User login
Caregiver Surveys on Firearms, Suicide Offer Pediatricians Prevention Opportunities
ORLANDO, FLORIDA — , according to researchers who presented their findings at the American Academy of Pediatrics (AAP) 2024 National Conference.
An estimated 4.6 million US homes with children have firearms that are loaded and unlocked, a risk factor for youth suicide, yet only about half of parents of suicidal children had been screened for gun ownership in the hospital even as most would be receptive to both firearm screening and counseling, found one study in Texas.
In another study in Colorado, nearly all firearm owners believed that securely storing guns reduces the risk for firearm injury or death, but owners were less likely than non-owners to believe suicide is preventable or that removing a gun from the home reduces the risk for injury or death.
“Previous studies have shown that when pediatricians discuss the importance of armed safe storage guidance with families, families are actually more likely to go home and store firearms safely — storing them locked, unloaded, and separate from the ammunition,” said study author Taylor Rosenbaum, MD, a former pediatric fellow at Baylor College of Medicine/Texas Children’s Hospital in Houston and now an assistant professor at Children’s Hospital University of Miami. “However, previous studies have also shown that pediatricians really are not discussing firearm safe storage with our patients and their families, and we see this both in the outpatient setting, but especially in the inpatient setting for youth suicides, which have risen since 2020 and now are the second leading cause of death for those who are 10-24 years old in the United States.”

Firearm Safety Is a Necessary Conversation
The leading cause of death among children and teens aged 1-19 years is actually firearms, which are also the most fatal method for suicide. While only 4% of all suicide attempts in youth are fatal, 90% of those attempted with a firearm are fatal, Dr. Rosenbaum said. In addition, she said, 80% of the guns used in attempted suicide by children and teens belonged to a family member, and an estimated 70% of firearm-related suicides in youth can be prevented with safe storage of guns.
“This really gives us, as pediatricians, something actionable to do during these hospitalizations” for suicidal ideation or attempts, Dr. Rosenbaum said. “We know that when pediatricians discuss the importance of firearm safe storage guidance with families, they’re more likely to store their firearm safely,” Dr. Rosenbaum said. “We also know that families are not being screened for firearm ownership, that caregivers of youth who are in the hospital for suicidal thoughts or actions want their healthcare team to be screening for firearms, to be giving them information on how to safely secure their firearms, and to be providing free firearm blocks.”
Nathan Boonstra, MD, a general pediatrician at Blank Children’s Hospital, Des Moines, Iowa, said these findings are encouraging in terms of the opportunity pediatricians have.
“There is so much politicization around even basic firearm safety that pediatricians might shy away from the topic, but this research is reassuring that parents are receptive to our advice on safe gun storage,” said Dr. Boonstra, who was not involved in any of this presented research. “It’s especially important for pediatricians to address home firearms when their patient has a history of suicidal ideation or an attempt.”
Reducing the Risk
The Colorado findings similarly reinforce the opportunity physicians have to help caregivers reduce suicide risk, according to Maya Haasz, MD, an associate professor of pediatrics and emergency medicine at the University of Colorado Anschutz Medical Campus, Aurora, Colorado.
“Only 60% of firearm owners believed that removing firearms from the home in times of mental health crisis can decrease the risk of suicide,” she said. “These findings are really concerning, but what we found on the flip side was that 93% of firearm owners actually believe that secure storage can overall decrease the risk for firearm injury and death. So overall, we are underestimating the risk for suicide in our community, and we’re also underestimating our ability to prevent it.”

That presents an opportunity, Dr. Haasz said, “to educate families both about the preventability of suicide but also to have specific strategies, like secure storage and temporary removable requirements from the home, that can prevent suicide.”
Dr. Boonstra found it “disheartening that so many children live in a house with an unlocked and even loaded firearm when the evidence is so clear that this is a significant risk factor for youth suicide,” he said. “It’s also disheartening, though not too surprising, that families with a firearm are less likely to think that youth suicide can be prevented.”
Survey Results
Dr. Rosenbaum’s team conducted the survey in Houston with caregivers whose children were 8-21 years old and hospitalized for suicidal ideation or attempts at a large children’s hospital and two nearby community hospitals between June 2023 and May 2024. The respondents were 46% White and 23% Black, and 47% of the population were Hispanic, all but three of whom were not gun owners.
Among 244 potential participants, only 150 were eligible and approached, and 100 of these completed the surveys, including 26% firearm owners and 68% non-owners. Most of the youth (74%) were aged 14-17 years, and about three in four respondents were their mothers. Only half of the respondents (51%) said the healthcare provider had asked them whether they owned a gun.
One of the key findings Dr. Rosenbaum highlighted was the receptiveness of firearm-owning caregivers to advice from healthcare providers about ownership. If the healthcare team advised parents not to have any guns in the home for the safety of their child with self-arm, 58% of the firearm owners would follow the advice and 27% would consider it, with none saying they would be offended by it.
Among the firearm owners, 81% said their guns were safely secured where they did not believe their child could access it, which meant one in five youth had unsecured access to firearms. Most of the gun owners (77%), like the non-owners (70%), were “not at all worried” about their child getting ahold of a gun in the home, though 11.5% of the firearm owners were “very worried” about it. Interestingly, more gun owners (19%) were very worried about their children accessing a gun outside their home, a concern shared by 37% of non-owners. Nearly twice as many gun owners (46%) as non-owners (25%) were not at all worried about their child getting a gun outside the home.
The vast majority of respondents — 88% of gun owners and 91% of non-owners — felt it was “very important for the healthcare team to ask parents of children with suicidal ideation/attempts about firearms in the home.” Similarly, high proportions believed it was important for the healthcare team to counsel those parents on safe gun storage. Although only 69% of firearm owners believed it was important to distribute firearm locks in the hospital, 81% would be interested in receiving a free one. Significantly more of the non-owners (80%; P = .02) believed free lock distribution was important, and 72% of non-owners would also be interested in one.
About half the respondents (55%) preferred to hear firearm counseling one-on-one from a provider, whereas 31% would like written information and 27% would be interested in a video. In terms of what information parents preferred to receive, a little over half of owners (54%) and non-owners (56%) were interested in how or when (50% and 40%, respectively) to discuss the topic with their child. Only about a third (35% owners and 37% non-owners) wanted information on how to discuss the topic with the parents of their child’s friends.
The survey’s biggest limitations after its small size were the selection bias of those willing to complete the survey and potential response bias from the self-reported data.
The study of Colorado caregivers, just published in Pediatrics, surveyed 512 Colorado caregivers in April-May 2023 to learn about their beliefs and perceptions regarding firearms, firearm storage and risk, and youth suicide (2024 Oct 1;154[4]:e2024066930. doi: 10.1542/peds.2024-066930). Just over half the respondents (52%) had grown up in a household with firearms, and 44% currently lived in a household with a gun. The sample was 43% men and 88% White, predominantly non-Hispanic (75%), with 11% living in rural areas and 19% who currently or previously served in the military. Most (79%) had a child age 12 or younger in the home.
Only about one in four caregivers (24%) correctly answered that suicide is the leading cause of firearm death in Colorado, with similar rates of correct responses among both firearm owners and non-firearm owners. Both groups were also similarly likely (64% overall) to be concerned about youth suicide in their community, though those from homes with firearms were less likely to be concerned about youth suicide in their own family (28%) than those from homes without firearms (39%; P = .013).
In addition, caregivers from homes with versus without firearms were considerably less likely to believe suicide can be prevented (48% vs 69%) and were less likely to believe that temporarily removing a firearm from the home reduces the risk for gun injury or death (60% vs 78%; P < .001 for both comparisons).
Firearm owners were also much less likely than non-owners to believe keeping a gun in the home makes it more dangerous (7% vs 29%) and over twice as likely to think keeping a firearm makes their home safer (52% vs 22%; P < .001). The vast majority of respondents (89%) believed secure storage of guns reduces the risk for injury or death, though the response was higher for firearm owners (93%) than for non-owners (86%; P < .001).
“Our finding that most firearm owners believe that secure firearm storage is protective against firearm injury is a promising messaging strategy,” the authors wrote. “It presents a preventive education opportunity for adults living with children who have mental health concerns, who may benefit most from secure in-home storage and/or temporary and voluntary storage of firearms away from home.”
Firearm Injuries
A separate study at the AAP conference underscored the devastating impact of firearm injuries even among those who survive, whether self-inflicted or not, and the potential for reducing healthcare treatment and costs from effective prevention efforts. A national analysis of pediatric inpatient data from 2017 to 2020 calculated how much greater the burden of healthcare treatment and costs is for firearm injuries of any kind compared with penetrating traumas and blunt traumas.
“As a surgical resident, I have seen these patients who make it into the trauma bed that we are then faced to care for,” said Colleen Nofi, DO, PhD, MBA, a general surgery resident at Cohen Children’s Medical Center at Northwell Health in New York. “Anecdotally, we understand that the devastation and injury caused by bullets far outweighs the injuries caused by other trauma mechanisms,” but the actual calculation of the burden hasn’t been studied.

Among 6615 firearm injuries, 9787 penetrating traumas and 66,003 blunt traumas examined from the National Inpatient Sample Healthcare Cost and Utilization Project Database, 11% of firearm traumas required a transfusion of red blood cells, compared with 1.4% of penetrating traumas and 3% of blunt traumas (P < .001). Patients with firearm injuries also had a longer length of stay — 10.8 days compared with 8.3 for patients with penetrating trauma and 9.8 for those with blunt trauma — and significantly higher rates of CPR, pericardiotomy, chest tube, exploratory laparotomy and/or thoracotomy, colorectal surgery, small bowel surgery, ostomy formation, splenectomy, hepatic resection, tracheostomy, and feeding tube placement.
Pulmonary complications were higher for firearm injuries (4.9%) than for penetrating trauma (0.6%) or blunt trauma (2.9%), and septicemia rates were also higher (1.7% vs 0.2% and 1%, respectively). Cardiac, neurologic, and urinary complications were also significantly and substantially higher for firearm injuries, 6.9% of which resulted in death compared with 0.2% of penetrating traumas and 1.2% of blunt traumas.
The costs from firearm injuries were also significantly higher than the costs from other traumas; “firearm injury remained independently predictive of greater hospital costs, even when controlling for injury severity as well as age, sex, race, insurance, region, hospital type, and household income.
“These findings underscore the urgent need for targeted prevention, supportive measures, and resource allocation to mitigate the devastating impact of firearm injuries on children and healthcare systems alike,” Dr. Nofi said.
The Colorado study was funded by the Colorado Department of Public Health and Environment and a National Institutes of Health grant to Dr. Haasz. The Texas study and the one from Northwell Health did not note any external funding. Dr. Haasz, Dr. Rosenbaum, Dr. Boonstra, and Dr. Nofi had no disclosures.
A version of this article appeared on Medscape.com.
ORLANDO, FLORIDA — , according to researchers who presented their findings at the American Academy of Pediatrics (AAP) 2024 National Conference.
An estimated 4.6 million US homes with children have firearms that are loaded and unlocked, a risk factor for youth suicide, yet only about half of parents of suicidal children had been screened for gun ownership in the hospital even as most would be receptive to both firearm screening and counseling, found one study in Texas.
In another study in Colorado, nearly all firearm owners believed that securely storing guns reduces the risk for firearm injury or death, but owners were less likely than non-owners to believe suicide is preventable or that removing a gun from the home reduces the risk for injury or death.
“Previous studies have shown that when pediatricians discuss the importance of armed safe storage guidance with families, families are actually more likely to go home and store firearms safely — storing them locked, unloaded, and separate from the ammunition,” said study author Taylor Rosenbaum, MD, a former pediatric fellow at Baylor College of Medicine/Texas Children’s Hospital in Houston and now an assistant professor at Children’s Hospital University of Miami. “However, previous studies have also shown that pediatricians really are not discussing firearm safe storage with our patients and their families, and we see this both in the outpatient setting, but especially in the inpatient setting for youth suicides, which have risen since 2020 and now are the second leading cause of death for those who are 10-24 years old in the United States.”
Firearm Safety Is a Necessary Conversation
The leading cause of death among children and teens aged 1-19 years is actually firearms, which are also the most fatal method for suicide. While only 4% of all suicide attempts in youth are fatal, 90% of those attempted with a firearm are fatal, Dr. Rosenbaum said. In addition, she said, 80% of the guns used in attempted suicide by children and teens belonged to a family member, and an estimated 70% of firearm-related suicides in youth can be prevented with safe storage of guns.
“This really gives us, as pediatricians, something actionable to do during these hospitalizations” for suicidal ideation or attempts, Dr. Rosenbaum said. “We know that when pediatricians discuss the importance of firearm safe storage guidance with families, they’re more likely to store their firearm safely,” Dr. Rosenbaum said. “We also know that families are not being screened for firearm ownership, that caregivers of youth who are in the hospital for suicidal thoughts or actions want their healthcare team to be screening for firearms, to be giving them information on how to safely secure their firearms, and to be providing free firearm blocks.”
Nathan Boonstra, MD, a general pediatrician at Blank Children’s Hospital, Des Moines, Iowa, said these findings are encouraging in terms of the opportunity pediatricians have.
“There is so much politicization around even basic firearm safety that pediatricians might shy away from the topic, but this research is reassuring that parents are receptive to our advice on safe gun storage,” said Dr. Boonstra, who was not involved in any of this presented research. “It’s especially important for pediatricians to address home firearms when their patient has a history of suicidal ideation or an attempt.”
Reducing the Risk
The Colorado findings similarly reinforce the opportunity physicians have to help caregivers reduce suicide risk, according to Maya Haasz, MD, an associate professor of pediatrics and emergency medicine at the University of Colorado Anschutz Medical Campus, Aurora, Colorado.
“Only 60% of firearm owners believed that removing firearms from the home in times of mental health crisis can decrease the risk of suicide,” she said. “These findings are really concerning, but what we found on the flip side was that 93% of firearm owners actually believe that secure storage can overall decrease the risk for firearm injury and death. So overall, we are underestimating the risk for suicide in our community, and we’re also underestimating our ability to prevent it.”
That presents an opportunity, Dr. Haasz said, “to educate families both about the preventability of suicide but also to have specific strategies, like secure storage and temporary removable requirements from the home, that can prevent suicide.”
Dr. Boonstra found it “disheartening that so many children live in a house with an unlocked and even loaded firearm when the evidence is so clear that this is a significant risk factor for youth suicide,” he said. “It’s also disheartening, though not too surprising, that families with a firearm are less likely to think that youth suicide can be prevented.”
Survey Results
Dr. Rosenbaum’s team conducted the survey in Houston with caregivers whose children were 8-21 years old and hospitalized for suicidal ideation or attempts at a large children’s hospital and two nearby community hospitals between June 2023 and May 2024. The respondents were 46% White and 23% Black, and 47% of the population were Hispanic, all but three of whom were not gun owners.
Among 244 potential participants, only 150 were eligible and approached, and 100 of these completed the surveys, including 26% firearm owners and 68% non-owners. Most of the youth (74%) were aged 14-17 years, and about three in four respondents were their mothers. Only half of the respondents (51%) said the healthcare provider had asked them whether they owned a gun.
One of the key findings Dr. Rosenbaum highlighted was the receptiveness of firearm-owning caregivers to advice from healthcare providers about ownership. If the healthcare team advised parents not to have any guns in the home for the safety of their child with self-arm, 58% of the firearm owners would follow the advice and 27% would consider it, with none saying they would be offended by it.
Among the firearm owners, 81% said their guns were safely secured where they did not believe their child could access it, which meant one in five youth had unsecured access to firearms. Most of the gun owners (77%), like the non-owners (70%), were “not at all worried” about their child getting ahold of a gun in the home, though 11.5% of the firearm owners were “very worried” about it. Interestingly, more gun owners (19%) were very worried about their children accessing a gun outside their home, a concern shared by 37% of non-owners. Nearly twice as many gun owners (46%) as non-owners (25%) were not at all worried about their child getting a gun outside the home.
The vast majority of respondents — 88% of gun owners and 91% of non-owners — felt it was “very important for the healthcare team to ask parents of children with suicidal ideation/attempts about firearms in the home.” Similarly, high proportions believed it was important for the healthcare team to counsel those parents on safe gun storage. Although only 69% of firearm owners believed it was important to distribute firearm locks in the hospital, 81% would be interested in receiving a free one. Significantly more of the non-owners (80%; P = .02) believed free lock distribution was important, and 72% of non-owners would also be interested in one.
About half the respondents (55%) preferred to hear firearm counseling one-on-one from a provider, whereas 31% would like written information and 27% would be interested in a video. In terms of what information parents preferred to receive, a little over half of owners (54%) and non-owners (56%) were interested in how or when (50% and 40%, respectively) to discuss the topic with their child. Only about a third (35% owners and 37% non-owners) wanted information on how to discuss the topic with the parents of their child’s friends.
The survey’s biggest limitations after its small size were the selection bias of those willing to complete the survey and potential response bias from the self-reported data.
The study of Colorado caregivers, just published in Pediatrics, surveyed 512 Colorado caregivers in April-May 2023 to learn about their beliefs and perceptions regarding firearms, firearm storage and risk, and youth suicide (2024 Oct 1;154[4]:e2024066930. doi: 10.1542/peds.2024-066930). Just over half the respondents (52%) had grown up in a household with firearms, and 44% currently lived in a household with a gun. The sample was 43% men and 88% White, predominantly non-Hispanic (75%), with 11% living in rural areas and 19% who currently or previously served in the military. Most (79%) had a child age 12 or younger in the home.
Only about one in four caregivers (24%) correctly answered that suicide is the leading cause of firearm death in Colorado, with similar rates of correct responses among both firearm owners and non-firearm owners. Both groups were also similarly likely (64% overall) to be concerned about youth suicide in their community, though those from homes with firearms were less likely to be concerned about youth suicide in their own family (28%) than those from homes without firearms (39%; P = .013).
In addition, caregivers from homes with versus without firearms were considerably less likely to believe suicide can be prevented (48% vs 69%) and were less likely to believe that temporarily removing a firearm from the home reduces the risk for gun injury or death (60% vs 78%; P < .001 for both comparisons).
Firearm owners were also much less likely than non-owners to believe keeping a gun in the home makes it more dangerous (7% vs 29%) and over twice as likely to think keeping a firearm makes their home safer (52% vs 22%; P < .001). The vast majority of respondents (89%) believed secure storage of guns reduces the risk for injury or death, though the response was higher for firearm owners (93%) than for non-owners (86%; P < .001).
“Our finding that most firearm owners believe that secure firearm storage is protective against firearm injury is a promising messaging strategy,” the authors wrote. “It presents a preventive education opportunity for adults living with children who have mental health concerns, who may benefit most from secure in-home storage and/or temporary and voluntary storage of firearms away from home.”
Firearm Injuries
A separate study at the AAP conference underscored the devastating impact of firearm injuries even among those who survive, whether self-inflicted or not, and the potential for reducing healthcare treatment and costs from effective prevention efforts. A national analysis of pediatric inpatient data from 2017 to 2020 calculated how much greater the burden of healthcare treatment and costs is for firearm injuries of any kind compared with penetrating traumas and blunt traumas.
“As a surgical resident, I have seen these patients who make it into the trauma bed that we are then faced to care for,” said Colleen Nofi, DO, PhD, MBA, a general surgery resident at Cohen Children’s Medical Center at Northwell Health in New York. “Anecdotally, we understand that the devastation and injury caused by bullets far outweighs the injuries caused by other trauma mechanisms,” but the actual calculation of the burden hasn’t been studied.
Among 6615 firearm injuries, 9787 penetrating traumas and 66,003 blunt traumas examined from the National Inpatient Sample Healthcare Cost and Utilization Project Database, 11% of firearm traumas required a transfusion of red blood cells, compared with 1.4% of penetrating traumas and 3% of blunt traumas (P < .001). Patients with firearm injuries also had a longer length of stay — 10.8 days compared with 8.3 for patients with penetrating trauma and 9.8 for those with blunt trauma — and significantly higher rates of CPR, pericardiotomy, chest tube, exploratory laparotomy and/or thoracotomy, colorectal surgery, small bowel surgery, ostomy formation, splenectomy, hepatic resection, tracheostomy, and feeding tube placement.
Pulmonary complications were higher for firearm injuries (4.9%) than for penetrating trauma (0.6%) or blunt trauma (2.9%), and septicemia rates were also higher (1.7% vs 0.2% and 1%, respectively). Cardiac, neurologic, and urinary complications were also significantly and substantially higher for firearm injuries, 6.9% of which resulted in death compared with 0.2% of penetrating traumas and 1.2% of blunt traumas.
The costs from firearm injuries were also significantly higher than the costs from other traumas; “firearm injury remained independently predictive of greater hospital costs, even when controlling for injury severity as well as age, sex, race, insurance, region, hospital type, and household income.
“These findings underscore the urgent need for targeted prevention, supportive measures, and resource allocation to mitigate the devastating impact of firearm injuries on children and healthcare systems alike,” Dr. Nofi said.
The Colorado study was funded by the Colorado Department of Public Health and Environment and a National Institutes of Health grant to Dr. Haasz. The Texas study and the one from Northwell Health did not note any external funding. Dr. Haasz, Dr. Rosenbaum, Dr. Boonstra, and Dr. Nofi had no disclosures.
A version of this article appeared on Medscape.com.
ORLANDO, FLORIDA — , according to researchers who presented their findings at the American Academy of Pediatrics (AAP) 2024 National Conference.
An estimated 4.6 million US homes with children have firearms that are loaded and unlocked, a risk factor for youth suicide, yet only about half of parents of suicidal children had been screened for gun ownership in the hospital even as most would be receptive to both firearm screening and counseling, found one study in Texas.
In another study in Colorado, nearly all firearm owners believed that securely storing guns reduces the risk for firearm injury or death, but owners were less likely than non-owners to believe suicide is preventable or that removing a gun from the home reduces the risk for injury or death.
“Previous studies have shown that when pediatricians discuss the importance of armed safe storage guidance with families, families are actually more likely to go home and store firearms safely — storing them locked, unloaded, and separate from the ammunition,” said study author Taylor Rosenbaum, MD, a former pediatric fellow at Baylor College of Medicine/Texas Children’s Hospital in Houston and now an assistant professor at Children’s Hospital University of Miami. “However, previous studies have also shown that pediatricians really are not discussing firearm safe storage with our patients and their families, and we see this both in the outpatient setting, but especially in the inpatient setting for youth suicides, which have risen since 2020 and now are the second leading cause of death for those who are 10-24 years old in the United States.”
Firearm Safety Is a Necessary Conversation
The leading cause of death among children and teens aged 1-19 years is actually firearms, which are also the most fatal method for suicide. While only 4% of all suicide attempts in youth are fatal, 90% of those attempted with a firearm are fatal, Dr. Rosenbaum said. In addition, she said, 80% of the guns used in attempted suicide by children and teens belonged to a family member, and an estimated 70% of firearm-related suicides in youth can be prevented with safe storage of guns.
“This really gives us, as pediatricians, something actionable to do during these hospitalizations” for suicidal ideation or attempts, Dr. Rosenbaum said. “We know that when pediatricians discuss the importance of firearm safe storage guidance with families, they’re more likely to store their firearm safely,” Dr. Rosenbaum said. “We also know that families are not being screened for firearm ownership, that caregivers of youth who are in the hospital for suicidal thoughts or actions want their healthcare team to be screening for firearms, to be giving them information on how to safely secure their firearms, and to be providing free firearm blocks.”
Nathan Boonstra, MD, a general pediatrician at Blank Children’s Hospital, Des Moines, Iowa, said these findings are encouraging in terms of the opportunity pediatricians have.
“There is so much politicization around even basic firearm safety that pediatricians might shy away from the topic, but this research is reassuring that parents are receptive to our advice on safe gun storage,” said Dr. Boonstra, who was not involved in any of this presented research. “It’s especially important for pediatricians to address home firearms when their patient has a history of suicidal ideation or an attempt.”
Reducing the Risk
The Colorado findings similarly reinforce the opportunity physicians have to help caregivers reduce suicide risk, according to Maya Haasz, MD, an associate professor of pediatrics and emergency medicine at the University of Colorado Anschutz Medical Campus, Aurora, Colorado.
“Only 60% of firearm owners believed that removing firearms from the home in times of mental health crisis can decrease the risk of suicide,” she said. “These findings are really concerning, but what we found on the flip side was that 93% of firearm owners actually believe that secure storage can overall decrease the risk for firearm injury and death. So overall, we are underestimating the risk for suicide in our community, and we’re also underestimating our ability to prevent it.”
That presents an opportunity, Dr. Haasz said, “to educate families both about the preventability of suicide but also to have specific strategies, like secure storage and temporary removable requirements from the home, that can prevent suicide.”
Dr. Boonstra found it “disheartening that so many children live in a house with an unlocked and even loaded firearm when the evidence is so clear that this is a significant risk factor for youth suicide,” he said. “It’s also disheartening, though not too surprising, that families with a firearm are less likely to think that youth suicide can be prevented.”
Survey Results
Dr. Rosenbaum’s team conducted the survey in Houston with caregivers whose children were 8-21 years old and hospitalized for suicidal ideation or attempts at a large children’s hospital and two nearby community hospitals between June 2023 and May 2024. The respondents were 46% White and 23% Black, and 47% of the population were Hispanic, all but three of whom were not gun owners.
Among 244 potential participants, only 150 were eligible and approached, and 100 of these completed the surveys, including 26% firearm owners and 68% non-owners. Most of the youth (74%) were aged 14-17 years, and about three in four respondents were their mothers. Only half of the respondents (51%) said the healthcare provider had asked them whether they owned a gun.
One of the key findings Dr. Rosenbaum highlighted was the receptiveness of firearm-owning caregivers to advice from healthcare providers about ownership. If the healthcare team advised parents not to have any guns in the home for the safety of their child with self-arm, 58% of the firearm owners would follow the advice and 27% would consider it, with none saying they would be offended by it.
Among the firearm owners, 81% said their guns were safely secured where they did not believe their child could access it, which meant one in five youth had unsecured access to firearms. Most of the gun owners (77%), like the non-owners (70%), were “not at all worried” about their child getting ahold of a gun in the home, though 11.5% of the firearm owners were “very worried” about it. Interestingly, more gun owners (19%) were very worried about their children accessing a gun outside their home, a concern shared by 37% of non-owners. Nearly twice as many gun owners (46%) as non-owners (25%) were not at all worried about their child getting a gun outside the home.
The vast majority of respondents — 88% of gun owners and 91% of non-owners — felt it was “very important for the healthcare team to ask parents of children with suicidal ideation/attempts about firearms in the home.” Similarly, high proportions believed it was important for the healthcare team to counsel those parents on safe gun storage. Although only 69% of firearm owners believed it was important to distribute firearm locks in the hospital, 81% would be interested in receiving a free one. Significantly more of the non-owners (80%; P = .02) believed free lock distribution was important, and 72% of non-owners would also be interested in one.
About half the respondents (55%) preferred to hear firearm counseling one-on-one from a provider, whereas 31% would like written information and 27% would be interested in a video. In terms of what information parents preferred to receive, a little over half of owners (54%) and non-owners (56%) were interested in how or when (50% and 40%, respectively) to discuss the topic with their child. Only about a third (35% owners and 37% non-owners) wanted information on how to discuss the topic with the parents of their child’s friends.
The survey’s biggest limitations after its small size were the selection bias of those willing to complete the survey and potential response bias from the self-reported data.
The study of Colorado caregivers, just published in Pediatrics, surveyed 512 Colorado caregivers in April-May 2023 to learn about their beliefs and perceptions regarding firearms, firearm storage and risk, and youth suicide (2024 Oct 1;154[4]:e2024066930. doi: 10.1542/peds.2024-066930). Just over half the respondents (52%) had grown up in a household with firearms, and 44% currently lived in a household with a gun. The sample was 43% men and 88% White, predominantly non-Hispanic (75%), with 11% living in rural areas and 19% who currently or previously served in the military. Most (79%) had a child age 12 or younger in the home.
Only about one in four caregivers (24%) correctly answered that suicide is the leading cause of firearm death in Colorado, with similar rates of correct responses among both firearm owners and non-firearm owners. Both groups were also similarly likely (64% overall) to be concerned about youth suicide in their community, though those from homes with firearms were less likely to be concerned about youth suicide in their own family (28%) than those from homes without firearms (39%; P = .013).
In addition, caregivers from homes with versus without firearms were considerably less likely to believe suicide can be prevented (48% vs 69%) and were less likely to believe that temporarily removing a firearm from the home reduces the risk for gun injury or death (60% vs 78%; P < .001 for both comparisons).
Firearm owners were also much less likely than non-owners to believe keeping a gun in the home makes it more dangerous (7% vs 29%) and over twice as likely to think keeping a firearm makes their home safer (52% vs 22%; P < .001). The vast majority of respondents (89%) believed secure storage of guns reduces the risk for injury or death, though the response was higher for firearm owners (93%) than for non-owners (86%; P < .001).
“Our finding that most firearm owners believe that secure firearm storage is protective against firearm injury is a promising messaging strategy,” the authors wrote. “It presents a preventive education opportunity for adults living with children who have mental health concerns, who may benefit most from secure in-home storage and/or temporary and voluntary storage of firearms away from home.”
Firearm Injuries
A separate study at the AAP conference underscored the devastating impact of firearm injuries even among those who survive, whether self-inflicted or not, and the potential for reducing healthcare treatment and costs from effective prevention efforts. A national analysis of pediatric inpatient data from 2017 to 2020 calculated how much greater the burden of healthcare treatment and costs is for firearm injuries of any kind compared with penetrating traumas and blunt traumas.
“As a surgical resident, I have seen these patients who make it into the trauma bed that we are then faced to care for,” said Colleen Nofi, DO, PhD, MBA, a general surgery resident at Cohen Children’s Medical Center at Northwell Health in New York. “Anecdotally, we understand that the devastation and injury caused by bullets far outweighs the injuries caused by other trauma mechanisms,” but the actual calculation of the burden hasn’t been studied.
Among 6615 firearm injuries, 9787 penetrating traumas and 66,003 blunt traumas examined from the National Inpatient Sample Healthcare Cost and Utilization Project Database, 11% of firearm traumas required a transfusion of red blood cells, compared with 1.4% of penetrating traumas and 3% of blunt traumas (P < .001). Patients with firearm injuries also had a longer length of stay — 10.8 days compared with 8.3 for patients with penetrating trauma and 9.8 for those with blunt trauma — and significantly higher rates of CPR, pericardiotomy, chest tube, exploratory laparotomy and/or thoracotomy, colorectal surgery, small bowel surgery, ostomy formation, splenectomy, hepatic resection, tracheostomy, and feeding tube placement.
Pulmonary complications were higher for firearm injuries (4.9%) than for penetrating trauma (0.6%) or blunt trauma (2.9%), and septicemia rates were also higher (1.7% vs 0.2% and 1%, respectively). Cardiac, neurologic, and urinary complications were also significantly and substantially higher for firearm injuries, 6.9% of which resulted in death compared with 0.2% of penetrating traumas and 1.2% of blunt traumas.
The costs from firearm injuries were also significantly higher than the costs from other traumas; “firearm injury remained independently predictive of greater hospital costs, even when controlling for injury severity as well as age, sex, race, insurance, region, hospital type, and household income.
“These findings underscore the urgent need for targeted prevention, supportive measures, and resource allocation to mitigate the devastating impact of firearm injuries on children and healthcare systems alike,” Dr. Nofi said.
The Colorado study was funded by the Colorado Department of Public Health and Environment and a National Institutes of Health grant to Dr. Haasz. The Texas study and the one from Northwell Health did not note any external funding. Dr. Haasz, Dr. Rosenbaum, Dr. Boonstra, and Dr. Nofi had no disclosures.
A version of this article appeared on Medscape.com.
From AAP 2024
FDA Approves Ustekinumab Biosimilar Otulfi
This is the fourth ustekinumab biosimilar approved in the United States. Like the reference product, ustekinumab-aauz is indicated for:
- Patients 6 years or older with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy
- Patients 6 years or older with active psoriatic arthritis
- Adult patients with moderately to severely active Crohn’s disease
- Adult patients with moderately to severely active ulcerative colitis
Ustekinumab-aauz, produced by a partnership between Fresenius Kabi and Formycon, has two formulations: subcutaneous injection (45 mg/0.5 mL or 90 mg/mL solution in a single-dose prefilled syringe) or intravenous infusion (130 mg/26 mL solution in a single-dose vial).
The biosimilar will launch in the United States “no later than February 22, 2025,” according to the press release, “in accordance with the patent settlement between Fresenius Kabi, Formycon, and Johnson & Johnson.”
Ustekinumab-aauz is Fresenius Kabi’s fourth biosimilar granted US approval, behind adalimumab-aacf (Idacio), tocilizumab-aazg (Tyenne), and pegfilgrastim-fpgk (Stimufend).
A version of this article first appeared on Medscape.com.
This is the fourth ustekinumab biosimilar approved in the United States. Like the reference product, ustekinumab-aauz is indicated for:
- Patients 6 years or older with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy
- Patients 6 years or older with active psoriatic arthritis
- Adult patients with moderately to severely active Crohn’s disease
- Adult patients with moderately to severely active ulcerative colitis
Ustekinumab-aauz, produced by a partnership between Fresenius Kabi and Formycon, has two formulations: subcutaneous injection (45 mg/0.5 mL or 90 mg/mL solution in a single-dose prefilled syringe) or intravenous infusion (130 mg/26 mL solution in a single-dose vial).
The biosimilar will launch in the United States “no later than February 22, 2025,” according to the press release, “in accordance with the patent settlement between Fresenius Kabi, Formycon, and Johnson & Johnson.”
Ustekinumab-aauz is Fresenius Kabi’s fourth biosimilar granted US approval, behind adalimumab-aacf (Idacio), tocilizumab-aazg (Tyenne), and pegfilgrastim-fpgk (Stimufend).
A version of this article first appeared on Medscape.com.
This is the fourth ustekinumab biosimilar approved in the United States. Like the reference product, ustekinumab-aauz is indicated for:
- Patients 6 years or older with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy
- Patients 6 years or older with active psoriatic arthritis
- Adult patients with moderately to severely active Crohn’s disease
- Adult patients with moderately to severely active ulcerative colitis
Ustekinumab-aauz, produced by a partnership between Fresenius Kabi and Formycon, has two formulations: subcutaneous injection (45 mg/0.5 mL or 90 mg/mL solution in a single-dose prefilled syringe) or intravenous infusion (130 mg/26 mL solution in a single-dose vial).
The biosimilar will launch in the United States “no later than February 22, 2025,” according to the press release, “in accordance with the patent settlement between Fresenius Kabi, Formycon, and Johnson & Johnson.”
Ustekinumab-aauz is Fresenius Kabi’s fourth biosimilar granted US approval, behind adalimumab-aacf (Idacio), tocilizumab-aazg (Tyenne), and pegfilgrastim-fpgk (Stimufend).
A version of this article first appeared on Medscape.com.
Time to Revisit the Standard Treatment Approach in Children With MS?
COPENHAGEN — However, only few of these medications are licensed for pediatric use, indicating it may be time to reconsider the standard treatment approach for this patient population.
Treatments for pediatric-onset MS have mostly been used off-label until the recent approvals of fingolimod, dimethyl fumarate, and teriflunomide. Typically, children with MS start with moderately effective therapies, while more potent options are reserved for those who don’t respond.
However, recent research suggests this may not be the most effective treatment strategy for this patient population. Several studies suggesting impressive treatment responses to highly effective therapies (HETs) in children were presented at the 2024 ECTRIMS annual meeting.
In one study, initiating monoclonal antibody treatment during childhood was associated with reduced disability into early adulthood and beyond.
“Our findings are a strong argument for rethinking current treatment guidelines,” said study investigator Sifat Sharmin, PhD, The University of Melbourne, Australia.
“By allowing earlier access to highly effective treatments, we can significantly enhance the quality of life for children with MS and reduce the burden of long-term disability,” she added.
In another presentation, Yael Hacohen, MD, Great Ormond Street Hospital, London, England, noted that the use of these more effective monoclonal antibody therapies in children with MS has been associated with some improvements in Expanded Disability Status Scale (EDSS) scores after 2 or 3 years of treatment.
Maybe this is a sign that “this is a population that can repair, in contrast to adult patients,” she wondered.
MS is primarily a disease of adults, but pediatric MS accounts for up to 5% of all cases. Children with MS tend to have much more active disease than adults, Dr. Hacohen explained. However, they also tend to recover from attacks more quickly with little disability, which sometimes causes diagnostic delays.
A pediatrician or family doctor will often dismiss pins and needles or blurred vision that only lasts a couple of days and won’t send the patient for an MRI, she said. But on MRI, pediatric patients with MS often have multiple lesions, even though they may have had very few symptoms. The EDSS may not change very much, but there can still be significant brain atrophy.
Over the past 20 years, there’s been an explosion of new disease-modifying treatments for MS, but these high-efficacy treatments, such as antibody therapies, are often not prescribed until the patient reaches the age of 18 years, both Dr. Sharmin and Dr. Hacohen pointed out.
“We need to get some of these medications approved for use in children,” Dr. Hacohen said.
Slowed Disability
In her presentation, Dr. Sharmin reported an observational study that included 282 patients younger than 18 years at MS onset identified from the French MS Registry, the Italian MS Register, and the Global MSBase Registry.
Of these, 110 (39%) had initiated therapy with ocrelizumab, rituximab, or natalizumab early in the disease course between ages 12 and 17 years and 172 (61%) had initiated treatment with one of these agents at ages 20-22 years.
The primary outcome was the difference in EDSS scores from baseline (at age 18 years) to ages 23-27 years between those who had started treatment with one of these agents early and those who had started late.
At the baseline of age 18 years, the median EDSS score was 1.5 in the early group and 1.3 in the late group. Median follow-up time was 10.8 years.
The data were adjusted for baseline differences in factors such as sex, age at symptom onset, time from onset to clinically definite MS, and the number of relapses (using inverse probability treatment weighting based on propensity scores).
Results showed that between ages 23 and 27 years, disability was a 0.57 step lower in the early group than in the late group. The mean absolute differences in EDSS from baseline were 0.40 in the early group and 0.95 in the late group. This benefit of early treatment persisted throughout the rest of the follow-up period.
The substantially lower risk of progressing to higher disability levels in the early treatment group was particularly evident in the moderate disability range, where further progression was reduced by up to 97%, Dr. Sharmin noted.
“Starting these highly effective therapies, before the onset of significant neurological impairments, appears crucial for preserving neurological function in children with relapsing-remitting MS over the long term,” she said.
These findings highlight the critical importance of early intervention in pediatric-onset MS, she concluded.
The researchers are planning further work to generate more evidence to support the proactive treatment of pediatric-onset MS, with a particular focus on assessing the long-term risks for immunosuppressive therapies in this population.
Ocrelizumab Experience in Children
Dr. Hacohen reported on a UK cohort of children with MS treated with ocrelizumab, with 66 patients having more than 12 months of follow-up. Of these, only four patients had relapses, and there was no evidence of disease activity in 94% patients.
“We’ve stopped doing relapse clinic because they really don’t relapse,” Dr. Hacohen reported.
“This has completely changed our practice in pediatric MS,” she said. Twice a year, patients come in to have pre-infusion bloods and clinical assessments and then return a month later for treatment.
“They only have to come to the hospital for 4 days a year, and the rest of the time, they can forget they have MS,” said Dr. Hacohen.
In terms of complications, one patient in the UK cohort developed enterovirus meningitis but recovered completely, and two patients had hypogammaglobulinemia and were changed to an extended interval or to a different agent.
Dr. Hacohen cautioned that hypogammaglobulinemia — a condition in which immunoglobulin levels are below normal — is “something that hypothetically we should maybe be more worried about in the pediatric population, particularly as these patients are more likely to be on anti-CD20 therapies for a much longer time.”
She said this complication tends to happen after about 4 or 5 years of treatment. “If we start seeing IgG levels dropping, we need to come up with a plan about extending the dosing interval. We need clinical trials to look at this.”
Dr. Hacohen also drew attention to the issue of vaccinations not being effective in patients on anti-CD20 antibody therapy, which could be a particular problem in children.
However, given that vaccinations do seem to be effective in patients taking natalizumab, pediatric patients with highly active disease could receive the drug for 3-6 months while receiving vaccines and then switched over to ocrelizumab, she said.
Giving natalizumab for such a short period is not believed to have a high risk of developing JCV antibodies, she added.
In another presentation, Brenda Banwell, MD, Johns Hopkins Children’s Center, Baltimore, reported new data from an early study (OPERETTA 1) with ocrelizumab in pediatric relapsing-remitting MS showing a safety profile similar to that observed in adults. The suggested dose is 300 mg for children under 35 kg and 600 mg for adults over 35 kg, administered every 24 weeks. These doses will be further investigated in the ongoing phase III OPERETTA 2 trial.
Dr. Sharmin received a postdoctoral fellowship from MS Australia. The OPERETTA studies were sponsored by F. Hoffmann-La Roche. Dr. Banwell served as a consultant to Roche. Dr. Hacohen reported no relevant disclosures.
A version of this article first appeared on Medscape.com.
COPENHAGEN — However, only few of these medications are licensed for pediatric use, indicating it may be time to reconsider the standard treatment approach for this patient population.
Treatments for pediatric-onset MS have mostly been used off-label until the recent approvals of fingolimod, dimethyl fumarate, and teriflunomide. Typically, children with MS start with moderately effective therapies, while more potent options are reserved for those who don’t respond.
However, recent research suggests this may not be the most effective treatment strategy for this patient population. Several studies suggesting impressive treatment responses to highly effective therapies (HETs) in children were presented at the 2024 ECTRIMS annual meeting.
In one study, initiating monoclonal antibody treatment during childhood was associated with reduced disability into early adulthood and beyond.
“Our findings are a strong argument for rethinking current treatment guidelines,” said study investigator Sifat Sharmin, PhD, The University of Melbourne, Australia.
“By allowing earlier access to highly effective treatments, we can significantly enhance the quality of life for children with MS and reduce the burden of long-term disability,” she added.
In another presentation, Yael Hacohen, MD, Great Ormond Street Hospital, London, England, noted that the use of these more effective monoclonal antibody therapies in children with MS has been associated with some improvements in Expanded Disability Status Scale (EDSS) scores after 2 or 3 years of treatment.
Maybe this is a sign that “this is a population that can repair, in contrast to adult patients,” she wondered.
MS is primarily a disease of adults, but pediatric MS accounts for up to 5% of all cases. Children with MS tend to have much more active disease than adults, Dr. Hacohen explained. However, they also tend to recover from attacks more quickly with little disability, which sometimes causes diagnostic delays.
A pediatrician or family doctor will often dismiss pins and needles or blurred vision that only lasts a couple of days and won’t send the patient for an MRI, she said. But on MRI, pediatric patients with MS often have multiple lesions, even though they may have had very few symptoms. The EDSS may not change very much, but there can still be significant brain atrophy.
Over the past 20 years, there’s been an explosion of new disease-modifying treatments for MS, but these high-efficacy treatments, such as antibody therapies, are often not prescribed until the patient reaches the age of 18 years, both Dr. Sharmin and Dr. Hacohen pointed out.
“We need to get some of these medications approved for use in children,” Dr. Hacohen said.
Slowed Disability
In her presentation, Dr. Sharmin reported an observational study that included 282 patients younger than 18 years at MS onset identified from the French MS Registry, the Italian MS Register, and the Global MSBase Registry.
Of these, 110 (39%) had initiated therapy with ocrelizumab, rituximab, or natalizumab early in the disease course between ages 12 and 17 years and 172 (61%) had initiated treatment with one of these agents at ages 20-22 years.
The primary outcome was the difference in EDSS scores from baseline (at age 18 years) to ages 23-27 years between those who had started treatment with one of these agents early and those who had started late.
At the baseline of age 18 years, the median EDSS score was 1.5 in the early group and 1.3 in the late group. Median follow-up time was 10.8 years.
The data were adjusted for baseline differences in factors such as sex, age at symptom onset, time from onset to clinically definite MS, and the number of relapses (using inverse probability treatment weighting based on propensity scores).
Results showed that between ages 23 and 27 years, disability was a 0.57 step lower in the early group than in the late group. The mean absolute differences in EDSS from baseline were 0.40 in the early group and 0.95 in the late group. This benefit of early treatment persisted throughout the rest of the follow-up period.
The substantially lower risk of progressing to higher disability levels in the early treatment group was particularly evident in the moderate disability range, where further progression was reduced by up to 97%, Dr. Sharmin noted.
“Starting these highly effective therapies, before the onset of significant neurological impairments, appears crucial for preserving neurological function in children with relapsing-remitting MS over the long term,” she said.
These findings highlight the critical importance of early intervention in pediatric-onset MS, she concluded.
The researchers are planning further work to generate more evidence to support the proactive treatment of pediatric-onset MS, with a particular focus on assessing the long-term risks for immunosuppressive therapies in this population.
Ocrelizumab Experience in Children
Dr. Hacohen reported on a UK cohort of children with MS treated with ocrelizumab, with 66 patients having more than 12 months of follow-up. Of these, only four patients had relapses, and there was no evidence of disease activity in 94% patients.
“We’ve stopped doing relapse clinic because they really don’t relapse,” Dr. Hacohen reported.
“This has completely changed our practice in pediatric MS,” she said. Twice a year, patients come in to have pre-infusion bloods and clinical assessments and then return a month later for treatment.
“They only have to come to the hospital for 4 days a year, and the rest of the time, they can forget they have MS,” said Dr. Hacohen.
In terms of complications, one patient in the UK cohort developed enterovirus meningitis but recovered completely, and two patients had hypogammaglobulinemia and were changed to an extended interval or to a different agent.
Dr. Hacohen cautioned that hypogammaglobulinemia — a condition in which immunoglobulin levels are below normal — is “something that hypothetically we should maybe be more worried about in the pediatric population, particularly as these patients are more likely to be on anti-CD20 therapies for a much longer time.”
She said this complication tends to happen after about 4 or 5 years of treatment. “If we start seeing IgG levels dropping, we need to come up with a plan about extending the dosing interval. We need clinical trials to look at this.”
Dr. Hacohen also drew attention to the issue of vaccinations not being effective in patients on anti-CD20 antibody therapy, which could be a particular problem in children.
However, given that vaccinations do seem to be effective in patients taking natalizumab, pediatric patients with highly active disease could receive the drug for 3-6 months while receiving vaccines and then switched over to ocrelizumab, she said.
Giving natalizumab for such a short period is not believed to have a high risk of developing JCV antibodies, she added.
In another presentation, Brenda Banwell, MD, Johns Hopkins Children’s Center, Baltimore, reported new data from an early study (OPERETTA 1) with ocrelizumab in pediatric relapsing-remitting MS showing a safety profile similar to that observed in adults. The suggested dose is 300 mg for children under 35 kg and 600 mg for adults over 35 kg, administered every 24 weeks. These doses will be further investigated in the ongoing phase III OPERETTA 2 trial.
Dr. Sharmin received a postdoctoral fellowship from MS Australia. The OPERETTA studies were sponsored by F. Hoffmann-La Roche. Dr. Banwell served as a consultant to Roche. Dr. Hacohen reported no relevant disclosures.
A version of this article first appeared on Medscape.com.
COPENHAGEN — However, only few of these medications are licensed for pediatric use, indicating it may be time to reconsider the standard treatment approach for this patient population.
Treatments for pediatric-onset MS have mostly been used off-label until the recent approvals of fingolimod, dimethyl fumarate, and teriflunomide. Typically, children with MS start with moderately effective therapies, while more potent options are reserved for those who don’t respond.
However, recent research suggests this may not be the most effective treatment strategy for this patient population. Several studies suggesting impressive treatment responses to highly effective therapies (HETs) in children were presented at the 2024 ECTRIMS annual meeting.
In one study, initiating monoclonal antibody treatment during childhood was associated with reduced disability into early adulthood and beyond.
“Our findings are a strong argument for rethinking current treatment guidelines,” said study investigator Sifat Sharmin, PhD, The University of Melbourne, Australia.
“By allowing earlier access to highly effective treatments, we can significantly enhance the quality of life for children with MS and reduce the burden of long-term disability,” she added.
In another presentation, Yael Hacohen, MD, Great Ormond Street Hospital, London, England, noted that the use of these more effective monoclonal antibody therapies in children with MS has been associated with some improvements in Expanded Disability Status Scale (EDSS) scores after 2 or 3 years of treatment.
Maybe this is a sign that “this is a population that can repair, in contrast to adult patients,” she wondered.
MS is primarily a disease of adults, but pediatric MS accounts for up to 5% of all cases. Children with MS tend to have much more active disease than adults, Dr. Hacohen explained. However, they also tend to recover from attacks more quickly with little disability, which sometimes causes diagnostic delays.
A pediatrician or family doctor will often dismiss pins and needles or blurred vision that only lasts a couple of days and won’t send the patient for an MRI, she said. But on MRI, pediatric patients with MS often have multiple lesions, even though they may have had very few symptoms. The EDSS may not change very much, but there can still be significant brain atrophy.
Over the past 20 years, there’s been an explosion of new disease-modifying treatments for MS, but these high-efficacy treatments, such as antibody therapies, are often not prescribed until the patient reaches the age of 18 years, both Dr. Sharmin and Dr. Hacohen pointed out.
“We need to get some of these medications approved for use in children,” Dr. Hacohen said.
Slowed Disability
In her presentation, Dr. Sharmin reported an observational study that included 282 patients younger than 18 years at MS onset identified from the French MS Registry, the Italian MS Register, and the Global MSBase Registry.
Of these, 110 (39%) had initiated therapy with ocrelizumab, rituximab, or natalizumab early in the disease course between ages 12 and 17 years and 172 (61%) had initiated treatment with one of these agents at ages 20-22 years.
The primary outcome was the difference in EDSS scores from baseline (at age 18 years) to ages 23-27 years between those who had started treatment with one of these agents early and those who had started late.
At the baseline of age 18 years, the median EDSS score was 1.5 in the early group and 1.3 in the late group. Median follow-up time was 10.8 years.
The data were adjusted for baseline differences in factors such as sex, age at symptom onset, time from onset to clinically definite MS, and the number of relapses (using inverse probability treatment weighting based on propensity scores).
Results showed that between ages 23 and 27 years, disability was a 0.57 step lower in the early group than in the late group. The mean absolute differences in EDSS from baseline were 0.40 in the early group and 0.95 in the late group. This benefit of early treatment persisted throughout the rest of the follow-up period.
The substantially lower risk of progressing to higher disability levels in the early treatment group was particularly evident in the moderate disability range, where further progression was reduced by up to 97%, Dr. Sharmin noted.
“Starting these highly effective therapies, before the onset of significant neurological impairments, appears crucial for preserving neurological function in children with relapsing-remitting MS over the long term,” she said.
These findings highlight the critical importance of early intervention in pediatric-onset MS, she concluded.
The researchers are planning further work to generate more evidence to support the proactive treatment of pediatric-onset MS, with a particular focus on assessing the long-term risks for immunosuppressive therapies in this population.
Ocrelizumab Experience in Children
Dr. Hacohen reported on a UK cohort of children with MS treated with ocrelizumab, with 66 patients having more than 12 months of follow-up. Of these, only four patients had relapses, and there was no evidence of disease activity in 94% patients.
“We’ve stopped doing relapse clinic because they really don’t relapse,” Dr. Hacohen reported.
“This has completely changed our practice in pediatric MS,” she said. Twice a year, patients come in to have pre-infusion bloods and clinical assessments and then return a month later for treatment.
“They only have to come to the hospital for 4 days a year, and the rest of the time, they can forget they have MS,” said Dr. Hacohen.
In terms of complications, one patient in the UK cohort developed enterovirus meningitis but recovered completely, and two patients had hypogammaglobulinemia and were changed to an extended interval or to a different agent.
Dr. Hacohen cautioned that hypogammaglobulinemia — a condition in which immunoglobulin levels are below normal — is “something that hypothetically we should maybe be more worried about in the pediatric population, particularly as these patients are more likely to be on anti-CD20 therapies for a much longer time.”
She said this complication tends to happen after about 4 or 5 years of treatment. “If we start seeing IgG levels dropping, we need to come up with a plan about extending the dosing interval. We need clinical trials to look at this.”
Dr. Hacohen also drew attention to the issue of vaccinations not being effective in patients on anti-CD20 antibody therapy, which could be a particular problem in children.
However, given that vaccinations do seem to be effective in patients taking natalizumab, pediatric patients with highly active disease could receive the drug for 3-6 months while receiving vaccines and then switched over to ocrelizumab, she said.
Giving natalizumab for such a short period is not believed to have a high risk of developing JCV antibodies, she added.
In another presentation, Brenda Banwell, MD, Johns Hopkins Children’s Center, Baltimore, reported new data from an early study (OPERETTA 1) with ocrelizumab in pediatric relapsing-remitting MS showing a safety profile similar to that observed in adults. The suggested dose is 300 mg for children under 35 kg and 600 mg for adults over 35 kg, administered every 24 weeks. These doses will be further investigated in the ongoing phase III OPERETTA 2 trial.
Dr. Sharmin received a postdoctoral fellowship from MS Australia. The OPERETTA studies were sponsored by F. Hoffmann-La Roche. Dr. Banwell served as a consultant to Roche. Dr. Hacohen reported no relevant disclosures.
A version of this article first appeared on Medscape.com.
FROM ECTRIMS 2024
Investigational Med for Tourette Syndrome Promising
PHILADELPHIA — , results of a new analysis suggest.
As previously reported, the first-in-class dopamine-1 (D1) receptor antagonist reduced the primary endpoint of tic severity scores by 30% compared with placebo among 149 patients in the 12-week, phase 2b D1AMOND trial.
What was unknown, however, is whether ecopipam would affect the comorbidities of attention-deficit/hyperactivity disorder (ADHD), anxiety, obsessive-compulsive disorder (OCD), and depression that were present in two thirds of participants.
The two key findings in this post hoc analysis were “first, that patients with a nonmotor diagnosis like depression or ADHD did not do any worse in terms of tic efficacy; and second, we didn’t find any evidence that any of the nonmotor symptoms of Tourette’s got worse with ecopipam,” said study investigator Donald Gilbert, MD, professor of pediatrics and neurology at University of Cincinnati Children’s Hospital Medical Center.
Dr. Gilbert presented the results at the International Congress of Parkinson’s Disease and Movement Disorders (MDS) 2024.
No Worsening of ADHD Symptoms
Tourette syndrome affects approximately 1 in 160 children between 5 and 17 years of age in the United States, data from the Tourette Association of America show. Research has shown that 85% of patients with Tourette syndrome will have a co-occurring psychiatric condition.
Guidelines recommend Comprehensive Behavioral Intervention for Tics (CBIT) as first-line treatment for Tourette syndrome, but cost and access are barriers. The only currently approved medications to treat Tourette syndrome are antipsychotics that act on the D2 receptor, but their use is limited by the potential for weight gain, metabolic changes, drug-induced movement disorders, and risk for suicidality, said Dr. Gilbert.
The D1AMOND study randomly assigned patients aged 6-17 years with Tourette syndrome and a Yale Global Tic Severity Total Tic Scale score of at least 20 to receive a target steady-state dose of 2 mg/kg/d of oral ecopipam or placebo for a 4-week titration period, followed by an 8-week treatment phase before being tapered off the study drug.
Patients were allowed to remain on medications without D2-receptor blocking activity for anxiety, OCD, and ADHD if the dosage was stable for 4 weeks before screening and not specifically prescribed for tics.
A mixed model for repeated measures was used to assess changes in several scales administered at baseline and at weeks 4, 6, 8, and 12: the Swanson, Nolan, and Pelham Teacher and Parent Rating Scale (SNAP-IV); Pediatric Anxiety Rating Scale; Children’s Yale-Brown Obsessive-Compulsive Scale (CY-BOCS), and Children’s Depression Rating Scale–Revised (CDRS-R).
In patients with a co-occurring psychiatric condition, no significant differences were found over time between ecopipam and placebo in terms of SNAP-IV (-4.4; P = .45), Pediatric Anxiety Rating Scale (1.0; P = .62), CDRS-R (-3.2; P = .65), or CY-BOCS (-0.7; P = .76) scores.
For ADHD, the most frequent comorbidity, scores trended lower in the ecopipam group but were not significantly different from those in the placebo group. “We found no evidence that ecopipam worsened ADHD symptoms,” Dr. Gilbert said.
No Weight Gain
Suicidal ideation was reported during the dosing period in eight patients in the placebo group and none in the ecopipam group. One patient treated with ecopipam had multiple depressive episodes and dropped out of the study on day 79. Ecopipam was discontinued in another patient because of anxiety.
Notably, there was more weight gain in the placebo group than in the ecopipam group (2.4 kg vs 1.8 kg) by 12 weeks. No shifts from baseline were seen in blood glucose, A1c, total cholesterol, or triglycerides in either group.
The lack of weight gain with ecopipam is important, Dr. Gilbert stressed. “Medicines that block D2 so often cause weight gain, and a lot of our patients, unfortunately, can be heavier already,” he explained. “We don’t want to make that worse or put them at a long-term risk of type 2 diabetes.”
For patients with more severe disease, we really “do need something else besides D2-blockers in our tool kit,” he added.
Commenting on the study, Tanya Simuni, MD, co-moderator of the session and director of the Parkinson’s Disease and Movement Disorders Center, Northwestern Feinberg School of Medicine, Chicago, said the aim of assessing D1-directed medications is to reduce the negative impact of traditional antipsychotics with a theoretical benefit on hyperkinetic movement.
But the most important thing that they’ve shown is that “there was no negative effect, no liability for the nonmotor manifestations of Tourette’s. That is important because Tourette’s is not a pure motor syndrome, and psychiatric manifestations in a lot of cases are associated with more disease-related quality of life impairment compared to the motor manifestations,” said Dr. Simuni.
That said, she noted, the “ideal drug would be the one that would have benefit for both motor and nonmotor domains.”
Multiple Agents in the Pipeline
“The neuropharmacology of Tourette syndrome has long remained stagnant, and most existing treatments often fail to balance efficacy with tolerability, underscoring the urgent need for newer therapeutics,” Christos Ganos, MD, professor of neurology, University of Toronto, said in a press release.
He noted that three studies have been published on ecopipam since 2014: an 8-week, open-label trial in adults with Tourette syndrome, a 4-week, placebo-controlled crossover trial in 38 children with Tourette syndrome, and the 12-week D1AMOND trial.
“These studies demonstrated clinically meaningful reductions in tics, without relevant safety concerns or changes in Tourette syndrome-typical neuropsychiatric measures, as also shown by the abstract highlighted here,” Dr. Ganos said.
“This emerging body of research provides a solid foundation for introducing ecopipam as a novel pharmacological agent to treat tics and may motivate further work, both on the pathophysiology and pharmacotherapy of tic disorders and their associations.”
A single-arm, phase 3 trial is currently underway at 58 centers in North America and Europe investigating the long-term safety and tolerability of ecopipam over 24 months in 150 children, adolescents, and adults with Tourette syndrome. The study is expected to be completed in 2027.
Several other new medications are also under investigation including the vesicular monoamine transporter (VMAT2) inhibitors tetrabenazine, deutetrabenazine, and valbenazine; the PEDE10A inhibitor gemlapodect; the allopregnanolone antagonist sepranolone; and SCI-110, which combines dronabinol (the synthetic form of tetrahydrocannabinol) and the endocannabinoid palmitoylethanolamide.
The study was funded by Emalex Biosciences. Dr. Gilbert’s institution received research support from Emalex Biosciences and PTC Therapeutics. Dr. Gilbert has received publishing royalties from a healthcare-related publication; compensation for serving as a medical expert with Teladoc; Advanced Medical; and the National Vaccine Injury Compensation Program, US Department of Health and Human Services. Simuni reports no relevant conflicts of interest. Dr. Ganos has received honoraria for educational activities from the Movement Disorder Society and academic research support from VolkswagenStiftung.
A version of this article first appeared on Medscape.com.
PHILADELPHIA — , results of a new analysis suggest.
As previously reported, the first-in-class dopamine-1 (D1) receptor antagonist reduced the primary endpoint of tic severity scores by 30% compared with placebo among 149 patients in the 12-week, phase 2b D1AMOND trial.
What was unknown, however, is whether ecopipam would affect the comorbidities of attention-deficit/hyperactivity disorder (ADHD), anxiety, obsessive-compulsive disorder (OCD), and depression that were present in two thirds of participants.
The two key findings in this post hoc analysis were “first, that patients with a nonmotor diagnosis like depression or ADHD did not do any worse in terms of tic efficacy; and second, we didn’t find any evidence that any of the nonmotor symptoms of Tourette’s got worse with ecopipam,” said study investigator Donald Gilbert, MD, professor of pediatrics and neurology at University of Cincinnati Children’s Hospital Medical Center.
Dr. Gilbert presented the results at the International Congress of Parkinson’s Disease and Movement Disorders (MDS) 2024.
No Worsening of ADHD Symptoms
Tourette syndrome affects approximately 1 in 160 children between 5 and 17 years of age in the United States, data from the Tourette Association of America show. Research has shown that 85% of patients with Tourette syndrome will have a co-occurring psychiatric condition.
Guidelines recommend Comprehensive Behavioral Intervention for Tics (CBIT) as first-line treatment for Tourette syndrome, but cost and access are barriers. The only currently approved medications to treat Tourette syndrome are antipsychotics that act on the D2 receptor, but their use is limited by the potential for weight gain, metabolic changes, drug-induced movement disorders, and risk for suicidality, said Dr. Gilbert.
The D1AMOND study randomly assigned patients aged 6-17 years with Tourette syndrome and a Yale Global Tic Severity Total Tic Scale score of at least 20 to receive a target steady-state dose of 2 mg/kg/d of oral ecopipam or placebo for a 4-week titration period, followed by an 8-week treatment phase before being tapered off the study drug.
Patients were allowed to remain on medications without D2-receptor blocking activity for anxiety, OCD, and ADHD if the dosage was stable for 4 weeks before screening and not specifically prescribed for tics.
A mixed model for repeated measures was used to assess changes in several scales administered at baseline and at weeks 4, 6, 8, and 12: the Swanson, Nolan, and Pelham Teacher and Parent Rating Scale (SNAP-IV); Pediatric Anxiety Rating Scale; Children’s Yale-Brown Obsessive-Compulsive Scale (CY-BOCS), and Children’s Depression Rating Scale–Revised (CDRS-R).
In patients with a co-occurring psychiatric condition, no significant differences were found over time between ecopipam and placebo in terms of SNAP-IV (-4.4; P = .45), Pediatric Anxiety Rating Scale (1.0; P = .62), CDRS-R (-3.2; P = .65), or CY-BOCS (-0.7; P = .76) scores.
For ADHD, the most frequent comorbidity, scores trended lower in the ecopipam group but were not significantly different from those in the placebo group. “We found no evidence that ecopipam worsened ADHD symptoms,” Dr. Gilbert said.
No Weight Gain
Suicidal ideation was reported during the dosing period in eight patients in the placebo group and none in the ecopipam group. One patient treated with ecopipam had multiple depressive episodes and dropped out of the study on day 79. Ecopipam was discontinued in another patient because of anxiety.
Notably, there was more weight gain in the placebo group than in the ecopipam group (2.4 kg vs 1.8 kg) by 12 weeks. No shifts from baseline were seen in blood glucose, A1c, total cholesterol, or triglycerides in either group.
The lack of weight gain with ecopipam is important, Dr. Gilbert stressed. “Medicines that block D2 so often cause weight gain, and a lot of our patients, unfortunately, can be heavier already,” he explained. “We don’t want to make that worse or put them at a long-term risk of type 2 diabetes.”
For patients with more severe disease, we really “do need something else besides D2-blockers in our tool kit,” he added.
Commenting on the study, Tanya Simuni, MD, co-moderator of the session and director of the Parkinson’s Disease and Movement Disorders Center, Northwestern Feinberg School of Medicine, Chicago, said the aim of assessing D1-directed medications is to reduce the negative impact of traditional antipsychotics with a theoretical benefit on hyperkinetic movement.
But the most important thing that they’ve shown is that “there was no negative effect, no liability for the nonmotor manifestations of Tourette’s. That is important because Tourette’s is not a pure motor syndrome, and psychiatric manifestations in a lot of cases are associated with more disease-related quality of life impairment compared to the motor manifestations,” said Dr. Simuni.
That said, she noted, the “ideal drug would be the one that would have benefit for both motor and nonmotor domains.”
Multiple Agents in the Pipeline
“The neuropharmacology of Tourette syndrome has long remained stagnant, and most existing treatments often fail to balance efficacy with tolerability, underscoring the urgent need for newer therapeutics,” Christos Ganos, MD, professor of neurology, University of Toronto, said in a press release.
He noted that three studies have been published on ecopipam since 2014: an 8-week, open-label trial in adults with Tourette syndrome, a 4-week, placebo-controlled crossover trial in 38 children with Tourette syndrome, and the 12-week D1AMOND trial.
“These studies demonstrated clinically meaningful reductions in tics, without relevant safety concerns or changes in Tourette syndrome-typical neuropsychiatric measures, as also shown by the abstract highlighted here,” Dr. Ganos said.
“This emerging body of research provides a solid foundation for introducing ecopipam as a novel pharmacological agent to treat tics and may motivate further work, both on the pathophysiology and pharmacotherapy of tic disorders and their associations.”
A single-arm, phase 3 trial is currently underway at 58 centers in North America and Europe investigating the long-term safety and tolerability of ecopipam over 24 months in 150 children, adolescents, and adults with Tourette syndrome. The study is expected to be completed in 2027.
Several other new medications are also under investigation including the vesicular monoamine transporter (VMAT2) inhibitors tetrabenazine, deutetrabenazine, and valbenazine; the PEDE10A inhibitor gemlapodect; the allopregnanolone antagonist sepranolone; and SCI-110, which combines dronabinol (the synthetic form of tetrahydrocannabinol) and the endocannabinoid palmitoylethanolamide.
The study was funded by Emalex Biosciences. Dr. Gilbert’s institution received research support from Emalex Biosciences and PTC Therapeutics. Dr. Gilbert has received publishing royalties from a healthcare-related publication; compensation for serving as a medical expert with Teladoc; Advanced Medical; and the National Vaccine Injury Compensation Program, US Department of Health and Human Services. Simuni reports no relevant conflicts of interest. Dr. Ganos has received honoraria for educational activities from the Movement Disorder Society and academic research support from VolkswagenStiftung.
A version of this article first appeared on Medscape.com.
PHILADELPHIA — , results of a new analysis suggest.
As previously reported, the first-in-class dopamine-1 (D1) receptor antagonist reduced the primary endpoint of tic severity scores by 30% compared with placebo among 149 patients in the 12-week, phase 2b D1AMOND trial.
What was unknown, however, is whether ecopipam would affect the comorbidities of attention-deficit/hyperactivity disorder (ADHD), anxiety, obsessive-compulsive disorder (OCD), and depression that were present in two thirds of participants.
The two key findings in this post hoc analysis were “first, that patients with a nonmotor diagnosis like depression or ADHD did not do any worse in terms of tic efficacy; and second, we didn’t find any evidence that any of the nonmotor symptoms of Tourette’s got worse with ecopipam,” said study investigator Donald Gilbert, MD, professor of pediatrics and neurology at University of Cincinnati Children’s Hospital Medical Center.
Dr. Gilbert presented the results at the International Congress of Parkinson’s Disease and Movement Disorders (MDS) 2024.
No Worsening of ADHD Symptoms
Tourette syndrome affects approximately 1 in 160 children between 5 and 17 years of age in the United States, data from the Tourette Association of America show. Research has shown that 85% of patients with Tourette syndrome will have a co-occurring psychiatric condition.
Guidelines recommend Comprehensive Behavioral Intervention for Tics (CBIT) as first-line treatment for Tourette syndrome, but cost and access are barriers. The only currently approved medications to treat Tourette syndrome are antipsychotics that act on the D2 receptor, but their use is limited by the potential for weight gain, metabolic changes, drug-induced movement disorders, and risk for suicidality, said Dr. Gilbert.
The D1AMOND study randomly assigned patients aged 6-17 years with Tourette syndrome and a Yale Global Tic Severity Total Tic Scale score of at least 20 to receive a target steady-state dose of 2 mg/kg/d of oral ecopipam or placebo for a 4-week titration period, followed by an 8-week treatment phase before being tapered off the study drug.
Patients were allowed to remain on medications without D2-receptor blocking activity for anxiety, OCD, and ADHD if the dosage was stable for 4 weeks before screening and not specifically prescribed for tics.
A mixed model for repeated measures was used to assess changes in several scales administered at baseline and at weeks 4, 6, 8, and 12: the Swanson, Nolan, and Pelham Teacher and Parent Rating Scale (SNAP-IV); Pediatric Anxiety Rating Scale; Children’s Yale-Brown Obsessive-Compulsive Scale (CY-BOCS), and Children’s Depression Rating Scale–Revised (CDRS-R).
In patients with a co-occurring psychiatric condition, no significant differences were found over time between ecopipam and placebo in terms of SNAP-IV (-4.4; P = .45), Pediatric Anxiety Rating Scale (1.0; P = .62), CDRS-R (-3.2; P = .65), or CY-BOCS (-0.7; P = .76) scores.
For ADHD, the most frequent comorbidity, scores trended lower in the ecopipam group but were not significantly different from those in the placebo group. “We found no evidence that ecopipam worsened ADHD symptoms,” Dr. Gilbert said.
No Weight Gain
Suicidal ideation was reported during the dosing period in eight patients in the placebo group and none in the ecopipam group. One patient treated with ecopipam had multiple depressive episodes and dropped out of the study on day 79. Ecopipam was discontinued in another patient because of anxiety.
Notably, there was more weight gain in the placebo group than in the ecopipam group (2.4 kg vs 1.8 kg) by 12 weeks. No shifts from baseline were seen in blood glucose, A1c, total cholesterol, or triglycerides in either group.
The lack of weight gain with ecopipam is important, Dr. Gilbert stressed. “Medicines that block D2 so often cause weight gain, and a lot of our patients, unfortunately, can be heavier already,” he explained. “We don’t want to make that worse or put them at a long-term risk of type 2 diabetes.”
For patients with more severe disease, we really “do need something else besides D2-blockers in our tool kit,” he added.
Commenting on the study, Tanya Simuni, MD, co-moderator of the session and director of the Parkinson’s Disease and Movement Disorders Center, Northwestern Feinberg School of Medicine, Chicago, said the aim of assessing D1-directed medications is to reduce the negative impact of traditional antipsychotics with a theoretical benefit on hyperkinetic movement.
But the most important thing that they’ve shown is that “there was no negative effect, no liability for the nonmotor manifestations of Tourette’s. That is important because Tourette’s is not a pure motor syndrome, and psychiatric manifestations in a lot of cases are associated with more disease-related quality of life impairment compared to the motor manifestations,” said Dr. Simuni.
That said, she noted, the “ideal drug would be the one that would have benefit for both motor and nonmotor domains.”
Multiple Agents in the Pipeline
“The neuropharmacology of Tourette syndrome has long remained stagnant, and most existing treatments often fail to balance efficacy with tolerability, underscoring the urgent need for newer therapeutics,” Christos Ganos, MD, professor of neurology, University of Toronto, said in a press release.
He noted that three studies have been published on ecopipam since 2014: an 8-week, open-label trial in adults with Tourette syndrome, a 4-week, placebo-controlled crossover trial in 38 children with Tourette syndrome, and the 12-week D1AMOND trial.
“These studies demonstrated clinically meaningful reductions in tics, without relevant safety concerns or changes in Tourette syndrome-typical neuropsychiatric measures, as also shown by the abstract highlighted here,” Dr. Ganos said.
“This emerging body of research provides a solid foundation for introducing ecopipam as a novel pharmacological agent to treat tics and may motivate further work, both on the pathophysiology and pharmacotherapy of tic disorders and their associations.”
A single-arm, phase 3 trial is currently underway at 58 centers in North America and Europe investigating the long-term safety and tolerability of ecopipam over 24 months in 150 children, adolescents, and adults with Tourette syndrome. The study is expected to be completed in 2027.
Several other new medications are also under investigation including the vesicular monoamine transporter (VMAT2) inhibitors tetrabenazine, deutetrabenazine, and valbenazine; the PEDE10A inhibitor gemlapodect; the allopregnanolone antagonist sepranolone; and SCI-110, which combines dronabinol (the synthetic form of tetrahydrocannabinol) and the endocannabinoid palmitoylethanolamide.
The study was funded by Emalex Biosciences. Dr. Gilbert’s institution received research support from Emalex Biosciences and PTC Therapeutics. Dr. Gilbert has received publishing royalties from a healthcare-related publication; compensation for serving as a medical expert with Teladoc; Advanced Medical; and the National Vaccine Injury Compensation Program, US Department of Health and Human Services. Simuni reports no relevant conflicts of interest. Dr. Ganos has received honoraria for educational activities from the Movement Disorder Society and academic research support from VolkswagenStiftung.
A version of this article first appeared on Medscape.com.
FROM MDS 2024
Pediatric Melanoma Outcomes by Race and Socioeconomic Factors
To the Editor:
Skin cancers are extremely common worldwide. Malignant melanomas comprise approximately 1 in 5 of these cancers. Exposure to UV radiation is postulated to be responsible for a global rise in melanoma cases over the past 50 years.1 Pediatric melanoma is a particularly rare condition that affects approximately 6 in every 1 million children.2 Melanoma incidence in children ranges by age, increasing by approximately 10-fold from age 1 to 4 years to age 15 to 19 years. Tumor ulceration is a feature more commonly seen among children younger than 10 years and is associated with worse outcomes. Tumor thickness and ulceration strongly predict sentinel lymph node metastases among children, which also is associated with a poor prognosis.3
A recent study evaluating stage IV melanoma survival rates in adolescents and young adults (AYAs) vs older adults found that survival is much worse among AYAs. Thicker tumors and public health insurance also were associated with worse survival rates for AYAs, while early detection was associated with better survival rates.4
Health disparities and their role in the prognosis of pediatric melanoma is another important factor. One study analyzed this relationship at the state level using Texas Cancer Registry data (1995-2009).5 Patients’ socioeconomic status (SES) and driving distance to the nearest pediatric cancer care center were included in the analysis. Hispanic children were found to be 3 times more likely to present with advanced disease than non-Hispanic White children. Although SES and distance to the nearest treatment center were not found to affect the melanoma stage at presentation, Hispanic ethnicity or being in the lowest SES quartile were correlated with a higher mortality risk.5
When considering specific subtypes of melanoma, acral lentiginous melanoma (ALM) is known to develop in patients with skin of color. A 2023 study by Holman et al6 reported that the percentage of melanomas that were ALMs ranged from 0.8% in non-Hispanic White individuals to 19.1% in Hispanic Black, American Indian/Alaska Native, and Asian/Pacific Islander individuals. However, ALM is rare in children. In a pooled cohort study with patient information retrieved from the nationwide Dutch Pathology Registry, only 1 child and 1 adolescent were found to have ALM across a total of 514 patients.7 We sought to analyze pediatric melanoma outcomes based on race and other barriers to appropriate care.
We conducted a search of the Surveillance, Epidemiology, and End Results (SEER) database from January 1995 to December 2016 for patients aged 21 years and younger with a primary melanoma diagnosis. The primary outcome was the 5-year survival rate. County-level SES variables were used to calculate a prosperity index. Kaplan-Meier analysis and Cox proportional hazards model were used to compare 5-year survival rates among the different racial/ethnic groups.
A sample of 2742 patients was identified during the study period and followed for 5 years. Eighty-two percent were White, 6% Hispanic, 2% Asian, 1% Black, and 5% classified as other/unknown race (data were missing for 4%). The cohort was predominantly female (61%). White patients were more likely to present with localized disease than any other race/ethnicity (83% vs 65% in Hispanic, 60% in Asian/Pacific Islander, and 45% in Black patients [P<.05]).
Black and Hispanic patients had the worst 5-year survival rates on bivariate analysis. On multivariate analysis, this finding remained significant for Hispanic patients when compared with White patients (hazard ratio, 2.37 [P<.05]). Increasing age, male sex, advanced stage at diagnosis, and failure to receive surgery were associated with increased odds of mortality.
Patients with regionalized and disseminated disease had increased odds of mortality (6.16 and 64.45, respectively; P<.05) compared with patients with localized disease. Socioeconomic status and urbanization were not found to influence 5-year survival rates.
Pediatric melanoma often presents a clinical challenge with special considerations. Pediatric-specific predisposing risk factors for melanoma and an atypical clinical presentation are some of the major concerns that necessitate a tailored approach to this malignancy, especially among different age groups, skin types, and racial and socioeconomic groups.5
Standard ABCDE criteria often are inadequate for accurate detection of pediatric melanomas. Initial lesions often manifest as raised, red, amelanotic lesions mimicking pyogenic granulomas. Lesions tend to be very small (<6 mm in diameter) and can be uniform in color, thereby making the melanoma more difficult to detect compared to the characteristic findings in adults.5 Bleeding or ulceration often can be a warning sign during physical examination.
With regard to incidence, pediatric melanoma is relatively rare. Since the 1970s, the incidence of pediatric melanoma has been increasing; however, a recent analysis of the SEER database showed a decreasing trend from 2000 to 2010.4
Our analysis of the SEER data showed an increased risk for pediatric melanoma in older adolescents. In addition, the incidence of pediatric melanoma was higher in females of all racial groups except Asian/Pacific Islander individuals. However, SES was not found to significantly influence the 5-year survival rate in pediatric melanoma.
White pediatric patients were more likely to present with localized disease compared with other races. Pediatric melanoma patients with regional disease had a 6-fold increase in mortality rate vs those with localized disease; those with disseminated disease had a 65-fold higher risk. Consistent with this, Black and Hispanic patients had the worst 5-year survival rates on bivariate analysis.
These findings suggest a relationship between race, melanoma spread, and disease severity. Patient education programs need to be directed specifically to minority groups to improve their knowledge on evolving skin lesions and sun protection practices. Physicians also need to have heightened suspicion and better knowledge of the unique traits of pediatric melanoma.5
Given the considerable influence these disparities can have on melanoma outcomes, further research is needed to characterize outcomes based on race and determine obstacles to appropriate care. Improved public outreach initiatives that accommodate specific cultural barriers (eg, language, traditional patterns of behavior) also are required to improve current circumstances.
- Arnold M, Singh D, Laversanne M, et al. Global burden of cutaneous melanoma in 2020 and projections to 2040. JAMA Dermatol. 2022;158:495-503.
- McCormack L, Hawryluk EB. Pediatric melanoma update. G Ital Dermatol Venereol. 2018;153:707-715.
- Saiyed FK, Hamilton EC, Austin MT. Pediatric melanoma: incidence, treatment, and prognosis. Pediatric Health Med Ther. 2017;8:39-45.
- Wojcik KY, Hawkins M, Anderson-Mellies A, et al. Melanoma survival by age group: population-based disparities for adolescent and young adult patients by stage, tumor thickness, and insurance type. J Am Acad Dermatol. 2023;88:831-840.
- Hamilton EC, Nguyen HT, Chang YC, et al. Health disparities influence childhood melanoma stage at diagnosis and outcome. J Pediatr. 2016;175:182-187.
- Holman DM, King JB, White A, et al. Acral lentiginous melanoma incidence by sex, race, ethnicity, and stage in the United States, 2010-2019. Prev Med. 2023;175:107692. doi:10.1016/j.ypmed.2023.107692
- El Sharouni MA, Rawson RV, Potter AJ, et al. Melanomas in children and adolescents: clinicopathologic features and survival outcomes. J Am Acad Dermatol. 2023;88:609-616. doi:10.1016/j.jaad.2022.08.067
To the Editor:
Skin cancers are extremely common worldwide. Malignant melanomas comprise approximately 1 in 5 of these cancers. Exposure to UV radiation is postulated to be responsible for a global rise in melanoma cases over the past 50 years.1 Pediatric melanoma is a particularly rare condition that affects approximately 6 in every 1 million children.2 Melanoma incidence in children ranges by age, increasing by approximately 10-fold from age 1 to 4 years to age 15 to 19 years. Tumor ulceration is a feature more commonly seen among children younger than 10 years and is associated with worse outcomes. Tumor thickness and ulceration strongly predict sentinel lymph node metastases among children, which also is associated with a poor prognosis.3
A recent study evaluating stage IV melanoma survival rates in adolescents and young adults (AYAs) vs older adults found that survival is much worse among AYAs. Thicker tumors and public health insurance also were associated with worse survival rates for AYAs, while early detection was associated with better survival rates.4
Health disparities and their role in the prognosis of pediatric melanoma is another important factor. One study analyzed this relationship at the state level using Texas Cancer Registry data (1995-2009).5 Patients’ socioeconomic status (SES) and driving distance to the nearest pediatric cancer care center were included in the analysis. Hispanic children were found to be 3 times more likely to present with advanced disease than non-Hispanic White children. Although SES and distance to the nearest treatment center were not found to affect the melanoma stage at presentation, Hispanic ethnicity or being in the lowest SES quartile were correlated with a higher mortality risk.5
When considering specific subtypes of melanoma, acral lentiginous melanoma (ALM) is known to develop in patients with skin of color. A 2023 study by Holman et al6 reported that the percentage of melanomas that were ALMs ranged from 0.8% in non-Hispanic White individuals to 19.1% in Hispanic Black, American Indian/Alaska Native, and Asian/Pacific Islander individuals. However, ALM is rare in children. In a pooled cohort study with patient information retrieved from the nationwide Dutch Pathology Registry, only 1 child and 1 adolescent were found to have ALM across a total of 514 patients.7 We sought to analyze pediatric melanoma outcomes based on race and other barriers to appropriate care.
We conducted a search of the Surveillance, Epidemiology, and End Results (SEER) database from January 1995 to December 2016 for patients aged 21 years and younger with a primary melanoma diagnosis. The primary outcome was the 5-year survival rate. County-level SES variables were used to calculate a prosperity index. Kaplan-Meier analysis and Cox proportional hazards model were used to compare 5-year survival rates among the different racial/ethnic groups.
A sample of 2742 patients was identified during the study period and followed for 5 years. Eighty-two percent were White, 6% Hispanic, 2% Asian, 1% Black, and 5% classified as other/unknown race (data were missing for 4%). The cohort was predominantly female (61%). White patients were more likely to present with localized disease than any other race/ethnicity (83% vs 65% in Hispanic, 60% in Asian/Pacific Islander, and 45% in Black patients [P<.05]).
Black and Hispanic patients had the worst 5-year survival rates on bivariate analysis. On multivariate analysis, this finding remained significant for Hispanic patients when compared with White patients (hazard ratio, 2.37 [P<.05]). Increasing age, male sex, advanced stage at diagnosis, and failure to receive surgery were associated with increased odds of mortality.
Patients with regionalized and disseminated disease had increased odds of mortality (6.16 and 64.45, respectively; P<.05) compared with patients with localized disease. Socioeconomic status and urbanization were not found to influence 5-year survival rates.
Pediatric melanoma often presents a clinical challenge with special considerations. Pediatric-specific predisposing risk factors for melanoma and an atypical clinical presentation are some of the major concerns that necessitate a tailored approach to this malignancy, especially among different age groups, skin types, and racial and socioeconomic groups.5
Standard ABCDE criteria often are inadequate for accurate detection of pediatric melanomas. Initial lesions often manifest as raised, red, amelanotic lesions mimicking pyogenic granulomas. Lesions tend to be very small (<6 mm in diameter) and can be uniform in color, thereby making the melanoma more difficult to detect compared to the characteristic findings in adults.5 Bleeding or ulceration often can be a warning sign during physical examination.
With regard to incidence, pediatric melanoma is relatively rare. Since the 1970s, the incidence of pediatric melanoma has been increasing; however, a recent analysis of the SEER database showed a decreasing trend from 2000 to 2010.4
Our analysis of the SEER data showed an increased risk for pediatric melanoma in older adolescents. In addition, the incidence of pediatric melanoma was higher in females of all racial groups except Asian/Pacific Islander individuals. However, SES was not found to significantly influence the 5-year survival rate in pediatric melanoma.
White pediatric patients were more likely to present with localized disease compared with other races. Pediatric melanoma patients with regional disease had a 6-fold increase in mortality rate vs those with localized disease; those with disseminated disease had a 65-fold higher risk. Consistent with this, Black and Hispanic patients had the worst 5-year survival rates on bivariate analysis.
These findings suggest a relationship between race, melanoma spread, and disease severity. Patient education programs need to be directed specifically to minority groups to improve their knowledge on evolving skin lesions and sun protection practices. Physicians also need to have heightened suspicion and better knowledge of the unique traits of pediatric melanoma.5
Given the considerable influence these disparities can have on melanoma outcomes, further research is needed to characterize outcomes based on race and determine obstacles to appropriate care. Improved public outreach initiatives that accommodate specific cultural barriers (eg, language, traditional patterns of behavior) also are required to improve current circumstances.
To the Editor:
Skin cancers are extremely common worldwide. Malignant melanomas comprise approximately 1 in 5 of these cancers. Exposure to UV radiation is postulated to be responsible for a global rise in melanoma cases over the past 50 years.1 Pediatric melanoma is a particularly rare condition that affects approximately 6 in every 1 million children.2 Melanoma incidence in children ranges by age, increasing by approximately 10-fold from age 1 to 4 years to age 15 to 19 years. Tumor ulceration is a feature more commonly seen among children younger than 10 years and is associated with worse outcomes. Tumor thickness and ulceration strongly predict sentinel lymph node metastases among children, which also is associated with a poor prognosis.3
A recent study evaluating stage IV melanoma survival rates in adolescents and young adults (AYAs) vs older adults found that survival is much worse among AYAs. Thicker tumors and public health insurance also were associated with worse survival rates for AYAs, while early detection was associated with better survival rates.4
Health disparities and their role in the prognosis of pediatric melanoma is another important factor. One study analyzed this relationship at the state level using Texas Cancer Registry data (1995-2009).5 Patients’ socioeconomic status (SES) and driving distance to the nearest pediatric cancer care center were included in the analysis. Hispanic children were found to be 3 times more likely to present with advanced disease than non-Hispanic White children. Although SES and distance to the nearest treatment center were not found to affect the melanoma stage at presentation, Hispanic ethnicity or being in the lowest SES quartile were correlated with a higher mortality risk.5
When considering specific subtypes of melanoma, acral lentiginous melanoma (ALM) is known to develop in patients with skin of color. A 2023 study by Holman et al6 reported that the percentage of melanomas that were ALMs ranged from 0.8% in non-Hispanic White individuals to 19.1% in Hispanic Black, American Indian/Alaska Native, and Asian/Pacific Islander individuals. However, ALM is rare in children. In a pooled cohort study with patient information retrieved from the nationwide Dutch Pathology Registry, only 1 child and 1 adolescent were found to have ALM across a total of 514 patients.7 We sought to analyze pediatric melanoma outcomes based on race and other barriers to appropriate care.
We conducted a search of the Surveillance, Epidemiology, and End Results (SEER) database from January 1995 to December 2016 for patients aged 21 years and younger with a primary melanoma diagnosis. The primary outcome was the 5-year survival rate. County-level SES variables were used to calculate a prosperity index. Kaplan-Meier analysis and Cox proportional hazards model were used to compare 5-year survival rates among the different racial/ethnic groups.
A sample of 2742 patients was identified during the study period and followed for 5 years. Eighty-two percent were White, 6% Hispanic, 2% Asian, 1% Black, and 5% classified as other/unknown race (data were missing for 4%). The cohort was predominantly female (61%). White patients were more likely to present with localized disease than any other race/ethnicity (83% vs 65% in Hispanic, 60% in Asian/Pacific Islander, and 45% in Black patients [P<.05]).
Black and Hispanic patients had the worst 5-year survival rates on bivariate analysis. On multivariate analysis, this finding remained significant for Hispanic patients when compared with White patients (hazard ratio, 2.37 [P<.05]). Increasing age, male sex, advanced stage at diagnosis, and failure to receive surgery were associated with increased odds of mortality.
Patients with regionalized and disseminated disease had increased odds of mortality (6.16 and 64.45, respectively; P<.05) compared with patients with localized disease. Socioeconomic status and urbanization were not found to influence 5-year survival rates.
Pediatric melanoma often presents a clinical challenge with special considerations. Pediatric-specific predisposing risk factors for melanoma and an atypical clinical presentation are some of the major concerns that necessitate a tailored approach to this malignancy, especially among different age groups, skin types, and racial and socioeconomic groups.5
Standard ABCDE criteria often are inadequate for accurate detection of pediatric melanomas. Initial lesions often manifest as raised, red, amelanotic lesions mimicking pyogenic granulomas. Lesions tend to be very small (<6 mm in diameter) and can be uniform in color, thereby making the melanoma more difficult to detect compared to the characteristic findings in adults.5 Bleeding or ulceration often can be a warning sign during physical examination.
With regard to incidence, pediatric melanoma is relatively rare. Since the 1970s, the incidence of pediatric melanoma has been increasing; however, a recent analysis of the SEER database showed a decreasing trend from 2000 to 2010.4
Our analysis of the SEER data showed an increased risk for pediatric melanoma in older adolescents. In addition, the incidence of pediatric melanoma was higher in females of all racial groups except Asian/Pacific Islander individuals. However, SES was not found to significantly influence the 5-year survival rate in pediatric melanoma.
White pediatric patients were more likely to present with localized disease compared with other races. Pediatric melanoma patients with regional disease had a 6-fold increase in mortality rate vs those with localized disease; those with disseminated disease had a 65-fold higher risk. Consistent with this, Black and Hispanic patients had the worst 5-year survival rates on bivariate analysis.
These findings suggest a relationship between race, melanoma spread, and disease severity. Patient education programs need to be directed specifically to minority groups to improve their knowledge on evolving skin lesions and sun protection practices. Physicians also need to have heightened suspicion and better knowledge of the unique traits of pediatric melanoma.5
Given the considerable influence these disparities can have on melanoma outcomes, further research is needed to characterize outcomes based on race and determine obstacles to appropriate care. Improved public outreach initiatives that accommodate specific cultural barriers (eg, language, traditional patterns of behavior) also are required to improve current circumstances.
- Arnold M, Singh D, Laversanne M, et al. Global burden of cutaneous melanoma in 2020 and projections to 2040. JAMA Dermatol. 2022;158:495-503.
- McCormack L, Hawryluk EB. Pediatric melanoma update. G Ital Dermatol Venereol. 2018;153:707-715.
- Saiyed FK, Hamilton EC, Austin MT. Pediatric melanoma: incidence, treatment, and prognosis. Pediatric Health Med Ther. 2017;8:39-45.
- Wojcik KY, Hawkins M, Anderson-Mellies A, et al. Melanoma survival by age group: population-based disparities for adolescent and young adult patients by stage, tumor thickness, and insurance type. J Am Acad Dermatol. 2023;88:831-840.
- Hamilton EC, Nguyen HT, Chang YC, et al. Health disparities influence childhood melanoma stage at diagnosis and outcome. J Pediatr. 2016;175:182-187.
- Holman DM, King JB, White A, et al. Acral lentiginous melanoma incidence by sex, race, ethnicity, and stage in the United States, 2010-2019. Prev Med. 2023;175:107692. doi:10.1016/j.ypmed.2023.107692
- El Sharouni MA, Rawson RV, Potter AJ, et al. Melanomas in children and adolescents: clinicopathologic features and survival outcomes. J Am Acad Dermatol. 2023;88:609-616. doi:10.1016/j.jaad.2022.08.067
- Arnold M, Singh D, Laversanne M, et al. Global burden of cutaneous melanoma in 2020 and projections to 2040. JAMA Dermatol. 2022;158:495-503.
- McCormack L, Hawryluk EB. Pediatric melanoma update. G Ital Dermatol Venereol. 2018;153:707-715.
- Saiyed FK, Hamilton EC, Austin MT. Pediatric melanoma: incidence, treatment, and prognosis. Pediatric Health Med Ther. 2017;8:39-45.
- Wojcik KY, Hawkins M, Anderson-Mellies A, et al. Melanoma survival by age group: population-based disparities for adolescent and young adult patients by stage, tumor thickness, and insurance type. J Am Acad Dermatol. 2023;88:831-840.
- Hamilton EC, Nguyen HT, Chang YC, et al. Health disparities influence childhood melanoma stage at diagnosis and outcome. J Pediatr. 2016;175:182-187.
- Holman DM, King JB, White A, et al. Acral lentiginous melanoma incidence by sex, race, ethnicity, and stage in the United States, 2010-2019. Prev Med. 2023;175:107692. doi:10.1016/j.ypmed.2023.107692
- El Sharouni MA, Rawson RV, Potter AJ, et al. Melanomas in children and adolescents: clinicopathologic features and survival outcomes. J Am Acad Dermatol. 2023;88:609-616. doi:10.1016/j.jaad.2022.08.067
Practice Points
- Pediatric melanoma is a unique clinical entity with a different clinical presentation than in adults.
- Thicker tumors and disseminated disease are associated with a worse prognosis, and these factors are more commonly seen in Black and Hispanic patients.
Diagnosing and Managing Duchenne Muscular Dystrophy: Tips for Practicing Clinicians
Duchenne muscular dystrophy (DMD) is a severe progressive inherited disease characterized by muscle wasting and ultimately culminating in death. It’s a common enough neuromuscular disorder that pediatricians and family practice physicians are likely to see at least a couple of patients with DMD over the course of their career,” John Brandsema, MD, Neuromuscular Section Head, Division of Neurology, Children’s Hospital of Philadelphia in Pennsylvania, said in an interview. Healthcare providers should therefore be familiar with the disorder so as to provide timely diagnosis and early intervention as well as practical and emotional support to the patient and family/caregivers as they traverse the challenging and often heartbreaking journey with this condition.
Pathophysiology and Disease Trajectory
DMD is caused by pathogenic variants in the X-linked DMD gene, leading to reduction in dystrophin, a protein that serves as a cytoskeletal integrator, stabilizing the plasma membrane of striated muscle cells. Dystrophin is critical for muscle membrane stability.2 In particular, mutations in the gene that encodes for dystrophin lead to dysfunction in Dp427m, which is the muscle isoform of dystrophin.3,4
DMD is one of several types of muscular dystrophies. All are progressive disorders. Over time, healthy muscle fibers disappear and are replaced by fibrotic tissue and fat, making the muscles “less able to generate force for everyday activity.”2 Ultimately, the skeletal muscle dysfunction affects not only the patient’s day-to-day mobility but other systems as well. Most patients with DMD eventually die of cardiac and/or respiratory failure between the ages of 20 and 40 years, with a median life expectancy of 22 years — although children born after 1990 have a somewhat higher median life expectancy (28 years), because of the improving standard of care.3,5
Typically, DMD first presents with developmental delays and weakness in skeletal leg muscles. As the disease goes through stages of progression, it starts involving upper extremities and other systems. (Table 1)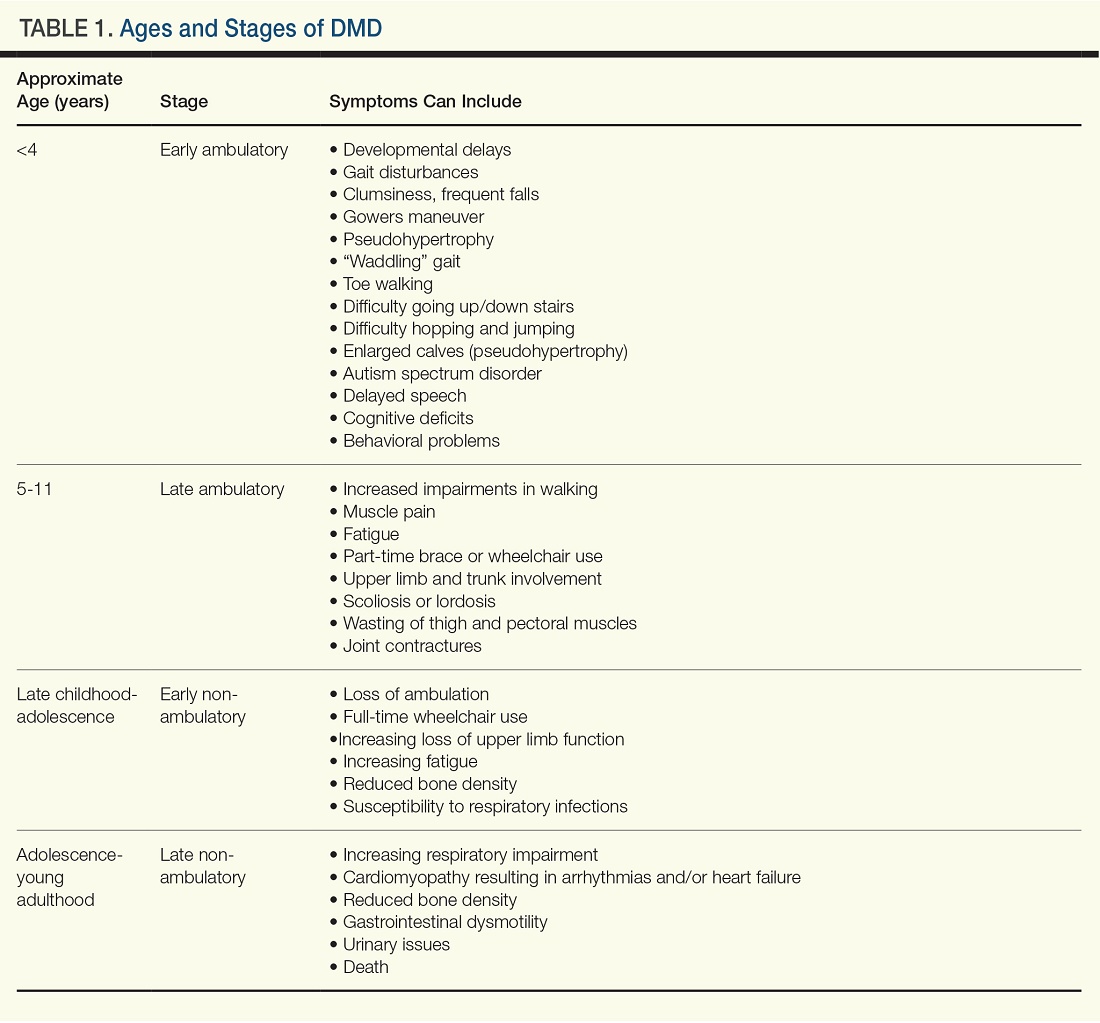
Genetic Causes of DMD
The DMD gene, located on the X chromosome, encodes for the production of dystrophin. Variants of this gene result in the lack of dystrophin protein, leading in turn to muscle fiber degeneration and the progressive symptoms of DMD. Because of the gene’s location on the X chromosome, males (who don’t have a second copy of the X chromosome) cannot compensate for the mutated gene, which is why the disease affects male children. Females with this mutation are carriers and typically do not develop the same severity of symptoms, although they might have milder muscle cramps, weakness, and cardiac issues.3
A female carrier with DMD (or any other X-linked disorder) has a 25% chance to have a carrier daughter, a 25% change of having a noncarrier daughter, a 25% chance of having an affected son, and a 25% chance of having a nonaffected son. A male with the disorder will pass the mutated gene on to his daughters who then become carriers. He cannot pass the disorder on to his sons because males inherit only the Y chromosome from their fathers.3
Diagnosing DMD
“It can take as long as 1-3 years for a child to be diagnosed with DMD,” Dr. Brandsema said. “Parents typically have concerns and know that something is ‘off’ about their child and they’re sent to various specialists, but it usually takes time for an accurate diagnosis to be made.” The mean age at diagnosis of DMD is between ages 4 and 5 years.6
Early identification of infants at risk for developing DMD can help move the needle toward earlier diagnosis. Newborn screening for DMD has been researched and piloted in several programs.6 In 2023, DMD was nominated for inclusion in the Recommended Universal Screening Panel (RUSP) for universal newborn screening. But in May 2024, the advisory committee on Heritable Disorders in Newborns and Children decided to postpone the vote to include DMD in the RUSP, requesting additional information to ensure an evidence-based decision.
In the absence of universal newborn screening for DMD, alternative approaches have been proposed to reduce the delay in clinical diagnosis and specialist referral, including increasing awareness among healthcare providers (eg, pediatricians, pediatric neurologists, and primary care physicians).6
The National Task Force for Early Identification of Childhood Neuromuscular Disorders delineates the steps necessary to identify pediatric muscle weakness and signs of neuromuscular disease. Primary care providers are encouraged to engage in regular developmental surveillance. A surveillance aid lays out the timetable for recommended visits, typical developmental milestones, and components of surveillance. Clinical evaluation includes a detailed patient history, family history, and physical examination.
If a neuromuscular condition is suspected, laboratory work should include creatinine phosphokinase (CK).6 Elevated serum CK points to leakage of CK through the muscle membrane, suggesting muscle damage. If CK is elevated, genetic testing should be performed; and, if negative, it should be followed by genetic sequencing that tests for small-scale mutations in the DMD gene. If that test is negative, a muscle biopsy should be performed to test for deep intronic mutations in the DMD gene.4
The diagnostic process and immediate steps after a confirmed DMD diagnosis is found in Figure 1.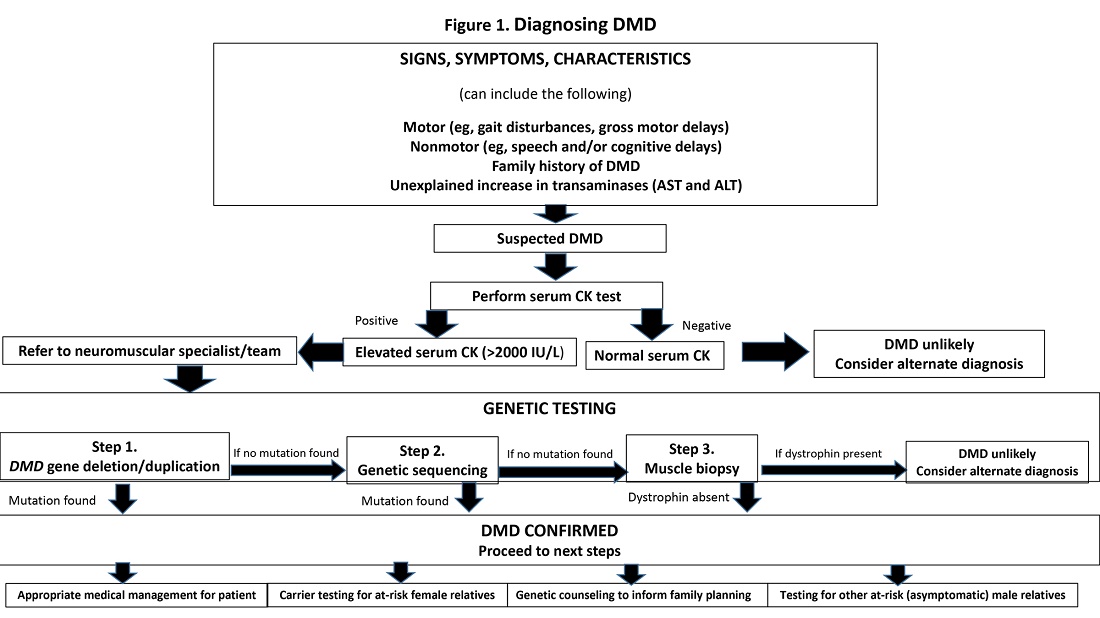
Targeting Inflammation in DMD
Traditionally, corticosteroids have been the only available medical treatment for DMD and they remain a cornerstone of DMD management. A meta-analysis found “moderate evidence” that corticosteroid therapy improves muscle strength and function in the short term (12 months), and strength up to 2 years.10
The two most common corticosteroids for DMD are prednisone and deflazacort. Deflazacort (Emflaza, PTC Therapeutics) was approved in 2017 to treat patients ages 5 years and older with DMD, subsequently expanded to 2 years and older. Deflazacort has been found to be more effective than prednisone in improving functional outcomes, delaying the onset of cardiomyopathy, and improving overall survival, with fewer adverse effects.11
In 2023, vamorolone (Agamree, Catalyst Pharmaceuticals) was approved by the Food and Drug Administration (FDA) to treat DMD patients (ages 2 years and older). Vamorolone is a dissociative steroidal anti-inflammatory that reduces bone morbidities and is regarded as a safer alternative than prednisone. A clinical trial comparing two doses of vamorolone with prednisone for 24 weeks found that vamorolone 6 mg/kg per day met the primary endpoint (time to stand velocity) and four sequential secondary motor function endpoints, with less bone morbidity, compared to prednisone.12 A more recent trial found improvements in motor outcomes at 48 weeks with a dose of 6 mg/kg per day of vamorolone. Bone morbidities of prednisone were reversed when the patient transitioned to vamorolone.13
“Steroid treatment has been proven to help, usually taken daily, although other schedules have been tried,” Dr. Brandsema said. However, all steroids are fraught with adverse effects and are suboptimal in the long term in reducing the disease burden.
The anti-inflammatory agent givinostat (Duvyzat, ITF Therapeutics), an oral histone deacetylase (HDAC) inhibitor, was approved in March 2024 for the treatment of DMD in patients 6 years of age and older. It is the first nonsteroidal drug to treat patients with all genetic variants of the disease, and it has a unique mechanism of action. Deficits in dystrophin can lead to increased HDAC activity in DMD, reducing the expression of genes involved in muscle regeneration. Givinostat therefore can help to counteract the pathogenic events downstream of dystrophin deficiency by inhibiting HDAC.14
Approval for givinostat was based on the phase 3 EPIDYS trial, which randomized 179 boys with DMD to receive either givinostat or placebo. Although results of a functional task worsened in both groups over the 12-month study period, the decline was significantly smaller with givinostat versus placebo. The most common adverse events were diarrhea and vomiting.14 Dr. Brandsema noted that monitoring of triglycerides and platelet count is required, as hypertriglyceridemia and thrombocytopenia can occur. This treatment was studied in tandem with corticosteroids as a combination approach to muscle stabilization.
New Pharmacotherapeutic Options: Exon-Skipping Agents
Today’s treatments have expanded beyond corticosteroids, with newer therapeutic options that include targeted exon-skipping therapies and, more recently, gene therapies. “These new treatment paradigms have changed the face of DMD treatment,” Dr. Brandsema said.

“Exon-skipping drugs in their current form have only a modest effect, but at least they’re a step in the right direction and a breakthrough, in terms of slowing disease progression,” Dr. Brandsema said.
Current exon-skipping agents use antisense phosphorodiamidate morpholino oligomers (PMOs) to restore a DMD open reading frame. Next-generation drugs called cell-penetrating peptide-conjugated PMOs (PPMOs) are being actively researched, Dr. Brandsema said. These agents have shown enhanced cellular uptake and more efficient dystrophin restoration, compared with unconjugated PMOs.15
There are currently four FDA-approved exon-skipping agents for DMD, all of which are administered via a weekly intravenous infusion: Casimersen (Amondys-45, SRP-4045), approved by the FDA in 2021; Eteplirsen (Exondys 51), approved in 2016; Golodirsen (Vyondys 53,SRP-4053), approved in 2019; and Vitolarsen (Viltepso), approved in 2020. They can be associated with multiple side effects, depending on the drug, including upper respiratory infection, fever, cough, rash, and gastrointestinal issues.16 These agents have the potential to help 30% of DMD patients, restoring low levels of dystrophin.16
Gene Transfer Therapies
Gene transfer therapies, a new class of agents, utilize a nonpathogenic viral vector (adeno-associated virus) to transfer specific genes to patients with DMD. Gene therapy involves overexpressing the micro-dystrophin gene to restore functional dystrophin expression.16
Multiple clinical trials of gene therapy are currently in progress. In 2023, delandistrogene moxeparvovec-rokl (Elevidys, Serepta) was granted accelerated FDA approval for ambulatory individuals with DMD between the ages of 4 and 5 years of age and a confirmed mutation in the DMD gene. It received expanded approval in June 2024 to include ambulatory and nonambulatory individuals aged 4 years and older with DMD and a confirmed mutation in the DMD gene (with the exception of exon 8 or 9 mutations).
The approval was based on preliminary data from two double-blind, placebo-controlled studies and two open-label studies, which enrolled a total of 218 male patients (including those who received placebo) with a confirmed disease-causing mutation in the DMD gene.
Delandistrogene moxeparvovec-rokl is delivered as a one-time infusion and has been associated with side effects and “a lot of potential issues,” Dr. Brandsema said. “We’ve seen cardiac effects, immune system effects, increased muscle inflammation and hepatic complications, and some people who became quite unwell were hospitalized for a long time.”
Fortunately, he added, “these seem to be rare but they do happen. Once the medication has been delivered, it’s permanently in the body, so you’re managing the side effects potentially on a long-term basis.”
It is critical to discuss the risks and benefits of this treatment with the family and caregivers and with the patient as well, if he old enough and able to participate in the decision-making progress. “We don’t want to give unrealistic expectations and we want people to be aware of the potential downside of this treatment,” he said. “This is a very complex discussion because the trajectory of the disease is so devastating and this treatment does hold out hope that other therapies don’t necessarily have.”
Nonpharmacologic Interventions
Physical therapy is a mainstay in DMD treatment, addressing protection of fragile muscles, preservation of strength, and prevention of muscle contractures.16 Given the respiratory impairments that occur with DMD progression, respiratory monitoring and therapy are essential; however, the number and type of evaluations and interventions vary with the stage of the disease, intensifying as the disease progresses.16 Similarly, cardiac monitoring should begin early, with patients screened for cardiac complications, and should intensify through the stages of disease progression.16
Bone health is compromised in patients with DMD, both as a result of corticosteroid treatment and as part of the disease itself. Fractures may be asymptomatic and may go unnoticed. Thus, bone health surveillance and maintenance are critical components of DMD management.16
Patients with DMD often experience gastrointestinal issues. They may experience weight gain because of lack of mobility and corticosteroid use in early stages, or weight loss as a result of diet or fluid imbalance, low bone density, or dysphagia in later stages. Patients should be closely followed by a nutritionist, a gastroenterologist as needed, and a physical therapist.16
Psychosocial support “should be developed and implemented across the lifespan in a manner that promotes thinking about the future and sets expectations that individuals will actively participate in their care and daily activities.”9 This includes psychological care, neuropsychological evaluations, and educational support.
Assisting Patients and Families Through the DMD Journey
DMD care is best delivered in a multidisciplinary setting, where physicians of relevant specialties, physical and occupational therapists, nutritionists, social workers, and genetic counselors collaborate. At Children’s Hospital of Philadelphia, DMD care is delivered through this collaborative model.
Unfortunately, Dr. Brandsema said, many patients don’t have this type of multidisciplinary resource available. “One specialist, such as a pulmonologist or neurologist, might have to be the sole source of care.” Or parents may have to ferry their child to multiple specialists in disparate locations, placing extra stress on an already-stressed family system.
“It’s helpful to connect the family with a comprehensive care center, if possible,” Dr. Brandsema advised. If that’s not available, then he suggests recommending educational opportunities and resources through national organizations such as the Muscular Dystrophy Association; Parent Project MD; NORD; Friends, Family and Duchenne; and Cure Duchenne. Families and caregivers, along with affected individuals, can get education and support from people who understand the day-to-day reality of living with this disease.
One of the major challenges that families face is navigating the high cost of treating DMD, especially the new medications, Dr. Brandsema said. “The authorization process can be intensive and long, and the family may need to take an active role, together with the provider team, in advocating for the patient to get access.”

Among her many responsibilities, Ms. Kaschak engages in care coordination tasks and management, helps patients and caregivers understand care plans, and provides psychosocial support and education about the disease process. She assists families in completing paperwork and navigating specialty authorizations, helping families understand and navigate the complex insurance process. “My role is to bridge gaps in care,” she said.
Dr. Brandsema noted that it’s important for couples to receive genetic counseling if they’re planning to have multiple children because there is a 50% chance that their next boy will be affected. About two thirds of mothers with children who have DMD are carriers, but many are not aware of it. Receiving counseling will enable them to understand their own risks of health complications, as well as the risk to future children.
Managing DMD Across the Lifespan
Another dimension of DMD care is providing resources and help to young people with DMD as they transition into adulthood. “In the past, we had limited treatment and mortality typically took place in the early 20s, so there weren’t a lot of patients who were adults. But as medication options have expanded and management of cardiac and respiratory failure has improved, we see a more significant proportion of adults who require adult-appropriate clinics — or, at the very least, specialists who are conversant in care or can provide care across the lifespan,” Dr. Brandsema said.
The DMD Care Considerations Working Group provides recommendations regarding care across the lifespan,9 as does the Adult North Star Network, of Muscular Dystrophy UK.17,18
Dr. Brandsema emphasized that, despite their disability, many adults with DMD “still engage with the community, and live life to its fullest.” It is to be hoped that, with ongoing research, earlier diagnosis, and improved treatment options, the future will look bright for people with DMD.
Dr. Brandsema has served as a consultant for Audentes, AveXis/Novartis, Biogen, Cytokinetics, Dyne, Edgewise, Fibrogen, Genentech, Marathon, Momenta/Janssen, NS Pharma, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Takeda, and WaVe. He is on the medical advisory council member for Cure SMA and is a site investigator for clinical trials with Alexion, Astellas, AveXis/Novartis, Biogen, Biohaven, Catabasis, CSL Behring, Cytokinetics, Dyne, Fibrogen, Genentech, Ionis, Lilly, ML Bio, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Summit, and WaVe. Ms. Kaschak has nothing to disclose.
References
1. Venugopal V and Pavlakis S. Duchenne Muscular Dystrophy. 2023 Jul 10. In: StatPearls [Internet]. Treasure Island, Florida: StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK482346/.
2. Gao QQ and McNally EM. Compr Physiol. 2015 Jul 1;5(3):1223-39. doi: 10.1002/cphy.c140048.
3. Duan D et al. Nat Rev Dis Primers. 2021 Feb 18;7(1):13. doi: 10.1038/s41572-021-00248-3.
4. Aartsma-Rus A et al. J Pediatr. 2019 Jan:204:305-313.e14. doi: 10.1016/j.jpeds.2018.10.043.
5. Broomfield J et al. Neurology. 2021 Dec 7;97(23):e2304-e2314. doi: 10.1212/WNL.0000000000012910.
6. Mercuri E et al. Front Pediatr. 2023 Nov 10:11:1276144. doi: 10.1212/WNL.0000000000012910.
7. Birnkrant DJ et al. Lancet Neurol. 2018 Mar;17(3):251-267. doi: 10.1016/S1474-4422(18)30024-3.
8. Birnkrant DJ et al. Lancet Neurol. 2018 Apr;17(4):347-361. doi: 10.1016/S1474-4422(18)30025-5.
9. Birnkrant DJ et al. Lancet Neurol. 2018 May;17(5):445-455. doi: 10.1016/S1474-4422(18)30026-7.
10. Matthews E et al. Cochrane Database Syst Rev. 2016 May 5;2016(5):CD003725. doi: 10.1002/14651858.CD003725.pub4.
11. Bylo M et al. Ann Pharmacother. 2020 Aug;54(8):788-794. doi: 10.1177/1060028019900500.
12. Guglieri M et al. JAMA Neurol. 2022 Oct 1;79(10):1005-1014. doi: 10.1001/jamaneurol.2022.2480.
13. Dang UJ et al. Neurology. 2024 Mar 12;102(5):e208112. doi: 10.1212/WNL.0000000000208112.
14. Mercuri E et al. Lancet Neurol. 2024 Apr;23(4):393-403. doi: 10.1016/S1474-4422(24)00036-X.
15. Gushchina LV et al. Mol Ther Nucleic Acids. 2022 Nov 9:30:479-492. doi: 10.1016/j.omtn.2022.10.025.
16. Patterson G et al. Eur J Pharmacol. 2023 May 15:947:175675. doi: 10.1016/j.ejphar.2023.175675.
17. Quinlivan R et al. J Neuromuscul Dis. 2021;8(6):899-926. doi: 10.3233/JND-200609.
18. Narayan S et al. J Neuromuscul Dis. 2022;9(3):365-381. doi: 10.3233/JND-210707.
Duchenne muscular dystrophy (DMD) is a severe progressive inherited disease characterized by muscle wasting and ultimately culminating in death. It’s a common enough neuromuscular disorder that pediatricians and family practice physicians are likely to see at least a couple of patients with DMD over the course of their career,” John Brandsema, MD, Neuromuscular Section Head, Division of Neurology, Children’s Hospital of Philadelphia in Pennsylvania, said in an interview. Healthcare providers should therefore be familiar with the disorder so as to provide timely diagnosis and early intervention as well as practical and emotional support to the patient and family/caregivers as they traverse the challenging and often heartbreaking journey with this condition.
Pathophysiology and Disease Trajectory
DMD is caused by pathogenic variants in the X-linked DMD gene, leading to reduction in dystrophin, a protein that serves as a cytoskeletal integrator, stabilizing the plasma membrane of striated muscle cells. Dystrophin is critical for muscle membrane stability.2 In particular, mutations in the gene that encodes for dystrophin lead to dysfunction in Dp427m, which is the muscle isoform of dystrophin.3,4
DMD is one of several types of muscular dystrophies. All are progressive disorders. Over time, healthy muscle fibers disappear and are replaced by fibrotic tissue and fat, making the muscles “less able to generate force for everyday activity.”2 Ultimately, the skeletal muscle dysfunction affects not only the patient’s day-to-day mobility but other systems as well. Most patients with DMD eventually die of cardiac and/or respiratory failure between the ages of 20 and 40 years, with a median life expectancy of 22 years — although children born after 1990 have a somewhat higher median life expectancy (28 years), because of the improving standard of care.3,5
Typically, DMD first presents with developmental delays and weakness in skeletal leg muscles. As the disease goes through stages of progression, it starts involving upper extremities and other systems. (Table 1)
Genetic Causes of DMD
The DMD gene, located on the X chromosome, encodes for the production of dystrophin. Variants of this gene result in the lack of dystrophin protein, leading in turn to muscle fiber degeneration and the progressive symptoms of DMD. Because of the gene’s location on the X chromosome, males (who don’t have a second copy of the X chromosome) cannot compensate for the mutated gene, which is why the disease affects male children. Females with this mutation are carriers and typically do not develop the same severity of symptoms, although they might have milder muscle cramps, weakness, and cardiac issues.3
A female carrier with DMD (or any other X-linked disorder) has a 25% chance to have a carrier daughter, a 25% change of having a noncarrier daughter, a 25% chance of having an affected son, and a 25% chance of having a nonaffected son. A male with the disorder will pass the mutated gene on to his daughters who then become carriers. He cannot pass the disorder on to his sons because males inherit only the Y chromosome from their fathers.3
Diagnosing DMD
“It can take as long as 1-3 years for a child to be diagnosed with DMD,” Dr. Brandsema said. “Parents typically have concerns and know that something is ‘off’ about their child and they’re sent to various specialists, but it usually takes time for an accurate diagnosis to be made.” The mean age at diagnosis of DMD is between ages 4 and 5 years.6
Early identification of infants at risk for developing DMD can help move the needle toward earlier diagnosis. Newborn screening for DMD has been researched and piloted in several programs.6 In 2023, DMD was nominated for inclusion in the Recommended Universal Screening Panel (RUSP) for universal newborn screening. But in May 2024, the advisory committee on Heritable Disorders in Newborns and Children decided to postpone the vote to include DMD in the RUSP, requesting additional information to ensure an evidence-based decision.
In the absence of universal newborn screening for DMD, alternative approaches have been proposed to reduce the delay in clinical diagnosis and specialist referral, including increasing awareness among healthcare providers (eg, pediatricians, pediatric neurologists, and primary care physicians).6
The National Task Force for Early Identification of Childhood Neuromuscular Disorders delineates the steps necessary to identify pediatric muscle weakness and signs of neuromuscular disease. Primary care providers are encouraged to engage in regular developmental surveillance. A surveillance aid lays out the timetable for recommended visits, typical developmental milestones, and components of surveillance. Clinical evaluation includes a detailed patient history, family history, and physical examination.
If a neuromuscular condition is suspected, laboratory work should include creatinine phosphokinase (CK).6 Elevated serum CK points to leakage of CK through the muscle membrane, suggesting muscle damage. If CK is elevated, genetic testing should be performed; and, if negative, it should be followed by genetic sequencing that tests for small-scale mutations in the DMD gene. If that test is negative, a muscle biopsy should be performed to test for deep intronic mutations in the DMD gene.4
The diagnostic process and immediate steps after a confirmed DMD diagnosis is found in Figure 1.
Targeting Inflammation in DMD
Traditionally, corticosteroids have been the only available medical treatment for DMD and they remain a cornerstone of DMD management. A meta-analysis found “moderate evidence” that corticosteroid therapy improves muscle strength and function in the short term (12 months), and strength up to 2 years.10
The two most common corticosteroids for DMD are prednisone and deflazacort. Deflazacort (Emflaza, PTC Therapeutics) was approved in 2017 to treat patients ages 5 years and older with DMD, subsequently expanded to 2 years and older. Deflazacort has been found to be more effective than prednisone in improving functional outcomes, delaying the onset of cardiomyopathy, and improving overall survival, with fewer adverse effects.11
In 2023, vamorolone (Agamree, Catalyst Pharmaceuticals) was approved by the Food and Drug Administration (FDA) to treat DMD patients (ages 2 years and older). Vamorolone is a dissociative steroidal anti-inflammatory that reduces bone morbidities and is regarded as a safer alternative than prednisone. A clinical trial comparing two doses of vamorolone with prednisone for 24 weeks found that vamorolone 6 mg/kg per day met the primary endpoint (time to stand velocity) and four sequential secondary motor function endpoints, with less bone morbidity, compared to prednisone.12 A more recent trial found improvements in motor outcomes at 48 weeks with a dose of 6 mg/kg per day of vamorolone. Bone morbidities of prednisone were reversed when the patient transitioned to vamorolone.13
“Steroid treatment has been proven to help, usually taken daily, although other schedules have been tried,” Dr. Brandsema said. However, all steroids are fraught with adverse effects and are suboptimal in the long term in reducing the disease burden.
The anti-inflammatory agent givinostat (Duvyzat, ITF Therapeutics), an oral histone deacetylase (HDAC) inhibitor, was approved in March 2024 for the treatment of DMD in patients 6 years of age and older. It is the first nonsteroidal drug to treat patients with all genetic variants of the disease, and it has a unique mechanism of action. Deficits in dystrophin can lead to increased HDAC activity in DMD, reducing the expression of genes involved in muscle regeneration. Givinostat therefore can help to counteract the pathogenic events downstream of dystrophin deficiency by inhibiting HDAC.14
Approval for givinostat was based on the phase 3 EPIDYS trial, which randomized 179 boys with DMD to receive either givinostat or placebo. Although results of a functional task worsened in both groups over the 12-month study period, the decline was significantly smaller with givinostat versus placebo. The most common adverse events were diarrhea and vomiting.14 Dr. Brandsema noted that monitoring of triglycerides and platelet count is required, as hypertriglyceridemia and thrombocytopenia can occur. This treatment was studied in tandem with corticosteroids as a combination approach to muscle stabilization.
New Pharmacotherapeutic Options: Exon-Skipping Agents
Today’s treatments have expanded beyond corticosteroids, with newer therapeutic options that include targeted exon-skipping therapies and, more recently, gene therapies. “These new treatment paradigms have changed the face of DMD treatment,” Dr. Brandsema said.
“Exon-skipping drugs in their current form have only a modest effect, but at least they’re a step in the right direction and a breakthrough, in terms of slowing disease progression,” Dr. Brandsema said.
Current exon-skipping agents use antisense phosphorodiamidate morpholino oligomers (PMOs) to restore a DMD open reading frame. Next-generation drugs called cell-penetrating peptide-conjugated PMOs (PPMOs) are being actively researched, Dr. Brandsema said. These agents have shown enhanced cellular uptake and more efficient dystrophin restoration, compared with unconjugated PMOs.15
There are currently four FDA-approved exon-skipping agents for DMD, all of which are administered via a weekly intravenous infusion: Casimersen (Amondys-45, SRP-4045), approved by the FDA in 2021; Eteplirsen (Exondys 51), approved in 2016; Golodirsen (Vyondys 53,SRP-4053), approved in 2019; and Vitolarsen (Viltepso), approved in 2020. They can be associated with multiple side effects, depending on the drug, including upper respiratory infection, fever, cough, rash, and gastrointestinal issues.16 These agents have the potential to help 30% of DMD patients, restoring low levels of dystrophin.16
Gene Transfer Therapies
Gene transfer therapies, a new class of agents, utilize a nonpathogenic viral vector (adeno-associated virus) to transfer specific genes to patients with DMD. Gene therapy involves overexpressing the micro-dystrophin gene to restore functional dystrophin expression.16
Multiple clinical trials of gene therapy are currently in progress. In 2023, delandistrogene moxeparvovec-rokl (Elevidys, Serepta) was granted accelerated FDA approval for ambulatory individuals with DMD between the ages of 4 and 5 years of age and a confirmed mutation in the DMD gene. It received expanded approval in June 2024 to include ambulatory and nonambulatory individuals aged 4 years and older with DMD and a confirmed mutation in the DMD gene (with the exception of exon 8 or 9 mutations).
The approval was based on preliminary data from two double-blind, placebo-controlled studies and two open-label studies, which enrolled a total of 218 male patients (including those who received placebo) with a confirmed disease-causing mutation in the DMD gene.
Delandistrogene moxeparvovec-rokl is delivered as a one-time infusion and has been associated with side effects and “a lot of potential issues,” Dr. Brandsema said. “We’ve seen cardiac effects, immune system effects, increased muscle inflammation and hepatic complications, and some people who became quite unwell were hospitalized for a long time.”
Fortunately, he added, “these seem to be rare but they do happen. Once the medication has been delivered, it’s permanently in the body, so you’re managing the side effects potentially on a long-term basis.”
It is critical to discuss the risks and benefits of this treatment with the family and caregivers and with the patient as well, if he old enough and able to participate in the decision-making progress. “We don’t want to give unrealistic expectations and we want people to be aware of the potential downside of this treatment,” he said. “This is a very complex discussion because the trajectory of the disease is so devastating and this treatment does hold out hope that other therapies don’t necessarily have.”
Nonpharmacologic Interventions
Physical therapy is a mainstay in DMD treatment, addressing protection of fragile muscles, preservation of strength, and prevention of muscle contractures.16 Given the respiratory impairments that occur with DMD progression, respiratory monitoring and therapy are essential; however, the number and type of evaluations and interventions vary with the stage of the disease, intensifying as the disease progresses.16 Similarly, cardiac monitoring should begin early, with patients screened for cardiac complications, and should intensify through the stages of disease progression.16
Bone health is compromised in patients with DMD, both as a result of corticosteroid treatment and as part of the disease itself. Fractures may be asymptomatic and may go unnoticed. Thus, bone health surveillance and maintenance are critical components of DMD management.16
Patients with DMD often experience gastrointestinal issues. They may experience weight gain because of lack of mobility and corticosteroid use in early stages, or weight loss as a result of diet or fluid imbalance, low bone density, or dysphagia in later stages. Patients should be closely followed by a nutritionist, a gastroenterologist as needed, and a physical therapist.16
Psychosocial support “should be developed and implemented across the lifespan in a manner that promotes thinking about the future and sets expectations that individuals will actively participate in their care and daily activities.”9 This includes psychological care, neuropsychological evaluations, and educational support.
Assisting Patients and Families Through the DMD Journey
DMD care is best delivered in a multidisciplinary setting, where physicians of relevant specialties, physical and occupational therapists, nutritionists, social workers, and genetic counselors collaborate. At Children’s Hospital of Philadelphia, DMD care is delivered through this collaborative model.
Unfortunately, Dr. Brandsema said, many patients don’t have this type of multidisciplinary resource available. “One specialist, such as a pulmonologist or neurologist, might have to be the sole source of care.” Or parents may have to ferry their child to multiple specialists in disparate locations, placing extra stress on an already-stressed family system.
“It’s helpful to connect the family with a comprehensive care center, if possible,” Dr. Brandsema advised. If that’s not available, then he suggests recommending educational opportunities and resources through national organizations such as the Muscular Dystrophy Association; Parent Project MD; NORD; Friends, Family and Duchenne; and Cure Duchenne. Families and caregivers, along with affected individuals, can get education and support from people who understand the day-to-day reality of living with this disease.
One of the major challenges that families face is navigating the high cost of treating DMD, especially the new medications, Dr. Brandsema said. “The authorization process can be intensive and long, and the family may need to take an active role, together with the provider team, in advocating for the patient to get access.”
Among her many responsibilities, Ms. Kaschak engages in care coordination tasks and management, helps patients and caregivers understand care plans, and provides psychosocial support and education about the disease process. She assists families in completing paperwork and navigating specialty authorizations, helping families understand and navigate the complex insurance process. “My role is to bridge gaps in care,” she said.
Dr. Brandsema noted that it’s important for couples to receive genetic counseling if they’re planning to have multiple children because there is a 50% chance that their next boy will be affected. About two thirds of mothers with children who have DMD are carriers, but many are not aware of it. Receiving counseling will enable them to understand their own risks of health complications, as well as the risk to future children.
Managing DMD Across the Lifespan
Another dimension of DMD care is providing resources and help to young people with DMD as they transition into adulthood. “In the past, we had limited treatment and mortality typically took place in the early 20s, so there weren’t a lot of patients who were adults. But as medication options have expanded and management of cardiac and respiratory failure has improved, we see a more significant proportion of adults who require adult-appropriate clinics — or, at the very least, specialists who are conversant in care or can provide care across the lifespan,” Dr. Brandsema said.
The DMD Care Considerations Working Group provides recommendations regarding care across the lifespan,9 as does the Adult North Star Network, of Muscular Dystrophy UK.17,18
Dr. Brandsema emphasized that, despite their disability, many adults with DMD “still engage with the community, and live life to its fullest.” It is to be hoped that, with ongoing research, earlier diagnosis, and improved treatment options, the future will look bright for people with DMD.
Dr. Brandsema has served as a consultant for Audentes, AveXis/Novartis, Biogen, Cytokinetics, Dyne, Edgewise, Fibrogen, Genentech, Marathon, Momenta/Janssen, NS Pharma, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Takeda, and WaVe. He is on the medical advisory council member for Cure SMA and is a site investigator for clinical trials with Alexion, Astellas, AveXis/Novartis, Biogen, Biohaven, Catabasis, CSL Behring, Cytokinetics, Dyne, Fibrogen, Genentech, Ionis, Lilly, ML Bio, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Summit, and WaVe. Ms. Kaschak has nothing to disclose.
References
1. Venugopal V and Pavlakis S. Duchenne Muscular Dystrophy. 2023 Jul 10. In: StatPearls [Internet]. Treasure Island, Florida: StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK482346/.
2. Gao QQ and McNally EM. Compr Physiol. 2015 Jul 1;5(3):1223-39. doi: 10.1002/cphy.c140048.
3. Duan D et al. Nat Rev Dis Primers. 2021 Feb 18;7(1):13. doi: 10.1038/s41572-021-00248-3.
4. Aartsma-Rus A et al. J Pediatr. 2019 Jan:204:305-313.e14. doi: 10.1016/j.jpeds.2018.10.043.
5. Broomfield J et al. Neurology. 2021 Dec 7;97(23):e2304-e2314. doi: 10.1212/WNL.0000000000012910.
6. Mercuri E et al. Front Pediatr. 2023 Nov 10:11:1276144. doi: 10.1212/WNL.0000000000012910.
7. Birnkrant DJ et al. Lancet Neurol. 2018 Mar;17(3):251-267. doi: 10.1016/S1474-4422(18)30024-3.
8. Birnkrant DJ et al. Lancet Neurol. 2018 Apr;17(4):347-361. doi: 10.1016/S1474-4422(18)30025-5.
9. Birnkrant DJ et al. Lancet Neurol. 2018 May;17(5):445-455. doi: 10.1016/S1474-4422(18)30026-7.
10. Matthews E et al. Cochrane Database Syst Rev. 2016 May 5;2016(5):CD003725. doi: 10.1002/14651858.CD003725.pub4.
11. Bylo M et al. Ann Pharmacother. 2020 Aug;54(8):788-794. doi: 10.1177/1060028019900500.
12. Guglieri M et al. JAMA Neurol. 2022 Oct 1;79(10):1005-1014. doi: 10.1001/jamaneurol.2022.2480.
13. Dang UJ et al. Neurology. 2024 Mar 12;102(5):e208112. doi: 10.1212/WNL.0000000000208112.
14. Mercuri E et al. Lancet Neurol. 2024 Apr;23(4):393-403. doi: 10.1016/S1474-4422(24)00036-X.
15. Gushchina LV et al. Mol Ther Nucleic Acids. 2022 Nov 9:30:479-492. doi: 10.1016/j.omtn.2022.10.025.
16. Patterson G et al. Eur J Pharmacol. 2023 May 15:947:175675. doi: 10.1016/j.ejphar.2023.175675.
17. Quinlivan R et al. J Neuromuscul Dis. 2021;8(6):899-926. doi: 10.3233/JND-200609.
18. Narayan S et al. J Neuromuscul Dis. 2022;9(3):365-381. doi: 10.3233/JND-210707.
Duchenne muscular dystrophy (DMD) is a severe progressive inherited disease characterized by muscle wasting and ultimately culminating in death. It’s a common enough neuromuscular disorder that pediatricians and family practice physicians are likely to see at least a couple of patients with DMD over the course of their career,” John Brandsema, MD, Neuromuscular Section Head, Division of Neurology, Children’s Hospital of Philadelphia in Pennsylvania, said in an interview. Healthcare providers should therefore be familiar with the disorder so as to provide timely diagnosis and early intervention as well as practical and emotional support to the patient and family/caregivers as they traverse the challenging and often heartbreaking journey with this condition.
Pathophysiology and Disease Trajectory
DMD is caused by pathogenic variants in the X-linked DMD gene, leading to reduction in dystrophin, a protein that serves as a cytoskeletal integrator, stabilizing the plasma membrane of striated muscle cells. Dystrophin is critical for muscle membrane stability.2 In particular, mutations in the gene that encodes for dystrophin lead to dysfunction in Dp427m, which is the muscle isoform of dystrophin.3,4
DMD is one of several types of muscular dystrophies. All are progressive disorders. Over time, healthy muscle fibers disappear and are replaced by fibrotic tissue and fat, making the muscles “less able to generate force for everyday activity.”2 Ultimately, the skeletal muscle dysfunction affects not only the patient’s day-to-day mobility but other systems as well. Most patients with DMD eventually die of cardiac and/or respiratory failure between the ages of 20 and 40 years, with a median life expectancy of 22 years — although children born after 1990 have a somewhat higher median life expectancy (28 years), because of the improving standard of care.3,5
Typically, DMD first presents with developmental delays and weakness in skeletal leg muscles. As the disease goes through stages of progression, it starts involving upper extremities and other systems. (Table 1)
Genetic Causes of DMD
The DMD gene, located on the X chromosome, encodes for the production of dystrophin. Variants of this gene result in the lack of dystrophin protein, leading in turn to muscle fiber degeneration and the progressive symptoms of DMD. Because of the gene’s location on the X chromosome, males (who don’t have a second copy of the X chromosome) cannot compensate for the mutated gene, which is why the disease affects male children. Females with this mutation are carriers and typically do not develop the same severity of symptoms, although they might have milder muscle cramps, weakness, and cardiac issues.3
A female carrier with DMD (or any other X-linked disorder) has a 25% chance to have a carrier daughter, a 25% change of having a noncarrier daughter, a 25% chance of having an affected son, and a 25% chance of having a nonaffected son. A male with the disorder will pass the mutated gene on to his daughters who then become carriers. He cannot pass the disorder on to his sons because males inherit only the Y chromosome from their fathers.3
Diagnosing DMD
“It can take as long as 1-3 years for a child to be diagnosed with DMD,” Dr. Brandsema said. “Parents typically have concerns and know that something is ‘off’ about their child and they’re sent to various specialists, but it usually takes time for an accurate diagnosis to be made.” The mean age at diagnosis of DMD is between ages 4 and 5 years.6
Early identification of infants at risk for developing DMD can help move the needle toward earlier diagnosis. Newborn screening for DMD has been researched and piloted in several programs.6 In 2023, DMD was nominated for inclusion in the Recommended Universal Screening Panel (RUSP) for universal newborn screening. But in May 2024, the advisory committee on Heritable Disorders in Newborns and Children decided to postpone the vote to include DMD in the RUSP, requesting additional information to ensure an evidence-based decision.
In the absence of universal newborn screening for DMD, alternative approaches have been proposed to reduce the delay in clinical diagnosis and specialist referral, including increasing awareness among healthcare providers (eg, pediatricians, pediatric neurologists, and primary care physicians).6
The National Task Force for Early Identification of Childhood Neuromuscular Disorders delineates the steps necessary to identify pediatric muscle weakness and signs of neuromuscular disease. Primary care providers are encouraged to engage in regular developmental surveillance. A surveillance aid lays out the timetable for recommended visits, typical developmental milestones, and components of surveillance. Clinical evaluation includes a detailed patient history, family history, and physical examination.
If a neuromuscular condition is suspected, laboratory work should include creatinine phosphokinase (CK).6 Elevated serum CK points to leakage of CK through the muscle membrane, suggesting muscle damage. If CK is elevated, genetic testing should be performed; and, if negative, it should be followed by genetic sequencing that tests for small-scale mutations in the DMD gene. If that test is negative, a muscle biopsy should be performed to test for deep intronic mutations in the DMD gene.4
The diagnostic process and immediate steps after a confirmed DMD diagnosis is found in Figure 1.
Targeting Inflammation in DMD
Traditionally, corticosteroids have been the only available medical treatment for DMD and they remain a cornerstone of DMD management. A meta-analysis found “moderate evidence” that corticosteroid therapy improves muscle strength and function in the short term (12 months), and strength up to 2 years.10
The two most common corticosteroids for DMD are prednisone and deflazacort. Deflazacort (Emflaza, PTC Therapeutics) was approved in 2017 to treat patients ages 5 years and older with DMD, subsequently expanded to 2 years and older. Deflazacort has been found to be more effective than prednisone in improving functional outcomes, delaying the onset of cardiomyopathy, and improving overall survival, with fewer adverse effects.11
In 2023, vamorolone (Agamree, Catalyst Pharmaceuticals) was approved by the Food and Drug Administration (FDA) to treat DMD patients (ages 2 years and older). Vamorolone is a dissociative steroidal anti-inflammatory that reduces bone morbidities and is regarded as a safer alternative than prednisone. A clinical trial comparing two doses of vamorolone with prednisone for 24 weeks found that vamorolone 6 mg/kg per day met the primary endpoint (time to stand velocity) and four sequential secondary motor function endpoints, with less bone morbidity, compared to prednisone.12 A more recent trial found improvements in motor outcomes at 48 weeks with a dose of 6 mg/kg per day of vamorolone. Bone morbidities of prednisone were reversed when the patient transitioned to vamorolone.13
“Steroid treatment has been proven to help, usually taken daily, although other schedules have been tried,” Dr. Brandsema said. However, all steroids are fraught with adverse effects and are suboptimal in the long term in reducing the disease burden.
The anti-inflammatory agent givinostat (Duvyzat, ITF Therapeutics), an oral histone deacetylase (HDAC) inhibitor, was approved in March 2024 for the treatment of DMD in patients 6 years of age and older. It is the first nonsteroidal drug to treat patients with all genetic variants of the disease, and it has a unique mechanism of action. Deficits in dystrophin can lead to increased HDAC activity in DMD, reducing the expression of genes involved in muscle regeneration. Givinostat therefore can help to counteract the pathogenic events downstream of dystrophin deficiency by inhibiting HDAC.14
Approval for givinostat was based on the phase 3 EPIDYS trial, which randomized 179 boys with DMD to receive either givinostat or placebo. Although results of a functional task worsened in both groups over the 12-month study period, the decline was significantly smaller with givinostat versus placebo. The most common adverse events were diarrhea and vomiting.14 Dr. Brandsema noted that monitoring of triglycerides and platelet count is required, as hypertriglyceridemia and thrombocytopenia can occur. This treatment was studied in tandem with corticosteroids as a combination approach to muscle stabilization.
New Pharmacotherapeutic Options: Exon-Skipping Agents
Today’s treatments have expanded beyond corticosteroids, with newer therapeutic options that include targeted exon-skipping therapies and, more recently, gene therapies. “These new treatment paradigms have changed the face of DMD treatment,” Dr. Brandsema said.
“Exon-skipping drugs in their current form have only a modest effect, but at least they’re a step in the right direction and a breakthrough, in terms of slowing disease progression,” Dr. Brandsema said.
Current exon-skipping agents use antisense phosphorodiamidate morpholino oligomers (PMOs) to restore a DMD open reading frame. Next-generation drugs called cell-penetrating peptide-conjugated PMOs (PPMOs) are being actively researched, Dr. Brandsema said. These agents have shown enhanced cellular uptake and more efficient dystrophin restoration, compared with unconjugated PMOs.15
There are currently four FDA-approved exon-skipping agents for DMD, all of which are administered via a weekly intravenous infusion: Casimersen (Amondys-45, SRP-4045), approved by the FDA in 2021; Eteplirsen (Exondys 51), approved in 2016; Golodirsen (Vyondys 53,SRP-4053), approved in 2019; and Vitolarsen (Viltepso), approved in 2020. They can be associated with multiple side effects, depending on the drug, including upper respiratory infection, fever, cough, rash, and gastrointestinal issues.16 These agents have the potential to help 30% of DMD patients, restoring low levels of dystrophin.16
Gene Transfer Therapies
Gene transfer therapies, a new class of agents, utilize a nonpathogenic viral vector (adeno-associated virus) to transfer specific genes to patients with DMD. Gene therapy involves overexpressing the micro-dystrophin gene to restore functional dystrophin expression.16
Multiple clinical trials of gene therapy are currently in progress. In 2023, delandistrogene moxeparvovec-rokl (Elevidys, Serepta) was granted accelerated FDA approval for ambulatory individuals with DMD between the ages of 4 and 5 years of age and a confirmed mutation in the DMD gene. It received expanded approval in June 2024 to include ambulatory and nonambulatory individuals aged 4 years and older with DMD and a confirmed mutation in the DMD gene (with the exception of exon 8 or 9 mutations).
The approval was based on preliminary data from two double-blind, placebo-controlled studies and two open-label studies, which enrolled a total of 218 male patients (including those who received placebo) with a confirmed disease-causing mutation in the DMD gene.
Delandistrogene moxeparvovec-rokl is delivered as a one-time infusion and has been associated with side effects and “a lot of potential issues,” Dr. Brandsema said. “We’ve seen cardiac effects, immune system effects, increased muscle inflammation and hepatic complications, and some people who became quite unwell were hospitalized for a long time.”
Fortunately, he added, “these seem to be rare but they do happen. Once the medication has been delivered, it’s permanently in the body, so you’re managing the side effects potentially on a long-term basis.”
It is critical to discuss the risks and benefits of this treatment with the family and caregivers and with the patient as well, if he old enough and able to participate in the decision-making progress. “We don’t want to give unrealistic expectations and we want people to be aware of the potential downside of this treatment,” he said. “This is a very complex discussion because the trajectory of the disease is so devastating and this treatment does hold out hope that other therapies don’t necessarily have.”
Nonpharmacologic Interventions
Physical therapy is a mainstay in DMD treatment, addressing protection of fragile muscles, preservation of strength, and prevention of muscle contractures.16 Given the respiratory impairments that occur with DMD progression, respiratory monitoring and therapy are essential; however, the number and type of evaluations and interventions vary with the stage of the disease, intensifying as the disease progresses.16 Similarly, cardiac monitoring should begin early, with patients screened for cardiac complications, and should intensify through the stages of disease progression.16
Bone health is compromised in patients with DMD, both as a result of corticosteroid treatment and as part of the disease itself. Fractures may be asymptomatic and may go unnoticed. Thus, bone health surveillance and maintenance are critical components of DMD management.16
Patients with DMD often experience gastrointestinal issues. They may experience weight gain because of lack of mobility and corticosteroid use in early stages, or weight loss as a result of diet or fluid imbalance, low bone density, or dysphagia in later stages. Patients should be closely followed by a nutritionist, a gastroenterologist as needed, and a physical therapist.16
Psychosocial support “should be developed and implemented across the lifespan in a manner that promotes thinking about the future and sets expectations that individuals will actively participate in their care and daily activities.”9 This includes psychological care, neuropsychological evaluations, and educational support.
Assisting Patients and Families Through the DMD Journey
DMD care is best delivered in a multidisciplinary setting, where physicians of relevant specialties, physical and occupational therapists, nutritionists, social workers, and genetic counselors collaborate. At Children’s Hospital of Philadelphia, DMD care is delivered through this collaborative model.
Unfortunately, Dr. Brandsema said, many patients don’t have this type of multidisciplinary resource available. “One specialist, such as a pulmonologist or neurologist, might have to be the sole source of care.” Or parents may have to ferry their child to multiple specialists in disparate locations, placing extra stress on an already-stressed family system.
“It’s helpful to connect the family with a comprehensive care center, if possible,” Dr. Brandsema advised. If that’s not available, then he suggests recommending educational opportunities and resources through national organizations such as the Muscular Dystrophy Association; Parent Project MD; NORD; Friends, Family and Duchenne; and Cure Duchenne. Families and caregivers, along with affected individuals, can get education and support from people who understand the day-to-day reality of living with this disease.
One of the major challenges that families face is navigating the high cost of treating DMD, especially the new medications, Dr. Brandsema said. “The authorization process can be intensive and long, and the family may need to take an active role, together with the provider team, in advocating for the patient to get access.”
Among her many responsibilities, Ms. Kaschak engages in care coordination tasks and management, helps patients and caregivers understand care plans, and provides psychosocial support and education about the disease process. She assists families in completing paperwork and navigating specialty authorizations, helping families understand and navigate the complex insurance process. “My role is to bridge gaps in care,” she said.
Dr. Brandsema noted that it’s important for couples to receive genetic counseling if they’re planning to have multiple children because there is a 50% chance that their next boy will be affected. About two thirds of mothers with children who have DMD are carriers, but many are not aware of it. Receiving counseling will enable them to understand their own risks of health complications, as well as the risk to future children.
Managing DMD Across the Lifespan
Another dimension of DMD care is providing resources and help to young people with DMD as they transition into adulthood. “In the past, we had limited treatment and mortality typically took place in the early 20s, so there weren’t a lot of patients who were adults. But as medication options have expanded and management of cardiac and respiratory failure has improved, we see a more significant proportion of adults who require adult-appropriate clinics — or, at the very least, specialists who are conversant in care or can provide care across the lifespan,” Dr. Brandsema said.
The DMD Care Considerations Working Group provides recommendations regarding care across the lifespan,9 as does the Adult North Star Network, of Muscular Dystrophy UK.17,18
Dr. Brandsema emphasized that, despite their disability, many adults with DMD “still engage with the community, and live life to its fullest.” It is to be hoped that, with ongoing research, earlier diagnosis, and improved treatment options, the future will look bright for people with DMD.
Dr. Brandsema has served as a consultant for Audentes, AveXis/Novartis, Biogen, Cytokinetics, Dyne, Edgewise, Fibrogen, Genentech, Marathon, Momenta/Janssen, NS Pharma, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Takeda, and WaVe. He is on the medical advisory council member for Cure SMA and is a site investigator for clinical trials with Alexion, Astellas, AveXis/Novartis, Biogen, Biohaven, Catabasis, CSL Behring, Cytokinetics, Dyne, Fibrogen, Genentech, Ionis, Lilly, ML Bio, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Summit, and WaVe. Ms. Kaschak has nothing to disclose.
References
1. Venugopal V and Pavlakis S. Duchenne Muscular Dystrophy. 2023 Jul 10. In: StatPearls [Internet]. Treasure Island, Florida: StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK482346/.
2. Gao QQ and McNally EM. Compr Physiol. 2015 Jul 1;5(3):1223-39. doi: 10.1002/cphy.c140048.
3. Duan D et al. Nat Rev Dis Primers. 2021 Feb 18;7(1):13. doi: 10.1038/s41572-021-00248-3.
4. Aartsma-Rus A et al. J Pediatr. 2019 Jan:204:305-313.e14. doi: 10.1016/j.jpeds.2018.10.043.
5. Broomfield J et al. Neurology. 2021 Dec 7;97(23):e2304-e2314. doi: 10.1212/WNL.0000000000012910.
6. Mercuri E et al. Front Pediatr. 2023 Nov 10:11:1276144. doi: 10.1212/WNL.0000000000012910.
7. Birnkrant DJ et al. Lancet Neurol. 2018 Mar;17(3):251-267. doi: 10.1016/S1474-4422(18)30024-3.
8. Birnkrant DJ et al. Lancet Neurol. 2018 Apr;17(4):347-361. doi: 10.1016/S1474-4422(18)30025-5.
9. Birnkrant DJ et al. Lancet Neurol. 2018 May;17(5):445-455. doi: 10.1016/S1474-4422(18)30026-7.
10. Matthews E et al. Cochrane Database Syst Rev. 2016 May 5;2016(5):CD003725. doi: 10.1002/14651858.CD003725.pub4.
11. Bylo M et al. Ann Pharmacother. 2020 Aug;54(8):788-794. doi: 10.1177/1060028019900500.
12. Guglieri M et al. JAMA Neurol. 2022 Oct 1;79(10):1005-1014. doi: 10.1001/jamaneurol.2022.2480.
13. Dang UJ et al. Neurology. 2024 Mar 12;102(5):e208112. doi: 10.1212/WNL.0000000000208112.
14. Mercuri E et al. Lancet Neurol. 2024 Apr;23(4):393-403. doi: 10.1016/S1474-4422(24)00036-X.
15. Gushchina LV et al. Mol Ther Nucleic Acids. 2022 Nov 9:30:479-492. doi: 10.1016/j.omtn.2022.10.025.
16. Patterson G et al. Eur J Pharmacol. 2023 May 15:947:175675. doi: 10.1016/j.ejphar.2023.175675.
17. Quinlivan R et al. J Neuromuscul Dis. 2021;8(6):899-926. doi: 10.3233/JND-200609.
18. Narayan S et al. J Neuromuscul Dis. 2022;9(3):365-381. doi: 10.3233/JND-210707.
Heat-Related Pediatric ED Visits More Than Double
ORLANDO – according to research presented at the annual meeting of the American Academy of Pediatrics (AAP).
“Our study really highlights the adverse effects that can come from extreme heat, and how increasing heat-related illness is affecting our children,” Taylor Merritt, MD, a pediatric resident at the University of Texas Southwestern Medical Center and Children’s Health in Dallas, said during a press briefing.

Underestimating the Problem?
Lori Byron, MD, a pediatrician from Red Lodge, Montana, who heads the AAP Chapter Climate Advocates program and was not involved in this research, was not surprised by the findings. “If anything, we’re vastly underestimating it because when people come in with heat exhaustion or heat smoke, that gets coded correctly, but when people come in with heart attacks, asthma attacks, strokes, and other exacerbations of chronic disease, it very rarely gets coded as a heat-related illness.”
Record-breaking summer temperatures from the changing climate have led to increased heat-related morbidity and mortality. Past research suggests that children and teens make up nearly half of all those affected by heat-related illnesses, she noted. 2023, for example, was the hottest year on record, and 2024 is predicted to be hotter, Dr. Merritt said.
A Sharp Increase in Cases
The retrospective study examined emergency department diagnoses during May-September from 2012-2023 at two large children’s hospitals within a north Texas pediatric health care system. The researchers compared heat-specific conditions with rhabdomyolysis encounters based on ICD-10 coding.
Heat-specific conditions include heatstroke/sunstroke, exertion heatstroke, heat syncope, heat crap, heat exhaustion, heat fatigue, heat edema, and exposure to excessive natural heat. Rhabdomyolysis encounters included both exertional and nonexertional rhabdomyolysis as well as non-traumatic rhabdomyolysis and elevated creatine kinase (CK) levels.
Among 542 heat-related encounters, 77% had heat-specific diagnoses and 24% had a rhabdomyolysis diagnosis. Combined, heat-related encounters increased 170% from 2012 to 2023, from 4.3 per 10,000 to 11.6 per 10,000 (P = .1). Summer months with higher peak temperatures were also associated with higher heat-related volume in the emergency department (P < .001).
Teenage boys were most likely to have rhabdomyolysis, with 82% of the cases occurring in boys and 70% in ages 12-18 (P < .001). “Compared to the rhabdomyolysis group, the heat-specific group was more likely to be younger, Hispanic, use government-based insurance, and live in an area with a lower Child Opportunity Index,” Dr. Merritt reported. “Most heat-specific encounters resulted in an ED discharge (96%), while most rhabdomyolysis encounters resulted in hospital admission (63%)” (P < .001).
”Thankfully, pediatric heat-related illness is still relatively rare,” Dr. Merritt said. “However, given the context of increasing temperatures, this is important for us all to know, anyone who cares for children, whether that be families or parents or pediatricians.”
Prevention Is Key
Dr. Byron noted that about half of AAP chapters now have climate committees, many of which have created educational materials on heat and wildfire smoke and on talking with athletes about risk of heat-related illnesses.
“A lot of the state high school sports associations are actually now adopting guidelines on when it’s safe to practice and when it’s safe to play for heat and for smoke, so that’s definitely something that we can talk to parents about and kids about,” Dr. Byron said. “Otherwise, you still have a lot of coaches and a lot of kids out there that think you’re just supposed to be tough and barrel through it.”
Rhabdomyolysis and heat stroke are both potentially deadly illnesses, so the biggest focus needs to be on prevention, Dr. Byron said. “Not just working with individuals in your office, but working within your school or within your state high school sports association is totally within the lane of a pediatrician to get involved.”
The research had no external funding. Dr. Merritt and Dr. Byron had no disclosures.
ORLANDO – according to research presented at the annual meeting of the American Academy of Pediatrics (AAP).
“Our study really highlights the adverse effects that can come from extreme heat, and how increasing heat-related illness is affecting our children,” Taylor Merritt, MD, a pediatric resident at the University of Texas Southwestern Medical Center and Children’s Health in Dallas, said during a press briefing.
Underestimating the Problem?
Lori Byron, MD, a pediatrician from Red Lodge, Montana, who heads the AAP Chapter Climate Advocates program and was not involved in this research, was not surprised by the findings. “If anything, we’re vastly underestimating it because when people come in with heat exhaustion or heat smoke, that gets coded correctly, but when people come in with heart attacks, asthma attacks, strokes, and other exacerbations of chronic disease, it very rarely gets coded as a heat-related illness.”
Record-breaking summer temperatures from the changing climate have led to increased heat-related morbidity and mortality. Past research suggests that children and teens make up nearly half of all those affected by heat-related illnesses, she noted. 2023, for example, was the hottest year on record, and 2024 is predicted to be hotter, Dr. Merritt said.
A Sharp Increase in Cases
The retrospective study examined emergency department diagnoses during May-September from 2012-2023 at two large children’s hospitals within a north Texas pediatric health care system. The researchers compared heat-specific conditions with rhabdomyolysis encounters based on ICD-10 coding.
Heat-specific conditions include heatstroke/sunstroke, exertion heatstroke, heat syncope, heat crap, heat exhaustion, heat fatigue, heat edema, and exposure to excessive natural heat. Rhabdomyolysis encounters included both exertional and nonexertional rhabdomyolysis as well as non-traumatic rhabdomyolysis and elevated creatine kinase (CK) levels.
Among 542 heat-related encounters, 77% had heat-specific diagnoses and 24% had a rhabdomyolysis diagnosis. Combined, heat-related encounters increased 170% from 2012 to 2023, from 4.3 per 10,000 to 11.6 per 10,000 (P = .1). Summer months with higher peak temperatures were also associated with higher heat-related volume in the emergency department (P < .001).
Teenage boys were most likely to have rhabdomyolysis, with 82% of the cases occurring in boys and 70% in ages 12-18 (P < .001). “Compared to the rhabdomyolysis group, the heat-specific group was more likely to be younger, Hispanic, use government-based insurance, and live in an area with a lower Child Opportunity Index,” Dr. Merritt reported. “Most heat-specific encounters resulted in an ED discharge (96%), while most rhabdomyolysis encounters resulted in hospital admission (63%)” (P < .001).
”Thankfully, pediatric heat-related illness is still relatively rare,” Dr. Merritt said. “However, given the context of increasing temperatures, this is important for us all to know, anyone who cares for children, whether that be families or parents or pediatricians.”
Prevention Is Key
Dr. Byron noted that about half of AAP chapters now have climate committees, many of which have created educational materials on heat and wildfire smoke and on talking with athletes about risk of heat-related illnesses.
“A lot of the state high school sports associations are actually now adopting guidelines on when it’s safe to practice and when it’s safe to play for heat and for smoke, so that’s definitely something that we can talk to parents about and kids about,” Dr. Byron said. “Otherwise, you still have a lot of coaches and a lot of kids out there that think you’re just supposed to be tough and barrel through it.”
Rhabdomyolysis and heat stroke are both potentially deadly illnesses, so the biggest focus needs to be on prevention, Dr. Byron said. “Not just working with individuals in your office, but working within your school or within your state high school sports association is totally within the lane of a pediatrician to get involved.”
The research had no external funding. Dr. Merritt and Dr. Byron had no disclosures.
ORLANDO – according to research presented at the annual meeting of the American Academy of Pediatrics (AAP).
“Our study really highlights the adverse effects that can come from extreme heat, and how increasing heat-related illness is affecting our children,” Taylor Merritt, MD, a pediatric resident at the University of Texas Southwestern Medical Center and Children’s Health in Dallas, said during a press briefing.
Underestimating the Problem?
Lori Byron, MD, a pediatrician from Red Lodge, Montana, who heads the AAP Chapter Climate Advocates program and was not involved in this research, was not surprised by the findings. “If anything, we’re vastly underestimating it because when people come in with heat exhaustion or heat smoke, that gets coded correctly, but when people come in with heart attacks, asthma attacks, strokes, and other exacerbations of chronic disease, it very rarely gets coded as a heat-related illness.”
Record-breaking summer temperatures from the changing climate have led to increased heat-related morbidity and mortality. Past research suggests that children and teens make up nearly half of all those affected by heat-related illnesses, she noted. 2023, for example, was the hottest year on record, and 2024 is predicted to be hotter, Dr. Merritt said.
A Sharp Increase in Cases
The retrospective study examined emergency department diagnoses during May-September from 2012-2023 at two large children’s hospitals within a north Texas pediatric health care system. The researchers compared heat-specific conditions with rhabdomyolysis encounters based on ICD-10 coding.
Heat-specific conditions include heatstroke/sunstroke, exertion heatstroke, heat syncope, heat crap, heat exhaustion, heat fatigue, heat edema, and exposure to excessive natural heat. Rhabdomyolysis encounters included both exertional and nonexertional rhabdomyolysis as well as non-traumatic rhabdomyolysis and elevated creatine kinase (CK) levels.
Among 542 heat-related encounters, 77% had heat-specific diagnoses and 24% had a rhabdomyolysis diagnosis. Combined, heat-related encounters increased 170% from 2012 to 2023, from 4.3 per 10,000 to 11.6 per 10,000 (P = .1). Summer months with higher peak temperatures were also associated with higher heat-related volume in the emergency department (P < .001).
Teenage boys were most likely to have rhabdomyolysis, with 82% of the cases occurring in boys and 70% in ages 12-18 (P < .001). “Compared to the rhabdomyolysis group, the heat-specific group was more likely to be younger, Hispanic, use government-based insurance, and live in an area with a lower Child Opportunity Index,” Dr. Merritt reported. “Most heat-specific encounters resulted in an ED discharge (96%), while most rhabdomyolysis encounters resulted in hospital admission (63%)” (P < .001).
”Thankfully, pediatric heat-related illness is still relatively rare,” Dr. Merritt said. “However, given the context of increasing temperatures, this is important for us all to know, anyone who cares for children, whether that be families or parents or pediatricians.”
Prevention Is Key
Dr. Byron noted that about half of AAP chapters now have climate committees, many of which have created educational materials on heat and wildfire smoke and on talking with athletes about risk of heat-related illnesses.
“A lot of the state high school sports associations are actually now adopting guidelines on when it’s safe to practice and when it’s safe to play for heat and for smoke, so that’s definitely something that we can talk to parents about and kids about,” Dr. Byron said. “Otherwise, you still have a lot of coaches and a lot of kids out there that think you’re just supposed to be tough and barrel through it.”
Rhabdomyolysis and heat stroke are both potentially deadly illnesses, so the biggest focus needs to be on prevention, Dr. Byron said. “Not just working with individuals in your office, but working within your school or within your state high school sports association is totally within the lane of a pediatrician to get involved.”
The research had no external funding. Dr. Merritt and Dr. Byron had no disclosures.
FROM AAP 2024
Newborn Screening Programs: What Do Clinicians Need to Know?
Newborn screening programs are public health services aimed at ensuring that the close to 4 million infants born each year in the United States are screened for certain serious disorders at birth. These disorders, albeit rare, are detected in roughly 12,500 newborn babies every year.
Newborn screening isn’t new, although it has expanded and transformed over the decades. The first newborn screening test was developed in the 1960s to detect phenylketonuria (PKU).1 Since then, the number of conditions screened for has increased, with programs in every US state and territory. “Newborn screening is well established now, not experimental or newfangled,” Wendy Chung, MD, PhD, professor of pediatrics, Harvard Medical School, Boston, Massachusetts, told Neurology Reviews.

In newborn screening, blood drawn from the baby’s heel is applied to specialized filter paper, which is then subjected to several analytical methods, including tandem mass spectrometry and molecular analyses to detect biomarkers for the diseases.2 More recently, genomic sequencing is being piloted as part of consented research studies.3
Newborn screening includes not only biochemical and genetic testing, but also includes noninvasive screening for hearing loss or for critical congenital heart disease using pulse oximetry. And newborn screening goes beyond analysis of a single drop of blood. Rather, “it’s an entire system, with the goal of identifying babies with genetic disorders who otherwise have no obvious symptoms,” said Dr. Chung. Left undetected and untreated, these conditions can be associated with serious adverse outcomes and even death.
Dr. Chung described newborn screening as a “one of the most successful public health programs, supporting health equity by screening almost every US baby after birth and then bringing timely treatments when relevant even before the baby develops symptoms of a disorder.” In this way, newborn screening has “saved lives and decreased disease burdens.”
There are at present 38 core conditions that the Department of Health and Human Services (HHS) regards as the most critical to screen for and 26 secondary conditions associated with these core disorders. This is called the Recommended Uniform Screening Panel (RUSP). Guidance regarding the most appropriate application of newborn screening tests, technologies and standards are provided by the Advisory Committee on Heritable Disorders in Newborns and Children (ACHDNC).
Each state “independently determines which screening tests are performed and what follow-up is provided.”4 Information about which tests are provided by which states can be found on the “Report Card” of the National Organization for Rare Diseases (NORD).
Challenges in Expanding the Current Newborn Screening
One of the major drawbacks in the current system is that “we don’t screen for enough diseases,” according to Zhanzhi Hu, PhD, of the Department of Systems Biology and the Department of Biomedical Information, Columbia University, New York City. “There are over 10,000 rare genetic diseases, but we’re currently screening for fewer than 100,” he told Neurology Reviews. Although in the United States, there are about 700-800 drugs approved for genetic diseases, “we can’t identify patients with these diseases early enough for the ideal window when treatments are most effective.”
Moreover, it’s a “lengthy process” to add new diseases to RUSP. “New conditions are added at the pace of less than one per year, on average — even for the hundreds of diseases for which there are treatments,” he said. “If we keep going at the current pace, we won’t be able to screen for those diseases for another few hundred years.”

Speeding up the pace of including new diseases in newborn screening is challenging because “we have more diseases than we have development dollars for,” Dr. Hu said. “Big pharmaceutical companies are reluctant to invest in rare diseases because the population is so small and it’s hard and expensive to develop such drugs. So if we can identify patients first, there will be more interest in developing treatments down the road.”
On the other hand, for trials to take place, these babies have to be identified in a timely manner — which requires testing. “Right now, we have a deadlock,” Dr. Hu said. “To nominate a disease, you need an approved treatment. But to get a treatment developed, you need to identify patients suitable for a clinical trial. If you have to wait for the symptoms to show up, the damage has already manifested and is irreversible. Our chance is to recognize the disease before symptom onset and then start treatment. I would call this a ‘chicken-and-egg’ problem.”
Dr. Hu is passionate about expanding newborn screening, and he has a very personal reason. Two of his children have a rare genetic disease. “My younger son, now 13 years old, was diagnosed at a much earlier age than my older son, although he had very few symptoms at the time, because his older brother was known to have the disease. As a result of this, his outcome was much better.” By contrast, Dr. Hu’s oldest son — now age 16 — wasn’t diagnosed until he became symptomatic.
His quest led him to join forces with Dr. Chung in conducting the Genomic Uniform-screening Against Rare Disease in All Newborns (Guardian) study, which screens newborns for more than 450 genetic conditions not currently screened as part of the standard newborn screening. To date, the study — which focuses on babies born in New York City — has screened about 11,000 infants.
“To accumulate enough evidence requires screening at least 100,000 babies because one requirement for nominating a disease for national inclusion in RUSP is an ‘N of 1’ study — meaning, to identify at least one positive patient using the proposed screening method in a prospective study,” Dr. Hu explained. “Most are rare diseases with an incidence rate of around one in 100,000. So getting to that magic number of 100,000 participants should enable us to hit that ‘N of 1’ for most diseases.”
The most challenging part, according to Dr. Hu, is the requirement of a prospective study, which means that you have to conduct a large-scale study enrolling tens of thousands of families and babies. If done for individual diseases (as has been the case in the past), “this is a huge cost and very inefficient.”
In reality, he added, the true incidence of these diseases is unclear. “Incidence rates are based on historical data rather than prospective studies. We’ve already seen some diseases show up more frequently than previously recorded, while others have shown up less frequently.”
For example, in the 11,000 babies screened to date, at least three girls with Rett syndrome have been identified, which is “quite a bit higher” than what has previously been identified in the literature (ie, one in 10,000-12,000 births). “This is a highly unmet need for these families because if you can initiate early treatment — at age 1, or even younger — the outcome will be better.”
He noted that there is at least one clinical trial underway for treating Rett syndrome, which has yielded “promising” data.5 “We’re hoping that by screening for diseases like Rett and identifying patients early, this will go hand-in-hand with clinical drug development. It can speed both the approval of the treatment and the addition to the newborn screening list,” Dr. Hu stated.
Screening and Drug Development Working in Tandem
Sequencing technologies have advanced and become more sophisticated as well as less costly, so interest in expanding newborn screening through newborn genome sequencing has increased. In fact, many states currently have incorporated genetic testing into newborn screening for conditions without biochemical markers. Additionally, newborn genomic sequencing is also used for further testing in infants with abnormal biochemical screening results.6
Genomic sequencing “identifies nucleotide changes that are the underlying etiology of monogenic disorders.”6 Its use could potentially enable identification of over 500 genetic disorders for which an newborn screening assay is not currently available, said Dr. Hu.
“Molecular DNA analysis has been integrated into newborn testing either as a first- or second-tier test for several conditions, including cystic fibrosis, severe combined immunodeficiency, and spinal muscular atrophy (SMA),” Dr. Hu said.
Dr. Hu pointed to SMA to illustrate the power and potential of newborn screening working hand-in-hand with the development of new treatments. SMA is a neurodegenerative disorder caused by mutations in SMN1, which encodes survival motor neuron protein (SMN).7 Deficiencies in SMN results in loss of motor neurons with muscle weakness and, often, early death.7A pilot study, on which Dr. Chung was the senior author, used both biochemical and genetic testing of close to 4000 newborns and found an SMA carrier frequency of 1.5%. One newborn was identified who had a homozygous SMN1 gene deletion and two copies of SMN2, strongly suggesting the presence of a severe type 1 SMA phenotype.8
At age 15 days, the baby was treated with nusinersen, an injection administered into the fluid surrounding the spinal cord, and the first FDA-approved genetic treatment for SMA. At the time of study publication, the baby was 12 months old, “meeting all developmental milestones and free of any respiratory issues,” the authors report.
“Screening for SMA — which was added to the RUSP in 2018 — has dramatically transformed what used to be the most common genetic cause of death in children under the age of 2,” Dr. Chung said. “Now, a once-and-done IV infusion of genetic therapy right after screening has transformed everything, taking what used to be a lethal condition and allowing children to grow up healthy.”
Advocating for Inclusion of Diseases With No Current Treatment
At present, any condition included in the RUSP is required to have a treatment, which can be dietary, surgical/procedural, or an FDA-approved drug-based agent. Unfortunately, a wide range of neurodevelopmental diseases still have no known treatments. But lack of availability of treatment shouldn’t invalidate a disease from being included in the RUSP, because even if there is no specific treatment for the condition itself, early intervention can still be initiated to prevent some of the manifestations of the condition, said Dr. Hu.
“For example, most patients with these diseases will sooner or later undergo seizures,” Dr. Hu remarked. “We know that repeated seizures can cause brain damage. If we can diagnose the disease before the seizures start to take place, we can put preventive seizure control interventions in place, even if there is no direct ‘treatment’ for the condition itself.”
Early identification can lead to early intervention, which can have other benefits, Dr. Hu noted. “If we train the brain at a young age, when the brain is most receptive, even though a disease may be progressive and will worsen, those abilities acquired earlier will last longer and remain in place longer. When these skills are acquired later, they’re forgotten sooner. This isn’t a ‘cure,’ but it will help with functional improvement.”
Moreover, parents are “interested in knowing that their child has a condition, even if no treatment is currently available for that disorder, according to our research,” Dr. Chung said. “We found that the parents we interviewed endorsed the nonmedical utility of having access to information, even in the absence of a ‘cure,’ so they could prepare for medical issues that might arise down the road and make informed choices.”9
Nina Gold, MD, director of Prenatal Medical Genetics and associate director for Research for Massachusetts General Brigham Personalized Medicine, Boston, obtained similar findings in her own research, which is currently under review for publication. “We conducted focus groups and one-on-one interviews with parents from diverse racial and socioeconomic backgrounds. At least one parent said they didn’t want to compare their child to other children if their child might have a different developmental trajectory. They stressed that the information would be helpful, even if there was no immediate clinical utility.”

Additionally, there are an “increasing number of fetal therapies for rare disorders, so information about a genetic disease in an older child can be helpful for parents who may go on to have another pregnancy,” Dr. Gold noted.
Dr. Hu detailed several other reasons for including a wider range of disorders in the RUSP. Doing so helps families avoid a “stressful and expensive diagnostic odyssey and also provides equitable access to a diagnosis.” And if these patients are identified early, “we can connect the family with clinical trials already underway or connect them to an organization such as the Accelerating Medicines Partnership (AMP) Program Bespoke Gene Therapy Consortium (AMP BGTC). Bespoke “brings together partners from the public, private, and nonprofit sectors to foster development of gene therapies intended to treat rare genetic diseases, which affect populations too small for viable commercial development.”
Next Steps Following Screening
Rebecca Sponberg, NP, of the Children’s Hospital of Orange County, UC Irvine School of Medicine, California, is part of a broader multidisciplinary team that interfaces with parents whose newborns have screened positive for a genetic disorder. The team also includes a biochemical geneticist, a pediatric neurologist, a pediatric endocrinologist, a genetic counselor, and a social worker.
Different states and locations have different procedures for receiving test results, said Dr. Chung. In some, pediatricians are the ones who receive the results, and they are tasked with the responsibility of making sure the children can start getting appropriate care. In particular, these pediatricians are associated with centers of excellence that specialize in working with families around these conditions. Other facilities have multidisciplinary teams.

Ms. Sponberg gave an example of how the process unfolded with X-linked adrenoleukodystrophy, a rare genetic disorder that affects the white matter of the nervous system and the adrenal cortex.10 “This is the most common peroxisomal disorder, affecting one in 20,000 males,” she said. “There are several different forms of the disorder, but males are most at risk for having the cerebral form, which can lead to neurological regression and hasten death. But the regression does not appear until 4 to 12 years of age.”
A baby who screens positive on the initial newborn screening has repeat testing; and if it’s confirmed, the family meets the entire team to help them understand what the disorder is, what to expect, and how it’s monitored and managed. “Children have to be followed closely with a brain MRI every 6 months to detect brain abnormalities quickly,” Ms. Sponberg explained “And we do regular bloodwork to look for adrenocortical insufficiency.”
A child who shows concerning changes on the MRI or abnormal blood test findings is immediately seen by the relevant specialist. “So far, our center has had one patient who had MRI changes consistent with the cerebral form of the disease and the patient was immediately able to receive a bone marrow transplant,” she reported. “We don’t think this child’s condition would have been picked up so quickly or treatment initiated so rapidly if we hadn’t known about it through newborn screening.”
Educating and Involving Families
Part of the role of clinicians is to provide education regarding newborn screening to families, according to Ms. Sponberg. “In my role, I have to call parents to tell them their child screened positive for a genetic condition and that we need to proceed with confirmatory testing,” she said. “We let them know if there’s a high concern that this might be a true positive for the condition, and we offer them information so they know what to expect.”
Unfortunately, Ms. Sponberg said, in the absence of education, some families are skeptical. “When I call families directly, some think it’s a scam and it can be hard to earn their trust. We need to do a better job educating families, especially our pregnant individuals, that testing will occur and if anything is abnormal, they will receive a call.”
References
1. Levy HL. Robert Guthrie and the Trials and Tribulations of Newborn Screening. Int J Neonatal Screen. 2021 Jan 19;7(1):5. doi: 10.3390/ijns7010005.
2. Chace DH et al. Clinical Chemistry and Dried Blood Spots: Increasing Laboratory Utilization by Improved Understanding of Quantitative Challenges. Bioanalysis. 2014;6(21):2791-2794. doi: 10.4155/bio.14.237.
3. Gold NB et al. Perspectives of Rare Disease Experts on Newborn Genome Sequencing. JAMA Netw Open. 2023 May 1;6(5):e2312231. doi: 10.1001/jamanetworkopen.2023.12231.
4. Weismiller DG. Expanded Newborn Screening: Information and Resources for the Family Physician. Am Fam Physician. 2017 Jun 1;95(11):703-709. https://www.aafp.org/pubs/afp/issues/2017/0601/p703.html.
5. Neul JL et al. Trofinetide for the Treatment of Rett Syndrome: A Randomized Phase 3 Study. Nat Med. 2023 Jun;29(6):1468-1475. doi: 10.1038/s41591-023-02398-1.
6. Chen T et al. Genomic Sequencing as a First-Tier Screening Test and Outcomes of Newborn Screening. JAMA Netw Open. 2023 Sep 5;6(9):e2331162. doi: 10.1001/jamanetworkopen.2023.31162.
7. Mercuri E et al. Spinal Muscular Atrophy. Nat Rev Dis Primers. 2022 Aug 4;8(1):52. doi: 10.1038/s41572-022-00380-8.
8. Kraszewski JN et al. Pilot Study of Population-Based Newborn Screening for Spinal Muscular Atrophy in New York State. Genet Med. 2018 Jun;20(6):608-613. doi: 10.1038/gim.2017.152.
9. Timmins GT et al. Diverse Parental Perspectives of the Social and Educational Needs for Expanding Newborn Screening Through Genomic Sequencing. Public Health Genomics. 2022 Sep 15:1-8. doi: 10.1159/000526382.
10. Turk BR et al. X-linked Adrenoleukodystrophy: Pathology, Pathophysiology, Diagnostic Testing, Newborn Screening and Therapies. Int J Dev Neurosci. 2020 Feb;80(1):52-72. doi: 10.1002/jdn.10003.
Newborn screening programs are public health services aimed at ensuring that the close to 4 million infants born each year in the United States are screened for certain serious disorders at birth. These disorders, albeit rare, are detected in roughly 12,500 newborn babies every year.
Newborn screening isn’t new, although it has expanded and transformed over the decades. The first newborn screening test was developed in the 1960s to detect phenylketonuria (PKU).1 Since then, the number of conditions screened for has increased, with programs in every US state and territory. “Newborn screening is well established now, not experimental or newfangled,” Wendy Chung, MD, PhD, professor of pediatrics, Harvard Medical School, Boston, Massachusetts, told Neurology Reviews.
In newborn screening, blood drawn from the baby’s heel is applied to specialized filter paper, which is then subjected to several analytical methods, including tandem mass spectrometry and molecular analyses to detect biomarkers for the diseases.2 More recently, genomic sequencing is being piloted as part of consented research studies.3
Newborn screening includes not only biochemical and genetic testing, but also includes noninvasive screening for hearing loss or for critical congenital heart disease using pulse oximetry. And newborn screening goes beyond analysis of a single drop of blood. Rather, “it’s an entire system, with the goal of identifying babies with genetic disorders who otherwise have no obvious symptoms,” said Dr. Chung. Left undetected and untreated, these conditions can be associated with serious adverse outcomes and even death.
Dr. Chung described newborn screening as a “one of the most successful public health programs, supporting health equity by screening almost every US baby after birth and then bringing timely treatments when relevant even before the baby develops symptoms of a disorder.” In this way, newborn screening has “saved lives and decreased disease burdens.”
There are at present 38 core conditions that the Department of Health and Human Services (HHS) regards as the most critical to screen for and 26 secondary conditions associated with these core disorders. This is called the Recommended Uniform Screening Panel (RUSP). Guidance regarding the most appropriate application of newborn screening tests, technologies and standards are provided by the Advisory Committee on Heritable Disorders in Newborns and Children (ACHDNC).
Each state “independently determines which screening tests are performed and what follow-up is provided.”4 Information about which tests are provided by which states can be found on the “Report Card” of the National Organization for Rare Diseases (NORD).
Challenges in Expanding the Current Newborn Screening
One of the major drawbacks in the current system is that “we don’t screen for enough diseases,” according to Zhanzhi Hu, PhD, of the Department of Systems Biology and the Department of Biomedical Information, Columbia University, New York City. “There are over 10,000 rare genetic diseases, but we’re currently screening for fewer than 100,” he told Neurology Reviews. Although in the United States, there are about 700-800 drugs approved for genetic diseases, “we can’t identify patients with these diseases early enough for the ideal window when treatments are most effective.”
Moreover, it’s a “lengthy process” to add new diseases to RUSP. “New conditions are added at the pace of less than one per year, on average — even for the hundreds of diseases for which there are treatments,” he said. “If we keep going at the current pace, we won’t be able to screen for those diseases for another few hundred years.”
Speeding up the pace of including new diseases in newborn screening is challenging because “we have more diseases than we have development dollars for,” Dr. Hu said. “Big pharmaceutical companies are reluctant to invest in rare diseases because the population is so small and it’s hard and expensive to develop such drugs. So if we can identify patients first, there will be more interest in developing treatments down the road.”
On the other hand, for trials to take place, these babies have to be identified in a timely manner — which requires testing. “Right now, we have a deadlock,” Dr. Hu said. “To nominate a disease, you need an approved treatment. But to get a treatment developed, you need to identify patients suitable for a clinical trial. If you have to wait for the symptoms to show up, the damage has already manifested and is irreversible. Our chance is to recognize the disease before symptom onset and then start treatment. I would call this a ‘chicken-and-egg’ problem.”
Dr. Hu is passionate about expanding newborn screening, and he has a very personal reason. Two of his children have a rare genetic disease. “My younger son, now 13 years old, was diagnosed at a much earlier age than my older son, although he had very few symptoms at the time, because his older brother was known to have the disease. As a result of this, his outcome was much better.” By contrast, Dr. Hu’s oldest son — now age 16 — wasn’t diagnosed until he became symptomatic.
His quest led him to join forces with Dr. Chung in conducting the Genomic Uniform-screening Against Rare Disease in All Newborns (Guardian) study, which screens newborns for more than 450 genetic conditions not currently screened as part of the standard newborn screening. To date, the study — which focuses on babies born in New York City — has screened about 11,000 infants.
“To accumulate enough evidence requires screening at least 100,000 babies because one requirement for nominating a disease for national inclusion in RUSP is an ‘N of 1’ study — meaning, to identify at least one positive patient using the proposed screening method in a prospective study,” Dr. Hu explained. “Most are rare diseases with an incidence rate of around one in 100,000. So getting to that magic number of 100,000 participants should enable us to hit that ‘N of 1’ for most diseases.”
The most challenging part, according to Dr. Hu, is the requirement of a prospective study, which means that you have to conduct a large-scale study enrolling tens of thousands of families and babies. If done for individual diseases (as has been the case in the past), “this is a huge cost and very inefficient.”
In reality, he added, the true incidence of these diseases is unclear. “Incidence rates are based on historical data rather than prospective studies. We’ve already seen some diseases show up more frequently than previously recorded, while others have shown up less frequently.”
For example, in the 11,000 babies screened to date, at least three girls with Rett syndrome have been identified, which is “quite a bit higher” than what has previously been identified in the literature (ie, one in 10,000-12,000 births). “This is a highly unmet need for these families because if you can initiate early treatment — at age 1, or even younger — the outcome will be better.”
He noted that there is at least one clinical trial underway for treating Rett syndrome, which has yielded “promising” data.5 “We’re hoping that by screening for diseases like Rett and identifying patients early, this will go hand-in-hand with clinical drug development. It can speed both the approval of the treatment and the addition to the newborn screening list,” Dr. Hu stated.
Screening and Drug Development Working in Tandem
Sequencing technologies have advanced and become more sophisticated as well as less costly, so interest in expanding newborn screening through newborn genome sequencing has increased. In fact, many states currently have incorporated genetic testing into newborn screening for conditions without biochemical markers. Additionally, newborn genomic sequencing is also used for further testing in infants with abnormal biochemical screening results.6
Genomic sequencing “identifies nucleotide changes that are the underlying etiology of monogenic disorders.”6 Its use could potentially enable identification of over 500 genetic disorders for which an newborn screening assay is not currently available, said Dr. Hu.
“Molecular DNA analysis has been integrated into newborn testing either as a first- or second-tier test for several conditions, including cystic fibrosis, severe combined immunodeficiency, and spinal muscular atrophy (SMA),” Dr. Hu said.
Dr. Hu pointed to SMA to illustrate the power and potential of newborn screening working hand-in-hand with the development of new treatments. SMA is a neurodegenerative disorder caused by mutations in SMN1, which encodes survival motor neuron protein (SMN).7 Deficiencies in SMN results in loss of motor neurons with muscle weakness and, often, early death.7A pilot study, on which Dr. Chung was the senior author, used both biochemical and genetic testing of close to 4000 newborns and found an SMA carrier frequency of 1.5%. One newborn was identified who had a homozygous SMN1 gene deletion and two copies of SMN2, strongly suggesting the presence of a severe type 1 SMA phenotype.8
At age 15 days, the baby was treated with nusinersen, an injection administered into the fluid surrounding the spinal cord, and the first FDA-approved genetic treatment for SMA. At the time of study publication, the baby was 12 months old, “meeting all developmental milestones and free of any respiratory issues,” the authors report.
“Screening for SMA — which was added to the RUSP in 2018 — has dramatically transformed what used to be the most common genetic cause of death in children under the age of 2,” Dr. Chung said. “Now, a once-and-done IV infusion of genetic therapy right after screening has transformed everything, taking what used to be a lethal condition and allowing children to grow up healthy.”
Advocating for Inclusion of Diseases With No Current Treatment
At present, any condition included in the RUSP is required to have a treatment, which can be dietary, surgical/procedural, or an FDA-approved drug-based agent. Unfortunately, a wide range of neurodevelopmental diseases still have no known treatments. But lack of availability of treatment shouldn’t invalidate a disease from being included in the RUSP, because even if there is no specific treatment for the condition itself, early intervention can still be initiated to prevent some of the manifestations of the condition, said Dr. Hu.
“For example, most patients with these diseases will sooner or later undergo seizures,” Dr. Hu remarked. “We know that repeated seizures can cause brain damage. If we can diagnose the disease before the seizures start to take place, we can put preventive seizure control interventions in place, even if there is no direct ‘treatment’ for the condition itself.”
Early identification can lead to early intervention, which can have other benefits, Dr. Hu noted. “If we train the brain at a young age, when the brain is most receptive, even though a disease may be progressive and will worsen, those abilities acquired earlier will last longer and remain in place longer. When these skills are acquired later, they’re forgotten sooner. This isn’t a ‘cure,’ but it will help with functional improvement.”
Moreover, parents are “interested in knowing that their child has a condition, even if no treatment is currently available for that disorder, according to our research,” Dr. Chung said. “We found that the parents we interviewed endorsed the nonmedical utility of having access to information, even in the absence of a ‘cure,’ so they could prepare for medical issues that might arise down the road and make informed choices.”9
Nina Gold, MD, director of Prenatal Medical Genetics and associate director for Research for Massachusetts General Brigham Personalized Medicine, Boston, obtained similar findings in her own research, which is currently under review for publication. “We conducted focus groups and one-on-one interviews with parents from diverse racial and socioeconomic backgrounds. At least one parent said they didn’t want to compare their child to other children if their child might have a different developmental trajectory. They stressed that the information would be helpful, even if there was no immediate clinical utility.”
Additionally, there are an “increasing number of fetal therapies for rare disorders, so information about a genetic disease in an older child can be helpful for parents who may go on to have another pregnancy,” Dr. Gold noted.
Dr. Hu detailed several other reasons for including a wider range of disorders in the RUSP. Doing so helps families avoid a “stressful and expensive diagnostic odyssey and also provides equitable access to a diagnosis.” And if these patients are identified early, “we can connect the family with clinical trials already underway or connect them to an organization such as the Accelerating Medicines Partnership (AMP) Program Bespoke Gene Therapy Consortium (AMP BGTC). Bespoke “brings together partners from the public, private, and nonprofit sectors to foster development of gene therapies intended to treat rare genetic diseases, which affect populations too small for viable commercial development.”
Next Steps Following Screening
Rebecca Sponberg, NP, of the Children’s Hospital of Orange County, UC Irvine School of Medicine, California, is part of a broader multidisciplinary team that interfaces with parents whose newborns have screened positive for a genetic disorder. The team also includes a biochemical geneticist, a pediatric neurologist, a pediatric endocrinologist, a genetic counselor, and a social worker.
Different states and locations have different procedures for receiving test results, said Dr. Chung. In some, pediatricians are the ones who receive the results, and they are tasked with the responsibility of making sure the children can start getting appropriate care. In particular, these pediatricians are associated with centers of excellence that specialize in working with families around these conditions. Other facilities have multidisciplinary teams.
Ms. Sponberg gave an example of how the process unfolded with X-linked adrenoleukodystrophy, a rare genetic disorder that affects the white matter of the nervous system and the adrenal cortex.10 “This is the most common peroxisomal disorder, affecting one in 20,000 males,” she said. “There are several different forms of the disorder, but males are most at risk for having the cerebral form, which can lead to neurological regression and hasten death. But the regression does not appear until 4 to 12 years of age.”
A baby who screens positive on the initial newborn screening has repeat testing; and if it’s confirmed, the family meets the entire team to help them understand what the disorder is, what to expect, and how it’s monitored and managed. “Children have to be followed closely with a brain MRI every 6 months to detect brain abnormalities quickly,” Ms. Sponberg explained “And we do regular bloodwork to look for adrenocortical insufficiency.”
A child who shows concerning changes on the MRI or abnormal blood test findings is immediately seen by the relevant specialist. “So far, our center has had one patient who had MRI changes consistent with the cerebral form of the disease and the patient was immediately able to receive a bone marrow transplant,” she reported. “We don’t think this child’s condition would have been picked up so quickly or treatment initiated so rapidly if we hadn’t known about it through newborn screening.”
Educating and Involving Families
Part of the role of clinicians is to provide education regarding newborn screening to families, according to Ms. Sponberg. “In my role, I have to call parents to tell them their child screened positive for a genetic condition and that we need to proceed with confirmatory testing,” she said. “We let them know if there’s a high concern that this might be a true positive for the condition, and we offer them information so they know what to expect.”
Unfortunately, Ms. Sponberg said, in the absence of education, some families are skeptical. “When I call families directly, some think it’s a scam and it can be hard to earn their trust. We need to do a better job educating families, especially our pregnant individuals, that testing will occur and if anything is abnormal, they will receive a call.”
References
1. Levy HL. Robert Guthrie and the Trials and Tribulations of Newborn Screening. Int J Neonatal Screen. 2021 Jan 19;7(1):5. doi: 10.3390/ijns7010005.
2. Chace DH et al. Clinical Chemistry and Dried Blood Spots: Increasing Laboratory Utilization by Improved Understanding of Quantitative Challenges. Bioanalysis. 2014;6(21):2791-2794. doi: 10.4155/bio.14.237.
3. Gold NB et al. Perspectives of Rare Disease Experts on Newborn Genome Sequencing. JAMA Netw Open. 2023 May 1;6(5):e2312231. doi: 10.1001/jamanetworkopen.2023.12231.
4. Weismiller DG. Expanded Newborn Screening: Information and Resources for the Family Physician. Am Fam Physician. 2017 Jun 1;95(11):703-709. https://www.aafp.org/pubs/afp/issues/2017/0601/p703.html.
5. Neul JL et al. Trofinetide for the Treatment of Rett Syndrome: A Randomized Phase 3 Study. Nat Med. 2023 Jun;29(6):1468-1475. doi: 10.1038/s41591-023-02398-1.
6. Chen T et al. Genomic Sequencing as a First-Tier Screening Test and Outcomes of Newborn Screening. JAMA Netw Open. 2023 Sep 5;6(9):e2331162. doi: 10.1001/jamanetworkopen.2023.31162.
7. Mercuri E et al. Spinal Muscular Atrophy. Nat Rev Dis Primers. 2022 Aug 4;8(1):52. doi: 10.1038/s41572-022-00380-8.
8. Kraszewski JN et al. Pilot Study of Population-Based Newborn Screening for Spinal Muscular Atrophy in New York State. Genet Med. 2018 Jun;20(6):608-613. doi: 10.1038/gim.2017.152.
9. Timmins GT et al. Diverse Parental Perspectives of the Social and Educational Needs for Expanding Newborn Screening Through Genomic Sequencing. Public Health Genomics. 2022 Sep 15:1-8. doi: 10.1159/000526382.
10. Turk BR et al. X-linked Adrenoleukodystrophy: Pathology, Pathophysiology, Diagnostic Testing, Newborn Screening and Therapies. Int J Dev Neurosci. 2020 Feb;80(1):52-72. doi: 10.1002/jdn.10003.
Newborn screening programs are public health services aimed at ensuring that the close to 4 million infants born each year in the United States are screened for certain serious disorders at birth. These disorders, albeit rare, are detected in roughly 12,500 newborn babies every year.
Newborn screening isn’t new, although it has expanded and transformed over the decades. The first newborn screening test was developed in the 1960s to detect phenylketonuria (PKU).1 Since then, the number of conditions screened for has increased, with programs in every US state and territory. “Newborn screening is well established now, not experimental or newfangled,” Wendy Chung, MD, PhD, professor of pediatrics, Harvard Medical School, Boston, Massachusetts, told Neurology Reviews.
In newborn screening, blood drawn from the baby’s heel is applied to specialized filter paper, which is then subjected to several analytical methods, including tandem mass spectrometry and molecular analyses to detect biomarkers for the diseases.2 More recently, genomic sequencing is being piloted as part of consented research studies.3
Newborn screening includes not only biochemical and genetic testing, but also includes noninvasive screening for hearing loss or for critical congenital heart disease using pulse oximetry. And newborn screening goes beyond analysis of a single drop of blood. Rather, “it’s an entire system, with the goal of identifying babies with genetic disorders who otherwise have no obvious symptoms,” said Dr. Chung. Left undetected and untreated, these conditions can be associated with serious adverse outcomes and even death.
Dr. Chung described newborn screening as a “one of the most successful public health programs, supporting health equity by screening almost every US baby after birth and then bringing timely treatments when relevant even before the baby develops symptoms of a disorder.” In this way, newborn screening has “saved lives and decreased disease burdens.”
There are at present 38 core conditions that the Department of Health and Human Services (HHS) regards as the most critical to screen for and 26 secondary conditions associated with these core disorders. This is called the Recommended Uniform Screening Panel (RUSP). Guidance regarding the most appropriate application of newborn screening tests, technologies and standards are provided by the Advisory Committee on Heritable Disorders in Newborns and Children (ACHDNC).
Each state “independently determines which screening tests are performed and what follow-up is provided.”4 Information about which tests are provided by which states can be found on the “Report Card” of the National Organization for Rare Diseases (NORD).
Challenges in Expanding the Current Newborn Screening
One of the major drawbacks in the current system is that “we don’t screen for enough diseases,” according to Zhanzhi Hu, PhD, of the Department of Systems Biology and the Department of Biomedical Information, Columbia University, New York City. “There are over 10,000 rare genetic diseases, but we’re currently screening for fewer than 100,” he told Neurology Reviews. Although in the United States, there are about 700-800 drugs approved for genetic diseases, “we can’t identify patients with these diseases early enough for the ideal window when treatments are most effective.”
Moreover, it’s a “lengthy process” to add new diseases to RUSP. “New conditions are added at the pace of less than one per year, on average — even for the hundreds of diseases for which there are treatments,” he said. “If we keep going at the current pace, we won’t be able to screen for those diseases for another few hundred years.”
Speeding up the pace of including new diseases in newborn screening is challenging because “we have more diseases than we have development dollars for,” Dr. Hu said. “Big pharmaceutical companies are reluctant to invest in rare diseases because the population is so small and it’s hard and expensive to develop such drugs. So if we can identify patients first, there will be more interest in developing treatments down the road.”
On the other hand, for trials to take place, these babies have to be identified in a timely manner — which requires testing. “Right now, we have a deadlock,” Dr. Hu said. “To nominate a disease, you need an approved treatment. But to get a treatment developed, you need to identify patients suitable for a clinical trial. If you have to wait for the symptoms to show up, the damage has already manifested and is irreversible. Our chance is to recognize the disease before symptom onset and then start treatment. I would call this a ‘chicken-and-egg’ problem.”
Dr. Hu is passionate about expanding newborn screening, and he has a very personal reason. Two of his children have a rare genetic disease. “My younger son, now 13 years old, was diagnosed at a much earlier age than my older son, although he had very few symptoms at the time, because his older brother was known to have the disease. As a result of this, his outcome was much better.” By contrast, Dr. Hu’s oldest son — now age 16 — wasn’t diagnosed until he became symptomatic.
His quest led him to join forces with Dr. Chung in conducting the Genomic Uniform-screening Against Rare Disease in All Newborns (Guardian) study, which screens newborns for more than 450 genetic conditions not currently screened as part of the standard newborn screening. To date, the study — which focuses on babies born in New York City — has screened about 11,000 infants.
“To accumulate enough evidence requires screening at least 100,000 babies because one requirement for nominating a disease for national inclusion in RUSP is an ‘N of 1’ study — meaning, to identify at least one positive patient using the proposed screening method in a prospective study,” Dr. Hu explained. “Most are rare diseases with an incidence rate of around one in 100,000. So getting to that magic number of 100,000 participants should enable us to hit that ‘N of 1’ for most diseases.”
The most challenging part, according to Dr. Hu, is the requirement of a prospective study, which means that you have to conduct a large-scale study enrolling tens of thousands of families and babies. If done for individual diseases (as has been the case in the past), “this is a huge cost and very inefficient.”
In reality, he added, the true incidence of these diseases is unclear. “Incidence rates are based on historical data rather than prospective studies. We’ve already seen some diseases show up more frequently than previously recorded, while others have shown up less frequently.”
For example, in the 11,000 babies screened to date, at least three girls with Rett syndrome have been identified, which is “quite a bit higher” than what has previously been identified in the literature (ie, one in 10,000-12,000 births). “This is a highly unmet need for these families because if you can initiate early treatment — at age 1, or even younger — the outcome will be better.”
He noted that there is at least one clinical trial underway for treating Rett syndrome, which has yielded “promising” data.5 “We’re hoping that by screening for diseases like Rett and identifying patients early, this will go hand-in-hand with clinical drug development. It can speed both the approval of the treatment and the addition to the newborn screening list,” Dr. Hu stated.
Screening and Drug Development Working in Tandem
Sequencing technologies have advanced and become more sophisticated as well as less costly, so interest in expanding newborn screening through newborn genome sequencing has increased. In fact, many states currently have incorporated genetic testing into newborn screening for conditions without biochemical markers. Additionally, newborn genomic sequencing is also used for further testing in infants with abnormal biochemical screening results.6
Genomic sequencing “identifies nucleotide changes that are the underlying etiology of monogenic disorders.”6 Its use could potentially enable identification of over 500 genetic disorders for which an newborn screening assay is not currently available, said Dr. Hu.
“Molecular DNA analysis has been integrated into newborn testing either as a first- or second-tier test for several conditions, including cystic fibrosis, severe combined immunodeficiency, and spinal muscular atrophy (SMA),” Dr. Hu said.
Dr. Hu pointed to SMA to illustrate the power and potential of newborn screening working hand-in-hand with the development of new treatments. SMA is a neurodegenerative disorder caused by mutations in SMN1, which encodes survival motor neuron protein (SMN).7 Deficiencies in SMN results in loss of motor neurons with muscle weakness and, often, early death.7A pilot study, on which Dr. Chung was the senior author, used both biochemical and genetic testing of close to 4000 newborns and found an SMA carrier frequency of 1.5%. One newborn was identified who had a homozygous SMN1 gene deletion and two copies of SMN2, strongly suggesting the presence of a severe type 1 SMA phenotype.8
At age 15 days, the baby was treated with nusinersen, an injection administered into the fluid surrounding the spinal cord, and the first FDA-approved genetic treatment for SMA. At the time of study publication, the baby was 12 months old, “meeting all developmental milestones and free of any respiratory issues,” the authors report.
“Screening for SMA — which was added to the RUSP in 2018 — has dramatically transformed what used to be the most common genetic cause of death in children under the age of 2,” Dr. Chung said. “Now, a once-and-done IV infusion of genetic therapy right after screening has transformed everything, taking what used to be a lethal condition and allowing children to grow up healthy.”
Advocating for Inclusion of Diseases With No Current Treatment
At present, any condition included in the RUSP is required to have a treatment, which can be dietary, surgical/procedural, or an FDA-approved drug-based agent. Unfortunately, a wide range of neurodevelopmental diseases still have no known treatments. But lack of availability of treatment shouldn’t invalidate a disease from being included in the RUSP, because even if there is no specific treatment for the condition itself, early intervention can still be initiated to prevent some of the manifestations of the condition, said Dr. Hu.
“For example, most patients with these diseases will sooner or later undergo seizures,” Dr. Hu remarked. “We know that repeated seizures can cause brain damage. If we can diagnose the disease before the seizures start to take place, we can put preventive seizure control interventions in place, even if there is no direct ‘treatment’ for the condition itself.”
Early identification can lead to early intervention, which can have other benefits, Dr. Hu noted. “If we train the brain at a young age, when the brain is most receptive, even though a disease may be progressive and will worsen, those abilities acquired earlier will last longer and remain in place longer. When these skills are acquired later, they’re forgotten sooner. This isn’t a ‘cure,’ but it will help with functional improvement.”
Moreover, parents are “interested in knowing that their child has a condition, even if no treatment is currently available for that disorder, according to our research,” Dr. Chung said. “We found that the parents we interviewed endorsed the nonmedical utility of having access to information, even in the absence of a ‘cure,’ so they could prepare for medical issues that might arise down the road and make informed choices.”9
Nina Gold, MD, director of Prenatal Medical Genetics and associate director for Research for Massachusetts General Brigham Personalized Medicine, Boston, obtained similar findings in her own research, which is currently under review for publication. “We conducted focus groups and one-on-one interviews with parents from diverse racial and socioeconomic backgrounds. At least one parent said they didn’t want to compare their child to other children if their child might have a different developmental trajectory. They stressed that the information would be helpful, even if there was no immediate clinical utility.”
Additionally, there are an “increasing number of fetal therapies for rare disorders, so information about a genetic disease in an older child can be helpful for parents who may go on to have another pregnancy,” Dr. Gold noted.
Dr. Hu detailed several other reasons for including a wider range of disorders in the RUSP. Doing so helps families avoid a “stressful and expensive diagnostic odyssey and also provides equitable access to a diagnosis.” And if these patients are identified early, “we can connect the family with clinical trials already underway or connect them to an organization such as the Accelerating Medicines Partnership (AMP) Program Bespoke Gene Therapy Consortium (AMP BGTC). Bespoke “brings together partners from the public, private, and nonprofit sectors to foster development of gene therapies intended to treat rare genetic diseases, which affect populations too small for viable commercial development.”
Next Steps Following Screening
Rebecca Sponberg, NP, of the Children’s Hospital of Orange County, UC Irvine School of Medicine, California, is part of a broader multidisciplinary team that interfaces with parents whose newborns have screened positive for a genetic disorder. The team also includes a biochemical geneticist, a pediatric neurologist, a pediatric endocrinologist, a genetic counselor, and a social worker.
Different states and locations have different procedures for receiving test results, said Dr. Chung. In some, pediatricians are the ones who receive the results, and they are tasked with the responsibility of making sure the children can start getting appropriate care. In particular, these pediatricians are associated with centers of excellence that specialize in working with families around these conditions. Other facilities have multidisciplinary teams.
Ms. Sponberg gave an example of how the process unfolded with X-linked adrenoleukodystrophy, a rare genetic disorder that affects the white matter of the nervous system and the adrenal cortex.10 “This is the most common peroxisomal disorder, affecting one in 20,000 males,” she said. “There are several different forms of the disorder, but males are most at risk for having the cerebral form, which can lead to neurological regression and hasten death. But the regression does not appear until 4 to 12 years of age.”
A baby who screens positive on the initial newborn screening has repeat testing; and if it’s confirmed, the family meets the entire team to help them understand what the disorder is, what to expect, and how it’s monitored and managed. “Children have to be followed closely with a brain MRI every 6 months to detect brain abnormalities quickly,” Ms. Sponberg explained “And we do regular bloodwork to look for adrenocortical insufficiency.”
A child who shows concerning changes on the MRI or abnormal blood test findings is immediately seen by the relevant specialist. “So far, our center has had one patient who had MRI changes consistent with the cerebral form of the disease and the patient was immediately able to receive a bone marrow transplant,” she reported. “We don’t think this child’s condition would have been picked up so quickly or treatment initiated so rapidly if we hadn’t known about it through newborn screening.”
Educating and Involving Families
Part of the role of clinicians is to provide education regarding newborn screening to families, according to Ms. Sponberg. “In my role, I have to call parents to tell them their child screened positive for a genetic condition and that we need to proceed with confirmatory testing,” she said. “We let them know if there’s a high concern that this might be a true positive for the condition, and we offer them information so they know what to expect.”
Unfortunately, Ms. Sponberg said, in the absence of education, some families are skeptical. “When I call families directly, some think it’s a scam and it can be hard to earn their trust. We need to do a better job educating families, especially our pregnant individuals, that testing will occur and if anything is abnormal, they will receive a call.”
References
1. Levy HL. Robert Guthrie and the Trials and Tribulations of Newborn Screening. Int J Neonatal Screen. 2021 Jan 19;7(1):5. doi: 10.3390/ijns7010005.
2. Chace DH et al. Clinical Chemistry and Dried Blood Spots: Increasing Laboratory Utilization by Improved Understanding of Quantitative Challenges. Bioanalysis. 2014;6(21):2791-2794. doi: 10.4155/bio.14.237.
3. Gold NB et al. Perspectives of Rare Disease Experts on Newborn Genome Sequencing. JAMA Netw Open. 2023 May 1;6(5):e2312231. doi: 10.1001/jamanetworkopen.2023.12231.
4. Weismiller DG. Expanded Newborn Screening: Information and Resources for the Family Physician. Am Fam Physician. 2017 Jun 1;95(11):703-709. https://www.aafp.org/pubs/afp/issues/2017/0601/p703.html.
5. Neul JL et al. Trofinetide for the Treatment of Rett Syndrome: A Randomized Phase 3 Study. Nat Med. 2023 Jun;29(6):1468-1475. doi: 10.1038/s41591-023-02398-1.
6. Chen T et al. Genomic Sequencing as a First-Tier Screening Test and Outcomes of Newborn Screening. JAMA Netw Open. 2023 Sep 5;6(9):e2331162. doi: 10.1001/jamanetworkopen.2023.31162.
7. Mercuri E et al. Spinal Muscular Atrophy. Nat Rev Dis Primers. 2022 Aug 4;8(1):52. doi: 10.1038/s41572-022-00380-8.
8. Kraszewski JN et al. Pilot Study of Population-Based Newborn Screening for Spinal Muscular Atrophy in New York State. Genet Med. 2018 Jun;20(6):608-613. doi: 10.1038/gim.2017.152.
9. Timmins GT et al. Diverse Parental Perspectives of the Social and Educational Needs for Expanding Newborn Screening Through Genomic Sequencing. Public Health Genomics. 2022 Sep 15:1-8. doi: 10.1159/000526382.
10. Turk BR et al. X-linked Adrenoleukodystrophy: Pathology, Pathophysiology, Diagnostic Testing, Newborn Screening and Therapies. Int J Dev Neurosci. 2020 Feb;80(1):52-72. doi: 10.1002/jdn.10003.
Aspects of the Skin Microbiome Remain Elusive
SAN DIEGO — Although it has been known for several years that
In one review of the topic, researchers from the National Institutes of Health wrote that the skin is composed of 1.8 million diverse habitats with an abundance of folds, invaginations, and specialized niches that support a wide range of microorganisms. “Many of these microorganisms are harmless and, in some cases, provide vital functions for us to live and they have not evolved over time,” Jill S. Waibel, MD, medical director of the Miami Dermatology and Laser Institute, said at the annual Masters of Aesthetics Symposium.

“This is complex ecosystem that we don’t really talk about,” she said. “There is wide topographical distribution of bacteria on skin sites. The bacteria we have on our head and neck area is different from that on our feet. There is also a lot of interpersonal variation of the skin microbiome, so one person may have a lot of one type of bacteria and not as much of another.”
A Shield From Foreign Pathogens
At its core, Dr. Waibel continued, the skin microbiome functions as an interface between the human body and the environment, a physical barrier that prevents the invasion of foreign pathogens. The skin also provides a home to commensal microbiota. She likened the skin’s landscape to that of the tundra: “It’s desiccated, has poor nutrients, and it’s very acidic, thus pathogens have a hard time living on it,” she said. “However, our skin microorganisms have adapted to utilize the sparse nutrients available on the skin. That’s why I tell my patients, ‘don’t use a sugar scrub because you’re potentially feeding these bad bacteria.’ ”
According to more recent research, the skin microbiota in healthy adults remains stable over time, despite environmental perturbations, and they have important roles in educating the innate and adaptive arms of the cutaneous immune system. “Some skin diseases are associated with an altered microbial state: dysbiosis,” said Dr. Waibel, subsection chief of dermatology at Baptist Health South Florida, Miami Beach. “Reversion of this may help prevent or treat the disease.”

She cited the following factors that influence the skin microbiome:
- Genetics affects the skin microbiome considerably. Individuals with autoimmune predispositions have different microbiota compared with those who don’t.
- Climate, pollution, and hygiene practices the other influencing factors. “Even clothing can impact the microbiome, by causing the transfer of microorganisms,” she said.
- Age and hormonal changes (particularly during puberty) and senescence alter the microbial landscape.
- Systemic health conditions such as diabetes mellitus and irritable bowel disease, as well as cutaneous conditions like psoriasis and atopic dermatitis can also disrupt the skin microbiome.
Ingredients contained in soaps, antibiotics, and cosmetics can also cause skin dysbiosis, Dr. Waibel said. However, the integrity of the skin’s microbiome following dermatological procedures such as excisions, dermabrasion, laser therapy, and other physical procedures is less understood, according to a recent review of the topic. Phototherapy appears to be the most extensively studied, “and shows an increase in microbial diversity post-treatment,” she said. “Light treatments have been found to kill bacteria by inducing DNA damage. More studies need to be performed on specific wavelengths of light used, conditions being treated and individual patient differences.”
According to the review’s authors, no change in the microbiome was observed in studies of debridement. “That was surprising, as it is a method to remove unhealthy tissue that often contains pathogenic bacteria,” Dr. Waibel said. “The big take-home message is that we need more research.”
Dr. Waibel disclosed that she has conducted clinical trials for several device and pharmaceutical companies.
A version of this article first appeared on Medscape.com.
SAN DIEGO — Although it has been known for several years that
In one review of the topic, researchers from the National Institutes of Health wrote that the skin is composed of 1.8 million diverse habitats with an abundance of folds, invaginations, and specialized niches that support a wide range of microorganisms. “Many of these microorganisms are harmless and, in some cases, provide vital functions for us to live and they have not evolved over time,” Jill S. Waibel, MD, medical director of the Miami Dermatology and Laser Institute, said at the annual Masters of Aesthetics Symposium.
“This is complex ecosystem that we don’t really talk about,” she said. “There is wide topographical distribution of bacteria on skin sites. The bacteria we have on our head and neck area is different from that on our feet. There is also a lot of interpersonal variation of the skin microbiome, so one person may have a lot of one type of bacteria and not as much of another.”
A Shield From Foreign Pathogens
At its core, Dr. Waibel continued, the skin microbiome functions as an interface between the human body and the environment, a physical barrier that prevents the invasion of foreign pathogens. The skin also provides a home to commensal microbiota. She likened the skin’s landscape to that of the tundra: “It’s desiccated, has poor nutrients, and it’s very acidic, thus pathogens have a hard time living on it,” she said. “However, our skin microorganisms have adapted to utilize the sparse nutrients available on the skin. That’s why I tell my patients, ‘don’t use a sugar scrub because you’re potentially feeding these bad bacteria.’ ”
According to more recent research, the skin microbiota in healthy adults remains stable over time, despite environmental perturbations, and they have important roles in educating the innate and adaptive arms of the cutaneous immune system. “Some skin diseases are associated with an altered microbial state: dysbiosis,” said Dr. Waibel, subsection chief of dermatology at Baptist Health South Florida, Miami Beach. “Reversion of this may help prevent or treat the disease.”
She cited the following factors that influence the skin microbiome:
- Genetics affects the skin microbiome considerably. Individuals with autoimmune predispositions have different microbiota compared with those who don’t.
- Climate, pollution, and hygiene practices the other influencing factors. “Even clothing can impact the microbiome, by causing the transfer of microorganisms,” she said.
- Age and hormonal changes (particularly during puberty) and senescence alter the microbial landscape.
- Systemic health conditions such as diabetes mellitus and irritable bowel disease, as well as cutaneous conditions like psoriasis and atopic dermatitis can also disrupt the skin microbiome.
Ingredients contained in soaps, antibiotics, and cosmetics can also cause skin dysbiosis, Dr. Waibel said. However, the integrity of the skin’s microbiome following dermatological procedures such as excisions, dermabrasion, laser therapy, and other physical procedures is less understood, according to a recent review of the topic. Phototherapy appears to be the most extensively studied, “and shows an increase in microbial diversity post-treatment,” she said. “Light treatments have been found to kill bacteria by inducing DNA damage. More studies need to be performed on specific wavelengths of light used, conditions being treated and individual patient differences.”
According to the review’s authors, no change in the microbiome was observed in studies of debridement. “That was surprising, as it is a method to remove unhealthy tissue that often contains pathogenic bacteria,” Dr. Waibel said. “The big take-home message is that we need more research.”
Dr. Waibel disclosed that she has conducted clinical trials for several device and pharmaceutical companies.
A version of this article first appeared on Medscape.com.
SAN DIEGO — Although it has been known for several years that
In one review of the topic, researchers from the National Institutes of Health wrote that the skin is composed of 1.8 million diverse habitats with an abundance of folds, invaginations, and specialized niches that support a wide range of microorganisms. “Many of these microorganisms are harmless and, in some cases, provide vital functions for us to live and they have not evolved over time,” Jill S. Waibel, MD, medical director of the Miami Dermatology and Laser Institute, said at the annual Masters of Aesthetics Symposium.
“This is complex ecosystem that we don’t really talk about,” she said. “There is wide topographical distribution of bacteria on skin sites. The bacteria we have on our head and neck area is different from that on our feet. There is also a lot of interpersonal variation of the skin microbiome, so one person may have a lot of one type of bacteria and not as much of another.”
A Shield From Foreign Pathogens
At its core, Dr. Waibel continued, the skin microbiome functions as an interface between the human body and the environment, a physical barrier that prevents the invasion of foreign pathogens. The skin also provides a home to commensal microbiota. She likened the skin’s landscape to that of the tundra: “It’s desiccated, has poor nutrients, and it’s very acidic, thus pathogens have a hard time living on it,” she said. “However, our skin microorganisms have adapted to utilize the sparse nutrients available on the skin. That’s why I tell my patients, ‘don’t use a sugar scrub because you’re potentially feeding these bad bacteria.’ ”
According to more recent research, the skin microbiota in healthy adults remains stable over time, despite environmental perturbations, and they have important roles in educating the innate and adaptive arms of the cutaneous immune system. “Some skin diseases are associated with an altered microbial state: dysbiosis,” said Dr. Waibel, subsection chief of dermatology at Baptist Health South Florida, Miami Beach. “Reversion of this may help prevent or treat the disease.”
She cited the following factors that influence the skin microbiome:
- Genetics affects the skin microbiome considerably. Individuals with autoimmune predispositions have different microbiota compared with those who don’t.
- Climate, pollution, and hygiene practices the other influencing factors. “Even clothing can impact the microbiome, by causing the transfer of microorganisms,” she said.
- Age and hormonal changes (particularly during puberty) and senescence alter the microbial landscape.
- Systemic health conditions such as diabetes mellitus and irritable bowel disease, as well as cutaneous conditions like psoriasis and atopic dermatitis can also disrupt the skin microbiome.
Ingredients contained in soaps, antibiotics, and cosmetics can also cause skin dysbiosis, Dr. Waibel said. However, the integrity of the skin’s microbiome following dermatological procedures such as excisions, dermabrasion, laser therapy, and other physical procedures is less understood, according to a recent review of the topic. Phototherapy appears to be the most extensively studied, “and shows an increase in microbial diversity post-treatment,” she said. “Light treatments have been found to kill bacteria by inducing DNA damage. More studies need to be performed on specific wavelengths of light used, conditions being treated and individual patient differences.”
According to the review’s authors, no change in the microbiome was observed in studies of debridement. “That was surprising, as it is a method to remove unhealthy tissue that often contains pathogenic bacteria,” Dr. Waibel said. “The big take-home message is that we need more research.”
Dr. Waibel disclosed that she has conducted clinical trials for several device and pharmaceutical companies.
A version of this article first appeared on Medscape.com.
FROM THE 2024 MASTERS OF AESTHETICS SYMPOSIUM
The Patient Encounter Is Changing
Over the last few decades the patient encounter has changed dramatically. Most recently fueled by the COVID pandemic, face-to-face events between patients and providers have become less frequent. The shift began years before with the slow acceptance of telemedicine by third-party payers.
, often leaving both providers and patients with more questions than answers. Even more recently, the explosive arrival of generative artificial intelligence (AI) has promised, some might say threatened, to add a whole new complexity and uncertainty to patient encounters regardless of the venue or platform.

Still, among the growing collection of options, I think it is fair to say that a live face-to-face encounter remains the gold standard in the opinions of both patients and providers. Patients may have become increasingly critical and vocal when they feel their provider appears rushed or is over focused on the desktop computer screen. However, given all of the options, I suspect that for the moment patients feel a face-to-face meeting continues to offer them the best chance of being heard and their concerns answered.
Even when the image on the video screen is sharp and the intelligibility of the audio feed is crystal clear, I bet most providers feel they can learn more about the patient during a live face-to-face encounter than a Zoom-style encounter.
Nonetheless, there are hints that face-to-face visits maybe losing their place in the pantheon of patient-provider encounters. A recent study from England found that there were a significant number of patients who were more forthcoming in reporting their preferences for social care-related quality of life when they were surveyed by internet rather than face-to-face. It is unclear what was behind this observation, however it may be that patients were embarrassed and viewed these questions about their social neediness as too sensitive to share face-to-face.
There is ample evidence of situations in which the internet can provide a level of anonymity that emboldens the user to say things that are cruel and hurtful, using words they might be afraid to voice in a live setting. This license to act in an uncivil manner is behind much of the harm generated by chat rooms and other social media sites. While in these cases the ability to hide behind the video screen is a negative, this study from England suggests that we should be looking for more opportunities to use this emboldening feature with certain individuals and populations who may be intimidated during a face-to-face encounter. It is likely a hybrid approach may be the most beneficial strategy tailored to the individual patient.
One advantage of a face-to-face visit is that each participant can read the body language of the other. This, of course, can be a disadvantage for the provider who has failed to master the art of disguising his “I’m running behind” stress level, when he should be replacing it with an “I’m ready to listen” posture.
Portals have opened up a whole other can of worms, particularly when the provider has failed to clearly delineate what sort of questions are appropriate for an online forum, not informed the patient who will be providing the answer, and a rough idea of when this will happen. It may take several trips up the learning curve for patients and providers to develop a style of writing that make optimal use of the portal format and make it fit the needs of the practice and the patients.
Regardless of what kind of visit platform we are talking about, a lot hinges on the providers choice of words. I recently reviewed some of the work of Jeffrey D. Robinson, PhD, a professor of communication at the Portland State University, Portland, Oregon. He offers the example of the difference between “some” and “any.” When the patient was asked “Is there something else you would like to address today” almost 80% of the patient’s unmet questions were addressed. However, when the question was “Is there anything else ...” very few of the patient’s unmet questions were addressed. Dr. Robinson has also found that when the question is posed early in the visit rather than at the end, it improves the chances of having the patient’s unmet concerns addressed.
I suspect that the face-to-face patient encounter will survive, but it will continue to lose its market share as other platforms emerge. We can be sure there will be change. We need look no further than generative AI to look for the next step. A well-crafted question could help the patient and the provider choose the most appropriate patient encounter format given the patient’s demographic, chief complaint, and prior history, and match this with the provider’s background and strengths.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at [email protected].
Over the last few decades the patient encounter has changed dramatically. Most recently fueled by the COVID pandemic, face-to-face events between patients and providers have become less frequent. The shift began years before with the slow acceptance of telemedicine by third-party payers.
, often leaving both providers and patients with more questions than answers. Even more recently, the explosive arrival of generative artificial intelligence (AI) has promised, some might say threatened, to add a whole new complexity and uncertainty to patient encounters regardless of the venue or platform.
Still, among the growing collection of options, I think it is fair to say that a live face-to-face encounter remains the gold standard in the opinions of both patients and providers. Patients may have become increasingly critical and vocal when they feel their provider appears rushed or is over focused on the desktop computer screen. However, given all of the options, I suspect that for the moment patients feel a face-to-face meeting continues to offer them the best chance of being heard and their concerns answered.
Even when the image on the video screen is sharp and the intelligibility of the audio feed is crystal clear, I bet most providers feel they can learn more about the patient during a live face-to-face encounter than a Zoom-style encounter.
Nonetheless, there are hints that face-to-face visits maybe losing their place in the pantheon of patient-provider encounters. A recent study from England found that there were a significant number of patients who were more forthcoming in reporting their preferences for social care-related quality of life when they were surveyed by internet rather than face-to-face. It is unclear what was behind this observation, however it may be that patients were embarrassed and viewed these questions about their social neediness as too sensitive to share face-to-face.
There is ample evidence of situations in which the internet can provide a level of anonymity that emboldens the user to say things that are cruel and hurtful, using words they might be afraid to voice in a live setting. This license to act in an uncivil manner is behind much of the harm generated by chat rooms and other social media sites. While in these cases the ability to hide behind the video screen is a negative, this study from England suggests that we should be looking for more opportunities to use this emboldening feature with certain individuals and populations who may be intimidated during a face-to-face encounter. It is likely a hybrid approach may be the most beneficial strategy tailored to the individual patient.
One advantage of a face-to-face visit is that each participant can read the body language of the other. This, of course, can be a disadvantage for the provider who has failed to master the art of disguising his “I’m running behind” stress level, when he should be replacing it with an “I’m ready to listen” posture.
Portals have opened up a whole other can of worms, particularly when the provider has failed to clearly delineate what sort of questions are appropriate for an online forum, not informed the patient who will be providing the answer, and a rough idea of when this will happen. It may take several trips up the learning curve for patients and providers to develop a style of writing that make optimal use of the portal format and make it fit the needs of the practice and the patients.
Regardless of what kind of visit platform we are talking about, a lot hinges on the providers choice of words. I recently reviewed some of the work of Jeffrey D. Robinson, PhD, a professor of communication at the Portland State University, Portland, Oregon. He offers the example of the difference between “some” and “any.” When the patient was asked “Is there something else you would like to address today” almost 80% of the patient’s unmet questions were addressed. However, when the question was “Is there anything else ...” very few of the patient’s unmet questions were addressed. Dr. Robinson has also found that when the question is posed early in the visit rather than at the end, it improves the chances of having the patient’s unmet concerns addressed.
I suspect that the face-to-face patient encounter will survive, but it will continue to lose its market share as other platforms emerge. We can be sure there will be change. We need look no further than generative AI to look for the next step. A well-crafted question could help the patient and the provider choose the most appropriate patient encounter format given the patient’s demographic, chief complaint, and prior history, and match this with the provider’s background and strengths.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at [email protected].
Over the last few decades the patient encounter has changed dramatically. Most recently fueled by the COVID pandemic, face-to-face events between patients and providers have become less frequent. The shift began years before with the slow acceptance of telemedicine by third-party payers.
, often leaving both providers and patients with more questions than answers. Even more recently, the explosive arrival of generative artificial intelligence (AI) has promised, some might say threatened, to add a whole new complexity and uncertainty to patient encounters regardless of the venue or platform.
Still, among the growing collection of options, I think it is fair to say that a live face-to-face encounter remains the gold standard in the opinions of both patients and providers. Patients may have become increasingly critical and vocal when they feel their provider appears rushed or is over focused on the desktop computer screen. However, given all of the options, I suspect that for the moment patients feel a face-to-face meeting continues to offer them the best chance of being heard and their concerns answered.
Even when the image on the video screen is sharp and the intelligibility of the audio feed is crystal clear, I bet most providers feel they can learn more about the patient during a live face-to-face encounter than a Zoom-style encounter.
Nonetheless, there are hints that face-to-face visits maybe losing their place in the pantheon of patient-provider encounters. A recent study from England found that there were a significant number of patients who were more forthcoming in reporting their preferences for social care-related quality of life when they were surveyed by internet rather than face-to-face. It is unclear what was behind this observation, however it may be that patients were embarrassed and viewed these questions about their social neediness as too sensitive to share face-to-face.
There is ample evidence of situations in which the internet can provide a level of anonymity that emboldens the user to say things that are cruel and hurtful, using words they might be afraid to voice in a live setting. This license to act in an uncivil manner is behind much of the harm generated by chat rooms and other social media sites. While in these cases the ability to hide behind the video screen is a negative, this study from England suggests that we should be looking for more opportunities to use this emboldening feature with certain individuals and populations who may be intimidated during a face-to-face encounter. It is likely a hybrid approach may be the most beneficial strategy tailored to the individual patient.
One advantage of a face-to-face visit is that each participant can read the body language of the other. This, of course, can be a disadvantage for the provider who has failed to master the art of disguising his “I’m running behind” stress level, when he should be replacing it with an “I’m ready to listen” posture.
Portals have opened up a whole other can of worms, particularly when the provider has failed to clearly delineate what sort of questions are appropriate for an online forum, not informed the patient who will be providing the answer, and a rough idea of when this will happen. It may take several trips up the learning curve for patients and providers to develop a style of writing that make optimal use of the portal format and make it fit the needs of the practice and the patients.
Regardless of what kind of visit platform we are talking about, a lot hinges on the providers choice of words. I recently reviewed some of the work of Jeffrey D. Robinson, PhD, a professor of communication at the Portland State University, Portland, Oregon. He offers the example of the difference between “some” and “any.” When the patient was asked “Is there something else you would like to address today” almost 80% of the patient’s unmet questions were addressed. However, when the question was “Is there anything else ...” very few of the patient’s unmet questions were addressed. Dr. Robinson has also found that when the question is posed early in the visit rather than at the end, it improves the chances of having the patient’s unmet concerns addressed.
I suspect that the face-to-face patient encounter will survive, but it will continue to lose its market share as other platforms emerge. We can be sure there will be change. We need look no further than generative AI to look for the next step. A well-crafted question could help the patient and the provider choose the most appropriate patient encounter format given the patient’s demographic, chief complaint, and prior history, and match this with the provider’s background and strengths.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at [email protected].