User login
Lichen Nitidus
Consensus Recommendations From the American Acne & Rosacea Society on the Management of Rosacea, Part 2: A Status Report on Topical Agents
Analysis of Multiple In‐Hospital CPR
Cardiopulmonary resuscitation (CPR) is a potentially lifesaving intervention associated with intense resource utilization and poor outcomes.[1, 2, 3] CPR is the default intervention for hospitalized patients in cardiopulmonary arrest in the United States. The most common measure of successful in‐hospital CPR reported in the literature is survival to (hospital) discharge, with most estimates between 13% and 37%.[3, 4, 5, 6] Poor rates of survival to discharge may be explained by use of CPR in patients for whom it was not originally intended, such as the very elderly with multiple illnesses or the terminally ill.[7, 8] Use of CPR in patients unlikely to benefit may be due to a physician's inability to estimate the probability of survival, desire to offer hope to patients, fear of litigation, and poor communication with patients about goals of care.[7, 8, 9, 10]
The general public has overly optimistic expectations about CPR; surveys have reported perceived survival after CPR of up to 90%.[11, 12, 13] Although objective information substantially affects patient preferences for resuscitation,[14] prognosis is rarely discussed during code status encounters[15, 16]; physician estimates of prognosis also are often inaccurate.[9, 17] With a scarcity of data describing the characteristics of patients undergoing multiple CPR attempts, and their outcomes, patients and their families could have false expectations about the likely outcomes from multiple CPR attempts, because physician counsel is not well‐informed.
In this study, we examine the epidemiology of in‐hospital CPR recipients stratified by the number of occurrences of CPR during a single hospitalization, along with their outcomes. We hypothesize that recipients of multiple CPR during a single hospitalization are an epidemiologically distinct group compared with those who receive CPR once during their hospitalization, and that their outcomes are worse.
METHODS
Data Source
We used unweighted data for the years 2000 to 2009 from the Healthcare Cost and Utilization ProjectNationwide Inpatient Sample (HCUP‐NIS). The NIS is the largest all‐payer inpatient‐care database in the United States, containing nationally representative information regarding up to 8 million hospital stays per year. Each year, NIS data consist of a 20% stratified sample of hospital discharges involving up to 1100 nonfederal hospitals from up to 44 states. The NIS utilizes International Classification of Diseases, Ninth Revision, Clinical Modification (ICD‐9‐CM) codes to capture up to 25 diagnoses and 15 procedures associated with the index hospitalization.[18]
Demographic, Clinical, and Hospital Characteristics of Cardiopulmonary Resuscitation Recipients
Adults (age 18 years) who underwent CPR (ICD‐9 procedure code 99.60) during their hospitalization were abstracted; this ICD‐9 code has been used previously to explore CPR epidemiology and outcomes.[3, 19, 20] Patients were divided into 2 groups, those who had 1 CPR attempt and those who had multiple (>1) CPR attempts, based on the number of times the ICD‐9 code for CPR was included in their hospitalization data. Patients who had cardiopulmonary arrest (ICD‐9 code 427.5 or 799.1) as a presenting diagnosis were excluded, as these indicate an out‐of‐hospital event.
Demographic variables included patient age, sex, race, median household income as defined annually in the NIS dataset, insurance status, admission source (skilled nursing facility or not; emergency room vs not), and type (elective vs nonelective; trauma vs nontrauma). Clinical variables included patient comorbidity as assessed by using the enhanced Charlson Comorbidity Index (CCI).[21] Rates of in‐hospital dialysis (ICD‐9 codes 39.95, V451, V561), tracheostomy (ICD‐9 codes 31.1, 31.2), in‐hospital neurologic compromise (coma, ICD‐9 code 780.01; semi‐coma, ICD‐9 code 780.09; persistent vegetative state, ICD‐9 code 780.03; anoxic brain injury, ICD‐9 code 348.1; and brain damage, ICD‐9 code 997.01), ventilator support (ICD‐9 code 967.02); and artificial nutrition (total parenteral nutrition, ICD‐9 code 99.15; enteral infusion of nutritional substances, ICD‐9 code 96.6) were assessed as potential indicators of clinical debilitation and/or intense healthcare resource utilization. Hospital variables were region in the United States (Northeast, Midwest, West, and South), location (urban vs nonurban), teaching status, and bed size (small, medium, and large), as defined annually in the NIS.[18]
Outcomes
Outcomes of interest were survival to discharge, discharge disposition, and cost of hospitalization.
Statistical Analysis
Sensitivity analyses were done to validate the use of the number of occurrences of CPR code 99.60 as a marker of multiple CPR, as well the association between multiple CPR and outcome. We computed the interval (in days) between the first and last CPR such that a result would not be computed if either value were missing. We found that 80.2% of patients who had CPR multiple times also had valid interval data between the first and last CPR. This was slightly higher than the 75.9% of patients with 1 CPR code who also had valid data for the interval (in days) between admission and CPR, indicating the reliability of using the number of CPR codes as a marker of multiple CPR attempts.
Bivariate analyses comparing characteristics and outcomes of interest for recipients of 1 CPR versus multiple CPR were performed using the [2] test for categorical variables and Student t test for continuous variables; differences in age and CCI score (analyzed as continuous variables) were assessed using the Mann‐Whitney U test because the distribution of data for these was not normal. Hospital length of stay and cost were natural log transformed to normalize distribution. Cost was calculated using HCUP‐NISadjusted, hospital‐specific cost‐to‐charge ratios; costs were adjusted for inflation, converting all costs to year 2009 dollar values using rates from the US Bureau of Labor Statistics.[22] Cost‐to‐charge ratios were first made available in the NIS datasets in year 2001; therefore, data for the year 2000 were excluded from all cost analyses. The aggregate cost of hospitalization at a population‐level was estimated using the discharge weight variable included in the NIS.
Separate multivariate logistic regression models were constructed to assess (1) factors independently associated with occurrence of multiple CPR, and (2) whether multiple CPR is independently associated with survival to discharge. Generalized estimating equations were used to account for hospital clustering. Odds ratios (OR) with 95% confidence intervals (CI) were computed for the final multivariate models. All P values <0.05 were considered significant; all tests were 2‐sided.
Data management and analysis were performed using SAS statistical software, version 9.3 (SAS Institute Inc, Cary, NC), and SPSS for Windows, version 18.0 (SPSS Inc, Chicago, IL). The HCUP‐NIS is a public database with no personally identifying information. This study was deemed exempt from institutional review board approval at our institution.
RESULTS
Of a total of 65,308,185 adults hospitalized between the years 2000 and 2009, there were 166,519 CPR recipients, yielding a CPR incidence of 2.5 per 1000 hospitalizations. Among CPR recipients, 96.6% (n=166,899) had 1 CPR and 3.4% (n=5620) had multiple CPR during their hospitalization (range, 111 CPR). When further stratified, 3% had 2 CPR attempts (n=4949) and 0.4% (n=671) had 3 CPR attempts.
Compared with patients who had 1 CPR, those who had multiple CPR were more often younger (median age, 71 vs 67 years), nonwhite, and in a low‐income quartile (all P<0.001; Table 1). Rates of admission from a nursing facility (3.3% for the 1‐CPR group vs 3.1% for the multiple‐CPR group, P=0.65) or as a trauma (0.3% for the 1‐CPR group and 0.4% for the multiple‐CPR group, P=0.34) were similar.
| Characteristic | 1 CPR (n=160,899), % | Multiple CPRs (n=5,620), % | P Value |
|---|---|---|---|
| |||
| Sex, F | 45.6 | 47.2 | 0.02 |
| Age, y, <65 | 37.3 | 42.5 | <0.001 |
| Race | <0.001 | ||
| White | 65.8 | 58.7 | |
| Black | 18.7 | 21.6 | |
| Other | 15.5 | 19.8 | |
| Income quartile | <0.001 | ||
| Low | 24.1 | 27.8 | |
| Medium‐low | 24.9 | 24.7 | |
| Medium | 23.2 | 22.9 | |
| High | 25.2 | 22.2 | |
| Unknown | 2.5 | 2.4 | |
| Insurance | <0.001 | ||
| Medicare | 65.1 | 61.8 | |
| Medicaid | 9.4 | 12.4 | |
| Private | 18.4 | 17.7 | |
| Other | 7.1 | 8.1 | |
| Admission source, ER | 67.9 | 72.0 | <0.001 |
| Admission type, elective | 10.0 | 7.1 | <0.001 |
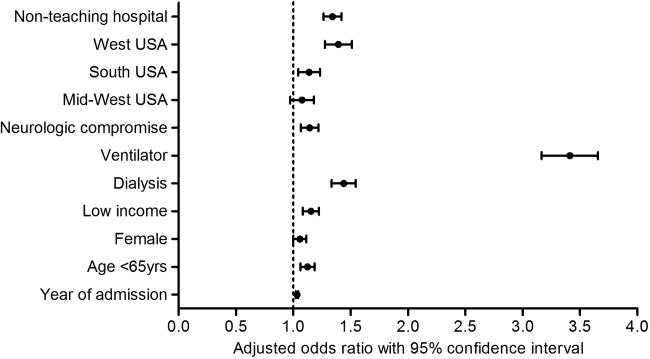
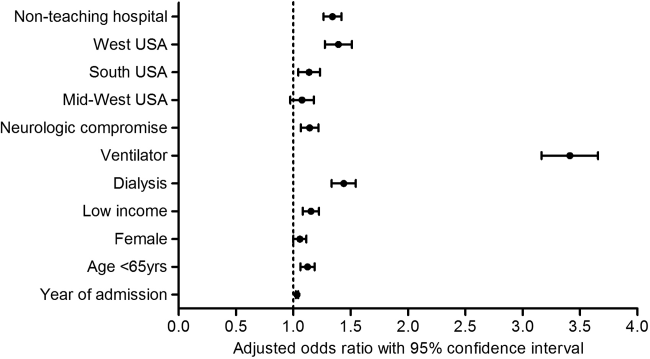
Patients who had multiple CPR had slightly higher mean CCI scores (2.7 vs 2.6, P=0.02). They had higher rates of neurologic compromise and aggressive interventions; they were also more commonly treated in nonteaching hospitals, and in the western region of the United States (Table 2). After multivariate analysis, several patient, clinical, and hospital factors were independently associated with occurrence of multiple CPR (Figure 1).
| Characteristic | 1 CPR (n=160,899), % | Multiple CPRs (n=5,620), % | P Value |
|---|---|---|---|
| |||
| Clinical | |||
| Charlson score 4 | 25.4 | 27.2 | 0.002 |
| MI | 24.9 | 28.5 | <0.001 |
| CHF | 38.3 | 43.3 | <0.001 |
| Cerebrovascular event | 8.5 | 7.1 | <0.001 |
| Metastatic malignancy | 10.6 | 8.7 | <0.001 |
| COPD | 26.0 | 26.0 | 0.945 |
| Neurologic impairment | 13.8 | 21.1 | <0.001 |
| Supplemental nutrition | 7.2 | 8.3 | 0.002 |
| Mechanical ventilator | 57.4 | 83.1 | <0.001 |
| Cardiac surgery | 2.6 | 2.0 | 0.007 |
| Hospital | |||
| Location, urban | 90.1 | 92.1 | <0.001 |
| Teaching status, no | 58.0 | 64.5 | <0.001 |
| Region | <0.001 | ||
| Northeast | 19.0 | 15.2 | |
| Midwest | 18.6 | 15.7 | |
| South | 37.4 | 37.1 | |
| West | 25.0 | 32.0 | |
| Bed size | 0.715 | ||
| Small | 10.2 | 9.8 | |
| Medium | 25.5 | 25.3 | |
| Large | 64.3 | 64.9 | |
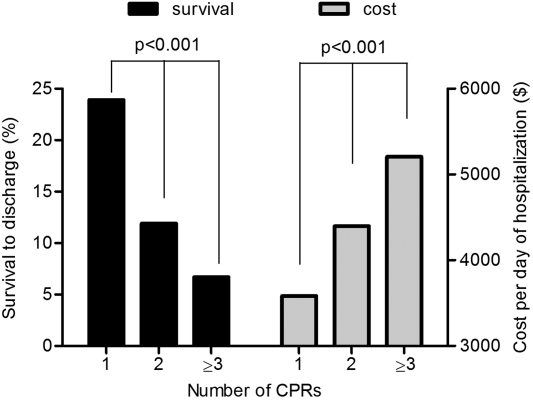
In bivariate analysis of survival, patients who had multiple CPR had lower rates of survival to discharge (11.3% vs 23.4%, P<0.001). Results were similar (11.6% for multiple CPR vs 22.5% for 1 CPR, P<0.001) when all patients who had CPR but did not have valid timing data were excluded in sensitivity analyses. Further stratification showed that survival to discharge decreased by >40% for each increase in CPR attempt (23.4%, 11.9%, and 6.7% for 1, 2, and 3 CPR attempts, respectively, P<0.001; Figure 2). After adjustment, multiple CPR versus 1 CPR during a hospitalization was independently associated with a lower likelihood of survival to discharge (adjusted OR: 0.41, 95% CI: 0.37‐0.44, P<0.001; Table 3).
| Characteristica | OR | 95% CI | P Value | |
|---|---|---|---|---|
| Lower | Upper | |||
| ||||
| Demographic | ||||
| Age <65 years | 1.339 | 1.304 | 1.375 | <0.001 |
| Sex, F | 1.128 | 1.099 | 1.157 | <0.001 |
| Race, nonwhite | 0.781 | 0.758 | 0.804 | <0.001 |
| Low income quartile | 0.887 | 0.858 | 0.915 | <0.001 |
| Year of admission | 1.051 | 1.046 | 1.056 | <0.001 |
| Clinical | ||||
| Multiple CPR | 0.406 | 0.371 | 0.445 | <0.001 |
| CCI score | 0.939 | 0.933 | 0.944 | <0.001 |
| Cardiac surgery | 1.785 | 1.720 | 1.853 | <0.001 |
| Hospital | ||||
| Region, Midwest | 1.472 | 1.405 | 1.543 | <0.001 |
| Region, South | 1.262 | 1.218 | 1.309 | 0.008 |
| Region, West | 1.452 | 1.398 | 1.509 | <0.001 |
| Location, urban | 0.876 | 0.837 | 0.917 | <0.001 |
Survivors with multiple CPR were less likely to be discharged home compared with survivors with 1 CPR (19.3% vs 29.9%, respectively, P<0.001); 1 in 15 survivors of multiple CPR were discharged to a hospice (6.8%) versus 1 in 23 1‐CPR survivors (4.3%; P=0.002). Mean length of stay was 5.8 versus 5.5 days for patients who had multiple CPR versus 1 CPR, respectively (P<0.001), and 16.0 versus 10.5 days for discharged survivors of multiple CPR versus 1 CPR (P<0.001). The average cost per day of hospitalization was higher for recipients of multiple CPR versus 1 CPR ($4484.60 vs $3581.40, P<0.001). The aggregate cost of hospitalization for 1‐time CPR recipients doubled between the years 2001 and 2009 (from $1.3 billion to $2.9 billion); that of recipients of multiple CPR attempts quadrupled in the same time frame (from $38.6 million to $160.7 million).
DISCUSSION
A number of studies have investigated the epidemiology of patients in whom CPR is attempted.[2, 3, 5, 20, 23, 24] Several pre‐, intra‐, and post‐resuscitation factors have been shown to affect the survival of resuscitated patients.[6, 7, 25, 26] To our knowledge, neither the epidemiology of hospitalized patients in whom resuscitation is attempted multiple times nor the prognostic value of multiple CPR attempts has been investigated. In this study, we found that multiple resuscitations are more commonly performed on younger, generally sicker patients; their outcomes are significantly compromised compared with patients who are resuscitated once during their hospitalization.
There was a steep decline in survival based on the number of resuscitation events. In multivariate analysis, patients who had multiple CPR were 2.5‐fold less likely to survive their hospitalization; survivors of multiple CPR also were more likely to be discharged to a hospice. Overall, this is indicative of clinical deterioration and prolongation of dying should a patient suffer multiple cardiopulmonary arrests during a hospitalization. The robust inverse relationship between multiple CPR and survival to discharge has implications for the development of prognostic models of outcomes following CPR, as previously designed prediction models of CPR outcomes such as the Cardiac Arrest Survival Post‐Resuscitation In‐hospital (CASPRI) score,[25] Pre‐Arrest Morbidity (PAM) score,[27] and Prognosis After Resuscitation (PAR) score[28] do not include multiple resuscitations as a variable of interest.
In‐hospital factors were found to be more important than patient factors, such as comorbidities or race, in determining the likelihood of multiple CPR attempts. Hospital teaching status and region remained significantly associated with likelihood of multiple CPR attempts. This is in agreement with studies that have described demographic and regional variation in utilization of do‐not‐resuscitate orders.[29, 30] These findings suggest substantial heterogeneity in the clinical culture and hospital practices across the United States regarding preemptive discussions about resuscitation. This means that where a patient receives care is a significant determinant of their probability of undergoing multiple CPR.
It is known that older patients are more likely to have advance directive orders[30, 31] and possibly document their wishes with regard to further resuscitation efforts. There also may be an inclination toward more aggressive care for younger adults compared with those of an advanced age. Uncertainty about a patient's goals of care likely feeds into an increased possibility of multiple resuscitation attempts; this may explain why neurologic compromise and being on ventilator support were independently associated with likelihood of multiple CPR, as these patients often have lost their ability to actively participate in decision‐making. The results of this study highlight the importance of engaging patients with a plausible risk of cardiopulmonary arrest about their goals for care and advance directives in a timely manner, regardless of age.
We found that the care of patients who undergo multiple resuscitations is associated with a higher cost of hospitalization than for patients in whom resuscitation is attempted once during their hospitalization. In addition, there was an exponential increase in aggregate cost over time for multiple CPR recipients compared with 1‐time CPR recipients. In a prior study, Ebell and Kruse showed an exponential inverse relationship between cost per surviving patient and rate of survival to discharge.[32] Considering that 93.3% of patients who had 3 resuscitation attempts died during their hospitalization, and that hospital‐level factors appear to play a significant role in likelihood of multiple CPR, consensus guidelines regarding the appropriateness of 3 resuscitation attempts during a single hospitalization may be relevant to aid the care of these patients.
Although the NIS is well‐validated,[18] there are some limitations. Whereas CPR incidence in this study (2.5 per 1000 hospitalizations) is within estimates (15 arrests per 1000 hospitalizations) reported in previous studies,[3, 5] potential undercoding of multiple CPR may explain why the multiple‐CPR rate in this study is lower than re‐arrest estimates provided in published studies.[2, 33] Indeed, accurate calculation of re‐arrest rates requires data on do‐not‐resuscitate orders instituted after successful resuscitation, which are not provided in the NIS. Information on patient‐provider discussions about CPR or prognosis is not included. Data regarding the underlying cause and type of arrest rhythm, rates of return to spontaneous circulation, length of code, patient location, critical‐care resources and length of critical‐care stay, availability of rapid‐response/code teams, time to defibrillation, use of therapeutic hypothermia, adherence to resuscitation guidelines, quality of CPR, and long‐term follow‐up are not included in the database. Presenting rhythms were not assessed, as there are no ICD‐9 codes for asystole and pulseless electrical activity. The NIS is de‐identified; therefore, chart review to assess the validity of codes is impossible. However, our sensitivity analyses indicate the reliability of using the number of occurrences of the CPR code as a marker of multiple CPR. The strength of our study lies in the use of data that provide a population‐level insight into the epidemiology of patients resuscitated multiple times during their hospitalization, and their outcomes.
Decision‐making about CPR is at the center of a complex debate that incorporates often divergent clinical, economic, ethical, and personal issues. As debate continues regarding when to not resuscitate,[34, 35, 36, 37] studies that explore the public perspective of survival thresholds for the provision of multiple resuscitations will be crucial. As competition for finite healthcare dollars escalates, stratified analyses of the cost implications of resuscitation care are essential. Studies are needed to examine the impact of a history of successful resuscitation in a previous hospitalization on outcomes following CPR in a subsequent hospitalization. Overall, our study fills an important knowledge gap in resuscitation practice and outcomes in the United States and highlights the importance of discussing resuscitation options between a patient and his or her family on hospital admission and, if needed, again after the first successful resuscitation attempt.
Disclosure
Nothing to report.
- , , , . Trends in inpatient treatment intensity among Medicare beneficiaries at the end of life. Health Serv Res. 2004;39:363–376.
- , , , et al. Cardiopulmonary resuscitation of adults in the hospital: a report of 14,720 cardiac arrests from the national registry of cardiopulmonary resuscitation. Resuscitation. 2003;58:297–308.
- , , , et al. Epidemiologic study of in‐hospital cardiopulmonary resuscitation in the elderly. N Engl J Med. 2009;361:22–31.
- , , , et al; National Registry of Cardiopulmonary Resuscitation Investigators. Survival from in‐hospital cardiac arrest during nights and weekends. JAMA. 2008;299:785–792.
- , , , . In‐hospital cardiac arrest: incidence, prognosis and possible measures to improve survival. Intensive Care Med. 2007;33:237–245.
- , , , . Predictors of survival following in‐hospital adult cardiopulmonary resuscitation. CMAJ. 2002;167:343–348.
- , . Pre‐arrest predictors of failure to survive after in‐hospital cardiopulmonary resuscitation: a meta‐analysis. Fam Pract. 2011;28:505–515.
- , . Cardiopulmonary resuscitation in older people—a review. Rev Clin Gerontol. 2010;20:20–29.
- , . Extent and determinants of error in doctors' prognoses in terminally ill patients: prospective cohort study. BMJ. 2000;320:469–472.
- , , . Physicians' confidence in discussing do not resuscitate orders with patients and surrogates. J Med Ethics. 2008;34:96–101.
- , . How misconceptions among elderly patients regarding survival outcomes of inpatient cardiopulmonary resuscitation affect do‐not‐resuscitate orders. J Am Osteopath Assoc. 2006;106:402–404.
- , , . Cardiopulmonary resuscitation on television—miracles and misinformation. N Engl J Med. 1996;334:1578–1582.
- , , . Public expectations of survival following cardiopulmonary resuscitation. Acad Emerg Med. 2000;7:48–53.
- , , , et al. The influence of the probability of survival on patients' preferences regarding cardiopulmonary resuscitation. N Engl J Med. 1994;330:545–549.
- , , , , . Code status discussions between attending hospitalist physicians and medical patients at hospital admission. J Gen Intern Med. 2011;26:359–366.
- , , . Hospital do‐not‐resuscitate orders: why they have failed and how to fix them. J Gen Intern Med. 2011;26:791–797.
- , , , . The inability of physicians to predict the outcome of in‐hospital resuscitation. J Gen Intern Med. 1996;11:16–22.
- Healthcare Cost and Utilization Project. Overview of the Nationwide Inpatient Sample. http://www.hcup‐us.ahrq.gov/nisoverview.jsp. Accessed June 24, 2013.
- , , , et al. Long‐term outcomes in elderly survivors of in‐hospital cardiac arrest. N Engl J Med. 2013;368:1019–1026.
- , , . Epidemiology and outcomes of in‐hospital cardiopulmonary resuscitation in the United States, 2000–2009. Resuscitation. 2013;84:1255–1260.
- , , , et al. Coding algorithms for defining comorbidities in ICD‐9‐CM and ICD‐10 administrative data. Med Care. 2005;43:1130–1139.
- US Department of Labor, Bureau of Labor Statistics. Inflation calculator. http://www.bls.gov/data/inflation_calculator.htm. Accessed June 24, 2013.
- , , , et al. Part 4: CPR overview. 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010;122:S676–S684.
- , , , et al. Choices of seriously ill patients about cardiopulmonary resuscitation: correlates and outcomes. Am J Med. 1996;100:128–137.
- , , , et al. A validated prediction tool for initial survivors of in‐hospital cardiac arrest. Arch Intern Med. 2012;172:947–953.
- , , , . Pre‐resuscitation factors associated with mortality in 49,130 cases of in‐hospital cardiac arrest: a report from the national registry for cardiopulmonary resuscitation. Resuscitation. 2010;81:302–311.
- , , , . Pre‐arrest morbidity and other correlates of survival after in‐hospital cardiopulmonary arrest. Am J Med. 1989;87:28–34.
- , . Prediction of failure to survive following in‐hospital cardiopulmonary resuscitation: comparison of two predictive instruments. Resuscitation. 1994;28:21–25.
- , . Regional and institutional variation in the initiation of early do‐not‐resuscitate orders. Arch Intern Med. 2005;165:1705–1712.
- , , , et al. Epidemiology of do‐not‐resuscitate orders: disparity by age, diagnosis, gender, race, and functional impairment. Arch Intern Med. 1995;155:2056–2062.
- , , , , , . Patients' understanding of advance directives and cardiopulmonary resuscitation. J Crit Care. 2005;20:26–34.
- , . A proposed model for the cost of cardiopulmonary resuscitation. Med Care. 1994;32:640–649.
- , , . Predictors of cardiopulmonary arrest outcome in a comprehensive cancer center intensive care unit. Scand J Trauma Resusc Emerg Med. 2013; 21:18.
- . A critic's assessment of our approach to cardiac arrest. N Engl J Med. 2011;364:374–375.
- . Should there be a choice for cardiopulmonary resuscitation when death is expected? Revisiting an old idea whose time is yet to come. J Palliat Med. 2002;5:107–116.
- . Clinical model for ethical cardiopulmonary resuscitation decision‐making. Intern Med J. 2013;43:77–83.
- , , . Avoiding the futility of resuscitation. Resuscitation. 2001;50:161–166.
Cardiopulmonary resuscitation (CPR) is a potentially lifesaving intervention associated with intense resource utilization and poor outcomes.[1, 2, 3] CPR is the default intervention for hospitalized patients in cardiopulmonary arrest in the United States. The most common measure of successful in‐hospital CPR reported in the literature is survival to (hospital) discharge, with most estimates between 13% and 37%.[3, 4, 5, 6] Poor rates of survival to discharge may be explained by use of CPR in patients for whom it was not originally intended, such as the very elderly with multiple illnesses or the terminally ill.[7, 8] Use of CPR in patients unlikely to benefit may be due to a physician's inability to estimate the probability of survival, desire to offer hope to patients, fear of litigation, and poor communication with patients about goals of care.[7, 8, 9, 10]
The general public has overly optimistic expectations about CPR; surveys have reported perceived survival after CPR of up to 90%.[11, 12, 13] Although objective information substantially affects patient preferences for resuscitation,[14] prognosis is rarely discussed during code status encounters[15, 16]; physician estimates of prognosis also are often inaccurate.[9, 17] With a scarcity of data describing the characteristics of patients undergoing multiple CPR attempts, and their outcomes, patients and their families could have false expectations about the likely outcomes from multiple CPR attempts, because physician counsel is not well‐informed.
In this study, we examine the epidemiology of in‐hospital CPR recipients stratified by the number of occurrences of CPR during a single hospitalization, along with their outcomes. We hypothesize that recipients of multiple CPR during a single hospitalization are an epidemiologically distinct group compared with those who receive CPR once during their hospitalization, and that their outcomes are worse.
METHODS
Data Source
We used unweighted data for the years 2000 to 2009 from the Healthcare Cost and Utilization ProjectNationwide Inpatient Sample (HCUP‐NIS). The NIS is the largest all‐payer inpatient‐care database in the United States, containing nationally representative information regarding up to 8 million hospital stays per year. Each year, NIS data consist of a 20% stratified sample of hospital discharges involving up to 1100 nonfederal hospitals from up to 44 states. The NIS utilizes International Classification of Diseases, Ninth Revision, Clinical Modification (ICD‐9‐CM) codes to capture up to 25 diagnoses and 15 procedures associated with the index hospitalization.[18]
Demographic, Clinical, and Hospital Characteristics of Cardiopulmonary Resuscitation Recipients
Adults (age 18 years) who underwent CPR (ICD‐9 procedure code 99.60) during their hospitalization were abstracted; this ICD‐9 code has been used previously to explore CPR epidemiology and outcomes.[3, 19, 20] Patients were divided into 2 groups, those who had 1 CPR attempt and those who had multiple (>1) CPR attempts, based on the number of times the ICD‐9 code for CPR was included in their hospitalization data. Patients who had cardiopulmonary arrest (ICD‐9 code 427.5 or 799.1) as a presenting diagnosis were excluded, as these indicate an out‐of‐hospital event.
Demographic variables included patient age, sex, race, median household income as defined annually in the NIS dataset, insurance status, admission source (skilled nursing facility or not; emergency room vs not), and type (elective vs nonelective; trauma vs nontrauma). Clinical variables included patient comorbidity as assessed by using the enhanced Charlson Comorbidity Index (CCI).[21] Rates of in‐hospital dialysis (ICD‐9 codes 39.95, V451, V561), tracheostomy (ICD‐9 codes 31.1, 31.2), in‐hospital neurologic compromise (coma, ICD‐9 code 780.01; semi‐coma, ICD‐9 code 780.09; persistent vegetative state, ICD‐9 code 780.03; anoxic brain injury, ICD‐9 code 348.1; and brain damage, ICD‐9 code 997.01), ventilator support (ICD‐9 code 967.02); and artificial nutrition (total parenteral nutrition, ICD‐9 code 99.15; enteral infusion of nutritional substances, ICD‐9 code 96.6) were assessed as potential indicators of clinical debilitation and/or intense healthcare resource utilization. Hospital variables were region in the United States (Northeast, Midwest, West, and South), location (urban vs nonurban), teaching status, and bed size (small, medium, and large), as defined annually in the NIS.[18]
Outcomes
Outcomes of interest were survival to discharge, discharge disposition, and cost of hospitalization.
Statistical Analysis
Sensitivity analyses were done to validate the use of the number of occurrences of CPR code 99.60 as a marker of multiple CPR, as well the association between multiple CPR and outcome. We computed the interval (in days) between the first and last CPR such that a result would not be computed if either value were missing. We found that 80.2% of patients who had CPR multiple times also had valid interval data between the first and last CPR. This was slightly higher than the 75.9% of patients with 1 CPR code who also had valid data for the interval (in days) between admission and CPR, indicating the reliability of using the number of CPR codes as a marker of multiple CPR attempts.
Bivariate analyses comparing characteristics and outcomes of interest for recipients of 1 CPR versus multiple CPR were performed using the [2] test for categorical variables and Student t test for continuous variables; differences in age and CCI score (analyzed as continuous variables) were assessed using the Mann‐Whitney U test because the distribution of data for these was not normal. Hospital length of stay and cost were natural log transformed to normalize distribution. Cost was calculated using HCUP‐NISadjusted, hospital‐specific cost‐to‐charge ratios; costs were adjusted for inflation, converting all costs to year 2009 dollar values using rates from the US Bureau of Labor Statistics.[22] Cost‐to‐charge ratios were first made available in the NIS datasets in year 2001; therefore, data for the year 2000 were excluded from all cost analyses. The aggregate cost of hospitalization at a population‐level was estimated using the discharge weight variable included in the NIS.
Separate multivariate logistic regression models were constructed to assess (1) factors independently associated with occurrence of multiple CPR, and (2) whether multiple CPR is independently associated with survival to discharge. Generalized estimating equations were used to account for hospital clustering. Odds ratios (OR) with 95% confidence intervals (CI) were computed for the final multivariate models. All P values <0.05 were considered significant; all tests were 2‐sided.
Data management and analysis were performed using SAS statistical software, version 9.3 (SAS Institute Inc, Cary, NC), and SPSS for Windows, version 18.0 (SPSS Inc, Chicago, IL). The HCUP‐NIS is a public database with no personally identifying information. This study was deemed exempt from institutional review board approval at our institution.
RESULTS
Of a total of 65,308,185 adults hospitalized between the years 2000 and 2009, there were 166,519 CPR recipients, yielding a CPR incidence of 2.5 per 1000 hospitalizations. Among CPR recipients, 96.6% (n=166,899) had 1 CPR and 3.4% (n=5620) had multiple CPR during their hospitalization (range, 111 CPR). When further stratified, 3% had 2 CPR attempts (n=4949) and 0.4% (n=671) had 3 CPR attempts.
Compared with patients who had 1 CPR, those who had multiple CPR were more often younger (median age, 71 vs 67 years), nonwhite, and in a low‐income quartile (all P<0.001; Table 1). Rates of admission from a nursing facility (3.3% for the 1‐CPR group vs 3.1% for the multiple‐CPR group, P=0.65) or as a trauma (0.3% for the 1‐CPR group and 0.4% for the multiple‐CPR group, P=0.34) were similar.
| Characteristic | 1 CPR (n=160,899), % | Multiple CPRs (n=5,620), % | P Value |
|---|---|---|---|
| |||
| Sex, F | 45.6 | 47.2 | 0.02 |
| Age, y, <65 | 37.3 | 42.5 | <0.001 |
| Race | <0.001 | ||
| White | 65.8 | 58.7 | |
| Black | 18.7 | 21.6 | |
| Other | 15.5 | 19.8 | |
| Income quartile | <0.001 | ||
| Low | 24.1 | 27.8 | |
| Medium‐low | 24.9 | 24.7 | |
| Medium | 23.2 | 22.9 | |
| High | 25.2 | 22.2 | |
| Unknown | 2.5 | 2.4 | |
| Insurance | <0.001 | ||
| Medicare | 65.1 | 61.8 | |
| Medicaid | 9.4 | 12.4 | |
| Private | 18.4 | 17.7 | |
| Other | 7.1 | 8.1 | |
| Admission source, ER | 67.9 | 72.0 | <0.001 |
| Admission type, elective | 10.0 | 7.1 | <0.001 |
Patients who had multiple CPR had slightly higher mean CCI scores (2.7 vs 2.6, P=0.02). They had higher rates of neurologic compromise and aggressive interventions; they were also more commonly treated in nonteaching hospitals, and in the western region of the United States (Table 2). After multivariate analysis, several patient, clinical, and hospital factors were independently associated with occurrence of multiple CPR (Figure 1).
| Characteristic | 1 CPR (n=160,899), % | Multiple CPRs (n=5,620), % | P Value |
|---|---|---|---|
| |||
| Clinical | |||
| Charlson score 4 | 25.4 | 27.2 | 0.002 |
| MI | 24.9 | 28.5 | <0.001 |
| CHF | 38.3 | 43.3 | <0.001 |
| Cerebrovascular event | 8.5 | 7.1 | <0.001 |
| Metastatic malignancy | 10.6 | 8.7 | <0.001 |
| COPD | 26.0 | 26.0 | 0.945 |
| Neurologic impairment | 13.8 | 21.1 | <0.001 |
| Supplemental nutrition | 7.2 | 8.3 | 0.002 |
| Mechanical ventilator | 57.4 | 83.1 | <0.001 |
| Cardiac surgery | 2.6 | 2.0 | 0.007 |
| Hospital | |||
| Location, urban | 90.1 | 92.1 | <0.001 |
| Teaching status, no | 58.0 | 64.5 | <0.001 |
| Region | <0.001 | ||
| Northeast | 19.0 | 15.2 | |
| Midwest | 18.6 | 15.7 | |
| South | 37.4 | 37.1 | |
| West | 25.0 | 32.0 | |
| Bed size | 0.715 | ||
| Small | 10.2 | 9.8 | |
| Medium | 25.5 | 25.3 | |
| Large | 64.3 | 64.9 | |
In bivariate analysis of survival, patients who had multiple CPR had lower rates of survival to discharge (11.3% vs 23.4%, P<0.001). Results were similar (11.6% for multiple CPR vs 22.5% for 1 CPR, P<0.001) when all patients who had CPR but did not have valid timing data were excluded in sensitivity analyses. Further stratification showed that survival to discharge decreased by >40% for each increase in CPR attempt (23.4%, 11.9%, and 6.7% for 1, 2, and 3 CPR attempts, respectively, P<0.001; Figure 2). After adjustment, multiple CPR versus 1 CPR during a hospitalization was independently associated with a lower likelihood of survival to discharge (adjusted OR: 0.41, 95% CI: 0.37‐0.44, P<0.001; Table 3).
| Characteristica | OR | 95% CI | P Value | |
|---|---|---|---|---|
| Lower | Upper | |||
| ||||
| Demographic | ||||
| Age <65 years | 1.339 | 1.304 | 1.375 | <0.001 |
| Sex, F | 1.128 | 1.099 | 1.157 | <0.001 |
| Race, nonwhite | 0.781 | 0.758 | 0.804 | <0.001 |
| Low income quartile | 0.887 | 0.858 | 0.915 | <0.001 |
| Year of admission | 1.051 | 1.046 | 1.056 | <0.001 |
| Clinical | ||||
| Multiple CPR | 0.406 | 0.371 | 0.445 | <0.001 |
| CCI score | 0.939 | 0.933 | 0.944 | <0.001 |
| Cardiac surgery | 1.785 | 1.720 | 1.853 | <0.001 |
| Hospital | ||||
| Region, Midwest | 1.472 | 1.405 | 1.543 | <0.001 |
| Region, South | 1.262 | 1.218 | 1.309 | 0.008 |
| Region, West | 1.452 | 1.398 | 1.509 | <0.001 |
| Location, urban | 0.876 | 0.837 | 0.917 | <0.001 |
Survivors with multiple CPR were less likely to be discharged home compared with survivors with 1 CPR (19.3% vs 29.9%, respectively, P<0.001); 1 in 15 survivors of multiple CPR were discharged to a hospice (6.8%) versus 1 in 23 1‐CPR survivors (4.3%; P=0.002). Mean length of stay was 5.8 versus 5.5 days for patients who had multiple CPR versus 1 CPR, respectively (P<0.001), and 16.0 versus 10.5 days for discharged survivors of multiple CPR versus 1 CPR (P<0.001). The average cost per day of hospitalization was higher for recipients of multiple CPR versus 1 CPR ($4484.60 vs $3581.40, P<0.001). The aggregate cost of hospitalization for 1‐time CPR recipients doubled between the years 2001 and 2009 (from $1.3 billion to $2.9 billion); that of recipients of multiple CPR attempts quadrupled in the same time frame (from $38.6 million to $160.7 million).
DISCUSSION
A number of studies have investigated the epidemiology of patients in whom CPR is attempted.[2, 3, 5, 20, 23, 24] Several pre‐, intra‐, and post‐resuscitation factors have been shown to affect the survival of resuscitated patients.[6, 7, 25, 26] To our knowledge, neither the epidemiology of hospitalized patients in whom resuscitation is attempted multiple times nor the prognostic value of multiple CPR attempts has been investigated. In this study, we found that multiple resuscitations are more commonly performed on younger, generally sicker patients; their outcomes are significantly compromised compared with patients who are resuscitated once during their hospitalization.
There was a steep decline in survival based on the number of resuscitation events. In multivariate analysis, patients who had multiple CPR were 2.5‐fold less likely to survive their hospitalization; survivors of multiple CPR also were more likely to be discharged to a hospice. Overall, this is indicative of clinical deterioration and prolongation of dying should a patient suffer multiple cardiopulmonary arrests during a hospitalization. The robust inverse relationship between multiple CPR and survival to discharge has implications for the development of prognostic models of outcomes following CPR, as previously designed prediction models of CPR outcomes such as the Cardiac Arrest Survival Post‐Resuscitation In‐hospital (CASPRI) score,[25] Pre‐Arrest Morbidity (PAM) score,[27] and Prognosis After Resuscitation (PAR) score[28] do not include multiple resuscitations as a variable of interest.
In‐hospital factors were found to be more important than patient factors, such as comorbidities or race, in determining the likelihood of multiple CPR attempts. Hospital teaching status and region remained significantly associated with likelihood of multiple CPR attempts. This is in agreement with studies that have described demographic and regional variation in utilization of do‐not‐resuscitate orders.[29, 30] These findings suggest substantial heterogeneity in the clinical culture and hospital practices across the United States regarding preemptive discussions about resuscitation. This means that where a patient receives care is a significant determinant of their probability of undergoing multiple CPR.
It is known that older patients are more likely to have advance directive orders[30, 31] and possibly document their wishes with regard to further resuscitation efforts. There also may be an inclination toward more aggressive care for younger adults compared with those of an advanced age. Uncertainty about a patient's goals of care likely feeds into an increased possibility of multiple resuscitation attempts; this may explain why neurologic compromise and being on ventilator support were independently associated with likelihood of multiple CPR, as these patients often have lost their ability to actively participate in decision‐making. The results of this study highlight the importance of engaging patients with a plausible risk of cardiopulmonary arrest about their goals for care and advance directives in a timely manner, regardless of age.
We found that the care of patients who undergo multiple resuscitations is associated with a higher cost of hospitalization than for patients in whom resuscitation is attempted once during their hospitalization. In addition, there was an exponential increase in aggregate cost over time for multiple CPR recipients compared with 1‐time CPR recipients. In a prior study, Ebell and Kruse showed an exponential inverse relationship between cost per surviving patient and rate of survival to discharge.[32] Considering that 93.3% of patients who had 3 resuscitation attempts died during their hospitalization, and that hospital‐level factors appear to play a significant role in likelihood of multiple CPR, consensus guidelines regarding the appropriateness of 3 resuscitation attempts during a single hospitalization may be relevant to aid the care of these patients.
Although the NIS is well‐validated,[18] there are some limitations. Whereas CPR incidence in this study (2.5 per 1000 hospitalizations) is within estimates (15 arrests per 1000 hospitalizations) reported in previous studies,[3, 5] potential undercoding of multiple CPR may explain why the multiple‐CPR rate in this study is lower than re‐arrest estimates provided in published studies.[2, 33] Indeed, accurate calculation of re‐arrest rates requires data on do‐not‐resuscitate orders instituted after successful resuscitation, which are not provided in the NIS. Information on patient‐provider discussions about CPR or prognosis is not included. Data regarding the underlying cause and type of arrest rhythm, rates of return to spontaneous circulation, length of code, patient location, critical‐care resources and length of critical‐care stay, availability of rapid‐response/code teams, time to defibrillation, use of therapeutic hypothermia, adherence to resuscitation guidelines, quality of CPR, and long‐term follow‐up are not included in the database. Presenting rhythms were not assessed, as there are no ICD‐9 codes for asystole and pulseless electrical activity. The NIS is de‐identified; therefore, chart review to assess the validity of codes is impossible. However, our sensitivity analyses indicate the reliability of using the number of occurrences of the CPR code as a marker of multiple CPR. The strength of our study lies in the use of data that provide a population‐level insight into the epidemiology of patients resuscitated multiple times during their hospitalization, and their outcomes.
Decision‐making about CPR is at the center of a complex debate that incorporates often divergent clinical, economic, ethical, and personal issues. As debate continues regarding when to not resuscitate,[34, 35, 36, 37] studies that explore the public perspective of survival thresholds for the provision of multiple resuscitations will be crucial. As competition for finite healthcare dollars escalates, stratified analyses of the cost implications of resuscitation care are essential. Studies are needed to examine the impact of a history of successful resuscitation in a previous hospitalization on outcomes following CPR in a subsequent hospitalization. Overall, our study fills an important knowledge gap in resuscitation practice and outcomes in the United States and highlights the importance of discussing resuscitation options between a patient and his or her family on hospital admission and, if needed, again after the first successful resuscitation attempt.
Disclosure
Nothing to report.
Cardiopulmonary resuscitation (CPR) is a potentially lifesaving intervention associated with intense resource utilization and poor outcomes.[1, 2, 3] CPR is the default intervention for hospitalized patients in cardiopulmonary arrest in the United States. The most common measure of successful in‐hospital CPR reported in the literature is survival to (hospital) discharge, with most estimates between 13% and 37%.[3, 4, 5, 6] Poor rates of survival to discharge may be explained by use of CPR in patients for whom it was not originally intended, such as the very elderly with multiple illnesses or the terminally ill.[7, 8] Use of CPR in patients unlikely to benefit may be due to a physician's inability to estimate the probability of survival, desire to offer hope to patients, fear of litigation, and poor communication with patients about goals of care.[7, 8, 9, 10]
The general public has overly optimistic expectations about CPR; surveys have reported perceived survival after CPR of up to 90%.[11, 12, 13] Although objective information substantially affects patient preferences for resuscitation,[14] prognosis is rarely discussed during code status encounters[15, 16]; physician estimates of prognosis also are often inaccurate.[9, 17] With a scarcity of data describing the characteristics of patients undergoing multiple CPR attempts, and their outcomes, patients and their families could have false expectations about the likely outcomes from multiple CPR attempts, because physician counsel is not well‐informed.
In this study, we examine the epidemiology of in‐hospital CPR recipients stratified by the number of occurrences of CPR during a single hospitalization, along with their outcomes. We hypothesize that recipients of multiple CPR during a single hospitalization are an epidemiologically distinct group compared with those who receive CPR once during their hospitalization, and that their outcomes are worse.
METHODS
Data Source
We used unweighted data for the years 2000 to 2009 from the Healthcare Cost and Utilization ProjectNationwide Inpatient Sample (HCUP‐NIS). The NIS is the largest all‐payer inpatient‐care database in the United States, containing nationally representative information regarding up to 8 million hospital stays per year. Each year, NIS data consist of a 20% stratified sample of hospital discharges involving up to 1100 nonfederal hospitals from up to 44 states. The NIS utilizes International Classification of Diseases, Ninth Revision, Clinical Modification (ICD‐9‐CM) codes to capture up to 25 diagnoses and 15 procedures associated with the index hospitalization.[18]
Demographic, Clinical, and Hospital Characteristics of Cardiopulmonary Resuscitation Recipients
Adults (age 18 years) who underwent CPR (ICD‐9 procedure code 99.60) during their hospitalization were abstracted; this ICD‐9 code has been used previously to explore CPR epidemiology and outcomes.[3, 19, 20] Patients were divided into 2 groups, those who had 1 CPR attempt and those who had multiple (>1) CPR attempts, based on the number of times the ICD‐9 code for CPR was included in their hospitalization data. Patients who had cardiopulmonary arrest (ICD‐9 code 427.5 or 799.1) as a presenting diagnosis were excluded, as these indicate an out‐of‐hospital event.
Demographic variables included patient age, sex, race, median household income as defined annually in the NIS dataset, insurance status, admission source (skilled nursing facility or not; emergency room vs not), and type (elective vs nonelective; trauma vs nontrauma). Clinical variables included patient comorbidity as assessed by using the enhanced Charlson Comorbidity Index (CCI).[21] Rates of in‐hospital dialysis (ICD‐9 codes 39.95, V451, V561), tracheostomy (ICD‐9 codes 31.1, 31.2), in‐hospital neurologic compromise (coma, ICD‐9 code 780.01; semi‐coma, ICD‐9 code 780.09; persistent vegetative state, ICD‐9 code 780.03; anoxic brain injury, ICD‐9 code 348.1; and brain damage, ICD‐9 code 997.01), ventilator support (ICD‐9 code 967.02); and artificial nutrition (total parenteral nutrition, ICD‐9 code 99.15; enteral infusion of nutritional substances, ICD‐9 code 96.6) were assessed as potential indicators of clinical debilitation and/or intense healthcare resource utilization. Hospital variables were region in the United States (Northeast, Midwest, West, and South), location (urban vs nonurban), teaching status, and bed size (small, medium, and large), as defined annually in the NIS.[18]
Outcomes
Outcomes of interest were survival to discharge, discharge disposition, and cost of hospitalization.
Statistical Analysis
Sensitivity analyses were done to validate the use of the number of occurrences of CPR code 99.60 as a marker of multiple CPR, as well the association between multiple CPR and outcome. We computed the interval (in days) between the first and last CPR such that a result would not be computed if either value were missing. We found that 80.2% of patients who had CPR multiple times also had valid interval data between the first and last CPR. This was slightly higher than the 75.9% of patients with 1 CPR code who also had valid data for the interval (in days) between admission and CPR, indicating the reliability of using the number of CPR codes as a marker of multiple CPR attempts.
Bivariate analyses comparing characteristics and outcomes of interest for recipients of 1 CPR versus multiple CPR were performed using the [2] test for categorical variables and Student t test for continuous variables; differences in age and CCI score (analyzed as continuous variables) were assessed using the Mann‐Whitney U test because the distribution of data for these was not normal. Hospital length of stay and cost were natural log transformed to normalize distribution. Cost was calculated using HCUP‐NISadjusted, hospital‐specific cost‐to‐charge ratios; costs were adjusted for inflation, converting all costs to year 2009 dollar values using rates from the US Bureau of Labor Statistics.[22] Cost‐to‐charge ratios were first made available in the NIS datasets in year 2001; therefore, data for the year 2000 were excluded from all cost analyses. The aggregate cost of hospitalization at a population‐level was estimated using the discharge weight variable included in the NIS.
Separate multivariate logistic regression models were constructed to assess (1) factors independently associated with occurrence of multiple CPR, and (2) whether multiple CPR is independently associated with survival to discharge. Generalized estimating equations were used to account for hospital clustering. Odds ratios (OR) with 95% confidence intervals (CI) were computed for the final multivariate models. All P values <0.05 were considered significant; all tests were 2‐sided.
Data management and analysis were performed using SAS statistical software, version 9.3 (SAS Institute Inc, Cary, NC), and SPSS for Windows, version 18.0 (SPSS Inc, Chicago, IL). The HCUP‐NIS is a public database with no personally identifying information. This study was deemed exempt from institutional review board approval at our institution.
RESULTS
Of a total of 65,308,185 adults hospitalized between the years 2000 and 2009, there were 166,519 CPR recipients, yielding a CPR incidence of 2.5 per 1000 hospitalizations. Among CPR recipients, 96.6% (n=166,899) had 1 CPR and 3.4% (n=5620) had multiple CPR during their hospitalization (range, 111 CPR). When further stratified, 3% had 2 CPR attempts (n=4949) and 0.4% (n=671) had 3 CPR attempts.
Compared with patients who had 1 CPR, those who had multiple CPR were more often younger (median age, 71 vs 67 years), nonwhite, and in a low‐income quartile (all P<0.001; Table 1). Rates of admission from a nursing facility (3.3% for the 1‐CPR group vs 3.1% for the multiple‐CPR group, P=0.65) or as a trauma (0.3% for the 1‐CPR group and 0.4% for the multiple‐CPR group, P=0.34) were similar.
| Characteristic | 1 CPR (n=160,899), % | Multiple CPRs (n=5,620), % | P Value |
|---|---|---|---|
| |||
| Sex, F | 45.6 | 47.2 | 0.02 |
| Age, y, <65 | 37.3 | 42.5 | <0.001 |
| Race | <0.001 | ||
| White | 65.8 | 58.7 | |
| Black | 18.7 | 21.6 | |
| Other | 15.5 | 19.8 | |
| Income quartile | <0.001 | ||
| Low | 24.1 | 27.8 | |
| Medium‐low | 24.9 | 24.7 | |
| Medium | 23.2 | 22.9 | |
| High | 25.2 | 22.2 | |
| Unknown | 2.5 | 2.4 | |
| Insurance | <0.001 | ||
| Medicare | 65.1 | 61.8 | |
| Medicaid | 9.4 | 12.4 | |
| Private | 18.4 | 17.7 | |
| Other | 7.1 | 8.1 | |
| Admission source, ER | 67.9 | 72.0 | <0.001 |
| Admission type, elective | 10.0 | 7.1 | <0.001 |
Patients who had multiple CPR had slightly higher mean CCI scores (2.7 vs 2.6, P=0.02). They had higher rates of neurologic compromise and aggressive interventions; they were also more commonly treated in nonteaching hospitals, and in the western region of the United States (Table 2). After multivariate analysis, several patient, clinical, and hospital factors were independently associated with occurrence of multiple CPR (Figure 1).
| Characteristic | 1 CPR (n=160,899), % | Multiple CPRs (n=5,620), % | P Value |
|---|---|---|---|
| |||
| Clinical | |||
| Charlson score 4 | 25.4 | 27.2 | 0.002 |
| MI | 24.9 | 28.5 | <0.001 |
| CHF | 38.3 | 43.3 | <0.001 |
| Cerebrovascular event | 8.5 | 7.1 | <0.001 |
| Metastatic malignancy | 10.6 | 8.7 | <0.001 |
| COPD | 26.0 | 26.0 | 0.945 |
| Neurologic impairment | 13.8 | 21.1 | <0.001 |
| Supplemental nutrition | 7.2 | 8.3 | 0.002 |
| Mechanical ventilator | 57.4 | 83.1 | <0.001 |
| Cardiac surgery | 2.6 | 2.0 | 0.007 |
| Hospital | |||
| Location, urban | 90.1 | 92.1 | <0.001 |
| Teaching status, no | 58.0 | 64.5 | <0.001 |
| Region | <0.001 | ||
| Northeast | 19.0 | 15.2 | |
| Midwest | 18.6 | 15.7 | |
| South | 37.4 | 37.1 | |
| West | 25.0 | 32.0 | |
| Bed size | 0.715 | ||
| Small | 10.2 | 9.8 | |
| Medium | 25.5 | 25.3 | |
| Large | 64.3 | 64.9 | |
In bivariate analysis of survival, patients who had multiple CPR had lower rates of survival to discharge (11.3% vs 23.4%, P<0.001). Results were similar (11.6% for multiple CPR vs 22.5% for 1 CPR, P<0.001) when all patients who had CPR but did not have valid timing data were excluded in sensitivity analyses. Further stratification showed that survival to discharge decreased by >40% for each increase in CPR attempt (23.4%, 11.9%, and 6.7% for 1, 2, and 3 CPR attempts, respectively, P<0.001; Figure 2). After adjustment, multiple CPR versus 1 CPR during a hospitalization was independently associated with a lower likelihood of survival to discharge (adjusted OR: 0.41, 95% CI: 0.37‐0.44, P<0.001; Table 3).
| Characteristica | OR | 95% CI | P Value | |
|---|---|---|---|---|
| Lower | Upper | |||
| ||||
| Demographic | ||||
| Age <65 years | 1.339 | 1.304 | 1.375 | <0.001 |
| Sex, F | 1.128 | 1.099 | 1.157 | <0.001 |
| Race, nonwhite | 0.781 | 0.758 | 0.804 | <0.001 |
| Low income quartile | 0.887 | 0.858 | 0.915 | <0.001 |
| Year of admission | 1.051 | 1.046 | 1.056 | <0.001 |
| Clinical | ||||
| Multiple CPR | 0.406 | 0.371 | 0.445 | <0.001 |
| CCI score | 0.939 | 0.933 | 0.944 | <0.001 |
| Cardiac surgery | 1.785 | 1.720 | 1.853 | <0.001 |
| Hospital | ||||
| Region, Midwest | 1.472 | 1.405 | 1.543 | <0.001 |
| Region, South | 1.262 | 1.218 | 1.309 | 0.008 |
| Region, West | 1.452 | 1.398 | 1.509 | <0.001 |
| Location, urban | 0.876 | 0.837 | 0.917 | <0.001 |
Survivors with multiple CPR were less likely to be discharged home compared with survivors with 1 CPR (19.3% vs 29.9%, respectively, P<0.001); 1 in 15 survivors of multiple CPR were discharged to a hospice (6.8%) versus 1 in 23 1‐CPR survivors (4.3%; P=0.002). Mean length of stay was 5.8 versus 5.5 days for patients who had multiple CPR versus 1 CPR, respectively (P<0.001), and 16.0 versus 10.5 days for discharged survivors of multiple CPR versus 1 CPR (P<0.001). The average cost per day of hospitalization was higher for recipients of multiple CPR versus 1 CPR ($4484.60 vs $3581.40, P<0.001). The aggregate cost of hospitalization for 1‐time CPR recipients doubled between the years 2001 and 2009 (from $1.3 billion to $2.9 billion); that of recipients of multiple CPR attempts quadrupled in the same time frame (from $38.6 million to $160.7 million).
DISCUSSION
A number of studies have investigated the epidemiology of patients in whom CPR is attempted.[2, 3, 5, 20, 23, 24] Several pre‐, intra‐, and post‐resuscitation factors have been shown to affect the survival of resuscitated patients.[6, 7, 25, 26] To our knowledge, neither the epidemiology of hospitalized patients in whom resuscitation is attempted multiple times nor the prognostic value of multiple CPR attempts has been investigated. In this study, we found that multiple resuscitations are more commonly performed on younger, generally sicker patients; their outcomes are significantly compromised compared with patients who are resuscitated once during their hospitalization.
There was a steep decline in survival based on the number of resuscitation events. In multivariate analysis, patients who had multiple CPR were 2.5‐fold less likely to survive their hospitalization; survivors of multiple CPR also were more likely to be discharged to a hospice. Overall, this is indicative of clinical deterioration and prolongation of dying should a patient suffer multiple cardiopulmonary arrests during a hospitalization. The robust inverse relationship between multiple CPR and survival to discharge has implications for the development of prognostic models of outcomes following CPR, as previously designed prediction models of CPR outcomes such as the Cardiac Arrest Survival Post‐Resuscitation In‐hospital (CASPRI) score,[25] Pre‐Arrest Morbidity (PAM) score,[27] and Prognosis After Resuscitation (PAR) score[28] do not include multiple resuscitations as a variable of interest.
In‐hospital factors were found to be more important than patient factors, such as comorbidities or race, in determining the likelihood of multiple CPR attempts. Hospital teaching status and region remained significantly associated with likelihood of multiple CPR attempts. This is in agreement with studies that have described demographic and regional variation in utilization of do‐not‐resuscitate orders.[29, 30] These findings suggest substantial heterogeneity in the clinical culture and hospital practices across the United States regarding preemptive discussions about resuscitation. This means that where a patient receives care is a significant determinant of their probability of undergoing multiple CPR.
It is known that older patients are more likely to have advance directive orders[30, 31] and possibly document their wishes with regard to further resuscitation efforts. There also may be an inclination toward more aggressive care for younger adults compared with those of an advanced age. Uncertainty about a patient's goals of care likely feeds into an increased possibility of multiple resuscitation attempts; this may explain why neurologic compromise and being on ventilator support were independently associated with likelihood of multiple CPR, as these patients often have lost their ability to actively participate in decision‐making. The results of this study highlight the importance of engaging patients with a plausible risk of cardiopulmonary arrest about their goals for care and advance directives in a timely manner, regardless of age.
We found that the care of patients who undergo multiple resuscitations is associated with a higher cost of hospitalization than for patients in whom resuscitation is attempted once during their hospitalization. In addition, there was an exponential increase in aggregate cost over time for multiple CPR recipients compared with 1‐time CPR recipients. In a prior study, Ebell and Kruse showed an exponential inverse relationship between cost per surviving patient and rate of survival to discharge.[32] Considering that 93.3% of patients who had 3 resuscitation attempts died during their hospitalization, and that hospital‐level factors appear to play a significant role in likelihood of multiple CPR, consensus guidelines regarding the appropriateness of 3 resuscitation attempts during a single hospitalization may be relevant to aid the care of these patients.
Although the NIS is well‐validated,[18] there are some limitations. Whereas CPR incidence in this study (2.5 per 1000 hospitalizations) is within estimates (15 arrests per 1000 hospitalizations) reported in previous studies,[3, 5] potential undercoding of multiple CPR may explain why the multiple‐CPR rate in this study is lower than re‐arrest estimates provided in published studies.[2, 33] Indeed, accurate calculation of re‐arrest rates requires data on do‐not‐resuscitate orders instituted after successful resuscitation, which are not provided in the NIS. Information on patient‐provider discussions about CPR or prognosis is not included. Data regarding the underlying cause and type of arrest rhythm, rates of return to spontaneous circulation, length of code, patient location, critical‐care resources and length of critical‐care stay, availability of rapid‐response/code teams, time to defibrillation, use of therapeutic hypothermia, adherence to resuscitation guidelines, quality of CPR, and long‐term follow‐up are not included in the database. Presenting rhythms were not assessed, as there are no ICD‐9 codes for asystole and pulseless electrical activity. The NIS is de‐identified; therefore, chart review to assess the validity of codes is impossible. However, our sensitivity analyses indicate the reliability of using the number of occurrences of the CPR code as a marker of multiple CPR. The strength of our study lies in the use of data that provide a population‐level insight into the epidemiology of patients resuscitated multiple times during their hospitalization, and their outcomes.
Decision‐making about CPR is at the center of a complex debate that incorporates often divergent clinical, economic, ethical, and personal issues. As debate continues regarding when to not resuscitate,[34, 35, 36, 37] studies that explore the public perspective of survival thresholds for the provision of multiple resuscitations will be crucial. As competition for finite healthcare dollars escalates, stratified analyses of the cost implications of resuscitation care are essential. Studies are needed to examine the impact of a history of successful resuscitation in a previous hospitalization on outcomes following CPR in a subsequent hospitalization. Overall, our study fills an important knowledge gap in resuscitation practice and outcomes in the United States and highlights the importance of discussing resuscitation options between a patient and his or her family on hospital admission and, if needed, again after the first successful resuscitation attempt.
Disclosure
Nothing to report.
- , , , . Trends in inpatient treatment intensity among Medicare beneficiaries at the end of life. Health Serv Res. 2004;39:363–376.
- , , , et al. Cardiopulmonary resuscitation of adults in the hospital: a report of 14,720 cardiac arrests from the national registry of cardiopulmonary resuscitation. Resuscitation. 2003;58:297–308.
- , , , et al. Epidemiologic study of in‐hospital cardiopulmonary resuscitation in the elderly. N Engl J Med. 2009;361:22–31.
- , , , et al; National Registry of Cardiopulmonary Resuscitation Investigators. Survival from in‐hospital cardiac arrest during nights and weekends. JAMA. 2008;299:785–792.
- , , , . In‐hospital cardiac arrest: incidence, prognosis and possible measures to improve survival. Intensive Care Med. 2007;33:237–245.
- , , , . Predictors of survival following in‐hospital adult cardiopulmonary resuscitation. CMAJ. 2002;167:343–348.
- , . Pre‐arrest predictors of failure to survive after in‐hospital cardiopulmonary resuscitation: a meta‐analysis. Fam Pract. 2011;28:505–515.
- , . Cardiopulmonary resuscitation in older people—a review. Rev Clin Gerontol. 2010;20:20–29.
- , . Extent and determinants of error in doctors' prognoses in terminally ill patients: prospective cohort study. BMJ. 2000;320:469–472.
- , , . Physicians' confidence in discussing do not resuscitate orders with patients and surrogates. J Med Ethics. 2008;34:96–101.
- , . How misconceptions among elderly patients regarding survival outcomes of inpatient cardiopulmonary resuscitation affect do‐not‐resuscitate orders. J Am Osteopath Assoc. 2006;106:402–404.
- , , . Cardiopulmonary resuscitation on television—miracles and misinformation. N Engl J Med. 1996;334:1578–1582.
- , , . Public expectations of survival following cardiopulmonary resuscitation. Acad Emerg Med. 2000;7:48–53.
- , , , et al. The influence of the probability of survival on patients' preferences regarding cardiopulmonary resuscitation. N Engl J Med. 1994;330:545–549.
- , , , , . Code status discussions between attending hospitalist physicians and medical patients at hospital admission. J Gen Intern Med. 2011;26:359–366.
- , , . Hospital do‐not‐resuscitate orders: why they have failed and how to fix them. J Gen Intern Med. 2011;26:791–797.
- , , , . The inability of physicians to predict the outcome of in‐hospital resuscitation. J Gen Intern Med. 1996;11:16–22.
- Healthcare Cost and Utilization Project. Overview of the Nationwide Inpatient Sample. http://www.hcup‐us.ahrq.gov/nisoverview.jsp. Accessed June 24, 2013.
- , , , et al. Long‐term outcomes in elderly survivors of in‐hospital cardiac arrest. N Engl J Med. 2013;368:1019–1026.
- , , . Epidemiology and outcomes of in‐hospital cardiopulmonary resuscitation in the United States, 2000–2009. Resuscitation. 2013;84:1255–1260.
- , , , et al. Coding algorithms for defining comorbidities in ICD‐9‐CM and ICD‐10 administrative data. Med Care. 2005;43:1130–1139.
- US Department of Labor, Bureau of Labor Statistics. Inflation calculator. http://www.bls.gov/data/inflation_calculator.htm. Accessed June 24, 2013.
- , , , et al. Part 4: CPR overview. 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010;122:S676–S684.
- , , , et al. Choices of seriously ill patients about cardiopulmonary resuscitation: correlates and outcomes. Am J Med. 1996;100:128–137.
- , , , et al. A validated prediction tool for initial survivors of in‐hospital cardiac arrest. Arch Intern Med. 2012;172:947–953.
- , , , . Pre‐resuscitation factors associated with mortality in 49,130 cases of in‐hospital cardiac arrest: a report from the national registry for cardiopulmonary resuscitation. Resuscitation. 2010;81:302–311.
- , , , . Pre‐arrest morbidity and other correlates of survival after in‐hospital cardiopulmonary arrest. Am J Med. 1989;87:28–34.
- , . Prediction of failure to survive following in‐hospital cardiopulmonary resuscitation: comparison of two predictive instruments. Resuscitation. 1994;28:21–25.
- , . Regional and institutional variation in the initiation of early do‐not‐resuscitate orders. Arch Intern Med. 2005;165:1705–1712.
- , , , et al. Epidemiology of do‐not‐resuscitate orders: disparity by age, diagnosis, gender, race, and functional impairment. Arch Intern Med. 1995;155:2056–2062.
- , , , , , . Patients' understanding of advance directives and cardiopulmonary resuscitation. J Crit Care. 2005;20:26–34.
- , . A proposed model for the cost of cardiopulmonary resuscitation. Med Care. 1994;32:640–649.
- , , . Predictors of cardiopulmonary arrest outcome in a comprehensive cancer center intensive care unit. Scand J Trauma Resusc Emerg Med. 2013; 21:18.
- . A critic's assessment of our approach to cardiac arrest. N Engl J Med. 2011;364:374–375.
- . Should there be a choice for cardiopulmonary resuscitation when death is expected? Revisiting an old idea whose time is yet to come. J Palliat Med. 2002;5:107–116.
- . Clinical model for ethical cardiopulmonary resuscitation decision‐making. Intern Med J. 2013;43:77–83.
- , , . Avoiding the futility of resuscitation. Resuscitation. 2001;50:161–166.
- , , , . Trends in inpatient treatment intensity among Medicare beneficiaries at the end of life. Health Serv Res. 2004;39:363–376.
- , , , et al. Cardiopulmonary resuscitation of adults in the hospital: a report of 14,720 cardiac arrests from the national registry of cardiopulmonary resuscitation. Resuscitation. 2003;58:297–308.
- , , , et al. Epidemiologic study of in‐hospital cardiopulmonary resuscitation in the elderly. N Engl J Med. 2009;361:22–31.
- , , , et al; National Registry of Cardiopulmonary Resuscitation Investigators. Survival from in‐hospital cardiac arrest during nights and weekends. JAMA. 2008;299:785–792.
- , , , . In‐hospital cardiac arrest: incidence, prognosis and possible measures to improve survival. Intensive Care Med. 2007;33:237–245.
- , , , . Predictors of survival following in‐hospital adult cardiopulmonary resuscitation. CMAJ. 2002;167:343–348.
- , . Pre‐arrest predictors of failure to survive after in‐hospital cardiopulmonary resuscitation: a meta‐analysis. Fam Pract. 2011;28:505–515.
- , . Cardiopulmonary resuscitation in older people—a review. Rev Clin Gerontol. 2010;20:20–29.
- , . Extent and determinants of error in doctors' prognoses in terminally ill patients: prospective cohort study. BMJ. 2000;320:469–472.
- , , . Physicians' confidence in discussing do not resuscitate orders with patients and surrogates. J Med Ethics. 2008;34:96–101.
- , . How misconceptions among elderly patients regarding survival outcomes of inpatient cardiopulmonary resuscitation affect do‐not‐resuscitate orders. J Am Osteopath Assoc. 2006;106:402–404.
- , , . Cardiopulmonary resuscitation on television—miracles and misinformation. N Engl J Med. 1996;334:1578–1582.
- , , . Public expectations of survival following cardiopulmonary resuscitation. Acad Emerg Med. 2000;7:48–53.
- , , , et al. The influence of the probability of survival on patients' preferences regarding cardiopulmonary resuscitation. N Engl J Med. 1994;330:545–549.
- , , , , . Code status discussions between attending hospitalist physicians and medical patients at hospital admission. J Gen Intern Med. 2011;26:359–366.
- , , . Hospital do‐not‐resuscitate orders: why they have failed and how to fix them. J Gen Intern Med. 2011;26:791–797.
- , , , . The inability of physicians to predict the outcome of in‐hospital resuscitation. J Gen Intern Med. 1996;11:16–22.
- Healthcare Cost and Utilization Project. Overview of the Nationwide Inpatient Sample. http://www.hcup‐us.ahrq.gov/nisoverview.jsp. Accessed June 24, 2013.
- , , , et al. Long‐term outcomes in elderly survivors of in‐hospital cardiac arrest. N Engl J Med. 2013;368:1019–1026.
- , , . Epidemiology and outcomes of in‐hospital cardiopulmonary resuscitation in the United States, 2000–2009. Resuscitation. 2013;84:1255–1260.
- , , , et al. Coding algorithms for defining comorbidities in ICD‐9‐CM and ICD‐10 administrative data. Med Care. 2005;43:1130–1139.
- US Department of Labor, Bureau of Labor Statistics. Inflation calculator. http://www.bls.gov/data/inflation_calculator.htm. Accessed June 24, 2013.
- , , , et al. Part 4: CPR overview. 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010;122:S676–S684.
- , , , et al. Choices of seriously ill patients about cardiopulmonary resuscitation: correlates and outcomes. Am J Med. 1996;100:128–137.
- , , , et al. A validated prediction tool for initial survivors of in‐hospital cardiac arrest. Arch Intern Med. 2012;172:947–953.
- , , , . Pre‐resuscitation factors associated with mortality in 49,130 cases of in‐hospital cardiac arrest: a report from the national registry for cardiopulmonary resuscitation. Resuscitation. 2010;81:302–311.
- , , , . Pre‐arrest morbidity and other correlates of survival after in‐hospital cardiopulmonary arrest. Am J Med. 1989;87:28–34.
- , . Prediction of failure to survive following in‐hospital cardiopulmonary resuscitation: comparison of two predictive instruments. Resuscitation. 1994;28:21–25.
- , . Regional and institutional variation in the initiation of early do‐not‐resuscitate orders. Arch Intern Med. 2005;165:1705–1712.
- , , , et al. Epidemiology of do‐not‐resuscitate orders: disparity by age, diagnosis, gender, race, and functional impairment. Arch Intern Med. 1995;155:2056–2062.
- , , , , , . Patients' understanding of advance directives and cardiopulmonary resuscitation. J Crit Care. 2005;20:26–34.
- , . A proposed model for the cost of cardiopulmonary resuscitation. Med Care. 1994;32:640–649.
- , , . Predictors of cardiopulmonary arrest outcome in a comprehensive cancer center intensive care unit. Scand J Trauma Resusc Emerg Med. 2013; 21:18.
- . A critic's assessment of our approach to cardiac arrest. N Engl J Med. 2011;364:374–375.
- . Should there be a choice for cardiopulmonary resuscitation when death is expected? Revisiting an old idea whose time is yet to come. J Palliat Med. 2002;5:107–116.
- . Clinical model for ethical cardiopulmonary resuscitation decision‐making. Intern Med J. 2013;43:77–83.
- , , . Avoiding the futility of resuscitation. Resuscitation. 2001;50:161–166.
© 2013 Society of Hospital Medicine
Time to Introduce Yourself to Patients
At the core of a good physician is mastery of critical communication skills. Good communication establishes rapport and can also heal patients. As communication is an essential ingredient of good physicianship, the recipe starts with a fundamental staplethe physician introduction. The physician introduction is step 2 of Kahn's etiquette‐based medicine checklist to promote good doctoring.[1] Although such rudimentary communication skills are cemented in kindergarten, sadly, more training is needed for doctors. In a recent Journal of Hospital Medicine study, interns failed to introduce themselves in 3 out of 5 inpatient encounters.[2]
Despite waning introductions, increasing importance is being placed on hospitalized patient's knowledge of their treating physician's name and role for patient safety. The Transitions of Care Consensus Policy Statement endorsed by 6 medical societies, including the Society of Hospital Medicine, recommend patients know who their treating physician is while caring for them at every step across the continuum, including hospitalization.[3] The Accreditation Council for Graduate Medical Education requires that patients be informed of who the supervising physician is and understand the roles of any trainees in their care.[4] Last, the death of young Lewis Blackman in South Carolina resulted in state legislation requiring clear identification of physicians and their roles for patients.[5] Given these recommendations, tools to remind physicians to introduce themselves and explain their role to patients are worth consideration. In this issue of the Journal of Hospital Medicine, the effectiveness of 2 interventions using physician photo tools is described.[6, 7]
Even though both studies advance our knowledge on the effectiveness of such interventions, nonrandom variable uptake by physicians represents a major common hurdle. Physician workload, competing priorities, and time pressures prevent physicians from distributing such tools. Consistent adopters of the cards likely already introduce themselves regularly. Interestingly, physicians likely withhold the cards from patients they perceive as unsatisfied, who ironically have the most to gain. System changes, such as increasing handoffs and transient coverage with resident duty hours, can also hamper tool effectiveness through the introduction of more physicians to remember, inherently decreasing the ability of patients to identify their treating physicians.[8]
Patient factors also affect the success of such interventions. Interestingly, patients' baseline ability to identify their physician ranged from 11% to 51% in these studies. Such differences can be readily attributed to previous disparities noted by age, race, gender, and education level in patient recall of their physician.[8] Future work should target interventions for these subgroups, while also accounting for the high prevalence of low health literacy, memory impairment, sleep loss, and poor vision among inpatients, all of which can hamper such interventions.[9, 10]
Although neither intervention improved overall patient satisfaction, patient satisfaction is influenced by a variety of factors unrelated to physician care, such as nursing or the environment. Given the inherent ceiling effect in patient satisfaction metrics, both studies were underpowered to show minor differences. It is also worth noting that complex social interventions depend on their context. Although some patients may enjoy receiving the cards, others may feel that it is not critical to their patient satisfaction. Using a realist evaluation would ask patients what they thought of the cards and why.[11] Like one of the authors, we noted that patients do like the cards, suggesting the problem is not the cards but the metrics of evaluation.[12]
In addition to robust evaluation metrics, future interventions should incorporate patient‐centered approaches to empower patients to ask their doctors about their name and role. With the request coming from patients, doctors are much more likely to comply. Using lessons from marketing and advertising, the hospital is full of artifacts, such as white boards, wristbands, remote controls, and monitors, that can be repurposed to advertise the doctor's name to the patient. Future advances can exploit new mobile technologies and repurpose old ones, such as the hospital television, to remind patients of their care team and other critical information. Regardless of what the future may bring, let's face itintroducing yourself properly to your patients is always good medicine.
- . Etiquette‐based medicine. N Engl J Med. 2008;358(19):1988–1989.
- , , , et al. Do internal medicine interns practice etiquette‐based communication? A critical look at the inpatient encounter. J Hosp Med. 2013;8(11):631–634.
- , , , et al. Transitions of Care Consensus policy statement: American College of Physicians, Society of General Internal Medicine, Society of Hospital Medicine, American Geriatrics Society, American College of Emergency Physicians, and Society for Academic Emergency Medicine. J Hosp Med. 2009;4(6):364–370.
- ACGME Common Program Requirements. Available at: http://www.acgme.org/acgmeweb/Portals/0/PFAssets/ProgramRequirements/CPRs2013.pdf. Accessed November 12, 2013.
- . The Informed Patient. Patients Get Power of Fast Response. Available at: http://online.wsj.com/news/articles/SB10001424052970204047504574384591232799668. Accessed November 12, 2013.
- , , . The impact of facecards on patients' knowledge, satisfaction, trust, and agreement with hospital physicians: a pilot study. J Hosp Med. 2014;9(3):137–141.
- , , . Effect of a face sheet tool on medical team provider identification and family satisfaction. J Hosp Med. 2014;9(3):186–188.
- , , , , , . Ability of hospitalized patients to identify their in‐hospital physicians. Arch Intern Med. 2009;169(2):199–201.
- , , , , . Noise and sleep among adult medical inpatients: far from a quiet night. Arch Intern Med. 2012;172(1):68–70.
- , , , , . More than meets the eye: relationship between low health literacy and poor vision in hospitalized patients. J Health Commun. 2013;18(suppl 1):197–204.
- , . Realist evaluation as a framework for the assessment of teaching about the improvement of care. J Nurs Educ. 2009;48(12):661–667.
- , , , et al. Improving inpatients' identification of their doctors: use of FACE cards. Jt Comm J Qual Patient Saf. 2009;35(12):613–619.
At the core of a good physician is mastery of critical communication skills. Good communication establishes rapport and can also heal patients. As communication is an essential ingredient of good physicianship, the recipe starts with a fundamental staplethe physician introduction. The physician introduction is step 2 of Kahn's etiquette‐based medicine checklist to promote good doctoring.[1] Although such rudimentary communication skills are cemented in kindergarten, sadly, more training is needed for doctors. In a recent Journal of Hospital Medicine study, interns failed to introduce themselves in 3 out of 5 inpatient encounters.[2]
Despite waning introductions, increasing importance is being placed on hospitalized patient's knowledge of their treating physician's name and role for patient safety. The Transitions of Care Consensus Policy Statement endorsed by 6 medical societies, including the Society of Hospital Medicine, recommend patients know who their treating physician is while caring for them at every step across the continuum, including hospitalization.[3] The Accreditation Council for Graduate Medical Education requires that patients be informed of who the supervising physician is and understand the roles of any trainees in their care.[4] Last, the death of young Lewis Blackman in South Carolina resulted in state legislation requiring clear identification of physicians and their roles for patients.[5] Given these recommendations, tools to remind physicians to introduce themselves and explain their role to patients are worth consideration. In this issue of the Journal of Hospital Medicine, the effectiveness of 2 interventions using physician photo tools is described.[6, 7]
Even though both studies advance our knowledge on the effectiveness of such interventions, nonrandom variable uptake by physicians represents a major common hurdle. Physician workload, competing priorities, and time pressures prevent physicians from distributing such tools. Consistent adopters of the cards likely already introduce themselves regularly. Interestingly, physicians likely withhold the cards from patients they perceive as unsatisfied, who ironically have the most to gain. System changes, such as increasing handoffs and transient coverage with resident duty hours, can also hamper tool effectiveness through the introduction of more physicians to remember, inherently decreasing the ability of patients to identify their treating physicians.[8]
Patient factors also affect the success of such interventions. Interestingly, patients' baseline ability to identify their physician ranged from 11% to 51% in these studies. Such differences can be readily attributed to previous disparities noted by age, race, gender, and education level in patient recall of their physician.[8] Future work should target interventions for these subgroups, while also accounting for the high prevalence of low health literacy, memory impairment, sleep loss, and poor vision among inpatients, all of which can hamper such interventions.[9, 10]
Although neither intervention improved overall patient satisfaction, patient satisfaction is influenced by a variety of factors unrelated to physician care, such as nursing or the environment. Given the inherent ceiling effect in patient satisfaction metrics, both studies were underpowered to show minor differences. It is also worth noting that complex social interventions depend on their context. Although some patients may enjoy receiving the cards, others may feel that it is not critical to their patient satisfaction. Using a realist evaluation would ask patients what they thought of the cards and why.[11] Like one of the authors, we noted that patients do like the cards, suggesting the problem is not the cards but the metrics of evaluation.[12]
In addition to robust evaluation metrics, future interventions should incorporate patient‐centered approaches to empower patients to ask their doctors about their name and role. With the request coming from patients, doctors are much more likely to comply. Using lessons from marketing and advertising, the hospital is full of artifacts, such as white boards, wristbands, remote controls, and monitors, that can be repurposed to advertise the doctor's name to the patient. Future advances can exploit new mobile technologies and repurpose old ones, such as the hospital television, to remind patients of their care team and other critical information. Regardless of what the future may bring, let's face itintroducing yourself properly to your patients is always good medicine.
At the core of a good physician is mastery of critical communication skills. Good communication establishes rapport and can also heal patients. As communication is an essential ingredient of good physicianship, the recipe starts with a fundamental staplethe physician introduction. The physician introduction is step 2 of Kahn's etiquette‐based medicine checklist to promote good doctoring.[1] Although such rudimentary communication skills are cemented in kindergarten, sadly, more training is needed for doctors. In a recent Journal of Hospital Medicine study, interns failed to introduce themselves in 3 out of 5 inpatient encounters.[2]
Despite waning introductions, increasing importance is being placed on hospitalized patient's knowledge of their treating physician's name and role for patient safety. The Transitions of Care Consensus Policy Statement endorsed by 6 medical societies, including the Society of Hospital Medicine, recommend patients know who their treating physician is while caring for them at every step across the continuum, including hospitalization.[3] The Accreditation Council for Graduate Medical Education requires that patients be informed of who the supervising physician is and understand the roles of any trainees in their care.[4] Last, the death of young Lewis Blackman in South Carolina resulted in state legislation requiring clear identification of physicians and their roles for patients.[5] Given these recommendations, tools to remind physicians to introduce themselves and explain their role to patients are worth consideration. In this issue of the Journal of Hospital Medicine, the effectiveness of 2 interventions using physician photo tools is described.[6, 7]
Even though both studies advance our knowledge on the effectiveness of such interventions, nonrandom variable uptake by physicians represents a major common hurdle. Physician workload, competing priorities, and time pressures prevent physicians from distributing such tools. Consistent adopters of the cards likely already introduce themselves regularly. Interestingly, physicians likely withhold the cards from patients they perceive as unsatisfied, who ironically have the most to gain. System changes, such as increasing handoffs and transient coverage with resident duty hours, can also hamper tool effectiveness through the introduction of more physicians to remember, inherently decreasing the ability of patients to identify their treating physicians.[8]
Patient factors also affect the success of such interventions. Interestingly, patients' baseline ability to identify their physician ranged from 11% to 51% in these studies. Such differences can be readily attributed to previous disparities noted by age, race, gender, and education level in patient recall of their physician.[8] Future work should target interventions for these subgroups, while also accounting for the high prevalence of low health literacy, memory impairment, sleep loss, and poor vision among inpatients, all of which can hamper such interventions.[9, 10]
Although neither intervention improved overall patient satisfaction, patient satisfaction is influenced by a variety of factors unrelated to physician care, such as nursing or the environment. Given the inherent ceiling effect in patient satisfaction metrics, both studies were underpowered to show minor differences. It is also worth noting that complex social interventions depend on their context. Although some patients may enjoy receiving the cards, others may feel that it is not critical to their patient satisfaction. Using a realist evaluation would ask patients what they thought of the cards and why.[11] Like one of the authors, we noted that patients do like the cards, suggesting the problem is not the cards but the metrics of evaluation.[12]
In addition to robust evaluation metrics, future interventions should incorporate patient‐centered approaches to empower patients to ask their doctors about their name and role. With the request coming from patients, doctors are much more likely to comply. Using lessons from marketing and advertising, the hospital is full of artifacts, such as white boards, wristbands, remote controls, and monitors, that can be repurposed to advertise the doctor's name to the patient. Future advances can exploit new mobile technologies and repurpose old ones, such as the hospital television, to remind patients of their care team and other critical information. Regardless of what the future may bring, let's face itintroducing yourself properly to your patients is always good medicine.
- . Etiquette‐based medicine. N Engl J Med. 2008;358(19):1988–1989.
- , , , et al. Do internal medicine interns practice etiquette‐based communication? A critical look at the inpatient encounter. J Hosp Med. 2013;8(11):631–634.
- , , , et al. Transitions of Care Consensus policy statement: American College of Physicians, Society of General Internal Medicine, Society of Hospital Medicine, American Geriatrics Society, American College of Emergency Physicians, and Society for Academic Emergency Medicine. J Hosp Med. 2009;4(6):364–370.
- ACGME Common Program Requirements. Available at: http://www.acgme.org/acgmeweb/Portals/0/PFAssets/ProgramRequirements/CPRs2013.pdf. Accessed November 12, 2013.
- . The Informed Patient. Patients Get Power of Fast Response. Available at: http://online.wsj.com/news/articles/SB10001424052970204047504574384591232799668. Accessed November 12, 2013.
- , , . The impact of facecards on patients' knowledge, satisfaction, trust, and agreement with hospital physicians: a pilot study. J Hosp Med. 2014;9(3):137–141.
- , , . Effect of a face sheet tool on medical team provider identification and family satisfaction. J Hosp Med. 2014;9(3):186–188.
- , , , , , . Ability of hospitalized patients to identify their in‐hospital physicians. Arch Intern Med. 2009;169(2):199–201.
- , , , , . Noise and sleep among adult medical inpatients: far from a quiet night. Arch Intern Med. 2012;172(1):68–70.
- , , , , . More than meets the eye: relationship between low health literacy and poor vision in hospitalized patients. J Health Commun. 2013;18(suppl 1):197–204.
- , . Realist evaluation as a framework for the assessment of teaching about the improvement of care. J Nurs Educ. 2009;48(12):661–667.
- , , , et al. Improving inpatients' identification of their doctors: use of FACE cards. Jt Comm J Qual Patient Saf. 2009;35(12):613–619.
- . Etiquette‐based medicine. N Engl J Med. 2008;358(19):1988–1989.
- , , , et al. Do internal medicine interns practice etiquette‐based communication? A critical look at the inpatient encounter. J Hosp Med. 2013;8(11):631–634.
- , , , et al. Transitions of Care Consensus policy statement: American College of Physicians, Society of General Internal Medicine, Society of Hospital Medicine, American Geriatrics Society, American College of Emergency Physicians, and Society for Academic Emergency Medicine. J Hosp Med. 2009;4(6):364–370.
- ACGME Common Program Requirements. Available at: http://www.acgme.org/acgmeweb/Portals/0/PFAssets/ProgramRequirements/CPRs2013.pdf. Accessed November 12, 2013.
- . The Informed Patient. Patients Get Power of Fast Response. Available at: http://online.wsj.com/news/articles/SB10001424052970204047504574384591232799668. Accessed November 12, 2013.
- , , . The impact of facecards on patients' knowledge, satisfaction, trust, and agreement with hospital physicians: a pilot study. J Hosp Med. 2014;9(3):137–141.
- , , . Effect of a face sheet tool on medical team provider identification and family satisfaction. J Hosp Med. 2014;9(3):186–188.
- , , , , , . Ability of hospitalized patients to identify their in‐hospital physicians. Arch Intern Med. 2009;169(2):199–201.
- , , , , . Noise and sleep among adult medical inpatients: far from a quiet night. Arch Intern Med. 2012;172(1):68–70.
- , , , , . More than meets the eye: relationship between low health literacy and poor vision in hospitalized patients. J Health Commun. 2013;18(suppl 1):197–204.
- , . Realist evaluation as a framework for the assessment of teaching about the improvement of care. J Nurs Educ. 2009;48(12):661–667.
- , , , et al. Improving inpatients' identification of their doctors: use of FACE cards. Jt Comm J Qual Patient Saf. 2009;35(12):613–619.
Prior Opioid use Among Veterans
Recent trends show a marked increase in outpatient use of chronic opioid therapy (COT) for chronic noncancer pain (CNCP)[1, 2] without decreases in reported CNCP,[3] raising concerns about the efficacy and risk‐to‐benefit ratio of opioids in this population.[4, 5, 6, 7, 8] Increasing rates of outpatient use likely are accompanied by increasing rates of opioid exposure among patients admitted to the hospital. To our knowledge there are no published data regarding the prevalence of COT during the months preceding hospitalization.
Opioid use has been linked to increased emergency room utilization[9, 10] and emergency hospitalization,[11] but associations between opioid use and inpatient metrics (eg, mortality, readmission) have not been explored. Furthermore, lack of knowledge about the prevalence of opioid use prior to hospitalization may impede efforts to improve inpatient pain management and satisfaction with care. Although there is reason to expect that strategies to safely and effectively treat acute pain during the inpatient stay differ between opioid‐nave patients and opioid‐exposed patients, evidence regarding treatment strategies is limited.[12, 13, 14] Opioid pain medications are associated with hospital adverse events, with both prior opioid exposure and lack of opioid use as proposed risk factors.[15] A better understanding of the prevalence and characteristics of hospitalized COT patients is fundamental to future work to achieve safer and more effective inpatient pain management.
The primary purpose of this study was to determine the prevalence of prior COT among hospitalized medical patients. Additionally, we aimed to characterize inpatients with occasional and chronic opioid therapy prior to admission in comparison to opioid‐nave inpatients, as differences between these groups may suggest directions for further investigation into the distinct needs or challenges of hospitalized opioid‐exposed patients.
METHODS
We used inpatient and outpatient administrative data from the Department of Veterans Affairs (VA) Healthcare System. The primary data source to identify acute medical admissions was the VA Patient Treatment File, a national administrative database of all inpatient admissions, including patient demographic characteristics, primary and secondary diagnoses (using International Classification of Diseases, 9th Revision, Clinical Modification [ICD‐9‐CM], codes), and hospitalization characteristics. Outpatient pharmacy data were from the VA Pharmacy Prescription Data Files. The VA Vital Status Files provided dates of death.
We identified all first acute medical admissions to 129 VA hospitals during fiscal years (FYs) 2009 to 2011 (October 2009September 2011). We defined first admissions as the initial medical hospitalization occurring following a minimum 365‐day hospitalization‐free period. Patients were required to demonstrate pharmacy use by receipt of any outpatient medication from the VA on 2 separate occasions within 270 days preceding the first admission, to avoid misclassification of patients who routinely obtained medications only from a non‐VA provider. Patients admitted from extended care facilities were excluded.
We grouped patients by opioid‐use status based on outpatient prescription records: (1) no opioid use, defined as no opioid prescriptions in the 6 months prior to hospitalization; (2) occasional opioid use, defined as patients who received any opioid prescription during the 6 months prior but did not meet definition of chronic use; and (3) chronic opioid therapy, defined as 90 or more days' supply of opioids received within 6 months preceding hospitalization. We did not specify continuous prescribing. Opioids included in the definition were codeine, dihydrocodeine, fentanyl (mucosal and topical), hydrocodone, hydromorphone, meperidine, methadone, morphine, oxycodone, oxymorphone, pentazocine, propoxyphene, tapentadol, and tramadol.[16, 17]
We compared groups by demographic variables including age, sex, race, income, rural vs urban residence (determined from Rural‐Urban Commuting Area codes), region based on hospital location; overall comorbidity using the Charlson Comorbidity Index (CCI);[18] and 10 selected conditions to characterize comorbidity (see Supporting Information, Appendix A, in the online version of this article). These 10 conditions were chosen based on probable associations with chronic opioid use or high prevalence among hospitalized veterans.[9, 19, 20]
We used a CNCP definition based on ICD‐9‐CM codes.[9] This definition did not include episodic conditions such as migraine[2] or a measure of pain intensity.[21] All conditions were determined from diagnoses coded during any encounter in the year prior to hospitalization, exclusive of the first (ie, index) admission. We also determined the frequency of palliative care use, defined as presence of ICD‐9‐CM code V667 during index hospitalization or within the past year. Patients with palliative care use (n=3070) were excluded from further analyses.
We compared opioid use groups by baseline characteristics using the [2] statistic to determine if the distribution was nonrandom. We used analysis of variance to compare hospital length of stay between groups. We used the [2] statistic to compare rates of 4 outcomes of interest: intensive care unit (ICU) admission during the index hospitalization, discharge disposition other than home, 30‐day readmission rate, and in‐hospital or 30‐day mortality.
To assess the association between opioid‐use status and the 4 outcomes of interest, we constructed 2 multivariable regression models; the first was adjusted only for admission diagnosis using the Clinical Classification Software (CCS),[22] and the second was adjusted for demographics, CCI, and the 10 selected comorbidities in addition to admission diagnosis.
The authors had full access to and take full responsibility for the integrity of the data. All analyses were conducted using SAS statistical software version 9.2 (SAS Institute, Cary, NC). The study was approved by the University of Iowa institutional review board and the Iowa City VA Health Care System Research and Development Committee.
RESULTS
Patient Demographics
Demographic characteristics of patients differed by opioid‐use group (Table 1). Hospitalized patients who received COT in the 6 months prior to admission tended to be younger than their comparators, more often female, white, have a rural residence, and live in the South or West.
| Variables | No Opioids, n=66,899 (54.5%) | Occasional Opioids, n=24,093 (19.6%) | Chronic Opioids, n=31,802 (25.9%) |
|---|---|---|---|
| |||
| Age, y, mean (SD) | 68.7 (12.8) | 66.5 (12.7) | 64.5 (11.5) |
| Age, n (%) | |||
| 59 (reference) | 15,170 (22.7) | 6,703 (27.8) | 10,334 (32.5) |
| 6065 | 15,076 (22.5) | 5,973 (24.8) | 8,983 (28.3) |
| 6677 | 17,226 (25.8) | 5,871 (24.4) | 7,453 (23.4) |
| 78 | 19,427 (29.0) | 5,546 (23.0) | 5,032 (15.8) |
| Male, n (%) | 64,673 (96.7) | 22,964 (95.3) | 30,200 (95.0) |
| Race, n (%) | |||
| White | 48,888 (73.1) | 17,358 (72.1) | 25,087 (78.9) |
| Black | 14,480 (21.6) | 5,553 (23.1) | 5,089 (16.0) |
| Other | 1,172 (1.8) | 450 (1.9) | 645 (2.0) |
| Unknown | 2,359 (3.5) | 732 (3.0) | 981 (3.1) |
| Income $20,000, n (%) | 40,414 (60.4) | 14,105 (58.5) | 18,945 (59.6) |
| Rural residence, n (%) | 16,697 (25.0) | 6,277 (26.1) | 9,356 (29.4) |
| Region, n (%) | |||
| Northeast | 15,053 (22.5) | 4,437 (18.4) | 5,231 (16.5) |
| South | 24,083 (36.0) | 9,390 (39.0) | 12,720 (40.0) |
| Midwest | 16,000 (23.9) | 5,714 (23.7) | 7,762 (24.4) |
| West | 11,763 (17.6) | 4,552 (18.9) | 6,089 (19.2) |
| Charlson Comorbidity Index, mean (SD) | 2.3 (2.0) | 2.6 (2.3) | 2.7 (2.3) |
| Comorbidities, n (%) | |||
| Cancer (not metastatic) | 11,818 (17.7) | 5,549 (23.0) | 6,874 (21.6) |
| Metastatic cancer | 866 (1.3) | 733 (3.0) | 1,104 (3.5) |
| Chronic pain | 25,748 (38.5) | 14,811 (61.5) | 23,894 (75.1) |
| COPD | 20,750 (31.0) | 7,876 (32.7) | 12,117 (38.1) |
| Diabetes, complicated | 10,917 (16.3) | 4,620 (19.2) | 6,304 (19.8) |
| Heart failure | 14,267 (21.3) | 5,035 (20.9) | 6,501 (20.4) |
| Renal disease | 11,311 (16.9) | 4,586 (19.0) | 4,981 (15.7) |
| Dementia | 2,180 (3.3) | 459 (1.9) | 453 (1.4) |
| Mental health other than PTSD | 33,390 (49.9) | 13,657 (56.7) | 20,726 (65.2) |
| PTSD | 7,216 (10.8) | 3,607 (15.0) | 5,938 (18.7) |
| Palliative care use, n (%) | 1,407 (2.1) | 639 (2.7) | 1,024 (3.2) |
Prevalence of Opioid Use
Among the cohort (N=122,794) of hospitalized veterans, 66,899 (54.5%) received no opioids from the VA during the 6‐month period prior to hospitalization; 31,802 (25.9%) received COT in the 6 months prior to admission. An additional 24,093 (19.6%) had occasional opioid therapy (Table 1). A total of 257,623 opioid prescriptions were provided to patients in the 6‐month period prior to their index hospitalization. Of these, 100,379 (39.0%) were for hydrocodone, 48,584 (18.9%) for oxycodone, 36,658 (14.2%) for tramadol, and 35,471 (13.8%) for morphine. These 4 medications accounted for 85.8% of total opioid prescriptions (see Supporting Information, Appendix B, in the online version of this article).
Among the COT group, 3610 (11.4%) received opioids 90 days, 10,110 (31.8%) received opioids between 91 and 179 days, and 18,082 (56.9%) patients received opioids 180 days in the prior 6 months (see Supporting Information, Appendix C, in the online version of this article).
Among the subset of patients with cancer (metastatic and nonmetastatic, n=26,944), 29.6% were prescribed COT, and 23.3% had occasional opioid use. Among the subset of patients with CNCP (n=64,453), 37.1% were prescribed COT, and 23.0% had occasional opioid use.
Comorbid Conditions
Compared to patients not receiving opioids, a larger proportion of patients receiving both occasional and chronic opioids had diagnoses of cancer and of CNCP. Diagnoses more common in COT patients included chronic obstructive pulmonary disease (COPD), complicated diabetes, post‐traumatic stress disorder (PTSD), and other mental health disorders. In contrast, COT patients were less likely than no‐opioid and occasional opioid patients to have heart failure (HF), renal disease, and dementia. Palliative care was used by 2.1% of patients in the no‐opioid group, and 3.2% of patients in the COT group (Table 1). Renal disease was most common among the occasional‐use group.
Unadjusted Hospitalization Outcomes
Unadjusted hospitalization outcomes differed between opioid‐exposure groups (Table 2). Patients receiving occasional or chronic opioids had shorter length of stay and lower rates of non‐home discharge than did patients without any opioid use. The rate of death during hospitalization or within 30 days did not differ between groups. The occasional‐use and COT groups had higher 30‐day readmission rates than did the no‐use group.
| No Opioids, n=65,492 | Occasional Opioids, n=23,454 | Chronic Opioids, n=30,778 | P | |
|---|---|---|---|---|
| ||||
| Hospital length of stay, d, mean (SD) | 4.7 (5.1) | 4.5 (4.8) | 4.5 (4.8) | 0.0003 |
| ICU stay, n (%) | 10,281 (15.7) | 3,299 (14.1) | 4,570 (14.9) | <0.0001 |
| Non‐home discharge, n (%) | 2,944 (4.5) | 997 (4.3) | 1,233 (4.0) | 0.0020 |
| 30‐day readmission, n (%) | 9,023 (13.8) | 3,629 (15.5) | 4,773 (15.5) | <0.0001 |
| Death during hospitalization or within 30 days, n (%) | 2,532 (3.9) | 863 (3.7) | 1,191 (3.9) | 0.4057 |
Multivariable Models
In the fully adjusted multivariable models, opioid exposure (in the form of either chronic or occasional use) had no significant association with ICU stay during index admission or non‐home discharge (Table 3). Both the occasional‐opioid use and COT groups were more likely to experience 30‐day hospital readmission, a relationship that remained consistent across the partially and fully adjusted models. The occasional‐opioid use group saw no increased mortality risk. In the model adjusted only for admission diagnosis, COT was not associated with increased mortality risk. When additionally adjusted for demographic variables, CCI, and selected comorbidities, however, COT was associated with increased risk of death during hospitalization or within 30 days (odds ratio: 1.19, 90% confidence interval: 1.10‐1.29).
| Occasional Opioid Use | Chronic Opioid Therapy | |||
|---|---|---|---|---|
| Model 1, OR (95% CI) | Model 2, OR (95% CI) | Model 1, OR (95% CI) | Model 2, OR (95% CI) | |
| ||||
| ICU stay | 0.94 (0.90‐0.99) | 0.95 (0.91‐1.00) | 1.00 (0.96‐1.04) | 1.01 (0.97‐1.05) |
| Non‐home discharge | 0.92 (0.85‐0.99) | 0.97 (0.90‐1.05) | 0.85 (0.80‐0.92) | 0.95 (0.88‐1.03) |
| 30‐day readmission | 1.14 (1.09‐1.19) | 1.14 (1.09‐1.19) | 1.14 (1.10‐1.19) | 1.15 (1.10‐1.20) |
| Death during hospitalization or within 30 days | 0.96 (0.88‐1.04) | 1.04 (0.95‐1.13) | 0.96 (0.90‐1.04) | 1.19 (1.10‐1.29) |
DISCUSSION
This observational study is, to our knowledge, the first to report prevalence of and characteristics associated with prior opioid use among hospitalized medical patients. The prevalence of any opioid use and of COT was substantially higher in this hospitalized cohort than reported in outpatient settings. The prevalence of any opioid use during 1 year (FY 2009) among all veterans with VA primary care use was 26.1%.[23] A study of incident prescribing rates among veterans with new diagnoses of noncancer‐related pain demonstrated 11% received an opioid prescription within 1 year.[24] Using a definition of 90 consecutive prescription days to define COT, Dobscha et al.[25] found that 5% of veterans with persistent elevated pain intensity and no previous opioid prescriptions subsequently received COT within 12 months. The high prevalence we found likely reflects cumulative effects of incident use as well as an increased symptom burden in a population defined by need for medical hospitalization.
Although a veteran population may not be generalizable to a nonveteran setting, we do note prior studies reporting prevalence of any opioid use in outpatient cohorts (in 2000 and 2005) of between 18% and 30%, with higher rates among women and patients over 65 years of age.[1, 2]
Our work was purposefully inclusive of cancer patients so that we might assess the degree to which cancer diagnoses accounted for prior opioid use in hospitalized patients. Surprisingly, the rate of COT for patients with cancer was lower than that for patients with CNCP, perhaps reflecting that a cancer condition defined in administrative data may not constitute a pain‐causing disease.
Recognition of the prevalence of opioid therapy is important as we work to understand and improve safety, satisfaction, utilization, and long‐term health outcomes associated with hospitalization. Our finding that over half of medical inpatients have preexisting CNCP diagnoses, and a not entirely overlapping proportion has prior opioid exposure, implies a need for future work to refine expectations and strategies for inpatient management, potentially tailored to prior opioid use and presence of CNCP.
A recent Joint Commission sentinel event alert[26] highlights opioid adverse events in the hospital and identifies both lack of previous opioid therapy and prior opioid therapy as factors increasing risk. ICU admission during the hospital stay may reflect adverse events such as opioid‐induced respiratory depression; in our study, patients with no opioid use prior to admission were more likely to have an ICU stay, although the effect was small. One might speculate that clinicians, accustomed to treating pain in opioid‐exposed patients, are using inappropriately large starting dosages of narcotics for inpatients without first assessing prior opioid exposure. Another possible explanation is that patients on COT are admitted to the hospital with less severe illness, potentially reflecting functional, social, or access limitations that compromise ability to manage illness in the outpatient setting. More detailed comparison of illness severity is beyond the scope of the present work.
Patient satisfaction with pain management is reflected in 2 of the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) questions, and is publically reported.[27] HCAHPS results also figure in the formula for the Centers for Medicare and Medicaid Services value‐based purchasing.[28] Preadmission pain is predictive of postoperative pain[29, 30] and may shape patient expectations; how preadmission opioid use modulates nonsurgical pain and satisfaction with management in the medical inpatient remains to be studied. The high prevalence of prior COT underscores the importance of understanding characteristics of patients on COT, and potential differences and disparities in pain management, when designing interventions to augment patient satisfaction with pain management.
Although the age distribution and patterns of comorbidities differed between the opioid‐use groups, opioid therapy remained a small but significant predictor of hospital readmission; this association was independent of CNCP diagnosis. Functional outcomes are recognized as important measures of efficacy of outpatient pain management strategies,[31] with some evidence that opioids are associated with worse functioning.[32, 33] Functional limitations, as well as inadequately or inappropriately treated pain, may drive both admissions and readmissions. Alternately, COT may be a marker for unmeasured factors that increase a patient's risk of returning to the hospital. Further work is needed to elucidate the relationship between COT and healthcare utilization associated with the inpatient stay.
Our finding that patients on COT have an increased mortality risk is concerning, given the rapid expansion in use of these medications. Although pain is increasingly prevalent toward end of life,[34] we did not observe an association between either CNCP (data not shown) or occasional opioid use and mortality. COT may complicate chronic disease through adverse drug effects including respiratory depression, apnea, or endocrine or immune alteration. Complex chronically ill patients with conditions such as COPD, HF, or diabetes may be particularly susceptible to these effects. Incident use of morphine is associated with increased mortality in acute coronary syndrome and HF[35, 36]: we are not aware of any work describing the relationship between prior opioid use and incident use during hospitalization in medical patients.
Limitations
Our work focuses on hospitalized veterans, a population that remains predominately male, limiting generalizability of the findings. Rates of mental health diagnoses and PTSD, associated with CNCP and COT,[24, 37] are higher in this population than would be expected in a general hospitalized population. Because our outcomes included readmission, and our definition of opioid exposure was designed to reflect outpatient prescribing, we included only patients without recent hospitalization. Therefore, our results may not be generalizable to patients with frequent and recurring hospitalization.
Our definition of opioid exposure depended on pharmacy dispensing records; we are not able to confirm if veterans were taking the medications as prescribed. Further, we were not able to capture data on opioids prescribed by non‐VA providers, which may have led to underestimation of prevalence.
Our definitions of COT and CNCP are imperfect, and should be noted when comparing to other studies. Because we did not specify continuous 90‐day prescribing, we may have misclassified occasional opioid therapy as COT in comparison to other authors. That continuous prescribing is equivalent to continuous use assumes that patients take medications exactly as prescribed. We used occasional opioid therapy as a comparison group, and detailed the distribution of days prescribed among the COT group (see Supporting Information, Appendix C, in the online version of this article), to augment interpretability of these results. Our CNCP diagnosis was less inclusive than others,[2] as we omitted episodic pain (eg, migraine and sprains) and human immunodeficiency virus‐related pain. As COT for CNCP conditions lacks a robust evidence base,[38] defining pain diagnoses using administrative data to reflect conditions for which COT is used in a guideline‐concordant way remains difficult.
Last, differences observed between opioid‐use groups may be due to an unmeasured confounder not captured by the variables we included. Specifically, we did not include other long‐term outpatient medications in our models. It is possible that COT is part of a larger context of inappropriate prescribing, rather than a single‐medication effect on outcomes studied.
CONCLUSION
Nearly 1 in 4 hospitalized veterans has current or recent COT at the time of hospital admission for nonsurgical conditions; nearly half have been prescribed any opioids. Practitioners designing interventions to improve pain management in the inpatient setting should account for prior opioid use. Patients who are on COT prior to hospitalization differ in age and comorbidities from their counterparts who are not on COT. Further elucidation of differences between opioid‐use groups may help providers address care needs during the transition to posthospitalization care. CNCP diagnoses and chronic opioid exposure are different entities and cannot serve as proxies in administrative data. Additional work on utilization and outcomes in specific patient populations may improve our understanding of the long‐term health effects of chronic opioid therapy.
Disclosures: Dr. Mosher is supported by the Veterans Administration (VA) Quality Scholars Fellowship, Office of Academic Affiliations, Department of Veterans Affairs. Dr. Cram is supported by a K24 award from NIAMS (AR062133) at the National Institutes of Health. The preliminary results of this article were presented at the Society of General Internal Medicine Annual Meeting in Denver, Colordao, April 2013. The views expressed in this article are those of the authors and do not necessarily represent the views of the Department of Veterans Affairs. Data are available to researchers with VA accreditation, the statistical code and the protocol are available to interested readers by contacting Dr. Mosher. The authors report no conflict of interest in regard to this study.
- , , , et al. Age and gender trends in long‐term opioid analgesic use for noncancer pain. Am J Public Health. 2010;100:2541–2547.
- , , , , , . Trends in use of opioids for non‐cancer pain conditions 2000–2005 in commercial and Medicaid insurance plans: the TROUP study. Pain. 2008;138:440–449.
- Institute of Medicine (US) Committee on Advancing Pain Research, Care, and Education. Relieving pain in America: a blueprint for transforming prevention, care, education, and research. Washington, DC: National Academies Press; 2011.
- , , , . Long‐term opioid therapy reconsidered. Ann Intern Med. 2011;155:325–328.
- , . What are we treating with long‐term opioid therapy? Arch Intern Med. 2012;172:433–434.
- , , , . Opioids for chronic noncancer pain: a meta‐analysis of effectiveness and side effects. CMAJ. 2006;174:1589–1594.
- , , , . Opioids in chronic non‐cancer pain: systematic review of efficacy and safety. Pain. 2004;112:372–380.
- , , , et al. A systematic review of randomized trials of long‐term opioid management for chronic non‐cancer pain. Pain Physician. 2011;14:91–121.
- , , , , , . Rates of adverse events of long‐acting opioids in a state Medicaid program. Ann Pharmacother. 2007;41:921–928.
- , , , et al. Emergency department visits among recipients of chronic opioid therapy. Arch Intern Med. 2010;170:1425–1432.
- , , , . Emergency hospitalizations for adverse drug events in older Americans. N Engl J Med. 2011;365:2002–2012.
- , . Assessment and management of acute pain in adult medical inpatients: a systematic review. Pain Med. 2009;10:1183–1199.
- , , , . Acute pain management in opioid‐tolerant patients: a growing challenge. Anaesth Intensive Care. 2011;39:804–823.
- , , , . Acute pain management of the chronic pain patient on opiates: a survey of caregivers at University of Washington Medical Center. Clin J Pain. 1994;10:133–138.
- The Joint Commission and the FDA take steps to curb adverse events related to the use and misuse of opioid drugs. ED Manag. 2012;24:112–116.
- , . Tramadol. CMAJ. 2013;185:E352.
- , , , , . Prescription opioid abuse in the United Kingdom. Br J Clin Pharmacol. 2013;76:823–824.
- , , , . Validation of a combined comorbidity index. J Clin Epidemiol. 1994;47:1245–1251.
- , , , , . Bringing the war back home: mental health disorders among 103,788 US veterans returning from Iraq and Afghanistan seen at Department of Veterans Affairs facilities. Arch Intern Med. 2007;167:476–482.
- , , , et al. Coding algorithms for defining comorbidities in ICD‐9‐CM and ICD‐10 administrative data. Med Care. 2005;43:1130–1139.
- , , , , , . Sex Differences in the medical care of VA patients with chronic non‐cancer pain [published online ahead of print June 26, 2013]. Pain Med. doi: 10.1111/pme.12177.
- Agency for Healthcare Research and Quality. Clinical Classifications Software (CCS) for ICD‐9‐CM. Available at: http://www.hcup‐us.ahrq.gov/toolssoftware/ccs/ccs.jsp. Accessed October 17, 2013.
- , , , et al. Predicting risk of hospitalization or death among patients receiving primary care in the Veterans Health Administration. Med Care. 2013;51:368–373.
- , , , et al. Association of mental health disorders with prescription opioids and high‐risk opioid use in US veterans of Iraq and Afghanistan. JAMA. 2012;307:940–947.
- , , , , . Correlates of prescription opioid initiation and long‐term opioid use in veterans with persistent pain. Clin J Pain. 2013;29:102–108.
- Safe use of opioids in hospitals. Sentinel Event Alert. 2012;49:1–5.
- Centers for Medicare (2):2–9.
- , , , , , . The risk of severe postoperative pain: modification and validation of a clinical prediction rule. Anesth Analg. 2008;107:1330–1339.
- , , , et al. Preoperative predictors of moderate to intense acute postoperative pain in patients undergoing abdominal surgery. Acta Anaesthesiol Scand. 2002;46:1265–1271.
- , , , , , . Successful and unsuccessful outcomes with long‐term opioid therapy: a survey of physicians' opinions. J Palliat Med. 2006;9:50–56.
- , , , . Opioid use among low back pain patients in primary care: is opioid prescription associated with disability at 6‐month follow‐up? Pain. 2013;154:1038–1044.
- , , , , ; Disability Risk Identification Study Cohort. Early opioid prescription and subsequent disability among workers with back injuries: the Disability Risk Identification Study Cohort. Spine (Phila Pa 1976). 2008;33:199–204.
- , , , et al. The epidemiology of pain during the last 2 years of life. Ann Intern Med. 2010;153:563–569.
- , , , et al. Association of intravenous morphine use and outcomes in acute coronary syndromes: results from the CRUSADE Quality Improvement Initiative. Am Heart J. 2005;149:1043–1049.
- , , , et al. Use of intravenous morphine for acute decompensated heart failure in patients with and without acute coronary syndromes. Acute Card Care. 2011;13:76–80.
- , , , et al. VA mental health services utilization in Iraq and Afghanistan veterans in the first year of receiving new mental health diagnoses. J Trauma Stress. 2010;23:5–16.
- , , , et al. Long‐term opioid management for chronic noncancer pain. Cochrane Database Syst Rev. 2010;(1):CD006605.
Recent trends show a marked increase in outpatient use of chronic opioid therapy (COT) for chronic noncancer pain (CNCP)[1, 2] without decreases in reported CNCP,[3] raising concerns about the efficacy and risk‐to‐benefit ratio of opioids in this population.[4, 5, 6, 7, 8] Increasing rates of outpatient use likely are accompanied by increasing rates of opioid exposure among patients admitted to the hospital. To our knowledge there are no published data regarding the prevalence of COT during the months preceding hospitalization.
Opioid use has been linked to increased emergency room utilization[9, 10] and emergency hospitalization,[11] but associations between opioid use and inpatient metrics (eg, mortality, readmission) have not been explored. Furthermore, lack of knowledge about the prevalence of opioid use prior to hospitalization may impede efforts to improve inpatient pain management and satisfaction with care. Although there is reason to expect that strategies to safely and effectively treat acute pain during the inpatient stay differ between opioid‐nave patients and opioid‐exposed patients, evidence regarding treatment strategies is limited.[12, 13, 14] Opioid pain medications are associated with hospital adverse events, with both prior opioid exposure and lack of opioid use as proposed risk factors.[15] A better understanding of the prevalence and characteristics of hospitalized COT patients is fundamental to future work to achieve safer and more effective inpatient pain management.
The primary purpose of this study was to determine the prevalence of prior COT among hospitalized medical patients. Additionally, we aimed to characterize inpatients with occasional and chronic opioid therapy prior to admission in comparison to opioid‐nave inpatients, as differences between these groups may suggest directions for further investigation into the distinct needs or challenges of hospitalized opioid‐exposed patients.
METHODS
We used inpatient and outpatient administrative data from the Department of Veterans Affairs (VA) Healthcare System. The primary data source to identify acute medical admissions was the VA Patient Treatment File, a national administrative database of all inpatient admissions, including patient demographic characteristics, primary and secondary diagnoses (using International Classification of Diseases, 9th Revision, Clinical Modification [ICD‐9‐CM], codes), and hospitalization characteristics. Outpatient pharmacy data were from the VA Pharmacy Prescription Data Files. The VA Vital Status Files provided dates of death.
We identified all first acute medical admissions to 129 VA hospitals during fiscal years (FYs) 2009 to 2011 (October 2009September 2011). We defined first admissions as the initial medical hospitalization occurring following a minimum 365‐day hospitalization‐free period. Patients were required to demonstrate pharmacy use by receipt of any outpatient medication from the VA on 2 separate occasions within 270 days preceding the first admission, to avoid misclassification of patients who routinely obtained medications only from a non‐VA provider. Patients admitted from extended care facilities were excluded.
We grouped patients by opioid‐use status based on outpatient prescription records: (1) no opioid use, defined as no opioid prescriptions in the 6 months prior to hospitalization; (2) occasional opioid use, defined as patients who received any opioid prescription during the 6 months prior but did not meet definition of chronic use; and (3) chronic opioid therapy, defined as 90 or more days' supply of opioids received within 6 months preceding hospitalization. We did not specify continuous prescribing. Opioids included in the definition were codeine, dihydrocodeine, fentanyl (mucosal and topical), hydrocodone, hydromorphone, meperidine, methadone, morphine, oxycodone, oxymorphone, pentazocine, propoxyphene, tapentadol, and tramadol.[16, 17]
We compared groups by demographic variables including age, sex, race, income, rural vs urban residence (determined from Rural‐Urban Commuting Area codes), region based on hospital location; overall comorbidity using the Charlson Comorbidity Index (CCI);[18] and 10 selected conditions to characterize comorbidity (see Supporting Information, Appendix A, in the online version of this article). These 10 conditions were chosen based on probable associations with chronic opioid use or high prevalence among hospitalized veterans.[9, 19, 20]
We used a CNCP definition based on ICD‐9‐CM codes.[9] This definition did not include episodic conditions such as migraine[2] or a measure of pain intensity.[21] All conditions were determined from diagnoses coded during any encounter in the year prior to hospitalization, exclusive of the first (ie, index) admission. We also determined the frequency of palliative care use, defined as presence of ICD‐9‐CM code V667 during index hospitalization or within the past year. Patients with palliative care use (n=3070) were excluded from further analyses.
We compared opioid use groups by baseline characteristics using the [2] statistic to determine if the distribution was nonrandom. We used analysis of variance to compare hospital length of stay between groups. We used the [2] statistic to compare rates of 4 outcomes of interest: intensive care unit (ICU) admission during the index hospitalization, discharge disposition other than home, 30‐day readmission rate, and in‐hospital or 30‐day mortality.
To assess the association between opioid‐use status and the 4 outcomes of interest, we constructed 2 multivariable regression models; the first was adjusted only for admission diagnosis using the Clinical Classification Software (CCS),[22] and the second was adjusted for demographics, CCI, and the 10 selected comorbidities in addition to admission diagnosis.
The authors had full access to and take full responsibility for the integrity of the data. All analyses were conducted using SAS statistical software version 9.2 (SAS Institute, Cary, NC). The study was approved by the University of Iowa institutional review board and the Iowa City VA Health Care System Research and Development Committee.
RESULTS
Patient Demographics
Demographic characteristics of patients differed by opioid‐use group (Table 1). Hospitalized patients who received COT in the 6 months prior to admission tended to be younger than their comparators, more often female, white, have a rural residence, and live in the South or West.
| Variables | No Opioids, n=66,899 (54.5%) | Occasional Opioids, n=24,093 (19.6%) | Chronic Opioids, n=31,802 (25.9%) |
|---|---|---|---|
| |||
| Age, y, mean (SD) | 68.7 (12.8) | 66.5 (12.7) | 64.5 (11.5) |
| Age, n (%) | |||
| 59 (reference) | 15,170 (22.7) | 6,703 (27.8) | 10,334 (32.5) |
| 6065 | 15,076 (22.5) | 5,973 (24.8) | 8,983 (28.3) |
| 6677 | 17,226 (25.8) | 5,871 (24.4) | 7,453 (23.4) |
| 78 | 19,427 (29.0) | 5,546 (23.0) | 5,032 (15.8) |
| Male, n (%) | 64,673 (96.7) | 22,964 (95.3) | 30,200 (95.0) |
| Race, n (%) | |||
| White | 48,888 (73.1) | 17,358 (72.1) | 25,087 (78.9) |
| Black | 14,480 (21.6) | 5,553 (23.1) | 5,089 (16.0) |
| Other | 1,172 (1.8) | 450 (1.9) | 645 (2.0) |
| Unknown | 2,359 (3.5) | 732 (3.0) | 981 (3.1) |
| Income $20,000, n (%) | 40,414 (60.4) | 14,105 (58.5) | 18,945 (59.6) |
| Rural residence, n (%) | 16,697 (25.0) | 6,277 (26.1) | 9,356 (29.4) |
| Region, n (%) | |||
| Northeast | 15,053 (22.5) | 4,437 (18.4) | 5,231 (16.5) |
| South | 24,083 (36.0) | 9,390 (39.0) | 12,720 (40.0) |
| Midwest | 16,000 (23.9) | 5,714 (23.7) | 7,762 (24.4) |
| West | 11,763 (17.6) | 4,552 (18.9) | 6,089 (19.2) |
| Charlson Comorbidity Index, mean (SD) | 2.3 (2.0) | 2.6 (2.3) | 2.7 (2.3) |
| Comorbidities, n (%) | |||
| Cancer (not metastatic) | 11,818 (17.7) | 5,549 (23.0) | 6,874 (21.6) |
| Metastatic cancer | 866 (1.3) | 733 (3.0) | 1,104 (3.5) |
| Chronic pain | 25,748 (38.5) | 14,811 (61.5) | 23,894 (75.1) |
| COPD | 20,750 (31.0) | 7,876 (32.7) | 12,117 (38.1) |
| Diabetes, complicated | 10,917 (16.3) | 4,620 (19.2) | 6,304 (19.8) |
| Heart failure | 14,267 (21.3) | 5,035 (20.9) | 6,501 (20.4) |
| Renal disease | 11,311 (16.9) | 4,586 (19.0) | 4,981 (15.7) |
| Dementia | 2,180 (3.3) | 459 (1.9) | 453 (1.4) |
| Mental health other than PTSD | 33,390 (49.9) | 13,657 (56.7) | 20,726 (65.2) |
| PTSD | 7,216 (10.8) | 3,607 (15.0) | 5,938 (18.7) |
| Palliative care use, n (%) | 1,407 (2.1) | 639 (2.7) | 1,024 (3.2) |
Prevalence of Opioid Use
Among the cohort (N=122,794) of hospitalized veterans, 66,899 (54.5%) received no opioids from the VA during the 6‐month period prior to hospitalization; 31,802 (25.9%) received COT in the 6 months prior to admission. An additional 24,093 (19.6%) had occasional opioid therapy (Table 1). A total of 257,623 opioid prescriptions were provided to patients in the 6‐month period prior to their index hospitalization. Of these, 100,379 (39.0%) were for hydrocodone, 48,584 (18.9%) for oxycodone, 36,658 (14.2%) for tramadol, and 35,471 (13.8%) for morphine. These 4 medications accounted for 85.8% of total opioid prescriptions (see Supporting Information, Appendix B, in the online version of this article).
Among the COT group, 3610 (11.4%) received opioids 90 days, 10,110 (31.8%) received opioids between 91 and 179 days, and 18,082 (56.9%) patients received opioids 180 days in the prior 6 months (see Supporting Information, Appendix C, in the online version of this article).
Among the subset of patients with cancer (metastatic and nonmetastatic, n=26,944), 29.6% were prescribed COT, and 23.3% had occasional opioid use. Among the subset of patients with CNCP (n=64,453), 37.1% were prescribed COT, and 23.0% had occasional opioid use.
Comorbid Conditions
Compared to patients not receiving opioids, a larger proportion of patients receiving both occasional and chronic opioids had diagnoses of cancer and of CNCP. Diagnoses more common in COT patients included chronic obstructive pulmonary disease (COPD), complicated diabetes, post‐traumatic stress disorder (PTSD), and other mental health disorders. In contrast, COT patients were less likely than no‐opioid and occasional opioid patients to have heart failure (HF), renal disease, and dementia. Palliative care was used by 2.1% of patients in the no‐opioid group, and 3.2% of patients in the COT group (Table 1). Renal disease was most common among the occasional‐use group.
Unadjusted Hospitalization Outcomes
Unadjusted hospitalization outcomes differed between opioid‐exposure groups (Table 2). Patients receiving occasional or chronic opioids had shorter length of stay and lower rates of non‐home discharge than did patients without any opioid use. The rate of death during hospitalization or within 30 days did not differ between groups. The occasional‐use and COT groups had higher 30‐day readmission rates than did the no‐use group.
| No Opioids, n=65,492 | Occasional Opioids, n=23,454 | Chronic Opioids, n=30,778 | P | |
|---|---|---|---|---|
| ||||
| Hospital length of stay, d, mean (SD) | 4.7 (5.1) | 4.5 (4.8) | 4.5 (4.8) | 0.0003 |
| ICU stay, n (%) | 10,281 (15.7) | 3,299 (14.1) | 4,570 (14.9) | <0.0001 |
| Non‐home discharge, n (%) | 2,944 (4.5) | 997 (4.3) | 1,233 (4.0) | 0.0020 |
| 30‐day readmission, n (%) | 9,023 (13.8) | 3,629 (15.5) | 4,773 (15.5) | <0.0001 |
| Death during hospitalization or within 30 days, n (%) | 2,532 (3.9) | 863 (3.7) | 1,191 (3.9) | 0.4057 |
Multivariable Models
In the fully adjusted multivariable models, opioid exposure (in the form of either chronic or occasional use) had no significant association with ICU stay during index admission or non‐home discharge (Table 3). Both the occasional‐opioid use and COT groups were more likely to experience 30‐day hospital readmission, a relationship that remained consistent across the partially and fully adjusted models. The occasional‐opioid use group saw no increased mortality risk. In the model adjusted only for admission diagnosis, COT was not associated with increased mortality risk. When additionally adjusted for demographic variables, CCI, and selected comorbidities, however, COT was associated with increased risk of death during hospitalization or within 30 days (odds ratio: 1.19, 90% confidence interval: 1.10‐1.29).
| Occasional Opioid Use | Chronic Opioid Therapy | |||
|---|---|---|---|---|
| Model 1, OR (95% CI) | Model 2, OR (95% CI) | Model 1, OR (95% CI) | Model 2, OR (95% CI) | |
| ||||
| ICU stay | 0.94 (0.90‐0.99) | 0.95 (0.91‐1.00) | 1.00 (0.96‐1.04) | 1.01 (0.97‐1.05) |
| Non‐home discharge | 0.92 (0.85‐0.99) | 0.97 (0.90‐1.05) | 0.85 (0.80‐0.92) | 0.95 (0.88‐1.03) |
| 30‐day readmission | 1.14 (1.09‐1.19) | 1.14 (1.09‐1.19) | 1.14 (1.10‐1.19) | 1.15 (1.10‐1.20) |
| Death during hospitalization or within 30 days | 0.96 (0.88‐1.04) | 1.04 (0.95‐1.13) | 0.96 (0.90‐1.04) | 1.19 (1.10‐1.29) |
DISCUSSION
This observational study is, to our knowledge, the first to report prevalence of and characteristics associated with prior opioid use among hospitalized medical patients. The prevalence of any opioid use and of COT was substantially higher in this hospitalized cohort than reported in outpatient settings. The prevalence of any opioid use during 1 year (FY 2009) among all veterans with VA primary care use was 26.1%.[23] A study of incident prescribing rates among veterans with new diagnoses of noncancer‐related pain demonstrated 11% received an opioid prescription within 1 year.[24] Using a definition of 90 consecutive prescription days to define COT, Dobscha et al.[25] found that 5% of veterans with persistent elevated pain intensity and no previous opioid prescriptions subsequently received COT within 12 months. The high prevalence we found likely reflects cumulative effects of incident use as well as an increased symptom burden in a population defined by need for medical hospitalization.
Although a veteran population may not be generalizable to a nonveteran setting, we do note prior studies reporting prevalence of any opioid use in outpatient cohorts (in 2000 and 2005) of between 18% and 30%, with higher rates among women and patients over 65 years of age.[1, 2]
Our work was purposefully inclusive of cancer patients so that we might assess the degree to which cancer diagnoses accounted for prior opioid use in hospitalized patients. Surprisingly, the rate of COT for patients with cancer was lower than that for patients with CNCP, perhaps reflecting that a cancer condition defined in administrative data may not constitute a pain‐causing disease.
Recognition of the prevalence of opioid therapy is important as we work to understand and improve safety, satisfaction, utilization, and long‐term health outcomes associated with hospitalization. Our finding that over half of medical inpatients have preexisting CNCP diagnoses, and a not entirely overlapping proportion has prior opioid exposure, implies a need for future work to refine expectations and strategies for inpatient management, potentially tailored to prior opioid use and presence of CNCP.
A recent Joint Commission sentinel event alert[26] highlights opioid adverse events in the hospital and identifies both lack of previous opioid therapy and prior opioid therapy as factors increasing risk. ICU admission during the hospital stay may reflect adverse events such as opioid‐induced respiratory depression; in our study, patients with no opioid use prior to admission were more likely to have an ICU stay, although the effect was small. One might speculate that clinicians, accustomed to treating pain in opioid‐exposed patients, are using inappropriately large starting dosages of narcotics for inpatients without first assessing prior opioid exposure. Another possible explanation is that patients on COT are admitted to the hospital with less severe illness, potentially reflecting functional, social, or access limitations that compromise ability to manage illness in the outpatient setting. More detailed comparison of illness severity is beyond the scope of the present work.
Patient satisfaction with pain management is reflected in 2 of the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) questions, and is publically reported.[27] HCAHPS results also figure in the formula for the Centers for Medicare and Medicaid Services value‐based purchasing.[28] Preadmission pain is predictive of postoperative pain[29, 30] and may shape patient expectations; how preadmission opioid use modulates nonsurgical pain and satisfaction with management in the medical inpatient remains to be studied. The high prevalence of prior COT underscores the importance of understanding characteristics of patients on COT, and potential differences and disparities in pain management, when designing interventions to augment patient satisfaction with pain management.
Although the age distribution and patterns of comorbidities differed between the opioid‐use groups, opioid therapy remained a small but significant predictor of hospital readmission; this association was independent of CNCP diagnosis. Functional outcomes are recognized as important measures of efficacy of outpatient pain management strategies,[31] with some evidence that opioids are associated with worse functioning.[32, 33] Functional limitations, as well as inadequately or inappropriately treated pain, may drive both admissions and readmissions. Alternately, COT may be a marker for unmeasured factors that increase a patient's risk of returning to the hospital. Further work is needed to elucidate the relationship between COT and healthcare utilization associated with the inpatient stay.
Our finding that patients on COT have an increased mortality risk is concerning, given the rapid expansion in use of these medications. Although pain is increasingly prevalent toward end of life,[34] we did not observe an association between either CNCP (data not shown) or occasional opioid use and mortality. COT may complicate chronic disease through adverse drug effects including respiratory depression, apnea, or endocrine or immune alteration. Complex chronically ill patients with conditions such as COPD, HF, or diabetes may be particularly susceptible to these effects. Incident use of morphine is associated with increased mortality in acute coronary syndrome and HF[35, 36]: we are not aware of any work describing the relationship between prior opioid use and incident use during hospitalization in medical patients.
Limitations
Our work focuses on hospitalized veterans, a population that remains predominately male, limiting generalizability of the findings. Rates of mental health diagnoses and PTSD, associated with CNCP and COT,[24, 37] are higher in this population than would be expected in a general hospitalized population. Because our outcomes included readmission, and our definition of opioid exposure was designed to reflect outpatient prescribing, we included only patients without recent hospitalization. Therefore, our results may not be generalizable to patients with frequent and recurring hospitalization.
Our definition of opioid exposure depended on pharmacy dispensing records; we are not able to confirm if veterans were taking the medications as prescribed. Further, we were not able to capture data on opioids prescribed by non‐VA providers, which may have led to underestimation of prevalence.
Our definitions of COT and CNCP are imperfect, and should be noted when comparing to other studies. Because we did not specify continuous 90‐day prescribing, we may have misclassified occasional opioid therapy as COT in comparison to other authors. That continuous prescribing is equivalent to continuous use assumes that patients take medications exactly as prescribed. We used occasional opioid therapy as a comparison group, and detailed the distribution of days prescribed among the COT group (see Supporting Information, Appendix C, in the online version of this article), to augment interpretability of these results. Our CNCP diagnosis was less inclusive than others,[2] as we omitted episodic pain (eg, migraine and sprains) and human immunodeficiency virus‐related pain. As COT for CNCP conditions lacks a robust evidence base,[38] defining pain diagnoses using administrative data to reflect conditions for which COT is used in a guideline‐concordant way remains difficult.
Last, differences observed between opioid‐use groups may be due to an unmeasured confounder not captured by the variables we included. Specifically, we did not include other long‐term outpatient medications in our models. It is possible that COT is part of a larger context of inappropriate prescribing, rather than a single‐medication effect on outcomes studied.
CONCLUSION
Nearly 1 in 4 hospitalized veterans has current or recent COT at the time of hospital admission for nonsurgical conditions; nearly half have been prescribed any opioids. Practitioners designing interventions to improve pain management in the inpatient setting should account for prior opioid use. Patients who are on COT prior to hospitalization differ in age and comorbidities from their counterparts who are not on COT. Further elucidation of differences between opioid‐use groups may help providers address care needs during the transition to posthospitalization care. CNCP diagnoses and chronic opioid exposure are different entities and cannot serve as proxies in administrative data. Additional work on utilization and outcomes in specific patient populations may improve our understanding of the long‐term health effects of chronic opioid therapy.
Disclosures: Dr. Mosher is supported by the Veterans Administration (VA) Quality Scholars Fellowship, Office of Academic Affiliations, Department of Veterans Affairs. Dr. Cram is supported by a K24 award from NIAMS (AR062133) at the National Institutes of Health. The preliminary results of this article were presented at the Society of General Internal Medicine Annual Meeting in Denver, Colordao, April 2013. The views expressed in this article are those of the authors and do not necessarily represent the views of the Department of Veterans Affairs. Data are available to researchers with VA accreditation, the statistical code and the protocol are available to interested readers by contacting Dr. Mosher. The authors report no conflict of interest in regard to this study.
Recent trends show a marked increase in outpatient use of chronic opioid therapy (COT) for chronic noncancer pain (CNCP)[1, 2] without decreases in reported CNCP,[3] raising concerns about the efficacy and risk‐to‐benefit ratio of opioids in this population.[4, 5, 6, 7, 8] Increasing rates of outpatient use likely are accompanied by increasing rates of opioid exposure among patients admitted to the hospital. To our knowledge there are no published data regarding the prevalence of COT during the months preceding hospitalization.
Opioid use has been linked to increased emergency room utilization[9, 10] and emergency hospitalization,[11] but associations between opioid use and inpatient metrics (eg, mortality, readmission) have not been explored. Furthermore, lack of knowledge about the prevalence of opioid use prior to hospitalization may impede efforts to improve inpatient pain management and satisfaction with care. Although there is reason to expect that strategies to safely and effectively treat acute pain during the inpatient stay differ between opioid‐nave patients and opioid‐exposed patients, evidence regarding treatment strategies is limited.[12, 13, 14] Opioid pain medications are associated with hospital adverse events, with both prior opioid exposure and lack of opioid use as proposed risk factors.[15] A better understanding of the prevalence and characteristics of hospitalized COT patients is fundamental to future work to achieve safer and more effective inpatient pain management.
The primary purpose of this study was to determine the prevalence of prior COT among hospitalized medical patients. Additionally, we aimed to characterize inpatients with occasional and chronic opioid therapy prior to admission in comparison to opioid‐nave inpatients, as differences between these groups may suggest directions for further investigation into the distinct needs or challenges of hospitalized opioid‐exposed patients.
METHODS
We used inpatient and outpatient administrative data from the Department of Veterans Affairs (VA) Healthcare System. The primary data source to identify acute medical admissions was the VA Patient Treatment File, a national administrative database of all inpatient admissions, including patient demographic characteristics, primary and secondary diagnoses (using International Classification of Diseases, 9th Revision, Clinical Modification [ICD‐9‐CM], codes), and hospitalization characteristics. Outpatient pharmacy data were from the VA Pharmacy Prescription Data Files. The VA Vital Status Files provided dates of death.
We identified all first acute medical admissions to 129 VA hospitals during fiscal years (FYs) 2009 to 2011 (October 2009September 2011). We defined first admissions as the initial medical hospitalization occurring following a minimum 365‐day hospitalization‐free period. Patients were required to demonstrate pharmacy use by receipt of any outpatient medication from the VA on 2 separate occasions within 270 days preceding the first admission, to avoid misclassification of patients who routinely obtained medications only from a non‐VA provider. Patients admitted from extended care facilities were excluded.
We grouped patients by opioid‐use status based on outpatient prescription records: (1) no opioid use, defined as no opioid prescriptions in the 6 months prior to hospitalization; (2) occasional opioid use, defined as patients who received any opioid prescription during the 6 months prior but did not meet definition of chronic use; and (3) chronic opioid therapy, defined as 90 or more days' supply of opioids received within 6 months preceding hospitalization. We did not specify continuous prescribing. Opioids included in the definition were codeine, dihydrocodeine, fentanyl (mucosal and topical), hydrocodone, hydromorphone, meperidine, methadone, morphine, oxycodone, oxymorphone, pentazocine, propoxyphene, tapentadol, and tramadol.[16, 17]
We compared groups by demographic variables including age, sex, race, income, rural vs urban residence (determined from Rural‐Urban Commuting Area codes), region based on hospital location; overall comorbidity using the Charlson Comorbidity Index (CCI);[18] and 10 selected conditions to characterize comorbidity (see Supporting Information, Appendix A, in the online version of this article). These 10 conditions were chosen based on probable associations with chronic opioid use or high prevalence among hospitalized veterans.[9, 19, 20]
We used a CNCP definition based on ICD‐9‐CM codes.[9] This definition did not include episodic conditions such as migraine[2] or a measure of pain intensity.[21] All conditions were determined from diagnoses coded during any encounter in the year prior to hospitalization, exclusive of the first (ie, index) admission. We also determined the frequency of palliative care use, defined as presence of ICD‐9‐CM code V667 during index hospitalization or within the past year. Patients with palliative care use (n=3070) were excluded from further analyses.
We compared opioid use groups by baseline characteristics using the [2] statistic to determine if the distribution was nonrandom. We used analysis of variance to compare hospital length of stay between groups. We used the [2] statistic to compare rates of 4 outcomes of interest: intensive care unit (ICU) admission during the index hospitalization, discharge disposition other than home, 30‐day readmission rate, and in‐hospital or 30‐day mortality.
To assess the association between opioid‐use status and the 4 outcomes of interest, we constructed 2 multivariable regression models; the first was adjusted only for admission diagnosis using the Clinical Classification Software (CCS),[22] and the second was adjusted for demographics, CCI, and the 10 selected comorbidities in addition to admission diagnosis.
The authors had full access to and take full responsibility for the integrity of the data. All analyses were conducted using SAS statistical software version 9.2 (SAS Institute, Cary, NC). The study was approved by the University of Iowa institutional review board and the Iowa City VA Health Care System Research and Development Committee.
RESULTS
Patient Demographics
Demographic characteristics of patients differed by opioid‐use group (Table 1). Hospitalized patients who received COT in the 6 months prior to admission tended to be younger than their comparators, more often female, white, have a rural residence, and live in the South or West.
| Variables | No Opioids, n=66,899 (54.5%) | Occasional Opioids, n=24,093 (19.6%) | Chronic Opioids, n=31,802 (25.9%) |
|---|---|---|---|
| |||
| Age, y, mean (SD) | 68.7 (12.8) | 66.5 (12.7) | 64.5 (11.5) |
| Age, n (%) | |||
| 59 (reference) | 15,170 (22.7) | 6,703 (27.8) | 10,334 (32.5) |
| 6065 | 15,076 (22.5) | 5,973 (24.8) | 8,983 (28.3) |
| 6677 | 17,226 (25.8) | 5,871 (24.4) | 7,453 (23.4) |
| 78 | 19,427 (29.0) | 5,546 (23.0) | 5,032 (15.8) |
| Male, n (%) | 64,673 (96.7) | 22,964 (95.3) | 30,200 (95.0) |
| Race, n (%) | |||
| White | 48,888 (73.1) | 17,358 (72.1) | 25,087 (78.9) |
| Black | 14,480 (21.6) | 5,553 (23.1) | 5,089 (16.0) |
| Other | 1,172 (1.8) | 450 (1.9) | 645 (2.0) |
| Unknown | 2,359 (3.5) | 732 (3.0) | 981 (3.1) |
| Income $20,000, n (%) | 40,414 (60.4) | 14,105 (58.5) | 18,945 (59.6) |
| Rural residence, n (%) | 16,697 (25.0) | 6,277 (26.1) | 9,356 (29.4) |
| Region, n (%) | |||
| Northeast | 15,053 (22.5) | 4,437 (18.4) | 5,231 (16.5) |
| South | 24,083 (36.0) | 9,390 (39.0) | 12,720 (40.0) |
| Midwest | 16,000 (23.9) | 5,714 (23.7) | 7,762 (24.4) |
| West | 11,763 (17.6) | 4,552 (18.9) | 6,089 (19.2) |
| Charlson Comorbidity Index, mean (SD) | 2.3 (2.0) | 2.6 (2.3) | 2.7 (2.3) |
| Comorbidities, n (%) | |||
| Cancer (not metastatic) | 11,818 (17.7) | 5,549 (23.0) | 6,874 (21.6) |
| Metastatic cancer | 866 (1.3) | 733 (3.0) | 1,104 (3.5) |
| Chronic pain | 25,748 (38.5) | 14,811 (61.5) | 23,894 (75.1) |
| COPD | 20,750 (31.0) | 7,876 (32.7) | 12,117 (38.1) |
| Diabetes, complicated | 10,917 (16.3) | 4,620 (19.2) | 6,304 (19.8) |
| Heart failure | 14,267 (21.3) | 5,035 (20.9) | 6,501 (20.4) |
| Renal disease | 11,311 (16.9) | 4,586 (19.0) | 4,981 (15.7) |
| Dementia | 2,180 (3.3) | 459 (1.9) | 453 (1.4) |
| Mental health other than PTSD | 33,390 (49.9) | 13,657 (56.7) | 20,726 (65.2) |
| PTSD | 7,216 (10.8) | 3,607 (15.0) | 5,938 (18.7) |
| Palliative care use, n (%) | 1,407 (2.1) | 639 (2.7) | 1,024 (3.2) |
Prevalence of Opioid Use
Among the cohort (N=122,794) of hospitalized veterans, 66,899 (54.5%) received no opioids from the VA during the 6‐month period prior to hospitalization; 31,802 (25.9%) received COT in the 6 months prior to admission. An additional 24,093 (19.6%) had occasional opioid therapy (Table 1). A total of 257,623 opioid prescriptions were provided to patients in the 6‐month period prior to their index hospitalization. Of these, 100,379 (39.0%) were for hydrocodone, 48,584 (18.9%) for oxycodone, 36,658 (14.2%) for tramadol, and 35,471 (13.8%) for morphine. These 4 medications accounted for 85.8% of total opioid prescriptions (see Supporting Information, Appendix B, in the online version of this article).
Among the COT group, 3610 (11.4%) received opioids 90 days, 10,110 (31.8%) received opioids between 91 and 179 days, and 18,082 (56.9%) patients received opioids 180 days in the prior 6 months (see Supporting Information, Appendix C, in the online version of this article).
Among the subset of patients with cancer (metastatic and nonmetastatic, n=26,944), 29.6% were prescribed COT, and 23.3% had occasional opioid use. Among the subset of patients with CNCP (n=64,453), 37.1% were prescribed COT, and 23.0% had occasional opioid use.
Comorbid Conditions
Compared to patients not receiving opioids, a larger proportion of patients receiving both occasional and chronic opioids had diagnoses of cancer and of CNCP. Diagnoses more common in COT patients included chronic obstructive pulmonary disease (COPD), complicated diabetes, post‐traumatic stress disorder (PTSD), and other mental health disorders. In contrast, COT patients were less likely than no‐opioid and occasional opioid patients to have heart failure (HF), renal disease, and dementia. Palliative care was used by 2.1% of patients in the no‐opioid group, and 3.2% of patients in the COT group (Table 1). Renal disease was most common among the occasional‐use group.
Unadjusted Hospitalization Outcomes
Unadjusted hospitalization outcomes differed between opioid‐exposure groups (Table 2). Patients receiving occasional or chronic opioids had shorter length of stay and lower rates of non‐home discharge than did patients without any opioid use. The rate of death during hospitalization or within 30 days did not differ between groups. The occasional‐use and COT groups had higher 30‐day readmission rates than did the no‐use group.
| No Opioids, n=65,492 | Occasional Opioids, n=23,454 | Chronic Opioids, n=30,778 | P | |
|---|---|---|---|---|
| ||||
| Hospital length of stay, d, mean (SD) | 4.7 (5.1) | 4.5 (4.8) | 4.5 (4.8) | 0.0003 |
| ICU stay, n (%) | 10,281 (15.7) | 3,299 (14.1) | 4,570 (14.9) | <0.0001 |
| Non‐home discharge, n (%) | 2,944 (4.5) | 997 (4.3) | 1,233 (4.0) | 0.0020 |
| 30‐day readmission, n (%) | 9,023 (13.8) | 3,629 (15.5) | 4,773 (15.5) | <0.0001 |
| Death during hospitalization or within 30 days, n (%) | 2,532 (3.9) | 863 (3.7) | 1,191 (3.9) | 0.4057 |
Multivariable Models
In the fully adjusted multivariable models, opioid exposure (in the form of either chronic or occasional use) had no significant association with ICU stay during index admission or non‐home discharge (Table 3). Both the occasional‐opioid use and COT groups were more likely to experience 30‐day hospital readmission, a relationship that remained consistent across the partially and fully adjusted models. The occasional‐opioid use group saw no increased mortality risk. In the model adjusted only for admission diagnosis, COT was not associated with increased mortality risk. When additionally adjusted for demographic variables, CCI, and selected comorbidities, however, COT was associated with increased risk of death during hospitalization or within 30 days (odds ratio: 1.19, 90% confidence interval: 1.10‐1.29).
| Occasional Opioid Use | Chronic Opioid Therapy | |||
|---|---|---|---|---|
| Model 1, OR (95% CI) | Model 2, OR (95% CI) | Model 1, OR (95% CI) | Model 2, OR (95% CI) | |
| ||||
| ICU stay | 0.94 (0.90‐0.99) | 0.95 (0.91‐1.00) | 1.00 (0.96‐1.04) | 1.01 (0.97‐1.05) |
| Non‐home discharge | 0.92 (0.85‐0.99) | 0.97 (0.90‐1.05) | 0.85 (0.80‐0.92) | 0.95 (0.88‐1.03) |
| 30‐day readmission | 1.14 (1.09‐1.19) | 1.14 (1.09‐1.19) | 1.14 (1.10‐1.19) | 1.15 (1.10‐1.20) |
| Death during hospitalization or within 30 days | 0.96 (0.88‐1.04) | 1.04 (0.95‐1.13) | 0.96 (0.90‐1.04) | 1.19 (1.10‐1.29) |
DISCUSSION
This observational study is, to our knowledge, the first to report prevalence of and characteristics associated with prior opioid use among hospitalized medical patients. The prevalence of any opioid use and of COT was substantially higher in this hospitalized cohort than reported in outpatient settings. The prevalence of any opioid use during 1 year (FY 2009) among all veterans with VA primary care use was 26.1%.[23] A study of incident prescribing rates among veterans with new diagnoses of noncancer‐related pain demonstrated 11% received an opioid prescription within 1 year.[24] Using a definition of 90 consecutive prescription days to define COT, Dobscha et al.[25] found that 5% of veterans with persistent elevated pain intensity and no previous opioid prescriptions subsequently received COT within 12 months. The high prevalence we found likely reflects cumulative effects of incident use as well as an increased symptom burden in a population defined by need for medical hospitalization.
Although a veteran population may not be generalizable to a nonveteran setting, we do note prior studies reporting prevalence of any opioid use in outpatient cohorts (in 2000 and 2005) of between 18% and 30%, with higher rates among women and patients over 65 years of age.[1, 2]
Our work was purposefully inclusive of cancer patients so that we might assess the degree to which cancer diagnoses accounted for prior opioid use in hospitalized patients. Surprisingly, the rate of COT for patients with cancer was lower than that for patients with CNCP, perhaps reflecting that a cancer condition defined in administrative data may not constitute a pain‐causing disease.
Recognition of the prevalence of opioid therapy is important as we work to understand and improve safety, satisfaction, utilization, and long‐term health outcomes associated with hospitalization. Our finding that over half of medical inpatients have preexisting CNCP diagnoses, and a not entirely overlapping proportion has prior opioid exposure, implies a need for future work to refine expectations and strategies for inpatient management, potentially tailored to prior opioid use and presence of CNCP.
A recent Joint Commission sentinel event alert[26] highlights opioid adverse events in the hospital and identifies both lack of previous opioid therapy and prior opioid therapy as factors increasing risk. ICU admission during the hospital stay may reflect adverse events such as opioid‐induced respiratory depression; in our study, patients with no opioid use prior to admission were more likely to have an ICU stay, although the effect was small. One might speculate that clinicians, accustomed to treating pain in opioid‐exposed patients, are using inappropriately large starting dosages of narcotics for inpatients without first assessing prior opioid exposure. Another possible explanation is that patients on COT are admitted to the hospital with less severe illness, potentially reflecting functional, social, or access limitations that compromise ability to manage illness in the outpatient setting. More detailed comparison of illness severity is beyond the scope of the present work.
Patient satisfaction with pain management is reflected in 2 of the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) questions, and is publically reported.[27] HCAHPS results also figure in the formula for the Centers for Medicare and Medicaid Services value‐based purchasing.[28] Preadmission pain is predictive of postoperative pain[29, 30] and may shape patient expectations; how preadmission opioid use modulates nonsurgical pain and satisfaction with management in the medical inpatient remains to be studied. The high prevalence of prior COT underscores the importance of understanding characteristics of patients on COT, and potential differences and disparities in pain management, when designing interventions to augment patient satisfaction with pain management.
Although the age distribution and patterns of comorbidities differed between the opioid‐use groups, opioid therapy remained a small but significant predictor of hospital readmission; this association was independent of CNCP diagnosis. Functional outcomes are recognized as important measures of efficacy of outpatient pain management strategies,[31] with some evidence that opioids are associated with worse functioning.[32, 33] Functional limitations, as well as inadequately or inappropriately treated pain, may drive both admissions and readmissions. Alternately, COT may be a marker for unmeasured factors that increase a patient's risk of returning to the hospital. Further work is needed to elucidate the relationship between COT and healthcare utilization associated with the inpatient stay.
Our finding that patients on COT have an increased mortality risk is concerning, given the rapid expansion in use of these medications. Although pain is increasingly prevalent toward end of life,[34] we did not observe an association between either CNCP (data not shown) or occasional opioid use and mortality. COT may complicate chronic disease through adverse drug effects including respiratory depression, apnea, or endocrine or immune alteration. Complex chronically ill patients with conditions such as COPD, HF, or diabetes may be particularly susceptible to these effects. Incident use of morphine is associated with increased mortality in acute coronary syndrome and HF[35, 36]: we are not aware of any work describing the relationship between prior opioid use and incident use during hospitalization in medical patients.
Limitations
Our work focuses on hospitalized veterans, a population that remains predominately male, limiting generalizability of the findings. Rates of mental health diagnoses and PTSD, associated with CNCP and COT,[24, 37] are higher in this population than would be expected in a general hospitalized population. Because our outcomes included readmission, and our definition of opioid exposure was designed to reflect outpatient prescribing, we included only patients without recent hospitalization. Therefore, our results may not be generalizable to patients with frequent and recurring hospitalization.
Our definition of opioid exposure depended on pharmacy dispensing records; we are not able to confirm if veterans were taking the medications as prescribed. Further, we were not able to capture data on opioids prescribed by non‐VA providers, which may have led to underestimation of prevalence.
Our definitions of COT and CNCP are imperfect, and should be noted when comparing to other studies. Because we did not specify continuous 90‐day prescribing, we may have misclassified occasional opioid therapy as COT in comparison to other authors. That continuous prescribing is equivalent to continuous use assumes that patients take medications exactly as prescribed. We used occasional opioid therapy as a comparison group, and detailed the distribution of days prescribed among the COT group (see Supporting Information, Appendix C, in the online version of this article), to augment interpretability of these results. Our CNCP diagnosis was less inclusive than others,[2] as we omitted episodic pain (eg, migraine and sprains) and human immunodeficiency virus‐related pain. As COT for CNCP conditions lacks a robust evidence base,[38] defining pain diagnoses using administrative data to reflect conditions for which COT is used in a guideline‐concordant way remains difficult.
Last, differences observed between opioid‐use groups may be due to an unmeasured confounder not captured by the variables we included. Specifically, we did not include other long‐term outpatient medications in our models. It is possible that COT is part of a larger context of inappropriate prescribing, rather than a single‐medication effect on outcomes studied.
CONCLUSION
Nearly 1 in 4 hospitalized veterans has current or recent COT at the time of hospital admission for nonsurgical conditions; nearly half have been prescribed any opioids. Practitioners designing interventions to improve pain management in the inpatient setting should account for prior opioid use. Patients who are on COT prior to hospitalization differ in age and comorbidities from their counterparts who are not on COT. Further elucidation of differences between opioid‐use groups may help providers address care needs during the transition to posthospitalization care. CNCP diagnoses and chronic opioid exposure are different entities and cannot serve as proxies in administrative data. Additional work on utilization and outcomes in specific patient populations may improve our understanding of the long‐term health effects of chronic opioid therapy.
Disclosures: Dr. Mosher is supported by the Veterans Administration (VA) Quality Scholars Fellowship, Office of Academic Affiliations, Department of Veterans Affairs. Dr. Cram is supported by a K24 award from NIAMS (AR062133) at the National Institutes of Health. The preliminary results of this article were presented at the Society of General Internal Medicine Annual Meeting in Denver, Colordao, April 2013. The views expressed in this article are those of the authors and do not necessarily represent the views of the Department of Veterans Affairs. Data are available to researchers with VA accreditation, the statistical code and the protocol are available to interested readers by contacting Dr. Mosher. The authors report no conflict of interest in regard to this study.
- , , , et al. Age and gender trends in long‐term opioid analgesic use for noncancer pain. Am J Public Health. 2010;100:2541–2547.
- , , , , , . Trends in use of opioids for non‐cancer pain conditions 2000–2005 in commercial and Medicaid insurance plans: the TROUP study. Pain. 2008;138:440–449.
- Institute of Medicine (US) Committee on Advancing Pain Research, Care, and Education. Relieving pain in America: a blueprint for transforming prevention, care, education, and research. Washington, DC: National Academies Press; 2011.
- , , , . Long‐term opioid therapy reconsidered. Ann Intern Med. 2011;155:325–328.
- , . What are we treating with long‐term opioid therapy? Arch Intern Med. 2012;172:433–434.
- , , , . Opioids for chronic noncancer pain: a meta‐analysis of effectiveness and side effects. CMAJ. 2006;174:1589–1594.
- , , , . Opioids in chronic non‐cancer pain: systematic review of efficacy and safety. Pain. 2004;112:372–380.
- , , , et al. A systematic review of randomized trials of long‐term opioid management for chronic non‐cancer pain. Pain Physician. 2011;14:91–121.
- , , , , , . Rates of adverse events of long‐acting opioids in a state Medicaid program. Ann Pharmacother. 2007;41:921–928.
- , , , et al. Emergency department visits among recipients of chronic opioid therapy. Arch Intern Med. 2010;170:1425–1432.
- , , , . Emergency hospitalizations for adverse drug events in older Americans. N Engl J Med. 2011;365:2002–2012.
- , . Assessment and management of acute pain in adult medical inpatients: a systematic review. Pain Med. 2009;10:1183–1199.
- , , , . Acute pain management in opioid‐tolerant patients: a growing challenge. Anaesth Intensive Care. 2011;39:804–823.
- , , , . Acute pain management of the chronic pain patient on opiates: a survey of caregivers at University of Washington Medical Center. Clin J Pain. 1994;10:133–138.
- The Joint Commission and the FDA take steps to curb adverse events related to the use and misuse of opioid drugs. ED Manag. 2012;24:112–116.
- , . Tramadol. CMAJ. 2013;185:E352.
- , , , , . Prescription opioid abuse in the United Kingdom. Br J Clin Pharmacol. 2013;76:823–824.
- , , , . Validation of a combined comorbidity index. J Clin Epidemiol. 1994;47:1245–1251.
- , , , , . Bringing the war back home: mental health disorders among 103,788 US veterans returning from Iraq and Afghanistan seen at Department of Veterans Affairs facilities. Arch Intern Med. 2007;167:476–482.
- , , , et al. Coding algorithms for defining comorbidities in ICD‐9‐CM and ICD‐10 administrative data. Med Care. 2005;43:1130–1139.
- , , , , , . Sex Differences in the medical care of VA patients with chronic non‐cancer pain [published online ahead of print June 26, 2013]. Pain Med. doi: 10.1111/pme.12177.
- Agency for Healthcare Research and Quality. Clinical Classifications Software (CCS) for ICD‐9‐CM. Available at: http://www.hcup‐us.ahrq.gov/toolssoftware/ccs/ccs.jsp. Accessed October 17, 2013.
- , , , et al. Predicting risk of hospitalization or death among patients receiving primary care in the Veterans Health Administration. Med Care. 2013;51:368–373.
- , , , et al. Association of mental health disorders with prescription opioids and high‐risk opioid use in US veterans of Iraq and Afghanistan. JAMA. 2012;307:940–947.
- , , , , . Correlates of prescription opioid initiation and long‐term opioid use in veterans with persistent pain. Clin J Pain. 2013;29:102–108.
- Safe use of opioids in hospitals. Sentinel Event Alert. 2012;49:1–5.
- Centers for Medicare (2):2–9.
- , , , , , . The risk of severe postoperative pain: modification and validation of a clinical prediction rule. Anesth Analg. 2008;107:1330–1339.
- , , , et al. Preoperative predictors of moderate to intense acute postoperative pain in patients undergoing abdominal surgery. Acta Anaesthesiol Scand. 2002;46:1265–1271.
- , , , , , . Successful and unsuccessful outcomes with long‐term opioid therapy: a survey of physicians' opinions. J Palliat Med. 2006;9:50–56.
- , , , . Opioid use among low back pain patients in primary care: is opioid prescription associated with disability at 6‐month follow‐up? Pain. 2013;154:1038–1044.
- , , , , ; Disability Risk Identification Study Cohort. Early opioid prescription and subsequent disability among workers with back injuries: the Disability Risk Identification Study Cohort. Spine (Phila Pa 1976). 2008;33:199–204.
- , , , et al. The epidemiology of pain during the last 2 years of life. Ann Intern Med. 2010;153:563–569.
- , , , et al. Association of intravenous morphine use and outcomes in acute coronary syndromes: results from the CRUSADE Quality Improvement Initiative. Am Heart J. 2005;149:1043–1049.
- , , , et al. Use of intravenous morphine for acute decompensated heart failure in patients with and without acute coronary syndromes. Acute Card Care. 2011;13:76–80.
- , , , et al. VA mental health services utilization in Iraq and Afghanistan veterans in the first year of receiving new mental health diagnoses. J Trauma Stress. 2010;23:5–16.
- , , , et al. Long‐term opioid management for chronic noncancer pain. Cochrane Database Syst Rev. 2010;(1):CD006605.
- , , , et al. Age and gender trends in long‐term opioid analgesic use for noncancer pain. Am J Public Health. 2010;100:2541–2547.
- , , , , , . Trends in use of opioids for non‐cancer pain conditions 2000–2005 in commercial and Medicaid insurance plans: the TROUP study. Pain. 2008;138:440–449.
- Institute of Medicine (US) Committee on Advancing Pain Research, Care, and Education. Relieving pain in America: a blueprint for transforming prevention, care, education, and research. Washington, DC: National Academies Press; 2011.
- , , , . Long‐term opioid therapy reconsidered. Ann Intern Med. 2011;155:325–328.
- , . What are we treating with long‐term opioid therapy? Arch Intern Med. 2012;172:433–434.
- , , , . Opioids for chronic noncancer pain: a meta‐analysis of effectiveness and side effects. CMAJ. 2006;174:1589–1594.
- , , , . Opioids in chronic non‐cancer pain: systematic review of efficacy and safety. Pain. 2004;112:372–380.
- , , , et al. A systematic review of randomized trials of long‐term opioid management for chronic non‐cancer pain. Pain Physician. 2011;14:91–121.
- , , , , , . Rates of adverse events of long‐acting opioids in a state Medicaid program. Ann Pharmacother. 2007;41:921–928.
- , , , et al. Emergency department visits among recipients of chronic opioid therapy. Arch Intern Med. 2010;170:1425–1432.
- , , , . Emergency hospitalizations for adverse drug events in older Americans. N Engl J Med. 2011;365:2002–2012.
- , . Assessment and management of acute pain in adult medical inpatients: a systematic review. Pain Med. 2009;10:1183–1199.
- , , , . Acute pain management in opioid‐tolerant patients: a growing challenge. Anaesth Intensive Care. 2011;39:804–823.
- , , , . Acute pain management of the chronic pain patient on opiates: a survey of caregivers at University of Washington Medical Center. Clin J Pain. 1994;10:133–138.
- The Joint Commission and the FDA take steps to curb adverse events related to the use and misuse of opioid drugs. ED Manag. 2012;24:112–116.
- , . Tramadol. CMAJ. 2013;185:E352.
- , , , , . Prescription opioid abuse in the United Kingdom. Br J Clin Pharmacol. 2013;76:823–824.
- , , , . Validation of a combined comorbidity index. J Clin Epidemiol. 1994;47:1245–1251.
- , , , , . Bringing the war back home: mental health disorders among 103,788 US veterans returning from Iraq and Afghanistan seen at Department of Veterans Affairs facilities. Arch Intern Med. 2007;167:476–482.
- , , , et al. Coding algorithms for defining comorbidities in ICD‐9‐CM and ICD‐10 administrative data. Med Care. 2005;43:1130–1139.
- , , , , , . Sex Differences in the medical care of VA patients with chronic non‐cancer pain [published online ahead of print June 26, 2013]. Pain Med. doi: 10.1111/pme.12177.
- Agency for Healthcare Research and Quality. Clinical Classifications Software (CCS) for ICD‐9‐CM. Available at: http://www.hcup‐us.ahrq.gov/toolssoftware/ccs/ccs.jsp. Accessed October 17, 2013.
- , , , et al. Predicting risk of hospitalization or death among patients receiving primary care in the Veterans Health Administration. Med Care. 2013;51:368–373.
- , , , et al. Association of mental health disorders with prescription opioids and high‐risk opioid use in US veterans of Iraq and Afghanistan. JAMA. 2012;307:940–947.
- , , , , . Correlates of prescription opioid initiation and long‐term opioid use in veterans with persistent pain. Clin J Pain. 2013;29:102–108.
- Safe use of opioids in hospitals. Sentinel Event Alert. 2012;49:1–5.
- Centers for Medicare (2):2–9.
- , , , , , . The risk of severe postoperative pain: modification and validation of a clinical prediction rule. Anesth Analg. 2008;107:1330–1339.
- , , , et al. Preoperative predictors of moderate to intense acute postoperative pain in patients undergoing abdominal surgery. Acta Anaesthesiol Scand. 2002;46:1265–1271.
- , , , , , . Successful and unsuccessful outcomes with long‐term opioid therapy: a survey of physicians' opinions. J Palliat Med. 2006;9:50–56.
- , , , . Opioid use among low back pain patients in primary care: is opioid prescription associated with disability at 6‐month follow‐up? Pain. 2013;154:1038–1044.
- , , , , ; Disability Risk Identification Study Cohort. Early opioid prescription and subsequent disability among workers with back injuries: the Disability Risk Identification Study Cohort. Spine (Phila Pa 1976). 2008;33:199–204.
- , , , et al. The epidemiology of pain during the last 2 years of life. Ann Intern Med. 2010;153:563–569.
- , , , et al. Association of intravenous morphine use and outcomes in acute coronary syndromes: results from the CRUSADE Quality Improvement Initiative. Am Heart J. 2005;149:1043–1049.
- , , , et al. Use of intravenous morphine for acute decompensated heart failure in patients with and without acute coronary syndromes. Acute Card Care. 2011;13:76–80.
- , , , et al. VA mental health services utilization in Iraq and Afghanistan veterans in the first year of receiving new mental health diagnoses. J Trauma Stress. 2010;23:5–16.
- , , , et al. Long‐term opioid management for chronic noncancer pain. Cochrane Database Syst Rev. 2010;(1):CD006605.
© 2013 Society of Hospital Medicine
USPSTF changes ABI screening recommendation
The U.S. Preventive Services Task Force (USPSTF) updated its earlier recommendations regarding the validity of using the ankle-brachial index (ABI) in the September Annals of Internal Medicine. In 2006, the USPSTF recommended against screening for PAD (D recommendation; Am Fam Physician 2006; 73:497).
The USPSTF now concludes that evidence is insufficient to make a recommendation. (I recommendation) and published both its systemic evidence review and recommendations.
The U.S. Preventive Services Task Force (USPSTF) updated its earlier recommendations regarding the validity of using the ankle-brachial index (ABI) in the September Annals of Internal Medicine. In 2006, the USPSTF recommended against screening for PAD (D recommendation; Am Fam Physician 2006; 73:497).
The USPSTF now concludes that evidence is insufficient to make a recommendation. (I recommendation) and published both its systemic evidence review and recommendations.
The U.S. Preventive Services Task Force (USPSTF) updated its earlier recommendations regarding the validity of using the ankle-brachial index (ABI) in the September Annals of Internal Medicine. In 2006, the USPSTF recommended against screening for PAD (D recommendation; Am Fam Physician 2006; 73:497).
The USPSTF now concludes that evidence is insufficient to make a recommendation. (I recommendation) and published both its systemic evidence review and recommendations.
Is your patient’s poor recall more than just a ‘senior moment’?
Memory and other cognitive complaints are common among the general population and become more prevalent with age.1 People who have significant emotional investment in their cognitive competence, mood disturbance, somatic symptoms, and anxiety or related disorders are likely to worry more about their cognitive functioning as they age.
Common complaints
Age-related complaints, typically beginning by age 50, often include problems retaining or retrieving names, difficulty recalling details of conversations and written materials, and hazy recollection of remote events and the time frame of recent life events. Common complaints involve difficulties with mental calculations, multi-tasking (including vulnerability to distraction), and problems keeping track of and organizing information. The most common complaint is difficulty with remembering the reason for entering a room.
More concerning are complaints involving recurrent lapses in judgment or forgetfulness with significant implications for everyday living (eg, physical safety, job performance, travel, and finances), especially when validated by friends or family members and coupled with decline in at least 1 activity of daily living, and poor insight.
Helping your forgetful patient
Office evaluation with brief cognitive screening instruments—namely, the Montreal Cognitive Assessment and the recent revision of the Mini-Mental State Examination—might help clarify the clinical presentation. Proceed with caution: Screening tests tap a limited number of neurocognitive functions and can generate a false-negative result among brighter and better educated patients and a false-positive result among the less intelligent and less educated.2 Applying age- and education-corrected norms can reduce misclassification but does not eliminate it.
Screening measures can facilitate decision-making regarding the need for more comprehensive psychometric assessment. Such evaluations sample a broader range of neurobehavioral domains, in greater depth, and provide a more nuanced picture of a patient’s neurocognition.
Findings on a battery of psychological and neuropsychological tests that might evoke concern include problems with incidental, anterograde, and recent memory that are not satisfactorily explained by: age and education or vocational training; estimated premorbid intelligence; residual neurodevelopmental disorders (attention, learning, and autistic-spectrum disorders); situational, sociocultural, and psychiatric factors; and motivational influences—notably, malingering.
Some difficulties with memory are highly associated with mild cognitive impairment or early dementia:
• anterograde memory (involving a reduced rate of verbal and nonverbal learning over repeated trials)
• poor retention
• accelerated forgetting of newly learned information
• failure to benefit from recognition and other mnemonic cues
• so-called source error confusion—a misattribution that involves difficulty differentiating target information from competing information, as reflected in confabulation errors and an elevated rate of intrusion errors.
Disclosure
Dr. Pollak reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Weiner MF, Garrett R, Bret ME. Neuropsychiatric assessment and diagnosis. In: Weiner MF, Lipton AM, eds. Clinical manual of Alzheimer disease and other dementias. Arlington, VA: American Psychiatric Publishing, Inc.; 2012: 3-46.
2. Strauss E, Sherman EMS, Spreen O. A compendium of neuropsychological tests: administration, norms and commentary: third edition. New York, NY: Oxford University Press; 2006.
Memory and other cognitive complaints are common among the general population and become more prevalent with age.1 People who have significant emotional investment in their cognitive competence, mood disturbance, somatic symptoms, and anxiety or related disorders are likely to worry more about their cognitive functioning as they age.
Common complaints
Age-related complaints, typically beginning by age 50, often include problems retaining or retrieving names, difficulty recalling details of conversations and written materials, and hazy recollection of remote events and the time frame of recent life events. Common complaints involve difficulties with mental calculations, multi-tasking (including vulnerability to distraction), and problems keeping track of and organizing information. The most common complaint is difficulty with remembering the reason for entering a room.
More concerning are complaints involving recurrent lapses in judgment or forgetfulness with significant implications for everyday living (eg, physical safety, job performance, travel, and finances), especially when validated by friends or family members and coupled with decline in at least 1 activity of daily living, and poor insight.
Helping your forgetful patient
Office evaluation with brief cognitive screening instruments—namely, the Montreal Cognitive Assessment and the recent revision of the Mini-Mental State Examination—might help clarify the clinical presentation. Proceed with caution: Screening tests tap a limited number of neurocognitive functions and can generate a false-negative result among brighter and better educated patients and a false-positive result among the less intelligent and less educated.2 Applying age- and education-corrected norms can reduce misclassification but does not eliminate it.
Screening measures can facilitate decision-making regarding the need for more comprehensive psychometric assessment. Such evaluations sample a broader range of neurobehavioral domains, in greater depth, and provide a more nuanced picture of a patient’s neurocognition.
Findings on a battery of psychological and neuropsychological tests that might evoke concern include problems with incidental, anterograde, and recent memory that are not satisfactorily explained by: age and education or vocational training; estimated premorbid intelligence; residual neurodevelopmental disorders (attention, learning, and autistic-spectrum disorders); situational, sociocultural, and psychiatric factors; and motivational influences—notably, malingering.
Some difficulties with memory are highly associated with mild cognitive impairment or early dementia:
• anterograde memory (involving a reduced rate of verbal and nonverbal learning over repeated trials)
• poor retention
• accelerated forgetting of newly learned information
• failure to benefit from recognition and other mnemonic cues
• so-called source error confusion—a misattribution that involves difficulty differentiating target information from competing information, as reflected in confabulation errors and an elevated rate of intrusion errors.
Disclosure
Dr. Pollak reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Memory and other cognitive complaints are common among the general population and become more prevalent with age.1 People who have significant emotional investment in their cognitive competence, mood disturbance, somatic symptoms, and anxiety or related disorders are likely to worry more about their cognitive functioning as they age.
Common complaints
Age-related complaints, typically beginning by age 50, often include problems retaining or retrieving names, difficulty recalling details of conversations and written materials, and hazy recollection of remote events and the time frame of recent life events. Common complaints involve difficulties with mental calculations, multi-tasking (including vulnerability to distraction), and problems keeping track of and organizing information. The most common complaint is difficulty with remembering the reason for entering a room.
More concerning are complaints involving recurrent lapses in judgment or forgetfulness with significant implications for everyday living (eg, physical safety, job performance, travel, and finances), especially when validated by friends or family members and coupled with decline in at least 1 activity of daily living, and poor insight.
Helping your forgetful patient
Office evaluation with brief cognitive screening instruments—namely, the Montreal Cognitive Assessment and the recent revision of the Mini-Mental State Examination—might help clarify the clinical presentation. Proceed with caution: Screening tests tap a limited number of neurocognitive functions and can generate a false-negative result among brighter and better educated patients and a false-positive result among the less intelligent and less educated.2 Applying age- and education-corrected norms can reduce misclassification but does not eliminate it.
Screening measures can facilitate decision-making regarding the need for more comprehensive psychometric assessment. Such evaluations sample a broader range of neurobehavioral domains, in greater depth, and provide a more nuanced picture of a patient’s neurocognition.
Findings on a battery of psychological and neuropsychological tests that might evoke concern include problems with incidental, anterograde, and recent memory that are not satisfactorily explained by: age and education or vocational training; estimated premorbid intelligence; residual neurodevelopmental disorders (attention, learning, and autistic-spectrum disorders); situational, sociocultural, and psychiatric factors; and motivational influences—notably, malingering.
Some difficulties with memory are highly associated with mild cognitive impairment or early dementia:
• anterograde memory (involving a reduced rate of verbal and nonverbal learning over repeated trials)
• poor retention
• accelerated forgetting of newly learned information
• failure to benefit from recognition and other mnemonic cues
• so-called source error confusion—a misattribution that involves difficulty differentiating target information from competing information, as reflected in confabulation errors and an elevated rate of intrusion errors.
Disclosure
Dr. Pollak reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Weiner MF, Garrett R, Bret ME. Neuropsychiatric assessment and diagnosis. In: Weiner MF, Lipton AM, eds. Clinical manual of Alzheimer disease and other dementias. Arlington, VA: American Psychiatric Publishing, Inc.; 2012: 3-46.
2. Strauss E, Sherman EMS, Spreen O. A compendium of neuropsychological tests: administration, norms and commentary: third edition. New York, NY: Oxford University Press; 2006.
1. Weiner MF, Garrett R, Bret ME. Neuropsychiatric assessment and diagnosis. In: Weiner MF, Lipton AM, eds. Clinical manual of Alzheimer disease and other dementias. Arlington, VA: American Psychiatric Publishing, Inc.; 2012: 3-46.
2. Strauss E, Sherman EMS, Spreen O. A compendium of neuropsychological tests: administration, norms and commentary: third edition. New York, NY: Oxford University Press; 2006.
Hearing voices, time traveling, and being hit with a high-heeled shoe
CASE Grief and confusion
Mr. P, age 47, is arrested for entering the apartment of a woman he does not know and tossing her belongings out the window. When he is assessed to determine if he can participate in his legal defense, examiners find an attentive, courteous man who is baffled by his own behavior.
Mr. P says that he had been “stressed out” after the recent death of his grandmother, with whom he was close. He says he entered the apartment because voices told him to do so. He has no recent history of substance abuse or psychiatric hospitalizations, but he had a similar episode of “confusion” years before, when another close family member died.
Mr. P is found not fit to stand trial and the charges are dropped. He accepts haloperidol, 10 mg/d, and benztropine, 2 mg/d, and is transferred to a hospital for psychiatric treatment.
On interview, Mr. P is well groomed, soft-spoken, and shy, without formal thought disorder. Physical exam and routine lab tests are within normal limits. He says that 18 months before his arrest, he and his frail grandmother moved to a large city in hopes that he would find a wife. Both depended on the grandmother’s Social Security benefits while he cared for her.
In the 2 months after she died, he reports that he felt sad and alone and slept poorly, but made efforts to find a job and keep his apartment. When his efforts failed and he lost the apartment, he stayed with various friends for a few days at a time, then spent several days in the subway before ending up on the streets.
His arrest on the current charge occurred 4 days after he began walking the streets.
a) continue haloperidol to treat psychotic symptoms
b) discontinue haloperidol and observe him
c) add an antidepressant to haloperidol
HISTORY Imagining nonsense
Mr. P cannot explain why he started “trashing” the woman’s apartment, but says he entered it because he thought it was his apartment. With embarrassment and regret, he admits he has been depressed and confused, “imagining things”—“foolish things,” he admits—such as being in a different “time zone.”
Contradicting his earlier statements, Mr. P now admits that he had “a few beers” and denies that he experienced auditory hallucinations, saying he only talks to himself. He now says that within 2 days after his arrest, he was “all over it.” Mr. P denies current symptoms, including hallucinations, but, when pressed, waffles, then admits to a strange belief: that some people, including him, can move from one “time zone” to another.
Mr. P says he was treated for psychiatric problems 4 years earlier when his parents were killed in a car crash. By his recollection, his reaction to their death was similar to his reaction to his grandmother’s death: He became upset and wandered the streets for a few days, “moving between time zones” and talking to himself but not experiencing hallucinations. After he was taken to a hospital and “given an injection,” he calmed down and was released. Within a few days he recovered and returned to supporting himself and caring for his grandmother. Mr. P says the idea of travelling between “time zones” is embarrassing and nonsensical but adds that he was affected in this way because he “bickered” with his mother.
Mr. P’s grandmother raised him until he was age 15, although he frequently visited his parents, who lived nearby and worked during the day. Mr. P initially denies substance abuse, then admits to smoking marijuana every day for about a year before admission. He also admits to cocaine abuse in his 20s. He denies a history of suicide attempts.
The author’s observations
Mr. P reported only 2 episodes of “confusion” (or psychosis) and strange behavior in his life, both precipitated by the loss of a loved one, and at least 1 while under the influence of alcohol and Cannabis. He gave an inconsistent and ambiguous history of auditory hallucinations associated with episodes of confusion. He believes that time travel is possible, an idea that he acknowledged is nonsense. This alone was not enough to warrant long-term antipsychotic treatment. The most likely diagnosis seemed to be brief psychotic episode induced by Cannabis and the stressors of homelessness and his grandmother’s death.
EVALUATION Changing stories
No longer taking haloperidol, Mr. P continues to deny hallucinations and depressed mood, but keeps to himself. Nine days after admission he becomes tearful after he informs his aunt of his grandmother’s death in a telephone call, then approaches a nurse and complains of sadness and auditory hallucinations.
Mr. P confesses that he denied hallucinations on admission because he feared he would remain in the hospital for years if he revealed the truth that he had been experiencing auditory hallucinations almost continuously from age 10. He reports that the voices distracted him when he worked; seem to be male; often spoke gibberish; and alternate between deprecating and positive and supportive. Mr. P is reluctant to disclose more about what the voices actually say, although he acknowledges that they are not commenting or conversing with him, and that he has never believed the voices were his own thoughts but did believe that they came from inside his brain.
With haloperidol, the voices stopped. They resumed, however, when haloperidol was discontinued.
When we ask what happened to him at age 10, Mr. P shrugs.
a) childhood onset schizophrenia
b) substance abuse
c) posttraumatic stress disorder (PTSD)
d) none
The author’s observations
In community samples of children and adolescents, auditory hallucinations are not rare and usually do not cause distress or dysfunction. In a study of 3,870 children age 7 and 8,1 9% endorsed auditory hallucinations. Most heard 1 voice, once a week or less, at low volume. In 85% of children who experienced hallucinations, they caused minimal or no suffering; 97% reported minimal or no interference with daily functioning. Among children who experienced auditory hallucinations at age 7 or 8, 24% continued to hallucinate 5 years later.2 Persistent hallucinations were associated with more problematic behaviors at baseline and follow up.
In a group of 12-year-old twins, 4.2% reported auditory hallucinations.3 In that study, hallucinations were not related to Cannabis use; rather, they were heritable and related to risk factors such as cognitive impairment; behavioral, emotional, and educational problems at age 5; and a history of physical abuse and self-harm at age 12. The authors noted that these are risk factors and correlates of schizophrenia, but are not specific to schizophrenia.
Hallucinations and delusions have been found in 4% to 8% of children and adolescents referred for psychiatric treatment,4 far more than the prevalence of childhood-onset schizophrenia (0.01% of children).5 Psychotic symptoms in children have been associated with bipolar disorder, but also with anxiety disorders, obsessive-compulsive disorder, PTSD, pervasive developmental disorder, conduct disorder, and substance abuse.4
Childhood-onset schizophrenia is rare and would require that Mr. P have a diagnosis of schizophrenia as an adult. It is possible that Mr. P’s childhood symptoms were related to substance abuse but he was not asked for this history because it seemed unlikely in a 10-year-old boy. A PTSD diagnosis requires a traumatic event, which Mr. P did not reveal. It is possible that at age 10 he did not have a psychiatric disorder.
a) PTSD
b) dissociative disorder
c) borderline personality disorder
d) chronic schizophrenia
e) no psychiatric diagnosis
Among adults in the general population, 10% to 15% report auditory hallucinations.6 Hallucinations could be caused by substance abuse or psychiatric conditions other than schizophrenia; however, in adults—as in children—auditory hallucinations can occur in the absence of these conditions (Table 1) and rarely cause distress or dysfunction.6 In Sommer and colleagues’6 study of 103 healthy persons, none who heard voices had disorganization or negative symptoms. Those who heard voices had significantly more schizotypal symptoms and more childhood trauma, including emotional, physical, and sexual abuse, than those who did not hear voices.6
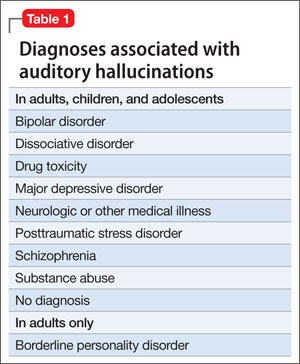
Conditions associated with hallucinations
PTSD is associated with auditory hallucinations and other psychotic symptoms.7 Most studies are of combat veterans with PTSD, in whom auditory hallucinations and delusions were associated with major depressive disorder, not a thought disorder or inappropriate affect.8 In a community sample,9 psychotic symptoms—particularly auditory hallucinations—were associated with PTSD. Subjects with PTSD and psychotic symptoms were more likely to have other psychiatric disorders, including major depressive disorder and substance use disorder, than patients with PTSD but no psychotic symptoms; however, the relationship between PTSD and psychosis remained after controlling for other psychiatric disorders.
Hallucinations can occur in persons with dissociative disorders in the absence of distinct personality states.10 Hallucinations have been seen transiently and chronically in persons with borderline personality disorder and can be associated with comorbid conditions such as substance abuse disorders, mood disorders, and PTSD.11
Mr. P lacked the reduced capacity for interpersonal relationships required for a schizotypal personality disorder diagnosis. A diagnosis of PTSD or dissociative disorder requires a history of trauma, which Mr. P did not report.
“Time travelling” with incomprehensible behavior could be interpreted as dissociation, but dissociative fugue or dissociative disorder not otherwise specified (NOS) cannot be diagnosed if symptoms might be the direct effect of a substance, such as Cannabis. Mr. P admitted to substance abuse. We can rule out borderline personality disorder because he did not display or admit to tempestuous interpersonal relationships.
A schizophrenia diagnosis requires the presence of auditory hallucinations that commented on his behavior or conversed among themselves, a second psychotic symptom for ≥1 month, or negative symptoms, which Mr. P lacked (unless belief in time travel is considered delusional).
Last, a physician might have considered malingering or a factitious disorder when Mr. P was found not able to participate in his own defense, but this seemed less likely after he revealed that he experienced auditory hallucinations since age 10.
HISTORY Bad beatings
With a few days of beginning risperidone, 4 mg/d, Mr. P reports that his hallucinations have stopped and he feels less sad. He reveals that, at age 10, when the hallucinations began, his mother hit him over the head with a high-heeled shoe, causing a scalp laceration that required a visit to the emergency room for suturing. His mother beat Mr. P for as long as he could remember. She beat him “bad” at least twice weekly, and he was taken to the hospital 7 or 8 times for injury, but she also beat him “constantly” with a belt buckle, sometimes striking his head. She instructed him to tell nobody.
The author’s observations
Auditory hallucinations in adults have been associated with childhood abuse, particularly childhood sexual abuse,12 in clinical and non-clinical samples.13 Some argue13 that child abuse itself causes hallucinations and other psychotic symptoms.
OUTCOME Depressed and sleepless
Mr. P admits that he had been smoking marijuana 2 to 3 times daily for a year. He also reports insomnia, sleeping approximately 4 hours a night and spending hours awake in bed thinking of his grandmother, with depressed mood and tearfulness. He denies suicidal ideas and hallucinations. He is treated for depressive disorder NOS first with amitriptyline, 50 mg at bedtime, for sleep, then paroxetine, 20 mg/d, for depressive symptoms, in addition to risperidone, 4 mg/d. Although Mr. P does not describe re-experiencing his childhood trauma, avoidance of stimuli associated with the trauma, or symptoms of increased arousal (except for insomnia), the treatment team did not ask, so it remains uncertain if he has PTSD (Table 2).

When Mr. P is discharged to a clinic, he smiles easily and is positive and supportive with other patients. He spruces up his appearance by wearing jewelry and works in the hospital kitchen.
Bottom Line
Chronic auditory hallucinations are associated with psychiatric illnesses other than chronic schizophrenia, particularly those resulting from trauma such as posttraumatic stress disorder. They can also occur in the absence of diagnosable psychiatric illness and rarely cause distress or functional impairment. Auditory hallucinations in adults have been associated with childhood abuse.
Related Resources
- Moskowitz A, Schafer I, Dorahy MJ. Psychosis, trauma and dissociation: emerging perspectives on severe psychopathology. West Sussex, UK: John Wiley and Sons, Ltd.; 2008.
- The International Hearing Voices Network. www.intervoiceonline.org.
Drug Brand Names
Amitriptyline • Elavil Paroxetine • Paxil
Benztropine • Cogentin Risperidone • Risperdal
Haloperidol • Haldol
Disclosure
Dr. Crowner reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Barthel-Velthuis AA, Jenner JA, van de Willige G, et al Prevalence and correlates of auditory vocal hallucinations in middle childhood. Br J Psychiatry. 2010;196(1):41-46.
2. Bartels-Velthuis AA, van de Willige G, Jenner JA, et al. Course of auditory vocal hallucinations in childhood: 5-year follow-up study. Br J Psychiatry. 2011;199(4):296-302.
3. Polanczyk G, Moffitt TE, Arsensault L, et al. Etiological and clinical features of childhood psychotic symptoms: results from a birth cohort. Arch Gen Psychiatry. 2010;67(4):328-338.
4. Biederman J, Pety C, Faracone SV, et al. Phenomenology of childhood psychosis: Findings from a large sample of psychiatrically referred youth. J Nerv Ment Dis 2004;192(9):607-614.
5. American Academy of Child and Adolescent Psychiatry. Practice parameters for the assessment and treatment of children and adolescents with schizophrenia. J Am Acad Child Adolesc Psychiatry. 2001;40(suppl 7):4SS-23S.
6. Sommer IEC, Daalman K, Rietkerk T, et al. Healthy individuals with auditory verbal hallucinations; Who are they? Psychiatric assessments of a selected sample of 103 subjects. Schizophr Bull. 2010;36(3):633-641.
7. Butler RW, Mueser KT, Sprock J, et al. Positive symptoms of psychosis in posttraumatic stress disorder. Biol Psychiatry. 1996;39:839-844.
8. David D, Kutcher GS, Jackson EI, et al Psychotic symptoms in combat-related posttraumatic stress disorder. J Clin Psychiatry. 1999;60(1):29-32.
9. Sareen J, Cox BJ, Goodwin RD, et al. Co-occurrence of posttraumatic stress disorder with positive psychotic symptoms in a nationally representative sample. J Trauma Stress. 2005;18(4):313-322.
10. Sar V, Akyuv G, Dogan O. Prevalence of dissociative disorders among women in the general population. Psychiatry Res. 2007;149:169-176.
11. Barnow S, Arens EA, Sieswerda S, et al. Borderline personality disorder and psychosis: a review. Curr Psychiatry Rep. 2010;12(3):186-195.
12. Bebbington P, Jonas S, Kuipers E, et al. Childhood sexual abuse and psychosis: data from a cross-sectional national psychiatric survey in England. Br J Psychiatry. 2011;199(1):29-37.
13. Read J, van Os J, Morrison AP, et al. Childhood trauma, psychosis and schizophrenia: a literature review with theoretical and clinical implications. Acta Psychiatr Scand. 2005;112(5):330-350.
CASE Grief and confusion
Mr. P, age 47, is arrested for entering the apartment of a woman he does not know and tossing her belongings out the window. When he is assessed to determine if he can participate in his legal defense, examiners find an attentive, courteous man who is baffled by his own behavior.
Mr. P says that he had been “stressed out” after the recent death of his grandmother, with whom he was close. He says he entered the apartment because voices told him to do so. He has no recent history of substance abuse or psychiatric hospitalizations, but he had a similar episode of “confusion” years before, when another close family member died.
Mr. P is found not fit to stand trial and the charges are dropped. He accepts haloperidol, 10 mg/d, and benztropine, 2 mg/d, and is transferred to a hospital for psychiatric treatment.
On interview, Mr. P is well groomed, soft-spoken, and shy, without formal thought disorder. Physical exam and routine lab tests are within normal limits. He says that 18 months before his arrest, he and his frail grandmother moved to a large city in hopes that he would find a wife. Both depended on the grandmother’s Social Security benefits while he cared for her.
In the 2 months after she died, he reports that he felt sad and alone and slept poorly, but made efforts to find a job and keep his apartment. When his efforts failed and he lost the apartment, he stayed with various friends for a few days at a time, then spent several days in the subway before ending up on the streets.
His arrest on the current charge occurred 4 days after he began walking the streets.
a) continue haloperidol to treat psychotic symptoms
b) discontinue haloperidol and observe him
c) add an antidepressant to haloperidol
HISTORY Imagining nonsense
Mr. P cannot explain why he started “trashing” the woman’s apartment, but says he entered it because he thought it was his apartment. With embarrassment and regret, he admits he has been depressed and confused, “imagining things”—“foolish things,” he admits—such as being in a different “time zone.”
Contradicting his earlier statements, Mr. P now admits that he had “a few beers” and denies that he experienced auditory hallucinations, saying he only talks to himself. He now says that within 2 days after his arrest, he was “all over it.” Mr. P denies current symptoms, including hallucinations, but, when pressed, waffles, then admits to a strange belief: that some people, including him, can move from one “time zone” to another.
Mr. P says he was treated for psychiatric problems 4 years earlier when his parents were killed in a car crash. By his recollection, his reaction to their death was similar to his reaction to his grandmother’s death: He became upset and wandered the streets for a few days, “moving between time zones” and talking to himself but not experiencing hallucinations. After he was taken to a hospital and “given an injection,” he calmed down and was released. Within a few days he recovered and returned to supporting himself and caring for his grandmother. Mr. P says the idea of travelling between “time zones” is embarrassing and nonsensical but adds that he was affected in this way because he “bickered” with his mother.
Mr. P’s grandmother raised him until he was age 15, although he frequently visited his parents, who lived nearby and worked during the day. Mr. P initially denies substance abuse, then admits to smoking marijuana every day for about a year before admission. He also admits to cocaine abuse in his 20s. He denies a history of suicide attempts.
The author’s observations
Mr. P reported only 2 episodes of “confusion” (or psychosis) and strange behavior in his life, both precipitated by the loss of a loved one, and at least 1 while under the influence of alcohol and Cannabis. He gave an inconsistent and ambiguous history of auditory hallucinations associated with episodes of confusion. He believes that time travel is possible, an idea that he acknowledged is nonsense. This alone was not enough to warrant long-term antipsychotic treatment. The most likely diagnosis seemed to be brief psychotic episode induced by Cannabis and the stressors of homelessness and his grandmother’s death.
EVALUATION Changing stories
No longer taking haloperidol, Mr. P continues to deny hallucinations and depressed mood, but keeps to himself. Nine days after admission he becomes tearful after he informs his aunt of his grandmother’s death in a telephone call, then approaches a nurse and complains of sadness and auditory hallucinations.
Mr. P confesses that he denied hallucinations on admission because he feared he would remain in the hospital for years if he revealed the truth that he had been experiencing auditory hallucinations almost continuously from age 10. He reports that the voices distracted him when he worked; seem to be male; often spoke gibberish; and alternate between deprecating and positive and supportive. Mr. P is reluctant to disclose more about what the voices actually say, although he acknowledges that they are not commenting or conversing with him, and that he has never believed the voices were his own thoughts but did believe that they came from inside his brain.
With haloperidol, the voices stopped. They resumed, however, when haloperidol was discontinued.
When we ask what happened to him at age 10, Mr. P shrugs.
a) childhood onset schizophrenia
b) substance abuse
c) posttraumatic stress disorder (PTSD)
d) none
The author’s observations
In community samples of children and adolescents, auditory hallucinations are not rare and usually do not cause distress or dysfunction. In a study of 3,870 children age 7 and 8,1 9% endorsed auditory hallucinations. Most heard 1 voice, once a week or less, at low volume. In 85% of children who experienced hallucinations, they caused minimal or no suffering; 97% reported minimal or no interference with daily functioning. Among children who experienced auditory hallucinations at age 7 or 8, 24% continued to hallucinate 5 years later.2 Persistent hallucinations were associated with more problematic behaviors at baseline and follow up.
In a group of 12-year-old twins, 4.2% reported auditory hallucinations.3 In that study, hallucinations were not related to Cannabis use; rather, they were heritable and related to risk factors such as cognitive impairment; behavioral, emotional, and educational problems at age 5; and a history of physical abuse and self-harm at age 12. The authors noted that these are risk factors and correlates of schizophrenia, but are not specific to schizophrenia.
Hallucinations and delusions have been found in 4% to 8% of children and adolescents referred for psychiatric treatment,4 far more than the prevalence of childhood-onset schizophrenia (0.01% of children).5 Psychotic symptoms in children have been associated with bipolar disorder, but also with anxiety disorders, obsessive-compulsive disorder, PTSD, pervasive developmental disorder, conduct disorder, and substance abuse.4
Childhood-onset schizophrenia is rare and would require that Mr. P have a diagnosis of schizophrenia as an adult. It is possible that Mr. P’s childhood symptoms were related to substance abuse but he was not asked for this history because it seemed unlikely in a 10-year-old boy. A PTSD diagnosis requires a traumatic event, which Mr. P did not reveal. It is possible that at age 10 he did not have a psychiatric disorder.
a) PTSD
b) dissociative disorder
c) borderline personality disorder
d) chronic schizophrenia
e) no psychiatric diagnosis
Among adults in the general population, 10% to 15% report auditory hallucinations.6 Hallucinations could be caused by substance abuse or psychiatric conditions other than schizophrenia; however, in adults—as in children—auditory hallucinations can occur in the absence of these conditions (Table 1) and rarely cause distress or dysfunction.6 In Sommer and colleagues’6 study of 103 healthy persons, none who heard voices had disorganization or negative symptoms. Those who heard voices had significantly more schizotypal symptoms and more childhood trauma, including emotional, physical, and sexual abuse, than those who did not hear voices.6
Conditions associated with hallucinations
PTSD is associated with auditory hallucinations and other psychotic symptoms.7 Most studies are of combat veterans with PTSD, in whom auditory hallucinations and delusions were associated with major depressive disorder, not a thought disorder or inappropriate affect.8 In a community sample,9 psychotic symptoms—particularly auditory hallucinations—were associated with PTSD. Subjects with PTSD and psychotic symptoms were more likely to have other psychiatric disorders, including major depressive disorder and substance use disorder, than patients with PTSD but no psychotic symptoms; however, the relationship between PTSD and psychosis remained after controlling for other psychiatric disorders.
Hallucinations can occur in persons with dissociative disorders in the absence of distinct personality states.10 Hallucinations have been seen transiently and chronically in persons with borderline personality disorder and can be associated with comorbid conditions such as substance abuse disorders, mood disorders, and PTSD.11
Mr. P lacked the reduced capacity for interpersonal relationships required for a schizotypal personality disorder diagnosis. A diagnosis of PTSD or dissociative disorder requires a history of trauma, which Mr. P did not report.
“Time travelling” with incomprehensible behavior could be interpreted as dissociation, but dissociative fugue or dissociative disorder not otherwise specified (NOS) cannot be diagnosed if symptoms might be the direct effect of a substance, such as Cannabis. Mr. P admitted to substance abuse. We can rule out borderline personality disorder because he did not display or admit to tempestuous interpersonal relationships.
A schizophrenia diagnosis requires the presence of auditory hallucinations that commented on his behavior or conversed among themselves, a second psychotic symptom for ≥1 month, or negative symptoms, which Mr. P lacked (unless belief in time travel is considered delusional).
Last, a physician might have considered malingering or a factitious disorder when Mr. P was found not able to participate in his own defense, but this seemed less likely after he revealed that he experienced auditory hallucinations since age 10.
HISTORY Bad beatings
With a few days of beginning risperidone, 4 mg/d, Mr. P reports that his hallucinations have stopped and he feels less sad. He reveals that, at age 10, when the hallucinations began, his mother hit him over the head with a high-heeled shoe, causing a scalp laceration that required a visit to the emergency room for suturing. His mother beat Mr. P for as long as he could remember. She beat him “bad” at least twice weekly, and he was taken to the hospital 7 or 8 times for injury, but she also beat him “constantly” with a belt buckle, sometimes striking his head. She instructed him to tell nobody.
The author’s observations
Auditory hallucinations in adults have been associated with childhood abuse, particularly childhood sexual abuse,12 in clinical and non-clinical samples.13 Some argue13 that child abuse itself causes hallucinations and other psychotic symptoms.
OUTCOME Depressed and sleepless
Mr. P admits that he had been smoking marijuana 2 to 3 times daily for a year. He also reports insomnia, sleeping approximately 4 hours a night and spending hours awake in bed thinking of his grandmother, with depressed mood and tearfulness. He denies suicidal ideas and hallucinations. He is treated for depressive disorder NOS first with amitriptyline, 50 mg at bedtime, for sleep, then paroxetine, 20 mg/d, for depressive symptoms, in addition to risperidone, 4 mg/d. Although Mr. P does not describe re-experiencing his childhood trauma, avoidance of stimuli associated with the trauma, or symptoms of increased arousal (except for insomnia), the treatment team did not ask, so it remains uncertain if he has PTSD (Table 2).
When Mr. P is discharged to a clinic, he smiles easily and is positive and supportive with other patients. He spruces up his appearance by wearing jewelry and works in the hospital kitchen.
Bottom Line
Chronic auditory hallucinations are associated with psychiatric illnesses other than chronic schizophrenia, particularly those resulting from trauma such as posttraumatic stress disorder. They can also occur in the absence of diagnosable psychiatric illness and rarely cause distress or functional impairment. Auditory hallucinations in adults have been associated with childhood abuse.
Related Resources
- Moskowitz A, Schafer I, Dorahy MJ. Psychosis, trauma and dissociation: emerging perspectives on severe psychopathology. West Sussex, UK: John Wiley and Sons, Ltd.; 2008.
- The International Hearing Voices Network. www.intervoiceonline.org.
Drug Brand Names
Amitriptyline • Elavil Paroxetine • Paxil
Benztropine • Cogentin Risperidone • Risperdal
Haloperidol • Haldol
Disclosure
Dr. Crowner reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE Grief and confusion
Mr. P, age 47, is arrested for entering the apartment of a woman he does not know and tossing her belongings out the window. When he is assessed to determine if he can participate in his legal defense, examiners find an attentive, courteous man who is baffled by his own behavior.
Mr. P says that he had been “stressed out” after the recent death of his grandmother, with whom he was close. He says he entered the apartment because voices told him to do so. He has no recent history of substance abuse or psychiatric hospitalizations, but he had a similar episode of “confusion” years before, when another close family member died.
Mr. P is found not fit to stand trial and the charges are dropped. He accepts haloperidol, 10 mg/d, and benztropine, 2 mg/d, and is transferred to a hospital for psychiatric treatment.
On interview, Mr. P is well groomed, soft-spoken, and shy, without formal thought disorder. Physical exam and routine lab tests are within normal limits. He says that 18 months before his arrest, he and his frail grandmother moved to a large city in hopes that he would find a wife. Both depended on the grandmother’s Social Security benefits while he cared for her.
In the 2 months after she died, he reports that he felt sad and alone and slept poorly, but made efforts to find a job and keep his apartment. When his efforts failed and he lost the apartment, he stayed with various friends for a few days at a time, then spent several days in the subway before ending up on the streets.
His arrest on the current charge occurred 4 days after he began walking the streets.
a) continue haloperidol to treat psychotic symptoms
b) discontinue haloperidol and observe him
c) add an antidepressant to haloperidol
HISTORY Imagining nonsense
Mr. P cannot explain why he started “trashing” the woman’s apartment, but says he entered it because he thought it was his apartment. With embarrassment and regret, he admits he has been depressed and confused, “imagining things”—“foolish things,” he admits—such as being in a different “time zone.”
Contradicting his earlier statements, Mr. P now admits that he had “a few beers” and denies that he experienced auditory hallucinations, saying he only talks to himself. He now says that within 2 days after his arrest, he was “all over it.” Mr. P denies current symptoms, including hallucinations, but, when pressed, waffles, then admits to a strange belief: that some people, including him, can move from one “time zone” to another.
Mr. P says he was treated for psychiatric problems 4 years earlier when his parents were killed in a car crash. By his recollection, his reaction to their death was similar to his reaction to his grandmother’s death: He became upset and wandered the streets for a few days, “moving between time zones” and talking to himself but not experiencing hallucinations. After he was taken to a hospital and “given an injection,” he calmed down and was released. Within a few days he recovered and returned to supporting himself and caring for his grandmother. Mr. P says the idea of travelling between “time zones” is embarrassing and nonsensical but adds that he was affected in this way because he “bickered” with his mother.
Mr. P’s grandmother raised him until he was age 15, although he frequently visited his parents, who lived nearby and worked during the day. Mr. P initially denies substance abuse, then admits to smoking marijuana every day for about a year before admission. He also admits to cocaine abuse in his 20s. He denies a history of suicide attempts.
The author’s observations
Mr. P reported only 2 episodes of “confusion” (or psychosis) and strange behavior in his life, both precipitated by the loss of a loved one, and at least 1 while under the influence of alcohol and Cannabis. He gave an inconsistent and ambiguous history of auditory hallucinations associated with episodes of confusion. He believes that time travel is possible, an idea that he acknowledged is nonsense. This alone was not enough to warrant long-term antipsychotic treatment. The most likely diagnosis seemed to be brief psychotic episode induced by Cannabis and the stressors of homelessness and his grandmother’s death.
EVALUATION Changing stories
No longer taking haloperidol, Mr. P continues to deny hallucinations and depressed mood, but keeps to himself. Nine days after admission he becomes tearful after he informs his aunt of his grandmother’s death in a telephone call, then approaches a nurse and complains of sadness and auditory hallucinations.
Mr. P confesses that he denied hallucinations on admission because he feared he would remain in the hospital for years if he revealed the truth that he had been experiencing auditory hallucinations almost continuously from age 10. He reports that the voices distracted him when he worked; seem to be male; often spoke gibberish; and alternate between deprecating and positive and supportive. Mr. P is reluctant to disclose more about what the voices actually say, although he acknowledges that they are not commenting or conversing with him, and that he has never believed the voices were his own thoughts but did believe that they came from inside his brain.
With haloperidol, the voices stopped. They resumed, however, when haloperidol was discontinued.
When we ask what happened to him at age 10, Mr. P shrugs.
a) childhood onset schizophrenia
b) substance abuse
c) posttraumatic stress disorder (PTSD)
d) none
The author’s observations
In community samples of children and adolescents, auditory hallucinations are not rare and usually do not cause distress or dysfunction. In a study of 3,870 children age 7 and 8,1 9% endorsed auditory hallucinations. Most heard 1 voice, once a week or less, at low volume. In 85% of children who experienced hallucinations, they caused minimal or no suffering; 97% reported minimal or no interference with daily functioning. Among children who experienced auditory hallucinations at age 7 or 8, 24% continued to hallucinate 5 years later.2 Persistent hallucinations were associated with more problematic behaviors at baseline and follow up.
In a group of 12-year-old twins, 4.2% reported auditory hallucinations.3 In that study, hallucinations were not related to Cannabis use; rather, they were heritable and related to risk factors such as cognitive impairment; behavioral, emotional, and educational problems at age 5; and a history of physical abuse and self-harm at age 12. The authors noted that these are risk factors and correlates of schizophrenia, but are not specific to schizophrenia.
Hallucinations and delusions have been found in 4% to 8% of children and adolescents referred for psychiatric treatment,4 far more than the prevalence of childhood-onset schizophrenia (0.01% of children).5 Psychotic symptoms in children have been associated with bipolar disorder, but also with anxiety disorders, obsessive-compulsive disorder, PTSD, pervasive developmental disorder, conduct disorder, and substance abuse.4
Childhood-onset schizophrenia is rare and would require that Mr. P have a diagnosis of schizophrenia as an adult. It is possible that Mr. P’s childhood symptoms were related to substance abuse but he was not asked for this history because it seemed unlikely in a 10-year-old boy. A PTSD diagnosis requires a traumatic event, which Mr. P did not reveal. It is possible that at age 10 he did not have a psychiatric disorder.
a) PTSD
b) dissociative disorder
c) borderline personality disorder
d) chronic schizophrenia
e) no psychiatric diagnosis
Among adults in the general population, 10% to 15% report auditory hallucinations.6 Hallucinations could be caused by substance abuse or psychiatric conditions other than schizophrenia; however, in adults—as in children—auditory hallucinations can occur in the absence of these conditions (Table 1) and rarely cause distress or dysfunction.6 In Sommer and colleagues’6 study of 103 healthy persons, none who heard voices had disorganization or negative symptoms. Those who heard voices had significantly more schizotypal symptoms and more childhood trauma, including emotional, physical, and sexual abuse, than those who did not hear voices.6
Conditions associated with hallucinations
PTSD is associated with auditory hallucinations and other psychotic symptoms.7 Most studies are of combat veterans with PTSD, in whom auditory hallucinations and delusions were associated with major depressive disorder, not a thought disorder or inappropriate affect.8 In a community sample,9 psychotic symptoms—particularly auditory hallucinations—were associated with PTSD. Subjects with PTSD and psychotic symptoms were more likely to have other psychiatric disorders, including major depressive disorder and substance use disorder, than patients with PTSD but no psychotic symptoms; however, the relationship between PTSD and psychosis remained after controlling for other psychiatric disorders.
Hallucinations can occur in persons with dissociative disorders in the absence of distinct personality states.10 Hallucinations have been seen transiently and chronically in persons with borderline personality disorder and can be associated with comorbid conditions such as substance abuse disorders, mood disorders, and PTSD.11
Mr. P lacked the reduced capacity for interpersonal relationships required for a schizotypal personality disorder diagnosis. A diagnosis of PTSD or dissociative disorder requires a history of trauma, which Mr. P did not report.
“Time travelling” with incomprehensible behavior could be interpreted as dissociation, but dissociative fugue or dissociative disorder not otherwise specified (NOS) cannot be diagnosed if symptoms might be the direct effect of a substance, such as Cannabis. Mr. P admitted to substance abuse. We can rule out borderline personality disorder because he did not display or admit to tempestuous interpersonal relationships.
A schizophrenia diagnosis requires the presence of auditory hallucinations that commented on his behavior or conversed among themselves, a second psychotic symptom for ≥1 month, or negative symptoms, which Mr. P lacked (unless belief in time travel is considered delusional).
Last, a physician might have considered malingering or a factitious disorder when Mr. P was found not able to participate in his own defense, but this seemed less likely after he revealed that he experienced auditory hallucinations since age 10.
HISTORY Bad beatings
With a few days of beginning risperidone, 4 mg/d, Mr. P reports that his hallucinations have stopped and he feels less sad. He reveals that, at age 10, when the hallucinations began, his mother hit him over the head with a high-heeled shoe, causing a scalp laceration that required a visit to the emergency room for suturing. His mother beat Mr. P for as long as he could remember. She beat him “bad” at least twice weekly, and he was taken to the hospital 7 or 8 times for injury, but she also beat him “constantly” with a belt buckle, sometimes striking his head. She instructed him to tell nobody.
The author’s observations
Auditory hallucinations in adults have been associated with childhood abuse, particularly childhood sexual abuse,12 in clinical and non-clinical samples.13 Some argue13 that child abuse itself causes hallucinations and other psychotic symptoms.
OUTCOME Depressed and sleepless
Mr. P admits that he had been smoking marijuana 2 to 3 times daily for a year. He also reports insomnia, sleeping approximately 4 hours a night and spending hours awake in bed thinking of his grandmother, with depressed mood and tearfulness. He denies suicidal ideas and hallucinations. He is treated for depressive disorder NOS first with amitriptyline, 50 mg at bedtime, for sleep, then paroxetine, 20 mg/d, for depressive symptoms, in addition to risperidone, 4 mg/d. Although Mr. P does not describe re-experiencing his childhood trauma, avoidance of stimuli associated with the trauma, or symptoms of increased arousal (except for insomnia), the treatment team did not ask, so it remains uncertain if he has PTSD (Table 2).
When Mr. P is discharged to a clinic, he smiles easily and is positive and supportive with other patients. He spruces up his appearance by wearing jewelry and works in the hospital kitchen.
Bottom Line
Chronic auditory hallucinations are associated with psychiatric illnesses other than chronic schizophrenia, particularly those resulting from trauma such as posttraumatic stress disorder. They can also occur in the absence of diagnosable psychiatric illness and rarely cause distress or functional impairment. Auditory hallucinations in adults have been associated with childhood abuse.
Related Resources
- Moskowitz A, Schafer I, Dorahy MJ. Psychosis, trauma and dissociation: emerging perspectives on severe psychopathology. West Sussex, UK: John Wiley and Sons, Ltd.; 2008.
- The International Hearing Voices Network. www.intervoiceonline.org.
Drug Brand Names
Amitriptyline • Elavil Paroxetine • Paxil
Benztropine • Cogentin Risperidone • Risperdal
Haloperidol • Haldol
Disclosure
Dr. Crowner reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Barthel-Velthuis AA, Jenner JA, van de Willige G, et al Prevalence and correlates of auditory vocal hallucinations in middle childhood. Br J Psychiatry. 2010;196(1):41-46.
2. Bartels-Velthuis AA, van de Willige G, Jenner JA, et al. Course of auditory vocal hallucinations in childhood: 5-year follow-up study. Br J Psychiatry. 2011;199(4):296-302.
3. Polanczyk G, Moffitt TE, Arsensault L, et al. Etiological and clinical features of childhood psychotic symptoms: results from a birth cohort. Arch Gen Psychiatry. 2010;67(4):328-338.
4. Biederman J, Pety C, Faracone SV, et al. Phenomenology of childhood psychosis: Findings from a large sample of psychiatrically referred youth. J Nerv Ment Dis 2004;192(9):607-614.
5. American Academy of Child and Adolescent Psychiatry. Practice parameters for the assessment and treatment of children and adolescents with schizophrenia. J Am Acad Child Adolesc Psychiatry. 2001;40(suppl 7):4SS-23S.
6. Sommer IEC, Daalman K, Rietkerk T, et al. Healthy individuals with auditory verbal hallucinations; Who are they? Psychiatric assessments of a selected sample of 103 subjects. Schizophr Bull. 2010;36(3):633-641.
7. Butler RW, Mueser KT, Sprock J, et al. Positive symptoms of psychosis in posttraumatic stress disorder. Biol Psychiatry. 1996;39:839-844.
8. David D, Kutcher GS, Jackson EI, et al Psychotic symptoms in combat-related posttraumatic stress disorder. J Clin Psychiatry. 1999;60(1):29-32.
9. Sareen J, Cox BJ, Goodwin RD, et al. Co-occurrence of posttraumatic stress disorder with positive psychotic symptoms in a nationally representative sample. J Trauma Stress. 2005;18(4):313-322.
10. Sar V, Akyuv G, Dogan O. Prevalence of dissociative disorders among women in the general population. Psychiatry Res. 2007;149:169-176.
11. Barnow S, Arens EA, Sieswerda S, et al. Borderline personality disorder and psychosis: a review. Curr Psychiatry Rep. 2010;12(3):186-195.
12. Bebbington P, Jonas S, Kuipers E, et al. Childhood sexual abuse and psychosis: data from a cross-sectional national psychiatric survey in England. Br J Psychiatry. 2011;199(1):29-37.
13. Read J, van Os J, Morrison AP, et al. Childhood trauma, psychosis and schizophrenia: a literature review with theoretical and clinical implications. Acta Psychiatr Scand. 2005;112(5):330-350.
1. Barthel-Velthuis AA, Jenner JA, van de Willige G, et al Prevalence and correlates of auditory vocal hallucinations in middle childhood. Br J Psychiatry. 2010;196(1):41-46.
2. Bartels-Velthuis AA, van de Willige G, Jenner JA, et al. Course of auditory vocal hallucinations in childhood: 5-year follow-up study. Br J Psychiatry. 2011;199(4):296-302.
3. Polanczyk G, Moffitt TE, Arsensault L, et al. Etiological and clinical features of childhood psychotic symptoms: results from a birth cohort. Arch Gen Psychiatry. 2010;67(4):328-338.
4. Biederman J, Pety C, Faracone SV, et al. Phenomenology of childhood psychosis: Findings from a large sample of psychiatrically referred youth. J Nerv Ment Dis 2004;192(9):607-614.
5. American Academy of Child and Adolescent Psychiatry. Practice parameters for the assessment and treatment of children and adolescents with schizophrenia. J Am Acad Child Adolesc Psychiatry. 2001;40(suppl 7):4SS-23S.
6. Sommer IEC, Daalman K, Rietkerk T, et al. Healthy individuals with auditory verbal hallucinations; Who are they? Psychiatric assessments of a selected sample of 103 subjects. Schizophr Bull. 2010;36(3):633-641.
7. Butler RW, Mueser KT, Sprock J, et al. Positive symptoms of psychosis in posttraumatic stress disorder. Biol Psychiatry. 1996;39:839-844.
8. David D, Kutcher GS, Jackson EI, et al Psychotic symptoms in combat-related posttraumatic stress disorder. J Clin Psychiatry. 1999;60(1):29-32.
9. Sareen J, Cox BJ, Goodwin RD, et al. Co-occurrence of posttraumatic stress disorder with positive psychotic symptoms in a nationally representative sample. J Trauma Stress. 2005;18(4):313-322.
10. Sar V, Akyuv G, Dogan O. Prevalence of dissociative disorders among women in the general population. Psychiatry Res. 2007;149:169-176.
11. Barnow S, Arens EA, Sieswerda S, et al. Borderline personality disorder and psychosis: a review. Curr Psychiatry Rep. 2010;12(3):186-195.
12. Bebbington P, Jonas S, Kuipers E, et al. Childhood sexual abuse and psychosis: data from a cross-sectional national psychiatric survey in England. Br J Psychiatry. 2011;199(1):29-37.
13. Read J, van Os J, Morrison AP, et al. Childhood trauma, psychosis and schizophrenia: a literature review with theoretical and clinical implications. Acta Psychiatr Scand. 2005;112(5):330-350.
Is he DISTRACTED? Considerations when diagnosing ADHD in an adult
Adult attention-deficit/hyperactivity disorder (ADHD) can be challenging to assess accurately. Adult ADHD differs significantly from childhood ADHD, in that hyperactivity often is absent or greatly diminished, comorbid disorders (depression or substance use) are common, and previously compensated attention deficits in school can manifest in the patient’s personal and professional life.1
The mnemonic DISTRACTED can help when recalling key components in assessing adult ADHD.2 Because ADHD is a developmental disorder—there are signs of onset in childhood—it is important to maintain a longitudinal view when asking about patterns of behavior or thinking.
Distractibility. Is there a pattern of getting “off track” in conversations or in school or work situations because of straying thoughts or daydreams? Is there a tendency to over-respond to extraneous stimuli (eg, cell phones, computers, television) that impedes the patient’s ability to converse, receive information, or follow directions?
Impulsivity. Does the patient have a history of saying things “off the cuff,” interrupting others, or “walking on” someone else’s words in a conversation? Is impulsivity evident in the person’s substance use or spending patterns?
School history. This domain is important in diagnosing ADHD in adults because there needs to be evidence that the disorder was present from an early age. How did the patient perform in school (ie, grades, organization, completion of homework assignments)? Was there a behavioral pattern that reflected hyperactivity (could not stay seated) or emotional dysregulation (frequent outbursts)?
Task completion. Does the patient have trouble finishing assignments at work, staying focused on a project that is considered boring, or completing a home project (eg, fixing a leaky faucet) in a timely fashion?
Rating scales. Rating scales should be used to help support the diagnosis, based on the patient’s history and life story. There are >12 scales that can be utilized in a
clinical setting3; the ADHD/Hyperactivity Disorder Self-Report Scale is a brief and easy measure of core ADHD symptoms.
Accidents. Adults with ADHD often are accident-prone because of inattention, hyperactivity, or impulsivity. Does the patient have a history of unintentionally hurting himself because he “wasn’t paying attention” (falls, burns), or was too impatient (traffic accidents or citations)?
Commitments. Does the patient fail to fulfill verbal obligations (by arriving late, forgetting to run errands)? Has this difficulty to commit created problems in relationships over time?
Time management. How difficult is it for the patient to stay organized while balancing work expectations, social obligations, and family needs? Is there a pattern of chaotic scheduling with regard to meals, work, or sleeping?
Employment. Has the patient changed jobs because the work becomes “too boring” or “uninteresting”? Is there a pattern of being terminated because of poor work quality based on time management or job performance?
Decisions. Adults with ADHD often make hasty, ill-informed choices or procrastinate so that they do not have to make a decision. Does the patient’s decision-making reveal a pattern of being too distracted to hear the information needed, or too impatient to consider all the details?
Remember: No single component of this mnemonic alone suffices to make a diagnosis of adult ADHD. However, these considerations will help clarify what lies behind your DISTRACTED patient’s search for self-understanding and appropriate medical care.
Disclosure
Dr. Christensen reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Barkley RA, Brown TE. Unrecognized attention-deficit/hyperactivity disorder in adults presenting with other psychiatric disorders. CNS Spectr. 2008;13(11):977-984.
2. Barkley R. Taking charge of adult ADHD. New York, NY: Guilford Press; 2010.
3. Attwell C. ADHD, rating scales, and your practice today. The Carlat Psychiatry Report. 2012;10(12):1,3,5-8.
Adult attention-deficit/hyperactivity disorder (ADHD) can be challenging to assess accurately. Adult ADHD differs significantly from childhood ADHD, in that hyperactivity often is absent or greatly diminished, comorbid disorders (depression or substance use) are common, and previously compensated attention deficits in school can manifest in the patient’s personal and professional life.1
The mnemonic DISTRACTED can help when recalling key components in assessing adult ADHD.2 Because ADHD is a developmental disorder—there are signs of onset in childhood—it is important to maintain a longitudinal view when asking about patterns of behavior or thinking.
Distractibility. Is there a pattern of getting “off track” in conversations or in school or work situations because of straying thoughts or daydreams? Is there a tendency to over-respond to extraneous stimuli (eg, cell phones, computers, television) that impedes the patient’s ability to converse, receive information, or follow directions?
Impulsivity. Does the patient have a history of saying things “off the cuff,” interrupting others, or “walking on” someone else’s words in a conversation? Is impulsivity evident in the person’s substance use or spending patterns?
School history. This domain is important in diagnosing ADHD in adults because there needs to be evidence that the disorder was present from an early age. How did the patient perform in school (ie, grades, organization, completion of homework assignments)? Was there a behavioral pattern that reflected hyperactivity (could not stay seated) or emotional dysregulation (frequent outbursts)?
Task completion. Does the patient have trouble finishing assignments at work, staying focused on a project that is considered boring, or completing a home project (eg, fixing a leaky faucet) in a timely fashion?
Rating scales. Rating scales should be used to help support the diagnosis, based on the patient’s history and life story. There are >12 scales that can be utilized in a
clinical setting3; the ADHD/Hyperactivity Disorder Self-Report Scale is a brief and easy measure of core ADHD symptoms.
Accidents. Adults with ADHD often are accident-prone because of inattention, hyperactivity, or impulsivity. Does the patient have a history of unintentionally hurting himself because he “wasn’t paying attention” (falls, burns), or was too impatient (traffic accidents or citations)?
Commitments. Does the patient fail to fulfill verbal obligations (by arriving late, forgetting to run errands)? Has this difficulty to commit created problems in relationships over time?
Time management. How difficult is it for the patient to stay organized while balancing work expectations, social obligations, and family needs? Is there a pattern of chaotic scheduling with regard to meals, work, or sleeping?
Employment. Has the patient changed jobs because the work becomes “too boring” or “uninteresting”? Is there a pattern of being terminated because of poor work quality based on time management or job performance?
Decisions. Adults with ADHD often make hasty, ill-informed choices or procrastinate so that they do not have to make a decision. Does the patient’s decision-making reveal a pattern of being too distracted to hear the information needed, or too impatient to consider all the details?
Remember: No single component of this mnemonic alone suffices to make a diagnosis of adult ADHD. However, these considerations will help clarify what lies behind your DISTRACTED patient’s search for self-understanding and appropriate medical care.
Disclosure
Dr. Christensen reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Adult attention-deficit/hyperactivity disorder (ADHD) can be challenging to assess accurately. Adult ADHD differs significantly from childhood ADHD, in that hyperactivity often is absent or greatly diminished, comorbid disorders (depression or substance use) are common, and previously compensated attention deficits in school can manifest in the patient’s personal and professional life.1
The mnemonic DISTRACTED can help when recalling key components in assessing adult ADHD.2 Because ADHD is a developmental disorder—there are signs of onset in childhood—it is important to maintain a longitudinal view when asking about patterns of behavior or thinking.
Distractibility. Is there a pattern of getting “off track” in conversations or in school or work situations because of straying thoughts or daydreams? Is there a tendency to over-respond to extraneous stimuli (eg, cell phones, computers, television) that impedes the patient’s ability to converse, receive information, or follow directions?
Impulsivity. Does the patient have a history of saying things “off the cuff,” interrupting others, or “walking on” someone else’s words in a conversation? Is impulsivity evident in the person’s substance use or spending patterns?
School history. This domain is important in diagnosing ADHD in adults because there needs to be evidence that the disorder was present from an early age. How did the patient perform in school (ie, grades, organization, completion of homework assignments)? Was there a behavioral pattern that reflected hyperactivity (could not stay seated) or emotional dysregulation (frequent outbursts)?
Task completion. Does the patient have trouble finishing assignments at work, staying focused on a project that is considered boring, or completing a home project (eg, fixing a leaky faucet) in a timely fashion?
Rating scales. Rating scales should be used to help support the diagnosis, based on the patient’s history and life story. There are >12 scales that can be utilized in a
clinical setting3; the ADHD/Hyperactivity Disorder Self-Report Scale is a brief and easy measure of core ADHD symptoms.
Accidents. Adults with ADHD often are accident-prone because of inattention, hyperactivity, or impulsivity. Does the patient have a history of unintentionally hurting himself because he “wasn’t paying attention” (falls, burns), or was too impatient (traffic accidents or citations)?
Commitments. Does the patient fail to fulfill verbal obligations (by arriving late, forgetting to run errands)? Has this difficulty to commit created problems in relationships over time?
Time management. How difficult is it for the patient to stay organized while balancing work expectations, social obligations, and family needs? Is there a pattern of chaotic scheduling with regard to meals, work, or sleeping?
Employment. Has the patient changed jobs because the work becomes “too boring” or “uninteresting”? Is there a pattern of being terminated because of poor work quality based on time management or job performance?
Decisions. Adults with ADHD often make hasty, ill-informed choices or procrastinate so that they do not have to make a decision. Does the patient’s decision-making reveal a pattern of being too distracted to hear the information needed, or too impatient to consider all the details?
Remember: No single component of this mnemonic alone suffices to make a diagnosis of adult ADHD. However, these considerations will help clarify what lies behind your DISTRACTED patient’s search for self-understanding and appropriate medical care.
Disclosure
Dr. Christensen reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Barkley RA, Brown TE. Unrecognized attention-deficit/hyperactivity disorder in adults presenting with other psychiatric disorders. CNS Spectr. 2008;13(11):977-984.
2. Barkley R. Taking charge of adult ADHD. New York, NY: Guilford Press; 2010.
3. Attwell C. ADHD, rating scales, and your practice today. The Carlat Psychiatry Report. 2012;10(12):1,3,5-8.
1. Barkley RA, Brown TE. Unrecognized attention-deficit/hyperactivity disorder in adults presenting with other psychiatric disorders. CNS Spectr. 2008;13(11):977-984.
2. Barkley R. Taking charge of adult ADHD. New York, NY: Guilford Press; 2010.
3. Attwell C. ADHD, rating scales, and your practice today. The Carlat Psychiatry Report. 2012;10(12):1,3,5-8.
Never ‘do nothing’ at end of life
Providing end-of-life care – is one of the toughest, most painful things we are called upon to do. Who among us has not had the gut-wrenching experience of informing a spouse of 50+ years that within a few short days, their life together will come to an abrupt end? No more anniversaries. No more anything.
I don’t think physicians can truly appreciate what patients’ loved ones go through when they are dying, until we become that loved one. I got my revelation when I was the caregiver and hospice physician for a very close relative who ultimately died from cancer in my home. I had asked an oncologist friend of mine to take on her case when she relocated to live with me. To my surprise, my relative found my colleague to be rather cold and unfeeling, just when she needed a compassionate physician the most.
I deeply understand the field of medicine, had care provided by a clinician/friend, and my relative still had a subpar experience, so what must it like for those without a medical background?
I recently spoke with a friend whose elderly aunt had just passed away. In addition to the grief she felt, she had to deal with frustration and anguish about how her aunt was treated in her final days. Her aunt’s DNI (do not intubate) status was mistakenly assumed by some on her health care team to mean "DNT" (do not treat). Basic care, such as intravenous fluids in the face of inadequate oral intake, was even neglected. To add insult to injury, the family – those who actually knew her belief system, feelings, and wishes – was not allowed to partner with the health care team to create the plan for her end-of-life care.
While we often wrestle with how to talk to family, including what we should and should not say, perhaps we should begin by learning a little about the background of the family members so we can tailor our conversations to a level appropriate to their level of understanding – great or small– of health care.
We can learn a lot by talking to friends about the experiences they have when a loved one dies. How were they and their family member treated by physicians and how did they respond to that treatment? What do they wish had happened differently? What made the transition from this life more difficult and what made it easier?
My friend’s words of wisdom for hospitalists center on communication and respect: "Each patient and family should be treated as if they are Kennedys or Annenbergs from the start."
Dr. Hester is a hospitalist with Baltimore-Washington Medical Center who has a passion for empowering patients to partner in their health care. She is the creator of the Patient Whiz, a patient-engagement app for iOS.
Providing end-of-life care – is one of the toughest, most painful things we are called upon to do. Who among us has not had the gut-wrenching experience of informing a spouse of 50+ years that within a few short days, their life together will come to an abrupt end? No more anniversaries. No more anything.
I don’t think physicians can truly appreciate what patients’ loved ones go through when they are dying, until we become that loved one. I got my revelation when I was the caregiver and hospice physician for a very close relative who ultimately died from cancer in my home. I had asked an oncologist friend of mine to take on her case when she relocated to live with me. To my surprise, my relative found my colleague to be rather cold and unfeeling, just when she needed a compassionate physician the most.
I deeply understand the field of medicine, had care provided by a clinician/friend, and my relative still had a subpar experience, so what must it like for those without a medical background?
I recently spoke with a friend whose elderly aunt had just passed away. In addition to the grief she felt, she had to deal with frustration and anguish about how her aunt was treated in her final days. Her aunt’s DNI (do not intubate) status was mistakenly assumed by some on her health care team to mean "DNT" (do not treat). Basic care, such as intravenous fluids in the face of inadequate oral intake, was even neglected. To add insult to injury, the family – those who actually knew her belief system, feelings, and wishes – was not allowed to partner with the health care team to create the plan for her end-of-life care.
While we often wrestle with how to talk to family, including what we should and should not say, perhaps we should begin by learning a little about the background of the family members so we can tailor our conversations to a level appropriate to their level of understanding – great or small– of health care.
We can learn a lot by talking to friends about the experiences they have when a loved one dies. How were they and their family member treated by physicians and how did they respond to that treatment? What do they wish had happened differently? What made the transition from this life more difficult and what made it easier?
My friend’s words of wisdom for hospitalists center on communication and respect: "Each patient and family should be treated as if they are Kennedys or Annenbergs from the start."
Dr. Hester is a hospitalist with Baltimore-Washington Medical Center who has a passion for empowering patients to partner in their health care. She is the creator of the Patient Whiz, a patient-engagement app for iOS.
Providing end-of-life care – is one of the toughest, most painful things we are called upon to do. Who among us has not had the gut-wrenching experience of informing a spouse of 50+ years that within a few short days, their life together will come to an abrupt end? No more anniversaries. No more anything.
I don’t think physicians can truly appreciate what patients’ loved ones go through when they are dying, until we become that loved one. I got my revelation when I was the caregiver and hospice physician for a very close relative who ultimately died from cancer in my home. I had asked an oncologist friend of mine to take on her case when she relocated to live with me. To my surprise, my relative found my colleague to be rather cold and unfeeling, just when she needed a compassionate physician the most.
I deeply understand the field of medicine, had care provided by a clinician/friend, and my relative still had a subpar experience, so what must it like for those without a medical background?
I recently spoke with a friend whose elderly aunt had just passed away. In addition to the grief she felt, she had to deal with frustration and anguish about how her aunt was treated in her final days. Her aunt’s DNI (do not intubate) status was mistakenly assumed by some on her health care team to mean "DNT" (do not treat). Basic care, such as intravenous fluids in the face of inadequate oral intake, was even neglected. To add insult to injury, the family – those who actually knew her belief system, feelings, and wishes – was not allowed to partner with the health care team to create the plan for her end-of-life care.
While we often wrestle with how to talk to family, including what we should and should not say, perhaps we should begin by learning a little about the background of the family members so we can tailor our conversations to a level appropriate to their level of understanding – great or small– of health care.
We can learn a lot by talking to friends about the experiences they have when a loved one dies. How were they and their family member treated by physicians and how did they respond to that treatment? What do they wish had happened differently? What made the transition from this life more difficult and what made it easier?
My friend’s words of wisdom for hospitalists center on communication and respect: "Each patient and family should be treated as if they are Kennedys or Annenbergs from the start."
Dr. Hester is a hospitalist with Baltimore-Washington Medical Center who has a passion for empowering patients to partner in their health care. She is the creator of the Patient Whiz, a patient-engagement app for iOS.
