User login
Emergency Ultrasound: Ultrasound-Guided Hip Arthrocentesis
Hip ultrasound has long been considered an effective diagnostic and interventional tool to identify hip effusions and perform guided arthrocentesis in patients with suspected septic arthritis. Although imaging and interventional techniques are typically performed by interventional radiologists, several case reports support the use of these techniques by the emergency physician (EP) in both pediatric and adult patients presenting with hip pain.1,2
Hip ultrasound permits rapid visualization of the joint space to assess the presence of a hip effusion, and provides the opportunity for the clinician to quickly perform hip arthrocentesis and to obtain synovial fluid for analysis—the current gold standard of diagnosis. The current literature shows treatment of effusion in the adult hip via ultrasound-guided interventional methods to be more convenient and less painful than traditional fluoroscopic-guided techniques, and to have the same procedural success rate.3 With the increasing utilization of point-of-care (POC) ultrasound in the ED, ultrasound-guided hip arthrocentesis has become a powerful tool in the EP’s armamentarium to aid in evaluating and treating patients in the ED presenting with hip pain.
Imaging Technique
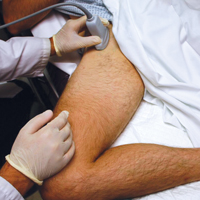
To perform an ultrasound-guided arthrocentesis, the patient should be placed in the supine position, with both knees bent and the hips externally rotated in the frog leg position (Figure 1). 

Arthrocentesis
When an effusion is present, arthrocentesis is warranted. To perform this procedure, the femoral vessels should be identified inferior to the inguinal ligament and avoided laterally. The hip should be prepared in a sterile fashion and a lubricated probe should be placed in a sterile dressing with a cord cover. The effusion should be visualized again, and the area should be anesthetized superficially and deeply with local anesthetic, aspirating prior to infusing at the deeper levels. An 18-gauge spinal needle affixed to a 20-mL syringe should be introduced and advanced while aspirating under direct visualization through the capsule of the hip into the effusion. The fluid is then aspirated and sent for laboratory analysis.
Summary
A delayed diagnosis of hip effusion and failure to initiate prompt treatment are the most common causes of late complications of septic arthritis.4 Point-of-care diagnostic and interventional ultrasound of the hip permit instant visualization and implementation of immediate diagnostic and therapeutic measures, which decrease morbidity in adult patients with septic arthritis. Hip arthrocentesis with subsequent synovial fluid analysis, the gold standard of diagnosis, has traditionally been performed by radiology services. Recent literature, however, has shown performance of these ultrasound-guided techniques by EPs to be safe and efficient, facilitating time to treatment.
1. Freeman K, Dewitz A, Baker WE. Ultrasound-guided hip arthrocentesis in the ED. Am J Emerg Med. 2007;25(1):80-86. doi:10.1016/j.ajem.2006.08.002.
2. Minardi JJ, Lander OM. Septic hip arthritis: diagnosis and arthrocentesis using bedside ultrasound. J Emerg Med. 2012;43(2):316-318. doi:10.1016/j.jemermed.2011.09.029.
3. Byrd JW, Potts EA, Allison RK, Jones KS. Ultrasound-guided hip injections: a comparative study with fluoroscopy-guided injections. Arthroscopy. 2014;30(1):42-46. doi:10.1016/j.arthro.2013.09.083.
4. Mascioli AA, Park AL. Infectious arthritis. In: Canale ST, Beaty JH eds. Campbell’s Operative Orthopaedics. Vol 1. 13th ed. Philadelphia, PA: Elsevier Mosby; 2013:749-772.
Hip ultrasound has long been considered an effective diagnostic and interventional tool to identify hip effusions and perform guided arthrocentesis in patients with suspected septic arthritis. Although imaging and interventional techniques are typically performed by interventional radiologists, several case reports support the use of these techniques by the emergency physician (EP) in both pediatric and adult patients presenting with hip pain.1,2
Hip ultrasound permits rapid visualization of the joint space to assess the presence of a hip effusion, and provides the opportunity for the clinician to quickly perform hip arthrocentesis and to obtain synovial fluid for analysis—the current gold standard of diagnosis. The current literature shows treatment of effusion in the adult hip via ultrasound-guided interventional methods to be more convenient and less painful than traditional fluoroscopic-guided techniques, and to have the same procedural success rate.3 With the increasing utilization of point-of-care (POC) ultrasound in the ED, ultrasound-guided hip arthrocentesis has become a powerful tool in the EP’s armamentarium to aid in evaluating and treating patients in the ED presenting with hip pain.
Imaging Technique
To perform an ultrasound-guided arthrocentesis, the patient should be placed in the supine position, with both knees bent and the hips externally rotated in the frog leg position (Figure 1).
Arthrocentesis
When an effusion is present, arthrocentesis is warranted. To perform this procedure, the femoral vessels should be identified inferior to the inguinal ligament and avoided laterally. The hip should be prepared in a sterile fashion and a lubricated probe should be placed in a sterile dressing with a cord cover. The effusion should be visualized again, and the area should be anesthetized superficially and deeply with local anesthetic, aspirating prior to infusing at the deeper levels. An 18-gauge spinal needle affixed to a 20-mL syringe should be introduced and advanced while aspirating under direct visualization through the capsule of the hip into the effusion. The fluid is then aspirated and sent for laboratory analysis.
Summary
A delayed diagnosis of hip effusion and failure to initiate prompt treatment are the most common causes of late complications of septic arthritis.4 Point-of-care diagnostic and interventional ultrasound of the hip permit instant visualization and implementation of immediate diagnostic and therapeutic measures, which decrease morbidity in adult patients with septic arthritis. Hip arthrocentesis with subsequent synovial fluid analysis, the gold standard of diagnosis, has traditionally been performed by radiology services. Recent literature, however, has shown performance of these ultrasound-guided techniques by EPs to be safe and efficient, facilitating time to treatment.
Hip ultrasound has long been considered an effective diagnostic and interventional tool to identify hip effusions and perform guided arthrocentesis in patients with suspected septic arthritis. Although imaging and interventional techniques are typically performed by interventional radiologists, several case reports support the use of these techniques by the emergency physician (EP) in both pediatric and adult patients presenting with hip pain.1,2
Hip ultrasound permits rapid visualization of the joint space to assess the presence of a hip effusion, and provides the opportunity for the clinician to quickly perform hip arthrocentesis and to obtain synovial fluid for analysis—the current gold standard of diagnosis. The current literature shows treatment of effusion in the adult hip via ultrasound-guided interventional methods to be more convenient and less painful than traditional fluoroscopic-guided techniques, and to have the same procedural success rate.3 With the increasing utilization of point-of-care (POC) ultrasound in the ED, ultrasound-guided hip arthrocentesis has become a powerful tool in the EP’s armamentarium to aid in evaluating and treating patients in the ED presenting with hip pain.
Imaging Technique
To perform an ultrasound-guided arthrocentesis, the patient should be placed in the supine position, with both knees bent and the hips externally rotated in the frog leg position (Figure 1).
Arthrocentesis
When an effusion is present, arthrocentesis is warranted. To perform this procedure, the femoral vessels should be identified inferior to the inguinal ligament and avoided laterally. The hip should be prepared in a sterile fashion and a lubricated probe should be placed in a sterile dressing with a cord cover. The effusion should be visualized again, and the area should be anesthetized superficially and deeply with local anesthetic, aspirating prior to infusing at the deeper levels. An 18-gauge spinal needle affixed to a 20-mL syringe should be introduced and advanced while aspirating under direct visualization through the capsule of the hip into the effusion. The fluid is then aspirated and sent for laboratory analysis.
Summary
A delayed diagnosis of hip effusion and failure to initiate prompt treatment are the most common causes of late complications of septic arthritis.4 Point-of-care diagnostic and interventional ultrasound of the hip permit instant visualization and implementation of immediate diagnostic and therapeutic measures, which decrease morbidity in adult patients with septic arthritis. Hip arthrocentesis with subsequent synovial fluid analysis, the gold standard of diagnosis, has traditionally been performed by radiology services. Recent literature, however, has shown performance of these ultrasound-guided techniques by EPs to be safe and efficient, facilitating time to treatment.
1. Freeman K, Dewitz A, Baker WE. Ultrasound-guided hip arthrocentesis in the ED. Am J Emerg Med. 2007;25(1):80-86. doi:10.1016/j.ajem.2006.08.002.
2. Minardi JJ, Lander OM. Septic hip arthritis: diagnosis and arthrocentesis using bedside ultrasound. J Emerg Med. 2012;43(2):316-318. doi:10.1016/j.jemermed.2011.09.029.
3. Byrd JW, Potts EA, Allison RK, Jones KS. Ultrasound-guided hip injections: a comparative study with fluoroscopy-guided injections. Arthroscopy. 2014;30(1):42-46. doi:10.1016/j.arthro.2013.09.083.
4. Mascioli AA, Park AL. Infectious arthritis. In: Canale ST, Beaty JH eds. Campbell’s Operative Orthopaedics. Vol 1. 13th ed. Philadelphia, PA: Elsevier Mosby; 2013:749-772.
1. Freeman K, Dewitz A, Baker WE. Ultrasound-guided hip arthrocentesis in the ED. Am J Emerg Med. 2007;25(1):80-86. doi:10.1016/j.ajem.2006.08.002.
2. Minardi JJ, Lander OM. Septic hip arthritis: diagnosis and arthrocentesis using bedside ultrasound. J Emerg Med. 2012;43(2):316-318. doi:10.1016/j.jemermed.2011.09.029.
3. Byrd JW, Potts EA, Allison RK, Jones KS. Ultrasound-guided hip injections: a comparative study with fluoroscopy-guided injections. Arthroscopy. 2014;30(1):42-46. doi:10.1016/j.arthro.2013.09.083.
4. Mascioli AA, Park AL. Infectious arthritis. In: Canale ST, Beaty JH eds. Campbell’s Operative Orthopaedics. Vol 1. 13th ed. Philadelphia, PA: Elsevier Mosby; 2013:749-772.
Pediatric ENT Complaints: An Update
Among all of the causes of ear, nose, and throat (ENT) complaints, acute otitis media (AOM), bacterial sinusitis, and streptococcal pharyngitis (SP) are the most common infections prompting pediatric presentation to the ED. Through a series of case scenarios, along with key questions to help guide the clinician’s work-up, this review covers the proper evaluation and management of pediatric ENT complaints.
Case Scenario 1
A 13-month-old girl presented to the ED with a 1-day history of fever and runny nose. According to her parents, the child had been continually pulling on her ears in apparent discomfort. During history-taking, the parents further informed the emergency physician (EP) that the patient started daycare 4 months earlier and had two elementary school-aged siblings. The patient’s medical history was significant for otitis media, but the parents stated she had not been on antibiotics for over 4 months.
On physical examination, the patient’s vital signs were: blood pressure (BP), 75/50 mm Hg; temperature (T), 101.3°F; slight tachycardia; and normal age-adjusted respiratory rate (RR). Oxygen saturation was 100% on room air. The lungs were clear to auscultation and heart sounds were normal and without murmur. The otolaryngologic examination revealed copious yellow discharge from both nostrils, non-erythematous posterior oropharynx, and erythema to the right tympanic membrane (TM). Questions to Guide the Work-Up: (1) What physical examination findings should be present for accurate diagnosis of otitis media? (2) Will this patient require antibiotics immediately, or is a “wait-and-see” approach indicated? (3) If treatment with antibiotic therapy is warranted, what are the appropriate therapeutic regimen and duration of therapy?
Otitis Media
Acute otitis media is one of the most common presentations in young children. Defined as the rapid onset of signs and symptoms of middle ear inflammation, in conjunction with middle ear effusion (MEE), AOM can develop secondary to a viral or bacterial infection. It is estimated that more than 80% of the pediatric population will experience at least one episode of AOM by age 3 years.1-3
Risk factors for AOM include upper respiratory infection (URI), daycare attendance, siblings, parental smoking, and formula-feeding versus breastfeeding. The patient’s history may include rapid-onset otalgia, fever, irritability, anorexia, and concurrent URI symptoms, as well as other nonspecific symptoms (eg, ear rubbing and/or pulling, crying, changes in behavior and sleep patterns).2-4 In general, otalgia and ear-rubbing in the nonverbal patient seem to have the best predictive value for AOM.3
Signs and Symptoms
A normal TM should be translucent and pearly gray, with visible landmarks of the manubrium of malleus and pars flaccida. A TM that is bulging, cloudy, and immobile is the most consistent finding in AOM, with bulging having a specificity of 97%. Redness of the tympanic membrane is not a useful predictor of AOM as this finding is noted in upward of 30% of pediatric patients on general examination but in <1% of AOM diagnoses in the absence of a bulging TM.
Diagnosis
Pneumatic otoscopy is the gold standard for diagnosing for MEE; however, this examination can be difficult in younger, often uncooperative, patients. A TM that does not perceptibly move with either positive or negative insufflation pressure greatly enhances the diagnostic accuracy for MEE over the use of visible eardrum characteristics alone.2-5
Acute otitis media is a clinical diagnosis and does not require imaging studies or laboratory evaluation unless more serious processes, such as skull fracture, mastoiditis, or intracranial abscess, are being considered.2,3
Treatment and Management
Analgesia. The first step in managing patients with AOM is to provide analgesia. In most cases, acetaminophen in patients over 2 months of age, or ibuprofen in patients over 6 months of age, are adequate choices for managing pain. When either of these analgesics is administered in the clinic/ED setting, patients should be monitored to assure adequate pain relief prior to discharge.
While topical agents such as combination antipyrine-benzocaine suspensions were commonly given in the past to alleviate the pain associated with AOM, there are limited data to support their effectiveness. As such, in July 2015, the US Food and Drug Administration ordered manufacturers to halt production on these unapproved prescription products.3,4,6 There are also no randomized controlled trials (RCTs) to support the use of decongestants or antihistamines for resolution of AOM or otalgia.3,7
Antibiotic Therapy. The most common bacteria associated with AOM are Streptococcus pneumonia, nontypeable Hemophilus influenza, and Moraxella catarrhalis. In 30% of patients, the causative etiology is viral. When the decision is made to treat AOM, high-dose amoxicillin is still considered the first-line treatment, despite ever evolving susceptibilities of bacteria. 
When a child is noted to have been treated with amoxicillin within a 30-day period or who has concurrent conjunctivitis, amoxicillin-clavulanate is considered the first-line treatment.2-4,7,8 The current American Academy of Pediatrics (AAP) guidelines recommend 10 days of antibiotic therapy for children younger than age 2 years, and 5 to 7 days for children older than age 2 years who have uncomplicated AOM. Intramuscular (IM) ceftriaxone is an acceptable first-line agent in a child who is unable to tolerate oral medications or who is suffering persistent emesis. Intramuscular ceftriaxone can be given as a single dose of 50 mg/kg, though the patient should be followed closely as studies show that a second dose may be necessary 5 to 7 days later to prevent infection recurrence. The IM dose of ceftriaxone 50 mg/kg can also be given if treatment with other antibiotics fails to resolve the AOM (failure is defined as no improvement in the patient’s condition 48 to 72 hours from treatment). In such cases, ceftriaxone is given in three consecutive doses.3,4,7
Wait-and-See Approach. Studies of patients whose AOM was confirmed via culture (19% were positive for S pneumoniae, 48% for H influenza, and 78% for M catarrhalis) showed bacterial clearance without antibiotic intervention.4 Based on these findings, the 2013 revised AAP evidence-based clinical practice guidelines indicate an initial watching-and-waiting period combined with pain management for patients older than 6 months of age who are diagnosed with unilateral AOM in the absence of severe symptoms (ie, fever is lower than 102.2˚F or patient has severe otalgia).4 A period of observation prior to treatment is also endorsed for children older than age 2 years who exhibit nonsevere symptoms—even if they have bilateral disease.4
Conversely, all patients younger than age 6 months and all children with severe symptoms should be treated with antibiotics at diagnosis.3,4 The wait-and-see approach, recommends an observation period of 24 to 48 hours for children in the lower risk group prior to antibiotic administration. Delayed antibiotic administration can be performed by a physician in an office/ED follow-up or as a safety-net antibiotic prescription (SNAP) sent home with the family on the initial ED encounter.2-4,8,9
Case 1 Resolution
Given this patient’s unilateral and nonsevere symptoms (minor otalgia, fever <102.2°F), age older than 6 months, and no recent antibiotic use), she was treated with oral ibuprofen. At discharge, the parents were given a 10-day SNAP prescription of high-dose amoxicillin (90 mg/kg/d, divided into two daily doses) and instructed to fill the prescription only if the patient’s otalgia did not improve in 1 or 2 days.
Case Scenario 2
A 5-year-old boy was presented for evaluation by his parents, who stated that their son had been sick since he had started kindergarten in the fall. The patient had a 10-day history of cough, thick runny nose, and facial pain, and a 1-day history of new-onset fever and headache. His parents further noted that the patient had been seen by his pediatrician several times over the past week. At each of these visits, the pediatrician had informed them that their son had a virus.
Vital signs on examination were: BP, 100/60 mm Hg; heart rate (HR), 112 beats/min; normal age-adjusted RR; and T, 102.6oF. Oxygen saturation was 100% on room air. The patient did not appear toxic, his lungs were clear on auscultation, and there were no other clinical signs suggestive of meningitis. The otolaryngologic examination revealed bilateral thick mucoid drainage and visible edema and erythema of the nasal turbinates. The patient was noted to have some facial pain in the maxillary area bilaterally.
Questions to Guide the Work-Up: (1) Does the patient have a prolonged URI or pediatric sinusitis, and what differentiates the two conditions? (2) What sinuses are present in a 5-year-old patient? (3) What treatment modalities are available for sinusitis? (4) Is imaging of the sinuses helpful in confirming the diagnosis?
Acute Bacterial Sinusitis
Rhinosinusitis is an inflammation of the mucosal lining of the nasal passages and paranasal sinuses. Most cases occur secondary to a viral URI and resolve spontaneously in 99% of the pediatric population.10,11
Acute bacterial sinusitis (ABS) is an inflammation of the same mucosal lining of the nasal passages secondary to bacterial overgrowth that lasts more than 10 days, with complete resolution by 30 days.12,13 When evaluating a pediatric patient for ABS, it is important to consider the sinus growth and development: If the sinus is not yet formed, it therefore cannot be the location of an ABS.13 The ethmoid and maxillary sinuses are present at birth, aerated within 4 months of life, and are fully developed by age 12 years. The sphenoid sinuses begin development around age 3 years, are aerated by age 7 or 8 years, and are fully developed by age 18 to 20 years. The frontal sinuses begin development around age 8 years and are aerated and fully developed by age 12 to 15 years.10,13,14 While most guidelines focus on children older than age 1 year (due to very small infantile sinuses), ABS does occur in children younger than age 1 year.12,14
Signs and Symptoms
Differentiation between a viral URI/rhinosinusitis and ABS is a challenge and can be based upon severity of symptoms as well as length of illness. Symptoms of ABS are typically present and persistent for more than 10 days, without improvement. Continuing illness and worsening of symptoms are identifying features of ABS given most viral URIs gradually resolve within a 10-day timeframe. Other common symptoms include milky/thick nasal discharge, fever, predominantly nocturnal cough, and headache. Other less common symptoms include facial pain, toothache, malodorous breath, and periorbital edema. On physical examination, erythema and edema of the turbinates, as well as reproducible pain over aerated sinuses, are suggestive of ABS.10-14
Diagnosis
In the acute care setting, diagnosis of ABS should be clinical in nature. Neither imaging nor laboratory work-up is generally required secondary to their poor diagnostic specificity for ABS. The bacteria involved in ABS are similar to those associated with AOM, with S pneumonia, nontypeable H influenza, and M catarrhalis being the predominant organisms.10-15
Treatment and Management
Treatment of ABS is generally recommended once the diagnosis is made, though this is based largely on expert opinion as there are limited RCTs available.13 However, available studies do show a more rapid improvement in children on antibiotic therapy than those on placebo.15,16
Antibiotic Therapy. Amoxicillin remains the antimicrobial agent of choice for first-line treatment of uncomplicated ABS forsituations in which antimicrobial resistance is not suspected. In communities with a high prevalence of nonsusceptible S pneumoniae (>10%, including intermediate- and high-level resistance), treatment may be initiated at 80 to 90 mg/kg/d in two divided doses, with a maximum of 2 g per dose.
Patients presenting with moderate to severe illness, as well as those who are younger than 2 years, attend childcare, or have recently been treated with an antimicrobial, may receive high-dose amoxicillin-clavulanate as initial therapy given the elevated beta-lactamase production of the common bacteria that cause ABS.
Second-line alternatives include azithromycin, cefdinir, and sulfamethoxizole-trimethoprim (Table 1). There are data to suggest higher rates of decreased susceptibility of S pneumonia and H influenza to third-generation cephalosporins, and the addition of clindamycin may be warranted when utilizing those medications. Treatment is recommended for 10 to 14 days, though improvement should be noted within 1 to 3 days.10-12,14-17
Adjuvant Therapy. Additional therapies include nasal irrigation, decongestants, antihistamines, and intranasal steroids; however, there are only anecdotal reports of their efficacy in providing symptom relief. Therefore, there are insufficient evidence-based data to support or refute the role of these adjuvant therapies in treating pediatric patients with ABS.9,13
Case 2 Resolution
The prolonged duration and severity of symptoms (high fever and headache) and the gradual worsening of the clinical course (ie, late-onset fever) in this patient all suggest ABS rather than a simple prolonged URI. The physical examination findings of inflamed turbinates and facial pain further increase the specificity for ABS. The patient was started on oral amoxicillin-clavulanate with planned treatment for 14 days. At discharge, his parents were instructed to follow-up with the patient’s pediatrician in 3 days to ensure a degree of clinical resolution.
Case Scenario 3
A 4-year-old boy was presented by his parents for evaluation of a 2-day history of a persistent and unimproved sore throat. The patient’s mother indicated that the child’s oral T upon returning home earlier from preschool was 101.2oF. She further noted that her 17-month-old daughter and 8-year-old son also experienced similar symptoms which had self-resolved. Triage vital signs were: T, 100.8oF, orally; BP, HR, and RR were all within normal limits. Oxygen saturation was 100% on room air.
On physical examination, the child was noted to have anterior cervical lymph nodes bilaterally and an erythematous oropharynx with exudate noted on both tonsils. There were no cutaneous abnormalities, nasal edema, erythema, or drainage. Based on the clinical examination, the EP was suspicious for SP.
Questions to Guide the Work-Up: (1) Is SP diagnosed based on clinical findings alone in this patient’s age group? (2) At what age in the pediatric population is it appropriate to perform a rapid streptococcal antigen test? (3) Are there medications other than antibiotics that are beneficial in treating symptomatic SP?
Streptococcal Pharyngitis
Streptococcal pharyngitis is a clinical condition caused by group A beta-hemolytic S pyogens. This bacterium is responsible for multiple conditions, including pharyngitis, skin infections, poststreptococcal glomerulonephritis, and rheumatic fever, as well as invasive syndromes. (This case focuses solely on SP).
Pharyngitis can occur secondary to a viral or bacterial infection, and SP is the most common cause of pediatric bacterial pharyngitis. It is estimated that children aged 5 to 15 years are more commonly diagnosed with SP, although approximately 24% of children younger than age 5 years with pharyngitis symptoms will be ultimately diagnosed with SP.
Signs and Symptoms
Typical symptoms include fever, pharyngitis, generalized abdominal pain, nausea, vomiting, headache, and absence of viral URI symptoms (eg, cough, nasal discharge). However, younger patients with SP may have clinical findings of prolonged nasal drainage and excoriated nares. Examination findings may include swollen and tender anterior cervical lymph nodes; generalized edema and erythema of the posterior pharynx; tonsillar exudates; and palatal petechiae.
Diagnosis
Centor Criteria. The Centor criteria were developed to assist practitioners in identifying patients with potential SP. Criteria for patients older than age 15 years include fever, absence of cough, tonsillar exudates, and tender anterior cervical lymphadenopathy. A modified Centor criteria was later established to include children older than age 5 years, with children between ages 5 and 15 years being the fifth variable in the modified score. In general, patients with a score of 4 or 5 (presence of each variable = 1 point) are most likely to test positive for SP on rapid antigen testing (RAT) or culture.18-20
Swab, Rapid Antigen Testing, and Culture. Swabbing the throat and RAT and/or culture should be performed in most children with suspected SP because the clinical features alone do not reliably discriminate SP from viral pharyngitis. Rapid antigen testing is only specific for group A beta-hemolytic streptococcal species, which is the only streptococcal species that is routinely treated with antibiotics in the setting of acute pharyngitis. It is unlikely for a patient with a score of 0 or 1 to have SP, and several sources suggest neither testing nor treating this cohort, but rather to consider an alternative diagnosis.18-20
Within the population of children and young adolescents, due to a RAT sensitivity of 70% to 90%, a negative result should always be backed-up by a throat culture, and treatment initiated if results of the culture are later found to be positive. As the current generation of RAT tests have a high specificity, a positive RAT does not necessitate a back-up culture, and treatment is indicated without further investigation.19,20
Routine RAT is not recommended in children younger than age 3 years as patients in this age group are at low-risk of developing rheumatic fever. One notable exception for these very young children would be if there are siblings in the home with confirmed SP, in which case, RAT should be considered in the clinical context of SP.21 Adolescents over age 15 years are another cohort with a low likelihood of developing rheumatic fever, though they can develop other poststreptococcal complications, such as glomerulonephritis.
The US Centers for Disease Control and Prevention/American Academy of Family Practitioners (AAFP) guidelines suggest that pharyngitis in older adolescents can be approached in a similar fashion to adults, with empiric therapy for a Centor score of 3 or 4, RAT (without the need for follow-up culture) for Centor score of 2, and neither testing nor treating patients with a score of 0 or 1.19
Treatment and Management
Streptococcal pharyngitis is treated mainly to prevent the poststreptococcal complications of rheumatic fever, though it will not prevent poststreptococcal glomerulonephritis. Treatment of SP also facilitates resolution of symptoms and return to baseline activities.
Antibiotic Therapy. Patients who have a positive RAT or a follow-up throat culture positive for group A streptococcus should be given antibiotics. The gold standard treatment is penicillin V orally for 10 days. 
Corticosteroid Therapy. The use of corticosteroids for symptom control of SP in pediatric patients is controversial. Although the Infectious Disease Society of America does not recommend corticosteroid therapy in the treatment of SP, several studies show such therapy (namely dexamethasone), improves pain in children and adolescents diagnosed with SP, but without significant change to the overall disease course.21,23-26
Case 3 Resolution
The patient had a modified Centor criteria score of 4, as well as siblings with similar symptoms. In following current guidelines, the EP performed a RAT and back-up culture. The RAT was negative in the ED, but the back-up culture was subsequently positive, and the child was started on a 10-day course of oral amoxicillin.
Conclusion
When evaluating pediatric patients presenting with ENT signs and symptoms such as ear pain and erythema, fever, sore throat, nasal congestion and discharge, a thorough physical examination and history-taking—including recent illness of any siblings—along with testing when indicated, is essential to guide the diagnosis and determine appropriate treatment and management. In addition to administering antibiotic therapy when such is warranted, the EP should provide appropriate analgesia to manage the patient’s pain and assure relief prior to discharge.
1. Rosenfeld RM, Shin JJ, Schwartz SR, et al. Clinical practice guideline: otitis media with effusion (Update). Otolaryngol Head Neck Surg. 2016;154(1 Suppl):S1-S41. doi:10.1177/0194599815623467.
2. Acute Otitis Media Guideline Team, Cincinnati Children’s Hospital Medical Center. Evidence-based care guideline for medical management of acute otitis media in children 2 months to 13 years of age. http://f.i-md.com/medinfo/material/4f4/4eb132ba44ae4ffe12a814f4/4eb132d744ae4ffe12a814f7.pdf. August 2006. Accessed December 29, 2016.
3. Nesbit CE, Powers MC. An evidence-based approach to managing acute otitis media. Pediatr Emerg Med Pract. 2013;10(4):1-26; quiz 26-27.
4. Lieberthal AS, Carroll AE, Chonmaitree T, et al. The diagnosis and management of acute otitis media. Pediatrics. 2013;131(3):e964-e999. doi:10.1542/peds.2012-3488.
5. American Academy of Family Physicians; American Academy of Otolaryngology-Head and Neck Surgery; American Academy of Pediatrics Subcommittee on Otitis Media With Effusion. Otitis media with effusion. Pediatrics. 2004;113(5):1412-1429.
6. US Food and Drug Administration Web site. FDA: Use only approved prescription ear drops. http://www.fda.gov/ForConsumers/ConsumerUpdates/-ucm453087.htm. Updated July 10, 2015. Accessed December 15, 2016.
7. Sack F. An evidence based approach to the management of uncomplicated acute otitis media in children. Int Pediatrics. 2005;20(1):44-46.
8. Johnson NC, Holger JS. Pediatric acute otitis media: the case for delayed antibiotic treatment. J Emerg Med. 2007;32(3):279-284. doi:10.1016/j.jemermed.2006.07.029.
9. Spiro DM, Tay KY, Arnold DH, Dziura JD, Baker MD, Shapiro ED. Wait-and-see prescription for the treatment of acute otitis media: a randomized controlled trial. JAMA. 2006;296(10):1235-1241. doi:10.1001/jama.296.10.1235.
10. Brook I. Management of acute rhinosinusitis in pediatric patients. Pediatr Emerg Med Pract. 2012;9(5):1-24.
11. Ferdman RM, Linzer JF Jr. The runny nose in the emergency department: rhinitis and sinusitis. Clin Pediatr Emerg Med. 2007;8(2):123-130.
12. Acute Bacterial Sinusitis Guideline Team, Cincinnati Children’s Hospital Medical Center: Evidence-based care guideline for medical management of acute bacterial sinusitis in children 1 through 18 years of age. http://www.antibioticos.msssi.gob.es/PDF/sinusitisguideline.pdf. July 7, 2006. Accessed December 29, 2016
13. Holt KR, Murdoch Cuenca M, Cuenca PJ, Johnston GM. acute pediatric sinusitis and “the 10-day rule.” Pediatr Emerg Med Pract. 2006;3(2):1-16.
14. American Academy of Pediatrics. Subcommittee on Management of Sinusitis and Committee on Quality Improvement. Clinical practice guideline: management of sinusitis. Pediatrics. 2001;108(3):798-808.
15. Wald ER, Nash D, Eickhoff J. Effectiveness of amoxicillin/clavulanate potassium in the treatment of acute bacterial sinusitis in children. Pediatrics. 2009;124(1):9-15. doi:10.1542/peds.2008-2902.
16. Arroll B, Kenealy T. Are antibiotics effective for acute purulent rhinitis? Systematic review and meta-analysis of placebo controlled randomised trials. BMJ. 2006;333(7562):279. doi:10.1136/bmj.38891.681215.AE.
17. McQuillan L, Crane LA, Kempe A. Diagnosis and management of acute sinusitis by pediatricians. Pediatrics. 2009;123(2):e193-e198.
18. Singer JI, Fontanette R. Recognizable and suspected group A beta-hemolytic streptococcal syndromes. Pediatr Emerg Med Rep. 2010;15(11):129-144.
19. Weglowski J. An evidence-based approach to the evaluation and treatment of pharyngitis in children. Pediatr Emerg Med Pract. 2011;8(12):1-28.
20. Gerber MA, Baltimore RS, Eaton CB, et al. Prevention of rheumatic fever and diagnosis and treatment of acute Streptococcal pharyngitis: a scientific statement from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee of the Council on Cardiovascular Disease in the Young, the Interdisciplinary Council on Functional Genomics and Translational Biology, and the Interdisciplinary Council on Quality of Care and Outcomes Research: endorsed by the American Academy of Pediatrics. Circulation. 2009;119(11):1541-1551. doi:10.1161/CIRCULATIONAHA.109.191959.
21. Shulman ST, Bisno AL, Clegg HW, et al. Clinical practice guideline for the diagnosis and management of group A streptococcal pharyngitis: 2012 update by the Infectious Diseases Society of America. Clin Infect Dis. 2012;55(10):1279-1282. doi:10.1093/cid/cis847.
22. Clegg HW, Ryan AG, Dallas SD, et al. Treatment of streptococcal pharyngitis with once-daily compared with twice-daily amoxicillin: a noninferiority trial. Pediatr Infect Dis J. 2006;25(9):761-767. doi:10.1097/01.inf.0000235678.46805.92.
23. Bulloch B, Kabani A, Tenenbein M. Oral dexamethasone for the treatment of pain in children with acute pharyngitis: a randomized, double-blind, placebo-controlled trial. Ann Emerg Med. 2003;41(5):601-608. doi:10.1067/mem.2003.136.
24. Niland ML, Bonsu BK, Nuss KE, Goodman DG. A pilot study of 1 versus 3 days of dexamethasone as add-on therapy in children with streptococcal pharyngitis. Pediatr Infect Dis J. 2006;25(6):477-481. doi:10.1097/01.inf.0000219469.95772.3f.
25. Wei JL, Kasperbauer JL, Weaver AL, Boggust AJ. Efficacy of single-dose dexamethasone as adjuvant therapy for acute pharyngitis. Laryngoscope. 2002;112(1):87-93. doi:10.1097/00005537-200201000-00016.
26. Hayward G, Thompson M, Heneghan C, Perera R, Del Mar C, Glasziou P. Corticosteroids for pain relief in sore throat: systematic review and meta-analysis. BMJ. 2009;339:b2976. doi:10.1136/bmj.b2976.
Among all of the causes of ear, nose, and throat (ENT) complaints, acute otitis media (AOM), bacterial sinusitis, and streptococcal pharyngitis (SP) are the most common infections prompting pediatric presentation to the ED. Through a series of case scenarios, along with key questions to help guide the clinician’s work-up, this review covers the proper evaluation and management of pediatric ENT complaints.
Case Scenario 1
A 13-month-old girl presented to the ED with a 1-day history of fever and runny nose. According to her parents, the child had been continually pulling on her ears in apparent discomfort. During history-taking, the parents further informed the emergency physician (EP) that the patient started daycare 4 months earlier and had two elementary school-aged siblings. The patient’s medical history was significant for otitis media, but the parents stated she had not been on antibiotics for over 4 months.
On physical examination, the patient’s vital signs were: blood pressure (BP), 75/50 mm Hg; temperature (T), 101.3°F; slight tachycardia; and normal age-adjusted respiratory rate (RR). Oxygen saturation was 100% on room air. The lungs were clear to auscultation and heart sounds were normal and without murmur. The otolaryngologic examination revealed copious yellow discharge from both nostrils, non-erythematous posterior oropharynx, and erythema to the right tympanic membrane (TM). Questions to Guide the Work-Up: (1) What physical examination findings should be present for accurate diagnosis of otitis media? (2) Will this patient require antibiotics immediately, or is a “wait-and-see” approach indicated? (3) If treatment with antibiotic therapy is warranted, what are the appropriate therapeutic regimen and duration of therapy?
Otitis Media
Acute otitis media is one of the most common presentations in young children. Defined as the rapid onset of signs and symptoms of middle ear inflammation, in conjunction with middle ear effusion (MEE), AOM can develop secondary to a viral or bacterial infection. It is estimated that more than 80% of the pediatric population will experience at least one episode of AOM by age 3 years.1-3
Risk factors for AOM include upper respiratory infection (URI), daycare attendance, siblings, parental smoking, and formula-feeding versus breastfeeding. The patient’s history may include rapid-onset otalgia, fever, irritability, anorexia, and concurrent URI symptoms, as well as other nonspecific symptoms (eg, ear rubbing and/or pulling, crying, changes in behavior and sleep patterns).2-4 In general, otalgia and ear-rubbing in the nonverbal patient seem to have the best predictive value for AOM.3
Signs and Symptoms
A normal TM should be translucent and pearly gray, with visible landmarks of the manubrium of malleus and pars flaccida. A TM that is bulging, cloudy, and immobile is the most consistent finding in AOM, with bulging having a specificity of 97%. Redness of the tympanic membrane is not a useful predictor of AOM as this finding is noted in upward of 30% of pediatric patients on general examination but in <1% of AOM diagnoses in the absence of a bulging TM.
Diagnosis
Pneumatic otoscopy is the gold standard for diagnosing for MEE; however, this examination can be difficult in younger, often uncooperative, patients. A TM that does not perceptibly move with either positive or negative insufflation pressure greatly enhances the diagnostic accuracy for MEE over the use of visible eardrum characteristics alone.2-5
Acute otitis media is a clinical diagnosis and does not require imaging studies or laboratory evaluation unless more serious processes, such as skull fracture, mastoiditis, or intracranial abscess, are being considered.2,3
Treatment and Management
Analgesia. The first step in managing patients with AOM is to provide analgesia. In most cases, acetaminophen in patients over 2 months of age, or ibuprofen in patients over 6 months of age, are adequate choices for managing pain. When either of these analgesics is administered in the clinic/ED setting, patients should be monitored to assure adequate pain relief prior to discharge.
While topical agents such as combination antipyrine-benzocaine suspensions were commonly given in the past to alleviate the pain associated with AOM, there are limited data to support their effectiveness. As such, in July 2015, the US Food and Drug Administration ordered manufacturers to halt production on these unapproved prescription products.3,4,6 There are also no randomized controlled trials (RCTs) to support the use of decongestants or antihistamines for resolution of AOM or otalgia.3,7
Antibiotic Therapy. The most common bacteria associated with AOM are Streptococcus pneumonia, nontypeable Hemophilus influenza, and Moraxella catarrhalis. In 30% of patients, the causative etiology is viral. When the decision is made to treat AOM, high-dose amoxicillin is still considered the first-line treatment, despite ever evolving susceptibilities of bacteria.
When a child is noted to have been treated with amoxicillin within a 30-day period or who has concurrent conjunctivitis, amoxicillin-clavulanate is considered the first-line treatment.2-4,7,8 The current American Academy of Pediatrics (AAP) guidelines recommend 10 days of antibiotic therapy for children younger than age 2 years, and 5 to 7 days for children older than age 2 years who have uncomplicated AOM. Intramuscular (IM) ceftriaxone is an acceptable first-line agent in a child who is unable to tolerate oral medications or who is suffering persistent emesis. Intramuscular ceftriaxone can be given as a single dose of 50 mg/kg, though the patient should be followed closely as studies show that a second dose may be necessary 5 to 7 days later to prevent infection recurrence. The IM dose of ceftriaxone 50 mg/kg can also be given if treatment with other antibiotics fails to resolve the AOM (failure is defined as no improvement in the patient’s condition 48 to 72 hours from treatment). In such cases, ceftriaxone is given in three consecutive doses.3,4,7
Wait-and-See Approach. Studies of patients whose AOM was confirmed via culture (19% were positive for S pneumoniae, 48% for H influenza, and 78% for M catarrhalis) showed bacterial clearance without antibiotic intervention.4 Based on these findings, the 2013 revised AAP evidence-based clinical practice guidelines indicate an initial watching-and-waiting period combined with pain management for patients older than 6 months of age who are diagnosed with unilateral AOM in the absence of severe symptoms (ie, fever is lower than 102.2˚F or patient has severe otalgia).4 A period of observation prior to treatment is also endorsed for children older than age 2 years who exhibit nonsevere symptoms—even if they have bilateral disease.4
Conversely, all patients younger than age 6 months and all children with severe symptoms should be treated with antibiotics at diagnosis.3,4 The wait-and-see approach, recommends an observation period of 24 to 48 hours for children in the lower risk group prior to antibiotic administration. Delayed antibiotic administration can be performed by a physician in an office/ED follow-up or as a safety-net antibiotic prescription (SNAP) sent home with the family on the initial ED encounter.2-4,8,9
Case 1 Resolution
Given this patient’s unilateral and nonsevere symptoms (minor otalgia, fever <102.2°F), age older than 6 months, and no recent antibiotic use), she was treated with oral ibuprofen. At discharge, the parents were given a 10-day SNAP prescription of high-dose amoxicillin (90 mg/kg/d, divided into two daily doses) and instructed to fill the prescription only if the patient’s otalgia did not improve in 1 or 2 days.
Case Scenario 2
A 5-year-old boy was presented for evaluation by his parents, who stated that their son had been sick since he had started kindergarten in the fall. The patient had a 10-day history of cough, thick runny nose, and facial pain, and a 1-day history of new-onset fever and headache. His parents further noted that the patient had been seen by his pediatrician several times over the past week. At each of these visits, the pediatrician had informed them that their son had a virus.
Vital signs on examination were: BP, 100/60 mm Hg; heart rate (HR), 112 beats/min; normal age-adjusted RR; and T, 102.6oF. Oxygen saturation was 100% on room air. The patient did not appear toxic, his lungs were clear on auscultation, and there were no other clinical signs suggestive of meningitis. The otolaryngologic examination revealed bilateral thick mucoid drainage and visible edema and erythema of the nasal turbinates. The patient was noted to have some facial pain in the maxillary area bilaterally.
Questions to Guide the Work-Up: (1) Does the patient have a prolonged URI or pediatric sinusitis, and what differentiates the two conditions? (2) What sinuses are present in a 5-year-old patient? (3) What treatment modalities are available for sinusitis? (4) Is imaging of the sinuses helpful in confirming the diagnosis?
Acute Bacterial Sinusitis
Rhinosinusitis is an inflammation of the mucosal lining of the nasal passages and paranasal sinuses. Most cases occur secondary to a viral URI and resolve spontaneously in 99% of the pediatric population.10,11
Acute bacterial sinusitis (ABS) is an inflammation of the same mucosal lining of the nasal passages secondary to bacterial overgrowth that lasts more than 10 days, with complete resolution by 30 days.12,13 When evaluating a pediatric patient for ABS, it is important to consider the sinus growth and development: If the sinus is not yet formed, it therefore cannot be the location of an ABS.13 The ethmoid and maxillary sinuses are present at birth, aerated within 4 months of life, and are fully developed by age 12 years. The sphenoid sinuses begin development around age 3 years, are aerated by age 7 or 8 years, and are fully developed by age 18 to 20 years. The frontal sinuses begin development around age 8 years and are aerated and fully developed by age 12 to 15 years.10,13,14 While most guidelines focus on children older than age 1 year (due to very small infantile sinuses), ABS does occur in children younger than age 1 year.12,14
Signs and Symptoms
Differentiation between a viral URI/rhinosinusitis and ABS is a challenge and can be based upon severity of symptoms as well as length of illness. Symptoms of ABS are typically present and persistent for more than 10 days, without improvement. Continuing illness and worsening of symptoms are identifying features of ABS given most viral URIs gradually resolve within a 10-day timeframe. Other common symptoms include milky/thick nasal discharge, fever, predominantly nocturnal cough, and headache. Other less common symptoms include facial pain, toothache, malodorous breath, and periorbital edema. On physical examination, erythema and edema of the turbinates, as well as reproducible pain over aerated sinuses, are suggestive of ABS.10-14
Diagnosis
In the acute care setting, diagnosis of ABS should be clinical in nature. Neither imaging nor laboratory work-up is generally required secondary to their poor diagnostic specificity for ABS. The bacteria involved in ABS are similar to those associated with AOM, with S pneumonia, nontypeable H influenza, and M catarrhalis being the predominant organisms.10-15
Treatment and Management
Treatment of ABS is generally recommended once the diagnosis is made, though this is based largely on expert opinion as there are limited RCTs available.13 However, available studies do show a more rapid improvement in children on antibiotic therapy than those on placebo.15,16
Antibiotic Therapy. Amoxicillin remains the antimicrobial agent of choice for first-line treatment of uncomplicated ABS forsituations in which antimicrobial resistance is not suspected. In communities with a high prevalence of nonsusceptible S pneumoniae (>10%, including intermediate- and high-level resistance), treatment may be initiated at 80 to 90 mg/kg/d in two divided doses, with a maximum of 2 g per dose.
Patients presenting with moderate to severe illness, as well as those who are younger than 2 years, attend childcare, or have recently been treated with an antimicrobial, may receive high-dose amoxicillin-clavulanate as initial therapy given the elevated beta-lactamase production of the common bacteria that cause ABS.
Second-line alternatives include azithromycin, cefdinir, and sulfamethoxizole-trimethoprim (Table 1). There are data to suggest higher rates of decreased susceptibility of S pneumonia and H influenza to third-generation cephalosporins, and the addition of clindamycin may be warranted when utilizing those medications. Treatment is recommended for 10 to 14 days, though improvement should be noted within 1 to 3 days.10-12,14-17
Adjuvant Therapy. Additional therapies include nasal irrigation, decongestants, antihistamines, and intranasal steroids; however, there are only anecdotal reports of their efficacy in providing symptom relief. Therefore, there are insufficient evidence-based data to support or refute the role of these adjuvant therapies in treating pediatric patients with ABS.9,13
Case 2 Resolution
The prolonged duration and severity of symptoms (high fever and headache) and the gradual worsening of the clinical course (ie, late-onset fever) in this patient all suggest ABS rather than a simple prolonged URI. The physical examination findings of inflamed turbinates and facial pain further increase the specificity for ABS. The patient was started on oral amoxicillin-clavulanate with planned treatment for 14 days. At discharge, his parents were instructed to follow-up with the patient’s pediatrician in 3 days to ensure a degree of clinical resolution.
Case Scenario 3
A 4-year-old boy was presented by his parents for evaluation of a 2-day history of a persistent and unimproved sore throat. The patient’s mother indicated that the child’s oral T upon returning home earlier from preschool was 101.2oF. She further noted that her 17-month-old daughter and 8-year-old son also experienced similar symptoms which had self-resolved. Triage vital signs were: T, 100.8oF, orally; BP, HR, and RR were all within normal limits. Oxygen saturation was 100% on room air.
On physical examination, the child was noted to have anterior cervical lymph nodes bilaterally and an erythematous oropharynx with exudate noted on both tonsils. There were no cutaneous abnormalities, nasal edema, erythema, or drainage. Based on the clinical examination, the EP was suspicious for SP.
Questions to Guide the Work-Up: (1) Is SP diagnosed based on clinical findings alone in this patient’s age group? (2) At what age in the pediatric population is it appropriate to perform a rapid streptococcal antigen test? (3) Are there medications other than antibiotics that are beneficial in treating symptomatic SP?
Streptococcal Pharyngitis
Streptococcal pharyngitis is a clinical condition caused by group A beta-hemolytic S pyogens. This bacterium is responsible for multiple conditions, including pharyngitis, skin infections, poststreptococcal glomerulonephritis, and rheumatic fever, as well as invasive syndromes. (This case focuses solely on SP).
Pharyngitis can occur secondary to a viral or bacterial infection, and SP is the most common cause of pediatric bacterial pharyngitis. It is estimated that children aged 5 to 15 years are more commonly diagnosed with SP, although approximately 24% of children younger than age 5 years with pharyngitis symptoms will be ultimately diagnosed with SP.
Signs and Symptoms
Typical symptoms include fever, pharyngitis, generalized abdominal pain, nausea, vomiting, headache, and absence of viral URI symptoms (eg, cough, nasal discharge). However, younger patients with SP may have clinical findings of prolonged nasal drainage and excoriated nares. Examination findings may include swollen and tender anterior cervical lymph nodes; generalized edema and erythema of the posterior pharynx; tonsillar exudates; and palatal petechiae.
Diagnosis
Centor Criteria. The Centor criteria were developed to assist practitioners in identifying patients with potential SP. Criteria for patients older than age 15 years include fever, absence of cough, tonsillar exudates, and tender anterior cervical lymphadenopathy. A modified Centor criteria was later established to include children older than age 5 years, with children between ages 5 and 15 years being the fifth variable in the modified score. In general, patients with a score of 4 or 5 (presence of each variable = 1 point) are most likely to test positive for SP on rapid antigen testing (RAT) or culture.18-20
Swab, Rapid Antigen Testing, and Culture. Swabbing the throat and RAT and/or culture should be performed in most children with suspected SP because the clinical features alone do not reliably discriminate SP from viral pharyngitis. Rapid antigen testing is only specific for group A beta-hemolytic streptococcal species, which is the only streptococcal species that is routinely treated with antibiotics in the setting of acute pharyngitis. It is unlikely for a patient with a score of 0 or 1 to have SP, and several sources suggest neither testing nor treating this cohort, but rather to consider an alternative diagnosis.18-20
Within the population of children and young adolescents, due to a RAT sensitivity of 70% to 90%, a negative result should always be backed-up by a throat culture, and treatment initiated if results of the culture are later found to be positive. As the current generation of RAT tests have a high specificity, a positive RAT does not necessitate a back-up culture, and treatment is indicated without further investigation.19,20
Routine RAT is not recommended in children younger than age 3 years as patients in this age group are at low-risk of developing rheumatic fever. One notable exception for these very young children would be if there are siblings in the home with confirmed SP, in which case, RAT should be considered in the clinical context of SP.21 Adolescents over age 15 years are another cohort with a low likelihood of developing rheumatic fever, though they can develop other poststreptococcal complications, such as glomerulonephritis.
The US Centers for Disease Control and Prevention/American Academy of Family Practitioners (AAFP) guidelines suggest that pharyngitis in older adolescents can be approached in a similar fashion to adults, with empiric therapy for a Centor score of 3 or 4, RAT (without the need for follow-up culture) for Centor score of 2, and neither testing nor treating patients with a score of 0 or 1.19
Treatment and Management
Streptococcal pharyngitis is treated mainly to prevent the poststreptococcal complications of rheumatic fever, though it will not prevent poststreptococcal glomerulonephritis. Treatment of SP also facilitates resolution of symptoms and return to baseline activities.
Antibiotic Therapy. Patients who have a positive RAT or a follow-up throat culture positive for group A streptococcus should be given antibiotics. The gold standard treatment is penicillin V orally for 10 days.
Corticosteroid Therapy. The use of corticosteroids for symptom control of SP in pediatric patients is controversial. Although the Infectious Disease Society of America does not recommend corticosteroid therapy in the treatment of SP, several studies show such therapy (namely dexamethasone), improves pain in children and adolescents diagnosed with SP, but without significant change to the overall disease course.21,23-26
Case 3 Resolution
The patient had a modified Centor criteria score of 4, as well as siblings with similar symptoms. In following current guidelines, the EP performed a RAT and back-up culture. The RAT was negative in the ED, but the back-up culture was subsequently positive, and the child was started on a 10-day course of oral amoxicillin.
Conclusion
When evaluating pediatric patients presenting with ENT signs and symptoms such as ear pain and erythema, fever, sore throat, nasal congestion and discharge, a thorough physical examination and history-taking—including recent illness of any siblings—along with testing when indicated, is essential to guide the diagnosis and determine appropriate treatment and management. In addition to administering antibiotic therapy when such is warranted, the EP should provide appropriate analgesia to manage the patient’s pain and assure relief prior to discharge.
Among all of the causes of ear, nose, and throat (ENT) complaints, acute otitis media (AOM), bacterial sinusitis, and streptococcal pharyngitis (SP) are the most common infections prompting pediatric presentation to the ED. Through a series of case scenarios, along with key questions to help guide the clinician’s work-up, this review covers the proper evaluation and management of pediatric ENT complaints.
Case Scenario 1
A 13-month-old girl presented to the ED with a 1-day history of fever and runny nose. According to her parents, the child had been continually pulling on her ears in apparent discomfort. During history-taking, the parents further informed the emergency physician (EP) that the patient started daycare 4 months earlier and had two elementary school-aged siblings. The patient’s medical history was significant for otitis media, but the parents stated she had not been on antibiotics for over 4 months.
On physical examination, the patient’s vital signs were: blood pressure (BP), 75/50 mm Hg; temperature (T), 101.3°F; slight tachycardia; and normal age-adjusted respiratory rate (RR). Oxygen saturation was 100% on room air. The lungs were clear to auscultation and heart sounds were normal and without murmur. The otolaryngologic examination revealed copious yellow discharge from both nostrils, non-erythematous posterior oropharynx, and erythema to the right tympanic membrane (TM). Questions to Guide the Work-Up: (1) What physical examination findings should be present for accurate diagnosis of otitis media? (2) Will this patient require antibiotics immediately, or is a “wait-and-see” approach indicated? (3) If treatment with antibiotic therapy is warranted, what are the appropriate therapeutic regimen and duration of therapy?
Otitis Media
Acute otitis media is one of the most common presentations in young children. Defined as the rapid onset of signs and symptoms of middle ear inflammation, in conjunction with middle ear effusion (MEE), AOM can develop secondary to a viral or bacterial infection. It is estimated that more than 80% of the pediatric population will experience at least one episode of AOM by age 3 years.1-3
Risk factors for AOM include upper respiratory infection (URI), daycare attendance, siblings, parental smoking, and formula-feeding versus breastfeeding. The patient’s history may include rapid-onset otalgia, fever, irritability, anorexia, and concurrent URI symptoms, as well as other nonspecific symptoms (eg, ear rubbing and/or pulling, crying, changes in behavior and sleep patterns).2-4 In general, otalgia and ear-rubbing in the nonverbal patient seem to have the best predictive value for AOM.3
Signs and Symptoms
A normal TM should be translucent and pearly gray, with visible landmarks of the manubrium of malleus and pars flaccida. A TM that is bulging, cloudy, and immobile is the most consistent finding in AOM, with bulging having a specificity of 97%. Redness of the tympanic membrane is not a useful predictor of AOM as this finding is noted in upward of 30% of pediatric patients on general examination but in <1% of AOM diagnoses in the absence of a bulging TM.
Diagnosis
Pneumatic otoscopy is the gold standard for diagnosing for MEE; however, this examination can be difficult in younger, often uncooperative, patients. A TM that does not perceptibly move with either positive or negative insufflation pressure greatly enhances the diagnostic accuracy for MEE over the use of visible eardrum characteristics alone.2-5
Acute otitis media is a clinical diagnosis and does not require imaging studies or laboratory evaluation unless more serious processes, such as skull fracture, mastoiditis, or intracranial abscess, are being considered.2,3
Treatment and Management
Analgesia. The first step in managing patients with AOM is to provide analgesia. In most cases, acetaminophen in patients over 2 months of age, or ibuprofen in patients over 6 months of age, are adequate choices for managing pain. When either of these analgesics is administered in the clinic/ED setting, patients should be monitored to assure adequate pain relief prior to discharge.
While topical agents such as combination antipyrine-benzocaine suspensions were commonly given in the past to alleviate the pain associated with AOM, there are limited data to support their effectiveness. As such, in July 2015, the US Food and Drug Administration ordered manufacturers to halt production on these unapproved prescription products.3,4,6 There are also no randomized controlled trials (RCTs) to support the use of decongestants or antihistamines for resolution of AOM or otalgia.3,7
Antibiotic Therapy. The most common bacteria associated with AOM are Streptococcus pneumonia, nontypeable Hemophilus influenza, and Moraxella catarrhalis. In 30% of patients, the causative etiology is viral. When the decision is made to treat AOM, high-dose amoxicillin is still considered the first-line treatment, despite ever evolving susceptibilities of bacteria.
When a child is noted to have been treated with amoxicillin within a 30-day period or who has concurrent conjunctivitis, amoxicillin-clavulanate is considered the first-line treatment.2-4,7,8 The current American Academy of Pediatrics (AAP) guidelines recommend 10 days of antibiotic therapy for children younger than age 2 years, and 5 to 7 days for children older than age 2 years who have uncomplicated AOM. Intramuscular (IM) ceftriaxone is an acceptable first-line agent in a child who is unable to tolerate oral medications or who is suffering persistent emesis. Intramuscular ceftriaxone can be given as a single dose of 50 mg/kg, though the patient should be followed closely as studies show that a second dose may be necessary 5 to 7 days later to prevent infection recurrence. The IM dose of ceftriaxone 50 mg/kg can also be given if treatment with other antibiotics fails to resolve the AOM (failure is defined as no improvement in the patient’s condition 48 to 72 hours from treatment). In such cases, ceftriaxone is given in three consecutive doses.3,4,7
Wait-and-See Approach. Studies of patients whose AOM was confirmed via culture (19% were positive for S pneumoniae, 48% for H influenza, and 78% for M catarrhalis) showed bacterial clearance without antibiotic intervention.4 Based on these findings, the 2013 revised AAP evidence-based clinical practice guidelines indicate an initial watching-and-waiting period combined with pain management for patients older than 6 months of age who are diagnosed with unilateral AOM in the absence of severe symptoms (ie, fever is lower than 102.2˚F or patient has severe otalgia).4 A period of observation prior to treatment is also endorsed for children older than age 2 years who exhibit nonsevere symptoms—even if they have bilateral disease.4
Conversely, all patients younger than age 6 months and all children with severe symptoms should be treated with antibiotics at diagnosis.3,4 The wait-and-see approach, recommends an observation period of 24 to 48 hours for children in the lower risk group prior to antibiotic administration. Delayed antibiotic administration can be performed by a physician in an office/ED follow-up or as a safety-net antibiotic prescription (SNAP) sent home with the family on the initial ED encounter.2-4,8,9
Case 1 Resolution
Given this patient’s unilateral and nonsevere symptoms (minor otalgia, fever <102.2°F), age older than 6 months, and no recent antibiotic use), she was treated with oral ibuprofen. At discharge, the parents were given a 10-day SNAP prescription of high-dose amoxicillin (90 mg/kg/d, divided into two daily doses) and instructed to fill the prescription only if the patient’s otalgia did not improve in 1 or 2 days.
Case Scenario 2
A 5-year-old boy was presented for evaluation by his parents, who stated that their son had been sick since he had started kindergarten in the fall. The patient had a 10-day history of cough, thick runny nose, and facial pain, and a 1-day history of new-onset fever and headache. His parents further noted that the patient had been seen by his pediatrician several times over the past week. At each of these visits, the pediatrician had informed them that their son had a virus.
Vital signs on examination were: BP, 100/60 mm Hg; heart rate (HR), 112 beats/min; normal age-adjusted RR; and T, 102.6oF. Oxygen saturation was 100% on room air. The patient did not appear toxic, his lungs were clear on auscultation, and there were no other clinical signs suggestive of meningitis. The otolaryngologic examination revealed bilateral thick mucoid drainage and visible edema and erythema of the nasal turbinates. The patient was noted to have some facial pain in the maxillary area bilaterally.
Questions to Guide the Work-Up: (1) Does the patient have a prolonged URI or pediatric sinusitis, and what differentiates the two conditions? (2) What sinuses are present in a 5-year-old patient? (3) What treatment modalities are available for sinusitis? (4) Is imaging of the sinuses helpful in confirming the diagnosis?
Acute Bacterial Sinusitis
Rhinosinusitis is an inflammation of the mucosal lining of the nasal passages and paranasal sinuses. Most cases occur secondary to a viral URI and resolve spontaneously in 99% of the pediatric population.10,11
Acute bacterial sinusitis (ABS) is an inflammation of the same mucosal lining of the nasal passages secondary to bacterial overgrowth that lasts more than 10 days, with complete resolution by 30 days.12,13 When evaluating a pediatric patient for ABS, it is important to consider the sinus growth and development: If the sinus is not yet formed, it therefore cannot be the location of an ABS.13 The ethmoid and maxillary sinuses are present at birth, aerated within 4 months of life, and are fully developed by age 12 years. The sphenoid sinuses begin development around age 3 years, are aerated by age 7 or 8 years, and are fully developed by age 18 to 20 years. The frontal sinuses begin development around age 8 years and are aerated and fully developed by age 12 to 15 years.10,13,14 While most guidelines focus on children older than age 1 year (due to very small infantile sinuses), ABS does occur in children younger than age 1 year.12,14
Signs and Symptoms
Differentiation between a viral URI/rhinosinusitis and ABS is a challenge and can be based upon severity of symptoms as well as length of illness. Symptoms of ABS are typically present and persistent for more than 10 days, without improvement. Continuing illness and worsening of symptoms are identifying features of ABS given most viral URIs gradually resolve within a 10-day timeframe. Other common symptoms include milky/thick nasal discharge, fever, predominantly nocturnal cough, and headache. Other less common symptoms include facial pain, toothache, malodorous breath, and periorbital edema. On physical examination, erythema and edema of the turbinates, as well as reproducible pain over aerated sinuses, are suggestive of ABS.10-14
Diagnosis
In the acute care setting, diagnosis of ABS should be clinical in nature. Neither imaging nor laboratory work-up is generally required secondary to their poor diagnostic specificity for ABS. The bacteria involved in ABS are similar to those associated with AOM, with S pneumonia, nontypeable H influenza, and M catarrhalis being the predominant organisms.10-15
Treatment and Management
Treatment of ABS is generally recommended once the diagnosis is made, though this is based largely on expert opinion as there are limited RCTs available.13 However, available studies do show a more rapid improvement in children on antibiotic therapy than those on placebo.15,16
Antibiotic Therapy. Amoxicillin remains the antimicrobial agent of choice for first-line treatment of uncomplicated ABS forsituations in which antimicrobial resistance is not suspected. In communities with a high prevalence of nonsusceptible S pneumoniae (>10%, including intermediate- and high-level resistance), treatment may be initiated at 80 to 90 mg/kg/d in two divided doses, with a maximum of 2 g per dose.
Patients presenting with moderate to severe illness, as well as those who are younger than 2 years, attend childcare, or have recently been treated with an antimicrobial, may receive high-dose amoxicillin-clavulanate as initial therapy given the elevated beta-lactamase production of the common bacteria that cause ABS.
Second-line alternatives include azithromycin, cefdinir, and sulfamethoxizole-trimethoprim (Table 1). There are data to suggest higher rates of decreased susceptibility of S pneumonia and H influenza to third-generation cephalosporins, and the addition of clindamycin may be warranted when utilizing those medications. Treatment is recommended for 10 to 14 days, though improvement should be noted within 1 to 3 days.10-12,14-17
Adjuvant Therapy. Additional therapies include nasal irrigation, decongestants, antihistamines, and intranasal steroids; however, there are only anecdotal reports of their efficacy in providing symptom relief. Therefore, there are insufficient evidence-based data to support or refute the role of these adjuvant therapies in treating pediatric patients with ABS.9,13
Case 2 Resolution
The prolonged duration and severity of symptoms (high fever and headache) and the gradual worsening of the clinical course (ie, late-onset fever) in this patient all suggest ABS rather than a simple prolonged URI. The physical examination findings of inflamed turbinates and facial pain further increase the specificity for ABS. The patient was started on oral amoxicillin-clavulanate with planned treatment for 14 days. At discharge, his parents were instructed to follow-up with the patient’s pediatrician in 3 days to ensure a degree of clinical resolution.
Case Scenario 3
A 4-year-old boy was presented by his parents for evaluation of a 2-day history of a persistent and unimproved sore throat. The patient’s mother indicated that the child’s oral T upon returning home earlier from preschool was 101.2oF. She further noted that her 17-month-old daughter and 8-year-old son also experienced similar symptoms which had self-resolved. Triage vital signs were: T, 100.8oF, orally; BP, HR, and RR were all within normal limits. Oxygen saturation was 100% on room air.
On physical examination, the child was noted to have anterior cervical lymph nodes bilaterally and an erythematous oropharynx with exudate noted on both tonsils. There were no cutaneous abnormalities, nasal edema, erythema, or drainage. Based on the clinical examination, the EP was suspicious for SP.
Questions to Guide the Work-Up: (1) Is SP diagnosed based on clinical findings alone in this patient’s age group? (2) At what age in the pediatric population is it appropriate to perform a rapid streptococcal antigen test? (3) Are there medications other than antibiotics that are beneficial in treating symptomatic SP?
Streptococcal Pharyngitis
Streptococcal pharyngitis is a clinical condition caused by group A beta-hemolytic S pyogens. This bacterium is responsible for multiple conditions, including pharyngitis, skin infections, poststreptococcal glomerulonephritis, and rheumatic fever, as well as invasive syndromes. (This case focuses solely on SP).
Pharyngitis can occur secondary to a viral or bacterial infection, and SP is the most common cause of pediatric bacterial pharyngitis. It is estimated that children aged 5 to 15 years are more commonly diagnosed with SP, although approximately 24% of children younger than age 5 years with pharyngitis symptoms will be ultimately diagnosed with SP.
Signs and Symptoms
Typical symptoms include fever, pharyngitis, generalized abdominal pain, nausea, vomiting, headache, and absence of viral URI symptoms (eg, cough, nasal discharge). However, younger patients with SP may have clinical findings of prolonged nasal drainage and excoriated nares. Examination findings may include swollen and tender anterior cervical lymph nodes; generalized edema and erythema of the posterior pharynx; tonsillar exudates; and palatal petechiae.
Diagnosis
Centor Criteria. The Centor criteria were developed to assist practitioners in identifying patients with potential SP. Criteria for patients older than age 15 years include fever, absence of cough, tonsillar exudates, and tender anterior cervical lymphadenopathy. A modified Centor criteria was later established to include children older than age 5 years, with children between ages 5 and 15 years being the fifth variable in the modified score. In general, patients with a score of 4 or 5 (presence of each variable = 1 point) are most likely to test positive for SP on rapid antigen testing (RAT) or culture.18-20
Swab, Rapid Antigen Testing, and Culture. Swabbing the throat and RAT and/or culture should be performed in most children with suspected SP because the clinical features alone do not reliably discriminate SP from viral pharyngitis. Rapid antigen testing is only specific for group A beta-hemolytic streptococcal species, which is the only streptococcal species that is routinely treated with antibiotics in the setting of acute pharyngitis. It is unlikely for a patient with a score of 0 or 1 to have SP, and several sources suggest neither testing nor treating this cohort, but rather to consider an alternative diagnosis.18-20
Within the population of children and young adolescents, due to a RAT sensitivity of 70% to 90%, a negative result should always be backed-up by a throat culture, and treatment initiated if results of the culture are later found to be positive. As the current generation of RAT tests have a high specificity, a positive RAT does not necessitate a back-up culture, and treatment is indicated without further investigation.19,20
Routine RAT is not recommended in children younger than age 3 years as patients in this age group are at low-risk of developing rheumatic fever. One notable exception for these very young children would be if there are siblings in the home with confirmed SP, in which case, RAT should be considered in the clinical context of SP.21 Adolescents over age 15 years are another cohort with a low likelihood of developing rheumatic fever, though they can develop other poststreptococcal complications, such as glomerulonephritis.
The US Centers for Disease Control and Prevention/American Academy of Family Practitioners (AAFP) guidelines suggest that pharyngitis in older adolescents can be approached in a similar fashion to adults, with empiric therapy for a Centor score of 3 or 4, RAT (without the need for follow-up culture) for Centor score of 2, and neither testing nor treating patients with a score of 0 or 1.19
Treatment and Management
Streptococcal pharyngitis is treated mainly to prevent the poststreptococcal complications of rheumatic fever, though it will not prevent poststreptococcal glomerulonephritis. Treatment of SP also facilitates resolution of symptoms and return to baseline activities.
Antibiotic Therapy. Patients who have a positive RAT or a follow-up throat culture positive for group A streptococcus should be given antibiotics. The gold standard treatment is penicillin V orally for 10 days.
Corticosteroid Therapy. The use of corticosteroids for symptom control of SP in pediatric patients is controversial. Although the Infectious Disease Society of America does not recommend corticosteroid therapy in the treatment of SP, several studies show such therapy (namely dexamethasone), improves pain in children and adolescents diagnosed with SP, but without significant change to the overall disease course.21,23-26
Case 3 Resolution
The patient had a modified Centor criteria score of 4, as well as siblings with similar symptoms. In following current guidelines, the EP performed a RAT and back-up culture. The RAT was negative in the ED, but the back-up culture was subsequently positive, and the child was started on a 10-day course of oral amoxicillin.
Conclusion
When evaluating pediatric patients presenting with ENT signs and symptoms such as ear pain and erythema, fever, sore throat, nasal congestion and discharge, a thorough physical examination and history-taking—including recent illness of any siblings—along with testing when indicated, is essential to guide the diagnosis and determine appropriate treatment and management. In addition to administering antibiotic therapy when such is warranted, the EP should provide appropriate analgesia to manage the patient’s pain and assure relief prior to discharge.
1. Rosenfeld RM, Shin JJ, Schwartz SR, et al. Clinical practice guideline: otitis media with effusion (Update). Otolaryngol Head Neck Surg. 2016;154(1 Suppl):S1-S41. doi:10.1177/0194599815623467.
2. Acute Otitis Media Guideline Team, Cincinnati Children’s Hospital Medical Center. Evidence-based care guideline for medical management of acute otitis media in children 2 months to 13 years of age. http://f.i-md.com/medinfo/material/4f4/4eb132ba44ae4ffe12a814f4/4eb132d744ae4ffe12a814f7.pdf. August 2006. Accessed December 29, 2016.
3. Nesbit CE, Powers MC. An evidence-based approach to managing acute otitis media. Pediatr Emerg Med Pract. 2013;10(4):1-26; quiz 26-27.
4. Lieberthal AS, Carroll AE, Chonmaitree T, et al. The diagnosis and management of acute otitis media. Pediatrics. 2013;131(3):e964-e999. doi:10.1542/peds.2012-3488.
5. American Academy of Family Physicians; American Academy of Otolaryngology-Head and Neck Surgery; American Academy of Pediatrics Subcommittee on Otitis Media With Effusion. Otitis media with effusion. Pediatrics. 2004;113(5):1412-1429.
6. US Food and Drug Administration Web site. FDA: Use only approved prescription ear drops. http://www.fda.gov/ForConsumers/ConsumerUpdates/-ucm453087.htm. Updated July 10, 2015. Accessed December 15, 2016.
7. Sack F. An evidence based approach to the management of uncomplicated acute otitis media in children. Int Pediatrics. 2005;20(1):44-46.
8. Johnson NC, Holger JS. Pediatric acute otitis media: the case for delayed antibiotic treatment. J Emerg Med. 2007;32(3):279-284. doi:10.1016/j.jemermed.2006.07.029.
9. Spiro DM, Tay KY, Arnold DH, Dziura JD, Baker MD, Shapiro ED. Wait-and-see prescription for the treatment of acute otitis media: a randomized controlled trial. JAMA. 2006;296(10):1235-1241. doi:10.1001/jama.296.10.1235.
10. Brook I. Management of acute rhinosinusitis in pediatric patients. Pediatr Emerg Med Pract. 2012;9(5):1-24.
11. Ferdman RM, Linzer JF Jr. The runny nose in the emergency department: rhinitis and sinusitis. Clin Pediatr Emerg Med. 2007;8(2):123-130.
12. Acute Bacterial Sinusitis Guideline Team, Cincinnati Children’s Hospital Medical Center: Evidence-based care guideline for medical management of acute bacterial sinusitis in children 1 through 18 years of age. http://www.antibioticos.msssi.gob.es/PDF/sinusitisguideline.pdf. July 7, 2006. Accessed December 29, 2016
13. Holt KR, Murdoch Cuenca M, Cuenca PJ, Johnston GM. acute pediatric sinusitis and “the 10-day rule.” Pediatr Emerg Med Pract. 2006;3(2):1-16.
14. American Academy of Pediatrics. Subcommittee on Management of Sinusitis and Committee on Quality Improvement. Clinical practice guideline: management of sinusitis. Pediatrics. 2001;108(3):798-808.
15. Wald ER, Nash D, Eickhoff J. Effectiveness of amoxicillin/clavulanate potassium in the treatment of acute bacterial sinusitis in children. Pediatrics. 2009;124(1):9-15. doi:10.1542/peds.2008-2902.
16. Arroll B, Kenealy T. Are antibiotics effective for acute purulent rhinitis? Systematic review and meta-analysis of placebo controlled randomised trials. BMJ. 2006;333(7562):279. doi:10.1136/bmj.38891.681215.AE.
17. McQuillan L, Crane LA, Kempe A. Diagnosis and management of acute sinusitis by pediatricians. Pediatrics. 2009;123(2):e193-e198.
18. Singer JI, Fontanette R. Recognizable and suspected group A beta-hemolytic streptococcal syndromes. Pediatr Emerg Med Rep. 2010;15(11):129-144.
19. Weglowski J. An evidence-based approach to the evaluation and treatment of pharyngitis in children. Pediatr Emerg Med Pract. 2011;8(12):1-28.
20. Gerber MA, Baltimore RS, Eaton CB, et al. Prevention of rheumatic fever and diagnosis and treatment of acute Streptococcal pharyngitis: a scientific statement from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee of the Council on Cardiovascular Disease in the Young, the Interdisciplinary Council on Functional Genomics and Translational Biology, and the Interdisciplinary Council on Quality of Care and Outcomes Research: endorsed by the American Academy of Pediatrics. Circulation. 2009;119(11):1541-1551. doi:10.1161/CIRCULATIONAHA.109.191959.
21. Shulman ST, Bisno AL, Clegg HW, et al. Clinical practice guideline for the diagnosis and management of group A streptococcal pharyngitis: 2012 update by the Infectious Diseases Society of America. Clin Infect Dis. 2012;55(10):1279-1282. doi:10.1093/cid/cis847.
22. Clegg HW, Ryan AG, Dallas SD, et al. Treatment of streptococcal pharyngitis with once-daily compared with twice-daily amoxicillin: a noninferiority trial. Pediatr Infect Dis J. 2006;25(9):761-767. doi:10.1097/01.inf.0000235678.46805.92.
23. Bulloch B, Kabani A, Tenenbein M. Oral dexamethasone for the treatment of pain in children with acute pharyngitis: a randomized, double-blind, placebo-controlled trial. Ann Emerg Med. 2003;41(5):601-608. doi:10.1067/mem.2003.136.
24. Niland ML, Bonsu BK, Nuss KE, Goodman DG. A pilot study of 1 versus 3 days of dexamethasone as add-on therapy in children with streptococcal pharyngitis. Pediatr Infect Dis J. 2006;25(6):477-481. doi:10.1097/01.inf.0000219469.95772.3f.
25. Wei JL, Kasperbauer JL, Weaver AL, Boggust AJ. Efficacy of single-dose dexamethasone as adjuvant therapy for acute pharyngitis. Laryngoscope. 2002;112(1):87-93. doi:10.1097/00005537-200201000-00016.
26. Hayward G, Thompson M, Heneghan C, Perera R, Del Mar C, Glasziou P. Corticosteroids for pain relief in sore throat: systematic review and meta-analysis. BMJ. 2009;339:b2976. doi:10.1136/bmj.b2976.
1. Rosenfeld RM, Shin JJ, Schwartz SR, et al. Clinical practice guideline: otitis media with effusion (Update). Otolaryngol Head Neck Surg. 2016;154(1 Suppl):S1-S41. doi:10.1177/0194599815623467.
2. Acute Otitis Media Guideline Team, Cincinnati Children’s Hospital Medical Center. Evidence-based care guideline for medical management of acute otitis media in children 2 months to 13 years of age. http://f.i-md.com/medinfo/material/4f4/4eb132ba44ae4ffe12a814f4/4eb132d744ae4ffe12a814f7.pdf. August 2006. Accessed December 29, 2016.
3. Nesbit CE, Powers MC. An evidence-based approach to managing acute otitis media. Pediatr Emerg Med Pract. 2013;10(4):1-26; quiz 26-27.
4. Lieberthal AS, Carroll AE, Chonmaitree T, et al. The diagnosis and management of acute otitis media. Pediatrics. 2013;131(3):e964-e999. doi:10.1542/peds.2012-3488.
5. American Academy of Family Physicians; American Academy of Otolaryngology-Head and Neck Surgery; American Academy of Pediatrics Subcommittee on Otitis Media With Effusion. Otitis media with effusion. Pediatrics. 2004;113(5):1412-1429.
6. US Food and Drug Administration Web site. FDA: Use only approved prescription ear drops. http://www.fda.gov/ForConsumers/ConsumerUpdates/-ucm453087.htm. Updated July 10, 2015. Accessed December 15, 2016.
7. Sack F. An evidence based approach to the management of uncomplicated acute otitis media in children. Int Pediatrics. 2005;20(1):44-46.
8. Johnson NC, Holger JS. Pediatric acute otitis media: the case for delayed antibiotic treatment. J Emerg Med. 2007;32(3):279-284. doi:10.1016/j.jemermed.2006.07.029.
9. Spiro DM, Tay KY, Arnold DH, Dziura JD, Baker MD, Shapiro ED. Wait-and-see prescription for the treatment of acute otitis media: a randomized controlled trial. JAMA. 2006;296(10):1235-1241. doi:10.1001/jama.296.10.1235.
10. Brook I. Management of acute rhinosinusitis in pediatric patients. Pediatr Emerg Med Pract. 2012;9(5):1-24.
11. Ferdman RM, Linzer JF Jr. The runny nose in the emergency department: rhinitis and sinusitis. Clin Pediatr Emerg Med. 2007;8(2):123-130.
12. Acute Bacterial Sinusitis Guideline Team, Cincinnati Children’s Hospital Medical Center: Evidence-based care guideline for medical management of acute bacterial sinusitis in children 1 through 18 years of age. http://www.antibioticos.msssi.gob.es/PDF/sinusitisguideline.pdf. July 7, 2006. Accessed December 29, 2016
13. Holt KR, Murdoch Cuenca M, Cuenca PJ, Johnston GM. acute pediatric sinusitis and “the 10-day rule.” Pediatr Emerg Med Pract. 2006;3(2):1-16.
14. American Academy of Pediatrics. Subcommittee on Management of Sinusitis and Committee on Quality Improvement. Clinical practice guideline: management of sinusitis. Pediatrics. 2001;108(3):798-808.
15. Wald ER, Nash D, Eickhoff J. Effectiveness of amoxicillin/clavulanate potassium in the treatment of acute bacterial sinusitis in children. Pediatrics. 2009;124(1):9-15. doi:10.1542/peds.2008-2902.
16. Arroll B, Kenealy T. Are antibiotics effective for acute purulent rhinitis? Systematic review and meta-analysis of placebo controlled randomised trials. BMJ. 2006;333(7562):279. doi:10.1136/bmj.38891.681215.AE.
17. McQuillan L, Crane LA, Kempe A. Diagnosis and management of acute sinusitis by pediatricians. Pediatrics. 2009;123(2):e193-e198.
18. Singer JI, Fontanette R. Recognizable and suspected group A beta-hemolytic streptococcal syndromes. Pediatr Emerg Med Rep. 2010;15(11):129-144.
19. Weglowski J. An evidence-based approach to the evaluation and treatment of pharyngitis in children. Pediatr Emerg Med Pract. 2011;8(12):1-28.
20. Gerber MA, Baltimore RS, Eaton CB, et al. Prevention of rheumatic fever and diagnosis and treatment of acute Streptococcal pharyngitis: a scientific statement from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee of the Council on Cardiovascular Disease in the Young, the Interdisciplinary Council on Functional Genomics and Translational Biology, and the Interdisciplinary Council on Quality of Care and Outcomes Research: endorsed by the American Academy of Pediatrics. Circulation. 2009;119(11):1541-1551. doi:10.1161/CIRCULATIONAHA.109.191959.
21. Shulman ST, Bisno AL, Clegg HW, et al. Clinical practice guideline for the diagnosis and management of group A streptococcal pharyngitis: 2012 update by the Infectious Diseases Society of America. Clin Infect Dis. 2012;55(10):1279-1282. doi:10.1093/cid/cis847.
22. Clegg HW, Ryan AG, Dallas SD, et al. Treatment of streptococcal pharyngitis with once-daily compared with twice-daily amoxicillin: a noninferiority trial. Pediatr Infect Dis J. 2006;25(9):761-767. doi:10.1097/01.inf.0000235678.46805.92.
23. Bulloch B, Kabani A, Tenenbein M. Oral dexamethasone for the treatment of pain in children with acute pharyngitis: a randomized, double-blind, placebo-controlled trial. Ann Emerg Med. 2003;41(5):601-608. doi:10.1067/mem.2003.136.
24. Niland ML, Bonsu BK, Nuss KE, Goodman DG. A pilot study of 1 versus 3 days of dexamethasone as add-on therapy in children with streptococcal pharyngitis. Pediatr Infect Dis J. 2006;25(6):477-481. doi:10.1097/01.inf.0000219469.95772.3f.
25. Wei JL, Kasperbauer JL, Weaver AL, Boggust AJ. Efficacy of single-dose dexamethasone as adjuvant therapy for acute pharyngitis. Laryngoscope. 2002;112(1):87-93. doi:10.1097/00005537-200201000-00016.
26. Hayward G, Thompson M, Heneghan C, Perera R, Del Mar C, Glasziou P. Corticosteroids for pain relief in sore throat: systematic review and meta-analysis. BMJ. 2009;339:b2976. doi:10.1136/bmj.b2976.
First-trimester blood glucose predicts congenital heart disease risk
NEW ORLEANS – A single, random, first-trimester maternal plasma glucose measurement is superior to an oral glucose tolerance test later in pregnancy as a predictor of congenital heart disease in newborns, Emmi Helle, MD, reported at the American Heart Association scientific sessions.
This finding from a large retrospective study, if confirmed in a prospective data set, is likely to be practice changing. At present, a 1-hour oral glucose tolerance test in the second or third trimester is considered the best means of identifying pregnant women who ought to undergo fetal echocardiography for prenatal diagnosis of congenital heart disease, noted Dr. Helle of Stanford (Calif.) University.
An elevated random plasma glucose value in the first trimester was broadly predictive of increased risk for a variety of congenital heart anomalies, not just, for example, cyanotic conditions.
Fetal heart development is completed during the first trimester, Dr. Helle observed.
Her study received a warm reception. Michael A. Portman, MD, singled it out in his final-day wrap-up of the meeting’s highlights in the field of congenital heart disease.
Several studies have demonstrated that prenatal diagnosis of congenital heart disease results in improved surgical outcomes in newborns. The question is, how to get the right women – those at increased risk – to diagnostic fetal echocardiography. Guidelines suggest but don’t mandate on the basis of weak evidence that an oral glucose tolerance test performed in the second or early third trimester may be a useful means of screening mothers for fetal imaging. Dr. Helle’s study points to a better way.
“Hopefully we can change our guidelines and make them more scientific for identification of mothers who should undergo fetal echocardiography,” said Dr. Portman, professor of pediatrics at the University of Washington, Seattle, and director of pediatric cardiovascular research at Seattle Children’s Hospital.
Dr. Helle and Dr. Portman reported having no relevant financial interests.
NEW ORLEANS – A single, random, first-trimester maternal plasma glucose measurement is superior to an oral glucose tolerance test later in pregnancy as a predictor of congenital heart disease in newborns, Emmi Helle, MD, reported at the American Heart Association scientific sessions.
This finding from a large retrospective study, if confirmed in a prospective data set, is likely to be practice changing. At present, a 1-hour oral glucose tolerance test in the second or third trimester is considered the best means of identifying pregnant women who ought to undergo fetal echocardiography for prenatal diagnosis of congenital heart disease, noted Dr. Helle of Stanford (Calif.) University.
An elevated random plasma glucose value in the first trimester was broadly predictive of increased risk for a variety of congenital heart anomalies, not just, for example, cyanotic conditions.
Fetal heart development is completed during the first trimester, Dr. Helle observed.
Her study received a warm reception. Michael A. Portman, MD, singled it out in his final-day wrap-up of the meeting’s highlights in the field of congenital heart disease.
Several studies have demonstrated that prenatal diagnosis of congenital heart disease results in improved surgical outcomes in newborns. The question is, how to get the right women – those at increased risk – to diagnostic fetal echocardiography. Guidelines suggest but don’t mandate on the basis of weak evidence that an oral glucose tolerance test performed in the second or early third trimester may be a useful means of screening mothers for fetal imaging. Dr. Helle’s study points to a better way.
“Hopefully we can change our guidelines and make them more scientific for identification of mothers who should undergo fetal echocardiography,” said Dr. Portman, professor of pediatrics at the University of Washington, Seattle, and director of pediatric cardiovascular research at Seattle Children’s Hospital.
Dr. Helle and Dr. Portman reported having no relevant financial interests.
NEW ORLEANS – A single, random, first-trimester maternal plasma glucose measurement is superior to an oral glucose tolerance test later in pregnancy as a predictor of congenital heart disease in newborns, Emmi Helle, MD, reported at the American Heart Association scientific sessions.
This finding from a large retrospective study, if confirmed in a prospective data set, is likely to be practice changing. At present, a 1-hour oral glucose tolerance test in the second or third trimester is considered the best means of identifying pregnant women who ought to undergo fetal echocardiography for prenatal diagnosis of congenital heart disease, noted Dr. Helle of Stanford (Calif.) University.
An elevated random plasma glucose value in the first trimester was broadly predictive of increased risk for a variety of congenital heart anomalies, not just, for example, cyanotic conditions.
Fetal heart development is completed during the first trimester, Dr. Helle observed.
Her study received a warm reception. Michael A. Portman, MD, singled it out in his final-day wrap-up of the meeting’s highlights in the field of congenital heart disease.
Several studies have demonstrated that prenatal diagnosis of congenital heart disease results in improved surgical outcomes in newborns. The question is, how to get the right women – those at increased risk – to diagnostic fetal echocardiography. Guidelines suggest but don’t mandate on the basis of weak evidence that an oral glucose tolerance test performed in the second or early third trimester may be a useful means of screening mothers for fetal imaging. Dr. Helle’s study points to a better way.
“Hopefully we can change our guidelines and make them more scientific for identification of mothers who should undergo fetal echocardiography,” said Dr. Portman, professor of pediatrics at the University of Washington, Seattle, and director of pediatric cardiovascular research at Seattle Children’s Hospital.
Dr. Helle and Dr. Portman reported having no relevant financial interests.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point:
Major finding: For every 10-mg/dL increase in maternal plasma glucose on a random first-trimester measurement, the risk of giving birth to a baby with congenital heart disease rose by 8%.
Data source: A retrospective study of 19,197 pregnancies, 811 of which resulted in congenital heart disease in the offspring.
Disclosures: The presenter reported having no financial conflicts of interest regarding the study.
Fewer people having problems with medical bills
The number of people under age 65 years who were in families having trouble paying medical bills dropped by more than 22% from 2011 to 2016, according to the National Center for Health Statistics.
For the first 6 months of 2016, there were 43.8 million people, or 16.2% of the population under age 65 years, who were in families that had problems paying medical bills in the past year, which was down from 56.5 million (21.3 % of the population) in 2011, the NCHS reported.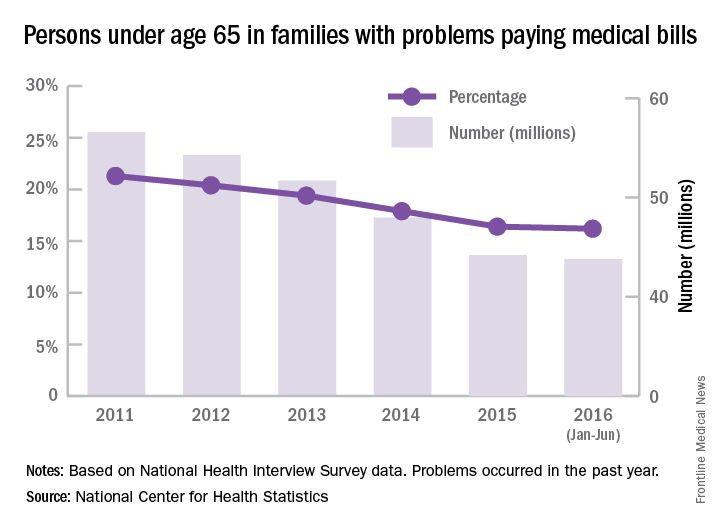
The drop was consistent across race/ethnicity lines, but not the start and endpoints. The percentage of non-Hispanic blacks in families having trouble paying their medical bills dropped from 27.3% in 2011 to 23% in 2016, although there was actually a small increase from 2015 to 2016. Hispanics dropped from 24.3% in 2011 to 17.4% in 2016, non-Hispanic whites dropped from 19.8% to 15.1%, and non-Hispanic Asians went from 11% to 6%, according to data collected from 579,379 people for the National Health Interview Survey.
The number of people under age 65 years who were in families having trouble paying medical bills dropped by more than 22% from 2011 to 2016, according to the National Center for Health Statistics.
For the first 6 months of 2016, there were 43.8 million people, or 16.2% of the population under age 65 years, who were in families that had problems paying medical bills in the past year, which was down from 56.5 million (21.3 % of the population) in 2011, the NCHS reported.
The drop was consistent across race/ethnicity lines, but not the start and endpoints. The percentage of non-Hispanic blacks in families having trouble paying their medical bills dropped from 27.3% in 2011 to 23% in 2016, although there was actually a small increase from 2015 to 2016. Hispanics dropped from 24.3% in 2011 to 17.4% in 2016, non-Hispanic whites dropped from 19.8% to 15.1%, and non-Hispanic Asians went from 11% to 6%, according to data collected from 579,379 people for the National Health Interview Survey.
The number of people under age 65 years who were in families having trouble paying medical bills dropped by more than 22% from 2011 to 2016, according to the National Center for Health Statistics.
For the first 6 months of 2016, there were 43.8 million people, or 16.2% of the population under age 65 years, who were in families that had problems paying medical bills in the past year, which was down from 56.5 million (21.3 % of the population) in 2011, the NCHS reported.
The drop was consistent across race/ethnicity lines, but not the start and endpoints. The percentage of non-Hispanic blacks in families having trouble paying their medical bills dropped from 27.3% in 2011 to 23% in 2016, although there was actually a small increase from 2015 to 2016. Hispanics dropped from 24.3% in 2011 to 17.4% in 2016, non-Hispanic whites dropped from 19.8% to 15.1%, and non-Hispanic Asians went from 11% to 6%, according to data collected from 579,379 people for the National Health Interview Survey.
Sorafenib survival benefit in HCC depends on etiology
The impact of sorafenib in treating patients with advanced hepatocellular cancer may be dependent on their hepatitis status, according to a meta-analysis of patient-level data from three large prospective clinical trials using sorafenib as the control treatment.
The analysis indicated that sorafenib had a favorable effect on overall survival for hepatocellular carcinoma patients who were positive for hepatitis C (HCV), but negative for hepatitis B (HBV) only.
For HCV-positive/HBV-negative patients with advanced unresectable hepatocellular carcinoma (aHCC) who received sorafenib, median unadjusted survival was 12.6 months, compared to 10.2 months for patients who received other treatments, yielding a log hazard ratio of –0.27 (95% confidence interval [CI] –0.46 to –0.06).
Though the study did not shed light on the reasons for this difference in overall survival (OS), the results were seen consistently in data from all trials, and sorafenib did not confer any significant survival benefit for individuals who were not HCV positive and HBV negative. “Irrespective of the mechanism, our data suggest that in future trials in aHCC, particularly where sorafenib is the control arm, there should be stratification according to etiology,” wrote Richard Jackson, MSc, and his coauthors (J Clin Oncol. 2017 Jan 3:JCO2016695197 [Epub ahead of print]).
Mr. Jackson, a medical statistician with the Institute of Translational Medicine at the University of Liverpool, England, and his collaborators examined data from 3,256 patients with aHCC. Of these, 1,643 (50%) received sorafenib. The remainder was aggregated into an “other treatment” group, pooling data from patients who received brivanib, sunitinib, and linifanib.
All patients were also divided into four etiologic subgroups: HBV-negative/HCV-negative; HBV-negative/HCV-positive; HBV-positive/HCV-negative; and HBV-positive/HCV-positive. Mr. Jackson and his colleagues then examined the pooled data to see what effect treatment type (sorafenib versus pooled comparator treatments) had on overall survival for each etiologic subgroup. They found no statistically significant survival benefit for sorafenib in the three etiologic subgroups that were not HCV positive/HBV negative, though the data showed a statistically insignificant trend favoring sorafenib. Race, when examined as a potential confounding factor, was not associated with a difference in OS benefit for sorafenib.
The sponsors of three large clinical trials of treatments for aHCC that used sorafenib as the control arm provided deidentified patient-level data to the study’s authors, who then undertook an individual patient data (IPD) meta-analysis. “IPD meta-analyses have a major advantage over aggregate meta-analyses in that they ensure consistent analytic techniques and allow for detailed inspection of interaction of subgroup effects that are not available in published evidence,” wrote Mr. Jackson and his coauthors.
In discussing their findings, the researchers called for more of the data-sharing that allowed their IPD meta-analysis, citing a proposal on the topic from the International Committee of Medical Journal Editors. “[O]ur study showed how the benefits of access to completed trial data are not necessarily confined to reanalysis of the original hypothesis tested by the trial. Here, by a meta-analysis, we arrive at an answer to a question that was not considered when the trials were conceived and could not have been answered by any of the trials individually,” they wrote.
The study data were provided by Bristol-Myers Squibb, Pfizer, and AbbVie from studies they sponsored. Mr. Jackson reported no conflicts of interest.
[email protected]
On Twitter @karioakes
The impact of sorafenib in treating patients with advanced hepatocellular cancer may be dependent on their hepatitis status, according to a meta-analysis of patient-level data from three large prospective clinical trials using sorafenib as the control treatment.
The analysis indicated that sorafenib had a favorable effect on overall survival for hepatocellular carcinoma patients who were positive for hepatitis C (HCV), but negative for hepatitis B (HBV) only.
For HCV-positive/HBV-negative patients with advanced unresectable hepatocellular carcinoma (aHCC) who received sorafenib, median unadjusted survival was 12.6 months, compared to 10.2 months for patients who received other treatments, yielding a log hazard ratio of –0.27 (95% confidence interval [CI] –0.46 to –0.06).
Though the study did not shed light on the reasons for this difference in overall survival (OS), the results were seen consistently in data from all trials, and sorafenib did not confer any significant survival benefit for individuals who were not HCV positive and HBV negative. “Irrespective of the mechanism, our data suggest that in future trials in aHCC, particularly where sorafenib is the control arm, there should be stratification according to etiology,” wrote Richard Jackson, MSc, and his coauthors (J Clin Oncol. 2017 Jan 3:JCO2016695197 [Epub ahead of print]).
Mr. Jackson, a medical statistician with the Institute of Translational Medicine at the University of Liverpool, England, and his collaborators examined data from 3,256 patients with aHCC. Of these, 1,643 (50%) received sorafenib. The remainder was aggregated into an “other treatment” group, pooling data from patients who received brivanib, sunitinib, and linifanib.
All patients were also divided into four etiologic subgroups: HBV-negative/HCV-negative; HBV-negative/HCV-positive; HBV-positive/HCV-negative; and HBV-positive/HCV-positive. Mr. Jackson and his colleagues then examined the pooled data to see what effect treatment type (sorafenib versus pooled comparator treatments) had on overall survival for each etiologic subgroup. They found no statistically significant survival benefit for sorafenib in the three etiologic subgroups that were not HCV positive/HBV negative, though the data showed a statistically insignificant trend favoring sorafenib. Race, when examined as a potential confounding factor, was not associated with a difference in OS benefit for sorafenib.
The sponsors of three large clinical trials of treatments for aHCC that used sorafenib as the control arm provided deidentified patient-level data to the study’s authors, who then undertook an individual patient data (IPD) meta-analysis. “IPD meta-analyses have a major advantage over aggregate meta-analyses in that they ensure consistent analytic techniques and allow for detailed inspection of interaction of subgroup effects that are not available in published evidence,” wrote Mr. Jackson and his coauthors.
In discussing their findings, the researchers called for more of the data-sharing that allowed their IPD meta-analysis, citing a proposal on the topic from the International Committee of Medical Journal Editors. “[O]ur study showed how the benefits of access to completed trial data are not necessarily confined to reanalysis of the original hypothesis tested by the trial. Here, by a meta-analysis, we arrive at an answer to a question that was not considered when the trials were conceived and could not have been answered by any of the trials individually,” they wrote.
The study data were provided by Bristol-Myers Squibb, Pfizer, and AbbVie from studies they sponsored. Mr. Jackson reported no conflicts of interest.
[email protected]
On Twitter @karioakes
The impact of sorafenib in treating patients with advanced hepatocellular cancer may be dependent on their hepatitis status, according to a meta-analysis of patient-level data from three large prospective clinical trials using sorafenib as the control treatment.
The analysis indicated that sorafenib had a favorable effect on overall survival for hepatocellular carcinoma patients who were positive for hepatitis C (HCV), but negative for hepatitis B (HBV) only.
For HCV-positive/HBV-negative patients with advanced unresectable hepatocellular carcinoma (aHCC) who received sorafenib, median unadjusted survival was 12.6 months, compared to 10.2 months for patients who received other treatments, yielding a log hazard ratio of –0.27 (95% confidence interval [CI] –0.46 to –0.06).
Though the study did not shed light on the reasons for this difference in overall survival (OS), the results were seen consistently in data from all trials, and sorafenib did not confer any significant survival benefit for individuals who were not HCV positive and HBV negative. “Irrespective of the mechanism, our data suggest that in future trials in aHCC, particularly where sorafenib is the control arm, there should be stratification according to etiology,” wrote Richard Jackson, MSc, and his coauthors (J Clin Oncol. 2017 Jan 3:JCO2016695197 [Epub ahead of print]).
Mr. Jackson, a medical statistician with the Institute of Translational Medicine at the University of Liverpool, England, and his collaborators examined data from 3,256 patients with aHCC. Of these, 1,643 (50%) received sorafenib. The remainder was aggregated into an “other treatment” group, pooling data from patients who received brivanib, sunitinib, and linifanib.
All patients were also divided into four etiologic subgroups: HBV-negative/HCV-negative; HBV-negative/HCV-positive; HBV-positive/HCV-negative; and HBV-positive/HCV-positive. Mr. Jackson and his colleagues then examined the pooled data to see what effect treatment type (sorafenib versus pooled comparator treatments) had on overall survival for each etiologic subgroup. They found no statistically significant survival benefit for sorafenib in the three etiologic subgroups that were not HCV positive/HBV negative, though the data showed a statistically insignificant trend favoring sorafenib. Race, when examined as a potential confounding factor, was not associated with a difference in OS benefit for sorafenib.
The sponsors of three large clinical trials of treatments for aHCC that used sorafenib as the control arm provided deidentified patient-level data to the study’s authors, who then undertook an individual patient data (IPD) meta-analysis. “IPD meta-analyses have a major advantage over aggregate meta-analyses in that they ensure consistent analytic techniques and allow for detailed inspection of interaction of subgroup effects that are not available in published evidence,” wrote Mr. Jackson and his coauthors.
In discussing their findings, the researchers called for more of the data-sharing that allowed their IPD meta-analysis, citing a proposal on the topic from the International Committee of Medical Journal Editors. “[O]ur study showed how the benefits of access to completed trial data are not necessarily confined to reanalysis of the original hypothesis tested by the trial. Here, by a meta-analysis, we arrive at an answer to a question that was not considered when the trials were conceived and could not have been answered by any of the trials individually,” they wrote.
The study data were provided by Bristol-Myers Squibb, Pfizer, and AbbVie from studies they sponsored. Mr. Jackson reported no conflicts of interest.
[email protected]
On Twitter @karioakes
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point:
Major finding: Median unadjusted survival time for HCV-positive/HBV-negative patients was 12.6 months on sorafenib, compared to 10.2 months for these patients who received other treatments.
Data source: Individual patient data meta-analysis of three randomized phase III clinical trials, examining data from 3,256 patients.
Disclosures: The study data were provided by Bristol-Myers Squibb, Pfizer, and AbbVie. Mr. Jackson reported no conflicts of interest.
Gastric cancer yields to growth hormone antagonist in lab
It sounds counterintuitive, but targeting a neuropeptide hormone produced in the hypothalamus may be an effective strategy for treating gastric cancer, the second most common cause of cancer deaths worldwide, investigators from China and the United States contend.
Growth hormone–releasing hormone (GHRH) and its receptor (GHRH-R) are found primarily in the anterior pituitary gland, but are also present in gastric cancers, other solid tumors, and lymphomas. Increased levels of GHRH-R in tumor samples from patients with gastric cancer are associated with poor outcomes, noted Andrew V. Schally, PhD, MD, DSc, of the University of Miami, and his colleagues at the Shantou (China) University Medical College.
Furthermore, an experimental peptide drug labeled MIA-602 that targets GHRH-R inhibited the growth of gastric cancer cell lines and human tumor xenografts in mice, the investigators reported in the journal PNAS.
“The GHRH receptor is both a biomarker that can confirm prognosis and a therapeutic target,” Dr. Schally said in a statement.
Elevated GHRH-R expression in tumors
GHRH-R antagonists such as MIA-602 work through downregulation of the p21-activated kinase 1 (PAK1)–mediated signal transducer and activator of transcription 3 (STAT3)/nuclear factor–kappaB (NF-kappaB) inflammatory pathway. This pathway is involved in the interplay between inflammatory processes and intracellular signaling thought to be the cause of gastric cancer tumorigenesis and progression, the investigators explained.
They first looked for GHRH-R expression in gastric cancer samples from 106 patients, using immunohistochemistry staining of primary tumors and adjacent normal tissues. They found that gastric cancer tissues “exhibited robust expression of GHRH-R, compared with normal tissues.”
In 50 samples, GHRH-R was determined to be overexpressed, and this overexpression was significantly associated with both greater tumor size (P = .031) and high pathologic tumor stage (P = .001). Increasing expression of GHRH-R was also significantly associated with worse overall survival (P less than .001).
They confirmed these findings in samples from a multinational cohort of patients, which again showed that the highest levels of GHRH-R expression were associated with poor overall survival (P less than .001).
The authors also looked at messenger RNA expression and gene copy number in 65 gastric cancer samples and 19 adjacent normal tissue samples, and found that GHRH-R mRNA was significantly higher in tumor tissues than normal control tissues (P less than .001).
MAI-602 in vitro and in vivo
To see whether MAI-602 could inhibit the growth of gastric cancer cells, the investigators tried it at various doses in three human gastric cancer cell lines, and found that it inhibited cells in a dose-dependent fashion, compared with vehicle used as a control (P less than .001).
In addition, the experimental agent “exhibited remarkable inhibitory effects on tumor growth in vivo” in mice with human tumor xenografts (P less than .001).
Finally, they showed that the cancer suppression effects of MAI-602 work through inhibition of STAT3/NF-kappaB inflammatory signaling. In vitro and in vivo, MAI-602 decreased the expression of both GHRH and GHRH-R, whereas as a GHRH-R agonist increased levels of both the hormone and its receptor. They also demonstrated that PAK1 appears to be a critical mediator of STAT3/NF-kappaB activity, and that MAI-602 works primarily by blocking PAK1-mediated inflammatory signaling.
“MIA-602 remarkably inhibits the growth of human in vitro and in vivo through the suppression of PAK1–STAT3/NF-kappaB signaling. Our study strongly highlights the therapeutic potential of GHRH-R antagonists in the treatment of gastric cancer patients. Knowledge gained in our study will shed light on how to select the appropriate patients for personalized cancer therapy using GHRH-R antagonists,” Dr. Schally and his coauthors wrote.
The study was supported by the Li Ka Shing Foundation, Chinese foundation, and government grants to individual researchers, as well as support from the the Medical Research Service of the U.S. Department of Veterans Affairs, South Florida Veterans Affairs Foundation for Research and Education, and the University of Miami.
It sounds counterintuitive, but targeting a neuropeptide hormone produced in the hypothalamus may be an effective strategy for treating gastric cancer, the second most common cause of cancer deaths worldwide, investigators from China and the United States contend.
Growth hormone–releasing hormone (GHRH) and its receptor (GHRH-R) are found primarily in the anterior pituitary gland, but are also present in gastric cancers, other solid tumors, and lymphomas. Increased levels of GHRH-R in tumor samples from patients with gastric cancer are associated with poor outcomes, noted Andrew V. Schally, PhD, MD, DSc, of the University of Miami, and his colleagues at the Shantou (China) University Medical College.
Furthermore, an experimental peptide drug labeled MIA-602 that targets GHRH-R inhibited the growth of gastric cancer cell lines and human tumor xenografts in mice, the investigators reported in the journal PNAS.
“The GHRH receptor is both a biomarker that can confirm prognosis and a therapeutic target,” Dr. Schally said in a statement.
Elevated GHRH-R expression in tumors
GHRH-R antagonists such as MIA-602 work through downregulation of the p21-activated kinase 1 (PAK1)–mediated signal transducer and activator of transcription 3 (STAT3)/nuclear factor–kappaB (NF-kappaB) inflammatory pathway. This pathway is involved in the interplay between inflammatory processes and intracellular signaling thought to be the cause of gastric cancer tumorigenesis and progression, the investigators explained.
They first looked for GHRH-R expression in gastric cancer samples from 106 patients, using immunohistochemistry staining of primary tumors and adjacent normal tissues. They found that gastric cancer tissues “exhibited robust expression of GHRH-R, compared with normal tissues.”
In 50 samples, GHRH-R was determined to be overexpressed, and this overexpression was significantly associated with both greater tumor size (P = .031) and high pathologic tumor stage (P = .001). Increasing expression of GHRH-R was also significantly associated with worse overall survival (P less than .001).
They confirmed these findings in samples from a multinational cohort of patients, which again showed that the highest levels of GHRH-R expression were associated with poor overall survival (P less than .001).
The authors also looked at messenger RNA expression and gene copy number in 65 gastric cancer samples and 19 adjacent normal tissue samples, and found that GHRH-R mRNA was significantly higher in tumor tissues than normal control tissues (P less than .001).
MAI-602 in vitro and in vivo
To see whether MAI-602 could inhibit the growth of gastric cancer cells, the investigators tried it at various doses in three human gastric cancer cell lines, and found that it inhibited cells in a dose-dependent fashion, compared with vehicle used as a control (P less than .001).
In addition, the experimental agent “exhibited remarkable inhibitory effects on tumor growth in vivo” in mice with human tumor xenografts (P less than .001).
Finally, they showed that the cancer suppression effects of MAI-602 work through inhibition of STAT3/NF-kappaB inflammatory signaling. In vitro and in vivo, MAI-602 decreased the expression of both GHRH and GHRH-R, whereas as a GHRH-R agonist increased levels of both the hormone and its receptor. They also demonstrated that PAK1 appears to be a critical mediator of STAT3/NF-kappaB activity, and that MAI-602 works primarily by blocking PAK1-mediated inflammatory signaling.
“MIA-602 remarkably inhibits the growth of human in vitro and in vivo through the suppression of PAK1–STAT3/NF-kappaB signaling. Our study strongly highlights the therapeutic potential of GHRH-R antagonists in the treatment of gastric cancer patients. Knowledge gained in our study will shed light on how to select the appropriate patients for personalized cancer therapy using GHRH-R antagonists,” Dr. Schally and his coauthors wrote.
The study was supported by the Li Ka Shing Foundation, Chinese foundation, and government grants to individual researchers, as well as support from the the Medical Research Service of the U.S. Department of Veterans Affairs, South Florida Veterans Affairs Foundation for Research and Education, and the University of Miami.
It sounds counterintuitive, but targeting a neuropeptide hormone produced in the hypothalamus may be an effective strategy for treating gastric cancer, the second most common cause of cancer deaths worldwide, investigators from China and the United States contend.
Growth hormone–releasing hormone (GHRH) and its receptor (GHRH-R) are found primarily in the anterior pituitary gland, but are also present in gastric cancers, other solid tumors, and lymphomas. Increased levels of GHRH-R in tumor samples from patients with gastric cancer are associated with poor outcomes, noted Andrew V. Schally, PhD, MD, DSc, of the University of Miami, and his colleagues at the Shantou (China) University Medical College.
Furthermore, an experimental peptide drug labeled MIA-602 that targets GHRH-R inhibited the growth of gastric cancer cell lines and human tumor xenografts in mice, the investigators reported in the journal PNAS.
“The GHRH receptor is both a biomarker that can confirm prognosis and a therapeutic target,” Dr. Schally said in a statement.
Elevated GHRH-R expression in tumors
GHRH-R antagonists such as MIA-602 work through downregulation of the p21-activated kinase 1 (PAK1)–mediated signal transducer and activator of transcription 3 (STAT3)/nuclear factor–kappaB (NF-kappaB) inflammatory pathway. This pathway is involved in the interplay between inflammatory processes and intracellular signaling thought to be the cause of gastric cancer tumorigenesis and progression, the investigators explained.
They first looked for GHRH-R expression in gastric cancer samples from 106 patients, using immunohistochemistry staining of primary tumors and adjacent normal tissues. They found that gastric cancer tissues “exhibited robust expression of GHRH-R, compared with normal tissues.”
In 50 samples, GHRH-R was determined to be overexpressed, and this overexpression was significantly associated with both greater tumor size (P = .031) and high pathologic tumor stage (P = .001). Increasing expression of GHRH-R was also significantly associated with worse overall survival (P less than .001).
They confirmed these findings in samples from a multinational cohort of patients, which again showed that the highest levels of GHRH-R expression were associated with poor overall survival (P less than .001).
The authors also looked at messenger RNA expression and gene copy number in 65 gastric cancer samples and 19 adjacent normal tissue samples, and found that GHRH-R mRNA was significantly higher in tumor tissues than normal control tissues (P less than .001).
MAI-602 in vitro and in vivo
To see whether MAI-602 could inhibit the growth of gastric cancer cells, the investigators tried it at various doses in three human gastric cancer cell lines, and found that it inhibited cells in a dose-dependent fashion, compared with vehicle used as a control (P less than .001).
In addition, the experimental agent “exhibited remarkable inhibitory effects on tumor growth in vivo” in mice with human tumor xenografts (P less than .001).
Finally, they showed that the cancer suppression effects of MAI-602 work through inhibition of STAT3/NF-kappaB inflammatory signaling. In vitro and in vivo, MAI-602 decreased the expression of both GHRH and GHRH-R, whereas as a GHRH-R agonist increased levels of both the hormone and its receptor. They also demonstrated that PAK1 appears to be a critical mediator of STAT3/NF-kappaB activity, and that MAI-602 works primarily by blocking PAK1-mediated inflammatory signaling.
“MIA-602 remarkably inhibits the growth of human in vitro and in vivo through the suppression of PAK1–STAT3/NF-kappaB signaling. Our study strongly highlights the therapeutic potential of GHRH-R antagonists in the treatment of gastric cancer patients. Knowledge gained in our study will shed light on how to select the appropriate patients for personalized cancer therapy using GHRH-R antagonists,” Dr. Schally and his coauthors wrote.
The study was supported by the Li Ka Shing Foundation, Chinese foundation, and government grants to individual researchers, as well as support from the the Medical Research Service of the U.S. Department of Veterans Affairs, South Florida Veterans Affairs Foundation for Research and Education, and the University of Miami.
FROM PNAS
Key clinical point:
Major finding: The experimental GHRH-R antagonist MAI-602 inhibited gastric cancer growth in cell lines and human tumor xenograft models.
Data source: Proof of concept experiments showing the relationship between GHRH-R and gastric cancer, and elucidation of a method for targeting GHRH-R with an investigational peptide compound.
Disclosures: The study was supported by the Li Ka Shing Foundation, Chinese foundation, and government grants to individual researchers, as well as support from the the Medical Research Service of the U.S. Department of Veterans Affairs, South Florida Veterans Affairs Foundation for Research and Education, and the University of Miami.
Painful Oral and Genital Ulcers
The Diagnosis: Pemphigus Vegetans
Pemphigus vegetans is a rare variant of pemphigus vulgaris. Clinically, pemphigus vegetans is characterized by vegetative lesions over the flexures, but any area of the skin may be involved. There have been case reports involving the scalp,1,2 mouth,3 and foot.4 There are 2 clinical subtypes: the Neumann type and the Hallopeau type.5 The Hallopeau type is relatively benign, requires lower doses of systemic corticosteroids, and has a prolonged remission, while the Neumann type necessitates higher doses of systemic corticosteroids and often presents with relapses and remissions.
The diagnosis of pemphigus vegetans is based on clinical suspicion and confirmed by histological examination and immunological findings. The diagnosis may be difficult, as its presentation varies and histopathological findings may resemble other conditions.
Systemic corticosteroids are the well-established drug of choice for treating pemphigus vegetans to induce remission and maintain healing before cautiously tapering down the dosage approximately 50% every 2 weeks.6 Adjuvant drugs used in conjunction with steroids for steroid-sparing purpose include azathioprine, cyclophosphamide, mycophenolate mofetil, methotrexate, and cyclosporine.6 Pulsed intravenous steroids,7 intravenous immunoglobulins,8 pulsed dexamethasone cyclophosphamide,9 and extracorporeal photopheresis10 are given for severe and recalcitrant disease.
Laboratory investigations of our patient showed a normal complete blood cell count and a normal renal and liver profile. Herpes simplex virus serology was positive for type 1 and type 2 IgM and IgG. Urethral swab was dry and negative for gonorrhea. Serology for chlamydia, toxoplasma, amoebiasis, and leishmaniasis was negative. Human immunodeficiency virus serology, hepatitis screening, rapid plasma reagin, Treponema pallidum hemagglutination, rheumatoid factor, and antinuclear antibody all were negative. The patient was given a course of oral acyclovir 400 mg 3 times daily and empirical treatment with oral doxycycline 100 mg twice daily for a week with no clinical response.
Two biopsies from the perianal ulcers showed inflamed squamous papillomata with no Donovan bodies. A third biopsy from an intact blister showed acantholytic cells in the suprabasal bullae with eosinophilic and lymphocytic infiltrates at the upper dermis. Direct immunofluorescence demonstrated intercellular C3 and IgG deposits.
The patient was started on oral prednisolone at 1 mg/kg daily and oral azathioprine 50 mg daily with resolution of the perianal, penile, and oral ulcers (Figures 1 and 2). He achieved good suppression of further eruption. At the patient's most recent follow-up (2.5 years after the initial presentation), he was in remission and was currently taking oral azathioprine 100 mg once daily and no oral corticosteroids.
- Danopoulou I, Stavropoulos P, Stratigos A, et al. Pemphigus vegetans confined to the scalp. Int J Dermatol. 2006;45:1008-1009.
- Mori M, Mariotti G, Grandi V, et al. Pemphigus vegetans of the scalp [published online October 22,2014]. J Eur Acad Dermatol Venereol. 2016;30:368-370.
- Augusto de Oliveira M, Martins E Martins F, Lourenço S, et al. Oral pemphigus vegetans: a case report. Dermatol Online J. 2012;18:10.
- Ma DL, Fang K. Hallopeau type of pemphigus vegetans confined to the right foot: case report. Chin Med J (Engl). 2009;122:588-590.
- Ahmed AR, Blose DA. Pemphigus vegetans. Neumann type and Hallopeau type. Int J Dermatol. 1984;23:135-141.
- Harman KE, Albert S, Black MM, et al. Guidelines for the management of pemphigus vulgaris. Br J Dermatol. 2003;149:926-937.
- Chryssomallis F, Dimitriades A, Chaidemenos GC, et al. Steroid-pulse therapy in pemphigus vulgaris long term follow-up. Int J Dermatol. 1995;34:438-442.
- Ahmed AR. Intravenous immunoglobulin therapy in the treatment of patients with pemphigus vulgaris unresponsive to conventional immunosuppressive treatment. J Am Acad Dermatol. 2001;45:679-690.
- Pasricha JS, Khaitan BK, Raman RS, et al. Dexamethasone-cyclophosphamide pulse therapy for pemphigus. Int J Dermatol. 1995;34:875-882.
- Rook AH, Jegasothy BV, Heald P, et al. Extracorporeal photochemotherapy for drug-resistant pemphigus vulgaris. Ann Int Med. 1990;112:303-305.
The Diagnosis: Pemphigus Vegetans
Pemphigus vegetans is a rare variant of pemphigus vulgaris. Clinically, pemphigus vegetans is characterized by vegetative lesions over the flexures, but any area of the skin may be involved. There have been case reports involving the scalp,1,2 mouth,3 and foot.4 There are 2 clinical subtypes: the Neumann type and the Hallopeau type.5 The Hallopeau type is relatively benign, requires lower doses of systemic corticosteroids, and has a prolonged remission, while the Neumann type necessitates higher doses of systemic corticosteroids and often presents with relapses and remissions.
The diagnosis of pemphigus vegetans is based on clinical suspicion and confirmed by histological examination and immunological findings. The diagnosis may be difficult, as its presentation varies and histopathological findings may resemble other conditions.
Systemic corticosteroids are the well-established drug of choice for treating pemphigus vegetans to induce remission and maintain healing before cautiously tapering down the dosage approximately 50% every 2 weeks.6 Adjuvant drugs used in conjunction with steroids for steroid-sparing purpose include azathioprine, cyclophosphamide, mycophenolate mofetil, methotrexate, and cyclosporine.6 Pulsed intravenous steroids,7 intravenous immunoglobulins,8 pulsed dexamethasone cyclophosphamide,9 and extracorporeal photopheresis10 are given for severe and recalcitrant disease.
Laboratory investigations of our patient showed a normal complete blood cell count and a normal renal and liver profile. Herpes simplex virus serology was positive for type 1 and type 2 IgM and IgG. Urethral swab was dry and negative for gonorrhea. Serology for chlamydia, toxoplasma, amoebiasis, and leishmaniasis was negative. Human immunodeficiency virus serology, hepatitis screening, rapid plasma reagin, Treponema pallidum hemagglutination, rheumatoid factor, and antinuclear antibody all were negative. The patient was given a course of oral acyclovir 400 mg 3 times daily and empirical treatment with oral doxycycline 100 mg twice daily for a week with no clinical response.
Two biopsies from the perianal ulcers showed inflamed squamous papillomata with no Donovan bodies. A third biopsy from an intact blister showed acantholytic cells in the suprabasal bullae with eosinophilic and lymphocytic infiltrates at the upper dermis. Direct immunofluorescence demonstrated intercellular C3 and IgG deposits.
The patient was started on oral prednisolone at 1 mg/kg daily and oral azathioprine 50 mg daily with resolution of the perianal, penile, and oral ulcers (Figures 1 and 2). He achieved good suppression of further eruption. At the patient's most recent follow-up (2.5 years after the initial presentation), he was in remission and was currently taking oral azathioprine 100 mg once daily and no oral corticosteroids.
The Diagnosis: Pemphigus Vegetans
Pemphigus vegetans is a rare variant of pemphigus vulgaris. Clinically, pemphigus vegetans is characterized by vegetative lesions over the flexures, but any area of the skin may be involved. There have been case reports involving the scalp,1,2 mouth,3 and foot.4 There are 2 clinical subtypes: the Neumann type and the Hallopeau type.5 The Hallopeau type is relatively benign, requires lower doses of systemic corticosteroids, and has a prolonged remission, while the Neumann type necessitates higher doses of systemic corticosteroids and often presents with relapses and remissions.
The diagnosis of pemphigus vegetans is based on clinical suspicion and confirmed by histological examination and immunological findings. The diagnosis may be difficult, as its presentation varies and histopathological findings may resemble other conditions.
Systemic corticosteroids are the well-established drug of choice for treating pemphigus vegetans to induce remission and maintain healing before cautiously tapering down the dosage approximately 50% every 2 weeks.6 Adjuvant drugs used in conjunction with steroids for steroid-sparing purpose include azathioprine, cyclophosphamide, mycophenolate mofetil, methotrexate, and cyclosporine.6 Pulsed intravenous steroids,7 intravenous immunoglobulins,8 pulsed dexamethasone cyclophosphamide,9 and extracorporeal photopheresis10 are given for severe and recalcitrant disease.
Laboratory investigations of our patient showed a normal complete blood cell count and a normal renal and liver profile. Herpes simplex virus serology was positive for type 1 and type 2 IgM and IgG. Urethral swab was dry and negative for gonorrhea. Serology for chlamydia, toxoplasma, amoebiasis, and leishmaniasis was negative. Human immunodeficiency virus serology, hepatitis screening, rapid plasma reagin, Treponema pallidum hemagglutination, rheumatoid factor, and antinuclear antibody all were negative. The patient was given a course of oral acyclovir 400 mg 3 times daily and empirical treatment with oral doxycycline 100 mg twice daily for a week with no clinical response.
Two biopsies from the perianal ulcers showed inflamed squamous papillomata with no Donovan bodies. A third biopsy from an intact blister showed acantholytic cells in the suprabasal bullae with eosinophilic and lymphocytic infiltrates at the upper dermis. Direct immunofluorescence demonstrated intercellular C3 and IgG deposits.
The patient was started on oral prednisolone at 1 mg/kg daily and oral azathioprine 50 mg daily with resolution of the perianal, penile, and oral ulcers (Figures 1 and 2). He achieved good suppression of further eruption. At the patient's most recent follow-up (2.5 years after the initial presentation), he was in remission and was currently taking oral azathioprine 100 mg once daily and no oral corticosteroids.
- Danopoulou I, Stavropoulos P, Stratigos A, et al. Pemphigus vegetans confined to the scalp. Int J Dermatol. 2006;45:1008-1009.
- Mori M, Mariotti G, Grandi V, et al. Pemphigus vegetans of the scalp [published online October 22,2014]. J Eur Acad Dermatol Venereol. 2016;30:368-370.
- Augusto de Oliveira M, Martins E Martins F, Lourenço S, et al. Oral pemphigus vegetans: a case report. Dermatol Online J. 2012;18:10.
- Ma DL, Fang K. Hallopeau type of pemphigus vegetans confined to the right foot: case report. Chin Med J (Engl). 2009;122:588-590.
- Ahmed AR, Blose DA. Pemphigus vegetans. Neumann type and Hallopeau type. Int J Dermatol. 1984;23:135-141.
- Harman KE, Albert S, Black MM, et al. Guidelines for the management of pemphigus vulgaris. Br J Dermatol. 2003;149:926-937.
- Chryssomallis F, Dimitriades A, Chaidemenos GC, et al. Steroid-pulse therapy in pemphigus vulgaris long term follow-up. Int J Dermatol. 1995;34:438-442.
- Ahmed AR. Intravenous immunoglobulin therapy in the treatment of patients with pemphigus vulgaris unresponsive to conventional immunosuppressive treatment. J Am Acad Dermatol. 2001;45:679-690.
- Pasricha JS, Khaitan BK, Raman RS, et al. Dexamethasone-cyclophosphamide pulse therapy for pemphigus. Int J Dermatol. 1995;34:875-882.
- Rook AH, Jegasothy BV, Heald P, et al. Extracorporeal photochemotherapy for drug-resistant pemphigus vulgaris. Ann Int Med. 1990;112:303-305.
- Danopoulou I, Stavropoulos P, Stratigos A, et al. Pemphigus vegetans confined to the scalp. Int J Dermatol. 2006;45:1008-1009.
- Mori M, Mariotti G, Grandi V, et al. Pemphigus vegetans of the scalp [published online October 22,2014]. J Eur Acad Dermatol Venereol. 2016;30:368-370.
- Augusto de Oliveira M, Martins E Martins F, Lourenço S, et al. Oral pemphigus vegetans: a case report. Dermatol Online J. 2012;18:10.
- Ma DL, Fang K. Hallopeau type of pemphigus vegetans confined to the right foot: case report. Chin Med J (Engl). 2009;122:588-590.
- Ahmed AR, Blose DA. Pemphigus vegetans. Neumann type and Hallopeau type. Int J Dermatol. 1984;23:135-141.
- Harman KE, Albert S, Black MM, et al. Guidelines for the management of pemphigus vulgaris. Br J Dermatol. 2003;149:926-937.
- Chryssomallis F, Dimitriades A, Chaidemenos GC, et al. Steroid-pulse therapy in pemphigus vulgaris long term follow-up. Int J Dermatol. 1995;34:438-442.
- Ahmed AR. Intravenous immunoglobulin therapy in the treatment of patients with pemphigus vulgaris unresponsive to conventional immunosuppressive treatment. J Am Acad Dermatol. 2001;45:679-690.
- Pasricha JS, Khaitan BK, Raman RS, et al. Dexamethasone-cyclophosphamide pulse therapy for pemphigus. Int J Dermatol. 1995;34:875-882.
- Rook AH, Jegasothy BV, Heald P, et al. Extracorporeal photochemotherapy for drug-resistant pemphigus vulgaris. Ann Int Med. 1990;112:303-305.
A 52-year-old man presented with persistent painful oral ulcers and penile and perianal erosions of 6 months' duration. He strongly denied engaging in high-risk sexual activities and had lost 10 kg over the last 6 months. He did not report taking any over-the-counter or alternative medications. On physical examination there were multiple fissures on the lower lip with erosive white plaques on the tongue and buccal mucosa. There were erosions over the foreskin and glans penis and a few erosive plaques on the perianal skin. Bilateral inguinal lymph nodes were enlarged.
Diagnostic laparoscopy identifies ovarian cancers amenable to PCS
For women with suspected advanced epithelial ovarian cancer, diagnostic laparoscopy can help to distinguish between patients who could benefit from primary cytoreductive surgery (PCS) and those who might have better outcomes with neoadjuvant chemotherapy and interval cytoreductive surgery, according to investigators in the Netherlands.
In a randomized controlled trial exploring whether initial diagnostic laparoscopy could spare some patients from undergoing futile PCS, the investigators found that only 10% of patients assigned to diagnostic laparoscopy prior to PCS underwent a subsequent futile laparotomy, defined as residual disease greater than 1 cm following surgery. In contrast, 39% of women assigned to primary PCS had disease that might have been better treated by chemotherapy and interval surgery,
“In women with a plan for PCS, these data suggest that performance of diagnostic laparoscopy first is reasonable and that if cytoreduction to [less than] 1 cm of residual disease seems feasible, to proceed with PCS,” wrote Marrije R. Buist, MD of Academic Medical Center, Amsterdam, and colleagues.
Among women with International Federation of Gynecology and Obstetrics (FIGO) stage IIIC to IV epithelial ovarian cancer, survival depends largely on the ability of surgery to either completely remove disease, or to leave at best less than 1 cm of residual disease. However, aggressive surgery in patients with more extensive disease is associated with significant morbidities, the authors noted.
“If at PCS, extensive disease is present, surgery could be ceased, and neoadjuvant chemotherapy with interval surgery could be a good alternative treatment. Therefore, the identification of patients with extensive disease who are likely to have [more than] 1 cm of residual tumor after PCS, defined as a futile laparotomy, is important,” they wrote.
To test this idea, the investigators, from eight cancer centers in the Netherlands, enrolled 201 patients with suspected FIGO stage IIB ovarian cancer or higher, and randomly assigned them to undergo either initial diagnostic laparoscopy or PCS.
They found that 10 of the 102 patients (10%) assigned to diagnostic laparoscopy went on to undergo PCS that revealed residual disease greater than 1 cm, compared with 39 of the 99 patients (39%) assigned to PCS. This difference translated into a relative risk for futile laparotomy of 0.25 for diagnostic laparoscopy compared with PCS (P less than .001).
Only 3 (3%) patients in the diagnostic laparoscopy group went on to have both PCS and interval surgery, compared with 28 (28%) patients initially assigned to PCS (P less than .001).
The Dutch Organization for Health Research and Development supported the study. All but one coauthor reported having no potential conflicts of interest.
For women with suspected advanced epithelial ovarian cancer, diagnostic laparoscopy can help to distinguish between patients who could benefit from primary cytoreductive surgery (PCS) and those who might have better outcomes with neoadjuvant chemotherapy and interval cytoreductive surgery, according to investigators in the Netherlands.
In a randomized controlled trial exploring whether initial diagnostic laparoscopy could spare some patients from undergoing futile PCS, the investigators found that only 10% of patients assigned to diagnostic laparoscopy prior to PCS underwent a subsequent futile laparotomy, defined as residual disease greater than 1 cm following surgery. In contrast, 39% of women assigned to primary PCS had disease that might have been better treated by chemotherapy and interval surgery,
“In women with a plan for PCS, these data suggest that performance of diagnostic laparoscopy first is reasonable and that if cytoreduction to [less than] 1 cm of residual disease seems feasible, to proceed with PCS,” wrote Marrije R. Buist, MD of Academic Medical Center, Amsterdam, and colleagues.
Among women with International Federation of Gynecology and Obstetrics (FIGO) stage IIIC to IV epithelial ovarian cancer, survival depends largely on the ability of surgery to either completely remove disease, or to leave at best less than 1 cm of residual disease. However, aggressive surgery in patients with more extensive disease is associated with significant morbidities, the authors noted.
“If at PCS, extensive disease is present, surgery could be ceased, and neoadjuvant chemotherapy with interval surgery could be a good alternative treatment. Therefore, the identification of patients with extensive disease who are likely to have [more than] 1 cm of residual tumor after PCS, defined as a futile laparotomy, is important,” they wrote.
To test this idea, the investigators, from eight cancer centers in the Netherlands, enrolled 201 patients with suspected FIGO stage IIB ovarian cancer or higher, and randomly assigned them to undergo either initial diagnostic laparoscopy or PCS.
They found that 10 of the 102 patients (10%) assigned to diagnostic laparoscopy went on to undergo PCS that revealed residual disease greater than 1 cm, compared with 39 of the 99 patients (39%) assigned to PCS. This difference translated into a relative risk for futile laparotomy of 0.25 for diagnostic laparoscopy compared with PCS (P less than .001).
Only 3 (3%) patients in the diagnostic laparoscopy group went on to have both PCS and interval surgery, compared with 28 (28%) patients initially assigned to PCS (P less than .001).
The Dutch Organization for Health Research and Development supported the study. All but one coauthor reported having no potential conflicts of interest.
For women with suspected advanced epithelial ovarian cancer, diagnostic laparoscopy can help to distinguish between patients who could benefit from primary cytoreductive surgery (PCS) and those who might have better outcomes with neoadjuvant chemotherapy and interval cytoreductive surgery, according to investigators in the Netherlands.
In a randomized controlled trial exploring whether initial diagnostic laparoscopy could spare some patients from undergoing futile PCS, the investigators found that only 10% of patients assigned to diagnostic laparoscopy prior to PCS underwent a subsequent futile laparotomy, defined as residual disease greater than 1 cm following surgery. In contrast, 39% of women assigned to primary PCS had disease that might have been better treated by chemotherapy and interval surgery,
“In women with a plan for PCS, these data suggest that performance of diagnostic laparoscopy first is reasonable and that if cytoreduction to [less than] 1 cm of residual disease seems feasible, to proceed with PCS,” wrote Marrije R. Buist, MD of Academic Medical Center, Amsterdam, and colleagues.
Among women with International Federation of Gynecology and Obstetrics (FIGO) stage IIIC to IV epithelial ovarian cancer, survival depends largely on the ability of surgery to either completely remove disease, or to leave at best less than 1 cm of residual disease. However, aggressive surgery in patients with more extensive disease is associated with significant morbidities, the authors noted.
“If at PCS, extensive disease is present, surgery could be ceased, and neoadjuvant chemotherapy with interval surgery could be a good alternative treatment. Therefore, the identification of patients with extensive disease who are likely to have [more than] 1 cm of residual tumor after PCS, defined as a futile laparotomy, is important,” they wrote.
To test this idea, the investigators, from eight cancer centers in the Netherlands, enrolled 201 patients with suspected FIGO stage IIB ovarian cancer or higher, and randomly assigned them to undergo either initial diagnostic laparoscopy or PCS.
They found that 10 of the 102 patients (10%) assigned to diagnostic laparoscopy went on to undergo PCS that revealed residual disease greater than 1 cm, compared with 39 of the 99 patients (39%) assigned to PCS. This difference translated into a relative risk for futile laparotomy of 0.25 for diagnostic laparoscopy compared with PCS (P less than .001).
Only 3 (3%) patients in the diagnostic laparoscopy group went on to have both PCS and interval surgery, compared with 28 (28%) patients initially assigned to PCS (P less than .001).
The Dutch Organization for Health Research and Development supported the study. All but one coauthor reported having no potential conflicts of interest.
Key clinical point: Diagnostic laparoscopy can help to identify patients with advanced ovarian cancer who can best benefit from primary surgery or chemotherapy.
Major finding: Ten percent of women assigned to diagnostic laparoscopy underwent futile laparotomy, vs. 39% assigned to primary cytoreductive surgery.
Data source: Randomized controlled trial of 201 women with suspected FIGO stage IIB or greater disease.
Disclosures The Dutch Organization for Health Research and Development supported the study. All but one coauthor reported having no potential conflicts of interest.
Law & Medicine: How case law shapes EMTALA
(This is the second installment of a three-part series.)
Question: Emergency Medical Treatment and Labor Act (EMTALA) litigation has yielded which of the following rules of law?
A. The statute is applicable only when a patient is physically in the hospital’s emergency department (ED).
B. Directing an ambulance away from the ED is a violation of EMTALA.
C. All patients presenting to the ED must have an appropriate medical screening exam conforming to customary standard of care.
D. It is not what is performed in any medical screening exam, but whether it is applied evenly to all patients similarly situated.
E. The U.S. Supreme Court has held that an improper motive behind an unstable transfer is a prerequisite to an EMTALA violation.
Answer: D. In 1986, Congress enacted the Emergency Medical Treatment and Labor Act to ensure that all patients who present themselves to the emergency department are appropriately screened for an emergent medical condition, and if one is present, that they be stabilized prior to transfer or discharge.
In the 3 decades since its enactment, the statute has, as expected, spawned numerous lawsuits. Parts two and three of this series on EMTALA summarize the salient findings and rules of law in several interesting and impactful cases. These cases are neither encyclopedic nor necessarily representative of the types of litigation commonly encountered.

EMTALA is about events in the emergency department. They begin with the patient coming to the ED seeking treatment, and the statute specifically refers to “any individual ... [who] comes to the emergency department and a request is made on the individual’s behalf for examination or treatment for a medical condition.”
But what if the patient has yet to arrive, e.g., in an ambulance en route, and was diverted elsewhere in an unstable condition? In Hawaii’s case of Arrington v. Wong (237 F.3d 1066 [9th Cir. 2001]), the court was faced with whether the requirement that a patient must first “come to” the hospital means his or her literal physical presence in the hospital.
On May 5, 1996, Harold Arrington developed dyspnea while driving to his job as a security guard. En route to the closest medical facility, the Queen’s Medical Center, the ambulance personnel contacted Dr. Norbert Wong, the physician on call, describing the patient as being in severe respiratory distress, speaking one to two words at a time, and breathing about 50 times a minute.
Although Queen’s was not on diversionary status at the time, Dr. Wong thought it was okay for the ambulance to go instead to Tripler Hospital, a more distant hospital, as the patient’s doctor worked there. Unfortunately, by the time the ambulance arrived at Tripler, Mr. Arrington’s condition had deteriorated, and he was pronounced dead shortly after arrival.
The lower court ruled for the defendant, holding that the statute demanded an actual physical presence in the ED, but the 9th Circuit Court of Appeals reversed. It held that under EMTALA, a hospital may divert an ambulance that has contacted its emergency department and is on the way to that hospital only if the hospital is in diversionary status, because the diverting hospital then has a valid, treatment-related reason for doing so.
Such an interpretation of the law works no hardship on the hospital and is consistent with the Centers for Medicare & Medicaid Services’ regulation that “only requires hospitals that offer emergency services to provide screening and stabilizing treatment within the scope of their capabilities.” The 9th Circuit felt that this was consistent with the purpose and language of the EMTALA statute.
The next case, Summers v. Baptist Medical Center (91 F. 3d 1132 [8th Cir. 1996]), addressed the screening aspect of EMTALA, specifically on the distinction between disparity and adequacy in screening procedures. In Summers, the plaintiff fell from a tree while hunting and sustained bilateral hemothoraces, vertebral, rib, and sternal fractures. Incredibly, the diagnosis in the first hospital ED was muscle spasms, and the diagnoses only became clear when he checked into a second hospital 2 days later.
Still, the court ruled that there was no EMTALA violation, and that allegations of substandard care should be addressed under a negligence theory in state courts and not under EMTALA. The court reasoned that under the statute, an “inappropriate” screening is one that is performed in a disparate manner to similarly situated patients, and the hospital itself is usually left to define for itself what is within its capabilities. It is up to the hospital itself to determine what its screening procedures will be and to apply them alike to all patients with comparable complaints.
Likewise, in Vickers v. Nash (78 F.3d 139 [4th Cir. 1996]), an intoxicated patient who sustained a head injury following a fight died 4 days later from an epidural hematoma that was missed. He did have his head laceration treated in the ED, and was observed for 11 hours before discharge. The court ruled that the plaintiffs had failed to prove there was disparate treatment, and that hospitals can only be expected to stabilize emergency medical conditions known to them at the time.
Finally, in its first and thus far only EMTALA case, the U.S. Supreme Court in 1999 looked at whether an improper motive was a prerequisite for a finding of an EMTALA violation regarding stabilization and transfer.
In Roberts v. Galen of Virginia (119 S. Court 685 [1999]), the patient, injured in a truck accident, required a splenectomy and ventilator support. After a prolonged hospital stay, she was about to be moved to a nearby nursing home when she developed a high fever from an infection, and had to be transferred to an acute care facility. Her guardian, Roberts, brought suit, asserting violations of EMTALA’s stabilization and transfer requirements. The hospital argued that no material deterioration of the condition was likely to result from or occur during the transfer, and the district court determined her transfer was not prompted by an improper motive.
On appeal, the 6th Circuit Court of Appeals affirmed, extending its earlier holding that a showing of improper motive was required to make out an inadequate screening claim under EMTALA.
The hospital raised a number of important defenses, which included the physician lacking actual knowledge that the patient had an emergency medical condition, that EMTALA did not apply to in-hospital treatment and discharge decisions, and denying that EMTALA imposes minimum substantive standards of medical care.
The U.S. Supreme Court granted certiorari on the single issue whether the improper motive test should apply to an allegedly wrongful transfer. Overturning the appeals court, it held that Section 1395dd(b) (stabilization and transfer) contained no express or implied “improper motive” requirement. The Supreme Court declined to resolve broader issues under the statute.
Dr. Tan is emeritus professor of medicine and former adjunct professor of law at the University of Hawaii, and currently directs the St. Francis International Center for Healthcare Ethics in Honolulu. This article is meant to be educational and does not constitute medical, ethical, or legal advice. Some of the articles in this series are adapted from the author’s 2006 book, “Medical Malpractice: Understanding the Law, Managing the Risk,” and his 2012 Halsbury treatise, “Medical Negligence and Professional Misconduct.” For additional information, readers may contact the author at [email protected].
(This is the second installment of a three-part series.)
Question: Emergency Medical Treatment and Labor Act (EMTALA) litigation has yielded which of the following rules of law?
A. The statute is applicable only when a patient is physically in the hospital’s emergency department (ED).
B. Directing an ambulance away from the ED is a violation of EMTALA.
C. All patients presenting to the ED must have an appropriate medical screening exam conforming to customary standard of care.
D. It is not what is performed in any medical screening exam, but whether it is applied evenly to all patients similarly situated.
E. The U.S. Supreme Court has held that an improper motive behind an unstable transfer is a prerequisite to an EMTALA violation.
Answer: D. In 1986, Congress enacted the Emergency Medical Treatment and Labor Act to ensure that all patients who present themselves to the emergency department are appropriately screened for an emergent medical condition, and if one is present, that they be stabilized prior to transfer or discharge.
In the 3 decades since its enactment, the statute has, as expected, spawned numerous lawsuits. Parts two and three of this series on EMTALA summarize the salient findings and rules of law in several interesting and impactful cases. These cases are neither encyclopedic nor necessarily representative of the types of litigation commonly encountered.
EMTALA is about events in the emergency department. They begin with the patient coming to the ED seeking treatment, and the statute specifically refers to “any individual ... [who] comes to the emergency department and a request is made on the individual’s behalf for examination or treatment for a medical condition.”
But what if the patient has yet to arrive, e.g., in an ambulance en route, and was diverted elsewhere in an unstable condition? In Hawaii’s case of Arrington v. Wong (237 F.3d 1066 [9th Cir. 2001]), the court was faced with whether the requirement that a patient must first “come to” the hospital means his or her literal physical presence in the hospital.
On May 5, 1996, Harold Arrington developed dyspnea while driving to his job as a security guard. En route to the closest medical facility, the Queen’s Medical Center, the ambulance personnel contacted Dr. Norbert Wong, the physician on call, describing the patient as being in severe respiratory distress, speaking one to two words at a time, and breathing about 50 times a minute.
Although Queen’s was not on diversionary status at the time, Dr. Wong thought it was okay for the ambulance to go instead to Tripler Hospital, a more distant hospital, as the patient’s doctor worked there. Unfortunately, by the time the ambulance arrived at Tripler, Mr. Arrington’s condition had deteriorated, and he was pronounced dead shortly after arrival.
The lower court ruled for the defendant, holding that the statute demanded an actual physical presence in the ED, but the 9th Circuit Court of Appeals reversed. It held that under EMTALA, a hospital may divert an ambulance that has contacted its emergency department and is on the way to that hospital only if the hospital is in diversionary status, because the diverting hospital then has a valid, treatment-related reason for doing so.
Such an interpretation of the law works no hardship on the hospital and is consistent with the Centers for Medicare & Medicaid Services’ regulation that “only requires hospitals that offer emergency services to provide screening and stabilizing treatment within the scope of their capabilities.” The 9th Circuit felt that this was consistent with the purpose and language of the EMTALA statute.
The next case, Summers v. Baptist Medical Center (91 F. 3d 1132 [8th Cir. 1996]), addressed the screening aspect of EMTALA, specifically on the distinction between disparity and adequacy in screening procedures. In Summers, the plaintiff fell from a tree while hunting and sustained bilateral hemothoraces, vertebral, rib, and sternal fractures. Incredibly, the diagnosis in the first hospital ED was muscle spasms, and the diagnoses only became clear when he checked into a second hospital 2 days later.
Still, the court ruled that there was no EMTALA violation, and that allegations of substandard care should be addressed under a negligence theory in state courts and not under EMTALA. The court reasoned that under the statute, an “inappropriate” screening is one that is performed in a disparate manner to similarly situated patients, and the hospital itself is usually left to define for itself what is within its capabilities. It is up to the hospital itself to determine what its screening procedures will be and to apply them alike to all patients with comparable complaints.
Likewise, in Vickers v. Nash (78 F.3d 139 [4th Cir. 1996]), an intoxicated patient who sustained a head injury following a fight died 4 days later from an epidural hematoma that was missed. He did have his head laceration treated in the ED, and was observed for 11 hours before discharge. The court ruled that the plaintiffs had failed to prove there was disparate treatment, and that hospitals can only be expected to stabilize emergency medical conditions known to them at the time.
Finally, in its first and thus far only EMTALA case, the U.S. Supreme Court in 1999 looked at whether an improper motive was a prerequisite for a finding of an EMTALA violation regarding stabilization and transfer.
In Roberts v. Galen of Virginia (119 S. Court 685 [1999]), the patient, injured in a truck accident, required a splenectomy and ventilator support. After a prolonged hospital stay, she was about to be moved to a nearby nursing home when she developed a high fever from an infection, and had to be transferred to an acute care facility. Her guardian, Roberts, brought suit, asserting violations of EMTALA’s stabilization and transfer requirements. The hospital argued that no material deterioration of the condition was likely to result from or occur during the transfer, and the district court determined her transfer was not prompted by an improper motive.
On appeal, the 6th Circuit Court of Appeals affirmed, extending its earlier holding that a showing of improper motive was required to make out an inadequate screening claim under EMTALA.
The hospital raised a number of important defenses, which included the physician lacking actual knowledge that the patient had an emergency medical condition, that EMTALA did not apply to in-hospital treatment and discharge decisions, and denying that EMTALA imposes minimum substantive standards of medical care.
The U.S. Supreme Court granted certiorari on the single issue whether the improper motive test should apply to an allegedly wrongful transfer. Overturning the appeals court, it held that Section 1395dd(b) (stabilization and transfer) contained no express or implied “improper motive” requirement. The Supreme Court declined to resolve broader issues under the statute.
Dr. Tan is emeritus professor of medicine and former adjunct professor of law at the University of Hawaii, and currently directs the St. Francis International Center for Healthcare Ethics in Honolulu. This article is meant to be educational and does not constitute medical, ethical, or legal advice. Some of the articles in this series are adapted from the author’s 2006 book, “Medical Malpractice: Understanding the Law, Managing the Risk,” and his 2012 Halsbury treatise, “Medical Negligence and Professional Misconduct.” For additional information, readers may contact the author at [email protected].
(This is the second installment of a three-part series.)
Question: Emergency Medical Treatment and Labor Act (EMTALA) litigation has yielded which of the following rules of law?
A. The statute is applicable only when a patient is physically in the hospital’s emergency department (ED).
B. Directing an ambulance away from the ED is a violation of EMTALA.
C. All patients presenting to the ED must have an appropriate medical screening exam conforming to customary standard of care.
D. It is not what is performed in any medical screening exam, but whether it is applied evenly to all patients similarly situated.
E. The U.S. Supreme Court has held that an improper motive behind an unstable transfer is a prerequisite to an EMTALA violation.
Answer: D. In 1986, Congress enacted the Emergency Medical Treatment and Labor Act to ensure that all patients who present themselves to the emergency department are appropriately screened for an emergent medical condition, and if one is present, that they be stabilized prior to transfer or discharge.
In the 3 decades since its enactment, the statute has, as expected, spawned numerous lawsuits. Parts two and three of this series on EMTALA summarize the salient findings and rules of law in several interesting and impactful cases. These cases are neither encyclopedic nor necessarily representative of the types of litigation commonly encountered.
EMTALA is about events in the emergency department. They begin with the patient coming to the ED seeking treatment, and the statute specifically refers to “any individual ... [who] comes to the emergency department and a request is made on the individual’s behalf for examination or treatment for a medical condition.”
But what if the patient has yet to arrive, e.g., in an ambulance en route, and was diverted elsewhere in an unstable condition? In Hawaii’s case of Arrington v. Wong (237 F.3d 1066 [9th Cir. 2001]), the court was faced with whether the requirement that a patient must first “come to” the hospital means his or her literal physical presence in the hospital.
On May 5, 1996, Harold Arrington developed dyspnea while driving to his job as a security guard. En route to the closest medical facility, the Queen’s Medical Center, the ambulance personnel contacted Dr. Norbert Wong, the physician on call, describing the patient as being in severe respiratory distress, speaking one to two words at a time, and breathing about 50 times a minute.
Although Queen’s was not on diversionary status at the time, Dr. Wong thought it was okay for the ambulance to go instead to Tripler Hospital, a more distant hospital, as the patient’s doctor worked there. Unfortunately, by the time the ambulance arrived at Tripler, Mr. Arrington’s condition had deteriorated, and he was pronounced dead shortly after arrival.
The lower court ruled for the defendant, holding that the statute demanded an actual physical presence in the ED, but the 9th Circuit Court of Appeals reversed. It held that under EMTALA, a hospital may divert an ambulance that has contacted its emergency department and is on the way to that hospital only if the hospital is in diversionary status, because the diverting hospital then has a valid, treatment-related reason for doing so.
Such an interpretation of the law works no hardship on the hospital and is consistent with the Centers for Medicare & Medicaid Services’ regulation that “only requires hospitals that offer emergency services to provide screening and stabilizing treatment within the scope of their capabilities.” The 9th Circuit felt that this was consistent with the purpose and language of the EMTALA statute.
The next case, Summers v. Baptist Medical Center (91 F. 3d 1132 [8th Cir. 1996]), addressed the screening aspect of EMTALA, specifically on the distinction between disparity and adequacy in screening procedures. In Summers, the plaintiff fell from a tree while hunting and sustained bilateral hemothoraces, vertebral, rib, and sternal fractures. Incredibly, the diagnosis in the first hospital ED was muscle spasms, and the diagnoses only became clear when he checked into a second hospital 2 days later.
Still, the court ruled that there was no EMTALA violation, and that allegations of substandard care should be addressed under a negligence theory in state courts and not under EMTALA. The court reasoned that under the statute, an “inappropriate” screening is one that is performed in a disparate manner to similarly situated patients, and the hospital itself is usually left to define for itself what is within its capabilities. It is up to the hospital itself to determine what its screening procedures will be and to apply them alike to all patients with comparable complaints.
Likewise, in Vickers v. Nash (78 F.3d 139 [4th Cir. 1996]), an intoxicated patient who sustained a head injury following a fight died 4 days later from an epidural hematoma that was missed. He did have his head laceration treated in the ED, and was observed for 11 hours before discharge. The court ruled that the plaintiffs had failed to prove there was disparate treatment, and that hospitals can only be expected to stabilize emergency medical conditions known to them at the time.
Finally, in its first and thus far only EMTALA case, the U.S. Supreme Court in 1999 looked at whether an improper motive was a prerequisite for a finding of an EMTALA violation regarding stabilization and transfer.
In Roberts v. Galen of Virginia (119 S. Court 685 [1999]), the patient, injured in a truck accident, required a splenectomy and ventilator support. After a prolonged hospital stay, she was about to be moved to a nearby nursing home when she developed a high fever from an infection, and had to be transferred to an acute care facility. Her guardian, Roberts, brought suit, asserting violations of EMTALA’s stabilization and transfer requirements. The hospital argued that no material deterioration of the condition was likely to result from or occur during the transfer, and the district court determined her transfer was not prompted by an improper motive.
On appeal, the 6th Circuit Court of Appeals affirmed, extending its earlier holding that a showing of improper motive was required to make out an inadequate screening claim under EMTALA.
The hospital raised a number of important defenses, which included the physician lacking actual knowledge that the patient had an emergency medical condition, that EMTALA did not apply to in-hospital treatment and discharge decisions, and denying that EMTALA imposes minimum substantive standards of medical care.
The U.S. Supreme Court granted certiorari on the single issue whether the improper motive test should apply to an allegedly wrongful transfer. Overturning the appeals court, it held that Section 1395dd(b) (stabilization and transfer) contained no express or implied “improper motive” requirement. The Supreme Court declined to resolve broader issues under the statute.
Dr. Tan is emeritus professor of medicine and former adjunct professor of law at the University of Hawaii, and currently directs the St. Francis International Center for Healthcare Ethics in Honolulu. This article is meant to be educational and does not constitute medical, ethical, or legal advice. Some of the articles in this series are adapted from the author’s 2006 book, “Medical Malpractice: Understanding the Law, Managing the Risk,” and his 2012 Halsbury treatise, “Medical Negligence and Professional Misconduct.” For additional information, readers may contact the author at [email protected].
What’s New for Federal Practitioner in 2017?
It has been a long time since a new year has brought as much uncertainty as 2017 promises to bring, making some federal employees excited and others apprehensive. Rumors abound of how the federal health care sector may change: Hiring freezes, manpower cuts, and privatization are all concerns of Federal Practitioner readers. As in the past, we will keep you up-to-date with in-depth interviews of leaders in federal health care, intelligent coverage of news stories impacting your practice, and clinical and research articles about new programs and initiatives.
Also this year, we are pleased to announce several new regular columns that we hope will inform and entertain you. The first is a column on mental health and traumatic brain injury in the DoD and the VA. We are privileged to have U.S. Army COL (Ret) Elspeth Cameron Ritchie, MD, MPH, edit this column. She is widely known and respected and brings her vast experience to the column as an active-duty psychiatrist coupled with her current position as a VA physician. Dr. Ritchie will author articles as well as edit those of her VA and DoD colleagues. Mental health touches almost every aspect of federal practice, and we all will learn our contributions and challenges in this rapidly moving specialty.
Whereas the mental health column looks toward the scientific future, the second column looks back to the humanistic past. We are thrilled that 2 physician-historians of military medicine, Robert Hierholzer, MD, a VA psychiatrist, and John Pierce, MD, a retired U.S. Army pediatrician share the writing and editing for this column, which will debut this spring.
Have you ever wondered who or how VA and military hospitals were named? These 2 historical writers have a wealth of interesting anecdotes and stories about VA and military medical centers. We hope you will enjoy reading the stories of military and veteran health care: the war heroes, devoted clinicians, and groundbreaking researchers who have left their mark on DoD and VA health care.
We also will be launching a new pilot study feature for clinicians and researchers who have a novel or valuable idea but have only a small number of participants or preliminary results. This will be a great way for new investigators, trainees, and young health care practitioners to present their work to the medical community.
These new editorial offerings are just a start—we also want to invite you, your colleagues, and learners to start your own new tradition of writing for Federal Practitioner. For those who have submitted articles in the past, please keep up the habit.We are eager to receive original research, review articles, and clinical cases from DoD, PHS, and VA mid-career and senior clinicians and researchers as well as articles describing innovative programs and modes of health care treatment and delivery. With a print circulation of more than 35,000 readers and very active online presence, consider Federal Practitioner for your next article!
This year my New Year’s resolution as editor-in-chief is to encourage readers to contact either Editor Reid Paul or me if you have an idea for an article you would like to write, a column you would like to see, or if you have an interest in serving as a peer reviewer or joining our Editorial Advisory Association. We want to hear from you about what you want and need from Federal Practitioner.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies.
It has been a long time since a new year has brought as much uncertainty as 2017 promises to bring, making some federal employees excited and others apprehensive. Rumors abound of how the federal health care sector may change: Hiring freezes, manpower cuts, and privatization are all concerns of Federal Practitioner readers. As in the past, we will keep you up-to-date with in-depth interviews of leaders in federal health care, intelligent coverage of news stories impacting your practice, and clinical and research articles about new programs and initiatives.
Also this year, we are pleased to announce several new regular columns that we hope will inform and entertain you. The first is a column on mental health and traumatic brain injury in the DoD and the VA. We are privileged to have U.S. Army COL (Ret) Elspeth Cameron Ritchie, MD, MPH, edit this column. She is widely known and respected and brings her vast experience to the column as an active-duty psychiatrist coupled with her current position as a VA physician. Dr. Ritchie will author articles as well as edit those of her VA and DoD colleagues. Mental health touches almost every aspect of federal practice, and we all will learn our contributions and challenges in this rapidly moving specialty.
Whereas the mental health column looks toward the scientific future, the second column looks back to the humanistic past. We are thrilled that 2 physician-historians of military medicine, Robert Hierholzer, MD, a VA psychiatrist, and John Pierce, MD, a retired U.S. Army pediatrician share the writing and editing for this column, which will debut this spring.
Have you ever wondered who or how VA and military hospitals were named? These 2 historical writers have a wealth of interesting anecdotes and stories about VA and military medical centers. We hope you will enjoy reading the stories of military and veteran health care: the war heroes, devoted clinicians, and groundbreaking researchers who have left their mark on DoD and VA health care.
We also will be launching a new pilot study feature for clinicians and researchers who have a novel or valuable idea but have only a small number of participants or preliminary results. This will be a great way for new investigators, trainees, and young health care practitioners to present their work to the medical community.
These new editorial offerings are just a start—we also want to invite you, your colleagues, and learners to start your own new tradition of writing for Federal Practitioner. For those who have submitted articles in the past, please keep up the habit.We are eager to receive original research, review articles, and clinical cases from DoD, PHS, and VA mid-career and senior clinicians and researchers as well as articles describing innovative programs and modes of health care treatment and delivery. With a print circulation of more than 35,000 readers and very active online presence, consider Federal Practitioner for your next article!
This year my New Year’s resolution as editor-in-chief is to encourage readers to contact either Editor Reid Paul or me if you have an idea for an article you would like to write, a column you would like to see, or if you have an interest in serving as a peer reviewer or joining our Editorial Advisory Association. We want to hear from you about what you want and need from Federal Practitioner.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies.
It has been a long time since a new year has brought as much uncertainty as 2017 promises to bring, making some federal employees excited and others apprehensive. Rumors abound of how the federal health care sector may change: Hiring freezes, manpower cuts, and privatization are all concerns of Federal Practitioner readers. As in the past, we will keep you up-to-date with in-depth interviews of leaders in federal health care, intelligent coverage of news stories impacting your practice, and clinical and research articles about new programs and initiatives.
Also this year, we are pleased to announce several new regular columns that we hope will inform and entertain you. The first is a column on mental health and traumatic brain injury in the DoD and the VA. We are privileged to have U.S. Army COL (Ret) Elspeth Cameron Ritchie, MD, MPH, edit this column. She is widely known and respected and brings her vast experience to the column as an active-duty psychiatrist coupled with her current position as a VA physician. Dr. Ritchie will author articles as well as edit those of her VA and DoD colleagues. Mental health touches almost every aspect of federal practice, and we all will learn our contributions and challenges in this rapidly moving specialty.
Whereas the mental health column looks toward the scientific future, the second column looks back to the humanistic past. We are thrilled that 2 physician-historians of military medicine, Robert Hierholzer, MD, a VA psychiatrist, and John Pierce, MD, a retired U.S. Army pediatrician share the writing and editing for this column, which will debut this spring.
Have you ever wondered who or how VA and military hospitals were named? These 2 historical writers have a wealth of interesting anecdotes and stories about VA and military medical centers. We hope you will enjoy reading the stories of military and veteran health care: the war heroes, devoted clinicians, and groundbreaking researchers who have left their mark on DoD and VA health care.
We also will be launching a new pilot study feature for clinicians and researchers who have a novel or valuable idea but have only a small number of participants or preliminary results. This will be a great way for new investigators, trainees, and young health care practitioners to present their work to the medical community.
These new editorial offerings are just a start—we also want to invite you, your colleagues, and learners to start your own new tradition of writing for Federal Practitioner. For those who have submitted articles in the past, please keep up the habit.We are eager to receive original research, review articles, and clinical cases from DoD, PHS, and VA mid-career and senior clinicians and researchers as well as articles describing innovative programs and modes of health care treatment and delivery. With a print circulation of more than 35,000 readers and very active online presence, consider Federal Practitioner for your next article!
This year my New Year’s resolution as editor-in-chief is to encourage readers to contact either Editor Reid Paul or me if you have an idea for an article you would like to write, a column you would like to see, or if you have an interest in serving as a peer reviewer or joining our Editorial Advisory Association. We want to hear from you about what you want and need from Federal Practitioner.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies.
