User login
Lower Extremity Injuries in Snowboarders
Epidemiology
The several studies of lower extremity injuries sustained while skiing and snowboarding have differed markedly with respect to patient demographics. Kim and colleagues1 compared snowboarding and skiing injuries over 18 seasons at a Vermont ski resort and found that the injury rate, assessed as mean number of days between injuries, was 400 for snowboarders and 345 for skiers. However, most snowboarding injuries were wrist injuries and generally of the upper extremity, whereas skiing injuries were mainly lower extremity injuries. Overall, young and inexperienced snowboarders had the highest injury rate. In a study on skiing and snowboarding injuries through 4 Utah seasons, Wasden and colleagues2 found that mean age at injury was 41 years for skiers and 23 years for snowboarders. This corroborates the finding from several studies1-3 that snowboarders tend to be younger. Snowboarding is a newer sport with many beginners. However, Ishimaru and colleagues4 found that lower extremity injuries may be associated with experienced snowboarders, who may be prone to take more risks and tackle more challenging slopes. Experienced snowboarders are also likely to sustain lower extremity injuries from falling, because of their risk-taking behavior.5
Although upper extremity injuries account for most snowboarding injuries, lower extremity injuries are a significant issue.6 Modern equipment and more challenging slopes have allowed snowboarders to attain great speeds going down slopes—leading to a surge in lower extremity injuries.7 Lower extremity injuries sustained during snowboarding are more likely to be on the leading side4; the ankle is the most frequent fracture site. Unlike snowboard equipment, modern ski equipment, including new boots and binding systems, is designed to reduce ankle injuries and lower leg fractures.6 The decline in foot, ankle, and tibia fractures can be attributed to taller and stiffer boots, which offer the lower extremities more protection.8
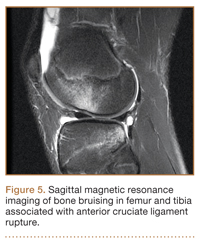
Mechanism of Injury
Talus Fractures
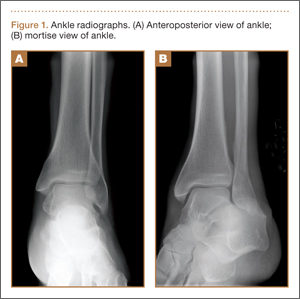
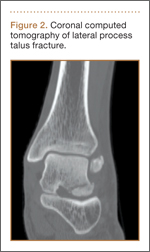
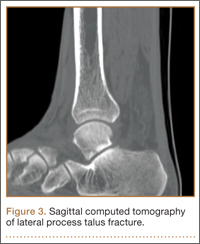
An increasingly common injury among snowboarders is a fracture of the lateral process of the talus; this injury accounts for 32% of snowboarders’ ankle fractures.6 The lateral process of the talus—wedge-shaped and covered in articular cartilage—is involved in the subtalar and ankle joints.9 A fracture here is often misdiagnosed as an ankle sprain (Figures 1–3).6,9,10 The exact mechanism of injury remains controversial, and several biomechanical factors seem to be involved. Funk and colleagues11 conducted a cadaveric study and concluded that eversion of an axially loaded, dorsiflexed ankle may be the primary injury mechanism for fracture. Furthermore, snowboarders have their feet in a position perpendicular to the board, and a fall parallel to the board could increase the eversion force on the ankle of the leading leg. Valderrabano and colleagues9 conducted a clinical study of 26 patients who sustained this injury from snowboarding. All the patients reported they had felt an axial impact from falling, jumping, or unexpectedly hitting a ground object, and 80% reported a rotational movement in the lower leg during the impact. The authors concluded that axial loading and dorsiflexion were not the only factors involved in lateral process talus fractures, and an external moment is necessary to cause this injury from a forward fall.9
Anterior Cruciate Ligament Injuries
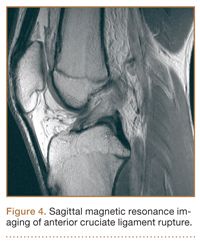
Although snowboarders’ lower extremity injuries are primarily ankle injuries, snowboarders are also at risk for serious knee issues when landing from jumps. In skiers, anterior cruciate ligament (ACL) injuries have 5 well-established mechanisms, all involving separation of the feet and a twisting force in the knee (Figures 4, 5): boot-induced anterior drawer mechanism, phantom-foot mechanism, valgus-external rotation, forceful quadriceps muscle contraction, and a combination of internal rotation and extension.8,12 A valgus–external rotation mechanism of knee injury occurs when external rotation of the tibia results from the skier catching the inside edge of the front of the ski. A valgus force acts on the knee as the lower leg is abducted during forward momentum. The torque created on the knee joint is amplified by the length of the knee and commonly results in an ACL injury or medial collateral ligament injury.6 Reports indicate that the phantom-foot mechanism is the most common mechanism of ACL injury among skiers.6,13,14 In this situation, internal rotation of the knee results when an off-balance skier falls backward, which causes the knee to hyperflex. The skier catches an inside edge on the snow, which creates a torque that rotates the tibia relative to the femur and results in injury to the ACL.6,14 A boot-induced anterior drawer mechanism occurs during a landing, when the tail of the ski lands first and in an off-balance position, resulting in a load transmitted through the skis to the skier; this load causes an anterior drawer of the ski boot and tibia relative to the femur, straining the ACL and causing ACL rupture.6,13,14 In the forceful quadriceps muscle contraction mechanism of ACL injury, a forceful quadriceps contraction occurs after a jump to prevent a backward fall. With the knee in flexion, this quadriceps contraction causes an anterior translation of the tibia, resulting in ACL rupture.13,14
The mechanism of injury differs in snowboarding, in which both feet remain attached to the board. Davies and colleagues15 examined 35 snowboarders who sustained ACL injuries after a flat landing from a jump and concluded that snowboarders preparing for a landing exhibit more quadriceps contraction, which increases the loading force on the ACL during landing. Furthermore, the snowboarder’s stance on the board, with the front foot slightly rotated relative to the board, results in a slight internal tibial rotation of the knee and establishes a posture that makes the snowboarder susceptible to injury. However, the lower incidence of knee injuries among snowboarders compared with skiers may be attributable to the fact that there is a limited amount of torque that can be generated on either knee as both feet are fixed to the board.16
The increased quadriceps force in anticipation of a landing, combined with the internal tibial rotation of the knee caused by the snowboarder’s stance, may be the primary mechanism of ACL rupture in snowboarders.15
Injury Prevention Strategies
Prevention strategies require an identification of injury risk factors for snowboarders. Hasler and colleagues7 conducted a study with 306 patients to identify variables that presented a risk for snowboarders. Low readiness for speed, bad weather, and bad visibility, as well as snow conditions, were found to be significant risk factors.
Skiers’ overall injury rate has decreased over the past 60 years, and this decrease has been attributed in part to improved ski technique and instruction.17,18 Improperly adjusted ski bindings are the culprit in many equipment-related lower extremity injuries, and beginners are at much higher risk for such injuries. Lessons and comprehensive safety training could reduce this injury rate.17,19 Several awareness video and training programs focusing on injury prevention have reduced knee sprains in ski patrollers compared with controls by 62% in 1 study; a similar program reduced injury by 30% in nonprofessional skiers.17 A study of injured snowboarders during a winter in Scotland found that 37% of the patients had no formal instruction or training in correct snowboarding and falling technique.20 Training programs for snowboarders could yield meaningful results in injury prevention and avoidance of risk-taking behavior among snowboarders.
Advances in equipment have also had an impact on the incidence of skiing injuries. Ski bindings protect skiers in 2 ways. First, the binding keeps the boot attached to the ski and prevents unintended release on difficult terrain. Second, the binding releases the boot from the ski during extreme conditions to prevent the skier from experiencing extreme forces or moments that could result in injury. Functional failure in ski bindings has been implicated in increased incidence of knee injuries and ligament rupture. In a study of injuries sustained by recreational alpine skiers in Japan, Urabe and colleagues21 found that 96% of those injured stated that the ski bindings had not released at time of incident. The effects of binding adjustment and maintenance among snowboarders have not been fully investigated, and there are no set guidelines for individual snowboarders on appropriate binding level. However, as there is a range of binding adjustment options available, snowboarders may have an optimum level that maximizes both mobility and protection from injury.22
Soft-shelled boots may also increase injury risk for snowboarders. Such boots allow for a wider range of ankle motion and offer little protection from extreme joint movements. Soft boots are generally preferred among snowboarders because they allow for increased mobility for sharp turns and maneuvers. However, modification of the stiffness of boots that limit ankle and foot joint mobility could reduce the incidence of ankle fractures and sprains among snowboarders.22
Summary
Snowboarding has become increasingly popular worldwide. It attracts a loyal group of amateur athletes and has developed into a billion-dollar industry with a growing rank of professionals. Although most snowboarding injuries are upper extremity injuries, the foot, ankle, and knee represent commonly injured areas among recreational and experienced snowboarders. Advances in ski equipment have significantly reduced the incidence of ankle injuries, but rising knee ligament injuries continue to pose a challenge. Foot and ankle injuries remain an issue in snowboarders despite advances in equipment and safety. New snowboard designs and boot and binding modifications may hold promise in decreasing the risk for injury in these athletes.
1. Kim S, Endres NK, Johnson RJ, Ettlinger CF, Shealy JE. Snowboarding injuries: trends over time and comparisons with alpine skiing injuries. Am J Sports Med. 2012;40(4):770-776.
2. Wasden CC, McIntosh SE, Keith DS, McCowan C. An analysis of skiing and snowboarding injuries on Utah slopes. J Trauma. 2009;67(5):1022-1026.
3. Rust DA, Gilmore CJ, Treme G. Injury patterns at a large western United States ski resort with and without snowboarders: the Taos experience. Am J Sports Med. 2013;41(3):652-656.
4. Ishimaru D, Ogawa H, Sumi H, Sumi Y, Shimizu K. Lower extremity injuries in snowboarding. J Trauma. 2011;70(3):E48-E52.
5. Torjussen J, Bahr R. Injuries among competitive snowboarders at the national elite level. Am J Sports Med. 2005;33(3):370-377.
6. Deady LH, Salonen D. Skiing and snowboarding injuries: a review with a focus on mechanism of injury. Radiol Clin North Am. 2010;48(6):1113-1124.
7. Hasler RM, Berov S, Banneker L, et al. Are there risk factors for snowboard injuries? A case–control multicentre study of 559 snowboarders. Br J Sports Med. 2010;44(11):816-821.
8. St-Onge N, Chevalier Y, Hagemeister N, Van De Putte M, De Guise J. Effect of ski binding parameters on knee biomechanics: a three-dimensional computational study. Med Sci Sports Exerc. 2004;36(7):1218-1225.
9. Valderrabano V, Perren T, Ryf C, Rillmann P, Hintermann B. Snowboarder’s talus fracture: treatment outcome of 20 cases after 3.5 years. Am J Sports Med. 2005;33(6):871-880.
10. von Knoch F, Reckord U, von Knoch M, Sommer C. Fracture of the lateral process of the talus in snowboarders. J Bone Joint Surg Br. 2007;89(6):772-777.
11. Funk JR, Srinivasan SC, Crandall JR. Snowboarder’s talus fractures experimentally produced by eversion and dorsiflexion. Am J Sports Med. 2003;31(6):921-928.
12. Pujol N, Blanchi MP, Chambat P. The incidence of anterior cruciate ligament injuries among competitive alpine skiers: a 25-year investigation. Am J Sports Med. 2007;35(7):1070-1074.
13. Hame SL, Oakes DA, Markolf KL. Injury to the anterior cruciate ligament during alpine skiing: a biomechanical analysis of tibial torque and knee flexion angle. Am J Sports Med. 2002;30(4):537-540.
14. Bere T, Flørenes TW, Krosshaug T, Nordsletten L, Bahr R. Events leading to anterior cruciate ligament injury in World Cup alpine skiing: a systematic video analysis of 20 cases. Br J Sports Med. 2011;45(16):1294-1302.
15. Davies H, Tietjens B, Van Sterkenburg M, Mehgan A. Anterior cruciate ligament injuries in snowboarders: a quadriceps-induced injury. Knee Surg Sports Traumatol Arthrosc. 2009;17(9):1048-1051.
16. Bladin C, McCrory P, Pogorzelski A. Snowboarding injuries: current trends and future directions. Sports Med. 2004;34(2):133-139.
17. Rossi MJ, Lubowitz JH, Guttmann D. The skier’s knee. Arthroscopy. 2003;19(1):75-84.
18. Pressman A, Johnson DH. A review of ski injuries resulting in combined injury to the anterior cruciate ligament and medial collateral ligaments. Arthroscopy. 2003;19(2):194-202.
19. Hildebrandt C, Mildner E, Hotter B, Kirschner W, Höbenreich C, Raschner C. Accident prevention on ski slopes—perceptions of safety and knowledge of existing rules. Accid Anal Prev. 2011;43(4):1421-1426.
20. Langran M, Selvaraj S. Increased injury risk among first-day skiers, snowboarders, and skiboarders. Am J Sports Med. 2004;32(1):96-103.
21. Urabe Y, Ochi M, Onari K, Ikuta Y. Anterior cruciate ligament injury in recreational alpine skiers: analysis of mechanisms and strategy for prevention. J Orthop Sci. 2002;7(1):1-5.
22. McAlpine PR. Biomechanical Analysis of Snowboard Jump Landings: A Focus on the Ankle Joint Complex [doctoral thesis]. Auckland, New Zealand: University of Auckland; 2010.
Epidemiology
The several studies of lower extremity injuries sustained while skiing and snowboarding have differed markedly with respect to patient demographics. Kim and colleagues1 compared snowboarding and skiing injuries over 18 seasons at a Vermont ski resort and found that the injury rate, assessed as mean number of days between injuries, was 400 for snowboarders and 345 for skiers. However, most snowboarding injuries were wrist injuries and generally of the upper extremity, whereas skiing injuries were mainly lower extremity injuries. Overall, young and inexperienced snowboarders had the highest injury rate. In a study on skiing and snowboarding injuries through 4 Utah seasons, Wasden and colleagues2 found that mean age at injury was 41 years for skiers and 23 years for snowboarders. This corroborates the finding from several studies1-3 that snowboarders tend to be younger. Snowboarding is a newer sport with many beginners. However, Ishimaru and colleagues4 found that lower extremity injuries may be associated with experienced snowboarders, who may be prone to take more risks and tackle more challenging slopes. Experienced snowboarders are also likely to sustain lower extremity injuries from falling, because of their risk-taking behavior.5
Although upper extremity injuries account for most snowboarding injuries, lower extremity injuries are a significant issue.6 Modern equipment and more challenging slopes have allowed snowboarders to attain great speeds going down slopes—leading to a surge in lower extremity injuries.7 Lower extremity injuries sustained during snowboarding are more likely to be on the leading side4; the ankle is the most frequent fracture site. Unlike snowboard equipment, modern ski equipment, including new boots and binding systems, is designed to reduce ankle injuries and lower leg fractures.6 The decline in foot, ankle, and tibia fractures can be attributed to taller and stiffer boots, which offer the lower extremities more protection.8
Mechanism of Injury
Talus Fractures
An increasingly common injury among snowboarders is a fracture of the lateral process of the talus; this injury accounts for 32% of snowboarders’ ankle fractures.6 The lateral process of the talus—wedge-shaped and covered in articular cartilage—is involved in the subtalar and ankle joints.9 A fracture here is often misdiagnosed as an ankle sprain (Figures 1–3).6,9,10 The exact mechanism of injury remains controversial, and several biomechanical factors seem to be involved. Funk and colleagues11 conducted a cadaveric study and concluded that eversion of an axially loaded, dorsiflexed ankle may be the primary injury mechanism for fracture. Furthermore, snowboarders have their feet in a position perpendicular to the board, and a fall parallel to the board could increase the eversion force on the ankle of the leading leg. Valderrabano and colleagues9 conducted a clinical study of 26 patients who sustained this injury from snowboarding. All the patients reported they had felt an axial impact from falling, jumping, or unexpectedly hitting a ground object, and 80% reported a rotational movement in the lower leg during the impact. The authors concluded that axial loading and dorsiflexion were not the only factors involved in lateral process talus fractures, and an external moment is necessary to cause this injury from a forward fall.9
Anterior Cruciate Ligament Injuries
Although snowboarders’ lower extremity injuries are primarily ankle injuries, snowboarders are also at risk for serious knee issues when landing from jumps. In skiers, anterior cruciate ligament (ACL) injuries have 5 well-established mechanisms, all involving separation of the feet and a twisting force in the knee (Figures 4, 5): boot-induced anterior drawer mechanism, phantom-foot mechanism, valgus-external rotation, forceful quadriceps muscle contraction, and a combination of internal rotation and extension.8,12 A valgus–external rotation mechanism of knee injury occurs when external rotation of the tibia results from the skier catching the inside edge of the front of the ski. A valgus force acts on the knee as the lower leg is abducted during forward momentum. The torque created on the knee joint is amplified by the length of the knee and commonly results in an ACL injury or medial collateral ligament injury.6 Reports indicate that the phantom-foot mechanism is the most common mechanism of ACL injury among skiers.6,13,14 In this situation, internal rotation of the knee results when an off-balance skier falls backward, which causes the knee to hyperflex. The skier catches an inside edge on the snow, which creates a torque that rotates the tibia relative to the femur and results in injury to the ACL.6,14 A boot-induced anterior drawer mechanism occurs during a landing, when the tail of the ski lands first and in an off-balance position, resulting in a load transmitted through the skis to the skier; this load causes an anterior drawer of the ski boot and tibia relative to the femur, straining the ACL and causing ACL rupture.6,13,14 In the forceful quadriceps muscle contraction mechanism of ACL injury, a forceful quadriceps contraction occurs after a jump to prevent a backward fall. With the knee in flexion, this quadriceps contraction causes an anterior translation of the tibia, resulting in ACL rupture.13,14
The mechanism of injury differs in snowboarding, in which both feet remain attached to the board. Davies and colleagues15 examined 35 snowboarders who sustained ACL injuries after a flat landing from a jump and concluded that snowboarders preparing for a landing exhibit more quadriceps contraction, which increases the loading force on the ACL during landing. Furthermore, the snowboarder’s stance on the board, with the front foot slightly rotated relative to the board, results in a slight internal tibial rotation of the knee and establishes a posture that makes the snowboarder susceptible to injury. However, the lower incidence of knee injuries among snowboarders compared with skiers may be attributable to the fact that there is a limited amount of torque that can be generated on either knee as both feet are fixed to the board.16
The increased quadriceps force in anticipation of a landing, combined with the internal tibial rotation of the knee caused by the snowboarder’s stance, may be the primary mechanism of ACL rupture in snowboarders.15
Injury Prevention Strategies
Prevention strategies require an identification of injury risk factors for snowboarders. Hasler and colleagues7 conducted a study with 306 patients to identify variables that presented a risk for snowboarders. Low readiness for speed, bad weather, and bad visibility, as well as snow conditions, were found to be significant risk factors.
Skiers’ overall injury rate has decreased over the past 60 years, and this decrease has been attributed in part to improved ski technique and instruction.17,18 Improperly adjusted ski bindings are the culprit in many equipment-related lower extremity injuries, and beginners are at much higher risk for such injuries. Lessons and comprehensive safety training could reduce this injury rate.17,19 Several awareness video and training programs focusing on injury prevention have reduced knee sprains in ski patrollers compared with controls by 62% in 1 study; a similar program reduced injury by 30% in nonprofessional skiers.17 A study of injured snowboarders during a winter in Scotland found that 37% of the patients had no formal instruction or training in correct snowboarding and falling technique.20 Training programs for snowboarders could yield meaningful results in injury prevention and avoidance of risk-taking behavior among snowboarders.
Advances in equipment have also had an impact on the incidence of skiing injuries. Ski bindings protect skiers in 2 ways. First, the binding keeps the boot attached to the ski and prevents unintended release on difficult terrain. Second, the binding releases the boot from the ski during extreme conditions to prevent the skier from experiencing extreme forces or moments that could result in injury. Functional failure in ski bindings has been implicated in increased incidence of knee injuries and ligament rupture. In a study of injuries sustained by recreational alpine skiers in Japan, Urabe and colleagues21 found that 96% of those injured stated that the ski bindings had not released at time of incident. The effects of binding adjustment and maintenance among snowboarders have not been fully investigated, and there are no set guidelines for individual snowboarders on appropriate binding level. However, as there is a range of binding adjustment options available, snowboarders may have an optimum level that maximizes both mobility and protection from injury.22
Soft-shelled boots may also increase injury risk for snowboarders. Such boots allow for a wider range of ankle motion and offer little protection from extreme joint movements. Soft boots are generally preferred among snowboarders because they allow for increased mobility for sharp turns and maneuvers. However, modification of the stiffness of boots that limit ankle and foot joint mobility could reduce the incidence of ankle fractures and sprains among snowboarders.22
Summary
Snowboarding has become increasingly popular worldwide. It attracts a loyal group of amateur athletes and has developed into a billion-dollar industry with a growing rank of professionals. Although most snowboarding injuries are upper extremity injuries, the foot, ankle, and knee represent commonly injured areas among recreational and experienced snowboarders. Advances in ski equipment have significantly reduced the incidence of ankle injuries, but rising knee ligament injuries continue to pose a challenge. Foot and ankle injuries remain an issue in snowboarders despite advances in equipment and safety. New snowboard designs and boot and binding modifications may hold promise in decreasing the risk for injury in these athletes.
Epidemiology
The several studies of lower extremity injuries sustained while skiing and snowboarding have differed markedly with respect to patient demographics. Kim and colleagues1 compared snowboarding and skiing injuries over 18 seasons at a Vermont ski resort and found that the injury rate, assessed as mean number of days between injuries, was 400 for snowboarders and 345 for skiers. However, most snowboarding injuries were wrist injuries and generally of the upper extremity, whereas skiing injuries were mainly lower extremity injuries. Overall, young and inexperienced snowboarders had the highest injury rate. In a study on skiing and snowboarding injuries through 4 Utah seasons, Wasden and colleagues2 found that mean age at injury was 41 years for skiers and 23 years for snowboarders. This corroborates the finding from several studies1-3 that snowboarders tend to be younger. Snowboarding is a newer sport with many beginners. However, Ishimaru and colleagues4 found that lower extremity injuries may be associated with experienced snowboarders, who may be prone to take more risks and tackle more challenging slopes. Experienced snowboarders are also likely to sustain lower extremity injuries from falling, because of their risk-taking behavior.5
Although upper extremity injuries account for most snowboarding injuries, lower extremity injuries are a significant issue.6 Modern equipment and more challenging slopes have allowed snowboarders to attain great speeds going down slopes—leading to a surge in lower extremity injuries.7 Lower extremity injuries sustained during snowboarding are more likely to be on the leading side4; the ankle is the most frequent fracture site. Unlike snowboard equipment, modern ski equipment, including new boots and binding systems, is designed to reduce ankle injuries and lower leg fractures.6 The decline in foot, ankle, and tibia fractures can be attributed to taller and stiffer boots, which offer the lower extremities more protection.8
Mechanism of Injury
Talus Fractures
An increasingly common injury among snowboarders is a fracture of the lateral process of the talus; this injury accounts for 32% of snowboarders’ ankle fractures.6 The lateral process of the talus—wedge-shaped and covered in articular cartilage—is involved in the subtalar and ankle joints.9 A fracture here is often misdiagnosed as an ankle sprain (Figures 1–3).6,9,10 The exact mechanism of injury remains controversial, and several biomechanical factors seem to be involved. Funk and colleagues11 conducted a cadaveric study and concluded that eversion of an axially loaded, dorsiflexed ankle may be the primary injury mechanism for fracture. Furthermore, snowboarders have their feet in a position perpendicular to the board, and a fall parallel to the board could increase the eversion force on the ankle of the leading leg. Valderrabano and colleagues9 conducted a clinical study of 26 patients who sustained this injury from snowboarding. All the patients reported they had felt an axial impact from falling, jumping, or unexpectedly hitting a ground object, and 80% reported a rotational movement in the lower leg during the impact. The authors concluded that axial loading and dorsiflexion were not the only factors involved in lateral process talus fractures, and an external moment is necessary to cause this injury from a forward fall.9
Anterior Cruciate Ligament Injuries
Although snowboarders’ lower extremity injuries are primarily ankle injuries, snowboarders are also at risk for serious knee issues when landing from jumps. In skiers, anterior cruciate ligament (ACL) injuries have 5 well-established mechanisms, all involving separation of the feet and a twisting force in the knee (Figures 4, 5): boot-induced anterior drawer mechanism, phantom-foot mechanism, valgus-external rotation, forceful quadriceps muscle contraction, and a combination of internal rotation and extension.8,12 A valgus–external rotation mechanism of knee injury occurs when external rotation of the tibia results from the skier catching the inside edge of the front of the ski. A valgus force acts on the knee as the lower leg is abducted during forward momentum. The torque created on the knee joint is amplified by the length of the knee and commonly results in an ACL injury or medial collateral ligament injury.6 Reports indicate that the phantom-foot mechanism is the most common mechanism of ACL injury among skiers.6,13,14 In this situation, internal rotation of the knee results when an off-balance skier falls backward, which causes the knee to hyperflex. The skier catches an inside edge on the snow, which creates a torque that rotates the tibia relative to the femur and results in injury to the ACL.6,14 A boot-induced anterior drawer mechanism occurs during a landing, when the tail of the ski lands first and in an off-balance position, resulting in a load transmitted through the skis to the skier; this load causes an anterior drawer of the ski boot and tibia relative to the femur, straining the ACL and causing ACL rupture.6,13,14 In the forceful quadriceps muscle contraction mechanism of ACL injury, a forceful quadriceps contraction occurs after a jump to prevent a backward fall. With the knee in flexion, this quadriceps contraction causes an anterior translation of the tibia, resulting in ACL rupture.13,14
The mechanism of injury differs in snowboarding, in which both feet remain attached to the board. Davies and colleagues15 examined 35 snowboarders who sustained ACL injuries after a flat landing from a jump and concluded that snowboarders preparing for a landing exhibit more quadriceps contraction, which increases the loading force on the ACL during landing. Furthermore, the snowboarder’s stance on the board, with the front foot slightly rotated relative to the board, results in a slight internal tibial rotation of the knee and establishes a posture that makes the snowboarder susceptible to injury. However, the lower incidence of knee injuries among snowboarders compared with skiers may be attributable to the fact that there is a limited amount of torque that can be generated on either knee as both feet are fixed to the board.16
The increased quadriceps force in anticipation of a landing, combined with the internal tibial rotation of the knee caused by the snowboarder’s stance, may be the primary mechanism of ACL rupture in snowboarders.15
Injury Prevention Strategies
Prevention strategies require an identification of injury risk factors for snowboarders. Hasler and colleagues7 conducted a study with 306 patients to identify variables that presented a risk for snowboarders. Low readiness for speed, bad weather, and bad visibility, as well as snow conditions, were found to be significant risk factors.
Skiers’ overall injury rate has decreased over the past 60 years, and this decrease has been attributed in part to improved ski technique and instruction.17,18 Improperly adjusted ski bindings are the culprit in many equipment-related lower extremity injuries, and beginners are at much higher risk for such injuries. Lessons and comprehensive safety training could reduce this injury rate.17,19 Several awareness video and training programs focusing on injury prevention have reduced knee sprains in ski patrollers compared with controls by 62% in 1 study; a similar program reduced injury by 30% in nonprofessional skiers.17 A study of injured snowboarders during a winter in Scotland found that 37% of the patients had no formal instruction or training in correct snowboarding and falling technique.20 Training programs for snowboarders could yield meaningful results in injury prevention and avoidance of risk-taking behavior among snowboarders.
Advances in equipment have also had an impact on the incidence of skiing injuries. Ski bindings protect skiers in 2 ways. First, the binding keeps the boot attached to the ski and prevents unintended release on difficult terrain. Second, the binding releases the boot from the ski during extreme conditions to prevent the skier from experiencing extreme forces or moments that could result in injury. Functional failure in ski bindings has been implicated in increased incidence of knee injuries and ligament rupture. In a study of injuries sustained by recreational alpine skiers in Japan, Urabe and colleagues21 found that 96% of those injured stated that the ski bindings had not released at time of incident. The effects of binding adjustment and maintenance among snowboarders have not been fully investigated, and there are no set guidelines for individual snowboarders on appropriate binding level. However, as there is a range of binding adjustment options available, snowboarders may have an optimum level that maximizes both mobility and protection from injury.22
Soft-shelled boots may also increase injury risk for snowboarders. Such boots allow for a wider range of ankle motion and offer little protection from extreme joint movements. Soft boots are generally preferred among snowboarders because they allow for increased mobility for sharp turns and maneuvers. However, modification of the stiffness of boots that limit ankle and foot joint mobility could reduce the incidence of ankle fractures and sprains among snowboarders.22
Summary
Snowboarding has become increasingly popular worldwide. It attracts a loyal group of amateur athletes and has developed into a billion-dollar industry with a growing rank of professionals. Although most snowboarding injuries are upper extremity injuries, the foot, ankle, and knee represent commonly injured areas among recreational and experienced snowboarders. Advances in ski equipment have significantly reduced the incidence of ankle injuries, but rising knee ligament injuries continue to pose a challenge. Foot and ankle injuries remain an issue in snowboarders despite advances in equipment and safety. New snowboard designs and boot and binding modifications may hold promise in decreasing the risk for injury in these athletes.
1. Kim S, Endres NK, Johnson RJ, Ettlinger CF, Shealy JE. Snowboarding injuries: trends over time and comparisons with alpine skiing injuries. Am J Sports Med. 2012;40(4):770-776.
2. Wasden CC, McIntosh SE, Keith DS, McCowan C. An analysis of skiing and snowboarding injuries on Utah slopes. J Trauma. 2009;67(5):1022-1026.
3. Rust DA, Gilmore CJ, Treme G. Injury patterns at a large western United States ski resort with and without snowboarders: the Taos experience. Am J Sports Med. 2013;41(3):652-656.
4. Ishimaru D, Ogawa H, Sumi H, Sumi Y, Shimizu K. Lower extremity injuries in snowboarding. J Trauma. 2011;70(3):E48-E52.
5. Torjussen J, Bahr R. Injuries among competitive snowboarders at the national elite level. Am J Sports Med. 2005;33(3):370-377.
6. Deady LH, Salonen D. Skiing and snowboarding injuries: a review with a focus on mechanism of injury. Radiol Clin North Am. 2010;48(6):1113-1124.
7. Hasler RM, Berov S, Banneker L, et al. Are there risk factors for snowboard injuries? A case–control multicentre study of 559 snowboarders. Br J Sports Med. 2010;44(11):816-821.
8. St-Onge N, Chevalier Y, Hagemeister N, Van De Putte M, De Guise J. Effect of ski binding parameters on knee biomechanics: a three-dimensional computational study. Med Sci Sports Exerc. 2004;36(7):1218-1225.
9. Valderrabano V, Perren T, Ryf C, Rillmann P, Hintermann B. Snowboarder’s talus fracture: treatment outcome of 20 cases after 3.5 years. Am J Sports Med. 2005;33(6):871-880.
10. von Knoch F, Reckord U, von Knoch M, Sommer C. Fracture of the lateral process of the talus in snowboarders. J Bone Joint Surg Br. 2007;89(6):772-777.
11. Funk JR, Srinivasan SC, Crandall JR. Snowboarder’s talus fractures experimentally produced by eversion and dorsiflexion. Am J Sports Med. 2003;31(6):921-928.
12. Pujol N, Blanchi MP, Chambat P. The incidence of anterior cruciate ligament injuries among competitive alpine skiers: a 25-year investigation. Am J Sports Med. 2007;35(7):1070-1074.
13. Hame SL, Oakes DA, Markolf KL. Injury to the anterior cruciate ligament during alpine skiing: a biomechanical analysis of tibial torque and knee flexion angle. Am J Sports Med. 2002;30(4):537-540.
14. Bere T, Flørenes TW, Krosshaug T, Nordsletten L, Bahr R. Events leading to anterior cruciate ligament injury in World Cup alpine skiing: a systematic video analysis of 20 cases. Br J Sports Med. 2011;45(16):1294-1302.
15. Davies H, Tietjens B, Van Sterkenburg M, Mehgan A. Anterior cruciate ligament injuries in snowboarders: a quadriceps-induced injury. Knee Surg Sports Traumatol Arthrosc. 2009;17(9):1048-1051.
16. Bladin C, McCrory P, Pogorzelski A. Snowboarding injuries: current trends and future directions. Sports Med. 2004;34(2):133-139.
17. Rossi MJ, Lubowitz JH, Guttmann D. The skier’s knee. Arthroscopy. 2003;19(1):75-84.
18. Pressman A, Johnson DH. A review of ski injuries resulting in combined injury to the anterior cruciate ligament and medial collateral ligaments. Arthroscopy. 2003;19(2):194-202.
19. Hildebrandt C, Mildner E, Hotter B, Kirschner W, Höbenreich C, Raschner C. Accident prevention on ski slopes—perceptions of safety and knowledge of existing rules. Accid Anal Prev. 2011;43(4):1421-1426.
20. Langran M, Selvaraj S. Increased injury risk among first-day skiers, snowboarders, and skiboarders. Am J Sports Med. 2004;32(1):96-103.
21. Urabe Y, Ochi M, Onari K, Ikuta Y. Anterior cruciate ligament injury in recreational alpine skiers: analysis of mechanisms and strategy for prevention. J Orthop Sci. 2002;7(1):1-5.
22. McAlpine PR. Biomechanical Analysis of Snowboard Jump Landings: A Focus on the Ankle Joint Complex [doctoral thesis]. Auckland, New Zealand: University of Auckland; 2010.
1. Kim S, Endres NK, Johnson RJ, Ettlinger CF, Shealy JE. Snowboarding injuries: trends over time and comparisons with alpine skiing injuries. Am J Sports Med. 2012;40(4):770-776.
2. Wasden CC, McIntosh SE, Keith DS, McCowan C. An analysis of skiing and snowboarding injuries on Utah slopes. J Trauma. 2009;67(5):1022-1026.
3. Rust DA, Gilmore CJ, Treme G. Injury patterns at a large western United States ski resort with and without snowboarders: the Taos experience. Am J Sports Med. 2013;41(3):652-656.
4. Ishimaru D, Ogawa H, Sumi H, Sumi Y, Shimizu K. Lower extremity injuries in snowboarding. J Trauma. 2011;70(3):E48-E52.
5. Torjussen J, Bahr R. Injuries among competitive snowboarders at the national elite level. Am J Sports Med. 2005;33(3):370-377.
6. Deady LH, Salonen D. Skiing and snowboarding injuries: a review with a focus on mechanism of injury. Radiol Clin North Am. 2010;48(6):1113-1124.
7. Hasler RM, Berov S, Banneker L, et al. Are there risk factors for snowboard injuries? A case–control multicentre study of 559 snowboarders. Br J Sports Med. 2010;44(11):816-821.
8. St-Onge N, Chevalier Y, Hagemeister N, Van De Putte M, De Guise J. Effect of ski binding parameters on knee biomechanics: a three-dimensional computational study. Med Sci Sports Exerc. 2004;36(7):1218-1225.
9. Valderrabano V, Perren T, Ryf C, Rillmann P, Hintermann B. Snowboarder’s talus fracture: treatment outcome of 20 cases after 3.5 years. Am J Sports Med. 2005;33(6):871-880.
10. von Knoch F, Reckord U, von Knoch M, Sommer C. Fracture of the lateral process of the talus in snowboarders. J Bone Joint Surg Br. 2007;89(6):772-777.
11. Funk JR, Srinivasan SC, Crandall JR. Snowboarder’s talus fractures experimentally produced by eversion and dorsiflexion. Am J Sports Med. 2003;31(6):921-928.
12. Pujol N, Blanchi MP, Chambat P. The incidence of anterior cruciate ligament injuries among competitive alpine skiers: a 25-year investigation. Am J Sports Med. 2007;35(7):1070-1074.
13. Hame SL, Oakes DA, Markolf KL. Injury to the anterior cruciate ligament during alpine skiing: a biomechanical analysis of tibial torque and knee flexion angle. Am J Sports Med. 2002;30(4):537-540.
14. Bere T, Flørenes TW, Krosshaug T, Nordsletten L, Bahr R. Events leading to anterior cruciate ligament injury in World Cup alpine skiing: a systematic video analysis of 20 cases. Br J Sports Med. 2011;45(16):1294-1302.
15. Davies H, Tietjens B, Van Sterkenburg M, Mehgan A. Anterior cruciate ligament injuries in snowboarders: a quadriceps-induced injury. Knee Surg Sports Traumatol Arthrosc. 2009;17(9):1048-1051.
16. Bladin C, McCrory P, Pogorzelski A. Snowboarding injuries: current trends and future directions. Sports Med. 2004;34(2):133-139.
17. Rossi MJ, Lubowitz JH, Guttmann D. The skier’s knee. Arthroscopy. 2003;19(1):75-84.
18. Pressman A, Johnson DH. A review of ski injuries resulting in combined injury to the anterior cruciate ligament and medial collateral ligaments. Arthroscopy. 2003;19(2):194-202.
19. Hildebrandt C, Mildner E, Hotter B, Kirschner W, Höbenreich C, Raschner C. Accident prevention on ski slopes—perceptions of safety and knowledge of existing rules. Accid Anal Prev. 2011;43(4):1421-1426.
20. Langran M, Selvaraj S. Increased injury risk among first-day skiers, snowboarders, and skiboarders. Am J Sports Med. 2004;32(1):96-103.
21. Urabe Y, Ochi M, Onari K, Ikuta Y. Anterior cruciate ligament injury in recreational alpine skiers: analysis of mechanisms and strategy for prevention. J Orthop Sci. 2002;7(1):1-5.
22. McAlpine PR. Biomechanical Analysis of Snowboard Jump Landings: A Focus on the Ankle Joint Complex [doctoral thesis]. Auckland, New Zealand: University of Auckland; 2010.
Statin Adverse Effects: Sorting out the Evidence
CASE
Mr L., a 57-year-old obese patient (BMI > 40) who had not been to a clinician in a decade, comes to see you after a health fair screening revealed dyslipidemia (LDL cholesterol, 188 mg/dL; HDL cholesterol, 32 mg/dL; total cholesterol, 240 mg/dL; triglycerides, 100 mg/dL). His blood pressure (BP) is 146/90 mm Hg, and his fasting glucose is 101 mg/dL. Labs drawn that day reveal an A1C of 5.9%, alanine aminotransferase (ALT) of 45 U/L, and aspartate aminotransferase (AST) of 62 U/L. In taking his history, you discover that Mr L. also has a notable family history of heart disease.
Mr L. agrees to take a low-dose statin, and you prescribe atorvastatin 10 mg and a thiazide diuretic. You advise the patient to contact you immediately if he develops significant myalgia, jaundice, dark urine, or symptoms of hyperglycemia such as excessive thirst or urination, and to schedule a follow-up visit in eight weeks.
Long recognized as the bedrock of hyperlipidemia therapy, statins achieved even greater prominence when the American College of Cardiology/American Heart Association (ACC/AHA) issued a new cholesterol guideline1 late last year. The ACC and AHA now recommend statins for a wider range of patients, often at a higher starting dose.
Based on the new recommendations, the use of statins is likely to rise.2 (A statin—rosuvastatin—is already the nation’s most widely prescribed medication.2) Thus, it is more important than ever for clinicians to know about the risks associated with statins and to be able to assess the benefits of therapy for individual patients.
A 2013 retrospective cohort study of more than 100,000 patients on statins found that 17% developed adverse effects (AEs). Therapy was withheld, at least temporarily, for 10% of study participants (60% of those experiencing AEs).3 At the same time, the authors of a large meta-analysis (135 randomized controlled trials [RCTs] and > 240,000 patients) reported that AEs associated with statins as a class were uncommon. The meta-analysis also found that the overall discontinuation rate for statin users—5.7%—was not significantly different from that of patients receiving placebo.4
Such discrepancies regarding particular risks, as well as the overall incidence of AEs and discontinuation rates, make the evidence difficult to sort out. We created this update with that in mind.
Continue for symptoms >>
MUSCULOSKELETAL SYMPTOMS ARE MOST COMMON
Musculoskeletal symptoms are the most common AEs reported by patients who are taking statins.5 These range from muscle weakness, fatigue, and pain to (rarely) rhabdomyolysis—a life-threatening condition characterized by severe muscle pain, muscle weakness, a 10-fold increase in creatine kinase (CK), and increased serum creatinine, often with myoglobinuria.5
Patients with myopathy—an umbrella term for any muscle disease—may report stiffness, weakness, tenderness, soreness, cramping, or heaviness. Symptoms are usually symmetrical and often involve the proximal limbs and trunk.6 Studies indicate that exercise increases the risk for statin-induced myalgia—muscle pain or weakness without an increase in CK—and that patients taking statins are more prone to exercise-related injury.7,8
A baseline CK is recommended for patients with an increased risk for muscular disorders.1 Risk factors include a personal or family history of statin intolerance or muscle disease, age older than 75, low levels of vitamin D, and concomitant use of medications that may increase the risk for myopathy (see Table 1).1 Routine monitoring of CK is not recommended, but CK levels should be obtained for those who exhibit muscle symptoms while on statin therapy.1
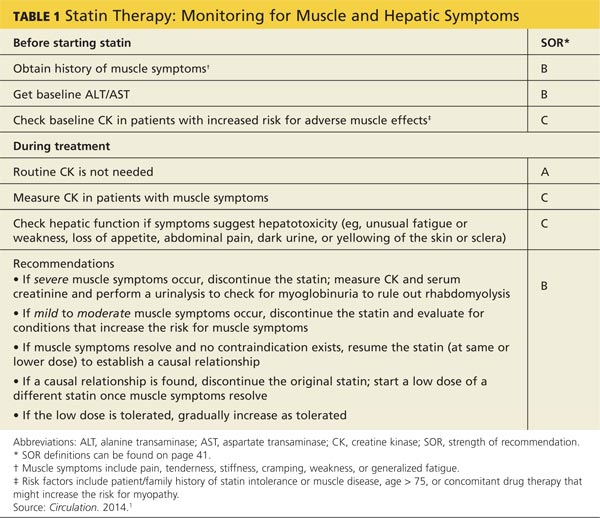
What the studies show
The incidence of myalgia reported in clinical studies is highly variable, ranging from less than 1% to 20%.1,9,10 The ACC/AHA guideline reports only one additional case of myopathy per 10,000 statin users compared with those on placebo and cites a rhabdomyolysis occurrence rate of less than 0.06% over five years.1
A 2006 systematic review estimated the absolute risk for rhabdomyolysis to be 3.4 per 100,000 person-years, but the incidence was 10 times higher for patients taking both a statin and gemfibrozil.11 (See Table 212,13 for more on drug interactions.) But both the meta-analysis cited earlier4 and a previous systematic review14 (35 RCTs and > 74,000 patients) found that statins as a class do not increase the incidence of myalgia or rhabdomyolysis.
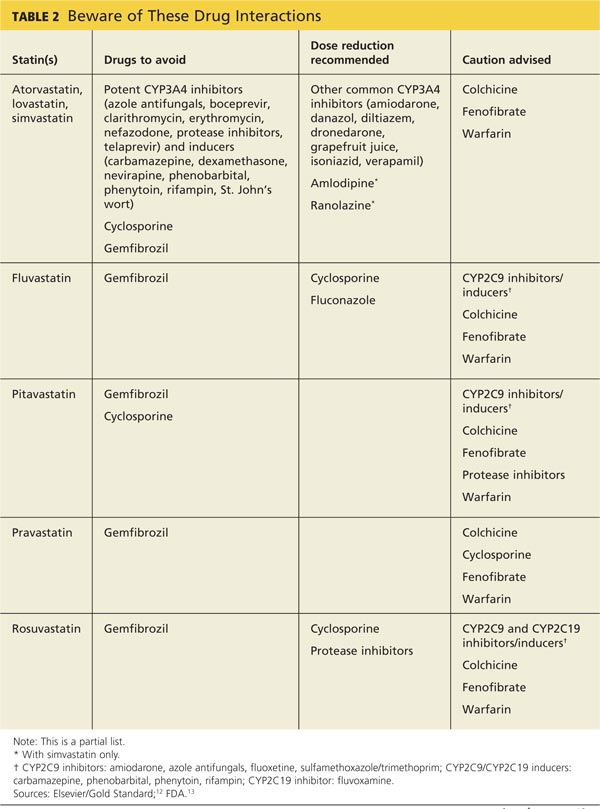
Differences in the way muscular disorders are defined has been suggested as one reason for the discrepancies.10 In addition, many clinical trials exclude patients at higher risk for statin-associated AEs, such as those with renal or hepatic insufficiency, prior muscular complaints, poorly controlled diabetes, or potential drug interactions.1
An FDA advisory. In a safety communication last updated in February 2012, the FDA cautioned against starting patients on the highest dose of simvastatin (80 mg).15 The warning is based on a large study (N = 12,064) that found an increased risk for myopathy (0.9%) and rhabdomyolysis (0.2%) in patients on the
80-mg dose versus those taking 20 mg (0.02% and 0%, respectively).16
With the ACC/AHA now recommending intensive therapy (atorvastatin 40-80 mg or rosuvastatin 20-40 mg) to achieve an LDL reduction greater than 50% for many patients,1 it is important to be aware that this risk is specific to simvastatin. A recent meta-analysis of studies directly comparing patients receiving intensive statin therapy with those on low to moderate doses did not find any increased risk in rhabdomyolysis associated with more intensive therapy when those taking 80-mg simvastatin were excluded.17
The bottom line: Although rhabdomyolysis is rare, its severity—a fatality rate of 10%11—makes it critical to educate patients about the disorder and instruct them to stop taking the statin and call the office immediately if they develop severe muscle pain or weakness.
Recommend CoQ10 for statin-induced myopathy
Although the exact mechanism of statin-induced myopathy is unknown, the most likely explanation is a depletion of coenzyme Q10 (CoQ10), which has negative effects on mitochondrial energy production.18 While studies using CoQ10 to treat this AE have been small and had mixed results, the overall evidence suggests that it decreases the development and/or severity of symptoms.18-20
In fact, CoQ10 supplementation is the only treatment that has shown promise in treating statin-induced muscle symptoms.18-20 Doses of about 100 mg bid have been found to be beneficial and safe; no clinically relevant AEs have been seen with doses lower than 300 mg/d.18,20,21 A large placebo-controlled study is currently evaluating a 600 mg/d dose of CoQ10 in patients with statin-induced myopathy.19
CASE
On his next visit, Mr L. reports a new ache in his left shoulder and upper back, which he describes as mild but annoying. He also tells you his memory seems to be getting worse and that he has developed an odd tingling in his hands. These symptoms began about a month after he started the medications, Mr L. says. He also began a new exercise program, but his BMI is unchanged.
On examination, you find the affected shoulder and upper back modestly and diffusely tender to palpation but with no decline in strength. Mr L.’s BP has fallen to
134/84 mm Hg, and his fasting glucose is 105 mg/dL. Lab tests reveal an LDL of 144 mg/dL and HDL of 36 mg/dL, A1C of 6.1%, ALT of 105 U/L, AST of 61 U/L, and a normal CK.
You recommend 100 mg CoQ10 bid. Because it is available only OTC, you advise the patient to look for a product whose purity and potency have been verified by an external source, such as the US Pharmacopeial Convention. You also prescribe metformin 500 mg bid for insulin resistance, refer the patient to a nutritionist and diabetes specialist, and order tests to evaluate his other symptoms.
Continue for hepatic effects >>
HEPATIC EFFECTS ARE RARE
Historically, statins have been linked to potential hepatotoxicity, with case reports of serum transaminase elevation, cholestasis, hepatitis, and acute liver failure. It is now recognized that hepatic AEs are rare and that statins are not associated with a risk for acute or chronic liver failure.1,11 In patients with coronary heart disease, the incidence of hepatotoxicity with statin use is reported to be less than 1.5% over the course of five years and appears to be dose-dependent.1
In 2012, the FDA revised the labeling for most statins, relaxing its earlier recommendations for monitoring of liver function, clarifying the risk for myopathy, and providing additional information about drug interactions.13
Checking transaminase levels before initiating therapy is recommended by both the ACC/AHA and the FDA.1,13 Routine monitoring is not necessary, the ACC/AHA guideline states, because RCTs have found little evidence of ALT/AST elevation.1 But here, too, evidence varies. An older meta-analysis (13 trials and nearly 50,000 participants) concluded that as a class, statins have no greater risk for transaminase elevations than placebo.22 But the 135-RCT meta-analysis4 found otherwise: Statins did increase the risk for transaminase elevation (odds ratio [OR], 1.51) compared with placebo, with differences associated with particular drugs and higher doses associated with more clinically significant elevations.4 It is important to note, however, that there was significant heterogeneity among the studies and no consistent definition of clinical significance.
The bottom line: Statins have been shown in multiple prospective studies to be safe for patients with chronic liver disease.22,23
STATIN USE AND DIABETES: IS THERE A LINK?
Recent studies have found an increased risk for new-onset type 2 diabetes in statin users, with a greater risk associated with higher-potency statins, including rosuvastatin and atorvastatin.4,24 Although the exact mechanism is not known, statins may modify insulin signaling in peripheral tissues or directly impair insulin secretion.
The ACC/AHA guideline reports an excess rate of diabetes of one per 1,000 patient-years for moderate-intensity therapy and three per 1,000 patient-years for high-intensity therapy.1 The 2013 meta-analysis found that the elevated risk for diabetes was relatively small (OR, 1.09).4 No difference among various statins was found.
In another meta-analysis—this one encompassing 17 RCTs and more than 110,000 patients—no statistically significant difference in the incidence of new-onset diabetes was seen based on either the specific statin being taken or the intensity of therapy (high vs moderate).24
The bottom line: Clinicians should monitor patients taking statins for signs and symptoms of hyperglycemia.
STATINS MAY BE RENOPROTECTIVE
Statin use has been found to be associated with an increased risk for tubular proteinuria—an effect that is both dose- and potency-dependent.25 Nonetheless, it has been suggested that statins may be a rare example of a drug class that is renoprotective in the long term, despite having an increased rate of proteinuria in the short term.25
The evidence? In prospective studies, statin therapy has been shown to slow the progression of kidney disease in diverse patient populations, including renal transplant recipients and those with chronic kidney disease (CKD).26,27
The Kidney Expert Panel of the National Lipid Association (NLA) has concluded that statins do not appear to cause significant proteinuria or acute kidney injury. The panel does not recommend routine monitoring for proteinuria or kidney function in statin users unless otherwise indicated but does recommend a lower dose for patients with CKD.28
The bottom line: Kidney Disease Improving Global Outcomes guidelines recommend that patients who have CKD, but are not on dialysis, be treated with statin therapy. Statins are contraindicated for patients on dialysis, as clinical trials have failed to show significant cardiovascular benefit.29
Continue for the risk of intracerebral hemorrhages >>
INTRACEREBRAL HEMORRHAGE: STATINS INCREASE RECURRENCE RISK
In recent years, there has been considerable concern about a statin-induced increased risk for intracerebral hemorrhage (ICH). In a major prospective study in which patients were put on high-dose statin therapy or placebo after an acute ischemic or hemorrhagic stroke, the overall incidence of a recurrent stroke was significantly lower in the statin group.30 Among those who’d had an ICH, however, the recurrence rate was 73% higher for patients taking statins.
A subanalysis that looked only at patients who’d had a hemorrhagic stroke as their initial event (n = 93) found that the absolute risk for recurrent ICH was 15.6% for patients randomized to atorvastatin versus 4.2% for those on placebo.31 Despite being based on a small subset of the original study group, multivariate analysis indicated the increased risk was statistically significant (hazard ratio [HR], 1.69).
A subsequent decision analysis study based on these results proposed that patients with a history of spontaneous deep ICH would need an exceedingly high 10-year cardiovascular event risk (> 40%) for the benefits of statin therapy to outweigh the risk.32 The risk is particularly high for those with a history of lobar ICH, which has an extremely high recurrence rate. However, subsequent retrospective and observational studies have found that patients who were already on statins when the ICH occurred had less severe strokes and more favorable outcomes, with a lower mortality rate at 90 days post-ICH.33-35
A 2010 ICH guideline from the AHA/American Stroke Association states that there is “insufficient data to recommend restrictions on use of statin agents” for patients who have had an ICH.36
The bottom line: Clinicians should carefully evaluate the anticipated cardiovascular risk for patients who have had a hemorrhagic stroke to determine whether statin therapy would be beneficial.
OTHER SERIOUS ADVERSE EFFECTS: WHICH REPORTS ARE ACCURATE?
Statin use has been associated with a number of other serious AEs. Some reports appear to be accurate; others do not hold up after a close look at the evidence.
Malignancy. A potential link between statins and an increased risk for malignancy has been considered for years. A large trial (N = 5,804) from 2002 found a correlation between pravastatin and an increased risk for new cancer diagnoses compared with placebo (HR, 1.25).37 But a 10-year follow-up did not substantiate this finding, and it is now believed that the original result may have been due to chance.38 Numerous other meta-analyses and systematic reviews have found no link between statin use and malignancy.39-41
Cataracts. Potential ocular effects have been widely studied and debated in recent years. Observational studies reporting an association between statin use and cataracts have had conflicting results, with some showing statins as protective42-45 and others finding an increased risk.46,47 However, a recent propensity-score matched analysis found that statin users do indeed have an increased risk for cataracts.48 The authors concluded that for primary prevention, the risk-benefit equation for statin use should include this added risk.48
In addition, a review of the databases of the National Registry of Drug-Induced Ocular Side Effects, the World Health Organization, and the FDA from 1987 to 2008 indicates that statin therapy may also cause diplopia, ptosis, and ophthalmoplegia.49
Peripheral neuropathy. Despite case reports of statin-induced peripheral neuropathy, the NLA’s Neurology Expert Panel states that statins do not appear to cause this condition. If a patient receiving statin therapy develops peripheral neuropathy, a full work-up for other causes should be initiated before modification of statin therapy is considered, the panel advises.28
Statins have also been linked to headache and dizziness, respiratory symptoms, gastrointestinal problems, and rash (see Table 3).50
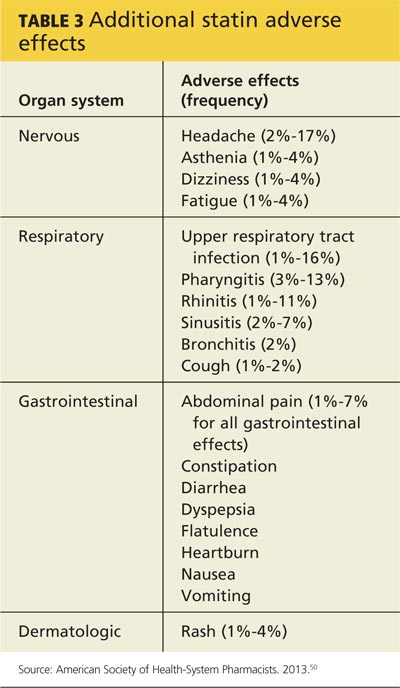
WHICH DRUG? POTENTIAL DIFFERENCES IN STATINS
A meta-analysis with more than 240,000 participants evaluated patients taking seven different statins (atorvastatin, fluvastatin, lovastatin, pravastatin, pitavastatin, rosuvastatin, and simvastatin), looking at AEs of the drugs both collectively and individually.4 As noted earlier, the overall discontinuation rate due to AEs for all statins was 5.7%. Discontinuation rates for each agent were not reported.4
The researchers did report, however, that atorvastatin and rosuvastatin had the highest discontinuation rates; atorvastatin and fluvastatin had the highest incidence of transaminase elevations (OR, 2.6 and 5.2, respectively); and pravastatin and simvastatin appeared to be the best-tolerated and safest statins, with the lowest discontinuation rates. However, higher doses of simvastatin (> 40 mg/d) significantly increased the risk for CK and transaminase elevations (OR, 4.1 and 2.8, respectively),4 as well as the risk for rhabdomyolysis when taken at the highest dose.15,16
Continue for safety concerns >>
ARE STATINS SAFE FOR THESE PATIENTS?
When considering statin therapy, there are some patient populations that warrant particular concern:
Women of childbearing age. Statins are contraindicated in women who are pregnant or breastfeeding1 and should not be initiated in women who are trying to conceive.
Children and adolescents (ages 8-18 years). Statins have been shown to be safe and effective for children and adolescents with familial hyperlipidemia. No effect on growth or maturation has been seen.51 As with adults, however, higher statin doses and the use of concomitant interacting drugs increase the risk for AEs.
Asians. The new ACC/AHA guideline suggests taking Asian ancestry into consideration when prescribing statins because Asians may be more sensitive to medications metabolized by the CYP450 system.1 However, there are no reports of an increased risk for AEs in Asian patients on statins.52
Patient factors that increase risk
Risk factors for statin-induced AEs include1
• Multiple and/or serious comorbidities (eg, hypothyroidism, impaired renal or hepatic function, rheumatic disorders)
• Unexplained ALT elevation more than 3x the upper limit of normal
• History of prior statin intolerance or concomitant use of drugs that affect statin metabolism
• Age older than 75
• Preexisting muscle disorders
• Low vitamin D levels.
If a patient who would clearly benefit from statin therapy develops an AE requiring discontinuation, a retrial—with the same drug or a different statin—is generally recommended once the symptoms resolve.1
CASE
The risk for elevated serum transaminases, insulin resistance, cognitive impairment, and neuropathy associated with statin use is minimal, and further evaluation revealed that Mr L.’s recent symptoms had other causes. The elevated transaminases were due to fatty liver disease, the cognitive impairment was secondary to sleep apnea (both linked to his obesity), and the tingling in his hands was the result of carpal tunnel syndrome caused by his exercise regimen.
When he returns in six months, Mr L. reports that he has been working with both a nutritionist and an athletic trainer. He has sustained a 15-lb weight loss. He is still taking atorvastatin 10 mg; after he began taking CoQ10, his muscle pain resolved. The patient’s cholesterol and transaminase levels are normal, and the cognitive impairment and peripheral neuropathy he reported at his last visit have improved significantly.
REFERENCES
1. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association task force on practice guidelines. Circulation. 2014;129:S1-S45.
2. Lowes R. Top 100 selling drugs through September reported. Medscape Med News. WebMD, LLC. 2013. www.medscape.com/viewarti cle/813571#3. Accessed October 19, 2014.
3. Zhang H, Plutzky J, Skentzos S, et al. Discontinuation of statins in routine care settings: a cohort study. Ann Intern Med. 2013;158:526-534.
4. Naci H, Brugts J, Ades T. Comparative tolerability and harms of individual statins: a study-level network meta-analysis of 246,955 participants from 135 randomized, controlled trials. Circ Cardiovasc Qual Outcomes. 2013;6:390-399.
5. Pasternak RC, Smith SC Jr, Bairey-Merz CN, et al; American College of Cardiology; American Heart Association; National Heart, Lung and Blood Institute. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. Circulation. 2002;106:1024-1028.
6. Eckel RH. Approach to the patient who is intolerant of statin therapy.
J Clin Endocrinol Metab. 2010;95:2015-2022.
7. Parker BA, Thompson PD. Effect of statins on skeletal muscle: exercise, myopathy, and muscle outcomes. Exerc Sport Sci Rev. 2012;40:188-194.
8. Mansi I, Frei CR, Pugh MJ, et al. Statins and musculoskeletal conditions, arthropathies, and injuries. JAMA Intern Med. 2013;173:1-10.
9. Bruckert E, Hayem G, Dejager S, et al. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—the PRIMO study. Cardiovasc Drugs Ther. 2005;19:403-414.
10. Fernandez G, Spatz ES, Jablecki C, et al. Statin myopathy: a common dilemma not reflected in clinical trials. Cleve Clin J Med. 2011;78:
393-403.
11. Law M, Rudnicka AR. Statin safety: a systematic review. Am J Cardiol. 2006;97:52C-60C.
12. Elsevier/Gold Standard. Gold Standard Drug Database. www.goldstand ard.com/product/gold-standard-drug-database/. Accessed October 19, 2014.
13. FDA. FDA drug safety communication: important safety label changes to cholesterol-lowering statin drugs. www.fda.gov/drugs/drugsafety/ucm293101.htm. Accessed October 19,2014.
14. Kashani A, Phillips CO, Foody JM, et al. Risks associated with statin therapy: a systematic overview of randomized clinical trials. Circulation. 2006;114:2788-2797.
15. FDA. FDA drug safety communication: ongoing safety review of high-dose Zocor (simvastatin) and increased risk of muscle injury. www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm204882.htm. Updated February 15, 2012. Accessed October 19, 2014.
16. Bowman L, Armitage J, Bulbulia R, et al; SEARCH Study Collaborative Group. Study of the effectiveness of additional reductions in cholesterol and homocysteine (SEARCH): characteristics of a randomized trial among 12064 myocardial infarction survivors. Am J Heart. 2007;154:815-823.
17. Mills EJ, O’Regan C, Eyawo O, et al. Intensive statin therapy compared with moderate dosing for prevention of cardiovascular events: a meta-analysis of >40,000 patients. Euro Heart J. 2011;32:1409-1415.
18. Bookstaver DA, Burkhalter NA, Hatzigeorgiou C. Effect of coenzyme Q10 supplementation on statin-induced myalgias. Am J Cardiol. 2012;110:
526-529.
19. Parker BA, Gregory SM, Lorson L, et al. A randomized trial of coenzyme Q10 in patients with statin myopathy: rationale and study design. J Clin Lipidol. 2013;7:187-193.
20. Fedacko J, Pella D, Fedackova P, et al. Coenzyme Q(10) and selenium in statin-associated myopathy treatment. Can J Physiol Pharmacol. 2013;91:165-170.
21. Jellin JM, Gregory PJ, et al. Natural Medicines Comprehensive Database. www.naturaldatabase.com.libproxy.uwyo.edu. Accessed October 19, 2014.
22. de Denus S, Spinler SA, Miller K, et al. Statins and liver toxicity: a meta-analysis. Pharmacotherapy. 2004;24:584-591.
23. Lewis JH. Clinical perspective: statins and the liver—harmful or helpful? Dig Dis Sci. 2012;57:1754-1763.
24. Navarese EP, Buffon A, Andreotti F, et al. Meta-analysis of impact of different types and doses of statins on new-onset diabetes mellitus. Am J Cardiol. 2013;111:1123-1130.
25. Agarwal R. Effects of statins on renal function. Am J Cardiol. 2006;97:748-755.
26. Fried LF, Orchard TJ, Lasiske BL. Effect of lipid reduction on the progression of renal disease: a meta-analysis. Kidney Int. 2001;59:260-269.
27. Fellström B, Holdaas H, Jardine AG, et al; Assessment of Lescol in Renal Transportation Study Investigators. Effect of fluvastatin on renal end points in the Assessment of Lescol in Renal Transplant (ALERT) Trial. Kidney Int. 2004;66:1549-1555.
28. McKenney JM, Davidson MH, Jacobson TA, et al; National Lipid Association Statin Safety Assessment Task Force. Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am J Cardiol. 2006;97:89C-94C.
29. KDIGO Clinical Practice Guideline for Lipid Management in Chronic Kidney Disease. Kidney Int. 2013;3(suppl):S259-S305.
30. Goldstein LB, Amarenco P, Szarek M, et al; SPARCL Investigators. Hemorrhagic stroke in the Stroke Prevention by Aggressive Reduction in Cholesterol Levels study. Neurology. 2008;70(24 pt 2):2364-2370.
31. Goldstein LB, Amarenco P, Lamonte M, et al; SPARCL investigators. Relative effects of statin therapy on stroke and cardiovascular events in men and women: secondary analysis of the Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) Study. Stroke. 2008;39:
2444-2448.
32. Westover MB, Bianchi MT, Eckman MH, et al. Statin use following intracerebral hemorrhage: a decision analysis. Arch Neurol. 2011;68:573-579.
33. Biffi A, Devan WJ, Anderson CD, et al. Statin use and outcome after intracerebral hemorrhage: case-control study and meta-analysis. Neurology. 2011;76:1581-1588.
34. Dowlatshahi D, Demchuck AM, Fang J, et al; Registry of the Canadian Stroke Network. Association of statins and statin discontinuation with poor outcome and survival after intracerebral hemorrhage. Stroke. 2012;43:1518-1523.
35. Bustamante A, Montaner J. Statin therapy should not be discontinued in patients with intracerebral hemorrhage. Stroke. 2013;44:2060-2061.
36. Morgenstern LB, Hemphill JC 3rd, Anderson C, et al; American Heart Association Stroke Council and Council on Cardiovascular Nursing. Guidelines for the management of spontaneous intracerebral hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2010;41:2108-2129.
37. Shepherd J, Blauw GJ, Murphy MB, et al; PROSPER study group. PROspective Study of Pravastatin in the Elderly at Risk. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet. 2002;360:1623-1630.
38. Jukema JW, Cannon CP, de Craen AJ, et al. The controversies of statin therapy: weighing the evidence. J Am Coll Cardiol. 2012;60:875-881.
39. Alberton M, Wu P, Druyts E, et al. Adverse events associated with individual statin treatments for cardiovascular disease: an indirect comparison meta-analysis. QJM. 2012;105:145-157.
40. Baigent C, Blackwell L, Emberson J, et al; Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670-1681.
41. Emberson JR, Kearney PM, Blackwell L, et al; Cholesterol Treatment Trialists’ (CTT) Collaboration. Lack of effect of lowering LDL cholesterol on cancer: meta-analysis of individual data from 175,000 people in 27 randomised trials of statin therapy. PLoS One. 2012;7:e29849.
42. Klein BE, Klein R, Lee KE, et al. Statin use and incident nuclear cataract. JAMA. 2006;295:2752-2758.
43. Fong DS, Poon KY. Recent statin use and cataract surgery. Am J Ophthalmol. 2012;153:222-228.e1.
44. Chodick G, Heymann AD, Flash S, et al. Persistence with statins and incident cataract: a population-based historical cohort study. Ann Epidemiol. 2010;20:136-142.
45. Tan JS, Mitchell P, Rochtchina E, et al. Statin use and the long-term risk of incident cataract: the Blue Mountains Eye Study. Am J Ophthalmol. 2007;143:687-689.
46. Machan CM, Hrynchak PK, Irving EL. Age-related cataract is associated with type 2 diabetes and statin use. Optom Vis Sci. 2012;89:1165-1171.
47. Hippisley-Cox J, Coupland C. Unintended effects of statins in men and women in England and Wales: population based cohort study using the QResearch database. BMJ. 2010;340:c2197.
48. Leuschen J, Mortensen EM, Frei CR, et al. Association of statin use with cataracts: a propensity score-matched analysis. JAMA Ophthalmol. 2013;131:1427-1434.
49. Fraunfelder FW, Richards AB. Diplopia, blepharoptosis, and ophthalmoplegia and 3-hydroxy-3-methyl-glutaryl-CoA reductase inhibitor use. Ophthalmology. 2008;115:2282-2285.
50. AHFS Drug Information 2013. Bethesda, MD: American Society of Health-System Pharmacists; 2013.
51. Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents; National Heart, Lung, and Blood Institute. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics. 2011;128(suppl 5):S213-S256.
52. Liao JK. Safety and efficacy of statins in Asians. Am J Cardiol. 2007;99:410-414.
CASE
Mr L., a 57-year-old obese patient (BMI > 40) who had not been to a clinician in a decade, comes to see you after a health fair screening revealed dyslipidemia (LDL cholesterol, 188 mg/dL; HDL cholesterol, 32 mg/dL; total cholesterol, 240 mg/dL; triglycerides, 100 mg/dL). His blood pressure (BP) is 146/90 mm Hg, and his fasting glucose is 101 mg/dL. Labs drawn that day reveal an A1C of 5.9%, alanine aminotransferase (ALT) of 45 U/L, and aspartate aminotransferase (AST) of 62 U/L. In taking his history, you discover that Mr L. also has a notable family history of heart disease.
Mr L. agrees to take a low-dose statin, and you prescribe atorvastatin 10 mg and a thiazide diuretic. You advise the patient to contact you immediately if he develops significant myalgia, jaundice, dark urine, or symptoms of hyperglycemia such as excessive thirst or urination, and to schedule a follow-up visit in eight weeks.
Long recognized as the bedrock of hyperlipidemia therapy, statins achieved even greater prominence when the American College of Cardiology/American Heart Association (ACC/AHA) issued a new cholesterol guideline1 late last year. The ACC and AHA now recommend statins for a wider range of patients, often at a higher starting dose.
Based on the new recommendations, the use of statins is likely to rise.2 (A statin—rosuvastatin—is already the nation’s most widely prescribed medication.2) Thus, it is more important than ever for clinicians to know about the risks associated with statins and to be able to assess the benefits of therapy for individual patients.
A 2013 retrospective cohort study of more than 100,000 patients on statins found that 17% developed adverse effects (AEs). Therapy was withheld, at least temporarily, for 10% of study participants (60% of those experiencing AEs).3 At the same time, the authors of a large meta-analysis (135 randomized controlled trials [RCTs] and > 240,000 patients) reported that AEs associated with statins as a class were uncommon. The meta-analysis also found that the overall discontinuation rate for statin users—5.7%—was not significantly different from that of patients receiving placebo.4
Such discrepancies regarding particular risks, as well as the overall incidence of AEs and discontinuation rates, make the evidence difficult to sort out. We created this update with that in mind.
Continue for symptoms >>
MUSCULOSKELETAL SYMPTOMS ARE MOST COMMON
Musculoskeletal symptoms are the most common AEs reported by patients who are taking statins.5 These range from muscle weakness, fatigue, and pain to (rarely) rhabdomyolysis—a life-threatening condition characterized by severe muscle pain, muscle weakness, a 10-fold increase in creatine kinase (CK), and increased serum creatinine, often with myoglobinuria.5
Patients with myopathy—an umbrella term for any muscle disease—may report stiffness, weakness, tenderness, soreness, cramping, or heaviness. Symptoms are usually symmetrical and often involve the proximal limbs and trunk.6 Studies indicate that exercise increases the risk for statin-induced myalgia—muscle pain or weakness without an increase in CK—and that patients taking statins are more prone to exercise-related injury.7,8
A baseline CK is recommended for patients with an increased risk for muscular disorders.1 Risk factors include a personal or family history of statin intolerance or muscle disease, age older than 75, low levels of vitamin D, and concomitant use of medications that may increase the risk for myopathy (see Table 1).1 Routine monitoring of CK is not recommended, but CK levels should be obtained for those who exhibit muscle symptoms while on statin therapy.1
What the studies show
The incidence of myalgia reported in clinical studies is highly variable, ranging from less than 1% to 20%.1,9,10 The ACC/AHA guideline reports only one additional case of myopathy per 10,000 statin users compared with those on placebo and cites a rhabdomyolysis occurrence rate of less than 0.06% over five years.1
A 2006 systematic review estimated the absolute risk for rhabdomyolysis to be 3.4 per 100,000 person-years, but the incidence was 10 times higher for patients taking both a statin and gemfibrozil.11 (See Table 212,13 for more on drug interactions.) But both the meta-analysis cited earlier4 and a previous systematic review14 (35 RCTs and > 74,000 patients) found that statins as a class do not increase the incidence of myalgia or rhabdomyolysis.
Differences in the way muscular disorders are defined has been suggested as one reason for the discrepancies.10 In addition, many clinical trials exclude patients at higher risk for statin-associated AEs, such as those with renal or hepatic insufficiency, prior muscular complaints, poorly controlled diabetes, or potential drug interactions.1
An FDA advisory. In a safety communication last updated in February 2012, the FDA cautioned against starting patients on the highest dose of simvastatin (80 mg).15 The warning is based on a large study (N = 12,064) that found an increased risk for myopathy (0.9%) and rhabdomyolysis (0.2%) in patients on the
80-mg dose versus those taking 20 mg (0.02% and 0%, respectively).16
With the ACC/AHA now recommending intensive therapy (atorvastatin 40-80 mg or rosuvastatin 20-40 mg) to achieve an LDL reduction greater than 50% for many patients,1 it is important to be aware that this risk is specific to simvastatin. A recent meta-analysis of studies directly comparing patients receiving intensive statin therapy with those on low to moderate doses did not find any increased risk in rhabdomyolysis associated with more intensive therapy when those taking 80-mg simvastatin were excluded.17
The bottom line: Although rhabdomyolysis is rare, its severity—a fatality rate of 10%11—makes it critical to educate patients about the disorder and instruct them to stop taking the statin and call the office immediately if they develop severe muscle pain or weakness.
Recommend CoQ10 for statin-induced myopathy
Although the exact mechanism of statin-induced myopathy is unknown, the most likely explanation is a depletion of coenzyme Q10 (CoQ10), which has negative effects on mitochondrial energy production.18 While studies using CoQ10 to treat this AE have been small and had mixed results, the overall evidence suggests that it decreases the development and/or severity of symptoms.18-20
In fact, CoQ10 supplementation is the only treatment that has shown promise in treating statin-induced muscle symptoms.18-20 Doses of about 100 mg bid have been found to be beneficial and safe; no clinically relevant AEs have been seen with doses lower than 300 mg/d.18,20,21 A large placebo-controlled study is currently evaluating a 600 mg/d dose of CoQ10 in patients with statin-induced myopathy.19
CASE
On his next visit, Mr L. reports a new ache in his left shoulder and upper back, which he describes as mild but annoying. He also tells you his memory seems to be getting worse and that he has developed an odd tingling in his hands. These symptoms began about a month after he started the medications, Mr L. says. He also began a new exercise program, but his BMI is unchanged.
On examination, you find the affected shoulder and upper back modestly and diffusely tender to palpation but with no decline in strength. Mr L.’s BP has fallen to
134/84 mm Hg, and his fasting glucose is 105 mg/dL. Lab tests reveal an LDL of 144 mg/dL and HDL of 36 mg/dL, A1C of 6.1%, ALT of 105 U/L, AST of 61 U/L, and a normal CK.
You recommend 100 mg CoQ10 bid. Because it is available only OTC, you advise the patient to look for a product whose purity and potency have been verified by an external source, such as the US Pharmacopeial Convention. You also prescribe metformin 500 mg bid for insulin resistance, refer the patient to a nutritionist and diabetes specialist, and order tests to evaluate his other symptoms.
Continue for hepatic effects >>
HEPATIC EFFECTS ARE RARE
Historically, statins have been linked to potential hepatotoxicity, with case reports of serum transaminase elevation, cholestasis, hepatitis, and acute liver failure. It is now recognized that hepatic AEs are rare and that statins are not associated with a risk for acute or chronic liver failure.1,11 In patients with coronary heart disease, the incidence of hepatotoxicity with statin use is reported to be less than 1.5% over the course of five years and appears to be dose-dependent.1
In 2012, the FDA revised the labeling for most statins, relaxing its earlier recommendations for monitoring of liver function, clarifying the risk for myopathy, and providing additional information about drug interactions.13
Checking transaminase levels before initiating therapy is recommended by both the ACC/AHA and the FDA.1,13 Routine monitoring is not necessary, the ACC/AHA guideline states, because RCTs have found little evidence of ALT/AST elevation.1 But here, too, evidence varies. An older meta-analysis (13 trials and nearly 50,000 participants) concluded that as a class, statins have no greater risk for transaminase elevations than placebo.22 But the 135-RCT meta-analysis4 found otherwise: Statins did increase the risk for transaminase elevation (odds ratio [OR], 1.51) compared with placebo, with differences associated with particular drugs and higher doses associated with more clinically significant elevations.4 It is important to note, however, that there was significant heterogeneity among the studies and no consistent definition of clinical significance.
The bottom line: Statins have been shown in multiple prospective studies to be safe for patients with chronic liver disease.22,23
STATIN USE AND DIABETES: IS THERE A LINK?
Recent studies have found an increased risk for new-onset type 2 diabetes in statin users, with a greater risk associated with higher-potency statins, including rosuvastatin and atorvastatin.4,24 Although the exact mechanism is not known, statins may modify insulin signaling in peripheral tissues or directly impair insulin secretion.
The ACC/AHA guideline reports an excess rate of diabetes of one per 1,000 patient-years for moderate-intensity therapy and three per 1,000 patient-years for high-intensity therapy.1 The 2013 meta-analysis found that the elevated risk for diabetes was relatively small (OR, 1.09).4 No difference among various statins was found.
In another meta-analysis—this one encompassing 17 RCTs and more than 110,000 patients—no statistically significant difference in the incidence of new-onset diabetes was seen based on either the specific statin being taken or the intensity of therapy (high vs moderate).24
The bottom line: Clinicians should monitor patients taking statins for signs and symptoms of hyperglycemia.
STATINS MAY BE RENOPROTECTIVE
Statin use has been found to be associated with an increased risk for tubular proteinuria—an effect that is both dose- and potency-dependent.25 Nonetheless, it has been suggested that statins may be a rare example of a drug class that is renoprotective in the long term, despite having an increased rate of proteinuria in the short term.25
The evidence? In prospective studies, statin therapy has been shown to slow the progression of kidney disease in diverse patient populations, including renal transplant recipients and those with chronic kidney disease (CKD).26,27
The Kidney Expert Panel of the National Lipid Association (NLA) has concluded that statins do not appear to cause significant proteinuria or acute kidney injury. The panel does not recommend routine monitoring for proteinuria or kidney function in statin users unless otherwise indicated but does recommend a lower dose for patients with CKD.28
The bottom line: Kidney Disease Improving Global Outcomes guidelines recommend that patients who have CKD, but are not on dialysis, be treated with statin therapy. Statins are contraindicated for patients on dialysis, as clinical trials have failed to show significant cardiovascular benefit.29
Continue for the risk of intracerebral hemorrhages >>
INTRACEREBRAL HEMORRHAGE: STATINS INCREASE RECURRENCE RISK
In recent years, there has been considerable concern about a statin-induced increased risk for intracerebral hemorrhage (ICH). In a major prospective study in which patients were put on high-dose statin therapy or placebo after an acute ischemic or hemorrhagic stroke, the overall incidence of a recurrent stroke was significantly lower in the statin group.30 Among those who’d had an ICH, however, the recurrence rate was 73% higher for patients taking statins.
A subanalysis that looked only at patients who’d had a hemorrhagic stroke as their initial event (n = 93) found that the absolute risk for recurrent ICH was 15.6% for patients randomized to atorvastatin versus 4.2% for those on placebo.31 Despite being based on a small subset of the original study group, multivariate analysis indicated the increased risk was statistically significant (hazard ratio [HR], 1.69).
A subsequent decision analysis study based on these results proposed that patients with a history of spontaneous deep ICH would need an exceedingly high 10-year cardiovascular event risk (> 40%) for the benefits of statin therapy to outweigh the risk.32 The risk is particularly high for those with a history of lobar ICH, which has an extremely high recurrence rate. However, subsequent retrospective and observational studies have found that patients who were already on statins when the ICH occurred had less severe strokes and more favorable outcomes, with a lower mortality rate at 90 days post-ICH.33-35
A 2010 ICH guideline from the AHA/American Stroke Association states that there is “insufficient data to recommend restrictions on use of statin agents” for patients who have had an ICH.36
The bottom line: Clinicians should carefully evaluate the anticipated cardiovascular risk for patients who have had a hemorrhagic stroke to determine whether statin therapy would be beneficial.
OTHER SERIOUS ADVERSE EFFECTS: WHICH REPORTS ARE ACCURATE?
Statin use has been associated with a number of other serious AEs. Some reports appear to be accurate; others do not hold up after a close look at the evidence.
Malignancy. A potential link between statins and an increased risk for malignancy has been considered for years. A large trial (N = 5,804) from 2002 found a correlation between pravastatin and an increased risk for new cancer diagnoses compared with placebo (HR, 1.25).37 But a 10-year follow-up did not substantiate this finding, and it is now believed that the original result may have been due to chance.38 Numerous other meta-analyses and systematic reviews have found no link between statin use and malignancy.39-41
Cataracts. Potential ocular effects have been widely studied and debated in recent years. Observational studies reporting an association between statin use and cataracts have had conflicting results, with some showing statins as protective42-45 and others finding an increased risk.46,47 However, a recent propensity-score matched analysis found that statin users do indeed have an increased risk for cataracts.48 The authors concluded that for primary prevention, the risk-benefit equation for statin use should include this added risk.48
In addition, a review of the databases of the National Registry of Drug-Induced Ocular Side Effects, the World Health Organization, and the FDA from 1987 to 2008 indicates that statin therapy may also cause diplopia, ptosis, and ophthalmoplegia.49
Peripheral neuropathy. Despite case reports of statin-induced peripheral neuropathy, the NLA’s Neurology Expert Panel states that statins do not appear to cause this condition. If a patient receiving statin therapy develops peripheral neuropathy, a full work-up for other causes should be initiated before modification of statin therapy is considered, the panel advises.28
Statins have also been linked to headache and dizziness, respiratory symptoms, gastrointestinal problems, and rash (see Table 3).50
WHICH DRUG? POTENTIAL DIFFERENCES IN STATINS
A meta-analysis with more than 240,000 participants evaluated patients taking seven different statins (atorvastatin, fluvastatin, lovastatin, pravastatin, pitavastatin, rosuvastatin, and simvastatin), looking at AEs of the drugs both collectively and individually.4 As noted earlier, the overall discontinuation rate due to AEs for all statins was 5.7%. Discontinuation rates for each agent were not reported.4
The researchers did report, however, that atorvastatin and rosuvastatin had the highest discontinuation rates; atorvastatin and fluvastatin had the highest incidence of transaminase elevations (OR, 2.6 and 5.2, respectively); and pravastatin and simvastatin appeared to be the best-tolerated and safest statins, with the lowest discontinuation rates. However, higher doses of simvastatin (> 40 mg/d) significantly increased the risk for CK and transaminase elevations (OR, 4.1 and 2.8, respectively),4 as well as the risk for rhabdomyolysis when taken at the highest dose.15,16
Continue for safety concerns >>
ARE STATINS SAFE FOR THESE PATIENTS?
When considering statin therapy, there are some patient populations that warrant particular concern:
Women of childbearing age. Statins are contraindicated in women who are pregnant or breastfeeding1 and should not be initiated in women who are trying to conceive.
Children and adolescents (ages 8-18 years). Statins have been shown to be safe and effective for children and adolescents with familial hyperlipidemia. No effect on growth or maturation has been seen.51 As with adults, however, higher statin doses and the use of concomitant interacting drugs increase the risk for AEs.
Asians. The new ACC/AHA guideline suggests taking Asian ancestry into consideration when prescribing statins because Asians may be more sensitive to medications metabolized by the CYP450 system.1 However, there are no reports of an increased risk for AEs in Asian patients on statins.52
Patient factors that increase risk
Risk factors for statin-induced AEs include1
• Multiple and/or serious comorbidities (eg, hypothyroidism, impaired renal or hepatic function, rheumatic disorders)
• Unexplained ALT elevation more than 3x the upper limit of normal
• History of prior statin intolerance or concomitant use of drugs that affect statin metabolism
• Age older than 75
• Preexisting muscle disorders
• Low vitamin D levels.
If a patient who would clearly benefit from statin therapy develops an AE requiring discontinuation, a retrial—with the same drug or a different statin—is generally recommended once the symptoms resolve.1
CASE
The risk for elevated serum transaminases, insulin resistance, cognitive impairment, and neuropathy associated with statin use is minimal, and further evaluation revealed that Mr L.’s recent symptoms had other causes. The elevated transaminases were due to fatty liver disease, the cognitive impairment was secondary to sleep apnea (both linked to his obesity), and the tingling in his hands was the result of carpal tunnel syndrome caused by his exercise regimen.
When he returns in six months, Mr L. reports that he has been working with both a nutritionist and an athletic trainer. He has sustained a 15-lb weight loss. He is still taking atorvastatin 10 mg; after he began taking CoQ10, his muscle pain resolved. The patient’s cholesterol and transaminase levels are normal, and the cognitive impairment and peripheral neuropathy he reported at his last visit have improved significantly.
REFERENCES
1. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association task force on practice guidelines. Circulation. 2014;129:S1-S45.
2. Lowes R. Top 100 selling drugs through September reported. Medscape Med News. WebMD, LLC. 2013. www.medscape.com/viewarti cle/813571#3. Accessed October 19, 2014.
3. Zhang H, Plutzky J, Skentzos S, et al. Discontinuation of statins in routine care settings: a cohort study. Ann Intern Med. 2013;158:526-534.
4. Naci H, Brugts J, Ades T. Comparative tolerability and harms of individual statins: a study-level network meta-analysis of 246,955 participants from 135 randomized, controlled trials. Circ Cardiovasc Qual Outcomes. 2013;6:390-399.
5. Pasternak RC, Smith SC Jr, Bairey-Merz CN, et al; American College of Cardiology; American Heart Association; National Heart, Lung and Blood Institute. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. Circulation. 2002;106:1024-1028.
6. Eckel RH. Approach to the patient who is intolerant of statin therapy.
J Clin Endocrinol Metab. 2010;95:2015-2022.
7. Parker BA, Thompson PD. Effect of statins on skeletal muscle: exercise, myopathy, and muscle outcomes. Exerc Sport Sci Rev. 2012;40:188-194.
8. Mansi I, Frei CR, Pugh MJ, et al. Statins and musculoskeletal conditions, arthropathies, and injuries. JAMA Intern Med. 2013;173:1-10.
9. Bruckert E, Hayem G, Dejager S, et al. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—the PRIMO study. Cardiovasc Drugs Ther. 2005;19:403-414.
10. Fernandez G, Spatz ES, Jablecki C, et al. Statin myopathy: a common dilemma not reflected in clinical trials. Cleve Clin J Med. 2011;78:
393-403.
11. Law M, Rudnicka AR. Statin safety: a systematic review. Am J Cardiol. 2006;97:52C-60C.
12. Elsevier/Gold Standard. Gold Standard Drug Database. www.goldstand ard.com/product/gold-standard-drug-database/. Accessed October 19, 2014.
13. FDA. FDA drug safety communication: important safety label changes to cholesterol-lowering statin drugs. www.fda.gov/drugs/drugsafety/ucm293101.htm. Accessed October 19,2014.
14. Kashani A, Phillips CO, Foody JM, et al. Risks associated with statin therapy: a systematic overview of randomized clinical trials. Circulation. 2006;114:2788-2797.
15. FDA. FDA drug safety communication: ongoing safety review of high-dose Zocor (simvastatin) and increased risk of muscle injury. www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm204882.htm. Updated February 15, 2012. Accessed October 19, 2014.
16. Bowman L, Armitage J, Bulbulia R, et al; SEARCH Study Collaborative Group. Study of the effectiveness of additional reductions in cholesterol and homocysteine (SEARCH): characteristics of a randomized trial among 12064 myocardial infarction survivors. Am J Heart. 2007;154:815-823.
17. Mills EJ, O’Regan C, Eyawo O, et al. Intensive statin therapy compared with moderate dosing for prevention of cardiovascular events: a meta-analysis of >40,000 patients. Euro Heart J. 2011;32:1409-1415.
18. Bookstaver DA, Burkhalter NA, Hatzigeorgiou C. Effect of coenzyme Q10 supplementation on statin-induced myalgias. Am J Cardiol. 2012;110:
526-529.
19. Parker BA, Gregory SM, Lorson L, et al. A randomized trial of coenzyme Q10 in patients with statin myopathy: rationale and study design. J Clin Lipidol. 2013;7:187-193.
20. Fedacko J, Pella D, Fedackova P, et al. Coenzyme Q(10) and selenium in statin-associated myopathy treatment. Can J Physiol Pharmacol. 2013;91:165-170.
21. Jellin JM, Gregory PJ, et al. Natural Medicines Comprehensive Database. www.naturaldatabase.com.libproxy.uwyo.edu. Accessed October 19, 2014.
22. de Denus S, Spinler SA, Miller K, et al. Statins and liver toxicity: a meta-analysis. Pharmacotherapy. 2004;24:584-591.
23. Lewis JH. Clinical perspective: statins and the liver—harmful or helpful? Dig Dis Sci. 2012;57:1754-1763.
24. Navarese EP, Buffon A, Andreotti F, et al. Meta-analysis of impact of different types and doses of statins on new-onset diabetes mellitus. Am J Cardiol. 2013;111:1123-1130.
25. Agarwal R. Effects of statins on renal function. Am J Cardiol. 2006;97:748-755.
26. Fried LF, Orchard TJ, Lasiske BL. Effect of lipid reduction on the progression of renal disease: a meta-analysis. Kidney Int. 2001;59:260-269.
27. Fellström B, Holdaas H, Jardine AG, et al; Assessment of Lescol in Renal Transportation Study Investigators. Effect of fluvastatin on renal end points in the Assessment of Lescol in Renal Transplant (ALERT) Trial. Kidney Int. 2004;66:1549-1555.
28. McKenney JM, Davidson MH, Jacobson TA, et al; National Lipid Association Statin Safety Assessment Task Force. Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am J Cardiol. 2006;97:89C-94C.
29. KDIGO Clinical Practice Guideline for Lipid Management in Chronic Kidney Disease. Kidney Int. 2013;3(suppl):S259-S305.
30. Goldstein LB, Amarenco P, Szarek M, et al; SPARCL Investigators. Hemorrhagic stroke in the Stroke Prevention by Aggressive Reduction in Cholesterol Levels study. Neurology. 2008;70(24 pt 2):2364-2370.
31. Goldstein LB, Amarenco P, Lamonte M, et al; SPARCL investigators. Relative effects of statin therapy on stroke and cardiovascular events in men and women: secondary analysis of the Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) Study. Stroke. 2008;39:
2444-2448.
32. Westover MB, Bianchi MT, Eckman MH, et al. Statin use following intracerebral hemorrhage: a decision analysis. Arch Neurol. 2011;68:573-579.
33. Biffi A, Devan WJ, Anderson CD, et al. Statin use and outcome after intracerebral hemorrhage: case-control study and meta-analysis. Neurology. 2011;76:1581-1588.
34. Dowlatshahi D, Demchuck AM, Fang J, et al; Registry of the Canadian Stroke Network. Association of statins and statin discontinuation with poor outcome and survival after intracerebral hemorrhage. Stroke. 2012;43:1518-1523.
35. Bustamante A, Montaner J. Statin therapy should not be discontinued in patients with intracerebral hemorrhage. Stroke. 2013;44:2060-2061.
36. Morgenstern LB, Hemphill JC 3rd, Anderson C, et al; American Heart Association Stroke Council and Council on Cardiovascular Nursing. Guidelines for the management of spontaneous intracerebral hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2010;41:2108-2129.
37. Shepherd J, Blauw GJ, Murphy MB, et al; PROSPER study group. PROspective Study of Pravastatin in the Elderly at Risk. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet. 2002;360:1623-1630.
38. Jukema JW, Cannon CP, de Craen AJ, et al. The controversies of statin therapy: weighing the evidence. J Am Coll Cardiol. 2012;60:875-881.
39. Alberton M, Wu P, Druyts E, et al. Adverse events associated with individual statin treatments for cardiovascular disease: an indirect comparison meta-analysis. QJM. 2012;105:145-157.
40. Baigent C, Blackwell L, Emberson J, et al; Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670-1681.
41. Emberson JR, Kearney PM, Blackwell L, et al; Cholesterol Treatment Trialists’ (CTT) Collaboration. Lack of effect of lowering LDL cholesterol on cancer: meta-analysis of individual data from 175,000 people in 27 randomised trials of statin therapy. PLoS One. 2012;7:e29849.
42. Klein BE, Klein R, Lee KE, et al. Statin use and incident nuclear cataract. JAMA. 2006;295:2752-2758.
43. Fong DS, Poon KY. Recent statin use and cataract surgery. Am J Ophthalmol. 2012;153:222-228.e1.
44. Chodick G, Heymann AD, Flash S, et al. Persistence with statins and incident cataract: a population-based historical cohort study. Ann Epidemiol. 2010;20:136-142.
45. Tan JS, Mitchell P, Rochtchina E, et al. Statin use and the long-term risk of incident cataract: the Blue Mountains Eye Study. Am J Ophthalmol. 2007;143:687-689.
46. Machan CM, Hrynchak PK, Irving EL. Age-related cataract is associated with type 2 diabetes and statin use. Optom Vis Sci. 2012;89:1165-1171.
47. Hippisley-Cox J, Coupland C. Unintended effects of statins in men and women in England and Wales: population based cohort study using the QResearch database. BMJ. 2010;340:c2197.
48. Leuschen J, Mortensen EM, Frei CR, et al. Association of statin use with cataracts: a propensity score-matched analysis. JAMA Ophthalmol. 2013;131:1427-1434.
49. Fraunfelder FW, Richards AB. Diplopia, blepharoptosis, and ophthalmoplegia and 3-hydroxy-3-methyl-glutaryl-CoA reductase inhibitor use. Ophthalmology. 2008;115:2282-2285.
50. AHFS Drug Information 2013. Bethesda, MD: American Society of Health-System Pharmacists; 2013.
51. Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents; National Heart, Lung, and Blood Institute. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics. 2011;128(suppl 5):S213-S256.
52. Liao JK. Safety and efficacy of statins in Asians. Am J Cardiol. 2007;99:410-414.
CASE
Mr L., a 57-year-old obese patient (BMI > 40) who had not been to a clinician in a decade, comes to see you after a health fair screening revealed dyslipidemia (LDL cholesterol, 188 mg/dL; HDL cholesterol, 32 mg/dL; total cholesterol, 240 mg/dL; triglycerides, 100 mg/dL). His blood pressure (BP) is 146/90 mm Hg, and his fasting glucose is 101 mg/dL. Labs drawn that day reveal an A1C of 5.9%, alanine aminotransferase (ALT) of 45 U/L, and aspartate aminotransferase (AST) of 62 U/L. In taking his history, you discover that Mr L. also has a notable family history of heart disease.
Mr L. agrees to take a low-dose statin, and you prescribe atorvastatin 10 mg and a thiazide diuretic. You advise the patient to contact you immediately if he develops significant myalgia, jaundice, dark urine, or symptoms of hyperglycemia such as excessive thirst or urination, and to schedule a follow-up visit in eight weeks.
Long recognized as the bedrock of hyperlipidemia therapy, statins achieved even greater prominence when the American College of Cardiology/American Heart Association (ACC/AHA) issued a new cholesterol guideline1 late last year. The ACC and AHA now recommend statins for a wider range of patients, often at a higher starting dose.
Based on the new recommendations, the use of statins is likely to rise.2 (A statin—rosuvastatin—is already the nation’s most widely prescribed medication.2) Thus, it is more important than ever for clinicians to know about the risks associated with statins and to be able to assess the benefits of therapy for individual patients.
A 2013 retrospective cohort study of more than 100,000 patients on statins found that 17% developed adverse effects (AEs). Therapy was withheld, at least temporarily, for 10% of study participants (60% of those experiencing AEs).3 At the same time, the authors of a large meta-analysis (135 randomized controlled trials [RCTs] and > 240,000 patients) reported that AEs associated with statins as a class were uncommon. The meta-analysis also found that the overall discontinuation rate for statin users—5.7%—was not significantly different from that of patients receiving placebo.4
Such discrepancies regarding particular risks, as well as the overall incidence of AEs and discontinuation rates, make the evidence difficult to sort out. We created this update with that in mind.
Continue for symptoms >>
MUSCULOSKELETAL SYMPTOMS ARE MOST COMMON
Musculoskeletal symptoms are the most common AEs reported by patients who are taking statins.5 These range from muscle weakness, fatigue, and pain to (rarely) rhabdomyolysis—a life-threatening condition characterized by severe muscle pain, muscle weakness, a 10-fold increase in creatine kinase (CK), and increased serum creatinine, often with myoglobinuria.5
Patients with myopathy—an umbrella term for any muscle disease—may report stiffness, weakness, tenderness, soreness, cramping, or heaviness. Symptoms are usually symmetrical and often involve the proximal limbs and trunk.6 Studies indicate that exercise increases the risk for statin-induced myalgia—muscle pain or weakness without an increase in CK—and that patients taking statins are more prone to exercise-related injury.7,8
A baseline CK is recommended for patients with an increased risk for muscular disorders.1 Risk factors include a personal or family history of statin intolerance or muscle disease, age older than 75, low levels of vitamin D, and concomitant use of medications that may increase the risk for myopathy (see Table 1).1 Routine monitoring of CK is not recommended, but CK levels should be obtained for those who exhibit muscle symptoms while on statin therapy.1
What the studies show
The incidence of myalgia reported in clinical studies is highly variable, ranging from less than 1% to 20%.1,9,10 The ACC/AHA guideline reports only one additional case of myopathy per 10,000 statin users compared with those on placebo and cites a rhabdomyolysis occurrence rate of less than 0.06% over five years.1
A 2006 systematic review estimated the absolute risk for rhabdomyolysis to be 3.4 per 100,000 person-years, but the incidence was 10 times higher for patients taking both a statin and gemfibrozil.11 (See Table 212,13 for more on drug interactions.) But both the meta-analysis cited earlier4 and a previous systematic review14 (35 RCTs and > 74,000 patients) found that statins as a class do not increase the incidence of myalgia or rhabdomyolysis.
Differences in the way muscular disorders are defined has been suggested as one reason for the discrepancies.10 In addition, many clinical trials exclude patients at higher risk for statin-associated AEs, such as those with renal or hepatic insufficiency, prior muscular complaints, poorly controlled diabetes, or potential drug interactions.1
An FDA advisory. In a safety communication last updated in February 2012, the FDA cautioned against starting patients on the highest dose of simvastatin (80 mg).15 The warning is based on a large study (N = 12,064) that found an increased risk for myopathy (0.9%) and rhabdomyolysis (0.2%) in patients on the
80-mg dose versus those taking 20 mg (0.02% and 0%, respectively).16
With the ACC/AHA now recommending intensive therapy (atorvastatin 40-80 mg or rosuvastatin 20-40 mg) to achieve an LDL reduction greater than 50% for many patients,1 it is important to be aware that this risk is specific to simvastatin. A recent meta-analysis of studies directly comparing patients receiving intensive statin therapy with those on low to moderate doses did not find any increased risk in rhabdomyolysis associated with more intensive therapy when those taking 80-mg simvastatin were excluded.17
The bottom line: Although rhabdomyolysis is rare, its severity—a fatality rate of 10%11—makes it critical to educate patients about the disorder and instruct them to stop taking the statin and call the office immediately if they develop severe muscle pain or weakness.
Recommend CoQ10 for statin-induced myopathy
Although the exact mechanism of statin-induced myopathy is unknown, the most likely explanation is a depletion of coenzyme Q10 (CoQ10), which has negative effects on mitochondrial energy production.18 While studies using CoQ10 to treat this AE have been small and had mixed results, the overall evidence suggests that it decreases the development and/or severity of symptoms.18-20
In fact, CoQ10 supplementation is the only treatment that has shown promise in treating statin-induced muscle symptoms.18-20 Doses of about 100 mg bid have been found to be beneficial and safe; no clinically relevant AEs have been seen with doses lower than 300 mg/d.18,20,21 A large placebo-controlled study is currently evaluating a 600 mg/d dose of CoQ10 in patients with statin-induced myopathy.19
CASE
On his next visit, Mr L. reports a new ache in his left shoulder and upper back, which he describes as mild but annoying. He also tells you his memory seems to be getting worse and that he has developed an odd tingling in his hands. These symptoms began about a month after he started the medications, Mr L. says. He also began a new exercise program, but his BMI is unchanged.
On examination, you find the affected shoulder and upper back modestly and diffusely tender to palpation but with no decline in strength. Mr L.’s BP has fallen to
134/84 mm Hg, and his fasting glucose is 105 mg/dL. Lab tests reveal an LDL of 144 mg/dL and HDL of 36 mg/dL, A1C of 6.1%, ALT of 105 U/L, AST of 61 U/L, and a normal CK.
You recommend 100 mg CoQ10 bid. Because it is available only OTC, you advise the patient to look for a product whose purity and potency have been verified by an external source, such as the US Pharmacopeial Convention. You also prescribe metformin 500 mg bid for insulin resistance, refer the patient to a nutritionist and diabetes specialist, and order tests to evaluate his other symptoms.
Continue for hepatic effects >>
HEPATIC EFFECTS ARE RARE
Historically, statins have been linked to potential hepatotoxicity, with case reports of serum transaminase elevation, cholestasis, hepatitis, and acute liver failure. It is now recognized that hepatic AEs are rare and that statins are not associated with a risk for acute or chronic liver failure.1,11 In patients with coronary heart disease, the incidence of hepatotoxicity with statin use is reported to be less than 1.5% over the course of five years and appears to be dose-dependent.1
In 2012, the FDA revised the labeling for most statins, relaxing its earlier recommendations for monitoring of liver function, clarifying the risk for myopathy, and providing additional information about drug interactions.13
Checking transaminase levels before initiating therapy is recommended by both the ACC/AHA and the FDA.1,13 Routine monitoring is not necessary, the ACC/AHA guideline states, because RCTs have found little evidence of ALT/AST elevation.1 But here, too, evidence varies. An older meta-analysis (13 trials and nearly 50,000 participants) concluded that as a class, statins have no greater risk for transaminase elevations than placebo.22 But the 135-RCT meta-analysis4 found otherwise: Statins did increase the risk for transaminase elevation (odds ratio [OR], 1.51) compared with placebo, with differences associated with particular drugs and higher doses associated with more clinically significant elevations.4 It is important to note, however, that there was significant heterogeneity among the studies and no consistent definition of clinical significance.
The bottom line: Statins have been shown in multiple prospective studies to be safe for patients with chronic liver disease.22,23
STATIN USE AND DIABETES: IS THERE A LINK?
Recent studies have found an increased risk for new-onset type 2 diabetes in statin users, with a greater risk associated with higher-potency statins, including rosuvastatin and atorvastatin.4,24 Although the exact mechanism is not known, statins may modify insulin signaling in peripheral tissues or directly impair insulin secretion.
The ACC/AHA guideline reports an excess rate of diabetes of one per 1,000 patient-years for moderate-intensity therapy and three per 1,000 patient-years for high-intensity therapy.1 The 2013 meta-analysis found that the elevated risk for diabetes was relatively small (OR, 1.09).4 No difference among various statins was found.
In another meta-analysis—this one encompassing 17 RCTs and more than 110,000 patients—no statistically significant difference in the incidence of new-onset diabetes was seen based on either the specific statin being taken or the intensity of therapy (high vs moderate).24
The bottom line: Clinicians should monitor patients taking statins for signs and symptoms of hyperglycemia.
STATINS MAY BE RENOPROTECTIVE
Statin use has been found to be associated with an increased risk for tubular proteinuria—an effect that is both dose- and potency-dependent.25 Nonetheless, it has been suggested that statins may be a rare example of a drug class that is renoprotective in the long term, despite having an increased rate of proteinuria in the short term.25
The evidence? In prospective studies, statin therapy has been shown to slow the progression of kidney disease in diverse patient populations, including renal transplant recipients and those with chronic kidney disease (CKD).26,27
The Kidney Expert Panel of the National Lipid Association (NLA) has concluded that statins do not appear to cause significant proteinuria or acute kidney injury. The panel does not recommend routine monitoring for proteinuria or kidney function in statin users unless otherwise indicated but does recommend a lower dose for patients with CKD.28
The bottom line: Kidney Disease Improving Global Outcomes guidelines recommend that patients who have CKD, but are not on dialysis, be treated with statin therapy. Statins are contraindicated for patients on dialysis, as clinical trials have failed to show significant cardiovascular benefit.29
Continue for the risk of intracerebral hemorrhages >>
INTRACEREBRAL HEMORRHAGE: STATINS INCREASE RECURRENCE RISK
In recent years, there has been considerable concern about a statin-induced increased risk for intracerebral hemorrhage (ICH). In a major prospective study in which patients were put on high-dose statin therapy or placebo after an acute ischemic or hemorrhagic stroke, the overall incidence of a recurrent stroke was significantly lower in the statin group.30 Among those who’d had an ICH, however, the recurrence rate was 73% higher for patients taking statins.
A subanalysis that looked only at patients who’d had a hemorrhagic stroke as their initial event (n = 93) found that the absolute risk for recurrent ICH was 15.6% for patients randomized to atorvastatin versus 4.2% for those on placebo.31 Despite being based on a small subset of the original study group, multivariate analysis indicated the increased risk was statistically significant (hazard ratio [HR], 1.69).
A subsequent decision analysis study based on these results proposed that patients with a history of spontaneous deep ICH would need an exceedingly high 10-year cardiovascular event risk (> 40%) for the benefits of statin therapy to outweigh the risk.32 The risk is particularly high for those with a history of lobar ICH, which has an extremely high recurrence rate. However, subsequent retrospective and observational studies have found that patients who were already on statins when the ICH occurred had less severe strokes and more favorable outcomes, with a lower mortality rate at 90 days post-ICH.33-35
A 2010 ICH guideline from the AHA/American Stroke Association states that there is “insufficient data to recommend restrictions on use of statin agents” for patients who have had an ICH.36
The bottom line: Clinicians should carefully evaluate the anticipated cardiovascular risk for patients who have had a hemorrhagic stroke to determine whether statin therapy would be beneficial.
OTHER SERIOUS ADVERSE EFFECTS: WHICH REPORTS ARE ACCURATE?
Statin use has been associated with a number of other serious AEs. Some reports appear to be accurate; others do not hold up after a close look at the evidence.
Malignancy. A potential link between statins and an increased risk for malignancy has been considered for years. A large trial (N = 5,804) from 2002 found a correlation between pravastatin and an increased risk for new cancer diagnoses compared with placebo (HR, 1.25).37 But a 10-year follow-up did not substantiate this finding, and it is now believed that the original result may have been due to chance.38 Numerous other meta-analyses and systematic reviews have found no link between statin use and malignancy.39-41
Cataracts. Potential ocular effects have been widely studied and debated in recent years. Observational studies reporting an association between statin use and cataracts have had conflicting results, with some showing statins as protective42-45 and others finding an increased risk.46,47 However, a recent propensity-score matched analysis found that statin users do indeed have an increased risk for cataracts.48 The authors concluded that for primary prevention, the risk-benefit equation for statin use should include this added risk.48
In addition, a review of the databases of the National Registry of Drug-Induced Ocular Side Effects, the World Health Organization, and the FDA from 1987 to 2008 indicates that statin therapy may also cause diplopia, ptosis, and ophthalmoplegia.49
Peripheral neuropathy. Despite case reports of statin-induced peripheral neuropathy, the NLA’s Neurology Expert Panel states that statins do not appear to cause this condition. If a patient receiving statin therapy develops peripheral neuropathy, a full work-up for other causes should be initiated before modification of statin therapy is considered, the panel advises.28
Statins have also been linked to headache and dizziness, respiratory symptoms, gastrointestinal problems, and rash (see Table 3).50
WHICH DRUG? POTENTIAL DIFFERENCES IN STATINS
A meta-analysis with more than 240,000 participants evaluated patients taking seven different statins (atorvastatin, fluvastatin, lovastatin, pravastatin, pitavastatin, rosuvastatin, and simvastatin), looking at AEs of the drugs both collectively and individually.4 As noted earlier, the overall discontinuation rate due to AEs for all statins was 5.7%. Discontinuation rates for each agent were not reported.4
The researchers did report, however, that atorvastatin and rosuvastatin had the highest discontinuation rates; atorvastatin and fluvastatin had the highest incidence of transaminase elevations (OR, 2.6 and 5.2, respectively); and pravastatin and simvastatin appeared to be the best-tolerated and safest statins, with the lowest discontinuation rates. However, higher doses of simvastatin (> 40 mg/d) significantly increased the risk for CK and transaminase elevations (OR, 4.1 and 2.8, respectively),4 as well as the risk for rhabdomyolysis when taken at the highest dose.15,16
Continue for safety concerns >>
ARE STATINS SAFE FOR THESE PATIENTS?
When considering statin therapy, there are some patient populations that warrant particular concern:
Women of childbearing age. Statins are contraindicated in women who are pregnant or breastfeeding1 and should not be initiated in women who are trying to conceive.
Children and adolescents (ages 8-18 years). Statins have been shown to be safe and effective for children and adolescents with familial hyperlipidemia. No effect on growth or maturation has been seen.51 As with adults, however, higher statin doses and the use of concomitant interacting drugs increase the risk for AEs.
Asians. The new ACC/AHA guideline suggests taking Asian ancestry into consideration when prescribing statins because Asians may be more sensitive to medications metabolized by the CYP450 system.1 However, there are no reports of an increased risk for AEs in Asian patients on statins.52
Patient factors that increase risk
Risk factors for statin-induced AEs include1
• Multiple and/or serious comorbidities (eg, hypothyroidism, impaired renal or hepatic function, rheumatic disorders)
• Unexplained ALT elevation more than 3x the upper limit of normal
• History of prior statin intolerance or concomitant use of drugs that affect statin metabolism
• Age older than 75
• Preexisting muscle disorders
• Low vitamin D levels.
If a patient who would clearly benefit from statin therapy develops an AE requiring discontinuation, a retrial—with the same drug or a different statin—is generally recommended once the symptoms resolve.1
CASE
The risk for elevated serum transaminases, insulin resistance, cognitive impairment, and neuropathy associated with statin use is minimal, and further evaluation revealed that Mr L.’s recent symptoms had other causes. The elevated transaminases were due to fatty liver disease, the cognitive impairment was secondary to sleep apnea (both linked to his obesity), and the tingling in his hands was the result of carpal tunnel syndrome caused by his exercise regimen.
When he returns in six months, Mr L. reports that he has been working with both a nutritionist and an athletic trainer. He has sustained a 15-lb weight loss. He is still taking atorvastatin 10 mg; after he began taking CoQ10, his muscle pain resolved. The patient’s cholesterol and transaminase levels are normal, and the cognitive impairment and peripheral neuropathy he reported at his last visit have improved significantly.
REFERENCES
1. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association task force on practice guidelines. Circulation. 2014;129:S1-S45.
2. Lowes R. Top 100 selling drugs through September reported. Medscape Med News. WebMD, LLC. 2013. www.medscape.com/viewarti cle/813571#3. Accessed October 19, 2014.
3. Zhang H, Plutzky J, Skentzos S, et al. Discontinuation of statins in routine care settings: a cohort study. Ann Intern Med. 2013;158:526-534.
4. Naci H, Brugts J, Ades T. Comparative tolerability and harms of individual statins: a study-level network meta-analysis of 246,955 participants from 135 randomized, controlled trials. Circ Cardiovasc Qual Outcomes. 2013;6:390-399.
5. Pasternak RC, Smith SC Jr, Bairey-Merz CN, et al; American College of Cardiology; American Heart Association; National Heart, Lung and Blood Institute. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. Circulation. 2002;106:1024-1028.
6. Eckel RH. Approach to the patient who is intolerant of statin therapy.
J Clin Endocrinol Metab. 2010;95:2015-2022.
7. Parker BA, Thompson PD. Effect of statins on skeletal muscle: exercise, myopathy, and muscle outcomes. Exerc Sport Sci Rev. 2012;40:188-194.
8. Mansi I, Frei CR, Pugh MJ, et al. Statins and musculoskeletal conditions, arthropathies, and injuries. JAMA Intern Med. 2013;173:1-10.
9. Bruckert E, Hayem G, Dejager S, et al. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—the PRIMO study. Cardiovasc Drugs Ther. 2005;19:403-414.
10. Fernandez G, Spatz ES, Jablecki C, et al. Statin myopathy: a common dilemma not reflected in clinical trials. Cleve Clin J Med. 2011;78:
393-403.
11. Law M, Rudnicka AR. Statin safety: a systematic review. Am J Cardiol. 2006;97:52C-60C.
12. Elsevier/Gold Standard. Gold Standard Drug Database. www.goldstand ard.com/product/gold-standard-drug-database/. Accessed October 19, 2014.
13. FDA. FDA drug safety communication: important safety label changes to cholesterol-lowering statin drugs. www.fda.gov/drugs/drugsafety/ucm293101.htm. Accessed October 19,2014.
14. Kashani A, Phillips CO, Foody JM, et al. Risks associated with statin therapy: a systematic overview of randomized clinical trials. Circulation. 2006;114:2788-2797.
15. FDA. FDA drug safety communication: ongoing safety review of high-dose Zocor (simvastatin) and increased risk of muscle injury. www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm204882.htm. Updated February 15, 2012. Accessed October 19, 2014.
16. Bowman L, Armitage J, Bulbulia R, et al; SEARCH Study Collaborative Group. Study of the effectiveness of additional reductions in cholesterol and homocysteine (SEARCH): characteristics of a randomized trial among 12064 myocardial infarction survivors. Am J Heart. 2007;154:815-823.
17. Mills EJ, O’Regan C, Eyawo O, et al. Intensive statin therapy compared with moderate dosing for prevention of cardiovascular events: a meta-analysis of >40,000 patients. Euro Heart J. 2011;32:1409-1415.
18. Bookstaver DA, Burkhalter NA, Hatzigeorgiou C. Effect of coenzyme Q10 supplementation on statin-induced myalgias. Am J Cardiol. 2012;110:
526-529.
19. Parker BA, Gregory SM, Lorson L, et al. A randomized trial of coenzyme Q10 in patients with statin myopathy: rationale and study design. J Clin Lipidol. 2013;7:187-193.
20. Fedacko J, Pella D, Fedackova P, et al. Coenzyme Q(10) and selenium in statin-associated myopathy treatment. Can J Physiol Pharmacol. 2013;91:165-170.
21. Jellin JM, Gregory PJ, et al. Natural Medicines Comprehensive Database. www.naturaldatabase.com.libproxy.uwyo.edu. Accessed October 19, 2014.
22. de Denus S, Spinler SA, Miller K, et al. Statins and liver toxicity: a meta-analysis. Pharmacotherapy. 2004;24:584-591.
23. Lewis JH. Clinical perspective: statins and the liver—harmful or helpful? Dig Dis Sci. 2012;57:1754-1763.
24. Navarese EP, Buffon A, Andreotti F, et al. Meta-analysis of impact of different types and doses of statins on new-onset diabetes mellitus. Am J Cardiol. 2013;111:1123-1130.
25. Agarwal R. Effects of statins on renal function. Am J Cardiol. 2006;97:748-755.
26. Fried LF, Orchard TJ, Lasiske BL. Effect of lipid reduction on the progression of renal disease: a meta-analysis. Kidney Int. 2001;59:260-269.
27. Fellström B, Holdaas H, Jardine AG, et al; Assessment of Lescol in Renal Transportation Study Investigators. Effect of fluvastatin on renal end points in the Assessment of Lescol in Renal Transplant (ALERT) Trial. Kidney Int. 2004;66:1549-1555.
28. McKenney JM, Davidson MH, Jacobson TA, et al; National Lipid Association Statin Safety Assessment Task Force. Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am J Cardiol. 2006;97:89C-94C.
29. KDIGO Clinical Practice Guideline for Lipid Management in Chronic Kidney Disease. Kidney Int. 2013;3(suppl):S259-S305.
30. Goldstein LB, Amarenco P, Szarek M, et al; SPARCL Investigators. Hemorrhagic stroke in the Stroke Prevention by Aggressive Reduction in Cholesterol Levels study. Neurology. 2008;70(24 pt 2):2364-2370.
31. Goldstein LB, Amarenco P, Lamonte M, et al; SPARCL investigators. Relative effects of statin therapy on stroke and cardiovascular events in men and women: secondary analysis of the Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) Study. Stroke. 2008;39:
2444-2448.
32. Westover MB, Bianchi MT, Eckman MH, et al. Statin use following intracerebral hemorrhage: a decision analysis. Arch Neurol. 2011;68:573-579.
33. Biffi A, Devan WJ, Anderson CD, et al. Statin use and outcome after intracerebral hemorrhage: case-control study and meta-analysis. Neurology. 2011;76:1581-1588.
34. Dowlatshahi D, Demchuck AM, Fang J, et al; Registry of the Canadian Stroke Network. Association of statins and statin discontinuation with poor outcome and survival after intracerebral hemorrhage. Stroke. 2012;43:1518-1523.
35. Bustamante A, Montaner J. Statin therapy should not be discontinued in patients with intracerebral hemorrhage. Stroke. 2013;44:2060-2061.
36. Morgenstern LB, Hemphill JC 3rd, Anderson C, et al; American Heart Association Stroke Council and Council on Cardiovascular Nursing. Guidelines for the management of spontaneous intracerebral hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2010;41:2108-2129.
37. Shepherd J, Blauw GJ, Murphy MB, et al; PROSPER study group. PROspective Study of Pravastatin in the Elderly at Risk. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet. 2002;360:1623-1630.
38. Jukema JW, Cannon CP, de Craen AJ, et al. The controversies of statin therapy: weighing the evidence. J Am Coll Cardiol. 2012;60:875-881.
39. Alberton M, Wu P, Druyts E, et al. Adverse events associated with individual statin treatments for cardiovascular disease: an indirect comparison meta-analysis. QJM. 2012;105:145-157.
40. Baigent C, Blackwell L, Emberson J, et al; Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670-1681.
41. Emberson JR, Kearney PM, Blackwell L, et al; Cholesterol Treatment Trialists’ (CTT) Collaboration. Lack of effect of lowering LDL cholesterol on cancer: meta-analysis of individual data from 175,000 people in 27 randomised trials of statin therapy. PLoS One. 2012;7:e29849.
42. Klein BE, Klein R, Lee KE, et al. Statin use and incident nuclear cataract. JAMA. 2006;295:2752-2758.
43. Fong DS, Poon KY. Recent statin use and cataract surgery. Am J Ophthalmol. 2012;153:222-228.e1.
44. Chodick G, Heymann AD, Flash S, et al. Persistence with statins and incident cataract: a population-based historical cohort study. Ann Epidemiol. 2010;20:136-142.
45. Tan JS, Mitchell P, Rochtchina E, et al. Statin use and the long-term risk of incident cataract: the Blue Mountains Eye Study. Am J Ophthalmol. 2007;143:687-689.
46. Machan CM, Hrynchak PK, Irving EL. Age-related cataract is associated with type 2 diabetes and statin use. Optom Vis Sci. 2012;89:1165-1171.
47. Hippisley-Cox J, Coupland C. Unintended effects of statins in men and women in England and Wales: population based cohort study using the QResearch database. BMJ. 2010;340:c2197.
48. Leuschen J, Mortensen EM, Frei CR, et al. Association of statin use with cataracts: a propensity score-matched analysis. JAMA Ophthalmol. 2013;131:1427-1434.
49. Fraunfelder FW, Richards AB. Diplopia, blepharoptosis, and ophthalmoplegia and 3-hydroxy-3-methyl-glutaryl-CoA reductase inhibitor use. Ophthalmology. 2008;115:2282-2285.
50. AHFS Drug Information 2013. Bethesda, MD: American Society of Health-System Pharmacists; 2013.
51. Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents; National Heart, Lung, and Blood Institute. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics. 2011;128(suppl 5):S213-S256.
52. Liao JK. Safety and efficacy of statins in Asians. Am J Cardiol. 2007;99:410-414.
Woman, 66, With Persistent Abdominal and Back Pain
A 66-year-old Latin American woman presented to the emergency department (ED) with persistent abdominal and back pain of about one month’s duration. She had visited another ED eight days earlier for similar symptoms and was discharged home with a mild opioid pain medication and a proton pump inhibitor. However, she said that she had received neither a diagnosis nor an explanation for her symptoms.
Medical history, obtained with the assistance of an interpreter because the patient was not fluent in English, included hypertension, coronary artery disease, and hyperlipidemia; these had gone untreated for at least two years. She denied any personal or family history of cancer or endocrine disorders. Surgical history included a cholecystectomy and a percutaneous coronary intervention for an unknown coronary artery lesion.
She had a 14-pack-year history of cigarette smoking. Her medications included only ibuprofen and hydrocodone, and she had no known drug allergies. The patient denied use of herbal preparations or vitamin supplements and unusual dietary practices.
Review of systems revealed occasional dizziness, constipation, decreased appetite, and some mild confusion noted by family members, but no fever, chills, palpitations, chest pain, shortness of breath, muscle spasm, or weakness. Vital signs were normal. Physical examination was remarkable for tenderness of the upper quadrants of the abdomen with deep palpation, without guarding or rebound. Bony tenderness at the right anterior costal margin of the rib cage was also noted.
Laboratory work-up revealed marked hypercalcemia (15.4 mg/dL), electrolyte abnormalities, anemia, impaired renal function, and elevated alkaline phosphatase and globulin levels (see Table 1). In addition, a plain abdominal x-ray series was negative for acute findings, but x-rays of the right ribs revealed a fracture of the sixth rib and osteopenia.

Continued >>
The patient was admitted to the hospital for treatment of hypercalcemia and hypokalemia and for work-up of elevated alkaline phosphatase and abdominal pain. Upon admission, serum ionized calcium measurement confirmed true hypercalcemia. Additional diagnostic tests were then ordered to help differentiate between parathyroid hormone (PTH)–mediated and non-PTH–mediated causes for the hypercalcemia (see Table 2).
The patient’s PTH level was normal and the urine fractional excretion of calcium level was high, ruling out familial hypocalciuric hypercalcemia (FHH), in which urine calcium level is low. A measurement of PTH-related protein (PTHrP), secreted by some cancers, was normal, suggesting exclusion of solid tumor malignancy. Vitamin D toxicity was ruled out because the patient’s 1,25-dihydroxyvitamin D level was low.
The patient continued to experience vague abdominal, back, and rib pain that seemed to migrate daily and worsened with movement. A skeletal x-ray was performed and revealed numerous lytic lesions of the skull (Figure 1), midright humerus (Figure 2), and distal left radius.
Continue for discussion >>
DISCUSSION
Hypercalcemia is a relatively common presentation in primary care. The most frequent causes are primary hyperparathyroidism and malignancy.1 One in 500 patients will be diagnosed incidentally with asymptomatic hypercalcemia caused by underlying hyperparathyroidism.1
Clinical manifestations of hypercalcemia can range from no symptoms to multisystem disease. Fatigue, nausea, vomiting, constipation, bone pain, osteoporosis, nephrolithiasis, mental status changes, hypertension, anemia, elevated creatinine, and cardiac arrhythmias are among the more common clinical conditions associated with hypercalcemia (hence the mnemonic “stones, bones, abdominal moans, and psychic groans” for its signs and symptoms).1
Diagnostic overview
Causes of hypercalcemia are numerous and can be broken down into two categories: PTH-mediated and non-PTH–mediated. PTH-mediated causes include primary and secondary hyperparathyroidism and FHH. Non-PTH–mediated causes include vitamin D toxicity, solid tumor malignancy with or without metastasis, multiple myeloma (MM) and other plasma cell dyscrasias, granulomatous disease such as sarcoid, and some medications.1
The differential diagnosis for hypercalcemia begins with measurement of the patient’s intact PTH level. An elevated or high-normal result indicates a PTH-mediated cause, so 24-hour measurement of excretion of urinary calcium is the next step. A low or low-normal PTH level (< 20 pg/mL), however, suggests the cause is non-PTH-mediated.2 The diagnostic approach in this situation is more challenging because testing to exclude or confirm various potential causes can be expensive and time-consuming. The degree of hypercalcemia, however, can aid in the diagnosis: Primary hyperparathyroidism is often associated with borderline or mild hypercalcemia, while calcium values > 13 mg/dL are more common in patients with malignancies.2
PTH elevates calcium levels in the blood when ionized (free) calcium levels are low by increasing gastrointestinal absorption, decreasing urinary excretion, and increasing bone resorption.3 With malignant tumors such as lung, breast, and renal cell, osteolytic metastases can destroy the bone, resulting in release of calcium. In other cases, solid-tumor cancers produce PTHrP, which increases serum calcium. This latter situation is referred to as humoral hypercalcemia of malignancy.3 Lymphoma and granulomatous disease, such as sarcoid, can be associated with excess production of 1,25-dihydroxyvitamin D.2 If vitamin D is elevated but PTH and PTHrP are normal, a chest x-ray should be obtained to evaluate the patient for sarcoid or lymphoma.
Hypercalcemia work-up
The work-up for suspected hypercalcemia begins with measurement of the patient’s calcium level. Because calcium is bound to albumin in the blood, the standard serum calcium test may not reflect the true calcium level. (If albumin is high, the calcium level will be high, and vice versa.)1 The true (serum ionized) calcium level (also known as corrected calcium level) should always be calculated to confirm true hypercalcemia. A formula commonly used to calculate the corrected calcium level is
Corrected calcium (mg/dL) = (measured calcium [mg/dL]) + 0.8 (4.0 – serum albumin [mg/dL])
Direct measurement of the serum ionized calcium level is not affected by the albumin level and can also confirm true hypercalcemia.4
Once hypercalcemia is confirmed, the next step is to measure the patient’s intact PTH level.
If intact PTH is elevated or high normal, consider primary hyperparathyroidism or FHH and confirm by obtaining the urine calcium level.
• If urine calcium level is high (> 200 mg/24 h), the diagnosis is primary hyperparathyroidism.
• If urine calcium level is low (< 100 mg/24 h), the diagnosis is FHH.
If intact PTH is low, consider non–PTH-mediated causes and confirm by obtaining PTHrP and vitamin D levels.
• If PTHrP level is elevated, scan for malignancy.
• If 1,25-dihydroxyvitamin D level is elevated, order a chest x-ray to rule out sarcoid or lymphoma.
• If both PTHrP and vitamin D levels are normal, order both serum protein electrophoresis (SPEP) and urine protein electrophoresis (UPEP) with immunofixation to rule out MM.
• If vitamin D level is elevated, check vitamin and herbal supplement use for excessive vitamin D intake.
Treatment of symptomatic hypercalcemia
The goals of treatment of symptomatic hypercalcemia are to reduce the serum calcium level to the normal range and to treat the underlying cause.1 Mild hypercalcemia (calcium level, 10-12 mg/dL) is typically asymptomatic and does not need to be treated. Moderate hypercalcemia (calcium level, 12-14 mg/dL) may not require treatment unless the patient is symptomatic and/or has had an acute rise in calcium level.1 In mild to moderate hypercalcemia, the serum calcium level should be monitored to establish a trend.
Treatment for symptomatic moderate and severe hypercalcemia (calcium level, > 14 mg/dL) typically involves a similar regimen:
• Volume expansion with isotonic saline at an initial rate of 2 to 4 L/d, which is then adjusted to achieve 200 mL/h of continuous urine output. IV furosemide can be used with caution (10-20 mg IV as needed) to promote diuresis if volume overload is a concern (furosemide promotes renal excretion of calcium).
• Administration of subcutaneous calcitonin (4-8 IU/kg, repeated every 6 h for 24 h). Calcitonin works rapidly to lower calcium levels in 4 to 6 h.
• Concurrent administration of IV bisphosphonate (zoledronic acid [4 mg over 15 min] or pamidronate [60-90 mg over 4 h]). Pamidronate is superior for reversal of malignancy-related hypercalcemia.1
Hypercalcemia and multiple myeloma
MM is a malignant neoplasm of plasma cells that accounts for approximately 1% of all cancers and about 10% of hematologic malignancies in the United States, with a median patient age of 70 at diagnosis.5-7 In MM, myeloma cells induce the secretion of cytokines and growth factors that alter plasma cells, activate osteoclasts, suppress osteoblasts, cause abnormal interactions between plasma cells and bone marrow, and stimulate aberrant angiogenesis.8 Osteoclastic bone resorption produces hypercalcemia as well as the lytic lesions seen on x-ray.7
Approximately 74% of patients present with typical MM symptoms of calcium elevation in the blood, renal insufficiency, anemia, and bone lesions, known as CRAB symptoms, but other myeloma-related manifestations may be present.9
Diagnostic criteria for MM include the following (all three must be present):10
• Monoclonal bone marrow plasma cells ≥ 10% and/or a biopsy-proven plasmacytoma
• Monoclonal protein in the serum and/or urine (if none is detected, disease is nonsecretory and diagnosis requires ≥ 30% bone marrow plasma cells and/or biopsy-proven plasmacytoma)
• Myeloma-related organ dysfunction, indicated by at least one of the CRAB symptoms.
In the absence of CRAB symptoms, an asymptomatic patient may have an MM precursor syndrome: monoclonal gammopathy of undetermined significance or smoldering (or indolent) MM.10
Treatment of multiple myeloma
In recent years, the use of induction therapy followed by autologous stem cell transplantation and the development of novel therapeutic agents have extended overall survival for patients with MM. These agents include proteasome inhibitors (bortezomib and the second-generation carfilzomib)11 and immunomodulators (thalidomide and the second-generation lenalidomide). Early diagnosis and treatment can improve progression-free survival as well as overall survival, including recovery of renal function for patients with renal failure.12 With survival ranging from one year or less—with aggressive disease—to 10 years or more for patients with responsive disease,7 there remains no cure for MM.
Outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
Further work-up included SPEP and UPEP with immunofixation, which revealed marked IgG free λ light chains with an M (monoclonal) component, making MM a very likely diagnosis. Confirmation by means of bone marrow biopsy was indicated, but the patient refused the procedure.
The patient’s hypercalcemia was treated by IV administration of calcitonin with isotonic saline. This reduced the serum calcium level from 15.4 mg/dL to 10.6 mg/dL within 48 hours. One dose of ergocalciferol (vitamin D2) was then administered to promote intestinal absorption of calcium and support bone mineralization, further lowering the patient’s serum calcium level to a normal 8.9 mg/dL. Hypokalemia was treated with oral potassium supplementation.
The patient, now stable, was referred to the hematology/oncology and bone mineral metabolism clinics and was discharged from the hospital. She did not keep those appointments and was lost to follow-up.
CONCLUSION
The most common causes of hypercalcemia are hyperparathyroidism and malignancy. Most cases do not require treatment unless the calcium level is >14 mg/dL and/or the patient is symptomatic. Red flag symptoms include weakness, abdominal pain, mental status changes, and coma.4 Primary care clinicians should suspect MM in older patients with laboratory findings of hypercalcemia, anemia, and renal dysfunction, with lytic lesions on x-ray.
REFERENCES
1. Carroll MF, Schade DS. A practical approach to hypercalcemia. Am Fam Physician. 2003;67(9):1959-1966.
2. Endres DB. Investigation of hypercalcemia. Clin Biochem. 2012;45(12):
954-963.
3. Moe SM. Disorders involving calcium, phosphorus, and magnesium. Prim Care. 2008;35(2):215-237.
4. Sharma B, Misicko NE. How should you evaluate elevated calcium in an asymptomatic patient? J Fam Pract. 2008;57(4):267-269.
5. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11-30.
6. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33.
7. Shaheen SP, Talwalkar SS, Medeiros LJ. Multiple myeloma and immunosecretory disorders: an update. Adv Anat Pathol. 2008;15(4):196-210.
8. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11): 1046-1060.
9. Talamo G, Farooq U, Zangari M, et al. Beyond the CRAB symptoms: a study of presenting clinical manifestations of multiple myeloma. Clin Lymphoma Myeloma Leuk. 2010;10(6):464-468.
10. Palumbo A, Sezer O, Kyle R, et al. International Myeloma Working Group guidelines for the management of multiple myeloma patients ineligible for standard high-dose chemotherapy with autologous stem cell transportation. Leukemia. 2009;23(10):1716-1730.
11. Kyprolis [package insert]. South San Francisco, CA: Onyx Pharmaceuticals, Inc; 2012.
12. Suyani E, Sucak GT, Erten Y, et al. Evaluation of multiple myeloma patients presenting with renal failure in a university hospital in the year 2010. Ren Fail. 2012;34(2):257-262.
A 66-year-old Latin American woman presented to the emergency department (ED) with persistent abdominal and back pain of about one month’s duration. She had visited another ED eight days earlier for similar symptoms and was discharged home with a mild opioid pain medication and a proton pump inhibitor. However, she said that she had received neither a diagnosis nor an explanation for her symptoms.
Medical history, obtained with the assistance of an interpreter because the patient was not fluent in English, included hypertension, coronary artery disease, and hyperlipidemia; these had gone untreated for at least two years. She denied any personal or family history of cancer or endocrine disorders. Surgical history included a cholecystectomy and a percutaneous coronary intervention for an unknown coronary artery lesion.
She had a 14-pack-year history of cigarette smoking. Her medications included only ibuprofen and hydrocodone, and she had no known drug allergies. The patient denied use of herbal preparations or vitamin supplements and unusual dietary practices.
Review of systems revealed occasional dizziness, constipation, decreased appetite, and some mild confusion noted by family members, but no fever, chills, palpitations, chest pain, shortness of breath, muscle spasm, or weakness. Vital signs were normal. Physical examination was remarkable for tenderness of the upper quadrants of the abdomen with deep palpation, without guarding or rebound. Bony tenderness at the right anterior costal margin of the rib cage was also noted.
Laboratory work-up revealed marked hypercalcemia (15.4 mg/dL), electrolyte abnormalities, anemia, impaired renal function, and elevated alkaline phosphatase and globulin levels (see Table 1). In addition, a plain abdominal x-ray series was negative for acute findings, but x-rays of the right ribs revealed a fracture of the sixth rib and osteopenia.
Continued >>
The patient was admitted to the hospital for treatment of hypercalcemia and hypokalemia and for work-up of elevated alkaline phosphatase and abdominal pain. Upon admission, serum ionized calcium measurement confirmed true hypercalcemia. Additional diagnostic tests were then ordered to help differentiate between parathyroid hormone (PTH)–mediated and non-PTH–mediated causes for the hypercalcemia (see Table 2).
The patient’s PTH level was normal and the urine fractional excretion of calcium level was high, ruling out familial hypocalciuric hypercalcemia (FHH), in which urine calcium level is low. A measurement of PTH-related protein (PTHrP), secreted by some cancers, was normal, suggesting exclusion of solid tumor malignancy. Vitamin D toxicity was ruled out because the patient’s 1,25-dihydroxyvitamin D level was low.
The patient continued to experience vague abdominal, back, and rib pain that seemed to migrate daily and worsened with movement. A skeletal x-ray was performed and revealed numerous lytic lesions of the skull (Figure 1), midright humerus (Figure 2), and distal left radius.
Continue for discussion >>
DISCUSSION
Hypercalcemia is a relatively common presentation in primary care. The most frequent causes are primary hyperparathyroidism and malignancy.1 One in 500 patients will be diagnosed incidentally with asymptomatic hypercalcemia caused by underlying hyperparathyroidism.1
Clinical manifestations of hypercalcemia can range from no symptoms to multisystem disease. Fatigue, nausea, vomiting, constipation, bone pain, osteoporosis, nephrolithiasis, mental status changes, hypertension, anemia, elevated creatinine, and cardiac arrhythmias are among the more common clinical conditions associated with hypercalcemia (hence the mnemonic “stones, bones, abdominal moans, and psychic groans” for its signs and symptoms).1
Diagnostic overview
Causes of hypercalcemia are numerous and can be broken down into two categories: PTH-mediated and non-PTH–mediated. PTH-mediated causes include primary and secondary hyperparathyroidism and FHH. Non-PTH–mediated causes include vitamin D toxicity, solid tumor malignancy with or without metastasis, multiple myeloma (MM) and other plasma cell dyscrasias, granulomatous disease such as sarcoid, and some medications.1
The differential diagnosis for hypercalcemia begins with measurement of the patient’s intact PTH level. An elevated or high-normal result indicates a PTH-mediated cause, so 24-hour measurement of excretion of urinary calcium is the next step. A low or low-normal PTH level (< 20 pg/mL), however, suggests the cause is non-PTH-mediated.2 The diagnostic approach in this situation is more challenging because testing to exclude or confirm various potential causes can be expensive and time-consuming. The degree of hypercalcemia, however, can aid in the diagnosis: Primary hyperparathyroidism is often associated with borderline or mild hypercalcemia, while calcium values > 13 mg/dL are more common in patients with malignancies.2
PTH elevates calcium levels in the blood when ionized (free) calcium levels are low by increasing gastrointestinal absorption, decreasing urinary excretion, and increasing bone resorption.3 With malignant tumors such as lung, breast, and renal cell, osteolytic metastases can destroy the bone, resulting in release of calcium. In other cases, solid-tumor cancers produce PTHrP, which increases serum calcium. This latter situation is referred to as humoral hypercalcemia of malignancy.3 Lymphoma and granulomatous disease, such as sarcoid, can be associated with excess production of 1,25-dihydroxyvitamin D.2 If vitamin D is elevated but PTH and PTHrP are normal, a chest x-ray should be obtained to evaluate the patient for sarcoid or lymphoma.
Hypercalcemia work-up
The work-up for suspected hypercalcemia begins with measurement of the patient’s calcium level. Because calcium is bound to albumin in the blood, the standard serum calcium test may not reflect the true calcium level. (If albumin is high, the calcium level will be high, and vice versa.)1 The true (serum ionized) calcium level (also known as corrected calcium level) should always be calculated to confirm true hypercalcemia. A formula commonly used to calculate the corrected calcium level is
Corrected calcium (mg/dL) = (measured calcium [mg/dL]) + 0.8 (4.0 – serum albumin [mg/dL])
Direct measurement of the serum ionized calcium level is not affected by the albumin level and can also confirm true hypercalcemia.4
Once hypercalcemia is confirmed, the next step is to measure the patient’s intact PTH level.
If intact PTH is elevated or high normal, consider primary hyperparathyroidism or FHH and confirm by obtaining the urine calcium level.
• If urine calcium level is high (> 200 mg/24 h), the diagnosis is primary hyperparathyroidism.
• If urine calcium level is low (< 100 mg/24 h), the diagnosis is FHH.
If intact PTH is low, consider non–PTH-mediated causes and confirm by obtaining PTHrP and vitamin D levels.
• If PTHrP level is elevated, scan for malignancy.
• If 1,25-dihydroxyvitamin D level is elevated, order a chest x-ray to rule out sarcoid or lymphoma.
• If both PTHrP and vitamin D levels are normal, order both serum protein electrophoresis (SPEP) and urine protein electrophoresis (UPEP) with immunofixation to rule out MM.
• If vitamin D level is elevated, check vitamin and herbal supplement use for excessive vitamin D intake.
Treatment of symptomatic hypercalcemia
The goals of treatment of symptomatic hypercalcemia are to reduce the serum calcium level to the normal range and to treat the underlying cause.1 Mild hypercalcemia (calcium level, 10-12 mg/dL) is typically asymptomatic and does not need to be treated. Moderate hypercalcemia (calcium level, 12-14 mg/dL) may not require treatment unless the patient is symptomatic and/or has had an acute rise in calcium level.1 In mild to moderate hypercalcemia, the serum calcium level should be monitored to establish a trend.
Treatment for symptomatic moderate and severe hypercalcemia (calcium level, > 14 mg/dL) typically involves a similar regimen:
• Volume expansion with isotonic saline at an initial rate of 2 to 4 L/d, which is then adjusted to achieve 200 mL/h of continuous urine output. IV furosemide can be used with caution (10-20 mg IV as needed) to promote diuresis if volume overload is a concern (furosemide promotes renal excretion of calcium).
• Administration of subcutaneous calcitonin (4-8 IU/kg, repeated every 6 h for 24 h). Calcitonin works rapidly to lower calcium levels in 4 to 6 h.
• Concurrent administration of IV bisphosphonate (zoledronic acid [4 mg over 15 min] or pamidronate [60-90 mg over 4 h]). Pamidronate is superior for reversal of malignancy-related hypercalcemia.1
Hypercalcemia and multiple myeloma
MM is a malignant neoplasm of plasma cells that accounts for approximately 1% of all cancers and about 10% of hematologic malignancies in the United States, with a median patient age of 70 at diagnosis.5-7 In MM, myeloma cells induce the secretion of cytokines and growth factors that alter plasma cells, activate osteoclasts, suppress osteoblasts, cause abnormal interactions between plasma cells and bone marrow, and stimulate aberrant angiogenesis.8 Osteoclastic bone resorption produces hypercalcemia as well as the lytic lesions seen on x-ray.7
Approximately 74% of patients present with typical MM symptoms of calcium elevation in the blood, renal insufficiency, anemia, and bone lesions, known as CRAB symptoms, but other myeloma-related manifestations may be present.9
Diagnostic criteria for MM include the following (all three must be present):10
• Monoclonal bone marrow plasma cells ≥ 10% and/or a biopsy-proven plasmacytoma
• Monoclonal protein in the serum and/or urine (if none is detected, disease is nonsecretory and diagnosis requires ≥ 30% bone marrow plasma cells and/or biopsy-proven plasmacytoma)
• Myeloma-related organ dysfunction, indicated by at least one of the CRAB symptoms.
In the absence of CRAB symptoms, an asymptomatic patient may have an MM precursor syndrome: monoclonal gammopathy of undetermined significance or smoldering (or indolent) MM.10
Treatment of multiple myeloma
In recent years, the use of induction therapy followed by autologous stem cell transplantation and the development of novel therapeutic agents have extended overall survival for patients with MM. These agents include proteasome inhibitors (bortezomib and the second-generation carfilzomib)11 and immunomodulators (thalidomide and the second-generation lenalidomide). Early diagnosis and treatment can improve progression-free survival as well as overall survival, including recovery of renal function for patients with renal failure.12 With survival ranging from one year or less—with aggressive disease—to 10 years or more for patients with responsive disease,7 there remains no cure for MM.
Outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
Further work-up included SPEP and UPEP with immunofixation, which revealed marked IgG free λ light chains with an M (monoclonal) component, making MM a very likely diagnosis. Confirmation by means of bone marrow biopsy was indicated, but the patient refused the procedure.
The patient’s hypercalcemia was treated by IV administration of calcitonin with isotonic saline. This reduced the serum calcium level from 15.4 mg/dL to 10.6 mg/dL within 48 hours. One dose of ergocalciferol (vitamin D2) was then administered to promote intestinal absorption of calcium and support bone mineralization, further lowering the patient’s serum calcium level to a normal 8.9 mg/dL. Hypokalemia was treated with oral potassium supplementation.
The patient, now stable, was referred to the hematology/oncology and bone mineral metabolism clinics and was discharged from the hospital. She did not keep those appointments and was lost to follow-up.
CONCLUSION
The most common causes of hypercalcemia are hyperparathyroidism and malignancy. Most cases do not require treatment unless the calcium level is >14 mg/dL and/or the patient is symptomatic. Red flag symptoms include weakness, abdominal pain, mental status changes, and coma.4 Primary care clinicians should suspect MM in older patients with laboratory findings of hypercalcemia, anemia, and renal dysfunction, with lytic lesions on x-ray.
REFERENCES
1. Carroll MF, Schade DS. A practical approach to hypercalcemia. Am Fam Physician. 2003;67(9):1959-1966.
2. Endres DB. Investigation of hypercalcemia. Clin Biochem. 2012;45(12):
954-963.
3. Moe SM. Disorders involving calcium, phosphorus, and magnesium. Prim Care. 2008;35(2):215-237.
4. Sharma B, Misicko NE. How should you evaluate elevated calcium in an asymptomatic patient? J Fam Pract. 2008;57(4):267-269.
5. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11-30.
6. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33.
7. Shaheen SP, Talwalkar SS, Medeiros LJ. Multiple myeloma and immunosecretory disorders: an update. Adv Anat Pathol. 2008;15(4):196-210.
8. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11): 1046-1060.
9. Talamo G, Farooq U, Zangari M, et al. Beyond the CRAB symptoms: a study of presenting clinical manifestations of multiple myeloma. Clin Lymphoma Myeloma Leuk. 2010;10(6):464-468.
10. Palumbo A, Sezer O, Kyle R, et al. International Myeloma Working Group guidelines for the management of multiple myeloma patients ineligible for standard high-dose chemotherapy with autologous stem cell transportation. Leukemia. 2009;23(10):1716-1730.
11. Kyprolis [package insert]. South San Francisco, CA: Onyx Pharmaceuticals, Inc; 2012.
12. Suyani E, Sucak GT, Erten Y, et al. Evaluation of multiple myeloma patients presenting with renal failure in a university hospital in the year 2010. Ren Fail. 2012;34(2):257-262.
A 66-year-old Latin American woman presented to the emergency department (ED) with persistent abdominal and back pain of about one month’s duration. She had visited another ED eight days earlier for similar symptoms and was discharged home with a mild opioid pain medication and a proton pump inhibitor. However, she said that she had received neither a diagnosis nor an explanation for her symptoms.
Medical history, obtained with the assistance of an interpreter because the patient was not fluent in English, included hypertension, coronary artery disease, and hyperlipidemia; these had gone untreated for at least two years. She denied any personal or family history of cancer or endocrine disorders. Surgical history included a cholecystectomy and a percutaneous coronary intervention for an unknown coronary artery lesion.
She had a 14-pack-year history of cigarette smoking. Her medications included only ibuprofen and hydrocodone, and she had no known drug allergies. The patient denied use of herbal preparations or vitamin supplements and unusual dietary practices.
Review of systems revealed occasional dizziness, constipation, decreased appetite, and some mild confusion noted by family members, but no fever, chills, palpitations, chest pain, shortness of breath, muscle spasm, or weakness. Vital signs were normal. Physical examination was remarkable for tenderness of the upper quadrants of the abdomen with deep palpation, without guarding or rebound. Bony tenderness at the right anterior costal margin of the rib cage was also noted.
Laboratory work-up revealed marked hypercalcemia (15.4 mg/dL), electrolyte abnormalities, anemia, impaired renal function, and elevated alkaline phosphatase and globulin levels (see Table 1). In addition, a plain abdominal x-ray series was negative for acute findings, but x-rays of the right ribs revealed a fracture of the sixth rib and osteopenia.
Continued >>
The patient was admitted to the hospital for treatment of hypercalcemia and hypokalemia and for work-up of elevated alkaline phosphatase and abdominal pain. Upon admission, serum ionized calcium measurement confirmed true hypercalcemia. Additional diagnostic tests were then ordered to help differentiate between parathyroid hormone (PTH)–mediated and non-PTH–mediated causes for the hypercalcemia (see Table 2).
The patient’s PTH level was normal and the urine fractional excretion of calcium level was high, ruling out familial hypocalciuric hypercalcemia (FHH), in which urine calcium level is low. A measurement of PTH-related protein (PTHrP), secreted by some cancers, was normal, suggesting exclusion of solid tumor malignancy. Vitamin D toxicity was ruled out because the patient’s 1,25-dihydroxyvitamin D level was low.
The patient continued to experience vague abdominal, back, and rib pain that seemed to migrate daily and worsened with movement. A skeletal x-ray was performed and revealed numerous lytic lesions of the skull (Figure 1), midright humerus (Figure 2), and distal left radius.
Continue for discussion >>
DISCUSSION
Hypercalcemia is a relatively common presentation in primary care. The most frequent causes are primary hyperparathyroidism and malignancy.1 One in 500 patients will be diagnosed incidentally with asymptomatic hypercalcemia caused by underlying hyperparathyroidism.1
Clinical manifestations of hypercalcemia can range from no symptoms to multisystem disease. Fatigue, nausea, vomiting, constipation, bone pain, osteoporosis, nephrolithiasis, mental status changes, hypertension, anemia, elevated creatinine, and cardiac arrhythmias are among the more common clinical conditions associated with hypercalcemia (hence the mnemonic “stones, bones, abdominal moans, and psychic groans” for its signs and symptoms).1
Diagnostic overview
Causes of hypercalcemia are numerous and can be broken down into two categories: PTH-mediated and non-PTH–mediated. PTH-mediated causes include primary and secondary hyperparathyroidism and FHH. Non-PTH–mediated causes include vitamin D toxicity, solid tumor malignancy with or without metastasis, multiple myeloma (MM) and other plasma cell dyscrasias, granulomatous disease such as sarcoid, and some medications.1
The differential diagnosis for hypercalcemia begins with measurement of the patient’s intact PTH level. An elevated or high-normal result indicates a PTH-mediated cause, so 24-hour measurement of excretion of urinary calcium is the next step. A low or low-normal PTH level (< 20 pg/mL), however, suggests the cause is non-PTH-mediated.2 The diagnostic approach in this situation is more challenging because testing to exclude or confirm various potential causes can be expensive and time-consuming. The degree of hypercalcemia, however, can aid in the diagnosis: Primary hyperparathyroidism is often associated with borderline or mild hypercalcemia, while calcium values > 13 mg/dL are more common in patients with malignancies.2
PTH elevates calcium levels in the blood when ionized (free) calcium levels are low by increasing gastrointestinal absorption, decreasing urinary excretion, and increasing bone resorption.3 With malignant tumors such as lung, breast, and renal cell, osteolytic metastases can destroy the bone, resulting in release of calcium. In other cases, solid-tumor cancers produce PTHrP, which increases serum calcium. This latter situation is referred to as humoral hypercalcemia of malignancy.3 Lymphoma and granulomatous disease, such as sarcoid, can be associated with excess production of 1,25-dihydroxyvitamin D.2 If vitamin D is elevated but PTH and PTHrP are normal, a chest x-ray should be obtained to evaluate the patient for sarcoid or lymphoma.
Hypercalcemia work-up
The work-up for suspected hypercalcemia begins with measurement of the patient’s calcium level. Because calcium is bound to albumin in the blood, the standard serum calcium test may not reflect the true calcium level. (If albumin is high, the calcium level will be high, and vice versa.)1 The true (serum ionized) calcium level (also known as corrected calcium level) should always be calculated to confirm true hypercalcemia. A formula commonly used to calculate the corrected calcium level is
Corrected calcium (mg/dL) = (measured calcium [mg/dL]) + 0.8 (4.0 – serum albumin [mg/dL])
Direct measurement of the serum ionized calcium level is not affected by the albumin level and can also confirm true hypercalcemia.4
Once hypercalcemia is confirmed, the next step is to measure the patient’s intact PTH level.
If intact PTH is elevated or high normal, consider primary hyperparathyroidism or FHH and confirm by obtaining the urine calcium level.
• If urine calcium level is high (> 200 mg/24 h), the diagnosis is primary hyperparathyroidism.
• If urine calcium level is low (< 100 mg/24 h), the diagnosis is FHH.
If intact PTH is low, consider non–PTH-mediated causes and confirm by obtaining PTHrP and vitamin D levels.
• If PTHrP level is elevated, scan for malignancy.
• If 1,25-dihydroxyvitamin D level is elevated, order a chest x-ray to rule out sarcoid or lymphoma.
• If both PTHrP and vitamin D levels are normal, order both serum protein electrophoresis (SPEP) and urine protein electrophoresis (UPEP) with immunofixation to rule out MM.
• If vitamin D level is elevated, check vitamin and herbal supplement use for excessive vitamin D intake.
Treatment of symptomatic hypercalcemia
The goals of treatment of symptomatic hypercalcemia are to reduce the serum calcium level to the normal range and to treat the underlying cause.1 Mild hypercalcemia (calcium level, 10-12 mg/dL) is typically asymptomatic and does not need to be treated. Moderate hypercalcemia (calcium level, 12-14 mg/dL) may not require treatment unless the patient is symptomatic and/or has had an acute rise in calcium level.1 In mild to moderate hypercalcemia, the serum calcium level should be monitored to establish a trend.
Treatment for symptomatic moderate and severe hypercalcemia (calcium level, > 14 mg/dL) typically involves a similar regimen:
• Volume expansion with isotonic saline at an initial rate of 2 to 4 L/d, which is then adjusted to achieve 200 mL/h of continuous urine output. IV furosemide can be used with caution (10-20 mg IV as needed) to promote diuresis if volume overload is a concern (furosemide promotes renal excretion of calcium).
• Administration of subcutaneous calcitonin (4-8 IU/kg, repeated every 6 h for 24 h). Calcitonin works rapidly to lower calcium levels in 4 to 6 h.
• Concurrent administration of IV bisphosphonate (zoledronic acid [4 mg over 15 min] or pamidronate [60-90 mg over 4 h]). Pamidronate is superior for reversal of malignancy-related hypercalcemia.1
Hypercalcemia and multiple myeloma
MM is a malignant neoplasm of plasma cells that accounts for approximately 1% of all cancers and about 10% of hematologic malignancies in the United States, with a median patient age of 70 at diagnosis.5-7 In MM, myeloma cells induce the secretion of cytokines and growth factors that alter plasma cells, activate osteoclasts, suppress osteoblasts, cause abnormal interactions between plasma cells and bone marrow, and stimulate aberrant angiogenesis.8 Osteoclastic bone resorption produces hypercalcemia as well as the lytic lesions seen on x-ray.7
Approximately 74% of patients present with typical MM symptoms of calcium elevation in the blood, renal insufficiency, anemia, and bone lesions, known as CRAB symptoms, but other myeloma-related manifestations may be present.9
Diagnostic criteria for MM include the following (all three must be present):10
• Monoclonal bone marrow plasma cells ≥ 10% and/or a biopsy-proven plasmacytoma
• Monoclonal protein in the serum and/or urine (if none is detected, disease is nonsecretory and diagnosis requires ≥ 30% bone marrow plasma cells and/or biopsy-proven plasmacytoma)
• Myeloma-related organ dysfunction, indicated by at least one of the CRAB symptoms.
In the absence of CRAB symptoms, an asymptomatic patient may have an MM precursor syndrome: monoclonal gammopathy of undetermined significance or smoldering (or indolent) MM.10
Treatment of multiple myeloma
In recent years, the use of induction therapy followed by autologous stem cell transplantation and the development of novel therapeutic agents have extended overall survival for patients with MM. These agents include proteasome inhibitors (bortezomib and the second-generation carfilzomib)11 and immunomodulators (thalidomide and the second-generation lenalidomide). Early diagnosis and treatment can improve progression-free survival as well as overall survival, including recovery of renal function for patients with renal failure.12 With survival ranging from one year or less—with aggressive disease—to 10 years or more for patients with responsive disease,7 there remains no cure for MM.
Outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
Further work-up included SPEP and UPEP with immunofixation, which revealed marked IgG free λ light chains with an M (monoclonal) component, making MM a very likely diagnosis. Confirmation by means of bone marrow biopsy was indicated, but the patient refused the procedure.
The patient’s hypercalcemia was treated by IV administration of calcitonin with isotonic saline. This reduced the serum calcium level from 15.4 mg/dL to 10.6 mg/dL within 48 hours. One dose of ergocalciferol (vitamin D2) was then administered to promote intestinal absorption of calcium and support bone mineralization, further lowering the patient’s serum calcium level to a normal 8.9 mg/dL. Hypokalemia was treated with oral potassium supplementation.
The patient, now stable, was referred to the hematology/oncology and bone mineral metabolism clinics and was discharged from the hospital. She did not keep those appointments and was lost to follow-up.
CONCLUSION
The most common causes of hypercalcemia are hyperparathyroidism and malignancy. Most cases do not require treatment unless the calcium level is >14 mg/dL and/or the patient is symptomatic. Red flag symptoms include weakness, abdominal pain, mental status changes, and coma.4 Primary care clinicians should suspect MM in older patients with laboratory findings of hypercalcemia, anemia, and renal dysfunction, with lytic lesions on x-ray.
REFERENCES
1. Carroll MF, Schade DS. A practical approach to hypercalcemia. Am Fam Physician. 2003;67(9):1959-1966.
2. Endres DB. Investigation of hypercalcemia. Clin Biochem. 2012;45(12):
954-963.
3. Moe SM. Disorders involving calcium, phosphorus, and magnesium. Prim Care. 2008;35(2):215-237.
4. Sharma B, Misicko NE. How should you evaluate elevated calcium in an asymptomatic patient? J Fam Pract. 2008;57(4):267-269.
5. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11-30.
6. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33.
7. Shaheen SP, Talwalkar SS, Medeiros LJ. Multiple myeloma and immunosecretory disorders: an update. Adv Anat Pathol. 2008;15(4):196-210.
8. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11): 1046-1060.
9. Talamo G, Farooq U, Zangari M, et al. Beyond the CRAB symptoms: a study of presenting clinical manifestations of multiple myeloma. Clin Lymphoma Myeloma Leuk. 2010;10(6):464-468.
10. Palumbo A, Sezer O, Kyle R, et al. International Myeloma Working Group guidelines for the management of multiple myeloma patients ineligible for standard high-dose chemotherapy with autologous stem cell transportation. Leukemia. 2009;23(10):1716-1730.
11. Kyprolis [package insert]. South San Francisco, CA: Onyx Pharmaceuticals, Inc; 2012.
12. Suyani E, Sucak GT, Erten Y, et al. Evaluation of multiple myeloma patients presenting with renal failure in a university hospital in the year 2010. Ren Fail. 2012;34(2):257-262.
The 2014-2015 Influenza Season: What You Need to Know
As clinicians and the CDC prepare for the upcoming influenza season, many of the immunization recommendations remain unchanged from last season. Vaccination continues to be recommended for everyone ages 6 months and older. However, for the first time, a specific vaccine is preferred for children ages 2 through 8 years. Here’s what you need to know about this change, as well as how to handle vaccination in patients who are, or might be, allergic to eggs.
USE LAIV FOR KIDS AGES 2 THROUGH 8 (IF AVAILABLE)
For the first time, the CDC’s Advisory Committee on Immunization Practices (ACIP) has stated a preference for a specific influenza vaccine for a specific age-group. It recommends using the live attenuated influenza vaccine (LAIV), which is a nasal spray, for children ages 2 through 8 years.1
A systematic review found evidence of increased efficacy of LAIV compared to inactivated influenza vaccine (IIV) in this age-group; both types of vaccine have similar rates of adverse reactions.2 This increased effectiveness results in 46 fewer cases of confirmed influenza per 1,000 children vaccinated (number needed to treat, 24). Although the evidence of LAIV’s increased effectiveness was found for children ages 2 to 6 years, ACIP extended this recommendation through age 8 because this is the age through which clinicians need to consider two doses of vaccine for a child previously unvaccinated with the influenza vaccine. Children younger than 2 should receive IIV3 or IIV4.3
ACIP realizes that due to programmatic constraints it would be difficult to vaccinate all children with LAIV this year; the committee emphasizes that this recommendation should be implemented when feasible this year but no later than the 2015-2016 influenza season. IIV is effective in children and should be given if LAIV is not available or is contraindicated. Vaccination should not be delayed in the hopes of receiving a supply of LAIV if IIV is available.1
LAIV should not be used in children younger than 2 or adults older than 49. This vaccine is contraindicated in children and adolescents who are taking chronic aspirin therapy, pregnant women, or persons who are immunosuppressed, have a history of egg allergy, or have taken influenza antiviral medications in the past 48 hours.1 LAIV also is not recommended for children ages 2 through 4 years who have asthma or have had a wheezing episode in the past 12 months.1
There are precautions for the use of LAIV in patients with chronic medical conditions that can place them at high risk for complications from influenza. These include chronic lung, heart, renal, neurologic, liver, blood, or metabolic disorders—particularly, asthma and diabetes.1
WHICH VACCINE FOR PATIENTS WHO ARE ALLERGIC TO EGGS?
Two influenza vaccines are now available that are not prepared in embryonated eggs: recombinant influenza vaccine (RIV3) and cell culture–based inactivated influenza vaccine (ccIIV3). Both are trivalent products that contain antigens from two influenza A viruses and one influenza B virus; they were introduced in time for the 2013-2014 flu season. The RIV3 is considered egg-free but ccIIV3 is not, although the amount of egg protein in the latter is miniscule (estimated at 5 × 10-8 mg/0.5 mL dose).1 Neither product is licensed for use in children younger than 18, and RIV3 is licensed only for those ages 18 through 49.
Patients who experience only hives after egg exposure can receive any of the flu vaccines except LAIV—and only because of a lack of data on this product, not because it has been shown to be less safe than the other vaccines. Patients who are unsure if they have an egg allergy or who only get hives when they eat eggs should be observed for at least 30 minutes1 following injection as a precaution. Those ages 18 through 49 who have a history of severe reactions to eggs should receive RIV3. Patients younger than 18 and older than 49 can receive IIV vaccines approved for their specific age-group.
Any patient who is severely allergic but who cannot receive an egg-free vaccine should be vaccinated by a clinician with experience managing severe allergic conditions. Although severe anaphylactic reactions to influenza vaccine are very rare, clinicians should be equipped and prepared to respond to a severe allergic reaction after providing influenza vaccine to anyone with a history of egg allergy.
Continue for additional tips and resources >>
ADDITIONAL TIPS AND RESOURCES
In addition to the LAIV, RIV3, and ccIIV3 vaccines described here, 10 other vaccines are available: five egg-based IIV3 products in standard-dose form, one IIV3 vaccine for intradermal use, one high-dose IIV3 product for patients ages 65 or older, and three standard-dose IIV4 products. More details on each of these vaccines are available on the CDC website (www.cdc.gov/mmwr/preview/mmwrhtml/rr6207a1.htm?s_cid=rr6207a1_w#Tab1).
Regardless of which type of flu vaccine they receive, children ages 6 months through 8 years should receive two doses, at least four weeks apart, unless they received
• One dose during the 2013-2014 season, or
• Two or more doses of seasonal influenza vaccine since July 2010, or
• Two or more doses of seasonal influenza vaccine before July 2010 and at least one dose of monovalent H1N1 vaccine, or
• At least one dose of seasonal influenza vaccine prior to July 2010 and one or more after.
Vaccine effectiveness. The CDC estimated that vaccine effectiveness during the 2013-2014 flu season was 66%.3 While this degree of effectiveness is important for minimizing morbidity and mortality from influenza each year, it’s important to appreciate the limitations of the vaccine and not rely on it as the only preventive intervention.
Other forms of prevention. We need to advise and practice good respiratory hygiene, frequent hand washing, self-isolation when sick, effective infection control practices at health care facilities, targeted early treatment with antivirals, and targeted pre- and postexposure antiviral chemoprevention. Details on each of these interventions, including recommendations on the use of antiviral medications, can be found on the CDC website (www.cdc.gov/flu).
REFERENCES
1. Grohskopf LA, Olsen SJ, Sokolow LZ, et al; Influenza Division, National Center for Immunization and Respiratory Diseases, CDC. Prevention and control of seasonal influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP)—United States 2014-2015 influenza season. MMWR Morb Mortal Wkly Rep. 2014;63: 691-697.
2. Grohskopf L, Olsen S, Sokolow L. Effectiveness of live-attenuated vs inactivated influenza vaccines for healthy children. Presented at: Meeting of the Advisory Committee on Immunization Practices; February 26, 2014; Atlanta, GA. www.cdc.gov/vaccines/acip/meetings/downloads/slides-2014-02/05-Flu-Grohskopf.pdf. Accessed October 19, 2014.
3. Flannery B. Interim estimates of 2013-14 seasonal influenza vaccine effectiveness. Presented at: Meeting of the Advisory Committee on Immunization Practices; February 26, 2014; Atlanta, GA. www.cdc.gov/vaccines/acip/meetings/downloads/slides-2014-02/04-Flu-Flannery.pdf. Accessed October 19, 2014.
As clinicians and the CDC prepare for the upcoming influenza season, many of the immunization recommendations remain unchanged from last season. Vaccination continues to be recommended for everyone ages 6 months and older. However, for the first time, a specific vaccine is preferred for children ages 2 through 8 years. Here’s what you need to know about this change, as well as how to handle vaccination in patients who are, or might be, allergic to eggs.
USE LAIV FOR KIDS AGES 2 THROUGH 8 (IF AVAILABLE)
For the first time, the CDC’s Advisory Committee on Immunization Practices (ACIP) has stated a preference for a specific influenza vaccine for a specific age-group. It recommends using the live attenuated influenza vaccine (LAIV), which is a nasal spray, for children ages 2 through 8 years.1
A systematic review found evidence of increased efficacy of LAIV compared to inactivated influenza vaccine (IIV) in this age-group; both types of vaccine have similar rates of adverse reactions.2 This increased effectiveness results in 46 fewer cases of confirmed influenza per 1,000 children vaccinated (number needed to treat, 24). Although the evidence of LAIV’s increased effectiveness was found for children ages 2 to 6 years, ACIP extended this recommendation through age 8 because this is the age through which clinicians need to consider two doses of vaccine for a child previously unvaccinated with the influenza vaccine. Children younger than 2 should receive IIV3 or IIV4.3
ACIP realizes that due to programmatic constraints it would be difficult to vaccinate all children with LAIV this year; the committee emphasizes that this recommendation should be implemented when feasible this year but no later than the 2015-2016 influenza season. IIV is effective in children and should be given if LAIV is not available or is contraindicated. Vaccination should not be delayed in the hopes of receiving a supply of LAIV if IIV is available.1
LAIV should not be used in children younger than 2 or adults older than 49. This vaccine is contraindicated in children and adolescents who are taking chronic aspirin therapy, pregnant women, or persons who are immunosuppressed, have a history of egg allergy, or have taken influenza antiviral medications in the past 48 hours.1 LAIV also is not recommended for children ages 2 through 4 years who have asthma or have had a wheezing episode in the past 12 months.1
There are precautions for the use of LAIV in patients with chronic medical conditions that can place them at high risk for complications from influenza. These include chronic lung, heart, renal, neurologic, liver, blood, or metabolic disorders—particularly, asthma and diabetes.1
WHICH VACCINE FOR PATIENTS WHO ARE ALLERGIC TO EGGS?
Two influenza vaccines are now available that are not prepared in embryonated eggs: recombinant influenza vaccine (RIV3) and cell culture–based inactivated influenza vaccine (ccIIV3). Both are trivalent products that contain antigens from two influenza A viruses and one influenza B virus; they were introduced in time for the 2013-2014 flu season. The RIV3 is considered egg-free but ccIIV3 is not, although the amount of egg protein in the latter is miniscule (estimated at 5 × 10-8 mg/0.5 mL dose).1 Neither product is licensed for use in children younger than 18, and RIV3 is licensed only for those ages 18 through 49.
Patients who experience only hives after egg exposure can receive any of the flu vaccines except LAIV—and only because of a lack of data on this product, not because it has been shown to be less safe than the other vaccines. Patients who are unsure if they have an egg allergy or who only get hives when they eat eggs should be observed for at least 30 minutes1 following injection as a precaution. Those ages 18 through 49 who have a history of severe reactions to eggs should receive RIV3. Patients younger than 18 and older than 49 can receive IIV vaccines approved for their specific age-group.
Any patient who is severely allergic but who cannot receive an egg-free vaccine should be vaccinated by a clinician with experience managing severe allergic conditions. Although severe anaphylactic reactions to influenza vaccine are very rare, clinicians should be equipped and prepared to respond to a severe allergic reaction after providing influenza vaccine to anyone with a history of egg allergy.
Continue for additional tips and resources >>
ADDITIONAL TIPS AND RESOURCES
In addition to the LAIV, RIV3, and ccIIV3 vaccines described here, 10 other vaccines are available: five egg-based IIV3 products in standard-dose form, one IIV3 vaccine for intradermal use, one high-dose IIV3 product for patients ages 65 or older, and three standard-dose IIV4 products. More details on each of these vaccines are available on the CDC website (www.cdc.gov/mmwr/preview/mmwrhtml/rr6207a1.htm?s_cid=rr6207a1_w#Tab1).
Regardless of which type of flu vaccine they receive, children ages 6 months through 8 years should receive two doses, at least four weeks apart, unless they received
• One dose during the 2013-2014 season, or
• Two or more doses of seasonal influenza vaccine since July 2010, or
• Two or more doses of seasonal influenza vaccine before July 2010 and at least one dose of monovalent H1N1 vaccine, or
• At least one dose of seasonal influenza vaccine prior to July 2010 and one or more after.
Vaccine effectiveness. The CDC estimated that vaccine effectiveness during the 2013-2014 flu season was 66%.3 While this degree of effectiveness is important for minimizing morbidity and mortality from influenza each year, it’s important to appreciate the limitations of the vaccine and not rely on it as the only preventive intervention.
Other forms of prevention. We need to advise and practice good respiratory hygiene, frequent hand washing, self-isolation when sick, effective infection control practices at health care facilities, targeted early treatment with antivirals, and targeted pre- and postexposure antiviral chemoprevention. Details on each of these interventions, including recommendations on the use of antiviral medications, can be found on the CDC website (www.cdc.gov/flu).
REFERENCES
1. Grohskopf LA, Olsen SJ, Sokolow LZ, et al; Influenza Division, National Center for Immunization and Respiratory Diseases, CDC. Prevention and control of seasonal influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP)—United States 2014-2015 influenza season. MMWR Morb Mortal Wkly Rep. 2014;63: 691-697.
2. Grohskopf L, Olsen S, Sokolow L. Effectiveness of live-attenuated vs inactivated influenza vaccines for healthy children. Presented at: Meeting of the Advisory Committee on Immunization Practices; February 26, 2014; Atlanta, GA. www.cdc.gov/vaccines/acip/meetings/downloads/slides-2014-02/05-Flu-Grohskopf.pdf. Accessed October 19, 2014.
3. Flannery B. Interim estimates of 2013-14 seasonal influenza vaccine effectiveness. Presented at: Meeting of the Advisory Committee on Immunization Practices; February 26, 2014; Atlanta, GA. www.cdc.gov/vaccines/acip/meetings/downloads/slides-2014-02/04-Flu-Flannery.pdf. Accessed October 19, 2014.
As clinicians and the CDC prepare for the upcoming influenza season, many of the immunization recommendations remain unchanged from last season. Vaccination continues to be recommended for everyone ages 6 months and older. However, for the first time, a specific vaccine is preferred for children ages 2 through 8 years. Here’s what you need to know about this change, as well as how to handle vaccination in patients who are, or might be, allergic to eggs.
USE LAIV FOR KIDS AGES 2 THROUGH 8 (IF AVAILABLE)
For the first time, the CDC’s Advisory Committee on Immunization Practices (ACIP) has stated a preference for a specific influenza vaccine for a specific age-group. It recommends using the live attenuated influenza vaccine (LAIV), which is a nasal spray, for children ages 2 through 8 years.1
A systematic review found evidence of increased efficacy of LAIV compared to inactivated influenza vaccine (IIV) in this age-group; both types of vaccine have similar rates of adverse reactions.2 This increased effectiveness results in 46 fewer cases of confirmed influenza per 1,000 children vaccinated (number needed to treat, 24). Although the evidence of LAIV’s increased effectiveness was found for children ages 2 to 6 years, ACIP extended this recommendation through age 8 because this is the age through which clinicians need to consider two doses of vaccine for a child previously unvaccinated with the influenza vaccine. Children younger than 2 should receive IIV3 or IIV4.3
ACIP realizes that due to programmatic constraints it would be difficult to vaccinate all children with LAIV this year; the committee emphasizes that this recommendation should be implemented when feasible this year but no later than the 2015-2016 influenza season. IIV is effective in children and should be given if LAIV is not available or is contraindicated. Vaccination should not be delayed in the hopes of receiving a supply of LAIV if IIV is available.1
LAIV should not be used in children younger than 2 or adults older than 49. This vaccine is contraindicated in children and adolescents who are taking chronic aspirin therapy, pregnant women, or persons who are immunosuppressed, have a history of egg allergy, or have taken influenza antiviral medications in the past 48 hours.1 LAIV also is not recommended for children ages 2 through 4 years who have asthma or have had a wheezing episode in the past 12 months.1
There are precautions for the use of LAIV in patients with chronic medical conditions that can place them at high risk for complications from influenza. These include chronic lung, heart, renal, neurologic, liver, blood, or metabolic disorders—particularly, asthma and diabetes.1
WHICH VACCINE FOR PATIENTS WHO ARE ALLERGIC TO EGGS?
Two influenza vaccines are now available that are not prepared in embryonated eggs: recombinant influenza vaccine (RIV3) and cell culture–based inactivated influenza vaccine (ccIIV3). Both are trivalent products that contain antigens from two influenza A viruses and one influenza B virus; they were introduced in time for the 2013-2014 flu season. The RIV3 is considered egg-free but ccIIV3 is not, although the amount of egg protein in the latter is miniscule (estimated at 5 × 10-8 mg/0.5 mL dose).1 Neither product is licensed for use in children younger than 18, and RIV3 is licensed only for those ages 18 through 49.
Patients who experience only hives after egg exposure can receive any of the flu vaccines except LAIV—and only because of a lack of data on this product, not because it has been shown to be less safe than the other vaccines. Patients who are unsure if they have an egg allergy or who only get hives when they eat eggs should be observed for at least 30 minutes1 following injection as a precaution. Those ages 18 through 49 who have a history of severe reactions to eggs should receive RIV3. Patients younger than 18 and older than 49 can receive IIV vaccines approved for their specific age-group.
Any patient who is severely allergic but who cannot receive an egg-free vaccine should be vaccinated by a clinician with experience managing severe allergic conditions. Although severe anaphylactic reactions to influenza vaccine are very rare, clinicians should be equipped and prepared to respond to a severe allergic reaction after providing influenza vaccine to anyone with a history of egg allergy.
Continue for additional tips and resources >>
ADDITIONAL TIPS AND RESOURCES
In addition to the LAIV, RIV3, and ccIIV3 vaccines described here, 10 other vaccines are available: five egg-based IIV3 products in standard-dose form, one IIV3 vaccine for intradermal use, one high-dose IIV3 product for patients ages 65 or older, and three standard-dose IIV4 products. More details on each of these vaccines are available on the CDC website (www.cdc.gov/mmwr/preview/mmwrhtml/rr6207a1.htm?s_cid=rr6207a1_w#Tab1).
Regardless of which type of flu vaccine they receive, children ages 6 months through 8 years should receive two doses, at least four weeks apart, unless they received
• One dose during the 2013-2014 season, or
• Two or more doses of seasonal influenza vaccine since July 2010, or
• Two or more doses of seasonal influenza vaccine before July 2010 and at least one dose of monovalent H1N1 vaccine, or
• At least one dose of seasonal influenza vaccine prior to July 2010 and one or more after.
Vaccine effectiveness. The CDC estimated that vaccine effectiveness during the 2013-2014 flu season was 66%.3 While this degree of effectiveness is important for minimizing morbidity and mortality from influenza each year, it’s important to appreciate the limitations of the vaccine and not rely on it as the only preventive intervention.
Other forms of prevention. We need to advise and practice good respiratory hygiene, frequent hand washing, self-isolation when sick, effective infection control practices at health care facilities, targeted early treatment with antivirals, and targeted pre- and postexposure antiviral chemoprevention. Details on each of these interventions, including recommendations on the use of antiviral medications, can be found on the CDC website (www.cdc.gov/flu).
REFERENCES
1. Grohskopf LA, Olsen SJ, Sokolow LZ, et al; Influenza Division, National Center for Immunization and Respiratory Diseases, CDC. Prevention and control of seasonal influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP)—United States 2014-2015 influenza season. MMWR Morb Mortal Wkly Rep. 2014;63: 691-697.
2. Grohskopf L, Olsen S, Sokolow L. Effectiveness of live-attenuated vs inactivated influenza vaccines for healthy children. Presented at: Meeting of the Advisory Committee on Immunization Practices; February 26, 2014; Atlanta, GA. www.cdc.gov/vaccines/acip/meetings/downloads/slides-2014-02/05-Flu-Grohskopf.pdf. Accessed October 19, 2014.
3. Flannery B. Interim estimates of 2013-14 seasonal influenza vaccine effectiveness. Presented at: Meeting of the Advisory Committee on Immunization Practices; February 26, 2014; Atlanta, GA. www.cdc.gov/vaccines/acip/meetings/downloads/slides-2014-02/04-Flu-Flannery.pdf. Accessed October 19, 2014.
How Effective Is Group Cognitive Behavioral Therapy to Treat PTSD?
Anxiety is a necessary and natural reaction to trauma, but, sometimes, anxiety symptoms become excessive and problematic, as experienced with posttraumatic stress disorder (PTSD). Some patients who struggle with PTSD endure a relentless apprehension so intense that it keeps them from participating in everyday activities, such as attending work and partaking in social activities. Associated anxiety symptoms severely impair everyday function and include increased heart rate, sweating, intrusive images, poor attention, fear, or insomnia. Posttraumatic stress disorder symptoms often lead to occupational dysfunction, relationship difficulty, and numerous other functional impairments.
Approximately 300,000 veterans meet the criteria for PTSD related to ongoing or recent wars.1 The veteran does not bear the personal and functional burden alone; however, the financial load is felt throughout society. One recent study suggests that for veterans diagnosed with PTSD, the first 2 years after deployment cost society an estimated $7,000 per individial.2 Current research suggests that this potentially debilitating disorder occurs in about 14% of Operation Iraqi Freedom/Operation Enduring Freedom combat troops, whereas the similar U.S. demographic population experiences PTSD at a rate of about 7%.1,3 The ongoing military trauma exposures are compelling the mental health community to establish efficient and effective treatment options.4,5
Several treatment strategies exist to reduce PTSD symptoms, but health care professionals must seek a balance between therapeutic benefit and cost. The treatment of PTSD is diverse and variable; however, in the most recent Clinical Practice Guideline (CPG) for PTSD, the VA and DoD specifically endorse some psychotherapeutic interventions while dissuading the use of others.6 Of note, the VA and DoD CPG strongly encourages Stress Inoculation Training (SIT) and similar cognitive therapies aimed at guiding patients through the process of consciously understanding the relationship between thoughts and feelings and then modifying thoughts to appropriately manage stressors.6 Meanwhile, group psychotherapy has been determined to be “somewhat helpful.”6 Even though cognitive- and group-based therapies have long been established as efficacious for numerous psychological disorders (depression, obsessive compulsive disorder, eating disorders, etc), neither the American Group Psychotherapy Association nor the VA and DoD CPG directly endorse the use of group cognitive behavioral therapy (GCBT) for the treatment of PTSD.6,7 However, both VA and DoD mental health providers commonly practice CBT and various group psychotherapies for the treatment of PTSD.
Despite the widespread use of CBT, there is a gap in the clinical understanding of the evidence supporting GCBT for PTSD. The goal of this synthesis was to understand the efficacy of treating PTSD symptoms with group psychotherapy. To begin this investigation, the following PICO (population, intervention, comparison, outcome) question was asked: In adults diagnosed with PTSD, how effective is group cognitive behavioral therapy in reducing PTSD-related symptoms?
Methods
Research articles addressing the use of GCBT in PTSD were obtained via database searches that took place during October 2012 (Table). Searched databases included the Cochrane Central Register of Controlled Trials, Cochrane Database of Systematic Reviews Randomized Controlled Trials, Psychological Information (PsycINFO), and Public Medicine (PubMed).
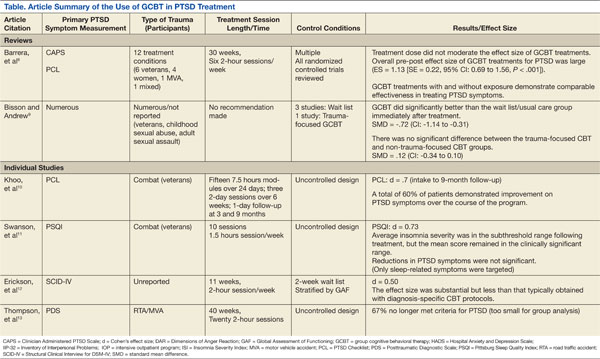
The PubMed database was searched using the following MeSH (medical subject heading) terms: “psychotherapy, group” and “stress disorders, post-traumatic” and “cognitive therapy.” Limitations were set to include only patients aged ≥ 18 years, results in English, those involving human subjects, and articles published within the past 5 years. A manual search of references was also conducted, and relevant articles were retained.
Articles that addressed primary substance abuse, other DSM Axis I disorders, intimate partner violence, or family issues were excluded from the evidence sample due to concerns of an alternate treatment focus. Articles with a focus on telehealth or alternative medicine were considered confounding to the scope of this review were also excluded. It was also noted that the term CBT is used collectively for an umbrella of treatments; however, treatments that focused on elements other than the components of CBT being delivered in a group were not included. To prevent duplication of the results, research from an inclusive review was not considered individually.
SUMMARY OF EVIDENCE
Six works fulfilled the PICO criteria and were of sufficient quality to be synthesized. Of the 6 articles retained for synthesis, 2 were high-level reviews. Both reviews supported the use of GCBT for PTSD treatment. Barrera and colleagues reported an overall large effect size regardless of the presence of exposure in-group among the 12 treatment conditions and 651 study participants.8 These researchers also reported that in-group exposure did not further traumatize other group members.
Similarly, although a notably older and smaller review, Bisson and Andrew reported a significant standard mean deviation between 4 GCBT treatment and wait list controls. These reviewers did not find a significant difference between trauma- and nontrauma-focused treatment groups. The reviews also noted that individual psychotherapy and/or pharmacotherapy was most often continued throughout the reviewed studies.8,9
The 4 other studies contribute substantively to this synthesis but arguably represent lower evidence quality. A large longitudinal study of 496 Australian veterans reported a large effect size that was sustained 9 months after treatment began.10 These researchers used an intensive outpatient program that included medication and other treatment modalities as the basis for GCBT delivery. They reported that the majority of the patients revealed improvement in PTSD symptoms.
Another study sampled a similar group of 10 combat veterans but focused particular attention on sleep-related PTSD symptoms of insomnia, nightmares, and sleep quality.11 Although these researchers were unable to report a significant difference in overall PTSD symptoms for the 8 subjects who completed the protocol, they did find a large effect size on insomnia severity and a medium effect size on sleep quality. Regular treatment, including medication, continued throughout this study.
Other researchers reported a medium effect size on PTSD symptoms while using GCBT in a heterogeneous group with various anxiety disorders, including obsessive compulsive disorder, generalized anxiety disorder, social phobia, panic disorders, and PTSD.12 Although reporting similar results as all other included studies, this study has some significant limitations, including a 26% dropout rate among the 152 participants. The final study included for synthesis reported a remarkable 67% elimination of the PTSD diagnosis among 6 motor vehicle accident survivors in the small, uncontrolled study.13 Concomitant treatments, including medications, were not reported in detail for these 4 studies except as mentioned.
As a whole, the 6 studies revealed some appreciable commonalities. Time since diagnosis did not seem to influence the results. Attrition was consistently found to be similar to other PTSD treatments. The reported session topics were loosely based on common CBT tenets (ie, education, challenging cognitions, and relaxation techniques) and were typically similar among treatment groups, including the use of homework.
DISCUSSION
As the diagnosis of PTSD increases to unfamiliar levels, GCBT has the potential to be helpful to clinicians and patients seeking alternatives to their current treatments.1,4,14 The reported results imply that GCBT can be useful in PTSD symptom reduction. This could be particularly useful to VA and military providers or rural providers operating with limited resources.
Treatment protocols are not well established and should be approached with care prior to the establishment of CBT treatment groups for those diagnosed with PTSD. Session overviews and descriptions, such as those mentioned in Thompson and colleagues, could provide a reference point for future use.13
Also worth considering, CBT can be an ambiguous term requiring deliberate definition within treatment protocols. As noted in the VA and DoD CPG, exposure- and trauma-focused treatment designs can be efficacious, but these elements do not seem to be required within the GCBT treatment setting.
The current research also suggests GCBT efficacy regardless of the index trauma. This does not suggest that heterogeneous groups were frequently studied nor can conclusions be drawn regarding heterogeneous treatment groups. Elements such as group size and session length are inconsistently reported and require specific consideration as well. There is a distinct lack of research directly comparing individual CBT with GCBT directly, which prohibits meaningful conclusions regarding PTSD symptom reduction. This research gap may well have influenced the recommendations within the VA and DoD CPG. Although some higher quality studies exist, many of the published reports on GCBT have noteworthy design flaw, such as inadequate controls and statistical analysis.
LIMITATIONS
There are some limitations to this literature synthesis. Although the search was limited to the past 5 years, the inclusion of reviews accounts for older evidence. As alluded to earlier, the lack of a standardized GCBT treatment protocol challenges results comparisons as well. The consequent treatment variations make direct interstudy comparison and synthesis difficult. Similarly, outcome measures varied between studies. Also, group psychotherapy is well established and accepted. Therefore, much of the supporting research was accomplished outside the parameters of this literature search. This empirical view of group psychotherapy among mental health providers may also contribute to the lack of available research.
It is also worth noting that studies finding neutral or negative results are often unpublished. This publication bias could account for the lack of available evidence. The research reports do not consistently report therapist qualifications; however, board certificates in group psychotherapy and CBT are undeniably variables available for debate. The inclusion of uncontrolled trials limits these findings as well. Although the above limitations are not exhaustive, they do provide necessary caveats to future generalizations.
FUTURE IMPLICATIONS
Perhaps the most important information to gain from future research is that of treatment outcomes. Studies that include a detailed outcome evaluation could reveal patient satisfaction, efficacy, and financial considerations. In the presence of adequate supportive data, GCBT could contribute outcome data regarding trauma survivor symptom normalization, peer support formation, access to care, treatment efficiency, and health care resources utilization. As noted in Barrera and colleagues, future analysis will require a greater volume of trials with an overall increase in methodological rigor.8
Current research has demonstrated a solid base from which to spawn specific treatment protocols. The available research is investigational in terms of treatment procedures. Replication of these studies could dictate treatment protocol and contribute substantively to future VA and DoD CPG updates. Future researchers should consider the use of a standard PTSD symptom assessment tool to make interstudy comparisons more meaningful. The length of treatment and exposure elements should be targeted specifically in future research as these components currently vary the most.
The military represents an obvious avenue for future research due to increased PTSD diagnosis in recent years. Although the etiology of the increase in PTSD is unclear and most likely multifactorial (decreased resilience, increased awareness, increased pursuit of secondary gains, etc), the need for treatment options is apparent.1 Group cohesion has been shown to be a core component of successful group psychotherapy, so military members who are accustomed to unit cohesion might represent a uniquely suitable population for this modality.15 Interestingly and for reasons not currently understood, veterans do not see effects of therapy as large as their civilian counterparts.8 This underscores the need for further evaluation of military-specific outcomes.
CONCLUSIONS
Although the available evidence is not robust, results do support the careful use of GCBT as an effective treatment for PTSD symptom reduction.8 Group psychotherapy has been generally regarded as an efficacious and cost-effective method to achieve similar outcomes to individual therapy. Increasing PTSD prevalence compels mental health care providers to explore all available treatment options. The potential for GCBT as an option is exciting, especially for mental health providers and those with limited resources. Rising health care standards and the current national fiscal situation is dictating a reevaluation of treatment options; so perhaps all health care providers will soon consider the use of GCBT.
As with any group assignment, the clinician should carefully consider the individual’s suitability and desire for group participation.16 With GCBT, providers could facilitate the relief of relentless apprehension and functional impairment for several patients simultaneously. Although there are many details left to explore regarding the use of GCBT for PTSD, the therapy’s foundation for use as a PTSD treatment is apparent.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Litz B, Schlenger W. Posttraumatic stress disorder in service members and new veterans of the Iraq and Afghanistan wars: A bibliography and critique. PTSD Res Q. 2009;20(1):1-3.
2. Tanielian T. Assessing combat exposure and post-traumatic stress disorder in troops and estimating the costs to society: Implications from the RAND Invisible Wounds of War Study. http://www.rand.org/pubs/testimonies/CT321.html. Published 2009. Accessed September 29, 2014.
3. Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):617-627.
4. Cukor J, Spitalnick J, Difede J, Rizzo A, Rothbaum BO. Emerging treatments for PTSD. Clinical Psychol Rev. 2009;29(8):715-726.
5. Hoge CW. Interventions for war-related posttraumatic stress disorder: Meeting veterans where they are. JAMA. 2011;306(5):549-551.
6. Veterans Health Administration, Department of Defense. VA/DoD Clinical Practice Guideline: Management of Post-Traumatic Stress, Version 2.0. Washington, DC: Veterans Health Administration and Department of Defense; 2010.
7. Burlingame GM, Fuhriman A, Mosier J. The differential effectiveness of group psychotherapy: A meta-analytic perspective. Group Dyn. 2003;7(1):3-12.
8. Barrera TL, Mott JM, Hofstein RF, Teng EJ. A meta-analytic review of exposure in group cognitive behavioral therapy for posttraumatic stress disorder. Clin Psychol Rev. 2013;33(1):24-32.
9. Bisson J, Andrew M. Psychological treatment of post-traumatic stress disorder (PTSD). Cochrane Database Syst Rev. 2007;(3):CD003388.
10. Khoo A, Dent MT, Oei TS. (2011). Group cognitive behaviour therapy for military service-related post-traumatic stress disorder: Effectiveness, sustainability and repeatability. Aust N Z J Psychiatry. 2011;45(8):663-672.
11. Swanson LM, Favorite TK, Horin E, Arnedt JT. A combined group treatment for nightmares and insomnia in combat veterans: A pilot study. J Trauma Stress. 2009;22(6):639-642.
12. Erickson DH, Janeck A, Tallman K. A cognitive-behavioral group for patients with various anxiety disorders. Psychiatr Serv. 2007;58(9):1205-1211.
13. Thompson AR, Wilde E, Boon K. The development of group CBT for the treatment of road-traffic-accident-related post-traumatic stress disorder: A pilot study. Cognitive Behav Therapist. 2010;2(1):32-42.
14. Slade T, Johnston A, Oakley-Browne MA, Andrews G, Whiteford H. 2007 National Survey of Mental Health and Wellbeing: Methods and key findings. Aust N Z J Psychiatry. 2009;43(7):594-605.
15. Crowe TP, Grenyer BF. Is therapist alliance or whole group cohesion more influential in group psychotherapy outcomes? Clin Psychol Psychother. 2008;15(4):239-246.
16. Leszcz M, Kobos JC. Evidence-based group psychotherapy: Using AGPA’s practice guidelines to enhance clinical effectiveness. J Clin Psychol. 2008;64(11):1238-1260.
Anxiety is a necessary and natural reaction to trauma, but, sometimes, anxiety symptoms become excessive and problematic, as experienced with posttraumatic stress disorder (PTSD). Some patients who struggle with PTSD endure a relentless apprehension so intense that it keeps them from participating in everyday activities, such as attending work and partaking in social activities. Associated anxiety symptoms severely impair everyday function and include increased heart rate, sweating, intrusive images, poor attention, fear, or insomnia. Posttraumatic stress disorder symptoms often lead to occupational dysfunction, relationship difficulty, and numerous other functional impairments.
Approximately 300,000 veterans meet the criteria for PTSD related to ongoing or recent wars.1 The veteran does not bear the personal and functional burden alone; however, the financial load is felt throughout society. One recent study suggests that for veterans diagnosed with PTSD, the first 2 years after deployment cost society an estimated $7,000 per individial.2 Current research suggests that this potentially debilitating disorder occurs in about 14% of Operation Iraqi Freedom/Operation Enduring Freedom combat troops, whereas the similar U.S. demographic population experiences PTSD at a rate of about 7%.1,3 The ongoing military trauma exposures are compelling the mental health community to establish efficient and effective treatment options.4,5
Several treatment strategies exist to reduce PTSD symptoms, but health care professionals must seek a balance between therapeutic benefit and cost. The treatment of PTSD is diverse and variable; however, in the most recent Clinical Practice Guideline (CPG) for PTSD, the VA and DoD specifically endorse some psychotherapeutic interventions while dissuading the use of others.6 Of note, the VA and DoD CPG strongly encourages Stress Inoculation Training (SIT) and similar cognitive therapies aimed at guiding patients through the process of consciously understanding the relationship between thoughts and feelings and then modifying thoughts to appropriately manage stressors.6 Meanwhile, group psychotherapy has been determined to be “somewhat helpful.”6 Even though cognitive- and group-based therapies have long been established as efficacious for numerous psychological disorders (depression, obsessive compulsive disorder, eating disorders, etc), neither the American Group Psychotherapy Association nor the VA and DoD CPG directly endorse the use of group cognitive behavioral therapy (GCBT) for the treatment of PTSD.6,7 However, both VA and DoD mental health providers commonly practice CBT and various group psychotherapies for the treatment of PTSD.
Despite the widespread use of CBT, there is a gap in the clinical understanding of the evidence supporting GCBT for PTSD. The goal of this synthesis was to understand the efficacy of treating PTSD symptoms with group psychotherapy. To begin this investigation, the following PICO (population, intervention, comparison, outcome) question was asked: In adults diagnosed with PTSD, how effective is group cognitive behavioral therapy in reducing PTSD-related symptoms?
Methods
Research articles addressing the use of GCBT in PTSD were obtained via database searches that took place during October 2012 (Table). Searched databases included the Cochrane Central Register of Controlled Trials, Cochrane Database of Systematic Reviews Randomized Controlled Trials, Psychological Information (PsycINFO), and Public Medicine (PubMed).
The PubMed database was searched using the following MeSH (medical subject heading) terms: “psychotherapy, group” and “stress disorders, post-traumatic” and “cognitive therapy.” Limitations were set to include only patients aged ≥ 18 years, results in English, those involving human subjects, and articles published within the past 5 years. A manual search of references was also conducted, and relevant articles were retained.
Articles that addressed primary substance abuse, other DSM Axis I disorders, intimate partner violence, or family issues were excluded from the evidence sample due to concerns of an alternate treatment focus. Articles with a focus on telehealth or alternative medicine were considered confounding to the scope of this review were also excluded. It was also noted that the term CBT is used collectively for an umbrella of treatments; however, treatments that focused on elements other than the components of CBT being delivered in a group were not included. To prevent duplication of the results, research from an inclusive review was not considered individually.
SUMMARY OF EVIDENCE
Six works fulfilled the PICO criteria and were of sufficient quality to be synthesized. Of the 6 articles retained for synthesis, 2 were high-level reviews. Both reviews supported the use of GCBT for PTSD treatment. Barrera and colleagues reported an overall large effect size regardless of the presence of exposure in-group among the 12 treatment conditions and 651 study participants.8 These researchers also reported that in-group exposure did not further traumatize other group members.
Similarly, although a notably older and smaller review, Bisson and Andrew reported a significant standard mean deviation between 4 GCBT treatment and wait list controls. These reviewers did not find a significant difference between trauma- and nontrauma-focused treatment groups. The reviews also noted that individual psychotherapy and/or pharmacotherapy was most often continued throughout the reviewed studies.8,9
The 4 other studies contribute substantively to this synthesis but arguably represent lower evidence quality. A large longitudinal study of 496 Australian veterans reported a large effect size that was sustained 9 months after treatment began.10 These researchers used an intensive outpatient program that included medication and other treatment modalities as the basis for GCBT delivery. They reported that the majority of the patients revealed improvement in PTSD symptoms.
Another study sampled a similar group of 10 combat veterans but focused particular attention on sleep-related PTSD symptoms of insomnia, nightmares, and sleep quality.11 Although these researchers were unable to report a significant difference in overall PTSD symptoms for the 8 subjects who completed the protocol, they did find a large effect size on insomnia severity and a medium effect size on sleep quality. Regular treatment, including medication, continued throughout this study.
Other researchers reported a medium effect size on PTSD symptoms while using GCBT in a heterogeneous group with various anxiety disorders, including obsessive compulsive disorder, generalized anxiety disorder, social phobia, panic disorders, and PTSD.12 Although reporting similar results as all other included studies, this study has some significant limitations, including a 26% dropout rate among the 152 participants. The final study included for synthesis reported a remarkable 67% elimination of the PTSD diagnosis among 6 motor vehicle accident survivors in the small, uncontrolled study.13 Concomitant treatments, including medications, were not reported in detail for these 4 studies except as mentioned.
As a whole, the 6 studies revealed some appreciable commonalities. Time since diagnosis did not seem to influence the results. Attrition was consistently found to be similar to other PTSD treatments. The reported session topics were loosely based on common CBT tenets (ie, education, challenging cognitions, and relaxation techniques) and were typically similar among treatment groups, including the use of homework.
DISCUSSION
As the diagnosis of PTSD increases to unfamiliar levels, GCBT has the potential to be helpful to clinicians and patients seeking alternatives to their current treatments.1,4,14 The reported results imply that GCBT can be useful in PTSD symptom reduction. This could be particularly useful to VA and military providers or rural providers operating with limited resources.
Treatment protocols are not well established and should be approached with care prior to the establishment of CBT treatment groups for those diagnosed with PTSD. Session overviews and descriptions, such as those mentioned in Thompson and colleagues, could provide a reference point for future use.13
Also worth considering, CBT can be an ambiguous term requiring deliberate definition within treatment protocols. As noted in the VA and DoD CPG, exposure- and trauma-focused treatment designs can be efficacious, but these elements do not seem to be required within the GCBT treatment setting.
The current research also suggests GCBT efficacy regardless of the index trauma. This does not suggest that heterogeneous groups were frequently studied nor can conclusions be drawn regarding heterogeneous treatment groups. Elements such as group size and session length are inconsistently reported and require specific consideration as well. There is a distinct lack of research directly comparing individual CBT with GCBT directly, which prohibits meaningful conclusions regarding PTSD symptom reduction. This research gap may well have influenced the recommendations within the VA and DoD CPG. Although some higher quality studies exist, many of the published reports on GCBT have noteworthy design flaw, such as inadequate controls and statistical analysis.
LIMITATIONS
There are some limitations to this literature synthesis. Although the search was limited to the past 5 years, the inclusion of reviews accounts for older evidence. As alluded to earlier, the lack of a standardized GCBT treatment protocol challenges results comparisons as well. The consequent treatment variations make direct interstudy comparison and synthesis difficult. Similarly, outcome measures varied between studies. Also, group psychotherapy is well established and accepted. Therefore, much of the supporting research was accomplished outside the parameters of this literature search. This empirical view of group psychotherapy among mental health providers may also contribute to the lack of available research.
It is also worth noting that studies finding neutral or negative results are often unpublished. This publication bias could account for the lack of available evidence. The research reports do not consistently report therapist qualifications; however, board certificates in group psychotherapy and CBT are undeniably variables available for debate. The inclusion of uncontrolled trials limits these findings as well. Although the above limitations are not exhaustive, they do provide necessary caveats to future generalizations.
FUTURE IMPLICATIONS
Perhaps the most important information to gain from future research is that of treatment outcomes. Studies that include a detailed outcome evaluation could reveal patient satisfaction, efficacy, and financial considerations. In the presence of adequate supportive data, GCBT could contribute outcome data regarding trauma survivor symptom normalization, peer support formation, access to care, treatment efficiency, and health care resources utilization. As noted in Barrera and colleagues, future analysis will require a greater volume of trials with an overall increase in methodological rigor.8
Current research has demonstrated a solid base from which to spawn specific treatment protocols. The available research is investigational in terms of treatment procedures. Replication of these studies could dictate treatment protocol and contribute substantively to future VA and DoD CPG updates. Future researchers should consider the use of a standard PTSD symptom assessment tool to make interstudy comparisons more meaningful. The length of treatment and exposure elements should be targeted specifically in future research as these components currently vary the most.
The military represents an obvious avenue for future research due to increased PTSD diagnosis in recent years. Although the etiology of the increase in PTSD is unclear and most likely multifactorial (decreased resilience, increased awareness, increased pursuit of secondary gains, etc), the need for treatment options is apparent.1 Group cohesion has been shown to be a core component of successful group psychotherapy, so military members who are accustomed to unit cohesion might represent a uniquely suitable population for this modality.15 Interestingly and for reasons not currently understood, veterans do not see effects of therapy as large as their civilian counterparts.8 This underscores the need for further evaluation of military-specific outcomes.
CONCLUSIONS
Although the available evidence is not robust, results do support the careful use of GCBT as an effective treatment for PTSD symptom reduction.8 Group psychotherapy has been generally regarded as an efficacious and cost-effective method to achieve similar outcomes to individual therapy. Increasing PTSD prevalence compels mental health care providers to explore all available treatment options. The potential for GCBT as an option is exciting, especially for mental health providers and those with limited resources. Rising health care standards and the current national fiscal situation is dictating a reevaluation of treatment options; so perhaps all health care providers will soon consider the use of GCBT.
As with any group assignment, the clinician should carefully consider the individual’s suitability and desire for group participation.16 With GCBT, providers could facilitate the relief of relentless apprehension and functional impairment for several patients simultaneously. Although there are many details left to explore regarding the use of GCBT for PTSD, the therapy’s foundation for use as a PTSD treatment is apparent.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Anxiety is a necessary and natural reaction to trauma, but, sometimes, anxiety symptoms become excessive and problematic, as experienced with posttraumatic stress disorder (PTSD). Some patients who struggle with PTSD endure a relentless apprehension so intense that it keeps them from participating in everyday activities, such as attending work and partaking in social activities. Associated anxiety symptoms severely impair everyday function and include increased heart rate, sweating, intrusive images, poor attention, fear, or insomnia. Posttraumatic stress disorder symptoms often lead to occupational dysfunction, relationship difficulty, and numerous other functional impairments.
Approximately 300,000 veterans meet the criteria for PTSD related to ongoing or recent wars.1 The veteran does not bear the personal and functional burden alone; however, the financial load is felt throughout society. One recent study suggests that for veterans diagnosed with PTSD, the first 2 years after deployment cost society an estimated $7,000 per individial.2 Current research suggests that this potentially debilitating disorder occurs in about 14% of Operation Iraqi Freedom/Operation Enduring Freedom combat troops, whereas the similar U.S. demographic population experiences PTSD at a rate of about 7%.1,3 The ongoing military trauma exposures are compelling the mental health community to establish efficient and effective treatment options.4,5
Several treatment strategies exist to reduce PTSD symptoms, but health care professionals must seek a balance between therapeutic benefit and cost. The treatment of PTSD is diverse and variable; however, in the most recent Clinical Practice Guideline (CPG) for PTSD, the VA and DoD specifically endorse some psychotherapeutic interventions while dissuading the use of others.6 Of note, the VA and DoD CPG strongly encourages Stress Inoculation Training (SIT) and similar cognitive therapies aimed at guiding patients through the process of consciously understanding the relationship between thoughts and feelings and then modifying thoughts to appropriately manage stressors.6 Meanwhile, group psychotherapy has been determined to be “somewhat helpful.”6 Even though cognitive- and group-based therapies have long been established as efficacious for numerous psychological disorders (depression, obsessive compulsive disorder, eating disorders, etc), neither the American Group Psychotherapy Association nor the VA and DoD CPG directly endorse the use of group cognitive behavioral therapy (GCBT) for the treatment of PTSD.6,7 However, both VA and DoD mental health providers commonly practice CBT and various group psychotherapies for the treatment of PTSD.
Despite the widespread use of CBT, there is a gap in the clinical understanding of the evidence supporting GCBT for PTSD. The goal of this synthesis was to understand the efficacy of treating PTSD symptoms with group psychotherapy. To begin this investigation, the following PICO (population, intervention, comparison, outcome) question was asked: In adults diagnosed with PTSD, how effective is group cognitive behavioral therapy in reducing PTSD-related symptoms?
Methods
Research articles addressing the use of GCBT in PTSD were obtained via database searches that took place during October 2012 (Table). Searched databases included the Cochrane Central Register of Controlled Trials, Cochrane Database of Systematic Reviews Randomized Controlled Trials, Psychological Information (PsycINFO), and Public Medicine (PubMed).
The PubMed database was searched using the following MeSH (medical subject heading) terms: “psychotherapy, group” and “stress disorders, post-traumatic” and “cognitive therapy.” Limitations were set to include only patients aged ≥ 18 years, results in English, those involving human subjects, and articles published within the past 5 years. A manual search of references was also conducted, and relevant articles were retained.
Articles that addressed primary substance abuse, other DSM Axis I disorders, intimate partner violence, or family issues were excluded from the evidence sample due to concerns of an alternate treatment focus. Articles with a focus on telehealth or alternative medicine were considered confounding to the scope of this review were also excluded. It was also noted that the term CBT is used collectively for an umbrella of treatments; however, treatments that focused on elements other than the components of CBT being delivered in a group were not included. To prevent duplication of the results, research from an inclusive review was not considered individually.
SUMMARY OF EVIDENCE
Six works fulfilled the PICO criteria and were of sufficient quality to be synthesized. Of the 6 articles retained for synthesis, 2 were high-level reviews. Both reviews supported the use of GCBT for PTSD treatment. Barrera and colleagues reported an overall large effect size regardless of the presence of exposure in-group among the 12 treatment conditions and 651 study participants.8 These researchers also reported that in-group exposure did not further traumatize other group members.
Similarly, although a notably older and smaller review, Bisson and Andrew reported a significant standard mean deviation between 4 GCBT treatment and wait list controls. These reviewers did not find a significant difference between trauma- and nontrauma-focused treatment groups. The reviews also noted that individual psychotherapy and/or pharmacotherapy was most often continued throughout the reviewed studies.8,9
The 4 other studies contribute substantively to this synthesis but arguably represent lower evidence quality. A large longitudinal study of 496 Australian veterans reported a large effect size that was sustained 9 months after treatment began.10 These researchers used an intensive outpatient program that included medication and other treatment modalities as the basis for GCBT delivery. They reported that the majority of the patients revealed improvement in PTSD symptoms.
Another study sampled a similar group of 10 combat veterans but focused particular attention on sleep-related PTSD symptoms of insomnia, nightmares, and sleep quality.11 Although these researchers were unable to report a significant difference in overall PTSD symptoms for the 8 subjects who completed the protocol, they did find a large effect size on insomnia severity and a medium effect size on sleep quality. Regular treatment, including medication, continued throughout this study.
Other researchers reported a medium effect size on PTSD symptoms while using GCBT in a heterogeneous group with various anxiety disorders, including obsessive compulsive disorder, generalized anxiety disorder, social phobia, panic disorders, and PTSD.12 Although reporting similar results as all other included studies, this study has some significant limitations, including a 26% dropout rate among the 152 participants. The final study included for synthesis reported a remarkable 67% elimination of the PTSD diagnosis among 6 motor vehicle accident survivors in the small, uncontrolled study.13 Concomitant treatments, including medications, were not reported in detail for these 4 studies except as mentioned.
As a whole, the 6 studies revealed some appreciable commonalities. Time since diagnosis did not seem to influence the results. Attrition was consistently found to be similar to other PTSD treatments. The reported session topics were loosely based on common CBT tenets (ie, education, challenging cognitions, and relaxation techniques) and were typically similar among treatment groups, including the use of homework.
DISCUSSION
As the diagnosis of PTSD increases to unfamiliar levels, GCBT has the potential to be helpful to clinicians and patients seeking alternatives to their current treatments.1,4,14 The reported results imply that GCBT can be useful in PTSD symptom reduction. This could be particularly useful to VA and military providers or rural providers operating with limited resources.
Treatment protocols are not well established and should be approached with care prior to the establishment of CBT treatment groups for those diagnosed with PTSD. Session overviews and descriptions, such as those mentioned in Thompson and colleagues, could provide a reference point for future use.13
Also worth considering, CBT can be an ambiguous term requiring deliberate definition within treatment protocols. As noted in the VA and DoD CPG, exposure- and trauma-focused treatment designs can be efficacious, but these elements do not seem to be required within the GCBT treatment setting.
The current research also suggests GCBT efficacy regardless of the index trauma. This does not suggest that heterogeneous groups were frequently studied nor can conclusions be drawn regarding heterogeneous treatment groups. Elements such as group size and session length are inconsistently reported and require specific consideration as well. There is a distinct lack of research directly comparing individual CBT with GCBT directly, which prohibits meaningful conclusions regarding PTSD symptom reduction. This research gap may well have influenced the recommendations within the VA and DoD CPG. Although some higher quality studies exist, many of the published reports on GCBT have noteworthy design flaw, such as inadequate controls and statistical analysis.
LIMITATIONS
There are some limitations to this literature synthesis. Although the search was limited to the past 5 years, the inclusion of reviews accounts for older evidence. As alluded to earlier, the lack of a standardized GCBT treatment protocol challenges results comparisons as well. The consequent treatment variations make direct interstudy comparison and synthesis difficult. Similarly, outcome measures varied between studies. Also, group psychotherapy is well established and accepted. Therefore, much of the supporting research was accomplished outside the parameters of this literature search. This empirical view of group psychotherapy among mental health providers may also contribute to the lack of available research.
It is also worth noting that studies finding neutral or negative results are often unpublished. This publication bias could account for the lack of available evidence. The research reports do not consistently report therapist qualifications; however, board certificates in group psychotherapy and CBT are undeniably variables available for debate. The inclusion of uncontrolled trials limits these findings as well. Although the above limitations are not exhaustive, they do provide necessary caveats to future generalizations.
FUTURE IMPLICATIONS
Perhaps the most important information to gain from future research is that of treatment outcomes. Studies that include a detailed outcome evaluation could reveal patient satisfaction, efficacy, and financial considerations. In the presence of adequate supportive data, GCBT could contribute outcome data regarding trauma survivor symptom normalization, peer support formation, access to care, treatment efficiency, and health care resources utilization. As noted in Barrera and colleagues, future analysis will require a greater volume of trials with an overall increase in methodological rigor.8
Current research has demonstrated a solid base from which to spawn specific treatment protocols. The available research is investigational in terms of treatment procedures. Replication of these studies could dictate treatment protocol and contribute substantively to future VA and DoD CPG updates. Future researchers should consider the use of a standard PTSD symptom assessment tool to make interstudy comparisons more meaningful. The length of treatment and exposure elements should be targeted specifically in future research as these components currently vary the most.
The military represents an obvious avenue for future research due to increased PTSD diagnosis in recent years. Although the etiology of the increase in PTSD is unclear and most likely multifactorial (decreased resilience, increased awareness, increased pursuit of secondary gains, etc), the need for treatment options is apparent.1 Group cohesion has been shown to be a core component of successful group psychotherapy, so military members who are accustomed to unit cohesion might represent a uniquely suitable population for this modality.15 Interestingly and for reasons not currently understood, veterans do not see effects of therapy as large as their civilian counterparts.8 This underscores the need for further evaluation of military-specific outcomes.
CONCLUSIONS
Although the available evidence is not robust, results do support the careful use of GCBT as an effective treatment for PTSD symptom reduction.8 Group psychotherapy has been generally regarded as an efficacious and cost-effective method to achieve similar outcomes to individual therapy. Increasing PTSD prevalence compels mental health care providers to explore all available treatment options. The potential for GCBT as an option is exciting, especially for mental health providers and those with limited resources. Rising health care standards and the current national fiscal situation is dictating a reevaluation of treatment options; so perhaps all health care providers will soon consider the use of GCBT.
As with any group assignment, the clinician should carefully consider the individual’s suitability and desire for group participation.16 With GCBT, providers could facilitate the relief of relentless apprehension and functional impairment for several patients simultaneously. Although there are many details left to explore regarding the use of GCBT for PTSD, the therapy’s foundation for use as a PTSD treatment is apparent.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Litz B, Schlenger W. Posttraumatic stress disorder in service members and new veterans of the Iraq and Afghanistan wars: A bibliography and critique. PTSD Res Q. 2009;20(1):1-3.
2. Tanielian T. Assessing combat exposure and post-traumatic stress disorder in troops and estimating the costs to society: Implications from the RAND Invisible Wounds of War Study. http://www.rand.org/pubs/testimonies/CT321.html. Published 2009. Accessed September 29, 2014.
3. Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):617-627.
4. Cukor J, Spitalnick J, Difede J, Rizzo A, Rothbaum BO. Emerging treatments for PTSD. Clinical Psychol Rev. 2009;29(8):715-726.
5. Hoge CW. Interventions for war-related posttraumatic stress disorder: Meeting veterans where they are. JAMA. 2011;306(5):549-551.
6. Veterans Health Administration, Department of Defense. VA/DoD Clinical Practice Guideline: Management of Post-Traumatic Stress, Version 2.0. Washington, DC: Veterans Health Administration and Department of Defense; 2010.
7. Burlingame GM, Fuhriman A, Mosier J. The differential effectiveness of group psychotherapy: A meta-analytic perspective. Group Dyn. 2003;7(1):3-12.
8. Barrera TL, Mott JM, Hofstein RF, Teng EJ. A meta-analytic review of exposure in group cognitive behavioral therapy for posttraumatic stress disorder. Clin Psychol Rev. 2013;33(1):24-32.
9. Bisson J, Andrew M. Psychological treatment of post-traumatic stress disorder (PTSD). Cochrane Database Syst Rev. 2007;(3):CD003388.
10. Khoo A, Dent MT, Oei TS. (2011). Group cognitive behaviour therapy for military service-related post-traumatic stress disorder: Effectiveness, sustainability and repeatability. Aust N Z J Psychiatry. 2011;45(8):663-672.
11. Swanson LM, Favorite TK, Horin E, Arnedt JT. A combined group treatment for nightmares and insomnia in combat veterans: A pilot study. J Trauma Stress. 2009;22(6):639-642.
12. Erickson DH, Janeck A, Tallman K. A cognitive-behavioral group for patients with various anxiety disorders. Psychiatr Serv. 2007;58(9):1205-1211.
13. Thompson AR, Wilde E, Boon K. The development of group CBT for the treatment of road-traffic-accident-related post-traumatic stress disorder: A pilot study. Cognitive Behav Therapist. 2010;2(1):32-42.
14. Slade T, Johnston A, Oakley-Browne MA, Andrews G, Whiteford H. 2007 National Survey of Mental Health and Wellbeing: Methods and key findings. Aust N Z J Psychiatry. 2009;43(7):594-605.
15. Crowe TP, Grenyer BF. Is therapist alliance or whole group cohesion more influential in group psychotherapy outcomes? Clin Psychol Psychother. 2008;15(4):239-246.
16. Leszcz M, Kobos JC. Evidence-based group psychotherapy: Using AGPA’s practice guidelines to enhance clinical effectiveness. J Clin Psychol. 2008;64(11):1238-1260.
1. Litz B, Schlenger W. Posttraumatic stress disorder in service members and new veterans of the Iraq and Afghanistan wars: A bibliography and critique. PTSD Res Q. 2009;20(1):1-3.
2. Tanielian T. Assessing combat exposure and post-traumatic stress disorder in troops and estimating the costs to society: Implications from the RAND Invisible Wounds of War Study. http://www.rand.org/pubs/testimonies/CT321.html. Published 2009. Accessed September 29, 2014.
3. Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):617-627.
4. Cukor J, Spitalnick J, Difede J, Rizzo A, Rothbaum BO. Emerging treatments for PTSD. Clinical Psychol Rev. 2009;29(8):715-726.
5. Hoge CW. Interventions for war-related posttraumatic stress disorder: Meeting veterans where they are. JAMA. 2011;306(5):549-551.
6. Veterans Health Administration, Department of Defense. VA/DoD Clinical Practice Guideline: Management of Post-Traumatic Stress, Version 2.0. Washington, DC: Veterans Health Administration and Department of Defense; 2010.
7. Burlingame GM, Fuhriman A, Mosier J. The differential effectiveness of group psychotherapy: A meta-analytic perspective. Group Dyn. 2003;7(1):3-12.
8. Barrera TL, Mott JM, Hofstein RF, Teng EJ. A meta-analytic review of exposure in group cognitive behavioral therapy for posttraumatic stress disorder. Clin Psychol Rev. 2013;33(1):24-32.
9. Bisson J, Andrew M. Psychological treatment of post-traumatic stress disorder (PTSD). Cochrane Database Syst Rev. 2007;(3):CD003388.
10. Khoo A, Dent MT, Oei TS. (2011). Group cognitive behaviour therapy for military service-related post-traumatic stress disorder: Effectiveness, sustainability and repeatability. Aust N Z J Psychiatry. 2011;45(8):663-672.
11. Swanson LM, Favorite TK, Horin E, Arnedt JT. A combined group treatment for nightmares and insomnia in combat veterans: A pilot study. J Trauma Stress. 2009;22(6):639-642.
12. Erickson DH, Janeck A, Tallman K. A cognitive-behavioral group for patients with various anxiety disorders. Psychiatr Serv. 2007;58(9):1205-1211.
13. Thompson AR, Wilde E, Boon K. The development of group CBT for the treatment of road-traffic-accident-related post-traumatic stress disorder: A pilot study. Cognitive Behav Therapist. 2010;2(1):32-42.
14. Slade T, Johnston A, Oakley-Browne MA, Andrews G, Whiteford H. 2007 National Survey of Mental Health and Wellbeing: Methods and key findings. Aust N Z J Psychiatry. 2009;43(7):594-605.
15. Crowe TP, Grenyer BF. Is therapist alliance or whole group cohesion more influential in group psychotherapy outcomes? Clin Psychol Psychother. 2008;15(4):239-246.
16. Leszcz M, Kobos JC. Evidence-based group psychotherapy: Using AGPA’s practice guidelines to enhance clinical effectiveness. J Clin Psychol. 2008;64(11):1238-1260.
Stent Thrombosis: A Disease for All Clinicians
Percutaneous coronary intervention (PCI) using coronary artery stent implantation is commonly used to treat symptomatic high-risk and unstable coronary artery disease (CAD). The use of stents has improved the safety and efficacy of PCI by reducing the need for repeat revascularization, reducing acute vessel closure requiring emergent coronary artery bypass graft surgery, and expanding the use of PCI to more complex diseases. Nevertheless, stents carry the risk of sudden thrombotic occlusion or stent thrombosis, particularly during the first several days or weeks after implantation. In turn, stent thrombosis can lead to acute myocardial infarction (MI) and a mortality rate > 25%.1,2
This article highlights 2 cases of patients with stent thrombosis and discusses its pathophysiology, clinical features, and risk-avoidance strategies. Given the high prevalence of CAD and ubiquitous PCI procedures in the U.S. health care system, it is essential that not only cardiologists, but all clinicians and health care providers who care for patients with coronary stents understand how to help prevent and manage this life-threatening clinical entity.1
Case 1
A 56-year-old man presented to his primary care physician with exertion-related angina. The patient had a history of type 2 diabetes mellitus, dyslipidemia, systemic hypertension, obesity, and CAD status post MI in 2002 treated with a bare metal stent (BMS) to the left circumflex coronary artery (LCx). A stress myocardial perfusion imaging with 99mTc-sestamibi revealed moderate reversible exercise-induced myocardial ischemia involving the inferior and inferoapical wall segments of the left ventricle with associated hypokinesia.
Coronary angiography revealed nonsignificant disease of the left anterior descending artery (LAD) and LCx, a patent LCx stent, and a 95% mid-right coronary artery (RCA) obstruction with delayed (TIMI grade 2) antegrade flow. The distal right posterior descending artery filled via left to right collaterals from the LAD.
Percutaneous coronary intervention was performed on the RCA lesion 8 days after the patient was started on dual antiplatelet therapy (DAPT) with aspirin 81 mg and clopidogrel 75 mg (including 300 mg loading dose on the day of the diagnostic angiogram). The mid RCA was treated with a drug-eluting stent (DES) and a BMS in a nonoverlapping fashion with an excellent angiographic result. The patient was instructed to continue DAPT with aspirin 325 mg daily and clopidogrel 75 mg daily for 12 months.
Three days post PCI, the patient arrived at the emergency department with angina of 1-hour duration associated with shortness of breath and diaphoresis. He reported strict adherence to DAPT.
Initial vital signs were normal. The electrocardiogram (ECG) showed ST segment elevation (1-2 mm) on leads III, aVF, and V5 to V6, suggestive of an acute inferolateral injury pattern for which emergent coronary angiography was performed. Angiography showed a 100% proximal RCA occlusion at the proximal edge of the most proximal stent with absence of any antegrade flow beyond the occlusion (TIMI grade 0 flow). This finding was diagnostic of definite angiographic subacute stent thrombosis. The patient underwent successful aspiration thrombectomy, balloon angioplasty, and restoration of normal TIMI grade 3 flow with a door-to-balloon time of 86 minutes.
Because stent thrombosis is relatively unexpected after an excellent angiographic result and DAPT adherence, the possibility of clopidogrel resistance was considered as a major contributor for the thrombotic event. Platelet aggregation tests showed adequate prolongation of collagen/epinephrine (COL-EPI) > 300 seconds (normal: 81-153 seconds), but inadequate prolongation of collagen/adenosindiphosphate (COL-ADP) of 109 seconds (normal: 53-105 seconds) while on clopidogrel. Therefore, the patient was switched to prasugrel.
The patient was discharged home after 5 days of observation at the cardiac care unit without any post-MI complications. During a follow-up appointment 1 month after discharge, he was clinically stable and free of cardiovascular symptoms. Workup performed for acquired or inherited thrombophilia was negative. He continued taking DAPT (daily aspirin 325 mg orally and prasugrel 10 mg orally) for 12 months. After completing 12 months of DAPT, he was maintained on aspirin 81 mg daily. At 24 months’ follow-up, he remained free of recurrent angina with no further cardiovascular events.
Case 2
An 84-year-old man with a medical history of dyslipidemia, paroxysmal atrial fibrillation, previous stroke, and peptic ulcer disease was brought to the emergency department following an episode of near syncope in the early morning hours. The patient revealed that he had experienced neck pain since midnight. The 12-lead ECG showed normal sinus rhythm with 2 mm ST segment elevation in leads II, III, aVF, V5-V6, and ST segment depression in V2, and Q waves in inferior leads. A right-sided ECG showed ST segment elevation in V4, suggestive of right ventricle infarction.
The patient remained hypotensive (83/49 mm Hg) despite isotonic fluid administration (about 1.5-2.0 liters of 0.9 normal saline at 999 mL/h). A dopamine drip for persistent hypotension was started, and he was taken emergently to the catheterization laboratory for primary PCI. Coronary angiography showed no significant left CAD and a 100% mid-RCA occlusion with faint left-to-right collaterals. After aspiration thrombectomy, bare metal RCA stenting was performed. Transient no-reflow was treated with intracoronary nicardipine and nitroglycerin. The patient continued to be in shock, and an intra-aortic balloon pump was inserted and 1:1 counterpulsation was initiated.
Following admission to the coronary care unit, the patient’s mean arterial pressure improved. Inotropes were weaned off 2 days after PCI, and the intra-aortic balloon pump was removed. During his stay, the post-MI course was uneventful except for an episode of asymptomatic paroxysmal atrial flutter and nonspecific back dermatitis attributed to a prolonged recumbent position.
The patient was transferred to the internal medicine ward for medical therapy optimization and the initiation of low-intensity cardiac rehabilitation. After 2 days on the ward, discharge planning was initiated. However, he developed an episode of atrial fibrillation with fast ventricular response. Metoprolol 5 mg IV bolus was given, and the ventricular rate was controlled. At that point, the dose of long-acting beta-blocker (metoprolol succinate) was optimized, he was started on full-dose anticoagulation (warfarin), and clopidogrel was discontinued. Two days later, the patient reported back pruritus, and an erythematous raised rash on his back spreading to the torso was noticed. An aspirin allergy was suspected as the trigger for the rash, thus aspirin was also discontinued.
Three days later, the patient developed recurrent neck pain (angina) with radiation to his shoulders and left arm. The ECG revealed re-elevation of the ST segment (inferior, posterior, and lateral leads). He received reloading of clopidogrel 600 mg and aspirin 325 mg. Also, an eptifibatide IV bolus followed by an infusion was given for immediate antiplatelet action. He was transferred for emergent coronary angiography with suspected subacute stent thrombosis.
Upon arrival to the catheterization lab, the patient was awake and alert but in mild respiratory distress. Intravenous dopamine was started due to hypotension (systolic blood pressure was about 85 mm Hg). Limited RCA angiography showed a large clot burden with a partially thrombosed stent and TIMI grade 3 flow. After intracoronary eptifibatide and nicardipine were given, successful aspiration thrombectomy was performed twice with partial removal of thrombus. In-stent high-pressure balloon angioplasty was performed and optimal stenting was confirmed by intravascular ultrasound (IVUS) criteria. However, a residual layered thrombus along the distal stent edge was noticed. The patient tolerated the procedure without complications.
Dual antiplatelet therapy with aspirin and clopidogrel for 12 months was recommended. The eptifibatide infusion was continued for 48 hours. The jaw pain, shortness of breath, and ECG changes disappeared, but the patient remained on vasopressors for the following 7 days.
Around 1 week after the stent thrombosis event, the patient was found pulseless. Advanced cardiopulmonary resuscitation was started. ST segment elevation in lead II was noted on the cardiac monitor. There was no return of spontaneous circulation after 20 minutes, and the patient was pronounced dead. The autopsy revealed a patent RCA stent without evidence of occlusion, a large transmural inferior MI, left ventricular rupture, and hemopericardium.
Discussion
Stent thrombosis is an uncommon complication after coronary stent implantation. Based on the Academic Research Consortium criteria, definite stent thrombosis is defined as a clinical event with symptoms suggestive of an acute coronary syndrome (ACS) with angiography or pathology that confirms the presence of stent thrombosis.2 Probable stent thrombosis is defined as an unexplained death within 30 days or MI involving the territory of the target vessel without angiographic confirmation of stent thrombosis.2 Finally, possible stent thrombosis is any unexplained death after 30 days.2
Based on timing, stent thrombosis is divided by acute (< 24 hours post stent implantation), subacute (24 hours to 30 days post stent implantation), late (> 30 days post stent implantation), and very late (> 12 months post stent implantation).3 However, most cases (up to 60%) occur within the first 30 days after placement, irrespective of stent type.4
The incidence of subacute stent thrombosis is reported to approach 1% during the first 30 days postprocedure but may be as high as 5% or 10% depending on associated clinical and angiographic variables (Table 1).5 The strongest clinical predictors of stent thrombosis are premature cessation of antiplatelet therapy, renal insufficiency, diabetes mellitus, and ACS.2,6 Lesion and procedural characteristics associated with increased risk of stent thrombosis include bifurcation lesions, longer stent length, multiple implanted stents, stent underexpansion, and/or stent malapposition.6-9 Stent type (drug or non–drug-eluting) has no impact on the risk of stent thrombosis during the first 30 days postprocedure.10,11

The clinical events related to late stent thrombosis, although rare, carry a mortality rate of up to 45%.12 The specific risk factors for late and very late stent thrombosis are less well defined but relate to delayed neointimal coverage, ongoing vessel inflammation, and the development of neoatherosclerosis within stents.13,14
Rationale for the Use of Dual Antiplatelet Regimen
Stent thrombosis is a platelet-mediated process related to a heightened state of systemic and intracoronary thrombogenicity and inflammation.15 Stent under-expansion enhances abnormal shear stress, which explains as many as 80% of these events.13,15,16 Stent thrombosis also has been frequently related to inadequate neointimal coverage.14 Angioscopic studies, especially with DES, suggest that stent endothelialization is delayed or incomplete, observing a correlation between the areas of uncovered stent surface and thrombosis.14,17
In the early days of coronary stenting, during the 1990s, the risk of acute and subacute stent thrombosis approached 20%.18,19 Initial attempts to reduce the risk included combining aspirin and warfarin, but at the expense of a marked increase in bleeding complications and prolonged hospital stays.20,21 In 1995, it became clear through the pivotal observations of Colombo and colleagues that incomplete expansion of the stent (documented by IVUS) was a major contributor to the risk of stent thrombosis.16 By using noncompliant balloons at high pressure (14-20 atmospheres) for stent postdilatation combined with DAPT (aspirin and ticlopidine), the high rates of early stent thrombosis were markedly reduced to the current level of 1% to 2%.16
Colombo and colleagues’ observations were prospectively evaluated in the Stent Anticoagulation Regimen Study (STARS) trial.22 Patients who underwent successful stenting were randomized to aspirin alone, aspirin and warfarin, or aspirin and ticlopidine. The STARS trial showed convincingly that the combination of aspirin and ticlopidine was superior to the other 2 regimens, reducing the stent thrombosis rate to only 0.5% (compared with 2.7% for aspirin and warfarin, and 3.6% for aspirin alone).22 Afterward, DAPT became the standard of care following coronary stenting.23
Although ticlopidine was the first widely used thienopyridine for the prevention of stent thrombosis, hematologic adverse events (AEs) (eg, neutropenia, thrombotic thrombocytopenia purpura) limited its use.24 Consequently, ticlopidine was replaced with clopidogrel, which seemed to offer similar efficacy but significantly fewer AEs.25
The current American College of Cardiology/American Heart Association/Society for Cardiovascular Angiography and Interventions (ACC/AHA/SCAI) guidelines for the prevention of ST after coronary stent implantation state that after PCI:
- Aspirin use should be continued indefinitely.
- The duration of adenosine diphosphate antagonists depends on the stent type (BMS or DES) and the indication for implantation (ACS or non-ACS).
a. Patients receiving a stent (BMS or DES) for ACS therapy should be given 1 of the following for at least 12 months:
i. Clopidogrel 75 mg daily
ii. Prasugrel 10 mg daily
iii. Ticagrelor 90 mg twice daily
b. In patients receiving DES for a non-ACS indication, clopidogrel should be given for at least 12 months if the patient is not at high risk for bleeding.
c. In patients receiving BMS for a non-ACS indication, clopidogrel should be given for a minimum of 1 month and ideally up to 12 months.23
Clopidogrel Hyporesponse
As shown in case 1, stent thrombosis may still occur in a patient on DAPT because of individual variability in platelet response to clopidogrel.5 Clopidogrel hyporesponse, also known as clopidogrel resistance, has been recognized as clinically significant because of its prevalence and association with poor outcomes.5 Its prevalence may range between 4% and 30%, although the definitions of clopidogrel hyporesponse varied between studies.26
Clopidogrel hyporesponse is defined as an inadequate inhibition of platelet function measured by nonspecific ex-vivo laboratory methods.27,28 The relationship between clopidogrel resistance (nonresponders), stent thrombosis, and ischemic events has been clearly established.5,29
Given the devastating consequences of stent thrombosis, efforts were directed to identify those patients at highest risk. One such effort has been focused on the measurement of platelet function, allowing for the identification of patients who do not respond adequately to antiplatelet therapy.15,28,30,31 However, the treatment of high-residual platelet reactivity as confirmed by laboratory assessment has not shown to clinically correlate with any benefit in the prevention of ST.6,15,29-31 Therefore, the current ACC/AHA/SCAI PCI guidelines do not recommend the routine clinical use of platelet function testing to screen patients treated with clopidogrel who are undergoing PCI.23
Clopidogrel is a prodrug, metabolized to its active form via the cytochrome P450 enzyme system before it can inhibit platelet function.32 Accordingly, certain genetic variation in enzyme activity, or polymorphisms, would be expected to influence its clinical effectiveness.33,34 The most common of these polymorphisms, CYP2C19*2, has been associated (in vitro) with reduced concentrations of active clopidogrel metabolites and with diminished platelet inhibition.35,36 As a result, the FDA has added a safety alert to the prescribing information for clopidogrel concerning how genetic differences in the metabolism of this agent can affect its effectiveness, ways to test for these genetic differences, and advice concerning alternative dosing strategies or use of other medications in poor metabolizers of clopidogrel.37 Although the routine clinical use of genetic testing to screen patients treated with clopidogrel who are undergoing PCI is not recommended, it may be considered in patients undergoing elective high-risk PCI procedures (eg, unprotected left main, last patent coronary artery, or bifurcating left main).23
The newer inhibitors of ADP-induced platelet activation, prasugrel and ticagrelor, are not prodrugs, and thus, their action is not affected by this genetic variability. Accordingly, these drugs have shown a more consistent, stronger, and faster inhibition of platelet aggregation compared with clopidogrel.36-39 In the pivotal trials (TRITON-TIMI 38 and PLATO), these agents have also been shown to be more effective in reducing the incidence of stent thrombosis.36,37,40,41 Therefore, in cases where clopidogrel resistance/hyporesponse is suspected in the setting of DAPT, such as stent thrombosis, guidelines recommend the use of 1 of these agents.23
Premature Discontinuation of Antiplatelet Therapy
As illustrated in case 2, premature discontinuation of antiplatelet therapy may be fatal, as it is associated with a marked increase in the risk of stent thrombosis. Indeed, premature discontinuation of DAPT is the leading independent predictor for stent thrombosis.12,42,43 Premature discontinuation of DAPT is defined when one or both agents (aspirin, ADP-antagonists) are suspended within 30 days of BMS placement or within 1 year of DES placement. In the case of DES, the first 6 months after implantation seem to be most critical. In a large observational study of patients treated with DES, stent thrombosis occurred in 29% of those patients in whom antiplatelet therapy was prematurely discontinued.12
In order to minimize the risk of premature DAPT discontinuation, one should address its causes. There are patient- and physician-related factors that may influence an early discontinuation of aspirin, thienopyridine, or both agents. Patient-related factors were identified in the PREMIER registry, including older age, not having completed high school, not being married, and/or not seeking health care because of costs.42 Another important but often overlooked factor that has an impact on adherence with prolonged DAPT post-DES implantation is nuisance or superficial bleeding.44 Physician-related factors include not providing discharge instructions for medication use and ill-advised instructions given by health care providers to discontinue therapy before procedures with a low risk of bleeding (eg, dental cleaning, cataract surgery, colonoscopy, skin biopsy).42
In addition, the perioperative management of DAPT during the first several weeks after coronary stenting has been shown to critically influence outcomes. In a study by Sharma and colleagues, fatal cases of stent thrombosis occurred after the discontinuation of antiplatelet therapy for noncardiac surgery among patients with BMS implantation within the past 90 days.43
In selected cases when a noncardiac procedure cannot be delayed for 1 year, recognizing the impact of the specific timing for the discontinuation of the antiplatelet regimen is essential. Kaluza and colleagues reported on 40 patients treated with BMS who underwent noncardiac surgery within 6 weeks of the stent implantation.45 Seven patients had an MI, of which 6 were fatal. Stent thrombosis was presumed to be the cause of all MIs. In 5 of 7 cases, ticlopidine was withheld before surgery.45
All clinicians should be aware of the following recommendations to avoid catastrophic cardiovascular complications related to premature discontinuation of DAPT during the perioperative setting:
- Elective procedures should be deferred until patients have completed an appropriate course of thienopyridine therapy (12 months after DES and a minimum of 4 weeks for BMS implantation).
- For those patients treated with DES who are to undergo a nonelective procedure that mandates discontinuation of thienopyridine therapy, the possibility of procedure postponement for completion of DAPT for at least 6 months should be judiciously deliberated. If the procedure cannot be postponed, aspirin should be continued if at all possible and the thienopyridine restarted as soon as possible after the procedure.42,46,47
Conclusion
Stent thrombosis is a rare but devastating complication of coronary stent implantation. Although it can occur at any time after stent placement, the majority of events occur within the first month. The use of optimal stenting techniques and adherence to DAPT are required to minimize the risk of stent thrombosis. Several clinical and procedural predictors have been related to an increased risk of stent thrombosis. The premature cessation of DAPT is the most important risk factor for stent thrombosis.
All physicians should ensure patients are properly and thoroughly educated about the reasons they are prescribed DAPT and the significant risks associated with prematurely discontinuing such therapy. All clinicians, especially noncardiologists, should realize the importance of close communication with a cardiologist or interventional cardiologist in situations when premature discontinuation is being considered for a specific reason.
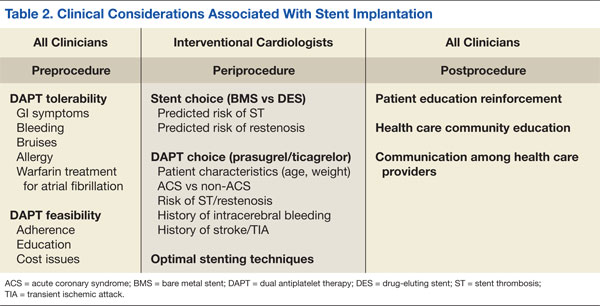
Table 2 summarizes a framework of the most relevant factors that should be taken into account before, during, and after stent implantation, both by interventional cardiologists, as well as by all clinicians involved in the care of the patient. Given current procedural volumes (> 1 million PCI procedures are performed in the U.S. annually) and because the risk of stent thrombosis is both time and treatment dependent, it is of paramount importance that, not only cardiologists, but all physicians know the impact of stent thrombosis in their patients and how to avoid situations that may increase its risk.1 Team-approach decisions about antiplatelet therapy after stent placement, especially within the first 12 months, and a patient-centered mind-set are indispensable to optimize patient outcomes.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Ryan J, Cohen DJ. Are drug-eluting stents cost-effective? It depends on whom you ask. Circulation. 2006;114(16):1736-1744.
2. Cutlip DE, Windecker S, Mehran R, et al; Academic Research Consortium. Clinical end points in coronary stent trials: A case for standardized definitions. Circulation. 2007;115(17):2344-2351.
3. Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med. 2001;345(7):494-502.
4. Palmerini T, Kirtane AJ, Serruys PW, et al. Stent thrombosis with everolimus-eluting stents: Meta-analysis of comparative randomized controlled trials. Circ Cardiovasc Interv. 2012;5(3):357-364.
5. Angiolillo DJ, Fernandez-Ortiz A, Bernardo E, et al. Variability in individual responsiveness to clopidogrel: Clinical implications, management, and future perspectives. J Am Coll Cardiol. 2007;49(14):1505-1516.
6. Moussa I, Di Mario C, Reimers B, Akiyama T, Tobis J, Colombo A. Subacute stent thrombosis in the era of intravascular ultrasound-guided coronary stenting without anticoagulation: Frequency, predictors and clinical outcome. J Am Coll Cardiol. 1997;29(1):6-12.
7. Fujii K, Carlier SG, Mintz GS, et al. Stent underexpansion and residual reference segment stenosis are related to stent thrombosis after sirolimus-eluting stent implantation: An intravascular ultrasound study. J Am Coll Cardiol. 2005;45(7):995-998.
8. Uren NG, Schwarzacher SP, Metz JA, et al; POST Registry Investigators. Predictors and outcomes of stent thrombosis: An intravascular ultrasound registry. Eur Heart J. 2002;23(2):124-132.
9. Cook S, Wenaweser P, Togni M, et al. Intravascular ultrasound in very late DES-stent thrombosis (abstr). J Am Coll Cardiol. 2006;47(suppl B):9B.
10. Moreno R, Fernández C, Hernández R, et al. Drug-eluting stent thrombosis: Results from a pooled analysis including 10 randomized studies. J Am Coll Cardiol. 2005;45(6):954-959.
11. Ellis SG, Colombo A, Grube E, et al. Incidence, timing, and correlates of stent thrombosis with the polymeric paclitaxel drug-eluting stent: A TAXUS II, IV, V, and VI meta-analysis of 3,445 patients followed for up to 3 years. J Am Coll Cardiol. 2007;49(10):1043-1051.
12. Iakovou I, Schmidt T, Bonizzoni E, et al. Incidence, predictors, and outcome of thrombosis after successful implantation of drug-eluting stents. JAMA. 2005;293(17):2126-2130.
13. Cutlip DE, Baim DS, Ho KK, et al. Stent thrombosis in the modern era: A pooled analysis of multicenter coronary stent clinical trials. Circulation. 2001;103(15):1967-1971.
14. Kotani J, Awata M, Nanto S, et al. Incomplete neointimal coverage of sirolimus-eluting stents: Angioscopic findings. J Am Coll Cardiol. 2006;47(10):2108–2111.
15. Cheneau E, Leborgne L, Mintz GS, et al. Predictors of subacute stent thrombosis: Results of a systematic intravascular ultrasound study. Circulation. 2003;108(1):43-47.
16. Colombo A, Hall P, Nakamura S, et al. Intracoronary stenting without anticoagulation achieved with intravascular ultrasound guidance. Circulation. 1995;91(6):1676-1688.
17. Oyabu J, Ueda Y, Ogasawara N, Okada K, Hirayama A, Kodama K. Angioscopic evaluation of neointima coverage: Sirolimus-drug eluting stent versus bare metal stent. Am Heart J. 2006;152(6):1168-1174.
18. Serruys PW, Strauss BH, Beatt KJ, et al. Angiographic follow-up after placement of a self-expanding coronary-artery stent. N Engl J Med. 1991;324(1):13-17.
19. Schatz RA, Baim DS, Leon M, et al. Clinical experience with the Palmaz-Schatz coronary stent. Initial results of a multicenter study. Circulation. 1991:83(1):148-161.
20. Fischman DL, Leon MB, Baim DS, et al. A randomized comparison of coronary-stent placement and balloon angioplasty in the treatment of coronary artery disease. Stent Restenosis Study Investigators. N Engl J Med. 1994;331(8):496-501.
21. Serruys PW, de Jaegere P, Kiemeneij F, et al. A comparison of balloon-expandable-stent implantation with balloon angioplasty in patients with coronary artery disease. Benestent Study Group. N Engl J Med. 1994;331(8):489-495.
22. Leon MD, Baim DS, Gordon P, et al. Clinical and angiographic results from the STent Anticoagulation Regimen Study (STARS) (abstr). Circulation. 1996;94(suppl I):I-685.
23. Levine GN, Bates ER, Blankenship JC, et al. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention: A report of the American College of Cardiology Foundation/American Heart Association Task Force of Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. J Am Coll Cardiol. 2011;58(24):e44-e122.
24. Bennett CL, Davidson CJ, Raisch DW, Weinberg PD, Bennett RH, Feldman MD. Thrombotic thrombocytopenic purpura associated with ticlopidine in the setting of coronary artery stents and stroke prevention. Arch Intern Med. 1999;159(21):2524-2528.
25. Bertrand ME, Rupprecht HJ, Urban P, Gershlick AH; CLASSICS Investigators. Double-blind study of the safety of clopidogrel with and without a loading dose in combination with aspirin compared with ticlopidine in combination with aspirin after coronary stenting: The clopidogrel aspirin stent international cooperative study (CLASSICS). Circulation. 2000;102(6):624-629.
26. Wang, TH, Bhatt DL, Topol EJ. Aspirin and clopidogrel resistance: An emerging clinical entity. Eur Heart J. 2006;27(6):647-654.
27. Vats HS, Hocking WG, Rezkalla SH. Suspected clopidogrel resistance in a patient with acute stent thrombosis. Nat Clin Pract Cardiovasc Med. 2006;3(4):226-230.
28. Gurbel PA, Becker RC, Mann KG, Steinhubl SR, Michelson AD. Platelet function monitoring in patients with coronary artery disease. J Am Coll Cardiol. 2007;50(19):1822-1834.
29. Fitzgerald DJ, Maree A. Aspirin and clopidogrel resistance. Hematology Am Soc Hematol Educ Program. 2007;2007(1):114-120.
30. Cattaneo M. Resistance to antiplatelet drugs: Molecular mechanisms and laboratory detection. J Thromb Haemost. 2007;5(suppl 1):230-237.
31. Trenk D, Hochholzer W, Fromm MF, et al. Cytochrome P450 2C19 681G>A polymorphism and high on-clopidogrel platelet reactivity associated with adverse 1-year clinical outcome of elective percutaneous coronary intervention with drug-eluting or bare-metal stents. J Am Coll Cardiol. 2008;51(20):1925-1934.
32. Kazui M, Nishiya Y, Ishizuka T, et al. Identification of the human cytochrome P450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab Dispos. 2010;38(1):92-99.
33. Hulot JS, Bura A, Villard E, et al. Cytochrome P450 2C19 loss-of-function polymorphism is a major determinant of clopidogrel responsiveness in healthy subjects. Blood. 2006;108(7):2244-2247.
34. Mega JL, Close SL, Wiviott SD, et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med. 2009;360(4):354-362.
35. U.S. Food and Drug Administration. Plavix (clopidogrel): Reduced effectiveness in patients who are poor metabolizers of the drug. U.S. Food and Drug Administration Website. http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm204256.htm. Updated September 6, 2013. Accessed September 4, 2014.
36. Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Intensive oral antiplatelet therapy for reduction of ischaemic events including stent thrombosis in patients with acute coronary syndromes treated with percutaneous coronary intervention and stenting in the TRITON-TIMI 38 trial: A subanalysis of a randomised trial. Lancet. 2008;371(9621):1353-1363.
37. Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361(11):1045-1057.
38. Wallentin L, Varenhorst C, James S, et al. Prasugrel achieves greater and faster P2Y12 receptor-mediated platelet inhibition than clopidogrel due to more efficient generation of its active metabolite in aspirin-treated patients with coronary artery disease. Eur Heart J. 2008;29(1):21-30.
39. Gurbel PA, Bliden KP, Butler K, et al. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: The ONSET/OFFSET study. Circulation. 2009;120(25):2577-2585.
40. Wiviott SD, Trenk D, Frelinger AL, et al; PRINCIPLE-TIMI 44 Investigators. Prasugrel compared with high loading- and maintenance-dose clopidogrel in patients with planned percutaneous coronary intervention: The Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation-Thrombolysis in Myocardial Infarction 44 trial. Circulation. 2007;116(25):2923-2932.
41. Gurbel PA, Bliden KP, Butler K, et al. Response to ticagrelor in clopidogrel nonresponders and responders and effect of switching therapies: The RESPOND study. Circulation. 2010;121(10):1188-1199.
42. Spertus, JA, Kettelkamp R, Vance C, et al. Prevalence, predictors, and outcomes of premature discontinuation of thienopyridine therapy after drug-eluting stent placement: Results from the PREMIER registry. Circulation. 2006;113(24):2803-2809.
43. Sharma AK, Ajani AE, Hamwi SM, et al. Major noncardiac surgery following coronary stenting: When is it safe to operate? Catheter Cardiovasc Interv. 2004;63(2):141-145.
44. Ben-Dor I, Torguson R, Scheinowitz M, et al. Incidence, correlates, and clinical impact of nuisance bleeding after antiplatelet therapy for patients with drug-eluting stents. Am Heart J. 2010;159(5):871-875.
45. Kaluza GL, Joseph J, Lee JR, Raizner ME, Raizner AE. Catastrophic outcomes of noncardiac surgery soon after coronary stenting. J Am Coll Cardiol. 2000;35(5):1288-1294.
46. Airoldi F, Colombo A, Morici N, et al. Incidence and predictors of drug-eluting stent thrombosis during and after discontinuation of thienopyridine treatment. Circulation. 2007;116(7):745-754.
47. Grines CL, Bonow RO, Casey DE Jr, et al; American Heart Association; American College of Cardiology; Society for Cardiovascular Angiography and Interventions; American College of Surgeons; American Dental Association; American College of Physicians. Prevention of premature discontinuation of dual antiplatelet therapy in patients with coronary artery stents: A science advisory from the American Heart Association, American College of Cardiology, Society for Cardiovascular Angiography and Interventions, American College of Surgeons, and American Dental Association, with representation from the American College of Physicians. J Am Coll Cardiol. 2007;49(6):734-739.
Percutaneous coronary intervention (PCI) using coronary artery stent implantation is commonly used to treat symptomatic high-risk and unstable coronary artery disease (CAD). The use of stents has improved the safety and efficacy of PCI by reducing the need for repeat revascularization, reducing acute vessel closure requiring emergent coronary artery bypass graft surgery, and expanding the use of PCI to more complex diseases. Nevertheless, stents carry the risk of sudden thrombotic occlusion or stent thrombosis, particularly during the first several days or weeks after implantation. In turn, stent thrombosis can lead to acute myocardial infarction (MI) and a mortality rate > 25%.1,2
This article highlights 2 cases of patients with stent thrombosis and discusses its pathophysiology, clinical features, and risk-avoidance strategies. Given the high prevalence of CAD and ubiquitous PCI procedures in the U.S. health care system, it is essential that not only cardiologists, but all clinicians and health care providers who care for patients with coronary stents understand how to help prevent and manage this life-threatening clinical entity.1
Case 1
A 56-year-old man presented to his primary care physician with exertion-related angina. The patient had a history of type 2 diabetes mellitus, dyslipidemia, systemic hypertension, obesity, and CAD status post MI in 2002 treated with a bare metal stent (BMS) to the left circumflex coronary artery (LCx). A stress myocardial perfusion imaging with 99mTc-sestamibi revealed moderate reversible exercise-induced myocardial ischemia involving the inferior and inferoapical wall segments of the left ventricle with associated hypokinesia.
Coronary angiography revealed nonsignificant disease of the left anterior descending artery (LAD) and LCx, a patent LCx stent, and a 95% mid-right coronary artery (RCA) obstruction with delayed (TIMI grade 2) antegrade flow. The distal right posterior descending artery filled via left to right collaterals from the LAD.
Percutaneous coronary intervention was performed on the RCA lesion 8 days after the patient was started on dual antiplatelet therapy (DAPT) with aspirin 81 mg and clopidogrel 75 mg (including 300 mg loading dose on the day of the diagnostic angiogram). The mid RCA was treated with a drug-eluting stent (DES) and a BMS in a nonoverlapping fashion with an excellent angiographic result. The patient was instructed to continue DAPT with aspirin 325 mg daily and clopidogrel 75 mg daily for 12 months.
Three days post PCI, the patient arrived at the emergency department with angina of 1-hour duration associated with shortness of breath and diaphoresis. He reported strict adherence to DAPT.
Initial vital signs were normal. The electrocardiogram (ECG) showed ST segment elevation (1-2 mm) on leads III, aVF, and V5 to V6, suggestive of an acute inferolateral injury pattern for which emergent coronary angiography was performed. Angiography showed a 100% proximal RCA occlusion at the proximal edge of the most proximal stent with absence of any antegrade flow beyond the occlusion (TIMI grade 0 flow). This finding was diagnostic of definite angiographic subacute stent thrombosis. The patient underwent successful aspiration thrombectomy, balloon angioplasty, and restoration of normal TIMI grade 3 flow with a door-to-balloon time of 86 minutes.
Because stent thrombosis is relatively unexpected after an excellent angiographic result and DAPT adherence, the possibility of clopidogrel resistance was considered as a major contributor for the thrombotic event. Platelet aggregation tests showed adequate prolongation of collagen/epinephrine (COL-EPI) > 300 seconds (normal: 81-153 seconds), but inadequate prolongation of collagen/adenosindiphosphate (COL-ADP) of 109 seconds (normal: 53-105 seconds) while on clopidogrel. Therefore, the patient was switched to prasugrel.
The patient was discharged home after 5 days of observation at the cardiac care unit without any post-MI complications. During a follow-up appointment 1 month after discharge, he was clinically stable and free of cardiovascular symptoms. Workup performed for acquired or inherited thrombophilia was negative. He continued taking DAPT (daily aspirin 325 mg orally and prasugrel 10 mg orally) for 12 months. After completing 12 months of DAPT, he was maintained on aspirin 81 mg daily. At 24 months’ follow-up, he remained free of recurrent angina with no further cardiovascular events.
Case 2
An 84-year-old man with a medical history of dyslipidemia, paroxysmal atrial fibrillation, previous stroke, and peptic ulcer disease was brought to the emergency department following an episode of near syncope in the early morning hours. The patient revealed that he had experienced neck pain since midnight. The 12-lead ECG showed normal sinus rhythm with 2 mm ST segment elevation in leads II, III, aVF, V5-V6, and ST segment depression in V2, and Q waves in inferior leads. A right-sided ECG showed ST segment elevation in V4, suggestive of right ventricle infarction.
The patient remained hypotensive (83/49 mm Hg) despite isotonic fluid administration (about 1.5-2.0 liters of 0.9 normal saline at 999 mL/h). A dopamine drip for persistent hypotension was started, and he was taken emergently to the catheterization laboratory for primary PCI. Coronary angiography showed no significant left CAD and a 100% mid-RCA occlusion with faint left-to-right collaterals. After aspiration thrombectomy, bare metal RCA stenting was performed. Transient no-reflow was treated with intracoronary nicardipine and nitroglycerin. The patient continued to be in shock, and an intra-aortic balloon pump was inserted and 1:1 counterpulsation was initiated.
Following admission to the coronary care unit, the patient’s mean arterial pressure improved. Inotropes were weaned off 2 days after PCI, and the intra-aortic balloon pump was removed. During his stay, the post-MI course was uneventful except for an episode of asymptomatic paroxysmal atrial flutter and nonspecific back dermatitis attributed to a prolonged recumbent position.
The patient was transferred to the internal medicine ward for medical therapy optimization and the initiation of low-intensity cardiac rehabilitation. After 2 days on the ward, discharge planning was initiated. However, he developed an episode of atrial fibrillation with fast ventricular response. Metoprolol 5 mg IV bolus was given, and the ventricular rate was controlled. At that point, the dose of long-acting beta-blocker (metoprolol succinate) was optimized, he was started on full-dose anticoagulation (warfarin), and clopidogrel was discontinued. Two days later, the patient reported back pruritus, and an erythematous raised rash on his back spreading to the torso was noticed. An aspirin allergy was suspected as the trigger for the rash, thus aspirin was also discontinued.
Three days later, the patient developed recurrent neck pain (angina) with radiation to his shoulders and left arm. The ECG revealed re-elevation of the ST segment (inferior, posterior, and lateral leads). He received reloading of clopidogrel 600 mg and aspirin 325 mg. Also, an eptifibatide IV bolus followed by an infusion was given for immediate antiplatelet action. He was transferred for emergent coronary angiography with suspected subacute stent thrombosis.
Upon arrival to the catheterization lab, the patient was awake and alert but in mild respiratory distress. Intravenous dopamine was started due to hypotension (systolic blood pressure was about 85 mm Hg). Limited RCA angiography showed a large clot burden with a partially thrombosed stent and TIMI grade 3 flow. After intracoronary eptifibatide and nicardipine were given, successful aspiration thrombectomy was performed twice with partial removal of thrombus. In-stent high-pressure balloon angioplasty was performed and optimal stenting was confirmed by intravascular ultrasound (IVUS) criteria. However, a residual layered thrombus along the distal stent edge was noticed. The patient tolerated the procedure without complications.
Dual antiplatelet therapy with aspirin and clopidogrel for 12 months was recommended. The eptifibatide infusion was continued for 48 hours. The jaw pain, shortness of breath, and ECG changes disappeared, but the patient remained on vasopressors for the following 7 days.
Around 1 week after the stent thrombosis event, the patient was found pulseless. Advanced cardiopulmonary resuscitation was started. ST segment elevation in lead II was noted on the cardiac monitor. There was no return of spontaneous circulation after 20 minutes, and the patient was pronounced dead. The autopsy revealed a patent RCA stent without evidence of occlusion, a large transmural inferior MI, left ventricular rupture, and hemopericardium.
Discussion
Stent thrombosis is an uncommon complication after coronary stent implantation. Based on the Academic Research Consortium criteria, definite stent thrombosis is defined as a clinical event with symptoms suggestive of an acute coronary syndrome (ACS) with angiography or pathology that confirms the presence of stent thrombosis.2 Probable stent thrombosis is defined as an unexplained death within 30 days or MI involving the territory of the target vessel without angiographic confirmation of stent thrombosis.2 Finally, possible stent thrombosis is any unexplained death after 30 days.2
Based on timing, stent thrombosis is divided by acute (< 24 hours post stent implantation), subacute (24 hours to 30 days post stent implantation), late (> 30 days post stent implantation), and very late (> 12 months post stent implantation).3 However, most cases (up to 60%) occur within the first 30 days after placement, irrespective of stent type.4
The incidence of subacute stent thrombosis is reported to approach 1% during the first 30 days postprocedure but may be as high as 5% or 10% depending on associated clinical and angiographic variables (Table 1).5 The strongest clinical predictors of stent thrombosis are premature cessation of antiplatelet therapy, renal insufficiency, diabetes mellitus, and ACS.2,6 Lesion and procedural characteristics associated with increased risk of stent thrombosis include bifurcation lesions, longer stent length, multiple implanted stents, stent underexpansion, and/or stent malapposition.6-9 Stent type (drug or non–drug-eluting) has no impact on the risk of stent thrombosis during the first 30 days postprocedure.10,11
The clinical events related to late stent thrombosis, although rare, carry a mortality rate of up to 45%.12 The specific risk factors for late and very late stent thrombosis are less well defined but relate to delayed neointimal coverage, ongoing vessel inflammation, and the development of neoatherosclerosis within stents.13,14
Rationale for the Use of Dual Antiplatelet Regimen
Stent thrombosis is a platelet-mediated process related to a heightened state of systemic and intracoronary thrombogenicity and inflammation.15 Stent under-expansion enhances abnormal shear stress, which explains as many as 80% of these events.13,15,16 Stent thrombosis also has been frequently related to inadequate neointimal coverage.14 Angioscopic studies, especially with DES, suggest that stent endothelialization is delayed or incomplete, observing a correlation between the areas of uncovered stent surface and thrombosis.14,17
In the early days of coronary stenting, during the 1990s, the risk of acute and subacute stent thrombosis approached 20%.18,19 Initial attempts to reduce the risk included combining aspirin and warfarin, but at the expense of a marked increase in bleeding complications and prolonged hospital stays.20,21 In 1995, it became clear through the pivotal observations of Colombo and colleagues that incomplete expansion of the stent (documented by IVUS) was a major contributor to the risk of stent thrombosis.16 By using noncompliant balloons at high pressure (14-20 atmospheres) for stent postdilatation combined with DAPT (aspirin and ticlopidine), the high rates of early stent thrombosis were markedly reduced to the current level of 1% to 2%.16
Colombo and colleagues’ observations were prospectively evaluated in the Stent Anticoagulation Regimen Study (STARS) trial.22 Patients who underwent successful stenting were randomized to aspirin alone, aspirin and warfarin, or aspirin and ticlopidine. The STARS trial showed convincingly that the combination of aspirin and ticlopidine was superior to the other 2 regimens, reducing the stent thrombosis rate to only 0.5% (compared with 2.7% for aspirin and warfarin, and 3.6% for aspirin alone).22 Afterward, DAPT became the standard of care following coronary stenting.23
Although ticlopidine was the first widely used thienopyridine for the prevention of stent thrombosis, hematologic adverse events (AEs) (eg, neutropenia, thrombotic thrombocytopenia purpura) limited its use.24 Consequently, ticlopidine was replaced with clopidogrel, which seemed to offer similar efficacy but significantly fewer AEs.25
The current American College of Cardiology/American Heart Association/Society for Cardiovascular Angiography and Interventions (ACC/AHA/SCAI) guidelines for the prevention of ST after coronary stent implantation state that after PCI:
- Aspirin use should be continued indefinitely.
- The duration of adenosine diphosphate antagonists depends on the stent type (BMS or DES) and the indication for implantation (ACS or non-ACS).
a. Patients receiving a stent (BMS or DES) for ACS therapy should be given 1 of the following for at least 12 months:
i. Clopidogrel 75 mg daily
ii. Prasugrel 10 mg daily
iii. Ticagrelor 90 mg twice daily
b. In patients receiving DES for a non-ACS indication, clopidogrel should be given for at least 12 months if the patient is not at high risk for bleeding.
c. In patients receiving BMS for a non-ACS indication, clopidogrel should be given for a minimum of 1 month and ideally up to 12 months.23
Clopidogrel Hyporesponse
As shown in case 1, stent thrombosis may still occur in a patient on DAPT because of individual variability in platelet response to clopidogrel.5 Clopidogrel hyporesponse, also known as clopidogrel resistance, has been recognized as clinically significant because of its prevalence and association with poor outcomes.5 Its prevalence may range between 4% and 30%, although the definitions of clopidogrel hyporesponse varied between studies.26
Clopidogrel hyporesponse is defined as an inadequate inhibition of platelet function measured by nonspecific ex-vivo laboratory methods.27,28 The relationship between clopidogrel resistance (nonresponders), stent thrombosis, and ischemic events has been clearly established.5,29
Given the devastating consequences of stent thrombosis, efforts were directed to identify those patients at highest risk. One such effort has been focused on the measurement of platelet function, allowing for the identification of patients who do not respond adequately to antiplatelet therapy.15,28,30,31 However, the treatment of high-residual platelet reactivity as confirmed by laboratory assessment has not shown to clinically correlate with any benefit in the prevention of ST.6,15,29-31 Therefore, the current ACC/AHA/SCAI PCI guidelines do not recommend the routine clinical use of platelet function testing to screen patients treated with clopidogrel who are undergoing PCI.23
Clopidogrel is a prodrug, metabolized to its active form via the cytochrome P450 enzyme system before it can inhibit platelet function.32 Accordingly, certain genetic variation in enzyme activity, or polymorphisms, would be expected to influence its clinical effectiveness.33,34 The most common of these polymorphisms, CYP2C19*2, has been associated (in vitro) with reduced concentrations of active clopidogrel metabolites and with diminished platelet inhibition.35,36 As a result, the FDA has added a safety alert to the prescribing information for clopidogrel concerning how genetic differences in the metabolism of this agent can affect its effectiveness, ways to test for these genetic differences, and advice concerning alternative dosing strategies or use of other medications in poor metabolizers of clopidogrel.37 Although the routine clinical use of genetic testing to screen patients treated with clopidogrel who are undergoing PCI is not recommended, it may be considered in patients undergoing elective high-risk PCI procedures (eg, unprotected left main, last patent coronary artery, or bifurcating left main).23
The newer inhibitors of ADP-induced platelet activation, prasugrel and ticagrelor, are not prodrugs, and thus, their action is not affected by this genetic variability. Accordingly, these drugs have shown a more consistent, stronger, and faster inhibition of platelet aggregation compared with clopidogrel.36-39 In the pivotal trials (TRITON-TIMI 38 and PLATO), these agents have also been shown to be more effective in reducing the incidence of stent thrombosis.36,37,40,41 Therefore, in cases where clopidogrel resistance/hyporesponse is suspected in the setting of DAPT, such as stent thrombosis, guidelines recommend the use of 1 of these agents.23
Premature Discontinuation of Antiplatelet Therapy
As illustrated in case 2, premature discontinuation of antiplatelet therapy may be fatal, as it is associated with a marked increase in the risk of stent thrombosis. Indeed, premature discontinuation of DAPT is the leading independent predictor for stent thrombosis.12,42,43 Premature discontinuation of DAPT is defined when one or both agents (aspirin, ADP-antagonists) are suspended within 30 days of BMS placement or within 1 year of DES placement. In the case of DES, the first 6 months after implantation seem to be most critical. In a large observational study of patients treated with DES, stent thrombosis occurred in 29% of those patients in whom antiplatelet therapy was prematurely discontinued.12
In order to minimize the risk of premature DAPT discontinuation, one should address its causes. There are patient- and physician-related factors that may influence an early discontinuation of aspirin, thienopyridine, or both agents. Patient-related factors were identified in the PREMIER registry, including older age, not having completed high school, not being married, and/or not seeking health care because of costs.42 Another important but often overlooked factor that has an impact on adherence with prolonged DAPT post-DES implantation is nuisance or superficial bleeding.44 Physician-related factors include not providing discharge instructions for medication use and ill-advised instructions given by health care providers to discontinue therapy before procedures with a low risk of bleeding (eg, dental cleaning, cataract surgery, colonoscopy, skin biopsy).42
In addition, the perioperative management of DAPT during the first several weeks after coronary stenting has been shown to critically influence outcomes. In a study by Sharma and colleagues, fatal cases of stent thrombosis occurred after the discontinuation of antiplatelet therapy for noncardiac surgery among patients with BMS implantation within the past 90 days.43
In selected cases when a noncardiac procedure cannot be delayed for 1 year, recognizing the impact of the specific timing for the discontinuation of the antiplatelet regimen is essential. Kaluza and colleagues reported on 40 patients treated with BMS who underwent noncardiac surgery within 6 weeks of the stent implantation.45 Seven patients had an MI, of which 6 were fatal. Stent thrombosis was presumed to be the cause of all MIs. In 5 of 7 cases, ticlopidine was withheld before surgery.45
All clinicians should be aware of the following recommendations to avoid catastrophic cardiovascular complications related to premature discontinuation of DAPT during the perioperative setting:
- Elective procedures should be deferred until patients have completed an appropriate course of thienopyridine therapy (12 months after DES and a minimum of 4 weeks for BMS implantation).
- For those patients treated with DES who are to undergo a nonelective procedure that mandates discontinuation of thienopyridine therapy, the possibility of procedure postponement for completion of DAPT for at least 6 months should be judiciously deliberated. If the procedure cannot be postponed, aspirin should be continued if at all possible and the thienopyridine restarted as soon as possible after the procedure.42,46,47
Conclusion
Stent thrombosis is a rare but devastating complication of coronary stent implantation. Although it can occur at any time after stent placement, the majority of events occur within the first month. The use of optimal stenting techniques and adherence to DAPT are required to minimize the risk of stent thrombosis. Several clinical and procedural predictors have been related to an increased risk of stent thrombosis. The premature cessation of DAPT is the most important risk factor for stent thrombosis.
All physicians should ensure patients are properly and thoroughly educated about the reasons they are prescribed DAPT and the significant risks associated with prematurely discontinuing such therapy. All clinicians, especially noncardiologists, should realize the importance of close communication with a cardiologist or interventional cardiologist in situations when premature discontinuation is being considered for a specific reason.
Table 2 summarizes a framework of the most relevant factors that should be taken into account before, during, and after stent implantation, both by interventional cardiologists, as well as by all clinicians involved in the care of the patient. Given current procedural volumes (> 1 million PCI procedures are performed in the U.S. annually) and because the risk of stent thrombosis is both time and treatment dependent, it is of paramount importance that, not only cardiologists, but all physicians know the impact of stent thrombosis in their patients and how to avoid situations that may increase its risk.1 Team-approach decisions about antiplatelet therapy after stent placement, especially within the first 12 months, and a patient-centered mind-set are indispensable to optimize patient outcomes.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Percutaneous coronary intervention (PCI) using coronary artery stent implantation is commonly used to treat symptomatic high-risk and unstable coronary artery disease (CAD). The use of stents has improved the safety and efficacy of PCI by reducing the need for repeat revascularization, reducing acute vessel closure requiring emergent coronary artery bypass graft surgery, and expanding the use of PCI to more complex diseases. Nevertheless, stents carry the risk of sudden thrombotic occlusion or stent thrombosis, particularly during the first several days or weeks after implantation. In turn, stent thrombosis can lead to acute myocardial infarction (MI) and a mortality rate > 25%.1,2
This article highlights 2 cases of patients with stent thrombosis and discusses its pathophysiology, clinical features, and risk-avoidance strategies. Given the high prevalence of CAD and ubiquitous PCI procedures in the U.S. health care system, it is essential that not only cardiologists, but all clinicians and health care providers who care for patients with coronary stents understand how to help prevent and manage this life-threatening clinical entity.1
Case 1
A 56-year-old man presented to his primary care physician with exertion-related angina. The patient had a history of type 2 diabetes mellitus, dyslipidemia, systemic hypertension, obesity, and CAD status post MI in 2002 treated with a bare metal stent (BMS) to the left circumflex coronary artery (LCx). A stress myocardial perfusion imaging with 99mTc-sestamibi revealed moderate reversible exercise-induced myocardial ischemia involving the inferior and inferoapical wall segments of the left ventricle with associated hypokinesia.
Coronary angiography revealed nonsignificant disease of the left anterior descending artery (LAD) and LCx, a patent LCx stent, and a 95% mid-right coronary artery (RCA) obstruction with delayed (TIMI grade 2) antegrade flow. The distal right posterior descending artery filled via left to right collaterals from the LAD.
Percutaneous coronary intervention was performed on the RCA lesion 8 days after the patient was started on dual antiplatelet therapy (DAPT) with aspirin 81 mg and clopidogrel 75 mg (including 300 mg loading dose on the day of the diagnostic angiogram). The mid RCA was treated with a drug-eluting stent (DES) and a BMS in a nonoverlapping fashion with an excellent angiographic result. The patient was instructed to continue DAPT with aspirin 325 mg daily and clopidogrel 75 mg daily for 12 months.
Three days post PCI, the patient arrived at the emergency department with angina of 1-hour duration associated with shortness of breath and diaphoresis. He reported strict adherence to DAPT.
Initial vital signs were normal. The electrocardiogram (ECG) showed ST segment elevation (1-2 mm) on leads III, aVF, and V5 to V6, suggestive of an acute inferolateral injury pattern for which emergent coronary angiography was performed. Angiography showed a 100% proximal RCA occlusion at the proximal edge of the most proximal stent with absence of any antegrade flow beyond the occlusion (TIMI grade 0 flow). This finding was diagnostic of definite angiographic subacute stent thrombosis. The patient underwent successful aspiration thrombectomy, balloon angioplasty, and restoration of normal TIMI grade 3 flow with a door-to-balloon time of 86 minutes.
Because stent thrombosis is relatively unexpected after an excellent angiographic result and DAPT adherence, the possibility of clopidogrel resistance was considered as a major contributor for the thrombotic event. Platelet aggregation tests showed adequate prolongation of collagen/epinephrine (COL-EPI) > 300 seconds (normal: 81-153 seconds), but inadequate prolongation of collagen/adenosindiphosphate (COL-ADP) of 109 seconds (normal: 53-105 seconds) while on clopidogrel. Therefore, the patient was switched to prasugrel.
The patient was discharged home after 5 days of observation at the cardiac care unit without any post-MI complications. During a follow-up appointment 1 month after discharge, he was clinically stable and free of cardiovascular symptoms. Workup performed for acquired or inherited thrombophilia was negative. He continued taking DAPT (daily aspirin 325 mg orally and prasugrel 10 mg orally) for 12 months. After completing 12 months of DAPT, he was maintained on aspirin 81 mg daily. At 24 months’ follow-up, he remained free of recurrent angina with no further cardiovascular events.
Case 2
An 84-year-old man with a medical history of dyslipidemia, paroxysmal atrial fibrillation, previous stroke, and peptic ulcer disease was brought to the emergency department following an episode of near syncope in the early morning hours. The patient revealed that he had experienced neck pain since midnight. The 12-lead ECG showed normal sinus rhythm with 2 mm ST segment elevation in leads II, III, aVF, V5-V6, and ST segment depression in V2, and Q waves in inferior leads. A right-sided ECG showed ST segment elevation in V4, suggestive of right ventricle infarction.
The patient remained hypotensive (83/49 mm Hg) despite isotonic fluid administration (about 1.5-2.0 liters of 0.9 normal saline at 999 mL/h). A dopamine drip for persistent hypotension was started, and he was taken emergently to the catheterization laboratory for primary PCI. Coronary angiography showed no significant left CAD and a 100% mid-RCA occlusion with faint left-to-right collaterals. After aspiration thrombectomy, bare metal RCA stenting was performed. Transient no-reflow was treated with intracoronary nicardipine and nitroglycerin. The patient continued to be in shock, and an intra-aortic balloon pump was inserted and 1:1 counterpulsation was initiated.
Following admission to the coronary care unit, the patient’s mean arterial pressure improved. Inotropes were weaned off 2 days after PCI, and the intra-aortic balloon pump was removed. During his stay, the post-MI course was uneventful except for an episode of asymptomatic paroxysmal atrial flutter and nonspecific back dermatitis attributed to a prolonged recumbent position.
The patient was transferred to the internal medicine ward for medical therapy optimization and the initiation of low-intensity cardiac rehabilitation. After 2 days on the ward, discharge planning was initiated. However, he developed an episode of atrial fibrillation with fast ventricular response. Metoprolol 5 mg IV bolus was given, and the ventricular rate was controlled. At that point, the dose of long-acting beta-blocker (metoprolol succinate) was optimized, he was started on full-dose anticoagulation (warfarin), and clopidogrel was discontinued. Two days later, the patient reported back pruritus, and an erythematous raised rash on his back spreading to the torso was noticed. An aspirin allergy was suspected as the trigger for the rash, thus aspirin was also discontinued.
Three days later, the patient developed recurrent neck pain (angina) with radiation to his shoulders and left arm. The ECG revealed re-elevation of the ST segment (inferior, posterior, and lateral leads). He received reloading of clopidogrel 600 mg and aspirin 325 mg. Also, an eptifibatide IV bolus followed by an infusion was given for immediate antiplatelet action. He was transferred for emergent coronary angiography with suspected subacute stent thrombosis.
Upon arrival to the catheterization lab, the patient was awake and alert but in mild respiratory distress. Intravenous dopamine was started due to hypotension (systolic blood pressure was about 85 mm Hg). Limited RCA angiography showed a large clot burden with a partially thrombosed stent and TIMI grade 3 flow. After intracoronary eptifibatide and nicardipine were given, successful aspiration thrombectomy was performed twice with partial removal of thrombus. In-stent high-pressure balloon angioplasty was performed and optimal stenting was confirmed by intravascular ultrasound (IVUS) criteria. However, a residual layered thrombus along the distal stent edge was noticed. The patient tolerated the procedure without complications.
Dual antiplatelet therapy with aspirin and clopidogrel for 12 months was recommended. The eptifibatide infusion was continued for 48 hours. The jaw pain, shortness of breath, and ECG changes disappeared, but the patient remained on vasopressors for the following 7 days.
Around 1 week after the stent thrombosis event, the patient was found pulseless. Advanced cardiopulmonary resuscitation was started. ST segment elevation in lead II was noted on the cardiac monitor. There was no return of spontaneous circulation after 20 minutes, and the patient was pronounced dead. The autopsy revealed a patent RCA stent without evidence of occlusion, a large transmural inferior MI, left ventricular rupture, and hemopericardium.
Discussion
Stent thrombosis is an uncommon complication after coronary stent implantation. Based on the Academic Research Consortium criteria, definite stent thrombosis is defined as a clinical event with symptoms suggestive of an acute coronary syndrome (ACS) with angiography or pathology that confirms the presence of stent thrombosis.2 Probable stent thrombosis is defined as an unexplained death within 30 days or MI involving the territory of the target vessel without angiographic confirmation of stent thrombosis.2 Finally, possible stent thrombosis is any unexplained death after 30 days.2
Based on timing, stent thrombosis is divided by acute (< 24 hours post stent implantation), subacute (24 hours to 30 days post stent implantation), late (> 30 days post stent implantation), and very late (> 12 months post stent implantation).3 However, most cases (up to 60%) occur within the first 30 days after placement, irrespective of stent type.4
The incidence of subacute stent thrombosis is reported to approach 1% during the first 30 days postprocedure but may be as high as 5% or 10% depending on associated clinical and angiographic variables (Table 1).5 The strongest clinical predictors of stent thrombosis are premature cessation of antiplatelet therapy, renal insufficiency, diabetes mellitus, and ACS.2,6 Lesion and procedural characteristics associated with increased risk of stent thrombosis include bifurcation lesions, longer stent length, multiple implanted stents, stent underexpansion, and/or stent malapposition.6-9 Stent type (drug or non–drug-eluting) has no impact on the risk of stent thrombosis during the first 30 days postprocedure.10,11
The clinical events related to late stent thrombosis, although rare, carry a mortality rate of up to 45%.12 The specific risk factors for late and very late stent thrombosis are less well defined but relate to delayed neointimal coverage, ongoing vessel inflammation, and the development of neoatherosclerosis within stents.13,14
Rationale for the Use of Dual Antiplatelet Regimen
Stent thrombosis is a platelet-mediated process related to a heightened state of systemic and intracoronary thrombogenicity and inflammation.15 Stent under-expansion enhances abnormal shear stress, which explains as many as 80% of these events.13,15,16 Stent thrombosis also has been frequently related to inadequate neointimal coverage.14 Angioscopic studies, especially with DES, suggest that stent endothelialization is delayed or incomplete, observing a correlation between the areas of uncovered stent surface and thrombosis.14,17
In the early days of coronary stenting, during the 1990s, the risk of acute and subacute stent thrombosis approached 20%.18,19 Initial attempts to reduce the risk included combining aspirin and warfarin, but at the expense of a marked increase in bleeding complications and prolonged hospital stays.20,21 In 1995, it became clear through the pivotal observations of Colombo and colleagues that incomplete expansion of the stent (documented by IVUS) was a major contributor to the risk of stent thrombosis.16 By using noncompliant balloons at high pressure (14-20 atmospheres) for stent postdilatation combined with DAPT (aspirin and ticlopidine), the high rates of early stent thrombosis were markedly reduced to the current level of 1% to 2%.16
Colombo and colleagues’ observations were prospectively evaluated in the Stent Anticoagulation Regimen Study (STARS) trial.22 Patients who underwent successful stenting were randomized to aspirin alone, aspirin and warfarin, or aspirin and ticlopidine. The STARS trial showed convincingly that the combination of aspirin and ticlopidine was superior to the other 2 regimens, reducing the stent thrombosis rate to only 0.5% (compared with 2.7% for aspirin and warfarin, and 3.6% for aspirin alone).22 Afterward, DAPT became the standard of care following coronary stenting.23
Although ticlopidine was the first widely used thienopyridine for the prevention of stent thrombosis, hematologic adverse events (AEs) (eg, neutropenia, thrombotic thrombocytopenia purpura) limited its use.24 Consequently, ticlopidine was replaced with clopidogrel, which seemed to offer similar efficacy but significantly fewer AEs.25
The current American College of Cardiology/American Heart Association/Society for Cardiovascular Angiography and Interventions (ACC/AHA/SCAI) guidelines for the prevention of ST after coronary stent implantation state that after PCI:
- Aspirin use should be continued indefinitely.
- The duration of adenosine diphosphate antagonists depends on the stent type (BMS or DES) and the indication for implantation (ACS or non-ACS).
a. Patients receiving a stent (BMS or DES) for ACS therapy should be given 1 of the following for at least 12 months:
i. Clopidogrel 75 mg daily
ii. Prasugrel 10 mg daily
iii. Ticagrelor 90 mg twice daily
b. In patients receiving DES for a non-ACS indication, clopidogrel should be given for at least 12 months if the patient is not at high risk for bleeding.
c. In patients receiving BMS for a non-ACS indication, clopidogrel should be given for a minimum of 1 month and ideally up to 12 months.23
Clopidogrel Hyporesponse
As shown in case 1, stent thrombosis may still occur in a patient on DAPT because of individual variability in platelet response to clopidogrel.5 Clopidogrel hyporesponse, also known as clopidogrel resistance, has been recognized as clinically significant because of its prevalence and association with poor outcomes.5 Its prevalence may range between 4% and 30%, although the definitions of clopidogrel hyporesponse varied between studies.26
Clopidogrel hyporesponse is defined as an inadequate inhibition of platelet function measured by nonspecific ex-vivo laboratory methods.27,28 The relationship between clopidogrel resistance (nonresponders), stent thrombosis, and ischemic events has been clearly established.5,29
Given the devastating consequences of stent thrombosis, efforts were directed to identify those patients at highest risk. One such effort has been focused on the measurement of platelet function, allowing for the identification of patients who do not respond adequately to antiplatelet therapy.15,28,30,31 However, the treatment of high-residual platelet reactivity as confirmed by laboratory assessment has not shown to clinically correlate with any benefit in the prevention of ST.6,15,29-31 Therefore, the current ACC/AHA/SCAI PCI guidelines do not recommend the routine clinical use of platelet function testing to screen patients treated with clopidogrel who are undergoing PCI.23
Clopidogrel is a prodrug, metabolized to its active form via the cytochrome P450 enzyme system before it can inhibit platelet function.32 Accordingly, certain genetic variation in enzyme activity, or polymorphisms, would be expected to influence its clinical effectiveness.33,34 The most common of these polymorphisms, CYP2C19*2, has been associated (in vitro) with reduced concentrations of active clopidogrel metabolites and with diminished platelet inhibition.35,36 As a result, the FDA has added a safety alert to the prescribing information for clopidogrel concerning how genetic differences in the metabolism of this agent can affect its effectiveness, ways to test for these genetic differences, and advice concerning alternative dosing strategies or use of other medications in poor metabolizers of clopidogrel.37 Although the routine clinical use of genetic testing to screen patients treated with clopidogrel who are undergoing PCI is not recommended, it may be considered in patients undergoing elective high-risk PCI procedures (eg, unprotected left main, last patent coronary artery, or bifurcating left main).23
The newer inhibitors of ADP-induced platelet activation, prasugrel and ticagrelor, are not prodrugs, and thus, their action is not affected by this genetic variability. Accordingly, these drugs have shown a more consistent, stronger, and faster inhibition of platelet aggregation compared with clopidogrel.36-39 In the pivotal trials (TRITON-TIMI 38 and PLATO), these agents have also been shown to be more effective in reducing the incidence of stent thrombosis.36,37,40,41 Therefore, in cases where clopidogrel resistance/hyporesponse is suspected in the setting of DAPT, such as stent thrombosis, guidelines recommend the use of 1 of these agents.23
Premature Discontinuation of Antiplatelet Therapy
As illustrated in case 2, premature discontinuation of antiplatelet therapy may be fatal, as it is associated with a marked increase in the risk of stent thrombosis. Indeed, premature discontinuation of DAPT is the leading independent predictor for stent thrombosis.12,42,43 Premature discontinuation of DAPT is defined when one or both agents (aspirin, ADP-antagonists) are suspended within 30 days of BMS placement or within 1 year of DES placement. In the case of DES, the first 6 months after implantation seem to be most critical. In a large observational study of patients treated with DES, stent thrombosis occurred in 29% of those patients in whom antiplatelet therapy was prematurely discontinued.12
In order to minimize the risk of premature DAPT discontinuation, one should address its causes. There are patient- and physician-related factors that may influence an early discontinuation of aspirin, thienopyridine, or both agents. Patient-related factors were identified in the PREMIER registry, including older age, not having completed high school, not being married, and/or not seeking health care because of costs.42 Another important but often overlooked factor that has an impact on adherence with prolonged DAPT post-DES implantation is nuisance or superficial bleeding.44 Physician-related factors include not providing discharge instructions for medication use and ill-advised instructions given by health care providers to discontinue therapy before procedures with a low risk of bleeding (eg, dental cleaning, cataract surgery, colonoscopy, skin biopsy).42
In addition, the perioperative management of DAPT during the first several weeks after coronary stenting has been shown to critically influence outcomes. In a study by Sharma and colleagues, fatal cases of stent thrombosis occurred after the discontinuation of antiplatelet therapy for noncardiac surgery among patients with BMS implantation within the past 90 days.43
In selected cases when a noncardiac procedure cannot be delayed for 1 year, recognizing the impact of the specific timing for the discontinuation of the antiplatelet regimen is essential. Kaluza and colleagues reported on 40 patients treated with BMS who underwent noncardiac surgery within 6 weeks of the stent implantation.45 Seven patients had an MI, of which 6 were fatal. Stent thrombosis was presumed to be the cause of all MIs. In 5 of 7 cases, ticlopidine was withheld before surgery.45
All clinicians should be aware of the following recommendations to avoid catastrophic cardiovascular complications related to premature discontinuation of DAPT during the perioperative setting:
- Elective procedures should be deferred until patients have completed an appropriate course of thienopyridine therapy (12 months after DES and a minimum of 4 weeks for BMS implantation).
- For those patients treated with DES who are to undergo a nonelective procedure that mandates discontinuation of thienopyridine therapy, the possibility of procedure postponement for completion of DAPT for at least 6 months should be judiciously deliberated. If the procedure cannot be postponed, aspirin should be continued if at all possible and the thienopyridine restarted as soon as possible after the procedure.42,46,47
Conclusion
Stent thrombosis is a rare but devastating complication of coronary stent implantation. Although it can occur at any time after stent placement, the majority of events occur within the first month. The use of optimal stenting techniques and adherence to DAPT are required to minimize the risk of stent thrombosis. Several clinical and procedural predictors have been related to an increased risk of stent thrombosis. The premature cessation of DAPT is the most important risk factor for stent thrombosis.
All physicians should ensure patients are properly and thoroughly educated about the reasons they are prescribed DAPT and the significant risks associated with prematurely discontinuing such therapy. All clinicians, especially noncardiologists, should realize the importance of close communication with a cardiologist or interventional cardiologist in situations when premature discontinuation is being considered for a specific reason.
Table 2 summarizes a framework of the most relevant factors that should be taken into account before, during, and after stent implantation, both by interventional cardiologists, as well as by all clinicians involved in the care of the patient. Given current procedural volumes (> 1 million PCI procedures are performed in the U.S. annually) and because the risk of stent thrombosis is both time and treatment dependent, it is of paramount importance that, not only cardiologists, but all physicians know the impact of stent thrombosis in their patients and how to avoid situations that may increase its risk.1 Team-approach decisions about antiplatelet therapy after stent placement, especially within the first 12 months, and a patient-centered mind-set are indispensable to optimize patient outcomes.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Ryan J, Cohen DJ. Are drug-eluting stents cost-effective? It depends on whom you ask. Circulation. 2006;114(16):1736-1744.
2. Cutlip DE, Windecker S, Mehran R, et al; Academic Research Consortium. Clinical end points in coronary stent trials: A case for standardized definitions. Circulation. 2007;115(17):2344-2351.
3. Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med. 2001;345(7):494-502.
4. Palmerini T, Kirtane AJ, Serruys PW, et al. Stent thrombosis with everolimus-eluting stents: Meta-analysis of comparative randomized controlled trials. Circ Cardiovasc Interv. 2012;5(3):357-364.
5. Angiolillo DJ, Fernandez-Ortiz A, Bernardo E, et al. Variability in individual responsiveness to clopidogrel: Clinical implications, management, and future perspectives. J Am Coll Cardiol. 2007;49(14):1505-1516.
6. Moussa I, Di Mario C, Reimers B, Akiyama T, Tobis J, Colombo A. Subacute stent thrombosis in the era of intravascular ultrasound-guided coronary stenting without anticoagulation: Frequency, predictors and clinical outcome. J Am Coll Cardiol. 1997;29(1):6-12.
7. Fujii K, Carlier SG, Mintz GS, et al. Stent underexpansion and residual reference segment stenosis are related to stent thrombosis after sirolimus-eluting stent implantation: An intravascular ultrasound study. J Am Coll Cardiol. 2005;45(7):995-998.
8. Uren NG, Schwarzacher SP, Metz JA, et al; POST Registry Investigators. Predictors and outcomes of stent thrombosis: An intravascular ultrasound registry. Eur Heart J. 2002;23(2):124-132.
9. Cook S, Wenaweser P, Togni M, et al. Intravascular ultrasound in very late DES-stent thrombosis (abstr). J Am Coll Cardiol. 2006;47(suppl B):9B.
10. Moreno R, Fernández C, Hernández R, et al. Drug-eluting stent thrombosis: Results from a pooled analysis including 10 randomized studies. J Am Coll Cardiol. 2005;45(6):954-959.
11. Ellis SG, Colombo A, Grube E, et al. Incidence, timing, and correlates of stent thrombosis with the polymeric paclitaxel drug-eluting stent: A TAXUS II, IV, V, and VI meta-analysis of 3,445 patients followed for up to 3 years. J Am Coll Cardiol. 2007;49(10):1043-1051.
12. Iakovou I, Schmidt T, Bonizzoni E, et al. Incidence, predictors, and outcome of thrombosis after successful implantation of drug-eluting stents. JAMA. 2005;293(17):2126-2130.
13. Cutlip DE, Baim DS, Ho KK, et al. Stent thrombosis in the modern era: A pooled analysis of multicenter coronary stent clinical trials. Circulation. 2001;103(15):1967-1971.
14. Kotani J, Awata M, Nanto S, et al. Incomplete neointimal coverage of sirolimus-eluting stents: Angioscopic findings. J Am Coll Cardiol. 2006;47(10):2108–2111.
15. Cheneau E, Leborgne L, Mintz GS, et al. Predictors of subacute stent thrombosis: Results of a systematic intravascular ultrasound study. Circulation. 2003;108(1):43-47.
16. Colombo A, Hall P, Nakamura S, et al. Intracoronary stenting without anticoagulation achieved with intravascular ultrasound guidance. Circulation. 1995;91(6):1676-1688.
17. Oyabu J, Ueda Y, Ogasawara N, Okada K, Hirayama A, Kodama K. Angioscopic evaluation of neointima coverage: Sirolimus-drug eluting stent versus bare metal stent. Am Heart J. 2006;152(6):1168-1174.
18. Serruys PW, Strauss BH, Beatt KJ, et al. Angiographic follow-up after placement of a self-expanding coronary-artery stent. N Engl J Med. 1991;324(1):13-17.
19. Schatz RA, Baim DS, Leon M, et al. Clinical experience with the Palmaz-Schatz coronary stent. Initial results of a multicenter study. Circulation. 1991:83(1):148-161.
20. Fischman DL, Leon MB, Baim DS, et al. A randomized comparison of coronary-stent placement and balloon angioplasty in the treatment of coronary artery disease. Stent Restenosis Study Investigators. N Engl J Med. 1994;331(8):496-501.
21. Serruys PW, de Jaegere P, Kiemeneij F, et al. A comparison of balloon-expandable-stent implantation with balloon angioplasty in patients with coronary artery disease. Benestent Study Group. N Engl J Med. 1994;331(8):489-495.
22. Leon MD, Baim DS, Gordon P, et al. Clinical and angiographic results from the STent Anticoagulation Regimen Study (STARS) (abstr). Circulation. 1996;94(suppl I):I-685.
23. Levine GN, Bates ER, Blankenship JC, et al. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention: A report of the American College of Cardiology Foundation/American Heart Association Task Force of Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. J Am Coll Cardiol. 2011;58(24):e44-e122.
24. Bennett CL, Davidson CJ, Raisch DW, Weinberg PD, Bennett RH, Feldman MD. Thrombotic thrombocytopenic purpura associated with ticlopidine in the setting of coronary artery stents and stroke prevention. Arch Intern Med. 1999;159(21):2524-2528.
25. Bertrand ME, Rupprecht HJ, Urban P, Gershlick AH; CLASSICS Investigators. Double-blind study of the safety of clopidogrel with and without a loading dose in combination with aspirin compared with ticlopidine in combination with aspirin after coronary stenting: The clopidogrel aspirin stent international cooperative study (CLASSICS). Circulation. 2000;102(6):624-629.
26. Wang, TH, Bhatt DL, Topol EJ. Aspirin and clopidogrel resistance: An emerging clinical entity. Eur Heart J. 2006;27(6):647-654.
27. Vats HS, Hocking WG, Rezkalla SH. Suspected clopidogrel resistance in a patient with acute stent thrombosis. Nat Clin Pract Cardiovasc Med. 2006;3(4):226-230.
28. Gurbel PA, Becker RC, Mann KG, Steinhubl SR, Michelson AD. Platelet function monitoring in patients with coronary artery disease. J Am Coll Cardiol. 2007;50(19):1822-1834.
29. Fitzgerald DJ, Maree A. Aspirin and clopidogrel resistance. Hematology Am Soc Hematol Educ Program. 2007;2007(1):114-120.
30. Cattaneo M. Resistance to antiplatelet drugs: Molecular mechanisms and laboratory detection. J Thromb Haemost. 2007;5(suppl 1):230-237.
31. Trenk D, Hochholzer W, Fromm MF, et al. Cytochrome P450 2C19 681G>A polymorphism and high on-clopidogrel platelet reactivity associated with adverse 1-year clinical outcome of elective percutaneous coronary intervention with drug-eluting or bare-metal stents. J Am Coll Cardiol. 2008;51(20):1925-1934.
32. Kazui M, Nishiya Y, Ishizuka T, et al. Identification of the human cytochrome P450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab Dispos. 2010;38(1):92-99.
33. Hulot JS, Bura A, Villard E, et al. Cytochrome P450 2C19 loss-of-function polymorphism is a major determinant of clopidogrel responsiveness in healthy subjects. Blood. 2006;108(7):2244-2247.
34. Mega JL, Close SL, Wiviott SD, et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med. 2009;360(4):354-362.
35. U.S. Food and Drug Administration. Plavix (clopidogrel): Reduced effectiveness in patients who are poor metabolizers of the drug. U.S. Food and Drug Administration Website. http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm204256.htm. Updated September 6, 2013. Accessed September 4, 2014.
36. Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Intensive oral antiplatelet therapy for reduction of ischaemic events including stent thrombosis in patients with acute coronary syndromes treated with percutaneous coronary intervention and stenting in the TRITON-TIMI 38 trial: A subanalysis of a randomised trial. Lancet. 2008;371(9621):1353-1363.
37. Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361(11):1045-1057.
38. Wallentin L, Varenhorst C, James S, et al. Prasugrel achieves greater and faster P2Y12 receptor-mediated platelet inhibition than clopidogrel due to more efficient generation of its active metabolite in aspirin-treated patients with coronary artery disease. Eur Heart J. 2008;29(1):21-30.
39. Gurbel PA, Bliden KP, Butler K, et al. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: The ONSET/OFFSET study. Circulation. 2009;120(25):2577-2585.
40. Wiviott SD, Trenk D, Frelinger AL, et al; PRINCIPLE-TIMI 44 Investigators. Prasugrel compared with high loading- and maintenance-dose clopidogrel in patients with planned percutaneous coronary intervention: The Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation-Thrombolysis in Myocardial Infarction 44 trial. Circulation. 2007;116(25):2923-2932.
41. Gurbel PA, Bliden KP, Butler K, et al. Response to ticagrelor in clopidogrel nonresponders and responders and effect of switching therapies: The RESPOND study. Circulation. 2010;121(10):1188-1199.
42. Spertus, JA, Kettelkamp R, Vance C, et al. Prevalence, predictors, and outcomes of premature discontinuation of thienopyridine therapy after drug-eluting stent placement: Results from the PREMIER registry. Circulation. 2006;113(24):2803-2809.
43. Sharma AK, Ajani AE, Hamwi SM, et al. Major noncardiac surgery following coronary stenting: When is it safe to operate? Catheter Cardiovasc Interv. 2004;63(2):141-145.
44. Ben-Dor I, Torguson R, Scheinowitz M, et al. Incidence, correlates, and clinical impact of nuisance bleeding after antiplatelet therapy for patients with drug-eluting stents. Am Heart J. 2010;159(5):871-875.
45. Kaluza GL, Joseph J, Lee JR, Raizner ME, Raizner AE. Catastrophic outcomes of noncardiac surgery soon after coronary stenting. J Am Coll Cardiol. 2000;35(5):1288-1294.
46. Airoldi F, Colombo A, Morici N, et al. Incidence and predictors of drug-eluting stent thrombosis during and after discontinuation of thienopyridine treatment. Circulation. 2007;116(7):745-754.
47. Grines CL, Bonow RO, Casey DE Jr, et al; American Heart Association; American College of Cardiology; Society for Cardiovascular Angiography and Interventions; American College of Surgeons; American Dental Association; American College of Physicians. Prevention of premature discontinuation of dual antiplatelet therapy in patients with coronary artery stents: A science advisory from the American Heart Association, American College of Cardiology, Society for Cardiovascular Angiography and Interventions, American College of Surgeons, and American Dental Association, with representation from the American College of Physicians. J Am Coll Cardiol. 2007;49(6):734-739.
1. Ryan J, Cohen DJ. Are drug-eluting stents cost-effective? It depends on whom you ask. Circulation. 2006;114(16):1736-1744.
2. Cutlip DE, Windecker S, Mehran R, et al; Academic Research Consortium. Clinical end points in coronary stent trials: A case for standardized definitions. Circulation. 2007;115(17):2344-2351.
3. Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med. 2001;345(7):494-502.
4. Palmerini T, Kirtane AJ, Serruys PW, et al. Stent thrombosis with everolimus-eluting stents: Meta-analysis of comparative randomized controlled trials. Circ Cardiovasc Interv. 2012;5(3):357-364.
5. Angiolillo DJ, Fernandez-Ortiz A, Bernardo E, et al. Variability in individual responsiveness to clopidogrel: Clinical implications, management, and future perspectives. J Am Coll Cardiol. 2007;49(14):1505-1516.
6. Moussa I, Di Mario C, Reimers B, Akiyama T, Tobis J, Colombo A. Subacute stent thrombosis in the era of intravascular ultrasound-guided coronary stenting without anticoagulation: Frequency, predictors and clinical outcome. J Am Coll Cardiol. 1997;29(1):6-12.
7. Fujii K, Carlier SG, Mintz GS, et al. Stent underexpansion and residual reference segment stenosis are related to stent thrombosis after sirolimus-eluting stent implantation: An intravascular ultrasound study. J Am Coll Cardiol. 2005;45(7):995-998.
8. Uren NG, Schwarzacher SP, Metz JA, et al; POST Registry Investigators. Predictors and outcomes of stent thrombosis: An intravascular ultrasound registry. Eur Heart J. 2002;23(2):124-132.
9. Cook S, Wenaweser P, Togni M, et al. Intravascular ultrasound in very late DES-stent thrombosis (abstr). J Am Coll Cardiol. 2006;47(suppl B):9B.
10. Moreno R, Fernández C, Hernández R, et al. Drug-eluting stent thrombosis: Results from a pooled analysis including 10 randomized studies. J Am Coll Cardiol. 2005;45(6):954-959.
11. Ellis SG, Colombo A, Grube E, et al. Incidence, timing, and correlates of stent thrombosis with the polymeric paclitaxel drug-eluting stent: A TAXUS II, IV, V, and VI meta-analysis of 3,445 patients followed for up to 3 years. J Am Coll Cardiol. 2007;49(10):1043-1051.
12. Iakovou I, Schmidt T, Bonizzoni E, et al. Incidence, predictors, and outcome of thrombosis after successful implantation of drug-eluting stents. JAMA. 2005;293(17):2126-2130.
13. Cutlip DE, Baim DS, Ho KK, et al. Stent thrombosis in the modern era: A pooled analysis of multicenter coronary stent clinical trials. Circulation. 2001;103(15):1967-1971.
14. Kotani J, Awata M, Nanto S, et al. Incomplete neointimal coverage of sirolimus-eluting stents: Angioscopic findings. J Am Coll Cardiol. 2006;47(10):2108–2111.
15. Cheneau E, Leborgne L, Mintz GS, et al. Predictors of subacute stent thrombosis: Results of a systematic intravascular ultrasound study. Circulation. 2003;108(1):43-47.
16. Colombo A, Hall P, Nakamura S, et al. Intracoronary stenting without anticoagulation achieved with intravascular ultrasound guidance. Circulation. 1995;91(6):1676-1688.
17. Oyabu J, Ueda Y, Ogasawara N, Okada K, Hirayama A, Kodama K. Angioscopic evaluation of neointima coverage: Sirolimus-drug eluting stent versus bare metal stent. Am Heart J. 2006;152(6):1168-1174.
18. Serruys PW, Strauss BH, Beatt KJ, et al. Angiographic follow-up after placement of a self-expanding coronary-artery stent. N Engl J Med. 1991;324(1):13-17.
19. Schatz RA, Baim DS, Leon M, et al. Clinical experience with the Palmaz-Schatz coronary stent. Initial results of a multicenter study. Circulation. 1991:83(1):148-161.
20. Fischman DL, Leon MB, Baim DS, et al. A randomized comparison of coronary-stent placement and balloon angioplasty in the treatment of coronary artery disease. Stent Restenosis Study Investigators. N Engl J Med. 1994;331(8):496-501.
21. Serruys PW, de Jaegere P, Kiemeneij F, et al. A comparison of balloon-expandable-stent implantation with balloon angioplasty in patients with coronary artery disease. Benestent Study Group. N Engl J Med. 1994;331(8):489-495.
22. Leon MD, Baim DS, Gordon P, et al. Clinical and angiographic results from the STent Anticoagulation Regimen Study (STARS) (abstr). Circulation. 1996;94(suppl I):I-685.
23. Levine GN, Bates ER, Blankenship JC, et al. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention: A report of the American College of Cardiology Foundation/American Heart Association Task Force of Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. J Am Coll Cardiol. 2011;58(24):e44-e122.
24. Bennett CL, Davidson CJ, Raisch DW, Weinberg PD, Bennett RH, Feldman MD. Thrombotic thrombocytopenic purpura associated with ticlopidine in the setting of coronary artery stents and stroke prevention. Arch Intern Med. 1999;159(21):2524-2528.
25. Bertrand ME, Rupprecht HJ, Urban P, Gershlick AH; CLASSICS Investigators. Double-blind study of the safety of clopidogrel with and without a loading dose in combination with aspirin compared with ticlopidine in combination with aspirin after coronary stenting: The clopidogrel aspirin stent international cooperative study (CLASSICS). Circulation. 2000;102(6):624-629.
26. Wang, TH, Bhatt DL, Topol EJ. Aspirin and clopidogrel resistance: An emerging clinical entity. Eur Heart J. 2006;27(6):647-654.
27. Vats HS, Hocking WG, Rezkalla SH. Suspected clopidogrel resistance in a patient with acute stent thrombosis. Nat Clin Pract Cardiovasc Med. 2006;3(4):226-230.
28. Gurbel PA, Becker RC, Mann KG, Steinhubl SR, Michelson AD. Platelet function monitoring in patients with coronary artery disease. J Am Coll Cardiol. 2007;50(19):1822-1834.
29. Fitzgerald DJ, Maree A. Aspirin and clopidogrel resistance. Hematology Am Soc Hematol Educ Program. 2007;2007(1):114-120.
30. Cattaneo M. Resistance to antiplatelet drugs: Molecular mechanisms and laboratory detection. J Thromb Haemost. 2007;5(suppl 1):230-237.
31. Trenk D, Hochholzer W, Fromm MF, et al. Cytochrome P450 2C19 681G>A polymorphism and high on-clopidogrel platelet reactivity associated with adverse 1-year clinical outcome of elective percutaneous coronary intervention with drug-eluting or bare-metal stents. J Am Coll Cardiol. 2008;51(20):1925-1934.
32. Kazui M, Nishiya Y, Ishizuka T, et al. Identification of the human cytochrome P450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab Dispos. 2010;38(1):92-99.
33. Hulot JS, Bura A, Villard E, et al. Cytochrome P450 2C19 loss-of-function polymorphism is a major determinant of clopidogrel responsiveness in healthy subjects. Blood. 2006;108(7):2244-2247.
34. Mega JL, Close SL, Wiviott SD, et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med. 2009;360(4):354-362.
35. U.S. Food and Drug Administration. Plavix (clopidogrel): Reduced effectiveness in patients who are poor metabolizers of the drug. U.S. Food and Drug Administration Website. http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm204256.htm. Updated September 6, 2013. Accessed September 4, 2014.
36. Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Intensive oral antiplatelet therapy for reduction of ischaemic events including stent thrombosis in patients with acute coronary syndromes treated with percutaneous coronary intervention and stenting in the TRITON-TIMI 38 trial: A subanalysis of a randomised trial. Lancet. 2008;371(9621):1353-1363.
37. Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361(11):1045-1057.
38. Wallentin L, Varenhorst C, James S, et al. Prasugrel achieves greater and faster P2Y12 receptor-mediated platelet inhibition than clopidogrel due to more efficient generation of its active metabolite in aspirin-treated patients with coronary artery disease. Eur Heart J. 2008;29(1):21-30.
39. Gurbel PA, Bliden KP, Butler K, et al. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: The ONSET/OFFSET study. Circulation. 2009;120(25):2577-2585.
40. Wiviott SD, Trenk D, Frelinger AL, et al; PRINCIPLE-TIMI 44 Investigators. Prasugrel compared with high loading- and maintenance-dose clopidogrel in patients with planned percutaneous coronary intervention: The Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation-Thrombolysis in Myocardial Infarction 44 trial. Circulation. 2007;116(25):2923-2932.
41. Gurbel PA, Bliden KP, Butler K, et al. Response to ticagrelor in clopidogrel nonresponders and responders and effect of switching therapies: The RESPOND study. Circulation. 2010;121(10):1188-1199.
42. Spertus, JA, Kettelkamp R, Vance C, et al. Prevalence, predictors, and outcomes of premature discontinuation of thienopyridine therapy after drug-eluting stent placement: Results from the PREMIER registry. Circulation. 2006;113(24):2803-2809.
43. Sharma AK, Ajani AE, Hamwi SM, et al. Major noncardiac surgery following coronary stenting: When is it safe to operate? Catheter Cardiovasc Interv. 2004;63(2):141-145.
44. Ben-Dor I, Torguson R, Scheinowitz M, et al. Incidence, correlates, and clinical impact of nuisance bleeding after antiplatelet therapy for patients with drug-eluting stents. Am Heart J. 2010;159(5):871-875.
45. Kaluza GL, Joseph J, Lee JR, Raizner ME, Raizner AE. Catastrophic outcomes of noncardiac surgery soon after coronary stenting. J Am Coll Cardiol. 2000;35(5):1288-1294.
46. Airoldi F, Colombo A, Morici N, et al. Incidence and predictors of drug-eluting stent thrombosis during and after discontinuation of thienopyridine treatment. Circulation. 2007;116(7):745-754.
47. Grines CL, Bonow RO, Casey DE Jr, et al; American Heart Association; American College of Cardiology; Society for Cardiovascular Angiography and Interventions; American College of Surgeons; American Dental Association; American College of Physicians. Prevention of premature discontinuation of dual antiplatelet therapy in patients with coronary artery stents: A science advisory from the American Heart Association, American College of Cardiology, Society for Cardiovascular Angiography and Interventions, American College of Surgeons, and American Dental Association, with representation from the American College of Physicians. J Am Coll Cardiol. 2007;49(6):734-739.
Imaging Use in Focal Rhabdomyolysis of the Left Shoulder
Rhabdomyolysis involves the breakdown of skeletal muscle with the release of intracellular contents into the extracellular space and circulation.1 Diffuse rhabdomyolysis has been found in athletes due to overexertion. However, focal rhabdomyolysis is rare.2,3 The clinical presentation of focal rhabdomyolysis is subtle and nonspecific, with swelling, vague pain, weakness, fatigue, and tea-colored urine.
Early recognition and prompt management are crucial to prevent complications such as compression syndrome, acute renal failure, disseminated intravascular coagulation, cardiac dysrhythmia, or even cardiac arrest. Sonography and magnetic resonance imaging (MRI) can, therefore, be a complementary part of the diagnosis and assessment of the extent of rhabdomyolysis.4-7
Case History
The patient was a 34-year-old white man with a history of polysubstance abuse who presented to the emergency department (ED) with numbness and weakness in the left arm and hand, pain in the left side of his neck, and 3 days of intermittent amnesia with confusion. He had used IV heroin about 2 weeks prior to admission and used tobacco and alcohol daily. He reported no current medications or known allergies. The patient was in a monogamous relationship with a same-sex partner.
On physical examination, vital signs were within normal limits. He was in distress, confused, and disoriented as to time and place. An extremity examination revealed 1/5 strength in the extensors of the left elbow, left wrist, and left fingers with normal strength noted in the right upper extremity as well as the lower extremities. No sensory deficits were noted. The patient’s skin was warm and dry. Remarkable laboratory findings included creatine kinase (CK) 1,744 U/L, creatinine (Cr) 1.9 mg/dL, ALT 1,065 U/L, AST 319 U/L, ALP 159 U/L. A urine toxicology screen was positive for cocaine and opiates, and the urine analysis dip was negative for red blood cells, white blood cells, and protein. A differential diagnosis favored a left arm inflammatory reaction to IV drugs, although rhabdomyolysis was questioned.
A neurology consult was obtained, and a bedside electroencephalography test was performed in the ED by the neurologist, showing mild left occipital slow wave abnormality with no epileptiform discharges. A chest X-ray and computed tomography (CT) scan of the head and cervical spine were unremarkable, other than incidental mild prominence of the ventricles.
Over the next 24 hours, the patient was hydrated with IV normal saline without bicarbonate. His altered mental status, urine output, and biochemical abnormalities returned to normal, except for the serum CK, which decreased to 917 U/L. He had minimal improvement in his left upper extremity nerve palsy symptoms; however, he was deemed to be stable for discharge with follow-up in the clinic.
Instead of a clinic follow-up, the patient returned to the ED 7 days later, with progressive weakness of the left arm, forearm, and wrist. The patient noted that his weakness was so significant that he had to move his left arm with his right arm. He also reported extremity swelling and increasing paresthesias involving the lateral aspect of his left arm and hand, dizziness, and left neck pain. A physical examination revealed 3/5 strength at the left deltoid and left triceps, and 0/5 strength in the left fingers and grip. Remeasurement of CK was 54 U/L and Cr was 0.9 mg/dL. Compartment pressures were not measured.
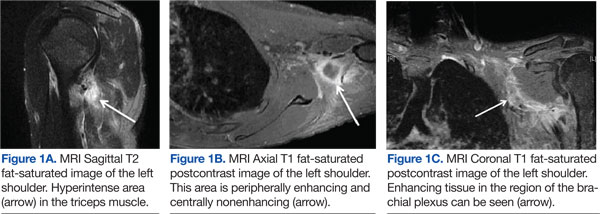
Magnetic resonance imaging using multiplanar spin echo T1 and fast spin T2 weighted and post-IV 16cc Omniscan contrast sequences of the left shoulder were performed, showing multiple patchy T2 hyperintense focal areas with peripheral enhancement in the muscles of the posterior shoulder and in the tissues adjacent to the brachial plexus in the neck and shoulder (Figures 1A, 1B, and 1C). Sonography with matrix array linear 6-15 MH3 transducer was performed, which demonstrated patchy focal hypoechoic areas of muscle with enlarged, thickened, and disrupted muscle, representing devitalized muscle without any drainable fluid collection or abscess (Figures 2A, 2B, 2C, and 2D).
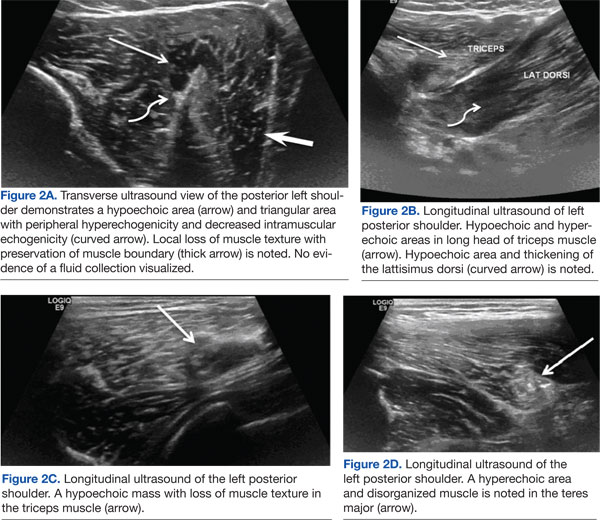
Magnetic resonance imaging and magnetic resonance angiogram scans of the brain and cervical spine with and without contrast were unremarkable. At that time, a definitive diagnosis was made of focal rhabdomyolysis and compressive neuropathy of the brachial plexus posterior cord, leading to brachial plexopathy of the left shoulder.
The patient was treated with hydration, a left arm sling, elevated left arm, and ibuprofen 600 mg qid to reduce inflammation. His swelling decreased markedly, and there was a slight improvement in pain and mobility at a 2-week neurology clinic follow-up. The patient lost contact after that.
Discussion
Rhabdomyolysis is caused by diverse etiologies. Most commonly, it is generalized and occurs due to overexertion, crush injury, steroid use, metabolic abnormalities, and certain medications and illicit drugs.1,2 The most likely etiology of rhabdomyolysis in patients presenting to the ED without significant trauma is of substance abuse, especially with ethanol, heroin, amphetamines, cocaine, and other sedatives or stimulants.1-3 The patient presented in this case study had a history of drug abuse, with a positive urine toxicology screen for cocaine and opiates. He had been intermittently confused and amnesic for 3 days prior to presentation, during which he may have been lying on his shoulder for a prolonged period.
Focal rhabdomyolysis and acute compression at the posterior shoulder leading to compressive brachial plexopathy is rare, with only 3 cases reported in the literature, all occurring with IV drug use.1-3 This patient had compression of the brachial plexus posterior cord from rhabdomyolysis and prolonged immobilization. Intravenous drug abusers may delay medical care due to perceived illicit drug effects and frequently present to the ED confused, agitated, or obtunded. Acute extremity swelling, a palpable lump, and pain can be due to various etiologies, such as trauma, fluid collection, muscle tear, myopathy, venous thrombosis, neoplasm, or rhabdomyolysis.
Diagnosis of nontraumatic rhabdomyolysis depends on clinical history and biochemical tests, such as serum CK and urine myoglobin.1,8 Creatine kinase is present in large quantities in the myocytes and is 100% sensitive as a marker for rhabdomyolysis.1,8 Creatine kinase may increase acutely > 1,000 U/L, suggesting muscle lysis and necrosis as etiology for pain as opposed to other causes such as hematomas, abscesses, or venous thrombi.1,9 Serum CK decreases rapidly at a rate of 39% per day, and it may normalize by the time a patient presents for medical care.1,10-12 Imaging plays a significant complimentary role. During the patient’s second ED presentation, the CK was normal at 54 U/L, whereas ultrasound and MRI findings were suggestive of focal muscle abnormalities.
Although there are diverse etiologies of rhabdomyolysis, the ultimate consequences of rhabdomyolysis are muscle cell membrane injury, metabolism malfunction, and destruction of the myofibril, resulting in inflammatory changes, such as muscle edema, hemorrhage, and myonecrosis and disruption of muscle fibers.1,2,8,9,13 This may cause an alteration in muscle size, shape, and echogenicity on sonography and abnormal signal intensity on MRI.13 The sensitivity of MRI in the detection of muscle involvement is higher than that of CT or ultrasound due to the high soft tissue contrast.4,13,14 Specificity of all 3 modalities is low and not reported.
Although the sensitivity of ultrasound is lower than that of MRI, use of ultrasound in neuromuscular evaluation has been increasing recently due to technical refinements. Ultrasound can be effectively used as a first-line screening modality, especially in an emergency.5 Magnetic resonance imaging best assesses the distribution and extension of the affected muscles, especially when fasciotomy is considered for treatment, and initially reveals edema, inflammation, and findings of myonecrosis; muscle atrophy and fatty degeneration occur later.4,13-15 Typical MRI findings include increased signal intensity on T2-weighted and STIR (short-tau inversion recovery) sequences and variable enhancement on T1 postcontrast images, as was seen in this case, which indicated edema, inflammation, and necrosis of the muscle tissue.
Shintani and colleagues described the reversibility of the MRI findings, showing that the high-intensity lesions seen on T2-weighted images resolved in parallel with the clinical course.14,16 Lu and colleagues investigated 10 patients with rhabdomyolysis and found 2 distinct imaging types: Type 1 shows homogenous signal changes and enhancement in the affected muscles, and Type 2 shows rim enhancement on contrast-enhanced MRI, a “stipple sign” indicating areas of myonecrosis.17 Magnetic resonance imaging signal alterations in the musculature can be nonspecific and overlap with those of inflammatory myopathies such as polymyositis, connective tissue diseases with inflammatory myositis, muscle infection, muscle infarction such as diabetic myonecrosis, muscle contusion, drug-induced myotoxicity, corticosteroids use, and use of cholesterol-lowering agents.18,19
Sonography is a useful screening modality for pain and swelling of the extremity, because it can detect a muscle tear, muscle sprain, and fluid collection, especially in emergent cases. There is scant literature about sonographic findings in rhabdomyolysis and compression nerve entrapment. The sonographic findings of rhabdomyolysis are local disorganization of the damaged muscle, decreased muscle echogenicity, and enlargement of the muscle, with preservation of the muscle boundaries.5-7
Intramuscular hyperechoic areas are seen due to hypercontractility of injured muscle. In this case, noted findings included patchy, irregular, hypoechoic areas, enlargement of the muscles and tendons, and irregular hyperechoic areas without focal defects. These findings differentiated an abnormality from a muscle tear or rupture, as these often show a focal muscle gap and focal defect, signifying the ruptured muscle retracting.
A study by Su and colleagues used the large number of crush injuries after an earthquake in China.5 The characteristic sonographic findings were edema and thickened disrupted striated muscle, good overall muscle continuity, vague muscle texture, and enhanced cloudy or ground-glass-like echo. There was no blood flow signal in the hypoechoic areas.6 Ultrasound was deemed a cost-effective, easily available modality by the authors.
Conclusion
Nontraumatic, focal rhabdomyolysis is rare and should be detected and differentiated from other causes of swelling, lump, pain, or other muscle disorders to prevent late complications. Sonography is an important screening diagnostic modality. MRI is used for assessment of the extent and distribution of injury. Awareness and familiarity with imaging findings can play a significant role, along with clinical and laboratory findings in the diagnosis and management of rhabdomyolysis.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
:
1. Richards JR. Rhabdomyolysis and drugs of abuse. J Emerg Med. 2000;19(1):51-56.
2. Farkash U, Shabshin N, Pritsch Perry M. Rhabdomyolysis of the deltoid muscle in a bodybuilder using anabolic-androgenic steroids: A case report. J Athl Train. 2009;44(1):98-100.
3. Mubarak SJ, Owen CA, Hargens AR, Garetto LP, Akeson WH. Acute compartment syndromes: Diagnosis and treatment with the aid of the wick catheter. J Bone Joint Surg Am. 1978;60(8):1091-1095.
4. Lamminen AE, Hekali PE, Tiula E, Suramo I, Korhola OA. Acute rhabdomyolysis: Evaluation with magnetic resonance imaging compared with computed tomography and ultrasonography. Br J Radiol. 1989;62(736):326-330.
5. Su BH, Qui L, Fu P, Luo Y, Tao Y, Peng YL. Ultrasonic appearance of rhabdomyolysis in patients with crush injury in the Wenchuan earthquake. Chin Med J (Engl). 2009;122(16):1872-1876.
6. Chiu Y-N, Wang T-G, Hsu C-Y, et al. Sonographic diagnosis of rhabdomyolysis. J Med Ultrasound. 2008;16(2):158-162.
7. Kaplan GN. Ultrasonic appearance of rhabdomyolysis. AJR Am J Roentgenol. 1980;134(2):375-377.
8. Spector R, Choudhury A, Cancilla P, Lakin R. Alcohol myopathy. Diagnosis by alcohol challenge. JAMA. 1979;242(15):1648-1649.
9. Gabow PA, Kaehny WD, Kelleher SP. The spectrum of rhabdomyolysis. Medicine (Baltimore). 1982;61(3):141-152.
10. Knochel JP. Mechanisms of rhabdomyolysis. Curr Opin Rheumatol. 1993;5(6):725-731.
11. Cadnapaphornchai P, Taher S, McDonald FD. Acute drug-association rhabdomyolysis: An examination of its diverse renal manifestations and complications. Am J Med Sci. 1980;280(2):66-72.
12. Curry SC, Chang D, Connor D. Drug and toxin-induced rhabdomyolysis. Ann Emerg Med. 1989;18(10):1068-1084.
13. May D, Disler DG, Jones EA, Balkissoon AA, Manaster BJ. Abnormal signal intensity in skeletal muscle at MR imaging: Patterns, pearls, and pitfalls. RadioGraphics. 2000;20(spec no):S295-S315.
14. Moratalla MB, Braun P, Fornas GM. Importance of MRI in the diagnosis and treatment of rhabdomyolysis. Eur J Radiol. 2008;65(2):311-315.
15. Beltran J, Rosenberg ZS. Diagnosis of compressive and entrapment neurorpathies of the upper extremity: Value of MR imaging. AJR Am J Roentgenol. 1994;163(3):525-531.
16. Shintani S, Shiigai T. Repeat MRI in acute rhabdomyolysis: Correlation with clinicopathological findings. J Comput Assist Tomogr. 1993;17(5):786-791.
17. Lu CH, Tsang YM, Yu CW, et al. Rhabdomyolysis: Magnetic resonance imaging and computed tomography findings. J Comput Assist Tomogr. 2007;31(3):368-374.
18. Schulze M, Kötter I, Ernemann U, et al. MRI findings in inflammatory muscle diseases and their noninflammatory mimics. AJR Am J Roentgenol. 2009;192(6):1708-1716.
19. Adams EM, Chow CK, Premkumar A, Plotz PH. The idiopathic inflammatory myopathies: Spectrum of MR imaging findings. Radiographics. 1995;15(3):563-574.
Rhabdomyolysis involves the breakdown of skeletal muscle with the release of intracellular contents into the extracellular space and circulation.1 Diffuse rhabdomyolysis has been found in athletes due to overexertion. However, focal rhabdomyolysis is rare.2,3 The clinical presentation of focal rhabdomyolysis is subtle and nonspecific, with swelling, vague pain, weakness, fatigue, and tea-colored urine.
Early recognition and prompt management are crucial to prevent complications such as compression syndrome, acute renal failure, disseminated intravascular coagulation, cardiac dysrhythmia, or even cardiac arrest. Sonography and magnetic resonance imaging (MRI) can, therefore, be a complementary part of the diagnosis and assessment of the extent of rhabdomyolysis.4-7
Case History
The patient was a 34-year-old white man with a history of polysubstance abuse who presented to the emergency department (ED) with numbness and weakness in the left arm and hand, pain in the left side of his neck, and 3 days of intermittent amnesia with confusion. He had used IV heroin about 2 weeks prior to admission and used tobacco and alcohol daily. He reported no current medications or known allergies. The patient was in a monogamous relationship with a same-sex partner.
On physical examination, vital signs were within normal limits. He was in distress, confused, and disoriented as to time and place. An extremity examination revealed 1/5 strength in the extensors of the left elbow, left wrist, and left fingers with normal strength noted in the right upper extremity as well as the lower extremities. No sensory deficits were noted. The patient’s skin was warm and dry. Remarkable laboratory findings included creatine kinase (CK) 1,744 U/L, creatinine (Cr) 1.9 mg/dL, ALT 1,065 U/L, AST 319 U/L, ALP 159 U/L. A urine toxicology screen was positive for cocaine and opiates, and the urine analysis dip was negative for red blood cells, white blood cells, and protein. A differential diagnosis favored a left arm inflammatory reaction to IV drugs, although rhabdomyolysis was questioned.
A neurology consult was obtained, and a bedside electroencephalography test was performed in the ED by the neurologist, showing mild left occipital slow wave abnormality with no epileptiform discharges. A chest X-ray and computed tomography (CT) scan of the head and cervical spine were unremarkable, other than incidental mild prominence of the ventricles.
Over the next 24 hours, the patient was hydrated with IV normal saline without bicarbonate. His altered mental status, urine output, and biochemical abnormalities returned to normal, except for the serum CK, which decreased to 917 U/L. He had minimal improvement in his left upper extremity nerve palsy symptoms; however, he was deemed to be stable for discharge with follow-up in the clinic.
Instead of a clinic follow-up, the patient returned to the ED 7 days later, with progressive weakness of the left arm, forearm, and wrist. The patient noted that his weakness was so significant that he had to move his left arm with his right arm. He also reported extremity swelling and increasing paresthesias involving the lateral aspect of his left arm and hand, dizziness, and left neck pain. A physical examination revealed 3/5 strength at the left deltoid and left triceps, and 0/5 strength in the left fingers and grip. Remeasurement of CK was 54 U/L and Cr was 0.9 mg/dL. Compartment pressures were not measured.
Magnetic resonance imaging using multiplanar spin echo T1 and fast spin T2 weighted and post-IV 16cc Omniscan contrast sequences of the left shoulder were performed, showing multiple patchy T2 hyperintense focal areas with peripheral enhancement in the muscles of the posterior shoulder and in the tissues adjacent to the brachial plexus in the neck and shoulder (Figures 1A, 1B, and 1C). Sonography with matrix array linear 6-15 MH3 transducer was performed, which demonstrated patchy focal hypoechoic areas of muscle with enlarged, thickened, and disrupted muscle, representing devitalized muscle without any drainable fluid collection or abscess (Figures 2A, 2B, 2C, and 2D).
Magnetic resonance imaging and magnetic resonance angiogram scans of the brain and cervical spine with and without contrast were unremarkable. At that time, a definitive diagnosis was made of focal rhabdomyolysis and compressive neuropathy of the brachial plexus posterior cord, leading to brachial plexopathy of the left shoulder.
The patient was treated with hydration, a left arm sling, elevated left arm, and ibuprofen 600 mg qid to reduce inflammation. His swelling decreased markedly, and there was a slight improvement in pain and mobility at a 2-week neurology clinic follow-up. The patient lost contact after that.
Discussion
Rhabdomyolysis is caused by diverse etiologies. Most commonly, it is generalized and occurs due to overexertion, crush injury, steroid use, metabolic abnormalities, and certain medications and illicit drugs.1,2 The most likely etiology of rhabdomyolysis in patients presenting to the ED without significant trauma is of substance abuse, especially with ethanol, heroin, amphetamines, cocaine, and other sedatives or stimulants.1-3 The patient presented in this case study had a history of drug abuse, with a positive urine toxicology screen for cocaine and opiates. He had been intermittently confused and amnesic for 3 days prior to presentation, during which he may have been lying on his shoulder for a prolonged period.
Focal rhabdomyolysis and acute compression at the posterior shoulder leading to compressive brachial plexopathy is rare, with only 3 cases reported in the literature, all occurring with IV drug use.1-3 This patient had compression of the brachial plexus posterior cord from rhabdomyolysis and prolonged immobilization. Intravenous drug abusers may delay medical care due to perceived illicit drug effects and frequently present to the ED confused, agitated, or obtunded. Acute extremity swelling, a palpable lump, and pain can be due to various etiologies, such as trauma, fluid collection, muscle tear, myopathy, venous thrombosis, neoplasm, or rhabdomyolysis.
Diagnosis of nontraumatic rhabdomyolysis depends on clinical history and biochemical tests, such as serum CK and urine myoglobin.1,8 Creatine kinase is present in large quantities in the myocytes and is 100% sensitive as a marker for rhabdomyolysis.1,8 Creatine kinase may increase acutely > 1,000 U/L, suggesting muscle lysis and necrosis as etiology for pain as opposed to other causes such as hematomas, abscesses, or venous thrombi.1,9 Serum CK decreases rapidly at a rate of 39% per day, and it may normalize by the time a patient presents for medical care.1,10-12 Imaging plays a significant complimentary role. During the patient’s second ED presentation, the CK was normal at 54 U/L, whereas ultrasound and MRI findings were suggestive of focal muscle abnormalities.
Although there are diverse etiologies of rhabdomyolysis, the ultimate consequences of rhabdomyolysis are muscle cell membrane injury, metabolism malfunction, and destruction of the myofibril, resulting in inflammatory changes, such as muscle edema, hemorrhage, and myonecrosis and disruption of muscle fibers.1,2,8,9,13 This may cause an alteration in muscle size, shape, and echogenicity on sonography and abnormal signal intensity on MRI.13 The sensitivity of MRI in the detection of muscle involvement is higher than that of CT or ultrasound due to the high soft tissue contrast.4,13,14 Specificity of all 3 modalities is low and not reported.
Although the sensitivity of ultrasound is lower than that of MRI, use of ultrasound in neuromuscular evaluation has been increasing recently due to technical refinements. Ultrasound can be effectively used as a first-line screening modality, especially in an emergency.5 Magnetic resonance imaging best assesses the distribution and extension of the affected muscles, especially when fasciotomy is considered for treatment, and initially reveals edema, inflammation, and findings of myonecrosis; muscle atrophy and fatty degeneration occur later.4,13-15 Typical MRI findings include increased signal intensity on T2-weighted and STIR (short-tau inversion recovery) sequences and variable enhancement on T1 postcontrast images, as was seen in this case, which indicated edema, inflammation, and necrosis of the muscle tissue.
Shintani and colleagues described the reversibility of the MRI findings, showing that the high-intensity lesions seen on T2-weighted images resolved in parallel with the clinical course.14,16 Lu and colleagues investigated 10 patients with rhabdomyolysis and found 2 distinct imaging types: Type 1 shows homogenous signal changes and enhancement in the affected muscles, and Type 2 shows rim enhancement on contrast-enhanced MRI, a “stipple sign” indicating areas of myonecrosis.17 Magnetic resonance imaging signal alterations in the musculature can be nonspecific and overlap with those of inflammatory myopathies such as polymyositis, connective tissue diseases with inflammatory myositis, muscle infection, muscle infarction such as diabetic myonecrosis, muscle contusion, drug-induced myotoxicity, corticosteroids use, and use of cholesterol-lowering agents.18,19
Sonography is a useful screening modality for pain and swelling of the extremity, because it can detect a muscle tear, muscle sprain, and fluid collection, especially in emergent cases. There is scant literature about sonographic findings in rhabdomyolysis and compression nerve entrapment. The sonographic findings of rhabdomyolysis are local disorganization of the damaged muscle, decreased muscle echogenicity, and enlargement of the muscle, with preservation of the muscle boundaries.5-7
Intramuscular hyperechoic areas are seen due to hypercontractility of injured muscle. In this case, noted findings included patchy, irregular, hypoechoic areas, enlargement of the muscles and tendons, and irregular hyperechoic areas without focal defects. These findings differentiated an abnormality from a muscle tear or rupture, as these often show a focal muscle gap and focal defect, signifying the ruptured muscle retracting.
A study by Su and colleagues used the large number of crush injuries after an earthquake in China.5 The characteristic sonographic findings were edema and thickened disrupted striated muscle, good overall muscle continuity, vague muscle texture, and enhanced cloudy or ground-glass-like echo. There was no blood flow signal in the hypoechoic areas.6 Ultrasound was deemed a cost-effective, easily available modality by the authors.
Conclusion
Nontraumatic, focal rhabdomyolysis is rare and should be detected and differentiated from other causes of swelling, lump, pain, or other muscle disorders to prevent late complications. Sonography is an important screening diagnostic modality. MRI is used for assessment of the extent and distribution of injury. Awareness and familiarity with imaging findings can play a significant role, along with clinical and laboratory findings in the diagnosis and management of rhabdomyolysis.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Rhabdomyolysis involves the breakdown of skeletal muscle with the release of intracellular contents into the extracellular space and circulation.1 Diffuse rhabdomyolysis has been found in athletes due to overexertion. However, focal rhabdomyolysis is rare.2,3 The clinical presentation of focal rhabdomyolysis is subtle and nonspecific, with swelling, vague pain, weakness, fatigue, and tea-colored urine.
Early recognition and prompt management are crucial to prevent complications such as compression syndrome, acute renal failure, disseminated intravascular coagulation, cardiac dysrhythmia, or even cardiac arrest. Sonography and magnetic resonance imaging (MRI) can, therefore, be a complementary part of the diagnosis and assessment of the extent of rhabdomyolysis.4-7
Case History
The patient was a 34-year-old white man with a history of polysubstance abuse who presented to the emergency department (ED) with numbness and weakness in the left arm and hand, pain in the left side of his neck, and 3 days of intermittent amnesia with confusion. He had used IV heroin about 2 weeks prior to admission and used tobacco and alcohol daily. He reported no current medications or known allergies. The patient was in a monogamous relationship with a same-sex partner.
On physical examination, vital signs were within normal limits. He was in distress, confused, and disoriented as to time and place. An extremity examination revealed 1/5 strength in the extensors of the left elbow, left wrist, and left fingers with normal strength noted in the right upper extremity as well as the lower extremities. No sensory deficits were noted. The patient’s skin was warm and dry. Remarkable laboratory findings included creatine kinase (CK) 1,744 U/L, creatinine (Cr) 1.9 mg/dL, ALT 1,065 U/L, AST 319 U/L, ALP 159 U/L. A urine toxicology screen was positive for cocaine and opiates, and the urine analysis dip was negative for red blood cells, white blood cells, and protein. A differential diagnosis favored a left arm inflammatory reaction to IV drugs, although rhabdomyolysis was questioned.
A neurology consult was obtained, and a bedside electroencephalography test was performed in the ED by the neurologist, showing mild left occipital slow wave abnormality with no epileptiform discharges. A chest X-ray and computed tomography (CT) scan of the head and cervical spine were unremarkable, other than incidental mild prominence of the ventricles.
Over the next 24 hours, the patient was hydrated with IV normal saline without bicarbonate. His altered mental status, urine output, and biochemical abnormalities returned to normal, except for the serum CK, which decreased to 917 U/L. He had minimal improvement in his left upper extremity nerve palsy symptoms; however, he was deemed to be stable for discharge with follow-up in the clinic.
Instead of a clinic follow-up, the patient returned to the ED 7 days later, with progressive weakness of the left arm, forearm, and wrist. The patient noted that his weakness was so significant that he had to move his left arm with his right arm. He also reported extremity swelling and increasing paresthesias involving the lateral aspect of his left arm and hand, dizziness, and left neck pain. A physical examination revealed 3/5 strength at the left deltoid and left triceps, and 0/5 strength in the left fingers and grip. Remeasurement of CK was 54 U/L and Cr was 0.9 mg/dL. Compartment pressures were not measured.
Magnetic resonance imaging using multiplanar spin echo T1 and fast spin T2 weighted and post-IV 16cc Omniscan contrast sequences of the left shoulder were performed, showing multiple patchy T2 hyperintense focal areas with peripheral enhancement in the muscles of the posterior shoulder and in the tissues adjacent to the brachial plexus in the neck and shoulder (Figures 1A, 1B, and 1C). Sonography with matrix array linear 6-15 MH3 transducer was performed, which demonstrated patchy focal hypoechoic areas of muscle with enlarged, thickened, and disrupted muscle, representing devitalized muscle without any drainable fluid collection or abscess (Figures 2A, 2B, 2C, and 2D).
Magnetic resonance imaging and magnetic resonance angiogram scans of the brain and cervical spine with and without contrast were unremarkable. At that time, a definitive diagnosis was made of focal rhabdomyolysis and compressive neuropathy of the brachial plexus posterior cord, leading to brachial plexopathy of the left shoulder.
The patient was treated with hydration, a left arm sling, elevated left arm, and ibuprofen 600 mg qid to reduce inflammation. His swelling decreased markedly, and there was a slight improvement in pain and mobility at a 2-week neurology clinic follow-up. The patient lost contact after that.
Discussion
Rhabdomyolysis is caused by diverse etiologies. Most commonly, it is generalized and occurs due to overexertion, crush injury, steroid use, metabolic abnormalities, and certain medications and illicit drugs.1,2 The most likely etiology of rhabdomyolysis in patients presenting to the ED without significant trauma is of substance abuse, especially with ethanol, heroin, amphetamines, cocaine, and other sedatives or stimulants.1-3 The patient presented in this case study had a history of drug abuse, with a positive urine toxicology screen for cocaine and opiates. He had been intermittently confused and amnesic for 3 days prior to presentation, during which he may have been lying on his shoulder for a prolonged period.
Focal rhabdomyolysis and acute compression at the posterior shoulder leading to compressive brachial plexopathy is rare, with only 3 cases reported in the literature, all occurring with IV drug use.1-3 This patient had compression of the brachial plexus posterior cord from rhabdomyolysis and prolonged immobilization. Intravenous drug abusers may delay medical care due to perceived illicit drug effects and frequently present to the ED confused, agitated, or obtunded. Acute extremity swelling, a palpable lump, and pain can be due to various etiologies, such as trauma, fluid collection, muscle tear, myopathy, venous thrombosis, neoplasm, or rhabdomyolysis.
Diagnosis of nontraumatic rhabdomyolysis depends on clinical history and biochemical tests, such as serum CK and urine myoglobin.1,8 Creatine kinase is present in large quantities in the myocytes and is 100% sensitive as a marker for rhabdomyolysis.1,8 Creatine kinase may increase acutely > 1,000 U/L, suggesting muscle lysis and necrosis as etiology for pain as opposed to other causes such as hematomas, abscesses, or venous thrombi.1,9 Serum CK decreases rapidly at a rate of 39% per day, and it may normalize by the time a patient presents for medical care.1,10-12 Imaging plays a significant complimentary role. During the patient’s second ED presentation, the CK was normal at 54 U/L, whereas ultrasound and MRI findings were suggestive of focal muscle abnormalities.
Although there are diverse etiologies of rhabdomyolysis, the ultimate consequences of rhabdomyolysis are muscle cell membrane injury, metabolism malfunction, and destruction of the myofibril, resulting in inflammatory changes, such as muscle edema, hemorrhage, and myonecrosis and disruption of muscle fibers.1,2,8,9,13 This may cause an alteration in muscle size, shape, and echogenicity on sonography and abnormal signal intensity on MRI.13 The sensitivity of MRI in the detection of muscle involvement is higher than that of CT or ultrasound due to the high soft tissue contrast.4,13,14 Specificity of all 3 modalities is low and not reported.
Although the sensitivity of ultrasound is lower than that of MRI, use of ultrasound in neuromuscular evaluation has been increasing recently due to technical refinements. Ultrasound can be effectively used as a first-line screening modality, especially in an emergency.5 Magnetic resonance imaging best assesses the distribution and extension of the affected muscles, especially when fasciotomy is considered for treatment, and initially reveals edema, inflammation, and findings of myonecrosis; muscle atrophy and fatty degeneration occur later.4,13-15 Typical MRI findings include increased signal intensity on T2-weighted and STIR (short-tau inversion recovery) sequences and variable enhancement on T1 postcontrast images, as was seen in this case, which indicated edema, inflammation, and necrosis of the muscle tissue.
Shintani and colleagues described the reversibility of the MRI findings, showing that the high-intensity lesions seen on T2-weighted images resolved in parallel with the clinical course.14,16 Lu and colleagues investigated 10 patients with rhabdomyolysis and found 2 distinct imaging types: Type 1 shows homogenous signal changes and enhancement in the affected muscles, and Type 2 shows rim enhancement on contrast-enhanced MRI, a “stipple sign” indicating areas of myonecrosis.17 Magnetic resonance imaging signal alterations in the musculature can be nonspecific and overlap with those of inflammatory myopathies such as polymyositis, connective tissue diseases with inflammatory myositis, muscle infection, muscle infarction such as diabetic myonecrosis, muscle contusion, drug-induced myotoxicity, corticosteroids use, and use of cholesterol-lowering agents.18,19
Sonography is a useful screening modality for pain and swelling of the extremity, because it can detect a muscle tear, muscle sprain, and fluid collection, especially in emergent cases. There is scant literature about sonographic findings in rhabdomyolysis and compression nerve entrapment. The sonographic findings of rhabdomyolysis are local disorganization of the damaged muscle, decreased muscle echogenicity, and enlargement of the muscle, with preservation of the muscle boundaries.5-7
Intramuscular hyperechoic areas are seen due to hypercontractility of injured muscle. In this case, noted findings included patchy, irregular, hypoechoic areas, enlargement of the muscles and tendons, and irregular hyperechoic areas without focal defects. These findings differentiated an abnormality from a muscle tear or rupture, as these often show a focal muscle gap and focal defect, signifying the ruptured muscle retracting.
A study by Su and colleagues used the large number of crush injuries after an earthquake in China.5 The characteristic sonographic findings were edema and thickened disrupted striated muscle, good overall muscle continuity, vague muscle texture, and enhanced cloudy or ground-glass-like echo. There was no blood flow signal in the hypoechoic areas.6 Ultrasound was deemed a cost-effective, easily available modality by the authors.
Conclusion
Nontraumatic, focal rhabdomyolysis is rare and should be detected and differentiated from other causes of swelling, lump, pain, or other muscle disorders to prevent late complications. Sonography is an important screening diagnostic modality. MRI is used for assessment of the extent and distribution of injury. Awareness and familiarity with imaging findings can play a significant role, along with clinical and laboratory findings in the diagnosis and management of rhabdomyolysis.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
:
1. Richards JR. Rhabdomyolysis and drugs of abuse. J Emerg Med. 2000;19(1):51-56.
2. Farkash U, Shabshin N, Pritsch Perry M. Rhabdomyolysis of the deltoid muscle in a bodybuilder using anabolic-androgenic steroids: A case report. J Athl Train. 2009;44(1):98-100.
3. Mubarak SJ, Owen CA, Hargens AR, Garetto LP, Akeson WH. Acute compartment syndromes: Diagnosis and treatment with the aid of the wick catheter. J Bone Joint Surg Am. 1978;60(8):1091-1095.
4. Lamminen AE, Hekali PE, Tiula E, Suramo I, Korhola OA. Acute rhabdomyolysis: Evaluation with magnetic resonance imaging compared with computed tomography and ultrasonography. Br J Radiol. 1989;62(736):326-330.
5. Su BH, Qui L, Fu P, Luo Y, Tao Y, Peng YL. Ultrasonic appearance of rhabdomyolysis in patients with crush injury in the Wenchuan earthquake. Chin Med J (Engl). 2009;122(16):1872-1876.
6. Chiu Y-N, Wang T-G, Hsu C-Y, et al. Sonographic diagnosis of rhabdomyolysis. J Med Ultrasound. 2008;16(2):158-162.
7. Kaplan GN. Ultrasonic appearance of rhabdomyolysis. AJR Am J Roentgenol. 1980;134(2):375-377.
8. Spector R, Choudhury A, Cancilla P, Lakin R. Alcohol myopathy. Diagnosis by alcohol challenge. JAMA. 1979;242(15):1648-1649.
9. Gabow PA, Kaehny WD, Kelleher SP. The spectrum of rhabdomyolysis. Medicine (Baltimore). 1982;61(3):141-152.
10. Knochel JP. Mechanisms of rhabdomyolysis. Curr Opin Rheumatol. 1993;5(6):725-731.
11. Cadnapaphornchai P, Taher S, McDonald FD. Acute drug-association rhabdomyolysis: An examination of its diverse renal manifestations and complications. Am J Med Sci. 1980;280(2):66-72.
12. Curry SC, Chang D, Connor D. Drug and toxin-induced rhabdomyolysis. Ann Emerg Med. 1989;18(10):1068-1084.
13. May D, Disler DG, Jones EA, Balkissoon AA, Manaster BJ. Abnormal signal intensity in skeletal muscle at MR imaging: Patterns, pearls, and pitfalls. RadioGraphics. 2000;20(spec no):S295-S315.
14. Moratalla MB, Braun P, Fornas GM. Importance of MRI in the diagnosis and treatment of rhabdomyolysis. Eur J Radiol. 2008;65(2):311-315.
15. Beltran J, Rosenberg ZS. Diagnosis of compressive and entrapment neurorpathies of the upper extremity: Value of MR imaging. AJR Am J Roentgenol. 1994;163(3):525-531.
16. Shintani S, Shiigai T. Repeat MRI in acute rhabdomyolysis: Correlation with clinicopathological findings. J Comput Assist Tomogr. 1993;17(5):786-791.
17. Lu CH, Tsang YM, Yu CW, et al. Rhabdomyolysis: Magnetic resonance imaging and computed tomography findings. J Comput Assist Tomogr. 2007;31(3):368-374.
18. Schulze M, Kötter I, Ernemann U, et al. MRI findings in inflammatory muscle diseases and their noninflammatory mimics. AJR Am J Roentgenol. 2009;192(6):1708-1716.
19. Adams EM, Chow CK, Premkumar A, Plotz PH. The idiopathic inflammatory myopathies: Spectrum of MR imaging findings. Radiographics. 1995;15(3):563-574.
:
1. Richards JR. Rhabdomyolysis and drugs of abuse. J Emerg Med. 2000;19(1):51-56.
2. Farkash U, Shabshin N, Pritsch Perry M. Rhabdomyolysis of the deltoid muscle in a bodybuilder using anabolic-androgenic steroids: A case report. J Athl Train. 2009;44(1):98-100.
3. Mubarak SJ, Owen CA, Hargens AR, Garetto LP, Akeson WH. Acute compartment syndromes: Diagnosis and treatment with the aid of the wick catheter. J Bone Joint Surg Am. 1978;60(8):1091-1095.
4. Lamminen AE, Hekali PE, Tiula E, Suramo I, Korhola OA. Acute rhabdomyolysis: Evaluation with magnetic resonance imaging compared with computed tomography and ultrasonography. Br J Radiol. 1989;62(736):326-330.
5. Su BH, Qui L, Fu P, Luo Y, Tao Y, Peng YL. Ultrasonic appearance of rhabdomyolysis in patients with crush injury in the Wenchuan earthquake. Chin Med J (Engl). 2009;122(16):1872-1876.
6. Chiu Y-N, Wang T-G, Hsu C-Y, et al. Sonographic diagnosis of rhabdomyolysis. J Med Ultrasound. 2008;16(2):158-162.
7. Kaplan GN. Ultrasonic appearance of rhabdomyolysis. AJR Am J Roentgenol. 1980;134(2):375-377.
8. Spector R, Choudhury A, Cancilla P, Lakin R. Alcohol myopathy. Diagnosis by alcohol challenge. JAMA. 1979;242(15):1648-1649.
9. Gabow PA, Kaehny WD, Kelleher SP. The spectrum of rhabdomyolysis. Medicine (Baltimore). 1982;61(3):141-152.
10. Knochel JP. Mechanisms of rhabdomyolysis. Curr Opin Rheumatol. 1993;5(6):725-731.
11. Cadnapaphornchai P, Taher S, McDonald FD. Acute drug-association rhabdomyolysis: An examination of its diverse renal manifestations and complications. Am J Med Sci. 1980;280(2):66-72.
12. Curry SC, Chang D, Connor D. Drug and toxin-induced rhabdomyolysis. Ann Emerg Med. 1989;18(10):1068-1084.
13. May D, Disler DG, Jones EA, Balkissoon AA, Manaster BJ. Abnormal signal intensity in skeletal muscle at MR imaging: Patterns, pearls, and pitfalls. RadioGraphics. 2000;20(spec no):S295-S315.
14. Moratalla MB, Braun P, Fornas GM. Importance of MRI in the diagnosis and treatment of rhabdomyolysis. Eur J Radiol. 2008;65(2):311-315.
15. Beltran J, Rosenberg ZS. Diagnosis of compressive and entrapment neurorpathies of the upper extremity: Value of MR imaging. AJR Am J Roentgenol. 1994;163(3):525-531.
16. Shintani S, Shiigai T. Repeat MRI in acute rhabdomyolysis: Correlation with clinicopathological findings. J Comput Assist Tomogr. 1993;17(5):786-791.
17. Lu CH, Tsang YM, Yu CW, et al. Rhabdomyolysis: Magnetic resonance imaging and computed tomography findings. J Comput Assist Tomogr. 2007;31(3):368-374.
18. Schulze M, Kötter I, Ernemann U, et al. MRI findings in inflammatory muscle diseases and their noninflammatory mimics. AJR Am J Roentgenol. 2009;192(6):1708-1716.
19. Adams EM, Chow CK, Premkumar A, Plotz PH. The idiopathic inflammatory myopathies: Spectrum of MR imaging findings. Radiographics. 1995;15(3):563-574.
2014 Update on ovarian cancer
Ovarian cancer remains the deadliest gynecologic malignancy in the United States, with more than 22,000 women newly diagnosed and more than 14,000 deaths each year. We have made slow progress in terms of survival with new drugs and applications, such as intraperitoneal chemotherapy combined with more aggressive cytoreductive efforts. Five-year survival rates have increased—from 36% to 44%—since the late 1970s.1 To make the leap from molecular genetics to successful screening, early diagnosis, and targeted treatment, we must first:
- Enhance our understanding of the changes that lead to ovarian cancer. Currently, malignant transformation of the fallopian tube epithelium is thought to result in high-grade papillary serous cancer.2 If this is indeed the pathologic origin of ovarian cancers, then early detection or even detection in the premalignant phase may be possible using tests of vaginal fluid. Are early detection, and even screening, possible and how would it effect treatment and survival?
- Develop new and powerful tools to detect molecular changes that might impact treatment and survival. Just a few years ago, initial sequencing of the human genome cost more than $100 million, but DNA sequencing technologies have evolved rapidly, with current estimates at less than a few thousand dollars per genome.3 Knowing the mutations responsible for an individual’s cancer would allow for targeted, individualized treatment plans. Would one patient benefit from neoadjuvant therapy while another needs primary surgical debulking?
In this article, we highlight the historical basis and recent developments in the field of ovarian cancer, focusing on:
- etiologic heterogeneity and molecular biology detection of small numbers of cancer cells in vaginal secretions and the blood stream.
- detection of small numbers of cancer cells in vaginal secretions and the blood stream.
What mutations are we looking for?
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609−615.
In last year’s Update, we discussed the role of The Cancer Genome Atlas (TCGA) project in endometrial cancer.4 For ovarian cancer, TCGA analyzed messenger RNA expression, microRNA expression, promoter methylation, and DNA copy number in 489 high-grade serous ovarian adenocarcinomas and the DNA sequences of exons from coding genes in 316 of these tumors.
Almost all tumors (96%) were characterized by mutations of the gene encoding TP53 in addition to statistically recurrent mutations in nine other loci, including NF1, BRCA1, BRCA2, RB1, and CDK12, although these were of low prevalence. Analyses also brought new insight regarding the survival impact of tumors containing BRCA1 or BRCA2 and CCNE1 mutations. Findings included NOTCH and FOXM1 signaling involvement in serous ovarian cancer pathophysiology as well as defective homologous recombination in approximately half of the tumors studied.
What this evidence means for practiceWith these mutations as our targets, we can screen vaginal secretions as well as blood for markers of ovarian cancer.
Ovarian and endometrial cancer cells detected in the vagina
Kinde I, Bettegowda C, Wang Y, et al. Evaluation of DNA from the Papanicolaou test to detect ovarian and endometrial cancers. Sci Transl Med. 2013;5(167):167ra4.
Erickson BK, Kinde I, Dobbin AC, et al. Detection of somatic TP53 mutations in tampons of patients with high-grade serous ovarian cancer [published online ahead of print October 2014]. Obstet Gynecol. 2014;124(5).
Ruth Graham, Papanicolaou’s cytology technician in the 1940s, first described ovarian cancer cells detected in vaginal/cervical cytology obtained from vaginal secretions.5 Current studies now demonstrate that we have technology capable of more than simple cytologic detection. We can isolate and evaluate these cancer cells in very small numbers.
Ovarian and endometrial cancer DNA identified in Pap specimenKinde and colleagues assembled a catalog of common mutations previously found in ovarian cancer as well as new data on 22 endometrial tumors. They tested 24 endometrial and 22 ovarian samples from patients with endometrial or ovarian cancers and confirmed that all 46 harbored at least some component of the common genetic changes in their catalog. Hypothesizing that the cancers likely shed cells from their surfaces, they sought to determine whether they could detect these cells among the cervical cells in a Pap smear.
These investigators used massively parallel sequencing to test DNA collected in modern liquid-based cytologic specimens for the same mutations found in the cancer cells. They found that 100% of the endometrial cancers and 41% of ovarian cancers were detectable by this method.
TP53 mutations in ovarian cancer cells detected in vaginally placed tamponWith similar technology, but a different collection method, Erickson and colleagues sought to detect tumor cells in the vagina of women with serous ovarian cancer by TP53 analysis of DNA samples collected via vaginal tampon.
Thirty-three women with pelvic masses suspicious for malignancy and scheduled to undergo diagnostic or therapeutic surgery were enrolled. Of the 25 patients who placed the tampon 8 to 12 hours prior to surgery; 13 had benign disease; three had nonovarian malignancies; and nine had serous adenocarcinoma of ovarian, tubal, or primary peritoneal origin. DNA from tumor specimens of eight patients with serous carcinoma and adequate DNA samples were analyzed for TP53 mutations. The corresponding DNA extracted from the tampon was then probed for the mutation identified in the tumor.
Mutational analysis of the tampon specimen DNA revealed no mutations in the tampon DNA of the three patients who had previously undergone tubal ligation, while mutations were observed in three of the five patients with intact tubes—producing a sensitivity of 60%. The fraction of mutant alleles in the tampon DNA was extremely low at 0.01% to 0.07%, requiring ultra-deep sequencing and increasing the importance of paired primary tumor specimens.
What this evidence means for practiceWhile sensitivity in a population of high-risk patients with intact tubes was found to be 60%, it is unclear what it would be in patients with less advanced disease. The ability of the test to detect mutations at exceptionally low limits is impressive; however, it increases the risk that a variant represents a sequencing error or a sample-to-sample contamination. This study is novel in its approach to diagnosis of ovarian cancer and is a stride toward screening, providing an opportunity to further validate the technology prior to widespread use and clinical application.
Circulating tumor cells—the future of cancer management?
Obermayr E, Castillo-Tong DC, Pils D, et al. Molecular characterization of circulating tumor cells in patients with ovarian cancer improves their prognostic significance: a study of the OVCAD consortium. Gynecol Oncol. 2013;128(1):15−21.
Similar in concept to noninvasive prenatal testing for fetal aneuploidy, high circulating tumor cell (CTC) numbers have been correlated with aggressive disease, increased metastasis, and decreased time to relapse. As with cancer cells in vaginal secretions, CTCs also may provide an opportunity for early detection and targeted treatment.6
While many CTC studies have used epithelial cell adhesion molecule (EpCAM)−based CTC detection, results have been found to be highly variable between tumor subtypes and phase of disease.7 Therefore, Obermayer and colleagues sought to identify novel markers for CTCs in patients with epithelial ovarian cancer and elucidate their impact on outcome.
Details of the studyMatched ovarian cancer tissues and peripheral blood leukocytes of 35 patients underwent microarray analysis to identify novel CTC markers. Gene expression of the novel markers as well as EpCAM were analyzed using blood samples taken from 39 healthy females and from 216 patients with ovarian cancer before primary treatment and 6 months after adjuvant chemotherapy. Overexpression of at least one gene, compared with the healthy control group, was considered CTC positivity.
CTCs were detected in 24.5% of the baseline and 20.4% of the follow-up samples, of which two-thirds showed overexpression of the cyclophilin C gene (PPIC), and just a few by EpCAM overexpression. PPIC-positive CTCs during follow-up were detected significantly more often in the platinum resistant group, and indicated poor outcome even when controlling for classical prognostic parameters.
What this evidence means for practiceThe study authors found that molecular characterization of CTC is superior to CTC enumeration. Ultimately, CTC diagnostics may lead to earlier detection and more personalized treatment of ovarian cancer.
Therefore, this technology could have great impact on screening for and the survival of a large subset of patients with ovarian cancer. In addition, the cells obtained preoperatively could help assess the risk of malignancy in an ovarian mass prior to surgery, or even help in treatment planning, as we enter an era in which we have the ability to assess cancers for prognosis and features of treatment response.
Share your thoughts on this article! Send your Letter to the Editor to [email protected].
1. Siegel R, Ma J, Zou Z, et al. Cancer Statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29.
2. Piek JM, van Diest PJ, Zweemer RP, et al. Dysplastic changes in prophylactically removed fallopian tubes of women predisposed to developing ovarian cancer. J Pathol. 2001;195(4):451–456.
3. Wetterstrand KA. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP). http://www.genome.gov/sequencingcosts. Updated July 18, 2014. Accessed September 21, 2014.
4. Kandoth C, Schultz N, Cherniack AD, et al; Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73.
5. Papanicolaou GN, Traut HF. The diagnostic value of vaginal smears in carcinoma of the uterus. Am J Obstet Gynecol. 1941;42:193–206.
6. Plaks V, Koopman CD, Werb Z. Cancer. Circulating tumor cells. Science. 2013;341(6151):1186–1188.
7. Sieuwerts AM, Kraan J, Bolt J, et al. Anti-epithelial cell adhesion molecule antibodies and the detection of circulating normal-like breast tumor cells. J Natl Cancer Inst. 2009;101(1):61–66.

Laura Divine, MD
Dr. Divine is a Fellow in the Department of Obstetrics and Gynecology, Division of Gynecologic Oncology, at Washington University School of Medicine, St. Louis, Missouri.

David G. Mutch, MD
Dr. Mutch is Ira C. and Judith Gall Professor of Obstetrics and Gynecology and Chief of Gynecologic Oncology at Washington University School of Medicine in St. Louis, Missouri. He serves on the OBG Management Board of Editors.
The authors report no financial relationships relevant to this article.
Laura Divine, MD
Dr. Divine is a Fellow in the Department of Obstetrics and Gynecology, Division of Gynecologic Oncology, at Washington University School of Medicine, St. Louis, Missouri.
David G. Mutch, MD
Dr. Mutch is Ira C. and Judith Gall Professor of Obstetrics and Gynecology and Chief of Gynecologic Oncology at Washington University School of Medicine in St. Louis, Missouri. He serves on the OBG Management Board of Editors.
The authors report no financial relationships relevant to this article.
Laura Divine, MD
Dr. Divine is a Fellow in the Department of Obstetrics and Gynecology, Division of Gynecologic Oncology, at Washington University School of Medicine, St. Louis, Missouri.
David G. Mutch, MD
Dr. Mutch is Ira C. and Judith Gall Professor of Obstetrics and Gynecology and Chief of Gynecologic Oncology at Washington University School of Medicine in St. Louis, Missouri. He serves on the OBG Management Board of Editors.
The authors report no financial relationships relevant to this article.
Ovarian cancer remains the deadliest gynecologic malignancy in the United States, with more than 22,000 women newly diagnosed and more than 14,000 deaths each year. We have made slow progress in terms of survival with new drugs and applications, such as intraperitoneal chemotherapy combined with more aggressive cytoreductive efforts. Five-year survival rates have increased—from 36% to 44%—since the late 1970s.1 To make the leap from molecular genetics to successful screening, early diagnosis, and targeted treatment, we must first:
- Enhance our understanding of the changes that lead to ovarian cancer. Currently, malignant transformation of the fallopian tube epithelium is thought to result in high-grade papillary serous cancer.2 If this is indeed the pathologic origin of ovarian cancers, then early detection or even detection in the premalignant phase may be possible using tests of vaginal fluid. Are early detection, and even screening, possible and how would it effect treatment and survival?
- Develop new and powerful tools to detect molecular changes that might impact treatment and survival. Just a few years ago, initial sequencing of the human genome cost more than $100 million, but DNA sequencing technologies have evolved rapidly, with current estimates at less than a few thousand dollars per genome.3 Knowing the mutations responsible for an individual’s cancer would allow for targeted, individualized treatment plans. Would one patient benefit from neoadjuvant therapy while another needs primary surgical debulking?
In this article, we highlight the historical basis and recent developments in the field of ovarian cancer, focusing on:
- etiologic heterogeneity and molecular biology detection of small numbers of cancer cells in vaginal secretions and the blood stream.
- detection of small numbers of cancer cells in vaginal secretions and the blood stream.
What mutations are we looking for?
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609−615.
In last year’s Update, we discussed the role of The Cancer Genome Atlas (TCGA) project in endometrial cancer.4 For ovarian cancer, TCGA analyzed messenger RNA expression, microRNA expression, promoter methylation, and DNA copy number in 489 high-grade serous ovarian adenocarcinomas and the DNA sequences of exons from coding genes in 316 of these tumors.
Almost all tumors (96%) were characterized by mutations of the gene encoding TP53 in addition to statistically recurrent mutations in nine other loci, including NF1, BRCA1, BRCA2, RB1, and CDK12, although these were of low prevalence. Analyses also brought new insight regarding the survival impact of tumors containing BRCA1 or BRCA2 and CCNE1 mutations. Findings included NOTCH and FOXM1 signaling involvement in serous ovarian cancer pathophysiology as well as defective homologous recombination in approximately half of the tumors studied.
What this evidence means for practiceWith these mutations as our targets, we can screen vaginal secretions as well as blood for markers of ovarian cancer.
Ovarian and endometrial cancer cells detected in the vagina
Kinde I, Bettegowda C, Wang Y, et al. Evaluation of DNA from the Papanicolaou test to detect ovarian and endometrial cancers. Sci Transl Med. 2013;5(167):167ra4.
Erickson BK, Kinde I, Dobbin AC, et al. Detection of somatic TP53 mutations in tampons of patients with high-grade serous ovarian cancer [published online ahead of print October 2014]. Obstet Gynecol. 2014;124(5).
Ruth Graham, Papanicolaou’s cytology technician in the 1940s, first described ovarian cancer cells detected in vaginal/cervical cytology obtained from vaginal secretions.5 Current studies now demonstrate that we have technology capable of more than simple cytologic detection. We can isolate and evaluate these cancer cells in very small numbers.
Ovarian and endometrial cancer DNA identified in Pap specimenKinde and colleagues assembled a catalog of common mutations previously found in ovarian cancer as well as new data on 22 endometrial tumors. They tested 24 endometrial and 22 ovarian samples from patients with endometrial or ovarian cancers and confirmed that all 46 harbored at least some component of the common genetic changes in their catalog. Hypothesizing that the cancers likely shed cells from their surfaces, they sought to determine whether they could detect these cells among the cervical cells in a Pap smear.
These investigators used massively parallel sequencing to test DNA collected in modern liquid-based cytologic specimens for the same mutations found in the cancer cells. They found that 100% of the endometrial cancers and 41% of ovarian cancers were detectable by this method.
TP53 mutations in ovarian cancer cells detected in vaginally placed tamponWith similar technology, but a different collection method, Erickson and colleagues sought to detect tumor cells in the vagina of women with serous ovarian cancer by TP53 analysis of DNA samples collected via vaginal tampon.
Thirty-three women with pelvic masses suspicious for malignancy and scheduled to undergo diagnostic or therapeutic surgery were enrolled. Of the 25 patients who placed the tampon 8 to 12 hours prior to surgery; 13 had benign disease; three had nonovarian malignancies; and nine had serous adenocarcinoma of ovarian, tubal, or primary peritoneal origin. DNA from tumor specimens of eight patients with serous carcinoma and adequate DNA samples were analyzed for TP53 mutations. The corresponding DNA extracted from the tampon was then probed for the mutation identified in the tumor.
Mutational analysis of the tampon specimen DNA revealed no mutations in the tampon DNA of the three patients who had previously undergone tubal ligation, while mutations were observed in three of the five patients with intact tubes—producing a sensitivity of 60%. The fraction of mutant alleles in the tampon DNA was extremely low at 0.01% to 0.07%, requiring ultra-deep sequencing and increasing the importance of paired primary tumor specimens.
What this evidence means for practiceWhile sensitivity in a population of high-risk patients with intact tubes was found to be 60%, it is unclear what it would be in patients with less advanced disease. The ability of the test to detect mutations at exceptionally low limits is impressive; however, it increases the risk that a variant represents a sequencing error or a sample-to-sample contamination. This study is novel in its approach to diagnosis of ovarian cancer and is a stride toward screening, providing an opportunity to further validate the technology prior to widespread use and clinical application.
Circulating tumor cells—the future of cancer management?
Obermayr E, Castillo-Tong DC, Pils D, et al. Molecular characterization of circulating tumor cells in patients with ovarian cancer improves their prognostic significance: a study of the OVCAD consortium. Gynecol Oncol. 2013;128(1):15−21.
Similar in concept to noninvasive prenatal testing for fetal aneuploidy, high circulating tumor cell (CTC) numbers have been correlated with aggressive disease, increased metastasis, and decreased time to relapse. As with cancer cells in vaginal secretions, CTCs also may provide an opportunity for early detection and targeted treatment.6
While many CTC studies have used epithelial cell adhesion molecule (EpCAM)−based CTC detection, results have been found to be highly variable between tumor subtypes and phase of disease.7 Therefore, Obermayer and colleagues sought to identify novel markers for CTCs in patients with epithelial ovarian cancer and elucidate their impact on outcome.
Details of the studyMatched ovarian cancer tissues and peripheral blood leukocytes of 35 patients underwent microarray analysis to identify novel CTC markers. Gene expression of the novel markers as well as EpCAM were analyzed using blood samples taken from 39 healthy females and from 216 patients with ovarian cancer before primary treatment and 6 months after adjuvant chemotherapy. Overexpression of at least one gene, compared with the healthy control group, was considered CTC positivity.
CTCs were detected in 24.5% of the baseline and 20.4% of the follow-up samples, of which two-thirds showed overexpression of the cyclophilin C gene (PPIC), and just a few by EpCAM overexpression. PPIC-positive CTCs during follow-up were detected significantly more often in the platinum resistant group, and indicated poor outcome even when controlling for classical prognostic parameters.
What this evidence means for practiceThe study authors found that molecular characterization of CTC is superior to CTC enumeration. Ultimately, CTC diagnostics may lead to earlier detection and more personalized treatment of ovarian cancer.
Therefore, this technology could have great impact on screening for and the survival of a large subset of patients with ovarian cancer. In addition, the cells obtained preoperatively could help assess the risk of malignancy in an ovarian mass prior to surgery, or even help in treatment planning, as we enter an era in which we have the ability to assess cancers for prognosis and features of treatment response.
Share your thoughts on this article! Send your Letter to the Editor to [email protected].
Ovarian cancer remains the deadliest gynecologic malignancy in the United States, with more than 22,000 women newly diagnosed and more than 14,000 deaths each year. We have made slow progress in terms of survival with new drugs and applications, such as intraperitoneal chemotherapy combined with more aggressive cytoreductive efforts. Five-year survival rates have increased—from 36% to 44%—since the late 1970s.1 To make the leap from molecular genetics to successful screening, early diagnosis, and targeted treatment, we must first:
- Enhance our understanding of the changes that lead to ovarian cancer. Currently, malignant transformation of the fallopian tube epithelium is thought to result in high-grade papillary serous cancer.2 If this is indeed the pathologic origin of ovarian cancers, then early detection or even detection in the premalignant phase may be possible using tests of vaginal fluid. Are early detection, and even screening, possible and how would it effect treatment and survival?
- Develop new and powerful tools to detect molecular changes that might impact treatment and survival. Just a few years ago, initial sequencing of the human genome cost more than $100 million, but DNA sequencing technologies have evolved rapidly, with current estimates at less than a few thousand dollars per genome.3 Knowing the mutations responsible for an individual’s cancer would allow for targeted, individualized treatment plans. Would one patient benefit from neoadjuvant therapy while another needs primary surgical debulking?
In this article, we highlight the historical basis and recent developments in the field of ovarian cancer, focusing on:
- etiologic heterogeneity and molecular biology detection of small numbers of cancer cells in vaginal secretions and the blood stream.
- detection of small numbers of cancer cells in vaginal secretions and the blood stream.
What mutations are we looking for?
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609−615.
In last year’s Update, we discussed the role of The Cancer Genome Atlas (TCGA) project in endometrial cancer.4 For ovarian cancer, TCGA analyzed messenger RNA expression, microRNA expression, promoter methylation, and DNA copy number in 489 high-grade serous ovarian adenocarcinomas and the DNA sequences of exons from coding genes in 316 of these tumors.
Almost all tumors (96%) were characterized by mutations of the gene encoding TP53 in addition to statistically recurrent mutations in nine other loci, including NF1, BRCA1, BRCA2, RB1, and CDK12, although these were of low prevalence. Analyses also brought new insight regarding the survival impact of tumors containing BRCA1 or BRCA2 and CCNE1 mutations. Findings included NOTCH and FOXM1 signaling involvement in serous ovarian cancer pathophysiology as well as defective homologous recombination in approximately half of the tumors studied.
What this evidence means for practiceWith these mutations as our targets, we can screen vaginal secretions as well as blood for markers of ovarian cancer.
Ovarian and endometrial cancer cells detected in the vagina
Kinde I, Bettegowda C, Wang Y, et al. Evaluation of DNA from the Papanicolaou test to detect ovarian and endometrial cancers. Sci Transl Med. 2013;5(167):167ra4.
Erickson BK, Kinde I, Dobbin AC, et al. Detection of somatic TP53 mutations in tampons of patients with high-grade serous ovarian cancer [published online ahead of print October 2014]. Obstet Gynecol. 2014;124(5).
Ruth Graham, Papanicolaou’s cytology technician in the 1940s, first described ovarian cancer cells detected in vaginal/cervical cytology obtained from vaginal secretions.5 Current studies now demonstrate that we have technology capable of more than simple cytologic detection. We can isolate and evaluate these cancer cells in very small numbers.
Ovarian and endometrial cancer DNA identified in Pap specimenKinde and colleagues assembled a catalog of common mutations previously found in ovarian cancer as well as new data on 22 endometrial tumors. They tested 24 endometrial and 22 ovarian samples from patients with endometrial or ovarian cancers and confirmed that all 46 harbored at least some component of the common genetic changes in their catalog. Hypothesizing that the cancers likely shed cells from their surfaces, they sought to determine whether they could detect these cells among the cervical cells in a Pap smear.
These investigators used massively parallel sequencing to test DNA collected in modern liquid-based cytologic specimens for the same mutations found in the cancer cells. They found that 100% of the endometrial cancers and 41% of ovarian cancers were detectable by this method.
TP53 mutations in ovarian cancer cells detected in vaginally placed tamponWith similar technology, but a different collection method, Erickson and colleagues sought to detect tumor cells in the vagina of women with serous ovarian cancer by TP53 analysis of DNA samples collected via vaginal tampon.
Thirty-three women with pelvic masses suspicious for malignancy and scheduled to undergo diagnostic or therapeutic surgery were enrolled. Of the 25 patients who placed the tampon 8 to 12 hours prior to surgery; 13 had benign disease; three had nonovarian malignancies; and nine had serous adenocarcinoma of ovarian, tubal, or primary peritoneal origin. DNA from tumor specimens of eight patients with serous carcinoma and adequate DNA samples were analyzed for TP53 mutations. The corresponding DNA extracted from the tampon was then probed for the mutation identified in the tumor.
Mutational analysis of the tampon specimen DNA revealed no mutations in the tampon DNA of the three patients who had previously undergone tubal ligation, while mutations were observed in three of the five patients with intact tubes—producing a sensitivity of 60%. The fraction of mutant alleles in the tampon DNA was extremely low at 0.01% to 0.07%, requiring ultra-deep sequencing and increasing the importance of paired primary tumor specimens.
What this evidence means for practiceWhile sensitivity in a population of high-risk patients with intact tubes was found to be 60%, it is unclear what it would be in patients with less advanced disease. The ability of the test to detect mutations at exceptionally low limits is impressive; however, it increases the risk that a variant represents a sequencing error or a sample-to-sample contamination. This study is novel in its approach to diagnosis of ovarian cancer and is a stride toward screening, providing an opportunity to further validate the technology prior to widespread use and clinical application.
Circulating tumor cells—the future of cancer management?
Obermayr E, Castillo-Tong DC, Pils D, et al. Molecular characterization of circulating tumor cells in patients with ovarian cancer improves their prognostic significance: a study of the OVCAD consortium. Gynecol Oncol. 2013;128(1):15−21.
Similar in concept to noninvasive prenatal testing for fetal aneuploidy, high circulating tumor cell (CTC) numbers have been correlated with aggressive disease, increased metastasis, and decreased time to relapse. As with cancer cells in vaginal secretions, CTCs also may provide an opportunity for early detection and targeted treatment.6
While many CTC studies have used epithelial cell adhesion molecule (EpCAM)−based CTC detection, results have been found to be highly variable between tumor subtypes and phase of disease.7 Therefore, Obermayer and colleagues sought to identify novel markers for CTCs in patients with epithelial ovarian cancer and elucidate their impact on outcome.
Details of the studyMatched ovarian cancer tissues and peripheral blood leukocytes of 35 patients underwent microarray analysis to identify novel CTC markers. Gene expression of the novel markers as well as EpCAM were analyzed using blood samples taken from 39 healthy females and from 216 patients with ovarian cancer before primary treatment and 6 months after adjuvant chemotherapy. Overexpression of at least one gene, compared with the healthy control group, was considered CTC positivity.
CTCs were detected in 24.5% of the baseline and 20.4% of the follow-up samples, of which two-thirds showed overexpression of the cyclophilin C gene (PPIC), and just a few by EpCAM overexpression. PPIC-positive CTCs during follow-up were detected significantly more often in the platinum resistant group, and indicated poor outcome even when controlling for classical prognostic parameters.
What this evidence means for practiceThe study authors found that molecular characterization of CTC is superior to CTC enumeration. Ultimately, CTC diagnostics may lead to earlier detection and more personalized treatment of ovarian cancer.
Therefore, this technology could have great impact on screening for and the survival of a large subset of patients with ovarian cancer. In addition, the cells obtained preoperatively could help assess the risk of malignancy in an ovarian mass prior to surgery, or even help in treatment planning, as we enter an era in which we have the ability to assess cancers for prognosis and features of treatment response.
Share your thoughts on this article! Send your Letter to the Editor to [email protected].
1. Siegel R, Ma J, Zou Z, et al. Cancer Statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29.
2. Piek JM, van Diest PJ, Zweemer RP, et al. Dysplastic changes in prophylactically removed fallopian tubes of women predisposed to developing ovarian cancer. J Pathol. 2001;195(4):451–456.
3. Wetterstrand KA. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP). http://www.genome.gov/sequencingcosts. Updated July 18, 2014. Accessed September 21, 2014.
4. Kandoth C, Schultz N, Cherniack AD, et al; Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73.
5. Papanicolaou GN, Traut HF. The diagnostic value of vaginal smears in carcinoma of the uterus. Am J Obstet Gynecol. 1941;42:193–206.
6. Plaks V, Koopman CD, Werb Z. Cancer. Circulating tumor cells. Science. 2013;341(6151):1186–1188.
7. Sieuwerts AM, Kraan J, Bolt J, et al. Anti-epithelial cell adhesion molecule antibodies and the detection of circulating normal-like breast tumor cells. J Natl Cancer Inst. 2009;101(1):61–66.
1. Siegel R, Ma J, Zou Z, et al. Cancer Statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29.
2. Piek JM, van Diest PJ, Zweemer RP, et al. Dysplastic changes in prophylactically removed fallopian tubes of women predisposed to developing ovarian cancer. J Pathol. 2001;195(4):451–456.
3. Wetterstrand KA. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP). http://www.genome.gov/sequencingcosts. Updated July 18, 2014. Accessed September 21, 2014.
4. Kandoth C, Schultz N, Cherniack AD, et al; Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73.
5. Papanicolaou GN, Traut HF. The diagnostic value of vaginal smears in carcinoma of the uterus. Am J Obstet Gynecol. 1941;42:193–206.
6. Plaks V, Koopman CD, Werb Z. Cancer. Circulating tumor cells. Science. 2013;341(6151):1186–1188.
7. Sieuwerts AM, Kraan J, Bolt J, et al. Anti-epithelial cell adhesion molecule antibodies and the detection of circulating normal-like breast tumor cells. J Natl Cancer Inst. 2009;101(1):61–66.
In this article:
What mutations are we looking for?
Ovarian and endometrial cancer cells detected in the vagina
Circulating tumor cells—the future of cancer management?
Global Ebola—Are We Prepared?
In this age of globalization, just a few hours of air travel separates even the most remote places in our world. Given this reality, the recent epidemic of Ebola virus disease (EVD) in West Africa (Figure 1) has arrived on the doorstep of Texas Health Presbyterian Hospital in Dallas. Ill and potentially infected US healthcare workers and missionaries brought home for treatment and quarantine, plus the travel of the general population through the affected countries of Sierra Leone, Liberia, Guinea, and Nigeria, require all acute-care providers to be cognizant that this deadly disease may present in any community ED in the United States. Awareness and knowledge of the appropriate steps to manage care safely and effectively, while mindfully preventing the potential for viral transmission, is paramount.
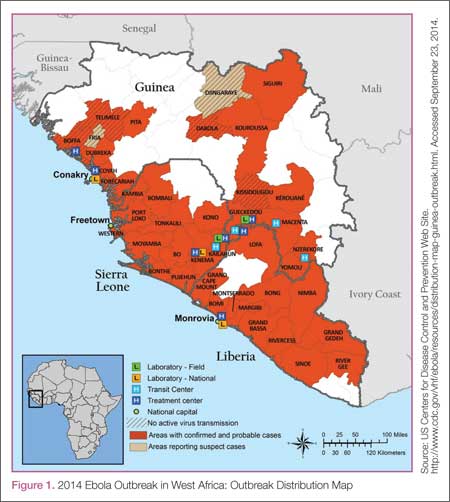
Hemorrhagic Fevers
Viral hemorrhagic fevers are manifestations of four distinct families of RNA viruses: arenaviruses, bunyaviruses, flaviviruses, and filoviruses. All of these families of viruses depend on a natural insect or animal (nonhuman) host and are thus restricted geographically to the regions where the endemic hosts reside. The viruses can only infect a human when one comes into direct contact with an infected host; this human becomes an infectious host when symptoms of disease develop and subsequently, the possibility of transmission to other close direct human contacts exists.
Ebola Virus Species
The family within which the Ebola virus species are classified is the filoviruses. Five species of Ebola filovirus have been isolated to date: Ebola virus (Zaire ebolavirus), Sudan virus (Sudan ebolavirus), Taï Forest virus (Taï Forest ebolavirus, formerly Côte d’Ivoire ebolavirus), and Bundibugyo virus (Bundibugyo ebolavirus). The fifth, Reston virus (Reston ebolavirus), has caused disease in nonhuman primates, but not in humans. This epidemic has been attributed to a variant of the Zaire species.2 Transmission through direct contact with body fluids of febrile live infected patients and the postmortem period continues in communities and healthcare sites, as lack of adequate personal protective equipment (PPE) and meticulous environmental hygiene remains a challenge in many of these settings.
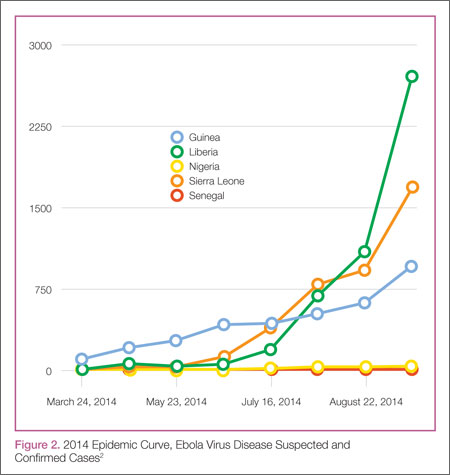
Clinical Presentation
In EVD, the onset of symptoms typically occurs abruptly at an average of 8 to 10 days postexposure and includes fever, headache, myalgia, and malaise; in some patients, an erythematous maculopapular rash involving the face, neck, trunk, and arms erupts by days 5 to 7.1 The nonspecific nature of these early signs and symptoms warrants caution in any patient known to have traveled in an endemic country with potential exposure to body fluids of infected patients—underscoring the importance of both obtaining a complete travel history to determine the potential for disease exposure in patients presenting with infectious-disease symptoms and effectively communicating this information to all ED staff.
In addition, such caution includes healthcare mission workers caring for Ebola patients, those involved in butchering infected animals for meat, and persons participating in traditional funeral rituals for those deceased from Ebola without the use of adequate PPE and/or environmental hygiene. Other more common infectious diseases with shared features of EVD must also be considered at this stage and include malaria, meningococcemia, measles, and typhoid fever, among others.
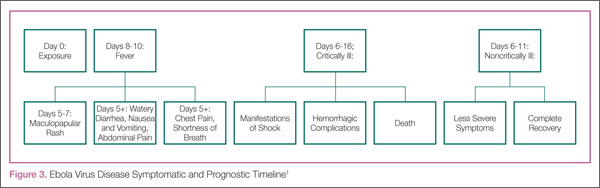
After the first 5 days of exposure, progression of symptoms may include severe watery diarrhea, nausea and vomiting, abdominal pain, shortness of breath, chest pain, headache, and/or confusion. Conjunctival injection may also develop. Not all patients will have signs of hemorrhagic fever with bleeding from the mouth, eyes, ears, in stool, or from internal organs, but petechiae, ecchymosis, and oozing from venipuncture sites may develop. Those at the highest risk of death show signs of sepsis, such as shock and multiorgan system failure, which may include hemorrhagic manifestations, early in the course of their illness. Patients with these complications typically expire between days 6 to16.1 Survivors of the disease tend to have fever with less severe symptoms for a period of several more days, and then begin to improve clinically between days 6 to 11 after onset of symptoms1 (Figure 3).
Ecology
A zoonotic filovirus transmissible from animal populations to humans causes EVD. Research strongly suggests that fruit bats are the reservoir and hosts for this filovirus. Direct human contact with bats or with wild animals that have been infected by bats initiates the human-to-human transmission of EVD.3
Pathogenesis
Through direct contact with mucous membranes, a break in the skin, or parenterally, Ebola enters and infects multiple cell types. Incubation periods appear shorter in infections acquired through direct injection (6 days) than for contact transmission (10 days).1 Emesis, urine, stool, sweat, semen, cerebrospinal fluid, breast milk, and saliva are actively capable of viral transmission. From point of entry, the virus migrates to the lymph nodes, then to liver, spleen, and adrenal glands. Hepatocellular necrosis leads to clotting-factor derangement and dysfunction resulting in coagulopathy and bleeding and potential liver failure. Necrosis of adrenal tissue may be present and results in impaired steroid synthesis and hypotension. The presence of the virus appears to incite a cytokine inflammatory storm causing microvascular leakage, with the end effect of multiorgan system failure, shock, coagulopathy, and lymphocytopenia from cellular apoptosis.1 With cellular death, immune system function is further disabled, more viral particles are released into the infected host, and body fluids remain infectious postmortem.
Laboratory Findings
Laboratory findings in viral hemorrhagic fevers can vary depending on the exact viral cause and the stage in the disease process. Leukocyte counts in early stages can reveal leukopenia and specifically lymphopenia, while in later stages leukocytosis with a left shift of neutrophils can predominate. Hemoglobin and hematocrit can show relative hemoconcentration, especially if renal manifestations of the disease occur. Thrombocytopenia also develops with viral hemorrhagic fevers, although in late stages thrombocytosis has been seen. Blood urea nitrogen (BUN) and creatinine (Cr) levels will rise with the occurrence of acute renal failure in late stages of the disease. Liver function studies, aspartate aminotransferase (AST) in particular, and alanine aminotransferase (ALT) have been found to rise in severe disease and in late stages due to multifocal hepatic necrosis, with AST typically greater than ALT. An association between elevated AST (~900 IU/L), BUN, Cr, albumin levels, and mortality has been statistically confirmed by McElroy et al4 in the Ebola outbreak in Uganda in 2000 to 2001; findings previously confirmed in the same geographic and temporal outbreak by Rollin et al.5 Survivors did not have nearly the same degree of elevation in liver enzymes, with AST levels averaging ~150 IU/L.5 The authors suggested that normalizing AST levels was perhaps indicative of acute recovery, but some patients still succumbed to complications of the illness.5
Coagulation studies, including prothrombin time, partial thromboplastin time, d-dimer, and fibrin split products will reflect disseminated intravascular coagulation (DIC) in those patients who develop hemorrhagic manifestations, which is common in late stages of the disease. Direct infection of vascular endothelial cells with damage to these cells has been shown to occur in the course of infection, yet nonhuman primate experimental studies and pathology examinations of Ebola victims have implied that DIC plays an important part in the hemorrhagic disease leading to the fatal shock syndrome seen in the most severe cases.6 Observed in 2000 during the care of Sudan species EVD patients in Uganda reported by Rollin et al,5 a distinct difference has been noted in quantitative d-dimer levels between survivors and fatal cases. Case fatalities showed a 4-fold increase in quantitative d-dimer levels (140,000 ng/mL) compared with survivors (44,000 ng/mL) during the acute phase of infection 6 to 8 days postsymptom onset.5
Acute phase reactants, high nitric oxide levels, cytokines, and higher viral loads have also been associated with fatal outcomes.4 McElroy et al4 measured the common acute phase reactant biomarker ferritin in patients of the 2000 Uganda outbreak and found levels to be higher in samples from patients who died and from patients with hemorrhagic complications and higher viral loads. Thus, these authors postulate ferritin is a potential marker for EVD severity.4
Commercially available assays for detection of viral particles are still in development and no point-of-care rapid detection testing is available. No test can reliably be used to diagnose viral hemorrhagic fever prior to symptom onset.7 Enzyme-linked immunosorbent assay and reverse transcriptase-polymerase chain reaction (RT-PCR) of viral particles or tissue cell cultures are available only through the CDC, and are the current most reliable methods to confirm the diagnosis of viral hemorrhagic fevers, including Ebola.
Patient Management in the ED
Standard infection-control procedures in place in US hospitals, when meticulously practiced, should be adequate to prevent transmission of EVD. As Ebola virus is only transmitted through direct contact with infected body fluids and secretions, PPE including mask, gloves, gown, shoe covers, and eye protection (goggles or face shield), should be appropriate. In donning PPE, remember that gloves should be the last item to pull in place and to pull off, turning the gown and gloves inside out. One’s hands should be washed before removing the mask and face shield/goggles, and they should be rewashed after completion of PPE removal. A tight fit of the glove over the elastic wristband of the gown is preferable and can ensure a better barrier to any biohazard. Meticulous hand hygiene after removal and proper disposal of PPE is paramount to successful contact protection. Diligent care should be taken in any procedure that might expose a healthcare provider to body fluids, such as blood draws, central line insertion, lumbar puncture and other invasive procedures. Standard contact isolation methods with the patient in a single room with the door closed is sufficient. Appropriate use of standard hospital disinfectants, including bleach solutions or hospital grade ammonium cleaners are already standard practice and easily implemented. It is recommended that procedures that produce aerosol particles should be avoided in patients with suspected infection; yet in some circumstances, the course of care may require such procedures. In this case, to minimize potential airborne spread, airborne and droplet precautions should also be initiated by placing the patient in a negative pressure room and implementing the use of properly fitted N-95 respirators for all present in the room.8
The mainstay of treatment for viral hemorrhagic fevers in the ED begins with initiation of contact isolation of the patient to prevent spread to the healthy. Supportive care involving aggressive intravenous fluid resuscitation to maintain blood pressure, oxygen support as needed, blood products as indicated for DIC, correction of electrolyte abnormalities, including dialysis for renal failure, is the only treatment widely available at this time. In patients who exhibit generalized edema as a result of hypoproteinemia from liver damage and third spacing, serum protein monitoring and replacement is indicated.9
An experimental treatment, ZMapp, which is a monoclonal antibody that is derived from mice and blocks Ebola from entering cells, has recently been used in combination with supportive treatment in six individuals infected with Ebola. However, ZMapp is still in early stages of development and testing is not widely available. No clinical trials have begun, but are planned. Two individuals treated with ZMapp have recovered in the United States; three healthcare workers are recovering in West Africa10,11; and one patient has died. It is not clear whether this medication is responsible wholly or in part for their recovery.
According to Dr Bruce Ribner, Director of Emory University Hospital’s Infectious Disease unit in an interview with Scientific American’s Dina Fine Maron on August 27, 2014, the two patients cared for at Emory have developed immunity to Zaire Ebolavirus.11 Continued outpatient monitoring is allowing study to help understand immunity to Ebola, and may lead to further treatment and vaccination development. Thus far, cross-protection against other Ebola viral strains for these recovered individuals is not as robust, indicating that this family of viruses are different enough that recovery from infection with one species may not be enough to confer immunity to a different species exposure. Blood transfusions from recovered patients have been described, but there is no clinical evidence to support any benefit from this therapy at this time.9

Personal protective equipment is a mainstay in the collection of specimens for viral-specific testing by the CDC, including full-face shield or goggles, masks to cover nose and mouth, gowns, and gloves. For routine laboratory testing and patient care, all of the above PPE is recommended, along with use of a biosafety cabinet or plexiglass splash guard, which is in accordance with Occupational Safety and Health Administration bloodborne pathogens standards.12
Specimens should be collected for Ebola testing only after the onset of symptoms, such as fever (see Table for supplemental information). In patients suspected of having viral exposure, it may take up to 3 days for the virus to reach detectable levels with RT-PCR. Consultation per hospital procedure with the local and/or state health department prior to specimen transport for testing to the CDC is mandatory. Public health officials will help ensure appropriate patient selection and proper procedures for specimen collection, and make arrangements for testing, which is only available through the CDC. The CDC will not accept any specimen without appropriate local/state health department consultation.
Ideally, preferred specimens for Ebola testing include 4 mL of whole blood properly preserved with EDTA, clot activator, sodium polyanethol sulfonate, or citrate in plastic collection tubes stored at 4˚C or frozen.12 Specimens should be placed in a durable, leak-proof container for transport within a facility; pneumatic tube systems should be avoided due to concerns for glass breakage or leaks. Hospitals should follow state or local health department policies and procedures for Ebola testing prior to notifying the CDC and initiating sample transportation.
The Dallas Index Case
Deplaning a flight from Liberia in Dallas, Texas on September 20, this index patient had no outward signs of illness, and thus no reason to cause any health concern. Joining in the community with friends and family, it was not until 4 days later that he reportedly developed a fever. Yet 2 more days passed before this patient initially sought ambulatory care at Dallas Health Presbyterian Hospital Emergency Department, during which time additional close contacts were exposed to infection. After evaluation, antibiotics were prescribed and the patient was released. Though this case is under investigation, according to a CNN report,13 the patient did inform a member of the ED nursing staff of his travel history, but this information was not communicated to the rest of the healthcare team. The presence and recognition of this patient’s travel history with disease symptoms heightens the level of suspicion for the possibility of EVD, and is the cornerstone of patient selection for Ebola testing.14
After the patient was discharged, another 2 days passed, during which time his condition deteriorated at home. Emergency medical services (EMS) transport was summoned to take the patient back to the hospital, expanding exposure to first responders, who appropriately utilized masks and gloves during transport. During this ED visit, his travel history was obtained and communicated, and he was appropriately isolated, supported, and admitted. These are early reported details of the case, and local public health officials continue to work with a team from the CDC to trace, isolate, and monitor all contacts with this patient (including the transporting emergency medical technicians) for evidence of further cases.15
This index case illustrates valuable lessons for all emergency care workers going forward. Ebola virus disease is now global, in the sense that it has proven its ability to present in a community far from its endemic home. Infectious diseases in their early stages present in nonspecific constellations of symptoms, and the key to rapid identification of EVD lies in careful attention to the recent travel history and exposure potential. Since patients may not offer this information for various reasons (eg, degree of symptoms, language barriers, fear, denial), it must be sought out lest more index patients be released into the public. As CDC Director Thomas Frieden related to CNN, “If someone’s been in West Africa within 21 days and they’ve got a fever, immediately isolate them and get them tested for Ebola.”13
Concerning the EMS transport of this patient, the ambulance used was disinfected per standard local protocols and remained in service for 2 days after this patient was transported. Though local officials are confident in the disinfection technique, it was pulled from service after the diagnosis was confirmed to ensure its full sterilization from Ebola virus16 before returning to full service. The rapid and robust public health response in progress will undoubtedly reveal further information over the coming days to weeks. To quote Michael Osterholm, PhD, MPH, director of the Center for Infectious Disease Research and Policy at the University of Minnesota, “We are going to see more cases show up around the world.”15
In addition to the index patient, it is important to keep in mind that secondary cases may present. If this occurs, the travel question must be altered to reflect the possibility that a secondary patient may not have been the traveler but rather one who was in close contact with an ill person who was in contact with him or her. Such a scenario would have a huge impact if that traveler did not seek treatment and would in turn require ED personnel to seek-out the information and report it to local public health officials.
Quelling the Panic
Even though Ebola is only spread through direct contact with infected bodily secretions, there is still significant public fear of the disease due to the high mortality rate and the graphic nature of symptoms in late stages of the disease. Daily monitoring of contacts of symptomatic Ebola patients for evidence of disease development—mainly fever—is sensible. Asymptomatic persons need not be confined or hospitalized unless fever develops, in which case contact isolation should occur until formal Ebola testing can rule-out the disease. Personal protective equipment, standardized hospital cleaning protocols with meticulous adherence, as well as quickly burying the deceased with adequate contact precautions, can all limit potential exposure and spread of the disease. Public health discussion and education about the virus and methods of transmission are needed so that individuals are not denied proper treatment or scared away from medical centers.
Importantly, communication by public health and medical experts on local and national levels should be with news media that embrace honest and careful reporting to avoid sensationalism and foster appropriate concern—ensuring that content is fundamental to curtailing panic and undue public fear.
For Internationally Traveling Clinicians
At present, the area endemic for Ebola remains confined to sub-Saharan Africa and West Africa. Clinicians should remain alert when traveling or treating patients in these areas. However, with the ease of international air travel, the potential for the spread of disease is recognized with many bordering nations now screening passengers from affected countries and some closing their borders to travelers from endemic areas. If a clinician encounters febrile patients in endemic areas, the differential diagnosis for any febrile illness must include Ebola, as well as malaria and other more common infectious agents. A thorough history about recent travel, ill contacts, and possible exposures should be sufficient in categorizing the risk of Ebola, but a high index of suspicion is necessary for prompt and proper treatment of those affected and to curtail spread of disease.
Despite the efforts of the national and local health systems and many nongovernmental organizations, including the World Health Organization, this epidemic continues to hold strong in the affected West African countries. Methods of containment of the virus are seemingly simple by modern standards, yet tragically beyond access for many on the ground. Lack of clean water sources in affected communities is a significant barrier to basic personal and environmental hygiene. Inadequate safe food sources and poaching encourages the hunt for primate bushmeat and thus presents a formidable local challenge.17 Lack of adequate PPE for healthcare workers, for those responsible for facility environmental hygiene, and for family members participating in traditional funeral rites for Ebola victims compounds the problem. Illness and deaths among exposed healthcare workers have led to the loss of significant numbers of nurses and doctors. This has caused legitimate fear in qualified individuals who subsequently decline to accept jobs caring for Ebola patients, which in turn increases the burden on those who remain. Additionally, some nongovernmental organizations have canceled scheduled aid trips to West Africa in response to the epidemic out of concern for the health of their workers. Meticulous management of environmental hygiene including sharps, surfaces, soiled linens, reusable medical equipment, waste products, and the preparations for burial of the deceased pose definite challenges to containment and prevention of transmission. Strict adherence to the use of PPE and hand hygiene is essential for all in contact with Ebola patients, pre- and postmortem. The lack of layperson comprehension and community understanding of the illness itself and the mechanism of viral transmission along with fear and mistrust for healthcare providers and nongovernmental medical missionaries are all serious barriers to the containment of disease spread. In fact, rumors that the virus does not truly exist, and that the illness is a result of biological warfare, cannibalistic rituals, or witchcraft add to the complexity of the situation.11
For these reasons, it is essential that efforts to control this epidemic in endemic healthcare facilities include effective health surveillance, infection-control programs, and community outreach fostering mutual trust-building, honest communication, and education. Success will require a multifaceted approach, and a global response will be needed to quiet this global threat. On September 16, the United States announced a robust response to deploy military engineers and medical personnel to assist in training healthcare workers and building care centers in Liberia. The United Nations, France, and the United Kingdom are also supporting this important effort to build stronger healthcare infrastructures in these vulnerable countries.16
Conclusion
This 2014 epidemic of EVD raises justifiable concerns regarding the impact of globalization. Though unlikely to pose a direct threat of epidemic proportion on US soil, the unanticipated occurrence of an index case may trigger a terrifying outbreak in any community, as it already has in the city of Dallas. Given that the early stages of EVD are indistinguishable from most other viral syndromes, the importance of reflection on individual and general healthcare facility adherence to standard infection control precautions and procedures warrant merit. Eliciting accurate travel histories and possible exposures are germane to narrowing the scope of possible etiologies of all infectious diseases. As an opportunity for improvement, this epidemic should incite elevated caution in the everyday handling of all patients with febrile illnesses and contact-transmissible infections including methicillin-resistant Staphylococcus aureus and Clostridium difficile, which affect a great number of US patients on a daily basis. Not only could this strategy prevent additional local outbreaks of EVD, but it would promote the safety of healthcare workers and the community served through attention to better infection preventive measures at the point of care, every time.
Dr McCammon is an assistant professor, department of emergency medicine, Eastern Virginia Medical School, Norfolk. Dr Chidester is an instructor, department of emergency medicine, and a fellow in international wilderness medicine, Eastern Virginia Medical School, Norfolk.
- Ebola virus disease information for clinicians in U.S. Healthcare Settings. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/vhf/ebola/hcp/clinician-information-us-healthcare-settings.html. Updated September 5, 2014. Accessed September 23, 2014.
- 2014 Ebola outbreak in West Africa. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/vhf/ebola/outbreaks/guinea/. Updated September 18, 2014. Accessed September 23, 2014.
- Virus ecology graphic. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/vhf/ebola/resources/virus-ecology.html. Updated August 1, 2014. Accessed September 23, 2014.
- McElroy AK, Erickson BR, Flietstra TD, et al. Ebola hemorrhagic fever: novel biomarker correlates of clinical outcome. J Infect Dis. 2014;210(4):558-566.
- Rollin PE, Bausch DG, Sanchez A. Blood chemistry measurements and D-Dimer levels associated with fatal and nonfatal outcomes in humans infected with Sudan Ebola virus. J Infect Dis. 2007;196(Suppl 2):S364-S371.
- Geisbert TW, Young HA, Jahrling PB, et al. Pathogenesis of Ebola hemorrhagic fever in primate models: evidence that hemorrhage is not a direct effect of virus-induced cytolysis of endothelial cells. Am J Pathol. 2003;163(6): 2371-2382.
- Blumberg L, Enria D, Bausch DG. Viral haemorrhagic fevers. In: Farrar J, Hotez, PJ, Junghanss T, Kang G, Lalloo D, White N, eds. Manson’s Tropical Diseases: Expert Consult. 23rd ed. Philadelphia, PA: Elsevier Saunders; 2014:171-194.
- Safe management of patients with Ebola virus disease (EVD) in U.S. hospitals. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/vhf/ebola/hcp/patient-management-us-hospitals.html. Updated September 5, 2014. Accessed September 23, 2014.
- Maron DF. Ebola doctor reveals how infected Americans were cured. Scientific American. http://www.scientificamerican.com/article/ebola-doctor-revealshow-infected-americans-were-cured/.
- Tribune wire reports. American Ebola patients treated with ZMapp experimental drug. Chicago Tribune. August 21, 2014. http://www.chicagotribune.com/lifestyles/health/chi-ebola-zmapp-20140821-story.html. Accessed September 23, 2014.
- Ebola crisis: doctors in Liberia ‘recovering after taking ZMapp’ [transcript]. BBC News. http://www.bbc.com/news/world-africa-28860204. August 19, 2014. Accessed September 23, 2014.
- Interim guidance for specimen collection, transport, testing, and submission for persons under investigation for Ebola virus disease in the United States. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/vhf/ebola/hcp/interim-guidance-specimen-collection-submission-patients-suspected-infection-ebola.html. Updated September 8, 2014. Accessed September 23, 2014.
- Catherine E. Shoichet CE, Fantz A, Yan H. Hospital ‘dropped the ball’ with Ebola patient’s travel history, NIH official says. CNN News. http://www.cnn.com/2014/10/01/health/ebola-us/index.html. Accessed October 2, 2014.
- Ebola (Ebola Virus Disease): Diagnosis. Centers for Disease Control and Prevention Web Site. http://www.cdc.gov/vhf/ebola/diagnosis/. Updated September 19, 2014. Accessed October 1, 2014.
- Gilblom K, Langreth R. Dallas hospital initially let Ebola patient go with drugs. Bloomberg News Web Site. http://www.bloomberg.com/news/2014-09-30/first-ebola-case-is-diagnosed-in-the-u-s-cdc-reports.html. Accessed October 1, 2014.
- Bates D, Szathmary Z and Boyle L. Up to twelve Americans could have Ebola: Fears grow in Dallas after first victim of deadly virus to reach U.S. remained at large for a week. September 30, 2014. Mail Online. http://www.dailymail.co.uk/news/article-2775608/CDC-confirms-Dallas-patient-isolation-testing-returning-region-plagued-Ebola-HAS-deadly-virus.html. Updated October 1, 2014. Accessed October 1, 2014.
- International affairs: bushmeat. U.S. Fish & Wildlife Service Web site. http://www.fws.gov/international/wildlife-without-borders/global-program/bushmeat.html. Accessed September 23, 2014.
In this age of globalization, just a few hours of air travel separates even the most remote places in our world. Given this reality, the recent epidemic of Ebola virus disease (EVD) in West Africa (Figure 1) has arrived on the doorstep of Texas Health Presbyterian Hospital in Dallas. Ill and potentially infected US healthcare workers and missionaries brought home for treatment and quarantine, plus the travel of the general population through the affected countries of Sierra Leone, Liberia, Guinea, and Nigeria, require all acute-care providers to be cognizant that this deadly disease may present in any community ED in the United States. Awareness and knowledge of the appropriate steps to manage care safely and effectively, while mindfully preventing the potential for viral transmission, is paramount.
Hemorrhagic Fevers
Viral hemorrhagic fevers are manifestations of four distinct families of RNA viruses: arenaviruses, bunyaviruses, flaviviruses, and filoviruses. All of these families of viruses depend on a natural insect or animal (nonhuman) host and are thus restricted geographically to the regions where the endemic hosts reside. The viruses can only infect a human when one comes into direct contact with an infected host; this human becomes an infectious host when symptoms of disease develop and subsequently, the possibility of transmission to other close direct human contacts exists.
Ebola Virus Species
The family within which the Ebola virus species are classified is the filoviruses. Five species of Ebola filovirus have been isolated to date: Ebola virus (Zaire ebolavirus), Sudan virus (Sudan ebolavirus), Taï Forest virus (Taï Forest ebolavirus, formerly Côte d’Ivoire ebolavirus), and Bundibugyo virus (Bundibugyo ebolavirus). The fifth, Reston virus (Reston ebolavirus), has caused disease in nonhuman primates, but not in humans. This epidemic has been attributed to a variant of the Zaire species.2 Transmission through direct contact with body fluids of febrile live infected patients and the postmortem period continues in communities and healthcare sites, as lack of adequate personal protective equipment (PPE) and meticulous environmental hygiene remains a challenge in many of these settings.
Clinical Presentation
In EVD, the onset of symptoms typically occurs abruptly at an average of 8 to 10 days postexposure and includes fever, headache, myalgia, and malaise; in some patients, an erythematous maculopapular rash involving the face, neck, trunk, and arms erupts by days 5 to 7.1 The nonspecific nature of these early signs and symptoms warrants caution in any patient known to have traveled in an endemic country with potential exposure to body fluids of infected patients—underscoring the importance of both obtaining a complete travel history to determine the potential for disease exposure in patients presenting with infectious-disease symptoms and effectively communicating this information to all ED staff.
In addition, such caution includes healthcare mission workers caring for Ebola patients, those involved in butchering infected animals for meat, and persons participating in traditional funeral rituals for those deceased from Ebola without the use of adequate PPE and/or environmental hygiene. Other more common infectious diseases with shared features of EVD must also be considered at this stage and include malaria, meningococcemia, measles, and typhoid fever, among others.
After the first 5 days of exposure, progression of symptoms may include severe watery diarrhea, nausea and vomiting, abdominal pain, shortness of breath, chest pain, headache, and/or confusion. Conjunctival injection may also develop. Not all patients will have signs of hemorrhagic fever with bleeding from the mouth, eyes, ears, in stool, or from internal organs, but petechiae, ecchymosis, and oozing from venipuncture sites may develop. Those at the highest risk of death show signs of sepsis, such as shock and multiorgan system failure, which may include hemorrhagic manifestations, early in the course of their illness. Patients with these complications typically expire between days 6 to16.1 Survivors of the disease tend to have fever with less severe symptoms for a period of several more days, and then begin to improve clinically between days 6 to 11 after onset of symptoms1 (Figure 3).
Ecology
A zoonotic filovirus transmissible from animal populations to humans causes EVD. Research strongly suggests that fruit bats are the reservoir and hosts for this filovirus. Direct human contact with bats or with wild animals that have been infected by bats initiates the human-to-human transmission of EVD.3
Pathogenesis
Through direct contact with mucous membranes, a break in the skin, or parenterally, Ebola enters and infects multiple cell types. Incubation periods appear shorter in infections acquired through direct injection (6 days) than for contact transmission (10 days).1 Emesis, urine, stool, sweat, semen, cerebrospinal fluid, breast milk, and saliva are actively capable of viral transmission. From point of entry, the virus migrates to the lymph nodes, then to liver, spleen, and adrenal glands. Hepatocellular necrosis leads to clotting-factor derangement and dysfunction resulting in coagulopathy and bleeding and potential liver failure. Necrosis of adrenal tissue may be present and results in impaired steroid synthesis and hypotension. The presence of the virus appears to incite a cytokine inflammatory storm causing microvascular leakage, with the end effect of multiorgan system failure, shock, coagulopathy, and lymphocytopenia from cellular apoptosis.1 With cellular death, immune system function is further disabled, more viral particles are released into the infected host, and body fluids remain infectious postmortem.
Laboratory Findings
Laboratory findings in viral hemorrhagic fevers can vary depending on the exact viral cause and the stage in the disease process. Leukocyte counts in early stages can reveal leukopenia and specifically lymphopenia, while in later stages leukocytosis with a left shift of neutrophils can predominate. Hemoglobin and hematocrit can show relative hemoconcentration, especially if renal manifestations of the disease occur. Thrombocytopenia also develops with viral hemorrhagic fevers, although in late stages thrombocytosis has been seen. Blood urea nitrogen (BUN) and creatinine (Cr) levels will rise with the occurrence of acute renal failure in late stages of the disease. Liver function studies, aspartate aminotransferase (AST) in particular, and alanine aminotransferase (ALT) have been found to rise in severe disease and in late stages due to multifocal hepatic necrosis, with AST typically greater than ALT. An association between elevated AST (~900 IU/L), BUN, Cr, albumin levels, and mortality has been statistically confirmed by McElroy et al4 in the Ebola outbreak in Uganda in 2000 to 2001; findings previously confirmed in the same geographic and temporal outbreak by Rollin et al.5 Survivors did not have nearly the same degree of elevation in liver enzymes, with AST levels averaging ~150 IU/L.5 The authors suggested that normalizing AST levels was perhaps indicative of acute recovery, but some patients still succumbed to complications of the illness.5
Coagulation studies, including prothrombin time, partial thromboplastin time, d-dimer, and fibrin split products will reflect disseminated intravascular coagulation (DIC) in those patients who develop hemorrhagic manifestations, which is common in late stages of the disease. Direct infection of vascular endothelial cells with damage to these cells has been shown to occur in the course of infection, yet nonhuman primate experimental studies and pathology examinations of Ebola victims have implied that DIC plays an important part in the hemorrhagic disease leading to the fatal shock syndrome seen in the most severe cases.6 Observed in 2000 during the care of Sudan species EVD patients in Uganda reported by Rollin et al,5 a distinct difference has been noted in quantitative d-dimer levels between survivors and fatal cases. Case fatalities showed a 4-fold increase in quantitative d-dimer levels (140,000 ng/mL) compared with survivors (44,000 ng/mL) during the acute phase of infection 6 to 8 days postsymptom onset.5
Acute phase reactants, high nitric oxide levels, cytokines, and higher viral loads have also been associated with fatal outcomes.4 McElroy et al4 measured the common acute phase reactant biomarker ferritin in patients of the 2000 Uganda outbreak and found levels to be higher in samples from patients who died and from patients with hemorrhagic complications and higher viral loads. Thus, these authors postulate ferritin is a potential marker for EVD severity.4
Commercially available assays for detection of viral particles are still in development and no point-of-care rapid detection testing is available. No test can reliably be used to diagnose viral hemorrhagic fever prior to symptom onset.7 Enzyme-linked immunosorbent assay and reverse transcriptase-polymerase chain reaction (RT-PCR) of viral particles or tissue cell cultures are available only through the CDC, and are the current most reliable methods to confirm the diagnosis of viral hemorrhagic fevers, including Ebola.
Patient Management in the ED
Standard infection-control procedures in place in US hospitals, when meticulously practiced, should be adequate to prevent transmission of EVD. As Ebola virus is only transmitted through direct contact with infected body fluids and secretions, PPE including mask, gloves, gown, shoe covers, and eye protection (goggles or face shield), should be appropriate. In donning PPE, remember that gloves should be the last item to pull in place and to pull off, turning the gown and gloves inside out. One’s hands should be washed before removing the mask and face shield/goggles, and they should be rewashed after completion of PPE removal. A tight fit of the glove over the elastic wristband of the gown is preferable and can ensure a better barrier to any biohazard. Meticulous hand hygiene after removal and proper disposal of PPE is paramount to successful contact protection. Diligent care should be taken in any procedure that might expose a healthcare provider to body fluids, such as blood draws, central line insertion, lumbar puncture and other invasive procedures. Standard contact isolation methods with the patient in a single room with the door closed is sufficient. Appropriate use of standard hospital disinfectants, including bleach solutions or hospital grade ammonium cleaners are already standard practice and easily implemented. It is recommended that procedures that produce aerosol particles should be avoided in patients with suspected infection; yet in some circumstances, the course of care may require such procedures. In this case, to minimize potential airborne spread, airborne and droplet precautions should also be initiated by placing the patient in a negative pressure room and implementing the use of properly fitted N-95 respirators for all present in the room.8
The mainstay of treatment for viral hemorrhagic fevers in the ED begins with initiation of contact isolation of the patient to prevent spread to the healthy. Supportive care involving aggressive intravenous fluid resuscitation to maintain blood pressure, oxygen support as needed, blood products as indicated for DIC, correction of electrolyte abnormalities, including dialysis for renal failure, is the only treatment widely available at this time. In patients who exhibit generalized edema as a result of hypoproteinemia from liver damage and third spacing, serum protein monitoring and replacement is indicated.9
An experimental treatment, ZMapp, which is a monoclonal antibody that is derived from mice and blocks Ebola from entering cells, has recently been used in combination with supportive treatment in six individuals infected with Ebola. However, ZMapp is still in early stages of development and testing is not widely available. No clinical trials have begun, but are planned. Two individuals treated with ZMapp have recovered in the United States; three healthcare workers are recovering in West Africa10,11; and one patient has died. It is not clear whether this medication is responsible wholly or in part for their recovery.
According to Dr Bruce Ribner, Director of Emory University Hospital’s Infectious Disease unit in an interview with Scientific American’s Dina Fine Maron on August 27, 2014, the two patients cared for at Emory have developed immunity to Zaire Ebolavirus.11 Continued outpatient monitoring is allowing study to help understand immunity to Ebola, and may lead to further treatment and vaccination development. Thus far, cross-protection against other Ebola viral strains for these recovered individuals is not as robust, indicating that this family of viruses are different enough that recovery from infection with one species may not be enough to confer immunity to a different species exposure. Blood transfusions from recovered patients have been described, but there is no clinical evidence to support any benefit from this therapy at this time.9
Personal protective equipment is a mainstay in the collection of specimens for viral-specific testing by the CDC, including full-face shield or goggles, masks to cover nose and mouth, gowns, and gloves. For routine laboratory testing and patient care, all of the above PPE is recommended, along with use of a biosafety cabinet or plexiglass splash guard, which is in accordance with Occupational Safety and Health Administration bloodborne pathogens standards.12
Specimens should be collected for Ebola testing only after the onset of symptoms, such as fever (see Table for supplemental information). In patients suspected of having viral exposure, it may take up to 3 days for the virus to reach detectable levels with RT-PCR. Consultation per hospital procedure with the local and/or state health department prior to specimen transport for testing to the CDC is mandatory. Public health officials will help ensure appropriate patient selection and proper procedures for specimen collection, and make arrangements for testing, which is only available through the CDC. The CDC will not accept any specimen without appropriate local/state health department consultation.
Ideally, preferred specimens for Ebola testing include 4 mL of whole blood properly preserved with EDTA, clot activator, sodium polyanethol sulfonate, or citrate in plastic collection tubes stored at 4˚C or frozen.12 Specimens should be placed in a durable, leak-proof container for transport within a facility; pneumatic tube systems should be avoided due to concerns for glass breakage or leaks. Hospitals should follow state or local health department policies and procedures for Ebola testing prior to notifying the CDC and initiating sample transportation.
The Dallas Index Case
Deplaning a flight from Liberia in Dallas, Texas on September 20, this index patient had no outward signs of illness, and thus no reason to cause any health concern. Joining in the community with friends and family, it was not until 4 days later that he reportedly developed a fever. Yet 2 more days passed before this patient initially sought ambulatory care at Dallas Health Presbyterian Hospital Emergency Department, during which time additional close contacts were exposed to infection. After evaluation, antibiotics were prescribed and the patient was released. Though this case is under investigation, according to a CNN report,13 the patient did inform a member of the ED nursing staff of his travel history, but this information was not communicated to the rest of the healthcare team. The presence and recognition of this patient’s travel history with disease symptoms heightens the level of suspicion for the possibility of EVD, and is the cornerstone of patient selection for Ebola testing.14
After the patient was discharged, another 2 days passed, during which time his condition deteriorated at home. Emergency medical services (EMS) transport was summoned to take the patient back to the hospital, expanding exposure to first responders, who appropriately utilized masks and gloves during transport. During this ED visit, his travel history was obtained and communicated, and he was appropriately isolated, supported, and admitted. These are early reported details of the case, and local public health officials continue to work with a team from the CDC to trace, isolate, and monitor all contacts with this patient (including the transporting emergency medical technicians) for evidence of further cases.15
This index case illustrates valuable lessons for all emergency care workers going forward. Ebola virus disease is now global, in the sense that it has proven its ability to present in a community far from its endemic home. Infectious diseases in their early stages present in nonspecific constellations of symptoms, and the key to rapid identification of EVD lies in careful attention to the recent travel history and exposure potential. Since patients may not offer this information for various reasons (eg, degree of symptoms, language barriers, fear, denial), it must be sought out lest more index patients be released into the public. As CDC Director Thomas Frieden related to CNN, “If someone’s been in West Africa within 21 days and they’ve got a fever, immediately isolate them and get them tested for Ebola.”13
Concerning the EMS transport of this patient, the ambulance used was disinfected per standard local protocols and remained in service for 2 days after this patient was transported. Though local officials are confident in the disinfection technique, it was pulled from service after the diagnosis was confirmed to ensure its full sterilization from Ebola virus16 before returning to full service. The rapid and robust public health response in progress will undoubtedly reveal further information over the coming days to weeks. To quote Michael Osterholm, PhD, MPH, director of the Center for Infectious Disease Research and Policy at the University of Minnesota, “We are going to see more cases show up around the world.”15
In addition to the index patient, it is important to keep in mind that secondary cases may present. If this occurs, the travel question must be altered to reflect the possibility that a secondary patient may not have been the traveler but rather one who was in close contact with an ill person who was in contact with him or her. Such a scenario would have a huge impact if that traveler did not seek treatment and would in turn require ED personnel to seek-out the information and report it to local public health officials.
Quelling the Panic
Even though Ebola is only spread through direct contact with infected bodily secretions, there is still significant public fear of the disease due to the high mortality rate and the graphic nature of symptoms in late stages of the disease. Daily monitoring of contacts of symptomatic Ebola patients for evidence of disease development—mainly fever—is sensible. Asymptomatic persons need not be confined or hospitalized unless fever develops, in which case contact isolation should occur until formal Ebola testing can rule-out the disease. Personal protective equipment, standardized hospital cleaning protocols with meticulous adherence, as well as quickly burying the deceased with adequate contact precautions, can all limit potential exposure and spread of the disease. Public health discussion and education about the virus and methods of transmission are needed so that individuals are not denied proper treatment or scared away from medical centers.
Importantly, communication by public health and medical experts on local and national levels should be with news media that embrace honest and careful reporting to avoid sensationalism and foster appropriate concern—ensuring that content is fundamental to curtailing panic and undue public fear.
For Internationally Traveling Clinicians
At present, the area endemic for Ebola remains confined to sub-Saharan Africa and West Africa. Clinicians should remain alert when traveling or treating patients in these areas. However, with the ease of international air travel, the potential for the spread of disease is recognized with many bordering nations now screening passengers from affected countries and some closing their borders to travelers from endemic areas. If a clinician encounters febrile patients in endemic areas, the differential diagnosis for any febrile illness must include Ebola, as well as malaria and other more common infectious agents. A thorough history about recent travel, ill contacts, and possible exposures should be sufficient in categorizing the risk of Ebola, but a high index of suspicion is necessary for prompt and proper treatment of those affected and to curtail spread of disease.
Despite the efforts of the national and local health systems and many nongovernmental organizations, including the World Health Organization, this epidemic continues to hold strong in the affected West African countries. Methods of containment of the virus are seemingly simple by modern standards, yet tragically beyond access for many on the ground. Lack of clean water sources in affected communities is a significant barrier to basic personal and environmental hygiene. Inadequate safe food sources and poaching encourages the hunt for primate bushmeat and thus presents a formidable local challenge.17 Lack of adequate PPE for healthcare workers, for those responsible for facility environmental hygiene, and for family members participating in traditional funeral rites for Ebola victims compounds the problem. Illness and deaths among exposed healthcare workers have led to the loss of significant numbers of nurses and doctors. This has caused legitimate fear in qualified individuals who subsequently decline to accept jobs caring for Ebola patients, which in turn increases the burden on those who remain. Additionally, some nongovernmental organizations have canceled scheduled aid trips to West Africa in response to the epidemic out of concern for the health of their workers. Meticulous management of environmental hygiene including sharps, surfaces, soiled linens, reusable medical equipment, waste products, and the preparations for burial of the deceased pose definite challenges to containment and prevention of transmission. Strict adherence to the use of PPE and hand hygiene is essential for all in contact with Ebola patients, pre- and postmortem. The lack of layperson comprehension and community understanding of the illness itself and the mechanism of viral transmission along with fear and mistrust for healthcare providers and nongovernmental medical missionaries are all serious barriers to the containment of disease spread. In fact, rumors that the virus does not truly exist, and that the illness is a result of biological warfare, cannibalistic rituals, or witchcraft add to the complexity of the situation.11
For these reasons, it is essential that efforts to control this epidemic in endemic healthcare facilities include effective health surveillance, infection-control programs, and community outreach fostering mutual trust-building, honest communication, and education. Success will require a multifaceted approach, and a global response will be needed to quiet this global threat. On September 16, the United States announced a robust response to deploy military engineers and medical personnel to assist in training healthcare workers and building care centers in Liberia. The United Nations, France, and the United Kingdom are also supporting this important effort to build stronger healthcare infrastructures in these vulnerable countries.16
Conclusion
This 2014 epidemic of EVD raises justifiable concerns regarding the impact of globalization. Though unlikely to pose a direct threat of epidemic proportion on US soil, the unanticipated occurrence of an index case may trigger a terrifying outbreak in any community, as it already has in the city of Dallas. Given that the early stages of EVD are indistinguishable from most other viral syndromes, the importance of reflection on individual and general healthcare facility adherence to standard infection control precautions and procedures warrant merit. Eliciting accurate travel histories and possible exposures are germane to narrowing the scope of possible etiologies of all infectious diseases. As an opportunity for improvement, this epidemic should incite elevated caution in the everyday handling of all patients with febrile illnesses and contact-transmissible infections including methicillin-resistant Staphylococcus aureus and Clostridium difficile, which affect a great number of US patients on a daily basis. Not only could this strategy prevent additional local outbreaks of EVD, but it would promote the safety of healthcare workers and the community served through attention to better infection preventive measures at the point of care, every time.
Dr McCammon is an assistant professor, department of emergency medicine, Eastern Virginia Medical School, Norfolk. Dr Chidester is an instructor, department of emergency medicine, and a fellow in international wilderness medicine, Eastern Virginia Medical School, Norfolk.
In this age of globalization, just a few hours of air travel separates even the most remote places in our world. Given this reality, the recent epidemic of Ebola virus disease (EVD) in West Africa (Figure 1) has arrived on the doorstep of Texas Health Presbyterian Hospital in Dallas. Ill and potentially infected US healthcare workers and missionaries brought home for treatment and quarantine, plus the travel of the general population through the affected countries of Sierra Leone, Liberia, Guinea, and Nigeria, require all acute-care providers to be cognizant that this deadly disease may present in any community ED in the United States. Awareness and knowledge of the appropriate steps to manage care safely and effectively, while mindfully preventing the potential for viral transmission, is paramount.
Hemorrhagic Fevers
Viral hemorrhagic fevers are manifestations of four distinct families of RNA viruses: arenaviruses, bunyaviruses, flaviviruses, and filoviruses. All of these families of viruses depend on a natural insect or animal (nonhuman) host and are thus restricted geographically to the regions where the endemic hosts reside. The viruses can only infect a human when one comes into direct contact with an infected host; this human becomes an infectious host when symptoms of disease develop and subsequently, the possibility of transmission to other close direct human contacts exists.
Ebola Virus Species
The family within which the Ebola virus species are classified is the filoviruses. Five species of Ebola filovirus have been isolated to date: Ebola virus (Zaire ebolavirus), Sudan virus (Sudan ebolavirus), Taï Forest virus (Taï Forest ebolavirus, formerly Côte d’Ivoire ebolavirus), and Bundibugyo virus (Bundibugyo ebolavirus). The fifth, Reston virus (Reston ebolavirus), has caused disease in nonhuman primates, but not in humans. This epidemic has been attributed to a variant of the Zaire species.2 Transmission through direct contact with body fluids of febrile live infected patients and the postmortem period continues in communities and healthcare sites, as lack of adequate personal protective equipment (PPE) and meticulous environmental hygiene remains a challenge in many of these settings.
Clinical Presentation
In EVD, the onset of symptoms typically occurs abruptly at an average of 8 to 10 days postexposure and includes fever, headache, myalgia, and malaise; in some patients, an erythematous maculopapular rash involving the face, neck, trunk, and arms erupts by days 5 to 7.1 The nonspecific nature of these early signs and symptoms warrants caution in any patient known to have traveled in an endemic country with potential exposure to body fluids of infected patients—underscoring the importance of both obtaining a complete travel history to determine the potential for disease exposure in patients presenting with infectious-disease symptoms and effectively communicating this information to all ED staff.
In addition, such caution includes healthcare mission workers caring for Ebola patients, those involved in butchering infected animals for meat, and persons participating in traditional funeral rituals for those deceased from Ebola without the use of adequate PPE and/or environmental hygiene. Other more common infectious diseases with shared features of EVD must also be considered at this stage and include malaria, meningococcemia, measles, and typhoid fever, among others.
After the first 5 days of exposure, progression of symptoms may include severe watery diarrhea, nausea and vomiting, abdominal pain, shortness of breath, chest pain, headache, and/or confusion. Conjunctival injection may also develop. Not all patients will have signs of hemorrhagic fever with bleeding from the mouth, eyes, ears, in stool, or from internal organs, but petechiae, ecchymosis, and oozing from venipuncture sites may develop. Those at the highest risk of death show signs of sepsis, such as shock and multiorgan system failure, which may include hemorrhagic manifestations, early in the course of their illness. Patients with these complications typically expire between days 6 to16.1 Survivors of the disease tend to have fever with less severe symptoms for a period of several more days, and then begin to improve clinically between days 6 to 11 after onset of symptoms1 (Figure 3).
Ecology
A zoonotic filovirus transmissible from animal populations to humans causes EVD. Research strongly suggests that fruit bats are the reservoir and hosts for this filovirus. Direct human contact with bats or with wild animals that have been infected by bats initiates the human-to-human transmission of EVD.3
Pathogenesis
Through direct contact with mucous membranes, a break in the skin, or parenterally, Ebola enters and infects multiple cell types. Incubation periods appear shorter in infections acquired through direct injection (6 days) than for contact transmission (10 days).1 Emesis, urine, stool, sweat, semen, cerebrospinal fluid, breast milk, and saliva are actively capable of viral transmission. From point of entry, the virus migrates to the lymph nodes, then to liver, spleen, and adrenal glands. Hepatocellular necrosis leads to clotting-factor derangement and dysfunction resulting in coagulopathy and bleeding and potential liver failure. Necrosis of adrenal tissue may be present and results in impaired steroid synthesis and hypotension. The presence of the virus appears to incite a cytokine inflammatory storm causing microvascular leakage, with the end effect of multiorgan system failure, shock, coagulopathy, and lymphocytopenia from cellular apoptosis.1 With cellular death, immune system function is further disabled, more viral particles are released into the infected host, and body fluids remain infectious postmortem.
Laboratory Findings
Laboratory findings in viral hemorrhagic fevers can vary depending on the exact viral cause and the stage in the disease process. Leukocyte counts in early stages can reveal leukopenia and specifically lymphopenia, while in later stages leukocytosis with a left shift of neutrophils can predominate. Hemoglobin and hematocrit can show relative hemoconcentration, especially if renal manifestations of the disease occur. Thrombocytopenia also develops with viral hemorrhagic fevers, although in late stages thrombocytosis has been seen. Blood urea nitrogen (BUN) and creatinine (Cr) levels will rise with the occurrence of acute renal failure in late stages of the disease. Liver function studies, aspartate aminotransferase (AST) in particular, and alanine aminotransferase (ALT) have been found to rise in severe disease and in late stages due to multifocal hepatic necrosis, with AST typically greater than ALT. An association between elevated AST (~900 IU/L), BUN, Cr, albumin levels, and mortality has been statistically confirmed by McElroy et al4 in the Ebola outbreak in Uganda in 2000 to 2001; findings previously confirmed in the same geographic and temporal outbreak by Rollin et al.5 Survivors did not have nearly the same degree of elevation in liver enzymes, with AST levels averaging ~150 IU/L.5 The authors suggested that normalizing AST levels was perhaps indicative of acute recovery, but some patients still succumbed to complications of the illness.5
Coagulation studies, including prothrombin time, partial thromboplastin time, d-dimer, and fibrin split products will reflect disseminated intravascular coagulation (DIC) in those patients who develop hemorrhagic manifestations, which is common in late stages of the disease. Direct infection of vascular endothelial cells with damage to these cells has been shown to occur in the course of infection, yet nonhuman primate experimental studies and pathology examinations of Ebola victims have implied that DIC plays an important part in the hemorrhagic disease leading to the fatal shock syndrome seen in the most severe cases.6 Observed in 2000 during the care of Sudan species EVD patients in Uganda reported by Rollin et al,5 a distinct difference has been noted in quantitative d-dimer levels between survivors and fatal cases. Case fatalities showed a 4-fold increase in quantitative d-dimer levels (140,000 ng/mL) compared with survivors (44,000 ng/mL) during the acute phase of infection 6 to 8 days postsymptom onset.5
Acute phase reactants, high nitric oxide levels, cytokines, and higher viral loads have also been associated with fatal outcomes.4 McElroy et al4 measured the common acute phase reactant biomarker ferritin in patients of the 2000 Uganda outbreak and found levels to be higher in samples from patients who died and from patients with hemorrhagic complications and higher viral loads. Thus, these authors postulate ferritin is a potential marker for EVD severity.4
Commercially available assays for detection of viral particles are still in development and no point-of-care rapid detection testing is available. No test can reliably be used to diagnose viral hemorrhagic fever prior to symptom onset.7 Enzyme-linked immunosorbent assay and reverse transcriptase-polymerase chain reaction (RT-PCR) of viral particles or tissue cell cultures are available only through the CDC, and are the current most reliable methods to confirm the diagnosis of viral hemorrhagic fevers, including Ebola.
Patient Management in the ED
Standard infection-control procedures in place in US hospitals, when meticulously practiced, should be adequate to prevent transmission of EVD. As Ebola virus is only transmitted through direct contact with infected body fluids and secretions, PPE including mask, gloves, gown, shoe covers, and eye protection (goggles or face shield), should be appropriate. In donning PPE, remember that gloves should be the last item to pull in place and to pull off, turning the gown and gloves inside out. One’s hands should be washed before removing the mask and face shield/goggles, and they should be rewashed after completion of PPE removal. A tight fit of the glove over the elastic wristband of the gown is preferable and can ensure a better barrier to any biohazard. Meticulous hand hygiene after removal and proper disposal of PPE is paramount to successful contact protection. Diligent care should be taken in any procedure that might expose a healthcare provider to body fluids, such as blood draws, central line insertion, lumbar puncture and other invasive procedures. Standard contact isolation methods with the patient in a single room with the door closed is sufficient. Appropriate use of standard hospital disinfectants, including bleach solutions or hospital grade ammonium cleaners are already standard practice and easily implemented. It is recommended that procedures that produce aerosol particles should be avoided in patients with suspected infection; yet in some circumstances, the course of care may require such procedures. In this case, to minimize potential airborne spread, airborne and droplet precautions should also be initiated by placing the patient in a negative pressure room and implementing the use of properly fitted N-95 respirators for all present in the room.8
The mainstay of treatment for viral hemorrhagic fevers in the ED begins with initiation of contact isolation of the patient to prevent spread to the healthy. Supportive care involving aggressive intravenous fluid resuscitation to maintain blood pressure, oxygen support as needed, blood products as indicated for DIC, correction of electrolyte abnormalities, including dialysis for renal failure, is the only treatment widely available at this time. In patients who exhibit generalized edema as a result of hypoproteinemia from liver damage and third spacing, serum protein monitoring and replacement is indicated.9
An experimental treatment, ZMapp, which is a monoclonal antibody that is derived from mice and blocks Ebola from entering cells, has recently been used in combination with supportive treatment in six individuals infected with Ebola. However, ZMapp is still in early stages of development and testing is not widely available. No clinical trials have begun, but are planned. Two individuals treated with ZMapp have recovered in the United States; three healthcare workers are recovering in West Africa10,11; and one patient has died. It is not clear whether this medication is responsible wholly or in part for their recovery.
According to Dr Bruce Ribner, Director of Emory University Hospital’s Infectious Disease unit in an interview with Scientific American’s Dina Fine Maron on August 27, 2014, the two patients cared for at Emory have developed immunity to Zaire Ebolavirus.11 Continued outpatient monitoring is allowing study to help understand immunity to Ebola, and may lead to further treatment and vaccination development. Thus far, cross-protection against other Ebola viral strains for these recovered individuals is not as robust, indicating that this family of viruses are different enough that recovery from infection with one species may not be enough to confer immunity to a different species exposure. Blood transfusions from recovered patients have been described, but there is no clinical evidence to support any benefit from this therapy at this time.9
Personal protective equipment is a mainstay in the collection of specimens for viral-specific testing by the CDC, including full-face shield or goggles, masks to cover nose and mouth, gowns, and gloves. For routine laboratory testing and patient care, all of the above PPE is recommended, along with use of a biosafety cabinet or plexiglass splash guard, which is in accordance with Occupational Safety and Health Administration bloodborne pathogens standards.12
Specimens should be collected for Ebola testing only after the onset of symptoms, such as fever (see Table for supplemental information). In patients suspected of having viral exposure, it may take up to 3 days for the virus to reach detectable levels with RT-PCR. Consultation per hospital procedure with the local and/or state health department prior to specimen transport for testing to the CDC is mandatory. Public health officials will help ensure appropriate patient selection and proper procedures for specimen collection, and make arrangements for testing, which is only available through the CDC. The CDC will not accept any specimen without appropriate local/state health department consultation.
Ideally, preferred specimens for Ebola testing include 4 mL of whole blood properly preserved with EDTA, clot activator, sodium polyanethol sulfonate, or citrate in plastic collection tubes stored at 4˚C or frozen.12 Specimens should be placed in a durable, leak-proof container for transport within a facility; pneumatic tube systems should be avoided due to concerns for glass breakage or leaks. Hospitals should follow state or local health department policies and procedures for Ebola testing prior to notifying the CDC and initiating sample transportation.
The Dallas Index Case
Deplaning a flight from Liberia in Dallas, Texas on September 20, this index patient had no outward signs of illness, and thus no reason to cause any health concern. Joining in the community with friends and family, it was not until 4 days later that he reportedly developed a fever. Yet 2 more days passed before this patient initially sought ambulatory care at Dallas Health Presbyterian Hospital Emergency Department, during which time additional close contacts were exposed to infection. After evaluation, antibiotics were prescribed and the patient was released. Though this case is under investigation, according to a CNN report,13 the patient did inform a member of the ED nursing staff of his travel history, but this information was not communicated to the rest of the healthcare team. The presence and recognition of this patient’s travel history with disease symptoms heightens the level of suspicion for the possibility of EVD, and is the cornerstone of patient selection for Ebola testing.14
After the patient was discharged, another 2 days passed, during which time his condition deteriorated at home. Emergency medical services (EMS) transport was summoned to take the patient back to the hospital, expanding exposure to first responders, who appropriately utilized masks and gloves during transport. During this ED visit, his travel history was obtained and communicated, and he was appropriately isolated, supported, and admitted. These are early reported details of the case, and local public health officials continue to work with a team from the CDC to trace, isolate, and monitor all contacts with this patient (including the transporting emergency medical technicians) for evidence of further cases.15
This index case illustrates valuable lessons for all emergency care workers going forward. Ebola virus disease is now global, in the sense that it has proven its ability to present in a community far from its endemic home. Infectious diseases in their early stages present in nonspecific constellations of symptoms, and the key to rapid identification of EVD lies in careful attention to the recent travel history and exposure potential. Since patients may not offer this information for various reasons (eg, degree of symptoms, language barriers, fear, denial), it must be sought out lest more index patients be released into the public. As CDC Director Thomas Frieden related to CNN, “If someone’s been in West Africa within 21 days and they’ve got a fever, immediately isolate them and get them tested for Ebola.”13
Concerning the EMS transport of this patient, the ambulance used was disinfected per standard local protocols and remained in service for 2 days after this patient was transported. Though local officials are confident in the disinfection technique, it was pulled from service after the diagnosis was confirmed to ensure its full sterilization from Ebola virus16 before returning to full service. The rapid and robust public health response in progress will undoubtedly reveal further information over the coming days to weeks. To quote Michael Osterholm, PhD, MPH, director of the Center for Infectious Disease Research and Policy at the University of Minnesota, “We are going to see more cases show up around the world.”15
In addition to the index patient, it is important to keep in mind that secondary cases may present. If this occurs, the travel question must be altered to reflect the possibility that a secondary patient may not have been the traveler but rather one who was in close contact with an ill person who was in contact with him or her. Such a scenario would have a huge impact if that traveler did not seek treatment and would in turn require ED personnel to seek-out the information and report it to local public health officials.
Quelling the Panic
Even though Ebola is only spread through direct contact with infected bodily secretions, there is still significant public fear of the disease due to the high mortality rate and the graphic nature of symptoms in late stages of the disease. Daily monitoring of contacts of symptomatic Ebola patients for evidence of disease development—mainly fever—is sensible. Asymptomatic persons need not be confined or hospitalized unless fever develops, in which case contact isolation should occur until formal Ebola testing can rule-out the disease. Personal protective equipment, standardized hospital cleaning protocols with meticulous adherence, as well as quickly burying the deceased with adequate contact precautions, can all limit potential exposure and spread of the disease. Public health discussion and education about the virus and methods of transmission are needed so that individuals are not denied proper treatment or scared away from medical centers.
Importantly, communication by public health and medical experts on local and national levels should be with news media that embrace honest and careful reporting to avoid sensationalism and foster appropriate concern—ensuring that content is fundamental to curtailing panic and undue public fear.
For Internationally Traveling Clinicians
At present, the area endemic for Ebola remains confined to sub-Saharan Africa and West Africa. Clinicians should remain alert when traveling or treating patients in these areas. However, with the ease of international air travel, the potential for the spread of disease is recognized with many bordering nations now screening passengers from affected countries and some closing their borders to travelers from endemic areas. If a clinician encounters febrile patients in endemic areas, the differential diagnosis for any febrile illness must include Ebola, as well as malaria and other more common infectious agents. A thorough history about recent travel, ill contacts, and possible exposures should be sufficient in categorizing the risk of Ebola, but a high index of suspicion is necessary for prompt and proper treatment of those affected and to curtail spread of disease.
Despite the efforts of the national and local health systems and many nongovernmental organizations, including the World Health Organization, this epidemic continues to hold strong in the affected West African countries. Methods of containment of the virus are seemingly simple by modern standards, yet tragically beyond access for many on the ground. Lack of clean water sources in affected communities is a significant barrier to basic personal and environmental hygiene. Inadequate safe food sources and poaching encourages the hunt for primate bushmeat and thus presents a formidable local challenge.17 Lack of adequate PPE for healthcare workers, for those responsible for facility environmental hygiene, and for family members participating in traditional funeral rites for Ebola victims compounds the problem. Illness and deaths among exposed healthcare workers have led to the loss of significant numbers of nurses and doctors. This has caused legitimate fear in qualified individuals who subsequently decline to accept jobs caring for Ebola patients, which in turn increases the burden on those who remain. Additionally, some nongovernmental organizations have canceled scheduled aid trips to West Africa in response to the epidemic out of concern for the health of their workers. Meticulous management of environmental hygiene including sharps, surfaces, soiled linens, reusable medical equipment, waste products, and the preparations for burial of the deceased pose definite challenges to containment and prevention of transmission. Strict adherence to the use of PPE and hand hygiene is essential for all in contact with Ebola patients, pre- and postmortem. The lack of layperson comprehension and community understanding of the illness itself and the mechanism of viral transmission along with fear and mistrust for healthcare providers and nongovernmental medical missionaries are all serious barriers to the containment of disease spread. In fact, rumors that the virus does not truly exist, and that the illness is a result of biological warfare, cannibalistic rituals, or witchcraft add to the complexity of the situation.11
For these reasons, it is essential that efforts to control this epidemic in endemic healthcare facilities include effective health surveillance, infection-control programs, and community outreach fostering mutual trust-building, honest communication, and education. Success will require a multifaceted approach, and a global response will be needed to quiet this global threat. On September 16, the United States announced a robust response to deploy military engineers and medical personnel to assist in training healthcare workers and building care centers in Liberia. The United Nations, France, and the United Kingdom are also supporting this important effort to build stronger healthcare infrastructures in these vulnerable countries.16
Conclusion
This 2014 epidemic of EVD raises justifiable concerns regarding the impact of globalization. Though unlikely to pose a direct threat of epidemic proportion on US soil, the unanticipated occurrence of an index case may trigger a terrifying outbreak in any community, as it already has in the city of Dallas. Given that the early stages of EVD are indistinguishable from most other viral syndromes, the importance of reflection on individual and general healthcare facility adherence to standard infection control precautions and procedures warrant merit. Eliciting accurate travel histories and possible exposures are germane to narrowing the scope of possible etiologies of all infectious diseases. As an opportunity for improvement, this epidemic should incite elevated caution in the everyday handling of all patients with febrile illnesses and contact-transmissible infections including methicillin-resistant Staphylococcus aureus and Clostridium difficile, which affect a great number of US patients on a daily basis. Not only could this strategy prevent additional local outbreaks of EVD, but it would promote the safety of healthcare workers and the community served through attention to better infection preventive measures at the point of care, every time.
Dr McCammon is an assistant professor, department of emergency medicine, Eastern Virginia Medical School, Norfolk. Dr Chidester is an instructor, department of emergency medicine, and a fellow in international wilderness medicine, Eastern Virginia Medical School, Norfolk.
- Ebola virus disease information for clinicians in U.S. Healthcare Settings. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/vhf/ebola/hcp/clinician-information-us-healthcare-settings.html. Updated September 5, 2014. Accessed September 23, 2014.
- 2014 Ebola outbreak in West Africa. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/vhf/ebola/outbreaks/guinea/. Updated September 18, 2014. Accessed September 23, 2014.
- Virus ecology graphic. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/vhf/ebola/resources/virus-ecology.html. Updated August 1, 2014. Accessed September 23, 2014.
- McElroy AK, Erickson BR, Flietstra TD, et al. Ebola hemorrhagic fever: novel biomarker correlates of clinical outcome. J Infect Dis. 2014;210(4):558-566.
- Rollin PE, Bausch DG, Sanchez A. Blood chemistry measurements and D-Dimer levels associated with fatal and nonfatal outcomes in humans infected with Sudan Ebola virus. J Infect Dis. 2007;196(Suppl 2):S364-S371.
- Geisbert TW, Young HA, Jahrling PB, et al. Pathogenesis of Ebola hemorrhagic fever in primate models: evidence that hemorrhage is not a direct effect of virus-induced cytolysis of endothelial cells. Am J Pathol. 2003;163(6): 2371-2382.
- Blumberg L, Enria D, Bausch DG. Viral haemorrhagic fevers. In: Farrar J, Hotez, PJ, Junghanss T, Kang G, Lalloo D, White N, eds. Manson’s Tropical Diseases: Expert Consult. 23rd ed. Philadelphia, PA: Elsevier Saunders; 2014:171-194.
- Safe management of patients with Ebola virus disease (EVD) in U.S. hospitals. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/vhf/ebola/hcp/patient-management-us-hospitals.html. Updated September 5, 2014. Accessed September 23, 2014.
- Maron DF. Ebola doctor reveals how infected Americans were cured. Scientific American. http://www.scientificamerican.com/article/ebola-doctor-revealshow-infected-americans-were-cured/.
- Tribune wire reports. American Ebola patients treated with ZMapp experimental drug. Chicago Tribune. August 21, 2014. http://www.chicagotribune.com/lifestyles/health/chi-ebola-zmapp-20140821-story.html. Accessed September 23, 2014.
- Ebola crisis: doctors in Liberia ‘recovering after taking ZMapp’ [transcript]. BBC News. http://www.bbc.com/news/world-africa-28860204. August 19, 2014. Accessed September 23, 2014.
- Interim guidance for specimen collection, transport, testing, and submission for persons under investigation for Ebola virus disease in the United States. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/vhf/ebola/hcp/interim-guidance-specimen-collection-submission-patients-suspected-infection-ebola.html. Updated September 8, 2014. Accessed September 23, 2014.
- Catherine E. Shoichet CE, Fantz A, Yan H. Hospital ‘dropped the ball’ with Ebola patient’s travel history, NIH official says. CNN News. http://www.cnn.com/2014/10/01/health/ebola-us/index.html. Accessed October 2, 2014.
- Ebola (Ebola Virus Disease): Diagnosis. Centers for Disease Control and Prevention Web Site. http://www.cdc.gov/vhf/ebola/diagnosis/. Updated September 19, 2014. Accessed October 1, 2014.
- Gilblom K, Langreth R. Dallas hospital initially let Ebola patient go with drugs. Bloomberg News Web Site. http://www.bloomberg.com/news/2014-09-30/first-ebola-case-is-diagnosed-in-the-u-s-cdc-reports.html. Accessed October 1, 2014.
- Bates D, Szathmary Z and Boyle L. Up to twelve Americans could have Ebola: Fears grow in Dallas after first victim of deadly virus to reach U.S. remained at large for a week. September 30, 2014. Mail Online. http://www.dailymail.co.uk/news/article-2775608/CDC-confirms-Dallas-patient-isolation-testing-returning-region-plagued-Ebola-HAS-deadly-virus.html. Updated October 1, 2014. Accessed October 1, 2014.
- International affairs: bushmeat. U.S. Fish & Wildlife Service Web site. http://www.fws.gov/international/wildlife-without-borders/global-program/bushmeat.html. Accessed September 23, 2014.
- Ebola virus disease information for clinicians in U.S. Healthcare Settings. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/vhf/ebola/hcp/clinician-information-us-healthcare-settings.html. Updated September 5, 2014. Accessed September 23, 2014.
- 2014 Ebola outbreak in West Africa. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/vhf/ebola/outbreaks/guinea/. Updated September 18, 2014. Accessed September 23, 2014.
- Virus ecology graphic. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/vhf/ebola/resources/virus-ecology.html. Updated August 1, 2014. Accessed September 23, 2014.
- McElroy AK, Erickson BR, Flietstra TD, et al. Ebola hemorrhagic fever: novel biomarker correlates of clinical outcome. J Infect Dis. 2014;210(4):558-566.
- Rollin PE, Bausch DG, Sanchez A. Blood chemistry measurements and D-Dimer levels associated with fatal and nonfatal outcomes in humans infected with Sudan Ebola virus. J Infect Dis. 2007;196(Suppl 2):S364-S371.
- Geisbert TW, Young HA, Jahrling PB, et al. Pathogenesis of Ebola hemorrhagic fever in primate models: evidence that hemorrhage is not a direct effect of virus-induced cytolysis of endothelial cells. Am J Pathol. 2003;163(6): 2371-2382.
- Blumberg L, Enria D, Bausch DG. Viral haemorrhagic fevers. In: Farrar J, Hotez, PJ, Junghanss T, Kang G, Lalloo D, White N, eds. Manson’s Tropical Diseases: Expert Consult. 23rd ed. Philadelphia, PA: Elsevier Saunders; 2014:171-194.
- Safe management of patients with Ebola virus disease (EVD) in U.S. hospitals. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/vhf/ebola/hcp/patient-management-us-hospitals.html. Updated September 5, 2014. Accessed September 23, 2014.
- Maron DF. Ebola doctor reveals how infected Americans were cured. Scientific American. http://www.scientificamerican.com/article/ebola-doctor-revealshow-infected-americans-were-cured/.
- Tribune wire reports. American Ebola patients treated with ZMapp experimental drug. Chicago Tribune. August 21, 2014. http://www.chicagotribune.com/lifestyles/health/chi-ebola-zmapp-20140821-story.html. Accessed September 23, 2014.
- Ebola crisis: doctors in Liberia ‘recovering after taking ZMapp’ [transcript]. BBC News. http://www.bbc.com/news/world-africa-28860204. August 19, 2014. Accessed September 23, 2014.
- Interim guidance for specimen collection, transport, testing, and submission for persons under investigation for Ebola virus disease in the United States. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/vhf/ebola/hcp/interim-guidance-specimen-collection-submission-patients-suspected-infection-ebola.html. Updated September 8, 2014. Accessed September 23, 2014.
- Catherine E. Shoichet CE, Fantz A, Yan H. Hospital ‘dropped the ball’ with Ebola patient’s travel history, NIH official says. CNN News. http://www.cnn.com/2014/10/01/health/ebola-us/index.html. Accessed October 2, 2014.
- Ebola (Ebola Virus Disease): Diagnosis. Centers for Disease Control and Prevention Web Site. http://www.cdc.gov/vhf/ebola/diagnosis/. Updated September 19, 2014. Accessed October 1, 2014.
- Gilblom K, Langreth R. Dallas hospital initially let Ebola patient go with drugs. Bloomberg News Web Site. http://www.bloomberg.com/news/2014-09-30/first-ebola-case-is-diagnosed-in-the-u-s-cdc-reports.html. Accessed October 1, 2014.
- Bates D, Szathmary Z and Boyle L. Up to twelve Americans could have Ebola: Fears grow in Dallas after first victim of deadly virus to reach U.S. remained at large for a week. September 30, 2014. Mail Online. http://www.dailymail.co.uk/news/article-2775608/CDC-confirms-Dallas-patient-isolation-testing-returning-region-plagued-Ebola-HAS-deadly-virus.html. Updated October 1, 2014. Accessed October 1, 2014.
- International affairs: bushmeat. U.S. Fish & Wildlife Service Web site. http://www.fws.gov/international/wildlife-without-borders/global-program/bushmeat.html. Accessed September 23, 2014.
Obesity, Brain Function, and Heart Failure
In healthy individuals, obesity has been linked to reduced cerebral blood flow and brain atrophy and to a higher risk of neurologic disorders, such as Alzheimer disease and vascular dementia, in patients with heart failure (HF), according to researchers from Kent State University in Ohio. Their study focused on the connection between HF, brain volume, and obesity. The researchers say it is time to consider obesity as an independent risk factor for structural brain changes in HF.
Their research supports the idea that common vascular risk factors, such as hypertension and diabetes, influence neurocognitive outcomes in HF through a negative impact on cerebral perfusion. The study is the first to examine the possible associations among body mass index (BMI), cerebral perfusion, and brain volume in the obese population.
The study participants (80 patients taking part in an ongoing study of neurocognitive outcomes in HF) were aged 50 to 85 years, with New York Heart Association class II, III, or IV HF. Exclusion criteria included a history of a significant neurologic disorder, such as dementia or stroke, severe head injury, severe psychiatric disorder, history of substance abuse, and renal failure. The participants underwent transcranial Doppler sonography and a brain MRI. The mean BMI was 29.89; 75% of the patients were overweight or obese.
Study findings suggested that increased BMI was associated with reduced white matter hyperintensities volume (P < .05). Body mass index also interacted with cerebral perfusion on total gray matter volume.
In discussing possible mechanisms, the researchers say their study and other work suggest that obesity and adiposity may introduce “unique pathophysiological mechanisms to produce brain changes in HF.” They note that obesity is associated with altered levels of the biomarkers leptin, ghrelin, and brain-derived neurotrophic factor, which play a role in regulating metabolism and weight. They also promote neuronal survival, neurogenesis, dendritic synaptic formations, and reduce apoptosis of neurons—all biologic processes that shape the cerebral structure. The researchers also cite that obesity can affect the brain through inflammatory processes.
However, the mechanisms remain unclear. The relationship between obesity and neurocognitive outcomes in HF is complex, they conclude, and likely involves other mechanisms beyond cerebral hemodynamics.
Source
Alosco ML, Brickman AM, Spitznagel MB, et al. BMC Obes. 2014;1:4.
doi: 10.1186/2052-9538-1-4.
In healthy individuals, obesity has been linked to reduced cerebral blood flow and brain atrophy and to a higher risk of neurologic disorders, such as Alzheimer disease and vascular dementia, in patients with heart failure (HF), according to researchers from Kent State University in Ohio. Their study focused on the connection between HF, brain volume, and obesity. The researchers say it is time to consider obesity as an independent risk factor for structural brain changes in HF.
Their research supports the idea that common vascular risk factors, such as hypertension and diabetes, influence neurocognitive outcomes in HF through a negative impact on cerebral perfusion. The study is the first to examine the possible associations among body mass index (BMI), cerebral perfusion, and brain volume in the obese population.
The study participants (80 patients taking part in an ongoing study of neurocognitive outcomes in HF) were aged 50 to 85 years, with New York Heart Association class II, III, or IV HF. Exclusion criteria included a history of a significant neurologic disorder, such as dementia or stroke, severe head injury, severe psychiatric disorder, history of substance abuse, and renal failure. The participants underwent transcranial Doppler sonography and a brain MRI. The mean BMI was 29.89; 75% of the patients were overweight or obese.
Study findings suggested that increased BMI was associated with reduced white matter hyperintensities volume (P < .05). Body mass index also interacted with cerebral perfusion on total gray matter volume.
In discussing possible mechanisms, the researchers say their study and other work suggest that obesity and adiposity may introduce “unique pathophysiological mechanisms to produce brain changes in HF.” They note that obesity is associated with altered levels of the biomarkers leptin, ghrelin, and brain-derived neurotrophic factor, which play a role in regulating metabolism and weight. They also promote neuronal survival, neurogenesis, dendritic synaptic formations, and reduce apoptosis of neurons—all biologic processes that shape the cerebral structure. The researchers also cite that obesity can affect the brain through inflammatory processes.
However, the mechanisms remain unclear. The relationship between obesity and neurocognitive outcomes in HF is complex, they conclude, and likely involves other mechanisms beyond cerebral hemodynamics.
Source
Alosco ML, Brickman AM, Spitznagel MB, et al. BMC Obes. 2014;1:4.
doi: 10.1186/2052-9538-1-4.
In healthy individuals, obesity has been linked to reduced cerebral blood flow and brain atrophy and to a higher risk of neurologic disorders, such as Alzheimer disease and vascular dementia, in patients with heart failure (HF), according to researchers from Kent State University in Ohio. Their study focused on the connection between HF, brain volume, and obesity. The researchers say it is time to consider obesity as an independent risk factor for structural brain changes in HF.
Their research supports the idea that common vascular risk factors, such as hypertension and diabetes, influence neurocognitive outcomes in HF through a negative impact on cerebral perfusion. The study is the first to examine the possible associations among body mass index (BMI), cerebral perfusion, and brain volume in the obese population.
The study participants (80 patients taking part in an ongoing study of neurocognitive outcomes in HF) were aged 50 to 85 years, with New York Heart Association class II, III, or IV HF. Exclusion criteria included a history of a significant neurologic disorder, such as dementia or stroke, severe head injury, severe psychiatric disorder, history of substance abuse, and renal failure. The participants underwent transcranial Doppler sonography and a brain MRI. The mean BMI was 29.89; 75% of the patients were overweight or obese.
Study findings suggested that increased BMI was associated with reduced white matter hyperintensities volume (P < .05). Body mass index also interacted with cerebral perfusion on total gray matter volume.
In discussing possible mechanisms, the researchers say their study and other work suggest that obesity and adiposity may introduce “unique pathophysiological mechanisms to produce brain changes in HF.” They note that obesity is associated with altered levels of the biomarkers leptin, ghrelin, and brain-derived neurotrophic factor, which play a role in regulating metabolism and weight. They also promote neuronal survival, neurogenesis, dendritic synaptic formations, and reduce apoptosis of neurons—all biologic processes that shape the cerebral structure. The researchers also cite that obesity can affect the brain through inflammatory processes.
However, the mechanisms remain unclear. The relationship between obesity and neurocognitive outcomes in HF is complex, they conclude, and likely involves other mechanisms beyond cerebral hemodynamics.
Source
Alosco ML, Brickman AM, Spitznagel MB, et al. BMC Obes. 2014;1:4.
doi: 10.1186/2052-9538-1-4.