User login
Childhood peanut allergy linked with other legume allergies
French children with peanut allergy tend to have reactions to other legumes, including soy, lentil, pea, bean, lupin, and fenugreek, and those other allergies often lead to anaphylactic reactions, a retrospective study from France reports.
“Among children allergic to peanut, at least two-thirds were sensitized to one other legume, and legume allergy was diagnosed in one-quarter of the sensitized patients,” wrote senior study author Amandine Divaret-Chauveau, MD, of Centre Hospitalier Universitaire de Nancy, Vandoeuvre-les-Nancy, and colleagues. The report is in Pediatric Allergy and Immunology.
People worldwide are eating more legumes these days, the authors noted. High in protein, low in unsaturated fats, with low production costs, legumes are important components of increasingly vegetarian, healthy, sustainable diets.
Food allergens are the most common childhood triggers of allergic reactions. Among children in France, legumes cause 14.6% of food-related anaphylactic reactions, with peanut as the main allergen, they added.
Dr. Divaret-Chauveau and colleagues assessed the prevalence and relevance of sensitization to legumes among all children and adolescents aged 1-17 years who had peanut allergy and had been admitted to one academic pediatric allergy department over roughly 3 years, beginning in early 2017. For the 195 study participants, peanut allergy had been confirmed, and they had been documented to have consumed or to have sensitization to at least one non-peanut legume; 69.7% were boys.
The researchers analyzed data on consumption history, skin prick tests, specific immunoglobulin E status, prior allergic reactions, and oral food challenges for each legume. They found the following:
- Among the 195 children with peanut allergy, 98.4% had at least one other atopic disease.
- Of the 195 children with peanut allergy, 122 (63.9%) were sensitized to at least one other legume. Of these 122 children, 66.3% were sensitized to fenugreek, 42.2% to lentil, 39.9% to soy, and 34.2% to lupin.
- Allergy to one or more legumes was confirmed for 27.9% of the 122 sensitized children, including 4.9% who had multiple legume allergies. Lentil, lupin, and pea were the main allergens.
- Of the 118 children also having a non-legume food allergy, the main food allergens were egg (57.6%), cow’s milk (33.0%), cashew (39.0%), pistachio (23.7%), and hazelnut (30.5%).
- Fifty percent of allergic reactions to non-peanut legumes were severe, often showing as asthma. Atopic comorbidities, including asthma, in most participants may have contributed to the severity of allergic reactions, the authors noted.
Allergy awareness needs to grow with plant-based diets
“The high prevalence of legume sensitization reported in our study highlights the need to explore legume consumption in children with PA [peanut allergy], and the need to investigate sensitization in the absence of consumption,” they added.
Jodi A. Shroba, MSN, APRN, CPNP, coordinator for the Food Allergy Program at Children’s Mercy Kansas City, in Missouri, told this news organization that few data are available in the literature regarding allergies to legumes other than peanut.
“It was interesting that these authors found such a high legume sensitization in their peanut-allergic patients,” Ms. Shroba, who was not involved in the study, said by email. “As more people are starting to eat plant-based diets, it is important that we better understand their allergenicity and cross-reactivity so we can better help guide patient management and education.”
Deborah Albright, MD, assistant professor of pediatrics at the University of Pittsburgh, agreed.
“As plant-based protein consumption broadens worldwide, awareness of the potential for cross-reactivity and co-allergy amongst legumes will become increasingly important,” she said by email.
“However, positive allergy tests do not reliably correlate with true food allergy; therefore, the diagnosis of legume co-allergy should be confirmed by the individual patient’s history, a formal food challenge, or both,” advised Dr. Albright. She was not involved in the study.
“Cross-sensitization to other legumes in patients with a single legume allergy is common; however, true clinical reactivity is often not present,” she added. “Also, legume allergy test sensitization rates and objective reactivity on food challenge can vary by region, depending on diet and pollen aeroallergen exposure.
“Systematic exploration of tolerance versus co-allergy to other legumes should be considered in patients allergic to peanut or other legumes,” Dr. Albright said.
The authors recommend further research and registry data collection of legume anaphylaxis.
Details regarding funding for the study were not provided. The authors, Ms. Shroba, and Dr. Albright report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
French children with peanut allergy tend to have reactions to other legumes, including soy, lentil, pea, bean, lupin, and fenugreek, and those other allergies often lead to anaphylactic reactions, a retrospective study from France reports.
“Among children allergic to peanut, at least two-thirds were sensitized to one other legume, and legume allergy was diagnosed in one-quarter of the sensitized patients,” wrote senior study author Amandine Divaret-Chauveau, MD, of Centre Hospitalier Universitaire de Nancy, Vandoeuvre-les-Nancy, and colleagues. The report is in Pediatric Allergy and Immunology.
People worldwide are eating more legumes these days, the authors noted. High in protein, low in unsaturated fats, with low production costs, legumes are important components of increasingly vegetarian, healthy, sustainable diets.
Food allergens are the most common childhood triggers of allergic reactions. Among children in France, legumes cause 14.6% of food-related anaphylactic reactions, with peanut as the main allergen, they added.
Dr. Divaret-Chauveau and colleagues assessed the prevalence and relevance of sensitization to legumes among all children and adolescents aged 1-17 years who had peanut allergy and had been admitted to one academic pediatric allergy department over roughly 3 years, beginning in early 2017. For the 195 study participants, peanut allergy had been confirmed, and they had been documented to have consumed or to have sensitization to at least one non-peanut legume; 69.7% were boys.
The researchers analyzed data on consumption history, skin prick tests, specific immunoglobulin E status, prior allergic reactions, and oral food challenges for each legume. They found the following:
- Among the 195 children with peanut allergy, 98.4% had at least one other atopic disease.
- Of the 195 children with peanut allergy, 122 (63.9%) were sensitized to at least one other legume. Of these 122 children, 66.3% were sensitized to fenugreek, 42.2% to lentil, 39.9% to soy, and 34.2% to lupin.
- Allergy to one or more legumes was confirmed for 27.9% of the 122 sensitized children, including 4.9% who had multiple legume allergies. Lentil, lupin, and pea were the main allergens.
- Of the 118 children also having a non-legume food allergy, the main food allergens were egg (57.6%), cow’s milk (33.0%), cashew (39.0%), pistachio (23.7%), and hazelnut (30.5%).
- Fifty percent of allergic reactions to non-peanut legumes were severe, often showing as asthma. Atopic comorbidities, including asthma, in most participants may have contributed to the severity of allergic reactions, the authors noted.
Allergy awareness needs to grow with plant-based diets
“The high prevalence of legume sensitization reported in our study highlights the need to explore legume consumption in children with PA [peanut allergy], and the need to investigate sensitization in the absence of consumption,” they added.
Jodi A. Shroba, MSN, APRN, CPNP, coordinator for the Food Allergy Program at Children’s Mercy Kansas City, in Missouri, told this news organization that few data are available in the literature regarding allergies to legumes other than peanut.
“It was interesting that these authors found such a high legume sensitization in their peanut-allergic patients,” Ms. Shroba, who was not involved in the study, said by email. “As more people are starting to eat plant-based diets, it is important that we better understand their allergenicity and cross-reactivity so we can better help guide patient management and education.”
Deborah Albright, MD, assistant professor of pediatrics at the University of Pittsburgh, agreed.
“As plant-based protein consumption broadens worldwide, awareness of the potential for cross-reactivity and co-allergy amongst legumes will become increasingly important,” she said by email.
“However, positive allergy tests do not reliably correlate with true food allergy; therefore, the diagnosis of legume co-allergy should be confirmed by the individual patient’s history, a formal food challenge, or both,” advised Dr. Albright. She was not involved in the study.
“Cross-sensitization to other legumes in patients with a single legume allergy is common; however, true clinical reactivity is often not present,” she added. “Also, legume allergy test sensitization rates and objective reactivity on food challenge can vary by region, depending on diet and pollen aeroallergen exposure.
“Systematic exploration of tolerance versus co-allergy to other legumes should be considered in patients allergic to peanut or other legumes,” Dr. Albright said.
The authors recommend further research and registry data collection of legume anaphylaxis.
Details regarding funding for the study were not provided. The authors, Ms. Shroba, and Dr. Albright report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
French children with peanut allergy tend to have reactions to other legumes, including soy, lentil, pea, bean, lupin, and fenugreek, and those other allergies often lead to anaphylactic reactions, a retrospective study from France reports.
“Among children allergic to peanut, at least two-thirds were sensitized to one other legume, and legume allergy was diagnosed in one-quarter of the sensitized patients,” wrote senior study author Amandine Divaret-Chauveau, MD, of Centre Hospitalier Universitaire de Nancy, Vandoeuvre-les-Nancy, and colleagues. The report is in Pediatric Allergy and Immunology.
People worldwide are eating more legumes these days, the authors noted. High in protein, low in unsaturated fats, with low production costs, legumes are important components of increasingly vegetarian, healthy, sustainable diets.
Food allergens are the most common childhood triggers of allergic reactions. Among children in France, legumes cause 14.6% of food-related anaphylactic reactions, with peanut as the main allergen, they added.
Dr. Divaret-Chauveau and colleagues assessed the prevalence and relevance of sensitization to legumes among all children and adolescents aged 1-17 years who had peanut allergy and had been admitted to one academic pediatric allergy department over roughly 3 years, beginning in early 2017. For the 195 study participants, peanut allergy had been confirmed, and they had been documented to have consumed or to have sensitization to at least one non-peanut legume; 69.7% were boys.
The researchers analyzed data on consumption history, skin prick tests, specific immunoglobulin E status, prior allergic reactions, and oral food challenges for each legume. They found the following:
- Among the 195 children with peanut allergy, 98.4% had at least one other atopic disease.
- Of the 195 children with peanut allergy, 122 (63.9%) were sensitized to at least one other legume. Of these 122 children, 66.3% were sensitized to fenugreek, 42.2% to lentil, 39.9% to soy, and 34.2% to lupin.
- Allergy to one or more legumes was confirmed for 27.9% of the 122 sensitized children, including 4.9% who had multiple legume allergies. Lentil, lupin, and pea were the main allergens.
- Of the 118 children also having a non-legume food allergy, the main food allergens were egg (57.6%), cow’s milk (33.0%), cashew (39.0%), pistachio (23.7%), and hazelnut (30.5%).
- Fifty percent of allergic reactions to non-peanut legumes were severe, often showing as asthma. Atopic comorbidities, including asthma, in most participants may have contributed to the severity of allergic reactions, the authors noted.
Allergy awareness needs to grow with plant-based diets
“The high prevalence of legume sensitization reported in our study highlights the need to explore legume consumption in children with PA [peanut allergy], and the need to investigate sensitization in the absence of consumption,” they added.
Jodi A. Shroba, MSN, APRN, CPNP, coordinator for the Food Allergy Program at Children’s Mercy Kansas City, in Missouri, told this news organization that few data are available in the literature regarding allergies to legumes other than peanut.
“It was interesting that these authors found such a high legume sensitization in their peanut-allergic patients,” Ms. Shroba, who was not involved in the study, said by email. “As more people are starting to eat plant-based diets, it is important that we better understand their allergenicity and cross-reactivity so we can better help guide patient management and education.”
Deborah Albright, MD, assistant professor of pediatrics at the University of Pittsburgh, agreed.
“As plant-based protein consumption broadens worldwide, awareness of the potential for cross-reactivity and co-allergy amongst legumes will become increasingly important,” she said by email.
“However, positive allergy tests do not reliably correlate with true food allergy; therefore, the diagnosis of legume co-allergy should be confirmed by the individual patient’s history, a formal food challenge, or both,” advised Dr. Albright. She was not involved in the study.
“Cross-sensitization to other legumes in patients with a single legume allergy is common; however, true clinical reactivity is often not present,” she added. “Also, legume allergy test sensitization rates and objective reactivity on food challenge can vary by region, depending on diet and pollen aeroallergen exposure.
“Systematic exploration of tolerance versus co-allergy to other legumes should be considered in patients allergic to peanut or other legumes,” Dr. Albright said.
The authors recommend further research and registry data collection of legume anaphylaxis.
Details regarding funding for the study were not provided. The authors, Ms. Shroba, and Dr. Albright report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Med groups urge feds to protect physicians from anti-trans violence
Several leading medical groups on Oct. 3 called on U.S. Attorney General Merrick Garland to investigate and prosecute those responsible for a recent spate of threats and attacks against hospitals and physicians who are providing gender-affirming care.
In an Oct. 3 letter, the American Academy of Pediatrics (AAP), the American Medical Association (AMA), and the Children’s Hospital Association detailed the risk posed by these threats to physicians, patients, and the federally protected right to health care.
The letter comes during a campaign of intimidation and misinformation that has disrupted gender-related care in Seattle, Akron, Ohio, Nashville, Tenn., and Boston in the past few weeks. Hospitals across the country and their ambulatory sites have been forced to substantially increase protection, and “some providers have needed 24/7 security,” according to the letter.
Not only do the threats bully physicians providing gender-affirming care and the patients who receive that care, but “they have also disrupted many other services to families seeking care,” the letter claims.
According to STAT, many hospitals that provide gender-affirming care have responded to the threats by removing information about the treatment from their websites.
At one hospital, a new mother was separated from her preterm infant because the facility’s neonatal intensive care unit was locked down as the result of a bomb threat. (It’s not clear whether that incident is the same as a similar threat that led to the arrest of a 37-year-old Massachusetts woman, who is facing criminal charges in the episode.)
“The attacks are rooted in an intentional campaign of disinformation” by high-profile social media users, according to the letter. The medical organizations have also called on major tech companies, including TikTok, Twitter, and Meta, to do more to prevent the coordination of disinformation campaigns and violence against health care providers and patients.
“We now urge your office to take swift action to investigate and prosecute all organizations, individuals, and entities responsible,” the letter states.
“We cannot stand by as threats of violence against our members and their patients proliferate with little consequence. We call on the Department of Justice to investigate these attacks and social media platforms to reduce the spread of the misinformation enabling them,” AAP President Moira Szilagyi, MD, PhD, FAAP, said in a press release.
In addition to physical threats at their workplace, providers face threats on their personal social media accounts and harassment via phone and email. The letter notes that these unchecked attacks are coming after health care workers spent 3 years working on the front lines of a pandemic.
“Individuals in all workplaces have the right to a safe environment, out of harm’s way and free of intimidation or reprisal,” AMA President Jack Resneck Jr, MD, said in a statement. “The AMA will continue to work with federal, state, and local law enforcement officials to develop and implement strategies that protect hard-working, law-abiding physicians and other health care workers from senseless acts of violence, abuse, and intimidation.”
A version of this article first appeared on Medscape.com.
Several leading medical groups on Oct. 3 called on U.S. Attorney General Merrick Garland to investigate and prosecute those responsible for a recent spate of threats and attacks against hospitals and physicians who are providing gender-affirming care.
In an Oct. 3 letter, the American Academy of Pediatrics (AAP), the American Medical Association (AMA), and the Children’s Hospital Association detailed the risk posed by these threats to physicians, patients, and the federally protected right to health care.
The letter comes during a campaign of intimidation and misinformation that has disrupted gender-related care in Seattle, Akron, Ohio, Nashville, Tenn., and Boston in the past few weeks. Hospitals across the country and their ambulatory sites have been forced to substantially increase protection, and “some providers have needed 24/7 security,” according to the letter.
Not only do the threats bully physicians providing gender-affirming care and the patients who receive that care, but “they have also disrupted many other services to families seeking care,” the letter claims.
According to STAT, many hospitals that provide gender-affirming care have responded to the threats by removing information about the treatment from their websites.
At one hospital, a new mother was separated from her preterm infant because the facility’s neonatal intensive care unit was locked down as the result of a bomb threat. (It’s not clear whether that incident is the same as a similar threat that led to the arrest of a 37-year-old Massachusetts woman, who is facing criminal charges in the episode.)
“The attacks are rooted in an intentional campaign of disinformation” by high-profile social media users, according to the letter. The medical organizations have also called on major tech companies, including TikTok, Twitter, and Meta, to do more to prevent the coordination of disinformation campaigns and violence against health care providers and patients.
“We now urge your office to take swift action to investigate and prosecute all organizations, individuals, and entities responsible,” the letter states.
“We cannot stand by as threats of violence against our members and their patients proliferate with little consequence. We call on the Department of Justice to investigate these attacks and social media platforms to reduce the spread of the misinformation enabling them,” AAP President Moira Szilagyi, MD, PhD, FAAP, said in a press release.
In addition to physical threats at their workplace, providers face threats on their personal social media accounts and harassment via phone and email. The letter notes that these unchecked attacks are coming after health care workers spent 3 years working on the front lines of a pandemic.
“Individuals in all workplaces have the right to a safe environment, out of harm’s way and free of intimidation or reprisal,” AMA President Jack Resneck Jr, MD, said in a statement. “The AMA will continue to work with federal, state, and local law enforcement officials to develop and implement strategies that protect hard-working, law-abiding physicians and other health care workers from senseless acts of violence, abuse, and intimidation.”
A version of this article first appeared on Medscape.com.
Several leading medical groups on Oct. 3 called on U.S. Attorney General Merrick Garland to investigate and prosecute those responsible for a recent spate of threats and attacks against hospitals and physicians who are providing gender-affirming care.
In an Oct. 3 letter, the American Academy of Pediatrics (AAP), the American Medical Association (AMA), and the Children’s Hospital Association detailed the risk posed by these threats to physicians, patients, and the federally protected right to health care.
The letter comes during a campaign of intimidation and misinformation that has disrupted gender-related care in Seattle, Akron, Ohio, Nashville, Tenn., and Boston in the past few weeks. Hospitals across the country and their ambulatory sites have been forced to substantially increase protection, and “some providers have needed 24/7 security,” according to the letter.
Not only do the threats bully physicians providing gender-affirming care and the patients who receive that care, but “they have also disrupted many other services to families seeking care,” the letter claims.
According to STAT, many hospitals that provide gender-affirming care have responded to the threats by removing information about the treatment from their websites.
At one hospital, a new mother was separated from her preterm infant because the facility’s neonatal intensive care unit was locked down as the result of a bomb threat. (It’s not clear whether that incident is the same as a similar threat that led to the arrest of a 37-year-old Massachusetts woman, who is facing criminal charges in the episode.)
“The attacks are rooted in an intentional campaign of disinformation” by high-profile social media users, according to the letter. The medical organizations have also called on major tech companies, including TikTok, Twitter, and Meta, to do more to prevent the coordination of disinformation campaigns and violence against health care providers and patients.
“We now urge your office to take swift action to investigate and prosecute all organizations, individuals, and entities responsible,” the letter states.
“We cannot stand by as threats of violence against our members and their patients proliferate with little consequence. We call on the Department of Justice to investigate these attacks and social media platforms to reduce the spread of the misinformation enabling them,” AAP President Moira Szilagyi, MD, PhD, FAAP, said in a press release.
In addition to physical threats at their workplace, providers face threats on their personal social media accounts and harassment via phone and email. The letter notes that these unchecked attacks are coming after health care workers spent 3 years working on the front lines of a pandemic.
“Individuals in all workplaces have the right to a safe environment, out of harm’s way and free of intimidation or reprisal,” AMA President Jack Resneck Jr, MD, said in a statement. “The AMA will continue to work with federal, state, and local law enforcement officials to develop and implement strategies that protect hard-working, law-abiding physicians and other health care workers from senseless acts of violence, abuse, and intimidation.”
A version of this article first appeared on Medscape.com.
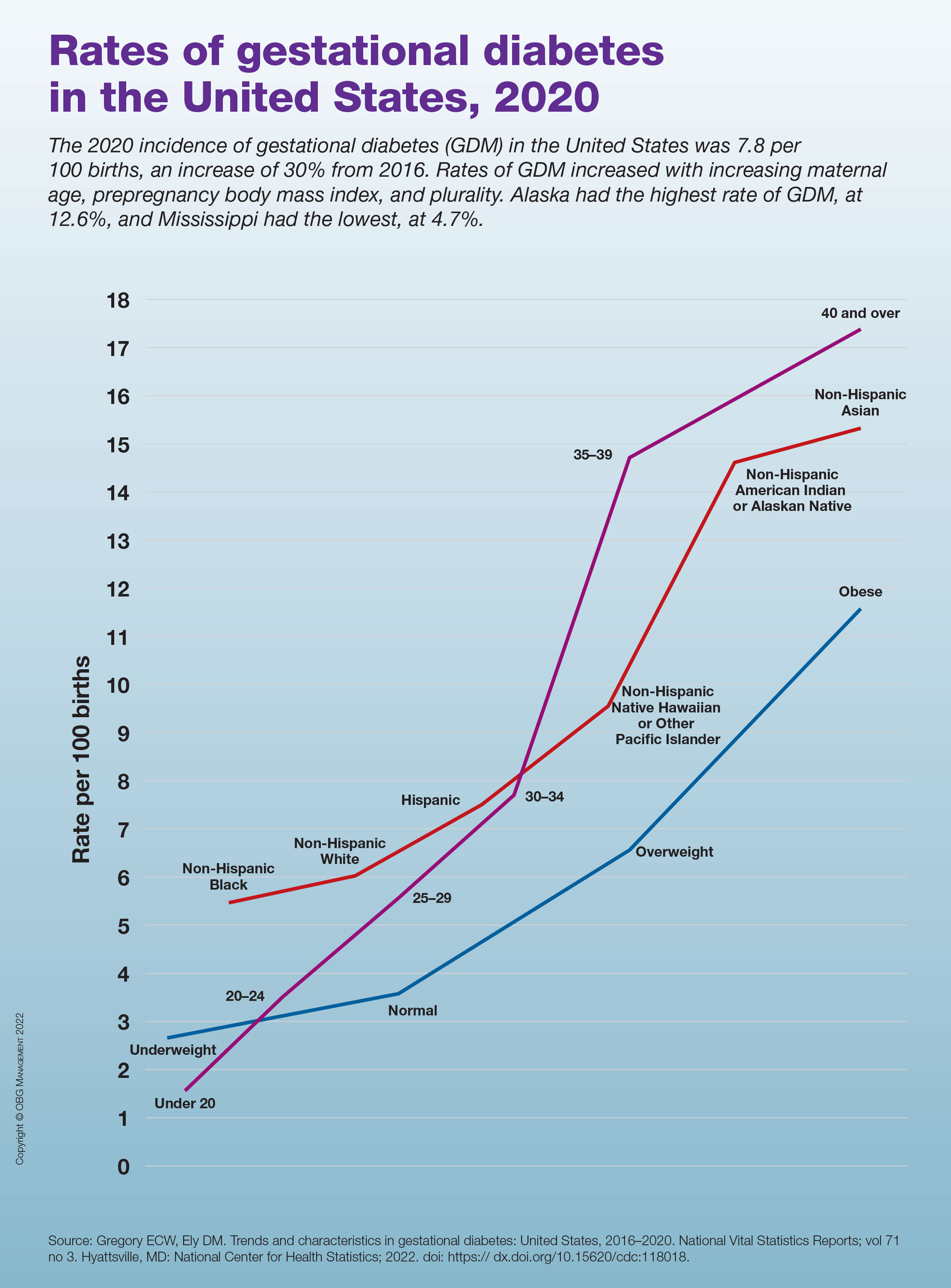
Rates of gestational diabetes in the United States, 2020
Commentary: Menstruation, sleep, and visual disturbances in migraine, October 2022

Lasmiditan—the first migraine treatment in the new ditan class— is a serotonin receptor agonist, similar to triptan medications. However, it is specific for the 5HT1F receptor rather than the 5HT1B/1D receptor. The main purpose of this specificity is that it leads to less vascular risk; specifically, this medication should be safer for populations at higher risk for vascular events, such as myocardial infarction and stroke.
Only one triptan, naratriptan, had previously been studied for the treatment of menstrual migraine, at a recommended dose of 2.5 mg twice daily as a bridge. A new study by MacGregor and colleagues looked at taking 50, 100, or 200 mg of lasmiditan vs placebo for an individual premenstrual attack.
The participants in the study were recruited from the intention-to-treat population of two prior studies for this drug, a phase 2 trial and a phase 3 trial. The menstrual calendars of the female participants were reviewed, followed by randomization into one of the four groups. Patients with chronic migraine were excluded from the study. The primary outcome was freedom from pain at 2 hours; secondary outcomes were freedom from the most bothersome symptom and reduction in pain severity.
Of the four populations followed, all three intervention groups noted significant results in freedom from pain at 2 hours compared with placebo. The 2-hour responder rate was 33.6% for the 200 mg group, 16.7% for the 50 mg and 100 mg groups, and 7.6% for the placebo group. Freedom from the most bothersome symptom and pain reduction were also significant in these populations.
Menstruation-associated migraine and worsening headache attacks due to patients' hormonal fluctuations are some of the most common issues and triggers that neurologists and headache specialists confront. Although the responder rates for freedom from pain at 2 hours were not very robust, lasmiditan does appear to be significantly effective in this population, and in those with menstrual triggers specifically. The field of headache medicine would be even better served by additional studies on both preventive and more acute medications in association with hormonal triggers.
Another very common trigger for migraine is changes in sleep patterns. An astute headache specialist will always ask about sleep quantity and quality during an initial assessment of a patient. Many headache centers have sleep-specific questionnaires that patients fill out during intake. The precise association between migraine and sleep deserves more elucidation. Duan and colleagues specifically set out to reveal whether differences in sleep quality affect migraine frequency; whether this is the same among different gender and age groups; and whether headache disability, severity, mood, and quality of life are related to underlying sleep changes independent of other factors.
A total of 134 participants with migraine and 70 without migraine or any other headache disorder were enrolled in the study. Sleep quality was assessed through The Pittsburgh Sleep Quality Index (PSQI) questionnaire. This is a commonly used self-reported questionnaire for assessing quality and quantity of sleep over the past month and is considered the standard of care in most sleep centers. The investigators here sought to determine the predictive value of the PSQI in regard to migraine. Migraine disability was assessed via the Migraine Disability Assessment (MIDAS) scale as well as the Headache Impact Test (HIT-6). Statistical analysis was performed with logistical regression, t test, and χ2 squared test.
There strongest correlations between poor sleep quality and risk of migraine were found in women, patients over 35 years old, and those with lower education levels. The results revealed that the migraine group had poorer sleep quality, as well as higher anxiety and depression scores, compared with the control group. A low PSQI score (eg, poorer sleep quality) was associated with higher migraine frequency; this was independent of body mass index (BMI), weekly exercise time, and smoking or drinking history. After participants were divided into good and poor sleep quality subgroups, the PSQI score was found to increase the odds ratio of migraine by a factor of 6.
The investigators were able to show the predictive quality of the PSQI score. Worse sleep quality was found to be associated with a higher MIDAS score and HIT-6 score as well as total pain burden, pain severity, decreased quality of life, depression, and anxiety. Although most headache specialists spend a significant amount of time discussing sleep as it relates to migraine, it may be worth considering following a sleep quality scale, such as the PSQI, over time as we monitor our patients. This may allow us to take a more proactive role and be able to prognosticate our patients' migraine journey somewhat better. Although sleep triggers and associations with migraine can be very difficult to discuss and treat, this study very clearly argues for the importance of focusing on sleep with our patients.
Although the most common aura our patients with migraine experience is visual, many patients with migraine will also experience non-aura visual changes. These can range from short-lasting episodes of blurred vision, such as transient visual obscurations, or other transient visual disturbances that do not fit the criteria of aura as defined by the International Classification of Headache Disorders (ICHD).
A prior study by Tsao and colleagues had revealed that almost half of headache patients experienced some headache-related visual change, the most common of which were short-lasting flickering lights or a movable, monochromatic scotoma. As opposed to visual aura, these transient disturbances were shorter in onset and duration and typically occurred during the headache phase of a migraine attack. In the current study, Tsao and colleagues sought to determine whether the presence of these findings was associated with a different headache burden from that typically found in migraine with aura.
The participants in this study were enrolled over a 10-year period from May 2010 to July 2020. They initially underwent a visual phenomenon questionnaire and then a thorough clinical interview to determine their headache diagnosis per ICHD criteria — specifically whether they had an underlying diagnosis of migraine with aura or migraine without aura. Participants were also separately diagnosed with chronic migraine or medication overuse headache. A visual rating scale was used in the initial questionnaires. This scale posed questions about the duration of symptoms; whether the symptoms develop gradually or suddenly; and whether the visual change was a scotoma, zigzag lines, or in a unilateral or bilateral visual field. A prior study by these investigators determined this visual rating scale to be highly sensitive and specific for diagnosing migraine with aura.
Participants were also given the MIDAS questionnaire and were assessed with the HIT-6 scale, a migraine photophobia score, and the Beck Depression Inventory. A total of 12,255 patients were enrolled, 9946 with migraine, who were subdivided on the basis of diagnosis of migraine with or without aura. Blurred vision was the most common visual complaint among all migraine patients. Patients who had transient visual disturbances that did not fit the criteria of migraine with aura were noted to have a statistically significant higher headache frequency, more severe headache-related disability, a higher likelihood of developing medication overuse headache, and a greater incidence of anxiety and depression.
An important distinction that all headache specialists make is whether their patients experience migraine with or without aura. The primary purpose for this distinction is to determine the appropriateness of specific medications (estrogen or vasoconstrictive medications), as migraine aura relates to vascular risk. We usually delve deeply into whether the visual symptoms that our patients experience do or do not fit into the ICHD criteria of migraine aura. We should not discard or think less of non-aura visual disturbances; these authors argue very clearly that these kinds of visual changes can be very relevant prognostically.
Lasmiditan—the first migraine treatment in the new ditan class— is a serotonin receptor agonist, similar to triptan medications. However, it is specific for the 5HT1F receptor rather than the 5HT1B/1D receptor. The main purpose of this specificity is that it leads to less vascular risk; specifically, this medication should be safer for populations at higher risk for vascular events, such as myocardial infarction and stroke.
Only one triptan, naratriptan, had previously been studied for the treatment of menstrual migraine, at a recommended dose of 2.5 mg twice daily as a bridge. A new study by MacGregor and colleagues looked at taking 50, 100, or 200 mg of lasmiditan vs placebo for an individual premenstrual attack.
The participants in the study were recruited from the intention-to-treat population of two prior studies for this drug, a phase 2 trial and a phase 3 trial. The menstrual calendars of the female participants were reviewed, followed by randomization into one of the four groups. Patients with chronic migraine were excluded from the study. The primary outcome was freedom from pain at 2 hours; secondary outcomes were freedom from the most bothersome symptom and reduction in pain severity.
Of the four populations followed, all three intervention groups noted significant results in freedom from pain at 2 hours compared with placebo. The 2-hour responder rate was 33.6% for the 200 mg group, 16.7% for the 50 mg and 100 mg groups, and 7.6% for the placebo group. Freedom from the most bothersome symptom and pain reduction were also significant in these populations.
Menstruation-associated migraine and worsening headache attacks due to patients' hormonal fluctuations are some of the most common issues and triggers that neurologists and headache specialists confront. Although the responder rates for freedom from pain at 2 hours were not very robust, lasmiditan does appear to be significantly effective in this population, and in those with menstrual triggers specifically. The field of headache medicine would be even better served by additional studies on both preventive and more acute medications in association with hormonal triggers.
Another very common trigger for migraine is changes in sleep patterns. An astute headache specialist will always ask about sleep quantity and quality during an initial assessment of a patient. Many headache centers have sleep-specific questionnaires that patients fill out during intake. The precise association between migraine and sleep deserves more elucidation. Duan and colleagues specifically set out to reveal whether differences in sleep quality affect migraine frequency; whether this is the same among different gender and age groups; and whether headache disability, severity, mood, and quality of life are related to underlying sleep changes independent of other factors.
A total of 134 participants with migraine and 70 without migraine or any other headache disorder were enrolled in the study. Sleep quality was assessed through The Pittsburgh Sleep Quality Index (PSQI) questionnaire. This is a commonly used self-reported questionnaire for assessing quality and quantity of sleep over the past month and is considered the standard of care in most sleep centers. The investigators here sought to determine the predictive value of the PSQI in regard to migraine. Migraine disability was assessed via the Migraine Disability Assessment (MIDAS) scale as well as the Headache Impact Test (HIT-6). Statistical analysis was performed with logistical regression, t test, and χ2 squared test.
There strongest correlations between poor sleep quality and risk of migraine were found in women, patients over 35 years old, and those with lower education levels. The results revealed that the migraine group had poorer sleep quality, as well as higher anxiety and depression scores, compared with the control group. A low PSQI score (eg, poorer sleep quality) was associated with higher migraine frequency; this was independent of body mass index (BMI), weekly exercise time, and smoking or drinking history. After participants were divided into good and poor sleep quality subgroups, the PSQI score was found to increase the odds ratio of migraine by a factor of 6.
The investigators were able to show the predictive quality of the PSQI score. Worse sleep quality was found to be associated with a higher MIDAS score and HIT-6 score as well as total pain burden, pain severity, decreased quality of life, depression, and anxiety. Although most headache specialists spend a significant amount of time discussing sleep as it relates to migraine, it may be worth considering following a sleep quality scale, such as the PSQI, over time as we monitor our patients. This may allow us to take a more proactive role and be able to prognosticate our patients' migraine journey somewhat better. Although sleep triggers and associations with migraine can be very difficult to discuss and treat, this study very clearly argues for the importance of focusing on sleep with our patients.
Although the most common aura our patients with migraine experience is visual, many patients with migraine will also experience non-aura visual changes. These can range from short-lasting episodes of blurred vision, such as transient visual obscurations, or other transient visual disturbances that do not fit the criteria of aura as defined by the International Classification of Headache Disorders (ICHD).
A prior study by Tsao and colleagues had revealed that almost half of headache patients experienced some headache-related visual change, the most common of which were short-lasting flickering lights or a movable, monochromatic scotoma. As opposed to visual aura, these transient disturbances were shorter in onset and duration and typically occurred during the headache phase of a migraine attack. In the current study, Tsao and colleagues sought to determine whether the presence of these findings was associated with a different headache burden from that typically found in migraine with aura.
The participants in this study were enrolled over a 10-year period from May 2010 to July 2020. They initially underwent a visual phenomenon questionnaire and then a thorough clinical interview to determine their headache diagnosis per ICHD criteria — specifically whether they had an underlying diagnosis of migraine with aura or migraine without aura. Participants were also separately diagnosed with chronic migraine or medication overuse headache. A visual rating scale was used in the initial questionnaires. This scale posed questions about the duration of symptoms; whether the symptoms develop gradually or suddenly; and whether the visual change was a scotoma, zigzag lines, or in a unilateral or bilateral visual field. A prior study by these investigators determined this visual rating scale to be highly sensitive and specific for diagnosing migraine with aura.
Participants were also given the MIDAS questionnaire and were assessed with the HIT-6 scale, a migraine photophobia score, and the Beck Depression Inventory. A total of 12,255 patients were enrolled, 9946 with migraine, who were subdivided on the basis of diagnosis of migraine with or without aura. Blurred vision was the most common visual complaint among all migraine patients. Patients who had transient visual disturbances that did not fit the criteria of migraine with aura were noted to have a statistically significant higher headache frequency, more severe headache-related disability, a higher likelihood of developing medication overuse headache, and a greater incidence of anxiety and depression.
An important distinction that all headache specialists make is whether their patients experience migraine with or without aura. The primary purpose for this distinction is to determine the appropriateness of specific medications (estrogen or vasoconstrictive medications), as migraine aura relates to vascular risk. We usually delve deeply into whether the visual symptoms that our patients experience do or do not fit into the ICHD criteria of migraine aura. We should not discard or think less of non-aura visual disturbances; these authors argue very clearly that these kinds of visual changes can be very relevant prognostically.
Lasmiditan—the first migraine treatment in the new ditan class— is a serotonin receptor agonist, similar to triptan medications. However, it is specific for the 5HT1F receptor rather than the 5HT1B/1D receptor. The main purpose of this specificity is that it leads to less vascular risk; specifically, this medication should be safer for populations at higher risk for vascular events, such as myocardial infarction and stroke.
Only one triptan, naratriptan, had previously been studied for the treatment of menstrual migraine, at a recommended dose of 2.5 mg twice daily as a bridge. A new study by MacGregor and colleagues looked at taking 50, 100, or 200 mg of lasmiditan vs placebo for an individual premenstrual attack.
The participants in the study were recruited from the intention-to-treat population of two prior studies for this drug, a phase 2 trial and a phase 3 trial. The menstrual calendars of the female participants were reviewed, followed by randomization into one of the four groups. Patients with chronic migraine were excluded from the study. The primary outcome was freedom from pain at 2 hours; secondary outcomes were freedom from the most bothersome symptom and reduction in pain severity.
Of the four populations followed, all three intervention groups noted significant results in freedom from pain at 2 hours compared with placebo. The 2-hour responder rate was 33.6% for the 200 mg group, 16.7% for the 50 mg and 100 mg groups, and 7.6% for the placebo group. Freedom from the most bothersome symptom and pain reduction were also significant in these populations.
Menstruation-associated migraine and worsening headache attacks due to patients' hormonal fluctuations are some of the most common issues and triggers that neurologists and headache specialists confront. Although the responder rates for freedom from pain at 2 hours were not very robust, lasmiditan does appear to be significantly effective in this population, and in those with menstrual triggers specifically. The field of headache medicine would be even better served by additional studies on both preventive and more acute medications in association with hormonal triggers.
Another very common trigger for migraine is changes in sleep patterns. An astute headache specialist will always ask about sleep quantity and quality during an initial assessment of a patient. Many headache centers have sleep-specific questionnaires that patients fill out during intake. The precise association between migraine and sleep deserves more elucidation. Duan and colleagues specifically set out to reveal whether differences in sleep quality affect migraine frequency; whether this is the same among different gender and age groups; and whether headache disability, severity, mood, and quality of life are related to underlying sleep changes independent of other factors.
A total of 134 participants with migraine and 70 without migraine or any other headache disorder were enrolled in the study. Sleep quality was assessed through The Pittsburgh Sleep Quality Index (PSQI) questionnaire. This is a commonly used self-reported questionnaire for assessing quality and quantity of sleep over the past month and is considered the standard of care in most sleep centers. The investigators here sought to determine the predictive value of the PSQI in regard to migraine. Migraine disability was assessed via the Migraine Disability Assessment (MIDAS) scale as well as the Headache Impact Test (HIT-6). Statistical analysis was performed with logistical regression, t test, and χ2 squared test.
There strongest correlations between poor sleep quality and risk of migraine were found in women, patients over 35 years old, and those with lower education levels. The results revealed that the migraine group had poorer sleep quality, as well as higher anxiety and depression scores, compared with the control group. A low PSQI score (eg, poorer sleep quality) was associated with higher migraine frequency; this was independent of body mass index (BMI), weekly exercise time, and smoking or drinking history. After participants were divided into good and poor sleep quality subgroups, the PSQI score was found to increase the odds ratio of migraine by a factor of 6.
The investigators were able to show the predictive quality of the PSQI score. Worse sleep quality was found to be associated with a higher MIDAS score and HIT-6 score as well as total pain burden, pain severity, decreased quality of life, depression, and anxiety. Although most headache specialists spend a significant amount of time discussing sleep as it relates to migraine, it may be worth considering following a sleep quality scale, such as the PSQI, over time as we monitor our patients. This may allow us to take a more proactive role and be able to prognosticate our patients' migraine journey somewhat better. Although sleep triggers and associations with migraine can be very difficult to discuss and treat, this study very clearly argues for the importance of focusing on sleep with our patients.
Although the most common aura our patients with migraine experience is visual, many patients with migraine will also experience non-aura visual changes. These can range from short-lasting episodes of blurred vision, such as transient visual obscurations, or other transient visual disturbances that do not fit the criteria of aura as defined by the International Classification of Headache Disorders (ICHD).
A prior study by Tsao and colleagues had revealed that almost half of headache patients experienced some headache-related visual change, the most common of which were short-lasting flickering lights or a movable, monochromatic scotoma. As opposed to visual aura, these transient disturbances were shorter in onset and duration and typically occurred during the headache phase of a migraine attack. In the current study, Tsao and colleagues sought to determine whether the presence of these findings was associated with a different headache burden from that typically found in migraine with aura.
The participants in this study were enrolled over a 10-year period from May 2010 to July 2020. They initially underwent a visual phenomenon questionnaire and then a thorough clinical interview to determine their headache diagnosis per ICHD criteria — specifically whether they had an underlying diagnosis of migraine with aura or migraine without aura. Participants were also separately diagnosed with chronic migraine or medication overuse headache. A visual rating scale was used in the initial questionnaires. This scale posed questions about the duration of symptoms; whether the symptoms develop gradually or suddenly; and whether the visual change was a scotoma, zigzag lines, or in a unilateral or bilateral visual field. A prior study by these investigators determined this visual rating scale to be highly sensitive and specific for diagnosing migraine with aura.
Participants were also given the MIDAS questionnaire and were assessed with the HIT-6 scale, a migraine photophobia score, and the Beck Depression Inventory. A total of 12,255 patients were enrolled, 9946 with migraine, who were subdivided on the basis of diagnosis of migraine with or without aura. Blurred vision was the most common visual complaint among all migraine patients. Patients who had transient visual disturbances that did not fit the criteria of migraine with aura were noted to have a statistically significant higher headache frequency, more severe headache-related disability, a higher likelihood of developing medication overuse headache, and a greater incidence of anxiety and depression.
An important distinction that all headache specialists make is whether their patients experience migraine with or without aura. The primary purpose for this distinction is to determine the appropriateness of specific medications (estrogen or vasoconstrictive medications), as migraine aura relates to vascular risk. We usually delve deeply into whether the visual symptoms that our patients experience do or do not fit into the ICHD criteria of migraine aura. We should not discard or think less of non-aura visual disturbances; these authors argue very clearly that these kinds of visual changes can be very relevant prognostically.
Adjuvant nivo+ipilimumab fails in kidney cancer, in contrast to pembro
PARIS – contrast with those from a previous trial that showed benefit with another agent.
The new results, from CheckMate 914, show that adjuvant treatment with the combination of nivolumab (Opdivo) plus ipilimumab (Yervoy) did not improve disease-free survival (DFS), compared with placebo.
The finding was presented at the annual meeting of the European Society for Medical Oncology.
CheckMate 914 “did not meet the primary endpoint,” study presenter Robert J. Motzer, MD, a medical oncologist at Memorial Sloan Kettering Cancer Center, New York, said at a press conference.
The results contrast with those seen with pembrolizumab (Keytruda) in the same setting, where the drug achieved a 32% reduction in risk of recurrence or death over placebo in KEYNOTE-564. This led to the U.S. Food and Drug Administration granting approval for the drug as adjuvant treatment following surgery in patients with renal cell carcinoma at intermediate or high risk for recurrence after nephrectomy or after nephrectomy and resection of metastatic lesions.
Another trial of adjuvant immunotherapy in renal cell carcinoma, also presented at ESMO 2022, the IMmotion010 trial with adjuvant atezolizumab (Tecentriq), also did not show any clinical benefit over placebo.
However, Dr. Motzer said that despite both of these new trials showing no benefit, “I don’t think it takes away from standard of care pembrolizumab” in this setting.
There is a great need for adjuvant therapy for patients who undergo surgery, Dr. Motzer commented. The standard treatment for stage I-III localized nonmetastatic renal cell carcinoma is radical or partial nephrectomy, but there remains a “substantial risk” of relapse after surgery, occurring in up to 50% of patients.
In the past, the standard of care for these patients would be watching and waiting and “hoping that the patient doesn’t relapse,” he said, and if they did, then “we would treat accordingly for metastatic disease.”
Differences between trials
When asked about the contrast between the latest trial with the adjuvant nivolumab-ipilimumab combination and the earlier trial with adjuvant pembrolizumab, Dr. Motzer told this news organization that there are differences in the designs of the two studies. “Although they are both global phase 2 trials ... [there are] some differences in the patient population.”
However, the “main differences” are the duration, intensity, and tolerability of the treatment regimens. “I suspect that’s impacted on the outcome of our trial,” he said, as “many of our patients didn’t complete even that 6 months of the more toxic immunotherapy [nivolumab-ipilimumab combination].”
Dr. Motzer also noted that, compared with the metastatic setting, patients “do not tolerate therapy as well” in the adjuvant setting. Consequently, the risk-benefit of a drug is “slightly different ... as we have to be much more concerned about toxicity.”
In addition, he said, “our trial also used these kind-of gross clinical features that were developed years ago” to select patients, but now “there’s other much more refined techniques” that look at the underlying biological signatures “to identify who responds to immunotherapy.”
“So I think we have to do a deep dive into the biology in this trial and in the Merck trial [of pembrolizumab] to see if we can better define who is going to relapse and who is going to benefit,” he said.
Commenting on the new results, Dominik Berthold, MD, Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland, also wondered whether differences in trial design and study populations could explain the divergent results between the CheckMate and KEYNOTE trials.
“Investigators will need to look in detail at subpopulations and biomarkers to guide treatment decisions and trial design for current and future patients,” he added.
Dr. Berthold said he agrees that pembrolizumab remains standard of care, but “I’m not really sure that we have really to offer all patients” the drug.
He explained that, on the one hand, there is the risk of over-treating many patients, depending on their stage, and on the other hand, “many patients who get pembrolizumab actually do progress.”
In addition, there is the question of the treatment sequence in patients who are already exposed to immunotherapy and when to start tyrosine kinase inhibitors, as well as the much broader issue of the lack of long-term overall survival data with pembrolizumab.
Dr. Berthold noted the issue of whether the high treatment discontinuation rate in CheckMate 914 affected the efficacy of nivolumab plus ipilimumab raises the question of whether, from an immunological point of view, 1 year of pembrolizumab is more effective than 3 months of the combination therapy.
“I think it might be one of the explanations,” he said, adding, however, that these are just “hypotheses” at this stage.
Details of the new results
Previous results with the nivolumab-ipilimumab combination, from the CheckMate 214 trial in patients with advanced renal cell carcinoma, had demonstrated that upfront nivolumab plus ipilimumab offered significantly longer treatment-free survival than the VEGF inhibitor sunitinib. The “striking results” from that trial indicated the combination was not only associated with a survival benefit, but also “high response rates, durable responses, complete responses, and even treatment responses that continue after treatment is discontinued,” Dr. Motzer commented.
So his team set out to test the combination in the adjuvant setting in the CheckMate 914 trial, designed in two parts: Part A, comparing nivolumab plus ipilimumab with placebo, and Part B, adding nivolumab monotherapy as another comparator.
Reporting on Part A of the trial, Dr. Motzer explained that they included 816 patients with renal cell carcinoma who had undergone radical or partial nephrectomy with negative surgical margins and had a predominantly clear cell histology.
They also selected patients based on their pathologic TNM staging, choosing “high-risk” individuals, Dr. Motzer explained, but who nevertheless had no evidence of residual disease or distant metastases following nephrectomy.
Between 4 and 12 weeks after surgery, patients were randomized to receive 12 doses of nivolumab plus four doses of ipilimumab or matched placebos for an expected treatment duration of 24 weeks.
The median age of patients was 58-59 years, and approximately 71% were men. By far the most common type of surgery was radical nephrectomy, with 93%, and Dr. Motzer noted that most patients (77%-78%) had pT3 disease without nodal involvement.
After a median follow-up of 37.0 months, there was no significant difference between groups in the primary endpoint of DFS, as assessed by blinded independent central review.
Median DFS was not reached for nivolumab plus ipilimumab versus 50.7 months for placebo, at a hazard ratio of 0.92 (P = .5347). At 24 months, DFS was 76.4% with the combination therapy versus 74.0% for placebo.
Subgroup analysis did not reveal any patient groups that significantly benefitted from the combination therapy, although there was a signal of greater benefit in those with other than pT3 disease.
While tumors with sarcomatoid features appeared to have a significant benefit from nivolumab plus ipilimumab therapy, they represented only 5% of the study population.
During his presentation, Dr. Motzer showed the median duration of therapy was 5.1 months in both groups, but only 57% of nivolumab plus ipilimumab patients completed all doses versus 89% of those assigned to placebo.
In addition, 33% of patients given nivolumab plus ipilimumab discontinued due to study drug toxicity and 29% had a treatment-related adverse event that led to treatment discontinuation. This compared with only 1% of patients for both outcomes with placebo.
The most common treatment-related adverse events in the combination therapy group were pruritus (27%), fatigue (25%), diarrhea (20%), rash (19%), hyperthyroidism (16%), and hypothyroidism (16%), and the vast majority of events were grade 1-2.
Dr. Motzer said that, following these negative results, they are “certainly digging deeper into the details to see which particular groups may have benefited and when toxicity occurred.
Then, more importantly, the team will look out for the results of Part B of the trial to assess the impact of nivolumab monotherapy. “I’m hoping it’s better tolerated,” he said.
Discussant James Larkin, MD, PhD, a consultant medical oncologist at The Royal Marsden, London, said the results from CheckMate 914 came “as a bit of a surprise.”
As did Dr. Motzer, Dr. Larkin singled out the high number of patients who could not complete the full dosing schedule and discontinued treatment.
He added that, while one has to be “cautious” when comparing trials, KEYNOTE-564 was “relatively similar” in design, and it’s “unlikely there’s any significant difference in activity” between the two drugs.
Dr. Larkin also believes data from Part B of CheckMate 914 will be “illuminating.”
There are nevertheless a number of outstanding questions about the results from Part A, he said, the main one being how to better select patients who might respond to the combination, which currently is not possible due to the lack of clinically relevant biomarkers.
The study was funded by Bristol Myers Squibb. Dr. Motzer has disclosed relationships with AstraZeneca, Aveo Pharmaceuticals, Bristol Myers Squibb, Eisai, EMD Serono, Exelixis, Genentech/Roche, Incyte, Lilly Oncology, Merck, Novartis, and Pfizer.
A version of this article first appeared on Medscape.com.
PARIS – contrast with those from a previous trial that showed benefit with another agent.
The new results, from CheckMate 914, show that adjuvant treatment with the combination of nivolumab (Opdivo) plus ipilimumab (Yervoy) did not improve disease-free survival (DFS), compared with placebo.
The finding was presented at the annual meeting of the European Society for Medical Oncology.
CheckMate 914 “did not meet the primary endpoint,” study presenter Robert J. Motzer, MD, a medical oncologist at Memorial Sloan Kettering Cancer Center, New York, said at a press conference.
The results contrast with those seen with pembrolizumab (Keytruda) in the same setting, where the drug achieved a 32% reduction in risk of recurrence or death over placebo in KEYNOTE-564. This led to the U.S. Food and Drug Administration granting approval for the drug as adjuvant treatment following surgery in patients with renal cell carcinoma at intermediate or high risk for recurrence after nephrectomy or after nephrectomy and resection of metastatic lesions.
Another trial of adjuvant immunotherapy in renal cell carcinoma, also presented at ESMO 2022, the IMmotion010 trial with adjuvant atezolizumab (Tecentriq), also did not show any clinical benefit over placebo.
However, Dr. Motzer said that despite both of these new trials showing no benefit, “I don’t think it takes away from standard of care pembrolizumab” in this setting.
There is a great need for adjuvant therapy for patients who undergo surgery, Dr. Motzer commented. The standard treatment for stage I-III localized nonmetastatic renal cell carcinoma is radical or partial nephrectomy, but there remains a “substantial risk” of relapse after surgery, occurring in up to 50% of patients.
In the past, the standard of care for these patients would be watching and waiting and “hoping that the patient doesn’t relapse,” he said, and if they did, then “we would treat accordingly for metastatic disease.”
Differences between trials
When asked about the contrast between the latest trial with the adjuvant nivolumab-ipilimumab combination and the earlier trial with adjuvant pembrolizumab, Dr. Motzer told this news organization that there are differences in the designs of the two studies. “Although they are both global phase 2 trials ... [there are] some differences in the patient population.”
However, the “main differences” are the duration, intensity, and tolerability of the treatment regimens. “I suspect that’s impacted on the outcome of our trial,” he said, as “many of our patients didn’t complete even that 6 months of the more toxic immunotherapy [nivolumab-ipilimumab combination].”
Dr. Motzer also noted that, compared with the metastatic setting, patients “do not tolerate therapy as well” in the adjuvant setting. Consequently, the risk-benefit of a drug is “slightly different ... as we have to be much more concerned about toxicity.”
In addition, he said, “our trial also used these kind-of gross clinical features that were developed years ago” to select patients, but now “there’s other much more refined techniques” that look at the underlying biological signatures “to identify who responds to immunotherapy.”
“So I think we have to do a deep dive into the biology in this trial and in the Merck trial [of pembrolizumab] to see if we can better define who is going to relapse and who is going to benefit,” he said.
Commenting on the new results, Dominik Berthold, MD, Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland, also wondered whether differences in trial design and study populations could explain the divergent results between the CheckMate and KEYNOTE trials.
“Investigators will need to look in detail at subpopulations and biomarkers to guide treatment decisions and trial design for current and future patients,” he added.
Dr. Berthold said he agrees that pembrolizumab remains standard of care, but “I’m not really sure that we have really to offer all patients” the drug.
He explained that, on the one hand, there is the risk of over-treating many patients, depending on their stage, and on the other hand, “many patients who get pembrolizumab actually do progress.”
In addition, there is the question of the treatment sequence in patients who are already exposed to immunotherapy and when to start tyrosine kinase inhibitors, as well as the much broader issue of the lack of long-term overall survival data with pembrolizumab.
Dr. Berthold noted the issue of whether the high treatment discontinuation rate in CheckMate 914 affected the efficacy of nivolumab plus ipilimumab raises the question of whether, from an immunological point of view, 1 year of pembrolizumab is more effective than 3 months of the combination therapy.
“I think it might be one of the explanations,” he said, adding, however, that these are just “hypotheses” at this stage.
Details of the new results
Previous results with the nivolumab-ipilimumab combination, from the CheckMate 214 trial in patients with advanced renal cell carcinoma, had demonstrated that upfront nivolumab plus ipilimumab offered significantly longer treatment-free survival than the VEGF inhibitor sunitinib. The “striking results” from that trial indicated the combination was not only associated with a survival benefit, but also “high response rates, durable responses, complete responses, and even treatment responses that continue after treatment is discontinued,” Dr. Motzer commented.
So his team set out to test the combination in the adjuvant setting in the CheckMate 914 trial, designed in two parts: Part A, comparing nivolumab plus ipilimumab with placebo, and Part B, adding nivolumab monotherapy as another comparator.
Reporting on Part A of the trial, Dr. Motzer explained that they included 816 patients with renal cell carcinoma who had undergone radical or partial nephrectomy with negative surgical margins and had a predominantly clear cell histology.
They also selected patients based on their pathologic TNM staging, choosing “high-risk” individuals, Dr. Motzer explained, but who nevertheless had no evidence of residual disease or distant metastases following nephrectomy.
Between 4 and 12 weeks after surgery, patients were randomized to receive 12 doses of nivolumab plus four doses of ipilimumab or matched placebos for an expected treatment duration of 24 weeks.
The median age of patients was 58-59 years, and approximately 71% were men. By far the most common type of surgery was radical nephrectomy, with 93%, and Dr. Motzer noted that most patients (77%-78%) had pT3 disease without nodal involvement.
After a median follow-up of 37.0 months, there was no significant difference between groups in the primary endpoint of DFS, as assessed by blinded independent central review.
Median DFS was not reached for nivolumab plus ipilimumab versus 50.7 months for placebo, at a hazard ratio of 0.92 (P = .5347). At 24 months, DFS was 76.4% with the combination therapy versus 74.0% for placebo.
Subgroup analysis did not reveal any patient groups that significantly benefitted from the combination therapy, although there was a signal of greater benefit in those with other than pT3 disease.
While tumors with sarcomatoid features appeared to have a significant benefit from nivolumab plus ipilimumab therapy, they represented only 5% of the study population.
During his presentation, Dr. Motzer showed the median duration of therapy was 5.1 months in both groups, but only 57% of nivolumab plus ipilimumab patients completed all doses versus 89% of those assigned to placebo.
In addition, 33% of patients given nivolumab plus ipilimumab discontinued due to study drug toxicity and 29% had a treatment-related adverse event that led to treatment discontinuation. This compared with only 1% of patients for both outcomes with placebo.
The most common treatment-related adverse events in the combination therapy group were pruritus (27%), fatigue (25%), diarrhea (20%), rash (19%), hyperthyroidism (16%), and hypothyroidism (16%), and the vast majority of events were grade 1-2.
Dr. Motzer said that, following these negative results, they are “certainly digging deeper into the details to see which particular groups may have benefited and when toxicity occurred.
Then, more importantly, the team will look out for the results of Part B of the trial to assess the impact of nivolumab monotherapy. “I’m hoping it’s better tolerated,” he said.
Discussant James Larkin, MD, PhD, a consultant medical oncologist at The Royal Marsden, London, said the results from CheckMate 914 came “as a bit of a surprise.”
As did Dr. Motzer, Dr. Larkin singled out the high number of patients who could not complete the full dosing schedule and discontinued treatment.
He added that, while one has to be “cautious” when comparing trials, KEYNOTE-564 was “relatively similar” in design, and it’s “unlikely there’s any significant difference in activity” between the two drugs.
Dr. Larkin also believes data from Part B of CheckMate 914 will be “illuminating.”
There are nevertheless a number of outstanding questions about the results from Part A, he said, the main one being how to better select patients who might respond to the combination, which currently is not possible due to the lack of clinically relevant biomarkers.
The study was funded by Bristol Myers Squibb. Dr. Motzer has disclosed relationships with AstraZeneca, Aveo Pharmaceuticals, Bristol Myers Squibb, Eisai, EMD Serono, Exelixis, Genentech/Roche, Incyte, Lilly Oncology, Merck, Novartis, and Pfizer.
A version of this article first appeared on Medscape.com.
PARIS – contrast with those from a previous trial that showed benefit with another agent.
The new results, from CheckMate 914, show that adjuvant treatment with the combination of nivolumab (Opdivo) plus ipilimumab (Yervoy) did not improve disease-free survival (DFS), compared with placebo.
The finding was presented at the annual meeting of the European Society for Medical Oncology.
CheckMate 914 “did not meet the primary endpoint,” study presenter Robert J. Motzer, MD, a medical oncologist at Memorial Sloan Kettering Cancer Center, New York, said at a press conference.
The results contrast with those seen with pembrolizumab (Keytruda) in the same setting, where the drug achieved a 32% reduction in risk of recurrence or death over placebo in KEYNOTE-564. This led to the U.S. Food and Drug Administration granting approval for the drug as adjuvant treatment following surgery in patients with renal cell carcinoma at intermediate or high risk for recurrence after nephrectomy or after nephrectomy and resection of metastatic lesions.
Another trial of adjuvant immunotherapy in renal cell carcinoma, also presented at ESMO 2022, the IMmotion010 trial with adjuvant atezolizumab (Tecentriq), also did not show any clinical benefit over placebo.
However, Dr. Motzer said that despite both of these new trials showing no benefit, “I don’t think it takes away from standard of care pembrolizumab” in this setting.
There is a great need for adjuvant therapy for patients who undergo surgery, Dr. Motzer commented. The standard treatment for stage I-III localized nonmetastatic renal cell carcinoma is radical or partial nephrectomy, but there remains a “substantial risk” of relapse after surgery, occurring in up to 50% of patients.
In the past, the standard of care for these patients would be watching and waiting and “hoping that the patient doesn’t relapse,” he said, and if they did, then “we would treat accordingly for metastatic disease.”
Differences between trials
When asked about the contrast between the latest trial with the adjuvant nivolumab-ipilimumab combination and the earlier trial with adjuvant pembrolizumab, Dr. Motzer told this news organization that there are differences in the designs of the two studies. “Although they are both global phase 2 trials ... [there are] some differences in the patient population.”
However, the “main differences” are the duration, intensity, and tolerability of the treatment regimens. “I suspect that’s impacted on the outcome of our trial,” he said, as “many of our patients didn’t complete even that 6 months of the more toxic immunotherapy [nivolumab-ipilimumab combination].”
Dr. Motzer also noted that, compared with the metastatic setting, patients “do not tolerate therapy as well” in the adjuvant setting. Consequently, the risk-benefit of a drug is “slightly different ... as we have to be much more concerned about toxicity.”
In addition, he said, “our trial also used these kind-of gross clinical features that were developed years ago” to select patients, but now “there’s other much more refined techniques” that look at the underlying biological signatures “to identify who responds to immunotherapy.”
“So I think we have to do a deep dive into the biology in this trial and in the Merck trial [of pembrolizumab] to see if we can better define who is going to relapse and who is going to benefit,” he said.
Commenting on the new results, Dominik Berthold, MD, Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland, also wondered whether differences in trial design and study populations could explain the divergent results between the CheckMate and KEYNOTE trials.
“Investigators will need to look in detail at subpopulations and biomarkers to guide treatment decisions and trial design for current and future patients,” he added.
Dr. Berthold said he agrees that pembrolizumab remains standard of care, but “I’m not really sure that we have really to offer all patients” the drug.
He explained that, on the one hand, there is the risk of over-treating many patients, depending on their stage, and on the other hand, “many patients who get pembrolizumab actually do progress.”
In addition, there is the question of the treatment sequence in patients who are already exposed to immunotherapy and when to start tyrosine kinase inhibitors, as well as the much broader issue of the lack of long-term overall survival data with pembrolizumab.
Dr. Berthold noted the issue of whether the high treatment discontinuation rate in CheckMate 914 affected the efficacy of nivolumab plus ipilimumab raises the question of whether, from an immunological point of view, 1 year of pembrolizumab is more effective than 3 months of the combination therapy.
“I think it might be one of the explanations,” he said, adding, however, that these are just “hypotheses” at this stage.
Details of the new results
Previous results with the nivolumab-ipilimumab combination, from the CheckMate 214 trial in patients with advanced renal cell carcinoma, had demonstrated that upfront nivolumab plus ipilimumab offered significantly longer treatment-free survival than the VEGF inhibitor sunitinib. The “striking results” from that trial indicated the combination was not only associated with a survival benefit, but also “high response rates, durable responses, complete responses, and even treatment responses that continue after treatment is discontinued,” Dr. Motzer commented.
So his team set out to test the combination in the adjuvant setting in the CheckMate 914 trial, designed in two parts: Part A, comparing nivolumab plus ipilimumab with placebo, and Part B, adding nivolumab monotherapy as another comparator.
Reporting on Part A of the trial, Dr. Motzer explained that they included 816 patients with renal cell carcinoma who had undergone radical or partial nephrectomy with negative surgical margins and had a predominantly clear cell histology.
They also selected patients based on their pathologic TNM staging, choosing “high-risk” individuals, Dr. Motzer explained, but who nevertheless had no evidence of residual disease or distant metastases following nephrectomy.
Between 4 and 12 weeks after surgery, patients were randomized to receive 12 doses of nivolumab plus four doses of ipilimumab or matched placebos for an expected treatment duration of 24 weeks.
The median age of patients was 58-59 years, and approximately 71% were men. By far the most common type of surgery was radical nephrectomy, with 93%, and Dr. Motzer noted that most patients (77%-78%) had pT3 disease without nodal involvement.
After a median follow-up of 37.0 months, there was no significant difference between groups in the primary endpoint of DFS, as assessed by blinded independent central review.
Median DFS was not reached for nivolumab plus ipilimumab versus 50.7 months for placebo, at a hazard ratio of 0.92 (P = .5347). At 24 months, DFS was 76.4% with the combination therapy versus 74.0% for placebo.
Subgroup analysis did not reveal any patient groups that significantly benefitted from the combination therapy, although there was a signal of greater benefit in those with other than pT3 disease.
While tumors with sarcomatoid features appeared to have a significant benefit from nivolumab plus ipilimumab therapy, they represented only 5% of the study population.
During his presentation, Dr. Motzer showed the median duration of therapy was 5.1 months in both groups, but only 57% of nivolumab plus ipilimumab patients completed all doses versus 89% of those assigned to placebo.
In addition, 33% of patients given nivolumab plus ipilimumab discontinued due to study drug toxicity and 29% had a treatment-related adverse event that led to treatment discontinuation. This compared with only 1% of patients for both outcomes with placebo.
The most common treatment-related adverse events in the combination therapy group were pruritus (27%), fatigue (25%), diarrhea (20%), rash (19%), hyperthyroidism (16%), and hypothyroidism (16%), and the vast majority of events were grade 1-2.
Dr. Motzer said that, following these negative results, they are “certainly digging deeper into the details to see which particular groups may have benefited and when toxicity occurred.
Then, more importantly, the team will look out for the results of Part B of the trial to assess the impact of nivolumab monotherapy. “I’m hoping it’s better tolerated,” he said.
Discussant James Larkin, MD, PhD, a consultant medical oncologist at The Royal Marsden, London, said the results from CheckMate 914 came “as a bit of a surprise.”
As did Dr. Motzer, Dr. Larkin singled out the high number of patients who could not complete the full dosing schedule and discontinued treatment.
He added that, while one has to be “cautious” when comparing trials, KEYNOTE-564 was “relatively similar” in design, and it’s “unlikely there’s any significant difference in activity” between the two drugs.
Dr. Larkin also believes data from Part B of CheckMate 914 will be “illuminating.”
There are nevertheless a number of outstanding questions about the results from Part A, he said, the main one being how to better select patients who might respond to the combination, which currently is not possible due to the lack of clinically relevant biomarkers.
The study was funded by Bristol Myers Squibb. Dr. Motzer has disclosed relationships with AstraZeneca, Aveo Pharmaceuticals, Bristol Myers Squibb, Eisai, EMD Serono, Exelixis, Genentech/Roche, Incyte, Lilly Oncology, Merck, Novartis, and Pfizer.
A version of this article first appeared on Medscape.com.
Bariatric surgery may up risk for epilepsy
Analyzing health records, investigators compared almost 17,000 patients who had undergone bariatric surgery with more than 620,000 individuals with obesity who had not undergone the surgery.
During a minimum 3-year follow-up period, the surgery group had a 45% higher risk of developing epilepsy than the nonsurgery group. Moreover, patients who had a stroke after their bariatric surgery were 14 times more likely to develop epilepsy than those who did not have a stroke.
“When considering having bariatric surgery, people should talk to their doctors about the benefits and risks,” senior investigator Jorge Burneo, MD, professor of neurology, biostatistics, and epidemiology and endowed chair in epilepsy at Western University, London, told this news organization.
“While there are many health benefits of weight loss, our findings suggest that epilepsy is a long-term risk of bariatric surgery for weight loss,” Dr. Burneo said.
The findings were published online in Neurology.
Unrecognized risk factor?
Bariatric surgery has become more common as global rates of obesity have increased. The surgery has been shown to reduce the risk for serious obesity-related conditions, the researchers note.
However, “in addition to the positive outcomes of bariatric surgery, several long-term neurological complications have also been identified,” they write.
One previous study reported increased epilepsy risk following gastric bypass. Those findings “suggest that bariatric surgery may be an unrecognized epilepsy risk factor; however, this possible association has not been thoroughly explored,” write the investigators.
Dr. Burneo said he conducted the study because he has seen patients with epilepsy in his clinic who were “without risk factors, with normal MRIs, who shared the history of having bariatric surgery before the development of epilepsy.”
The researchers’ primary objective was to “assess whether epilepsy risk is elevated following bariatric surgery for weight loss relative to a nonsurgical cohort of patients who are obese,” he noted.
The study used linked administrative health databases in Ontario, Canada. Patients were accrued from July 1, 2010, to Dec. 31, 2016, and were followed until Dec. 31, 2019. The analysis included 639,472 participants, 2.7% of whom had undergone bariatric surgery.
The “exposed” cohort consisted of all Ontario residents aged 18 years or older who had undergone bariatric surgery during the 6-year period (n = 16,958; 65.1% women; mean age, 47.4 years), while the “unexposed” cohort consisted of patients hospitalized with a diagnosis of obesity who had not undergone bariatric surgery (n = 622,514; 62.8% women; mean age, 47.6 years).
Patients with a history of seizures, epilepsy, epilepsy risk factors, prior brain surgery, psychiatric disorders, or drug or alcohol abuse/dependence were excluded from the analysis.
The researchers collected data on patients’ sociodemographic characteristics at the index date, as well as Charlson Comorbidity Index scores during the 2 years prior to index, and data regarding several specific comorbidities, such as diabetes mellitus, hypertension, sleep apnea, depression/anxiety, and cardiovascular factors.
The exposed and unexposed cohorts were followed for a median period of 5.8 and 5.9 person-years, respectively.
‘Unclear’ mechanisms
Before weighting, 0.4% of participants in the exposed cohort (n = 73) developed epilepsy, versus 0.2% of participants in the unexposed cohort (n = 1,260) by the end of the follow-up period.
In the weighted cohorts, there were 50.1 epilepsy diagnoses per 100,000 person-years, versus 34.1 per 100,000 person-years (rate difference, 16 per 100,000 person-years).
The multivariable analysis of the weighted cohort showed the hazard ratio for epilepsy cases that were associated with bariatric surgery was 1.45 (95% confidence interval, 1.35-1.56), after adjusting for sleep apnea and including stroke as a time-varying covariate.
Having a stroke during the follow-up period increased epilepsy 14-fold in the exposed cohort (HR, 14.03; 95% CI, 4.25-46.25).
The investigators note that they were unable to measure obesity status or body mass index throughout the study and that some obesity-related comorbidities “may affect epilepsy risk.”
In addition, Dr. Burneo reported that the study did not investigate potential causes and mechanisms of the association between bariatric surgery and epilepsy risk.
Hypotheses “include potential nutritional deficiencies, receipt of general anesthesia, or other unclear causes,” he said.
“Future research should investigate epilepsy as a potential long-term complication of bariatric surgery, exploring the possible effects of this procedure,” Dr. Burneo added.
Risk-benefit discussion
In a comment, Jacqueline French, MD, professor of neurology at NYU Grossman School of Medicine, and director of NYU’s Epilepsy Study Consortium, said she was “not 100% surprised by the findings” because she has seen in her clinical practice “a number of patients who developed epilepsy after bariatric surgery or had a history of bariatric surgery at the time they developed epilepsy.”
On the other hand, she has also seen patients who did not have a history of bariatric surgery and who developed epilepsy.
“I’m unable to tell if there is an association, although I’ve had it at the back of my head as a thought and wondered about it,” said Dr. French, who is also the chief medical and innovation officer at the Epilepsy Foundation. She was not involved with the study.
She noted that possible mechanisms underlying the association are that gastric bypass surgery leads to a “significant alteration” in nutrient absorption. Moreover, “we now know that the microbiome is associated with epilepsy” and that changes occur in the gut microbiome after bariatric surgery, Dr. French said.
There are two take-home messages for practicing clinicians, she added.
“Although the risk [of developing epilepsy] is very low, it should be presented as part of the risks and benefits to patients considering bariatric surgery,” she said.
“It’s equally important to follow up on the potential differences in these patients who go on to develop epilepsy following bariatric surgery,” said Dr. French. “Is there a certain metabolic profile or some nutrient previously absorbed that now is not absorbed that might predispose people to risk?”
This would be “enormously important to know because it might not just pertain to these people but to a whole other cohort of people who develop epilepsy,” Dr. French concluded.
The study was funded by the Ontario Ministry of Health and Ministry of Long-Term Care and by the Jack Cowin Endowed Chair in Epilepsy Research at Western University. Dr. Burneo holds the Jack Cowin Endowed Chair in Epilepsy Research at Western University. The other investigators and Dr. French have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Analyzing health records, investigators compared almost 17,000 patients who had undergone bariatric surgery with more than 620,000 individuals with obesity who had not undergone the surgery.
During a minimum 3-year follow-up period, the surgery group had a 45% higher risk of developing epilepsy than the nonsurgery group. Moreover, patients who had a stroke after their bariatric surgery were 14 times more likely to develop epilepsy than those who did not have a stroke.
“When considering having bariatric surgery, people should talk to their doctors about the benefits and risks,” senior investigator Jorge Burneo, MD, professor of neurology, biostatistics, and epidemiology and endowed chair in epilepsy at Western University, London, told this news organization.
“While there are many health benefits of weight loss, our findings suggest that epilepsy is a long-term risk of bariatric surgery for weight loss,” Dr. Burneo said.
The findings were published online in Neurology.
Unrecognized risk factor?
Bariatric surgery has become more common as global rates of obesity have increased. The surgery has been shown to reduce the risk for serious obesity-related conditions, the researchers note.
However, “in addition to the positive outcomes of bariatric surgery, several long-term neurological complications have also been identified,” they write.
One previous study reported increased epilepsy risk following gastric bypass. Those findings “suggest that bariatric surgery may be an unrecognized epilepsy risk factor; however, this possible association has not been thoroughly explored,” write the investigators.
Dr. Burneo said he conducted the study because he has seen patients with epilepsy in his clinic who were “without risk factors, with normal MRIs, who shared the history of having bariatric surgery before the development of epilepsy.”
The researchers’ primary objective was to “assess whether epilepsy risk is elevated following bariatric surgery for weight loss relative to a nonsurgical cohort of patients who are obese,” he noted.
The study used linked administrative health databases in Ontario, Canada. Patients were accrued from July 1, 2010, to Dec. 31, 2016, and were followed until Dec. 31, 2019. The analysis included 639,472 participants, 2.7% of whom had undergone bariatric surgery.
The “exposed” cohort consisted of all Ontario residents aged 18 years or older who had undergone bariatric surgery during the 6-year period (n = 16,958; 65.1% women; mean age, 47.4 years), while the “unexposed” cohort consisted of patients hospitalized with a diagnosis of obesity who had not undergone bariatric surgery (n = 622,514; 62.8% women; mean age, 47.6 years).
Patients with a history of seizures, epilepsy, epilepsy risk factors, prior brain surgery, psychiatric disorders, or drug or alcohol abuse/dependence were excluded from the analysis.
The researchers collected data on patients’ sociodemographic characteristics at the index date, as well as Charlson Comorbidity Index scores during the 2 years prior to index, and data regarding several specific comorbidities, such as diabetes mellitus, hypertension, sleep apnea, depression/anxiety, and cardiovascular factors.
The exposed and unexposed cohorts were followed for a median period of 5.8 and 5.9 person-years, respectively.
‘Unclear’ mechanisms
Before weighting, 0.4% of participants in the exposed cohort (n = 73) developed epilepsy, versus 0.2% of participants in the unexposed cohort (n = 1,260) by the end of the follow-up period.
In the weighted cohorts, there were 50.1 epilepsy diagnoses per 100,000 person-years, versus 34.1 per 100,000 person-years (rate difference, 16 per 100,000 person-years).
The multivariable analysis of the weighted cohort showed the hazard ratio for epilepsy cases that were associated with bariatric surgery was 1.45 (95% confidence interval, 1.35-1.56), after adjusting for sleep apnea and including stroke as a time-varying covariate.
Having a stroke during the follow-up period increased epilepsy 14-fold in the exposed cohort (HR, 14.03; 95% CI, 4.25-46.25).
The investigators note that they were unable to measure obesity status or body mass index throughout the study and that some obesity-related comorbidities “may affect epilepsy risk.”
In addition, Dr. Burneo reported that the study did not investigate potential causes and mechanisms of the association between bariatric surgery and epilepsy risk.
Hypotheses “include potential nutritional deficiencies, receipt of general anesthesia, or other unclear causes,” he said.
“Future research should investigate epilepsy as a potential long-term complication of bariatric surgery, exploring the possible effects of this procedure,” Dr. Burneo added.
Risk-benefit discussion
In a comment, Jacqueline French, MD, professor of neurology at NYU Grossman School of Medicine, and director of NYU’s Epilepsy Study Consortium, said she was “not 100% surprised by the findings” because she has seen in her clinical practice “a number of patients who developed epilepsy after bariatric surgery or had a history of bariatric surgery at the time they developed epilepsy.”
On the other hand, she has also seen patients who did not have a history of bariatric surgery and who developed epilepsy.
“I’m unable to tell if there is an association, although I’ve had it at the back of my head as a thought and wondered about it,” said Dr. French, who is also the chief medical and innovation officer at the Epilepsy Foundation. She was not involved with the study.
She noted that possible mechanisms underlying the association are that gastric bypass surgery leads to a “significant alteration” in nutrient absorption. Moreover, “we now know that the microbiome is associated with epilepsy” and that changes occur in the gut microbiome after bariatric surgery, Dr. French said.
There are two take-home messages for practicing clinicians, she added.
“Although the risk [of developing epilepsy] is very low, it should be presented as part of the risks and benefits to patients considering bariatric surgery,” she said.
“It’s equally important to follow up on the potential differences in these patients who go on to develop epilepsy following bariatric surgery,” said Dr. French. “Is there a certain metabolic profile or some nutrient previously absorbed that now is not absorbed that might predispose people to risk?”
This would be “enormously important to know because it might not just pertain to these people but to a whole other cohort of people who develop epilepsy,” Dr. French concluded.
The study was funded by the Ontario Ministry of Health and Ministry of Long-Term Care and by the Jack Cowin Endowed Chair in Epilepsy Research at Western University. Dr. Burneo holds the Jack Cowin Endowed Chair in Epilepsy Research at Western University. The other investigators and Dr. French have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Analyzing health records, investigators compared almost 17,000 patients who had undergone bariatric surgery with more than 620,000 individuals with obesity who had not undergone the surgery.
During a minimum 3-year follow-up period, the surgery group had a 45% higher risk of developing epilepsy than the nonsurgery group. Moreover, patients who had a stroke after their bariatric surgery were 14 times more likely to develop epilepsy than those who did not have a stroke.
“When considering having bariatric surgery, people should talk to their doctors about the benefits and risks,” senior investigator Jorge Burneo, MD, professor of neurology, biostatistics, and epidemiology and endowed chair in epilepsy at Western University, London, told this news organization.
“While there are many health benefits of weight loss, our findings suggest that epilepsy is a long-term risk of bariatric surgery for weight loss,” Dr. Burneo said.
The findings were published online in Neurology.
Unrecognized risk factor?
Bariatric surgery has become more common as global rates of obesity have increased. The surgery has been shown to reduce the risk for serious obesity-related conditions, the researchers note.
However, “in addition to the positive outcomes of bariatric surgery, several long-term neurological complications have also been identified,” they write.
One previous study reported increased epilepsy risk following gastric bypass. Those findings “suggest that bariatric surgery may be an unrecognized epilepsy risk factor; however, this possible association has not been thoroughly explored,” write the investigators.
Dr. Burneo said he conducted the study because he has seen patients with epilepsy in his clinic who were “without risk factors, with normal MRIs, who shared the history of having bariatric surgery before the development of epilepsy.”
The researchers’ primary objective was to “assess whether epilepsy risk is elevated following bariatric surgery for weight loss relative to a nonsurgical cohort of patients who are obese,” he noted.
The study used linked administrative health databases in Ontario, Canada. Patients were accrued from July 1, 2010, to Dec. 31, 2016, and were followed until Dec. 31, 2019. The analysis included 639,472 participants, 2.7% of whom had undergone bariatric surgery.
The “exposed” cohort consisted of all Ontario residents aged 18 years or older who had undergone bariatric surgery during the 6-year period (n = 16,958; 65.1% women; mean age, 47.4 years), while the “unexposed” cohort consisted of patients hospitalized with a diagnosis of obesity who had not undergone bariatric surgery (n = 622,514; 62.8% women; mean age, 47.6 years).
Patients with a history of seizures, epilepsy, epilepsy risk factors, prior brain surgery, psychiatric disorders, or drug or alcohol abuse/dependence were excluded from the analysis.
The researchers collected data on patients’ sociodemographic characteristics at the index date, as well as Charlson Comorbidity Index scores during the 2 years prior to index, and data regarding several specific comorbidities, such as diabetes mellitus, hypertension, sleep apnea, depression/anxiety, and cardiovascular factors.
The exposed and unexposed cohorts were followed for a median period of 5.8 and 5.9 person-years, respectively.
‘Unclear’ mechanisms
Before weighting, 0.4% of participants in the exposed cohort (n = 73) developed epilepsy, versus 0.2% of participants in the unexposed cohort (n = 1,260) by the end of the follow-up period.
In the weighted cohorts, there were 50.1 epilepsy diagnoses per 100,000 person-years, versus 34.1 per 100,000 person-years (rate difference, 16 per 100,000 person-years).
The multivariable analysis of the weighted cohort showed the hazard ratio for epilepsy cases that were associated with bariatric surgery was 1.45 (95% confidence interval, 1.35-1.56), after adjusting for sleep apnea and including stroke as a time-varying covariate.
Having a stroke during the follow-up period increased epilepsy 14-fold in the exposed cohort (HR, 14.03; 95% CI, 4.25-46.25).
The investigators note that they were unable to measure obesity status or body mass index throughout the study and that some obesity-related comorbidities “may affect epilepsy risk.”
In addition, Dr. Burneo reported that the study did not investigate potential causes and mechanisms of the association between bariatric surgery and epilepsy risk.
Hypotheses “include potential nutritional deficiencies, receipt of general anesthesia, or other unclear causes,” he said.
“Future research should investigate epilepsy as a potential long-term complication of bariatric surgery, exploring the possible effects of this procedure,” Dr. Burneo added.
Risk-benefit discussion
In a comment, Jacqueline French, MD, professor of neurology at NYU Grossman School of Medicine, and director of NYU’s Epilepsy Study Consortium, said she was “not 100% surprised by the findings” because she has seen in her clinical practice “a number of patients who developed epilepsy after bariatric surgery or had a history of bariatric surgery at the time they developed epilepsy.”
On the other hand, she has also seen patients who did not have a history of bariatric surgery and who developed epilepsy.
“I’m unable to tell if there is an association, although I’ve had it at the back of my head as a thought and wondered about it,” said Dr. French, who is also the chief medical and innovation officer at the Epilepsy Foundation. She was not involved with the study.
She noted that possible mechanisms underlying the association are that gastric bypass surgery leads to a “significant alteration” in nutrient absorption. Moreover, “we now know that the microbiome is associated with epilepsy” and that changes occur in the gut microbiome after bariatric surgery, Dr. French said.
There are two take-home messages for practicing clinicians, she added.
“Although the risk [of developing epilepsy] is very low, it should be presented as part of the risks and benefits to patients considering bariatric surgery,” she said.
“It’s equally important to follow up on the potential differences in these patients who go on to develop epilepsy following bariatric surgery,” said Dr. French. “Is there a certain metabolic profile or some nutrient previously absorbed that now is not absorbed that might predispose people to risk?”
This would be “enormously important to know because it might not just pertain to these people but to a whole other cohort of people who develop epilepsy,” Dr. French concluded.
The study was funded by the Ontario Ministry of Health and Ministry of Long-Term Care and by the Jack Cowin Endowed Chair in Epilepsy Research at Western University. Dr. Burneo holds the Jack Cowin Endowed Chair in Epilepsy Research at Western University. The other investigators and Dr. French have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM NEUROLOGY
How to handle pesky molluscum contagiosum lesions
.
“If you don’t treat them, they’re going to spread,” Dr. Smith, who practices dermatology in Fort Mill, S.C., said at Medscape Live’s annual Coastal Dermatology Symposium. “They’re going to be itchy, they can spread on the patient themselves and then to others, and they can cause scarring. The prevalence is anywhere from 5% to 11%. That means there are 6 million patients out there, just waiting to come into your clinics.”

To date, no treatment has been approved by the Food and Drug Administration for MC, although a laundry list of agents have been tried, including cantharidin; cryotherapy; curettage with and without imiquimod; sinecatechins ointment, 15%; imiquimod; and retinoids. And there are several treatments that are being investigated.
A 2017 Cochrane review of 22 studies involving 1,650 patients demonstrated that no single intervention has been consistently effective in treating MC. “Most of the studies were actually very low quality,” said Dr. Smith, who was not involved with the analysis. “The one high quality study showed that imiquimod did not work any better than its vehicle.”
Investigational treatments
One of the products in the pipeline is VP-102, a proprietary drug-device combination of cantharidin 0.7% administered through a single-use precision applicator, which has been evaluated in phase 3 studies of patients with molluscum aged 2 years and older. It features a visualization agent so that the person applying the drug can see which lesions have been treated. It also contains a bittering agent to mitigate oral ingestion by children.
VP-102, which is being developed by Verrica Pharmaceuticals, is applied once every 21 days in up to 4 applications, and multiple lesions can be treated with one applicator. “It’s a stable concentration with a good shelf life, and two phase 3 randomized studies have shown about a 50% complete clearance of new and existing lesions at day 84,” Dr. Smith said. Those studies enrolled children and adults.
A separate analysis of the same data presented at a meeting in 2019 showed that 77% of patients treated with VP-102 achieved greater than 75% clearance, while 65.8% achieved more than 90% clearance.
The new kid on the block is a gel formulation of a nitric oxide–releasing medication, berdazimer 10.3%, a first-in-class topical treatment being developed by Novan, which can be applied at home. In a multicenter study published in JAMA Dermatology, researchers randomized 444 patients to berdazimer gel 10.3% and 447 to a placebo gel, applied once daily in a thin layer on all MC lesions for 12 weeks. The study was conducted at 55 clinics across the United States between Sept. 1, 2020, and July 21, 2021. The mean age of the patients was about 6.5 years and participants had 3-70 raised MC lesions; those with sexually transmitted MC or MC in the periocular area were excluded. The primary endpoint was complete clearance of MC lesions after 12 weeks of treatment.
At 12 weeks, significantly more patients treated with berdazimer gel achieved complete clearance than those on vehicle (32.4% vs. 19.7%; P < .001). A total of 64 (14.4%) patients in the berdazimer group discontinued treatment because of MC clearance, compared with 40 patients (8.9%) in the vehicle group.
More recently, investigators evaluated autoinoculation vs. 35% trichloroacetic acid (TCA) for the treatment of MC. Autoinoculation involves puncturing the perilesional and lesional skin 5-7 times with an insulin syringe. “This gets a little bit of the virus into the dermis, and you hope to elicit an immune response,” explained Dr. Smith, who was not involved with the study. At 3 months, 80% of patients in the autoinoculation group achieved complete clearance, compared with 62% of those in the TCA group, while recurrence at 6 months was 3% vs. 40%, respectively.
Manual extraction of MC lesions is another option. “I love to pop the cores out with my thumbs,” Dr. Smith said. “You have to pick the patients who can tolerate this, and the MC lesions need to be ripe and ready.”
For ophthalmic lesions, watchful waiting is advisable unless the MC lesions are symptomatic or bothersome or large lesions form on the lid margin, which may cause ocular irritation or even a corneal abrasion. “If a patient presents with a multisite infection that includes ocular lesions, treat lesions on other parts of the body and keep your fingers crossed that a systemic immune response occurs,” she said.
The desired immune response is known as the “BOTE” sign (the beginning of the end), which heralds the clearance of the molluscum infection. This often appears as reddening of all the MC lesions and occasionally as a granulomatous “id-like” reaction especially on the extensor elbows and knees. “When this happens, it often scares the patients,” Dr. Smith said. But she explains that this is a positive development, and that “this means that the lesions are about to self-resolve.”
Dr. Smith disclosed that she serves as a speaker or a member of the speakers bureau for Amgen, CeraVe, EPI, Galderma, InCyte, Lilly, Pfizer, Regeneron, Sanofi Genzyme, and Sun. She also serves as an advisor or consultant for Janssen, Lilly, Regeneron, and Sanofi Genzyme.
Medscape Live and this news organization are owned by the same parent company.
.
“If you don’t treat them, they’re going to spread,” Dr. Smith, who practices dermatology in Fort Mill, S.C., said at Medscape Live’s annual Coastal Dermatology Symposium. “They’re going to be itchy, they can spread on the patient themselves and then to others, and they can cause scarring. The prevalence is anywhere from 5% to 11%. That means there are 6 million patients out there, just waiting to come into your clinics.”
To date, no treatment has been approved by the Food and Drug Administration for MC, although a laundry list of agents have been tried, including cantharidin; cryotherapy; curettage with and without imiquimod; sinecatechins ointment, 15%; imiquimod; and retinoids. And there are several treatments that are being investigated.
A 2017 Cochrane review of 22 studies involving 1,650 patients demonstrated that no single intervention has been consistently effective in treating MC. “Most of the studies were actually very low quality,” said Dr. Smith, who was not involved with the analysis. “The one high quality study showed that imiquimod did not work any better than its vehicle.”
Investigational treatments
One of the products in the pipeline is VP-102, a proprietary drug-device combination of cantharidin 0.7% administered through a single-use precision applicator, which has been evaluated in phase 3 studies of patients with molluscum aged 2 years and older. It features a visualization agent so that the person applying the drug can see which lesions have been treated. It also contains a bittering agent to mitigate oral ingestion by children.
VP-102, which is being developed by Verrica Pharmaceuticals, is applied once every 21 days in up to 4 applications, and multiple lesions can be treated with one applicator. “It’s a stable concentration with a good shelf life, and two phase 3 randomized studies have shown about a 50% complete clearance of new and existing lesions at day 84,” Dr. Smith said. Those studies enrolled children and adults.
A separate analysis of the same data presented at a meeting in 2019 showed that 77% of patients treated with VP-102 achieved greater than 75% clearance, while 65.8% achieved more than 90% clearance.
The new kid on the block is a gel formulation of a nitric oxide–releasing medication, berdazimer 10.3%, a first-in-class topical treatment being developed by Novan, which can be applied at home. In a multicenter study published in JAMA Dermatology, researchers randomized 444 patients to berdazimer gel 10.3% and 447 to a placebo gel, applied once daily in a thin layer on all MC lesions for 12 weeks. The study was conducted at 55 clinics across the United States between Sept. 1, 2020, and July 21, 2021. The mean age of the patients was about 6.5 years and participants had 3-70 raised MC lesions; those with sexually transmitted MC or MC in the periocular area were excluded. The primary endpoint was complete clearance of MC lesions after 12 weeks of treatment.
At 12 weeks, significantly more patients treated with berdazimer gel achieved complete clearance than those on vehicle (32.4% vs. 19.7%; P < .001). A total of 64 (14.4%) patients in the berdazimer group discontinued treatment because of MC clearance, compared with 40 patients (8.9%) in the vehicle group.
More recently, investigators evaluated autoinoculation vs. 35% trichloroacetic acid (TCA) for the treatment of MC. Autoinoculation involves puncturing the perilesional and lesional skin 5-7 times with an insulin syringe. “This gets a little bit of the virus into the dermis, and you hope to elicit an immune response,” explained Dr. Smith, who was not involved with the study. At 3 months, 80% of patients in the autoinoculation group achieved complete clearance, compared with 62% of those in the TCA group, while recurrence at 6 months was 3% vs. 40%, respectively.
Manual extraction of MC lesions is another option. “I love to pop the cores out with my thumbs,” Dr. Smith said. “You have to pick the patients who can tolerate this, and the MC lesions need to be ripe and ready.”
For ophthalmic lesions, watchful waiting is advisable unless the MC lesions are symptomatic or bothersome or large lesions form on the lid margin, which may cause ocular irritation or even a corneal abrasion. “If a patient presents with a multisite infection that includes ocular lesions, treat lesions on other parts of the body and keep your fingers crossed that a systemic immune response occurs,” she said.
The desired immune response is known as the “BOTE” sign (the beginning of the end), which heralds the clearance of the molluscum infection. This often appears as reddening of all the MC lesions and occasionally as a granulomatous “id-like” reaction especially on the extensor elbows and knees. “When this happens, it often scares the patients,” Dr. Smith said. But she explains that this is a positive development, and that “this means that the lesions are about to self-resolve.”
Dr. Smith disclosed that she serves as a speaker or a member of the speakers bureau for Amgen, CeraVe, EPI, Galderma, InCyte, Lilly, Pfizer, Regeneron, Sanofi Genzyme, and Sun. She also serves as an advisor or consultant for Janssen, Lilly, Regeneron, and Sanofi Genzyme.
Medscape Live and this news organization are owned by the same parent company.
.
“If you don’t treat them, they’re going to spread,” Dr. Smith, who practices dermatology in Fort Mill, S.C., said at Medscape Live’s annual Coastal Dermatology Symposium. “They’re going to be itchy, they can spread on the patient themselves and then to others, and they can cause scarring. The prevalence is anywhere from 5% to 11%. That means there are 6 million patients out there, just waiting to come into your clinics.”
To date, no treatment has been approved by the Food and Drug Administration for MC, although a laundry list of agents have been tried, including cantharidin; cryotherapy; curettage with and without imiquimod; sinecatechins ointment, 15%; imiquimod; and retinoids. And there are several treatments that are being investigated.
A 2017 Cochrane review of 22 studies involving 1,650 patients demonstrated that no single intervention has been consistently effective in treating MC. “Most of the studies were actually very low quality,” said Dr. Smith, who was not involved with the analysis. “The one high quality study showed that imiquimod did not work any better than its vehicle.”
Investigational treatments
One of the products in the pipeline is VP-102, a proprietary drug-device combination of cantharidin 0.7% administered through a single-use precision applicator, which has been evaluated in phase 3 studies of patients with molluscum aged 2 years and older. It features a visualization agent so that the person applying the drug can see which lesions have been treated. It also contains a bittering agent to mitigate oral ingestion by children.
VP-102, which is being developed by Verrica Pharmaceuticals, is applied once every 21 days in up to 4 applications, and multiple lesions can be treated with one applicator. “It’s a stable concentration with a good shelf life, and two phase 3 randomized studies have shown about a 50% complete clearance of new and existing lesions at day 84,” Dr. Smith said. Those studies enrolled children and adults.
A separate analysis of the same data presented at a meeting in 2019 showed that 77% of patients treated with VP-102 achieved greater than 75% clearance, while 65.8% achieved more than 90% clearance.
The new kid on the block is a gel formulation of a nitric oxide–releasing medication, berdazimer 10.3%, a first-in-class topical treatment being developed by Novan, which can be applied at home. In a multicenter study published in JAMA Dermatology, researchers randomized 444 patients to berdazimer gel 10.3% and 447 to a placebo gel, applied once daily in a thin layer on all MC lesions for 12 weeks. The study was conducted at 55 clinics across the United States between Sept. 1, 2020, and July 21, 2021. The mean age of the patients was about 6.5 years and participants had 3-70 raised MC lesions; those with sexually transmitted MC or MC in the periocular area were excluded. The primary endpoint was complete clearance of MC lesions after 12 weeks of treatment.
At 12 weeks, significantly more patients treated with berdazimer gel achieved complete clearance than those on vehicle (32.4% vs. 19.7%; P < .001). A total of 64 (14.4%) patients in the berdazimer group discontinued treatment because of MC clearance, compared with 40 patients (8.9%) in the vehicle group.
More recently, investigators evaluated autoinoculation vs. 35% trichloroacetic acid (TCA) for the treatment of MC. Autoinoculation involves puncturing the perilesional and lesional skin 5-7 times with an insulin syringe. “This gets a little bit of the virus into the dermis, and you hope to elicit an immune response,” explained Dr. Smith, who was not involved with the study. At 3 months, 80% of patients in the autoinoculation group achieved complete clearance, compared with 62% of those in the TCA group, while recurrence at 6 months was 3% vs. 40%, respectively.
Manual extraction of MC lesions is another option. “I love to pop the cores out with my thumbs,” Dr. Smith said. “You have to pick the patients who can tolerate this, and the MC lesions need to be ripe and ready.”
For ophthalmic lesions, watchful waiting is advisable unless the MC lesions are symptomatic or bothersome or large lesions form on the lid margin, which may cause ocular irritation or even a corneal abrasion. “If a patient presents with a multisite infection that includes ocular lesions, treat lesions on other parts of the body and keep your fingers crossed that a systemic immune response occurs,” she said.
The desired immune response is known as the “BOTE” sign (the beginning of the end), which heralds the clearance of the molluscum infection. This often appears as reddening of all the MC lesions and occasionally as a granulomatous “id-like” reaction especially on the extensor elbows and knees. “When this happens, it often scares the patients,” Dr. Smith said. But she explains that this is a positive development, and that “this means that the lesions are about to self-resolve.”
Dr. Smith disclosed that she serves as a speaker or a member of the speakers bureau for Amgen, CeraVe, EPI, Galderma, InCyte, Lilly, Pfizer, Regeneron, Sanofi Genzyme, and Sun. She also serves as an advisor or consultant for Janssen, Lilly, Regeneron, and Sanofi Genzyme.
Medscape Live and this news organization are owned by the same parent company.
FROM MEDSCAPE LIVE COASTAL DERM
Breakthrough COVID studies lend support to use of new boosters in immunosuppressed patients
People with immune-mediated inflammatory diseases who are taking immunosuppressants don’t mount as strong of an immune defense against the Omicron variant as they did against the original SARS-CoV-2 wild-type virus, according to two studies published in Annals of the Rheumatic Diseases. One of the studies further showed that vaccinated individuals taking immunosuppressants have poorer cross-neutralizing responses to Omicron than do healthy vaccinated individuals, even after three doses of the COVID-19 mRNA vaccines.
“We carefully suggest that if Omicron-specific vaccination can be administered, it may be an effective way to reduce the risk of breakthrough infections in patients with autoimmune rheumatic disease,” Sang Tae Choi, MD, PhD, of the University College of Medicine, Seoul, Korea, and one of the authors of the study on cross-neutralizing protection, told this news organization. “However, further research is needed on Omicron-specific vaccine effectiveness in patients with immune dysfunctions. We believe that these study results can be of great benefit in determining the strategy of vaccination in the future.”
The earlier study, published in July, examined the ability of COVID-19 vaccines to induce cross-reactive antibody responses against Omicron infections in patients with autoimmune rheumatic diseases (ARDs). The observational study involved 149 patients with ARDs and 94 health care workers as controls, all of whom provided blood samples a median 15 weeks after their second COVID vaccine dose or a median 8 weeks after their third dose. A little more than two-thirds of the patients (68.5%) had received a third mRNA vaccine dose. None of the participants previously had COVID-19.
The researchers compared the rate of breakthrough infections with the Omicron variant to the neutralizing responses in patients’ blood, specifically the cross-neutralizing antibody responses because the original mRNA vaccines targeted a different variant than Omicron. Breakthrough infections were assessed by survey questions.
“Our findings suggested that neither primary series vaccinations nor booster doses are sufficient to induce Omicron-neutralizing responses above the threshold in patients with ARDs, although responses were noticeably increased following the third dose of an mRNA vaccine,” write Woo-Joong Kim, of the Chung-Ang University College of Medicine, Seoul, Korea, and his colleagues. “This impairment of cross-neutralization responses across most of our patients contrasts starkly with a potent elicitation of the Omicron-neutralizing responses after the third vaccination in healthy recipients.”
The average neutralizing responses against the original SARS-CoV-2 strain were similar in both groups: 76% in patients with ARDs and 72% in health care workers after the second dose. The mean response after a third dose was 97% in health care workers and 88% in patients.
The average cross-neutralizing response against the Omicron variant was far lower, particularly in those with rheumatic disease: only 11.5%, which rose to 27% after the third dose. Only 39% of the patient sera showed neutralization of Omicron, even after the third dose. Meanwhile, the mean cross-neutralizing response in health care workers was 18% after the second dose and 50% after the third.
When the researchers compared seropositivity rates against the original virus to neutralizing responses against Omicron, the association between these was stronger in health care workers than in those with ARDs. In fact, among patients with ARDs who seroconverted, only 41% showed any response against Omicron. Among all the patients, most of those who didn’t respond to Omicron (93.5%) had initially seroconverted.
The researchers also looked at the ability to neutralize Omicron on the basis of disease in those who received three doses of the vaccine. About half of those with lupus (52%) showed any neutralization against Omicron, compared with 25% of those with rheumatoid arthritis, 37.5% of those with ankylosing spondylitis, 33% of those with Behçet snydrome, and all of those with adult-onset Still’s disease.
The rate of breakthrough infections was lower in patients (19%) than in health care workers (33%). A similar pattern was seen in the more recent study published Sept. 5. Researchers used data from a prospective cohort study in the Netherlands to examine incidence and severity of Omicron breakthrough infections in patients with immune-mediated inflammatory diseases. The researchers compared infection rates and severity among 1,593 vaccinated patients with inflammatory disease who were taking immunosuppressants and 579 vaccinated controls (418 patients with inflammatory disease not on immunosuppressants and 161 healthy controls).
One in five patients with inflammatory disease (21%) were taking immunosuppressants that substantially impair antibodies, such as anti-CD20 therapy, S1P modulators, or mycophenolate mofetil combination therapy, and 48% of these patients seroconverted after primary vaccination, compared with 96% of patients taking other immunosuppressants and 98% of controls.
Breakthrough infection rates were similar between the control group (31%) and those taking immunosuppressants (30%). Only three participants had severe disease requiring hospitalization: one control and two patients taking immunosuppressants.
“In both studies, the controls had similar or higher rates of breakthrough infections, compared with the immunosuppressed,” noted Alfred Kim, MD, an assistant professor of medicine at Washington University, St Louis, but he added, “one has to consider differences in mitigation strategies, such as masking, that may explain these findings.” That is, patients taking immunosuppressants may be taking fewer risks in the community or have fewer potential exposures, especially in the Korean study, wherein the controls were health care workers.
A greater disparity in infections occurred when considering seroconversion rates. Breakthrough incidence was 38% among those taking immunosuppressants who did not seroconvert, compared with 29% among those who did. A similar trend was seen in breakthrough incidence between those taking strongly antibody-impairing immunosuppressants (36% breakthrough rate) and those taking other immunosuppressants (28%).

Among those taking immunosuppressants who seroconverted, a primary series of vaccination reduced the risk of a breakthrough infection by 29%. Protection became more robust with a booster or prior infection, both of which reduced breakthrough infection risk by 39% in those taking immunosuppressants who seroconverted.
“We demonstrate in patients with immune-mediated inflammatory diseases on immunosuppressants that additional vaccinations are associated with decreased risk of SARS-CoV-2 Omicron breakthrough infections,” wrote Eileen W. Stalman, MD, PhD, of Amsterdam UMC in the Netherlands, and her colleagues.
Though neither study broke down immune response or breakthrough infection based on individual medications, Kim said that previous research allows one to extrapolate “that prior culprits of poor vaccine responses [such as B-cell depleting drugs, mycophenolate, and TNF [tumor necrosis factor] inhibitors will continue to bear the greatest burden in breakthrough infection, including Omicron.”
Overall, he found the data from both studies relatively consistent with one another.
“Those on immunosuppression, particularly mechanisms that have been established as risk factors for poor vaccine responses, are at risk of breakthrough infection during the era of Omicron,” Dr. Kim said.
The earlier study from Korea also found that “the median time between the third-dose vaccination and the date of confirmed breakthrough infection in patients with ARDs was significantly shorter, compared with that in health care workers” at just 93 days in patients versus 122 days in health care workers. They postulated that this population’s limited neutralization of Omicron explained this short-lived protection.
Most of the patients with breakthrough infections (74%) in that study showed no neutralization against Omicron, including the only two hospitalized patients, both of whom had strong responses against the original SARS-CoV-2 strain. The significant decline over time of neutralization against Omicron suggested “the potential for a substantial loss of the protection from breakthrough infection,” the authors write.
“The third dose of an mRNA vaccine could improve the cross-neutralization of the SARS-CoV-2 Omicron variant in patients with autoimmune rheumatic disease [although] more than half of the patients failed to generate Omicron-neutralizing antibodies,” Tae Choi said in an interview. “Our study sheds light on the relative deficiency of the Omicron-specific neutralizing responses in patients with autoimmune rheumatic disease and their anticipated vulnerability to breakthrough infection.”
The message for clinicians, Dr. Kim said, is to “continue to urge our patients to maintain additional and boosting doses per guidance, use pre-exposure prophylaxis such as Evusheld, and continue other mitigation strategies as they have done.”
The Dutch study was funded by The Netherlands Organization for Health Research and Development; the Korean study used no external funding.
The authors of the Korean study had no disclosures. The Dutch study’s authors reported a wide range of disclosures involving more than a dozen pharmaceutical companies but not including Pfizer or Moderna. Dr. Kim’s industry disclosures include Alexion, ANI, AstraZeneca, Aurinia, Exagen, Foghorn Therapeutics, GlaxoSmithKline, Kypha, and Pfizer.
A version of this article first appeared on Medscape.com.
People with immune-mediated inflammatory diseases who are taking immunosuppressants don’t mount as strong of an immune defense against the Omicron variant as they did against the original SARS-CoV-2 wild-type virus, according to two studies published in Annals of the Rheumatic Diseases. One of the studies further showed that vaccinated individuals taking immunosuppressants have poorer cross-neutralizing responses to Omicron than do healthy vaccinated individuals, even after three doses of the COVID-19 mRNA vaccines.
“We carefully suggest that if Omicron-specific vaccination can be administered, it may be an effective way to reduce the risk of breakthrough infections in patients with autoimmune rheumatic disease,” Sang Tae Choi, MD, PhD, of the University College of Medicine, Seoul, Korea, and one of the authors of the study on cross-neutralizing protection, told this news organization. “However, further research is needed on Omicron-specific vaccine effectiveness in patients with immune dysfunctions. We believe that these study results can be of great benefit in determining the strategy of vaccination in the future.”
The earlier study, published in July, examined the ability of COVID-19 vaccines to induce cross-reactive antibody responses against Omicron infections in patients with autoimmune rheumatic diseases (ARDs). The observational study involved 149 patients with ARDs and 94 health care workers as controls, all of whom provided blood samples a median 15 weeks after their second COVID vaccine dose or a median 8 weeks after their third dose. A little more than two-thirds of the patients (68.5%) had received a third mRNA vaccine dose. None of the participants previously had COVID-19.
The researchers compared the rate of breakthrough infections with the Omicron variant to the neutralizing responses in patients’ blood, specifically the cross-neutralizing antibody responses because the original mRNA vaccines targeted a different variant than Omicron. Breakthrough infections were assessed by survey questions.
“Our findings suggested that neither primary series vaccinations nor booster doses are sufficient to induce Omicron-neutralizing responses above the threshold in patients with ARDs, although responses were noticeably increased following the third dose of an mRNA vaccine,” write Woo-Joong Kim, of the Chung-Ang University College of Medicine, Seoul, Korea, and his colleagues. “This impairment of cross-neutralization responses across most of our patients contrasts starkly with a potent elicitation of the Omicron-neutralizing responses after the third vaccination in healthy recipients.”
The average neutralizing responses against the original SARS-CoV-2 strain were similar in both groups: 76% in patients with ARDs and 72% in health care workers after the second dose. The mean response after a third dose was 97% in health care workers and 88% in patients.
The average cross-neutralizing response against the Omicron variant was far lower, particularly in those with rheumatic disease: only 11.5%, which rose to 27% after the third dose. Only 39% of the patient sera showed neutralization of Omicron, even after the third dose. Meanwhile, the mean cross-neutralizing response in health care workers was 18% after the second dose and 50% after the third.
When the researchers compared seropositivity rates against the original virus to neutralizing responses against Omicron, the association between these was stronger in health care workers than in those with ARDs. In fact, among patients with ARDs who seroconverted, only 41% showed any response against Omicron. Among all the patients, most of those who didn’t respond to Omicron (93.5%) had initially seroconverted.
The researchers also looked at the ability to neutralize Omicron on the basis of disease in those who received three doses of the vaccine. About half of those with lupus (52%) showed any neutralization against Omicron, compared with 25% of those with rheumatoid arthritis, 37.5% of those with ankylosing spondylitis, 33% of those with Behçet snydrome, and all of those with adult-onset Still’s disease.
The rate of breakthrough infections was lower in patients (19%) than in health care workers (33%). A similar pattern was seen in the more recent study published Sept. 5. Researchers used data from a prospective cohort study in the Netherlands to examine incidence and severity of Omicron breakthrough infections in patients with immune-mediated inflammatory diseases. The researchers compared infection rates and severity among 1,593 vaccinated patients with inflammatory disease who were taking immunosuppressants and 579 vaccinated controls (418 patients with inflammatory disease not on immunosuppressants and 161 healthy controls).
One in five patients with inflammatory disease (21%) were taking immunosuppressants that substantially impair antibodies, such as anti-CD20 therapy, S1P modulators, or mycophenolate mofetil combination therapy, and 48% of these patients seroconverted after primary vaccination, compared with 96% of patients taking other immunosuppressants and 98% of controls.
Breakthrough infection rates were similar between the control group (31%) and those taking immunosuppressants (30%). Only three participants had severe disease requiring hospitalization: one control and two patients taking immunosuppressants.
“In both studies, the controls had similar or higher rates of breakthrough infections, compared with the immunosuppressed,” noted Alfred Kim, MD, an assistant professor of medicine at Washington University, St Louis, but he added, “one has to consider differences in mitigation strategies, such as masking, that may explain these findings.” That is, patients taking immunosuppressants may be taking fewer risks in the community or have fewer potential exposures, especially in the Korean study, wherein the controls were health care workers.
A greater disparity in infections occurred when considering seroconversion rates. Breakthrough incidence was 38% among those taking immunosuppressants who did not seroconvert, compared with 29% among those who did. A similar trend was seen in breakthrough incidence between those taking strongly antibody-impairing immunosuppressants (36% breakthrough rate) and those taking other immunosuppressants (28%).
Among those taking immunosuppressants who seroconverted, a primary series of vaccination reduced the risk of a breakthrough infection by 29%. Protection became more robust with a booster or prior infection, both of which reduced breakthrough infection risk by 39% in those taking immunosuppressants who seroconverted.
“We demonstrate in patients with immune-mediated inflammatory diseases on immunosuppressants that additional vaccinations are associated with decreased risk of SARS-CoV-2 Omicron breakthrough infections,” wrote Eileen W. Stalman, MD, PhD, of Amsterdam UMC in the Netherlands, and her colleagues.
Though neither study broke down immune response or breakthrough infection based on individual medications, Kim said that previous research allows one to extrapolate “that prior culprits of poor vaccine responses [such as B-cell depleting drugs, mycophenolate, and TNF [tumor necrosis factor] inhibitors will continue to bear the greatest burden in breakthrough infection, including Omicron.”
Overall, he found the data from both studies relatively consistent with one another.
“Those on immunosuppression, particularly mechanisms that have been established as risk factors for poor vaccine responses, are at risk of breakthrough infection during the era of Omicron,” Dr. Kim said.
The earlier study from Korea also found that “the median time between the third-dose vaccination and the date of confirmed breakthrough infection in patients with ARDs was significantly shorter, compared with that in health care workers” at just 93 days in patients versus 122 days in health care workers. They postulated that this population’s limited neutralization of Omicron explained this short-lived protection.
Most of the patients with breakthrough infections (74%) in that study showed no neutralization against Omicron, including the only two hospitalized patients, both of whom had strong responses against the original SARS-CoV-2 strain. The significant decline over time of neutralization against Omicron suggested “the potential for a substantial loss of the protection from breakthrough infection,” the authors write.
“The third dose of an mRNA vaccine could improve the cross-neutralization of the SARS-CoV-2 Omicron variant in patients with autoimmune rheumatic disease [although] more than half of the patients failed to generate Omicron-neutralizing antibodies,” Tae Choi said in an interview. “Our study sheds light on the relative deficiency of the Omicron-specific neutralizing responses in patients with autoimmune rheumatic disease and their anticipated vulnerability to breakthrough infection.”
The message for clinicians, Dr. Kim said, is to “continue to urge our patients to maintain additional and boosting doses per guidance, use pre-exposure prophylaxis such as Evusheld, and continue other mitigation strategies as they have done.”
The Dutch study was funded by The Netherlands Organization for Health Research and Development; the Korean study used no external funding.
The authors of the Korean study had no disclosures. The Dutch study’s authors reported a wide range of disclosures involving more than a dozen pharmaceutical companies but not including Pfizer or Moderna. Dr. Kim’s industry disclosures include Alexion, ANI, AstraZeneca, Aurinia, Exagen, Foghorn Therapeutics, GlaxoSmithKline, Kypha, and Pfizer.
A version of this article first appeared on Medscape.com.
People with immune-mediated inflammatory diseases who are taking immunosuppressants don’t mount as strong of an immune defense against the Omicron variant as they did against the original SARS-CoV-2 wild-type virus, according to two studies published in Annals of the Rheumatic Diseases. One of the studies further showed that vaccinated individuals taking immunosuppressants have poorer cross-neutralizing responses to Omicron than do healthy vaccinated individuals, even after three doses of the COVID-19 mRNA vaccines.
“We carefully suggest that if Omicron-specific vaccination can be administered, it may be an effective way to reduce the risk of breakthrough infections in patients with autoimmune rheumatic disease,” Sang Tae Choi, MD, PhD, of the University College of Medicine, Seoul, Korea, and one of the authors of the study on cross-neutralizing protection, told this news organization. “However, further research is needed on Omicron-specific vaccine effectiveness in patients with immune dysfunctions. We believe that these study results can be of great benefit in determining the strategy of vaccination in the future.”
The earlier study, published in July, examined the ability of COVID-19 vaccines to induce cross-reactive antibody responses against Omicron infections in patients with autoimmune rheumatic diseases (ARDs). The observational study involved 149 patients with ARDs and 94 health care workers as controls, all of whom provided blood samples a median 15 weeks after their second COVID vaccine dose or a median 8 weeks after their third dose. A little more than two-thirds of the patients (68.5%) had received a third mRNA vaccine dose. None of the participants previously had COVID-19.
The researchers compared the rate of breakthrough infections with the Omicron variant to the neutralizing responses in patients’ blood, specifically the cross-neutralizing antibody responses because the original mRNA vaccines targeted a different variant than Omicron. Breakthrough infections were assessed by survey questions.
“Our findings suggested that neither primary series vaccinations nor booster doses are sufficient to induce Omicron-neutralizing responses above the threshold in patients with ARDs, although responses were noticeably increased following the third dose of an mRNA vaccine,” write Woo-Joong Kim, of the Chung-Ang University College of Medicine, Seoul, Korea, and his colleagues. “This impairment of cross-neutralization responses across most of our patients contrasts starkly with a potent elicitation of the Omicron-neutralizing responses after the third vaccination in healthy recipients.”
The average neutralizing responses against the original SARS-CoV-2 strain were similar in both groups: 76% in patients with ARDs and 72% in health care workers after the second dose. The mean response after a third dose was 97% in health care workers and 88% in patients.
The average cross-neutralizing response against the Omicron variant was far lower, particularly in those with rheumatic disease: only 11.5%, which rose to 27% after the third dose. Only 39% of the patient sera showed neutralization of Omicron, even after the third dose. Meanwhile, the mean cross-neutralizing response in health care workers was 18% after the second dose and 50% after the third.
When the researchers compared seropositivity rates against the original virus to neutralizing responses against Omicron, the association between these was stronger in health care workers than in those with ARDs. In fact, among patients with ARDs who seroconverted, only 41% showed any response against Omicron. Among all the patients, most of those who didn’t respond to Omicron (93.5%) had initially seroconverted.
The researchers also looked at the ability to neutralize Omicron on the basis of disease in those who received three doses of the vaccine. About half of those with lupus (52%) showed any neutralization against Omicron, compared with 25% of those with rheumatoid arthritis, 37.5% of those with ankylosing spondylitis, 33% of those with Behçet snydrome, and all of those with adult-onset Still’s disease.
The rate of breakthrough infections was lower in patients (19%) than in health care workers (33%). A similar pattern was seen in the more recent study published Sept. 5. Researchers used data from a prospective cohort study in the Netherlands to examine incidence and severity of Omicron breakthrough infections in patients with immune-mediated inflammatory diseases. The researchers compared infection rates and severity among 1,593 vaccinated patients with inflammatory disease who were taking immunosuppressants and 579 vaccinated controls (418 patients with inflammatory disease not on immunosuppressants and 161 healthy controls).
One in five patients with inflammatory disease (21%) were taking immunosuppressants that substantially impair antibodies, such as anti-CD20 therapy, S1P modulators, or mycophenolate mofetil combination therapy, and 48% of these patients seroconverted after primary vaccination, compared with 96% of patients taking other immunosuppressants and 98% of controls.
Breakthrough infection rates were similar between the control group (31%) and those taking immunosuppressants (30%). Only three participants had severe disease requiring hospitalization: one control and two patients taking immunosuppressants.
“In both studies, the controls had similar or higher rates of breakthrough infections, compared with the immunosuppressed,” noted Alfred Kim, MD, an assistant professor of medicine at Washington University, St Louis, but he added, “one has to consider differences in mitigation strategies, such as masking, that may explain these findings.” That is, patients taking immunosuppressants may be taking fewer risks in the community or have fewer potential exposures, especially in the Korean study, wherein the controls were health care workers.
A greater disparity in infections occurred when considering seroconversion rates. Breakthrough incidence was 38% among those taking immunosuppressants who did not seroconvert, compared with 29% among those who did. A similar trend was seen in breakthrough incidence between those taking strongly antibody-impairing immunosuppressants (36% breakthrough rate) and those taking other immunosuppressants (28%).
Among those taking immunosuppressants who seroconverted, a primary series of vaccination reduced the risk of a breakthrough infection by 29%. Protection became more robust with a booster or prior infection, both of which reduced breakthrough infection risk by 39% in those taking immunosuppressants who seroconverted.
“We demonstrate in patients with immune-mediated inflammatory diseases on immunosuppressants that additional vaccinations are associated with decreased risk of SARS-CoV-2 Omicron breakthrough infections,” wrote Eileen W. Stalman, MD, PhD, of Amsterdam UMC in the Netherlands, and her colleagues.
Though neither study broke down immune response or breakthrough infection based on individual medications, Kim said that previous research allows one to extrapolate “that prior culprits of poor vaccine responses [such as B-cell depleting drugs, mycophenolate, and TNF [tumor necrosis factor] inhibitors will continue to bear the greatest burden in breakthrough infection, including Omicron.”
Overall, he found the data from both studies relatively consistent with one another.
“Those on immunosuppression, particularly mechanisms that have been established as risk factors for poor vaccine responses, are at risk of breakthrough infection during the era of Omicron,” Dr. Kim said.
The earlier study from Korea also found that “the median time between the third-dose vaccination and the date of confirmed breakthrough infection in patients with ARDs was significantly shorter, compared with that in health care workers” at just 93 days in patients versus 122 days in health care workers. They postulated that this population’s limited neutralization of Omicron explained this short-lived protection.
Most of the patients with breakthrough infections (74%) in that study showed no neutralization against Omicron, including the only two hospitalized patients, both of whom had strong responses against the original SARS-CoV-2 strain. The significant decline over time of neutralization against Omicron suggested “the potential for a substantial loss of the protection from breakthrough infection,” the authors write.
“The third dose of an mRNA vaccine could improve the cross-neutralization of the SARS-CoV-2 Omicron variant in patients with autoimmune rheumatic disease [although] more than half of the patients failed to generate Omicron-neutralizing antibodies,” Tae Choi said in an interview. “Our study sheds light on the relative deficiency of the Omicron-specific neutralizing responses in patients with autoimmune rheumatic disease and their anticipated vulnerability to breakthrough infection.”
The message for clinicians, Dr. Kim said, is to “continue to urge our patients to maintain additional and boosting doses per guidance, use pre-exposure prophylaxis such as Evusheld, and continue other mitigation strategies as they have done.”
The Dutch study was funded by The Netherlands Organization for Health Research and Development; the Korean study used no external funding.
The authors of the Korean study had no disclosures. The Dutch study’s authors reported a wide range of disclosures involving more than a dozen pharmaceutical companies but not including Pfizer or Moderna. Dr. Kim’s industry disclosures include Alexion, ANI, AstraZeneca, Aurinia, Exagen, Foghorn Therapeutics, GlaxoSmithKline, Kypha, and Pfizer.
A version of this article first appeared on Medscape.com.
Don’t make children with head lice leave school, report says
The American Academy of Pediatrics says children with head lice don’t need to be sent home from school.
Head lice infestations aren’t really a health hazard because of low transmission rates, a new report from the academy says, and sending students home “may stigmatize children suspected of having head lice.” The group says schools should instead offer education programs to help families understand how to manage head lice.
“Head lice are an unpleasant part of the human experience, but they can be successfully managed and are no reason for a child to miss school,” Dawn Nolt, MD, lead author of the report on head lice, said in a news release.
The report advises schools to abandon “no-nit” policies, which call for a student to be lice-free before being allowed back in class.
“A child or adolescent should not be restricted from school attendance because of head lice, given the low contagion within classrooms. ‘No-nit’ policies that exclude children or adolescents until all nits are removed may violate a child’s or adolescent’s civil liberties and are best addressed with legal counsel for schools,” the report says.
The report notes that lice almost always spread through head-to-head contact, not by “jumping” from one person to another. It’s possible for lice to spread by touching the belongings of a person with lice, such as combs or sports helmets, but the chances of that happening are very low, the academy said.
“Lice found on combs are likely to be injured or dead, and a louse is not likely to leave a healthy head unless there is a heavy infestation,” the report says.
The report lists new medications for treatment and gives an algorithm for managing head lice cases.
“The ideal treatment of head lice should be safe, free of toxic chemicals, readily available, simple to apply, effective, and inexpensive,” the report says.
This is the first updated guidance on head lice from the American Academy of Pediatrics since 2015. The CDC also says students with head lice don’t need to be sent home.
“Students diagnosed with live head lice do not need to be sent home early from school; they can go home at the end of the day, be treated, and return to class after appropriate treatment has begun. Nits may persist after treatment, but successful treatment should kill crawling lice,” the CDC says.
A version of this article first appeared on WebMD.com.
The American Academy of Pediatrics says children with head lice don’t need to be sent home from school.
Head lice infestations aren’t really a health hazard because of low transmission rates, a new report from the academy says, and sending students home “may stigmatize children suspected of having head lice.” The group says schools should instead offer education programs to help families understand how to manage head lice.
“Head lice are an unpleasant part of the human experience, but they can be successfully managed and are no reason for a child to miss school,” Dawn Nolt, MD, lead author of the report on head lice, said in a news release.
The report advises schools to abandon “no-nit” policies, which call for a student to be lice-free before being allowed back in class.
“A child or adolescent should not be restricted from school attendance because of head lice, given the low contagion within classrooms. ‘No-nit’ policies that exclude children or adolescents until all nits are removed may violate a child’s or adolescent’s civil liberties and are best addressed with legal counsel for schools,” the report says.
The report notes that lice almost always spread through head-to-head contact, not by “jumping” from one person to another. It’s possible for lice to spread by touching the belongings of a person with lice, such as combs or sports helmets, but the chances of that happening are very low, the academy said.
“Lice found on combs are likely to be injured or dead, and a louse is not likely to leave a healthy head unless there is a heavy infestation,” the report says.
The report lists new medications for treatment and gives an algorithm for managing head lice cases.
“The ideal treatment of head lice should be safe, free of toxic chemicals, readily available, simple to apply, effective, and inexpensive,” the report says.
This is the first updated guidance on head lice from the American Academy of Pediatrics since 2015. The CDC also says students with head lice don’t need to be sent home.
“Students diagnosed with live head lice do not need to be sent home early from school; they can go home at the end of the day, be treated, and return to class after appropriate treatment has begun. Nits may persist after treatment, but successful treatment should kill crawling lice,” the CDC says.
A version of this article first appeared on WebMD.com.
The American Academy of Pediatrics says children with head lice don’t need to be sent home from school.
Head lice infestations aren’t really a health hazard because of low transmission rates, a new report from the academy says, and sending students home “may stigmatize children suspected of having head lice.” The group says schools should instead offer education programs to help families understand how to manage head lice.
“Head lice are an unpleasant part of the human experience, but they can be successfully managed and are no reason for a child to miss school,” Dawn Nolt, MD, lead author of the report on head lice, said in a news release.
The report advises schools to abandon “no-nit” policies, which call for a student to be lice-free before being allowed back in class.
“A child or adolescent should not be restricted from school attendance because of head lice, given the low contagion within classrooms. ‘No-nit’ policies that exclude children or adolescents until all nits are removed may violate a child’s or adolescent’s civil liberties and are best addressed with legal counsel for schools,” the report says.
The report notes that lice almost always spread through head-to-head contact, not by “jumping” from one person to another. It’s possible for lice to spread by touching the belongings of a person with lice, such as combs or sports helmets, but the chances of that happening are very low, the academy said.
“Lice found on combs are likely to be injured or dead, and a louse is not likely to leave a healthy head unless there is a heavy infestation,” the report says.
The report lists new medications for treatment and gives an algorithm for managing head lice cases.
“The ideal treatment of head lice should be safe, free of toxic chemicals, readily available, simple to apply, effective, and inexpensive,” the report says.
This is the first updated guidance on head lice from the American Academy of Pediatrics since 2015. The CDC also says students with head lice don’t need to be sent home.
“Students diagnosed with live head lice do not need to be sent home early from school; they can go home at the end of the day, be treated, and return to class after appropriate treatment has begun. Nits may persist after treatment, but successful treatment should kill crawling lice,” the CDC says.
A version of this article first appeared on WebMD.com.
Balanced fat intake links with less type 2 diabetes
Researchers published the study covered in this summary on Preprints with The Lancet as a preprint that has not yet been peer reviewed.
Key takeaways
- Adults in China who consumed a “balanced,” moderate ratio (middle three quintiles) of animal-to-vegetable cooking oil had a lower rate of developing type 2 diabetes during a median follow-up of 8.6 years, compared with those who consumed the lowest ratio (first quintile), after multivariable adjustment using prospectively collected data.
- The results also indicate that increasing animal cooking oil (such as lard, tallow, or butter) and vegetable cooking oil (such as peanut or soybean oil) consumption were each positively associated with a higher rate of developing type 2 diabetes.
- Those who consumed the highest ratio (fifth quintile) of animal-to-vegetable cooking oil had a nonsignificant difference in their rate of developing type 2 diabetes, compared with those in the first quintile.
Why this matters
- The findings suggest that consuming a diet with a “balanced” moderate intake of animal and vegetable oil might lower the risk of type 2 diabetes, which would reduce disease burden and health care expenditures.
- The results imply that using a single source of cooking oil, either animal or vegetable, contributes to the incidence of type 2 diabetes.
- This is the first large epidemiological study showing a relationship between the ratio of animal- and vegetable-derived fats in people’s diets and their risk for incident type 2 diabetes.
Study design
- The researchers used data collected prospectively starting in 2010-2012 from 7,274 adult residents of Guizhou province, China, with follow-up assessment in 2020 after a median of 8.6 years.
- At baseline, participants underwent an oral glucose tolerance test and provided information on demographics, family medical history, and personal medical history, including whether they had been diagnosed with type 2 diabetes or were taking antihyperglycemic medications. The study did not include anyone with a history of diabetes.
- Data on intake of animal and vegetable cooking oil came from a dietary questionnaire.
- The authors calculated hazard ratios for development of type 2 diabetes after adjusting for multiple potential confounders.
Key results
- The study cohort averaged 44 years old, and 53% were women.
- During a median follow-up of 8.6 years, 747 people developed type 2 diabetes.
- Compared with those who had the lowest intake of animal cooking oil (first quintile), those with the highest intake (fifth quintile) had a significant 28% increased relative rate for developing type 2 diabetes after adjustment for several potential confounders.
- Compared with those with the lowest intake of vegetable cooking oil, those with the highest intake had a significant 56% increased rate of developing type 2 diabetes after adjustment.
- Compared with adults with the lowest animal-to-vegetable cooking oil ratio (first quintile), those in the second, third, and fourth quintiles for this ratio had significantly lower adjusted relative rates of developing type 2 diabetes, with adjusted hazard ratios of 0.79, 0.65, and 0.68, respectively. Those in the highest quintile (fifth quintile) did not have a significantly different risk, compared with the first quintile.
- The protective effect of a balanced ratio of animal-to-vegetable cooking oils was stronger in people who lived in rural districts and in those who had obesity.
Limitations
- The dietary information came from participants’ self-reports, which may have produced biased data.
- The study only included information about animal and vegetable cooking oil consumed at home.
- There may have been residual confounding from variables not included in the study.
- The time of diagnosis of type 2 diabetes may have been inaccurate because follow-up occurred only once.
- The study may have underestimated the incidence of type 2 diabetes because of a lack of information about hemoglobin A1c levels at follow-up.
Disclosures
- The study did not receive commercial funding.
- The authors reported no financial disclosures.
This is a summary of a preprint article “The consumption ratio of animal cooking oil to vegetable cooking oil and reduced risk of type 2 diabetes mellitus: A prospective cohort study in Southwest China” written by researchers primarily from Zunyi Medical University, China, on Preprints with The Lancet. This study has not yet been peer reviewed. The full text of the study can be found on papers.ssrn.com.
A version of this article first appeared on Medscape.com.
Researchers published the study covered in this summary on Preprints with The Lancet as a preprint that has not yet been peer reviewed.
Key takeaways
- Adults in China who consumed a “balanced,” moderate ratio (middle three quintiles) of animal-to-vegetable cooking oil had a lower rate of developing type 2 diabetes during a median follow-up of 8.6 years, compared with those who consumed the lowest ratio (first quintile), after multivariable adjustment using prospectively collected data.
- The results also indicate that increasing animal cooking oil (such as lard, tallow, or butter) and vegetable cooking oil (such as peanut or soybean oil) consumption were each positively associated with a higher rate of developing type 2 diabetes.
- Those who consumed the highest ratio (fifth quintile) of animal-to-vegetable cooking oil had a nonsignificant difference in their rate of developing type 2 diabetes, compared with those in the first quintile.
Why this matters
- The findings suggest that consuming a diet with a “balanced” moderate intake of animal and vegetable oil might lower the risk of type 2 diabetes, which would reduce disease burden and health care expenditures.
- The results imply that using a single source of cooking oil, either animal or vegetable, contributes to the incidence of type 2 diabetes.
- This is the first large epidemiological study showing a relationship between the ratio of animal- and vegetable-derived fats in people’s diets and their risk for incident type 2 diabetes.
Study design
- The researchers used data collected prospectively starting in 2010-2012 from 7,274 adult residents of Guizhou province, China, with follow-up assessment in 2020 after a median of 8.6 years.
- At baseline, participants underwent an oral glucose tolerance test and provided information on demographics, family medical history, and personal medical history, including whether they had been diagnosed with type 2 diabetes or were taking antihyperglycemic medications. The study did not include anyone with a history of diabetes.
- Data on intake of animal and vegetable cooking oil came from a dietary questionnaire.
- The authors calculated hazard ratios for development of type 2 diabetes after adjusting for multiple potential confounders.
Key results
- The study cohort averaged 44 years old, and 53% were women.
- During a median follow-up of 8.6 years, 747 people developed type 2 diabetes.
- Compared with those who had the lowest intake of animal cooking oil (first quintile), those with the highest intake (fifth quintile) had a significant 28% increased relative rate for developing type 2 diabetes after adjustment for several potential confounders.
- Compared with those with the lowest intake of vegetable cooking oil, those with the highest intake had a significant 56% increased rate of developing type 2 diabetes after adjustment.
- Compared with adults with the lowest animal-to-vegetable cooking oil ratio (first quintile), those in the second, third, and fourth quintiles for this ratio had significantly lower adjusted relative rates of developing type 2 diabetes, with adjusted hazard ratios of 0.79, 0.65, and 0.68, respectively. Those in the highest quintile (fifth quintile) did not have a significantly different risk, compared with the first quintile.
- The protective effect of a balanced ratio of animal-to-vegetable cooking oils was stronger in people who lived in rural districts and in those who had obesity.
Limitations
- The dietary information came from participants’ self-reports, which may have produced biased data.
- The study only included information about animal and vegetable cooking oil consumed at home.
- There may have been residual confounding from variables not included in the study.
- The time of diagnosis of type 2 diabetes may have been inaccurate because follow-up occurred only once.
- The study may have underestimated the incidence of type 2 diabetes because of a lack of information about hemoglobin A1c levels at follow-up.
Disclosures
- The study did not receive commercial funding.
- The authors reported no financial disclosures.
This is a summary of a preprint article “The consumption ratio of animal cooking oil to vegetable cooking oil and reduced risk of type 2 diabetes mellitus: A prospective cohort study in Southwest China” written by researchers primarily from Zunyi Medical University, China, on Preprints with The Lancet. This study has not yet been peer reviewed. The full text of the study can be found on papers.ssrn.com.
A version of this article first appeared on Medscape.com.
Researchers published the study covered in this summary on Preprints with The Lancet as a preprint that has not yet been peer reviewed.
Key takeaways
- Adults in China who consumed a “balanced,” moderate ratio (middle three quintiles) of animal-to-vegetable cooking oil had a lower rate of developing type 2 diabetes during a median follow-up of 8.6 years, compared with those who consumed the lowest ratio (first quintile), after multivariable adjustment using prospectively collected data.
- The results also indicate that increasing animal cooking oil (such as lard, tallow, or butter) and vegetable cooking oil (such as peanut or soybean oil) consumption were each positively associated with a higher rate of developing type 2 diabetes.
- Those who consumed the highest ratio (fifth quintile) of animal-to-vegetable cooking oil had a nonsignificant difference in their rate of developing type 2 diabetes, compared with those in the first quintile.
Why this matters
- The findings suggest that consuming a diet with a “balanced” moderate intake of animal and vegetable oil might lower the risk of type 2 diabetes, which would reduce disease burden and health care expenditures.
- The results imply that using a single source of cooking oil, either animal or vegetable, contributes to the incidence of type 2 diabetes.
- This is the first large epidemiological study showing a relationship between the ratio of animal- and vegetable-derived fats in people’s diets and their risk for incident type 2 diabetes.
Study design
- The researchers used data collected prospectively starting in 2010-2012 from 7,274 adult residents of Guizhou province, China, with follow-up assessment in 2020 after a median of 8.6 years.
- At baseline, participants underwent an oral glucose tolerance test and provided information on demographics, family medical history, and personal medical history, including whether they had been diagnosed with type 2 diabetes or were taking antihyperglycemic medications. The study did not include anyone with a history of diabetes.
- Data on intake of animal and vegetable cooking oil came from a dietary questionnaire.
- The authors calculated hazard ratios for development of type 2 diabetes after adjusting for multiple potential confounders.
Key results
- The study cohort averaged 44 years old, and 53% were women.
- During a median follow-up of 8.6 years, 747 people developed type 2 diabetes.
- Compared with those who had the lowest intake of animal cooking oil (first quintile), those with the highest intake (fifth quintile) had a significant 28% increased relative rate for developing type 2 diabetes after adjustment for several potential confounders.
- Compared with those with the lowest intake of vegetable cooking oil, those with the highest intake had a significant 56% increased rate of developing type 2 diabetes after adjustment.
- Compared with adults with the lowest animal-to-vegetable cooking oil ratio (first quintile), those in the second, third, and fourth quintiles for this ratio had significantly lower adjusted relative rates of developing type 2 diabetes, with adjusted hazard ratios of 0.79, 0.65, and 0.68, respectively. Those in the highest quintile (fifth quintile) did not have a significantly different risk, compared with the first quintile.
- The protective effect of a balanced ratio of animal-to-vegetable cooking oils was stronger in people who lived in rural districts and in those who had obesity.
Limitations
- The dietary information came from participants’ self-reports, which may have produced biased data.
- The study only included information about animal and vegetable cooking oil consumed at home.
- There may have been residual confounding from variables not included in the study.
- The time of diagnosis of type 2 diabetes may have been inaccurate because follow-up occurred only once.
- The study may have underestimated the incidence of type 2 diabetes because of a lack of information about hemoglobin A1c levels at follow-up.
Disclosures
- The study did not receive commercial funding.
- The authors reported no financial disclosures.
This is a summary of a preprint article “The consumption ratio of animal cooking oil to vegetable cooking oil and reduced risk of type 2 diabetes mellitus: A prospective cohort study in Southwest China” written by researchers primarily from Zunyi Medical University, China, on Preprints with The Lancet. This study has not yet been peer reviewed. The full text of the study can be found on papers.ssrn.com.
A version of this article first appeared on Medscape.com.