of 21,500 administrators and leaders of physician group practices. Dr. Singer, who learned of the award last month, is the first hospitalist honoree.
The award recognizes physician executives who have exhibited outstanding leadership and achieved exceptional performance in healthcare delivery.
Dr. Singer, 50, who is founder, CEO, and chief medical officer of IPC: The Hospitalist Company, took the North Hollywood, Calif.-based company public in 2008. A founding member of SHM, Dr. Singer's award represents a milestone in the growing acceptance of HM as a medical business and of its business model, says Dan Fuller, president of IN Compass Health of Alpharetta, Ga.
“We’re seeing more attention to hospitalist compensation models, payment incentives, and recognition of the need to align these with both productivity and quality,” says Dr. Fuller, a member of SHM's Practice Management Committee. The award “is great for Adam, but really exciting for our movement.”
Steven Deitelzweig, MD, chair of hospital medicine for Ochsner Health System in New Orleans and chair of SHM’s Practice Management Committee, says the MGMA honor reflects growing recognition of the role hospitalists will play in the changing business of hospital care under Affordable Care Act reforms. “Hospitalists will be the ones senior hospital leaders come to for help in figuring out healthcare reform,” he says.
Dr. Singer says the award recognizes his success, as a physician, and as an entrepreneur, and is a "signpost for hospital medicine as a whole. They're recognizing HM not just as a medical specialty that has emerged in recent years, but our success as a business," he says. "HM is a business, at least as we practice it at IPC. We've shown that it can be profitable as a standalone medical service, without requiring subsidization."
Hospitalist leader Adam Singer, MD, is to be formally named physician executive of the year on Oct. 26 by the Medical Group Management Association (MGMA), a national association of 21,500 administrators and leaders of physician group practices. Dr. Singer, who learned of the award last month, is the first hospitalist honoree.
The award recognizes physician executives who have exhibited outstanding leadership and achieved exceptional performance in healthcare delivery.
Dr. Singer, 50, who is founder, CEO, and chief medical officer of IPC: The Hospitalist Company, took the North Hollywood, Calif.-based company public in 2008. A founding member of SHM, Dr. Singer's award represents a milestone in the growing acceptance of HM as a medical business and of its business model, says Dan Fuller, president of IN Compass Health of Alpharetta, Ga.
“We’re seeing more attention to hospitalist compensation models, payment incentives, and recognition of the need to align these with both productivity and quality,” says Dr. Fuller, a member of SHM's Practice Management Committee. The award “is great for Adam, but really exciting for our movement.”
Steven Deitelzweig, MD, chair of hospital medicine for Ochsner Health System in New Orleans and chair of SHM’s Practice Management Committee, says the MGMA honor reflects growing recognition of the role hospitalists will play in the changing business of hospital care under Affordable Care Act reforms. “Hospitalists will be the ones senior hospital leaders come to for help in figuring out healthcare reform,” he says.
Dr. Singer says the award recognizes his success, as a physician, and as an entrepreneur, and is a "signpost for hospital medicine as a whole. They're recognizing HM not just as a medical specialty that has emerged in recent years, but our success as a business," he says. "HM is a business, at least as we practice it at IPC. We've shown that it can be profitable as a standalone medical service, without requiring subsidization."