User login
Multipart collaboration brings data-driven care management tools to patients
One Drop announced on Oct. 31 a multipart collaboration with Fitbit to bring enhanced data-driven care management tools to the diabetes community.
One Drop users will now be able to sync Fitbit intraday data to their One Drop accounts. The first initiative will be to help users better understand the impact of physical activity on blood glucose management. The app software also will analyze user-generated health data points with the goal of gaining deeper insights and improving health outcomes for all people with diabetes worldwide. It can potentially allow users to see how their physical activity impacts blood glucose levels. Users can review these data with their very own Certified Diabetes Educator as they work together to meet personalized health goals.
In a study published in JMIR Diabetes in August 2017, results showed a 1.1%-1.3% absolute reduction in hemoglobin A1C in just 4 months in patients using One Drop. It is noted that this was a more significant reduction than other published research suggested was possible using a mobile care management app.
“By integrating Fitbit data and creating an app for Fitbit Ionic, we will be able to provide our users and their health care providers with more data and deeper insights to better manage their diabetes,” said Jeff Dachis, CEO and founder of One Drop, in a press release.
Read the full press release here.
One Drop announced on Oct. 31 a multipart collaboration with Fitbit to bring enhanced data-driven care management tools to the diabetes community.
One Drop users will now be able to sync Fitbit intraday data to their One Drop accounts. The first initiative will be to help users better understand the impact of physical activity on blood glucose management. The app software also will analyze user-generated health data points with the goal of gaining deeper insights and improving health outcomes for all people with diabetes worldwide. It can potentially allow users to see how their physical activity impacts blood glucose levels. Users can review these data with their very own Certified Diabetes Educator as they work together to meet personalized health goals.
In a study published in JMIR Diabetes in August 2017, results showed a 1.1%-1.3% absolute reduction in hemoglobin A1C in just 4 months in patients using One Drop. It is noted that this was a more significant reduction than other published research suggested was possible using a mobile care management app.
“By integrating Fitbit data and creating an app for Fitbit Ionic, we will be able to provide our users and their health care providers with more data and deeper insights to better manage their diabetes,” said Jeff Dachis, CEO and founder of One Drop, in a press release.
Read the full press release here.
One Drop announced on Oct. 31 a multipart collaboration with Fitbit to bring enhanced data-driven care management tools to the diabetes community.
One Drop users will now be able to sync Fitbit intraday data to their One Drop accounts. The first initiative will be to help users better understand the impact of physical activity on blood glucose management. The app software also will analyze user-generated health data points with the goal of gaining deeper insights and improving health outcomes for all people with diabetes worldwide. It can potentially allow users to see how their physical activity impacts blood glucose levels. Users can review these data with their very own Certified Diabetes Educator as they work together to meet personalized health goals.
In a study published in JMIR Diabetes in August 2017, results showed a 1.1%-1.3% absolute reduction in hemoglobin A1C in just 4 months in patients using One Drop. It is noted that this was a more significant reduction than other published research suggested was possible using a mobile care management app.
“By integrating Fitbit data and creating an app for Fitbit Ionic, we will be able to provide our users and their health care providers with more data and deeper insights to better manage their diabetes,” said Jeff Dachis, CEO and founder of One Drop, in a press release.
Read the full press release here.
VIDEO: Rosacea patients no longer considered in ‘buckets’
LAS VEGAS – Clinicians are starting to see and treat rosacea differently, Julie Harper, MD, said in a video interview at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.
“For a long time, we thought about putting rosacea patients into buckets,” based on the predominant type of rosacea they had, such as papulopustular, ocular, or erythematotelangiectatic rosacea, but “what we find is that people have pieces and parts of all of those,” she commented.
In the interview, Dr. Harper, a dermatologist in private practice in Birmingham, Ala., emphasized the importance of directing treatment to all aspects of an individual patient’s rosacea, using combinations of treatments that are approved by the Food and Drug Administration, “or at least proven to be effective for these different parts” of the disease. “That’s something that’s really new in our thinking,” she said.
Dr. Harper disclosed relationships with multiple companies including Allergan, Bayer, Galderma, La Roche-Posay, Promius, and Valeant.
SDEF and this news organization are owned by the same parent company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
LAS VEGAS – Clinicians are starting to see and treat rosacea differently, Julie Harper, MD, said in a video interview at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.
“For a long time, we thought about putting rosacea patients into buckets,” based on the predominant type of rosacea they had, such as papulopustular, ocular, or erythematotelangiectatic rosacea, but “what we find is that people have pieces and parts of all of those,” she commented.
In the interview, Dr. Harper, a dermatologist in private practice in Birmingham, Ala., emphasized the importance of directing treatment to all aspects of an individual patient’s rosacea, using combinations of treatments that are approved by the Food and Drug Administration, “or at least proven to be effective for these different parts” of the disease. “That’s something that’s really new in our thinking,” she said.
Dr. Harper disclosed relationships with multiple companies including Allergan, Bayer, Galderma, La Roche-Posay, Promius, and Valeant.
SDEF and this news organization are owned by the same parent company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
LAS VEGAS – Clinicians are starting to see and treat rosacea differently, Julie Harper, MD, said in a video interview at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.
“For a long time, we thought about putting rosacea patients into buckets,” based on the predominant type of rosacea they had, such as papulopustular, ocular, or erythematotelangiectatic rosacea, but “what we find is that people have pieces and parts of all of those,” she commented.
In the interview, Dr. Harper, a dermatologist in private practice in Birmingham, Ala., emphasized the importance of directing treatment to all aspects of an individual patient’s rosacea, using combinations of treatments that are approved by the Food and Drug Administration, “or at least proven to be effective for these different parts” of the disease. “That’s something that’s really new in our thinking,” she said.
Dr. Harper disclosed relationships with multiple companies including Allergan, Bayer, Galderma, La Roche-Posay, Promius, and Valeant.
SDEF and this news organization are owned by the same parent company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT SDEF LAS VEGAS DERMATOLOGY SEMINAR

Use of MenACWY-CRM vaccine in 2- to 10-year-olds raised no safety concerns
, reported Sara Yee Tartof, PhD, of Kaiser Permanente Southern California, Pasadena, and her associates.
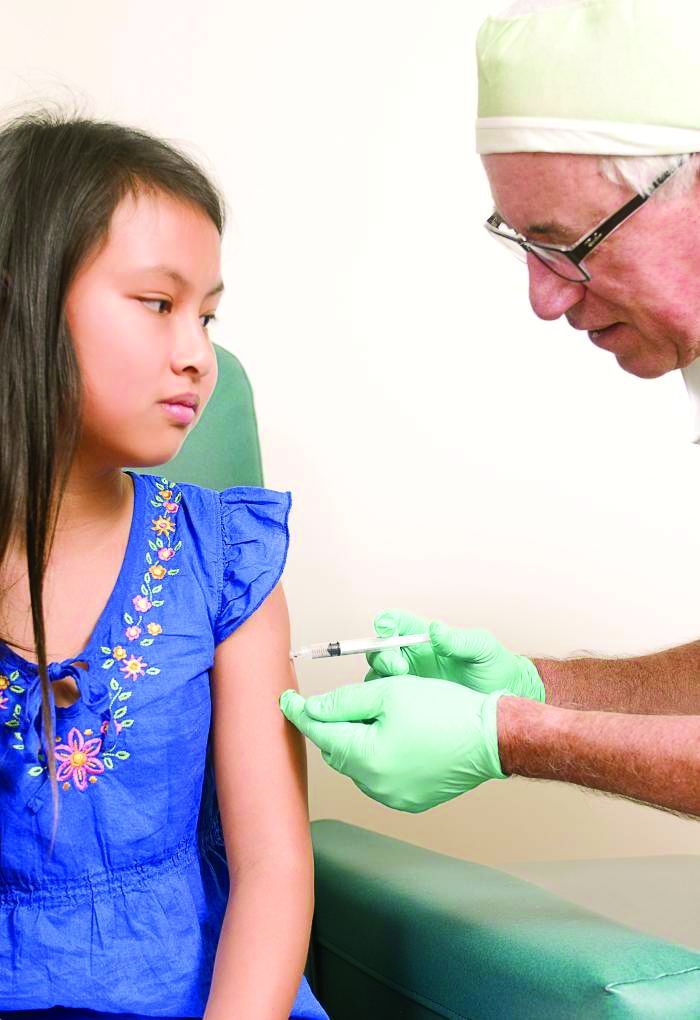
The study was undertaken to evaluate the safety of the MenACWY-CRM vaccine in the care of children aged 2-10 years in the real world. The other quadrivalent meningococcal conjugate vaccine, MenACWY-D (Menactra), was not used in this study.
In a retrospective, observational study of 327 children aged 2-10 years when they received the MenACWY-CRM vaccine as part of routine clinical care, there was only one event of interest, which was a child who developed asthma during the 1-year observation period, and that occurred 237 days after vaccination with both the MenACWY-CRM and a typhoid vaccine. “A causal link between the two events is unlikely,” Dr. Tartof and her associates wrote.
Most of the serious medically attended events were considered to be unrelated to MenACWY-CRM vaccination, or symptom onset occurred a long time after vaccination. “The remaining events were pneumonia, bronchitis, cough, febrile convulsion, and vomiting identified within 30 days of vaccination among four children,” the investigators reported. “Pneumonia, bronchitis, and cough were diagnosed in the same child; cough, febrile convulsion, and vomiting were diagnosed separately in the other 3 children.
“It appears that many MenACWY-CRM recipients in our study population received the vaccine because of travel to high-risk areas, possible exposure to meningitis, or as routine vaccinations received early as part of the ACIP-recommended routine vaccination for those 11-12 years old,” Dr. Tartof and her associates wrote.
Read more in the Pediatric Infectious Diseases Journal (2017 Nov 1. doi: 10.1097/INF.0000000000001696).
, reported Sara Yee Tartof, PhD, of Kaiser Permanente Southern California, Pasadena, and her associates.
The study was undertaken to evaluate the safety of the MenACWY-CRM vaccine in the care of children aged 2-10 years in the real world. The other quadrivalent meningococcal conjugate vaccine, MenACWY-D (Menactra), was not used in this study.
In a retrospective, observational study of 327 children aged 2-10 years when they received the MenACWY-CRM vaccine as part of routine clinical care, there was only one event of interest, which was a child who developed asthma during the 1-year observation period, and that occurred 237 days after vaccination with both the MenACWY-CRM and a typhoid vaccine. “A causal link between the two events is unlikely,” Dr. Tartof and her associates wrote.
Most of the serious medically attended events were considered to be unrelated to MenACWY-CRM vaccination, or symptom onset occurred a long time after vaccination. “The remaining events were pneumonia, bronchitis, cough, febrile convulsion, and vomiting identified within 30 days of vaccination among four children,” the investigators reported. “Pneumonia, bronchitis, and cough were diagnosed in the same child; cough, febrile convulsion, and vomiting were diagnosed separately in the other 3 children.
“It appears that many MenACWY-CRM recipients in our study population received the vaccine because of travel to high-risk areas, possible exposure to meningitis, or as routine vaccinations received early as part of the ACIP-recommended routine vaccination for those 11-12 years old,” Dr. Tartof and her associates wrote.
Read more in the Pediatric Infectious Diseases Journal (2017 Nov 1. doi: 10.1097/INF.0000000000001696).
, reported Sara Yee Tartof, PhD, of Kaiser Permanente Southern California, Pasadena, and her associates.
The study was undertaken to evaluate the safety of the MenACWY-CRM vaccine in the care of children aged 2-10 years in the real world. The other quadrivalent meningococcal conjugate vaccine, MenACWY-D (Menactra), was not used in this study.
In a retrospective, observational study of 327 children aged 2-10 years when they received the MenACWY-CRM vaccine as part of routine clinical care, there was only one event of interest, which was a child who developed asthma during the 1-year observation period, and that occurred 237 days after vaccination with both the MenACWY-CRM and a typhoid vaccine. “A causal link between the two events is unlikely,” Dr. Tartof and her associates wrote.
Most of the serious medically attended events were considered to be unrelated to MenACWY-CRM vaccination, or symptom onset occurred a long time after vaccination. “The remaining events were pneumonia, bronchitis, cough, febrile convulsion, and vomiting identified within 30 days of vaccination among four children,” the investigators reported. “Pneumonia, bronchitis, and cough were diagnosed in the same child; cough, febrile convulsion, and vomiting were diagnosed separately in the other 3 children.
“It appears that many MenACWY-CRM recipients in our study population received the vaccine because of travel to high-risk areas, possible exposure to meningitis, or as routine vaccinations received early as part of the ACIP-recommended routine vaccination for those 11-12 years old,” Dr. Tartof and her associates wrote.
Read more in the Pediatric Infectious Diseases Journal (2017 Nov 1. doi: 10.1097/INF.0000000000001696).
FROM PEDIATRIC INFECTIOUS DISEASE JOURNAL
Early diagnosis of tuberous sclerosis may be possible in infants
with an echocardiogram for cardiac rhabdomyomas and a skin examination for hypomelanotic macules, noninvasive tests that do not require sedation, said Peter E. Davis, MD, of Boston Children’s Hospital, and his associates on behalf of the Tuberous Sclerosis Complex Autism Center of Excellence Research Network.
“Early TSC diagnosis in infants opens a window of opportunity to treat before the onset of epilepsy or other neurodevelopmental disorders and allows for close surveillance for sequelae of TSC,” the researchers said. “However, this window may be as small as a few months.”
In two concurrent, prospective, longitudinal, observational studies at five medical centers of 130 infants meeting a genetic or clinical diagnosis of TSC, cardiac rhabdomyomas, and hypomelanotic macules were the most common initial presenting features, occurring in 59% and 39% of infants, respectively; 85% of infants had either or both. In terms of prevalence, hypomelanotic macules and tubers or other cortical dysplasias were the most prevalent TSC features both occurring at 94%, followed by subependymal nodules (SENs) at 90%, and cardiac rhabdomyomas at 82%. Every infant had at least one of these diagnostic criteria, and 61% had all four of them, the investigators reported.
Neuroimaging results were available in 115 infants, of whom 94% had tubers or cortical dysplasias, 90% had SENs, and 89% had both; 6% had subependymal giant cell astrocytomas. Seizure onset occurred in 15% of infants before or when other TSC criteria were recorded, “suggesting that seizure was an initial presenting symptom.” Seizure onset occurred within 3 months after initial presentation in 17% of infants, within 6 months in 39%, and within 12 months in 57%. Ultimately, 57% of the infants had infantile spasms, 55% had focal seizures, and 12% had another seizure type, Dr. Davis and his associates said.
“Early, prospective use of EEGs may enable risk stratification in studies of epilepsy prevention in infants with TSC. The antiepileptic medication vigabatrin is particularly effective in treating infantile spasms in TSC and has mTOR [mechanistic target of the rapamycin]–inhibiting effects. Vigabatrin is currently in clinical trials to determine its efficacy at preventing epilepsy in patients with TSC,” they said, although the drug is not currently recommended for infants. “mTOR inhibitors have been successfully used to treat multiple TSC manifestations and have shown some efficacy as adjunctive treatment of refractory epilepsy,” they wrote, adding that studies are needed “to determine the safety and efficacy of mTOR inhibitors in this age group.”
Read more at Pediatrics. 2017;140(6):e20164040 (doi: 10.1542/peds.2016-4040).
with an echocardiogram for cardiac rhabdomyomas and a skin examination for hypomelanotic macules, noninvasive tests that do not require sedation, said Peter E. Davis, MD, of Boston Children’s Hospital, and his associates on behalf of the Tuberous Sclerosis Complex Autism Center of Excellence Research Network.
“Early TSC diagnosis in infants opens a window of opportunity to treat before the onset of epilepsy or other neurodevelopmental disorders and allows for close surveillance for sequelae of TSC,” the researchers said. “However, this window may be as small as a few months.”
In two concurrent, prospective, longitudinal, observational studies at five medical centers of 130 infants meeting a genetic or clinical diagnosis of TSC, cardiac rhabdomyomas, and hypomelanotic macules were the most common initial presenting features, occurring in 59% and 39% of infants, respectively; 85% of infants had either or both. In terms of prevalence, hypomelanotic macules and tubers or other cortical dysplasias were the most prevalent TSC features both occurring at 94%, followed by subependymal nodules (SENs) at 90%, and cardiac rhabdomyomas at 82%. Every infant had at least one of these diagnostic criteria, and 61% had all four of them, the investigators reported.
Neuroimaging results were available in 115 infants, of whom 94% had tubers or cortical dysplasias, 90% had SENs, and 89% had both; 6% had subependymal giant cell astrocytomas. Seizure onset occurred in 15% of infants before or when other TSC criteria were recorded, “suggesting that seizure was an initial presenting symptom.” Seizure onset occurred within 3 months after initial presentation in 17% of infants, within 6 months in 39%, and within 12 months in 57%. Ultimately, 57% of the infants had infantile spasms, 55% had focal seizures, and 12% had another seizure type, Dr. Davis and his associates said.
“Early, prospective use of EEGs may enable risk stratification in studies of epilepsy prevention in infants with TSC. The antiepileptic medication vigabatrin is particularly effective in treating infantile spasms in TSC and has mTOR [mechanistic target of the rapamycin]–inhibiting effects. Vigabatrin is currently in clinical trials to determine its efficacy at preventing epilepsy in patients with TSC,” they said, although the drug is not currently recommended for infants. “mTOR inhibitors have been successfully used to treat multiple TSC manifestations and have shown some efficacy as adjunctive treatment of refractory epilepsy,” they wrote, adding that studies are needed “to determine the safety and efficacy of mTOR inhibitors in this age group.”
Read more at Pediatrics. 2017;140(6):e20164040 (doi: 10.1542/peds.2016-4040).
with an echocardiogram for cardiac rhabdomyomas and a skin examination for hypomelanotic macules, noninvasive tests that do not require sedation, said Peter E. Davis, MD, of Boston Children’s Hospital, and his associates on behalf of the Tuberous Sclerosis Complex Autism Center of Excellence Research Network.
“Early TSC diagnosis in infants opens a window of opportunity to treat before the onset of epilepsy or other neurodevelopmental disorders and allows for close surveillance for sequelae of TSC,” the researchers said. “However, this window may be as small as a few months.”
In two concurrent, prospective, longitudinal, observational studies at five medical centers of 130 infants meeting a genetic or clinical diagnosis of TSC, cardiac rhabdomyomas, and hypomelanotic macules were the most common initial presenting features, occurring in 59% and 39% of infants, respectively; 85% of infants had either or both. In terms of prevalence, hypomelanotic macules and tubers or other cortical dysplasias were the most prevalent TSC features both occurring at 94%, followed by subependymal nodules (SENs) at 90%, and cardiac rhabdomyomas at 82%. Every infant had at least one of these diagnostic criteria, and 61% had all four of them, the investigators reported.
Neuroimaging results were available in 115 infants, of whom 94% had tubers or cortical dysplasias, 90% had SENs, and 89% had both; 6% had subependymal giant cell astrocytomas. Seizure onset occurred in 15% of infants before or when other TSC criteria were recorded, “suggesting that seizure was an initial presenting symptom.” Seizure onset occurred within 3 months after initial presentation in 17% of infants, within 6 months in 39%, and within 12 months in 57%. Ultimately, 57% of the infants had infantile spasms, 55% had focal seizures, and 12% had another seizure type, Dr. Davis and his associates said.
“Early, prospective use of EEGs may enable risk stratification in studies of epilepsy prevention in infants with TSC. The antiepileptic medication vigabatrin is particularly effective in treating infantile spasms in TSC and has mTOR [mechanistic target of the rapamycin]–inhibiting effects. Vigabatrin is currently in clinical trials to determine its efficacy at preventing epilepsy in patients with TSC,” they said, although the drug is not currently recommended for infants. “mTOR inhibitors have been successfully used to treat multiple TSC manifestations and have shown some efficacy as adjunctive treatment of refractory epilepsy,” they wrote, adding that studies are needed “to determine the safety and efficacy of mTOR inhibitors in this age group.”
Read more at Pediatrics. 2017;140(6):e20164040 (doi: 10.1542/peds.2016-4040).
FROM PEDIATRICS
Applying Choosing Wisely principles to telemetry and catheter use
The Choosing Wisely recommendations for hospitalists have launched numerous research projects. One dealing with telemetry and catheter use was published in September’s American Journal of Medicine.
After reviewing the literature on how people were implementing these recommendations, the researchers noticed most projects “1) narrowly focused on only one of the recommendations; 2) often used intrusive interventions that appeared to be burdensome and not adaptable to physician workflow; and 3) were expensive to implement,” said lead author Charlie M. Wray, DO, MS, of the Division of Hospital Medicine, San Francisco Veterans Affairs Medical Center, and the University of California, San Francisco. “We set out to design a project that could minimize these aspects while hopefully decreasing the use of telemetry and Foley catheters.”
The researchers created a “silent” reminder that was posted on a widely used screen within their EHR and was only activated when the user clicked on it. “Additionally, we wanted to make sure that this intervention made its way to teaching rounds and the patients’ bedsides,” Dr. Wray said. “So, when the attendings and residents would print out their daily census, it would contain the reminders, which allowed the team to quickly review which patients were actively using telemetry or had a Foley and discuss, at a team-level, whose telemetry or Foley could be stopped.”
The project demonstrated a trend toward less telemetry use, less time spent on telemetry, fewer catheters ordered, and more selective utilization of catheters in sicker patients.
“We believe that our project shows that the bundling of interventions has the potential to impart an effect on a greater proportion of the population than those that focus on a single issue,” Dr. Wray said. “Second, future interventions that look to utilize EHR-based clinical reminders should consider utilizing a ‘silent’ design that is prominent but doesn’t intrude upon practitioners workflow.”
You don’t need to be at a large academic institution to implement this idea, he added. “A few hours with your IT expert and a champion who is willing to take the lead could easily implement this project and hopefully see similar outcomes.”
Reference
Wray, Charlie M. et al. Improving value by reducing unnecessary telemetry and urinary catheter utilization in hospitalized patients. Am J Med. 2017 Sep;130(9):1037-41.
The Choosing Wisely recommendations for hospitalists have launched numerous research projects. One dealing with telemetry and catheter use was published in September’s American Journal of Medicine.
After reviewing the literature on how people were implementing these recommendations, the researchers noticed most projects “1) narrowly focused on only one of the recommendations; 2) often used intrusive interventions that appeared to be burdensome and not adaptable to physician workflow; and 3) were expensive to implement,” said lead author Charlie M. Wray, DO, MS, of the Division of Hospital Medicine, San Francisco Veterans Affairs Medical Center, and the University of California, San Francisco. “We set out to design a project that could minimize these aspects while hopefully decreasing the use of telemetry and Foley catheters.”
The researchers created a “silent” reminder that was posted on a widely used screen within their EHR and was only activated when the user clicked on it. “Additionally, we wanted to make sure that this intervention made its way to teaching rounds and the patients’ bedsides,” Dr. Wray said. “So, when the attendings and residents would print out their daily census, it would contain the reminders, which allowed the team to quickly review which patients were actively using telemetry or had a Foley and discuss, at a team-level, whose telemetry or Foley could be stopped.”
The project demonstrated a trend toward less telemetry use, less time spent on telemetry, fewer catheters ordered, and more selective utilization of catheters in sicker patients.
“We believe that our project shows that the bundling of interventions has the potential to impart an effect on a greater proportion of the population than those that focus on a single issue,” Dr. Wray said. “Second, future interventions that look to utilize EHR-based clinical reminders should consider utilizing a ‘silent’ design that is prominent but doesn’t intrude upon practitioners workflow.”
You don’t need to be at a large academic institution to implement this idea, he added. “A few hours with your IT expert and a champion who is willing to take the lead could easily implement this project and hopefully see similar outcomes.”
Reference
Wray, Charlie M. et al. Improving value by reducing unnecessary telemetry and urinary catheter utilization in hospitalized patients. Am J Med. 2017 Sep;130(9):1037-41.
The Choosing Wisely recommendations for hospitalists have launched numerous research projects. One dealing with telemetry and catheter use was published in September’s American Journal of Medicine.
After reviewing the literature on how people were implementing these recommendations, the researchers noticed most projects “1) narrowly focused on only one of the recommendations; 2) often used intrusive interventions that appeared to be burdensome and not adaptable to physician workflow; and 3) were expensive to implement,” said lead author Charlie M. Wray, DO, MS, of the Division of Hospital Medicine, San Francisco Veterans Affairs Medical Center, and the University of California, San Francisco. “We set out to design a project that could minimize these aspects while hopefully decreasing the use of telemetry and Foley catheters.”
The researchers created a “silent” reminder that was posted on a widely used screen within their EHR and was only activated when the user clicked on it. “Additionally, we wanted to make sure that this intervention made its way to teaching rounds and the patients’ bedsides,” Dr. Wray said. “So, when the attendings and residents would print out their daily census, it would contain the reminders, which allowed the team to quickly review which patients were actively using telemetry or had a Foley and discuss, at a team-level, whose telemetry or Foley could be stopped.”
The project demonstrated a trend toward less telemetry use, less time spent on telemetry, fewer catheters ordered, and more selective utilization of catheters in sicker patients.
“We believe that our project shows that the bundling of interventions has the potential to impart an effect on a greater proportion of the population than those that focus on a single issue,” Dr. Wray said. “Second, future interventions that look to utilize EHR-based clinical reminders should consider utilizing a ‘silent’ design that is prominent but doesn’t intrude upon practitioners workflow.”
You don’t need to be at a large academic institution to implement this idea, he added. “A few hours with your IT expert and a champion who is willing to take the lead could easily implement this project and hopefully see similar outcomes.”
Reference
Wray, Charlie M. et al. Improving value by reducing unnecessary telemetry and urinary catheter utilization in hospitalized patients. Am J Med. 2017 Sep;130(9):1037-41.
Are microRNAs the key to an endometriosis biomarker?
SAN ANTONIO – The results of a prospective study showed that, in patients without a surgical endometriosis diagnosis, a serum test of microRNA (miRNA) levels yielded a very high predictive value when endometriosis was later surgically confirmed.
“This is the first prospective study performed within a diverse population that identifies miRNAs can reliably be used to differentiate between endometriosis and other gynecologic pathologies,” Sarah Moustafa, MD, said at the annual meeting of the American Society for Reproductive Medicine.

Dr. Moustafa and her collaborators found that the area under the receiver operating characteristic curve (AUC) for the combination of miRNAs 125, 451, and 3613 was 0.917. The AUC of another combination was even higher: the miRNAs let-7b, 150, 342, and 451 yielded an AUC of 0.977 for endometriosis. These figures support that the miRNA combinations give “an excellent diagnostic potential for endometriosis,” Dr. Moustafa said.
The study enrolled 86 women of reproductive age who were scheduled to have a laparotomy or laparoscopy for benign gynecologic reasons. Pregnant patients and those with malignancy were excluded. Patients had blood drawn before their surgeries to have blinded miRNA analysis via quantitative reverse transcriptase polymerase chain reaction.
Dr. Moustafa and her colleagues looked at the diagnostic value of individual miRNAs and also examined combinations of promising miRNAs by seeing which levels were elevated or depressed in patients who were later found to have surgically diagnosed endometriosis. The study built on previous retrospective work that had identified some candidate miRNAs.
In this prospective study, women with endometriosis had significantly lower serum levels of miRNAs 3613 and let-7b and significantly higher serum levels of miRNAs 150, 125b, 451, and 342. The investigators also analyzed the data to see if there were miRNA level differences between women who were on hormones and those who were not, and also to see if cycle timing affected results; neither of these factors affected miRNA levels, Dr. Moustafa reported.
When the enrolled patients had surgery, 36 were found to have endometriosis and 50 had a variety of other diagnoses, with uterine fibroids (48%) and no abnormal pathology (28%) predominating.
“A noninvasive diagnostic test does not currently exist” but is sorely needed, Dr. Moustafa said.
There’s a long gap from the onset of endometriosis symptoms to diagnosis, with one study showing that symptoms can be present for an average of 6.7-11 years before surgical diagnosis occurs. In one survey of more than 7,000 women, nearly half (46%) saw more than five physicians before they got the correct diagnosis, said Dr. Moustafa, a resident in the obstetrics, gynecology, and reproductive sciences department at Yale University, New Haven, Conn.
In a discussion following the presentation, attendee Steve Young, MD, PhD, professor of reproductive endocrinology and fertility at the University of North Carolina, Chapel Hill, said it would be useful to have a biomarker that could be a surrogate for disease burden. “You could look at miRNAs after surgery. Do you see changes back toward normal?” Dr. Young said.
Dr. Moustafa responded that the research team is in the process of collecting postoperative data to see whether levels change when the disease burden is diminished by surgery.
Next steps, she said, include looking for miRNAs in saliva to explore whether an even less invasive test might be possible. Also, there are suggestions that miRNA 125 might be a marker for more severe disease, so the team is investigating this association as well.
Whether the combination miRNA screen is ready for prime time is still an open question, she said. “Any time a new screening test is developed we have to have caution.” Current thinking, she said, points toward the utility of miRNA screening for patients with unexplained fertility, for example.
“At this point, the pendulum is so far on the side of underdiagnosis that we think patients would benefit from a screening test,” Dr. Moustafa added.
The study was supported by OvaScience. Dr. Moustafa reported having no relevant financial disclosures.
[email protected]
On Twitter @karioakes
SAN ANTONIO – The results of a prospective study showed that, in patients without a surgical endometriosis diagnosis, a serum test of microRNA (miRNA) levels yielded a very high predictive value when endometriosis was later surgically confirmed.
“This is the first prospective study performed within a diverse population that identifies miRNAs can reliably be used to differentiate between endometriosis and other gynecologic pathologies,” Sarah Moustafa, MD, said at the annual meeting of the American Society for Reproductive Medicine.
Dr. Moustafa and her collaborators found that the area under the receiver operating characteristic curve (AUC) for the combination of miRNAs 125, 451, and 3613 was 0.917. The AUC of another combination was even higher: the miRNAs let-7b, 150, 342, and 451 yielded an AUC of 0.977 for endometriosis. These figures support that the miRNA combinations give “an excellent diagnostic potential for endometriosis,” Dr. Moustafa said.
The study enrolled 86 women of reproductive age who were scheduled to have a laparotomy or laparoscopy for benign gynecologic reasons. Pregnant patients and those with malignancy were excluded. Patients had blood drawn before their surgeries to have blinded miRNA analysis via quantitative reverse transcriptase polymerase chain reaction.
Dr. Moustafa and her colleagues looked at the diagnostic value of individual miRNAs and also examined combinations of promising miRNAs by seeing which levels were elevated or depressed in patients who were later found to have surgically diagnosed endometriosis. The study built on previous retrospective work that had identified some candidate miRNAs.
In this prospective study, women with endometriosis had significantly lower serum levels of miRNAs 3613 and let-7b and significantly higher serum levels of miRNAs 150, 125b, 451, and 342. The investigators also analyzed the data to see if there were miRNA level differences between women who were on hormones and those who were not, and also to see if cycle timing affected results; neither of these factors affected miRNA levels, Dr. Moustafa reported.
When the enrolled patients had surgery, 36 were found to have endometriosis and 50 had a variety of other diagnoses, with uterine fibroids (48%) and no abnormal pathology (28%) predominating.
“A noninvasive diagnostic test does not currently exist” but is sorely needed, Dr. Moustafa said.
There’s a long gap from the onset of endometriosis symptoms to diagnosis, with one study showing that symptoms can be present for an average of 6.7-11 years before surgical diagnosis occurs. In one survey of more than 7,000 women, nearly half (46%) saw more than five physicians before they got the correct diagnosis, said Dr. Moustafa, a resident in the obstetrics, gynecology, and reproductive sciences department at Yale University, New Haven, Conn.
In a discussion following the presentation, attendee Steve Young, MD, PhD, professor of reproductive endocrinology and fertility at the University of North Carolina, Chapel Hill, said it would be useful to have a biomarker that could be a surrogate for disease burden. “You could look at miRNAs after surgery. Do you see changes back toward normal?” Dr. Young said.
Dr. Moustafa responded that the research team is in the process of collecting postoperative data to see whether levels change when the disease burden is diminished by surgery.
Next steps, she said, include looking for miRNAs in saliva to explore whether an even less invasive test might be possible. Also, there are suggestions that miRNA 125 might be a marker for more severe disease, so the team is investigating this association as well.
Whether the combination miRNA screen is ready for prime time is still an open question, she said. “Any time a new screening test is developed we have to have caution.” Current thinking, she said, points toward the utility of miRNA screening for patients with unexplained fertility, for example.
“At this point, the pendulum is so far on the side of underdiagnosis that we think patients would benefit from a screening test,” Dr. Moustafa added.
The study was supported by OvaScience. Dr. Moustafa reported having no relevant financial disclosures.
[email protected]
On Twitter @karioakes
SAN ANTONIO – The results of a prospective study showed that, in patients without a surgical endometriosis diagnosis, a serum test of microRNA (miRNA) levels yielded a very high predictive value when endometriosis was later surgically confirmed.
“This is the first prospective study performed within a diverse population that identifies miRNAs can reliably be used to differentiate between endometriosis and other gynecologic pathologies,” Sarah Moustafa, MD, said at the annual meeting of the American Society for Reproductive Medicine.
Dr. Moustafa and her collaborators found that the area under the receiver operating characteristic curve (AUC) for the combination of miRNAs 125, 451, and 3613 was 0.917. The AUC of another combination was even higher: the miRNAs let-7b, 150, 342, and 451 yielded an AUC of 0.977 for endometriosis. These figures support that the miRNA combinations give “an excellent diagnostic potential for endometriosis,” Dr. Moustafa said.
The study enrolled 86 women of reproductive age who were scheduled to have a laparotomy or laparoscopy for benign gynecologic reasons. Pregnant patients and those with malignancy were excluded. Patients had blood drawn before their surgeries to have blinded miRNA analysis via quantitative reverse transcriptase polymerase chain reaction.
Dr. Moustafa and her colleagues looked at the diagnostic value of individual miRNAs and also examined combinations of promising miRNAs by seeing which levels were elevated or depressed in patients who were later found to have surgically diagnosed endometriosis. The study built on previous retrospective work that had identified some candidate miRNAs.
In this prospective study, women with endometriosis had significantly lower serum levels of miRNAs 3613 and let-7b and significantly higher serum levels of miRNAs 150, 125b, 451, and 342. The investigators also analyzed the data to see if there were miRNA level differences between women who were on hormones and those who were not, and also to see if cycle timing affected results; neither of these factors affected miRNA levels, Dr. Moustafa reported.
When the enrolled patients had surgery, 36 were found to have endometriosis and 50 had a variety of other diagnoses, with uterine fibroids (48%) and no abnormal pathology (28%) predominating.
“A noninvasive diagnostic test does not currently exist” but is sorely needed, Dr. Moustafa said.
There’s a long gap from the onset of endometriosis symptoms to diagnosis, with one study showing that symptoms can be present for an average of 6.7-11 years before surgical diagnosis occurs. In one survey of more than 7,000 women, nearly half (46%) saw more than five physicians before they got the correct diagnosis, said Dr. Moustafa, a resident in the obstetrics, gynecology, and reproductive sciences department at Yale University, New Haven, Conn.
In a discussion following the presentation, attendee Steve Young, MD, PhD, professor of reproductive endocrinology and fertility at the University of North Carolina, Chapel Hill, said it would be useful to have a biomarker that could be a surrogate for disease burden. “You could look at miRNAs after surgery. Do you see changes back toward normal?” Dr. Young said.
Dr. Moustafa responded that the research team is in the process of collecting postoperative data to see whether levels change when the disease burden is diminished by surgery.
Next steps, she said, include looking for miRNAs in saliva to explore whether an even less invasive test might be possible. Also, there are suggestions that miRNA 125 might be a marker for more severe disease, so the team is investigating this association as well.
Whether the combination miRNA screen is ready for prime time is still an open question, she said. “Any time a new screening test is developed we have to have caution.” Current thinking, she said, points toward the utility of miRNA screening for patients with unexplained fertility, for example.
“At this point, the pendulum is so far on the side of underdiagnosis that we think patients would benefit from a screening test,” Dr. Moustafa added.
The study was supported by OvaScience. Dr. Moustafa reported having no relevant financial disclosures.
[email protected]
On Twitter @karioakes
AT ASRM 2017
Key clinical point:
Major finding: The areas under the receiver operating curve for the most promising miRNA combinations were 0.917 and 0.977.
Data source: Blinded, prospective study of 86 women slated to have diagnostic pelvic laparoscopy or laparotomy.
Disclosures: Dr. Moustafa reported no relevant disclosures. OvaScience supported the study.

Solitary Tender Nodule on the Back
The Diagnosis: Solitary Fibrous Tumor
Solitary fibrous tumors (SFTs), as first described by Klemperer and Rabin1 in 1931, are relatively uncommon mesenchymal neoplasms that occur primarily in the pleura. This lesion is now known to affect many other extrathoracic sites, such as the liver, kidney, adrenal glands, thyroid, central nervous system, and soft tissue, with rare examples originating from the skin.2 Okamura et al3 reported the first known case of cutaneous SFT in 1997, with most of the literature limited to case reports. Erdag et al2 described one of the largest case series of primary cutaneous SFTs. These lesions can occur across a wide age range but tend to primarily affect middle-aged adults. Solitary fibrous tumors have been known to have no sex predilection; however, Erdag et al2 found a male predominance with a male to female ratio of 4 to 1.
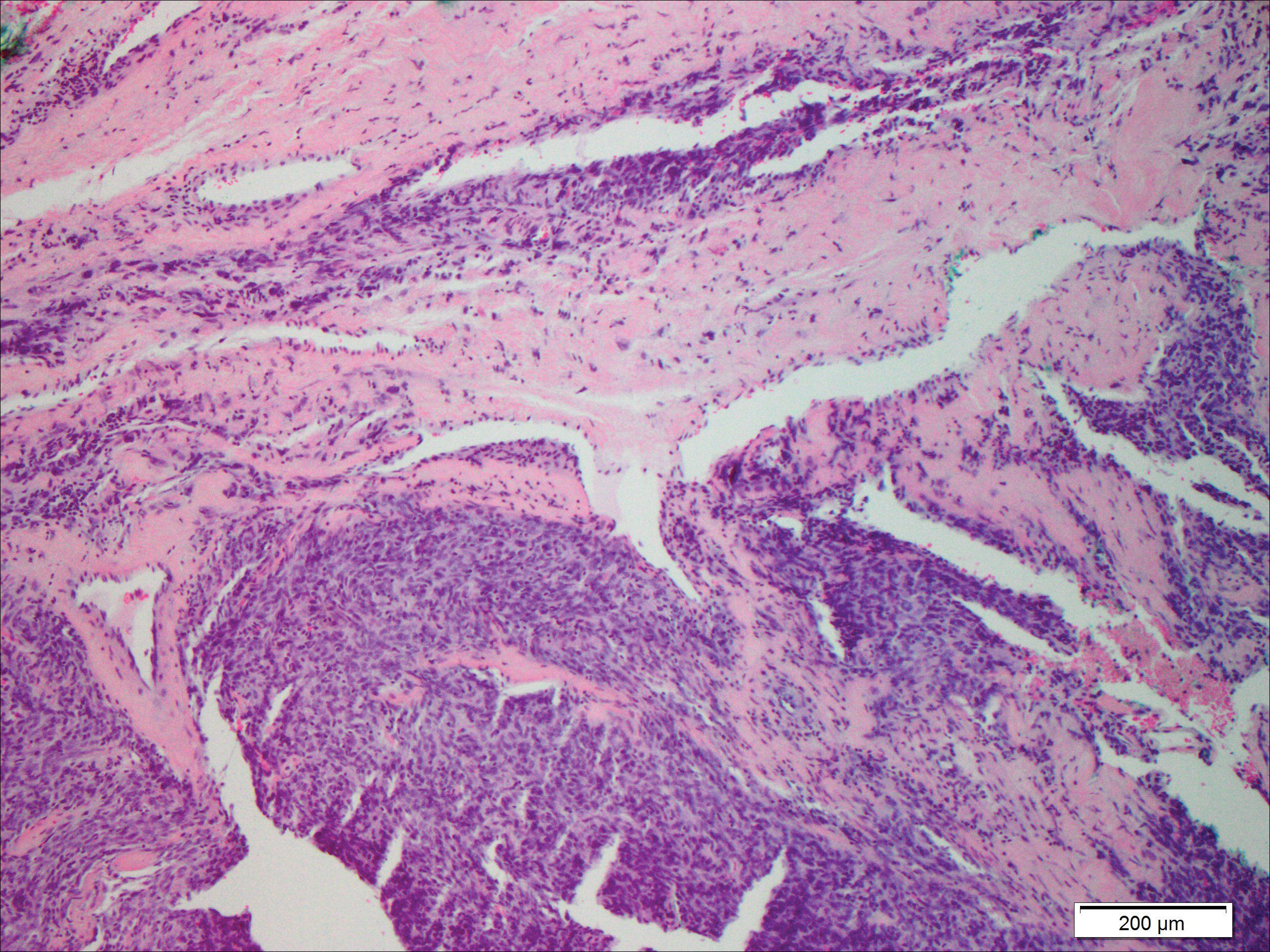
Histopathologically, a cutaneous SFT is known to appear as a well-circumscribed nodular spindle cell proliferation arranged in interlacing fascicles with an abundant hyalinized collagen stroma (quiz image). Alternating hypocellular and hypercellular areas can be seen. Supporting vasculature often is relatively prominent, represented by angulated and branching staghorn blood vessels (Figure 1).2 A common histopathologic finding of SFTs is a patternless pattern, which suggests that the tumor can have a variety of morphologic appearances (eg, storiform, fascicular, neural, herringbone growth patterns), making histologic diagnosis difficult (quiz image).4 Therefore, immunohistochemistry plays a large role in the diagnosis of this tumor. The most important positive markers include CD34, CD99, B-cell lymphoma 2 (BCL-2), and signal transducer and activator of transcription 6 (STAT6).5 Nuclear STAT6 staining is an immunomarker for NGFI-A binding protein 2 (NAB2)-STAT6 gene fusion, which is specific for SFT.5,6 Vivero et al7 also reported glutamate receptor, inotropic, AMPA 2 (GRIA2) as a useful immunostain in SFT, though it is also expressed in dermatofibrosarcoma protuberans (DFSP). In this case, the clinical and histopathologic findings best supported a diagnosis of SFT. Some consider hemangiopericytomas to be examples of SFTs; however, true hemangiopericytomas lack the thick hyalinized collagen and hypercellular areas seen in SFT.
A cellular dermatofibroma generally presents as a single round, reddish brown papule or nodule approximately 0.5 to 1 cm in diameter that is firm to palpation with a central depression or dimple created over the lesion from the lateral pressure. Cellular dermatofibromas mostly occur in middle-aged adults, with the most common locations on the legs and on the sides of the trunk. They are thought to arise after injuries to the skin. On histopathologic examination, cellular dermatofibromas typically exhibit a proliferation of fibrohistiocytic cells with collagen trapping, often at the periphery of the tumor (Figure 2). Although cellular dermatofibromas appear clinically different than SFTs, they often mimic SFTs histopathologically. Immunostaining also can be helpful in differentiating cellular dermatofibromas in which cells stain positive for factor XIIIa. CD34 staining is negative.

Dermatofibrosarcoma protuberans usually appears as one or multiple firm, red to violaceous nodules or plaques. They most often occur on the trunk in middle-aged adults. Histopathologically, DFSP presents with a dense, hypercellular, spindle cell proliferation that demonstrates a typical storiform pattern. The tumor generally infiltrates into the deep dermis and subcutaneous adipose layer with characteristic adipocyte entrapment (Figure 3). Positive CD34 and negative factor XIIIa staining helps to differentiate DFSP from a cellular dermatofibroma. Immunohistochemically, it is more difficult to distinguish DFSP from SFT, as both are CD34+ spindle cell neoplasms that also stain positive for CD99 and BCL-2.2 GRIA2 positivity also is seen in both SFT and DFSP.7 However, differentiation can be made on morphologic grounds alone, as DFSP has ill-defined tumor borders with adnexal and fat entrapment and SFT tends to be more circumscribed with prominent arborizing hyalinized vessels.8
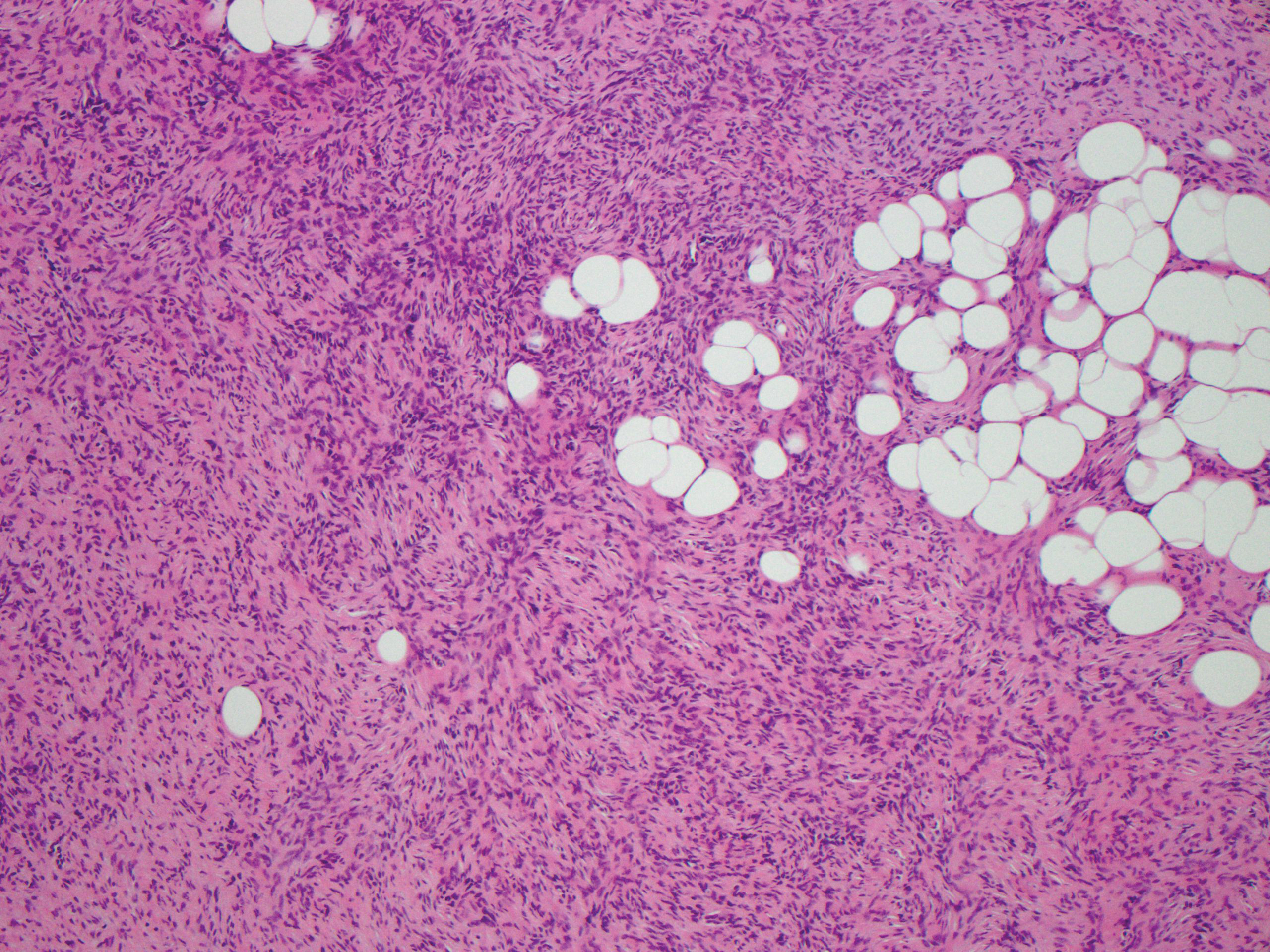
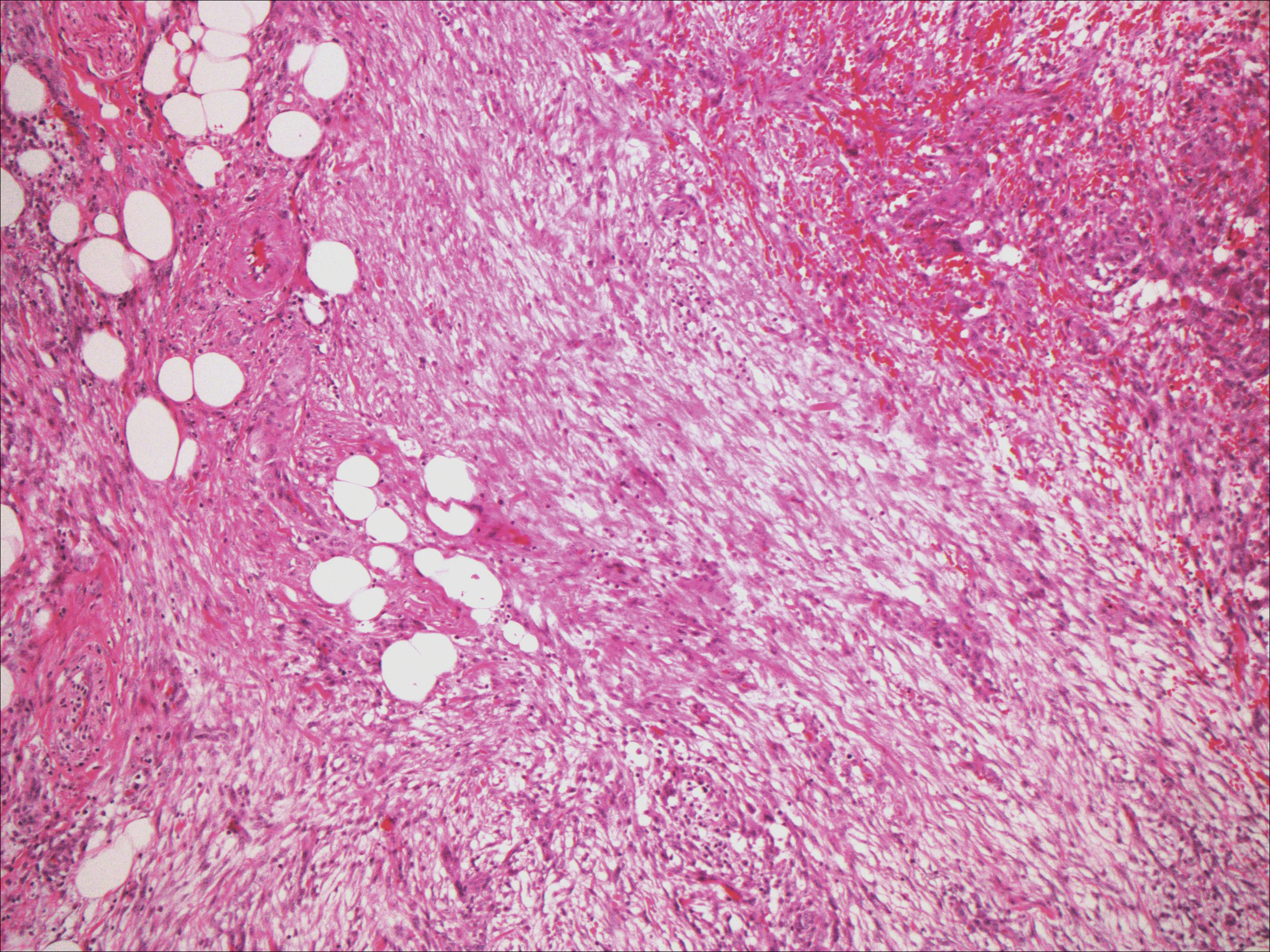
Spindle cell lipoma (SCL) is an asymptomatic subcutaneous tumor commonly located on the back, neck, and shoulders in older patients, typically men. It often presents as a solitary lesion, though multiple lesions may occur. It is a well-circumscribed tumor of mature adipose tissue with areas of spindle cell proliferation and ropey collagen bundles (Figure 4). In early lesions, the spindle cell areas are myxoid with the presence of many mast cells.9 The spindle cells stain positive for CD34. Although spindle cell lipoma would be included in both the clinical and histopathologic differential diagnosis for SFT, its histopathologic features often are enough to differentiate SCL, which is highlighted by the aforementioned features as well as a relatively low cellularity and lack of ectatic vessels.8 However, discerning tumor variants, such as low-fat pseudoangiomatous SCL and lipomatous or myxoid SFT, might prove more challenging.
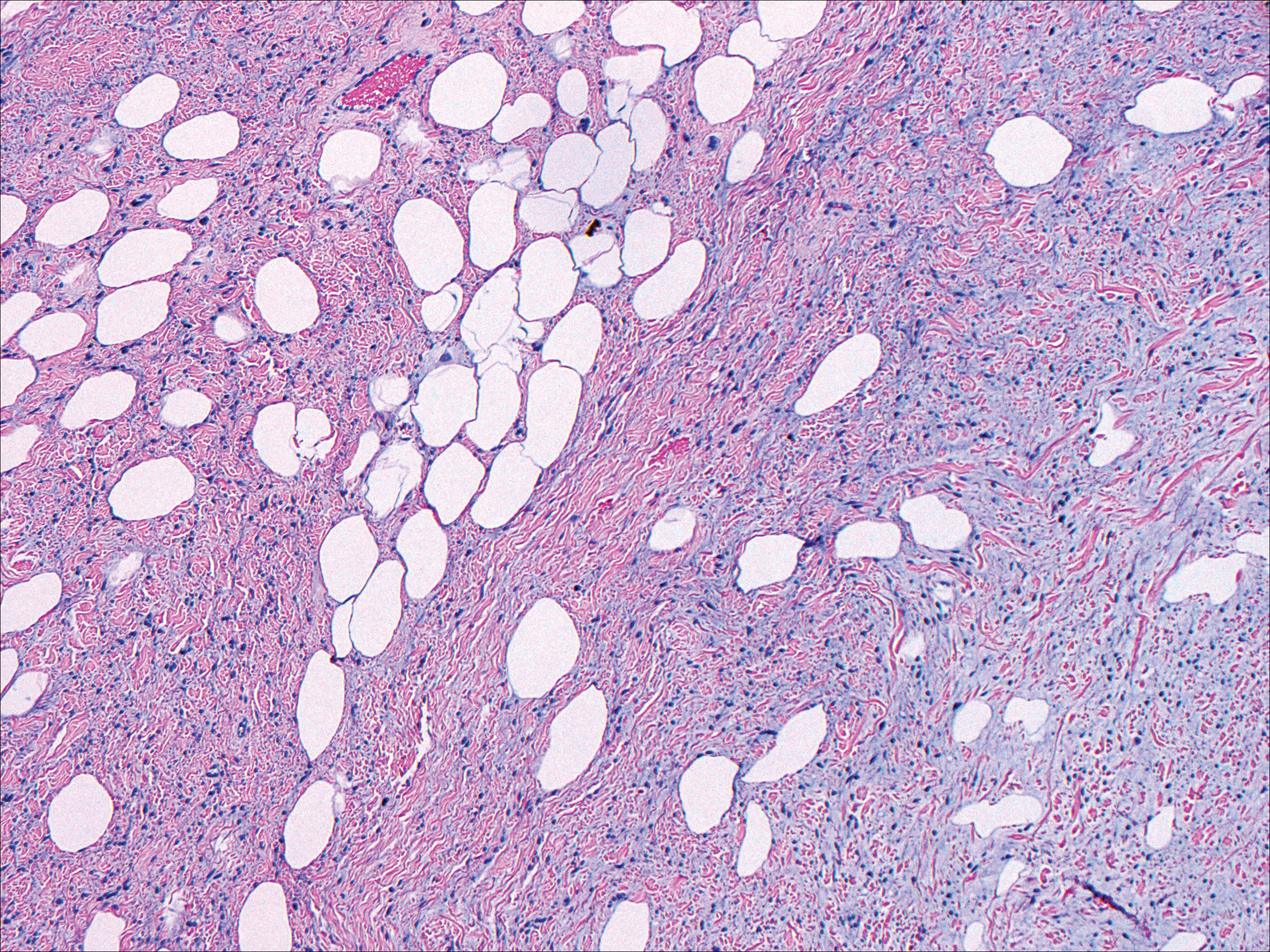
Nodular fasciitis typically presents as a rapidly growing subcutaneous nodule that may be tender. It is a benign reactive process usually affecting the arms and trunk of young to middle-aged adults, though it commonly involves the head and neck region in children.10 The tumor histopathologically appears as a well-circumscribed subcutaneous or fascial nodule with an angulated appearance. Spindle-shaped and stellate fibroblasts are loosely arranged in an edematous myxomatous stroma with a feathered appearance (Figure 5). Extravasated erythrocytes often are present. With time, collagen bundles become thicker and hyalinized. Immunohistochemical studies demonstrate positivity for vimentin, calponin, muscle-specific actin, and smooth muscle actin. Desmin, CD34, cytokeratin, and S-100 typically are negative.10-12 Therefore, CD34 staining is one of the main differentiating factors between nodular fasciitis and SFTs.
- Klemperer P, Rabin CB. Primary neoplasms of the pleura: a report of five cases. Arch Pathol. 1931;11:385-412.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
- Okamura JM, Barr RJ, Battifora H. Solitary fibrous tumor of the skin. Am J Dermatopathol. 1997;19:515-518.
- Lee JY, Park SE, Shin SJ, et al. Solitary fibrous tumor with myxoid stromal change. Am J Dermatopathol. 2015;37:570-573.
- Geramizadeh B, Marzban M, Churg A. Role of immunohistochemistry in the diagnosis of solitary fibrous tumor, a review. Iran J Pathol. 2016;11:195-293.
- Creytens D, Ferdinande L, Dorpe JV. Histopathologically malignant solitary fibrous tumor of the skin: a report of an unusual case. J Cutan Pathol. 2016;43:629-631.
- Vivero M, Doyle LA, Fletcher CD, et al. GRIA2 is a novel diagnostic marker for solitary fibrous tumour identified through gene expression profiling. Histopathology. 2014;65:71-80.
- Wood L, Fountaine TJ, Rosamilia L, et al. Cutaneous CD34 spindle cell neoplasms: histopathologic features distinguish spindle cell lipoma, solitary fibrous tumor, and dermatofibrosarcoma protuberans. Am J Dermatopathol. 2010;32:764-768.
- Khatib Y, Khade AL, Shah VB, et al. Cytohistological features of spindle cell lipoma--a case report with differential diagnosis. J Clin Diagn Res. 2017;11:10-11.
- Kumar E, Patel NR, Demicco EG, et al. Cutaneous nodular fasciitis with genetic analysis: a case series. J Cutan Pathol. 2016;43:1143-1149.
- Bracey TS, Wharton S, Smith ME. Nodular 'fasciitis' presenting as a cutaneous polyp. J Cutan Pathol. 2009;36:980-982.
- Perez-Montiel MD, Plaza JA, Dominguez-Malagon H, et al. Differential expression of smooth muscle myosin, smooth muscle actin, h-caldesmon, and calponin in the diagnosis of myofibroblastic and smooth muscle lesions of skin and soft tissue. Am J Dermatopathol. 2006;28:105-111.
The Diagnosis: Solitary Fibrous Tumor
Solitary fibrous tumors (SFTs), as first described by Klemperer and Rabin1 in 1931, are relatively uncommon mesenchymal neoplasms that occur primarily in the pleura. This lesion is now known to affect many other extrathoracic sites, such as the liver, kidney, adrenal glands, thyroid, central nervous system, and soft tissue, with rare examples originating from the skin.2 Okamura et al3 reported the first known case of cutaneous SFT in 1997, with most of the literature limited to case reports. Erdag et al2 described one of the largest case series of primary cutaneous SFTs. These lesions can occur across a wide age range but tend to primarily affect middle-aged adults. Solitary fibrous tumors have been known to have no sex predilection; however, Erdag et al2 found a male predominance with a male to female ratio of 4 to 1.
Histopathologically, a cutaneous SFT is known to appear as a well-circumscribed nodular spindle cell proliferation arranged in interlacing fascicles with an abundant hyalinized collagen stroma (quiz image). Alternating hypocellular and hypercellular areas can be seen. Supporting vasculature often is relatively prominent, represented by angulated and branching staghorn blood vessels (Figure 1).2 A common histopathologic finding of SFTs is a patternless pattern, which suggests that the tumor can have a variety of morphologic appearances (eg, storiform, fascicular, neural, herringbone growth patterns), making histologic diagnosis difficult (quiz image).4 Therefore, immunohistochemistry plays a large role in the diagnosis of this tumor. The most important positive markers include CD34, CD99, B-cell lymphoma 2 (BCL-2), and signal transducer and activator of transcription 6 (STAT6).5 Nuclear STAT6 staining is an immunomarker for NGFI-A binding protein 2 (NAB2)-STAT6 gene fusion, which is specific for SFT.5,6 Vivero et al7 also reported glutamate receptor, inotropic, AMPA 2 (GRIA2) as a useful immunostain in SFT, though it is also expressed in dermatofibrosarcoma protuberans (DFSP). In this case, the clinical and histopathologic findings best supported a diagnosis of SFT. Some consider hemangiopericytomas to be examples of SFTs; however, true hemangiopericytomas lack the thick hyalinized collagen and hypercellular areas seen in SFT.
A cellular dermatofibroma generally presents as a single round, reddish brown papule or nodule approximately 0.5 to 1 cm in diameter that is firm to palpation with a central depression or dimple created over the lesion from the lateral pressure. Cellular dermatofibromas mostly occur in middle-aged adults, with the most common locations on the legs and on the sides of the trunk. They are thought to arise after injuries to the skin. On histopathologic examination, cellular dermatofibromas typically exhibit a proliferation of fibrohistiocytic cells with collagen trapping, often at the periphery of the tumor (Figure 2). Although cellular dermatofibromas appear clinically different than SFTs, they often mimic SFTs histopathologically. Immunostaining also can be helpful in differentiating cellular dermatofibromas in which cells stain positive for factor XIIIa. CD34 staining is negative.
Dermatofibrosarcoma protuberans usually appears as one or multiple firm, red to violaceous nodules or plaques. They most often occur on the trunk in middle-aged adults. Histopathologically, DFSP presents with a dense, hypercellular, spindle cell proliferation that demonstrates a typical storiform pattern. The tumor generally infiltrates into the deep dermis and subcutaneous adipose layer with characteristic adipocyte entrapment (Figure 3). Positive CD34 and negative factor XIIIa staining helps to differentiate DFSP from a cellular dermatofibroma. Immunohistochemically, it is more difficult to distinguish DFSP from SFT, as both are CD34+ spindle cell neoplasms that also stain positive for CD99 and BCL-2.2 GRIA2 positivity also is seen in both SFT and DFSP.7 However, differentiation can be made on morphologic grounds alone, as DFSP has ill-defined tumor borders with adnexal and fat entrapment and SFT tends to be more circumscribed with prominent arborizing hyalinized vessels.8
Spindle cell lipoma (SCL) is an asymptomatic subcutaneous tumor commonly located on the back, neck, and shoulders in older patients, typically men. It often presents as a solitary lesion, though multiple lesions may occur. It is a well-circumscribed tumor of mature adipose tissue with areas of spindle cell proliferation and ropey collagen bundles (Figure 4). In early lesions, the spindle cell areas are myxoid with the presence of many mast cells.9 The spindle cells stain positive for CD34. Although spindle cell lipoma would be included in both the clinical and histopathologic differential diagnosis for SFT, its histopathologic features often are enough to differentiate SCL, which is highlighted by the aforementioned features as well as a relatively low cellularity and lack of ectatic vessels.8 However, discerning tumor variants, such as low-fat pseudoangiomatous SCL and lipomatous or myxoid SFT, might prove more challenging.
Nodular fasciitis typically presents as a rapidly growing subcutaneous nodule that may be tender. It is a benign reactive process usually affecting the arms and trunk of young to middle-aged adults, though it commonly involves the head and neck region in children.10 The tumor histopathologically appears as a well-circumscribed subcutaneous or fascial nodule with an angulated appearance. Spindle-shaped and stellate fibroblasts are loosely arranged in an edematous myxomatous stroma with a feathered appearance (Figure 5). Extravasated erythrocytes often are present. With time, collagen bundles become thicker and hyalinized. Immunohistochemical studies demonstrate positivity for vimentin, calponin, muscle-specific actin, and smooth muscle actin. Desmin, CD34, cytokeratin, and S-100 typically are negative.10-12 Therefore, CD34 staining is one of the main differentiating factors between nodular fasciitis and SFTs.
The Diagnosis: Solitary Fibrous Tumor
Solitary fibrous tumors (SFTs), as first described by Klemperer and Rabin1 in 1931, are relatively uncommon mesenchymal neoplasms that occur primarily in the pleura. This lesion is now known to affect many other extrathoracic sites, such as the liver, kidney, adrenal glands, thyroid, central nervous system, and soft tissue, with rare examples originating from the skin.2 Okamura et al3 reported the first known case of cutaneous SFT in 1997, with most of the literature limited to case reports. Erdag et al2 described one of the largest case series of primary cutaneous SFTs. These lesions can occur across a wide age range but tend to primarily affect middle-aged adults. Solitary fibrous tumors have been known to have no sex predilection; however, Erdag et al2 found a male predominance with a male to female ratio of 4 to 1.
Histopathologically, a cutaneous SFT is known to appear as a well-circumscribed nodular spindle cell proliferation arranged in interlacing fascicles with an abundant hyalinized collagen stroma (quiz image). Alternating hypocellular and hypercellular areas can be seen. Supporting vasculature often is relatively prominent, represented by angulated and branching staghorn blood vessels (Figure 1).2 A common histopathologic finding of SFTs is a patternless pattern, which suggests that the tumor can have a variety of morphologic appearances (eg, storiform, fascicular, neural, herringbone growth patterns), making histologic diagnosis difficult (quiz image).4 Therefore, immunohistochemistry plays a large role in the diagnosis of this tumor. The most important positive markers include CD34, CD99, B-cell lymphoma 2 (BCL-2), and signal transducer and activator of transcription 6 (STAT6).5 Nuclear STAT6 staining is an immunomarker for NGFI-A binding protein 2 (NAB2)-STAT6 gene fusion, which is specific for SFT.5,6 Vivero et al7 also reported glutamate receptor, inotropic, AMPA 2 (GRIA2) as a useful immunostain in SFT, though it is also expressed in dermatofibrosarcoma protuberans (DFSP). In this case, the clinical and histopathologic findings best supported a diagnosis of SFT. Some consider hemangiopericytomas to be examples of SFTs; however, true hemangiopericytomas lack the thick hyalinized collagen and hypercellular areas seen in SFT.
A cellular dermatofibroma generally presents as a single round, reddish brown papule or nodule approximately 0.5 to 1 cm in diameter that is firm to palpation with a central depression or dimple created over the lesion from the lateral pressure. Cellular dermatofibromas mostly occur in middle-aged adults, with the most common locations on the legs and on the sides of the trunk. They are thought to arise after injuries to the skin. On histopathologic examination, cellular dermatofibromas typically exhibit a proliferation of fibrohistiocytic cells with collagen trapping, often at the periphery of the tumor (Figure 2). Although cellular dermatofibromas appear clinically different than SFTs, they often mimic SFTs histopathologically. Immunostaining also can be helpful in differentiating cellular dermatofibromas in which cells stain positive for factor XIIIa. CD34 staining is negative.
Dermatofibrosarcoma protuberans usually appears as one or multiple firm, red to violaceous nodules or plaques. They most often occur on the trunk in middle-aged adults. Histopathologically, DFSP presents with a dense, hypercellular, spindle cell proliferation that demonstrates a typical storiform pattern. The tumor generally infiltrates into the deep dermis and subcutaneous adipose layer with characteristic adipocyte entrapment (Figure 3). Positive CD34 and negative factor XIIIa staining helps to differentiate DFSP from a cellular dermatofibroma. Immunohistochemically, it is more difficult to distinguish DFSP from SFT, as both are CD34+ spindle cell neoplasms that also stain positive for CD99 and BCL-2.2 GRIA2 positivity also is seen in both SFT and DFSP.7 However, differentiation can be made on morphologic grounds alone, as DFSP has ill-defined tumor borders with adnexal and fat entrapment and SFT tends to be more circumscribed with prominent arborizing hyalinized vessels.8
Spindle cell lipoma (SCL) is an asymptomatic subcutaneous tumor commonly located on the back, neck, and shoulders in older patients, typically men. It often presents as a solitary lesion, though multiple lesions may occur. It is a well-circumscribed tumor of mature adipose tissue with areas of spindle cell proliferation and ropey collagen bundles (Figure 4). In early lesions, the spindle cell areas are myxoid with the presence of many mast cells.9 The spindle cells stain positive for CD34. Although spindle cell lipoma would be included in both the clinical and histopathologic differential diagnosis for SFT, its histopathologic features often are enough to differentiate SCL, which is highlighted by the aforementioned features as well as a relatively low cellularity and lack of ectatic vessels.8 However, discerning tumor variants, such as low-fat pseudoangiomatous SCL and lipomatous or myxoid SFT, might prove more challenging.
Nodular fasciitis typically presents as a rapidly growing subcutaneous nodule that may be tender. It is a benign reactive process usually affecting the arms and trunk of young to middle-aged adults, though it commonly involves the head and neck region in children.10 The tumor histopathologically appears as a well-circumscribed subcutaneous or fascial nodule with an angulated appearance. Spindle-shaped and stellate fibroblasts are loosely arranged in an edematous myxomatous stroma with a feathered appearance (Figure 5). Extravasated erythrocytes often are present. With time, collagen bundles become thicker and hyalinized. Immunohistochemical studies demonstrate positivity for vimentin, calponin, muscle-specific actin, and smooth muscle actin. Desmin, CD34, cytokeratin, and S-100 typically are negative.10-12 Therefore, CD34 staining is one of the main differentiating factors between nodular fasciitis and SFTs.
- Klemperer P, Rabin CB. Primary neoplasms of the pleura: a report of five cases. Arch Pathol. 1931;11:385-412.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
- Okamura JM, Barr RJ, Battifora H. Solitary fibrous tumor of the skin. Am J Dermatopathol. 1997;19:515-518.
- Lee JY, Park SE, Shin SJ, et al. Solitary fibrous tumor with myxoid stromal change. Am J Dermatopathol. 2015;37:570-573.
- Geramizadeh B, Marzban M, Churg A. Role of immunohistochemistry in the diagnosis of solitary fibrous tumor, a review. Iran J Pathol. 2016;11:195-293.
- Creytens D, Ferdinande L, Dorpe JV. Histopathologically malignant solitary fibrous tumor of the skin: a report of an unusual case. J Cutan Pathol. 2016;43:629-631.
- Vivero M, Doyle LA, Fletcher CD, et al. GRIA2 is a novel diagnostic marker for solitary fibrous tumour identified through gene expression profiling. Histopathology. 2014;65:71-80.
- Wood L, Fountaine TJ, Rosamilia L, et al. Cutaneous CD34 spindle cell neoplasms: histopathologic features distinguish spindle cell lipoma, solitary fibrous tumor, and dermatofibrosarcoma protuberans. Am J Dermatopathol. 2010;32:764-768.
- Khatib Y, Khade AL, Shah VB, et al. Cytohistological features of spindle cell lipoma--a case report with differential diagnosis. J Clin Diagn Res. 2017;11:10-11.
- Kumar E, Patel NR, Demicco EG, et al. Cutaneous nodular fasciitis with genetic analysis: a case series. J Cutan Pathol. 2016;43:1143-1149.
- Bracey TS, Wharton S, Smith ME. Nodular 'fasciitis' presenting as a cutaneous polyp. J Cutan Pathol. 2009;36:980-982.
- Perez-Montiel MD, Plaza JA, Dominguez-Malagon H, et al. Differential expression of smooth muscle myosin, smooth muscle actin, h-caldesmon, and calponin in the diagnosis of myofibroblastic and smooth muscle lesions of skin and soft tissue. Am J Dermatopathol. 2006;28:105-111.
- Klemperer P, Rabin CB. Primary neoplasms of the pleura: a report of five cases. Arch Pathol. 1931;11:385-412.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
- Okamura JM, Barr RJ, Battifora H. Solitary fibrous tumor of the skin. Am J Dermatopathol. 1997;19:515-518.
- Lee JY, Park SE, Shin SJ, et al. Solitary fibrous tumor with myxoid stromal change. Am J Dermatopathol. 2015;37:570-573.
- Geramizadeh B, Marzban M, Churg A. Role of immunohistochemistry in the diagnosis of solitary fibrous tumor, a review. Iran J Pathol. 2016;11:195-293.
- Creytens D, Ferdinande L, Dorpe JV. Histopathologically malignant solitary fibrous tumor of the skin: a report of an unusual case. J Cutan Pathol. 2016;43:629-631.
- Vivero M, Doyle LA, Fletcher CD, et al. GRIA2 is a novel diagnostic marker for solitary fibrous tumour identified through gene expression profiling. Histopathology. 2014;65:71-80.
- Wood L, Fountaine TJ, Rosamilia L, et al. Cutaneous CD34 spindle cell neoplasms: histopathologic features distinguish spindle cell lipoma, solitary fibrous tumor, and dermatofibrosarcoma protuberans. Am J Dermatopathol. 2010;32:764-768.
- Khatib Y, Khade AL, Shah VB, et al. Cytohistological features of spindle cell lipoma--a case report with differential diagnosis. J Clin Diagn Res. 2017;11:10-11.
- Kumar E, Patel NR, Demicco EG, et al. Cutaneous nodular fasciitis with genetic analysis: a case series. J Cutan Pathol. 2016;43:1143-1149.
- Bracey TS, Wharton S, Smith ME. Nodular 'fasciitis' presenting as a cutaneous polyp. J Cutan Pathol. 2009;36:980-982.
- Perez-Montiel MD, Plaza JA, Dominguez-Malagon H, et al. Differential expression of smooth muscle myosin, smooth muscle actin, h-caldesmon, and calponin in the diagnosis of myofibroblastic and smooth muscle lesions of skin and soft tissue. Am J Dermatopathol. 2006;28:105-111.
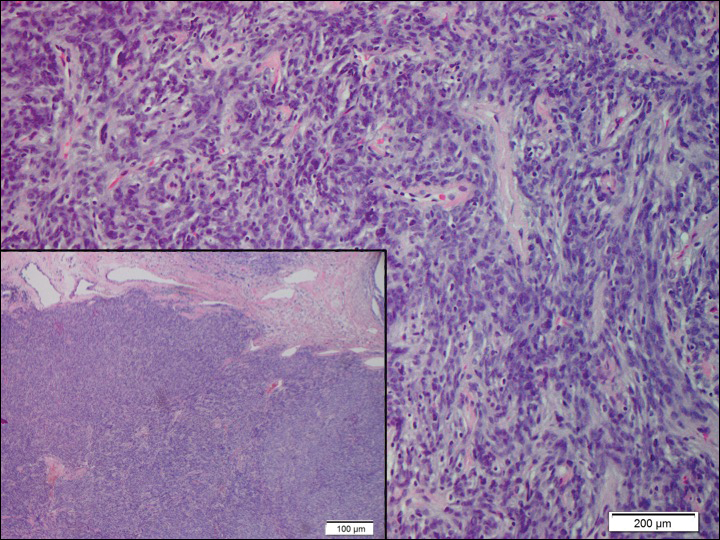
A 73-year-old man presented with a tender nodule on the back that had recently increased in size. On physical examination, a solitary 4-cm nodule was noted in the right trapezius region. The patient denied any personal or family history of similar lesions or a penchant for cysts. Due to the symptomatic nature of the lesion, surgical excision was performed.
ACOG: VBAC is safe for many women
Women and their , according to an updated practice bulletin from the American College of Obstetricians and Gynecologists.
Trial of labor after cesarean delivery (TOLAC) results in a successful birth in 60%-80% of cases, sparing mothers from major abdominal surgery and reducing the risk of hemorrhage, thromboses, and infection, the authors of the practice bulletin wrote. “The preponderance of evidence suggests that most women with one previous cesarean delivery with a low-transverse incision are candidates for and should be counseled about and offered TOLAC,” they said (Obstet Gynecol. 2017 Nov;130[5]:e217-33. doi: 10.1097/AOG.0000000000002398).
Rates of cesarean delivery in the United States jumped from 5% to nearly 32% between 1970 and 2016. Although rates of VBAC rose between the mid-1980s and the mid-1990s, cases of uterine rupture and other complications spurred fears of malpractice litigation and reversed this trend. VBAC rates were more than 28% in 1996 but fell to 8.5% by 2006, according to the practice bulletin.
To reduce the risk of uterine rupture, avoid misoprostol for cervical ripening and labor induction in women with a prior cesarean delivery, ACOG recommended.
“No evidence suggests that epidural analgesia is a causal risk factor for unsuccessful TOLAC,” the authors added. “Therefore, epidural analgesia for labor may be used as part of TOLAC, and adequate pain relief may encourage more women to choose TOLAC.”
Women with two prior low-transverse cesareans also are potential candidates for TOLAC, depending on other predictors of successful VBAC. Factors that reduce the chances of a successful TOLAC include advanced maternal age, high body mass index, high birth weight, gestational age of more than 40 weeks at delivery, and preeclampsia at the time of delivery, according to the practice bulletin.
To reduce the risk of adverse outcomes of complications, TOLAC should not occur at home and should only occur at level I facilities (or higher) that can perform an emergency cesarean delivery if the mother or fetus is in jeopardy.
The practice bulletin recommends continuous fetal heart rate monitoring during TOLAC and notes several additional categories of TOLAC candidates. Obstetricians and patients should discuss the potential risks and benefits of both TOLAC and elective repeat cesarean delivery, and that discussion should be documented in the medical record, ACOG recommended.
Women and their , according to an updated practice bulletin from the American College of Obstetricians and Gynecologists.
Trial of labor after cesarean delivery (TOLAC) results in a successful birth in 60%-80% of cases, sparing mothers from major abdominal surgery and reducing the risk of hemorrhage, thromboses, and infection, the authors of the practice bulletin wrote. “The preponderance of evidence suggests that most women with one previous cesarean delivery with a low-transverse incision are candidates for and should be counseled about and offered TOLAC,” they said (Obstet Gynecol. 2017 Nov;130[5]:e217-33. doi: 10.1097/AOG.0000000000002398).
Rates of cesarean delivery in the United States jumped from 5% to nearly 32% between 1970 and 2016. Although rates of VBAC rose between the mid-1980s and the mid-1990s, cases of uterine rupture and other complications spurred fears of malpractice litigation and reversed this trend. VBAC rates were more than 28% in 1996 but fell to 8.5% by 2006, according to the practice bulletin.
To reduce the risk of uterine rupture, avoid misoprostol for cervical ripening and labor induction in women with a prior cesarean delivery, ACOG recommended.
“No evidence suggests that epidural analgesia is a causal risk factor for unsuccessful TOLAC,” the authors added. “Therefore, epidural analgesia for labor may be used as part of TOLAC, and adequate pain relief may encourage more women to choose TOLAC.”
Women with two prior low-transverse cesareans also are potential candidates for TOLAC, depending on other predictors of successful VBAC. Factors that reduce the chances of a successful TOLAC include advanced maternal age, high body mass index, high birth weight, gestational age of more than 40 weeks at delivery, and preeclampsia at the time of delivery, according to the practice bulletin.
To reduce the risk of adverse outcomes of complications, TOLAC should not occur at home and should only occur at level I facilities (or higher) that can perform an emergency cesarean delivery if the mother or fetus is in jeopardy.
The practice bulletin recommends continuous fetal heart rate monitoring during TOLAC and notes several additional categories of TOLAC candidates. Obstetricians and patients should discuss the potential risks and benefits of both TOLAC and elective repeat cesarean delivery, and that discussion should be documented in the medical record, ACOG recommended.
Women and their , according to an updated practice bulletin from the American College of Obstetricians and Gynecologists.
Trial of labor after cesarean delivery (TOLAC) results in a successful birth in 60%-80% of cases, sparing mothers from major abdominal surgery and reducing the risk of hemorrhage, thromboses, and infection, the authors of the practice bulletin wrote. “The preponderance of evidence suggests that most women with one previous cesarean delivery with a low-transverse incision are candidates for and should be counseled about and offered TOLAC,” they said (Obstet Gynecol. 2017 Nov;130[5]:e217-33. doi: 10.1097/AOG.0000000000002398).
Rates of cesarean delivery in the United States jumped from 5% to nearly 32% between 1970 and 2016. Although rates of VBAC rose between the mid-1980s and the mid-1990s, cases of uterine rupture and other complications spurred fears of malpractice litigation and reversed this trend. VBAC rates were more than 28% in 1996 but fell to 8.5% by 2006, according to the practice bulletin.
To reduce the risk of uterine rupture, avoid misoprostol for cervical ripening and labor induction in women with a prior cesarean delivery, ACOG recommended.
“No evidence suggests that epidural analgesia is a causal risk factor for unsuccessful TOLAC,” the authors added. “Therefore, epidural analgesia for labor may be used as part of TOLAC, and adequate pain relief may encourage more women to choose TOLAC.”
Women with two prior low-transverse cesareans also are potential candidates for TOLAC, depending on other predictors of successful VBAC. Factors that reduce the chances of a successful TOLAC include advanced maternal age, high body mass index, high birth weight, gestational age of more than 40 weeks at delivery, and preeclampsia at the time of delivery, according to the practice bulletin.
To reduce the risk of adverse outcomes of complications, TOLAC should not occur at home and should only occur at level I facilities (or higher) that can perform an emergency cesarean delivery if the mother or fetus is in jeopardy.
The practice bulletin recommends continuous fetal heart rate monitoring during TOLAC and notes several additional categories of TOLAC candidates. Obstetricians and patients should discuss the potential risks and benefits of both TOLAC and elective repeat cesarean delivery, and that discussion should be documented in the medical record, ACOG recommended.
FROM OBSTETRICS & GYNECOLOGY
Black Eschars on the Face and Body
The Diagnosis: Lymphomatoid Papulosis
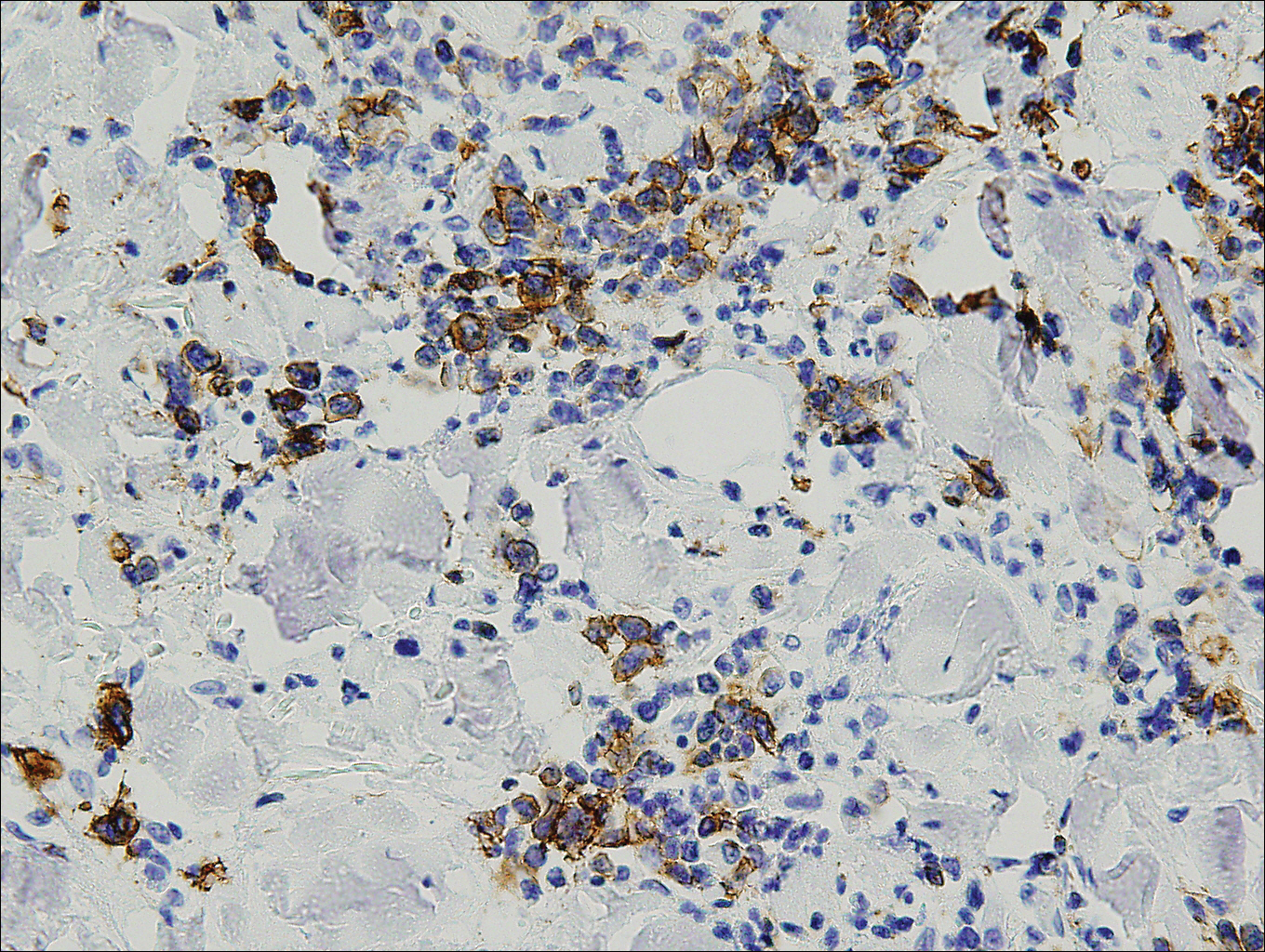
Histopathologic and immunohistochemical examination of the ulcer revealed a dense nodular and diffuse infiltrate in the papillary and reticular dermis comprised predominantly of atypical, CD30+, small T cells and large lymphoid cells admixed with neutrophils and eosinophils (Figures 1 and 2). Tissue cultures and infectious stains were negative. The complete blood cell count, metabolic panel, serum lactate dehydrogenase level, and peripheral blood flow cytometry were normal. Correlation of the lesions' self-healing nature with the histopathologic and immunohistochemical findings led to a diagnosis of lymphomatoid papulosis (LyP). In light of this diagnosis, a shave biopsy was obtained of one of the patient's poikilodermatous patches and was found to be consistent with poikilodermatous mycosis fungoides (MF).
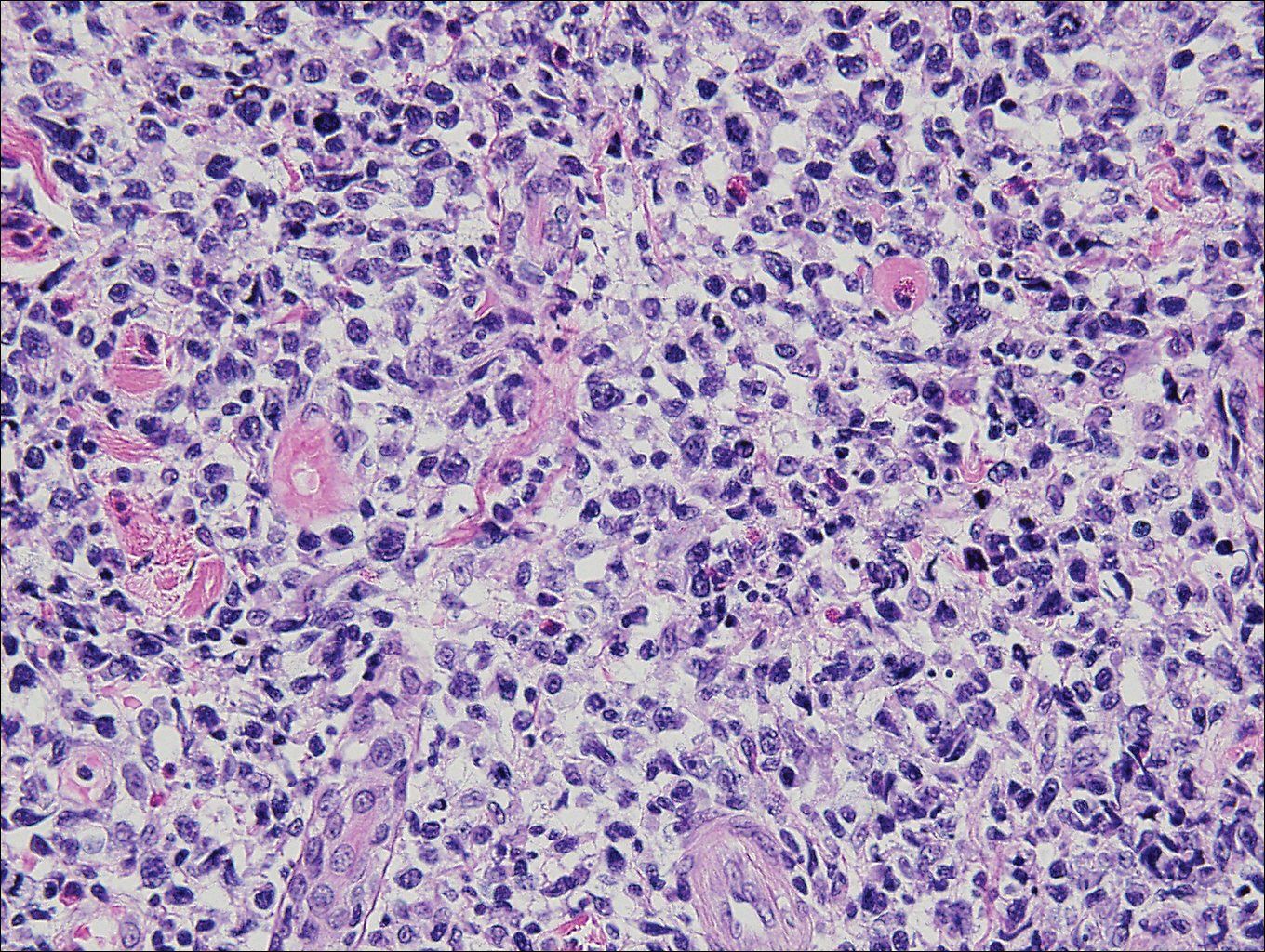
At 4-month follow-up, the patient reported that she continued to develop crops of 1 to 3 LyP lesions each month. She continued to deny systemic concerns, and the poikilodermatous MF appeared unchanged. As part of a hematologic workup, a positron emission tomography-computed tomography scan revealed glucose-avid lymph nodes in the axillary, supraclavicular, abdominal, and inguinal regions. These findings raised concern for possible lymphomatous involvement of the patient's MF. Systemic therapy may be required pending further surveillance.
Lymphomatoid papulosis is a chronic papulonecrotic disease characterized clinically by recurrent crops of self-healing papules. Histopathologically, LyP features a perivascular infiltrate with atypical dermal T cells. Macaulay1 first described LyP in 1968 in a 41-year-old woman with a several-year history of continuously self-resolving crops of necrotic papules, noting the paradox between the patient's benign clinical course and malignant-seeming histology featuring "an alarming infiltrate of anaplastic cells." Since this report, LyP has continued to spur debate regarding its malignant potential but is now recognized as an indolent cutaneous T-cell lymphoma with an excellent prognosis.2
There are several histopathologic subtypes of LyP, the most common of which are type A, resembling Hodgkin lymphoma; type B, resembling MF; type C, resembling primary cutaneous anaplastic large cell lymphoma (C-ALCL); and type D, resembling aggressive epidermotropic CD8+ cutaneous T-cell lymphoma.2
The multifocal ulcers and eschars of LyP may appropriately raise suspicion for an infectious process, as in the present case. Numerous reports show that LyP may be initially misdiagnosed as an infection, such as cellulitis,3 furunculosis,4 parapoxvirus Orf,5 and ecthyma.6 Furthermore, several cutaneous infections have histopathologic features indistinguishable from LyP.7 For example, herpes simplex virus infection, molluscum contagiosum, Milker nodule, syphilis, and leishmaniasis may contain an appreciable number of large CD30+ T cells, which is compatible with both LyP type C and C-ALCL.7 As in the present case, the final diagnosis rests on clinicopathologic correlation, with LyP often distinguished by its invariable self-resolution, unlike its numerous infectious mimickers. The self-regressing nature of LyP also helps differentiate LyP occurring in the setting of MF from MF that has underwent CD30+ large cell transformation. In addition, the diagnosis of MF-associated LyP is favored over transformed MF when, as in the present case, CD30+ lesions develop on skin distinct from MF-affected skin.
Although isolated LyP is benign, 18% (11/61) of patients will subsequently develop lymphoma. More commonly, lymphomas may precede or occur concomitantly with the onset of LyP. In a retrospective study of 84 LyP patients, for example, 40% (34/84) had prior or concomitant lymphoma.8 Owing to the well-established link between LyP and lymphoma, there is appropriate emphasis on close monitoring of these patients. In addition, a careful history and physical examination are necessary to evaluate for a preceding, previously undiagnosed lymphoma. In point of fact, our patient had undiagnosed poikilodermatous MF prior to developing LyP, which was proven by biopsy at the time of LyP diagnosis. A distinct clinical variant of MF, poikilodermatous MF is characterized by hyperpigmented and hypopigmented patches, atrophy, and telangiectasia. A study of 49 patients with poikilodermatous MF found that this variant had an earlier age of onset compared with other types of MF. The study also showed that 18% (9/49) of patients had coexistent LyP, suggesting that poikilodermatous MF and LyP may be more frequently associated than previously believed.9
Treatment of LyP is unnecessary beyond basic wound care to avoid bacterial superinfection.2,10 Therapy for poikilodermatous MF, similar to other types of MF, is based on disease stage. Topical therapy may be utilized for localized disease, while systemic therapies are reserved for recalcitrant cases and internal involvement.9
Acknowledgments
We thank David L. Ramsay, MD, for obtaining aspects of the patient's history, and Shane A. Meehan, MD, and Adnan Mir, MD, PhD, as well as Cynthia M. Magro, MD, (all from New York, New York) for performing the histopathologic and immunohistochemical analyses.
- Macaulay WL. Lymphomatoid papulosis. a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Meena M, Martin PA, Abouseif C, et al. Lymphomatoid papulosis type C of the eyelid in a young girl: a case report and review of literature. Orbit. 2014;3:395-398.
- Dinotta F, Lacarrubba F, Micali G. Sixteen-year-old girl with papules and nodules on the face and upper limbs. Pediatr Dermatol. 2014;31:103-104.
- Eminger LA, Shinohara MM, Kim EJ, et al. Clinicopathologic challenge: acral lymphomatoid papulosis. Int J Dermatol. 2012;51:531-534.
- Harder D, Kuhn A, Mahrle G. Lymphomatoid papulosis resembling ecthyma. a case report. Z Hautkr. 1989;64:593-595.
- Werner B, Massone C, Kerl H, et al. Large CD30-positive cells in benign, atypical lymphoid infiltrates of the skin. J Cutan Pathol. 2008;35:1100-1107.
- Kunishige JH, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-581.
- Abbott RA, Sahni D, Robson A, et al. Poikilodermatous mycosis fungoides: a study of its clinicopathological, immunophenotypic, and prognostic features. J Am Acad Dermatol. 2011;65:313-319.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
The Diagnosis: Lymphomatoid Papulosis
Histopathologic and immunohistochemical examination of the ulcer revealed a dense nodular and diffuse infiltrate in the papillary and reticular dermis comprised predominantly of atypical, CD30+, small T cells and large lymphoid cells admixed with neutrophils and eosinophils (Figures 1 and 2). Tissue cultures and infectious stains were negative. The complete blood cell count, metabolic panel, serum lactate dehydrogenase level, and peripheral blood flow cytometry were normal. Correlation of the lesions' self-healing nature with the histopathologic and immunohistochemical findings led to a diagnosis of lymphomatoid papulosis (LyP). In light of this diagnosis, a shave biopsy was obtained of one of the patient's poikilodermatous patches and was found to be consistent with poikilodermatous mycosis fungoides (MF).
At 4-month follow-up, the patient reported that she continued to develop crops of 1 to 3 LyP lesions each month. She continued to deny systemic concerns, and the poikilodermatous MF appeared unchanged. As part of a hematologic workup, a positron emission tomography-computed tomography scan revealed glucose-avid lymph nodes in the axillary, supraclavicular, abdominal, and inguinal regions. These findings raised concern for possible lymphomatous involvement of the patient's MF. Systemic therapy may be required pending further surveillance.
Lymphomatoid papulosis is a chronic papulonecrotic disease characterized clinically by recurrent crops of self-healing papules. Histopathologically, LyP features a perivascular infiltrate with atypical dermal T cells. Macaulay1 first described LyP in 1968 in a 41-year-old woman with a several-year history of continuously self-resolving crops of necrotic papules, noting the paradox between the patient's benign clinical course and malignant-seeming histology featuring "an alarming infiltrate of anaplastic cells." Since this report, LyP has continued to spur debate regarding its malignant potential but is now recognized as an indolent cutaneous T-cell lymphoma with an excellent prognosis.2
There are several histopathologic subtypes of LyP, the most common of which are type A, resembling Hodgkin lymphoma; type B, resembling MF; type C, resembling primary cutaneous anaplastic large cell lymphoma (C-ALCL); and type D, resembling aggressive epidermotropic CD8+ cutaneous T-cell lymphoma.2
The multifocal ulcers and eschars of LyP may appropriately raise suspicion for an infectious process, as in the present case. Numerous reports show that LyP may be initially misdiagnosed as an infection, such as cellulitis,3 furunculosis,4 parapoxvirus Orf,5 and ecthyma.6 Furthermore, several cutaneous infections have histopathologic features indistinguishable from LyP.7 For example, herpes simplex virus infection, molluscum contagiosum, Milker nodule, syphilis, and leishmaniasis may contain an appreciable number of large CD30+ T cells, which is compatible with both LyP type C and C-ALCL.7 As in the present case, the final diagnosis rests on clinicopathologic correlation, with LyP often distinguished by its invariable self-resolution, unlike its numerous infectious mimickers. The self-regressing nature of LyP also helps differentiate LyP occurring in the setting of MF from MF that has underwent CD30+ large cell transformation. In addition, the diagnosis of MF-associated LyP is favored over transformed MF when, as in the present case, CD30+ lesions develop on skin distinct from MF-affected skin.
Although isolated LyP is benign, 18% (11/61) of patients will subsequently develop lymphoma. More commonly, lymphomas may precede or occur concomitantly with the onset of LyP. In a retrospective study of 84 LyP patients, for example, 40% (34/84) had prior or concomitant lymphoma.8 Owing to the well-established link between LyP and lymphoma, there is appropriate emphasis on close monitoring of these patients. In addition, a careful history and physical examination are necessary to evaluate for a preceding, previously undiagnosed lymphoma. In point of fact, our patient had undiagnosed poikilodermatous MF prior to developing LyP, which was proven by biopsy at the time of LyP diagnosis. A distinct clinical variant of MF, poikilodermatous MF is characterized by hyperpigmented and hypopigmented patches, atrophy, and telangiectasia. A study of 49 patients with poikilodermatous MF found that this variant had an earlier age of onset compared with other types of MF. The study also showed that 18% (9/49) of patients had coexistent LyP, suggesting that poikilodermatous MF and LyP may be more frequently associated than previously believed.9
Treatment of LyP is unnecessary beyond basic wound care to avoid bacterial superinfection.2,10 Therapy for poikilodermatous MF, similar to other types of MF, is based on disease stage. Topical therapy may be utilized for localized disease, while systemic therapies are reserved for recalcitrant cases and internal involvement.9
Acknowledgments
We thank David L. Ramsay, MD, for obtaining aspects of the patient's history, and Shane A. Meehan, MD, and Adnan Mir, MD, PhD, as well as Cynthia M. Magro, MD, (all from New York, New York) for performing the histopathologic and immunohistochemical analyses.
The Diagnosis: Lymphomatoid Papulosis
Histopathologic and immunohistochemical examination of the ulcer revealed a dense nodular and diffuse infiltrate in the papillary and reticular dermis comprised predominantly of atypical, CD30+, small T cells and large lymphoid cells admixed with neutrophils and eosinophils (Figures 1 and 2). Tissue cultures and infectious stains were negative. The complete blood cell count, metabolic panel, serum lactate dehydrogenase level, and peripheral blood flow cytometry were normal. Correlation of the lesions' self-healing nature with the histopathologic and immunohistochemical findings led to a diagnosis of lymphomatoid papulosis (LyP). In light of this diagnosis, a shave biopsy was obtained of one of the patient's poikilodermatous patches and was found to be consistent with poikilodermatous mycosis fungoides (MF).
At 4-month follow-up, the patient reported that she continued to develop crops of 1 to 3 LyP lesions each month. She continued to deny systemic concerns, and the poikilodermatous MF appeared unchanged. As part of a hematologic workup, a positron emission tomography-computed tomography scan revealed glucose-avid lymph nodes in the axillary, supraclavicular, abdominal, and inguinal regions. These findings raised concern for possible lymphomatous involvement of the patient's MF. Systemic therapy may be required pending further surveillance.
Lymphomatoid papulosis is a chronic papulonecrotic disease characterized clinically by recurrent crops of self-healing papules. Histopathologically, LyP features a perivascular infiltrate with atypical dermal T cells. Macaulay1 first described LyP in 1968 in a 41-year-old woman with a several-year history of continuously self-resolving crops of necrotic papules, noting the paradox between the patient's benign clinical course and malignant-seeming histology featuring "an alarming infiltrate of anaplastic cells." Since this report, LyP has continued to spur debate regarding its malignant potential but is now recognized as an indolent cutaneous T-cell lymphoma with an excellent prognosis.2
There are several histopathologic subtypes of LyP, the most common of which are type A, resembling Hodgkin lymphoma; type B, resembling MF; type C, resembling primary cutaneous anaplastic large cell lymphoma (C-ALCL); and type D, resembling aggressive epidermotropic CD8+ cutaneous T-cell lymphoma.2
The multifocal ulcers and eschars of LyP may appropriately raise suspicion for an infectious process, as in the present case. Numerous reports show that LyP may be initially misdiagnosed as an infection, such as cellulitis,3 furunculosis,4 parapoxvirus Orf,5 and ecthyma.6 Furthermore, several cutaneous infections have histopathologic features indistinguishable from LyP.7 For example, herpes simplex virus infection, molluscum contagiosum, Milker nodule, syphilis, and leishmaniasis may contain an appreciable number of large CD30+ T cells, which is compatible with both LyP type C and C-ALCL.7 As in the present case, the final diagnosis rests on clinicopathologic correlation, with LyP often distinguished by its invariable self-resolution, unlike its numerous infectious mimickers. The self-regressing nature of LyP also helps differentiate LyP occurring in the setting of MF from MF that has underwent CD30+ large cell transformation. In addition, the diagnosis of MF-associated LyP is favored over transformed MF when, as in the present case, CD30+ lesions develop on skin distinct from MF-affected skin.
Although isolated LyP is benign, 18% (11/61) of patients will subsequently develop lymphoma. More commonly, lymphomas may precede or occur concomitantly with the onset of LyP. In a retrospective study of 84 LyP patients, for example, 40% (34/84) had prior or concomitant lymphoma.8 Owing to the well-established link between LyP and lymphoma, there is appropriate emphasis on close monitoring of these patients. In addition, a careful history and physical examination are necessary to evaluate for a preceding, previously undiagnosed lymphoma. In point of fact, our patient had undiagnosed poikilodermatous MF prior to developing LyP, which was proven by biopsy at the time of LyP diagnosis. A distinct clinical variant of MF, poikilodermatous MF is characterized by hyperpigmented and hypopigmented patches, atrophy, and telangiectasia. A study of 49 patients with poikilodermatous MF found that this variant had an earlier age of onset compared with other types of MF. The study also showed that 18% (9/49) of patients had coexistent LyP, suggesting that poikilodermatous MF and LyP may be more frequently associated than previously believed.9
Treatment of LyP is unnecessary beyond basic wound care to avoid bacterial superinfection.2,10 Therapy for poikilodermatous MF, similar to other types of MF, is based on disease stage. Topical therapy may be utilized for localized disease, while systemic therapies are reserved for recalcitrant cases and internal involvement.9
Acknowledgments
We thank David L. Ramsay, MD, for obtaining aspects of the patient's history, and Shane A. Meehan, MD, and Adnan Mir, MD, PhD, as well as Cynthia M. Magro, MD, (all from New York, New York) for performing the histopathologic and immunohistochemical analyses.
- Macaulay WL. Lymphomatoid papulosis. a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Meena M, Martin PA, Abouseif C, et al. Lymphomatoid papulosis type C of the eyelid in a young girl: a case report and review of literature. Orbit. 2014;3:395-398.
- Dinotta F, Lacarrubba F, Micali G. Sixteen-year-old girl with papules and nodules on the face and upper limbs. Pediatr Dermatol. 2014;31:103-104.
- Eminger LA, Shinohara MM, Kim EJ, et al. Clinicopathologic challenge: acral lymphomatoid papulosis. Int J Dermatol. 2012;51:531-534.
- Harder D, Kuhn A, Mahrle G. Lymphomatoid papulosis resembling ecthyma. a case report. Z Hautkr. 1989;64:593-595.
- Werner B, Massone C, Kerl H, et al. Large CD30-positive cells in benign, atypical lymphoid infiltrates of the skin. J Cutan Pathol. 2008;35:1100-1107.
- Kunishige JH, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-581.
- Abbott RA, Sahni D, Robson A, et al. Poikilodermatous mycosis fungoides: a study of its clinicopathological, immunophenotypic, and prognostic features. J Am Acad Dermatol. 2011;65:313-319.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
- Macaulay WL. Lymphomatoid papulosis. a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Meena M, Martin PA, Abouseif C, et al. Lymphomatoid papulosis type C of the eyelid in a young girl: a case report and review of literature. Orbit. 2014;3:395-398.
- Dinotta F, Lacarrubba F, Micali G. Sixteen-year-old girl with papules and nodules on the face and upper limbs. Pediatr Dermatol. 2014;31:103-104.
- Eminger LA, Shinohara MM, Kim EJ, et al. Clinicopathologic challenge: acral lymphomatoid papulosis. Int J Dermatol. 2012;51:531-534.
- Harder D, Kuhn A, Mahrle G. Lymphomatoid papulosis resembling ecthyma. a case report. Z Hautkr. 1989;64:593-595.
- Werner B, Massone C, Kerl H, et al. Large CD30-positive cells in benign, atypical lymphoid infiltrates of the skin. J Cutan Pathol. 2008;35:1100-1107.
- Kunishige JH, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-581.
- Abbott RA, Sahni D, Robson A, et al. Poikilodermatous mycosis fungoides: a study of its clinicopathological, immunophenotypic, and prognostic features. J Am Acad Dermatol. 2011;65:313-319.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
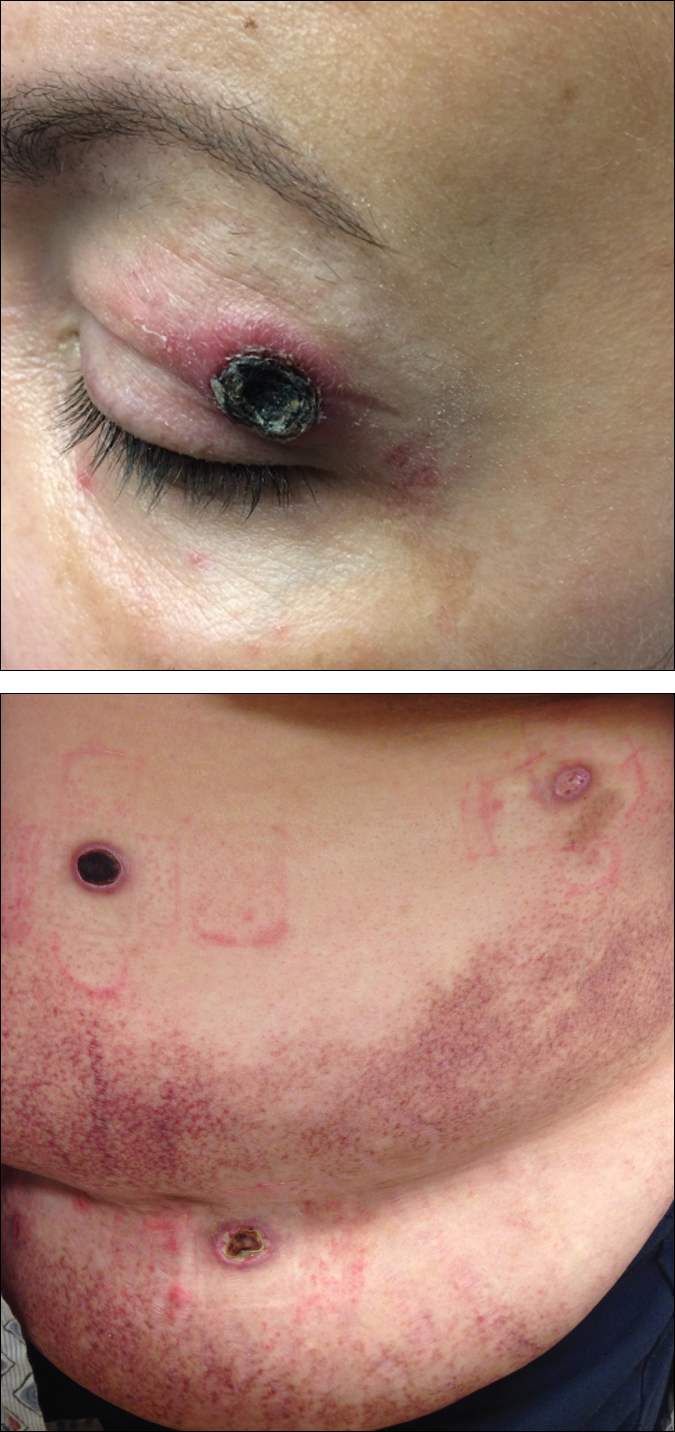
A 50-year-old woman presented for evaluation of black eschars on the face and body. Over the preceding 8 weeks she had developed several asymptomatic papules that gradually enlarged, ulcerated, and formed a black eschar, prior to gradually self-resolving over the course of several weeks. During this time, new lesions were forming. The resulting skin revealed dyspigmentation and scar formation. Prior to presentation, antimicrobial therapy had been initiated for a presumed infectious etiology; however, the eruption continued to progress. The patient denied sick contacts, livestock exposure, or recent travel. A complete review of systems, including fever, chills, or lymphadenopathy, was negative. Physical examination revealed 6 circular necrotic ulcers with an overlying black eschar on the face (top), trunk (bottom), hands, and thighs, all in various stages of healing. In addition, large, reticulated, poikilodermatous patches were incidentally noted in areas free of ulcers and eschars on the trunk (bottom) and bilateral arms and legs. Upon questioning, the patient said these patches had been present for more than 30 years. A punch biopsy from an ulcer on the chest was obtained and sent for histopathologic and immunohistochemical examination.
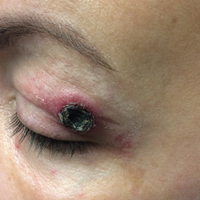
PCV13 vaccination safe, effective in 6- to 17-year-olds
Positive immunogenicity results and lack of adverse events with receipt of the 13-valent pneumococcal conjugate vaccine by 200 children and adolescents in India supports extension of the indication to that population, reported Sharad Agarkhedkar, MD, of the Padmashree Dr. D. Y. Patil Medical College in Pune, India, and associates.
In India, the PCV13 vaccine is indicated for children aged 6 months to 5 years and for adults older than 50 years. The study assessed the safety and immunogenicity of the vaccine in children aged 6-17 years to consider expanding the indication to that age group, the investigators said.
In an open-label study of 200 children and adolescents who received PCV13, 113 were 6 years to less than 10 years of age and 87 were aged 10-17 years; 54% were female. Antibody-mediated opsonophagocytic activity geometric mean titers were significantly higher for all vaccine serotypes 1 month after vaccination with PCV13, and there was no significant difference in psonophagocytic activity geometric mean titers between the two age groups. , the investigators said.
There were no acute reactions within 20 minutes of immunization. No adverse effects or severe adverse effects were recalled by caregivers who were questioned by the investigators 28-42 days following vaccination.
Read more in the Pediatric Infectious Diseases Journal (2017 Nov 1;36[11]:e282-5. doi: 10.1097/INF.0000000000001695).
Positive immunogenicity results and lack of adverse events with receipt of the 13-valent pneumococcal conjugate vaccine by 200 children and adolescents in India supports extension of the indication to that population, reported Sharad Agarkhedkar, MD, of the Padmashree Dr. D. Y. Patil Medical College in Pune, India, and associates.
In India, the PCV13 vaccine is indicated for children aged 6 months to 5 years and for adults older than 50 years. The study assessed the safety and immunogenicity of the vaccine in children aged 6-17 years to consider expanding the indication to that age group, the investigators said.
In an open-label study of 200 children and adolescents who received PCV13, 113 were 6 years to less than 10 years of age and 87 were aged 10-17 years; 54% were female. Antibody-mediated opsonophagocytic activity geometric mean titers were significantly higher for all vaccine serotypes 1 month after vaccination with PCV13, and there was no significant difference in psonophagocytic activity geometric mean titers between the two age groups. , the investigators said.
There were no acute reactions within 20 minutes of immunization. No adverse effects or severe adverse effects were recalled by caregivers who were questioned by the investigators 28-42 days following vaccination.
Read more in the Pediatric Infectious Diseases Journal (2017 Nov 1;36[11]:e282-5. doi: 10.1097/INF.0000000000001695).
Positive immunogenicity results and lack of adverse events with receipt of the 13-valent pneumococcal conjugate vaccine by 200 children and adolescents in India supports extension of the indication to that population, reported Sharad Agarkhedkar, MD, of the Padmashree Dr. D. Y. Patil Medical College in Pune, India, and associates.
In India, the PCV13 vaccine is indicated for children aged 6 months to 5 years and for adults older than 50 years. The study assessed the safety and immunogenicity of the vaccine in children aged 6-17 years to consider expanding the indication to that age group, the investigators said.
In an open-label study of 200 children and adolescents who received PCV13, 113 were 6 years to less than 10 years of age and 87 were aged 10-17 years; 54% were female. Antibody-mediated opsonophagocytic activity geometric mean titers were significantly higher for all vaccine serotypes 1 month after vaccination with PCV13, and there was no significant difference in psonophagocytic activity geometric mean titers between the two age groups. , the investigators said.
There were no acute reactions within 20 minutes of immunization. No adverse effects or severe adverse effects were recalled by caregivers who were questioned by the investigators 28-42 days following vaccination.
Read more in the Pediatric Infectious Diseases Journal (2017 Nov 1;36[11]:e282-5. doi: 10.1097/INF.0000000000001695).
FROM PEDIATRIC INFECTIOUS DISEASE JOURNAL