User login
Cancer disparities: One of the most pressing public health issues
“The burden of cancer is not shouldered equally by all segments of the U.S. population,” the AACR adds. “The adverse differences in cancer burden that exist among certain population groups are one of the most pressing public health challenges that we face in the United States.”
AACR president Antoni Ribas, MD, PhD, gave some examples of these disparities at a September 16 Congressional briefing that focused on the inaugural AACR Cancer Disparities Progress Report 2020.
He noted that:
- Black men have more than double the rate of death from prostate cancer compared with men of other racial and ethnic groups.
- Hispanic children are 24% more likely to develop leukemia than non-Hispanic children.
- Non-Hispanic Black children and adolescents with cancer are more than 50% more likely to die from the cancer than non-Hispanic white children and adolescents with cancer.
- Women of low socioeconomic status with early stage ovarian cancer are 50% less likely to receive recommended care than are women of high socioeconomic status.
- In addition to racial and ethnic minority groups, other populations that bear a disproportionate burden when it comes to cancer include individuals lacking adequate health insurance coverage, immigrants, those with disabilities, residents in rural areas, and members of the lesbian, gay, bisexual, and transgender communities.
“It is absolutely unacceptable that advances in cancer care and treatment are not benefiting everyone equally,” Ribas commented.
Making progress against cancer
Progress being made against cancer was highlighted in another publication, the annual AACR Cancer Progress Report 2020.
U.S. cancer deaths declined by 29% between 1991 and 2017, translating to nearly 3 million cancer deaths avoided, the report notes. In addition, 5-year survival rates for all cancers combined increased from 49% in the mid-1970s to 70% for patients diagnosed from 2010-2016.
Between August 2019 and July 31 of this year, the U.S. Food and Drug Administration approved 20 new anticancer drugs for various cancer types and 15 new indications for previously approved cancer drugs, marking the highest number of approvals in one 12-month period since AACR started producing these reports 10 years ago.
A continuing reduction in the cigarette smoking rate among US adults, which is now below 14%, is contributing greatly to declines in lung cancer rates, which have largely driven the improvements in cancer survival, the AACR noted.
This report also notes that progress has been made toward reducing cancer disparities. Overall disparities in cancer death rates among racial and ethnic groups are less pronounced now than they have been in the past two decades. For example, the overall cancer death rate for African American patients was 33% higher than for White patients in 1990 but just 14% higher in 2016.
However, both reports agree that more must be done to reduce cancer disparities even further.
They highlight initiatives that are underway, including:
- The draft guidance issued by the FDA to promote diversification of clinical trial populations.
- The National Institutes of Health’s (NIH’s) Continuing Umbrella of Research Experiences (CURE) program supporting underrepresented students and scientists along their academic and research career pathway.
- The Centers for Disease Control and Prevention’s Racial and Ethnic Approaches to Community Health (REACH) program, a grant-making program focused on encouraging preventive behaviors in underserved communities.
- The NIH’s All of Us program, which is gathering information from the genomes of 1 million healthy individuals with a focus on recruitment from historically underrepresented populations.
Ribas also announced that AACR has established a task force to focus on racial inequalities in cancer research.
Eliminating disparities would save money, argued John D. Carpten, PhD, from the University of Southern California, Los Angeles, who chaired the steering committee that developed the AACR Cancer Disparities Progress Report.
Carpten noted research showing that eliminating disparities for racial and ethnic minorities between 2003 and 2006 would have reduced health care costs by more than $1 trillion in the United States. This underscores the potentially far-reaching impact of efforts to eliminate disparities, he said.
“Without a doubt, socioeconomics and inequities in access to quality care represent major factors influencing cancer health disparities, and these disparities will persist until we address these issues” he said.
Both progress reports culminate in a call to action, largely focused on the need for “unwavering, bipartisan support from Congress, in the form of robust and sustained annual increases in funding for the NIH, NCI [National Cancer Institute], and FDA,” which is vital for accelerating the pace of progress.
The challenge is now compounded by the ongoing COVID-19 pandemic: Both progress reports note that racial and ethnic minorities, including African Americans, are not only affected disproportionately by cancer, but also by COVID-19, further highlighting the “stark inequities in health care.”
Ribas further called for action from national leadership and the scientific community.
“During this unprecedented time in our nation’s history, there is also a need for our nation’s leaders to take on a much bigger role in confronting and combating the structural and systemic racism that contributes to health disparities,” he said. The “pervasive racism and social injustices” that have contributed to disparities in both COVID-19 and cancer underscore the need for “the scientific community to step up and partner with Congress to assess and address this issue within the research community.”
This article first appeared on Medscape.com.
“The burden of cancer is not shouldered equally by all segments of the U.S. population,” the AACR adds. “The adverse differences in cancer burden that exist among certain population groups are one of the most pressing public health challenges that we face in the United States.”
AACR president Antoni Ribas, MD, PhD, gave some examples of these disparities at a September 16 Congressional briefing that focused on the inaugural AACR Cancer Disparities Progress Report 2020.
He noted that:
- Black men have more than double the rate of death from prostate cancer compared with men of other racial and ethnic groups.
- Hispanic children are 24% more likely to develop leukemia than non-Hispanic children.
- Non-Hispanic Black children and adolescents with cancer are more than 50% more likely to die from the cancer than non-Hispanic white children and adolescents with cancer.
- Women of low socioeconomic status with early stage ovarian cancer are 50% less likely to receive recommended care than are women of high socioeconomic status.
- In addition to racial and ethnic minority groups, other populations that bear a disproportionate burden when it comes to cancer include individuals lacking adequate health insurance coverage, immigrants, those with disabilities, residents in rural areas, and members of the lesbian, gay, bisexual, and transgender communities.
“It is absolutely unacceptable that advances in cancer care and treatment are not benefiting everyone equally,” Ribas commented.
Making progress against cancer
Progress being made against cancer was highlighted in another publication, the annual AACR Cancer Progress Report 2020.
U.S. cancer deaths declined by 29% between 1991 and 2017, translating to nearly 3 million cancer deaths avoided, the report notes. In addition, 5-year survival rates for all cancers combined increased from 49% in the mid-1970s to 70% for patients diagnosed from 2010-2016.
Between August 2019 and July 31 of this year, the U.S. Food and Drug Administration approved 20 new anticancer drugs for various cancer types and 15 new indications for previously approved cancer drugs, marking the highest number of approvals in one 12-month period since AACR started producing these reports 10 years ago.
A continuing reduction in the cigarette smoking rate among US adults, which is now below 14%, is contributing greatly to declines in lung cancer rates, which have largely driven the improvements in cancer survival, the AACR noted.
This report also notes that progress has been made toward reducing cancer disparities. Overall disparities in cancer death rates among racial and ethnic groups are less pronounced now than they have been in the past two decades. For example, the overall cancer death rate for African American patients was 33% higher than for White patients in 1990 but just 14% higher in 2016.
However, both reports agree that more must be done to reduce cancer disparities even further.
They highlight initiatives that are underway, including:
- The draft guidance issued by the FDA to promote diversification of clinical trial populations.
- The National Institutes of Health’s (NIH’s) Continuing Umbrella of Research Experiences (CURE) program supporting underrepresented students and scientists along their academic and research career pathway.
- The Centers for Disease Control and Prevention’s Racial and Ethnic Approaches to Community Health (REACH) program, a grant-making program focused on encouraging preventive behaviors in underserved communities.
- The NIH’s All of Us program, which is gathering information from the genomes of 1 million healthy individuals with a focus on recruitment from historically underrepresented populations.
Ribas also announced that AACR has established a task force to focus on racial inequalities in cancer research.
Eliminating disparities would save money, argued John D. Carpten, PhD, from the University of Southern California, Los Angeles, who chaired the steering committee that developed the AACR Cancer Disparities Progress Report.
Carpten noted research showing that eliminating disparities for racial and ethnic minorities between 2003 and 2006 would have reduced health care costs by more than $1 trillion in the United States. This underscores the potentially far-reaching impact of efforts to eliminate disparities, he said.
“Without a doubt, socioeconomics and inequities in access to quality care represent major factors influencing cancer health disparities, and these disparities will persist until we address these issues” he said.
Both progress reports culminate in a call to action, largely focused on the need for “unwavering, bipartisan support from Congress, in the form of robust and sustained annual increases in funding for the NIH, NCI [National Cancer Institute], and FDA,” which is vital for accelerating the pace of progress.
The challenge is now compounded by the ongoing COVID-19 pandemic: Both progress reports note that racial and ethnic minorities, including African Americans, are not only affected disproportionately by cancer, but also by COVID-19, further highlighting the “stark inequities in health care.”
Ribas further called for action from national leadership and the scientific community.
“During this unprecedented time in our nation’s history, there is also a need for our nation’s leaders to take on a much bigger role in confronting and combating the structural and systemic racism that contributes to health disparities,” he said. The “pervasive racism and social injustices” that have contributed to disparities in both COVID-19 and cancer underscore the need for “the scientific community to step up and partner with Congress to assess and address this issue within the research community.”
This article first appeared on Medscape.com.
“The burden of cancer is not shouldered equally by all segments of the U.S. population,” the AACR adds. “The adverse differences in cancer burden that exist among certain population groups are one of the most pressing public health challenges that we face in the United States.”
AACR president Antoni Ribas, MD, PhD, gave some examples of these disparities at a September 16 Congressional briefing that focused on the inaugural AACR Cancer Disparities Progress Report 2020.
He noted that:
- Black men have more than double the rate of death from prostate cancer compared with men of other racial and ethnic groups.
- Hispanic children are 24% more likely to develop leukemia than non-Hispanic children.
- Non-Hispanic Black children and adolescents with cancer are more than 50% more likely to die from the cancer than non-Hispanic white children and adolescents with cancer.
- Women of low socioeconomic status with early stage ovarian cancer are 50% less likely to receive recommended care than are women of high socioeconomic status.
- In addition to racial and ethnic minority groups, other populations that bear a disproportionate burden when it comes to cancer include individuals lacking adequate health insurance coverage, immigrants, those with disabilities, residents in rural areas, and members of the lesbian, gay, bisexual, and transgender communities.
“It is absolutely unacceptable that advances in cancer care and treatment are not benefiting everyone equally,” Ribas commented.
Making progress against cancer
Progress being made against cancer was highlighted in another publication, the annual AACR Cancer Progress Report 2020.
U.S. cancer deaths declined by 29% between 1991 and 2017, translating to nearly 3 million cancer deaths avoided, the report notes. In addition, 5-year survival rates for all cancers combined increased from 49% in the mid-1970s to 70% for patients diagnosed from 2010-2016.
Between August 2019 and July 31 of this year, the U.S. Food and Drug Administration approved 20 new anticancer drugs for various cancer types and 15 new indications for previously approved cancer drugs, marking the highest number of approvals in one 12-month period since AACR started producing these reports 10 years ago.
A continuing reduction in the cigarette smoking rate among US adults, which is now below 14%, is contributing greatly to declines in lung cancer rates, which have largely driven the improvements in cancer survival, the AACR noted.
This report also notes that progress has been made toward reducing cancer disparities. Overall disparities in cancer death rates among racial and ethnic groups are less pronounced now than they have been in the past two decades. For example, the overall cancer death rate for African American patients was 33% higher than for White patients in 1990 but just 14% higher in 2016.
However, both reports agree that more must be done to reduce cancer disparities even further.
They highlight initiatives that are underway, including:
- The draft guidance issued by the FDA to promote diversification of clinical trial populations.
- The National Institutes of Health’s (NIH’s) Continuing Umbrella of Research Experiences (CURE) program supporting underrepresented students and scientists along their academic and research career pathway.
- The Centers for Disease Control and Prevention’s Racial and Ethnic Approaches to Community Health (REACH) program, a grant-making program focused on encouraging preventive behaviors in underserved communities.
- The NIH’s All of Us program, which is gathering information from the genomes of 1 million healthy individuals with a focus on recruitment from historically underrepresented populations.
Ribas also announced that AACR has established a task force to focus on racial inequalities in cancer research.
Eliminating disparities would save money, argued John D. Carpten, PhD, from the University of Southern California, Los Angeles, who chaired the steering committee that developed the AACR Cancer Disparities Progress Report.
Carpten noted research showing that eliminating disparities for racial and ethnic minorities between 2003 and 2006 would have reduced health care costs by more than $1 trillion in the United States. This underscores the potentially far-reaching impact of efforts to eliminate disparities, he said.
“Without a doubt, socioeconomics and inequities in access to quality care represent major factors influencing cancer health disparities, and these disparities will persist until we address these issues” he said.
Both progress reports culminate in a call to action, largely focused on the need for “unwavering, bipartisan support from Congress, in the form of robust and sustained annual increases in funding for the NIH, NCI [National Cancer Institute], and FDA,” which is vital for accelerating the pace of progress.
The challenge is now compounded by the ongoing COVID-19 pandemic: Both progress reports note that racial and ethnic minorities, including African Americans, are not only affected disproportionately by cancer, but also by COVID-19, further highlighting the “stark inequities in health care.”
Ribas further called for action from national leadership and the scientific community.
“During this unprecedented time in our nation’s history, there is also a need for our nation’s leaders to take on a much bigger role in confronting and combating the structural and systemic racism that contributes to health disparities,” he said. The “pervasive racism and social injustices” that have contributed to disparities in both COVID-19 and cancer underscore the need for “the scientific community to step up and partner with Congress to assess and address this issue within the research community.”
This article first appeared on Medscape.com.
FDA adds polyarticular-course JIA to approved indications for tofacitinib
The Food and Drug Administration has (pJIA).

The approval, announced Sept. 28 by tofacitinib’s manufacturer, Pfizer, marks the first JAK inhibitor to be approved for the condition in the United States and is the fourth indication to be approved for the drug after approvals in adult patients with moderate to severe rheumatoid arthritis following methotrexate failure, active psoriatic arthritis after disease-modifying antirheumatic drug failure, and moderate to severe ulcerative colitis after failure on a tumor necrosis factor inhibitor.
The agency based its approval on a phase 3, multinational, randomized, double-blind, controlled withdrawal study that had an 18-week, open-label, run-in phase involving 225 patients who twice daily took either a 5-mg tablet or, in patients under 40 kg, a weight-based lower dose in the form of a 1 mg/mL oral solution, according to the company press release. A total of 173 patients from this phase met JIA American College of Rheumatology 30 response criteria, defined as 30% or greater improvement in three of six JIA core set variables and worsening in no more than one of the core set variables; they were then randomized in part 2 of the study to continue the same dose of tofacitinib or receive placebo until 44 weeks. By the end of this period, 31% who received tofacitinib had a disease flare, compared with 55% on placebo (P = .0007). Disease flare was defined as a 30% or greater worsening in at least three of the six variables of the JIA core set, with no more than one of the remaining JIA core response variables improving by 30% or more after randomization.
The types of adverse drug reactions in patients with pJIA were consistent with those seen in adult rheumatoid arthritis patients, according to Pfizer. Serious adverse drug reactions have most commonly been serious infections that may lead to hospitalization or death, and most patients who developed these infections were taking concomitant immunosuppressants, such as methotrexate or corticosteroids. Common adverse drug reactions reported in 2% or more of patients during the first 3 months in controlled clinical trials in patients with rheumatoid arthritis taking tofacitinib at 5 mg twice daily were upper respiratory tract infection, nasopharyngitis, diarrhea, headache, and hypertension.
While the 5-mg tablet formulation is already available, Pfizer said it expects the oral solution to be available by the end of the first quarter in 2021.
Prescribing information can be found on the FDA website.
The Food and Drug Administration has (pJIA).
The approval, announced Sept. 28 by tofacitinib’s manufacturer, Pfizer, marks the first JAK inhibitor to be approved for the condition in the United States and is the fourth indication to be approved for the drug after approvals in adult patients with moderate to severe rheumatoid arthritis following methotrexate failure, active psoriatic arthritis after disease-modifying antirheumatic drug failure, and moderate to severe ulcerative colitis after failure on a tumor necrosis factor inhibitor.
The agency based its approval on a phase 3, multinational, randomized, double-blind, controlled withdrawal study that had an 18-week, open-label, run-in phase involving 225 patients who twice daily took either a 5-mg tablet or, in patients under 40 kg, a weight-based lower dose in the form of a 1 mg/mL oral solution, according to the company press release. A total of 173 patients from this phase met JIA American College of Rheumatology 30 response criteria, defined as 30% or greater improvement in three of six JIA core set variables and worsening in no more than one of the core set variables; they were then randomized in part 2 of the study to continue the same dose of tofacitinib or receive placebo until 44 weeks. By the end of this period, 31% who received tofacitinib had a disease flare, compared with 55% on placebo (P = .0007). Disease flare was defined as a 30% or greater worsening in at least three of the six variables of the JIA core set, with no more than one of the remaining JIA core response variables improving by 30% or more after randomization.
The types of adverse drug reactions in patients with pJIA were consistent with those seen in adult rheumatoid arthritis patients, according to Pfizer. Serious adverse drug reactions have most commonly been serious infections that may lead to hospitalization or death, and most patients who developed these infections were taking concomitant immunosuppressants, such as methotrexate or corticosteroids. Common adverse drug reactions reported in 2% or more of patients during the first 3 months in controlled clinical trials in patients with rheumatoid arthritis taking tofacitinib at 5 mg twice daily were upper respiratory tract infection, nasopharyngitis, diarrhea, headache, and hypertension.
While the 5-mg tablet formulation is already available, Pfizer said it expects the oral solution to be available by the end of the first quarter in 2021.
Prescribing information can be found on the FDA website.
The Food and Drug Administration has (pJIA).
The approval, announced Sept. 28 by tofacitinib’s manufacturer, Pfizer, marks the first JAK inhibitor to be approved for the condition in the United States and is the fourth indication to be approved for the drug after approvals in adult patients with moderate to severe rheumatoid arthritis following methotrexate failure, active psoriatic arthritis after disease-modifying antirheumatic drug failure, and moderate to severe ulcerative colitis after failure on a tumor necrosis factor inhibitor.
The agency based its approval on a phase 3, multinational, randomized, double-blind, controlled withdrawal study that had an 18-week, open-label, run-in phase involving 225 patients who twice daily took either a 5-mg tablet or, in patients under 40 kg, a weight-based lower dose in the form of a 1 mg/mL oral solution, according to the company press release. A total of 173 patients from this phase met JIA American College of Rheumatology 30 response criteria, defined as 30% or greater improvement in three of six JIA core set variables and worsening in no more than one of the core set variables; they were then randomized in part 2 of the study to continue the same dose of tofacitinib or receive placebo until 44 weeks. By the end of this period, 31% who received tofacitinib had a disease flare, compared with 55% on placebo (P = .0007). Disease flare was defined as a 30% or greater worsening in at least three of the six variables of the JIA core set, with no more than one of the remaining JIA core response variables improving by 30% or more after randomization.
The types of adverse drug reactions in patients with pJIA were consistent with those seen in adult rheumatoid arthritis patients, according to Pfizer. Serious adverse drug reactions have most commonly been serious infections that may lead to hospitalization or death, and most patients who developed these infections were taking concomitant immunosuppressants, such as methotrexate or corticosteroids. Common adverse drug reactions reported in 2% or more of patients during the first 3 months in controlled clinical trials in patients with rheumatoid arthritis taking tofacitinib at 5 mg twice daily were upper respiratory tract infection, nasopharyngitis, diarrhea, headache, and hypertension.
While the 5-mg tablet formulation is already available, Pfizer said it expects the oral solution to be available by the end of the first quarter in 2021.
Prescribing information can be found on the FDA website.
COVID-19 prompts ‘democratization’ of cancer trials

The pandemic has taught researchers how to decentralize trials, which should not only improve patient satisfaction but increase trial accrual by providing access to typically underserved populations, Patricia M. LoRusso, DO, of Yale University, New Haven, Conn., said at the meeting.
Dr. LoRusso was one of six panelists who participated in a forum about changes to cancer trials that were prompted by the pandemic. The forum was moderated by Keith T. Flaherty, MD, of Massachusetts General Hospital in Boston.
Dr. Flaherty asked the panelists to explain adjustments their organizations have made in response to the pandemic, discuss accomplishments, and speculate on future challenges and priorities.
Trial, administrative, and patient-care modifications
COVID-19 put some cancer trials on hold. For others, the pandemic forced sponsors and study chairs to reduce trial complexity and identify nonessential aspects of the studies, according to panelist José Baselga, MD, PhD, of AstraZeneca.
Specifically, exploratory objectives were subjugated to patient safety and a focus on the primary endpoints of each trial.
Once the critical data were identified, study chairs were asked to determine whether data could be obtained through technologies that could substitute for face-to-face contact between patients and staff – for example, patient-reported outcome tools and at-home digital monitoring.
Modifications prompted by the pandemic include the following:
- On-site auditing was suspended.
- Oral investigational agents were shipped directly to patients.
- “Remote” informed consent (telephone or video consenting) was permitted.
- Local providers could perform study-related services, with oversight by the research site.
- Minor deviations from the written protocols were allowed, provided the deviations did not affect patient care or data integrity.
“Obviously, the pandemic has been horrible, but what it has allowed us to do, as investigators in the clinical research landscape, … is to change our focus somewhat and realize, first and foremost, the patient is at the center of this,” Dr. LoRusso said.
Operational accomplishments and benefits
The pandemic caused a 40% decline in accrual to studies supported by the National Cancer Institute’s (NCI) Clinical Trials Network (NCTN) from mid-March to early April, according to James H. Doroshow, MD, of NCI.
However, after modifications to administrative and regulatory procedures, accrual to NCTN trials recovered to approximately 80% of prepandemic levels, Dr. Doroshow said.
The pandemic prompted investigators to leverage tools and technology they had not previously used frequently or at all, the panelists pointed out.
Investigators discovered perforce that telehealth could be used for almost all trial-related assessments. In lieu of physical examination, patients could send pictures of rashes and use electronic devices to monitor blood sugar values and vital signs.
Digital radiographic studies were performed at sites that were most convenient for patients, downloaded, and reinterpreted at the study institution. Visiting nurses and neighborhood laboratories enabled less-frequent in-person visits for assessments.
These adjustments have been particularly important for geographically and/or socioeconomically disadvantaged patients, the panelists said.
Overall, there was agreement among the panelists that shared values and trust among regulatory authorities, sponsors, investigators, and clinicians were impressive in their urgency, sincerity, and patient centricity.
“This pandemic … has forced us to think differently and be nimble and creative to our approach to maintaining our overriding goals while at the same time bringing these innovative therapies forward for patients with cancer and other serious and life-threatening diseases as quickly as possible,” said panelist Kristen M. Hege, MD, of Bristol-Myers Squibb.
In fact, Dr. Hege noted, some cancer-related therapies (e.g., BTK inhibitors, JAK inhibitors, and immunomodulatory agents) were “repurposed” rapidly and tested against COVID-related complications.
Streamlining trial regulatory processes
In addition to changing ongoing trials, the pandemic has affected how new research projects are launched.
One new study that came together quickly in response to the pandemic is the NCI COVID-19 in Cancer Patients Study (NCCAPS). NCCAPS is a natural history study with biospecimens and an imaging library. It was approved in just 5 weeks and is active in 650 sites, with “gangbusters” accrual, Dr. Doroshow said.
The rapidness of NCCAPS’ design and implementation should prompt the revision of previously accepted timelines for trial activation and lead to streamlined future processes.
Another project that was launched quickly in response to the pandemic is the COVID-19 evidence accelerator, according to Paul G. Kluetz, MD, of the Food and Drug Administration.
The COVID-19 evidence accelerator integrates real-world evidence into a database to provide investigators and health systems with the ability to gather information, design rapid turnaround queries, and share results. The evidence accelerator can provide study chairs with information that may have relevance to the safety of participants in clinical trials.
Future directions and challenges
The panelists agreed that pandemic-related modifications in processes will not only accelerate trial approval and activation but should facilitate higher study accrual, increase the diversity of protocol participants, and decrease the costs associated with clinical trial conduct.
With that in mind, the NCI is planning randomized clinical trials in which “process A” is compared with “process B,” Dr. Doroshow said. The goal is to determine which modifications are most likely to make trials available to patients without compromising data integrity or patient safety.
“How much less data do you need to have an outcome that will be similar?” Dr. Doroshow asked. “How many fewer visits, how many fewer tests, how much can you save? Physicians, clinical trialists, all of us respond to data, and if you get the same outcome at a third of the cost, then everybody benefits.”
Nonetheless, we will need to be vigilant for unintended vulnerabilities from well-intended efforts, according to Dr. Kluetz. Study chairs, sponsors, and regulatory agencies will need to be attentive to whether there are important differences in scan quality or interpretation, missing data that influence trial outcomes, and so on.
Dr. Hege pointed out that differences among data sources may be less important when treatments generate large effects but may be vitally important when the relative differences among treatments are small.
On a practical level, decentralizing clinical research may negatively impact the finances of tertiary care centers, which could threaten the required infrastructure for clinical trials, a few panelists noted.
The relative balance of NCI-, industry-, and investigator-initiated trials may require adjustment so that research income is adequate to maintain the costs associated with cancer clinical trials.
Shared goals and democratization
The pandemic has required all stakeholders in clinical research to rely on relationships of trust and shared goals, said Caroline Robert, MD, PhD, of Institut Gustave Roussy in Villejuif, France.
Dr. Kluetz summarized those goals as improving trial efficiencies, decreasing patient burden, decentralizing trials, and maintaining trial integrity.
A decentralized clinical trials operational model could lead to better generalizability of study outcomes, normalization of life for patients on studies, and lower costs of trial conduct. As such, decentralization would promote democratization.
Coupled with ongoing efforts to reduce eligibility criteria in cancer trials, the pandemic has brought operational solutions that should be perpetuated and has reminded us of the interlocking and mutually supportive relationships on which clinical research success depends.
Dr. Doroshow and Dr. Kluetz disclosed no conflicts of interest. All other panelists disclosed financial relationships, including employment, with a range of companies.
Dr. Lyss was a community-based medical oncologist and clinical researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers, as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
SOURCE: Flaherty KT et al. AACR: COVID-19 and Cancer, Regulatory and Operational Implications of Cancer Clinical Trial Changes During COVID-19.
The pandemic has taught researchers how to decentralize trials, which should not only improve patient satisfaction but increase trial accrual by providing access to typically underserved populations, Patricia M. LoRusso, DO, of Yale University, New Haven, Conn., said at the meeting.
Dr. LoRusso was one of six panelists who participated in a forum about changes to cancer trials that were prompted by the pandemic. The forum was moderated by Keith T. Flaherty, MD, of Massachusetts General Hospital in Boston.
Dr. Flaherty asked the panelists to explain adjustments their organizations have made in response to the pandemic, discuss accomplishments, and speculate on future challenges and priorities.
Trial, administrative, and patient-care modifications
COVID-19 put some cancer trials on hold. For others, the pandemic forced sponsors and study chairs to reduce trial complexity and identify nonessential aspects of the studies, according to panelist José Baselga, MD, PhD, of AstraZeneca.
Specifically, exploratory objectives were subjugated to patient safety and a focus on the primary endpoints of each trial.
Once the critical data were identified, study chairs were asked to determine whether data could be obtained through technologies that could substitute for face-to-face contact between patients and staff – for example, patient-reported outcome tools and at-home digital monitoring.
Modifications prompted by the pandemic include the following:
- On-site auditing was suspended.
- Oral investigational agents were shipped directly to patients.
- “Remote” informed consent (telephone or video consenting) was permitted.
- Local providers could perform study-related services, with oversight by the research site.
- Minor deviations from the written protocols were allowed, provided the deviations did not affect patient care or data integrity.
“Obviously, the pandemic has been horrible, but what it has allowed us to do, as investigators in the clinical research landscape, … is to change our focus somewhat and realize, first and foremost, the patient is at the center of this,” Dr. LoRusso said.
Operational accomplishments and benefits
The pandemic caused a 40% decline in accrual to studies supported by the National Cancer Institute’s (NCI) Clinical Trials Network (NCTN) from mid-March to early April, according to James H. Doroshow, MD, of NCI.
However, after modifications to administrative and regulatory procedures, accrual to NCTN trials recovered to approximately 80% of prepandemic levels, Dr. Doroshow said.
The pandemic prompted investigators to leverage tools and technology they had not previously used frequently or at all, the panelists pointed out.
Investigators discovered perforce that telehealth could be used for almost all trial-related assessments. In lieu of physical examination, patients could send pictures of rashes and use electronic devices to monitor blood sugar values and vital signs.
Digital radiographic studies were performed at sites that were most convenient for patients, downloaded, and reinterpreted at the study institution. Visiting nurses and neighborhood laboratories enabled less-frequent in-person visits for assessments.
These adjustments have been particularly important for geographically and/or socioeconomically disadvantaged patients, the panelists said.
Overall, there was agreement among the panelists that shared values and trust among regulatory authorities, sponsors, investigators, and clinicians were impressive in their urgency, sincerity, and patient centricity.
“This pandemic … has forced us to think differently and be nimble and creative to our approach to maintaining our overriding goals while at the same time bringing these innovative therapies forward for patients with cancer and other serious and life-threatening diseases as quickly as possible,” said panelist Kristen M. Hege, MD, of Bristol-Myers Squibb.
In fact, Dr. Hege noted, some cancer-related therapies (e.g., BTK inhibitors, JAK inhibitors, and immunomodulatory agents) were “repurposed” rapidly and tested against COVID-related complications.
Streamlining trial regulatory processes
In addition to changing ongoing trials, the pandemic has affected how new research projects are launched.
One new study that came together quickly in response to the pandemic is the NCI COVID-19 in Cancer Patients Study (NCCAPS). NCCAPS is a natural history study with biospecimens and an imaging library. It was approved in just 5 weeks and is active in 650 sites, with “gangbusters” accrual, Dr. Doroshow said.
The rapidness of NCCAPS’ design and implementation should prompt the revision of previously accepted timelines for trial activation and lead to streamlined future processes.
Another project that was launched quickly in response to the pandemic is the COVID-19 evidence accelerator, according to Paul G. Kluetz, MD, of the Food and Drug Administration.
The COVID-19 evidence accelerator integrates real-world evidence into a database to provide investigators and health systems with the ability to gather information, design rapid turnaround queries, and share results. The evidence accelerator can provide study chairs with information that may have relevance to the safety of participants in clinical trials.
Future directions and challenges
The panelists agreed that pandemic-related modifications in processes will not only accelerate trial approval and activation but should facilitate higher study accrual, increase the diversity of protocol participants, and decrease the costs associated with clinical trial conduct.
With that in mind, the NCI is planning randomized clinical trials in which “process A” is compared with “process B,” Dr. Doroshow said. The goal is to determine which modifications are most likely to make trials available to patients without compromising data integrity or patient safety.
“How much less data do you need to have an outcome that will be similar?” Dr. Doroshow asked. “How many fewer visits, how many fewer tests, how much can you save? Physicians, clinical trialists, all of us respond to data, and if you get the same outcome at a third of the cost, then everybody benefits.”
Nonetheless, we will need to be vigilant for unintended vulnerabilities from well-intended efforts, according to Dr. Kluetz. Study chairs, sponsors, and regulatory agencies will need to be attentive to whether there are important differences in scan quality or interpretation, missing data that influence trial outcomes, and so on.
Dr. Hege pointed out that differences among data sources may be less important when treatments generate large effects but may be vitally important when the relative differences among treatments are small.
On a practical level, decentralizing clinical research may negatively impact the finances of tertiary care centers, which could threaten the required infrastructure for clinical trials, a few panelists noted.
The relative balance of NCI-, industry-, and investigator-initiated trials may require adjustment so that research income is adequate to maintain the costs associated with cancer clinical trials.
Shared goals and democratization
The pandemic has required all stakeholders in clinical research to rely on relationships of trust and shared goals, said Caroline Robert, MD, PhD, of Institut Gustave Roussy in Villejuif, France.
Dr. Kluetz summarized those goals as improving trial efficiencies, decreasing patient burden, decentralizing trials, and maintaining trial integrity.
A decentralized clinical trials operational model could lead to better generalizability of study outcomes, normalization of life for patients on studies, and lower costs of trial conduct. As such, decentralization would promote democratization.
Coupled with ongoing efforts to reduce eligibility criteria in cancer trials, the pandemic has brought operational solutions that should be perpetuated and has reminded us of the interlocking and mutually supportive relationships on which clinical research success depends.
Dr. Doroshow and Dr. Kluetz disclosed no conflicts of interest. All other panelists disclosed financial relationships, including employment, with a range of companies.
Dr. Lyss was a community-based medical oncologist and clinical researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers, as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
SOURCE: Flaherty KT et al. AACR: COVID-19 and Cancer, Regulatory and Operational Implications of Cancer Clinical Trial Changes During COVID-19.
The pandemic has taught researchers how to decentralize trials, which should not only improve patient satisfaction but increase trial accrual by providing access to typically underserved populations, Patricia M. LoRusso, DO, of Yale University, New Haven, Conn., said at the meeting.
Dr. LoRusso was one of six panelists who participated in a forum about changes to cancer trials that were prompted by the pandemic. The forum was moderated by Keith T. Flaherty, MD, of Massachusetts General Hospital in Boston.
Dr. Flaherty asked the panelists to explain adjustments their organizations have made in response to the pandemic, discuss accomplishments, and speculate on future challenges and priorities.
Trial, administrative, and patient-care modifications
COVID-19 put some cancer trials on hold. For others, the pandemic forced sponsors and study chairs to reduce trial complexity and identify nonessential aspects of the studies, according to panelist José Baselga, MD, PhD, of AstraZeneca.
Specifically, exploratory objectives were subjugated to patient safety and a focus on the primary endpoints of each trial.
Once the critical data were identified, study chairs were asked to determine whether data could be obtained through technologies that could substitute for face-to-face contact between patients and staff – for example, patient-reported outcome tools and at-home digital monitoring.
Modifications prompted by the pandemic include the following:
- On-site auditing was suspended.
- Oral investigational agents were shipped directly to patients.
- “Remote” informed consent (telephone or video consenting) was permitted.
- Local providers could perform study-related services, with oversight by the research site.
- Minor deviations from the written protocols were allowed, provided the deviations did not affect patient care or data integrity.
“Obviously, the pandemic has been horrible, but what it has allowed us to do, as investigators in the clinical research landscape, … is to change our focus somewhat and realize, first and foremost, the patient is at the center of this,” Dr. LoRusso said.
Operational accomplishments and benefits
The pandemic caused a 40% decline in accrual to studies supported by the National Cancer Institute’s (NCI) Clinical Trials Network (NCTN) from mid-March to early April, according to James H. Doroshow, MD, of NCI.
However, after modifications to administrative and regulatory procedures, accrual to NCTN trials recovered to approximately 80% of prepandemic levels, Dr. Doroshow said.
The pandemic prompted investigators to leverage tools and technology they had not previously used frequently or at all, the panelists pointed out.
Investigators discovered perforce that telehealth could be used for almost all trial-related assessments. In lieu of physical examination, patients could send pictures of rashes and use electronic devices to monitor blood sugar values and vital signs.
Digital radiographic studies were performed at sites that were most convenient for patients, downloaded, and reinterpreted at the study institution. Visiting nurses and neighborhood laboratories enabled less-frequent in-person visits for assessments.
These adjustments have been particularly important for geographically and/or socioeconomically disadvantaged patients, the panelists said.
Overall, there was agreement among the panelists that shared values and trust among regulatory authorities, sponsors, investigators, and clinicians were impressive in their urgency, sincerity, and patient centricity.
“This pandemic … has forced us to think differently and be nimble and creative to our approach to maintaining our overriding goals while at the same time bringing these innovative therapies forward for patients with cancer and other serious and life-threatening diseases as quickly as possible,” said panelist Kristen M. Hege, MD, of Bristol-Myers Squibb.
In fact, Dr. Hege noted, some cancer-related therapies (e.g., BTK inhibitors, JAK inhibitors, and immunomodulatory agents) were “repurposed” rapidly and tested against COVID-related complications.
Streamlining trial regulatory processes
In addition to changing ongoing trials, the pandemic has affected how new research projects are launched.
One new study that came together quickly in response to the pandemic is the NCI COVID-19 in Cancer Patients Study (NCCAPS). NCCAPS is a natural history study with biospecimens and an imaging library. It was approved in just 5 weeks and is active in 650 sites, with “gangbusters” accrual, Dr. Doroshow said.
The rapidness of NCCAPS’ design and implementation should prompt the revision of previously accepted timelines for trial activation and lead to streamlined future processes.
Another project that was launched quickly in response to the pandemic is the COVID-19 evidence accelerator, according to Paul G. Kluetz, MD, of the Food and Drug Administration.
The COVID-19 evidence accelerator integrates real-world evidence into a database to provide investigators and health systems with the ability to gather information, design rapid turnaround queries, and share results. The evidence accelerator can provide study chairs with information that may have relevance to the safety of participants in clinical trials.
Future directions and challenges
The panelists agreed that pandemic-related modifications in processes will not only accelerate trial approval and activation but should facilitate higher study accrual, increase the diversity of protocol participants, and decrease the costs associated with clinical trial conduct.
With that in mind, the NCI is planning randomized clinical trials in which “process A” is compared with “process B,” Dr. Doroshow said. The goal is to determine which modifications are most likely to make trials available to patients without compromising data integrity or patient safety.
“How much less data do you need to have an outcome that will be similar?” Dr. Doroshow asked. “How many fewer visits, how many fewer tests, how much can you save? Physicians, clinical trialists, all of us respond to data, and if you get the same outcome at a third of the cost, then everybody benefits.”
Nonetheless, we will need to be vigilant for unintended vulnerabilities from well-intended efforts, according to Dr. Kluetz. Study chairs, sponsors, and regulatory agencies will need to be attentive to whether there are important differences in scan quality or interpretation, missing data that influence trial outcomes, and so on.
Dr. Hege pointed out that differences among data sources may be less important when treatments generate large effects but may be vitally important when the relative differences among treatments are small.
On a practical level, decentralizing clinical research may negatively impact the finances of tertiary care centers, which could threaten the required infrastructure for clinical trials, a few panelists noted.
The relative balance of NCI-, industry-, and investigator-initiated trials may require adjustment so that research income is adequate to maintain the costs associated with cancer clinical trials.
Shared goals and democratization
The pandemic has required all stakeholders in clinical research to rely on relationships of trust and shared goals, said Caroline Robert, MD, PhD, of Institut Gustave Roussy in Villejuif, France.
Dr. Kluetz summarized those goals as improving trial efficiencies, decreasing patient burden, decentralizing trials, and maintaining trial integrity.
A decentralized clinical trials operational model could lead to better generalizability of study outcomes, normalization of life for patients on studies, and lower costs of trial conduct. As such, decentralization would promote democratization.
Coupled with ongoing efforts to reduce eligibility criteria in cancer trials, the pandemic has brought operational solutions that should be perpetuated and has reminded us of the interlocking and mutually supportive relationships on which clinical research success depends.
Dr. Doroshow and Dr. Kluetz disclosed no conflicts of interest. All other panelists disclosed financial relationships, including employment, with a range of companies.
Dr. Lyss was a community-based medical oncologist and clinical researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers, as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
SOURCE: Flaherty KT et al. AACR: COVID-19 and Cancer, Regulatory and Operational Implications of Cancer Clinical Trial Changes During COVID-19.
FROM AACR: COVID-19 and Cancer
Dystrophic Calcinosis Cutis: Treatment With Intravenous Sodium Thiosulfate
To the Editor:
Severe dystrophic calcinosis cutis is a debilitating disease with no universally accepted therapeutic options. This case demonstrates the benefit of intravenous (IV) sodium thiosulfate in alleviating the calcified lesions as well as the associated pain and disability. This application of IV sodium thiosulfate with a favorable outcome is new and should be considered for the treatment of generalized dystrophic calcinosis cutis, especially when topical, procedural, or surgical options are not feasible.
A 54-year-old woman with a history of well-controlled dermatomyositis and systemic lupus erythematosus presented with diffuse, hard, calcified lesions on the legs, arms, clavicular region, and neck that had slowly progressed over at least a 10-year period (Figure 1). The lesions were consistent with dystrophic calcinosis cutis. The patient was started on 12.5 g of IV sodium thiosulfate 3 times weekly infused over 30 minutes. Drastic diminution of the cutaneous calcification was observed at 3-month follow-up (Figure 2). She reported decreased pain and burning as well as increased overall functionality and improved sleep. The patient completed 8 months of therapy, but the treatment was stopped secondary to suspicion of a lupuslike flare, and the lesions recurred with more widespread involvement, including the trunk, tendons, bony prominences, and supraclavicular soft tissue. The patient reported burning pain and pruritus that resulted in impairment of daily activities such as getting dressed. Sodium thiosulfate was restarted once weekly, which again resulted in reduction of the dystrophic calcinosis cutis.
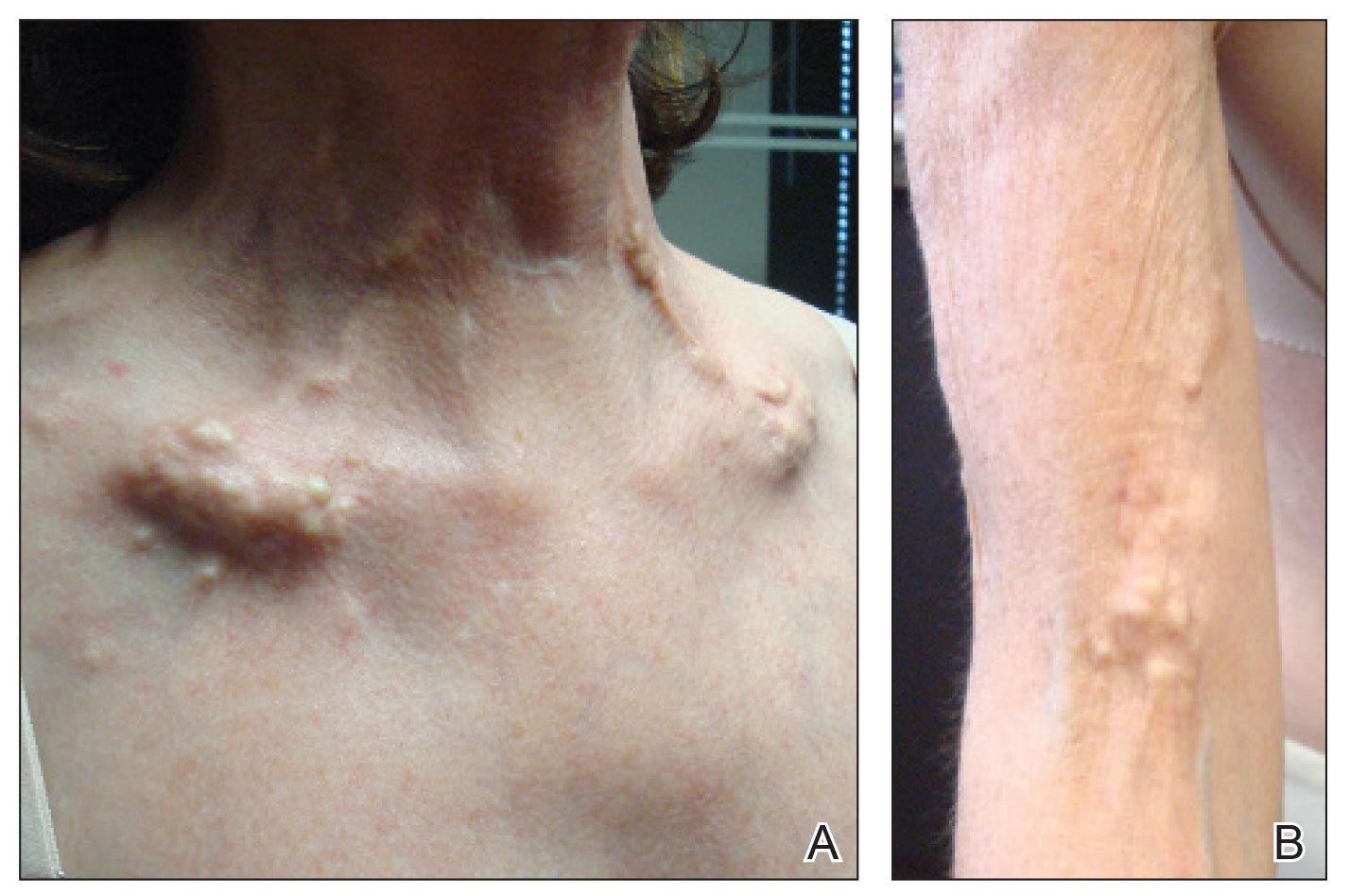
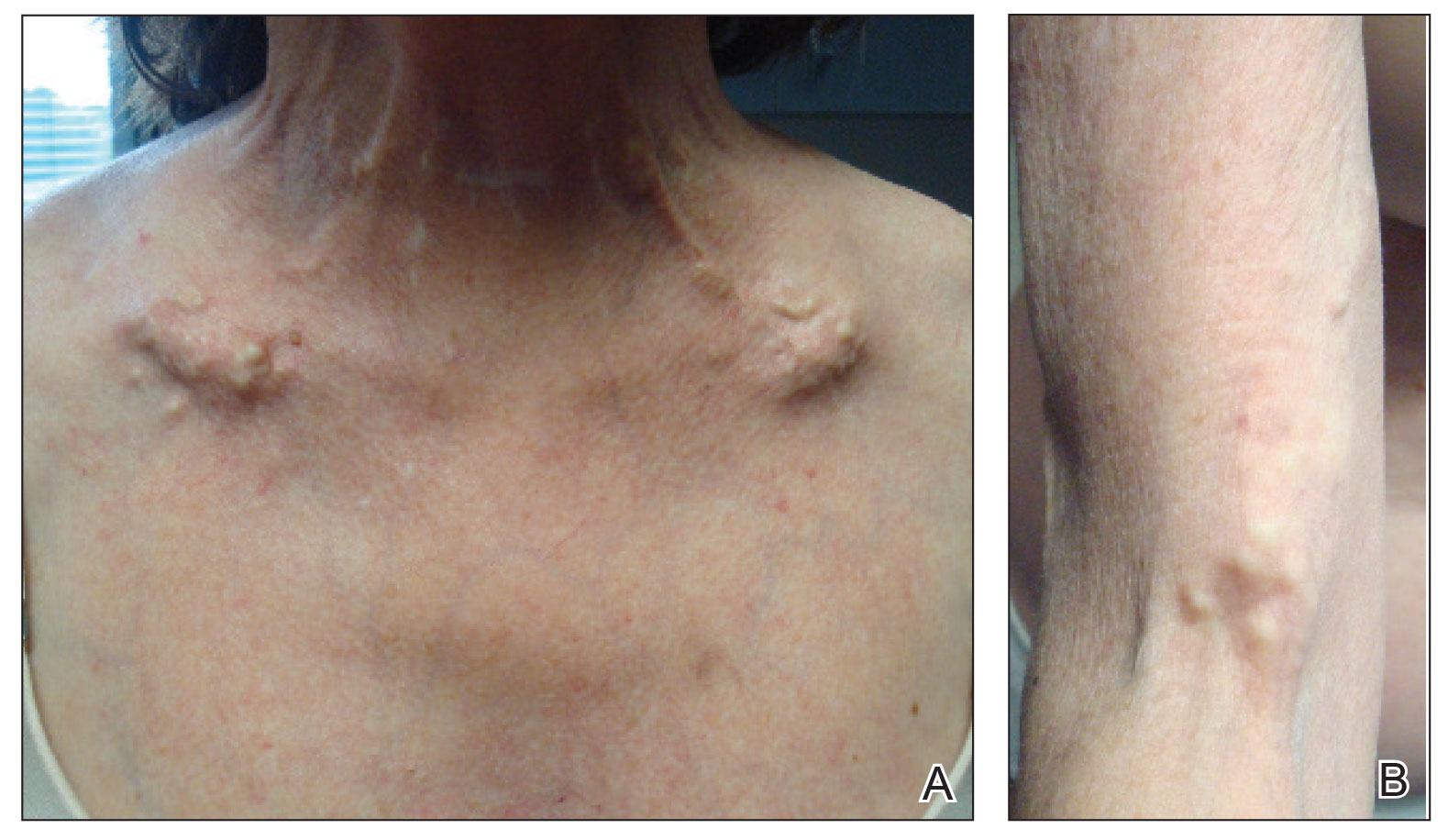
Dystrophic calcinosis cutis is a debilitating disease that results in considerable morbidity and pain with major implications on quality of life. The pathophysiology is unclear; calcium and phosphate serum levels generally are normal. A proposed mechanism is that chronic inflammation causes tissue damage and defective collagen synthesis, resulting in a distorted architecture that facilitates calcium deposition in the skin and subcutaneous tissues.1 Dystrophic calcinosis cutis most commonly is associated with systemic sclerosis and dermatomyositis but also can be seen in systemic lupus erythematosus, panniculitis, and other connective tissue diseases. It also can occur with skin neoplasms, collagen and elastin disorders, porphyria cutanea tarda, and pancreatic panniculitis.1 Progression of dystrophic calcinosis cutis usually is independent of the associated disease status.
Treatment is based on anecdotal evidence from case reports, as there is no universally accepted pharmacologic or procedural intervention available for dystrophic calcinosis cutis. Medications that have been reported to be helpful to varying degrees include diltiazem, colchicine, minocycline, IV immunoglobulin, ceftriaxone, aluminum hydroxide, probenecid, alendronic acid, etidronate disodium, warfarin, intralesional corticosteroids, and sodium thiosulfate. Procedural interventions also have been reported, such as surgical excision, extracorporeal shock wave lithotripsy, and CO2 and erbium: YAG lasers.1 Surgical excision of dystrophic calcinosis cutis is widely implemented but outcomes are poor. Moreover, in patients with widely diffuse calcinosis, targeted procedural therapy is impractical.
Intravenous sodium thiosulfate has been widely used for the treatment of calciphylaxis secondary to end-stage renal failure and tumoral calcinosis.2 It also has been reported to be effective in iatrogenic calcinosis cutis secondary to extravasation of calcium-containing solutions in a patient with T-cell acute lymphoblastic leukemia.3 However, reports of its use in treating dystrophic calcinosis cutis are limited. Intravenous sodium thiosulfate—10 g 3 times weekly for 2 weeks, followed by 15 g twice weekly for the next 3 months—was used with abatacept for treatment of dystrophic calcinosis cutis in a patient with juvenile dermatomyositis.4 Other formulations of sodium thiosulfate have been reported to result in clearance of calcified lesions, including a topical application compounded in zinc oxide5 and intradermal injection at the base of a nodule.6 We used 12.5 g over 30 minutes 3 times weekly; however, the dose can be increased to 25 g over 60 minutes if 3 to 4 treatments are tolerated, with nausea being the only notable side effect. Its mechanism of action in treating dystrophic calcinosis cutis is unclear, but it likely is due to its ability to chelate and dissolve calcium deposits. Topical and intradermal therapy is impractical for widespread, dystrophic calcinosis cutis as in our patient.
Our case highlights the successful use of IV sodium thiosulfate as a stand-alone treatment modality for generalized dystrophic calcinosis cutis in an adult patient. Both our patient and a child in a previously reported case who received the same treatment4 had dermatomyositis, but we suspect IV sodium thiosulfate also may be effective for dystrophic calcinosis cutis associated with other diseases. Sodium thiosulfate should be considered as a treatment for patients who experience tremendous pain and disability. It is safe, inexpensive, and easy to administer and is especially helpful in patients for whom topical, intradermal, or procedural therapy is not possible.
- Gutierrez A Jr, Wetter DA. Calcinosis cutis in autoimmune connective tissue diseases. Dermatol Ther. 2012;25:195-206.
- Mageau A, Guigonis V, Ratzimbasafy V, et al. Intravenous sodium thiosulfate for treating tumoral calcinosis associated with systemic disorders: report of four cases. Joint Bone Spine. 2017;84:341-344.
Raffaella C, Annapaola C, Tullio I, et al. Successful treatment of severe iatrogenic calcinosis cutis with intravenous sodium thiosulfate in a child affected by T-acute lymphoblastic leukemia. Pediatr Dermatol. 2009;26:311-315. - Arabshahi B, Silverman RA, Jones OY, et al. Abatacept and sodium thiosulfate for treatment of recalcitrant juvenile dermatomyositis complicated by ulceration and calcinosis. J Pediatrics. 2012;160:520-522.
- Bair B, Fivenson D. A novel treatment for ulcerative calcinosis cutis. J Drugs Dermatol. 2011;10:1042-1044.
- Smith GP. Intradermal sodium thiosulfate for exophytic calcinosis cutis of connective tissue disease. J Am Acad Dermatol. 2013;69:E146-E147.
To the Editor:
Severe dystrophic calcinosis cutis is a debilitating disease with no universally accepted therapeutic options. This case demonstrates the benefit of intravenous (IV) sodium thiosulfate in alleviating the calcified lesions as well as the associated pain and disability. This application of IV sodium thiosulfate with a favorable outcome is new and should be considered for the treatment of generalized dystrophic calcinosis cutis, especially when topical, procedural, or surgical options are not feasible.
A 54-year-old woman with a history of well-controlled dermatomyositis and systemic lupus erythematosus presented with diffuse, hard, calcified lesions on the legs, arms, clavicular region, and neck that had slowly progressed over at least a 10-year period (Figure 1). The lesions were consistent with dystrophic calcinosis cutis. The patient was started on 12.5 g of IV sodium thiosulfate 3 times weekly infused over 30 minutes. Drastic diminution of the cutaneous calcification was observed at 3-month follow-up (Figure 2). She reported decreased pain and burning as well as increased overall functionality and improved sleep. The patient completed 8 months of therapy, but the treatment was stopped secondary to suspicion of a lupuslike flare, and the lesions recurred with more widespread involvement, including the trunk, tendons, bony prominences, and supraclavicular soft tissue. The patient reported burning pain and pruritus that resulted in impairment of daily activities such as getting dressed. Sodium thiosulfate was restarted once weekly, which again resulted in reduction of the dystrophic calcinosis cutis.
Dystrophic calcinosis cutis is a debilitating disease that results in considerable morbidity and pain with major implications on quality of life. The pathophysiology is unclear; calcium and phosphate serum levels generally are normal. A proposed mechanism is that chronic inflammation causes tissue damage and defective collagen synthesis, resulting in a distorted architecture that facilitates calcium deposition in the skin and subcutaneous tissues.1 Dystrophic calcinosis cutis most commonly is associated with systemic sclerosis and dermatomyositis but also can be seen in systemic lupus erythematosus, panniculitis, and other connective tissue diseases. It also can occur with skin neoplasms, collagen and elastin disorders, porphyria cutanea tarda, and pancreatic panniculitis.1 Progression of dystrophic calcinosis cutis usually is independent of the associated disease status.
Treatment is based on anecdotal evidence from case reports, as there is no universally accepted pharmacologic or procedural intervention available for dystrophic calcinosis cutis. Medications that have been reported to be helpful to varying degrees include diltiazem, colchicine, minocycline, IV immunoglobulin, ceftriaxone, aluminum hydroxide, probenecid, alendronic acid, etidronate disodium, warfarin, intralesional corticosteroids, and sodium thiosulfate. Procedural interventions also have been reported, such as surgical excision, extracorporeal shock wave lithotripsy, and CO2 and erbium: YAG lasers.1 Surgical excision of dystrophic calcinosis cutis is widely implemented but outcomes are poor. Moreover, in patients with widely diffuse calcinosis, targeted procedural therapy is impractical.
Intravenous sodium thiosulfate has been widely used for the treatment of calciphylaxis secondary to end-stage renal failure and tumoral calcinosis.2 It also has been reported to be effective in iatrogenic calcinosis cutis secondary to extravasation of calcium-containing solutions in a patient with T-cell acute lymphoblastic leukemia.3 However, reports of its use in treating dystrophic calcinosis cutis are limited. Intravenous sodium thiosulfate—10 g 3 times weekly for 2 weeks, followed by 15 g twice weekly for the next 3 months—was used with abatacept for treatment of dystrophic calcinosis cutis in a patient with juvenile dermatomyositis.4 Other formulations of sodium thiosulfate have been reported to result in clearance of calcified lesions, including a topical application compounded in zinc oxide5 and intradermal injection at the base of a nodule.6 We used 12.5 g over 30 minutes 3 times weekly; however, the dose can be increased to 25 g over 60 minutes if 3 to 4 treatments are tolerated, with nausea being the only notable side effect. Its mechanism of action in treating dystrophic calcinosis cutis is unclear, but it likely is due to its ability to chelate and dissolve calcium deposits. Topical and intradermal therapy is impractical for widespread, dystrophic calcinosis cutis as in our patient.
Our case highlights the successful use of IV sodium thiosulfate as a stand-alone treatment modality for generalized dystrophic calcinosis cutis in an adult patient. Both our patient and a child in a previously reported case who received the same treatment4 had dermatomyositis, but we suspect IV sodium thiosulfate also may be effective for dystrophic calcinosis cutis associated with other diseases. Sodium thiosulfate should be considered as a treatment for patients who experience tremendous pain and disability. It is safe, inexpensive, and easy to administer and is especially helpful in patients for whom topical, intradermal, or procedural therapy is not possible.
To the Editor:
Severe dystrophic calcinosis cutis is a debilitating disease with no universally accepted therapeutic options. This case demonstrates the benefit of intravenous (IV) sodium thiosulfate in alleviating the calcified lesions as well as the associated pain and disability. This application of IV sodium thiosulfate with a favorable outcome is new and should be considered for the treatment of generalized dystrophic calcinosis cutis, especially when topical, procedural, or surgical options are not feasible.
A 54-year-old woman with a history of well-controlled dermatomyositis and systemic lupus erythematosus presented with diffuse, hard, calcified lesions on the legs, arms, clavicular region, and neck that had slowly progressed over at least a 10-year period (Figure 1). The lesions were consistent with dystrophic calcinosis cutis. The patient was started on 12.5 g of IV sodium thiosulfate 3 times weekly infused over 30 minutes. Drastic diminution of the cutaneous calcification was observed at 3-month follow-up (Figure 2). She reported decreased pain and burning as well as increased overall functionality and improved sleep. The patient completed 8 months of therapy, but the treatment was stopped secondary to suspicion of a lupuslike flare, and the lesions recurred with more widespread involvement, including the trunk, tendons, bony prominences, and supraclavicular soft tissue. The patient reported burning pain and pruritus that resulted in impairment of daily activities such as getting dressed. Sodium thiosulfate was restarted once weekly, which again resulted in reduction of the dystrophic calcinosis cutis.
Dystrophic calcinosis cutis is a debilitating disease that results in considerable morbidity and pain with major implications on quality of life. The pathophysiology is unclear; calcium and phosphate serum levels generally are normal. A proposed mechanism is that chronic inflammation causes tissue damage and defective collagen synthesis, resulting in a distorted architecture that facilitates calcium deposition in the skin and subcutaneous tissues.1 Dystrophic calcinosis cutis most commonly is associated with systemic sclerosis and dermatomyositis but also can be seen in systemic lupus erythematosus, panniculitis, and other connective tissue diseases. It also can occur with skin neoplasms, collagen and elastin disorders, porphyria cutanea tarda, and pancreatic panniculitis.1 Progression of dystrophic calcinosis cutis usually is independent of the associated disease status.
Treatment is based on anecdotal evidence from case reports, as there is no universally accepted pharmacologic or procedural intervention available for dystrophic calcinosis cutis. Medications that have been reported to be helpful to varying degrees include diltiazem, colchicine, minocycline, IV immunoglobulin, ceftriaxone, aluminum hydroxide, probenecid, alendronic acid, etidronate disodium, warfarin, intralesional corticosteroids, and sodium thiosulfate. Procedural interventions also have been reported, such as surgical excision, extracorporeal shock wave lithotripsy, and CO2 and erbium: YAG lasers.1 Surgical excision of dystrophic calcinosis cutis is widely implemented but outcomes are poor. Moreover, in patients with widely diffuse calcinosis, targeted procedural therapy is impractical.
Intravenous sodium thiosulfate has been widely used for the treatment of calciphylaxis secondary to end-stage renal failure and tumoral calcinosis.2 It also has been reported to be effective in iatrogenic calcinosis cutis secondary to extravasation of calcium-containing solutions in a patient with T-cell acute lymphoblastic leukemia.3 However, reports of its use in treating dystrophic calcinosis cutis are limited. Intravenous sodium thiosulfate—10 g 3 times weekly for 2 weeks, followed by 15 g twice weekly for the next 3 months—was used with abatacept for treatment of dystrophic calcinosis cutis in a patient with juvenile dermatomyositis.4 Other formulations of sodium thiosulfate have been reported to result in clearance of calcified lesions, including a topical application compounded in zinc oxide5 and intradermal injection at the base of a nodule.6 We used 12.5 g over 30 minutes 3 times weekly; however, the dose can be increased to 25 g over 60 minutes if 3 to 4 treatments are tolerated, with nausea being the only notable side effect. Its mechanism of action in treating dystrophic calcinosis cutis is unclear, but it likely is due to its ability to chelate and dissolve calcium deposits. Topical and intradermal therapy is impractical for widespread, dystrophic calcinosis cutis as in our patient.
Our case highlights the successful use of IV sodium thiosulfate as a stand-alone treatment modality for generalized dystrophic calcinosis cutis in an adult patient. Both our patient and a child in a previously reported case who received the same treatment4 had dermatomyositis, but we suspect IV sodium thiosulfate also may be effective for dystrophic calcinosis cutis associated with other diseases. Sodium thiosulfate should be considered as a treatment for patients who experience tremendous pain and disability. It is safe, inexpensive, and easy to administer and is especially helpful in patients for whom topical, intradermal, or procedural therapy is not possible.
- Gutierrez A Jr, Wetter DA. Calcinosis cutis in autoimmune connective tissue diseases. Dermatol Ther. 2012;25:195-206.
- Mageau A, Guigonis V, Ratzimbasafy V, et al. Intravenous sodium thiosulfate for treating tumoral calcinosis associated with systemic disorders: report of four cases. Joint Bone Spine. 2017;84:341-344.
Raffaella C, Annapaola C, Tullio I, et al. Successful treatment of severe iatrogenic calcinosis cutis with intravenous sodium thiosulfate in a child affected by T-acute lymphoblastic leukemia. Pediatr Dermatol. 2009;26:311-315. - Arabshahi B, Silverman RA, Jones OY, et al. Abatacept and sodium thiosulfate for treatment of recalcitrant juvenile dermatomyositis complicated by ulceration and calcinosis. J Pediatrics. 2012;160:520-522.
- Bair B, Fivenson D. A novel treatment for ulcerative calcinosis cutis. J Drugs Dermatol. 2011;10:1042-1044.
- Smith GP. Intradermal sodium thiosulfate for exophytic calcinosis cutis of connective tissue disease. J Am Acad Dermatol. 2013;69:E146-E147.
- Gutierrez A Jr, Wetter DA. Calcinosis cutis in autoimmune connective tissue diseases. Dermatol Ther. 2012;25:195-206.
- Mageau A, Guigonis V, Ratzimbasafy V, et al. Intravenous sodium thiosulfate for treating tumoral calcinosis associated with systemic disorders: report of four cases. Joint Bone Spine. 2017;84:341-344.
Raffaella C, Annapaola C, Tullio I, et al. Successful treatment of severe iatrogenic calcinosis cutis with intravenous sodium thiosulfate in a child affected by T-acute lymphoblastic leukemia. Pediatr Dermatol. 2009;26:311-315. - Arabshahi B, Silverman RA, Jones OY, et al. Abatacept and sodium thiosulfate for treatment of recalcitrant juvenile dermatomyositis complicated by ulceration and calcinosis. J Pediatrics. 2012;160:520-522.
- Bair B, Fivenson D. A novel treatment for ulcerative calcinosis cutis. J Drugs Dermatol. 2011;10:1042-1044.
- Smith GP. Intradermal sodium thiosulfate for exophytic calcinosis cutis of connective tissue disease. J Am Acad Dermatol. 2013;69:E146-E147.
Practice Points
- Dystrophic calcinosis cutis is a potentially debilitating condition with limited effective therapies.
- Consider intravenous sodium thiosulfate in patients with diffuse and severe dystrophic calcinosis cutis.
Management of Classic Ulcerative Pyoderma Gangrenosum
Pyoderma gangrenosum (PG) is a rare, chronic, ulcerative, neutrophilic dermatosis of unclear etiology. Large, multicentered, randomized controlled trials (RCTs) are challenging due to the rarity of PG and the lack of a diagnostic confirmatory test; therefore, evidence-based guidelines for diagnosis and treatment are not well established. Current management of PG primarily is guided by case series, small clinical trials, and expert opinion.1-4 We conducted a survey of expert medical dermatologists to highlight best practices in diagnostic and therapeutic approaches to PG.
Methods
The Society of Dermatology Hospitalists (SDH) Scientific Task Force gathered expert opinions from members of the SDH and Rheumatologic Dermatology Society (RDS) regarding PG workup and treatment through an online survey of 15 items (eTable 1). Subscribers of the SDH and RDS LISTSERVs were invited via email to participate in the survey from January 2016 to February 2016. Anonymous survey responses were collected and collated using SurveyMonkey. The survey results identified expert recommendations for evaluation, diagnosis, and treatment of PG and are reported as the sum of the percentage of respondents who answered always (almost 100% of the time) or often (more than half the time) following a particular course of action. A subanalysis was performed defining 2 groups of respondents based on the number of cases of PG treated per year (≥10 vs <10). Survey responses between each group were compared using χ2 analysis with statistical significance set at P=.05.

Results
Fifty-one respondents completed the survey out of 140 surveyed (36% response rate). All respondents were dermatologists, and 96% (49/51) were affiliated with an academic institution. Among the respondents, the number of PG cases managed per year ranged from 2 to 35.
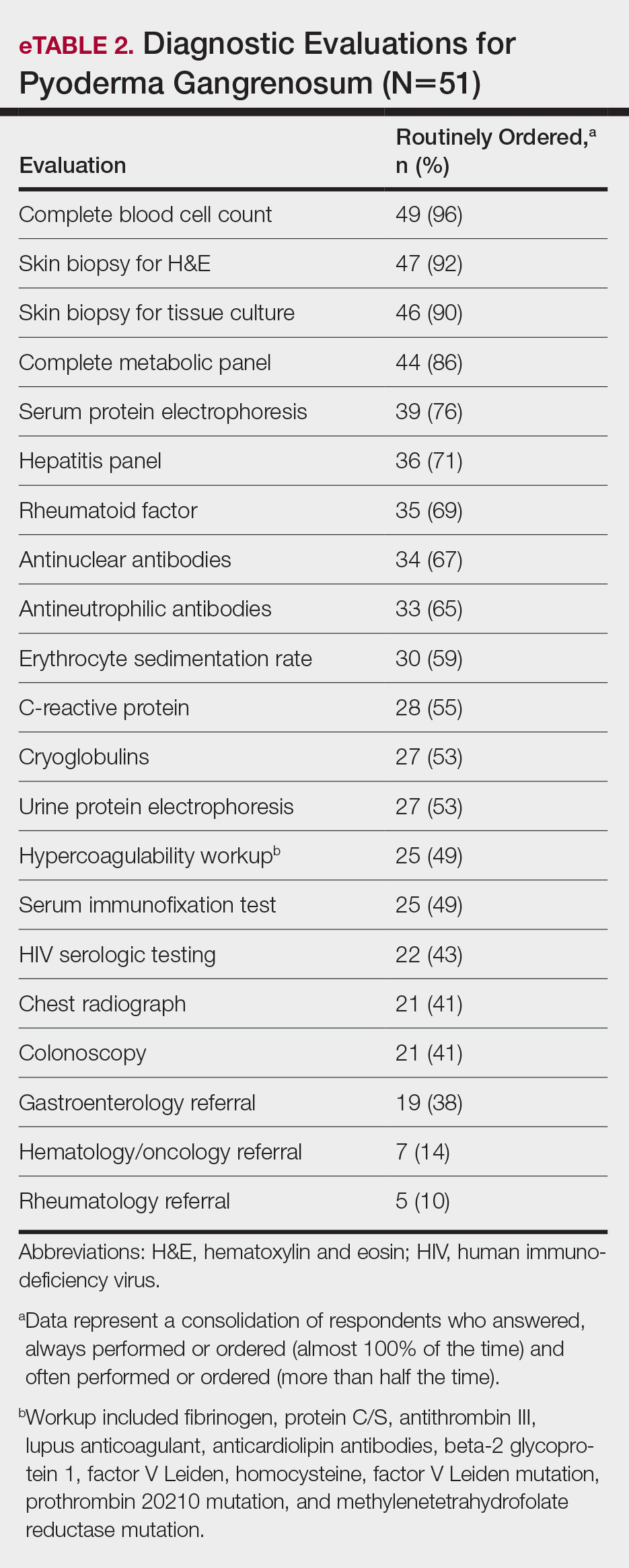
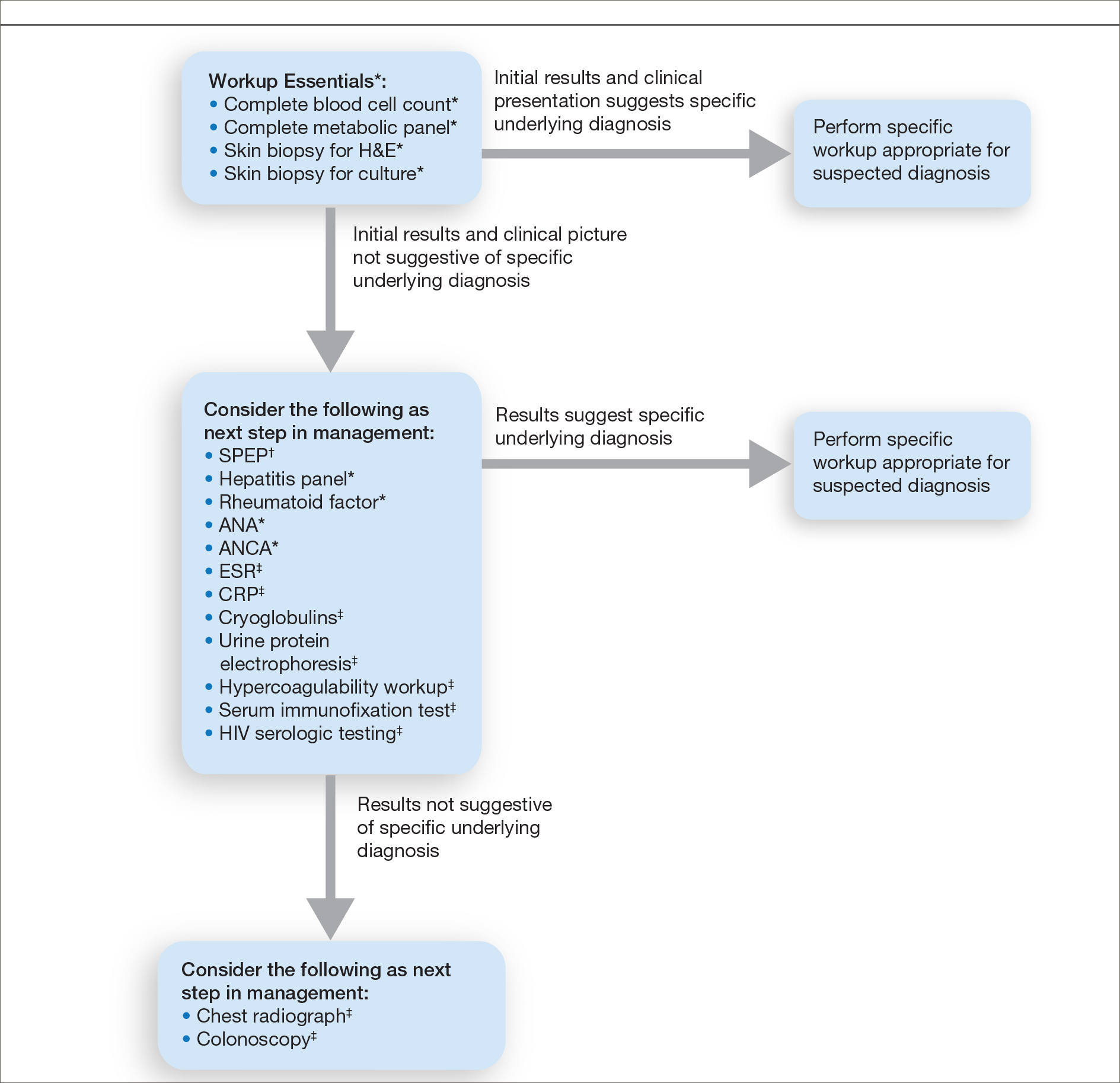
Respondents consistently ordered skin biopsies (92% [47/51]) and tissue cultures (90% [46/51]), as well as certain ancillary tests, including complete blood cell count (96% [49/51]), complete metabolic panel (86% [44/51]), serum protein electrophoresis (76% [39/51]), and hepatitis panel (71% [36/51]). Other frequently ordered studies were rheumatoid factor (69% [35/51]), antinuclear antibodies (67% [34/51]), and antineutrophilic antibodies (65% [33/51]). Respondents frequently ordered erythrocyte sedimentation rate (59% [30/51]), C-reactive protein (55% [28/51]), cryoglobulins (53% [27/51]), urine protein electrophoresis (53% [27/51]), hypercoagulability workup (49% [25/51]), and serum immunofixation test (49% [25/51]). Human immunodeficiency virus testing (43% [22/51]), chest radiograph (41% [21/51]), colonoscopy (41% [21/51]) and referral to other specialties for workup—gastroenterology (38% [19/51]), hematology/oncology (14% [7/51]), and rheumatology (10% [5/51])—were less frequently ordered (eTable 2).
Systemic corticosteroids were reported as first-line therapy by most respondents (94% [48/51]), followed by topical immunomodulatory therapies (63% [32/51]). Topical corticosteroids (75% [38/51]) were the most common first-line topical agents. Thirty-nine percent of respondents (20/51) prescribed topical calcineurin inhibitors as first-line topical therapy. Additional therapies frequently used included systemic cyclosporine (47% [24/51]), antineutrophilic agents (41% [21/51]), and biologic agents (37% [19/51]). Fifty-seven percent of respondents (29/51) supported using combination topical and systemic therapy (Table).
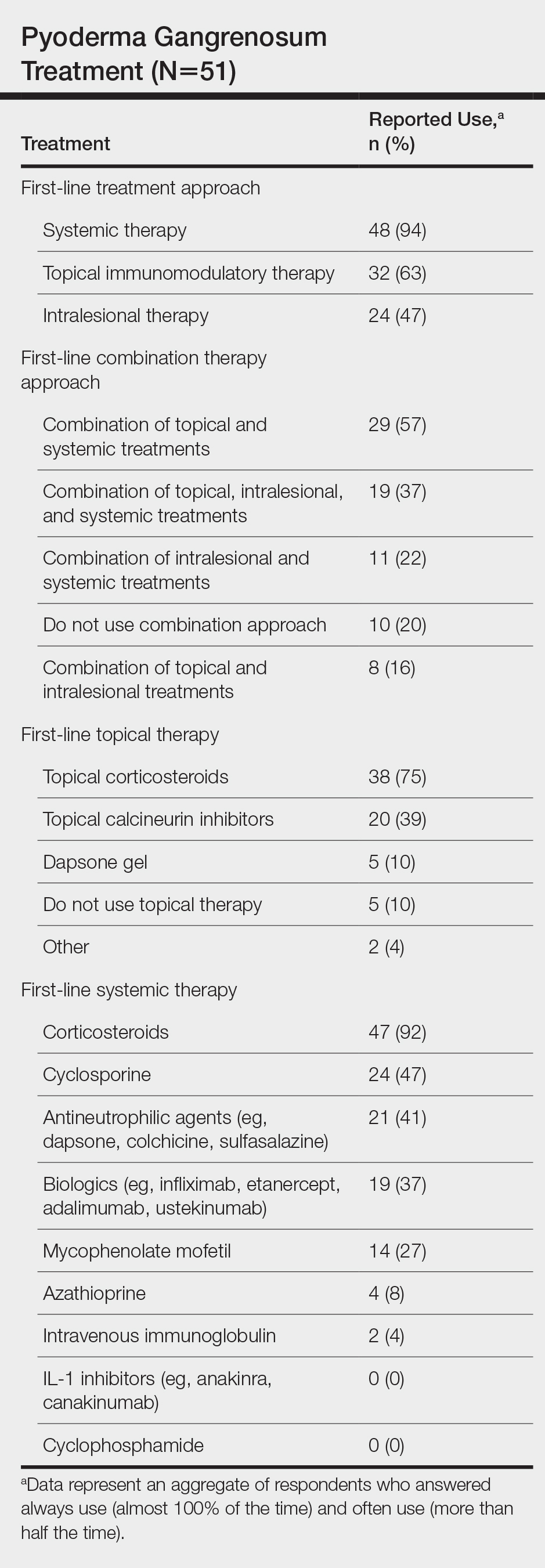
A wide variety of wound care practices were reported in the management of PG. Seventy-six percent of respondents (39/51) favored petroleum-impregnated gauze, 69% (35/51) used nonadhesive dressings, and 43% (22/51) added antimicrobial therapy for PG wound care (eTable 3). In the subanalysis, there were no significant differences in the majority of answer responses in patients treating 10 or more PG cases per year vs fewer than 10 PG cases, except with regard to the practice of combination therapy. Those treating more than 10 cases of PG per year more frequently reported use of combination therapies compared to respondents treating fewer than 10 cases (P=.04).
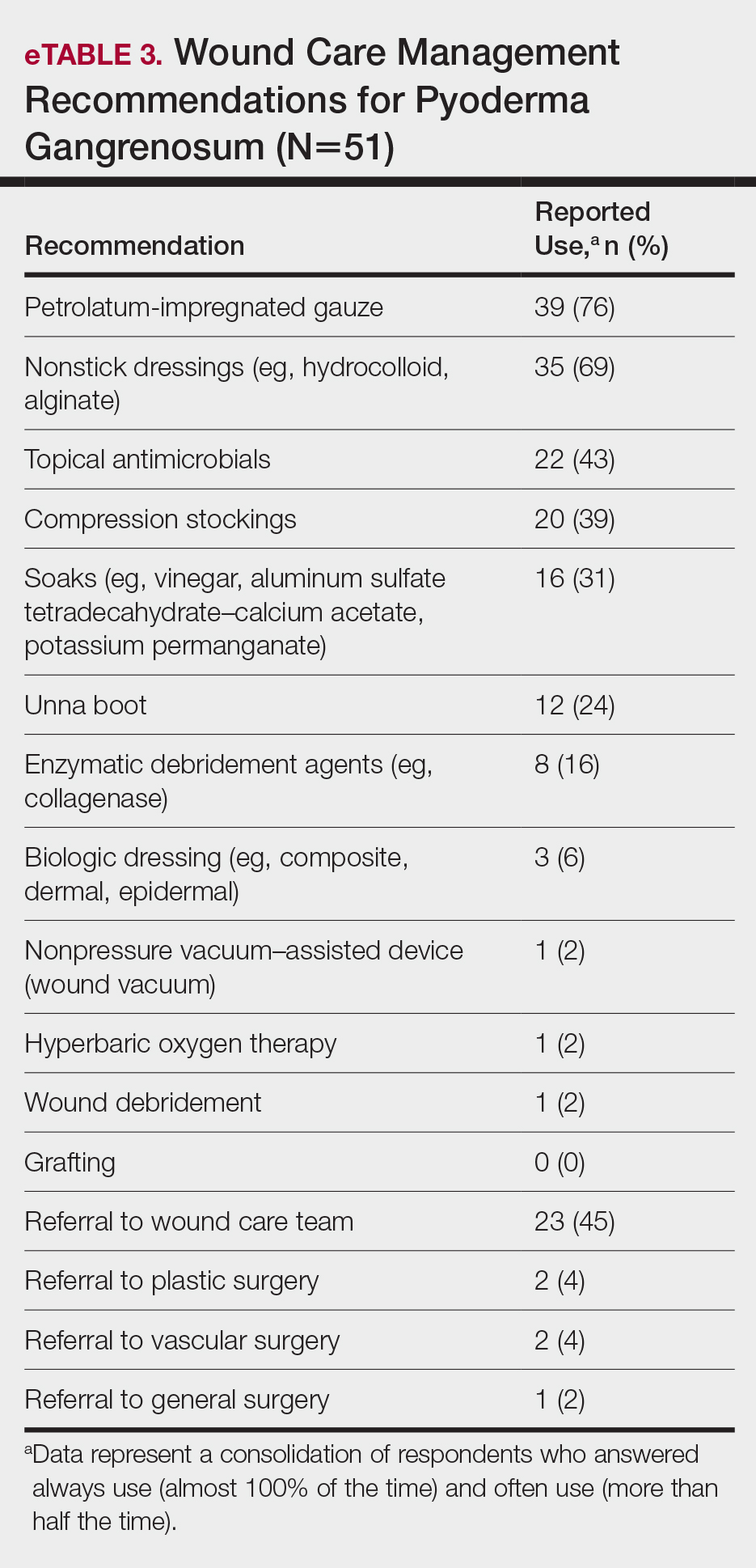
Comment
Skin biopsies and tissue cultures were strongly recommended (>90% survey respondents) for the initial evaluation of lesions suspected to be PG to evaluate for typical histopathologic changes that appear early in the disease, to rule out PG mimickers such as infectious or vascular causes, and to prevent the detrimental effects of inappropriate treatment and delayed diagnosis.5
Suspected PG warrants a reasonable search for related conditions because more than 50% of PG cases are associated with comorbidities such as rheumatoid arthritis, inflammatory bowel disease, and hematologic disease/malignancy.6,7 A complete blood cell count and comprehensive metabolic panel were recommended by most respondents, aiding in the preliminary screening for hematologic and infectious causes as well as detecting liver and kidney dysfunction associated with systemic conditions. Additionally, exclusion of infection or malignancy may be particularly important if the patient will undergo systemic immunosuppression. In challenging PG cases when initial findings are inconclusive and the clinical presentation does not direct workup (eg, colonoscopy to evaluate gastrointestinal tract symptoms), serum protein electrophoresis, hepatitis panel, rheumatoid factor, antinuclear antibodies, and antineutrophilic antibody tests also were frequently ordered by respondents to further evaluate for underlying or associated conditions.
This consensus regarding skin biopsies and certain ancillary tests is consistent with the proposed diagnostic criteria for classic ulcerative PG in which the absence or exclusion of other relevant causes of cutaneous ulcers is required based on the criteria.8 The importance of ensuring an accurate diagnosis is paramount, as a 10% misdiagnosis rate has been documented in the literature.5
Importantly, a stepwise diagnostic workup for PG is proposed based on survey results, which may limit unnecessary testing and the associated costs to the health care system (Figure 1). Selection of additional testing is guided by initial test results and features of the patient’s clinical presentation, including age, review of systems, and associated comorbidities. Available data suggest that underlying inflammatory bowel disease is more frequent in PG patients who are younger than 65 years, whereas those who are 65 years and older are more likely to have inflammatory arthritis, cancer, or an underlying hematologic disorder.9
Treatment of PG should address both the inflammatory and wound components of the disease (Figure 2).7 In our survey results, systemic corticosteroids were identified as an important first-line therapy supported by reasonable evidence and were favored for their rapid response and minimal cost.1,10,11 Many respondents endorsed the use of systemic therapy in combination with topical steroids or calcineurin inhibitors. Combination therapy may provide more immediate control of rapidly progressing disease while minimizing adverse effects of long-term systemic corticosteroid use. A survey of German wound experts similarly endorsed frequent use of topical calcineurin inhibitors and combination systemic and topical glucocorticoid therapy as common therapeutic approaches.1
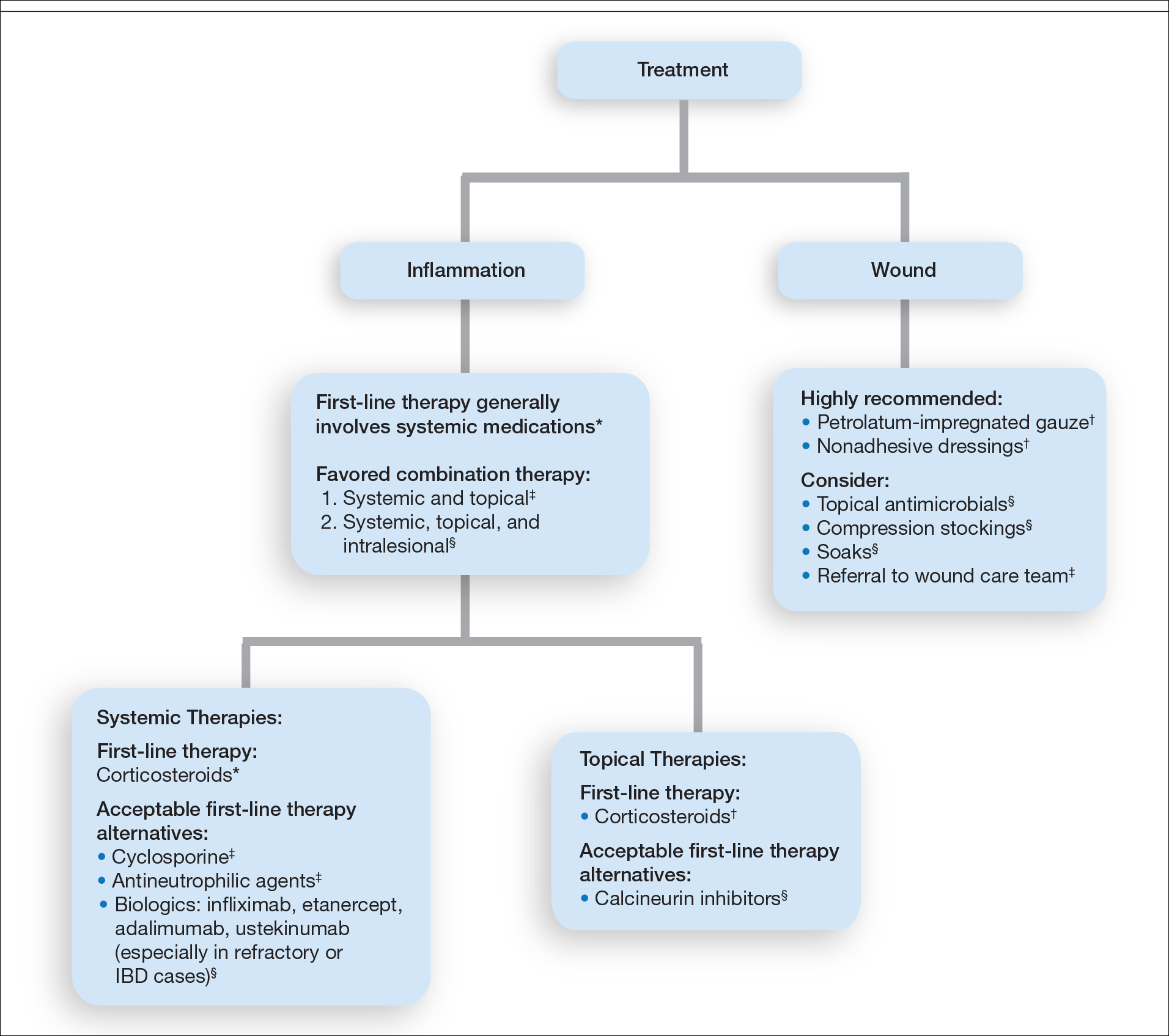
Importantly, treatments may vary depending on patient characteristics, comorbidities, and underlying disease, which underscores the need for individualized treatment approaches. Alternative first-line systemic treatments favored by respondents were cyclosporine, biologic medications, and antineutrophilic agents such as dapsone. Cyclosporine has demonstrated comparable efficacy to systemic glucocorticoids in one RCT and is considered an important steroid-sparing alternative for PG treatment.2 Biologic agents, especially tumor necrosis factor inhibitors, may be effective in treating cases of refractory PG or for concomitant inflammatory bowel disease management, as demonstrated by a small RCT documenting improvement of PG following infliximab infusion.3
Respondents strongly recommended petrolatum-impregnated gauze and other nonadhesive dressings, including alginate and hydrocolloid dressings, as part of PG wound care. Topical antimicrobials and compression stockings also were recommended by respondents. These practices aim to promote moist environments for healing, avoid maceration, prevent superinfection, optimize wound healing, and minimize damage from adhesive injury.12 Wound debridement and grafting generally were not recommended. However, pathergy is not a universal phenomenon in PG, and wounds that are no longer in the inflammatory phase may benefit from gentle debridement of necrotic tissue and/or grafting in select cases.10
Conclusion
An approach to modifying PG management based on clinical presentation and the practice of combination therapy with multiple systemic agents in refractory PG cases was not addressed in our survey. The low response rate is a limitation; however, the opinions of 51 medical dermatologist experts who regularly manage PG (in contrast to papers based on individualized clinical experience) can provide important clinical guidance until more scientific evidence is established.
Acknowledgments
We would like to thank the SDH and RDS membership for their participation in this survey. We especially acknowledge the other members of the SDH Scientific Task Force for their feedback: Misha Rosenbach, MD (Philadelphia, Pennsylvania); Robert G. Micheletti, MD (Philadelphia, Pennsylvania); Karolyn Wanat, MD (Milwaukee, Wisconsin); Amy Chen, MD (Cromwell, Connecticut); and A. Rambi Cardones, MD (Durham, North Carolina).
- Al Ghazal P, Dissemond J. Therapy of pyoderma gangrenosum in Germany: results of a survey among wound experts. J Dtsch Dermatol Ges . 2015;13:317-324.
- Ormerod AD, Thomas KS, Craig FE, et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: results of the STOP GAP randomised controlled trial. BMJ. 2015;350:h2958.
- Brooklyn TN, Dunnill MG, Shetty A, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55:505-509.
- Al Ghazal P, Klode J, Dissemond J. Diagnostic criteria for pyoderma gangrenosum: results of a survey among dermatologic wound experts in Germany. J Dtsch Dermatol Ges. 2014;12:1129-1131.
- Weenig RH, Davis MD, Dahl PR, et al. Skin ulcers misdiagnosed as pyoderma gangrenosum. N Engl J Med. 2002;347:1412-1418.
- Powell FC, Su WP, Perry HO. Pyoderma gangrenosum: classification and management. J Am Acad Dermatol. 1996;34:395-409.
- Bennett ML, Jackson JM, Jorizzo JL, et al. Pyoderma gangrenosum: a comparison of typical and atypical forms with an emphasis on time to remission. case review of 86 patients from 2 institutions. Medicine. 2000;79:37-46.
- Su WP, Davis MD, Weening RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Aschyan H, Butler DC, Nelson CA, et al. The association of age with clinical presentation and comorbidities of pyoderma gangrenosum. JAMA Dermatol. 2018;154:409-413.
- Binus AM, Qureshi AA, Li VW, et al. Pyoderma gangrenosum: a retrospective review of patient characteristics, comorbidities and therapy in 103 patients. Br J Dermatol. 2011;165:1244-1250.
- Reichrath J, Bens G, Bonowitz A, et al. Treatment recommendations for pyoderma gangrenosum: an evidence-based review of the literature based on more than 350 patients. J Am Acad Dermatol. 2005;53:273-283.
- Miller J, Yentzer BA, Clark A, et al. Pyoderma gangrenosum: a review and update on new therapies. J Am Acad Dermatol. 2010;62:646-654.
Pyoderma gangrenosum (PG) is a rare, chronic, ulcerative, neutrophilic dermatosis of unclear etiology. Large, multicentered, randomized controlled trials (RCTs) are challenging due to the rarity of PG and the lack of a diagnostic confirmatory test; therefore, evidence-based guidelines for diagnosis and treatment are not well established. Current management of PG primarily is guided by case series, small clinical trials, and expert opinion.1-4 We conducted a survey of expert medical dermatologists to highlight best practices in diagnostic and therapeutic approaches to PG.
Methods
The Society of Dermatology Hospitalists (SDH) Scientific Task Force gathered expert opinions from members of the SDH and Rheumatologic Dermatology Society (RDS) regarding PG workup and treatment through an online survey of 15 items (eTable 1). Subscribers of the SDH and RDS LISTSERVs were invited via email to participate in the survey from January 2016 to February 2016. Anonymous survey responses were collected and collated using SurveyMonkey. The survey results identified expert recommendations for evaluation, diagnosis, and treatment of PG and are reported as the sum of the percentage of respondents who answered always (almost 100% of the time) or often (more than half the time) following a particular course of action. A subanalysis was performed defining 2 groups of respondents based on the number of cases of PG treated per year (≥10 vs <10). Survey responses between each group were compared using χ2 analysis with statistical significance set at P=.05.
Results
Fifty-one respondents completed the survey out of 140 surveyed (36% response rate). All respondents were dermatologists, and 96% (49/51) were affiliated with an academic institution. Among the respondents, the number of PG cases managed per year ranged from 2 to 35.
Respondents consistently ordered skin biopsies (92% [47/51]) and tissue cultures (90% [46/51]), as well as certain ancillary tests, including complete blood cell count (96% [49/51]), complete metabolic panel (86% [44/51]), serum protein electrophoresis (76% [39/51]), and hepatitis panel (71% [36/51]). Other frequently ordered studies were rheumatoid factor (69% [35/51]), antinuclear antibodies (67% [34/51]), and antineutrophilic antibodies (65% [33/51]). Respondents frequently ordered erythrocyte sedimentation rate (59% [30/51]), C-reactive protein (55% [28/51]), cryoglobulins (53% [27/51]), urine protein electrophoresis (53% [27/51]), hypercoagulability workup (49% [25/51]), and serum immunofixation test (49% [25/51]). Human immunodeficiency virus testing (43% [22/51]), chest radiograph (41% [21/51]), colonoscopy (41% [21/51]) and referral to other specialties for workup—gastroenterology (38% [19/51]), hematology/oncology (14% [7/51]), and rheumatology (10% [5/51])—were less frequently ordered (eTable 2).
Systemic corticosteroids were reported as first-line therapy by most respondents (94% [48/51]), followed by topical immunomodulatory therapies (63% [32/51]). Topical corticosteroids (75% [38/51]) were the most common first-line topical agents. Thirty-nine percent of respondents (20/51) prescribed topical calcineurin inhibitors as first-line topical therapy. Additional therapies frequently used included systemic cyclosporine (47% [24/51]), antineutrophilic agents (41% [21/51]), and biologic agents (37% [19/51]). Fifty-seven percent of respondents (29/51) supported using combination topical and systemic therapy (Table).
A wide variety of wound care practices were reported in the management of PG. Seventy-six percent of respondents (39/51) favored petroleum-impregnated gauze, 69% (35/51) used nonadhesive dressings, and 43% (22/51) added antimicrobial therapy for PG wound care (eTable 3). In the subanalysis, there were no significant differences in the majority of answer responses in patients treating 10 or more PG cases per year vs fewer than 10 PG cases, except with regard to the practice of combination therapy. Those treating more than 10 cases of PG per year more frequently reported use of combination therapies compared to respondents treating fewer than 10 cases (P=.04).
Comment
Skin biopsies and tissue cultures were strongly recommended (>90% survey respondents) for the initial evaluation of lesions suspected to be PG to evaluate for typical histopathologic changes that appear early in the disease, to rule out PG mimickers such as infectious or vascular causes, and to prevent the detrimental effects of inappropriate treatment and delayed diagnosis.5
Suspected PG warrants a reasonable search for related conditions because more than 50% of PG cases are associated with comorbidities such as rheumatoid arthritis, inflammatory bowel disease, and hematologic disease/malignancy.6,7 A complete blood cell count and comprehensive metabolic panel were recommended by most respondents, aiding in the preliminary screening for hematologic and infectious causes as well as detecting liver and kidney dysfunction associated with systemic conditions. Additionally, exclusion of infection or malignancy may be particularly important if the patient will undergo systemic immunosuppression. In challenging PG cases when initial findings are inconclusive and the clinical presentation does not direct workup (eg, colonoscopy to evaluate gastrointestinal tract symptoms), serum protein electrophoresis, hepatitis panel, rheumatoid factor, antinuclear antibodies, and antineutrophilic antibody tests also were frequently ordered by respondents to further evaluate for underlying or associated conditions.
This consensus regarding skin biopsies and certain ancillary tests is consistent with the proposed diagnostic criteria for classic ulcerative PG in which the absence or exclusion of other relevant causes of cutaneous ulcers is required based on the criteria.8 The importance of ensuring an accurate diagnosis is paramount, as a 10% misdiagnosis rate has been documented in the literature.5
Importantly, a stepwise diagnostic workup for PG is proposed based on survey results, which may limit unnecessary testing and the associated costs to the health care system (Figure 1). Selection of additional testing is guided by initial test results and features of the patient’s clinical presentation, including age, review of systems, and associated comorbidities. Available data suggest that underlying inflammatory bowel disease is more frequent in PG patients who are younger than 65 years, whereas those who are 65 years and older are more likely to have inflammatory arthritis, cancer, or an underlying hematologic disorder.9
Treatment of PG should address both the inflammatory and wound components of the disease (Figure 2).7 In our survey results, systemic corticosteroids were identified as an important first-line therapy supported by reasonable evidence and were favored for their rapid response and minimal cost.1,10,11 Many respondents endorsed the use of systemic therapy in combination with topical steroids or calcineurin inhibitors. Combination therapy may provide more immediate control of rapidly progressing disease while minimizing adverse effects of long-term systemic corticosteroid use. A survey of German wound experts similarly endorsed frequent use of topical calcineurin inhibitors and combination systemic and topical glucocorticoid therapy as common therapeutic approaches.1
Importantly, treatments may vary depending on patient characteristics, comorbidities, and underlying disease, which underscores the need for individualized treatment approaches. Alternative first-line systemic treatments favored by respondents were cyclosporine, biologic medications, and antineutrophilic agents such as dapsone. Cyclosporine has demonstrated comparable efficacy to systemic glucocorticoids in one RCT and is considered an important steroid-sparing alternative for PG treatment.2 Biologic agents, especially tumor necrosis factor inhibitors, may be effective in treating cases of refractory PG or for concomitant inflammatory bowel disease management, as demonstrated by a small RCT documenting improvement of PG following infliximab infusion.3
Respondents strongly recommended petrolatum-impregnated gauze and other nonadhesive dressings, including alginate and hydrocolloid dressings, as part of PG wound care. Topical antimicrobials and compression stockings also were recommended by respondents. These practices aim to promote moist environments for healing, avoid maceration, prevent superinfection, optimize wound healing, and minimize damage from adhesive injury.12 Wound debridement and grafting generally were not recommended. However, pathergy is not a universal phenomenon in PG, and wounds that are no longer in the inflammatory phase may benefit from gentle debridement of necrotic tissue and/or grafting in select cases.10
Conclusion
An approach to modifying PG management based on clinical presentation and the practice of combination therapy with multiple systemic agents in refractory PG cases was not addressed in our survey. The low response rate is a limitation; however, the opinions of 51 medical dermatologist experts who regularly manage PG (in contrast to papers based on individualized clinical experience) can provide important clinical guidance until more scientific evidence is established.
Acknowledgments
We would like to thank the SDH and RDS membership for their participation in this survey. We especially acknowledge the other members of the SDH Scientific Task Force for their feedback: Misha Rosenbach, MD (Philadelphia, Pennsylvania); Robert G. Micheletti, MD (Philadelphia, Pennsylvania); Karolyn Wanat, MD (Milwaukee, Wisconsin); Amy Chen, MD (Cromwell, Connecticut); and A. Rambi Cardones, MD (Durham, North Carolina).
Pyoderma gangrenosum (PG) is a rare, chronic, ulcerative, neutrophilic dermatosis of unclear etiology. Large, multicentered, randomized controlled trials (RCTs) are challenging due to the rarity of PG and the lack of a diagnostic confirmatory test; therefore, evidence-based guidelines for diagnosis and treatment are not well established. Current management of PG primarily is guided by case series, small clinical trials, and expert opinion.1-4 We conducted a survey of expert medical dermatologists to highlight best practices in diagnostic and therapeutic approaches to PG.
Methods
The Society of Dermatology Hospitalists (SDH) Scientific Task Force gathered expert opinions from members of the SDH and Rheumatologic Dermatology Society (RDS) regarding PG workup and treatment through an online survey of 15 items (eTable 1). Subscribers of the SDH and RDS LISTSERVs were invited via email to participate in the survey from January 2016 to February 2016. Anonymous survey responses were collected and collated using SurveyMonkey. The survey results identified expert recommendations for evaluation, diagnosis, and treatment of PG and are reported as the sum of the percentage of respondents who answered always (almost 100% of the time) or often (more than half the time) following a particular course of action. A subanalysis was performed defining 2 groups of respondents based on the number of cases of PG treated per year (≥10 vs <10). Survey responses between each group were compared using χ2 analysis with statistical significance set at P=.05.
Results
Fifty-one respondents completed the survey out of 140 surveyed (36% response rate). All respondents were dermatologists, and 96% (49/51) were affiliated with an academic institution. Among the respondents, the number of PG cases managed per year ranged from 2 to 35.
Respondents consistently ordered skin biopsies (92% [47/51]) and tissue cultures (90% [46/51]), as well as certain ancillary tests, including complete blood cell count (96% [49/51]), complete metabolic panel (86% [44/51]), serum protein electrophoresis (76% [39/51]), and hepatitis panel (71% [36/51]). Other frequently ordered studies were rheumatoid factor (69% [35/51]), antinuclear antibodies (67% [34/51]), and antineutrophilic antibodies (65% [33/51]). Respondents frequently ordered erythrocyte sedimentation rate (59% [30/51]), C-reactive protein (55% [28/51]), cryoglobulins (53% [27/51]), urine protein electrophoresis (53% [27/51]), hypercoagulability workup (49% [25/51]), and serum immunofixation test (49% [25/51]). Human immunodeficiency virus testing (43% [22/51]), chest radiograph (41% [21/51]), colonoscopy (41% [21/51]) and referral to other specialties for workup—gastroenterology (38% [19/51]), hematology/oncology (14% [7/51]), and rheumatology (10% [5/51])—were less frequently ordered (eTable 2).
Systemic corticosteroids were reported as first-line therapy by most respondents (94% [48/51]), followed by topical immunomodulatory therapies (63% [32/51]). Topical corticosteroids (75% [38/51]) were the most common first-line topical agents. Thirty-nine percent of respondents (20/51) prescribed topical calcineurin inhibitors as first-line topical therapy. Additional therapies frequently used included systemic cyclosporine (47% [24/51]), antineutrophilic agents (41% [21/51]), and biologic agents (37% [19/51]). Fifty-seven percent of respondents (29/51) supported using combination topical and systemic therapy (Table).
A wide variety of wound care practices were reported in the management of PG. Seventy-six percent of respondents (39/51) favored petroleum-impregnated gauze, 69% (35/51) used nonadhesive dressings, and 43% (22/51) added antimicrobial therapy for PG wound care (eTable 3). In the subanalysis, there were no significant differences in the majority of answer responses in patients treating 10 or more PG cases per year vs fewer than 10 PG cases, except with regard to the practice of combination therapy. Those treating more than 10 cases of PG per year more frequently reported use of combination therapies compared to respondents treating fewer than 10 cases (P=.04).
Comment
Skin biopsies and tissue cultures were strongly recommended (>90% survey respondents) for the initial evaluation of lesions suspected to be PG to evaluate for typical histopathologic changes that appear early in the disease, to rule out PG mimickers such as infectious or vascular causes, and to prevent the detrimental effects of inappropriate treatment and delayed diagnosis.5
Suspected PG warrants a reasonable search for related conditions because more than 50% of PG cases are associated with comorbidities such as rheumatoid arthritis, inflammatory bowel disease, and hematologic disease/malignancy.6,7 A complete blood cell count and comprehensive metabolic panel were recommended by most respondents, aiding in the preliminary screening for hematologic and infectious causes as well as detecting liver and kidney dysfunction associated with systemic conditions. Additionally, exclusion of infection or malignancy may be particularly important if the patient will undergo systemic immunosuppression. In challenging PG cases when initial findings are inconclusive and the clinical presentation does not direct workup (eg, colonoscopy to evaluate gastrointestinal tract symptoms), serum protein electrophoresis, hepatitis panel, rheumatoid factor, antinuclear antibodies, and antineutrophilic antibody tests also were frequently ordered by respondents to further evaluate for underlying or associated conditions.
This consensus regarding skin biopsies and certain ancillary tests is consistent with the proposed diagnostic criteria for classic ulcerative PG in which the absence or exclusion of other relevant causes of cutaneous ulcers is required based on the criteria.8 The importance of ensuring an accurate diagnosis is paramount, as a 10% misdiagnosis rate has been documented in the literature.5
Importantly, a stepwise diagnostic workup for PG is proposed based on survey results, which may limit unnecessary testing and the associated costs to the health care system (Figure 1). Selection of additional testing is guided by initial test results and features of the patient’s clinical presentation, including age, review of systems, and associated comorbidities. Available data suggest that underlying inflammatory bowel disease is more frequent in PG patients who are younger than 65 years, whereas those who are 65 years and older are more likely to have inflammatory arthritis, cancer, or an underlying hematologic disorder.9
Treatment of PG should address both the inflammatory and wound components of the disease (Figure 2).7 In our survey results, systemic corticosteroids were identified as an important first-line therapy supported by reasonable evidence and were favored for their rapid response and minimal cost.1,10,11 Many respondents endorsed the use of systemic therapy in combination with topical steroids or calcineurin inhibitors. Combination therapy may provide more immediate control of rapidly progressing disease while minimizing adverse effects of long-term systemic corticosteroid use. A survey of German wound experts similarly endorsed frequent use of topical calcineurin inhibitors and combination systemic and topical glucocorticoid therapy as common therapeutic approaches.1
Importantly, treatments may vary depending on patient characteristics, comorbidities, and underlying disease, which underscores the need for individualized treatment approaches. Alternative first-line systemic treatments favored by respondents were cyclosporine, biologic medications, and antineutrophilic agents such as dapsone. Cyclosporine has demonstrated comparable efficacy to systemic glucocorticoids in one RCT and is considered an important steroid-sparing alternative for PG treatment.2 Biologic agents, especially tumor necrosis factor inhibitors, may be effective in treating cases of refractory PG or for concomitant inflammatory bowel disease management, as demonstrated by a small RCT documenting improvement of PG following infliximab infusion.3
Respondents strongly recommended petrolatum-impregnated gauze and other nonadhesive dressings, including alginate and hydrocolloid dressings, as part of PG wound care. Topical antimicrobials and compression stockings also were recommended by respondents. These practices aim to promote moist environments for healing, avoid maceration, prevent superinfection, optimize wound healing, and minimize damage from adhesive injury.12 Wound debridement and grafting generally were not recommended. However, pathergy is not a universal phenomenon in PG, and wounds that are no longer in the inflammatory phase may benefit from gentle debridement of necrotic tissue and/or grafting in select cases.10
Conclusion
An approach to modifying PG management based on clinical presentation and the practice of combination therapy with multiple systemic agents in refractory PG cases was not addressed in our survey. The low response rate is a limitation; however, the opinions of 51 medical dermatologist experts who regularly manage PG (in contrast to papers based on individualized clinical experience) can provide important clinical guidance until more scientific evidence is established.
Acknowledgments
We would like to thank the SDH and RDS membership for their participation in this survey. We especially acknowledge the other members of the SDH Scientific Task Force for their feedback: Misha Rosenbach, MD (Philadelphia, Pennsylvania); Robert G. Micheletti, MD (Philadelphia, Pennsylvania); Karolyn Wanat, MD (Milwaukee, Wisconsin); Amy Chen, MD (Cromwell, Connecticut); and A. Rambi Cardones, MD (Durham, North Carolina).
- Al Ghazal P, Dissemond J. Therapy of pyoderma gangrenosum in Germany: results of a survey among wound experts. J Dtsch Dermatol Ges . 2015;13:317-324.
- Ormerod AD, Thomas KS, Craig FE, et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: results of the STOP GAP randomised controlled trial. BMJ. 2015;350:h2958.
- Brooklyn TN, Dunnill MG, Shetty A, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55:505-509.
- Al Ghazal P, Klode J, Dissemond J. Diagnostic criteria for pyoderma gangrenosum: results of a survey among dermatologic wound experts in Germany. J Dtsch Dermatol Ges. 2014;12:1129-1131.
- Weenig RH, Davis MD, Dahl PR, et al. Skin ulcers misdiagnosed as pyoderma gangrenosum. N Engl J Med. 2002;347:1412-1418.
- Powell FC, Su WP, Perry HO. Pyoderma gangrenosum: classification and management. J Am Acad Dermatol. 1996;34:395-409.
- Bennett ML, Jackson JM, Jorizzo JL, et al. Pyoderma gangrenosum: a comparison of typical and atypical forms with an emphasis on time to remission. case review of 86 patients from 2 institutions. Medicine. 2000;79:37-46.
- Su WP, Davis MD, Weening RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Aschyan H, Butler DC, Nelson CA, et al. The association of age with clinical presentation and comorbidities of pyoderma gangrenosum. JAMA Dermatol. 2018;154:409-413.
- Binus AM, Qureshi AA, Li VW, et al. Pyoderma gangrenosum: a retrospective review of patient characteristics, comorbidities and therapy in 103 patients. Br J Dermatol. 2011;165:1244-1250.
- Reichrath J, Bens G, Bonowitz A, et al. Treatment recommendations for pyoderma gangrenosum: an evidence-based review of the literature based on more than 350 patients. J Am Acad Dermatol. 2005;53:273-283.
- Miller J, Yentzer BA, Clark A, et al. Pyoderma gangrenosum: a review and update on new therapies. J Am Acad Dermatol. 2010;62:646-654.
- Al Ghazal P, Dissemond J. Therapy of pyoderma gangrenosum in Germany: results of a survey among wound experts. J Dtsch Dermatol Ges . 2015;13:317-324.
- Ormerod AD, Thomas KS, Craig FE, et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: results of the STOP GAP randomised controlled trial. BMJ. 2015;350:h2958.
- Brooklyn TN, Dunnill MG, Shetty A, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55:505-509.
- Al Ghazal P, Klode J, Dissemond J. Diagnostic criteria for pyoderma gangrenosum: results of a survey among dermatologic wound experts in Germany. J Dtsch Dermatol Ges. 2014;12:1129-1131.
- Weenig RH, Davis MD, Dahl PR, et al. Skin ulcers misdiagnosed as pyoderma gangrenosum. N Engl J Med. 2002;347:1412-1418.
- Powell FC, Su WP, Perry HO. Pyoderma gangrenosum: classification and management. J Am Acad Dermatol. 1996;34:395-409.
- Bennett ML, Jackson JM, Jorizzo JL, et al. Pyoderma gangrenosum: a comparison of typical and atypical forms with an emphasis on time to remission. case review of 86 patients from 2 institutions. Medicine. 2000;79:37-46.
- Su WP, Davis MD, Weening RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Aschyan H, Butler DC, Nelson CA, et al. The association of age with clinical presentation and comorbidities of pyoderma gangrenosum. JAMA Dermatol. 2018;154:409-413.
- Binus AM, Qureshi AA, Li VW, et al. Pyoderma gangrenosum: a retrospective review of patient characteristics, comorbidities and therapy in 103 patients. Br J Dermatol. 2011;165:1244-1250.
- Reichrath J, Bens G, Bonowitz A, et al. Treatment recommendations for pyoderma gangrenosum: an evidence-based review of the literature based on more than 350 patients. J Am Acad Dermatol. 2005;53:273-283.
- Miller J, Yentzer BA, Clark A, et al. Pyoderma gangrenosum: a review and update on new therapies. J Am Acad Dermatol. 2010;62:646-654.
Practice Points
- The diagnosis of pyoderma gangrenosum (PG) poses a challenge in clinical practice that could be minimized by following a stepwise algorithm based on initial test results (including skin biopsies) and features of the patient’s clinical presentation.
- As there is no US Food and Drug Administration–approved treatment for PG, a stepwise algorithm approach in combination with the clinical experience addressing inflammation and wound care is essential to reach control and remission of PG.
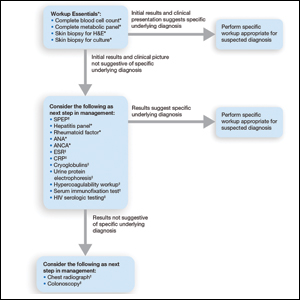
Drug combo slows functional decline in ALS
, according to results of the phase 2/3 CENTAUR study.

Patients with a fast-progressing form of ALS who were treated with AMX0035 “retained higher levels of physical function over 6 months compared with those who received placebo,” reported principal investigator Sabrina Paganoni, MD, PhD, of the Sean M. Healey and AMG Center for ALS at Massachusetts General Hospital, Boston.
“This is very hopeful news for people affected by ALS, especially because we were able to see a treatment effect in a relatively short period of time,” Dr. Paganoni said.
The study was published online Sept. 3 in the New England Journal of Medicine.
In this study, AMX0035 demonstrated a “clinically meaningful benefit and a favorable safety profile for people living with ALS,” Josh Cohen, co-CEO, chairman, and cofounder at Amylyx, said in a news release. The company is “working collaboratively and expeditiously with agencies worldwide to bring this potential new treatment option forward.”
“The data ... makes a clear and compelling case that AMX0035 should be made available to people with ALS as soon as possible,” Calaneet Balas, president and CEO of The ALS Association, said in the release.
The CENTAUR trial
Sodium phenylbutyrate and taurursodiol have been found to reduce neuronal death in experimental models. AMX0035 combines 3 g sodium phenylbutyrate and 1 g of taurursodiol.
The CENTAUR trial tested AMX0035 against placebo in 137 ALS patients with symptom onset within the prior 18 months, with 89 patients in the AMX0035 group and 48 in the placebo group. AMX0035 or matching placebo were administered once daily for 3 weeks and then twice daily for a planned duration of 24 weeks.
In a modified intention-to-treat analysis, the mean rate of change in the Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (ALSFRS-R) score was −1.24 points per month with AMX0035 and −1.66 points per month with placebo (difference, 0.42 points per month; 95% CI, 0.03 - 0.81; P = .03). After 24 weeks, patients treated with AMX0035 scored on average 2.32 points higher on the ALSFRS-R than their peers on placebo group (P = .03).
“The score, consisting of four subdomains, showed a change that was most prominent for the fine-motor subscale and less apparent for the other subscales,” the investigators said.
Treatment with AMX0035 led to slowing of disease progression in a population in which many participants were receiving riluzole (Tiglutik), edaravone (Radicava) or both, they pointed out.
The secondary outcomes were rate of decline in isometric muscle strength and breathing function; change in plasma phosphorylated axonal neurofilament H subunit (pNF-H) levels; and the time to composite events of death, tracheostomy, permanent ventilation, and hospitalization. These outcomes did not differ significantly between the two groups.
Open-label extension ongoing
AMX0035 was generally well tolerated. Nearly all patients in both groups had one or more adverse events. Events occurring at 2% or greater frequency in the AMX0035 group were primarily gastrointestinal (diarrhea, nausea, salivary hypersecretion, and abdominal discomfort). Serious adverse events were more common in the placebo group (19% vs. 12%). The incidence of respiratory serious adverse events was 8% in the placebo group and 3% in the AMX0035 group.
More patients on active treatment than placebo (19% vs. 8%) stopped the trial regimen early owing to adverse events. The most common adverse events leading to discontinuation of the trial regimen were diarrhea and respiratory failure.
The trial was “too short for us to detect an effect on survival,” Dr. Paganoni said in an interview. Most of the participants who completed the trial elected to enroll in an open-label extension study and receive AMX0035 long-term. “This is important because it will teach us about the impact of AMX0035 on survival,” said Dr. Paganoni.
Interim data from the ongoing open-label extension study are being submitted to a peer-reviewed journal shortly and will be published in the coming months.
A cause for hope
“There has been understandable frustration with the slow pace of development of therapy for ALS,” Michael Benatar, MD, PhD, University of Miami, and Michael McDermott, PhD, University of Rochester (N.Y.), said in an accompanying editorial.
“Despite dozens of trials, few pharmacologic agents have emerged that affect functional decline or survival – and all only modestly so. Although the effects of sodium phenylbutyrate–taurursodiol are similarly modest, the incremental gains that they provide in the battle against ALS are a cause for hope,” they wrote.
They caution, however, that this study was enriched for patients with more rapidly progressive disease, which “raises questions about generalizability to the broader population of patients with ALS.
“Although the patients who were enrolled in the trial may not be biologically different from the broader population of patients with ALS, the magnitude of therapeutic effect may be smaller in the latter,” Dr. Benatar and Dr. McDermott noted.
They said that in light of “residual questions about efficacy and the ability of patients to continue taking the drug,” they agree with the authors’ conclusion that “longer and larger trials are needed to evaluate the efficacy and safety of sodium phenylbutyrate–taurursodiol in persons with ALS.”
Given these “tantalizing preliminary data,” Dr. Benatar and Dr. McDermott said they look forward to “a confirmatory phase 3 trial.”
The study was supported by Amylyx Pharmaceuticals, the ALS Finding a Cure Foundation, and the ALS Association. Dr. Paganoni has received grants from Revalesio, Ra Pharma, Biohaven, Clene, and Prilenia. A complete list of disclosures for authors and editorialists is available with the original article.
A version of this article originally appeared on Medscape.com.
, according to results of the phase 2/3 CENTAUR study.
Patients with a fast-progressing form of ALS who were treated with AMX0035 “retained higher levels of physical function over 6 months compared with those who received placebo,” reported principal investigator Sabrina Paganoni, MD, PhD, of the Sean M. Healey and AMG Center for ALS at Massachusetts General Hospital, Boston.
“This is very hopeful news for people affected by ALS, especially because we were able to see a treatment effect in a relatively short period of time,” Dr. Paganoni said.
The study was published online Sept. 3 in the New England Journal of Medicine.
In this study, AMX0035 demonstrated a “clinically meaningful benefit and a favorable safety profile for people living with ALS,” Josh Cohen, co-CEO, chairman, and cofounder at Amylyx, said in a news release. The company is “working collaboratively and expeditiously with agencies worldwide to bring this potential new treatment option forward.”
“The data ... makes a clear and compelling case that AMX0035 should be made available to people with ALS as soon as possible,” Calaneet Balas, president and CEO of The ALS Association, said in the release.
The CENTAUR trial
Sodium phenylbutyrate and taurursodiol have been found to reduce neuronal death in experimental models. AMX0035 combines 3 g sodium phenylbutyrate and 1 g of taurursodiol.
The CENTAUR trial tested AMX0035 against placebo in 137 ALS patients with symptom onset within the prior 18 months, with 89 patients in the AMX0035 group and 48 in the placebo group. AMX0035 or matching placebo were administered once daily for 3 weeks and then twice daily for a planned duration of 24 weeks.
In a modified intention-to-treat analysis, the mean rate of change in the Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (ALSFRS-R) score was −1.24 points per month with AMX0035 and −1.66 points per month with placebo (difference, 0.42 points per month; 95% CI, 0.03 - 0.81; P = .03). After 24 weeks, patients treated with AMX0035 scored on average 2.32 points higher on the ALSFRS-R than their peers on placebo group (P = .03).
“The score, consisting of four subdomains, showed a change that was most prominent for the fine-motor subscale and less apparent for the other subscales,” the investigators said.
Treatment with AMX0035 led to slowing of disease progression in a population in which many participants were receiving riluzole (Tiglutik), edaravone (Radicava) or both, they pointed out.
The secondary outcomes were rate of decline in isometric muscle strength and breathing function; change in plasma phosphorylated axonal neurofilament H subunit (pNF-H) levels; and the time to composite events of death, tracheostomy, permanent ventilation, and hospitalization. These outcomes did not differ significantly between the two groups.
Open-label extension ongoing
AMX0035 was generally well tolerated. Nearly all patients in both groups had one or more adverse events. Events occurring at 2% or greater frequency in the AMX0035 group were primarily gastrointestinal (diarrhea, nausea, salivary hypersecretion, and abdominal discomfort). Serious adverse events were more common in the placebo group (19% vs. 12%). The incidence of respiratory serious adverse events was 8% in the placebo group and 3% in the AMX0035 group.
More patients on active treatment than placebo (19% vs. 8%) stopped the trial regimen early owing to adverse events. The most common adverse events leading to discontinuation of the trial regimen were diarrhea and respiratory failure.
The trial was “too short for us to detect an effect on survival,” Dr. Paganoni said in an interview. Most of the participants who completed the trial elected to enroll in an open-label extension study and receive AMX0035 long-term. “This is important because it will teach us about the impact of AMX0035 on survival,” said Dr. Paganoni.
Interim data from the ongoing open-label extension study are being submitted to a peer-reviewed journal shortly and will be published in the coming months.
A cause for hope
“There has been understandable frustration with the slow pace of development of therapy for ALS,” Michael Benatar, MD, PhD, University of Miami, and Michael McDermott, PhD, University of Rochester (N.Y.), said in an accompanying editorial.
“Despite dozens of trials, few pharmacologic agents have emerged that affect functional decline or survival – and all only modestly so. Although the effects of sodium phenylbutyrate–taurursodiol are similarly modest, the incremental gains that they provide in the battle against ALS are a cause for hope,” they wrote.
They caution, however, that this study was enriched for patients with more rapidly progressive disease, which “raises questions about generalizability to the broader population of patients with ALS.
“Although the patients who were enrolled in the trial may not be biologically different from the broader population of patients with ALS, the magnitude of therapeutic effect may be smaller in the latter,” Dr. Benatar and Dr. McDermott noted.
They said that in light of “residual questions about efficacy and the ability of patients to continue taking the drug,” they agree with the authors’ conclusion that “longer and larger trials are needed to evaluate the efficacy and safety of sodium phenylbutyrate–taurursodiol in persons with ALS.”
Given these “tantalizing preliminary data,” Dr. Benatar and Dr. McDermott said they look forward to “a confirmatory phase 3 trial.”
The study was supported by Amylyx Pharmaceuticals, the ALS Finding a Cure Foundation, and the ALS Association. Dr. Paganoni has received grants from Revalesio, Ra Pharma, Biohaven, Clene, and Prilenia. A complete list of disclosures for authors and editorialists is available with the original article.
A version of this article originally appeared on Medscape.com.
, according to results of the phase 2/3 CENTAUR study.
Patients with a fast-progressing form of ALS who were treated with AMX0035 “retained higher levels of physical function over 6 months compared with those who received placebo,” reported principal investigator Sabrina Paganoni, MD, PhD, of the Sean M. Healey and AMG Center for ALS at Massachusetts General Hospital, Boston.
“This is very hopeful news for people affected by ALS, especially because we were able to see a treatment effect in a relatively short period of time,” Dr. Paganoni said.
The study was published online Sept. 3 in the New England Journal of Medicine.
In this study, AMX0035 demonstrated a “clinically meaningful benefit and a favorable safety profile for people living with ALS,” Josh Cohen, co-CEO, chairman, and cofounder at Amylyx, said in a news release. The company is “working collaboratively and expeditiously with agencies worldwide to bring this potential new treatment option forward.”
“The data ... makes a clear and compelling case that AMX0035 should be made available to people with ALS as soon as possible,” Calaneet Balas, president and CEO of The ALS Association, said in the release.
The CENTAUR trial
Sodium phenylbutyrate and taurursodiol have been found to reduce neuronal death in experimental models. AMX0035 combines 3 g sodium phenylbutyrate and 1 g of taurursodiol.
The CENTAUR trial tested AMX0035 against placebo in 137 ALS patients with symptom onset within the prior 18 months, with 89 patients in the AMX0035 group and 48 in the placebo group. AMX0035 or matching placebo were administered once daily for 3 weeks and then twice daily for a planned duration of 24 weeks.
In a modified intention-to-treat analysis, the mean rate of change in the Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (ALSFRS-R) score was −1.24 points per month with AMX0035 and −1.66 points per month with placebo (difference, 0.42 points per month; 95% CI, 0.03 - 0.81; P = .03). After 24 weeks, patients treated with AMX0035 scored on average 2.32 points higher on the ALSFRS-R than their peers on placebo group (P = .03).
“The score, consisting of four subdomains, showed a change that was most prominent for the fine-motor subscale and less apparent for the other subscales,” the investigators said.
Treatment with AMX0035 led to slowing of disease progression in a population in which many participants were receiving riluzole (Tiglutik), edaravone (Radicava) or both, they pointed out.
The secondary outcomes were rate of decline in isometric muscle strength and breathing function; change in plasma phosphorylated axonal neurofilament H subunit (pNF-H) levels; and the time to composite events of death, tracheostomy, permanent ventilation, and hospitalization. These outcomes did not differ significantly between the two groups.
Open-label extension ongoing
AMX0035 was generally well tolerated. Nearly all patients in both groups had one or more adverse events. Events occurring at 2% or greater frequency in the AMX0035 group were primarily gastrointestinal (diarrhea, nausea, salivary hypersecretion, and abdominal discomfort). Serious adverse events were more common in the placebo group (19% vs. 12%). The incidence of respiratory serious adverse events was 8% in the placebo group and 3% in the AMX0035 group.
More patients on active treatment than placebo (19% vs. 8%) stopped the trial regimen early owing to adverse events. The most common adverse events leading to discontinuation of the trial regimen were diarrhea and respiratory failure.
The trial was “too short for us to detect an effect on survival,” Dr. Paganoni said in an interview. Most of the participants who completed the trial elected to enroll in an open-label extension study and receive AMX0035 long-term. “This is important because it will teach us about the impact of AMX0035 on survival,” said Dr. Paganoni.
Interim data from the ongoing open-label extension study are being submitted to a peer-reviewed journal shortly and will be published in the coming months.
A cause for hope
“There has been understandable frustration with the slow pace of development of therapy for ALS,” Michael Benatar, MD, PhD, University of Miami, and Michael McDermott, PhD, University of Rochester (N.Y.), said in an accompanying editorial.
“Despite dozens of trials, few pharmacologic agents have emerged that affect functional decline or survival – and all only modestly so. Although the effects of sodium phenylbutyrate–taurursodiol are similarly modest, the incremental gains that they provide in the battle against ALS are a cause for hope,” they wrote.
They caution, however, that this study was enriched for patients with more rapidly progressive disease, which “raises questions about generalizability to the broader population of patients with ALS.
“Although the patients who were enrolled in the trial may not be biologically different from the broader population of patients with ALS, the magnitude of therapeutic effect may be smaller in the latter,” Dr. Benatar and Dr. McDermott noted.
They said that in light of “residual questions about efficacy and the ability of patients to continue taking the drug,” they agree with the authors’ conclusion that “longer and larger trials are needed to evaluate the efficacy and safety of sodium phenylbutyrate–taurursodiol in persons with ALS.”
Given these “tantalizing preliminary data,” Dr. Benatar and Dr. McDermott said they look forward to “a confirmatory phase 3 trial.”
The study was supported by Amylyx Pharmaceuticals, the ALS Finding a Cure Foundation, and the ALS Association. Dr. Paganoni has received grants from Revalesio, Ra Pharma, Biohaven, Clene, and Prilenia. A complete list of disclosures for authors and editorialists is available with the original article.
A version of this article originally appeared on Medscape.com.
From New England Journal of Medicine
TNF inhibitors linked to inflammatory CNS events
, new research suggests
The nested case-control study included more than 200 participants with diseases such as rheumatoid arthritis, psoriasis, and Crohn’s disease. Results showed that exposure to TNF inhibitors was significantly associated with increased risk for demyelinating CNS events, such as multiple sclerosis, and nondemyelinating events, such as meningitis and encephalitis.
Interestingly, disease-specific secondary analyses showed that the strongest association for inflammatory events was in patients with rheumatoid arthritis.
Lead author Amy Kunchok, MD, of Mayo Clinic, Rochester, Minn., noted that “these are highly effective therapies for patients” and that these CNS events are likely uncommon.
“Our study has observed an association, but this does not imply causality. Therefore, we are not cautioning against using these therapies in appropriate patients,” Dr. Kunchok said in an interview.
“Rather, we recommend that clinicians assessing patients with both inflammatory demyelinating and nondemyelinating CNS events consider a detailed evaluation of the medication history, particularly in patients with coexistent autoimmune diseases who may have a current or past history of biological therapies,” she said.
The findings were published in JAMA Neurology.
Poorly understood
TNF inhibitors “are common therapies for certain autoimmune diseases,” the investigators noted.
Previously, a link between exposure to these inhibitors and inflammatory CNS events “has been postulated but is poorly understood,” they wrote.
In the current study, they examined records for 106 patients who were treated at Mayo clinics in Minnesota, Arizona, or Florida from January 2003 through February 2019. All participants had been diagnosed with an autoimmune disease that the Food and Drug Administration has listed as an indication for TNF inhibitor use. This included rheumatoid arthritis (n = 48), ankylosing spondylitis (n = 4), psoriasis and psoriatic arthritis (n = 21), Crohn’s disease (n = 27), and ulcerative colitis (n = 6). Their records also showed diagnostic codes for the inflammatory demyelinating CNS events of relapsing-remitting or primary progressive MS, clinically isolated syndrome, radiologically isolated syndrome, neuromyelitis optica spectrum disorder, and transverse myelitis or for the inflammatory nondemyelinating CNS events of meningitis, meningoencephalitis, encephalitis, neurosarcoidosis, and CNS vasculitis. The investigators also included 106 age-, sex-, and autoimmune disease–matched participants 1:1 to act as the control group.
In the total study population, 64% were women and the median age at disease onset was 52 years. In addition, 60% of the patient group and 40% of the control group were exposed to TNF inhibitors.
Novel finding?
Results showed that TNF inhibitor exposure was significantly linked to increased risk for developing any inflammatory CNS event (adjusted odds ratio, 3.01; 95% CI, 1.55-5.82; P = .001). When the outcomes were stratified by class of inflammatory event, these results were similar. The aOR was 3.09 (95% CI, 1.19-8.04; P = .02) for inflammatory demyelinating CNS events and was 2.97 (95% CI, 1.15-7.65; P = .02) for inflammatory nondemyelinating events.
Dr. Kunchok noted that the association between the inhibitors and nondemyelinating events was “a novel finding from this study.”
In secondary analyses, patients with rheumatoid arthritis and exposure to TNF inhibitors had the strongest association with any inflammatory CNS event (aOR, 4.82; 95% CI, 1.62-14.36; P = .005).
A pooled cohort comprising only the participants with the other autoimmune diseases did not show a significant association between exposure to TNF inhibitors and development of CNS events (P = .09).
“Because of the lack of power, further stratification by individual autoimmune diseases was not analyzed,” the investigators reported.
Although the overall findings showed that exposure to TNF inhibitors was linked to increased risk for inflammatory events, whether this association “represents de novo or exacerbated inflammatory pathways requires further research,” the authors wrote.
Dr. Kunchok added that more research, especially population-based studies, is also needed to examine the incidence of these inflammatory CNS events in patients exposed to TNF-alpha inhibitors.
Adds to the literature
In an accompanying editorial, Jeffrey M. Gelfand, MD, department of neurology at the University of California, San Francisco, and Jinoos Yazdany, MD, Zuckerberg San Francisco General Hospital at UCSF, noted that although the study adds to the literature, the magnitude of the risk found “remains unclear.”
“Randomized clinical trials are not suited to the study of rare adverse events,” Dr. Gelfand and Dr. Yazdany wrote. They agree with Dr. Kunchok that “next steps should include population-based observational studies that control for disease severity.”
Still, the current study provides additional evidence of rare adverse events in patients receiving TNF inhibitors, they noted. So how should prescribers proceed?
“As with all treatments, the risk-benefit ratio for the individual patient’s situation must be weighed and appropriate counseling must be given to facilitate shared decision-making discussions,” wrote the editorialists.
“Given what is known about the risk of harm, avoiding TNF inhibitors is advisable in patients with known MS,” they wrote.
In addition, neurologic consultation can be helpful for clarifying diagnoses and providing advice on monitoring strategies for TNF inhibitor treatment in those with possible MS or other demyelinating conditions, noted the editorialists.
“In patients who develop new concerning neurological symptoms while receiving TNF inhibitor treatment, timely evaluation is indicated, including consideration of neuroinflammatory, infectious, and neurological diagnoses that may be unrelated to treatment,” they added.
“Broader awareness of risks that studies such as this one by Kunchok et al provide can ... encourage timelier recognition of potential TNF inhibitor–associated neuroinflammatory events and may improve outcomes for patients,” Dr. Gelfand and Dr. Yazdany concluded.
The study was funded by a grant from the National Center for Advancing Translational Sciences. Dr. Kunchok reports having received research funding from Biogen outside this study. A full list of disclosures for the other study authors is in the original article. Dr. Gelfand reports having received g rants for a clinical trial from Genentech and consulting fees from Biogen, Alexion, Theranica, Impel Neuropharma, Advanced Clinical, Biohaven, and Satsuma. Dr. Yazdany reports having received grants from Pfizer and consulting fees from AstraZeneca and Eli Lilly outside the submitted work.
A version of this article originally appeared on Medscape.com.
, new research suggests
The nested case-control study included more than 200 participants with diseases such as rheumatoid arthritis, psoriasis, and Crohn’s disease. Results showed that exposure to TNF inhibitors was significantly associated with increased risk for demyelinating CNS events, such as multiple sclerosis, and nondemyelinating events, such as meningitis and encephalitis.
Interestingly, disease-specific secondary analyses showed that the strongest association for inflammatory events was in patients with rheumatoid arthritis.
Lead author Amy Kunchok, MD, of Mayo Clinic, Rochester, Minn., noted that “these are highly effective therapies for patients” and that these CNS events are likely uncommon.
“Our study has observed an association, but this does not imply causality. Therefore, we are not cautioning against using these therapies in appropriate patients,” Dr. Kunchok said in an interview.
“Rather, we recommend that clinicians assessing patients with both inflammatory demyelinating and nondemyelinating CNS events consider a detailed evaluation of the medication history, particularly in patients with coexistent autoimmune diseases who may have a current or past history of biological therapies,” she said.
The findings were published in JAMA Neurology.
Poorly understood
TNF inhibitors “are common therapies for certain autoimmune diseases,” the investigators noted.
Previously, a link between exposure to these inhibitors and inflammatory CNS events “has been postulated but is poorly understood,” they wrote.
In the current study, they examined records for 106 patients who were treated at Mayo clinics in Minnesota, Arizona, or Florida from January 2003 through February 2019. All participants had been diagnosed with an autoimmune disease that the Food and Drug Administration has listed as an indication for TNF inhibitor use. This included rheumatoid arthritis (n = 48), ankylosing spondylitis (n = 4), psoriasis and psoriatic arthritis (n = 21), Crohn’s disease (n = 27), and ulcerative colitis (n = 6). Their records also showed diagnostic codes for the inflammatory demyelinating CNS events of relapsing-remitting or primary progressive MS, clinically isolated syndrome, radiologically isolated syndrome, neuromyelitis optica spectrum disorder, and transverse myelitis or for the inflammatory nondemyelinating CNS events of meningitis, meningoencephalitis, encephalitis, neurosarcoidosis, and CNS vasculitis. The investigators also included 106 age-, sex-, and autoimmune disease–matched participants 1:1 to act as the control group.
In the total study population, 64% were women and the median age at disease onset was 52 years. In addition, 60% of the patient group and 40% of the control group were exposed to TNF inhibitors.
Novel finding?
Results showed that TNF inhibitor exposure was significantly linked to increased risk for developing any inflammatory CNS event (adjusted odds ratio, 3.01; 95% CI, 1.55-5.82; P = .001). When the outcomes were stratified by class of inflammatory event, these results were similar. The aOR was 3.09 (95% CI, 1.19-8.04; P = .02) for inflammatory demyelinating CNS events and was 2.97 (95% CI, 1.15-7.65; P = .02) for inflammatory nondemyelinating events.
Dr. Kunchok noted that the association between the inhibitors and nondemyelinating events was “a novel finding from this study.”
In secondary analyses, patients with rheumatoid arthritis and exposure to TNF inhibitors had the strongest association with any inflammatory CNS event (aOR, 4.82; 95% CI, 1.62-14.36; P = .005).
A pooled cohort comprising only the participants with the other autoimmune diseases did not show a significant association between exposure to TNF inhibitors and development of CNS events (P = .09).
“Because of the lack of power, further stratification by individual autoimmune diseases was not analyzed,” the investigators reported.
Although the overall findings showed that exposure to TNF inhibitors was linked to increased risk for inflammatory events, whether this association “represents de novo or exacerbated inflammatory pathways requires further research,” the authors wrote.
Dr. Kunchok added that more research, especially population-based studies, is also needed to examine the incidence of these inflammatory CNS events in patients exposed to TNF-alpha inhibitors.
Adds to the literature
In an accompanying editorial, Jeffrey M. Gelfand, MD, department of neurology at the University of California, San Francisco, and Jinoos Yazdany, MD, Zuckerberg San Francisco General Hospital at UCSF, noted that although the study adds to the literature, the magnitude of the risk found “remains unclear.”
“Randomized clinical trials are not suited to the study of rare adverse events,” Dr. Gelfand and Dr. Yazdany wrote. They agree with Dr. Kunchok that “next steps should include population-based observational studies that control for disease severity.”
Still, the current study provides additional evidence of rare adverse events in patients receiving TNF inhibitors, they noted. So how should prescribers proceed?
“As with all treatments, the risk-benefit ratio for the individual patient’s situation must be weighed and appropriate counseling must be given to facilitate shared decision-making discussions,” wrote the editorialists.
“Given what is known about the risk of harm, avoiding TNF inhibitors is advisable in patients with known MS,” they wrote.
In addition, neurologic consultation can be helpful for clarifying diagnoses and providing advice on monitoring strategies for TNF inhibitor treatment in those with possible MS or other demyelinating conditions, noted the editorialists.
“In patients who develop new concerning neurological symptoms while receiving TNF inhibitor treatment, timely evaluation is indicated, including consideration of neuroinflammatory, infectious, and neurological diagnoses that may be unrelated to treatment,” they added.
“Broader awareness of risks that studies such as this one by Kunchok et al provide can ... encourage timelier recognition of potential TNF inhibitor–associated neuroinflammatory events and may improve outcomes for patients,” Dr. Gelfand and Dr. Yazdany concluded.
The study was funded by a grant from the National Center for Advancing Translational Sciences. Dr. Kunchok reports having received research funding from Biogen outside this study. A full list of disclosures for the other study authors is in the original article. Dr. Gelfand reports having received g rants for a clinical trial from Genentech and consulting fees from Biogen, Alexion, Theranica, Impel Neuropharma, Advanced Clinical, Biohaven, and Satsuma. Dr. Yazdany reports having received grants from Pfizer and consulting fees from AstraZeneca and Eli Lilly outside the submitted work.
A version of this article originally appeared on Medscape.com.
, new research suggests
The nested case-control study included more than 200 participants with diseases such as rheumatoid arthritis, psoriasis, and Crohn’s disease. Results showed that exposure to TNF inhibitors was significantly associated with increased risk for demyelinating CNS events, such as multiple sclerosis, and nondemyelinating events, such as meningitis and encephalitis.
Interestingly, disease-specific secondary analyses showed that the strongest association for inflammatory events was in patients with rheumatoid arthritis.
Lead author Amy Kunchok, MD, of Mayo Clinic, Rochester, Minn., noted that “these are highly effective therapies for patients” and that these CNS events are likely uncommon.
“Our study has observed an association, but this does not imply causality. Therefore, we are not cautioning against using these therapies in appropriate patients,” Dr. Kunchok said in an interview.
“Rather, we recommend that clinicians assessing patients with both inflammatory demyelinating and nondemyelinating CNS events consider a detailed evaluation of the medication history, particularly in patients with coexistent autoimmune diseases who may have a current or past history of biological therapies,” she said.
The findings were published in JAMA Neurology.
Poorly understood
TNF inhibitors “are common therapies for certain autoimmune diseases,” the investigators noted.
Previously, a link between exposure to these inhibitors and inflammatory CNS events “has been postulated but is poorly understood,” they wrote.
In the current study, they examined records for 106 patients who were treated at Mayo clinics in Minnesota, Arizona, or Florida from January 2003 through February 2019. All participants had been diagnosed with an autoimmune disease that the Food and Drug Administration has listed as an indication for TNF inhibitor use. This included rheumatoid arthritis (n = 48), ankylosing spondylitis (n = 4), psoriasis and psoriatic arthritis (n = 21), Crohn’s disease (n = 27), and ulcerative colitis (n = 6). Their records also showed diagnostic codes for the inflammatory demyelinating CNS events of relapsing-remitting or primary progressive MS, clinically isolated syndrome, radiologically isolated syndrome, neuromyelitis optica spectrum disorder, and transverse myelitis or for the inflammatory nondemyelinating CNS events of meningitis, meningoencephalitis, encephalitis, neurosarcoidosis, and CNS vasculitis. The investigators also included 106 age-, sex-, and autoimmune disease–matched participants 1:1 to act as the control group.
In the total study population, 64% were women and the median age at disease onset was 52 years. In addition, 60% of the patient group and 40% of the control group were exposed to TNF inhibitors.
Novel finding?
Results showed that TNF inhibitor exposure was significantly linked to increased risk for developing any inflammatory CNS event (adjusted odds ratio, 3.01; 95% CI, 1.55-5.82; P = .001). When the outcomes were stratified by class of inflammatory event, these results were similar. The aOR was 3.09 (95% CI, 1.19-8.04; P = .02) for inflammatory demyelinating CNS events and was 2.97 (95% CI, 1.15-7.65; P = .02) for inflammatory nondemyelinating events.
Dr. Kunchok noted that the association between the inhibitors and nondemyelinating events was “a novel finding from this study.”
In secondary analyses, patients with rheumatoid arthritis and exposure to TNF inhibitors had the strongest association with any inflammatory CNS event (aOR, 4.82; 95% CI, 1.62-14.36; P = .005).
A pooled cohort comprising only the participants with the other autoimmune diseases did not show a significant association between exposure to TNF inhibitors and development of CNS events (P = .09).
“Because of the lack of power, further stratification by individual autoimmune diseases was not analyzed,” the investigators reported.
Although the overall findings showed that exposure to TNF inhibitors was linked to increased risk for inflammatory events, whether this association “represents de novo or exacerbated inflammatory pathways requires further research,” the authors wrote.
Dr. Kunchok added that more research, especially population-based studies, is also needed to examine the incidence of these inflammatory CNS events in patients exposed to TNF-alpha inhibitors.
Adds to the literature
In an accompanying editorial, Jeffrey M. Gelfand, MD, department of neurology at the University of California, San Francisco, and Jinoos Yazdany, MD, Zuckerberg San Francisco General Hospital at UCSF, noted that although the study adds to the literature, the magnitude of the risk found “remains unclear.”
“Randomized clinical trials are not suited to the study of rare adverse events,” Dr. Gelfand and Dr. Yazdany wrote. They agree with Dr. Kunchok that “next steps should include population-based observational studies that control for disease severity.”
Still, the current study provides additional evidence of rare adverse events in patients receiving TNF inhibitors, they noted. So how should prescribers proceed?
“As with all treatments, the risk-benefit ratio for the individual patient’s situation must be weighed and appropriate counseling must be given to facilitate shared decision-making discussions,” wrote the editorialists.
“Given what is known about the risk of harm, avoiding TNF inhibitors is advisable in patients with known MS,” they wrote.
In addition, neurologic consultation can be helpful for clarifying diagnoses and providing advice on monitoring strategies for TNF inhibitor treatment in those with possible MS or other demyelinating conditions, noted the editorialists.
“In patients who develop new concerning neurological symptoms while receiving TNF inhibitor treatment, timely evaluation is indicated, including consideration of neuroinflammatory, infectious, and neurological diagnoses that may be unrelated to treatment,” they added.
“Broader awareness of risks that studies such as this one by Kunchok et al provide can ... encourage timelier recognition of potential TNF inhibitor–associated neuroinflammatory events and may improve outcomes for patients,” Dr. Gelfand and Dr. Yazdany concluded.
The study was funded by a grant from the National Center for Advancing Translational Sciences. Dr. Kunchok reports having received research funding from Biogen outside this study. A full list of disclosures for the other study authors is in the original article. Dr. Gelfand reports having received g rants for a clinical trial from Genentech and consulting fees from Biogen, Alexion, Theranica, Impel Neuropharma, Advanced Clinical, Biohaven, and Satsuma. Dr. Yazdany reports having received grants from Pfizer and consulting fees from AstraZeneca and Eli Lilly outside the submitted work.
A version of this article originally appeared on Medscape.com.
Novel oral drug improves sunlight tolerance in patients with erythropoietic protoporphyria
in a multicenter, phase 2, randomized trial, Kirstine Belongie, PhD, reported at the virtual annual meeting of the American Academy of Dermatology.
Based upon these favorable phase 2 results, a pivotal phase 3 clinical trial is now underway, added Dr. Belongie of Mitsubishi Tanabe Pharma Development America, in Jersey City, N.J., the study sponsor.
Erythropoietic protoporphyria (EPP) is the most common cutaneous porphyria as well as the most common porphyria of any type in children. It’s a rare but devastating disorder, with an incidence estimated at 1 in 75,000-200,000. It involves acute cutaneous photosensitivity to sunlight, which takes the form of incapacitating burning pain that lasts 3-7 days and is then followed by erythema and edema.
“These phototoxic reactions are extremely painful and cause the patients to have extreme fear of the sun. They do everything they can to avoid the sun. It leads to a highly impaired quality of life that’s restricted to the indoors,” she explained.
Current first-line therapy is sun avoidance, the use of zinc oxide sunblock, and protective clothing. It’s inadequate for most patients. “There is a tremendously high unmet medical need for treatment options, especially in the pediatric population,” Dr. Belongie observed.
Patients with EPP experience prodromal symptoms – tingling, itching, and burning – which serve as a signal to get out of the sun immediately. As demonstrated in the phase 2 trial, dersimelagon prolongs the time to onset of these prodromal symptoms by increasing melanin density in the skin in a dose-dependent fashion.
The phase 2 study included 102 EPP patients, with an average age 40 years, at 9 sites, who were randomized double blind to 16 weeks of dersimelagon at 100 mg or 300 mg once daily or placebo. The goal was to increase their pain-free sunlight exposure time.
The primary endpoint was change from baseline to week 16 in the average daily time to first prodromal symptoms. There was a 20-minute increase with placebo, a 74-minute gain with dersimelagon at 100 mg, and an 83-minute gain with dersimelagon at 300 mg. The difference between active medication and placebo became significant at week 6.
Treatment-emergent adverse events leading to study discontinuation occurred in one patient on dersimelagon at 100 mg/day, five patients on the higher dose, and none on placebo. Dr. Belangie said that the drug was well tolerated, with roughly 90% of adverse events being mild or moderate in severity. The frequency of adverse events was dose-related. The most common were headache, nausea, and diarrhea, occurring in 29%, 46%, and 23%, respectively, of patients on dersimelagon at 300 mg/day, compared with 18%, 12% and 12% of those on placebo.
Consistent with the drug’s mechanism of action, there was also a dose-related increase in hyperpigmentation side effects. New freckles were documented in 15% and 31% of patients on low- and high-dose dersimelagon, skin hyperpigmentation in 9% and 31%, and melanocytic nevi in 12% and 20%.
The ongoing double-blind, international, phase 3 trial includes not only patients with EPP, but also individuals with X-linked porphyria, which has similar clinical symptoms. The trial is double blind for the first 26 weeks, followed by another 26 weeks of open-label treatment.
EPP is an inherited metabolic disorder caused by a genetic mutation resulting in deficient activity of the enzyme ferrochelatase. This leads to accumulation of protoporphyrin IX in erythrocytes, skin, and the liver. The excess protoporphyrin is excreted in bile and can cause hepatobiliary disease. Indeed, up to 5% of patients with EPP develop liver failure.
In October 2019, the Food and Drug Administration approved afamelanotide (Scenesse), also a melanocortin-1 receptor agonist, to increase pain-free light exposure in adults with a history of phototoxic reactions EPP; this was the first FDA-approved treatment for helping EPP patients increase their exposure to light, according to the agency. It is administered as an implant every 2 months.
Dr. Belangie is employed by the study sponsor.
in a multicenter, phase 2, randomized trial, Kirstine Belongie, PhD, reported at the virtual annual meeting of the American Academy of Dermatology.
Based upon these favorable phase 2 results, a pivotal phase 3 clinical trial is now underway, added Dr. Belongie of Mitsubishi Tanabe Pharma Development America, in Jersey City, N.J., the study sponsor.
Erythropoietic protoporphyria (EPP) is the most common cutaneous porphyria as well as the most common porphyria of any type in children. It’s a rare but devastating disorder, with an incidence estimated at 1 in 75,000-200,000. It involves acute cutaneous photosensitivity to sunlight, which takes the form of incapacitating burning pain that lasts 3-7 days and is then followed by erythema and edema.
“These phototoxic reactions are extremely painful and cause the patients to have extreme fear of the sun. They do everything they can to avoid the sun. It leads to a highly impaired quality of life that’s restricted to the indoors,” she explained.
Current first-line therapy is sun avoidance, the use of zinc oxide sunblock, and protective clothing. It’s inadequate for most patients. “There is a tremendously high unmet medical need for treatment options, especially in the pediatric population,” Dr. Belongie observed.
Patients with EPP experience prodromal symptoms – tingling, itching, and burning – which serve as a signal to get out of the sun immediately. As demonstrated in the phase 2 trial, dersimelagon prolongs the time to onset of these prodromal symptoms by increasing melanin density in the skin in a dose-dependent fashion.
The phase 2 study included 102 EPP patients, with an average age 40 years, at 9 sites, who were randomized double blind to 16 weeks of dersimelagon at 100 mg or 300 mg once daily or placebo. The goal was to increase their pain-free sunlight exposure time.
The primary endpoint was change from baseline to week 16 in the average daily time to first prodromal symptoms. There was a 20-minute increase with placebo, a 74-minute gain with dersimelagon at 100 mg, and an 83-minute gain with dersimelagon at 300 mg. The difference between active medication and placebo became significant at week 6.
Treatment-emergent adverse events leading to study discontinuation occurred in one patient on dersimelagon at 100 mg/day, five patients on the higher dose, and none on placebo. Dr. Belangie said that the drug was well tolerated, with roughly 90% of adverse events being mild or moderate in severity. The frequency of adverse events was dose-related. The most common were headache, nausea, and diarrhea, occurring in 29%, 46%, and 23%, respectively, of patients on dersimelagon at 300 mg/day, compared with 18%, 12% and 12% of those on placebo.
Consistent with the drug’s mechanism of action, there was also a dose-related increase in hyperpigmentation side effects. New freckles were documented in 15% and 31% of patients on low- and high-dose dersimelagon, skin hyperpigmentation in 9% and 31%, and melanocytic nevi in 12% and 20%.
The ongoing double-blind, international, phase 3 trial includes not only patients with EPP, but also individuals with X-linked porphyria, which has similar clinical symptoms. The trial is double blind for the first 26 weeks, followed by another 26 weeks of open-label treatment.
EPP is an inherited metabolic disorder caused by a genetic mutation resulting in deficient activity of the enzyme ferrochelatase. This leads to accumulation of protoporphyrin IX in erythrocytes, skin, and the liver. The excess protoporphyrin is excreted in bile and can cause hepatobiliary disease. Indeed, up to 5% of patients with EPP develop liver failure.
In October 2019, the Food and Drug Administration approved afamelanotide (Scenesse), also a melanocortin-1 receptor agonist, to increase pain-free light exposure in adults with a history of phototoxic reactions EPP; this was the first FDA-approved treatment for helping EPP patients increase their exposure to light, according to the agency. It is administered as an implant every 2 months.
Dr. Belangie is employed by the study sponsor.
in a multicenter, phase 2, randomized trial, Kirstine Belongie, PhD, reported at the virtual annual meeting of the American Academy of Dermatology.
Based upon these favorable phase 2 results, a pivotal phase 3 clinical trial is now underway, added Dr. Belongie of Mitsubishi Tanabe Pharma Development America, in Jersey City, N.J., the study sponsor.
Erythropoietic protoporphyria (EPP) is the most common cutaneous porphyria as well as the most common porphyria of any type in children. It’s a rare but devastating disorder, with an incidence estimated at 1 in 75,000-200,000. It involves acute cutaneous photosensitivity to sunlight, which takes the form of incapacitating burning pain that lasts 3-7 days and is then followed by erythema and edema.
“These phototoxic reactions are extremely painful and cause the patients to have extreme fear of the sun. They do everything they can to avoid the sun. It leads to a highly impaired quality of life that’s restricted to the indoors,” she explained.
Current first-line therapy is sun avoidance, the use of zinc oxide sunblock, and protective clothing. It’s inadequate for most patients. “There is a tremendously high unmet medical need for treatment options, especially in the pediatric population,” Dr. Belongie observed.
Patients with EPP experience prodromal symptoms – tingling, itching, and burning – which serve as a signal to get out of the sun immediately. As demonstrated in the phase 2 trial, dersimelagon prolongs the time to onset of these prodromal symptoms by increasing melanin density in the skin in a dose-dependent fashion.
The phase 2 study included 102 EPP patients, with an average age 40 years, at 9 sites, who were randomized double blind to 16 weeks of dersimelagon at 100 mg or 300 mg once daily or placebo. The goal was to increase their pain-free sunlight exposure time.
The primary endpoint was change from baseline to week 16 in the average daily time to first prodromal symptoms. There was a 20-minute increase with placebo, a 74-minute gain with dersimelagon at 100 mg, and an 83-minute gain with dersimelagon at 300 mg. The difference between active medication and placebo became significant at week 6.
Treatment-emergent adverse events leading to study discontinuation occurred in one patient on dersimelagon at 100 mg/day, five patients on the higher dose, and none on placebo. Dr. Belangie said that the drug was well tolerated, with roughly 90% of adverse events being mild or moderate in severity. The frequency of adverse events was dose-related. The most common were headache, nausea, and diarrhea, occurring in 29%, 46%, and 23%, respectively, of patients on dersimelagon at 300 mg/day, compared with 18%, 12% and 12% of those on placebo.
Consistent with the drug’s mechanism of action, there was also a dose-related increase in hyperpigmentation side effects. New freckles were documented in 15% and 31% of patients on low- and high-dose dersimelagon, skin hyperpigmentation in 9% and 31%, and melanocytic nevi in 12% and 20%.
The ongoing double-blind, international, phase 3 trial includes not only patients with EPP, but also individuals with X-linked porphyria, which has similar clinical symptoms. The trial is double blind for the first 26 weeks, followed by another 26 weeks of open-label treatment.
EPP is an inherited metabolic disorder caused by a genetic mutation resulting in deficient activity of the enzyme ferrochelatase. This leads to accumulation of protoporphyrin IX in erythrocytes, skin, and the liver. The excess protoporphyrin is excreted in bile and can cause hepatobiliary disease. Indeed, up to 5% of patients with EPP develop liver failure.
In October 2019, the Food and Drug Administration approved afamelanotide (Scenesse), also a melanocortin-1 receptor agonist, to increase pain-free light exposure in adults with a history of phototoxic reactions EPP; this was the first FDA-approved treatment for helping EPP patients increase their exposure to light, according to the agency. It is administered as an implant every 2 months.
Dr. Belangie is employed by the study sponsor.
FROM AAD 2020
COVID-19 linked to development of myasthenia gravis
Myasthenia gravis should be added to the growing list of potential neurological sequelae associated with COVID-19, new research suggests. Clinicians from Italy have described what they believe are
“I think it is possible that there could be many more cases,” said lead author Domenico Restivo, MD, of the Garibaldi Hospital, Catania, Italy. “In fact, myasthenia gravis could be underestimated especially in the course of COVID-19 infection in which a specific muscular weakness is frequently present. For this reason, this association is easy to miss if not top of mind,” Dr. Restivo said.
None of the three patients had previous neurologic or autoimmune disorders. In all three cases, symptoms of myasthenia gravis appeared within 5-7 days after onset of fever caused by SARS-CoV-2 infection. The time from presumed SARS-CoV-2 infection to myasthenia gravis symptoms “is consistent with the time from infection to symptoms in other neurologic disorders triggered by infections,” the investigators reported.
The findings were published online August 10 in Annals of Internal Medicine.
First patients
The first patient described in the report was a 64-year-old man who had a fever as high as 39° C (102.2° F) for 4 days. Five days after fever onset, he developed diplopia and muscle fatigue. The patient’s neurologic examination was “unremarkable.” Computed tomography (CT) of the thorax excluded thymoma, and findings on chest radiograph were normal. He tested positive for SARS-CoV-2 on nasopharyngeal swab and real-time reverse transcriptase polymerase chain reaction (RT-PCR).
The patient’s symptoms led the investigators to suspect myasthenia gravis. Repetitive stimulation of the patient’s facial nerve showed a 57% decrement, confirming involvement of the postsynaptic neuromuscular junction. The concentration of AChR antibodies in serum was also elevated (22.8 pmol/L; reference range, <0.4 pmol/L). The patient was treated with pyridostigmine bromide and prednisone and had a response “typical for someone with myasthenia gravis,” the researchers wrote.
The second patient was a 68-year-old man who had a fever as high as 38.8° C (101.8° F) for 7 days. On day 7, he developed muscle fatigue, diplopia, and dysphagia. Findings of a chest CT and neurologic exam were normal. Nasopharyngeal swab and RT-PCR testing for COVID-19 were positive. As with the first patient, myasthenia gravis was suspected because of the patient’s symptoms. Repetitive nerve stimulation revealed a postsynaptic deficit of neuromuscular transmission of the facial (52%) and ulnar (21%) nerves. His serum AChR antibody level was elevated (27.6 pmol/L). The patient improved after one cycle of intravenous immunoglobulin treatment.
Possible mechanisms
The third patient was a 71-year-old woman with cough and a fever up to 38.6° C (101.5° F) for 6 days. She initially tested negative for SARS-CoV-2 on nasopharyngeal swab and RT-PCR. Five days after her symptoms began, she developed bilateral ocular ptosis, diplopia, and hypophonia. CT of the thorax excluded thymoma but showed bilateral interstitial pneumonia. On day 6, she developed dysphagia and respiratory failure, and was transferred to the ICU where she received mechanical ventilation.
Repetitive nerve stimulation revealed a postsynaptic deficit of neuromuscular transmission of the ulnar nerve (56%), and her serum AChR antibody level was elevated (35.6 pmol/L). Five days later, a second nasopharyngeal swab test for SARS-CoV-2 was positive. The patient improved following plasmapheresis treatment and was successfully extubated.
The investigators noted that this patient received hydroxychloroquine the day after the onset of neurologic symptoms, but the drug was withdrawn a day later, so they do not believe that it caused the symptoms of myasthenia gravis.
The observations in these three patients are “consistent with reports of other infections that induce autoimmune disorders, as well as with the growing evidence of other neurologic disorders with presumed autoimmune mechanisms after COVID-19 onset,” the researchers wrote.
They offered several possible explanations for the link between COVID-19 and myasthenia gravis. “Antibodies that are directed against SARS-CoV-2 proteins may cross-react with AChR subunits, because the virus has epitopes that are similar to components of the neuromuscular junction; this is known to occur in other neurologic autoimmune disorders after infection. Alternatively, COVID-19 infection may break immunologic self-tolerance,” the investigators wrote.
“The main message for clinicians is that myasthenia gravis, as well as other neurological disorders associated with autoimmunity, could occur in the course of SARS-CoV-2 infection,” Dr. Restivo said. Prompt recognition of the disease “could lead to a drug treatment that limits its evolution as quickly as possible,” he added.
An “unmasking”
Commenting on the findings, Anthony Geraci, MD, director of neuromuscular medicine, Northwell Health, Great Neck, N.Y., said these case reports of myasthenia gravis after SARS-CoV-2 infection are “not unique or novel as there has been a long understanding that seropositive [AChR antibody-positive] myasthenia gravis can and is frequently ‘unmasked’ in the setting” of several viral and bacterial infections.
“Antibodies in myasthenia gravis are of a type that take several weeks to develop to measurable levels as in the reported cases by Restivo et al., giving strong support to the notion that subclinical myasthenia gravis can be immunologically upregulated in the setting of viral infection and this is a far more likely explanation of the observed association reported,” added Dr. Geraci, who was not involved with the research.
He noted that, at his institution, “we have also observed ocular myasthenia gravis emerge in patients with SARS-CoV-2 infection, with similar double vision and lid droop, as we have seen similarly in patients with Zika, West Nile, and other viral infections, as well as a multiplicity of bacterial infections.”
“Most of our observed patients have responded to treatment much the same as reported by the three cases from Restivo and colleagues,” Dr.Geraci reported.
The authors of the study disclosed no conflicts of interest.
A version of this article originally appeared on Medscape.com.
Myasthenia gravis should be added to the growing list of potential neurological sequelae associated with COVID-19, new research suggests. Clinicians from Italy have described what they believe are
“I think it is possible that there could be many more cases,” said lead author Domenico Restivo, MD, of the Garibaldi Hospital, Catania, Italy. “In fact, myasthenia gravis could be underestimated especially in the course of COVID-19 infection in which a specific muscular weakness is frequently present. For this reason, this association is easy to miss if not top of mind,” Dr. Restivo said.
None of the three patients had previous neurologic or autoimmune disorders. In all three cases, symptoms of myasthenia gravis appeared within 5-7 days after onset of fever caused by SARS-CoV-2 infection. The time from presumed SARS-CoV-2 infection to myasthenia gravis symptoms “is consistent with the time from infection to symptoms in other neurologic disorders triggered by infections,” the investigators reported.
The findings were published online August 10 in Annals of Internal Medicine.
First patients
The first patient described in the report was a 64-year-old man who had a fever as high as 39° C (102.2° F) for 4 days. Five days after fever onset, he developed diplopia and muscle fatigue. The patient’s neurologic examination was “unremarkable.” Computed tomography (CT) of the thorax excluded thymoma, and findings on chest radiograph were normal. He tested positive for SARS-CoV-2 on nasopharyngeal swab and real-time reverse transcriptase polymerase chain reaction (RT-PCR).
The patient’s symptoms led the investigators to suspect myasthenia gravis. Repetitive stimulation of the patient’s facial nerve showed a 57% decrement, confirming involvement of the postsynaptic neuromuscular junction. The concentration of AChR antibodies in serum was also elevated (22.8 pmol/L; reference range, <0.4 pmol/L). The patient was treated with pyridostigmine bromide and prednisone and had a response “typical for someone with myasthenia gravis,” the researchers wrote.
The second patient was a 68-year-old man who had a fever as high as 38.8° C (101.8° F) for 7 days. On day 7, he developed muscle fatigue, diplopia, and dysphagia. Findings of a chest CT and neurologic exam were normal. Nasopharyngeal swab and RT-PCR testing for COVID-19 were positive. As with the first patient, myasthenia gravis was suspected because of the patient’s symptoms. Repetitive nerve stimulation revealed a postsynaptic deficit of neuromuscular transmission of the facial (52%) and ulnar (21%) nerves. His serum AChR antibody level was elevated (27.6 pmol/L). The patient improved after one cycle of intravenous immunoglobulin treatment.
Possible mechanisms
The third patient was a 71-year-old woman with cough and a fever up to 38.6° C (101.5° F) for 6 days. She initially tested negative for SARS-CoV-2 on nasopharyngeal swab and RT-PCR. Five days after her symptoms began, she developed bilateral ocular ptosis, diplopia, and hypophonia. CT of the thorax excluded thymoma but showed bilateral interstitial pneumonia. On day 6, she developed dysphagia and respiratory failure, and was transferred to the ICU where she received mechanical ventilation.
Repetitive nerve stimulation revealed a postsynaptic deficit of neuromuscular transmission of the ulnar nerve (56%), and her serum AChR antibody level was elevated (35.6 pmol/L). Five days later, a second nasopharyngeal swab test for SARS-CoV-2 was positive. The patient improved following plasmapheresis treatment and was successfully extubated.
The investigators noted that this patient received hydroxychloroquine the day after the onset of neurologic symptoms, but the drug was withdrawn a day later, so they do not believe that it caused the symptoms of myasthenia gravis.
The observations in these three patients are “consistent with reports of other infections that induce autoimmune disorders, as well as with the growing evidence of other neurologic disorders with presumed autoimmune mechanisms after COVID-19 onset,” the researchers wrote.
They offered several possible explanations for the link between COVID-19 and myasthenia gravis. “Antibodies that are directed against SARS-CoV-2 proteins may cross-react with AChR subunits, because the virus has epitopes that are similar to components of the neuromuscular junction; this is known to occur in other neurologic autoimmune disorders after infection. Alternatively, COVID-19 infection may break immunologic self-tolerance,” the investigators wrote.
“The main message for clinicians is that myasthenia gravis, as well as other neurological disorders associated with autoimmunity, could occur in the course of SARS-CoV-2 infection,” Dr. Restivo said. Prompt recognition of the disease “could lead to a drug treatment that limits its evolution as quickly as possible,” he added.
An “unmasking”
Commenting on the findings, Anthony Geraci, MD, director of neuromuscular medicine, Northwell Health, Great Neck, N.Y., said these case reports of myasthenia gravis after SARS-CoV-2 infection are “not unique or novel as there has been a long understanding that seropositive [AChR antibody-positive] myasthenia gravis can and is frequently ‘unmasked’ in the setting” of several viral and bacterial infections.
“Antibodies in myasthenia gravis are of a type that take several weeks to develop to measurable levels as in the reported cases by Restivo et al., giving strong support to the notion that subclinical myasthenia gravis can be immunologically upregulated in the setting of viral infection and this is a far more likely explanation of the observed association reported,” added Dr. Geraci, who was not involved with the research.
He noted that, at his institution, “we have also observed ocular myasthenia gravis emerge in patients with SARS-CoV-2 infection, with similar double vision and lid droop, as we have seen similarly in patients with Zika, West Nile, and other viral infections, as well as a multiplicity of bacterial infections.”
“Most of our observed patients have responded to treatment much the same as reported by the three cases from Restivo and colleagues,” Dr.Geraci reported.
The authors of the study disclosed no conflicts of interest.
A version of this article originally appeared on Medscape.com.
Myasthenia gravis should be added to the growing list of potential neurological sequelae associated with COVID-19, new research suggests. Clinicians from Italy have described what they believe are
“I think it is possible that there could be many more cases,” said lead author Domenico Restivo, MD, of the Garibaldi Hospital, Catania, Italy. “In fact, myasthenia gravis could be underestimated especially in the course of COVID-19 infection in which a specific muscular weakness is frequently present. For this reason, this association is easy to miss if not top of mind,” Dr. Restivo said.
None of the three patients had previous neurologic or autoimmune disorders. In all three cases, symptoms of myasthenia gravis appeared within 5-7 days after onset of fever caused by SARS-CoV-2 infection. The time from presumed SARS-CoV-2 infection to myasthenia gravis symptoms “is consistent with the time from infection to symptoms in other neurologic disorders triggered by infections,” the investigators reported.
The findings were published online August 10 in Annals of Internal Medicine.
First patients
The first patient described in the report was a 64-year-old man who had a fever as high as 39° C (102.2° F) for 4 days. Five days after fever onset, he developed diplopia and muscle fatigue. The patient’s neurologic examination was “unremarkable.” Computed tomography (CT) of the thorax excluded thymoma, and findings on chest radiograph were normal. He tested positive for SARS-CoV-2 on nasopharyngeal swab and real-time reverse transcriptase polymerase chain reaction (RT-PCR).
The patient’s symptoms led the investigators to suspect myasthenia gravis. Repetitive stimulation of the patient’s facial nerve showed a 57% decrement, confirming involvement of the postsynaptic neuromuscular junction. The concentration of AChR antibodies in serum was also elevated (22.8 pmol/L; reference range, <0.4 pmol/L). The patient was treated with pyridostigmine bromide and prednisone and had a response “typical for someone with myasthenia gravis,” the researchers wrote.
The second patient was a 68-year-old man who had a fever as high as 38.8° C (101.8° F) for 7 days. On day 7, he developed muscle fatigue, diplopia, and dysphagia. Findings of a chest CT and neurologic exam were normal. Nasopharyngeal swab and RT-PCR testing for COVID-19 were positive. As with the first patient, myasthenia gravis was suspected because of the patient’s symptoms. Repetitive nerve stimulation revealed a postsynaptic deficit of neuromuscular transmission of the facial (52%) and ulnar (21%) nerves. His serum AChR antibody level was elevated (27.6 pmol/L). The patient improved after one cycle of intravenous immunoglobulin treatment.
Possible mechanisms
The third patient was a 71-year-old woman with cough and a fever up to 38.6° C (101.5° F) for 6 days. She initially tested negative for SARS-CoV-2 on nasopharyngeal swab and RT-PCR. Five days after her symptoms began, she developed bilateral ocular ptosis, diplopia, and hypophonia. CT of the thorax excluded thymoma but showed bilateral interstitial pneumonia. On day 6, she developed dysphagia and respiratory failure, and was transferred to the ICU where she received mechanical ventilation.
Repetitive nerve stimulation revealed a postsynaptic deficit of neuromuscular transmission of the ulnar nerve (56%), and her serum AChR antibody level was elevated (35.6 pmol/L). Five days later, a second nasopharyngeal swab test for SARS-CoV-2 was positive. The patient improved following plasmapheresis treatment and was successfully extubated.
The investigators noted that this patient received hydroxychloroquine the day after the onset of neurologic symptoms, but the drug was withdrawn a day later, so they do not believe that it caused the symptoms of myasthenia gravis.
The observations in these three patients are “consistent with reports of other infections that induce autoimmune disorders, as well as with the growing evidence of other neurologic disorders with presumed autoimmune mechanisms after COVID-19 onset,” the researchers wrote.
They offered several possible explanations for the link between COVID-19 and myasthenia gravis. “Antibodies that are directed against SARS-CoV-2 proteins may cross-react with AChR subunits, because the virus has epitopes that are similar to components of the neuromuscular junction; this is known to occur in other neurologic autoimmune disorders after infection. Alternatively, COVID-19 infection may break immunologic self-tolerance,” the investigators wrote.
“The main message for clinicians is that myasthenia gravis, as well as other neurological disorders associated with autoimmunity, could occur in the course of SARS-CoV-2 infection,” Dr. Restivo said. Prompt recognition of the disease “could lead to a drug treatment that limits its evolution as quickly as possible,” he added.
An “unmasking”
Commenting on the findings, Anthony Geraci, MD, director of neuromuscular medicine, Northwell Health, Great Neck, N.Y., said these case reports of myasthenia gravis after SARS-CoV-2 infection are “not unique or novel as there has been a long understanding that seropositive [AChR antibody-positive] myasthenia gravis can and is frequently ‘unmasked’ in the setting” of several viral and bacterial infections.
“Antibodies in myasthenia gravis are of a type that take several weeks to develop to measurable levels as in the reported cases by Restivo et al., giving strong support to the notion that subclinical myasthenia gravis can be immunologically upregulated in the setting of viral infection and this is a far more likely explanation of the observed association reported,” added Dr. Geraci, who was not involved with the research.
He noted that, at his institution, “we have also observed ocular myasthenia gravis emerge in patients with SARS-CoV-2 infection, with similar double vision and lid droop, as we have seen similarly in patients with Zika, West Nile, and other viral infections, as well as a multiplicity of bacterial infections.”
“Most of our observed patients have responded to treatment much the same as reported by the three cases from Restivo and colleagues,” Dr.Geraci reported.
The authors of the study disclosed no conflicts of interest.
A version of this article originally appeared on Medscape.com.
Canakinumab (Ilaris) tapering tested in systemic JIA trial
A trial that tested two ways of tapering canakinumab (Ilaris) monotherapy in children with systemic juvenile idiopathic arthritis (sJIA) showed that both approaches might be feasible, but the lack of a control arm means that more data are needed before putting either into routine clinical practice.

Pierre Quartier, MD, and associates reported in Arthritis & Rheumatology. That’s with the proviso that children are taking canakinumab at the recommended dose of 4 mg/kg every 4 weeks and achieve clinical remission before any tapering is started.
The researchers found that 70%-80% of children who were in complete clinical remission (CR) maintained this for 6 months after the first dose-tapering step had been taken. However, at least one-fifth of study participants experienced a disease flare during treatment withdrawal, and only one-third were able to discontinue treatment altogether, which suggests continued treatment is needed.
“The results are a step in the right direction,” commented Athimaleipet Ramanan, MBBS, a consultant pediatric rheumatologist who was not involved in the study. They “offer the first vision of whether we can actually taper a medication” in sJIA because “until now we have not had any evidence for this,” he added.
“What we really need to know, which the study doesn’t tell you, is: Is decreasing the dose better than stopping?” said Dr. Ramanan, of the Bristol (England) Royal Hospital for Children.
Another thing that is important to know is: Does reducing the dose lead to the development of anti-drug antibodies? he said.

“There were some concerns that, when you give less of a monoclonal antibody, you might get more neutralizing anti-drug antibodies or you might make a drug more immunogenic,” he said. These data perhaps suggest that this isn’t the case because only one child on one occasion had detectable non-neutralizing anti-drug antibodies during the entire study, and that was 11 weeks after the last dose of canakinumab had been given.
Study results and design
Canakinumab is a monoclonal antibody that inhibits interleukin-1 (IL-1) that has been approved in the United States and Europe for the treatment of sJIA since 2013. Reducing a child’s exposure to canakinumab once their disease is under control is an attractive proposition given the treat-to-target approach used increasingly throughout modern rheumatologic practice. It could also help reduce the cost of what is an expensive treatment, compared with other available options for sJIA such as the interleukin-6 inhibitor tocilizumab (Actemra) or another IL-1 inhibitor anakinra (Kineret), which is not FDA-approved for use in sJIA and requires weekly injections.
“We don’t want to the disease to reappear, to flare, but we also don’t want to overuse treatments in these patients,” explained Dr. Quartier of Hôpital Necker-Enfants Malades, Assistance Publique-Hôpitaux de Paris in an interview.
The study he coconducted, given the acronym B-SPECIFIC-4 Patients, was an open-label study that consisted of two parts. In part 1, 182 children were given subcutaneous canakinumab 4 mg/kg every 4 weeks. In part 2, 76 children who were in complete CR after canakinumab treatment were randomized to one of two tapering strategies. In one arm, the dose of canakinumab was reduced from 4 mg/kg to 2 mg/kg given every 4 weeks before eventually discontinuing treatment, and in other arm the duration between dosing was increased by 4-week intervals, from 4 to 8 weeks, then 12 weeks, then discontinuation.
If children were taking glucocorticoids or methotrexate, the treating physicians were encouraged to stop these medications if possible, with 34%-39% and 42%-59% of children, respectively, being able to do so. The rate depended on whether children had received canakinumab before entering the study because some had been recruited from a long-term extension study while others were naive to the biologic; all had inactive disease at study entry.
This was the first study to evaluate the effectiveness of canakinumab in enabling the discontinuation of methotrexate, but the primary objective was to assess whether more than 40% of randomized patients in either discontinuation arm remained in CR for 24 weeks after the first step of discontinuation. This was achieved in 71% of children who were in the dose-reduction arm and in 84% of children in the dose-prolongation arm (P ≤ .0001 for each arm vs. 40%).
Prior exposure to canakinumab did not seem to affect the maintenance of CR, but it was also found that more children who maintained CR at their second but not their first attempt at tapering were in CR than those who were still in CR at the first step (76% and 89% in the dose-reduction and dose-prolongation arms, respectively).
Among the two dosing regimens, failure occurred in 18% of children in the dose-reduction arm at the first step, 10% at the second, and 8% at the third, whereas 2.7% of children in the dose-prolongation arm experienced regimen failure at the first step, 6.1% at the second, and 15% at the third.
No substantial difference between the two tapering approaches was observed because the study was not powered to look at this. There was also no control arm, such as a group continuing treatment while the other groups tapered, or as Dr. Ramanan had pointed out, stopping canakinumab altogether.
“As long as the treatment was continued, even at very low dosage, most patients remained in inactive disease,” Dr. Quartier said. He added, however, that only a minority of patients could stop treatment completely. Treatment should not be stopped abruptly, he advised, because this was associated with a substantial number of disease flares that needed treatment to be reinstated.
These findings suggest that “a certain level of sustained inhibition of the IL-1 pathway seems important for the maintenance of CR in most sJIA patients,” Dr. Quartier and coinvestigators wrote in their article.
They added: “We believe that these results are relevant for clinical practice, particularly for designing personalized tapering strategies that can allow an adequate control of disease while minimizing the side effects of certain medications, notably glucocorticoids.”
The study was funded by Novartis in collaboration with the Pediatric Rheumatology International Trials Organization and the Pediatric Rheumatology Collaborative Study Group. Dr. Quartier was an investigator for the trial and has received research and consultancy fees from Novartis, among other pharmaceutical companies. Two of his coauthors are employees of Novartis. Dr. Ramanan has acted as an investigator in prior canakinumab trials and has received consultancy fees from Novartis and multiple other companies.
SOURCE: Quartier P et al. Arthritis Rheumatol. 2020 Aug 11. doi: 10.1002/art.41488.
A trial that tested two ways of tapering canakinumab (Ilaris) monotherapy in children with systemic juvenile idiopathic arthritis (sJIA) showed that both approaches might be feasible, but the lack of a control arm means that more data are needed before putting either into routine clinical practice.
Pierre Quartier, MD, and associates reported in Arthritis & Rheumatology. That’s with the proviso that children are taking canakinumab at the recommended dose of 4 mg/kg every 4 weeks and achieve clinical remission before any tapering is started.
The researchers found that 70%-80% of children who were in complete clinical remission (CR) maintained this for 6 months after the first dose-tapering step had been taken. However, at least one-fifth of study participants experienced a disease flare during treatment withdrawal, and only one-third were able to discontinue treatment altogether, which suggests continued treatment is needed.
“The results are a step in the right direction,” commented Athimaleipet Ramanan, MBBS, a consultant pediatric rheumatologist who was not involved in the study. They “offer the first vision of whether we can actually taper a medication” in sJIA because “until now we have not had any evidence for this,” he added.
“What we really need to know, which the study doesn’t tell you, is: Is decreasing the dose better than stopping?” said Dr. Ramanan, of the Bristol (England) Royal Hospital for Children.
Another thing that is important to know is: Does reducing the dose lead to the development of anti-drug antibodies? he said.
“There were some concerns that, when you give less of a monoclonal antibody, you might get more neutralizing anti-drug antibodies or you might make a drug more immunogenic,” he said. These data perhaps suggest that this isn’t the case because only one child on one occasion had detectable non-neutralizing anti-drug antibodies during the entire study, and that was 11 weeks after the last dose of canakinumab had been given.
Study results and design
Canakinumab is a monoclonal antibody that inhibits interleukin-1 (IL-1) that has been approved in the United States and Europe for the treatment of sJIA since 2013. Reducing a child’s exposure to canakinumab once their disease is under control is an attractive proposition given the treat-to-target approach used increasingly throughout modern rheumatologic practice. It could also help reduce the cost of what is an expensive treatment, compared with other available options for sJIA such as the interleukin-6 inhibitor tocilizumab (Actemra) or another IL-1 inhibitor anakinra (Kineret), which is not FDA-approved for use in sJIA and requires weekly injections.
“We don’t want to the disease to reappear, to flare, but we also don’t want to overuse treatments in these patients,” explained Dr. Quartier of Hôpital Necker-Enfants Malades, Assistance Publique-Hôpitaux de Paris in an interview.
The study he coconducted, given the acronym B-SPECIFIC-4 Patients, was an open-label study that consisted of two parts. In part 1, 182 children were given subcutaneous canakinumab 4 mg/kg every 4 weeks. In part 2, 76 children who were in complete CR after canakinumab treatment were randomized to one of two tapering strategies. In one arm, the dose of canakinumab was reduced from 4 mg/kg to 2 mg/kg given every 4 weeks before eventually discontinuing treatment, and in other arm the duration between dosing was increased by 4-week intervals, from 4 to 8 weeks, then 12 weeks, then discontinuation.
If children were taking glucocorticoids or methotrexate, the treating physicians were encouraged to stop these medications if possible, with 34%-39% and 42%-59% of children, respectively, being able to do so. The rate depended on whether children had received canakinumab before entering the study because some had been recruited from a long-term extension study while others were naive to the biologic; all had inactive disease at study entry.
This was the first study to evaluate the effectiveness of canakinumab in enabling the discontinuation of methotrexate, but the primary objective was to assess whether more than 40% of randomized patients in either discontinuation arm remained in CR for 24 weeks after the first step of discontinuation. This was achieved in 71% of children who were in the dose-reduction arm and in 84% of children in the dose-prolongation arm (P ≤ .0001 for each arm vs. 40%).
Prior exposure to canakinumab did not seem to affect the maintenance of CR, but it was also found that more children who maintained CR at their second but not their first attempt at tapering were in CR than those who were still in CR at the first step (76% and 89% in the dose-reduction and dose-prolongation arms, respectively).
Among the two dosing regimens, failure occurred in 18% of children in the dose-reduction arm at the first step, 10% at the second, and 8% at the third, whereas 2.7% of children in the dose-prolongation arm experienced regimen failure at the first step, 6.1% at the second, and 15% at the third.
No substantial difference between the two tapering approaches was observed because the study was not powered to look at this. There was also no control arm, such as a group continuing treatment while the other groups tapered, or as Dr. Ramanan had pointed out, stopping canakinumab altogether.
“As long as the treatment was continued, even at very low dosage, most patients remained in inactive disease,” Dr. Quartier said. He added, however, that only a minority of patients could stop treatment completely. Treatment should not be stopped abruptly, he advised, because this was associated with a substantial number of disease flares that needed treatment to be reinstated.
These findings suggest that “a certain level of sustained inhibition of the IL-1 pathway seems important for the maintenance of CR in most sJIA patients,” Dr. Quartier and coinvestigators wrote in their article.
They added: “We believe that these results are relevant for clinical practice, particularly for designing personalized tapering strategies that can allow an adequate control of disease while minimizing the side effects of certain medications, notably glucocorticoids.”
The study was funded by Novartis in collaboration with the Pediatric Rheumatology International Trials Organization and the Pediatric Rheumatology Collaborative Study Group. Dr. Quartier was an investigator for the trial and has received research and consultancy fees from Novartis, among other pharmaceutical companies. Two of his coauthors are employees of Novartis. Dr. Ramanan has acted as an investigator in prior canakinumab trials and has received consultancy fees from Novartis and multiple other companies.
SOURCE: Quartier P et al. Arthritis Rheumatol. 2020 Aug 11. doi: 10.1002/art.41488.
A trial that tested two ways of tapering canakinumab (Ilaris) monotherapy in children with systemic juvenile idiopathic arthritis (sJIA) showed that both approaches might be feasible, but the lack of a control arm means that more data are needed before putting either into routine clinical practice.
Pierre Quartier, MD, and associates reported in Arthritis & Rheumatology. That’s with the proviso that children are taking canakinumab at the recommended dose of 4 mg/kg every 4 weeks and achieve clinical remission before any tapering is started.
The researchers found that 70%-80% of children who were in complete clinical remission (CR) maintained this for 6 months after the first dose-tapering step had been taken. However, at least one-fifth of study participants experienced a disease flare during treatment withdrawal, and only one-third were able to discontinue treatment altogether, which suggests continued treatment is needed.
“The results are a step in the right direction,” commented Athimaleipet Ramanan, MBBS, a consultant pediatric rheumatologist who was not involved in the study. They “offer the first vision of whether we can actually taper a medication” in sJIA because “until now we have not had any evidence for this,” he added.
“What we really need to know, which the study doesn’t tell you, is: Is decreasing the dose better than stopping?” said Dr. Ramanan, of the Bristol (England) Royal Hospital for Children.
Another thing that is important to know is: Does reducing the dose lead to the development of anti-drug antibodies? he said.
“There were some concerns that, when you give less of a monoclonal antibody, you might get more neutralizing anti-drug antibodies or you might make a drug more immunogenic,” he said. These data perhaps suggest that this isn’t the case because only one child on one occasion had detectable non-neutralizing anti-drug antibodies during the entire study, and that was 11 weeks after the last dose of canakinumab had been given.
Study results and design
Canakinumab is a monoclonal antibody that inhibits interleukin-1 (IL-1) that has been approved in the United States and Europe for the treatment of sJIA since 2013. Reducing a child’s exposure to canakinumab once their disease is under control is an attractive proposition given the treat-to-target approach used increasingly throughout modern rheumatologic practice. It could also help reduce the cost of what is an expensive treatment, compared with other available options for sJIA such as the interleukin-6 inhibitor tocilizumab (Actemra) or another IL-1 inhibitor anakinra (Kineret), which is not FDA-approved for use in sJIA and requires weekly injections.
“We don’t want to the disease to reappear, to flare, but we also don’t want to overuse treatments in these patients,” explained Dr. Quartier of Hôpital Necker-Enfants Malades, Assistance Publique-Hôpitaux de Paris in an interview.
The study he coconducted, given the acronym B-SPECIFIC-4 Patients, was an open-label study that consisted of two parts. In part 1, 182 children were given subcutaneous canakinumab 4 mg/kg every 4 weeks. In part 2, 76 children who were in complete CR after canakinumab treatment were randomized to one of two tapering strategies. In one arm, the dose of canakinumab was reduced from 4 mg/kg to 2 mg/kg given every 4 weeks before eventually discontinuing treatment, and in other arm the duration between dosing was increased by 4-week intervals, from 4 to 8 weeks, then 12 weeks, then discontinuation.
If children were taking glucocorticoids or methotrexate, the treating physicians were encouraged to stop these medications if possible, with 34%-39% and 42%-59% of children, respectively, being able to do so. The rate depended on whether children had received canakinumab before entering the study because some had been recruited from a long-term extension study while others were naive to the biologic; all had inactive disease at study entry.
This was the first study to evaluate the effectiveness of canakinumab in enabling the discontinuation of methotrexate, but the primary objective was to assess whether more than 40% of randomized patients in either discontinuation arm remained in CR for 24 weeks after the first step of discontinuation. This was achieved in 71% of children who were in the dose-reduction arm and in 84% of children in the dose-prolongation arm (P ≤ .0001 for each arm vs. 40%).
Prior exposure to canakinumab did not seem to affect the maintenance of CR, but it was also found that more children who maintained CR at their second but not their first attempt at tapering were in CR than those who were still in CR at the first step (76% and 89% in the dose-reduction and dose-prolongation arms, respectively).
Among the two dosing regimens, failure occurred in 18% of children in the dose-reduction arm at the first step, 10% at the second, and 8% at the third, whereas 2.7% of children in the dose-prolongation arm experienced regimen failure at the first step, 6.1% at the second, and 15% at the third.
No substantial difference between the two tapering approaches was observed because the study was not powered to look at this. There was also no control arm, such as a group continuing treatment while the other groups tapered, or as Dr. Ramanan had pointed out, stopping canakinumab altogether.
“As long as the treatment was continued, even at very low dosage, most patients remained in inactive disease,” Dr. Quartier said. He added, however, that only a minority of patients could stop treatment completely. Treatment should not be stopped abruptly, he advised, because this was associated with a substantial number of disease flares that needed treatment to be reinstated.
These findings suggest that “a certain level of sustained inhibition of the IL-1 pathway seems important for the maintenance of CR in most sJIA patients,” Dr. Quartier and coinvestigators wrote in their article.
They added: “We believe that these results are relevant for clinical practice, particularly for designing personalized tapering strategies that can allow an adequate control of disease while minimizing the side effects of certain medications, notably glucocorticoids.”
The study was funded by Novartis in collaboration with the Pediatric Rheumatology International Trials Organization and the Pediatric Rheumatology Collaborative Study Group. Dr. Quartier was an investigator for the trial and has received research and consultancy fees from Novartis, among other pharmaceutical companies. Two of his coauthors are employees of Novartis. Dr. Ramanan has acted as an investigator in prior canakinumab trials and has received consultancy fees from Novartis and multiple other companies.
SOURCE: Quartier P et al. Arthritis Rheumatol. 2020 Aug 11. doi: 10.1002/art.41488.
FROM ARTHRITIS & RHEUMATOLOGY