User login
Urban Legends

Registration for the Focused Practice in Hospital Medicine (FPHM) Maintenance of Certification (MOC) through the American Board of Internal Medicine (ABIM) opened March 15. Since then, hundreds of board-certified IM physicians have registered to complete their MOC through the FPHM. For those of you who haven’t gone through MOC yet, it is required every 10 years in order to maintain your board certification.
As a member of the committee tasked with helping ABIM develop the FPHM, as well as write the FPHM examination, I’m frequently asked questions about this process, especially since FPHM was featured on the May 2010 cover of this magazine. Some of the questions stem from the perplexity associated with this significant change to the MOC process. Others arise from misinformation and apprehension, and could rightly be called urban legends. Here is a sample of those questions, with their respective veracity.
Does This Certification Mean I’ll No Longer be an Internist?
No. I clearly remember the day I found out I passed the ABIM certification exam; it represented the culmination of years of work, the pinnacle. All those long hours of study, late-night admissions, and exam preparation had finally paid off. I was a board-certified internist! I cherish my board certification and hold it out as recognition of my mastery of the field of IM. As such, I certainly understand the concern that entering the FPHM will somehow result in “losing” IM certification. However, this just isn’t true.
First, all diplomates—ABIM terminology for those enrolled in the board (re)certification process—in the FPHM are certified as internists. This is simply, as the name suggests, recognition of focused practice in HM—the core certification is still in IM. We are still internists—just internists who have focused our practice to hospital care. The formal board designation will read: ABIM Board Certified in Internal Medicine with a Focused Practice in Hospital Medicine.
Will Hospital Credentialing Boards Recognize This Certification?
Yes. The FPHM is certification in IM by the ABIM. This carries the same weight and meaning as the regular IM MOC. All credentialing boards that recognize the ABIM MOC in IM will recognize the FPHM.
Is the FPHM MOC More Rigorous Than the Regular IM MOC?
Yes, and it was intentional. It is recognized that hospitalists do things that make them “special” by acquiring and refining skills learned experientially outside of a supervised training program. Thus, one can attain FPHM board designation only after three years of practice as a hospitalist. The problem is that this is largely unsupervised time (unlike a fellowship), so the threshold to ensure we have achieved a level of competency has to be established through the MOC process.
As such, the bar for the MOC for FPHM has been set higher. The upshot is that to maintain designation of FPHM, hospitalists are required to achieve 60 self-evaluation points every three years, compared with 100 points every 10 years for IM MOC. Forty of those 60 points must come in the form of Practice Improvement Modules, or PIMs. While more rigorous, it only makes sense that a group committed to the improvement of healthcare quality would commit to higher levels of quality assurance.
Will the FPHM Confer Subspecialty Status to Hospitalists?
No. This is where there has been the greatest deal of controversy surrounding the FPHM program. FPHM is not a subspecialty certification. Rather, it is MOC for IM physicians who focus their practice in the hospital setting. The American Board of Medical Specialties (ABMS), the group that oversees the ABIM, is very clear that only training-based subspecialties can be deemed board-certified subspecialties.
Still, the new FPHM will differentiate hospitalists from nonhospitalists. To me, this is semantics, and the recognition that comes with the FPHM is enough to recognize what I do as “special.”
Will the Examination for the FPHM Reflect What I Do?
Yes. I’ve had several colleagues tell me they’ve heard that the examination will contain elements not found on the standard MOC test. This is true, but it also is the major benefit of the new test. As reported on the ABIM website, the FPHM MOC exam will consist of roughly 45% inpatient medicine, 15% consultative and comanagement work, 15% transitions and ambulatory medicine, 15% patient safety/quality improvement, 5% epidemiology, and 5% ethics and end-of-life care. In this respect, it better reflects what most of us spend most of our time doing—inpatient care, consultative medicine, and transitions of care. Those three areas comprise nearly 75% of the examination. The one area that is new is the focus on patient safety and quality. However, for a field built on the promise of improving the quality, safety, and efficiency of healthcare, this is a welcome change.
Will I Be Able to Prepare for the FPHM Exam?
Yes. Because the FPHM utilizes a different exam than the standard IM MOC, some physicians are concerned that they there are no avenues for preparation, but this is not true. While the exam will weigh various portions of the test differently (e.g., less ambulatory and more inpatient content), the inpatient content will be similar to what is currently on the standard MOC test. After all, heart failure, cellulitis, and pulmonary embolism are the same, regardless of the test it shows up on.
The difference is that there will be more of it—a good thing for the practicing hospitalist. Many of the standard IM test preparation options will help you prepare for the FPHM exam. The ABIM also offers HM knowledge modules as part of the enrollment fee, which, while not meant to be preparation for the exam, can help you identify gaps in your knowledge. The blueprint for the exam is on the ABIM website and can give you clues to areas in which to prepare.
It is true that there will be patient safety and QI content on the HM test, which might not be available for study in typical board review books. However, much of this is the kind of information hospitalists live every day, such as handoffs, transitions of care, and infection control.
Should I Fear the Change?
Humans are intrinsically wired to dislike change. It’s that old saw about choosing the devil we know rather than the devil we don’t. My guess is that much of the concern around the new process stems from this human sentiment—it’s just easier to not do the FPHM and go down the standard IM MOC route. We must avoid this temptation.
The FPHM is a key step in solidifying HM’s status in the healthcare milieu. It gives us credibility in a way no other designation can. It also allows those of us who are serious about an HM career to differentiate ourselves from those who masquerade as hospitalists. And, most importantly, it allows us to demonstrate to our patients our sincere commitment to improving the quality of the inpatient systems that envelop them at their sickest moments.
For that reason alone, I’m enrolling in the FPHM MOC. TH
Dr. Glasheen is physician editor of The Hospitalist.
Registration for the Focused Practice in Hospital Medicine (FPHM) Maintenance of Certification (MOC) through the American Board of Internal Medicine (ABIM) opened March 15. Since then, hundreds of board-certified IM physicians have registered to complete their MOC through the FPHM. For those of you who haven’t gone through MOC yet, it is required every 10 years in order to maintain your board certification.
As a member of the committee tasked with helping ABIM develop the FPHM, as well as write the FPHM examination, I’m frequently asked questions about this process, especially since FPHM was featured on the May 2010 cover of this magazine. Some of the questions stem from the perplexity associated with this significant change to the MOC process. Others arise from misinformation and apprehension, and could rightly be called urban legends. Here is a sample of those questions, with their respective veracity.
Does This Certification Mean I’ll No Longer be an Internist?
No. I clearly remember the day I found out I passed the ABIM certification exam; it represented the culmination of years of work, the pinnacle. All those long hours of study, late-night admissions, and exam preparation had finally paid off. I was a board-certified internist! I cherish my board certification and hold it out as recognition of my mastery of the field of IM. As such, I certainly understand the concern that entering the FPHM will somehow result in “losing” IM certification. However, this just isn’t true.
First, all diplomates—ABIM terminology for those enrolled in the board (re)certification process—in the FPHM are certified as internists. This is simply, as the name suggests, recognition of focused practice in HM—the core certification is still in IM. We are still internists—just internists who have focused our practice to hospital care. The formal board designation will read: ABIM Board Certified in Internal Medicine with a Focused Practice in Hospital Medicine.
Will Hospital Credentialing Boards Recognize This Certification?
Yes. The FPHM is certification in IM by the ABIM. This carries the same weight and meaning as the regular IM MOC. All credentialing boards that recognize the ABIM MOC in IM will recognize the FPHM.
Is the FPHM MOC More Rigorous Than the Regular IM MOC?
Yes, and it was intentional. It is recognized that hospitalists do things that make them “special” by acquiring and refining skills learned experientially outside of a supervised training program. Thus, one can attain FPHM board designation only after three years of practice as a hospitalist. The problem is that this is largely unsupervised time (unlike a fellowship), so the threshold to ensure we have achieved a level of competency has to be established through the MOC process.
As such, the bar for the MOC for FPHM has been set higher. The upshot is that to maintain designation of FPHM, hospitalists are required to achieve 60 self-evaluation points every three years, compared with 100 points every 10 years for IM MOC. Forty of those 60 points must come in the form of Practice Improvement Modules, or PIMs. While more rigorous, it only makes sense that a group committed to the improvement of healthcare quality would commit to higher levels of quality assurance.
Will the FPHM Confer Subspecialty Status to Hospitalists?
No. This is where there has been the greatest deal of controversy surrounding the FPHM program. FPHM is not a subspecialty certification. Rather, it is MOC for IM physicians who focus their practice in the hospital setting. The American Board of Medical Specialties (ABMS), the group that oversees the ABIM, is very clear that only training-based subspecialties can be deemed board-certified subspecialties.
Still, the new FPHM will differentiate hospitalists from nonhospitalists. To me, this is semantics, and the recognition that comes with the FPHM is enough to recognize what I do as “special.”
Will the Examination for the FPHM Reflect What I Do?
Yes. I’ve had several colleagues tell me they’ve heard that the examination will contain elements not found on the standard MOC test. This is true, but it also is the major benefit of the new test. As reported on the ABIM website, the FPHM MOC exam will consist of roughly 45% inpatient medicine, 15% consultative and comanagement work, 15% transitions and ambulatory medicine, 15% patient safety/quality improvement, 5% epidemiology, and 5% ethics and end-of-life care. In this respect, it better reflects what most of us spend most of our time doing—inpatient care, consultative medicine, and transitions of care. Those three areas comprise nearly 75% of the examination. The one area that is new is the focus on patient safety and quality. However, for a field built on the promise of improving the quality, safety, and efficiency of healthcare, this is a welcome change.
Will I Be Able to Prepare for the FPHM Exam?
Yes. Because the FPHM utilizes a different exam than the standard IM MOC, some physicians are concerned that they there are no avenues for preparation, but this is not true. While the exam will weigh various portions of the test differently (e.g., less ambulatory and more inpatient content), the inpatient content will be similar to what is currently on the standard MOC test. After all, heart failure, cellulitis, and pulmonary embolism are the same, regardless of the test it shows up on.
The difference is that there will be more of it—a good thing for the practicing hospitalist. Many of the standard IM test preparation options will help you prepare for the FPHM exam. The ABIM also offers HM knowledge modules as part of the enrollment fee, which, while not meant to be preparation for the exam, can help you identify gaps in your knowledge. The blueprint for the exam is on the ABIM website and can give you clues to areas in which to prepare.
It is true that there will be patient safety and QI content on the HM test, which might not be available for study in typical board review books. However, much of this is the kind of information hospitalists live every day, such as handoffs, transitions of care, and infection control.
Should I Fear the Change?
Humans are intrinsically wired to dislike change. It’s that old saw about choosing the devil we know rather than the devil we don’t. My guess is that much of the concern around the new process stems from this human sentiment—it’s just easier to not do the FPHM and go down the standard IM MOC route. We must avoid this temptation.
The FPHM is a key step in solidifying HM’s status in the healthcare milieu. It gives us credibility in a way no other designation can. It also allows those of us who are serious about an HM career to differentiate ourselves from those who masquerade as hospitalists. And, most importantly, it allows us to demonstrate to our patients our sincere commitment to improving the quality of the inpatient systems that envelop them at their sickest moments.
For that reason alone, I’m enrolling in the FPHM MOC. TH
Dr. Glasheen is physician editor of The Hospitalist.
Registration for the Focused Practice in Hospital Medicine (FPHM) Maintenance of Certification (MOC) through the American Board of Internal Medicine (ABIM) opened March 15. Since then, hundreds of board-certified IM physicians have registered to complete their MOC through the FPHM. For those of you who haven’t gone through MOC yet, it is required every 10 years in order to maintain your board certification.
As a member of the committee tasked with helping ABIM develop the FPHM, as well as write the FPHM examination, I’m frequently asked questions about this process, especially since FPHM was featured on the May 2010 cover of this magazine. Some of the questions stem from the perplexity associated with this significant change to the MOC process. Others arise from misinformation and apprehension, and could rightly be called urban legends. Here is a sample of those questions, with their respective veracity.
Does This Certification Mean I’ll No Longer be an Internist?
No. I clearly remember the day I found out I passed the ABIM certification exam; it represented the culmination of years of work, the pinnacle. All those long hours of study, late-night admissions, and exam preparation had finally paid off. I was a board-certified internist! I cherish my board certification and hold it out as recognition of my mastery of the field of IM. As such, I certainly understand the concern that entering the FPHM will somehow result in “losing” IM certification. However, this just isn’t true.
First, all diplomates—ABIM terminology for those enrolled in the board (re)certification process—in the FPHM are certified as internists. This is simply, as the name suggests, recognition of focused practice in HM—the core certification is still in IM. We are still internists—just internists who have focused our practice to hospital care. The formal board designation will read: ABIM Board Certified in Internal Medicine with a Focused Practice in Hospital Medicine.
Will Hospital Credentialing Boards Recognize This Certification?
Yes. The FPHM is certification in IM by the ABIM. This carries the same weight and meaning as the regular IM MOC. All credentialing boards that recognize the ABIM MOC in IM will recognize the FPHM.
Is the FPHM MOC More Rigorous Than the Regular IM MOC?
Yes, and it was intentional. It is recognized that hospitalists do things that make them “special” by acquiring and refining skills learned experientially outside of a supervised training program. Thus, one can attain FPHM board designation only after three years of practice as a hospitalist. The problem is that this is largely unsupervised time (unlike a fellowship), so the threshold to ensure we have achieved a level of competency has to be established through the MOC process.
As such, the bar for the MOC for FPHM has been set higher. The upshot is that to maintain designation of FPHM, hospitalists are required to achieve 60 self-evaluation points every three years, compared with 100 points every 10 years for IM MOC. Forty of those 60 points must come in the form of Practice Improvement Modules, or PIMs. While more rigorous, it only makes sense that a group committed to the improvement of healthcare quality would commit to higher levels of quality assurance.
Will the FPHM Confer Subspecialty Status to Hospitalists?
No. This is where there has been the greatest deal of controversy surrounding the FPHM program. FPHM is not a subspecialty certification. Rather, it is MOC for IM physicians who focus their practice in the hospital setting. The American Board of Medical Specialties (ABMS), the group that oversees the ABIM, is very clear that only training-based subspecialties can be deemed board-certified subspecialties.
Still, the new FPHM will differentiate hospitalists from nonhospitalists. To me, this is semantics, and the recognition that comes with the FPHM is enough to recognize what I do as “special.”
Will the Examination for the FPHM Reflect What I Do?
Yes. I’ve had several colleagues tell me they’ve heard that the examination will contain elements not found on the standard MOC test. This is true, but it also is the major benefit of the new test. As reported on the ABIM website, the FPHM MOC exam will consist of roughly 45% inpatient medicine, 15% consultative and comanagement work, 15% transitions and ambulatory medicine, 15% patient safety/quality improvement, 5% epidemiology, and 5% ethics and end-of-life care. In this respect, it better reflects what most of us spend most of our time doing—inpatient care, consultative medicine, and transitions of care. Those three areas comprise nearly 75% of the examination. The one area that is new is the focus on patient safety and quality. However, for a field built on the promise of improving the quality, safety, and efficiency of healthcare, this is a welcome change.
Will I Be Able to Prepare for the FPHM Exam?
Yes. Because the FPHM utilizes a different exam than the standard IM MOC, some physicians are concerned that they there are no avenues for preparation, but this is not true. While the exam will weigh various portions of the test differently (e.g., less ambulatory and more inpatient content), the inpatient content will be similar to what is currently on the standard MOC test. After all, heart failure, cellulitis, and pulmonary embolism are the same, regardless of the test it shows up on.
The difference is that there will be more of it—a good thing for the practicing hospitalist. Many of the standard IM test preparation options will help you prepare for the FPHM exam. The ABIM also offers HM knowledge modules as part of the enrollment fee, which, while not meant to be preparation for the exam, can help you identify gaps in your knowledge. The blueprint for the exam is on the ABIM website and can give you clues to areas in which to prepare.
It is true that there will be patient safety and QI content on the HM test, which might not be available for study in typical board review books. However, much of this is the kind of information hospitalists live every day, such as handoffs, transitions of care, and infection control.
Should I Fear the Change?
Humans are intrinsically wired to dislike change. It’s that old saw about choosing the devil we know rather than the devil we don’t. My guess is that much of the concern around the new process stems from this human sentiment—it’s just easier to not do the FPHM and go down the standard IM MOC route. We must avoid this temptation.
The FPHM is a key step in solidifying HM’s status in the healthcare milieu. It gives us credibility in a way no other designation can. It also allows those of us who are serious about an HM career to differentiate ourselves from those who masquerade as hospitalists. And, most importantly, it allows us to demonstrate to our patients our sincere commitment to improving the quality of the inpatient systems that envelop them at their sickest moments.
For that reason alone, I’m enrolling in the FPHM MOC. TH
Dr. Glasheen is physician editor of The Hospitalist.
ONLINE EXCLUSIVE: Audio interviews with transitions of care experts
ONLINE EXCLUSIVE: Simulator Training Program Aims to Improve Hospital Handoffs
Few medical students receive formal training in how to perform patient handoffs effectively and efficiently, says Vineet Arora, MD, FHM, who chairs an SHM task force on improving handoffs. Most pick it up on the job, but Dr. Arora and her colleagues at the University of Chicago—where she is associate director of the internal-medicine residency program—and at the University of Michigan have been exploring ways to improve the process through education.1 Handoffs and care transitions are a major focus for hospital quality-improvement (QI) efforts nationally.
Dr. Arora’s group created an “observed simulation handoff experience” for medical students and residents, “offering an air of authenticity to the experience without the high-risk environment of learning on live patients,” she explains. Students who have completed an interactive training session perform the simulation at a computer station. They are provided with two types of information: static data about the mock patient—including such information as diagnosis, primary-care physician, and code status from a history and physical report—and video clips offering “a virtual, real-time barrage of constant updates” about the patient’s changing clinical status.
“They are given some time to extract and synthesize the important data for a handoff, and then they go in and perform the handoff in person to a ‘standardized receiver,’ ” Dr. Arora explains. The receiver is a resident or other clinician familiar both with the case and how the handoff should go, and who then provides a standardized evaluation, grade, and feedback.
“The goal is to teach students the triggers that need to be incorporated into an effective handoff,” Dr. Arora says. Her group also reviewed concepts of good handoffs from the medical literature and worked with a psychology expert in human communication.
“How do you teach people to give good handoffs? We don’t know all of the answers, but we think you have to start somewhere,” she says. “Technology can be a great facilitator to make handoffs go better. It’s not a perfect substitute for face-to-face, interactive handoffs, but it can dramatically inform care transitions.”
Larry Beresford is a freelance writer based in Oakland, Calif.
1. Farnan JM, Paro JA, Rodriguez RM, et al. Hand-off education and evaluation: piloting the observed simulated hand-off experience (OSHE). J Gen Intern Med. 2010;25(2):129-134.
Few medical students receive formal training in how to perform patient handoffs effectively and efficiently, says Vineet Arora, MD, FHM, who chairs an SHM task force on improving handoffs. Most pick it up on the job, but Dr. Arora and her colleagues at the University of Chicago—where she is associate director of the internal-medicine residency program—and at the University of Michigan have been exploring ways to improve the process through education.1 Handoffs and care transitions are a major focus for hospital quality-improvement (QI) efforts nationally.
Dr. Arora’s group created an “observed simulation handoff experience” for medical students and residents, “offering an air of authenticity to the experience without the high-risk environment of learning on live patients,” she explains. Students who have completed an interactive training session perform the simulation at a computer station. They are provided with two types of information: static data about the mock patient—including such information as diagnosis, primary-care physician, and code status from a history and physical report—and video clips offering “a virtual, real-time barrage of constant updates” about the patient’s changing clinical status.
“They are given some time to extract and synthesize the important data for a handoff, and then they go in and perform the handoff in person to a ‘standardized receiver,’ ” Dr. Arora explains. The receiver is a resident or other clinician familiar both with the case and how the handoff should go, and who then provides a standardized evaluation, grade, and feedback.
“The goal is to teach students the triggers that need to be incorporated into an effective handoff,” Dr. Arora says. Her group also reviewed concepts of good handoffs from the medical literature and worked with a psychology expert in human communication.
“How do you teach people to give good handoffs? We don’t know all of the answers, but we think you have to start somewhere,” she says. “Technology can be a great facilitator to make handoffs go better. It’s not a perfect substitute for face-to-face, interactive handoffs, but it can dramatically inform care transitions.”
Larry Beresford is a freelance writer based in Oakland, Calif.
1. Farnan JM, Paro JA, Rodriguez RM, et al. Hand-off education and evaluation: piloting the observed simulated hand-off experience (OSHE). J Gen Intern Med. 2010;25(2):129-134.
Few medical students receive formal training in how to perform patient handoffs effectively and efficiently, says Vineet Arora, MD, FHM, who chairs an SHM task force on improving handoffs. Most pick it up on the job, but Dr. Arora and her colleagues at the University of Chicago—where she is associate director of the internal-medicine residency program—and at the University of Michigan have been exploring ways to improve the process through education.1 Handoffs and care transitions are a major focus for hospital quality-improvement (QI) efforts nationally.
Dr. Arora’s group created an “observed simulation handoff experience” for medical students and residents, “offering an air of authenticity to the experience without the high-risk environment of learning on live patients,” she explains. Students who have completed an interactive training session perform the simulation at a computer station. They are provided with two types of information: static data about the mock patient—including such information as diagnosis, primary-care physician, and code status from a history and physical report—and video clips offering “a virtual, real-time barrage of constant updates” about the patient’s changing clinical status.
“They are given some time to extract and synthesize the important data for a handoff, and then they go in and perform the handoff in person to a ‘standardized receiver,’ ” Dr. Arora explains. The receiver is a resident or other clinician familiar both with the case and how the handoff should go, and who then provides a standardized evaluation, grade, and feedback.
“The goal is to teach students the triggers that need to be incorporated into an effective handoff,” Dr. Arora says. Her group also reviewed concepts of good handoffs from the medical literature and worked with a psychology expert in human communication.
“How do you teach people to give good handoffs? We don’t know all of the answers, but we think you have to start somewhere,” she says. “Technology can be a great facilitator to make handoffs go better. It’s not a perfect substitute for face-to-face, interactive handoffs, but it can dramatically inform care transitions.”
Larry Beresford is a freelance writer based in Oakland, Calif.
1. Farnan JM, Paro JA, Rodriguez RM, et al. Hand-off education and evaluation: piloting the observed simulated hand-off experience (OSHE). J Gen Intern Med. 2010;25(2):129-134.
Proceedings of the 2009 Heart-Brain Summit

Supplement Editor:
Marc S. Penn, MD, PhD
Contents
Introduction: Heart-brain medicine: Update 2009
Marc S. Penn, MD, PhD, and Earl E. Bakken, MD (HonC), DSc (3 Hon), DHL (2 Hon)
Depression and Heart Disease
Depression and heart failure: An overview of what we know and don't know
Marc A. Silver, MD
The American Heart Association science advisory on depression and coronary heart disease: An exploration of the issues raised
J. Thomas Bigger, MD, and Alexander H. Glassman, MD
Depression and cardiovascular disease: Selected findings, controversies, and clinical implications from 2009
Karina W. Davidson, PhD, and Maya Rom Korin, PhD
Pioneer Lecture
Recovery of consciousness after severe brain injury: The role of arousal regulation mechanisms and some speculation on the heart-brain interface
Nicholas D. Schiff, MD
Heart-Brain Medicine Publications: The Year in Review
Neuroscience and heart-brain medicine: The year in review
David S. Goldstein, MD, PhD
Pathophysiologic mechanisms linking impaired cardiovascular health and neurologic dysfunction: The year in review
Ki E. Park, MD, and Carl J. Pepine, MD
Biomedical engineering in heart-brain medicine: A review
Peter G. Katona, ScD
Novel Findings in Heart-Brain Medicine
Sudden death in epilepsy, surgery, and seizure outcomes: The interface between heart and brain
Lara Jehi, MD
Biofeedback in the Treatment of Disease
Biofeedback in the treatment of heart failure
Michael G. McKee, PhD, and Christine S. Moravec, PhD
Biofeedback in the treatment of epilepsy
M. Barry Sterman, PhD
The effects of biofeedback in diabetes and essential hypertension
Angele McGrady, PhD, MEd, LPCC
Biofeedback in headache: An overview of approaches and evidence
Frank Andrasik, PhD
Device-Based Therapies
Use of deep brain stimulation in treatment-resistant depression
Donald A. Malone, Jr, MD
Poster Abstracts
Abstract 1: Potential role of the cardiac protease corin in energy metabolism
Jingjing Jiang, Yujie Cui, Wei Wang, and Qingyu Wu
Abstract 2: Anxiety and type D personality in ICD patients: Impact of shocks
Mina K. Chung, MD; Melanie Panko, RN; Tina Gupta; Scott Bea, PhD; Karen Broer, PhD; Diana Bauer; Denise Kosty-Sweeney, RN; Betty Ching, RN; Suzanne Pedersen, PhD; Sam Sears, PhD; and Leo Pozuelo, MD
Abstract 3: Microglia activation and neuroprotection during CNS preconditioning
Walid Jalabi, Ranjan Dutta, Yongming Jin, Gerson Criste, Xinghua Yin, Grahame J. Kidd, and Bruce D. Trapp
Abstract 4: Brain MRI correlates of atrial fibrillation
Stephen E. Jones, MD, PhD; Thomas Callahan, MD; Kamal Chémali, MD; Michael Phillips, MD; David Van Wagoner, PhD; and Walid Saliba, MD
Abstract 5: Identification and characterization of autonomic dysfunction in migraineurs with and without auras: Phase I
Mark Stillman, MD
Abstract 6: Sudden unexpected death in epilepsy: Finding the missing cardiac links
Lara Jehi, MD; Kanjana Unnongswe, MD; Thomas Callahan, MD; Liang Li, PhD; and Imad Najm, MD
Abstract 7: Cardiomyopathy after subarachnoid hemorrhage is mediated by neutrophils
J. Javier Provencio, Shari Moore, and Saksith Smithason
Abstract 8: Mindfulness, yoga, and cardiovascular disease
Didier Allexandre, Emily Fox, Mladen Golubic, Tom Morledge, and Joan E.B. Fox
Abstract 9: Multidisciplinary research in biofeedback
Christine S. Moravec, PhD; Michael G. McKee, PhD; James B. Young, MD; Betul Hatipoglu, MD; Leopoldo Pozuelo, MD; Leslie Cho, MD; Gordon Blackburn, MD; Francois Bethoux, MD; Mary Rensel, MD; Katherine Hoercher, RN; J. Javier Provencio, MD; and Marc S. Penn, MD
Abstract 10: Complex regional pain syndrome (CRPS I): A systemic disease of the autonomic nervous system
Kamal Chemali, MD; Robert Shields, MD; Lan Zhou, MD, PhD; Salim Hayek, MD, PhD; and Thomas Chelimsky, MD
Abstract 11: Biofeedback in the treatment of heart failure
Dana L. Frank, BA; Lamees Khorshid, PsyD; Jerome Kiffer, MA; Christine S. Moravec, PhD; and Michael G. McKee, PhD
Abstract 12: Change in depressive symptom status predicts health-related quality of life in patients with heart failure
Rebecca L. Dekker, MSN, RN, PhD candidate; Terry A. Lennie, PhD, RN; Nancy Albert, PhD, CCNS; Barbara Riegel, DNSc, RN; Misook L. Chung, PhD, RN; Seongkum Heo, PhD, RN; Eun Kyeung Song, PhD, RN; Jia-Rong Wu, PhD, RN; and Debra K. Moser, DNSc, RN
Abstract 13: Entropy of EKG time series distinguishes epileptic from nonepileptic patients
Rebecca O’Dwyer, Ulrich Zurcher, Brian Vyhnalek, Miron Kaufman, and Richard Burgess
Abstract 14: Evaluation of cardiac autonomic balance in major depression treated with different antidepressant therapies: A study with heart rate variability measures
K. Udupa, K.R. Kishore, J. Thirthalli, B.N. Gangadhar, T.R. Raju, and T.N. Sathyaprabha
Abstract 15: Proinflammatory status in major depression: Effects of escitalopram
John Piletz, PhD; Angelos Halaris, MD; Erin Tobin, MS; Edwin Meresh, MD; Jawed Fareed, PhD; Omer Iqbal, MD; Debra Hoppenstead, PhD; and James Sinacore, PhD
Abstract 16: Heart rate variability in depression: Effect of escitalopram
Angelos Halaris, MD; John Piletz, PhD; Erin Tobin, MA; Edwin Meresh, MD; James Sinacore, PhD; and Christopher Lowden
Abstract 17: Effects of omega-3/6 dietary ratio variation after a myocardial infarction in a rat model
Guy Rousseau, Isabelle Rondeau, Sandrine Picard, Thierno Madjou Bah, Louis Roy, and Roger Godbout
Abstract 18: The effects of tai chi on the heart and the brain
Qian Luo, Xi Cheng, and Xi Zha
Abstract 19: A randomized controlled trial of the effect of hostility reduction on cardiac autonomic regulation
Richard P. Sloan, PhD; Peter A. Shapiro, MD; Ethan E. Gorenstein, PhD; Felice A. Tager, PhD; Catherine E. Monk, PhD; Paula S. McKinley, PhD; Michael M. Myers, PhD; Emilia Bagiella, PhD; Ivy Chen, MST; Richard Steinman, BA; and J. Thomas Bigger, Jr., MD
Supplement Editor:
Marc S. Penn, MD, PhD
Contents
Introduction: Heart-brain medicine: Update 2009
Marc S. Penn, MD, PhD, and Earl E. Bakken, MD (HonC), DSc (3 Hon), DHL (2 Hon)
Depression and Heart Disease
Depression and heart failure: An overview of what we know and don't know
Marc A. Silver, MD
The American Heart Association science advisory on depression and coronary heart disease: An exploration of the issues raised
J. Thomas Bigger, MD, and Alexander H. Glassman, MD
Depression and cardiovascular disease: Selected findings, controversies, and clinical implications from 2009
Karina W. Davidson, PhD, and Maya Rom Korin, PhD
Pioneer Lecture
Recovery of consciousness after severe brain injury: The role of arousal regulation mechanisms and some speculation on the heart-brain interface
Nicholas D. Schiff, MD
Heart-Brain Medicine Publications: The Year in Review
Neuroscience and heart-brain medicine: The year in review
David S. Goldstein, MD, PhD
Pathophysiologic mechanisms linking impaired cardiovascular health and neurologic dysfunction: The year in review
Ki E. Park, MD, and Carl J. Pepine, MD
Biomedical engineering in heart-brain medicine: A review
Peter G. Katona, ScD
Novel Findings in Heart-Brain Medicine
Sudden death in epilepsy, surgery, and seizure outcomes: The interface between heart and brain
Lara Jehi, MD
Biofeedback in the Treatment of Disease
Biofeedback in the treatment of heart failure
Michael G. McKee, PhD, and Christine S. Moravec, PhD
Biofeedback in the treatment of epilepsy
M. Barry Sterman, PhD
The effects of biofeedback in diabetes and essential hypertension
Angele McGrady, PhD, MEd, LPCC
Biofeedback in headache: An overview of approaches and evidence
Frank Andrasik, PhD
Device-Based Therapies
Use of deep brain stimulation in treatment-resistant depression
Donald A. Malone, Jr, MD
Poster Abstracts
Abstract 1: Potential role of the cardiac protease corin in energy metabolism
Jingjing Jiang, Yujie Cui, Wei Wang, and Qingyu Wu
Abstract 2: Anxiety and type D personality in ICD patients: Impact of shocks
Mina K. Chung, MD; Melanie Panko, RN; Tina Gupta; Scott Bea, PhD; Karen Broer, PhD; Diana Bauer; Denise Kosty-Sweeney, RN; Betty Ching, RN; Suzanne Pedersen, PhD; Sam Sears, PhD; and Leo Pozuelo, MD
Abstract 3: Microglia activation and neuroprotection during CNS preconditioning
Walid Jalabi, Ranjan Dutta, Yongming Jin, Gerson Criste, Xinghua Yin, Grahame J. Kidd, and Bruce D. Trapp
Abstract 4: Brain MRI correlates of atrial fibrillation
Stephen E. Jones, MD, PhD; Thomas Callahan, MD; Kamal Chémali, MD; Michael Phillips, MD; David Van Wagoner, PhD; and Walid Saliba, MD
Abstract 5: Identification and characterization of autonomic dysfunction in migraineurs with and without auras: Phase I
Mark Stillman, MD
Abstract 6: Sudden unexpected death in epilepsy: Finding the missing cardiac links
Lara Jehi, MD; Kanjana Unnongswe, MD; Thomas Callahan, MD; Liang Li, PhD; and Imad Najm, MD
Abstract 7: Cardiomyopathy after subarachnoid hemorrhage is mediated by neutrophils
J. Javier Provencio, Shari Moore, and Saksith Smithason
Abstract 8: Mindfulness, yoga, and cardiovascular disease
Didier Allexandre, Emily Fox, Mladen Golubic, Tom Morledge, and Joan E.B. Fox
Abstract 9: Multidisciplinary research in biofeedback
Christine S. Moravec, PhD; Michael G. McKee, PhD; James B. Young, MD; Betul Hatipoglu, MD; Leopoldo Pozuelo, MD; Leslie Cho, MD; Gordon Blackburn, MD; Francois Bethoux, MD; Mary Rensel, MD; Katherine Hoercher, RN; J. Javier Provencio, MD; and Marc S. Penn, MD
Abstract 10: Complex regional pain syndrome (CRPS I): A systemic disease of the autonomic nervous system
Kamal Chemali, MD; Robert Shields, MD; Lan Zhou, MD, PhD; Salim Hayek, MD, PhD; and Thomas Chelimsky, MD
Abstract 11: Biofeedback in the treatment of heart failure
Dana L. Frank, BA; Lamees Khorshid, PsyD; Jerome Kiffer, MA; Christine S. Moravec, PhD; and Michael G. McKee, PhD
Abstract 12: Change in depressive symptom status predicts health-related quality of life in patients with heart failure
Rebecca L. Dekker, MSN, RN, PhD candidate; Terry A. Lennie, PhD, RN; Nancy Albert, PhD, CCNS; Barbara Riegel, DNSc, RN; Misook L. Chung, PhD, RN; Seongkum Heo, PhD, RN; Eun Kyeung Song, PhD, RN; Jia-Rong Wu, PhD, RN; and Debra K. Moser, DNSc, RN
Abstract 13: Entropy of EKG time series distinguishes epileptic from nonepileptic patients
Rebecca O’Dwyer, Ulrich Zurcher, Brian Vyhnalek, Miron Kaufman, and Richard Burgess
Abstract 14: Evaluation of cardiac autonomic balance in major depression treated with different antidepressant therapies: A study with heart rate variability measures
K. Udupa, K.R. Kishore, J. Thirthalli, B.N. Gangadhar, T.R. Raju, and T.N. Sathyaprabha
Abstract 15: Proinflammatory status in major depression: Effects of escitalopram
John Piletz, PhD; Angelos Halaris, MD; Erin Tobin, MS; Edwin Meresh, MD; Jawed Fareed, PhD; Omer Iqbal, MD; Debra Hoppenstead, PhD; and James Sinacore, PhD
Abstract 16: Heart rate variability in depression: Effect of escitalopram
Angelos Halaris, MD; John Piletz, PhD; Erin Tobin, MA; Edwin Meresh, MD; James Sinacore, PhD; and Christopher Lowden
Abstract 17: Effects of omega-3/6 dietary ratio variation after a myocardial infarction in a rat model
Guy Rousseau, Isabelle Rondeau, Sandrine Picard, Thierno Madjou Bah, Louis Roy, and Roger Godbout
Abstract 18: The effects of tai chi on the heart and the brain
Qian Luo, Xi Cheng, and Xi Zha
Abstract 19: A randomized controlled trial of the effect of hostility reduction on cardiac autonomic regulation
Richard P. Sloan, PhD; Peter A. Shapiro, MD; Ethan E. Gorenstein, PhD; Felice A. Tager, PhD; Catherine E. Monk, PhD; Paula S. McKinley, PhD; Michael M. Myers, PhD; Emilia Bagiella, PhD; Ivy Chen, MST; Richard Steinman, BA; and J. Thomas Bigger, Jr., MD
Supplement Editor:
Marc S. Penn, MD, PhD
Contents
Introduction: Heart-brain medicine: Update 2009
Marc S. Penn, MD, PhD, and Earl E. Bakken, MD (HonC), DSc (3 Hon), DHL (2 Hon)
Depression and Heart Disease
Depression and heart failure: An overview of what we know and don't know
Marc A. Silver, MD
The American Heart Association science advisory on depression and coronary heart disease: An exploration of the issues raised
J. Thomas Bigger, MD, and Alexander H. Glassman, MD
Depression and cardiovascular disease: Selected findings, controversies, and clinical implications from 2009
Karina W. Davidson, PhD, and Maya Rom Korin, PhD
Pioneer Lecture
Recovery of consciousness after severe brain injury: The role of arousal regulation mechanisms and some speculation on the heart-brain interface
Nicholas D. Schiff, MD
Heart-Brain Medicine Publications: The Year in Review
Neuroscience and heart-brain medicine: The year in review
David S. Goldstein, MD, PhD
Pathophysiologic mechanisms linking impaired cardiovascular health and neurologic dysfunction: The year in review
Ki E. Park, MD, and Carl J. Pepine, MD
Biomedical engineering in heart-brain medicine: A review
Peter G. Katona, ScD
Novel Findings in Heart-Brain Medicine
Sudden death in epilepsy, surgery, and seizure outcomes: The interface between heart and brain
Lara Jehi, MD
Biofeedback in the Treatment of Disease
Biofeedback in the treatment of heart failure
Michael G. McKee, PhD, and Christine S. Moravec, PhD
Biofeedback in the treatment of epilepsy
M. Barry Sterman, PhD
The effects of biofeedback in diabetes and essential hypertension
Angele McGrady, PhD, MEd, LPCC
Biofeedback in headache: An overview of approaches and evidence
Frank Andrasik, PhD
Device-Based Therapies
Use of deep brain stimulation in treatment-resistant depression
Donald A. Malone, Jr, MD
Poster Abstracts
Abstract 1: Potential role of the cardiac protease corin in energy metabolism
Jingjing Jiang, Yujie Cui, Wei Wang, and Qingyu Wu
Abstract 2: Anxiety and type D personality in ICD patients: Impact of shocks
Mina K. Chung, MD; Melanie Panko, RN; Tina Gupta; Scott Bea, PhD; Karen Broer, PhD; Diana Bauer; Denise Kosty-Sweeney, RN; Betty Ching, RN; Suzanne Pedersen, PhD; Sam Sears, PhD; and Leo Pozuelo, MD
Abstract 3: Microglia activation and neuroprotection during CNS preconditioning
Walid Jalabi, Ranjan Dutta, Yongming Jin, Gerson Criste, Xinghua Yin, Grahame J. Kidd, and Bruce D. Trapp
Abstract 4: Brain MRI correlates of atrial fibrillation
Stephen E. Jones, MD, PhD; Thomas Callahan, MD; Kamal Chémali, MD; Michael Phillips, MD; David Van Wagoner, PhD; and Walid Saliba, MD
Abstract 5: Identification and characterization of autonomic dysfunction in migraineurs with and without auras: Phase I
Mark Stillman, MD
Abstract 6: Sudden unexpected death in epilepsy: Finding the missing cardiac links
Lara Jehi, MD; Kanjana Unnongswe, MD; Thomas Callahan, MD; Liang Li, PhD; and Imad Najm, MD
Abstract 7: Cardiomyopathy after subarachnoid hemorrhage is mediated by neutrophils
J. Javier Provencio, Shari Moore, and Saksith Smithason
Abstract 8: Mindfulness, yoga, and cardiovascular disease
Didier Allexandre, Emily Fox, Mladen Golubic, Tom Morledge, and Joan E.B. Fox
Abstract 9: Multidisciplinary research in biofeedback
Christine S. Moravec, PhD; Michael G. McKee, PhD; James B. Young, MD; Betul Hatipoglu, MD; Leopoldo Pozuelo, MD; Leslie Cho, MD; Gordon Blackburn, MD; Francois Bethoux, MD; Mary Rensel, MD; Katherine Hoercher, RN; J. Javier Provencio, MD; and Marc S. Penn, MD
Abstract 10: Complex regional pain syndrome (CRPS I): A systemic disease of the autonomic nervous system
Kamal Chemali, MD; Robert Shields, MD; Lan Zhou, MD, PhD; Salim Hayek, MD, PhD; and Thomas Chelimsky, MD
Abstract 11: Biofeedback in the treatment of heart failure
Dana L. Frank, BA; Lamees Khorshid, PsyD; Jerome Kiffer, MA; Christine S. Moravec, PhD; and Michael G. McKee, PhD
Abstract 12: Change in depressive symptom status predicts health-related quality of life in patients with heart failure
Rebecca L. Dekker, MSN, RN, PhD candidate; Terry A. Lennie, PhD, RN; Nancy Albert, PhD, CCNS; Barbara Riegel, DNSc, RN; Misook L. Chung, PhD, RN; Seongkum Heo, PhD, RN; Eun Kyeung Song, PhD, RN; Jia-Rong Wu, PhD, RN; and Debra K. Moser, DNSc, RN
Abstract 13: Entropy of EKG time series distinguishes epileptic from nonepileptic patients
Rebecca O’Dwyer, Ulrich Zurcher, Brian Vyhnalek, Miron Kaufman, and Richard Burgess
Abstract 14: Evaluation of cardiac autonomic balance in major depression treated with different antidepressant therapies: A study with heart rate variability measures
K. Udupa, K.R. Kishore, J. Thirthalli, B.N. Gangadhar, T.R. Raju, and T.N. Sathyaprabha
Abstract 15: Proinflammatory status in major depression: Effects of escitalopram
John Piletz, PhD; Angelos Halaris, MD; Erin Tobin, MS; Edwin Meresh, MD; Jawed Fareed, PhD; Omer Iqbal, MD; Debra Hoppenstead, PhD; and James Sinacore, PhD
Abstract 16: Heart rate variability in depression: Effect of escitalopram
Angelos Halaris, MD; John Piletz, PhD; Erin Tobin, MA; Edwin Meresh, MD; James Sinacore, PhD; and Christopher Lowden
Abstract 17: Effects of omega-3/6 dietary ratio variation after a myocardial infarction in a rat model
Guy Rousseau, Isabelle Rondeau, Sandrine Picard, Thierno Madjou Bah, Louis Roy, and Roger Godbout
Abstract 18: The effects of tai chi on the heart and the brain
Qian Luo, Xi Cheng, and Xi Zha
Abstract 19: A randomized controlled trial of the effect of hostility reduction on cardiac autonomic regulation
Richard P. Sloan, PhD; Peter A. Shapiro, MD; Ethan E. Gorenstein, PhD; Felice A. Tager, PhD; Catherine E. Monk, PhD; Paula S. McKinley, PhD; Michael M. Myers, PhD; Emilia Bagiella, PhD; Ivy Chen, MST; Richard Steinman, BA; and J. Thomas Bigger, Jr., MD

Late-onset male hypogonadism and testosterone replacement therapy in primary care
Hypogonadism is a highly prevalent condition that is under-diagnosed and under-treated despite available effective therapies. Low levels of testosterone result in losses of lean body mass and bone mass, decreases in libido and sexual function and is associated with insulin resistance, metabolic syndrome, diabetes, and other chronic comorbid conditions. It is important for physicians to be aware of how testosterone levels can be related to these conditions, which are seen in primary care practices on a daily basis.
This CME supplement discusses the definition, epidemiology and key signs and symptoms of late-onset hypogonadism, the role of lab measurements, factors to consider in selecting patients for testosterone replacement therapy, and how to identify the best treatment strategy for each patient.
Hypogonadism is a highly prevalent condition that is under-diagnosed and under-treated despite available effective therapies. Low levels of testosterone result in losses of lean body mass and bone mass, decreases in libido and sexual function and is associated with insulin resistance, metabolic syndrome, diabetes, and other chronic comorbid conditions. It is important for physicians to be aware of how testosterone levels can be related to these conditions, which are seen in primary care practices on a daily basis.
This CME supplement discusses the definition, epidemiology and key signs and symptoms of late-onset hypogonadism, the role of lab measurements, factors to consider in selecting patients for testosterone replacement therapy, and how to identify the best treatment strategy for each patient.
Hypogonadism is a highly prevalent condition that is under-diagnosed and under-treated despite available effective therapies. Low levels of testosterone result in losses of lean body mass and bone mass, decreases in libido and sexual function and is associated with insulin resistance, metabolic syndrome, diabetes, and other chronic comorbid conditions. It is important for physicians to be aware of how testosterone levels can be related to these conditions, which are seen in primary care practices on a daily basis.
This CME supplement discusses the definition, epidemiology and key signs and symptoms of late-onset hypogonadism, the role of lab measurements, factors to consider in selecting patients for testosterone replacement therapy, and how to identify the best treatment strategy for each patient.

Coenzyme Q10: A therapy for hypertension and statin-induced myalgia?
Coenzyme Q10 supplements have been purported to be effective for treating a variety of disorders,1,2 in particular hypertension and statin-induced myalgia.
Several studies3–7 found that coenzyme Q10 supplementation significantly lowered blood pressure in hypertensive patients. Moreover, some trials have demonstrated that statin therapy reduces serum or muscle levels of coenzyme Q10,8–14 prompting investigations to determine whether coenzyme Q10 deficiency is related to statin-induced muscle pain.15–17
In this review, we discuss the efficacy and safety of coenzyme Q10 supplementation in patients with hypertension and those taking statins, and some of the caveats about using supplements that are not approved by the US Food and Drug Administration (FDA), as well as the bioavailability and quality of available formulations.
WHAT IS COENZYME Q10?
Coenzyme Q10, also known as coenzyme Q, ubidecarenone, and ubiquinone, is found in all human cells, with the highest concentrations in the heart, liver, kidney, and pancreas.1,2 It is a potent antioxidant, a membrane stabilizer, and an integral cofactor in the mitochondrial respiratory chain, helping to generate adenosine triphosphate, the major cellular energy source.1,2,18 It may also regulate genes associated with cell metabolism.19
RATIONALE FOR SUPPLEMENTATION
Coenzyme Q10 supplementation has been used, recommended, or studied in heart failure, hypertension, parkinsonism, mitochondrial encephalomyopathies, and other ailments.
In hypertension
Depending on the class, various antihypertensive drugs can have adverse effects such as depression, cough, and cardiac and renal dysfunction. 20,21 Furthermore, many patients need to take more than one drug to control their blood pressure, increasing their risk of side effects. Some researchers believe coenzyme Q10 supplementation may reduce the need to take multiple antihypertensive drugs.5
Coenzyme Q10 appears to lower blood pressure. The exact mechanism is not known, but one theory is that it reduces peripheral resistance by preserving nitric oxide.21 Nitric oxide relaxes peripheral arteries, lowering blood pressure. In some forms of hypertension, superoxide radicals that inactivate nitric oxide are overproduced; coenzyme Q10, with its antioxidant effects, may prevent the inactivation of nitric oxide by these free radicals. Alternatively, coenzyme Q10 may boost the production of the prostaglandin prostacyclin (PGI2) a potent vasodilator and inhibitor of platelet aggregation, or it may enhance the sensitivity of arterial smooth muscle to PGI2, or both.1,22
In patients taking statins
Hydroxymethylglutaryl coenzyme A reductase inhibitors (statins), first-line agents for lowering cholesterol levels to prevent cardiovascular disease, are some of the most commonly prescribed medications.23,24 However, statin therapy carries a risk of myopathy, which can range from muscle aches to rhabdomyolysis. 23,24
In a clinical advisory,25 the American College of Cardiology, the American Heart Association, and the National Heart, Lung, and Blood Institute recommend that patients on statin therapy who experience muscle soreness, tenderness, or pain with serum creatine kinase levels 3 to 10 times the upper limit of normal should have their creatine kinase level checked weekly. If the level is 3 to 10 times the upper limit of normal, statin therapy may be continued, but if it exceeds 10 times the upper limit, then statins and other potential offending agents (eg, niacin, fibrate) need to be discontinued.
Statins inhibit the synthesis of cholesterol by reducing the production of mevalonate, a precursor of both cholesterol and coenzyme Q10. Since both cholesterol and coenzyme Q10 are produced by the same pathway, it is not surprising that statins have been reported to reduce serum and muscle coenzyme Q10 levels.9–14 However, one study did not report a significant reduction of coenzyme Q10 levels in muscle tissue in patients treated with simvastatin 20 mg for 6 months.26
Nonetheless, researchers have hypothesized that a reduction in coenzyme Q10 levels in muscle tissue causes mitochondrial dysfunction, which could increase the risk of statininduced myopathy,13–17 and some believe that treatment with coenzyme Q10 may reduce myalgic symptoms and allow patients to remain on statin therapy.13,24
Researchers have investigated the potential of coenzyme Q10 supplementation to reduce or prevent statin-induced myopathy.15–17 (More on this below.)
Interestingly, a randomized, placebo-controlled trial27 found that 6 months of daily therapy with simvastatin (Zocor) 20 mg or pravastatin (Pravachol) 40 mg lowered systolic and diastolic blood pressure significantly in patients with no documented history of cardiovascular disease or diabetes. A possible mechanism of statin-induced blood pressure reduction is the up-regulation of endothelial nitric oxide synthetase, a potent vasodilator. Coenzyme Q10 levels were not assessed during this study. Whether coenzyme Q10 supplementation used to treat statin-induced myalgia enhances or inhibits the antihypertensive effects of statins is not yet known.
EVIDENCE OF EFFECTIVENESS IN HYPERTENSION
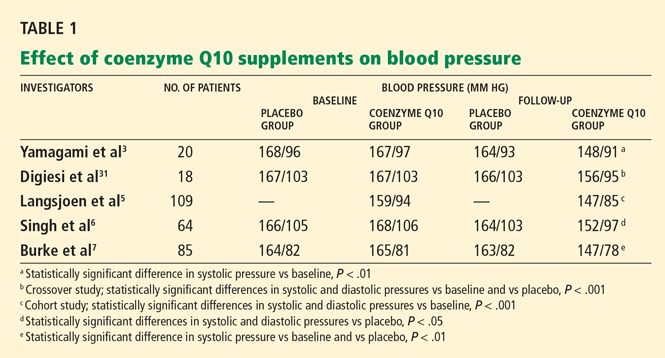
Rosenfeldt et al28 performed a meta-analysis and found that some trials documented statistically significant reductions in diastolic or systolic blood pressure or both, while others reported negligible effects.3,29 In one small trial,30 blood pressures actually went up in patients taking coenzyme Q10. Coenzyme Q10 dosages and length of therapy varied from study to study in the meta-analysis. Only minor adverse effects such as gastrointestinal upset and headache were reported.
Yamagami et al3 randomly assigned 20 patients with hypertension and a low coenzyme Q10 level to receive 100 mg of coenzyme Q10 or placebo daily for 12 weeks. Patients continued their usual antihypertensive regimen during the study period. Blood pressures, coenzyme Q10 levels, and antihypertensive drugs used were comparable between the study groups.
After 12 weeks of therapy, the mean coenzyme Q10 level in the active-treatment group had more than doubled, from 0.704 to 1.597 μg/mL. This group also experienced a statistically significant drop in systolic blood pressure, from 167 mm Hg at baseline to 148 mm Hg at 12 weeks. In the placebo group, the systolic blood pressure was 168 mm Hg at baseline and 164 mm Hg at 12 weeks; the change was not statistically significant. Diastolic pressure was not significantly lower at 12 weeks than at baseline in either group.
The authors concluded that coenzyme Q10 supplementation brought a mild reduction in high blood pressure in patients who had low coenzyme Q10 serum levels.
Digiesi et al31 randomized 18 patients with essential hypertension to receive either coenzyme Q10 100 mg or placebo daily for 10 weeks. All antihypertensive therapy was discontinued at baseline. After the first 10 weeks, patients went through a 2-week washout period and then were switched to the opposite therapy for an additional 10 weeks. Mean baseline blood pressure values were 167 mm Hg systolic and 103 mm Hg diastolic.
Those taking the supplement had a statistically significant decrease in systolic and diastolic pressures (P < .001). The antihypertensive effect was noted in the 3rd or 4th week of active treatment and persisted for the duration of therapy. The effects dissipated 7 to 10 days after coenzyme Q10 was stopped.
Langsjoen et al5 evaluated the effects of adding coenzyme Q10 to the antihypertensive drug regimen of 109 patients who had a primary diagnosis of essential hypertension in a prospective observational study. Patients with hypertension as a secondary diagnosis and other cardiovascular diseases were excluded. Variable doses of coenzyme Q10 were given, adjusted according to clinical response and to achieve serum levels greater than 2.0 μg/mL. The average dose was 225 mg/day; the mean serum level attained was 3.02 μg/mL.
Over several months, patients taking the supplement had a reduction in mean systolic pressure from 159 mm Hg at baseline to 147 mm Hg (P < .001), and a reduction in mean diastolic pressure from 94 to 85 mm Hg (P < .001). Thirty-seven percent of patients were able to discontinue one antihypertensive drug, 11% discontinued two drugs, and 4% were able to stop taking three drugs. However, 46% remained on the same antihypertensive regimen, and 3% needed an additional drug.
Singh et al6 randomized 64 patients who had coronary artery disease and who had been on antihypertensive drugs for more than 1 year to receive either B-complex vitamins or coenzyme Q10 (hydrosoluble Q-Gel) 60 mg orally once daily for 8 weeks. Five patients were not available for follow-up; therefore, only 59 patients were evaluated. Fifty-five (93%) of the 59 patients were taking only one antihypertensive drug. Initial antihypertensive drug use was similar between study groups and was continued throughout the trial.
After 8 weeks of therapy, the coenzyme Q10 group had significantly lower systolic and diastolic blood pressure than the placebo group (P < .05 for both). There was also a statistically significant decrease in the dosage of antihypertensive drugs in the coenzyme Q10 group but not in the placebo group (P < .05), reflecting coenzyme Q10’s additive antihypertensive effect.
Burke et al7 randomized 41 men and 35 women with isolated systolic hypertension (systolic pressure 150–170 mm Hg, diastolic pressure < 90 mm Hg) to receive a twice-daily dose of 60 mg of emulsified coenzyme Q10 (hydrosoluble Q-Gel) with 150 IU of vitamin E or placebo containing vitamin E alone for 12 weeks. The study also included 5 men and 4 women with normal blood pressure, all of whom received coenzyme Q10. A total of 80 patients completed treatment. The primary goal of the study was to determine the efficacy of coenzyme Q10 in the treatment of isolated systolic hypertension in patients without comorbid conditions. Blood pressures were monitored twice a week during the trial, by the same nurse.
After 12 weeks of treatment, the mean reduction in systolic pressure in hypertensive patients on coenzyme Q10 was 17.8 ± 7.3 mm Hg. There were no significant changes in diastolic pressure in any study group with treatment. Patients with isolated systolic hypertension who were taking coenzyme Q10 had a statistically significant reduction in systolic pressure compared with baseline and placebo (P < .01 for both). Approximately 55% of patients on coenzyme Q10 achieved a reduction in systolic pressure of 4 mm Hg or greater, while 45% did not respond to therapy. The mean plasma coenzyme Q10 level of the treatment group increased from 0.47 ± 0.19 μg/mL to 2.69 ± 0.54 μg/mL after 12 weeks; however, the study did not have the statistical power to demonstrate a relationship between coenzyme Q10 levels and changes in blood pressure. Twenty-seven (34%) of the 80 patients were taking a statin while on coenzyme Q10 therapy.
STUDIES IN STATIN-INDUCED MYOPATHY
Thibault et al32 and Kim et al33 reported that patients taking lovastatin (Mevacor) at dosages as high as 35 mg/kg/day to inhibit tumor growth achieved symptomatic relief of statin-induced musculoskeletal toxicity after coenzyme Q10 supplementation.
Caso et al15 performed a small pilot study in 32 patients to determine if coenzyme Q10 supplementation would improve myalgic symptoms in patients treated with statins. In this double-blind, randomized trial, patients received either coenzyme Q10 100 mg/day or vitamin E 400 IU/day for 30 days. The extent of muscle pain and its interference with daily activities were determined before and after therapy using the Brief Pain Inventory Questionnaire. The statins were atorvastatin (Lipitor) 10 mg or 20 mg, lovastatin 40 mg, pravastatin 40 mg, and simvastatin 10, 20, 40, and 80 mg. Five patients in the coenzyme Q10 group and four patients in the vitamin E group were taking nonsteroidal anti-inflammatory drugs before and during the trial. The intensity of muscle pain and its interference with daily activities were similar between study groups before the start of therapy.
After 30 days of treatment with coenzyme Q10, the pain intensity had decreased significantly from baseline (P < .001). In contrast, no change in pain intensity from baseline was noted in patients receiving vitamin E. The Pain Severity Score was significantly different between study groups, favoring the coenzyme Q10 group (P < .001). Sixteen of 18 patients on coenzyme Q10 reported a reduction in pain, while only 3 of 14 patients on vitamin E reported a similar response. Also, the interference of pain with daily activities significantly improved with coenzyme Q10 (P < .02), whereas vitamin E did not have a significant impact on this.
Young et al17 randomized 44 patients with prior statin-induced myalgia to receive increasing doses of simvastatin (10–40 mg/day) in combination with either coenzyme Q10 (Q-Gel) 200 mg/day or placebo. The primary goal was to determine if coenzyme Q10 supplementation would help improve statin tolerance in patients with a history of statininduced myalgia. Plasma coenzyme Q10 and lipid levels were measured at baseline and at the end of the study. The intensity of myalgia was assessed with a visual analogue scale.
At 12 weeks, the coenzyme Q10 plasma level was significantly higher in the treatment group than in the placebo group (P < .001). However, no differences were noted between groups in the number of patients who tolerated the 40-mg/day simvastatin dose (P = .34) or in the number of patients who remained on any simvastatin dose (P = .47). Additionally, myalgia scores did not differ between groups (P = .63). The authors acknowledged that there were only small increases in the myalgia pain scores reported in either group. Therefore, patients in the treatment group may not have experienced sufficiently severe muscle pain to have benefited from coenzyme Q10 supplementation.
IS COENZYME Q10 SAFE?
Studies have indicated that these supplements are well tolerated, with relatively few adverse effects or potential drug interactions.1,2,34
The FDA does not routinely assess the purity or quality of over-the-counter coenzyme Q10 products.35 However, the United States Pharmacopeia (USP) does test dietary supplements to make sure that they are not mislabeled and that they do not contain contaminants. 36
A USP-verified dietary supplement should:
- Contain the exact ingredients listed on the label in the listed potency and amounts
- Not include harmful levels of certain contaminants such as lead, mercury, pesticides, or bacteria
- Appropriately disintegrate and release its contents into the body within a specified period of time
- Be produced using the FDA’s current Good Manufacturing Practices.36
Side effects, contraindications, warnings
Coenzyme Q10 is a relatively safe dietary supplement. It is contraindicated in patients who are allergic to it or to any of its components.2 Most clinical trials have not reported significant adverse effects that necessitated stopping therapy.34 However, gastrointestinal effects such as abdominal discomfort, nausea, vomiting, diarrhea, and anorexia have occurred.1,2,34 Allergic rash and headache have also been reported.1,2,34 In addition, coenzyme Q10’s antiplatelet effect may increase the risk of bleeding. 37,38 It undergoes biotransformation in the liver and is eliminated primarily via the biliary tract,39 so it can accumulate in patients with hepatic impairment or biliary obstruction.
Interactions with drugs
Coenzyme Q10’s effects on platelet function may increase the risk of bleeding in patients taking antiplatelet drugs such as aspirin or clopidogrel (Plavix).37,38 On the other hand, since it acts like vitamin K, it may counteract the anticoagulant effects of warfarin (Coumadin). 1,2,40
Coenzyme Q10 may have an additive antihypertensive effect when given with antihypertensive drugs.41
Coenzyme Q10 may improve beta-cell function and enhance insulin sensitivity, which may reduce insulin requirements for diabetic patients.42,43
SLOWLY ABSORBED
Coenzyme Q10 is absorbed slowly from the gastrointestinal tract, possibly because it has a high molecular weight and is not very watersoluble. 39
One pharmacokinetic study found that after a single 100-mg oral dose of coenzyme Q10, the mean peak plasma levels of about 1 μg/mL occurred between 5 and 10 hours (mean 6.5 hours).44 Coenzyme Q10 100 mg given orally three times daily produced a mean steadystate plasma level of 5.4 μg/mL; about 90% of this steady-state concentration was achieved after 4 days.39
Some formulations have significantly better oral bioavailability and therefore produce higher plasma levels. Soft-gel capsules, especially those with vegetable oil or vitamin E, may have better absorption.43
A pharmacokinetic study showed that the area under the curve of the plasma coenzyme Q10 concentration was more than twice as high with Q-Gel soft-gel capsules, a completely solubilized formulation, than with softgel capsules with an oil suspension, powderfilled hard-shell capsules, or regular tablets.45 Another study reported that colloidal-Q10, a formulation contained in VESIsorb (a novel drug delivery system sold as CoQsource) had greater bioavailability than solubilized and oil-based preparations.46 Commercially available solubilized preparations containing ubiquinol, a metabolized form of coenzyme Q10, have been shown to produce higher serum levels than solubilized products.47
Of note: unless the manufacturer claims that its product is water-soluble, the USP does not evaluate its dissolution rate.48 Therefore, USP-verified coenzyme Q10 products that are not water-soluble may have lower bioavailability than their solubilized counterparts.
Dry dosage forms of coenzyme Q10 (eg, tablets, capsules) may be more readily absorbed if taken with a fatty meal.43
SLOWLY ELIMINATED
Taken orally, coenzyme Q10 has a low clearance rate, with an elimination half-life of about 34 hours.39
After absorption, exogenous coenzyme Q10 is taken up by chylomicrons that transport it to the liver, where it is incorporated into verylow-density lipoproteins. It is then distributed to various organs, including the adrenal glands, spleen, kidneys, lungs, and heart. Coenzyme Q10 is eliminated primarily via the biliary tract. About 60% of an oral dose is eliminated in the feces during chronic oral administration.39
TWICE-DAILY DOSING
A typical daily dose of coenzyme Q10 for treating hypertension is 120 to 200 mg, usually given orally in two divided doses.1 For statininduced myopathy, 100 to 200 mg orally daily has been used.1
Coenzyme Q10 is given in divided doses to enhance its absorption and to minimize gastrointestinal effects.1,43 Taking it with a fatty meal may also increase its absorption.43
Since solubilized forms of coenzyme Q10 and ubiquinol have significantly greater bioavailability than nonsolubilized forms, the therapeutic dose of these formulations may be lower.47
MONITORING DURING TREATMENT
Without supplementation, the mean serum level of endogenous coenzyme Q10 has been reported to be 0.99 ± 0.30 mg/L (range 0.55– 1.87).18 Serum levels above 2 μg/mL have been associated with significant reductions in blood pressure.5,7,28
The possible effects of coenzyme Q10 on blood pressure, blood glucose levels, serum creatine kinase levels, and myopathic symptoms should be kept in mind when monitoring patients who have hypertension,41 diabetes,41,42 or statin-induced myalgia.15,17 Coenzyme Q10’s possible potentiating effects on antiplatelet drugs and its inhibitory effect on warfarin should be kept in mind as well.
COST VARIES
Coenzyme Q10 is available in different dosage forms (eg, regular and rapid-release softgel capsules, regular and chewable tablets, chewable wafers, and liquid) from a variety of manufacturers. Products come in different strengths, typically ranging from 30 to 400 mg. USP-verified formulations are listed at www.usp.org/USPVerified/dietarySupplements/under “Verified Supplements.” Only USP-verified products that claim to be water-soluble meet USP dissolution requirements.
The cost varies, depending on the vendor. In general, dosage forms with greater bioavailability, such as Q-Gel and ubiquinol supplements, are more expensive. For example, a regimen of 60 mg twice daily of regular-release coenzyme Q capsules may cost approximately $20 per month, compared with $60 per month for the same supply of Q-Gel Ultra capsules. However, in some cases, supplements that produce higher serum levels may be more cost-effective.
CURRENT ROLE IN THERAPY
As an antihypertensive adjunct
Several small clinical trials have shown that coenzyme Q10 supplementation can lower blood pressure. The supplements were reported to be safe and well tolerated. Moreover, some patients with essential hypertension who were taking coenzyme Q10 were able to discontinue one or more antihypertensive drugs. A significant reduction in blood pressure with use of coenzyme Q10 would be expected to reduce the adverse consequences of hypertension in the same manner as conventional antihypertensive agents.
However, no large, double-blind, randomized study has evaluated the impact of coenzyme Q10 when taken with other antihypertensive drugs (eg, angiotensin-converting enzyme inhibitors, beta-blockers, diuretics) on specific clinical end points such as the incidence of stroke or death from a major cardiac event. Furthermore, its effects on cardiac function, exercise tolerance, and quality of life have not been determined.
The bottom line. In some cases, it seems reasonable to recommend this product as an adjunct to conventional antihypertensive therapy. Larger, well-designed clinical trials of coenzyme Q10’s antihypertensive effects on specific clinical end points such as the risk of stroke or myocardial infarction are needed to define its true therapeutic value.
As a treatment for statin-induced myalgia
Clinical evidence supporting coenzyme Q10’s use in the treatment of statin-induced myopathy is limited. Whether coenzyme Q10 is depleted from muscle tissue during statin therapy has not been confirmed. Supplementation helped reduce the severity of musculoskeletal effects of megadoses of lovastatin. However, clinical trials of coenzyme Q10 in the treatment of myalgia associated with antilipidemic statin doses did not consistently report significant improvement. Nevertheless, coenzyme Q10 has been shown to be relatively safe, with few adverse effects.
The bottom line. In some cases, coenzyme Q could be considered as a possible treatment for statin-induced myalgia, pending large-scale studies to determine if it is truly effective for this purpose.
- Jelin JM, Gregory PJ, et al. Natural medicines comprehensive database/compiled by the editors of Pharmacist’s Letter, Prescriber’s Letter. 11th ed. Stockton, CA: Therapeutic Research Faculty; 2009:452–457.
- Fetrow CW, Avila JR. Professional’s Handbook of Complementary & Alternative Medicines. 2nd ed. Springhouse, PA: Springhouse; 2001:211–215.
- Yamagami T, Takagi M, Akagami H, et al. Effect of coenzyme Q10 on essential hypertension, a double-blind controlled study. In:Folkers K, Yamamura Y, editors. Biomedical and Clinical Aspects of Coenzyme Q10: Proceedings of the Fifth International Symposium on the Biomedical and Clinical Aspects of Coenzyme Q10, vol 5. Amsterdam: Elsevier Science Publishers; 1986:337–343.
- Digiesi V, Cantini F, Oradei A, et al. Coenzyme Q10 in essential hypertension. Mol Aspects Med 1994; 15(suppl):S257–S263.
- Langsjoen P, Langsjoen P, Willis R, Folkers K. Treatment of essential hypertension with coenzyme Q10. Mol Aspects Med 1994; 15(suppl):S265–S272.
- Singh RB, Niaz MA, Rastogi SS, Shukla PK, Thakur AS. Effect of hydrosoluble coenzyme Q10 on blood pressures and insulin resistance in hypertensive patients with coronary artery disease. J Hum Hypertens 1999; 13:203–208.
- Burke BE, Neuenschwander R, Olson RD. Randomized, double-blind, placebo-controlled trial of coenzyme Q10 in isolated systolic hypertension. South Med J 2001; 94:1112–1117.
- De Pinieux G, Chariot P, Ammi-Saïd M, et al. Lipidlowering drugs and mitochondrial function: effects of HMG-CoA reductase inhibitors on serum ubiquinone and blood lactate/pyruvate ratio. Br J Clin Pharmacol 1996; 42:333–337.
- Mortensen SA, Leth A, Agner E, Rohde M. Dose-related decrease of serum coenzyme Q10 during treatment with HMG-CoA reductase inhibitors. Mol Aspects Med 1997; 18(suppl):S137–S144.
- Ghirlanda G, Oradei A, Manto A, et al. Evidence of plasma CoQ10-lowering effect by HMG-CoA reductase inhibitors: a double-blind, placebo-controlled study. J Clin Pharmacol 1993; 33:226–229.
- Folkers K, Langsjoen P, Willis R, et al. Lovastatin decreases coenzyme Q10 levels in humans. Proc Natl Acad Sci U S A 1990; 87:8931–8934.
- Watts GF, Castelluccio C, Rice-Evans C, Taub NA, Baum H, Quinn PJ. Plasma coenzyme Q10 (ubiquinone) concentrations in patients treated with simvastatin. J Clin Pathol 1993; 46:1055–1057.
- Lamperti C, Naini AB, Lucchini V, et al. Muscle coenzyme Q10 level in statin-related myopathy. Arch Neurol 2005; 62:1709–1712.
- Päivä H, Thelen KM, Van Coster R, et al. High-dose statins and skeletal muscle metabolism in humans: a randomized, controlled trial. Clin Pharmacol Ther 2005; 78:60–68.
- Caso G, Kelly P, McNurlan MA, Lawson WE. Effect of coenzyme Q10 on myopathic symptoms in patients treated with statins. Am J Cardiol 2007; 99:1409–1412.
- Marcoff L, Thompson PD. The role of coenzyme Q10 in statin-associated myopathy: a systematic review. J Am Coll Cardiol 2007; 49:2231–2237.
- Young JM, Florkowski CM, Molyneux SL, et al. Effect of coenzyme Q(10) supplementation on simvastatin-induced myalgia. Am J Cardiol 2007; 100:1400–1403.
- Berthold HK, Naini A, Di Mauro S, et al. Effect of ezetimibe and/or simvastatin on coenzyme Q10 levels in plasma: a randomised trial. Drug Saf 2006; 29:703–712.
- Groneberg DA, Kindermann B, Althammer M, et al. Coenzyme Q10 affects expression of genes involved in cell signalling, metabolism and transport in human CaCo-2 cells. Int J Biochem Cell Biol 2005; 37:1208–1218.
- Hadj A, Pepe S, Rosenfeldt F. The clinical application of metabolic therapy for cardiovascular disease. Heart Lung Circ 2007; 16(suppl 3):S56–S64.
- Pepe S, Marasco SF, Haas SJ, Sheeran FL, Krum H, Rosenfeldt FL. Coenzyme Q10 in cardiovascular disease. Mitochondrion 2007; 7(suppl 1):S154–S167.
- Lönnrot K, Pörsti I, Alho H, Wu X, Hervonen A, Tolvanen JP. Control of arterial tone after long-term coenzyme Q10 supplementation in senescent rats. Br J Pharmacol 1998; 124:1500–1506.
- Sewright KA, Clarkson PM, Thompson PD. Statin myopathy: incidence, risk factors, and pathophysiology. Curr Atheroscler Rep 2007; 9:389–396.
- Radcliffe KA, Campbell WW. Statin myopathy. Curr Neurol Neurosci Rep 2008; 8:66–72.
- Pasternak RC, Smith SC, Bairey-Merz CN, Grundy SM, Cleeman JI, Lenfant C. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. Circulation 2002; 106:1024–1028.
- Laaksonen R, Jokelainen K, Laakso J, et al. The effect of simvastatin treatment on natural antioxidants in low-density lipoproteins and high-energy phosphates and ubiquinone in skeletal muscle. Am J Cardiol 1996; 77:851–854.
- Golomb BA, Dimsdale JE, White HL, Ritchie JB, Criqui MH. Reduction in blood pressure with statins: results from the UCSD Statin Study, a randomized trial. Arch Intern Med 2008; 168:721–727.
- Rosenfeldt FL, Haas SJ, Krum H, et al. Coenzyme Q10 in the treatment of hypertension: a meta-analysis of the clinical trials. J Hum Hypertens 2007; 21:297–306.
- Yamagami T, Shibata N, Folkers K. Bioenergetics in clinical medicine. Studies on coenzyme Q10 and essential hypertension. Res Commun Chem Pathol Pharmacol 1975; 11:273–288.
- Yamagami T, Shibata N, Folkers K. Study of coenzyme Q10. In:Folkers K, Yamamura Y, editors. Biomedical and clinical aspects of coenzyme Q10: proceedings of the International Symposium on Coenzyme Q10, held at Lake Yamanaka, Japan, September 16/17, 1976, a Naito Foundation symposium. Amsterdam: Elsevier Scientific Publishing Company; 1977:231–242.
- Digiesi V, Cantini F, Brodbeck B. Effect of coenzyme Q10 on essential arterial hypertension. Curr Ther Res; 1990; 47:841–845.
- Thibault A, Samid D, Tompkins AC, et al. Phase I study of lovastatin, an inhibitor of the mevalonate pathway, in patients with cancer. Clin Cancer Res 1996; 2:483–491.
- Kim WS, Kim MM, Choi HJ, et al. Phase II study of high-dose lova-statin in patients with advanced gastric adenocarcinoma. Invest New Drugs 2001; 19:81–83.
- Hidaka T, Fujii K, Funahashi I, Fukutomi N, Hosoe K. Safety assessment of coenzyme Q10 (CoQ10). Biofactors 2008; 32:199–208.
- US Food and Drug Administration. Consumer Information on Dietary Supplements. Overview of Dietary Supplements. http://www.fda.gov/Food/DietarySupplements/ConsumerInformation/. Accessed May 25, 2010.
- US Pharmacopeia. The USP Dietary Supplement Verification Program http://www.usp.org/USPVerified/dietary-Supplements/. Accessed May 25, 2010.
- Serebruany VL, Ordonez JV, Herzog WR, et al. Dietary coenzyme Q10 supplementation alters platelet size and inhibits human vitronectin (CD51/CD61) receptor expression. J Cardiovasc Pharmacol 1997; 29:16–22.
- A close look at coenzyme Q10 and policosanol. Do these supplements live up to their claims for improving heart health? Harv Heart Lett 2002; 13:6.
- Greenberg S, Frishman WH. Co-enzyme Q10: a new drug for cardiovascular disease. J Clin Pharmacol 1990; 30:596–608.
- Singh U, Devaraj S, Jialal I. Coenzyme Q10 supplementation and heart failure. Nutr Rev 2007; 65:286–293.
- Bonakdar RA, Guarneri E. Coenzyme Q10. Am Fam Physician 2005; 72:1065–1070.
- Hodgson JM, Watts GF, Playford DA, Burke V, Croft KD. Coenzyme Q10 improves blood pressure and glycaemic control: a controlled trial in subjects with type 2 diabetes. Eur J Clin Nutr 2002; 56:1137–1142.
- Pepping J. Coenzyme Q10. Am J Health Syst Pharm 1999; 56:519–521.
- Tomono Y, Hasegawa J, Seki T, Motegi K, Morishita N. Pharmacokinetic study of deuterium-labelled coenzyme Q10 in man. Int J Clin Pharmacol Ther Toxicol 1986; 24:536–541.
- Chopra RK, Goldman R, Sinatra ST, Bhagavan HN. Relative bioavailability of coenzyme Q10 formulations in human subjects. Int J Vitam Nutr Res 1998; 68:109–113.
- Liu ZX, Artmann C. Relative bioavailability comparison of different coenzyme Q10 formulations with a novel delivery system. Altern Ther Health Med 2009; 15:42–46.
- Bhagavan HN, Chopra RK. Plasma coenzyme Q10 response to oral ingestion of coenzyme Q10 formulations. Mitochondrion 2007; 7(suppl 1):S78–S88.
- The United States Pharmacopeia. Ubidecarenone Capsules Monograph. 32nd Revision. Baltimore: United Book Press, 2009:1080.
Coenzyme Q10 supplements have been purported to be effective for treating a variety of disorders,1,2 in particular hypertension and statin-induced myalgia.
Several studies3–7 found that coenzyme Q10 supplementation significantly lowered blood pressure in hypertensive patients. Moreover, some trials have demonstrated that statin therapy reduces serum or muscle levels of coenzyme Q10,8–14 prompting investigations to determine whether coenzyme Q10 deficiency is related to statin-induced muscle pain.15–17
In this review, we discuss the efficacy and safety of coenzyme Q10 supplementation in patients with hypertension and those taking statins, and some of the caveats about using supplements that are not approved by the US Food and Drug Administration (FDA), as well as the bioavailability and quality of available formulations.
WHAT IS COENZYME Q10?
Coenzyme Q10, also known as coenzyme Q, ubidecarenone, and ubiquinone, is found in all human cells, with the highest concentrations in the heart, liver, kidney, and pancreas.1,2 It is a potent antioxidant, a membrane stabilizer, and an integral cofactor in the mitochondrial respiratory chain, helping to generate adenosine triphosphate, the major cellular energy source.1,2,18 It may also regulate genes associated with cell metabolism.19
RATIONALE FOR SUPPLEMENTATION
Coenzyme Q10 supplementation has been used, recommended, or studied in heart failure, hypertension, parkinsonism, mitochondrial encephalomyopathies, and other ailments.
In hypertension
Depending on the class, various antihypertensive drugs can have adverse effects such as depression, cough, and cardiac and renal dysfunction. 20,21 Furthermore, many patients need to take more than one drug to control their blood pressure, increasing their risk of side effects. Some researchers believe coenzyme Q10 supplementation may reduce the need to take multiple antihypertensive drugs.5
Coenzyme Q10 appears to lower blood pressure. The exact mechanism is not known, but one theory is that it reduces peripheral resistance by preserving nitric oxide.21 Nitric oxide relaxes peripheral arteries, lowering blood pressure. In some forms of hypertension, superoxide radicals that inactivate nitric oxide are overproduced; coenzyme Q10, with its antioxidant effects, may prevent the inactivation of nitric oxide by these free radicals. Alternatively, coenzyme Q10 may boost the production of the prostaglandin prostacyclin (PGI2) a potent vasodilator and inhibitor of platelet aggregation, or it may enhance the sensitivity of arterial smooth muscle to PGI2, or both.1,22
In patients taking statins
Hydroxymethylglutaryl coenzyme A reductase inhibitors (statins), first-line agents for lowering cholesterol levels to prevent cardiovascular disease, are some of the most commonly prescribed medications.23,24 However, statin therapy carries a risk of myopathy, which can range from muscle aches to rhabdomyolysis. 23,24
In a clinical advisory,25 the American College of Cardiology, the American Heart Association, and the National Heart, Lung, and Blood Institute recommend that patients on statin therapy who experience muscle soreness, tenderness, or pain with serum creatine kinase levels 3 to 10 times the upper limit of normal should have their creatine kinase level checked weekly. If the level is 3 to 10 times the upper limit of normal, statin therapy may be continued, but if it exceeds 10 times the upper limit, then statins and other potential offending agents (eg, niacin, fibrate) need to be discontinued.
Statins inhibit the synthesis of cholesterol by reducing the production of mevalonate, a precursor of both cholesterol and coenzyme Q10. Since both cholesterol and coenzyme Q10 are produced by the same pathway, it is not surprising that statins have been reported to reduce serum and muscle coenzyme Q10 levels.9–14 However, one study did not report a significant reduction of coenzyme Q10 levels in muscle tissue in patients treated with simvastatin 20 mg for 6 months.26
Nonetheless, researchers have hypothesized that a reduction in coenzyme Q10 levels in muscle tissue causes mitochondrial dysfunction, which could increase the risk of statininduced myopathy,13–17 and some believe that treatment with coenzyme Q10 may reduce myalgic symptoms and allow patients to remain on statin therapy.13,24
Researchers have investigated the potential of coenzyme Q10 supplementation to reduce or prevent statin-induced myopathy.15–17 (More on this below.)
Interestingly, a randomized, placebo-controlled trial27 found that 6 months of daily therapy with simvastatin (Zocor) 20 mg or pravastatin (Pravachol) 40 mg lowered systolic and diastolic blood pressure significantly in patients with no documented history of cardiovascular disease or diabetes. A possible mechanism of statin-induced blood pressure reduction is the up-regulation of endothelial nitric oxide synthetase, a potent vasodilator. Coenzyme Q10 levels were not assessed during this study. Whether coenzyme Q10 supplementation used to treat statin-induced myalgia enhances or inhibits the antihypertensive effects of statins is not yet known.
EVIDENCE OF EFFECTIVENESS IN HYPERTENSION
Rosenfeldt et al28 performed a meta-analysis and found that some trials documented statistically significant reductions in diastolic or systolic blood pressure or both, while others reported negligible effects.3,29 In one small trial,30 blood pressures actually went up in patients taking coenzyme Q10. Coenzyme Q10 dosages and length of therapy varied from study to study in the meta-analysis. Only minor adverse effects such as gastrointestinal upset and headache were reported.
Yamagami et al3 randomly assigned 20 patients with hypertension and a low coenzyme Q10 level to receive 100 mg of coenzyme Q10 or placebo daily for 12 weeks. Patients continued their usual antihypertensive regimen during the study period. Blood pressures, coenzyme Q10 levels, and antihypertensive drugs used were comparable between the study groups.
After 12 weeks of therapy, the mean coenzyme Q10 level in the active-treatment group had more than doubled, from 0.704 to 1.597 μg/mL. This group also experienced a statistically significant drop in systolic blood pressure, from 167 mm Hg at baseline to 148 mm Hg at 12 weeks. In the placebo group, the systolic blood pressure was 168 mm Hg at baseline and 164 mm Hg at 12 weeks; the change was not statistically significant. Diastolic pressure was not significantly lower at 12 weeks than at baseline in either group.
The authors concluded that coenzyme Q10 supplementation brought a mild reduction in high blood pressure in patients who had low coenzyme Q10 serum levels.
Digiesi et al31 randomized 18 patients with essential hypertension to receive either coenzyme Q10 100 mg or placebo daily for 10 weeks. All antihypertensive therapy was discontinued at baseline. After the first 10 weeks, patients went through a 2-week washout period and then were switched to the opposite therapy for an additional 10 weeks. Mean baseline blood pressure values were 167 mm Hg systolic and 103 mm Hg diastolic.
Those taking the supplement had a statistically significant decrease in systolic and diastolic pressures (P < .001). The antihypertensive effect was noted in the 3rd or 4th week of active treatment and persisted for the duration of therapy. The effects dissipated 7 to 10 days after coenzyme Q10 was stopped.
Langsjoen et al5 evaluated the effects of adding coenzyme Q10 to the antihypertensive drug regimen of 109 patients who had a primary diagnosis of essential hypertension in a prospective observational study. Patients with hypertension as a secondary diagnosis and other cardiovascular diseases were excluded. Variable doses of coenzyme Q10 were given, adjusted according to clinical response and to achieve serum levels greater than 2.0 μg/mL. The average dose was 225 mg/day; the mean serum level attained was 3.02 μg/mL.
Over several months, patients taking the supplement had a reduction in mean systolic pressure from 159 mm Hg at baseline to 147 mm Hg (P < .001), and a reduction in mean diastolic pressure from 94 to 85 mm Hg (P < .001). Thirty-seven percent of patients were able to discontinue one antihypertensive drug, 11% discontinued two drugs, and 4% were able to stop taking three drugs. However, 46% remained on the same antihypertensive regimen, and 3% needed an additional drug.
Singh et al6 randomized 64 patients who had coronary artery disease and who had been on antihypertensive drugs for more than 1 year to receive either B-complex vitamins or coenzyme Q10 (hydrosoluble Q-Gel) 60 mg orally once daily for 8 weeks. Five patients were not available for follow-up; therefore, only 59 patients were evaluated. Fifty-five (93%) of the 59 patients were taking only one antihypertensive drug. Initial antihypertensive drug use was similar between study groups and was continued throughout the trial.
After 8 weeks of therapy, the coenzyme Q10 group had significantly lower systolic and diastolic blood pressure than the placebo group (P < .05 for both). There was also a statistically significant decrease in the dosage of antihypertensive drugs in the coenzyme Q10 group but not in the placebo group (P < .05), reflecting coenzyme Q10’s additive antihypertensive effect.
Burke et al7 randomized 41 men and 35 women with isolated systolic hypertension (systolic pressure 150–170 mm Hg, diastolic pressure < 90 mm Hg) to receive a twice-daily dose of 60 mg of emulsified coenzyme Q10 (hydrosoluble Q-Gel) with 150 IU of vitamin E or placebo containing vitamin E alone for 12 weeks. The study also included 5 men and 4 women with normal blood pressure, all of whom received coenzyme Q10. A total of 80 patients completed treatment. The primary goal of the study was to determine the efficacy of coenzyme Q10 in the treatment of isolated systolic hypertension in patients without comorbid conditions. Blood pressures were monitored twice a week during the trial, by the same nurse.
After 12 weeks of treatment, the mean reduction in systolic pressure in hypertensive patients on coenzyme Q10 was 17.8 ± 7.3 mm Hg. There were no significant changes in diastolic pressure in any study group with treatment. Patients with isolated systolic hypertension who were taking coenzyme Q10 had a statistically significant reduction in systolic pressure compared with baseline and placebo (P < .01 for both). Approximately 55% of patients on coenzyme Q10 achieved a reduction in systolic pressure of 4 mm Hg or greater, while 45% did not respond to therapy. The mean plasma coenzyme Q10 level of the treatment group increased from 0.47 ± 0.19 μg/mL to 2.69 ± 0.54 μg/mL after 12 weeks; however, the study did not have the statistical power to demonstrate a relationship between coenzyme Q10 levels and changes in blood pressure. Twenty-seven (34%) of the 80 patients were taking a statin while on coenzyme Q10 therapy.
STUDIES IN STATIN-INDUCED MYOPATHY
Thibault et al32 and Kim et al33 reported that patients taking lovastatin (Mevacor) at dosages as high as 35 mg/kg/day to inhibit tumor growth achieved symptomatic relief of statin-induced musculoskeletal toxicity after coenzyme Q10 supplementation.
Caso et al15 performed a small pilot study in 32 patients to determine if coenzyme Q10 supplementation would improve myalgic symptoms in patients treated with statins. In this double-blind, randomized trial, patients received either coenzyme Q10 100 mg/day or vitamin E 400 IU/day for 30 days. The extent of muscle pain and its interference with daily activities were determined before and after therapy using the Brief Pain Inventory Questionnaire. The statins were atorvastatin (Lipitor) 10 mg or 20 mg, lovastatin 40 mg, pravastatin 40 mg, and simvastatin 10, 20, 40, and 80 mg. Five patients in the coenzyme Q10 group and four patients in the vitamin E group were taking nonsteroidal anti-inflammatory drugs before and during the trial. The intensity of muscle pain and its interference with daily activities were similar between study groups before the start of therapy.
After 30 days of treatment with coenzyme Q10, the pain intensity had decreased significantly from baseline (P < .001). In contrast, no change in pain intensity from baseline was noted in patients receiving vitamin E. The Pain Severity Score was significantly different between study groups, favoring the coenzyme Q10 group (P < .001). Sixteen of 18 patients on coenzyme Q10 reported a reduction in pain, while only 3 of 14 patients on vitamin E reported a similar response. Also, the interference of pain with daily activities significantly improved with coenzyme Q10 (P < .02), whereas vitamin E did not have a significant impact on this.
Young et al17 randomized 44 patients with prior statin-induced myalgia to receive increasing doses of simvastatin (10–40 mg/day) in combination with either coenzyme Q10 (Q-Gel) 200 mg/day or placebo. The primary goal was to determine if coenzyme Q10 supplementation would help improve statin tolerance in patients with a history of statininduced myalgia. Plasma coenzyme Q10 and lipid levels were measured at baseline and at the end of the study. The intensity of myalgia was assessed with a visual analogue scale.
At 12 weeks, the coenzyme Q10 plasma level was significantly higher in the treatment group than in the placebo group (P < .001). However, no differences were noted between groups in the number of patients who tolerated the 40-mg/day simvastatin dose (P = .34) or in the number of patients who remained on any simvastatin dose (P = .47). Additionally, myalgia scores did not differ between groups (P = .63). The authors acknowledged that there were only small increases in the myalgia pain scores reported in either group. Therefore, patients in the treatment group may not have experienced sufficiently severe muscle pain to have benefited from coenzyme Q10 supplementation.
IS COENZYME Q10 SAFE?
Studies have indicated that these supplements are well tolerated, with relatively few adverse effects or potential drug interactions.1,2,34
The FDA does not routinely assess the purity or quality of over-the-counter coenzyme Q10 products.35 However, the United States Pharmacopeia (USP) does test dietary supplements to make sure that they are not mislabeled and that they do not contain contaminants. 36
A USP-verified dietary supplement should:
- Contain the exact ingredients listed on the label in the listed potency and amounts
- Not include harmful levels of certain contaminants such as lead, mercury, pesticides, or bacteria
- Appropriately disintegrate and release its contents into the body within a specified period of time
- Be produced using the FDA’s current Good Manufacturing Practices.36
Side effects, contraindications, warnings
Coenzyme Q10 is a relatively safe dietary supplement. It is contraindicated in patients who are allergic to it or to any of its components.2 Most clinical trials have not reported significant adverse effects that necessitated stopping therapy.34 However, gastrointestinal effects such as abdominal discomfort, nausea, vomiting, diarrhea, and anorexia have occurred.1,2,34 Allergic rash and headache have also been reported.1,2,34 In addition, coenzyme Q10’s antiplatelet effect may increase the risk of bleeding. 37,38 It undergoes biotransformation in the liver and is eliminated primarily via the biliary tract,39 so it can accumulate in patients with hepatic impairment or biliary obstruction.
Interactions with drugs
Coenzyme Q10’s effects on platelet function may increase the risk of bleeding in patients taking antiplatelet drugs such as aspirin or clopidogrel (Plavix).37,38 On the other hand, since it acts like vitamin K, it may counteract the anticoagulant effects of warfarin (Coumadin). 1,2,40
Coenzyme Q10 may have an additive antihypertensive effect when given with antihypertensive drugs.41
Coenzyme Q10 may improve beta-cell function and enhance insulin sensitivity, which may reduce insulin requirements for diabetic patients.42,43
SLOWLY ABSORBED
Coenzyme Q10 is absorbed slowly from the gastrointestinal tract, possibly because it has a high molecular weight and is not very watersoluble. 39
One pharmacokinetic study found that after a single 100-mg oral dose of coenzyme Q10, the mean peak plasma levels of about 1 μg/mL occurred between 5 and 10 hours (mean 6.5 hours).44 Coenzyme Q10 100 mg given orally three times daily produced a mean steadystate plasma level of 5.4 μg/mL; about 90% of this steady-state concentration was achieved after 4 days.39
Some formulations have significantly better oral bioavailability and therefore produce higher plasma levels. Soft-gel capsules, especially those with vegetable oil or vitamin E, may have better absorption.43
A pharmacokinetic study showed that the area under the curve of the plasma coenzyme Q10 concentration was more than twice as high with Q-Gel soft-gel capsules, a completely solubilized formulation, than with softgel capsules with an oil suspension, powderfilled hard-shell capsules, or regular tablets.45 Another study reported that colloidal-Q10, a formulation contained in VESIsorb (a novel drug delivery system sold as CoQsource) had greater bioavailability than solubilized and oil-based preparations.46 Commercially available solubilized preparations containing ubiquinol, a metabolized form of coenzyme Q10, have been shown to produce higher serum levels than solubilized products.47
Of note: unless the manufacturer claims that its product is water-soluble, the USP does not evaluate its dissolution rate.48 Therefore, USP-verified coenzyme Q10 products that are not water-soluble may have lower bioavailability than their solubilized counterparts.
Dry dosage forms of coenzyme Q10 (eg, tablets, capsules) may be more readily absorbed if taken with a fatty meal.43
SLOWLY ELIMINATED
Taken orally, coenzyme Q10 has a low clearance rate, with an elimination half-life of about 34 hours.39
After absorption, exogenous coenzyme Q10 is taken up by chylomicrons that transport it to the liver, where it is incorporated into verylow-density lipoproteins. It is then distributed to various organs, including the adrenal glands, spleen, kidneys, lungs, and heart. Coenzyme Q10 is eliminated primarily via the biliary tract. About 60% of an oral dose is eliminated in the feces during chronic oral administration.39
TWICE-DAILY DOSING
A typical daily dose of coenzyme Q10 for treating hypertension is 120 to 200 mg, usually given orally in two divided doses.1 For statininduced myopathy, 100 to 200 mg orally daily has been used.1
Coenzyme Q10 is given in divided doses to enhance its absorption and to minimize gastrointestinal effects.1,43 Taking it with a fatty meal may also increase its absorption.43
Since solubilized forms of coenzyme Q10 and ubiquinol have significantly greater bioavailability than nonsolubilized forms, the therapeutic dose of these formulations may be lower.47
MONITORING DURING TREATMENT
Without supplementation, the mean serum level of endogenous coenzyme Q10 has been reported to be 0.99 ± 0.30 mg/L (range 0.55– 1.87).18 Serum levels above 2 μg/mL have been associated with significant reductions in blood pressure.5,7,28
The possible effects of coenzyme Q10 on blood pressure, blood glucose levels, serum creatine kinase levels, and myopathic symptoms should be kept in mind when monitoring patients who have hypertension,41 diabetes,41,42 or statin-induced myalgia.15,17 Coenzyme Q10’s possible potentiating effects on antiplatelet drugs and its inhibitory effect on warfarin should be kept in mind as well.
COST VARIES
Coenzyme Q10 is available in different dosage forms (eg, regular and rapid-release softgel capsules, regular and chewable tablets, chewable wafers, and liquid) from a variety of manufacturers. Products come in different strengths, typically ranging from 30 to 400 mg. USP-verified formulations are listed at www.usp.org/USPVerified/dietarySupplements/under “Verified Supplements.” Only USP-verified products that claim to be water-soluble meet USP dissolution requirements.
The cost varies, depending on the vendor. In general, dosage forms with greater bioavailability, such as Q-Gel and ubiquinol supplements, are more expensive. For example, a regimen of 60 mg twice daily of regular-release coenzyme Q capsules may cost approximately $20 per month, compared with $60 per month for the same supply of Q-Gel Ultra capsules. However, in some cases, supplements that produce higher serum levels may be more cost-effective.
CURRENT ROLE IN THERAPY
As an antihypertensive adjunct
Several small clinical trials have shown that coenzyme Q10 supplementation can lower blood pressure. The supplements were reported to be safe and well tolerated. Moreover, some patients with essential hypertension who were taking coenzyme Q10 were able to discontinue one or more antihypertensive drugs. A significant reduction in blood pressure with use of coenzyme Q10 would be expected to reduce the adverse consequences of hypertension in the same manner as conventional antihypertensive agents.
However, no large, double-blind, randomized study has evaluated the impact of coenzyme Q10 when taken with other antihypertensive drugs (eg, angiotensin-converting enzyme inhibitors, beta-blockers, diuretics) on specific clinical end points such as the incidence of stroke or death from a major cardiac event. Furthermore, its effects on cardiac function, exercise tolerance, and quality of life have not been determined.
The bottom line. In some cases, it seems reasonable to recommend this product as an adjunct to conventional antihypertensive therapy. Larger, well-designed clinical trials of coenzyme Q10’s antihypertensive effects on specific clinical end points such as the risk of stroke or myocardial infarction are needed to define its true therapeutic value.
As a treatment for statin-induced myalgia
Clinical evidence supporting coenzyme Q10’s use in the treatment of statin-induced myopathy is limited. Whether coenzyme Q10 is depleted from muscle tissue during statin therapy has not been confirmed. Supplementation helped reduce the severity of musculoskeletal effects of megadoses of lovastatin. However, clinical trials of coenzyme Q10 in the treatment of myalgia associated with antilipidemic statin doses did not consistently report significant improvement. Nevertheless, coenzyme Q10 has been shown to be relatively safe, with few adverse effects.
The bottom line. In some cases, coenzyme Q could be considered as a possible treatment for statin-induced myalgia, pending large-scale studies to determine if it is truly effective for this purpose.
Coenzyme Q10 supplements have been purported to be effective for treating a variety of disorders,1,2 in particular hypertension and statin-induced myalgia.
Several studies3–7 found that coenzyme Q10 supplementation significantly lowered blood pressure in hypertensive patients. Moreover, some trials have demonstrated that statin therapy reduces serum or muscle levels of coenzyme Q10,8–14 prompting investigations to determine whether coenzyme Q10 deficiency is related to statin-induced muscle pain.15–17
In this review, we discuss the efficacy and safety of coenzyme Q10 supplementation in patients with hypertension and those taking statins, and some of the caveats about using supplements that are not approved by the US Food and Drug Administration (FDA), as well as the bioavailability and quality of available formulations.
WHAT IS COENZYME Q10?
Coenzyme Q10, also known as coenzyme Q, ubidecarenone, and ubiquinone, is found in all human cells, with the highest concentrations in the heart, liver, kidney, and pancreas.1,2 It is a potent antioxidant, a membrane stabilizer, and an integral cofactor in the mitochondrial respiratory chain, helping to generate adenosine triphosphate, the major cellular energy source.1,2,18 It may also regulate genes associated with cell metabolism.19
RATIONALE FOR SUPPLEMENTATION
Coenzyme Q10 supplementation has been used, recommended, or studied in heart failure, hypertension, parkinsonism, mitochondrial encephalomyopathies, and other ailments.
In hypertension
Depending on the class, various antihypertensive drugs can have adverse effects such as depression, cough, and cardiac and renal dysfunction. 20,21 Furthermore, many patients need to take more than one drug to control their blood pressure, increasing their risk of side effects. Some researchers believe coenzyme Q10 supplementation may reduce the need to take multiple antihypertensive drugs.5
Coenzyme Q10 appears to lower blood pressure. The exact mechanism is not known, but one theory is that it reduces peripheral resistance by preserving nitric oxide.21 Nitric oxide relaxes peripheral arteries, lowering blood pressure. In some forms of hypertension, superoxide radicals that inactivate nitric oxide are overproduced; coenzyme Q10, with its antioxidant effects, may prevent the inactivation of nitric oxide by these free radicals. Alternatively, coenzyme Q10 may boost the production of the prostaglandin prostacyclin (PGI2) a potent vasodilator and inhibitor of platelet aggregation, or it may enhance the sensitivity of arterial smooth muscle to PGI2, or both.1,22
In patients taking statins
Hydroxymethylglutaryl coenzyme A reductase inhibitors (statins), first-line agents for lowering cholesterol levels to prevent cardiovascular disease, are some of the most commonly prescribed medications.23,24 However, statin therapy carries a risk of myopathy, which can range from muscle aches to rhabdomyolysis. 23,24
In a clinical advisory,25 the American College of Cardiology, the American Heart Association, and the National Heart, Lung, and Blood Institute recommend that patients on statin therapy who experience muscle soreness, tenderness, or pain with serum creatine kinase levels 3 to 10 times the upper limit of normal should have their creatine kinase level checked weekly. If the level is 3 to 10 times the upper limit of normal, statin therapy may be continued, but if it exceeds 10 times the upper limit, then statins and other potential offending agents (eg, niacin, fibrate) need to be discontinued.
Statins inhibit the synthesis of cholesterol by reducing the production of mevalonate, a precursor of both cholesterol and coenzyme Q10. Since both cholesterol and coenzyme Q10 are produced by the same pathway, it is not surprising that statins have been reported to reduce serum and muscle coenzyme Q10 levels.9–14 However, one study did not report a significant reduction of coenzyme Q10 levels in muscle tissue in patients treated with simvastatin 20 mg for 6 months.26
Nonetheless, researchers have hypothesized that a reduction in coenzyme Q10 levels in muscle tissue causes mitochondrial dysfunction, which could increase the risk of statininduced myopathy,13–17 and some believe that treatment with coenzyme Q10 may reduce myalgic symptoms and allow patients to remain on statin therapy.13,24
Researchers have investigated the potential of coenzyme Q10 supplementation to reduce or prevent statin-induced myopathy.15–17 (More on this below.)
Interestingly, a randomized, placebo-controlled trial27 found that 6 months of daily therapy with simvastatin (Zocor) 20 mg or pravastatin (Pravachol) 40 mg lowered systolic and diastolic blood pressure significantly in patients with no documented history of cardiovascular disease or diabetes. A possible mechanism of statin-induced blood pressure reduction is the up-regulation of endothelial nitric oxide synthetase, a potent vasodilator. Coenzyme Q10 levels were not assessed during this study. Whether coenzyme Q10 supplementation used to treat statin-induced myalgia enhances or inhibits the antihypertensive effects of statins is not yet known.
EVIDENCE OF EFFECTIVENESS IN HYPERTENSION
Rosenfeldt et al28 performed a meta-analysis and found that some trials documented statistically significant reductions in diastolic or systolic blood pressure or both, while others reported negligible effects.3,29 In one small trial,30 blood pressures actually went up in patients taking coenzyme Q10. Coenzyme Q10 dosages and length of therapy varied from study to study in the meta-analysis. Only minor adverse effects such as gastrointestinal upset and headache were reported.
Yamagami et al3 randomly assigned 20 patients with hypertension and a low coenzyme Q10 level to receive 100 mg of coenzyme Q10 or placebo daily for 12 weeks. Patients continued their usual antihypertensive regimen during the study period. Blood pressures, coenzyme Q10 levels, and antihypertensive drugs used were comparable between the study groups.
After 12 weeks of therapy, the mean coenzyme Q10 level in the active-treatment group had more than doubled, from 0.704 to 1.597 μg/mL. This group also experienced a statistically significant drop in systolic blood pressure, from 167 mm Hg at baseline to 148 mm Hg at 12 weeks. In the placebo group, the systolic blood pressure was 168 mm Hg at baseline and 164 mm Hg at 12 weeks; the change was not statistically significant. Diastolic pressure was not significantly lower at 12 weeks than at baseline in either group.
The authors concluded that coenzyme Q10 supplementation brought a mild reduction in high blood pressure in patients who had low coenzyme Q10 serum levels.
Digiesi et al31 randomized 18 patients with essential hypertension to receive either coenzyme Q10 100 mg or placebo daily for 10 weeks. All antihypertensive therapy was discontinued at baseline. After the first 10 weeks, patients went through a 2-week washout period and then were switched to the opposite therapy for an additional 10 weeks. Mean baseline blood pressure values were 167 mm Hg systolic and 103 mm Hg diastolic.
Those taking the supplement had a statistically significant decrease in systolic and diastolic pressures (P < .001). The antihypertensive effect was noted in the 3rd or 4th week of active treatment and persisted for the duration of therapy. The effects dissipated 7 to 10 days after coenzyme Q10 was stopped.
Langsjoen et al5 evaluated the effects of adding coenzyme Q10 to the antihypertensive drug regimen of 109 patients who had a primary diagnosis of essential hypertension in a prospective observational study. Patients with hypertension as a secondary diagnosis and other cardiovascular diseases were excluded. Variable doses of coenzyme Q10 were given, adjusted according to clinical response and to achieve serum levels greater than 2.0 μg/mL. The average dose was 225 mg/day; the mean serum level attained was 3.02 μg/mL.
Over several months, patients taking the supplement had a reduction in mean systolic pressure from 159 mm Hg at baseline to 147 mm Hg (P < .001), and a reduction in mean diastolic pressure from 94 to 85 mm Hg (P < .001). Thirty-seven percent of patients were able to discontinue one antihypertensive drug, 11% discontinued two drugs, and 4% were able to stop taking three drugs. However, 46% remained on the same antihypertensive regimen, and 3% needed an additional drug.
Singh et al6 randomized 64 patients who had coronary artery disease and who had been on antihypertensive drugs for more than 1 year to receive either B-complex vitamins or coenzyme Q10 (hydrosoluble Q-Gel) 60 mg orally once daily for 8 weeks. Five patients were not available for follow-up; therefore, only 59 patients were evaluated. Fifty-five (93%) of the 59 patients were taking only one antihypertensive drug. Initial antihypertensive drug use was similar between study groups and was continued throughout the trial.
After 8 weeks of therapy, the coenzyme Q10 group had significantly lower systolic and diastolic blood pressure than the placebo group (P < .05 for both). There was also a statistically significant decrease in the dosage of antihypertensive drugs in the coenzyme Q10 group but not in the placebo group (P < .05), reflecting coenzyme Q10’s additive antihypertensive effect.
Burke et al7 randomized 41 men and 35 women with isolated systolic hypertension (systolic pressure 150–170 mm Hg, diastolic pressure < 90 mm Hg) to receive a twice-daily dose of 60 mg of emulsified coenzyme Q10 (hydrosoluble Q-Gel) with 150 IU of vitamin E or placebo containing vitamin E alone for 12 weeks. The study also included 5 men and 4 women with normal blood pressure, all of whom received coenzyme Q10. A total of 80 patients completed treatment. The primary goal of the study was to determine the efficacy of coenzyme Q10 in the treatment of isolated systolic hypertension in patients without comorbid conditions. Blood pressures were monitored twice a week during the trial, by the same nurse.
After 12 weeks of treatment, the mean reduction in systolic pressure in hypertensive patients on coenzyme Q10 was 17.8 ± 7.3 mm Hg. There were no significant changes in diastolic pressure in any study group with treatment. Patients with isolated systolic hypertension who were taking coenzyme Q10 had a statistically significant reduction in systolic pressure compared with baseline and placebo (P < .01 for both). Approximately 55% of patients on coenzyme Q10 achieved a reduction in systolic pressure of 4 mm Hg or greater, while 45% did not respond to therapy. The mean plasma coenzyme Q10 level of the treatment group increased from 0.47 ± 0.19 μg/mL to 2.69 ± 0.54 μg/mL after 12 weeks; however, the study did not have the statistical power to demonstrate a relationship between coenzyme Q10 levels and changes in blood pressure. Twenty-seven (34%) of the 80 patients were taking a statin while on coenzyme Q10 therapy.
STUDIES IN STATIN-INDUCED MYOPATHY
Thibault et al32 and Kim et al33 reported that patients taking lovastatin (Mevacor) at dosages as high as 35 mg/kg/day to inhibit tumor growth achieved symptomatic relief of statin-induced musculoskeletal toxicity after coenzyme Q10 supplementation.
Caso et al15 performed a small pilot study in 32 patients to determine if coenzyme Q10 supplementation would improve myalgic symptoms in patients treated with statins. In this double-blind, randomized trial, patients received either coenzyme Q10 100 mg/day or vitamin E 400 IU/day for 30 days. The extent of muscle pain and its interference with daily activities were determined before and after therapy using the Brief Pain Inventory Questionnaire. The statins were atorvastatin (Lipitor) 10 mg or 20 mg, lovastatin 40 mg, pravastatin 40 mg, and simvastatin 10, 20, 40, and 80 mg. Five patients in the coenzyme Q10 group and four patients in the vitamin E group were taking nonsteroidal anti-inflammatory drugs before and during the trial. The intensity of muscle pain and its interference with daily activities were similar between study groups before the start of therapy.
After 30 days of treatment with coenzyme Q10, the pain intensity had decreased significantly from baseline (P < .001). In contrast, no change in pain intensity from baseline was noted in patients receiving vitamin E. The Pain Severity Score was significantly different between study groups, favoring the coenzyme Q10 group (P < .001). Sixteen of 18 patients on coenzyme Q10 reported a reduction in pain, while only 3 of 14 patients on vitamin E reported a similar response. Also, the interference of pain with daily activities significantly improved with coenzyme Q10 (P < .02), whereas vitamin E did not have a significant impact on this.
Young et al17 randomized 44 patients with prior statin-induced myalgia to receive increasing doses of simvastatin (10–40 mg/day) in combination with either coenzyme Q10 (Q-Gel) 200 mg/day or placebo. The primary goal was to determine if coenzyme Q10 supplementation would help improve statin tolerance in patients with a history of statininduced myalgia. Plasma coenzyme Q10 and lipid levels were measured at baseline and at the end of the study. The intensity of myalgia was assessed with a visual analogue scale.
At 12 weeks, the coenzyme Q10 plasma level was significantly higher in the treatment group than in the placebo group (P < .001). However, no differences were noted between groups in the number of patients who tolerated the 40-mg/day simvastatin dose (P = .34) or in the number of patients who remained on any simvastatin dose (P = .47). Additionally, myalgia scores did not differ between groups (P = .63). The authors acknowledged that there were only small increases in the myalgia pain scores reported in either group. Therefore, patients in the treatment group may not have experienced sufficiently severe muscle pain to have benefited from coenzyme Q10 supplementation.
IS COENZYME Q10 SAFE?
Studies have indicated that these supplements are well tolerated, with relatively few adverse effects or potential drug interactions.1,2,34
The FDA does not routinely assess the purity or quality of over-the-counter coenzyme Q10 products.35 However, the United States Pharmacopeia (USP) does test dietary supplements to make sure that they are not mislabeled and that they do not contain contaminants. 36
A USP-verified dietary supplement should:
- Contain the exact ingredients listed on the label in the listed potency and amounts
- Not include harmful levels of certain contaminants such as lead, mercury, pesticides, or bacteria
- Appropriately disintegrate and release its contents into the body within a specified period of time
- Be produced using the FDA’s current Good Manufacturing Practices.36
Side effects, contraindications, warnings
Coenzyme Q10 is a relatively safe dietary supplement. It is contraindicated in patients who are allergic to it or to any of its components.2 Most clinical trials have not reported significant adverse effects that necessitated stopping therapy.34 However, gastrointestinal effects such as abdominal discomfort, nausea, vomiting, diarrhea, and anorexia have occurred.1,2,34 Allergic rash and headache have also been reported.1,2,34 In addition, coenzyme Q10’s antiplatelet effect may increase the risk of bleeding. 37,38 It undergoes biotransformation in the liver and is eliminated primarily via the biliary tract,39 so it can accumulate in patients with hepatic impairment or biliary obstruction.
Interactions with drugs
Coenzyme Q10’s effects on platelet function may increase the risk of bleeding in patients taking antiplatelet drugs such as aspirin or clopidogrel (Plavix).37,38 On the other hand, since it acts like vitamin K, it may counteract the anticoagulant effects of warfarin (Coumadin). 1,2,40
Coenzyme Q10 may have an additive antihypertensive effect when given with antihypertensive drugs.41
Coenzyme Q10 may improve beta-cell function and enhance insulin sensitivity, which may reduce insulin requirements for diabetic patients.42,43
SLOWLY ABSORBED
Coenzyme Q10 is absorbed slowly from the gastrointestinal tract, possibly because it has a high molecular weight and is not very watersoluble. 39
One pharmacokinetic study found that after a single 100-mg oral dose of coenzyme Q10, the mean peak plasma levels of about 1 μg/mL occurred between 5 and 10 hours (mean 6.5 hours).44 Coenzyme Q10 100 mg given orally three times daily produced a mean steadystate plasma level of 5.4 μg/mL; about 90% of this steady-state concentration was achieved after 4 days.39
Some formulations have significantly better oral bioavailability and therefore produce higher plasma levels. Soft-gel capsules, especially those with vegetable oil or vitamin E, may have better absorption.43
A pharmacokinetic study showed that the area under the curve of the plasma coenzyme Q10 concentration was more than twice as high with Q-Gel soft-gel capsules, a completely solubilized formulation, than with softgel capsules with an oil suspension, powderfilled hard-shell capsules, or regular tablets.45 Another study reported that colloidal-Q10, a formulation contained in VESIsorb (a novel drug delivery system sold as CoQsource) had greater bioavailability than solubilized and oil-based preparations.46 Commercially available solubilized preparations containing ubiquinol, a metabolized form of coenzyme Q10, have been shown to produce higher serum levels than solubilized products.47
Of note: unless the manufacturer claims that its product is water-soluble, the USP does not evaluate its dissolution rate.48 Therefore, USP-verified coenzyme Q10 products that are not water-soluble may have lower bioavailability than their solubilized counterparts.
Dry dosage forms of coenzyme Q10 (eg, tablets, capsules) may be more readily absorbed if taken with a fatty meal.43
SLOWLY ELIMINATED
Taken orally, coenzyme Q10 has a low clearance rate, with an elimination half-life of about 34 hours.39
After absorption, exogenous coenzyme Q10 is taken up by chylomicrons that transport it to the liver, where it is incorporated into verylow-density lipoproteins. It is then distributed to various organs, including the adrenal glands, spleen, kidneys, lungs, and heart. Coenzyme Q10 is eliminated primarily via the biliary tract. About 60% of an oral dose is eliminated in the feces during chronic oral administration.39
TWICE-DAILY DOSING
A typical daily dose of coenzyme Q10 for treating hypertension is 120 to 200 mg, usually given orally in two divided doses.1 For statininduced myopathy, 100 to 200 mg orally daily has been used.1
Coenzyme Q10 is given in divided doses to enhance its absorption and to minimize gastrointestinal effects.1,43 Taking it with a fatty meal may also increase its absorption.43
Since solubilized forms of coenzyme Q10 and ubiquinol have significantly greater bioavailability than nonsolubilized forms, the therapeutic dose of these formulations may be lower.47
MONITORING DURING TREATMENT
Without supplementation, the mean serum level of endogenous coenzyme Q10 has been reported to be 0.99 ± 0.30 mg/L (range 0.55– 1.87).18 Serum levels above 2 μg/mL have been associated with significant reductions in blood pressure.5,7,28
The possible effects of coenzyme Q10 on blood pressure, blood glucose levels, serum creatine kinase levels, and myopathic symptoms should be kept in mind when monitoring patients who have hypertension,41 diabetes,41,42 or statin-induced myalgia.15,17 Coenzyme Q10’s possible potentiating effects on antiplatelet drugs and its inhibitory effect on warfarin should be kept in mind as well.
COST VARIES
Coenzyme Q10 is available in different dosage forms (eg, regular and rapid-release softgel capsules, regular and chewable tablets, chewable wafers, and liquid) from a variety of manufacturers. Products come in different strengths, typically ranging from 30 to 400 mg. USP-verified formulations are listed at www.usp.org/USPVerified/dietarySupplements/under “Verified Supplements.” Only USP-verified products that claim to be water-soluble meet USP dissolution requirements.
The cost varies, depending on the vendor. In general, dosage forms with greater bioavailability, such as Q-Gel and ubiquinol supplements, are more expensive. For example, a regimen of 60 mg twice daily of regular-release coenzyme Q capsules may cost approximately $20 per month, compared with $60 per month for the same supply of Q-Gel Ultra capsules. However, in some cases, supplements that produce higher serum levels may be more cost-effective.
CURRENT ROLE IN THERAPY
As an antihypertensive adjunct
Several small clinical trials have shown that coenzyme Q10 supplementation can lower blood pressure. The supplements were reported to be safe and well tolerated. Moreover, some patients with essential hypertension who were taking coenzyme Q10 were able to discontinue one or more antihypertensive drugs. A significant reduction in blood pressure with use of coenzyme Q10 would be expected to reduce the adverse consequences of hypertension in the same manner as conventional antihypertensive agents.
However, no large, double-blind, randomized study has evaluated the impact of coenzyme Q10 when taken with other antihypertensive drugs (eg, angiotensin-converting enzyme inhibitors, beta-blockers, diuretics) on specific clinical end points such as the incidence of stroke or death from a major cardiac event. Furthermore, its effects on cardiac function, exercise tolerance, and quality of life have not been determined.
The bottom line. In some cases, it seems reasonable to recommend this product as an adjunct to conventional antihypertensive therapy. Larger, well-designed clinical trials of coenzyme Q10’s antihypertensive effects on specific clinical end points such as the risk of stroke or myocardial infarction are needed to define its true therapeutic value.
As a treatment for statin-induced myalgia
Clinical evidence supporting coenzyme Q10’s use in the treatment of statin-induced myopathy is limited. Whether coenzyme Q10 is depleted from muscle tissue during statin therapy has not been confirmed. Supplementation helped reduce the severity of musculoskeletal effects of megadoses of lovastatin. However, clinical trials of coenzyme Q10 in the treatment of myalgia associated with antilipidemic statin doses did not consistently report significant improvement. Nevertheless, coenzyme Q10 has been shown to be relatively safe, with few adverse effects.
The bottom line. In some cases, coenzyme Q could be considered as a possible treatment for statin-induced myalgia, pending large-scale studies to determine if it is truly effective for this purpose.
- Jelin JM, Gregory PJ, et al. Natural medicines comprehensive database/compiled by the editors of Pharmacist’s Letter, Prescriber’s Letter. 11th ed. Stockton, CA: Therapeutic Research Faculty; 2009:452–457.
- Fetrow CW, Avila JR. Professional’s Handbook of Complementary & Alternative Medicines. 2nd ed. Springhouse, PA: Springhouse; 2001:211–215.
- Yamagami T, Takagi M, Akagami H, et al. Effect of coenzyme Q10 on essential hypertension, a double-blind controlled study. In:Folkers K, Yamamura Y, editors. Biomedical and Clinical Aspects of Coenzyme Q10: Proceedings of the Fifth International Symposium on the Biomedical and Clinical Aspects of Coenzyme Q10, vol 5. Amsterdam: Elsevier Science Publishers; 1986:337–343.
- Digiesi V, Cantini F, Oradei A, et al. Coenzyme Q10 in essential hypertension. Mol Aspects Med 1994; 15(suppl):S257–S263.
- Langsjoen P, Langsjoen P, Willis R, Folkers K. Treatment of essential hypertension with coenzyme Q10. Mol Aspects Med 1994; 15(suppl):S265–S272.
- Singh RB, Niaz MA, Rastogi SS, Shukla PK, Thakur AS. Effect of hydrosoluble coenzyme Q10 on blood pressures and insulin resistance in hypertensive patients with coronary artery disease. J Hum Hypertens 1999; 13:203–208.
- Burke BE, Neuenschwander R, Olson RD. Randomized, double-blind, placebo-controlled trial of coenzyme Q10 in isolated systolic hypertension. South Med J 2001; 94:1112–1117.
- De Pinieux G, Chariot P, Ammi-Saïd M, et al. Lipidlowering drugs and mitochondrial function: effects of HMG-CoA reductase inhibitors on serum ubiquinone and blood lactate/pyruvate ratio. Br J Clin Pharmacol 1996; 42:333–337.
- Mortensen SA, Leth A, Agner E, Rohde M. Dose-related decrease of serum coenzyme Q10 during treatment with HMG-CoA reductase inhibitors. Mol Aspects Med 1997; 18(suppl):S137–S144.
- Ghirlanda G, Oradei A, Manto A, et al. Evidence of plasma CoQ10-lowering effect by HMG-CoA reductase inhibitors: a double-blind, placebo-controlled study. J Clin Pharmacol 1993; 33:226–229.
- Folkers K, Langsjoen P, Willis R, et al. Lovastatin decreases coenzyme Q10 levels in humans. Proc Natl Acad Sci U S A 1990; 87:8931–8934.
- Watts GF, Castelluccio C, Rice-Evans C, Taub NA, Baum H, Quinn PJ. Plasma coenzyme Q10 (ubiquinone) concentrations in patients treated with simvastatin. J Clin Pathol 1993; 46:1055–1057.
- Lamperti C, Naini AB, Lucchini V, et al. Muscle coenzyme Q10 level in statin-related myopathy. Arch Neurol 2005; 62:1709–1712.
- Päivä H, Thelen KM, Van Coster R, et al. High-dose statins and skeletal muscle metabolism in humans: a randomized, controlled trial. Clin Pharmacol Ther 2005; 78:60–68.
- Caso G, Kelly P, McNurlan MA, Lawson WE. Effect of coenzyme Q10 on myopathic symptoms in patients treated with statins. Am J Cardiol 2007; 99:1409–1412.
- Marcoff L, Thompson PD. The role of coenzyme Q10 in statin-associated myopathy: a systematic review. J Am Coll Cardiol 2007; 49:2231–2237.
- Young JM, Florkowski CM, Molyneux SL, et al. Effect of coenzyme Q(10) supplementation on simvastatin-induced myalgia. Am J Cardiol 2007; 100:1400–1403.
- Berthold HK, Naini A, Di Mauro S, et al. Effect of ezetimibe and/or simvastatin on coenzyme Q10 levels in plasma: a randomised trial. Drug Saf 2006; 29:703–712.
- Groneberg DA, Kindermann B, Althammer M, et al. Coenzyme Q10 affects expression of genes involved in cell signalling, metabolism and transport in human CaCo-2 cells. Int J Biochem Cell Biol 2005; 37:1208–1218.
- Hadj A, Pepe S, Rosenfeldt F. The clinical application of metabolic therapy for cardiovascular disease. Heart Lung Circ 2007; 16(suppl 3):S56–S64.
- Pepe S, Marasco SF, Haas SJ, Sheeran FL, Krum H, Rosenfeldt FL. Coenzyme Q10 in cardiovascular disease. Mitochondrion 2007; 7(suppl 1):S154–S167.
- Lönnrot K, Pörsti I, Alho H, Wu X, Hervonen A, Tolvanen JP. Control of arterial tone after long-term coenzyme Q10 supplementation in senescent rats. Br J Pharmacol 1998; 124:1500–1506.
- Sewright KA, Clarkson PM, Thompson PD. Statin myopathy: incidence, risk factors, and pathophysiology. Curr Atheroscler Rep 2007; 9:389–396.
- Radcliffe KA, Campbell WW. Statin myopathy. Curr Neurol Neurosci Rep 2008; 8:66–72.
- Pasternak RC, Smith SC, Bairey-Merz CN, Grundy SM, Cleeman JI, Lenfant C. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. Circulation 2002; 106:1024–1028.
- Laaksonen R, Jokelainen K, Laakso J, et al. The effect of simvastatin treatment on natural antioxidants in low-density lipoproteins and high-energy phosphates and ubiquinone in skeletal muscle. Am J Cardiol 1996; 77:851–854.
- Golomb BA, Dimsdale JE, White HL, Ritchie JB, Criqui MH. Reduction in blood pressure with statins: results from the UCSD Statin Study, a randomized trial. Arch Intern Med 2008; 168:721–727.
- Rosenfeldt FL, Haas SJ, Krum H, et al. Coenzyme Q10 in the treatment of hypertension: a meta-analysis of the clinical trials. J Hum Hypertens 2007; 21:297–306.
- Yamagami T, Shibata N, Folkers K. Bioenergetics in clinical medicine. Studies on coenzyme Q10 and essential hypertension. Res Commun Chem Pathol Pharmacol 1975; 11:273–288.
- Yamagami T, Shibata N, Folkers K. Study of coenzyme Q10. In:Folkers K, Yamamura Y, editors. Biomedical and clinical aspects of coenzyme Q10: proceedings of the International Symposium on Coenzyme Q10, held at Lake Yamanaka, Japan, September 16/17, 1976, a Naito Foundation symposium. Amsterdam: Elsevier Scientific Publishing Company; 1977:231–242.
- Digiesi V, Cantini F, Brodbeck B. Effect of coenzyme Q10 on essential arterial hypertension. Curr Ther Res; 1990; 47:841–845.
- Thibault A, Samid D, Tompkins AC, et al. Phase I study of lovastatin, an inhibitor of the mevalonate pathway, in patients with cancer. Clin Cancer Res 1996; 2:483–491.
- Kim WS, Kim MM, Choi HJ, et al. Phase II study of high-dose lova-statin in patients with advanced gastric adenocarcinoma. Invest New Drugs 2001; 19:81–83.
- Hidaka T, Fujii K, Funahashi I, Fukutomi N, Hosoe K. Safety assessment of coenzyme Q10 (CoQ10). Biofactors 2008; 32:199–208.
- US Food and Drug Administration. Consumer Information on Dietary Supplements. Overview of Dietary Supplements. http://www.fda.gov/Food/DietarySupplements/ConsumerInformation/. Accessed May 25, 2010.
- US Pharmacopeia. The USP Dietary Supplement Verification Program http://www.usp.org/USPVerified/dietary-Supplements/. Accessed May 25, 2010.
- Serebruany VL, Ordonez JV, Herzog WR, et al. Dietary coenzyme Q10 supplementation alters platelet size and inhibits human vitronectin (CD51/CD61) receptor expression. J Cardiovasc Pharmacol 1997; 29:16–22.
- A close look at coenzyme Q10 and policosanol. Do these supplements live up to their claims for improving heart health? Harv Heart Lett 2002; 13:6.
- Greenberg S, Frishman WH. Co-enzyme Q10: a new drug for cardiovascular disease. J Clin Pharmacol 1990; 30:596–608.
- Singh U, Devaraj S, Jialal I. Coenzyme Q10 supplementation and heart failure. Nutr Rev 2007; 65:286–293.
- Bonakdar RA, Guarneri E. Coenzyme Q10. Am Fam Physician 2005; 72:1065–1070.
- Hodgson JM, Watts GF, Playford DA, Burke V, Croft KD. Coenzyme Q10 improves blood pressure and glycaemic control: a controlled trial in subjects with type 2 diabetes. Eur J Clin Nutr 2002; 56:1137–1142.
- Pepping J. Coenzyme Q10. Am J Health Syst Pharm 1999; 56:519–521.
- Tomono Y, Hasegawa J, Seki T, Motegi K, Morishita N. Pharmacokinetic study of deuterium-labelled coenzyme Q10 in man. Int J Clin Pharmacol Ther Toxicol 1986; 24:536–541.
- Chopra RK, Goldman R, Sinatra ST, Bhagavan HN. Relative bioavailability of coenzyme Q10 formulations in human subjects. Int J Vitam Nutr Res 1998; 68:109–113.
- Liu ZX, Artmann C. Relative bioavailability comparison of different coenzyme Q10 formulations with a novel delivery system. Altern Ther Health Med 2009; 15:42–46.
- Bhagavan HN, Chopra RK. Plasma coenzyme Q10 response to oral ingestion of coenzyme Q10 formulations. Mitochondrion 2007; 7(suppl 1):S78–S88.
- The United States Pharmacopeia. Ubidecarenone Capsules Monograph. 32nd Revision. Baltimore: United Book Press, 2009:1080.
- Jelin JM, Gregory PJ, et al. Natural medicines comprehensive database/compiled by the editors of Pharmacist’s Letter, Prescriber’s Letter. 11th ed. Stockton, CA: Therapeutic Research Faculty; 2009:452–457.
- Fetrow CW, Avila JR. Professional’s Handbook of Complementary & Alternative Medicines. 2nd ed. Springhouse, PA: Springhouse; 2001:211–215.
- Yamagami T, Takagi M, Akagami H, et al. Effect of coenzyme Q10 on essential hypertension, a double-blind controlled study. In:Folkers K, Yamamura Y, editors. Biomedical and Clinical Aspects of Coenzyme Q10: Proceedings of the Fifth International Symposium on the Biomedical and Clinical Aspects of Coenzyme Q10, vol 5. Amsterdam: Elsevier Science Publishers; 1986:337–343.
- Digiesi V, Cantini F, Oradei A, et al. Coenzyme Q10 in essential hypertension. Mol Aspects Med 1994; 15(suppl):S257–S263.
- Langsjoen P, Langsjoen P, Willis R, Folkers K. Treatment of essential hypertension with coenzyme Q10. Mol Aspects Med 1994; 15(suppl):S265–S272.
- Singh RB, Niaz MA, Rastogi SS, Shukla PK, Thakur AS. Effect of hydrosoluble coenzyme Q10 on blood pressures and insulin resistance in hypertensive patients with coronary artery disease. J Hum Hypertens 1999; 13:203–208.
- Burke BE, Neuenschwander R, Olson RD. Randomized, double-blind, placebo-controlled trial of coenzyme Q10 in isolated systolic hypertension. South Med J 2001; 94:1112–1117.
- De Pinieux G, Chariot P, Ammi-Saïd M, et al. Lipidlowering drugs and mitochondrial function: effects of HMG-CoA reductase inhibitors on serum ubiquinone and blood lactate/pyruvate ratio. Br J Clin Pharmacol 1996; 42:333–337.
- Mortensen SA, Leth A, Agner E, Rohde M. Dose-related decrease of serum coenzyme Q10 during treatment with HMG-CoA reductase inhibitors. Mol Aspects Med 1997; 18(suppl):S137–S144.
- Ghirlanda G, Oradei A, Manto A, et al. Evidence of plasma CoQ10-lowering effect by HMG-CoA reductase inhibitors: a double-blind, placebo-controlled study. J Clin Pharmacol 1993; 33:226–229.
- Folkers K, Langsjoen P, Willis R, et al. Lovastatin decreases coenzyme Q10 levels in humans. Proc Natl Acad Sci U S A 1990; 87:8931–8934.
- Watts GF, Castelluccio C, Rice-Evans C, Taub NA, Baum H, Quinn PJ. Plasma coenzyme Q10 (ubiquinone) concentrations in patients treated with simvastatin. J Clin Pathol 1993; 46:1055–1057.
- Lamperti C, Naini AB, Lucchini V, et al. Muscle coenzyme Q10 level in statin-related myopathy. Arch Neurol 2005; 62:1709–1712.
- Päivä H, Thelen KM, Van Coster R, et al. High-dose statins and skeletal muscle metabolism in humans: a randomized, controlled trial. Clin Pharmacol Ther 2005; 78:60–68.
- Caso G, Kelly P, McNurlan MA, Lawson WE. Effect of coenzyme Q10 on myopathic symptoms in patients treated with statins. Am J Cardiol 2007; 99:1409–1412.
- Marcoff L, Thompson PD. The role of coenzyme Q10 in statin-associated myopathy: a systematic review. J Am Coll Cardiol 2007; 49:2231–2237.
- Young JM, Florkowski CM, Molyneux SL, et al. Effect of coenzyme Q(10) supplementation on simvastatin-induced myalgia. Am J Cardiol 2007; 100:1400–1403.
- Berthold HK, Naini A, Di Mauro S, et al. Effect of ezetimibe and/or simvastatin on coenzyme Q10 levels in plasma: a randomised trial. Drug Saf 2006; 29:703–712.
- Groneberg DA, Kindermann B, Althammer M, et al. Coenzyme Q10 affects expression of genes involved in cell signalling, metabolism and transport in human CaCo-2 cells. Int J Biochem Cell Biol 2005; 37:1208–1218.
- Hadj A, Pepe S, Rosenfeldt F. The clinical application of metabolic therapy for cardiovascular disease. Heart Lung Circ 2007; 16(suppl 3):S56–S64.
- Pepe S, Marasco SF, Haas SJ, Sheeran FL, Krum H, Rosenfeldt FL. Coenzyme Q10 in cardiovascular disease. Mitochondrion 2007; 7(suppl 1):S154–S167.
- Lönnrot K, Pörsti I, Alho H, Wu X, Hervonen A, Tolvanen JP. Control of arterial tone after long-term coenzyme Q10 supplementation in senescent rats. Br J Pharmacol 1998; 124:1500–1506.
- Sewright KA, Clarkson PM, Thompson PD. Statin myopathy: incidence, risk factors, and pathophysiology. Curr Atheroscler Rep 2007; 9:389–396.
- Radcliffe KA, Campbell WW. Statin myopathy. Curr Neurol Neurosci Rep 2008; 8:66–72.
- Pasternak RC, Smith SC, Bairey-Merz CN, Grundy SM, Cleeman JI, Lenfant C. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. Circulation 2002; 106:1024–1028.
- Laaksonen R, Jokelainen K, Laakso J, et al. The effect of simvastatin treatment on natural antioxidants in low-density lipoproteins and high-energy phosphates and ubiquinone in skeletal muscle. Am J Cardiol 1996; 77:851–854.
- Golomb BA, Dimsdale JE, White HL, Ritchie JB, Criqui MH. Reduction in blood pressure with statins: results from the UCSD Statin Study, a randomized trial. Arch Intern Med 2008; 168:721–727.
- Rosenfeldt FL, Haas SJ, Krum H, et al. Coenzyme Q10 in the treatment of hypertension: a meta-analysis of the clinical trials. J Hum Hypertens 2007; 21:297–306.
- Yamagami T, Shibata N, Folkers K. Bioenergetics in clinical medicine. Studies on coenzyme Q10 and essential hypertension. Res Commun Chem Pathol Pharmacol 1975; 11:273–288.
- Yamagami T, Shibata N, Folkers K. Study of coenzyme Q10. In:Folkers K, Yamamura Y, editors. Biomedical and clinical aspects of coenzyme Q10: proceedings of the International Symposium on Coenzyme Q10, held at Lake Yamanaka, Japan, September 16/17, 1976, a Naito Foundation symposium. Amsterdam: Elsevier Scientific Publishing Company; 1977:231–242.
- Digiesi V, Cantini F, Brodbeck B. Effect of coenzyme Q10 on essential arterial hypertension. Curr Ther Res; 1990; 47:841–845.
- Thibault A, Samid D, Tompkins AC, et al. Phase I study of lovastatin, an inhibitor of the mevalonate pathway, in patients with cancer. Clin Cancer Res 1996; 2:483–491.
- Kim WS, Kim MM, Choi HJ, et al. Phase II study of high-dose lova-statin in patients with advanced gastric adenocarcinoma. Invest New Drugs 2001; 19:81–83.
- Hidaka T, Fujii K, Funahashi I, Fukutomi N, Hosoe K. Safety assessment of coenzyme Q10 (CoQ10). Biofactors 2008; 32:199–208.
- US Food and Drug Administration. Consumer Information on Dietary Supplements. Overview of Dietary Supplements. http://www.fda.gov/Food/DietarySupplements/ConsumerInformation/. Accessed May 25, 2010.
- US Pharmacopeia. The USP Dietary Supplement Verification Program http://www.usp.org/USPVerified/dietary-Supplements/. Accessed May 25, 2010.
- Serebruany VL, Ordonez JV, Herzog WR, et al. Dietary coenzyme Q10 supplementation alters platelet size and inhibits human vitronectin (CD51/CD61) receptor expression. J Cardiovasc Pharmacol 1997; 29:16–22.
- A close look at coenzyme Q10 and policosanol. Do these supplements live up to their claims for improving heart health? Harv Heart Lett 2002; 13:6.
- Greenberg S, Frishman WH. Co-enzyme Q10: a new drug for cardiovascular disease. J Clin Pharmacol 1990; 30:596–608.
- Singh U, Devaraj S, Jialal I. Coenzyme Q10 supplementation and heart failure. Nutr Rev 2007; 65:286–293.
- Bonakdar RA, Guarneri E. Coenzyme Q10. Am Fam Physician 2005; 72:1065–1070.
- Hodgson JM, Watts GF, Playford DA, Burke V, Croft KD. Coenzyme Q10 improves blood pressure and glycaemic control: a controlled trial in subjects with type 2 diabetes. Eur J Clin Nutr 2002; 56:1137–1142.
- Pepping J. Coenzyme Q10. Am J Health Syst Pharm 1999; 56:519–521.
- Tomono Y, Hasegawa J, Seki T, Motegi K, Morishita N. Pharmacokinetic study of deuterium-labelled coenzyme Q10 in man. Int J Clin Pharmacol Ther Toxicol 1986; 24:536–541.
- Chopra RK, Goldman R, Sinatra ST, Bhagavan HN. Relative bioavailability of coenzyme Q10 formulations in human subjects. Int J Vitam Nutr Res 1998; 68:109–113.
- Liu ZX, Artmann C. Relative bioavailability comparison of different coenzyme Q10 formulations with a novel delivery system. Altern Ther Health Med 2009; 15:42–46.
- Bhagavan HN, Chopra RK. Plasma coenzyme Q10 response to oral ingestion of coenzyme Q10 formulations. Mitochondrion 2007; 7(suppl 1):S78–S88.
- The United States Pharmacopeia. Ubidecarenone Capsules Monograph. 32nd Revision. Baltimore: United Book Press, 2009:1080.
KEY POINTS
- In some clinical trials, coenzyme Q10 supplements significantly lowered diastolic and systolic blood pressure.
- Statins may lower coenzyme Q10 serum levels, and some investigators have evaluated the relationship between coenzyme Q10 deficiency and statin-related myalgia, but more evidence is needed to support the use of coenzyme Q10 supplements to prevent or treat myalgia.
- Coenzyme Q10 supplementation appears to be relatively safe. Most clinical trials have not reported significant side effects that necessitated stopping therapy. Gastrointestinal effects include abdominal discomfort, nausea, vomiting, diarrhea, and anorexia. Allergic rash and headache have also been reported.
Escaping the heat in the EMR pool

In reply, based on his experience, Dr. Tom Abelson defends EMRs, predicting that their current weaknesses will be overcome as technology improves, and the faster we embrace this technologic advance, the faster fixes for initial limitations will be developed. We are, he says, just learning to swim.
The EMR is a tool, but a tool performs at the skill level of the user. The EMR is more powerful than the paper record it replaces, offering the promise of being searchable, interactive, and able to prompt us to perform in predefined ways, and also linking us at the point of care with reference materials. But the information contained in the EMR can be no better than what we enter. An EMR cannot supplant our need to think and act as physicians defending our patients’ best interests.
Our skill in using the EMR is evolving. Thus far, cut-and-paste and other electronic shortcuts are rampant and are a detriment to quality care, but these are examples of misuse and are not an intrinsic fault of the tool. These physician behaviors can be curtailed.
The EMR cannot be read like a book. Events, consultations, and nursing interventions do not readily unfold in chronologic order. Suggestions of consultants can be missed, and perhaps due to limited typing skills, clinical reasoning is not fully explained (was it always clearly explained on paper?). The notes, however, are legible.
We must not be enticed to let EMRs overly influence our billing practices. Rather, the EMR should be a tool to improve the accuracy of the record of the patient encounter.
The EMR can come between the doctor and patient, but it need not. We need to be a bit more attentive to the patient, push back intermittently from the keyboard, and make eye contact. We need to engage the patient with his or her EMR on screen—show some trended lab results or radiographic images and, when they leave, present them with a typed set of legible instructions and their drug list (noting that all this information, and more, can be available to the next physician that they see, by cyberlink or via fax).
For those of you adopting an EMR system, I suggest you make sure you get adequate, continued access to on-site support from your vendor. The tool must be trained to swim in your pool.
In reply, based on his experience, Dr. Tom Abelson defends EMRs, predicting that their current weaknesses will be overcome as technology improves, and the faster we embrace this technologic advance, the faster fixes for initial limitations will be developed. We are, he says, just learning to swim.
The EMR is a tool, but a tool performs at the skill level of the user. The EMR is more powerful than the paper record it replaces, offering the promise of being searchable, interactive, and able to prompt us to perform in predefined ways, and also linking us at the point of care with reference materials. But the information contained in the EMR can be no better than what we enter. An EMR cannot supplant our need to think and act as physicians defending our patients’ best interests.
Our skill in using the EMR is evolving. Thus far, cut-and-paste and other electronic shortcuts are rampant and are a detriment to quality care, but these are examples of misuse and are not an intrinsic fault of the tool. These physician behaviors can be curtailed.
The EMR cannot be read like a book. Events, consultations, and nursing interventions do not readily unfold in chronologic order. Suggestions of consultants can be missed, and perhaps due to limited typing skills, clinical reasoning is not fully explained (was it always clearly explained on paper?). The notes, however, are legible.
We must not be enticed to let EMRs overly influence our billing practices. Rather, the EMR should be a tool to improve the accuracy of the record of the patient encounter.
The EMR can come between the doctor and patient, but it need not. We need to be a bit more attentive to the patient, push back intermittently from the keyboard, and make eye contact. We need to engage the patient with his or her EMR on screen—show some trended lab results or radiographic images and, when they leave, present them with a typed set of legible instructions and their drug list (noting that all this information, and more, can be available to the next physician that they see, by cyberlink or via fax).
For those of you adopting an EMR system, I suggest you make sure you get adequate, continued access to on-site support from your vendor. The tool must be trained to swim in your pool.
In reply, based on his experience, Dr. Tom Abelson defends EMRs, predicting that their current weaknesses will be overcome as technology improves, and the faster we embrace this technologic advance, the faster fixes for initial limitations will be developed. We are, he says, just learning to swim.
The EMR is a tool, but a tool performs at the skill level of the user. The EMR is more powerful than the paper record it replaces, offering the promise of being searchable, interactive, and able to prompt us to perform in predefined ways, and also linking us at the point of care with reference materials. But the information contained in the EMR can be no better than what we enter. An EMR cannot supplant our need to think and act as physicians defending our patients’ best interests.
Our skill in using the EMR is evolving. Thus far, cut-and-paste and other electronic shortcuts are rampant and are a detriment to quality care, but these are examples of misuse and are not an intrinsic fault of the tool. These physician behaviors can be curtailed.
The EMR cannot be read like a book. Events, consultations, and nursing interventions do not readily unfold in chronologic order. Suggestions of consultants can be missed, and perhaps due to limited typing skills, clinical reasoning is not fully explained (was it always clearly explained on paper?). The notes, however, are legible.
We must not be enticed to let EMRs overly influence our billing practices. Rather, the EMR should be a tool to improve the accuracy of the record of the patient encounter.
The EMR can come between the doctor and patient, but it need not. We need to be a bit more attentive to the patient, push back intermittently from the keyboard, and make eye contact. We need to engage the patient with his or her EMR on screen—show some trended lab results or radiographic images and, when they leave, present them with a typed set of legible instructions and their drug list (noting that all this information, and more, can be available to the next physician that they see, by cyberlink or via fax).
For those of you adopting an EMR system, I suggest you make sure you get adequate, continued access to on-site support from your vendor. The tool must be trained to swim in your pool.
The electronic medical record: Learning to swim
While Dr. J. Timothy hanlon raises compelling issues about electronic medical records (EMRs),1 I think that his goslow approach can lead to lost opportunities. The push toward implementing EMR systems does not amount to a dangerous dive into a shallow pool. Rather, we should be optimistic that we are just learning to swim.
Addressing these concerns during these early years of the EMR is constructive and necessary. But his commentary may leave physicians wondering what to do. Should the EMR be scrapped until it is fully developed outside the clinical realm? Is that goal even attainable? Are physicians who use EMR systems putting patient care and information security at risk? I believe there is a more positive way to look at each of the issues.
No new technology comes into the world 100% formed and vetted. The nature of progress is evolution based on experience, as unforeseen problems are corrected. Skepticism and vigilance are warranted, but so is optimism.
CONNECTIVITY WILL IMPROVE
Dr. Hanlon notes that many EMR systems are available and that, at this point, they do not communicate with one another. That is often true. But Google, Microsoft, and others are working on the issues of connectivity and “portability” of the EMR. It is reasonable to expect that eventually there will be winners and losers, just as when VHS won out over Beta in the early days of video recording. More efficient sharing of information will eventually be possible. It would be counterproductive to legislate a single EMR system nationwide before a number of EMRs can be fully tried and tested in the trenches of patient care.
In the meantime, it is no harder—in fact, it is easier than ever—for one physician to send information to another, either by printing it out and mailing or faxing it or by e-mailing it. Furthermore, that information is much more legible than the handwritten records we continue to receive from physicians who do not use EMRs.
Therefore, while one can criticize the current lack of complete intersystem communication, this is only a temporary limitation, and developing complete interconnectivity of EMR systems is a key goal.
STAYING VIGILANT ABOUT SECURITY
The security of EMRs has always been a concern. However, improvements in security have prevented a large-scale privacy breach since an incident in 2006 in which a laptop computer containing information on 26.5 million people was stolen from the home of a Veterans Administration employee.2 For instance, Cleveland Clinic recently encrypted all of its laptop computers containing patient data, to protect patient information should a laptop be lost or stolen. This is only one of many security innovations that are being implemented. While we must remain highly vigilant and continue to improve security, we must remember that the paper chart is not immune to privacy breaches either, and when paper charts were stolen, the medical record was irretrievably lost and was not reproducible. This is not the case with the EMR.
QUALITY OF CARE IS PARAMOUNT
As Dr. Hanlon accurately notes, evidence that the EMR improves the quality of care is mixed so far. He is concerned that most of the studies showing improved outcomes came from “benchmark” institutions, and that the results may not be broadly applicable. Such pessimism is unwarranted, given that the EMR is in its relative infancy and the motivation to improve quality of care is paramount, especially in this era of health care reform. While benchmark institutions are in an ideal position to do the studies on quality, there is no reason to assume that the results will not be applicable to other institutions as well.
EDUCATION: AN AREA FOR INNOVATION
Dr. Hanlon notes that research on EMRs for medical education is in its infancy. But infants grow rapidly. While it may be true that students might have to learn to use different EMR systems at different institutions, these students have grown up with rapidly changing computer systems and can learn and adapt at a remarkable rate. Therefore, education is a wonderful area for innovation and research on the EMR. It is not a reason to fear the EMR or the present diversity of EMR systems.
ACCURACY CAN BE IMPROVED
Dr. Hanlon is correct that the problem of cutting and pasting of previous notes, potentially propagating an initial error (so-called high-risk copying3) is profound within the EMR. But I prefer to look at this as an area for innovation— such as nonerasable tags to identify copied material.
While errors in medication lists are possible, especially if practitioners use cut-and-paste methods and thus perpetuate a previous error, systems and workflows are being developed to overcome such problems. Some of these include special alerts when certain high-risk drugs are ordered, drop-down menus with drug dosing included, and links to databases that allow quick access to information on drug interactions.
And again, medication errors are not unique to the EMR. They also occur in paper charts as a result of photocopying, illegible handwriting, and transcription errors.
Compared with the paper chart, the EMR is more legible, and the ability to instantaneously transfer unchanged important and valid information potentially enhances the completeness and logic of a given note and provides the physician more time to spend evaluating (and looking at) the patient. So, rather than focusing on the negatives of the current problem of cutting and pasting, I prefer to focus on how to improve it. That is, how can we make the information in the EMR more accurate, catch errors, and then make the latest information easily accessible to users?
STAYING FOCUSED ON THE PATIENT, EVEN WITH A COMPUTER IN THE ROOM
A major complaint by patients and caregivers is that using an EMR makes the physician focus on a computer screen rather than looking at the patient. This concern is valid, but I think we can learn to stay focused on the patient, even with a computer in the examination room, and still take advantage of everything technology has to offer.
This issue will disappear in less than one generation. Young people are remarkably able to multitask while typing. They are able to talk with their patients while typing and to look them in the eyes. And typing letter by letter will become obsolete as soon as voice-recognition software and ways to edit its output accurately are perfected. Many of us at Cleveland Clinic use a combination of templates, typing, and voice-recognition dictation, and find this to be effective.
When we tell our grandchildren that we used to type each individual letter on a page, they will be as amazed as we are to hear that cars used to be started with an external crank.
DOCTOR-DOCTOR COMMUNICATION IS ENHANCED
I agree that template notes written by physicians who cannot type very well can lack the substance and color found in a well-reported medical history and examination. But voice-recognition transcription can help flesh out key parts of the history, differential diagnosis, and management plan. Further, the note can be produced on the spot, the patient can check the note for accuracy, and the conclusions can be shared instantaneously with all involved caregivers. Doctor-doctor communication is thus enhanced.
EVERY REASON TO MOVE FORWARD
Dr. Hanlon is also concerned about EMRs and the potential for “billing creep” and outright fraud. But fraud is as old as billing. What is required is continued vigilance and system controls, which actually might be more effective in an EMR system than in a paper billing system. Integrity will be neither enhanced nor diminished by digitization, unfortunately.
In summary, while Dr. Hanlon sees reason to slow down the move to EMRs, I see every reason to move forward. The problems he describes are part of the growing pains of any new technology. He is right that we cannot move blindly, ignoring the challenges of this technology. But slowing down will only delay its benefits.
- Hanlon JT. The electronic medical record: diving into a shallow pool? Cleve Clin J Med 2010; 77:408–411.
- Lemos R. Veterans Affairs warns of massive privacy breach. SecurityFocus 2006 (May 22). http://www.securityfocus.com/news/11393. Accessed May 19, 2010.
- Hammond KW, Helbig ST, Benson CC, Brathwaite-Sketoe BM. Are Electronic Medical Records Trustworthy? Observations on Copying, Pasting and Duplication. AMIA Annu Symp Proc 2003 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1480345/. Accessed May 19, 2010.
While Dr. J. Timothy hanlon raises compelling issues about electronic medical records (EMRs),1 I think that his goslow approach can lead to lost opportunities. The push toward implementing EMR systems does not amount to a dangerous dive into a shallow pool. Rather, we should be optimistic that we are just learning to swim.
Addressing these concerns during these early years of the EMR is constructive and necessary. But his commentary may leave physicians wondering what to do. Should the EMR be scrapped until it is fully developed outside the clinical realm? Is that goal even attainable? Are physicians who use EMR systems putting patient care and information security at risk? I believe there is a more positive way to look at each of the issues.
No new technology comes into the world 100% formed and vetted. The nature of progress is evolution based on experience, as unforeseen problems are corrected. Skepticism and vigilance are warranted, but so is optimism.
CONNECTIVITY WILL IMPROVE
Dr. Hanlon notes that many EMR systems are available and that, at this point, they do not communicate with one another. That is often true. But Google, Microsoft, and others are working on the issues of connectivity and “portability” of the EMR. It is reasonable to expect that eventually there will be winners and losers, just as when VHS won out over Beta in the early days of video recording. More efficient sharing of information will eventually be possible. It would be counterproductive to legislate a single EMR system nationwide before a number of EMRs can be fully tried and tested in the trenches of patient care.
In the meantime, it is no harder—in fact, it is easier than ever—for one physician to send information to another, either by printing it out and mailing or faxing it or by e-mailing it. Furthermore, that information is much more legible than the handwritten records we continue to receive from physicians who do not use EMRs.
Therefore, while one can criticize the current lack of complete intersystem communication, this is only a temporary limitation, and developing complete interconnectivity of EMR systems is a key goal.
STAYING VIGILANT ABOUT SECURITY
The security of EMRs has always been a concern. However, improvements in security have prevented a large-scale privacy breach since an incident in 2006 in which a laptop computer containing information on 26.5 million people was stolen from the home of a Veterans Administration employee.2 For instance, Cleveland Clinic recently encrypted all of its laptop computers containing patient data, to protect patient information should a laptop be lost or stolen. This is only one of many security innovations that are being implemented. While we must remain highly vigilant and continue to improve security, we must remember that the paper chart is not immune to privacy breaches either, and when paper charts were stolen, the medical record was irretrievably lost and was not reproducible. This is not the case with the EMR.
QUALITY OF CARE IS PARAMOUNT
As Dr. Hanlon accurately notes, evidence that the EMR improves the quality of care is mixed so far. He is concerned that most of the studies showing improved outcomes came from “benchmark” institutions, and that the results may not be broadly applicable. Such pessimism is unwarranted, given that the EMR is in its relative infancy and the motivation to improve quality of care is paramount, especially in this era of health care reform. While benchmark institutions are in an ideal position to do the studies on quality, there is no reason to assume that the results will not be applicable to other institutions as well.
EDUCATION: AN AREA FOR INNOVATION
Dr. Hanlon notes that research on EMRs for medical education is in its infancy. But infants grow rapidly. While it may be true that students might have to learn to use different EMR systems at different institutions, these students have grown up with rapidly changing computer systems and can learn and adapt at a remarkable rate. Therefore, education is a wonderful area for innovation and research on the EMR. It is not a reason to fear the EMR or the present diversity of EMR systems.
ACCURACY CAN BE IMPROVED
Dr. Hanlon is correct that the problem of cutting and pasting of previous notes, potentially propagating an initial error (so-called high-risk copying3) is profound within the EMR. But I prefer to look at this as an area for innovation— such as nonerasable tags to identify copied material.
While errors in medication lists are possible, especially if practitioners use cut-and-paste methods and thus perpetuate a previous error, systems and workflows are being developed to overcome such problems. Some of these include special alerts when certain high-risk drugs are ordered, drop-down menus with drug dosing included, and links to databases that allow quick access to information on drug interactions.
And again, medication errors are not unique to the EMR. They also occur in paper charts as a result of photocopying, illegible handwriting, and transcription errors.
Compared with the paper chart, the EMR is more legible, and the ability to instantaneously transfer unchanged important and valid information potentially enhances the completeness and logic of a given note and provides the physician more time to spend evaluating (and looking at) the patient. So, rather than focusing on the negatives of the current problem of cutting and pasting, I prefer to focus on how to improve it. That is, how can we make the information in the EMR more accurate, catch errors, and then make the latest information easily accessible to users?
STAYING FOCUSED ON THE PATIENT, EVEN WITH A COMPUTER IN THE ROOM
A major complaint by patients and caregivers is that using an EMR makes the physician focus on a computer screen rather than looking at the patient. This concern is valid, but I think we can learn to stay focused on the patient, even with a computer in the examination room, and still take advantage of everything technology has to offer.
This issue will disappear in less than one generation. Young people are remarkably able to multitask while typing. They are able to talk with their patients while typing and to look them in the eyes. And typing letter by letter will become obsolete as soon as voice-recognition software and ways to edit its output accurately are perfected. Many of us at Cleveland Clinic use a combination of templates, typing, and voice-recognition dictation, and find this to be effective.
When we tell our grandchildren that we used to type each individual letter on a page, they will be as amazed as we are to hear that cars used to be started with an external crank.
DOCTOR-DOCTOR COMMUNICATION IS ENHANCED
I agree that template notes written by physicians who cannot type very well can lack the substance and color found in a well-reported medical history and examination. But voice-recognition transcription can help flesh out key parts of the history, differential diagnosis, and management plan. Further, the note can be produced on the spot, the patient can check the note for accuracy, and the conclusions can be shared instantaneously with all involved caregivers. Doctor-doctor communication is thus enhanced.
EVERY REASON TO MOVE FORWARD
Dr. Hanlon is also concerned about EMRs and the potential for “billing creep” and outright fraud. But fraud is as old as billing. What is required is continued vigilance and system controls, which actually might be more effective in an EMR system than in a paper billing system. Integrity will be neither enhanced nor diminished by digitization, unfortunately.
In summary, while Dr. Hanlon sees reason to slow down the move to EMRs, I see every reason to move forward. The problems he describes are part of the growing pains of any new technology. He is right that we cannot move blindly, ignoring the challenges of this technology. But slowing down will only delay its benefits.
While Dr. J. Timothy hanlon raises compelling issues about electronic medical records (EMRs),1 I think that his goslow approach can lead to lost opportunities. The push toward implementing EMR systems does not amount to a dangerous dive into a shallow pool. Rather, we should be optimistic that we are just learning to swim.
Addressing these concerns during these early years of the EMR is constructive and necessary. But his commentary may leave physicians wondering what to do. Should the EMR be scrapped until it is fully developed outside the clinical realm? Is that goal even attainable? Are physicians who use EMR systems putting patient care and information security at risk? I believe there is a more positive way to look at each of the issues.
No new technology comes into the world 100% formed and vetted. The nature of progress is evolution based on experience, as unforeseen problems are corrected. Skepticism and vigilance are warranted, but so is optimism.
CONNECTIVITY WILL IMPROVE
Dr. Hanlon notes that many EMR systems are available and that, at this point, they do not communicate with one another. That is often true. But Google, Microsoft, and others are working on the issues of connectivity and “portability” of the EMR. It is reasonable to expect that eventually there will be winners and losers, just as when VHS won out over Beta in the early days of video recording. More efficient sharing of information will eventually be possible. It would be counterproductive to legislate a single EMR system nationwide before a number of EMRs can be fully tried and tested in the trenches of patient care.
In the meantime, it is no harder—in fact, it is easier than ever—for one physician to send information to another, either by printing it out and mailing or faxing it or by e-mailing it. Furthermore, that information is much more legible than the handwritten records we continue to receive from physicians who do not use EMRs.
Therefore, while one can criticize the current lack of complete intersystem communication, this is only a temporary limitation, and developing complete interconnectivity of EMR systems is a key goal.
STAYING VIGILANT ABOUT SECURITY
The security of EMRs has always been a concern. However, improvements in security have prevented a large-scale privacy breach since an incident in 2006 in which a laptop computer containing information on 26.5 million people was stolen from the home of a Veterans Administration employee.2 For instance, Cleveland Clinic recently encrypted all of its laptop computers containing patient data, to protect patient information should a laptop be lost or stolen. This is only one of many security innovations that are being implemented. While we must remain highly vigilant and continue to improve security, we must remember that the paper chart is not immune to privacy breaches either, and when paper charts were stolen, the medical record was irretrievably lost and was not reproducible. This is not the case with the EMR.
QUALITY OF CARE IS PARAMOUNT
As Dr. Hanlon accurately notes, evidence that the EMR improves the quality of care is mixed so far. He is concerned that most of the studies showing improved outcomes came from “benchmark” institutions, and that the results may not be broadly applicable. Such pessimism is unwarranted, given that the EMR is in its relative infancy and the motivation to improve quality of care is paramount, especially in this era of health care reform. While benchmark institutions are in an ideal position to do the studies on quality, there is no reason to assume that the results will not be applicable to other institutions as well.
EDUCATION: AN AREA FOR INNOVATION
Dr. Hanlon notes that research on EMRs for medical education is in its infancy. But infants grow rapidly. While it may be true that students might have to learn to use different EMR systems at different institutions, these students have grown up with rapidly changing computer systems and can learn and adapt at a remarkable rate. Therefore, education is a wonderful area for innovation and research on the EMR. It is not a reason to fear the EMR or the present diversity of EMR systems.
ACCURACY CAN BE IMPROVED
Dr. Hanlon is correct that the problem of cutting and pasting of previous notes, potentially propagating an initial error (so-called high-risk copying3) is profound within the EMR. But I prefer to look at this as an area for innovation— such as nonerasable tags to identify copied material.
While errors in medication lists are possible, especially if practitioners use cut-and-paste methods and thus perpetuate a previous error, systems and workflows are being developed to overcome such problems. Some of these include special alerts when certain high-risk drugs are ordered, drop-down menus with drug dosing included, and links to databases that allow quick access to information on drug interactions.
And again, medication errors are not unique to the EMR. They also occur in paper charts as a result of photocopying, illegible handwriting, and transcription errors.
Compared with the paper chart, the EMR is more legible, and the ability to instantaneously transfer unchanged important and valid information potentially enhances the completeness and logic of a given note and provides the physician more time to spend evaluating (and looking at) the patient. So, rather than focusing on the negatives of the current problem of cutting and pasting, I prefer to focus on how to improve it. That is, how can we make the information in the EMR more accurate, catch errors, and then make the latest information easily accessible to users?
STAYING FOCUSED ON THE PATIENT, EVEN WITH A COMPUTER IN THE ROOM
A major complaint by patients and caregivers is that using an EMR makes the physician focus on a computer screen rather than looking at the patient. This concern is valid, but I think we can learn to stay focused on the patient, even with a computer in the examination room, and still take advantage of everything technology has to offer.
This issue will disappear in less than one generation. Young people are remarkably able to multitask while typing. They are able to talk with their patients while typing and to look them in the eyes. And typing letter by letter will become obsolete as soon as voice-recognition software and ways to edit its output accurately are perfected. Many of us at Cleveland Clinic use a combination of templates, typing, and voice-recognition dictation, and find this to be effective.
When we tell our grandchildren that we used to type each individual letter on a page, they will be as amazed as we are to hear that cars used to be started with an external crank.
DOCTOR-DOCTOR COMMUNICATION IS ENHANCED
I agree that template notes written by physicians who cannot type very well can lack the substance and color found in a well-reported medical history and examination. But voice-recognition transcription can help flesh out key parts of the history, differential diagnosis, and management plan. Further, the note can be produced on the spot, the patient can check the note for accuracy, and the conclusions can be shared instantaneously with all involved caregivers. Doctor-doctor communication is thus enhanced.
EVERY REASON TO MOVE FORWARD
Dr. Hanlon is also concerned about EMRs and the potential for “billing creep” and outright fraud. But fraud is as old as billing. What is required is continued vigilance and system controls, which actually might be more effective in an EMR system than in a paper billing system. Integrity will be neither enhanced nor diminished by digitization, unfortunately.
In summary, while Dr. Hanlon sees reason to slow down the move to EMRs, I see every reason to move forward. The problems he describes are part of the growing pains of any new technology. He is right that we cannot move blindly, ignoring the challenges of this technology. But slowing down will only delay its benefits.
- Hanlon JT. The electronic medical record: diving into a shallow pool? Cleve Clin J Med 2010; 77:408–411.
- Lemos R. Veterans Affairs warns of massive privacy breach. SecurityFocus 2006 (May 22). http://www.securityfocus.com/news/11393. Accessed May 19, 2010.
- Hammond KW, Helbig ST, Benson CC, Brathwaite-Sketoe BM. Are Electronic Medical Records Trustworthy? Observations on Copying, Pasting and Duplication. AMIA Annu Symp Proc 2003 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1480345/. Accessed May 19, 2010.
- Hanlon JT. The electronic medical record: diving into a shallow pool? Cleve Clin J Med 2010; 77:408–411.
- Lemos R. Veterans Affairs warns of massive privacy breach. SecurityFocus 2006 (May 22). http://www.securityfocus.com/news/11393. Accessed May 19, 2010.
- Hammond KW, Helbig ST, Benson CC, Brathwaite-Sketoe BM. Are Electronic Medical Records Trustworthy? Observations on Copying, Pasting and Duplication. AMIA Annu Symp Proc 2003 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1480345/. Accessed May 19, 2010.
Bariatric surgery for type 2 diabetes: Weighing the impact for obese patients
Evidence is mounting for the use of bariatric surgery to treat type 2 diabetes mellitus in patients whose body mass index (BMI) is 35 kg/m2 or higher. In obese patients who also have type 2 diabetes, bariatric surgery sends it into remission (defined as normoglycemic control without the need for diabetic medications) in more than three-fourths of cases, with higher rates with the Roux-en-Y gastric bypass procedure than with the laparoscopic adjustable gastric banding procedure.
However, data on the effects of this surgery on type 2 diabetes come primarily from observational studies that lacked appropriate control groups, and the relative benefit of bariatric surgery vs aggressive medical antidiabetic therapy is not yet known. Needed are randomized trials comparing the two types of therapy (and the various types of bariatric surgery) in diabetic patients with less-severe obesity.
Further, why would bariatric surgery help with diabetes, and why would one procedure do it better than another? To be honest, we are not sure, but evidence points not only to weight loss but also to better insulin sensitivity and to alterations in levels of hormones secreted by the gut that increase insulin secretion.
OBESITY PROMOTES DIABETES; WEIGHT LOSS COUNTERACTS IT
Type 2 diabetes mellitus is a complex metabolic disease characterized by insulin resistance and progressive failure of pancreatic beta cells, resulting in hyperglycemia.1,2
Obesity, a potent risk factor for type 2 diabetes, contributes to its development by inducing insulin resistance and inflammation, which in turn impair glucose regulation.3,4 Fat deposits in the abdomen, muscles, and liver contribute to elevations of circulating free fatty acids and adipocyte-derived cytokines that mediate insulin resistance and inflammatory pathways.5
In the Diabetes Prevention Program,6 modest weight loss (5% to 10% of body weight) through diet and exercise reduced the incidence of type 2 diabetes, and in the ongoing Action for Health in Diabetes (Look AHEAD) study of the National Institutes of Health, it improved glucose homeostasis.7,8
The current medical approach to type 2 diabetes includes advising the patient to lose weight through lifestyle modification, and prescribing drugs that restore glycemic control by reducing insulin resistance (biguanides, glitazones) and improving insulin secretion (incretin mimetics and analogues and sulfonylureas). 9,10
However, several factors make type 2 diabetes challenging to treat in obese people. Patients who lose weight via behavioral changes and weight-loss drugs tend to gain the weight back. Antidiabetic drugs pose the risk of hypoglycemia. Moreover, although many new classes of drugs have been developed to treat type 2 diabetes, most patients fail to achieve the American Diabetes Association goal for glycemic control, ie, a hemoglobin A1c level lower than 7%.11
BARIATRIC PROCEDURES AND THEIR EFFECT ON DIABETES CONTROL
After bariatric surgery, patients lose more weight than with traditional weight-loss methods—up to 25% of their total body weight. Furthermore, of those with type 2 diabetes, 87% achieve at least better glucose control and need fewer antidiabetic medications,12 and an average of 78% achieve normal glycemic control without taking any antidiabetic medications at all.12,13
But not all bariatric procedures have the same effect on weight and diabetes: certain procedures have a greater effect.
The two major types are classified as gastric restrictive procedures and intestinal bypass procedures. The classification was initially based on the presumed mechanism of weight loss.
Gastric restrictive procedures (laparoscopic adjustable gastric banding, sleeve gastrectomy, vertical gastroplasty) limit gastric volume and, hence, restrict the intake of calories by inducing satiety. Afterward, patients lose approximately 10% to 20% of their total body weight.
Furthermore, multiple studies, including a randomized controlled trial14 (more about this below), have shown remission of type 2 diabetes with laparoscopic adjustable gastric banding but not with conventional medical therapy. The effect is primarily mediated by weight loss and improved insulin sensitivity, both of which occur several months following surgery. Of note, however: in this trial,14 all the patients had diabetes of short duration, less than 2 years.
Hence, different procedures have different effects on diabetes.12 The speed at which type 2 diabetes goes into remission differs with restrictive vs malabsorptive procedures. After Roux-en-Y gastric bypass and biliopancreatic diversion, diabetes remits within days, even before the patient has lost much weight.15 This does not happen after gastric restrictive procedures.12,16
Observational studies of the effect of Roux-en-Y surgery on diabetes
Several observational studies have evaluated the benefit of Roux-en-Y surgery for patients with type 2 diabetes mellitus.
Pories et al15 followed 608 severely obese patients, of whom 165 (27%) had type 2 diabetes or impaired glucose tolerance.
At a mean follow-up of 7.6 years after surgery, 83% of the diabetic patients were off their antidiabetic drugs, and 99% of those with impaired glucose tolerance were normoglycemic, with normal fasting glucose and hemoglobin A1c levels. Marked improvements in hyperlipidemia, hypertension, fertility, osteoarthritis, and obstructive sleep apnea were also noted.
Schauer et al17 observed similar results in 1,160 morbidly obese patients, of whom 240 (21%) had type 2 diabetes or impaired fasting glucose.
After laparoscopic Roux-en-Y gastric bypass surgery, fasting glucose and hemoglobin A1c levels returned to normal levels in 83% of cases and were markedly improved in the remaining 17%. Significantly (80%) fewer patients needed oral antidiabetic agents or insulin (79% fewer). Patients most likely to achieve complete remission of diabetes were those with the shortest duration of diabetes (< 5 years), the mildest severity of diabetes (diet-controlled), and the greatest weight loss after surgery. The rate of diabetes remission in patients who had been diabetic for 5 years or less was 95%, compared with 75% in those who had been diabetic for 6 to 10 years and 54% in those who had been diabetic for more than 10 years (P < .001).
The Swedish Obese Subjects (SOS) study18 prospectively followed 1,703 patients, of whom 118 had type 2 diabetes, for 10 years after various bariatric surgery procedures (primarily vertical gastroplasty). In a control group that received medical therapy, 77 patients had type 2 diabetes. Medical therapy was ill-defined with respect to aggressiveness and adherence to intervention with lifestyle and pharmacotherapy.
At 2 years, the surgical group had lost a mean of 28 kg, glycemic control had improved in the diabetic patients, and many of them had been able to stop taking oral hypoglycemic drugs or insulin. In contrast, the need for these agents increased in the medically treated patients. The proportion treated by diet alone rose from 59% to 73% in the surgical group, but declined from 55% to 34% in the nonsurgical group.13
In these studies, surgery also reduced the risk of progressing from impaired glucose tolerance to type 2 diabetes; the risk was 30 times lower in the study by Pories et al.15 In the SOS study,18 the frequency of diabetes was 30 times lower at 2 years and five times lower at 8 years after surgery.
Studies of biliopancreatic diversion
Data on the effects of biliopancreatic diversion, a primarily malabsorptive procedure, are limited to European studies.
Scopinaro et al19,20 reported long-term follow-up data on 312 patients with type 2 diabetes who underwent biliopancreatic diversion; 310 patients (99%) achieved normal fasting glucose values by 1 year after surgery. At 10 years after surgery, 98% of the patients were still in complete remission of diabetes, defined as normal glucose values without the use of antidiabetic medications.
Others have noted similar findings.21,22
Limitations of the studies
Although these data seem encouraging, these studies had major limitations.
The patients were mostly white women with severe obesity, ie, a BMI greater than 40 kg/m2, which is not representative of patients with type 2 diabetes in the community. Only about 20% had glucose intolerance or overt type 2 diabetes mellitus. Would other groups benefit, particularly men and those with lesssevere obesity?
Moreover, these studies were observational, with no randomized control groups. Many reports consisted of large case series. It is not clear how specific bariatric procedures were chosen or what criteria were used for performing bariatric surgery. A lack of complete follow-up data is also a concern.
Needed are large randomized trials evaluating the effects of various bariatric procedures in a less obese cohort with type 2 diabetes, ie, typical patients seen in the community. Moreover, surgery has not been compared directly with more vigorous medical weight-loss strategies, such as those used in the Diabetes Prevention Project6 and the Look AHEAD trial.7,8
A randomized controlled trial of gastric banding
The only randomized controlled trial to date that compared standard medical diabetes therapy with bariatric surgery was conducted by Dixon et al.14
Sixty patients with type 2 diabetes (duration < 2 years and mean hemoglobin A1c 7.7%) were randomized either to receive medical management as defined by the American Diabetes Association guidelines or to undergo laparoscopic adjustable gastric banding.
At 2 years, the rate of remission (defined as hemoglobin A1c < 6.2% and a normal fasting glucose level) was 13% in the medical treatment group vs 73% in the surgery group (P < .001). Patients receiving medical treatment had lost a mean of 1.7% of their body weight, vs 20.7% in the surgical patients (P < .001). Weight loss was strongly associated with remission of type 2 diabetes after surgery.
This study was controversial in that the medical intervention in this trial was not as aggressive as in the Diabetes Prevention Project and Look AHEAD trials.
INDICATIONS FOR BARIATRIC SURGERY IN PATIENTS WITH DIABETES
According to guidelines from the National Institutes of Health,23 the current indications for bariatric surgery include a BMI of 40 kg/m2 or higher, or a BMI between 35 and 40 kg/m2 with at least two obesity-related comorbidities. Diabetes is considered a key comorbidity that justifies the risk of surgery. The guidelines suggest that bariatric surgery be discussed with all severely obese patients (BMI > 35 kg/m2) with type 2 diabetes who have not been able to lose weight with other weight-control approaches.
Since type 2 diabetes mellitus is a progressive disease characterized by relentless deterioration of beta-cell function, many endocrinologists favor aggressive weight-loss approaches early in the course of the disease. We believe that bariatric surgery should be considered early, as it may help preserve pancreatic betacell function and slow the progression of microvascular and macrovascular complications.
HOW DOES BARIATRIC SURGERY IMPROVE TYPE 2 DIABETES?

Hypothesis 1: Weight loss increases insulin sensitivity
The enforced caloric restriction, negative energy balance, and weight loss after bariatric surgery reduce insulin resistance. Consequently, the beta cells can rest because they don’t need to produce as much insulin. These effects have been observed after both gastric restrictive procedures and gastric bypass procedures.
Hypothesis 2: Less lipotoxicity, inflammation
Another theory is that bariatric surgery lessens insulin resistance by reducing “lipotoxicity,” a condition related to dysregulated fatty acid flux, lipid metabolites in tissues, and direct and indirect effects of hormones secreted by adipocytes.
The strongest evidence for this theory comes from Bikman et al,26 who found that insulin sensitivity increased after Roux-en-Y surgery more than expected from weight loss alone. One year after surgery, even though they remained anthropometrically obese (BMI > 30 kg/m2), the patients had insulin sensitivity levels similar to those in a control group of lean people (BMI < 25 kg/m2).
Insulin sensitivity begins to improve within 1 week of intestinal bypass procedures,15,27 suggesting that these procedures are doing something more than simply forcing weight loss via caloric restriction, as gastric restrictive procedures do.
Hypothesis 3: An effect on gut hormones

The “hindgut hypothesis” raised by Cummings et al24 suggests that accelerated transit of concentrated nutrients (particularly glucose) to the distal intestine results in increased production of insulinotropic and appetite-controlling substances, which account for the reversal of hyperglycemia and obesity.
In contrast, the “foregut hypothesis” raised by Rubino et al28 suggests that nutrient interactions in the duodenum are diabetogenic and, hence, bypassing the duodenum would reverse this defect. Their conclusions come from experiments in rodents that underwent jejunoileal bypass and subsequent refeeding through the bypassed intestine.
GUT HORMONES AND OTHER PEPTIDES ALTERED BY BARIATRIC SURGERY
Incretin hormones: GLP-1, GIP
Gastrointestinal hormones that increase insulin release after a meal are known as incretins. Of interest, they have this effect only when glucose is ingested orally—not when it is infused intravenously.29,30
Glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP) account for 50% to 60% of nutrient-related insulin secretion. In addition to stimulating insulin, GLP-1 suppresses glucagon and slows gastric emptying, which delays digestion and reduces postprandial glycemia. GLP-1 also acts on the hypothalamus to induce satiety.
Laferrère et al31 and others32,33 documented robust increases in postprandial levels of GLP-1 within 4 weeks after Roux-en-Y surgery. GLP-1 levels did not increase with comparable weight loss induced by diet.
Rubino et al28,34 documented similar findings that occurred prior to marked weight loss, suggesting that the benefit of Roux-en-Y surgery on remission of diabetes may not be completely attributable to reduced caloric intake and weight loss. Insulin secretion is generally reduced after gastric restrictive procedures (eg, laparoscopic adjustable gastric banding) and biliopancreatic diversion,35 and is increased after Roux-en-Y gastric bypass.32,33,36
Noninsulinotropic peptides: Ghrelin, peptide YY
Noninsulinotropic gut peptides that are altered after Roux-en-Y surgery include ghrelin and peptide YY.
Ghrelin, a hormone derived from the gastric fundus, stimulates appetite. Ghrelin concentrations are lower after Roux-en-Y surgery, indicating that suppression of hunger signals helps sustain weight loss. In contrast, ghrelin levels increase with diet-induced weight loss.37 However, the data on ghrelin levels at various times after bariatric surgical procedures are not consistent.33,38
Peptide YY, like GLP-1, is secreted by L cells of the distal small intestine and is responsible for increasing satiety and delaying gastric emptying after meals. Numerous studies have consistently documented increases in postprandial peptide YY and GLP-1 levels after gastric bypass.32,33,39–41
ACUTE EFFECTS OF BARIATRIC SURGERY ON INSULIN SECRETION, SENSITIVITY
Bariatric surgery alters both insulin secretion and insulin sensitivity, thus improving glucose regulation.
The relationship between insulin secretion and sensitivity is a hyperbolic curve, so that any change in insulin sensitivity is balanced by a reciprocal and proportionate change in insulin secretion. The development of type 2 diabetes is characterized by a reduction in insulin secretion (decompensation) relative to the severity of insulin resistance.
In the first 6 weeks after Roux-en-Y gastric bypass or biliopancreatic diversion, insulin sensitivity improves while insulin secretion increases disproportionately, associated with a robust increase in GLP-1, and resulting in normal glucose homeostasis.16,31,42
In contrast, patients who lose weight by dieting or undergoing gastric restrictive procedures show a modest increase in insulin sensitivity and a compensatory reduction in insulin secretion, termed “beta-cell rest.”16,31,42
RISKS OF BARIATRIC SURGERY
Short-term risks
An important concern about using bariatric surgery to treat type 2 diabetes is the risk of morbidity and death associated with these procedures.
Buchwald et al13 performed a meta-analysis of 136 bariatric studies that included 22,094 patients. The 30-day operative death rates were 1.1% with biliopancreatic diversion, 0.5% with Roux-en-Y surgery, and 0.1% with restrictive procedures.
Laparoscopic adjustable gastric banding is considered the safest of the current bariatric procedures. It does not involve bowel anastomosis, and the risks of major hemorrhage, gastric perforation, and pulmonary embolism are less than 1%. Late complications requiring reoperation include band slippage or prolapse (5%–10%) and band erosion (1%–3%). The entire intestinal tract is left intact, so subsequent nutritional deficiencies are rare.43
Roux-en-Y gastric bypass carries an overall risk of major complications of 10% to 15%. Anastomotic leak (1%–5%), pulmonary embolism (< 1%), and hemorrhage (1%–4%) can be life-threatening but are rare if the staff are experienced. Late complications such as ulcer or stricture formation at the gastrojejunostomy site occur in 5% to 10% of cases and are managed nonoperatively.
Nutritional deficiencies

Protein-calorie malnutrition is recognized by signs such as edema, hypoalbuminemia, anemia, and hair loss. To minimize this problem after Roux-en-Y surgery, we suggest that patients take in 60 to 80 g of protein and 700 to 800 kcal a day.
Vitamin deficiencies can lead to Wernicke encephalopathy (due to thiamine deficiency), peripheral neuropathy (due to vitamin B12 deficiency),45,46 and metabolic bone disease (due to long-term deficiencies of vitamin D and calcium). Often, vitamin deficiencies are present before surgery and require prompt supplementation to avoid exacerbation of these deficiencies afterward.
Biliopancreatic diversion procedures are performed at relatively few centers worldwide, largely because of the massive amounts of protein, fat, and carbohydrate malabsorption they cause. Long-term deficiencies of fat-soluble vitamins, iron, calcium, and vitamins B12 and D have been reported in one-third to one-half of patients undergoing these procedures, and nutritional supplementation is mandatory.43 Protein-calorie malnutrition occurs in 7% of cases, and 2% of patients require operative revision to lengthen the common channel.
In rare cases, severe hypoglycemia has been noted after Roux-en-Y surgery and is associated with prandial hyperinsulinemia related to elevated GLP-1 levels.36,47 Neuroglycopenia and seizures have been reported in severe cases. Initial treatment of hypoglycemia involves dietary modification targeting carbohydrate restriction, the use of alpha glucosidase inhibitors such as acarbose (Precose), and referral to an endocrinologist for further management.
Long-term death rates
Death rates after bariatric surgery must be weighed against the long-term cardiovascular risks of continued obesity and type 2 diabetes.
Strong evidence now exists that bariatric surgery increases life expectancy48 and that this is largely attributable to reduction in cardiovascular risk factors such as diabetes and cancer. Recent studies have found that the long-term death rate is 32% to 73% lower for patients undergoing bariatric surgery than in matched controls who do not undergo surgery.49 A decrease in the death rate related to diabetes has played an important role in these results.
Acknowledgments: We acknowledge support from the National Institutes of Health, Multidisciplinary Clinical Research Career Development Programs Grant 5K12RR023264 (SRK), National Center for Research Resources, CTSA 1UL1RR024989, and research grants from Ethicon Endo-Surgery (PS,SRK).
- DeFronzo RA. Pathogenesis of type 2 diabetes mellitus. Med Clin North Am 2004; 88:787–835.
- Kashyap SR, Defronzo RA. The insulin resistance syndrome: physiological considerations. Diab Vasc Dis Res 2007; 4:13–19.
- Mokdad AH, Ford ES, Bowman BA, et al. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA 2003; 289:76–79.
- Unger RH. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology 2003; 144:5159–5165.
- Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 2002; 51:2005–2011.
- Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346:393–403.
- Look AHEAD Research Group; Pi-Sunyer X, Blackburn G, Brancati FL, et al. Reduction in weight and cardiovascular disease risk factors in individuals with type 2 diabetes: one-year results of the look AHEAD trial. Diabetes Care 2007; 30:1374–1383.
- Look AHEAD Research Group; Wadden TA, West DS, Delahanty L, et al. The Look AHEAD study: a description of the lifestyle intervention and the evidence supporting it. Obesity (Silver Spring) 2006; 14:737–752.
- Nathan DM. Clinical practice. Initial management of glycemia in type 2 diabetes mellitus. N Engl J Med 2002; 347:1342–1349.
- Nathan DM, Buse JB, Davidson MB, et al. Management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: update regarding thiazolidinediones: a consensus statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2008; 31:173–175.
- Spann SJ, Nutting PA, Galliher JM, et al. Management of type 2 diabetes in the primary care setting: a practice-based research network study. Ann Fam Med 2006; 4:23–31.
- Buchwald H, Estok R, Fahrbach K, et al. Weight and type 2 diabetes after bariatric surgery: systematic review and meta-analysis. Am J Med 2009; 122:248–256.
- Buchwald H, Avidor Y, Braunwald E, et al. Bariatric surgery: a systematic review and meta-analysis. JAMA 2004; 292:1724–1737.
- Dixon JB, O’Brien PE, Playfair J, et al. Adjustable gastric banding and conventional therapy for type 2 diabetes. JAMA 2008; 299:316–323.
- Pories WJ, Swanson MS, MacDonald KG, et al. Who would have thought it? An operation proves to be the most effective therapy for adult-onset diabetes mellitus. Ann Surg 1995; 222:339–350.
- Kashyap SR, Daud S, Kelly KR, et al. Acute effects of gastric bypass versus gastric restrictive surgery on beta-cell function and insulinotropic hormones in severely obese patients with type 2 diabetes. Int J Obes (Lond) 2009; epub ahead of print
- Schauer PR, Burguera B, Ikramuddin S, et al. Effect of laparoscopic Roux-en Y gastric bypass on type 2 diabetes mellitus. Ann Surg 2003; 238:467–484.
- Sjöström L, Lindroos AK, Peltonen M, et al; Swedish Obese Subjects Study Scientific Group. Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. N Engl J Med 2004; 351:2683–2693.
- Scopinaro N, Marinari GM, Camerini GB, Papadia FS, Adami GF. Specific effects of biliopancreatic diversion on the major components of metabolic syndrome: a long-term follow-up study. Diabetes Care 2005; 28:2406–2411.
- Scopinaro N, Papadia F, Marinari G, Camerini G, Adami G. Long-term control of type 2 diabetes mellitus and the other major components of the metabolic syndrome after biliopancreatic diversion in patients with BMI < 35 kg/m2. Obes Surg 2007; 17:185–192.
- Alexandrides TK, Skroubis G, Kalfarentzos F. Resolution of diabetes mellitus and metabolic syndrome following Roux-en-Y gastric bypass and a variant of biliopancreatic diversion in patients with morbid obesity. Obes Surg 2007; 17:176–184.
- Chiellini C, Rubino F, Castagneto M, Nanni G, Mingrone G. The effect of bilio-pancreatic diversion on type 2 diabetes in patients with BMI < 35 kg/m2. Diabetologia 2009; 52:1027–1030.
- Consensus Development Conference Panel. NIH conference. Gastrointestinal surgery for severe obesity. Ann Intern Med 1991; 115:956–961.
- Cummings DE, Overduin J, Foster-Schubert KE. Gastric bypass for obesity: mechanisms of weight loss and diabetes resolution. J Clin Endocrinol Metab 2004; 89:2608–2615.
- Cummings DE, Flum DR. Gastrointestinal surgery as a treatment for diabetes. JAMA 2008; 299:341–343.
- Bikman BT, Zheng D, Pories WJ, et al. Mechanism for improved insulin sensitivity after gastric bypass surgery. J Clin Endocrinol Metab 2008; 93:4656–4663.
- Guidone C, Manco M, Valera-Mora E, et al. Mechanisms of recovery from type 2 diabetes after malabsorptive bariatric surgery. Diabetes 2006; 55:2025–2031.
- Rubino F, Forgione A, Cummings DE, et al. The mechanism of diabetes control after gastrointestinal bypass surgery reveals a role of the proximal small intestine in the pathophysiology of type 2 diabetes. Ann Surg 2006; 244:741–749.
- Vilsbøll T, Krarup T, Madsbad S, Holst JJ. Both GLP-1 and GIP are insulinotropic at basal and postprandial glucose levels and contribute nearly equally to the incretin effect of a meal in healthy subjects. Regul Pept 2003; 114:115–121.
- Vollmer K, Holst JJ, Baller B, et al. Predictors of incretin concentrations in subjects with normal, impaired, and diabetic glucose tolerance. Diabetes 2008; 57:678–687.
- Laferrère B, Teixeira J, McGinty J, et al. Effect of weight loss by gastric bypass surgery versus hypocaloric diet on glucose and incretin levels in patients with type 2 diabetes. J Clin Endocrinol Metab 2008; 93:2479–2485.
- Korner J, Bessler M, Inabnet W, Taveras C, Holst JJ. Exaggerated glucagon-like peptide-1 and blunted glucose-dependent insulinotropic peptide secretion are associated with Roux-en-Y gastric bypass but not adjustable gastric banding. Surg Obes Relat Dis 2007; 3:597–601.
- le Roux CW, Aylwin SJ, Batterham RL, et al. Gut hormone profiles following bariatric surgery favor an anorectic state, facilitate weight loss, and improve metabolic parameters. Ann Surg 2006; 243:108–114.
- Rubino F, Gagner M, Gentileschi P, et al. The early effect of the Roux-en-Y gastric bypass on hormones involved in body weight regulation and glucose metabolism. Ann Surg 2004; 240:236–242.
- Salinari S, Bertuzzi A, Asnaghi S, Guidone C, Manco M, Mingrone G. First-phase insulin secretion restoration and differential response to glucose load depending on the route of administration in type 2 diabetic subjects after bariatric surgery. Diabetes Care 2009; 32:375–380.
- Goldfine AB, Mun EC, Devine E, et al. Patients with neuroglycopenia after gastric bypass surgery have exaggerated incretin and insulin secretory responses to a mixed meal. J Clin Endocrinol Metab 2007; 92:4678–4685.
- Cummings DE, Weigle DS, Frayo RS, et al. Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N Engl J Med 2002; 346:1623–1630.
- Chandarana K, Drew ME, Emmanuel J, et al. Subject standardization, acclimatization, and sample processing affect gut hormone levels and appetite in humans. Gastroenterology 2009; 136:2115–2126.
- Korner J, Inabnet W, Febres G, et al. Prospective study of gut hormone and metabolic changes after adjustable gastric banding and Roux-en-Y gastric bypass. Int J Obes (Lond) 2009; 33:786–795.
- Boey D, Sainsbury A, Herzog H. The role of peptide YY in regulating glucose homeostasis. Peptides 2007; 28:390–395.
- Hanusch-Enserer U, Ghatei MA, Cauza E, Bloom SR, Prager R, Roden M. Relation of fasting plasma peptide YY to glucose metabolism and cardiovascular risk factors after restrictive bariatric surgery. Wien Klin Wochenschr 2007; 119:291–296.
- Laferrère B, Heshka S, Wang K, et al. Incretin levels and effect are markedly enhanced 1 month after Roux-en-Y gastric bypass surgery in obese patients with type 2 diabetes. Diabetes Care 2007; 30:1709–1716.
- Tucker ON, Szomstein S, Rosenthal RJ. Nutritional consequences of weight-loss surgery. Med Clin North Am 2007; 91:499–514.
- Davies DJ, Baxter JM, Baxter JN. Nutritional deficiencies after bariatric surgery. Obes Surg 2007; 17:1150–1158.
- Angstadt JD, Bodziner RA. Peripheral polyneuropathy from thiamine deficiency following laparoscopic Roux-en-Y gastric bypass. Obes Surg 2005; 15:890–892.
- Ritz P, Becouarn G, Douay O, Sallé A, Topart P, Rohmer V. Gastric bypass is not associated with protein malnutrition in morbidly obese patients. Obes Surg 2009; 19:840–844.
- Service GJ, Thompson GB, Service FJ, Andrews JC, Collazo-Clavell ML, Lloyd RV. Hyperinsulinemic hypoglycemia with nesidioblastosis after gastric-bypass surgery. N Engl J Med 2005; 353:249–254.
- Sjöström L, Narbro K, Sjöström CD, et al;Swedish Obese Subjects Study. Effects of bariatric surgery on mortality in Swedish obese subjects. N Engl J Med 2007; 357:741–752.
- Adams TD, Gress RE, Smith SC, et al. Long-term mortality after gastric bypass surgery. N Engl J Med 2007; 357:753–761.
Evidence is mounting for the use of bariatric surgery to treat type 2 diabetes mellitus in patients whose body mass index (BMI) is 35 kg/m2 or higher. In obese patients who also have type 2 diabetes, bariatric surgery sends it into remission (defined as normoglycemic control without the need for diabetic medications) in more than three-fourths of cases, with higher rates with the Roux-en-Y gastric bypass procedure than with the laparoscopic adjustable gastric banding procedure.
However, data on the effects of this surgery on type 2 diabetes come primarily from observational studies that lacked appropriate control groups, and the relative benefit of bariatric surgery vs aggressive medical antidiabetic therapy is not yet known. Needed are randomized trials comparing the two types of therapy (and the various types of bariatric surgery) in diabetic patients with less-severe obesity.
Further, why would bariatric surgery help with diabetes, and why would one procedure do it better than another? To be honest, we are not sure, but evidence points not only to weight loss but also to better insulin sensitivity and to alterations in levels of hormones secreted by the gut that increase insulin secretion.
OBESITY PROMOTES DIABETES; WEIGHT LOSS COUNTERACTS IT
Type 2 diabetes mellitus is a complex metabolic disease characterized by insulin resistance and progressive failure of pancreatic beta cells, resulting in hyperglycemia.1,2
Obesity, a potent risk factor for type 2 diabetes, contributes to its development by inducing insulin resistance and inflammation, which in turn impair glucose regulation.3,4 Fat deposits in the abdomen, muscles, and liver contribute to elevations of circulating free fatty acids and adipocyte-derived cytokines that mediate insulin resistance and inflammatory pathways.5
In the Diabetes Prevention Program,6 modest weight loss (5% to 10% of body weight) through diet and exercise reduced the incidence of type 2 diabetes, and in the ongoing Action for Health in Diabetes (Look AHEAD) study of the National Institutes of Health, it improved glucose homeostasis.7,8
The current medical approach to type 2 diabetes includes advising the patient to lose weight through lifestyle modification, and prescribing drugs that restore glycemic control by reducing insulin resistance (biguanides, glitazones) and improving insulin secretion (incretin mimetics and analogues and sulfonylureas). 9,10
However, several factors make type 2 diabetes challenging to treat in obese people. Patients who lose weight via behavioral changes and weight-loss drugs tend to gain the weight back. Antidiabetic drugs pose the risk of hypoglycemia. Moreover, although many new classes of drugs have been developed to treat type 2 diabetes, most patients fail to achieve the American Diabetes Association goal for glycemic control, ie, a hemoglobin A1c level lower than 7%.11
BARIATRIC PROCEDURES AND THEIR EFFECT ON DIABETES CONTROL
After bariatric surgery, patients lose more weight than with traditional weight-loss methods—up to 25% of their total body weight. Furthermore, of those with type 2 diabetes, 87% achieve at least better glucose control and need fewer antidiabetic medications,12 and an average of 78% achieve normal glycemic control without taking any antidiabetic medications at all.12,13
But not all bariatric procedures have the same effect on weight and diabetes: certain procedures have a greater effect.
The two major types are classified as gastric restrictive procedures and intestinal bypass procedures. The classification was initially based on the presumed mechanism of weight loss.
Gastric restrictive procedures (laparoscopic adjustable gastric banding, sleeve gastrectomy, vertical gastroplasty) limit gastric volume and, hence, restrict the intake of calories by inducing satiety. Afterward, patients lose approximately 10% to 20% of their total body weight.
Furthermore, multiple studies, including a randomized controlled trial14 (more about this below), have shown remission of type 2 diabetes with laparoscopic adjustable gastric banding but not with conventional medical therapy. The effect is primarily mediated by weight loss and improved insulin sensitivity, both of which occur several months following surgery. Of note, however: in this trial,14 all the patients had diabetes of short duration, less than 2 years.
Hence, different procedures have different effects on diabetes.12 The speed at which type 2 diabetes goes into remission differs with restrictive vs malabsorptive procedures. After Roux-en-Y gastric bypass and biliopancreatic diversion, diabetes remits within days, even before the patient has lost much weight.15 This does not happen after gastric restrictive procedures.12,16
Observational studies of the effect of Roux-en-Y surgery on diabetes
Several observational studies have evaluated the benefit of Roux-en-Y surgery for patients with type 2 diabetes mellitus.
Pories et al15 followed 608 severely obese patients, of whom 165 (27%) had type 2 diabetes or impaired glucose tolerance.
At a mean follow-up of 7.6 years after surgery, 83% of the diabetic patients were off their antidiabetic drugs, and 99% of those with impaired glucose tolerance were normoglycemic, with normal fasting glucose and hemoglobin A1c levels. Marked improvements in hyperlipidemia, hypertension, fertility, osteoarthritis, and obstructive sleep apnea were also noted.
Schauer et al17 observed similar results in 1,160 morbidly obese patients, of whom 240 (21%) had type 2 diabetes or impaired fasting glucose.
After laparoscopic Roux-en-Y gastric bypass surgery, fasting glucose and hemoglobin A1c levels returned to normal levels in 83% of cases and were markedly improved in the remaining 17%. Significantly (80%) fewer patients needed oral antidiabetic agents or insulin (79% fewer). Patients most likely to achieve complete remission of diabetes were those with the shortest duration of diabetes (< 5 years), the mildest severity of diabetes (diet-controlled), and the greatest weight loss after surgery. The rate of diabetes remission in patients who had been diabetic for 5 years or less was 95%, compared with 75% in those who had been diabetic for 6 to 10 years and 54% in those who had been diabetic for more than 10 years (P < .001).
The Swedish Obese Subjects (SOS) study18 prospectively followed 1,703 patients, of whom 118 had type 2 diabetes, for 10 years after various bariatric surgery procedures (primarily vertical gastroplasty). In a control group that received medical therapy, 77 patients had type 2 diabetes. Medical therapy was ill-defined with respect to aggressiveness and adherence to intervention with lifestyle and pharmacotherapy.
At 2 years, the surgical group had lost a mean of 28 kg, glycemic control had improved in the diabetic patients, and many of them had been able to stop taking oral hypoglycemic drugs or insulin. In contrast, the need for these agents increased in the medically treated patients. The proportion treated by diet alone rose from 59% to 73% in the surgical group, but declined from 55% to 34% in the nonsurgical group.13
In these studies, surgery also reduced the risk of progressing from impaired glucose tolerance to type 2 diabetes; the risk was 30 times lower in the study by Pories et al.15 In the SOS study,18 the frequency of diabetes was 30 times lower at 2 years and five times lower at 8 years after surgery.
Studies of biliopancreatic diversion
Data on the effects of biliopancreatic diversion, a primarily malabsorptive procedure, are limited to European studies.
Scopinaro et al19,20 reported long-term follow-up data on 312 patients with type 2 diabetes who underwent biliopancreatic diversion; 310 patients (99%) achieved normal fasting glucose values by 1 year after surgery. At 10 years after surgery, 98% of the patients were still in complete remission of diabetes, defined as normal glucose values without the use of antidiabetic medications.
Others have noted similar findings.21,22
Limitations of the studies
Although these data seem encouraging, these studies had major limitations.
The patients were mostly white women with severe obesity, ie, a BMI greater than 40 kg/m2, which is not representative of patients with type 2 diabetes in the community. Only about 20% had glucose intolerance or overt type 2 diabetes mellitus. Would other groups benefit, particularly men and those with lesssevere obesity?
Moreover, these studies were observational, with no randomized control groups. Many reports consisted of large case series. It is not clear how specific bariatric procedures were chosen or what criteria were used for performing bariatric surgery. A lack of complete follow-up data is also a concern.
Needed are large randomized trials evaluating the effects of various bariatric procedures in a less obese cohort with type 2 diabetes, ie, typical patients seen in the community. Moreover, surgery has not been compared directly with more vigorous medical weight-loss strategies, such as those used in the Diabetes Prevention Project6 and the Look AHEAD trial.7,8
A randomized controlled trial of gastric banding
The only randomized controlled trial to date that compared standard medical diabetes therapy with bariatric surgery was conducted by Dixon et al.14
Sixty patients with type 2 diabetes (duration < 2 years and mean hemoglobin A1c 7.7%) were randomized either to receive medical management as defined by the American Diabetes Association guidelines or to undergo laparoscopic adjustable gastric banding.
At 2 years, the rate of remission (defined as hemoglobin A1c < 6.2% and a normal fasting glucose level) was 13% in the medical treatment group vs 73% in the surgery group (P < .001). Patients receiving medical treatment had lost a mean of 1.7% of their body weight, vs 20.7% in the surgical patients (P < .001). Weight loss was strongly associated with remission of type 2 diabetes after surgery.
This study was controversial in that the medical intervention in this trial was not as aggressive as in the Diabetes Prevention Project and Look AHEAD trials.
INDICATIONS FOR BARIATRIC SURGERY IN PATIENTS WITH DIABETES
According to guidelines from the National Institutes of Health,23 the current indications for bariatric surgery include a BMI of 40 kg/m2 or higher, or a BMI between 35 and 40 kg/m2 with at least two obesity-related comorbidities. Diabetes is considered a key comorbidity that justifies the risk of surgery. The guidelines suggest that bariatric surgery be discussed with all severely obese patients (BMI > 35 kg/m2) with type 2 diabetes who have not been able to lose weight with other weight-control approaches.
Since type 2 diabetes mellitus is a progressive disease characterized by relentless deterioration of beta-cell function, many endocrinologists favor aggressive weight-loss approaches early in the course of the disease. We believe that bariatric surgery should be considered early, as it may help preserve pancreatic betacell function and slow the progression of microvascular and macrovascular complications.
HOW DOES BARIATRIC SURGERY IMPROVE TYPE 2 DIABETES?
Hypothesis 1: Weight loss increases insulin sensitivity
The enforced caloric restriction, negative energy balance, and weight loss after bariatric surgery reduce insulin resistance. Consequently, the beta cells can rest because they don’t need to produce as much insulin. These effects have been observed after both gastric restrictive procedures and gastric bypass procedures.
Hypothesis 2: Less lipotoxicity, inflammation
Another theory is that bariatric surgery lessens insulin resistance by reducing “lipotoxicity,” a condition related to dysregulated fatty acid flux, lipid metabolites in tissues, and direct and indirect effects of hormones secreted by adipocytes.
The strongest evidence for this theory comes from Bikman et al,26 who found that insulin sensitivity increased after Roux-en-Y surgery more than expected from weight loss alone. One year after surgery, even though they remained anthropometrically obese (BMI > 30 kg/m2), the patients had insulin sensitivity levels similar to those in a control group of lean people (BMI < 25 kg/m2).
Insulin sensitivity begins to improve within 1 week of intestinal bypass procedures,15,27 suggesting that these procedures are doing something more than simply forcing weight loss via caloric restriction, as gastric restrictive procedures do.
Hypothesis 3: An effect on gut hormones
The “hindgut hypothesis” raised by Cummings et al24 suggests that accelerated transit of concentrated nutrients (particularly glucose) to the distal intestine results in increased production of insulinotropic and appetite-controlling substances, which account for the reversal of hyperglycemia and obesity.
In contrast, the “foregut hypothesis” raised by Rubino et al28 suggests that nutrient interactions in the duodenum are diabetogenic and, hence, bypassing the duodenum would reverse this defect. Their conclusions come from experiments in rodents that underwent jejunoileal bypass and subsequent refeeding through the bypassed intestine.
GUT HORMONES AND OTHER PEPTIDES ALTERED BY BARIATRIC SURGERY
Incretin hormones: GLP-1, GIP
Gastrointestinal hormones that increase insulin release after a meal are known as incretins. Of interest, they have this effect only when glucose is ingested orally—not when it is infused intravenously.29,30
Glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP) account for 50% to 60% of nutrient-related insulin secretion. In addition to stimulating insulin, GLP-1 suppresses glucagon and slows gastric emptying, which delays digestion and reduces postprandial glycemia. GLP-1 also acts on the hypothalamus to induce satiety.
Laferrère et al31 and others32,33 documented robust increases in postprandial levels of GLP-1 within 4 weeks after Roux-en-Y surgery. GLP-1 levels did not increase with comparable weight loss induced by diet.
Rubino et al28,34 documented similar findings that occurred prior to marked weight loss, suggesting that the benefit of Roux-en-Y surgery on remission of diabetes may not be completely attributable to reduced caloric intake and weight loss. Insulin secretion is generally reduced after gastric restrictive procedures (eg, laparoscopic adjustable gastric banding) and biliopancreatic diversion,35 and is increased after Roux-en-Y gastric bypass.32,33,36
Noninsulinotropic peptides: Ghrelin, peptide YY
Noninsulinotropic gut peptides that are altered after Roux-en-Y surgery include ghrelin and peptide YY.
Ghrelin, a hormone derived from the gastric fundus, stimulates appetite. Ghrelin concentrations are lower after Roux-en-Y surgery, indicating that suppression of hunger signals helps sustain weight loss. In contrast, ghrelin levels increase with diet-induced weight loss.37 However, the data on ghrelin levels at various times after bariatric surgical procedures are not consistent.33,38
Peptide YY, like GLP-1, is secreted by L cells of the distal small intestine and is responsible for increasing satiety and delaying gastric emptying after meals. Numerous studies have consistently documented increases in postprandial peptide YY and GLP-1 levels after gastric bypass.32,33,39–41
ACUTE EFFECTS OF BARIATRIC SURGERY ON INSULIN SECRETION, SENSITIVITY
Bariatric surgery alters both insulin secretion and insulin sensitivity, thus improving glucose regulation.
The relationship between insulin secretion and sensitivity is a hyperbolic curve, so that any change in insulin sensitivity is balanced by a reciprocal and proportionate change in insulin secretion. The development of type 2 diabetes is characterized by a reduction in insulin secretion (decompensation) relative to the severity of insulin resistance.
In the first 6 weeks after Roux-en-Y gastric bypass or biliopancreatic diversion, insulin sensitivity improves while insulin secretion increases disproportionately, associated with a robust increase in GLP-1, and resulting in normal glucose homeostasis.16,31,42
In contrast, patients who lose weight by dieting or undergoing gastric restrictive procedures show a modest increase in insulin sensitivity and a compensatory reduction in insulin secretion, termed “beta-cell rest.”16,31,42
RISKS OF BARIATRIC SURGERY
Short-term risks
An important concern about using bariatric surgery to treat type 2 diabetes is the risk of morbidity and death associated with these procedures.
Buchwald et al13 performed a meta-analysis of 136 bariatric studies that included 22,094 patients. The 30-day operative death rates were 1.1% with biliopancreatic diversion, 0.5% with Roux-en-Y surgery, and 0.1% with restrictive procedures.
Laparoscopic adjustable gastric banding is considered the safest of the current bariatric procedures. It does not involve bowel anastomosis, and the risks of major hemorrhage, gastric perforation, and pulmonary embolism are less than 1%. Late complications requiring reoperation include band slippage or prolapse (5%–10%) and band erosion (1%–3%). The entire intestinal tract is left intact, so subsequent nutritional deficiencies are rare.43
Roux-en-Y gastric bypass carries an overall risk of major complications of 10% to 15%. Anastomotic leak (1%–5%), pulmonary embolism (< 1%), and hemorrhage (1%–4%) can be life-threatening but are rare if the staff are experienced. Late complications such as ulcer or stricture formation at the gastrojejunostomy site occur in 5% to 10% of cases and are managed nonoperatively.
Nutritional deficiencies
Protein-calorie malnutrition is recognized by signs such as edema, hypoalbuminemia, anemia, and hair loss. To minimize this problem after Roux-en-Y surgery, we suggest that patients take in 60 to 80 g of protein and 700 to 800 kcal a day.
Vitamin deficiencies can lead to Wernicke encephalopathy (due to thiamine deficiency), peripheral neuropathy (due to vitamin B12 deficiency),45,46 and metabolic bone disease (due to long-term deficiencies of vitamin D and calcium). Often, vitamin deficiencies are present before surgery and require prompt supplementation to avoid exacerbation of these deficiencies afterward.
Biliopancreatic diversion procedures are performed at relatively few centers worldwide, largely because of the massive amounts of protein, fat, and carbohydrate malabsorption they cause. Long-term deficiencies of fat-soluble vitamins, iron, calcium, and vitamins B12 and D have been reported in one-third to one-half of patients undergoing these procedures, and nutritional supplementation is mandatory.43 Protein-calorie malnutrition occurs in 7% of cases, and 2% of patients require operative revision to lengthen the common channel.
In rare cases, severe hypoglycemia has been noted after Roux-en-Y surgery and is associated with prandial hyperinsulinemia related to elevated GLP-1 levels.36,47 Neuroglycopenia and seizures have been reported in severe cases. Initial treatment of hypoglycemia involves dietary modification targeting carbohydrate restriction, the use of alpha glucosidase inhibitors such as acarbose (Precose), and referral to an endocrinologist for further management.
Long-term death rates
Death rates after bariatric surgery must be weighed against the long-term cardiovascular risks of continued obesity and type 2 diabetes.
Strong evidence now exists that bariatric surgery increases life expectancy48 and that this is largely attributable to reduction in cardiovascular risk factors such as diabetes and cancer. Recent studies have found that the long-term death rate is 32% to 73% lower for patients undergoing bariatric surgery than in matched controls who do not undergo surgery.49 A decrease in the death rate related to diabetes has played an important role in these results.
Acknowledgments: We acknowledge support from the National Institutes of Health, Multidisciplinary Clinical Research Career Development Programs Grant 5K12RR023264 (SRK), National Center for Research Resources, CTSA 1UL1RR024989, and research grants from Ethicon Endo-Surgery (PS,SRK).
Evidence is mounting for the use of bariatric surgery to treat type 2 diabetes mellitus in patients whose body mass index (BMI) is 35 kg/m2 or higher. In obese patients who also have type 2 diabetes, bariatric surgery sends it into remission (defined as normoglycemic control without the need for diabetic medications) in more than three-fourths of cases, with higher rates with the Roux-en-Y gastric bypass procedure than with the laparoscopic adjustable gastric banding procedure.
However, data on the effects of this surgery on type 2 diabetes come primarily from observational studies that lacked appropriate control groups, and the relative benefit of bariatric surgery vs aggressive medical antidiabetic therapy is not yet known. Needed are randomized trials comparing the two types of therapy (and the various types of bariatric surgery) in diabetic patients with less-severe obesity.
Further, why would bariatric surgery help with diabetes, and why would one procedure do it better than another? To be honest, we are not sure, but evidence points not only to weight loss but also to better insulin sensitivity and to alterations in levels of hormones secreted by the gut that increase insulin secretion.
OBESITY PROMOTES DIABETES; WEIGHT LOSS COUNTERACTS IT
Type 2 diabetes mellitus is a complex metabolic disease characterized by insulin resistance and progressive failure of pancreatic beta cells, resulting in hyperglycemia.1,2
Obesity, a potent risk factor for type 2 diabetes, contributes to its development by inducing insulin resistance and inflammation, which in turn impair glucose regulation.3,4 Fat deposits in the abdomen, muscles, and liver contribute to elevations of circulating free fatty acids and adipocyte-derived cytokines that mediate insulin resistance and inflammatory pathways.5
In the Diabetes Prevention Program,6 modest weight loss (5% to 10% of body weight) through diet and exercise reduced the incidence of type 2 diabetes, and in the ongoing Action for Health in Diabetes (Look AHEAD) study of the National Institutes of Health, it improved glucose homeostasis.7,8
The current medical approach to type 2 diabetes includes advising the patient to lose weight through lifestyle modification, and prescribing drugs that restore glycemic control by reducing insulin resistance (biguanides, glitazones) and improving insulin secretion (incretin mimetics and analogues and sulfonylureas). 9,10
However, several factors make type 2 diabetes challenging to treat in obese people. Patients who lose weight via behavioral changes and weight-loss drugs tend to gain the weight back. Antidiabetic drugs pose the risk of hypoglycemia. Moreover, although many new classes of drugs have been developed to treat type 2 diabetes, most patients fail to achieve the American Diabetes Association goal for glycemic control, ie, a hemoglobin A1c level lower than 7%.11
BARIATRIC PROCEDURES AND THEIR EFFECT ON DIABETES CONTROL
After bariatric surgery, patients lose more weight than with traditional weight-loss methods—up to 25% of their total body weight. Furthermore, of those with type 2 diabetes, 87% achieve at least better glucose control and need fewer antidiabetic medications,12 and an average of 78% achieve normal glycemic control without taking any antidiabetic medications at all.12,13
But not all bariatric procedures have the same effect on weight and diabetes: certain procedures have a greater effect.
The two major types are classified as gastric restrictive procedures and intestinal bypass procedures. The classification was initially based on the presumed mechanism of weight loss.
Gastric restrictive procedures (laparoscopic adjustable gastric banding, sleeve gastrectomy, vertical gastroplasty) limit gastric volume and, hence, restrict the intake of calories by inducing satiety. Afterward, patients lose approximately 10% to 20% of their total body weight.
Furthermore, multiple studies, including a randomized controlled trial14 (more about this below), have shown remission of type 2 diabetes with laparoscopic adjustable gastric banding but not with conventional medical therapy. The effect is primarily mediated by weight loss and improved insulin sensitivity, both of which occur several months following surgery. Of note, however: in this trial,14 all the patients had diabetes of short duration, less than 2 years.
Hence, different procedures have different effects on diabetes.12 The speed at which type 2 diabetes goes into remission differs with restrictive vs malabsorptive procedures. After Roux-en-Y gastric bypass and biliopancreatic diversion, diabetes remits within days, even before the patient has lost much weight.15 This does not happen after gastric restrictive procedures.12,16
Observational studies of the effect of Roux-en-Y surgery on diabetes
Several observational studies have evaluated the benefit of Roux-en-Y surgery for patients with type 2 diabetes mellitus.
Pories et al15 followed 608 severely obese patients, of whom 165 (27%) had type 2 diabetes or impaired glucose tolerance.
At a mean follow-up of 7.6 years after surgery, 83% of the diabetic patients were off their antidiabetic drugs, and 99% of those with impaired glucose tolerance were normoglycemic, with normal fasting glucose and hemoglobin A1c levels. Marked improvements in hyperlipidemia, hypertension, fertility, osteoarthritis, and obstructive sleep apnea were also noted.
Schauer et al17 observed similar results in 1,160 morbidly obese patients, of whom 240 (21%) had type 2 diabetes or impaired fasting glucose.
After laparoscopic Roux-en-Y gastric bypass surgery, fasting glucose and hemoglobin A1c levels returned to normal levels in 83% of cases and were markedly improved in the remaining 17%. Significantly (80%) fewer patients needed oral antidiabetic agents or insulin (79% fewer). Patients most likely to achieve complete remission of diabetes were those with the shortest duration of diabetes (< 5 years), the mildest severity of diabetes (diet-controlled), and the greatest weight loss after surgery. The rate of diabetes remission in patients who had been diabetic for 5 years or less was 95%, compared with 75% in those who had been diabetic for 6 to 10 years and 54% in those who had been diabetic for more than 10 years (P < .001).
The Swedish Obese Subjects (SOS) study18 prospectively followed 1,703 patients, of whom 118 had type 2 diabetes, for 10 years after various bariatric surgery procedures (primarily vertical gastroplasty). In a control group that received medical therapy, 77 patients had type 2 diabetes. Medical therapy was ill-defined with respect to aggressiveness and adherence to intervention with lifestyle and pharmacotherapy.
At 2 years, the surgical group had lost a mean of 28 kg, glycemic control had improved in the diabetic patients, and many of them had been able to stop taking oral hypoglycemic drugs or insulin. In contrast, the need for these agents increased in the medically treated patients. The proportion treated by diet alone rose from 59% to 73% in the surgical group, but declined from 55% to 34% in the nonsurgical group.13
In these studies, surgery also reduced the risk of progressing from impaired glucose tolerance to type 2 diabetes; the risk was 30 times lower in the study by Pories et al.15 In the SOS study,18 the frequency of diabetes was 30 times lower at 2 years and five times lower at 8 years after surgery.
Studies of biliopancreatic diversion
Data on the effects of biliopancreatic diversion, a primarily malabsorptive procedure, are limited to European studies.
Scopinaro et al19,20 reported long-term follow-up data on 312 patients with type 2 diabetes who underwent biliopancreatic diversion; 310 patients (99%) achieved normal fasting glucose values by 1 year after surgery. At 10 years after surgery, 98% of the patients were still in complete remission of diabetes, defined as normal glucose values without the use of antidiabetic medications.
Others have noted similar findings.21,22
Limitations of the studies
Although these data seem encouraging, these studies had major limitations.
The patients were mostly white women with severe obesity, ie, a BMI greater than 40 kg/m2, which is not representative of patients with type 2 diabetes in the community. Only about 20% had glucose intolerance or overt type 2 diabetes mellitus. Would other groups benefit, particularly men and those with lesssevere obesity?
Moreover, these studies were observational, with no randomized control groups. Many reports consisted of large case series. It is not clear how specific bariatric procedures were chosen or what criteria were used for performing bariatric surgery. A lack of complete follow-up data is also a concern.
Needed are large randomized trials evaluating the effects of various bariatric procedures in a less obese cohort with type 2 diabetes, ie, typical patients seen in the community. Moreover, surgery has not been compared directly with more vigorous medical weight-loss strategies, such as those used in the Diabetes Prevention Project6 and the Look AHEAD trial.7,8
A randomized controlled trial of gastric banding
The only randomized controlled trial to date that compared standard medical diabetes therapy with bariatric surgery was conducted by Dixon et al.14
Sixty patients with type 2 diabetes (duration < 2 years and mean hemoglobin A1c 7.7%) were randomized either to receive medical management as defined by the American Diabetes Association guidelines or to undergo laparoscopic adjustable gastric banding.
At 2 years, the rate of remission (defined as hemoglobin A1c < 6.2% and a normal fasting glucose level) was 13% in the medical treatment group vs 73% in the surgery group (P < .001). Patients receiving medical treatment had lost a mean of 1.7% of their body weight, vs 20.7% in the surgical patients (P < .001). Weight loss was strongly associated with remission of type 2 diabetes after surgery.
This study was controversial in that the medical intervention in this trial was not as aggressive as in the Diabetes Prevention Project and Look AHEAD trials.
INDICATIONS FOR BARIATRIC SURGERY IN PATIENTS WITH DIABETES
According to guidelines from the National Institutes of Health,23 the current indications for bariatric surgery include a BMI of 40 kg/m2 or higher, or a BMI between 35 and 40 kg/m2 with at least two obesity-related comorbidities. Diabetes is considered a key comorbidity that justifies the risk of surgery. The guidelines suggest that bariatric surgery be discussed with all severely obese patients (BMI > 35 kg/m2) with type 2 diabetes who have not been able to lose weight with other weight-control approaches.
Since type 2 diabetes mellitus is a progressive disease characterized by relentless deterioration of beta-cell function, many endocrinologists favor aggressive weight-loss approaches early in the course of the disease. We believe that bariatric surgery should be considered early, as it may help preserve pancreatic betacell function and slow the progression of microvascular and macrovascular complications.
HOW DOES BARIATRIC SURGERY IMPROVE TYPE 2 DIABETES?
Hypothesis 1: Weight loss increases insulin sensitivity
The enforced caloric restriction, negative energy balance, and weight loss after bariatric surgery reduce insulin resistance. Consequently, the beta cells can rest because they don’t need to produce as much insulin. These effects have been observed after both gastric restrictive procedures and gastric bypass procedures.
Hypothesis 2: Less lipotoxicity, inflammation
Another theory is that bariatric surgery lessens insulin resistance by reducing “lipotoxicity,” a condition related to dysregulated fatty acid flux, lipid metabolites in tissues, and direct and indirect effects of hormones secreted by adipocytes.
The strongest evidence for this theory comes from Bikman et al,26 who found that insulin sensitivity increased after Roux-en-Y surgery more than expected from weight loss alone. One year after surgery, even though they remained anthropometrically obese (BMI > 30 kg/m2), the patients had insulin sensitivity levels similar to those in a control group of lean people (BMI < 25 kg/m2).
Insulin sensitivity begins to improve within 1 week of intestinal bypass procedures,15,27 suggesting that these procedures are doing something more than simply forcing weight loss via caloric restriction, as gastric restrictive procedures do.
Hypothesis 3: An effect on gut hormones
The “hindgut hypothesis” raised by Cummings et al24 suggests that accelerated transit of concentrated nutrients (particularly glucose) to the distal intestine results in increased production of insulinotropic and appetite-controlling substances, which account for the reversal of hyperglycemia and obesity.
In contrast, the “foregut hypothesis” raised by Rubino et al28 suggests that nutrient interactions in the duodenum are diabetogenic and, hence, bypassing the duodenum would reverse this defect. Their conclusions come from experiments in rodents that underwent jejunoileal bypass and subsequent refeeding through the bypassed intestine.
GUT HORMONES AND OTHER PEPTIDES ALTERED BY BARIATRIC SURGERY
Incretin hormones: GLP-1, GIP
Gastrointestinal hormones that increase insulin release after a meal are known as incretins. Of interest, they have this effect only when glucose is ingested orally—not when it is infused intravenously.29,30
Glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP) account for 50% to 60% of nutrient-related insulin secretion. In addition to stimulating insulin, GLP-1 suppresses glucagon and slows gastric emptying, which delays digestion and reduces postprandial glycemia. GLP-1 also acts on the hypothalamus to induce satiety.
Laferrère et al31 and others32,33 documented robust increases in postprandial levels of GLP-1 within 4 weeks after Roux-en-Y surgery. GLP-1 levels did not increase with comparable weight loss induced by diet.
Rubino et al28,34 documented similar findings that occurred prior to marked weight loss, suggesting that the benefit of Roux-en-Y surgery on remission of diabetes may not be completely attributable to reduced caloric intake and weight loss. Insulin secretion is generally reduced after gastric restrictive procedures (eg, laparoscopic adjustable gastric banding) and biliopancreatic diversion,35 and is increased after Roux-en-Y gastric bypass.32,33,36
Noninsulinotropic peptides: Ghrelin, peptide YY
Noninsulinotropic gut peptides that are altered after Roux-en-Y surgery include ghrelin and peptide YY.
Ghrelin, a hormone derived from the gastric fundus, stimulates appetite. Ghrelin concentrations are lower after Roux-en-Y surgery, indicating that suppression of hunger signals helps sustain weight loss. In contrast, ghrelin levels increase with diet-induced weight loss.37 However, the data on ghrelin levels at various times after bariatric surgical procedures are not consistent.33,38
Peptide YY, like GLP-1, is secreted by L cells of the distal small intestine and is responsible for increasing satiety and delaying gastric emptying after meals. Numerous studies have consistently documented increases in postprandial peptide YY and GLP-1 levels after gastric bypass.32,33,39–41
ACUTE EFFECTS OF BARIATRIC SURGERY ON INSULIN SECRETION, SENSITIVITY
Bariatric surgery alters both insulin secretion and insulin sensitivity, thus improving glucose regulation.
The relationship between insulin secretion and sensitivity is a hyperbolic curve, so that any change in insulin sensitivity is balanced by a reciprocal and proportionate change in insulin secretion. The development of type 2 diabetes is characterized by a reduction in insulin secretion (decompensation) relative to the severity of insulin resistance.
In the first 6 weeks after Roux-en-Y gastric bypass or biliopancreatic diversion, insulin sensitivity improves while insulin secretion increases disproportionately, associated with a robust increase in GLP-1, and resulting in normal glucose homeostasis.16,31,42
In contrast, patients who lose weight by dieting or undergoing gastric restrictive procedures show a modest increase in insulin sensitivity and a compensatory reduction in insulin secretion, termed “beta-cell rest.”16,31,42
RISKS OF BARIATRIC SURGERY
Short-term risks
An important concern about using bariatric surgery to treat type 2 diabetes is the risk of morbidity and death associated with these procedures.
Buchwald et al13 performed a meta-analysis of 136 bariatric studies that included 22,094 patients. The 30-day operative death rates were 1.1% with biliopancreatic diversion, 0.5% with Roux-en-Y surgery, and 0.1% with restrictive procedures.
Laparoscopic adjustable gastric banding is considered the safest of the current bariatric procedures. It does not involve bowel anastomosis, and the risks of major hemorrhage, gastric perforation, and pulmonary embolism are less than 1%. Late complications requiring reoperation include band slippage or prolapse (5%–10%) and band erosion (1%–3%). The entire intestinal tract is left intact, so subsequent nutritional deficiencies are rare.43
Roux-en-Y gastric bypass carries an overall risk of major complications of 10% to 15%. Anastomotic leak (1%–5%), pulmonary embolism (< 1%), and hemorrhage (1%–4%) can be life-threatening but are rare if the staff are experienced. Late complications such as ulcer or stricture formation at the gastrojejunostomy site occur in 5% to 10% of cases and are managed nonoperatively.
Nutritional deficiencies
Protein-calorie malnutrition is recognized by signs such as edema, hypoalbuminemia, anemia, and hair loss. To minimize this problem after Roux-en-Y surgery, we suggest that patients take in 60 to 80 g of protein and 700 to 800 kcal a day.
Vitamin deficiencies can lead to Wernicke encephalopathy (due to thiamine deficiency), peripheral neuropathy (due to vitamin B12 deficiency),45,46 and metabolic bone disease (due to long-term deficiencies of vitamin D and calcium). Often, vitamin deficiencies are present before surgery and require prompt supplementation to avoid exacerbation of these deficiencies afterward.
Biliopancreatic diversion procedures are performed at relatively few centers worldwide, largely because of the massive amounts of protein, fat, and carbohydrate malabsorption they cause. Long-term deficiencies of fat-soluble vitamins, iron, calcium, and vitamins B12 and D have been reported in one-third to one-half of patients undergoing these procedures, and nutritional supplementation is mandatory.43 Protein-calorie malnutrition occurs in 7% of cases, and 2% of patients require operative revision to lengthen the common channel.
In rare cases, severe hypoglycemia has been noted after Roux-en-Y surgery and is associated with prandial hyperinsulinemia related to elevated GLP-1 levels.36,47 Neuroglycopenia and seizures have been reported in severe cases. Initial treatment of hypoglycemia involves dietary modification targeting carbohydrate restriction, the use of alpha glucosidase inhibitors such as acarbose (Precose), and referral to an endocrinologist for further management.
Long-term death rates
Death rates after bariatric surgery must be weighed against the long-term cardiovascular risks of continued obesity and type 2 diabetes.
Strong evidence now exists that bariatric surgery increases life expectancy48 and that this is largely attributable to reduction in cardiovascular risk factors such as diabetes and cancer. Recent studies have found that the long-term death rate is 32% to 73% lower for patients undergoing bariatric surgery than in matched controls who do not undergo surgery.49 A decrease in the death rate related to diabetes has played an important role in these results.
Acknowledgments: We acknowledge support from the National Institutes of Health, Multidisciplinary Clinical Research Career Development Programs Grant 5K12RR023264 (SRK), National Center for Research Resources, CTSA 1UL1RR024989, and research grants from Ethicon Endo-Surgery (PS,SRK).
- DeFronzo RA. Pathogenesis of type 2 diabetes mellitus. Med Clin North Am 2004; 88:787–835.
- Kashyap SR, Defronzo RA. The insulin resistance syndrome: physiological considerations. Diab Vasc Dis Res 2007; 4:13–19.
- Mokdad AH, Ford ES, Bowman BA, et al. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA 2003; 289:76–79.
- Unger RH. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology 2003; 144:5159–5165.
- Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 2002; 51:2005–2011.
- Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346:393–403.
- Look AHEAD Research Group; Pi-Sunyer X, Blackburn G, Brancati FL, et al. Reduction in weight and cardiovascular disease risk factors in individuals with type 2 diabetes: one-year results of the look AHEAD trial. Diabetes Care 2007; 30:1374–1383.
- Look AHEAD Research Group; Wadden TA, West DS, Delahanty L, et al. The Look AHEAD study: a description of the lifestyle intervention and the evidence supporting it. Obesity (Silver Spring) 2006; 14:737–752.
- Nathan DM. Clinical practice. Initial management of glycemia in type 2 diabetes mellitus. N Engl J Med 2002; 347:1342–1349.
- Nathan DM, Buse JB, Davidson MB, et al. Management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: update regarding thiazolidinediones: a consensus statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2008; 31:173–175.
- Spann SJ, Nutting PA, Galliher JM, et al. Management of type 2 diabetes in the primary care setting: a practice-based research network study. Ann Fam Med 2006; 4:23–31.
- Buchwald H, Estok R, Fahrbach K, et al. Weight and type 2 diabetes after bariatric surgery: systematic review and meta-analysis. Am J Med 2009; 122:248–256.
- Buchwald H, Avidor Y, Braunwald E, et al. Bariatric surgery: a systematic review and meta-analysis. JAMA 2004; 292:1724–1737.
- Dixon JB, O’Brien PE, Playfair J, et al. Adjustable gastric banding and conventional therapy for type 2 diabetes. JAMA 2008; 299:316–323.
- Pories WJ, Swanson MS, MacDonald KG, et al. Who would have thought it? An operation proves to be the most effective therapy for adult-onset diabetes mellitus. Ann Surg 1995; 222:339–350.
- Kashyap SR, Daud S, Kelly KR, et al. Acute effects of gastric bypass versus gastric restrictive surgery on beta-cell function and insulinotropic hormones in severely obese patients with type 2 diabetes. Int J Obes (Lond) 2009; epub ahead of print
- Schauer PR, Burguera B, Ikramuddin S, et al. Effect of laparoscopic Roux-en Y gastric bypass on type 2 diabetes mellitus. Ann Surg 2003; 238:467–484.
- Sjöström L, Lindroos AK, Peltonen M, et al; Swedish Obese Subjects Study Scientific Group. Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. N Engl J Med 2004; 351:2683–2693.
- Scopinaro N, Marinari GM, Camerini GB, Papadia FS, Adami GF. Specific effects of biliopancreatic diversion on the major components of metabolic syndrome: a long-term follow-up study. Diabetes Care 2005; 28:2406–2411.
- Scopinaro N, Papadia F, Marinari G, Camerini G, Adami G. Long-term control of type 2 diabetes mellitus and the other major components of the metabolic syndrome after biliopancreatic diversion in patients with BMI < 35 kg/m2. Obes Surg 2007; 17:185–192.
- Alexandrides TK, Skroubis G, Kalfarentzos F. Resolution of diabetes mellitus and metabolic syndrome following Roux-en-Y gastric bypass and a variant of biliopancreatic diversion in patients with morbid obesity. Obes Surg 2007; 17:176–184.
- Chiellini C, Rubino F, Castagneto M, Nanni G, Mingrone G. The effect of bilio-pancreatic diversion on type 2 diabetes in patients with BMI < 35 kg/m2. Diabetologia 2009; 52:1027–1030.
- Consensus Development Conference Panel. NIH conference. Gastrointestinal surgery for severe obesity. Ann Intern Med 1991; 115:956–961.
- Cummings DE, Overduin J, Foster-Schubert KE. Gastric bypass for obesity: mechanisms of weight loss and diabetes resolution. J Clin Endocrinol Metab 2004; 89:2608–2615.
- Cummings DE, Flum DR. Gastrointestinal surgery as a treatment for diabetes. JAMA 2008; 299:341–343.
- Bikman BT, Zheng D, Pories WJ, et al. Mechanism for improved insulin sensitivity after gastric bypass surgery. J Clin Endocrinol Metab 2008; 93:4656–4663.
- Guidone C, Manco M, Valera-Mora E, et al. Mechanisms of recovery from type 2 diabetes after malabsorptive bariatric surgery. Diabetes 2006; 55:2025–2031.
- Rubino F, Forgione A, Cummings DE, et al. The mechanism of diabetes control after gastrointestinal bypass surgery reveals a role of the proximal small intestine in the pathophysiology of type 2 diabetes. Ann Surg 2006; 244:741–749.
- Vilsbøll T, Krarup T, Madsbad S, Holst JJ. Both GLP-1 and GIP are insulinotropic at basal and postprandial glucose levels and contribute nearly equally to the incretin effect of a meal in healthy subjects. Regul Pept 2003; 114:115–121.
- Vollmer K, Holst JJ, Baller B, et al. Predictors of incretin concentrations in subjects with normal, impaired, and diabetic glucose tolerance. Diabetes 2008; 57:678–687.
- Laferrère B, Teixeira J, McGinty J, et al. Effect of weight loss by gastric bypass surgery versus hypocaloric diet on glucose and incretin levels in patients with type 2 diabetes. J Clin Endocrinol Metab 2008; 93:2479–2485.
- Korner J, Bessler M, Inabnet W, Taveras C, Holst JJ. Exaggerated glucagon-like peptide-1 and blunted glucose-dependent insulinotropic peptide secretion are associated with Roux-en-Y gastric bypass but not adjustable gastric banding. Surg Obes Relat Dis 2007; 3:597–601.
- le Roux CW, Aylwin SJ, Batterham RL, et al. Gut hormone profiles following bariatric surgery favor an anorectic state, facilitate weight loss, and improve metabolic parameters. Ann Surg 2006; 243:108–114.
- Rubino F, Gagner M, Gentileschi P, et al. The early effect of the Roux-en-Y gastric bypass on hormones involved in body weight regulation and glucose metabolism. Ann Surg 2004; 240:236–242.
- Salinari S, Bertuzzi A, Asnaghi S, Guidone C, Manco M, Mingrone G. First-phase insulin secretion restoration and differential response to glucose load depending on the route of administration in type 2 diabetic subjects after bariatric surgery. Diabetes Care 2009; 32:375–380.
- Goldfine AB, Mun EC, Devine E, et al. Patients with neuroglycopenia after gastric bypass surgery have exaggerated incretin and insulin secretory responses to a mixed meal. J Clin Endocrinol Metab 2007; 92:4678–4685.
- Cummings DE, Weigle DS, Frayo RS, et al. Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N Engl J Med 2002; 346:1623–1630.
- Chandarana K, Drew ME, Emmanuel J, et al. Subject standardization, acclimatization, and sample processing affect gut hormone levels and appetite in humans. Gastroenterology 2009; 136:2115–2126.
- Korner J, Inabnet W, Febres G, et al. Prospective study of gut hormone and metabolic changes after adjustable gastric banding and Roux-en-Y gastric bypass. Int J Obes (Lond) 2009; 33:786–795.
- Boey D, Sainsbury A, Herzog H. The role of peptide YY in regulating glucose homeostasis. Peptides 2007; 28:390–395.
- Hanusch-Enserer U, Ghatei MA, Cauza E, Bloom SR, Prager R, Roden M. Relation of fasting plasma peptide YY to glucose metabolism and cardiovascular risk factors after restrictive bariatric surgery. Wien Klin Wochenschr 2007; 119:291–296.
- Laferrère B, Heshka S, Wang K, et al. Incretin levels and effect are markedly enhanced 1 month after Roux-en-Y gastric bypass surgery in obese patients with type 2 diabetes. Diabetes Care 2007; 30:1709–1716.
- Tucker ON, Szomstein S, Rosenthal RJ. Nutritional consequences of weight-loss surgery. Med Clin North Am 2007; 91:499–514.
- Davies DJ, Baxter JM, Baxter JN. Nutritional deficiencies after bariatric surgery. Obes Surg 2007; 17:1150–1158.
- Angstadt JD, Bodziner RA. Peripheral polyneuropathy from thiamine deficiency following laparoscopic Roux-en-Y gastric bypass. Obes Surg 2005; 15:890–892.
- Ritz P, Becouarn G, Douay O, Sallé A, Topart P, Rohmer V. Gastric bypass is not associated with protein malnutrition in morbidly obese patients. Obes Surg 2009; 19:840–844.
- Service GJ, Thompson GB, Service FJ, Andrews JC, Collazo-Clavell ML, Lloyd RV. Hyperinsulinemic hypoglycemia with nesidioblastosis after gastric-bypass surgery. N Engl J Med 2005; 353:249–254.
- Sjöström L, Narbro K, Sjöström CD, et al;Swedish Obese Subjects Study. Effects of bariatric surgery on mortality in Swedish obese subjects. N Engl J Med 2007; 357:741–752.
- Adams TD, Gress RE, Smith SC, et al. Long-term mortality after gastric bypass surgery. N Engl J Med 2007; 357:753–761.
- DeFronzo RA. Pathogenesis of type 2 diabetes mellitus. Med Clin North Am 2004; 88:787–835.
- Kashyap SR, Defronzo RA. The insulin resistance syndrome: physiological considerations. Diab Vasc Dis Res 2007; 4:13–19.
- Mokdad AH, Ford ES, Bowman BA, et al. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA 2003; 289:76–79.
- Unger RH. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology 2003; 144:5159–5165.
- Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 2002; 51:2005–2011.
- Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346:393–403.
- Look AHEAD Research Group; Pi-Sunyer X, Blackburn G, Brancati FL, et al. Reduction in weight and cardiovascular disease risk factors in individuals with type 2 diabetes: one-year results of the look AHEAD trial. Diabetes Care 2007; 30:1374–1383.
- Look AHEAD Research Group; Wadden TA, West DS, Delahanty L, et al. The Look AHEAD study: a description of the lifestyle intervention and the evidence supporting it. Obesity (Silver Spring) 2006; 14:737–752.
- Nathan DM. Clinical practice. Initial management of glycemia in type 2 diabetes mellitus. N Engl J Med 2002; 347:1342–1349.
- Nathan DM, Buse JB, Davidson MB, et al. Management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: update regarding thiazolidinediones: a consensus statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2008; 31:173–175.
- Spann SJ, Nutting PA, Galliher JM, et al. Management of type 2 diabetes in the primary care setting: a practice-based research network study. Ann Fam Med 2006; 4:23–31.
- Buchwald H, Estok R, Fahrbach K, et al. Weight and type 2 diabetes after bariatric surgery: systematic review and meta-analysis. Am J Med 2009; 122:248–256.
- Buchwald H, Avidor Y, Braunwald E, et al. Bariatric surgery: a systematic review and meta-analysis. JAMA 2004; 292:1724–1737.
- Dixon JB, O’Brien PE, Playfair J, et al. Adjustable gastric banding and conventional therapy for type 2 diabetes. JAMA 2008; 299:316–323.
- Pories WJ, Swanson MS, MacDonald KG, et al. Who would have thought it? An operation proves to be the most effective therapy for adult-onset diabetes mellitus. Ann Surg 1995; 222:339–350.
- Kashyap SR, Daud S, Kelly KR, et al. Acute effects of gastric bypass versus gastric restrictive surgery on beta-cell function and insulinotropic hormones in severely obese patients with type 2 diabetes. Int J Obes (Lond) 2009; epub ahead of print
- Schauer PR, Burguera B, Ikramuddin S, et al. Effect of laparoscopic Roux-en Y gastric bypass on type 2 diabetes mellitus. Ann Surg 2003; 238:467–484.
- Sjöström L, Lindroos AK, Peltonen M, et al; Swedish Obese Subjects Study Scientific Group. Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. N Engl J Med 2004; 351:2683–2693.
- Scopinaro N, Marinari GM, Camerini GB, Papadia FS, Adami GF. Specific effects of biliopancreatic diversion on the major components of metabolic syndrome: a long-term follow-up study. Diabetes Care 2005; 28:2406–2411.
- Scopinaro N, Papadia F, Marinari G, Camerini G, Adami G. Long-term control of type 2 diabetes mellitus and the other major components of the metabolic syndrome after biliopancreatic diversion in patients with BMI < 35 kg/m2. Obes Surg 2007; 17:185–192.
- Alexandrides TK, Skroubis G, Kalfarentzos F. Resolution of diabetes mellitus and metabolic syndrome following Roux-en-Y gastric bypass and a variant of biliopancreatic diversion in patients with morbid obesity. Obes Surg 2007; 17:176–184.
- Chiellini C, Rubino F, Castagneto M, Nanni G, Mingrone G. The effect of bilio-pancreatic diversion on type 2 diabetes in patients with BMI < 35 kg/m2. Diabetologia 2009; 52:1027–1030.
- Consensus Development Conference Panel. NIH conference. Gastrointestinal surgery for severe obesity. Ann Intern Med 1991; 115:956–961.
- Cummings DE, Overduin J, Foster-Schubert KE. Gastric bypass for obesity: mechanisms of weight loss and diabetes resolution. J Clin Endocrinol Metab 2004; 89:2608–2615.
- Cummings DE, Flum DR. Gastrointestinal surgery as a treatment for diabetes. JAMA 2008; 299:341–343.
- Bikman BT, Zheng D, Pories WJ, et al. Mechanism for improved insulin sensitivity after gastric bypass surgery. J Clin Endocrinol Metab 2008; 93:4656–4663.
- Guidone C, Manco M, Valera-Mora E, et al. Mechanisms of recovery from type 2 diabetes after malabsorptive bariatric surgery. Diabetes 2006; 55:2025–2031.
- Rubino F, Forgione A, Cummings DE, et al. The mechanism of diabetes control after gastrointestinal bypass surgery reveals a role of the proximal small intestine in the pathophysiology of type 2 diabetes. Ann Surg 2006; 244:741–749.
- Vilsbøll T, Krarup T, Madsbad S, Holst JJ. Both GLP-1 and GIP are insulinotropic at basal and postprandial glucose levels and contribute nearly equally to the incretin effect of a meal in healthy subjects. Regul Pept 2003; 114:115–121.
- Vollmer K, Holst JJ, Baller B, et al. Predictors of incretin concentrations in subjects with normal, impaired, and diabetic glucose tolerance. Diabetes 2008; 57:678–687.
- Laferrère B, Teixeira J, McGinty J, et al. Effect of weight loss by gastric bypass surgery versus hypocaloric diet on glucose and incretin levels in patients with type 2 diabetes. J Clin Endocrinol Metab 2008; 93:2479–2485.
- Korner J, Bessler M, Inabnet W, Taveras C, Holst JJ. Exaggerated glucagon-like peptide-1 and blunted glucose-dependent insulinotropic peptide secretion are associated with Roux-en-Y gastric bypass but not adjustable gastric banding. Surg Obes Relat Dis 2007; 3:597–601.
- le Roux CW, Aylwin SJ, Batterham RL, et al. Gut hormone profiles following bariatric surgery favor an anorectic state, facilitate weight loss, and improve metabolic parameters. Ann Surg 2006; 243:108–114.
- Rubino F, Gagner M, Gentileschi P, et al. The early effect of the Roux-en-Y gastric bypass on hormones involved in body weight regulation and glucose metabolism. Ann Surg 2004; 240:236–242.
- Salinari S, Bertuzzi A, Asnaghi S, Guidone C, Manco M, Mingrone G. First-phase insulin secretion restoration and differential response to glucose load depending on the route of administration in type 2 diabetic subjects after bariatric surgery. Diabetes Care 2009; 32:375–380.
- Goldfine AB, Mun EC, Devine E, et al. Patients with neuroglycopenia after gastric bypass surgery have exaggerated incretin and insulin secretory responses to a mixed meal. J Clin Endocrinol Metab 2007; 92:4678–4685.
- Cummings DE, Weigle DS, Frayo RS, et al. Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N Engl J Med 2002; 346:1623–1630.
- Chandarana K, Drew ME, Emmanuel J, et al. Subject standardization, acclimatization, and sample processing affect gut hormone levels and appetite in humans. Gastroenterology 2009; 136:2115–2126.
- Korner J, Inabnet W, Febres G, et al. Prospective study of gut hormone and metabolic changes after adjustable gastric banding and Roux-en-Y gastric bypass. Int J Obes (Lond) 2009; 33:786–795.
- Boey D, Sainsbury A, Herzog H. The role of peptide YY in regulating glucose homeostasis. Peptides 2007; 28:390–395.
- Hanusch-Enserer U, Ghatei MA, Cauza E, Bloom SR, Prager R, Roden M. Relation of fasting plasma peptide YY to glucose metabolism and cardiovascular risk factors after restrictive bariatric surgery. Wien Klin Wochenschr 2007; 119:291–296.
- Laferrère B, Heshka S, Wang K, et al. Incretin levels and effect are markedly enhanced 1 month after Roux-en-Y gastric bypass surgery in obese patients with type 2 diabetes. Diabetes Care 2007; 30:1709–1716.
- Tucker ON, Szomstein S, Rosenthal RJ. Nutritional consequences of weight-loss surgery. Med Clin North Am 2007; 91:499–514.
- Davies DJ, Baxter JM, Baxter JN. Nutritional deficiencies after bariatric surgery. Obes Surg 2007; 17:1150–1158.
- Angstadt JD, Bodziner RA. Peripheral polyneuropathy from thiamine deficiency following laparoscopic Roux-en-Y gastric bypass. Obes Surg 2005; 15:890–892.
- Ritz P, Becouarn G, Douay O, Sallé A, Topart P, Rohmer V. Gastric bypass is not associated with protein malnutrition in morbidly obese patients. Obes Surg 2009; 19:840–844.
- Service GJ, Thompson GB, Service FJ, Andrews JC, Collazo-Clavell ML, Lloyd RV. Hyperinsulinemic hypoglycemia with nesidioblastosis after gastric-bypass surgery. N Engl J Med 2005; 353:249–254.
- Sjöström L, Narbro K, Sjöström CD, et al;Swedish Obese Subjects Study. Effects of bariatric surgery on mortality in Swedish obese subjects. N Engl J Med 2007; 357:741–752.
- Adams TD, Gress RE, Smith SC, et al. Long-term mortality after gastric bypass surgery. N Engl J Med 2007; 357:753–761.
KEY POINTS
- After Roux-en-Y gastric bypass and biliopancreatic diversion, normoglycemia is restored within days, even before the patient has lost much weight.
- Alterations in postprandial levels of intestine-derived hormones (glucagon-like peptide 1, peptide YY, and ghrelin) contribute to the robust metabolic benefits of intestinal bypass procedures.
- Nutritional deficiencies are common after bariatric surgery, and long-term follow-up is mandatory for surveillance of metabolic status.
- Although curing diabetes cannot yet be considered a goal of bariatric surgery, it may be a serendipitous benefit.

The electronic medical record: Diving into a shallow pool?
The rush to adopt the electronic medical record (EMR) has accelerated since the signing of the Health Information Technology for Economic and Clinical Health (HITECH) Act, part of the American Recovery and Reinvestment (ie, the Stimulus) Act of 2009. The HITECH Act provides incentives for physicians to adopt EMRs. However, I fear that our mad rush to complete adoption of the hodgepodge of currently available EMR systems may have unforeseen and unintended consequences. A skeptical look at several unresolved issues is warranted.
SO FAR, ELECTRONIC SYSTEMS ARE NOT INTERCONNECTABLE
More than 300 EMR systems are available, but only about two dozen account for most systems in use.1 So far, these systems are not interconnectable, ie, they are unable to share information, so patients seen by different physicians may still have a fragmented electronic record.
EMRs can also be inefficient to use. Many systems require logging on to a separate, password-protected system to view images. These problems are likely to go away over time with Internet-based solutions under development by Google and others, but the current lack of interconnectivity leaves much to be desired.
ELECTRONIC RECORDS ARE AT RISK
EMRs are at considerable security risk. About 13% of medical offices in the United States are using some form of EMR.2 A 1995 Harris poll revealed that 70% of Americans were concerned about the security of EMR systems.3 In 2007, the New York Times reported that more than 250,000 patients each year are victims of medical identity theft.4 A New Zealand survey revealed that 73.3% of patients were “highly concerned” about security and privacy.5 Even more troubling to physicians is the reported 13% incidence of patients withholding medical information because of security concerns. Furthermore, multiple breaches of electronic records have already been reported, including an extensive breach of the Veterans Administration system.6
DO ELECTRONIC RECORDS IMPROVE OR WORSEN THE QUALITY OF CAR E?
Proponents have repeatedly touted that EMRs improve the quality of medical care, and these claims have been used to accelerate the adoption of the EMR. The contention that EMRs improve the accuracy of billing, coding, and administrative functions is supported by considerable data; however, the evidence of the effect of EMRs on quality of care is mixed, with some information suggesting quality may not improve.
In an analysis of 750,000 patient records for a 2-year period as part of the National Ambulatory Care Survey, Linder et al7 found that the EMR was superior in one quality area, worse in another area, and the same as paper-based records in 14 other areas. They pointed out that previous studies showing improved outcomes were mainly from large institutions with internally developed EMR systems, and that outcomes reported from these “benchmark” institutions may not be broadly applicable.7 Linder et al concluded that use of electronic records “was generally not associated with improved quality of ambulatory care,”7 and that increased use of EMRs does not imply an automatic improvement in quality of care.7
Crosson et al8 evaluated diabetes care in a cross-sectional analysis of 50 ambulatory care practices from 2003 and 2004 and reported that “after controlling for potential practice- and patient-level confounders and for the clustering of patients within practices, patients with diabetes in practices that did not have an EMR were significantly more likely to have received care that met the guidelines for processes of care, treatment, and intermediate outcomes.”8
The Palo Alto Medical Foundation reported on the sources and types of discrepancies between EMR-listed medications and actual patient medications and found that 79.8% of the time the errors were generated by the EMR system.9 And an outpatient study that videotaped medical encounters to evaluate the accuracy of EMR in an area in which accuracy would be expected (medication lists) found that fewer than one-fifth of exchanges “ended with clear conclusions by both parties regarding prescribed medication regimens.”10 Never mind the lingering questions regarding our ability to define quality: these data provide at least some cause for concern and caution in our rush to adopt innovation in health care without proper consideration of the possible unintended consequences.
WHAT EFFECT ON MEDICAL EDUCATION?
Almost no information is available on the effects of the EMR on the process of medical student education. One could postulate and hope that embedded diagnostic algorithms and drug interaction software would facilitate the education process.
In a paper in Academic Psychiatry, Keenan et al noted that research on EMRs for education is in its infancy.11 A 2008 study of the effects of EMR on third-year medical students’ clinical experience found that students reported significant concerns about the potential impact of EMRs on their ability to conduct the doctor-patient encounter.12 Furthermore, 48% reported spending less time with patients face to face because of the EMR, and 34% reported less time talking to patients.12 In today’s world of off-site rotations and with nearly two dozen EMR systems in outpatient use alone, it is likely that a considerable amount of medical students’ time and effort is expended learning how to use different systems, which may detract from their actual medical experience.
Lastly, a survey of Canadian and US medical schools13 found that only 44% of schools had a policy regarding medical students’ documentation of progress notes in the EMR during ambulatory internal medicine clerkships. In an era when the medical student has been relegated to an observer in the education process, 14 the EMR has introduced yet another poorly understood variable in student education, which clearly begs for a thorough evaluation as the use of EMRs becomes more widespread. How can we maximize rather than dilute student education through the vehicle of electronic records?
ACCURACY VS COPYING AND PASTING
A recent Veterans Adminstration study found that 99% of progress notes in EMRs that were examined contained copied or duplicated text.15 Ten percent of 98,753 examined records contained an instance of what was considered “high-risk copying.” Weir et al16 manually reviewed a set of 60 inpatient charts at the Salt Lake City VA Health Care System and found an average of one factual error introduced into the electronic record per episode of copying.16 The clinical accuracy of the EMR is therefore questionable. Physicians pressed for time are more likely to introduce errors in the EMR, and the information put into the EMR is unlikely to be questioned—and may well be perpetuated by copy-and-paste methodology.
A THIRD PARTY IN THE EXAMINATION ROOM
Considerable information is available about the effect of the EMR on doctor-patient interaction. Margalit et al17 studied videotapes of physician encounters and noted that physicians spent an average of 25% (in some cases as much as 42%) of each visit gazing at the computer screen. They also noted that screengazing seemed to be particularly disruptive to psychological and emotional exchange.
Ventres et al18 reported that in the examination room the EMR is “much like a third party to a conversation”18 and contended that the widespread use of EMRs would have intended and unintended consquences on the cognitive and social dimensions of the physician-patient encounter. They concluded that these issues demand thoughtful consideration as the use of the EMR proliferates, “not only to forestall problems but to maximize the effectiveness of this burgeoning medical technology.”18
DEVOID OF REAL MEDICAL THOUGHT
Notwithstanding data errors and the cutting and pasting of prior notes in the EMR, we still know very little about how the EMR affects how doctors express their thoughts and communicate with one another. My particular concern is with menu-driven or templatedriven notes: they produce reams of important data, and they help ensure that coding requirements are met. But this way of writing notes about a patient is devoid of real medical thought. To describe a patient in templatedriven fashion as “an 88-year-old white male” pales next to a personalized description such as “an 88-year-old World War II B-17 bomber pilot shot down three times over Europe.”
A colleague of mine recently lamented, “I can no longer make use of my partners’ templated notes, as they convey no real information.” I do believe we should be concerned about the undesirable effects that such changes in record-keeping may produce.
LET’S CHECK THE WATER BEFORE DIVING IN
What should we do as we face these issues?
First, we should be aware that governmental and financial pressures and the availability of new technology are pushing us rapidly into new, poorly understood territory. This awareness is critical, as it at least permits a more open mind and allows the potential for honest dialogue, rather than just following directives from above.
Second, we should recognize the gaps in our understanding of the overall effects of the EMR on medicine as a profession and begin to more critically study these effects: ie, we need to be proactive rather than reactive. Denying that we lack answers to key questions about EMRs is clearly counterproductive.
We live in the electronic age. EMRs will continue to proliferate, and they have the potential to be cost-effective, care-enhancing, and time-saving. Obviously, there is no turning back the clock. However, the issues I have raised here—and other issues such as additional physician time,1 potential “billing creep,” and the opportunity for outright fraud (rarely discussed in physician circles), not to mention cost—are deeply concerning and worthy of notice and careful consideration.
My thoughts here are meant to serve as a call to reassess the possible unintended consequences of the federally mandated rush toward an as-yet poorly integrated system of EMRs. Perhaps we should check the water first, lest we find we are diving into a shallow pool.
- Prosser K. Sonoma County Medical Association. The true costs of EMRs. Sonoma Medicine Spring 2009. http://www.scma.org/magazine/articles/?articleid=398. Accessed April 13, 2010.
- DesRoches CM, Campbell EG, Rao SR, et al. Electronic health records in ambulatory care—a national survey of physicians. N Engl J Med 2008; 359:50–60.
- Cummings J. The benefits of electronic medical records sound good, but privacy could become a difficult issue. Harris Interactive Feb 8, 2007. http://news.harrisinteractive.com/profiles/investor/fullpage.asp?f=1&BzID=1963&to=cp&Nav=0&LangID=1&s=0&ID=11259. Accessed May 31, 2010.
- Konrad W. Medical problems could include identity theft. The New York Times. June 12, 2009.
- Chhanabhi P, Holt A. Consumers are ready to accept the transition to online and electronic records if they can be assured of the security measures. MedGenMed 2007; 9:8.
- Lemos R. Veterans Affairs warns of massive privacy breach. SecurityFocus 2006 (May 22). http://www.securityfocus.com/news/11393. Accessed May 31, 2010.
- Linder JA, Ma J, Bates DW, Middleton B, Stafford RS. Electronic health record use and the quality of ambulatory care in the United States. Arch Intern Med 2007; 167:1400–1405.
- Crosson JC, Ohman-Strickland PA, Hahn KA, et al. Electronic medical records and diabetes quality of care: results from a sample of family medicine practices. Ann Fam Med 2007; 5:209–215.
- Orrico KB. Sources and types of discrepancies between electronic medical records and actual outpatient medication use. J Manag Care Pharm 2008; 14:626–631.
- Arar NH, Wen L, McGrath J, Steinbach R, Pugh JA. Communicating about medications during primary care outpatient visits: the role of electronic medical records. Inform Prim Care 2005; 13:13–22.
- Keenan CR, Nguyen HH, Srinivasan M. Electronic medical records and their impact on resident and medical student education. Acad Psychiatry 2006; 30:522–527.
- Rouf E, Chumley HS, Dobbie AE. Electronic health records in outpatient clinics: perspectives of third year medical students. BMC Med Educ 2008; 8:13.
- Mintz M, Narvarte HJ, O’Brien KE, Papp KK, Thomas M, Durning SJ. Use of electronic medical records by physicians and students in academic internal medicine settings. Acad Med 2009; 84:1698–1704.
- Alpert JS, Mandell BF. Back to the future: medical students can matter again. Am J Med 2009; 122:971–972.
- Hammond KW, Helbig ST, Benson CC, Brathwaite-Sketoe BM. Are electronic medical records trustworthy? Observations on copying, pasting and duplication. AMIA Annu Symp Proc 2003:269–273.
- Weir CR, Hurdle JF, Felgar MA, Hoffman JM, Roth B, Nebeker JR. Direct text entry in electronic progress notes. An evaluation of input errors. Methods Inf Med 2003; 42:61–67.
- Margalit RS, Roter D, Dunevant MA, Larson S, Reis S. Electronic medical record use and physician-patient communication: an observational study of Israeli primary care encounters. Patient Educ Couns 2006; 61:134–141.
- Ventres W, Kooienga S, Vuckovic N, Marlin R, Nygren P, Stewart V. Physicians, patients, and the electronic health record: an ethnographic analysis. Ann Fam Med 2006; 4:124–131.
The rush to adopt the electronic medical record (EMR) has accelerated since the signing of the Health Information Technology for Economic and Clinical Health (HITECH) Act, part of the American Recovery and Reinvestment (ie, the Stimulus) Act of 2009. The HITECH Act provides incentives for physicians to adopt EMRs. However, I fear that our mad rush to complete adoption of the hodgepodge of currently available EMR systems may have unforeseen and unintended consequences. A skeptical look at several unresolved issues is warranted.
SO FAR, ELECTRONIC SYSTEMS ARE NOT INTERCONNECTABLE
More than 300 EMR systems are available, but only about two dozen account for most systems in use.1 So far, these systems are not interconnectable, ie, they are unable to share information, so patients seen by different physicians may still have a fragmented electronic record.
EMRs can also be inefficient to use. Many systems require logging on to a separate, password-protected system to view images. These problems are likely to go away over time with Internet-based solutions under development by Google and others, but the current lack of interconnectivity leaves much to be desired.
ELECTRONIC RECORDS ARE AT RISK
EMRs are at considerable security risk. About 13% of medical offices in the United States are using some form of EMR.2 A 1995 Harris poll revealed that 70% of Americans were concerned about the security of EMR systems.3 In 2007, the New York Times reported that more than 250,000 patients each year are victims of medical identity theft.4 A New Zealand survey revealed that 73.3% of patients were “highly concerned” about security and privacy.5 Even more troubling to physicians is the reported 13% incidence of patients withholding medical information because of security concerns. Furthermore, multiple breaches of electronic records have already been reported, including an extensive breach of the Veterans Administration system.6
DO ELECTRONIC RECORDS IMPROVE OR WORSEN THE QUALITY OF CAR E?
Proponents have repeatedly touted that EMRs improve the quality of medical care, and these claims have been used to accelerate the adoption of the EMR. The contention that EMRs improve the accuracy of billing, coding, and administrative functions is supported by considerable data; however, the evidence of the effect of EMRs on quality of care is mixed, with some information suggesting quality may not improve.
In an analysis of 750,000 patient records for a 2-year period as part of the National Ambulatory Care Survey, Linder et al7 found that the EMR was superior in one quality area, worse in another area, and the same as paper-based records in 14 other areas. They pointed out that previous studies showing improved outcomes were mainly from large institutions with internally developed EMR systems, and that outcomes reported from these “benchmark” institutions may not be broadly applicable.7 Linder et al concluded that use of electronic records “was generally not associated with improved quality of ambulatory care,”7 and that increased use of EMRs does not imply an automatic improvement in quality of care.7
Crosson et al8 evaluated diabetes care in a cross-sectional analysis of 50 ambulatory care practices from 2003 and 2004 and reported that “after controlling for potential practice- and patient-level confounders and for the clustering of patients within practices, patients with diabetes in practices that did not have an EMR were significantly more likely to have received care that met the guidelines for processes of care, treatment, and intermediate outcomes.”8
The Palo Alto Medical Foundation reported on the sources and types of discrepancies between EMR-listed medications and actual patient medications and found that 79.8% of the time the errors were generated by the EMR system.9 And an outpatient study that videotaped medical encounters to evaluate the accuracy of EMR in an area in which accuracy would be expected (medication lists) found that fewer than one-fifth of exchanges “ended with clear conclusions by both parties regarding prescribed medication regimens.”10 Never mind the lingering questions regarding our ability to define quality: these data provide at least some cause for concern and caution in our rush to adopt innovation in health care without proper consideration of the possible unintended consequences.
WHAT EFFECT ON MEDICAL EDUCATION?
Almost no information is available on the effects of the EMR on the process of medical student education. One could postulate and hope that embedded diagnostic algorithms and drug interaction software would facilitate the education process.
In a paper in Academic Psychiatry, Keenan et al noted that research on EMRs for education is in its infancy.11 A 2008 study of the effects of EMR on third-year medical students’ clinical experience found that students reported significant concerns about the potential impact of EMRs on their ability to conduct the doctor-patient encounter.12 Furthermore, 48% reported spending less time with patients face to face because of the EMR, and 34% reported less time talking to patients.12 In today’s world of off-site rotations and with nearly two dozen EMR systems in outpatient use alone, it is likely that a considerable amount of medical students’ time and effort is expended learning how to use different systems, which may detract from their actual medical experience.
Lastly, a survey of Canadian and US medical schools13 found that only 44% of schools had a policy regarding medical students’ documentation of progress notes in the EMR during ambulatory internal medicine clerkships. In an era when the medical student has been relegated to an observer in the education process, 14 the EMR has introduced yet another poorly understood variable in student education, which clearly begs for a thorough evaluation as the use of EMRs becomes more widespread. How can we maximize rather than dilute student education through the vehicle of electronic records?
ACCURACY VS COPYING AND PASTING
A recent Veterans Adminstration study found that 99% of progress notes in EMRs that were examined contained copied or duplicated text.15 Ten percent of 98,753 examined records contained an instance of what was considered “high-risk copying.” Weir et al16 manually reviewed a set of 60 inpatient charts at the Salt Lake City VA Health Care System and found an average of one factual error introduced into the electronic record per episode of copying.16 The clinical accuracy of the EMR is therefore questionable. Physicians pressed for time are more likely to introduce errors in the EMR, and the information put into the EMR is unlikely to be questioned—and may well be perpetuated by copy-and-paste methodology.
A THIRD PARTY IN THE EXAMINATION ROOM
Considerable information is available about the effect of the EMR on doctor-patient interaction. Margalit et al17 studied videotapes of physician encounters and noted that physicians spent an average of 25% (in some cases as much as 42%) of each visit gazing at the computer screen. They also noted that screengazing seemed to be particularly disruptive to psychological and emotional exchange.
Ventres et al18 reported that in the examination room the EMR is “much like a third party to a conversation”18 and contended that the widespread use of EMRs would have intended and unintended consquences on the cognitive and social dimensions of the physician-patient encounter. They concluded that these issues demand thoughtful consideration as the use of the EMR proliferates, “not only to forestall problems but to maximize the effectiveness of this burgeoning medical technology.”18
DEVOID OF REAL MEDICAL THOUGHT
Notwithstanding data errors and the cutting and pasting of prior notes in the EMR, we still know very little about how the EMR affects how doctors express their thoughts and communicate with one another. My particular concern is with menu-driven or templatedriven notes: they produce reams of important data, and they help ensure that coding requirements are met. But this way of writing notes about a patient is devoid of real medical thought. To describe a patient in templatedriven fashion as “an 88-year-old white male” pales next to a personalized description such as “an 88-year-old World War II B-17 bomber pilot shot down three times over Europe.”
A colleague of mine recently lamented, “I can no longer make use of my partners’ templated notes, as they convey no real information.” I do believe we should be concerned about the undesirable effects that such changes in record-keeping may produce.
LET’S CHECK THE WATER BEFORE DIVING IN
What should we do as we face these issues?
First, we should be aware that governmental and financial pressures and the availability of new technology are pushing us rapidly into new, poorly understood territory. This awareness is critical, as it at least permits a more open mind and allows the potential for honest dialogue, rather than just following directives from above.
Second, we should recognize the gaps in our understanding of the overall effects of the EMR on medicine as a profession and begin to more critically study these effects: ie, we need to be proactive rather than reactive. Denying that we lack answers to key questions about EMRs is clearly counterproductive.
We live in the electronic age. EMRs will continue to proliferate, and they have the potential to be cost-effective, care-enhancing, and time-saving. Obviously, there is no turning back the clock. However, the issues I have raised here—and other issues such as additional physician time,1 potential “billing creep,” and the opportunity for outright fraud (rarely discussed in physician circles), not to mention cost—are deeply concerning and worthy of notice and careful consideration.
My thoughts here are meant to serve as a call to reassess the possible unintended consequences of the federally mandated rush toward an as-yet poorly integrated system of EMRs. Perhaps we should check the water first, lest we find we are diving into a shallow pool.
The rush to adopt the electronic medical record (EMR) has accelerated since the signing of the Health Information Technology for Economic and Clinical Health (HITECH) Act, part of the American Recovery and Reinvestment (ie, the Stimulus) Act of 2009. The HITECH Act provides incentives for physicians to adopt EMRs. However, I fear that our mad rush to complete adoption of the hodgepodge of currently available EMR systems may have unforeseen and unintended consequences. A skeptical look at several unresolved issues is warranted.
SO FAR, ELECTRONIC SYSTEMS ARE NOT INTERCONNECTABLE
More than 300 EMR systems are available, but only about two dozen account for most systems in use.1 So far, these systems are not interconnectable, ie, they are unable to share information, so patients seen by different physicians may still have a fragmented electronic record.
EMRs can also be inefficient to use. Many systems require logging on to a separate, password-protected system to view images. These problems are likely to go away over time with Internet-based solutions under development by Google and others, but the current lack of interconnectivity leaves much to be desired.
ELECTRONIC RECORDS ARE AT RISK
EMRs are at considerable security risk. About 13% of medical offices in the United States are using some form of EMR.2 A 1995 Harris poll revealed that 70% of Americans were concerned about the security of EMR systems.3 In 2007, the New York Times reported that more than 250,000 patients each year are victims of medical identity theft.4 A New Zealand survey revealed that 73.3% of patients were “highly concerned” about security and privacy.5 Even more troubling to physicians is the reported 13% incidence of patients withholding medical information because of security concerns. Furthermore, multiple breaches of electronic records have already been reported, including an extensive breach of the Veterans Administration system.6
DO ELECTRONIC RECORDS IMPROVE OR WORSEN THE QUALITY OF CAR E?
Proponents have repeatedly touted that EMRs improve the quality of medical care, and these claims have been used to accelerate the adoption of the EMR. The contention that EMRs improve the accuracy of billing, coding, and administrative functions is supported by considerable data; however, the evidence of the effect of EMRs on quality of care is mixed, with some information suggesting quality may not improve.
In an analysis of 750,000 patient records for a 2-year period as part of the National Ambulatory Care Survey, Linder et al7 found that the EMR was superior in one quality area, worse in another area, and the same as paper-based records in 14 other areas. They pointed out that previous studies showing improved outcomes were mainly from large institutions with internally developed EMR systems, and that outcomes reported from these “benchmark” institutions may not be broadly applicable.7 Linder et al concluded that use of electronic records “was generally not associated with improved quality of ambulatory care,”7 and that increased use of EMRs does not imply an automatic improvement in quality of care.7
Crosson et al8 evaluated diabetes care in a cross-sectional analysis of 50 ambulatory care practices from 2003 and 2004 and reported that “after controlling for potential practice- and patient-level confounders and for the clustering of patients within practices, patients with diabetes in practices that did not have an EMR were significantly more likely to have received care that met the guidelines for processes of care, treatment, and intermediate outcomes.”8
The Palo Alto Medical Foundation reported on the sources and types of discrepancies between EMR-listed medications and actual patient medications and found that 79.8% of the time the errors were generated by the EMR system.9 And an outpatient study that videotaped medical encounters to evaluate the accuracy of EMR in an area in which accuracy would be expected (medication lists) found that fewer than one-fifth of exchanges “ended with clear conclusions by both parties regarding prescribed medication regimens.”10 Never mind the lingering questions regarding our ability to define quality: these data provide at least some cause for concern and caution in our rush to adopt innovation in health care without proper consideration of the possible unintended consequences.
WHAT EFFECT ON MEDICAL EDUCATION?
Almost no information is available on the effects of the EMR on the process of medical student education. One could postulate and hope that embedded diagnostic algorithms and drug interaction software would facilitate the education process.
In a paper in Academic Psychiatry, Keenan et al noted that research on EMRs for education is in its infancy.11 A 2008 study of the effects of EMR on third-year medical students’ clinical experience found that students reported significant concerns about the potential impact of EMRs on their ability to conduct the doctor-patient encounter.12 Furthermore, 48% reported spending less time with patients face to face because of the EMR, and 34% reported less time talking to patients.12 In today’s world of off-site rotations and with nearly two dozen EMR systems in outpatient use alone, it is likely that a considerable amount of medical students’ time and effort is expended learning how to use different systems, which may detract from their actual medical experience.
Lastly, a survey of Canadian and US medical schools13 found that only 44% of schools had a policy regarding medical students’ documentation of progress notes in the EMR during ambulatory internal medicine clerkships. In an era when the medical student has been relegated to an observer in the education process, 14 the EMR has introduced yet another poorly understood variable in student education, which clearly begs for a thorough evaluation as the use of EMRs becomes more widespread. How can we maximize rather than dilute student education through the vehicle of electronic records?
ACCURACY VS COPYING AND PASTING
A recent Veterans Adminstration study found that 99% of progress notes in EMRs that were examined contained copied or duplicated text.15 Ten percent of 98,753 examined records contained an instance of what was considered “high-risk copying.” Weir et al16 manually reviewed a set of 60 inpatient charts at the Salt Lake City VA Health Care System and found an average of one factual error introduced into the electronic record per episode of copying.16 The clinical accuracy of the EMR is therefore questionable. Physicians pressed for time are more likely to introduce errors in the EMR, and the information put into the EMR is unlikely to be questioned—and may well be perpetuated by copy-and-paste methodology.
A THIRD PARTY IN THE EXAMINATION ROOM
Considerable information is available about the effect of the EMR on doctor-patient interaction. Margalit et al17 studied videotapes of physician encounters and noted that physicians spent an average of 25% (in some cases as much as 42%) of each visit gazing at the computer screen. They also noted that screengazing seemed to be particularly disruptive to psychological and emotional exchange.
Ventres et al18 reported that in the examination room the EMR is “much like a third party to a conversation”18 and contended that the widespread use of EMRs would have intended and unintended consquences on the cognitive and social dimensions of the physician-patient encounter. They concluded that these issues demand thoughtful consideration as the use of the EMR proliferates, “not only to forestall problems but to maximize the effectiveness of this burgeoning medical technology.”18
DEVOID OF REAL MEDICAL THOUGHT
Notwithstanding data errors and the cutting and pasting of prior notes in the EMR, we still know very little about how the EMR affects how doctors express their thoughts and communicate with one another. My particular concern is with menu-driven or templatedriven notes: they produce reams of important data, and they help ensure that coding requirements are met. But this way of writing notes about a patient is devoid of real medical thought. To describe a patient in templatedriven fashion as “an 88-year-old white male” pales next to a personalized description such as “an 88-year-old World War II B-17 bomber pilot shot down three times over Europe.”
A colleague of mine recently lamented, “I can no longer make use of my partners’ templated notes, as they convey no real information.” I do believe we should be concerned about the undesirable effects that such changes in record-keeping may produce.
LET’S CHECK THE WATER BEFORE DIVING IN
What should we do as we face these issues?
First, we should be aware that governmental and financial pressures and the availability of new technology are pushing us rapidly into new, poorly understood territory. This awareness is critical, as it at least permits a more open mind and allows the potential for honest dialogue, rather than just following directives from above.
Second, we should recognize the gaps in our understanding of the overall effects of the EMR on medicine as a profession and begin to more critically study these effects: ie, we need to be proactive rather than reactive. Denying that we lack answers to key questions about EMRs is clearly counterproductive.
We live in the electronic age. EMRs will continue to proliferate, and they have the potential to be cost-effective, care-enhancing, and time-saving. Obviously, there is no turning back the clock. However, the issues I have raised here—and other issues such as additional physician time,1 potential “billing creep,” and the opportunity for outright fraud (rarely discussed in physician circles), not to mention cost—are deeply concerning and worthy of notice and careful consideration.
My thoughts here are meant to serve as a call to reassess the possible unintended consequences of the federally mandated rush toward an as-yet poorly integrated system of EMRs. Perhaps we should check the water first, lest we find we are diving into a shallow pool.
- Prosser K. Sonoma County Medical Association. The true costs of EMRs. Sonoma Medicine Spring 2009. http://www.scma.org/magazine/articles/?articleid=398. Accessed April 13, 2010.
- DesRoches CM, Campbell EG, Rao SR, et al. Electronic health records in ambulatory care—a national survey of physicians. N Engl J Med 2008; 359:50–60.
- Cummings J. The benefits of electronic medical records sound good, but privacy could become a difficult issue. Harris Interactive Feb 8, 2007. http://news.harrisinteractive.com/profiles/investor/fullpage.asp?f=1&BzID=1963&to=cp&Nav=0&LangID=1&s=0&ID=11259. Accessed May 31, 2010.
- Konrad W. Medical problems could include identity theft. The New York Times. June 12, 2009.
- Chhanabhi P, Holt A. Consumers are ready to accept the transition to online and electronic records if they can be assured of the security measures. MedGenMed 2007; 9:8.
- Lemos R. Veterans Affairs warns of massive privacy breach. SecurityFocus 2006 (May 22). http://www.securityfocus.com/news/11393. Accessed May 31, 2010.
- Linder JA, Ma J, Bates DW, Middleton B, Stafford RS. Electronic health record use and the quality of ambulatory care in the United States. Arch Intern Med 2007; 167:1400–1405.
- Crosson JC, Ohman-Strickland PA, Hahn KA, et al. Electronic medical records and diabetes quality of care: results from a sample of family medicine practices. Ann Fam Med 2007; 5:209–215.
- Orrico KB. Sources and types of discrepancies between electronic medical records and actual outpatient medication use. J Manag Care Pharm 2008; 14:626–631.
- Arar NH, Wen L, McGrath J, Steinbach R, Pugh JA. Communicating about medications during primary care outpatient visits: the role of electronic medical records. Inform Prim Care 2005; 13:13–22.
- Keenan CR, Nguyen HH, Srinivasan M. Electronic medical records and their impact on resident and medical student education. Acad Psychiatry 2006; 30:522–527.
- Rouf E, Chumley HS, Dobbie AE. Electronic health records in outpatient clinics: perspectives of third year medical students. BMC Med Educ 2008; 8:13.
- Mintz M, Narvarte HJ, O’Brien KE, Papp KK, Thomas M, Durning SJ. Use of electronic medical records by physicians and students in academic internal medicine settings. Acad Med 2009; 84:1698–1704.
- Alpert JS, Mandell BF. Back to the future: medical students can matter again. Am J Med 2009; 122:971–972.
- Hammond KW, Helbig ST, Benson CC, Brathwaite-Sketoe BM. Are electronic medical records trustworthy? Observations on copying, pasting and duplication. AMIA Annu Symp Proc 2003:269–273.
- Weir CR, Hurdle JF, Felgar MA, Hoffman JM, Roth B, Nebeker JR. Direct text entry in electronic progress notes. An evaluation of input errors. Methods Inf Med 2003; 42:61–67.
- Margalit RS, Roter D, Dunevant MA, Larson S, Reis S. Electronic medical record use and physician-patient communication: an observational study of Israeli primary care encounters. Patient Educ Couns 2006; 61:134–141.
- Ventres W, Kooienga S, Vuckovic N, Marlin R, Nygren P, Stewart V. Physicians, patients, and the electronic health record: an ethnographic analysis. Ann Fam Med 2006; 4:124–131.
- Prosser K. Sonoma County Medical Association. The true costs of EMRs. Sonoma Medicine Spring 2009. http://www.scma.org/magazine/articles/?articleid=398. Accessed April 13, 2010.
- DesRoches CM, Campbell EG, Rao SR, et al. Electronic health records in ambulatory care—a national survey of physicians. N Engl J Med 2008; 359:50–60.
- Cummings J. The benefits of electronic medical records sound good, but privacy could become a difficult issue. Harris Interactive Feb 8, 2007. http://news.harrisinteractive.com/profiles/investor/fullpage.asp?f=1&BzID=1963&to=cp&Nav=0&LangID=1&s=0&ID=11259. Accessed May 31, 2010.
- Konrad W. Medical problems could include identity theft. The New York Times. June 12, 2009.
- Chhanabhi P, Holt A. Consumers are ready to accept the transition to online and electronic records if they can be assured of the security measures. MedGenMed 2007; 9:8.
- Lemos R. Veterans Affairs warns of massive privacy breach. SecurityFocus 2006 (May 22). http://www.securityfocus.com/news/11393. Accessed May 31, 2010.
- Linder JA, Ma J, Bates DW, Middleton B, Stafford RS. Electronic health record use and the quality of ambulatory care in the United States. Arch Intern Med 2007; 167:1400–1405.
- Crosson JC, Ohman-Strickland PA, Hahn KA, et al. Electronic medical records and diabetes quality of care: results from a sample of family medicine practices. Ann Fam Med 2007; 5:209–215.
- Orrico KB. Sources and types of discrepancies between electronic medical records and actual outpatient medication use. J Manag Care Pharm 2008; 14:626–631.
- Arar NH, Wen L, McGrath J, Steinbach R, Pugh JA. Communicating about medications during primary care outpatient visits: the role of electronic medical records. Inform Prim Care 2005; 13:13–22.
- Keenan CR, Nguyen HH, Srinivasan M. Electronic medical records and their impact on resident and medical student education. Acad Psychiatry 2006; 30:522–527.
- Rouf E, Chumley HS, Dobbie AE. Electronic health records in outpatient clinics: perspectives of third year medical students. BMC Med Educ 2008; 8:13.
- Mintz M, Narvarte HJ, O’Brien KE, Papp KK, Thomas M, Durning SJ. Use of electronic medical records by physicians and students in academic internal medicine settings. Acad Med 2009; 84:1698–1704.
- Alpert JS, Mandell BF. Back to the future: medical students can matter again. Am J Med 2009; 122:971–972.
- Hammond KW, Helbig ST, Benson CC, Brathwaite-Sketoe BM. Are electronic medical records trustworthy? Observations on copying, pasting and duplication. AMIA Annu Symp Proc 2003:269–273.
- Weir CR, Hurdle JF, Felgar MA, Hoffman JM, Roth B, Nebeker JR. Direct text entry in electronic progress notes. An evaluation of input errors. Methods Inf Med 2003; 42:61–67.
- Margalit RS, Roter D, Dunevant MA, Larson S, Reis S. Electronic medical record use and physician-patient communication: an observational study of Israeli primary care encounters. Patient Educ Couns 2006; 61:134–141.
- Ventres W, Kooienga S, Vuckovic N, Marlin R, Nygren P, Stewart V. Physicians, patients, and the electronic health record: an ethnographic analysis. Ann Fam Med 2006; 4:124–131.