User login
Enlarging Pigmented Lesion on the Thigh
The Diagnosis: Localized Cutaneous Argyria
The differential diagnosis of an enlarging pigmented lesion is broad, including various neoplasms, pigmented deep fungal infections, and cutaneous deposits secondary to systemic or topical medications or other exogenous substances. In our patient, identification of black particulate material on biopsy prompted further questioning. After the sinus tract persisted for 6 months, our patient’s infectious disease physician started applying silver nitrate at 3-week intervals to minimize drainage, exudate, and granulation tissue formation. After 3 months, marked pigmentation of the skin around the sinus tract was noted.
Argyria is a rare skin disorder that results from deposition of silver via localized exposure or systemic ingestion. Discoloration can either be reversible or irreversible, usually dependent on the length of silver exposure.1 Affected individuals exhibit blue-gray pigmentation of the skin that may be localized or diffuse. Photoactivated reduction of silver salts leads to conversion to elemental silver in the skin.2 Although argyria is most common on sun-exposed areas, the mucosae and nails may be involved in systemic cases. The etiology of argyria includes occupational exposure by ingestion of dust or traumatic cutaneous exposure in jewelry manufacturing, mining, or photographic or radiograph manufacturing. Other sources of localized argyria include prolonged contact with topical silver nitrate or silver sulfadiazine for wound care, silver-coated jewelry or piercings, acupuncture, tooth restoration procedures using dental amalgam, silver-containing surgical implants, or other silver-containing medications or wound dressings. Discontinuing contact with the source of silver minimizes further pigmentation, and excision of deposits may be helpful in some instances.3
Histopathologic findings in argyria may be subtle and diverse. Small particulate material may be apparent on careful examination at high magnification only, and the depth of deposition can depend on the etiology of absorption or implantation as well as the length of exposure. Short-term exposure may be associated with deposition of dark, brown-black, coarse granules confined to the stratum corneum.1 Frequently, cases of argyria reveal small, extracellular, brown-black, pigmented granules in a bandlike distribution primarily around vasculature, eccrine glands, perineural tissue, hair follicles, or arrector pili muscles or free in the dermis around collagen bundles. The granules can be highlighted by dark-field microscopy that will display scattered, refractile, white particles, described as a “stars in heaven” pattern.3 Rare ochre-colored collagen bundles have been reported in some cases, described as a pseudo-ochronosis pattern of argyria.4
Given the clinical history in our patient, a melanocytic lesion was considered but was excluded based on the histopathologic findings. Regressed melanoma clinically may resemble cutaneous silver deposition, as tumoral melanosis can be associated with an intense blue-black presentation. Histopathology will reveal an absence of melanocytes with residual coarse melanin in melanophages (Figure 1) rather than the particulate material associated with silver deposition. Although argyria can be associated with increased melanin in the basal epidermal keratinocytes and melanophages in the papillary dermis, silver granules can be distinguished by their uniform appearance and location throughout the skin (dermis, around vasculature/adnexal structures vs melanin in melanophages and basal epidermal keratinocytes).3,5,6

Blue nevi typically present as well-circumscribed, blue to gray or even dark brown lesions most often located on the arms, legs, head, and neck. Histopathology reveals spindle-shaped dendritic melanocytes dissecting through collagen bundles in the dermis with melanophages (Figure 2). Pigmentation may vary from extensive to little or even none. Blue nevi are demarcated and may be associated with dermal sclerosis.7

Drug-induced hyperpigmentation has a variable presentation both clinically and histologically depending on the type of drug implicated. Tetracyclines, particularly minocycline, are known culprits of drug-induced pigmentation, which can present as blue-gray to brown discoloration in at least 3 classically described patterns: (1) blue-black pigmentation around scars or prior inflammatory sites, (2) blue-black pigmentation on the shins or upper extremities, or (3) brown pigmentation in photosensitive areas. Histopathology reveals brown-black granules intracellularly in macrophages or fibroblasts or localized around vessels or eccrine glands (Figure 3). Special stains such as Perls Prussian blue or Fontana-Masson may highlight the pigmented granules. Widespread pigmentation in other organs, such as the thyroid, and history of long-standing tetracycline use are helpful clues to distinguish drug-induced pigmentation from other entities.8

Tattoo ink reaction frequently presents as an irregular pigmented lesion that can have associated features of inflammation including rash, erythema, and swelling. Histopathology reveals small clumped pigment in the dermis localized either extracellularly preferentially around vascular structures and collagen fibers or intracellularly in macrophages or fibroblasts (Figure 4). Considering the pigment is foreign material, a mixed inflammatory infiltrate can be present or more rarely the presence of pigment may induce pseudoepitheliomatous hyperplasia. The inflammatory reaction pattern on histology can vary, but granulomatous and lichenoid patterns frequently have been described. Other helpful clues to suggest tattoo pigment include refractile granules under polarized light and multiple pigmented colors.3

Dermal melanocytosis also may be considered, which consists of blue-gray irregular macules to patches on the skin that are frequently present at birth but may develop later in life. Histopathology reveals pigmented dendritic to spindle-shaped dermal melanocytes and melanophages dissecting between collagen fibers localized to the deep dermis. In addition, some hematologic or vascular disorders, including resolving hemorrhage or cyanosis, may be considered in the clinical differential. Deposition disorders such as chrysiasis and ochronosis could exhibit clinical or histopathologic similarities.3,8
Occasionally, prolonged use of topical silver nitrate may result in a pigmented lesion that mimics a melanocytic neoplasm or other pigmented lesions. However, these conditions can be readily differentiated by their characteristic histopathologic findings along with detailed clinical history.
- Ondrasik RM, Jordan P, Sriharan A. A clinical mimicker of melanoma with distinctive histopathology: topical silver nitrate exposure. J Cutan Pathol. 2020;47:1205-1210.
- Gill P, Richards K, Cho WC, et al. Localized cutaneous argyria: review of a rare clinical mimicker of melanocytic lesions. Ann Diagn Pathol. 2021;54:151776.
- Molina-Ruiz AM, Cerroni L, Kutzner H, et al. Cutaneous deposits. Am J Dermatopathol. 2014;36:1-48.
- Lee J, Korgavkar K, DiMarco C, et al. Localized argyria with pseudoochronosis. J Cutan Pathol. 2020;47:671-674.
- El Sharouni MA, Aivazian K, Witkamp AJ, et al. Association of histologic regression with a favorable outcome in patients with stage 1 and stage 2 cutaneous melanoma. JAMA Dermatol. 2021;157:166-173.
- Staser K, Chen D, Solus J, et al. Extensive tumoral melanosis associated with ipilimumab-treated melanoma. Br J Dermatol. 2016;175:391-393.
- Sugianto JZ, Ralston JS, Metcalf JS, et al. Blue nevus and “malignant blue nevus”: a concise review. Semin Diagn Pathol. 2016;33:219-224.
- Wang RF, Ko D, Friedman BJ, et al. Disorders of hyperpigmentation. part I. pathogenesis and clinical features of common pigmentary disorders. J Am Acad Dermatol. 2023;88:271-288.
The Diagnosis: Localized Cutaneous Argyria
The differential diagnosis of an enlarging pigmented lesion is broad, including various neoplasms, pigmented deep fungal infections, and cutaneous deposits secondary to systemic or topical medications or other exogenous substances. In our patient, identification of black particulate material on biopsy prompted further questioning. After the sinus tract persisted for 6 months, our patient’s infectious disease physician started applying silver nitrate at 3-week intervals to minimize drainage, exudate, and granulation tissue formation. After 3 months, marked pigmentation of the skin around the sinus tract was noted.
Argyria is a rare skin disorder that results from deposition of silver via localized exposure or systemic ingestion. Discoloration can either be reversible or irreversible, usually dependent on the length of silver exposure.1 Affected individuals exhibit blue-gray pigmentation of the skin that may be localized or diffuse. Photoactivated reduction of silver salts leads to conversion to elemental silver in the skin.2 Although argyria is most common on sun-exposed areas, the mucosae and nails may be involved in systemic cases. The etiology of argyria includes occupational exposure by ingestion of dust or traumatic cutaneous exposure in jewelry manufacturing, mining, or photographic or radiograph manufacturing. Other sources of localized argyria include prolonged contact with topical silver nitrate or silver sulfadiazine for wound care, silver-coated jewelry or piercings, acupuncture, tooth restoration procedures using dental amalgam, silver-containing surgical implants, or other silver-containing medications or wound dressings. Discontinuing contact with the source of silver minimizes further pigmentation, and excision of deposits may be helpful in some instances.3
Histopathologic findings in argyria may be subtle and diverse. Small particulate material may be apparent on careful examination at high magnification only, and the depth of deposition can depend on the etiology of absorption or implantation as well as the length of exposure. Short-term exposure may be associated with deposition of dark, brown-black, coarse granules confined to the stratum corneum.1 Frequently, cases of argyria reveal small, extracellular, brown-black, pigmented granules in a bandlike distribution primarily around vasculature, eccrine glands, perineural tissue, hair follicles, or arrector pili muscles or free in the dermis around collagen bundles. The granules can be highlighted by dark-field microscopy that will display scattered, refractile, white particles, described as a “stars in heaven” pattern.3 Rare ochre-colored collagen bundles have been reported in some cases, described as a pseudo-ochronosis pattern of argyria.4
Given the clinical history in our patient, a melanocytic lesion was considered but was excluded based on the histopathologic findings. Regressed melanoma clinically may resemble cutaneous silver deposition, as tumoral melanosis can be associated with an intense blue-black presentation. Histopathology will reveal an absence of melanocytes with residual coarse melanin in melanophages (Figure 1) rather than the particulate material associated with silver deposition. Although argyria can be associated with increased melanin in the basal epidermal keratinocytes and melanophages in the papillary dermis, silver granules can be distinguished by their uniform appearance and location throughout the skin (dermis, around vasculature/adnexal structures vs melanin in melanophages and basal epidermal keratinocytes).3,5,6
Blue nevi typically present as well-circumscribed, blue to gray or even dark brown lesions most often located on the arms, legs, head, and neck. Histopathology reveals spindle-shaped dendritic melanocytes dissecting through collagen bundles in the dermis with melanophages (Figure 2). Pigmentation may vary from extensive to little or even none. Blue nevi are demarcated and may be associated with dermal sclerosis.7
Drug-induced hyperpigmentation has a variable presentation both clinically and histologically depending on the type of drug implicated. Tetracyclines, particularly minocycline, are known culprits of drug-induced pigmentation, which can present as blue-gray to brown discoloration in at least 3 classically described patterns: (1) blue-black pigmentation around scars or prior inflammatory sites, (2) blue-black pigmentation on the shins or upper extremities, or (3) brown pigmentation in photosensitive areas. Histopathology reveals brown-black granules intracellularly in macrophages or fibroblasts or localized around vessels or eccrine glands (Figure 3). Special stains such as Perls Prussian blue or Fontana-Masson may highlight the pigmented granules. Widespread pigmentation in other organs, such as the thyroid, and history of long-standing tetracycline use are helpful clues to distinguish drug-induced pigmentation from other entities.8
Tattoo ink reaction frequently presents as an irregular pigmented lesion that can have associated features of inflammation including rash, erythema, and swelling. Histopathology reveals small clumped pigment in the dermis localized either extracellularly preferentially around vascular structures and collagen fibers or intracellularly in macrophages or fibroblasts (Figure 4). Considering the pigment is foreign material, a mixed inflammatory infiltrate can be present or more rarely the presence of pigment may induce pseudoepitheliomatous hyperplasia. The inflammatory reaction pattern on histology can vary, but granulomatous and lichenoid patterns frequently have been described. Other helpful clues to suggest tattoo pigment include refractile granules under polarized light and multiple pigmented colors.3
Dermal melanocytosis also may be considered, which consists of blue-gray irregular macules to patches on the skin that are frequently present at birth but may develop later in life. Histopathology reveals pigmented dendritic to spindle-shaped dermal melanocytes and melanophages dissecting between collagen fibers localized to the deep dermis. In addition, some hematologic or vascular disorders, including resolving hemorrhage or cyanosis, may be considered in the clinical differential. Deposition disorders such as chrysiasis and ochronosis could exhibit clinical or histopathologic similarities.3,8
Occasionally, prolonged use of topical silver nitrate may result in a pigmented lesion that mimics a melanocytic neoplasm or other pigmented lesions. However, these conditions can be readily differentiated by their characteristic histopathologic findings along with detailed clinical history.
The Diagnosis: Localized Cutaneous Argyria
The differential diagnosis of an enlarging pigmented lesion is broad, including various neoplasms, pigmented deep fungal infections, and cutaneous deposits secondary to systemic or topical medications or other exogenous substances. In our patient, identification of black particulate material on biopsy prompted further questioning. After the sinus tract persisted for 6 months, our patient’s infectious disease physician started applying silver nitrate at 3-week intervals to minimize drainage, exudate, and granulation tissue formation. After 3 months, marked pigmentation of the skin around the sinus tract was noted.
Argyria is a rare skin disorder that results from deposition of silver via localized exposure or systemic ingestion. Discoloration can either be reversible or irreversible, usually dependent on the length of silver exposure.1 Affected individuals exhibit blue-gray pigmentation of the skin that may be localized or diffuse. Photoactivated reduction of silver salts leads to conversion to elemental silver in the skin.2 Although argyria is most common on sun-exposed areas, the mucosae and nails may be involved in systemic cases. The etiology of argyria includes occupational exposure by ingestion of dust or traumatic cutaneous exposure in jewelry manufacturing, mining, or photographic or radiograph manufacturing. Other sources of localized argyria include prolonged contact with topical silver nitrate or silver sulfadiazine for wound care, silver-coated jewelry or piercings, acupuncture, tooth restoration procedures using dental amalgam, silver-containing surgical implants, or other silver-containing medications or wound dressings. Discontinuing contact with the source of silver minimizes further pigmentation, and excision of deposits may be helpful in some instances.3
Histopathologic findings in argyria may be subtle and diverse. Small particulate material may be apparent on careful examination at high magnification only, and the depth of deposition can depend on the etiology of absorption or implantation as well as the length of exposure. Short-term exposure may be associated with deposition of dark, brown-black, coarse granules confined to the stratum corneum.1 Frequently, cases of argyria reveal small, extracellular, brown-black, pigmented granules in a bandlike distribution primarily around vasculature, eccrine glands, perineural tissue, hair follicles, or arrector pili muscles or free in the dermis around collagen bundles. The granules can be highlighted by dark-field microscopy that will display scattered, refractile, white particles, described as a “stars in heaven” pattern.3 Rare ochre-colored collagen bundles have been reported in some cases, described as a pseudo-ochronosis pattern of argyria.4
Given the clinical history in our patient, a melanocytic lesion was considered but was excluded based on the histopathologic findings. Regressed melanoma clinically may resemble cutaneous silver deposition, as tumoral melanosis can be associated with an intense blue-black presentation. Histopathology will reveal an absence of melanocytes with residual coarse melanin in melanophages (Figure 1) rather than the particulate material associated with silver deposition. Although argyria can be associated with increased melanin in the basal epidermal keratinocytes and melanophages in the papillary dermis, silver granules can be distinguished by their uniform appearance and location throughout the skin (dermis, around vasculature/adnexal structures vs melanin in melanophages and basal epidermal keratinocytes).3,5,6
Blue nevi typically present as well-circumscribed, blue to gray or even dark brown lesions most often located on the arms, legs, head, and neck. Histopathology reveals spindle-shaped dendritic melanocytes dissecting through collagen bundles in the dermis with melanophages (Figure 2). Pigmentation may vary from extensive to little or even none. Blue nevi are demarcated and may be associated with dermal sclerosis.7
Drug-induced hyperpigmentation has a variable presentation both clinically and histologically depending on the type of drug implicated. Tetracyclines, particularly minocycline, are known culprits of drug-induced pigmentation, which can present as blue-gray to brown discoloration in at least 3 classically described patterns: (1) blue-black pigmentation around scars or prior inflammatory sites, (2) blue-black pigmentation on the shins or upper extremities, or (3) brown pigmentation in photosensitive areas. Histopathology reveals brown-black granules intracellularly in macrophages or fibroblasts or localized around vessels or eccrine glands (Figure 3). Special stains such as Perls Prussian blue or Fontana-Masson may highlight the pigmented granules. Widespread pigmentation in other organs, such as the thyroid, and history of long-standing tetracycline use are helpful clues to distinguish drug-induced pigmentation from other entities.8
Tattoo ink reaction frequently presents as an irregular pigmented lesion that can have associated features of inflammation including rash, erythema, and swelling. Histopathology reveals small clumped pigment in the dermis localized either extracellularly preferentially around vascular structures and collagen fibers or intracellularly in macrophages or fibroblasts (Figure 4). Considering the pigment is foreign material, a mixed inflammatory infiltrate can be present or more rarely the presence of pigment may induce pseudoepitheliomatous hyperplasia. The inflammatory reaction pattern on histology can vary, but granulomatous and lichenoid patterns frequently have been described. Other helpful clues to suggest tattoo pigment include refractile granules under polarized light and multiple pigmented colors.3
Dermal melanocytosis also may be considered, which consists of blue-gray irregular macules to patches on the skin that are frequently present at birth but may develop later in life. Histopathology reveals pigmented dendritic to spindle-shaped dermal melanocytes and melanophages dissecting between collagen fibers localized to the deep dermis. In addition, some hematologic or vascular disorders, including resolving hemorrhage or cyanosis, may be considered in the clinical differential. Deposition disorders such as chrysiasis and ochronosis could exhibit clinical or histopathologic similarities.3,8
Occasionally, prolonged use of topical silver nitrate may result in a pigmented lesion that mimics a melanocytic neoplasm or other pigmented lesions. However, these conditions can be readily differentiated by their characteristic histopathologic findings along with detailed clinical history.
- Ondrasik RM, Jordan P, Sriharan A. A clinical mimicker of melanoma with distinctive histopathology: topical silver nitrate exposure. J Cutan Pathol. 2020;47:1205-1210.
- Gill P, Richards K, Cho WC, et al. Localized cutaneous argyria: review of a rare clinical mimicker of melanocytic lesions. Ann Diagn Pathol. 2021;54:151776.
- Molina-Ruiz AM, Cerroni L, Kutzner H, et al. Cutaneous deposits. Am J Dermatopathol. 2014;36:1-48.
- Lee J, Korgavkar K, DiMarco C, et al. Localized argyria with pseudoochronosis. J Cutan Pathol. 2020;47:671-674.
- El Sharouni MA, Aivazian K, Witkamp AJ, et al. Association of histologic regression with a favorable outcome in patients with stage 1 and stage 2 cutaneous melanoma. JAMA Dermatol. 2021;157:166-173.
- Staser K, Chen D, Solus J, et al. Extensive tumoral melanosis associated with ipilimumab-treated melanoma. Br J Dermatol. 2016;175:391-393.
- Sugianto JZ, Ralston JS, Metcalf JS, et al. Blue nevus and “malignant blue nevus”: a concise review. Semin Diagn Pathol. 2016;33:219-224.
- Wang RF, Ko D, Friedman BJ, et al. Disorders of hyperpigmentation. part I. pathogenesis and clinical features of common pigmentary disorders. J Am Acad Dermatol. 2023;88:271-288.
- Ondrasik RM, Jordan P, Sriharan A. A clinical mimicker of melanoma with distinctive histopathology: topical silver nitrate exposure. J Cutan Pathol. 2020;47:1205-1210.
- Gill P, Richards K, Cho WC, et al. Localized cutaneous argyria: review of a rare clinical mimicker of melanocytic lesions. Ann Diagn Pathol. 2021;54:151776.
- Molina-Ruiz AM, Cerroni L, Kutzner H, et al. Cutaneous deposits. Am J Dermatopathol. 2014;36:1-48.
- Lee J, Korgavkar K, DiMarco C, et al. Localized argyria with pseudoochronosis. J Cutan Pathol. 2020;47:671-674.
- El Sharouni MA, Aivazian K, Witkamp AJ, et al. Association of histologic regression with a favorable outcome in patients with stage 1 and stage 2 cutaneous melanoma. JAMA Dermatol. 2021;157:166-173.
- Staser K, Chen D, Solus J, et al. Extensive tumoral melanosis associated with ipilimumab-treated melanoma. Br J Dermatol. 2016;175:391-393.
- Sugianto JZ, Ralston JS, Metcalf JS, et al. Blue nevus and “malignant blue nevus”: a concise review. Semin Diagn Pathol. 2016;33:219-224.
- Wang RF, Ko D, Friedman BJ, et al. Disorders of hyperpigmentation. part I. pathogenesis and clinical features of common pigmentary disorders. J Am Acad Dermatol. 2023;88:271-288.
An 80-year-old man presented with a pigmented lesion on the left lateral thigh near the knee that had been gradually enlarging over several weeks (top [inset]). He underwent a left knee replacement surgery for advanced osteoarthritis many months prior that was complicated by postoperative Staphylococcus aureus infection with sinus tract formation that was persistent for 6 months and treated with a topical medication. A pigmented lesion developed near the opening of the sinus tract. His medical history was remarkable for extensive actinic damage as well as multiple actinic keratoses treated with cryotherapy but no history of melanoma. An excisional biopsy was performed (top and bottom).
.")


Cancer Screening for Dermatomyositis: A Survey of Indirect Costs, Burden, and Patient Willingness to Pay
Dermatomyositis (DM) is an uncommon idiopathic inflammatory myopathy (IIM) characterized by muscle inflammation; proximal muscle weakness; and dermatologic findings, such as the heliotrope eruption and Gottron papules.1-3 Dermatomyositis is associated with an increased malignancy risk compared to other IIMs, with a 13% to 42% lifetime risk for malignancy development.4,5 The incidence for malignancy peaks during the first year following diagnosis and falls gradually over 5 years but remains increased compared to the general population.6-11 Adenocarcinoma represents the majority of cancers associated with DM, particularly of the ovaries, lungs, breasts, gastrointestinal tract, pancreas, bladder, and prostate. The lymphatic system (non-Hodgkin lymphoma) also is overrepresented among cancers in DM.12
Because of the increased malignancy risk and cancer-related mortality in patients with DM, cancer screening generally is recommended following diagnosis.13,14 However, consensus guidelines for screening modalities and frequency currently do not exist, resulting in widely varying practice patterns.15 Some experts advocate for a conventional cancer screening panel (CSP), as summarized in Table 1.15-18 These tests may be repeated annually for 3 to 5 years following the diagnosis of DM. Although the use of myositis-specific antibodies (MSAs) recently has helped to risk-stratify DM patients, up to half of patients are MSA negative,19 and broad malignancy screening remains essential. Individualized discussions with patients about their risk factors, screening options, and risks and benefits of screening also are strongly encouraged.19-22 Studies of the direct costs and effectiveness of streamlined screening with positron emission tomography/computed tomography (PET/CT) compared with a CSP have shown similar efficacy and lower out-of-pocket costs for patients receiving PET/CT imaging.16-18

The goal of our study was to further characterize patients’ perspectives and experience of cancer screening in DM as well as indirect costs, both of which must be taken into consideration when developing consensus guidelines for DM malignancy screening. Inclusion of patient voice is essential given the similar efficacy of both screening methods. We assessed the indirect costs (eg, travel, lost work or wages, childcare) of a CSP in patients with DM. We theorized that the large quantity of tests involved in a CSP, which are performed at various locations on multiple days over the course of several years, may have substantial costs to patients beyond the co-pay and deductible. We also sought to measure patients’ perception of the burden associated with an annual CSP, which we defined to participants as the inconvenience or unpleasantness experienced by the patient, compared with an annual whole-body PET/CT. Finally, we examined the relative value of these screening methods to patients using a willingness-to-pay (WTP) analysis.
Materials and Methods
Patient Eligibility—Our study included Penn State Health (Hershey, Pennsylvania) patients 18 years or older with a recent diagnosis of DM—International Classification of Diseases, Ninth Revision code 710.3 or International Classification of Diseases, Tenth Revision codes M33.10 or M33.90—who were undergoing or had recently completed a CSP. Patients were excluded from the study if they had a concurrent or preceding diagnosis of malignancy (excluding nonmelanoma skin cancers) or had another IIM. The institutional review board at Penn State Health College of Medicine approved the study. Data for all patients were prospectively obtained.
Survey Design—A survey was generated to assess the burden and indirect costs associated with a CSP, which was modified from work done by Tchuenche et al23 and Teni et al.24 Focus groups were held in 2018 and 2019 with patients who met our inclusion criteria with the purpose of refining the survey instrument based on patient input. A summary explanation of research was provided to all participants, and informed consent was obtained. Patients were compensated for their time for focus groups. Audio of each focus group was then transcribed and analyzed for common themes. Following focus group feedback, a finalized survey was generated for assessing burden and indirect costs (survey instrument provided in the Supplementary Information). REDCap (Vanderbilt University), a secure web application, was used to construct the finalized survey and to collect and manage data.25
Patients who fit our inclusion criteria were identified and recruited in multiple ways. Patients with appointments at the Penn State Milton S. Hershey Medical Center Department of Dermatology were presented with the opportunity to participate, Penn State Health records with the appropriate billing codes were collected and patients were contacted, and an advertisement for the study was posted on StudyFinder. Surveys constructed on REDCap were then sent electronically to patients who agreed to participate in the study. A second summary explanation of research was included on the first page of the survey to describe the process.
The survey had 3 main sections. The first section collected demographic information. In the second section, we surveyed patients regarding the various aspects of a CSP that focus groups identified as burdensome. In addition, patients were asked to compare their feelings regarding an annual CSP vs whole-body PET/CT for a 3-year period utilizing a rating scale of strongly disagree, somewhat disagree, somewhat agree, and strongly agree. This section also included a willingness-to-pay (WTP) analysis for each modality. We defined WTP as the maximum out-of-pocket cost that the patient would be willing to pay to receive testing, which was measured in a hypothetical scenario where neither whole-body PET/CT nor CSP was covered by insurance.26 Although WTP may be influenced by external factors such as patient income, it can serve as a numerical measure of how much the patient values each service. Furthermore, these external factors become less relevant when comparing the relative value of 2 separate tests, as such factors apply equally in both scenarios. In the third section of the survey, patients were queried regarding various indirect costs associated with a CSP. Descriptions for a CSP and whole-body PET/CT, including risks and benefits, were provided to allow patients to make informed decisions.
Statistical Analysis—Because of the rarity of DM and the subsequently limited sample size, summary and descriptive statistics were utilized to characterize the sample and identify patterns in the results. Continuous variables are presented with means and standard deviations, and proportions are presented with frequencies and percentages. All analyses were done using SAS Version 9.4 (SAS Institute Inc).

Results
Patient Demographics—Fifty-four patients were identified using StudyFinder, physician referral, and search of the electronic health record. Nine patients agreed to take part in the focus groups, and 27 offered email addresses to be contacted for the survey. Of those 27 patients, 16 (59.3%) fit our inclusion criteria and completed the survey. Patient demographics are detailed in Table 2. The mean age was 55 years, and most patients were White (88% [14/16]), female (81% [13/16]), and had at least a bachelor’s degree (69% [11/16]). Most patients (69% [11/16]) had an annual income of less than $50,000, and half (50% [8/16]) were employed. All patients had been diagnosed with DM in or after 2013. Two patients were diagnosed with basal cell carcinoma during or after cancer screening.

Patient Preference for Screening and WTP—A majority (81% [13/16]) of patients desired some form of screening for occult malignancy following the diagnosis of DM, even in the hypothetical situation in which screening did not provide survival benefit (Figure 1). Twenty-five percent (4/16) of patients expressed that a CSP was burdensome, and 12.5% of patients (2/16) missed a CSP appointment; all of these patients rescheduled or were planning to reschedule. Assuming that both screening methods had similar predictive value in detecting malignancy, all 16 patients felt annual whole-body PET/CT for a 3-year period would be less burdensome than a CSP, and most (73% [11/15]) felt that it would decrease the likelihood of missed appointments. Overall, 93% (13/14) of patients preferred whole-body PET/CT over a CSP when given the choice between the 2 options (Figure 2). This preference was consistent with the patients’ WTP for these tests; patients reliably reported that they would pay more for annual whole-body PET/CT than for a CSP (Figure 3). Specifically, 75% (12/16) and 38% (6/16) of patients were willing to spend $250 or more and $1000 or more for annual whole-body PET/CT, respectively, compared with 56% (9/16) and 19% (3/16), respectively, for an annual CSP. Many patients (38% [6/16]) reported that they would not be willing to pay any out-of-pocket cost for a CSP compared with 13% (2/16) for PET/CT.Indirect Costs of Screening for Patients—Indirect costs incurred by patients undergoing a CSP are summarized in Table 3. Specifically, a large percentage of employed patients missed work (63% [5/8]) or had family miss work (38% [3/8]), necessitating the use of vacation and/or sick days to attend CSP appointments. A subset (25% [2/8]) lost income (average, $1500), and 1 patient reported that a family member lost income due to attending a CSP appointment. Most (75% [12/16]) patients also incurred substantial transportation costs (average, $243), with 1 patient spending $1000. No patients incurred child or elder care costs. One patient paid a small sum for lodging/meals while traveling to attend a CSP appointment.

Comment
Patients with DM have an increased incidence of malignancy, thus cancer screening serves a crucial role in the detection of occult disease.13 Up to half of DM patients are MSA negative, and most cancers in these patients are found with blind screening. Whole-body PET/CT has emerged as an alternative to a CSP. Evidence suggests that it has similar efficacy in detecting malignancy and may be particularly useful for identifying malignancies not routinely screened for in a CSP. In a prospective study of patients diagnosed with DM and polymyositis (N=55), whole-body PET/CT had a positive predictive value of 85.7% and negative predictive value for detecting occult malignancy of 93.8% compared with 77.8% and 95.7%, respectively, for a CSP.17
 and a conventional cancer screening panel (n=14).")
The results of our study showed that cancer screening is important to patients diagnosed with DM and that most of these patients desire some form of cancer screening. This finding held true even when patients were presented with a hypothetical situation in which screening was proven to have no survival benefit. Based on focus group data, this desire was likely driven by the fear generated by not knowing whether cancer is present, as reported by the following DM patients:
“I mean [cancer screening] is peace of mind. It is ultimately worth it. You know, better than . . . not doing the screenings and finding 3 years down the road that you have, you know, a serious problem . . . you had the cancer, and you didn’t have the screenings.” (DM patient 1)
 vs a conventional cancer screening panel (CSP) in patients with dermatomyositis (DM)(N=16).")
“I would rather know than not know, even if it is bad news, just tell me. The sooner the better, and give me the whole spiel . . . maybe all the screenings don’t need to be done, done so much, so often afterwards if the initial ones are ok, but I think too, for peace of mind, I would rather know it all up front.” (DM patient 2)
Further, when presented with the hypothetical situation that insurance would not cover screenings, a few patients remarked they would relocate to obtain them:
“I would find a place where the screenings were done. I’d move.” (DM patient 4)
“If it was just sky high and [insurance companies] weren’t willing to negotiate, I would consider moving.” (DM patient 3).
Sentiments such as these emphasize the importance and value that DM patients place on being screened for cancer and also may explain why only 25% of patients felt a CSP was burdensome and only 13% reported missing appointments, all of whom planned on making them up at a later time.
When presented with the choice of a CSP or annual whole-body PET/CT for a 3-year period following the diagnosis of DM, all patients expressed that whole-body PET/CT would be less burdensome. Most preferred annual whole-body PET/CT despite the slightly increased radiation exposure associated and thought that it would limit missed appointments. Accordingly, more patients responded that they would pay more money out-of-pocket for annual whole-body PET/CT. Given that WTP can function as a numerical measure of value, our results showed that patients placed a higher value on whole-body PET/CT compared with a CSP. The indirect costs associated with a CSP also were substantial, particularly regarding missed work, use of vacation and/or sick days, and travel expenses, which is particularly important because most patients reported an annual income less than $50,000.
The direct costs of a CSP and whole-body PET/CT have been studied. Specifically, Kundrick et al18 found that whole-body PET/CT was less expensive for patients (by approximately $111) out-of-pocket compared with a CSP, though cost to insurance companies was slightly greater. The present study adds to these findings by better illustrating the burden and indirect costs that patients experience while undergoing a CSP and by characterizing the patient’s perception and preference of these 2 screening methods.
Limitations of our study include a small sample size willing to complete the survey. There also was a predominance of White and female participants, partially attributed to the greater number of female patients who develop DM compared to male patients. However, this still may limit applicability of this study to males and patients of other races. Another limitation includes recall bias on survey responses, particularly regarding indirect costs incurred with a CSP. A final limitation was that only patients with a recent diagnosis of DM who were actively undergoing screening or had recently completed malignancy screening were included in the study. Given that these patients were receiving (or had completed) exclusively a CSP, patients were comparing their personal experience with a described experience. In addition, only 2 patients were diagnosed with cancer—both with basal cell carcinoma diagnosed on physical examination—which may have influenced their perception of a CSP, given that nothing was found on an extensive number of tests. However, these patients still greatly valued their screening, as evidenced in the survey.
Conclusion
- Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971-982. doi:10.1016/S0140-6736(03)14368-1
- Schmidt J. Current classification and management of inflammatory myopathies. J Neuromuscul Dis. 2018;5:109-129. doi:10.3233/JND-180308
- Lazarou IN, Guerne PA. Classification, diagnosis, and management of idiopathic inflammatory myopathies. J Rheumatol. 201;40:550-564. doi:10.3899/jrheum.120682
- Wang J, Guo G, Chen G, et al. Meta-analysis of the association of dermatomyositis and polymyositis with cancer. Br J Dermatol. 2013;169:838-847. doi:10.1111/bjd.12564
- Zampieri S, Valente M, Adami N, et al. Polymyositis, dermatomyositis and malignancy: a further intriguing link. Autoimmun Rev. 2010;9:449-453. doi:10.1016/j.autrev.2009.12.005
- Sigurgeirsson B, Lindelöf B, Edhag O, et al. Risk of cancer in patients with dermatomyositis or polymyositis. a population-based study. N Engl J Med. 1992;326:363-367. doi:10.1056/nejm199202063260602
- Chen YJ, Wu CY, Huang YL, et al. Cancer risks of dermatomyositis and polymyositis: a nationwide cohort study in Taiwan. Arthritis Res Ther. 2010;12:R70. doi:10.1186/ar2987
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case-control study. Br J Dermatol. 2001;144:825-831. doi:10.1046/j.1365-2133.2001.04140.x
- Targoff IN, Mamyrova G, Trieu EP, et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum. 2006;54:3682-3689. doi:10.1002/art.22164
- Chow WH, Gridley G, Mellemkjær L, et al. Cancer risk following polymyositis and dermatomyositis: a nationwide cohort study in Denmark. Cancer Causes Control. 1995;6:9-13. doi:10.1007/BF00051675
- Buchbinder R, Forbes A, Hall S, et al. Incidence of malignant disease in biopsy-proven inflammatory myopathy: a population-based cohort study. Ann Intern Med. 2001;134:1087-1095. doi:10.7326/0003-4819-134-12-200106190-00008
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100. doi:10.1016/S0140-6736(00)03540-6
- Leatham H, Schadt C, Chisolm S, et al. Evidence supports blind screening for internal malignancy in dermatomyositis: data from 2 large US dermatology cohorts. Medicine (Baltimore). 2018;97:E9639. doi:10.1097/MD.0000000000009639
- Sparsa A, Liozon E, Herrmann F, et al. Routine vs extensive malignancy search for adult dermatomyositis and polymyositis: a study of 40 patients. Arch Dermatol. 2002;138:885-890.
- Dutton K, Soden M. Malignancy screening in autoimmune myositis among Australian rheumatologists. Intern Med J. 2017;47:1367-1375. doi:10.1111/imj.13556
- Selva-O’Callaghan A, Martinez-Gómez X, Trallero-Araguás E, et al. The diagnostic work-up of cancer-associated myositis. Curr Opin Rheumatol. 2018;30:630-636. doi:10.1097/BOR.0000000000000535
- Selva-O’Callaghan A, Grau JM, Gámez-Cenzano C, et al. Conventional cancer screening versus PET/CT in dermatomyositis/polymyositis. Am J Med. 2010;123:558-562. doi:10.1016/j.amjmed.2009.11.012
- Kundrick A, Kirby J, Ba D, et al. Positron emission tomography costs less to patients than conventional screening for malignancy in dermatomyositis. Semin Arthritis Rheum. 2019;49:140-144. doi:10.1016/j.semarthrit.2018.10.021
- Satoh M, Tanaka S, Ceribelli A, et al. A comprehensive overview on myositis-specific antibodies: new and old biomarkers in idiopathic inflammatory myopathy. Clin Rev Allergy Immunol. 2017;52:1-19. doi:10.1007/s12016-015-8510-y
- Vaughan H, Rugo HS, Haemel A. Risk-based screening for cancer in patients with dermatomyositis: toward a more individualized approach. JAMA Dermatol. 2022;158:244-247. doi:10.1001/jamadermatol.2021.5841
- Khanna U, Galimberti F, Li Y, et al. Dermatomyositis and malignancy: should all patients with dermatomyositis undergo malignancy screening? Ann Transl Med. 2021;9:432. doi:10.21037/atm-20-5215
- Oldroyd AGS, Allard AB, Callen JP, et al. Corrigendum to: A systematic review and meta-analysis to inform cancer screening guidelines in idiopathic inflammatory myopathies. Rheumatology (Oxford). 2021;60:5483. doi:10.1093/rheumatology/keab616
- Tchuenche M, Haté V, McPherson D, et al. Estimating client out-of-pocket costs for accessing voluntary medical male circumcision in South Africa. PLoS One. 2016;11:E0164147. doi:10.1371/journal.pone.0164147
- Teni FS, Gebresillassie BM, Birru EM, et al. Costs incurred by outpatients at a university hospital in northwestern Ethiopia: a cross-sectional study. BMC Health Serv Res. 2018;18:842. doi:10.1186/s12913-018-3628-2
- Harris PA, Taylor R, Thielke R, et al. Research electronic data capture (REDCap)—a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377-381. doi:10.1016/j.jbi.2008.08.010
- Bala MV, Mauskopf JA, Wood LL. Willingness to pay as a measure of health benefits. Pharmacoeconomics. 1999;15:9-18. doi:10.2165/00019053-199915010-00002
Dermatomyositis (DM) is an uncommon idiopathic inflammatory myopathy (IIM) characterized by muscle inflammation; proximal muscle weakness; and dermatologic findings, such as the heliotrope eruption and Gottron papules.1-3 Dermatomyositis is associated with an increased malignancy risk compared to other IIMs, with a 13% to 42% lifetime risk for malignancy development.4,5 The incidence for malignancy peaks during the first year following diagnosis and falls gradually over 5 years but remains increased compared to the general population.6-11 Adenocarcinoma represents the majority of cancers associated with DM, particularly of the ovaries, lungs, breasts, gastrointestinal tract, pancreas, bladder, and prostate. The lymphatic system (non-Hodgkin lymphoma) also is overrepresented among cancers in DM.12
Because of the increased malignancy risk and cancer-related mortality in patients with DM, cancer screening generally is recommended following diagnosis.13,14 However, consensus guidelines for screening modalities and frequency currently do not exist, resulting in widely varying practice patterns.15 Some experts advocate for a conventional cancer screening panel (CSP), as summarized in Table 1.15-18 These tests may be repeated annually for 3 to 5 years following the diagnosis of DM. Although the use of myositis-specific antibodies (MSAs) recently has helped to risk-stratify DM patients, up to half of patients are MSA negative,19 and broad malignancy screening remains essential. Individualized discussions with patients about their risk factors, screening options, and risks and benefits of screening also are strongly encouraged.19-22 Studies of the direct costs and effectiveness of streamlined screening with positron emission tomography/computed tomography (PET/CT) compared with a CSP have shown similar efficacy and lower out-of-pocket costs for patients receiving PET/CT imaging.16-18
The goal of our study was to further characterize patients’ perspectives and experience of cancer screening in DM as well as indirect costs, both of which must be taken into consideration when developing consensus guidelines for DM malignancy screening. Inclusion of patient voice is essential given the similar efficacy of both screening methods. We assessed the indirect costs (eg, travel, lost work or wages, childcare) of a CSP in patients with DM. We theorized that the large quantity of tests involved in a CSP, which are performed at various locations on multiple days over the course of several years, may have substantial costs to patients beyond the co-pay and deductible. We also sought to measure patients’ perception of the burden associated with an annual CSP, which we defined to participants as the inconvenience or unpleasantness experienced by the patient, compared with an annual whole-body PET/CT. Finally, we examined the relative value of these screening methods to patients using a willingness-to-pay (WTP) analysis.
Materials and Methods
Patient Eligibility—Our study included Penn State Health (Hershey, Pennsylvania) patients 18 years or older with a recent diagnosis of DM—International Classification of Diseases, Ninth Revision code 710.3 or International Classification of Diseases, Tenth Revision codes M33.10 or M33.90—who were undergoing or had recently completed a CSP. Patients were excluded from the study if they had a concurrent or preceding diagnosis of malignancy (excluding nonmelanoma skin cancers) or had another IIM. The institutional review board at Penn State Health College of Medicine approved the study. Data for all patients were prospectively obtained.
Survey Design—A survey was generated to assess the burden and indirect costs associated with a CSP, which was modified from work done by Tchuenche et al23 and Teni et al.24 Focus groups were held in 2018 and 2019 with patients who met our inclusion criteria with the purpose of refining the survey instrument based on patient input. A summary explanation of research was provided to all participants, and informed consent was obtained. Patients were compensated for their time for focus groups. Audio of each focus group was then transcribed and analyzed for common themes. Following focus group feedback, a finalized survey was generated for assessing burden and indirect costs (survey instrument provided in the Supplementary Information). REDCap (Vanderbilt University), a secure web application, was used to construct the finalized survey and to collect and manage data.25
Patients who fit our inclusion criteria were identified and recruited in multiple ways. Patients with appointments at the Penn State Milton S. Hershey Medical Center Department of Dermatology were presented with the opportunity to participate, Penn State Health records with the appropriate billing codes were collected and patients were contacted, and an advertisement for the study was posted on StudyFinder. Surveys constructed on REDCap were then sent electronically to patients who agreed to participate in the study. A second summary explanation of research was included on the first page of the survey to describe the process.
The survey had 3 main sections. The first section collected demographic information. In the second section, we surveyed patients regarding the various aspects of a CSP that focus groups identified as burdensome. In addition, patients were asked to compare their feelings regarding an annual CSP vs whole-body PET/CT for a 3-year period utilizing a rating scale of strongly disagree, somewhat disagree, somewhat agree, and strongly agree. This section also included a willingness-to-pay (WTP) analysis for each modality. We defined WTP as the maximum out-of-pocket cost that the patient would be willing to pay to receive testing, which was measured in a hypothetical scenario where neither whole-body PET/CT nor CSP was covered by insurance.26 Although WTP may be influenced by external factors such as patient income, it can serve as a numerical measure of how much the patient values each service. Furthermore, these external factors become less relevant when comparing the relative value of 2 separate tests, as such factors apply equally in both scenarios. In the third section of the survey, patients were queried regarding various indirect costs associated with a CSP. Descriptions for a CSP and whole-body PET/CT, including risks and benefits, were provided to allow patients to make informed decisions.
Statistical Analysis—Because of the rarity of DM and the subsequently limited sample size, summary and descriptive statistics were utilized to characterize the sample and identify patterns in the results. Continuous variables are presented with means and standard deviations, and proportions are presented with frequencies and percentages. All analyses were done using SAS Version 9.4 (SAS Institute Inc).
Results
Patient Demographics—Fifty-four patients were identified using StudyFinder, physician referral, and search of the electronic health record. Nine patients agreed to take part in the focus groups, and 27 offered email addresses to be contacted for the survey. Of those 27 patients, 16 (59.3%) fit our inclusion criteria and completed the survey. Patient demographics are detailed in Table 2. The mean age was 55 years, and most patients were White (88% [14/16]), female (81% [13/16]), and had at least a bachelor’s degree (69% [11/16]). Most patients (69% [11/16]) had an annual income of less than $50,000, and half (50% [8/16]) were employed. All patients had been diagnosed with DM in or after 2013. Two patients were diagnosed with basal cell carcinoma during or after cancer screening.
Patient Preference for Screening and WTP—A majority (81% [13/16]) of patients desired some form of screening for occult malignancy following the diagnosis of DM, even in the hypothetical situation in which screening did not provide survival benefit (Figure 1). Twenty-five percent (4/16) of patients expressed that a CSP was burdensome, and 12.5% of patients (2/16) missed a CSP appointment; all of these patients rescheduled or were planning to reschedule. Assuming that both screening methods had similar predictive value in detecting malignancy, all 16 patients felt annual whole-body PET/CT for a 3-year period would be less burdensome than a CSP, and most (73% [11/15]) felt that it would decrease the likelihood of missed appointments. Overall, 93% (13/14) of patients preferred whole-body PET/CT over a CSP when given the choice between the 2 options (Figure 2). This preference was consistent with the patients’ WTP for these tests; patients reliably reported that they would pay more for annual whole-body PET/CT than for a CSP (Figure 3). Specifically, 75% (12/16) and 38% (6/16) of patients were willing to spend $250 or more and $1000 or more for annual whole-body PET/CT, respectively, compared with 56% (9/16) and 19% (3/16), respectively, for an annual CSP. Many patients (38% [6/16]) reported that they would not be willing to pay any out-of-pocket cost for a CSP compared with 13% (2/16) for PET/CT.Indirect Costs of Screening for Patients—Indirect costs incurred by patients undergoing a CSP are summarized in Table 3. Specifically, a large percentage of employed patients missed work (63% [5/8]) or had family miss work (38% [3/8]), necessitating the use of vacation and/or sick days to attend CSP appointments. A subset (25% [2/8]) lost income (average, $1500), and 1 patient reported that a family member lost income due to attending a CSP appointment. Most (75% [12/16]) patients also incurred substantial transportation costs (average, $243), with 1 patient spending $1000. No patients incurred child or elder care costs. One patient paid a small sum for lodging/meals while traveling to attend a CSP appointment.
Comment
Patients with DM have an increased incidence of malignancy, thus cancer screening serves a crucial role in the detection of occult disease.13 Up to half of DM patients are MSA negative, and most cancers in these patients are found with blind screening. Whole-body PET/CT has emerged as an alternative to a CSP. Evidence suggests that it has similar efficacy in detecting malignancy and may be particularly useful for identifying malignancies not routinely screened for in a CSP. In a prospective study of patients diagnosed with DM and polymyositis (N=55), whole-body PET/CT had a positive predictive value of 85.7% and negative predictive value for detecting occult malignancy of 93.8% compared with 77.8% and 95.7%, respectively, for a CSP.17
The results of our study showed that cancer screening is important to patients diagnosed with DM and that most of these patients desire some form of cancer screening. This finding held true even when patients were presented with a hypothetical situation in which screening was proven to have no survival benefit. Based on focus group data, this desire was likely driven by the fear generated by not knowing whether cancer is present, as reported by the following DM patients:
“I mean [cancer screening] is peace of mind. It is ultimately worth it. You know, better than . . . not doing the screenings and finding 3 years down the road that you have, you know, a serious problem . . . you had the cancer, and you didn’t have the screenings.” (DM patient 1)
“I would rather know than not know, even if it is bad news, just tell me. The sooner the better, and give me the whole spiel . . . maybe all the screenings don’t need to be done, done so much, so often afterwards if the initial ones are ok, but I think too, for peace of mind, I would rather know it all up front.” (DM patient 2)
Further, when presented with the hypothetical situation that insurance would not cover screenings, a few patients remarked they would relocate to obtain them:
“I would find a place where the screenings were done. I’d move.” (DM patient 4)
“If it was just sky high and [insurance companies] weren’t willing to negotiate, I would consider moving.” (DM patient 3).
Sentiments such as these emphasize the importance and value that DM patients place on being screened for cancer and also may explain why only 25% of patients felt a CSP was burdensome and only 13% reported missing appointments, all of whom planned on making them up at a later time.
When presented with the choice of a CSP or annual whole-body PET/CT for a 3-year period following the diagnosis of DM, all patients expressed that whole-body PET/CT would be less burdensome. Most preferred annual whole-body PET/CT despite the slightly increased radiation exposure associated and thought that it would limit missed appointments. Accordingly, more patients responded that they would pay more money out-of-pocket for annual whole-body PET/CT. Given that WTP can function as a numerical measure of value, our results showed that patients placed a higher value on whole-body PET/CT compared with a CSP. The indirect costs associated with a CSP also were substantial, particularly regarding missed work, use of vacation and/or sick days, and travel expenses, which is particularly important because most patients reported an annual income less than $50,000.
The direct costs of a CSP and whole-body PET/CT have been studied. Specifically, Kundrick et al18 found that whole-body PET/CT was less expensive for patients (by approximately $111) out-of-pocket compared with a CSP, though cost to insurance companies was slightly greater. The present study adds to these findings by better illustrating the burden and indirect costs that patients experience while undergoing a CSP and by characterizing the patient’s perception and preference of these 2 screening methods.
Limitations of our study include a small sample size willing to complete the survey. There also was a predominance of White and female participants, partially attributed to the greater number of female patients who develop DM compared to male patients. However, this still may limit applicability of this study to males and patients of other races. Another limitation includes recall bias on survey responses, particularly regarding indirect costs incurred with a CSP. A final limitation was that only patients with a recent diagnosis of DM who were actively undergoing screening or had recently completed malignancy screening were included in the study. Given that these patients were receiving (or had completed) exclusively a CSP, patients were comparing their personal experience with a described experience. In addition, only 2 patients were diagnosed with cancer—both with basal cell carcinoma diagnosed on physical examination—which may have influenced their perception of a CSP, given that nothing was found on an extensive number of tests. However, these patients still greatly valued their screening, as evidenced in the survey.
Conclusion
Dermatomyositis (DM) is an uncommon idiopathic inflammatory myopathy (IIM) characterized by muscle inflammation; proximal muscle weakness; and dermatologic findings, such as the heliotrope eruption and Gottron papules.1-3 Dermatomyositis is associated with an increased malignancy risk compared to other IIMs, with a 13% to 42% lifetime risk for malignancy development.4,5 The incidence for malignancy peaks during the first year following diagnosis and falls gradually over 5 years but remains increased compared to the general population.6-11 Adenocarcinoma represents the majority of cancers associated with DM, particularly of the ovaries, lungs, breasts, gastrointestinal tract, pancreas, bladder, and prostate. The lymphatic system (non-Hodgkin lymphoma) also is overrepresented among cancers in DM.12
Because of the increased malignancy risk and cancer-related mortality in patients with DM, cancer screening generally is recommended following diagnosis.13,14 However, consensus guidelines for screening modalities and frequency currently do not exist, resulting in widely varying practice patterns.15 Some experts advocate for a conventional cancer screening panel (CSP), as summarized in Table 1.15-18 These tests may be repeated annually for 3 to 5 years following the diagnosis of DM. Although the use of myositis-specific antibodies (MSAs) recently has helped to risk-stratify DM patients, up to half of patients are MSA negative,19 and broad malignancy screening remains essential. Individualized discussions with patients about their risk factors, screening options, and risks and benefits of screening also are strongly encouraged.19-22 Studies of the direct costs and effectiveness of streamlined screening with positron emission tomography/computed tomography (PET/CT) compared with a CSP have shown similar efficacy and lower out-of-pocket costs for patients receiving PET/CT imaging.16-18
The goal of our study was to further characterize patients’ perspectives and experience of cancer screening in DM as well as indirect costs, both of which must be taken into consideration when developing consensus guidelines for DM malignancy screening. Inclusion of patient voice is essential given the similar efficacy of both screening methods. We assessed the indirect costs (eg, travel, lost work or wages, childcare) of a CSP in patients with DM. We theorized that the large quantity of tests involved in a CSP, which are performed at various locations on multiple days over the course of several years, may have substantial costs to patients beyond the co-pay and deductible. We also sought to measure patients’ perception of the burden associated with an annual CSP, which we defined to participants as the inconvenience or unpleasantness experienced by the patient, compared with an annual whole-body PET/CT. Finally, we examined the relative value of these screening methods to patients using a willingness-to-pay (WTP) analysis.
Materials and Methods
Patient Eligibility—Our study included Penn State Health (Hershey, Pennsylvania) patients 18 years or older with a recent diagnosis of DM—International Classification of Diseases, Ninth Revision code 710.3 or International Classification of Diseases, Tenth Revision codes M33.10 or M33.90—who were undergoing or had recently completed a CSP. Patients were excluded from the study if they had a concurrent or preceding diagnosis of malignancy (excluding nonmelanoma skin cancers) or had another IIM. The institutional review board at Penn State Health College of Medicine approved the study. Data for all patients were prospectively obtained.
Survey Design—A survey was generated to assess the burden and indirect costs associated with a CSP, which was modified from work done by Tchuenche et al23 and Teni et al.24 Focus groups were held in 2018 and 2019 with patients who met our inclusion criteria with the purpose of refining the survey instrument based on patient input. A summary explanation of research was provided to all participants, and informed consent was obtained. Patients were compensated for their time for focus groups. Audio of each focus group was then transcribed and analyzed for common themes. Following focus group feedback, a finalized survey was generated for assessing burden and indirect costs (survey instrument provided in the Supplementary Information). REDCap (Vanderbilt University), a secure web application, was used to construct the finalized survey and to collect and manage data.25
Patients who fit our inclusion criteria were identified and recruited in multiple ways. Patients with appointments at the Penn State Milton S. Hershey Medical Center Department of Dermatology were presented with the opportunity to participate, Penn State Health records with the appropriate billing codes were collected and patients were contacted, and an advertisement for the study was posted on StudyFinder. Surveys constructed on REDCap were then sent electronically to patients who agreed to participate in the study. A second summary explanation of research was included on the first page of the survey to describe the process.
The survey had 3 main sections. The first section collected demographic information. In the second section, we surveyed patients regarding the various aspects of a CSP that focus groups identified as burdensome. In addition, patients were asked to compare their feelings regarding an annual CSP vs whole-body PET/CT for a 3-year period utilizing a rating scale of strongly disagree, somewhat disagree, somewhat agree, and strongly agree. This section also included a willingness-to-pay (WTP) analysis for each modality. We defined WTP as the maximum out-of-pocket cost that the patient would be willing to pay to receive testing, which was measured in a hypothetical scenario where neither whole-body PET/CT nor CSP was covered by insurance.26 Although WTP may be influenced by external factors such as patient income, it can serve as a numerical measure of how much the patient values each service. Furthermore, these external factors become less relevant when comparing the relative value of 2 separate tests, as such factors apply equally in both scenarios. In the third section of the survey, patients were queried regarding various indirect costs associated with a CSP. Descriptions for a CSP and whole-body PET/CT, including risks and benefits, were provided to allow patients to make informed decisions.
Statistical Analysis—Because of the rarity of DM and the subsequently limited sample size, summary and descriptive statistics were utilized to characterize the sample and identify patterns in the results. Continuous variables are presented with means and standard deviations, and proportions are presented with frequencies and percentages. All analyses were done using SAS Version 9.4 (SAS Institute Inc).
Results
Patient Demographics—Fifty-four patients were identified using StudyFinder, physician referral, and search of the electronic health record. Nine patients agreed to take part in the focus groups, and 27 offered email addresses to be contacted for the survey. Of those 27 patients, 16 (59.3%) fit our inclusion criteria and completed the survey. Patient demographics are detailed in Table 2. The mean age was 55 years, and most patients were White (88% [14/16]), female (81% [13/16]), and had at least a bachelor’s degree (69% [11/16]). Most patients (69% [11/16]) had an annual income of less than $50,000, and half (50% [8/16]) were employed. All patients had been diagnosed with DM in or after 2013. Two patients were diagnosed with basal cell carcinoma during or after cancer screening.
Patient Preference for Screening and WTP—A majority (81% [13/16]) of patients desired some form of screening for occult malignancy following the diagnosis of DM, even in the hypothetical situation in which screening did not provide survival benefit (Figure 1). Twenty-five percent (4/16) of patients expressed that a CSP was burdensome, and 12.5% of patients (2/16) missed a CSP appointment; all of these patients rescheduled or were planning to reschedule. Assuming that both screening methods had similar predictive value in detecting malignancy, all 16 patients felt annual whole-body PET/CT for a 3-year period would be less burdensome than a CSP, and most (73% [11/15]) felt that it would decrease the likelihood of missed appointments. Overall, 93% (13/14) of patients preferred whole-body PET/CT over a CSP when given the choice between the 2 options (Figure 2). This preference was consistent with the patients’ WTP for these tests; patients reliably reported that they would pay more for annual whole-body PET/CT than for a CSP (Figure 3). Specifically, 75% (12/16) and 38% (6/16) of patients were willing to spend $250 or more and $1000 or more for annual whole-body PET/CT, respectively, compared with 56% (9/16) and 19% (3/16), respectively, for an annual CSP. Many patients (38% [6/16]) reported that they would not be willing to pay any out-of-pocket cost for a CSP compared with 13% (2/16) for PET/CT.Indirect Costs of Screening for Patients—Indirect costs incurred by patients undergoing a CSP are summarized in Table 3. Specifically, a large percentage of employed patients missed work (63% [5/8]) or had family miss work (38% [3/8]), necessitating the use of vacation and/or sick days to attend CSP appointments. A subset (25% [2/8]) lost income (average, $1500), and 1 patient reported that a family member lost income due to attending a CSP appointment. Most (75% [12/16]) patients also incurred substantial transportation costs (average, $243), with 1 patient spending $1000. No patients incurred child or elder care costs. One patient paid a small sum for lodging/meals while traveling to attend a CSP appointment.
Comment
Patients with DM have an increased incidence of malignancy, thus cancer screening serves a crucial role in the detection of occult disease.13 Up to half of DM patients are MSA negative, and most cancers in these patients are found with blind screening. Whole-body PET/CT has emerged as an alternative to a CSP. Evidence suggests that it has similar efficacy in detecting malignancy and may be particularly useful for identifying malignancies not routinely screened for in a CSP. In a prospective study of patients diagnosed with DM and polymyositis (N=55), whole-body PET/CT had a positive predictive value of 85.7% and negative predictive value for detecting occult malignancy of 93.8% compared with 77.8% and 95.7%, respectively, for a CSP.17
The results of our study showed that cancer screening is important to patients diagnosed with DM and that most of these patients desire some form of cancer screening. This finding held true even when patients were presented with a hypothetical situation in which screening was proven to have no survival benefit. Based on focus group data, this desire was likely driven by the fear generated by not knowing whether cancer is present, as reported by the following DM patients:
“I mean [cancer screening] is peace of mind. It is ultimately worth it. You know, better than . . . not doing the screenings and finding 3 years down the road that you have, you know, a serious problem . . . you had the cancer, and you didn’t have the screenings.” (DM patient 1)
“I would rather know than not know, even if it is bad news, just tell me. The sooner the better, and give me the whole spiel . . . maybe all the screenings don’t need to be done, done so much, so often afterwards if the initial ones are ok, but I think too, for peace of mind, I would rather know it all up front.” (DM patient 2)
Further, when presented with the hypothetical situation that insurance would not cover screenings, a few patients remarked they would relocate to obtain them:
“I would find a place where the screenings were done. I’d move.” (DM patient 4)
“If it was just sky high and [insurance companies] weren’t willing to negotiate, I would consider moving.” (DM patient 3).
Sentiments such as these emphasize the importance and value that DM patients place on being screened for cancer and also may explain why only 25% of patients felt a CSP was burdensome and only 13% reported missing appointments, all of whom planned on making them up at a later time.
When presented with the choice of a CSP or annual whole-body PET/CT for a 3-year period following the diagnosis of DM, all patients expressed that whole-body PET/CT would be less burdensome. Most preferred annual whole-body PET/CT despite the slightly increased radiation exposure associated and thought that it would limit missed appointments. Accordingly, more patients responded that they would pay more money out-of-pocket for annual whole-body PET/CT. Given that WTP can function as a numerical measure of value, our results showed that patients placed a higher value on whole-body PET/CT compared with a CSP. The indirect costs associated with a CSP also were substantial, particularly regarding missed work, use of vacation and/or sick days, and travel expenses, which is particularly important because most patients reported an annual income less than $50,000.
The direct costs of a CSP and whole-body PET/CT have been studied. Specifically, Kundrick et al18 found that whole-body PET/CT was less expensive for patients (by approximately $111) out-of-pocket compared with a CSP, though cost to insurance companies was slightly greater. The present study adds to these findings by better illustrating the burden and indirect costs that patients experience while undergoing a CSP and by characterizing the patient’s perception and preference of these 2 screening methods.
Limitations of our study include a small sample size willing to complete the survey. There also was a predominance of White and female participants, partially attributed to the greater number of female patients who develop DM compared to male patients. However, this still may limit applicability of this study to males and patients of other races. Another limitation includes recall bias on survey responses, particularly regarding indirect costs incurred with a CSP. A final limitation was that only patients with a recent diagnosis of DM who were actively undergoing screening or had recently completed malignancy screening were included in the study. Given that these patients were receiving (or had completed) exclusively a CSP, patients were comparing their personal experience with a described experience. In addition, only 2 patients were diagnosed with cancer—both with basal cell carcinoma diagnosed on physical examination—which may have influenced their perception of a CSP, given that nothing was found on an extensive number of tests. However, these patients still greatly valued their screening, as evidenced in the survey.
Conclusion
- Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971-982. doi:10.1016/S0140-6736(03)14368-1
- Schmidt J. Current classification and management of inflammatory myopathies. J Neuromuscul Dis. 2018;5:109-129. doi:10.3233/JND-180308
- Lazarou IN, Guerne PA. Classification, diagnosis, and management of idiopathic inflammatory myopathies. J Rheumatol. 201;40:550-564. doi:10.3899/jrheum.120682
- Wang J, Guo G, Chen G, et al. Meta-analysis of the association of dermatomyositis and polymyositis with cancer. Br J Dermatol. 2013;169:838-847. doi:10.1111/bjd.12564
- Zampieri S, Valente M, Adami N, et al. Polymyositis, dermatomyositis and malignancy: a further intriguing link. Autoimmun Rev. 2010;9:449-453. doi:10.1016/j.autrev.2009.12.005
- Sigurgeirsson B, Lindelöf B, Edhag O, et al. Risk of cancer in patients with dermatomyositis or polymyositis. a population-based study. N Engl J Med. 1992;326:363-367. doi:10.1056/nejm199202063260602
- Chen YJ, Wu CY, Huang YL, et al. Cancer risks of dermatomyositis and polymyositis: a nationwide cohort study in Taiwan. Arthritis Res Ther. 2010;12:R70. doi:10.1186/ar2987
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case-control study. Br J Dermatol. 2001;144:825-831. doi:10.1046/j.1365-2133.2001.04140.x
- Targoff IN, Mamyrova G, Trieu EP, et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum. 2006;54:3682-3689. doi:10.1002/art.22164
- Chow WH, Gridley G, Mellemkjær L, et al. Cancer risk following polymyositis and dermatomyositis: a nationwide cohort study in Denmark. Cancer Causes Control. 1995;6:9-13. doi:10.1007/BF00051675
- Buchbinder R, Forbes A, Hall S, et al. Incidence of malignant disease in biopsy-proven inflammatory myopathy: a population-based cohort study. Ann Intern Med. 2001;134:1087-1095. doi:10.7326/0003-4819-134-12-200106190-00008
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100. doi:10.1016/S0140-6736(00)03540-6
- Leatham H, Schadt C, Chisolm S, et al. Evidence supports blind screening for internal malignancy in dermatomyositis: data from 2 large US dermatology cohorts. Medicine (Baltimore). 2018;97:E9639. doi:10.1097/MD.0000000000009639
- Sparsa A, Liozon E, Herrmann F, et al. Routine vs extensive malignancy search for adult dermatomyositis and polymyositis: a study of 40 patients. Arch Dermatol. 2002;138:885-890.
- Dutton K, Soden M. Malignancy screening in autoimmune myositis among Australian rheumatologists. Intern Med J. 2017;47:1367-1375. doi:10.1111/imj.13556
- Selva-O’Callaghan A, Martinez-Gómez X, Trallero-Araguás E, et al. The diagnostic work-up of cancer-associated myositis. Curr Opin Rheumatol. 2018;30:630-636. doi:10.1097/BOR.0000000000000535
- Selva-O’Callaghan A, Grau JM, Gámez-Cenzano C, et al. Conventional cancer screening versus PET/CT in dermatomyositis/polymyositis. Am J Med. 2010;123:558-562. doi:10.1016/j.amjmed.2009.11.012
- Kundrick A, Kirby J, Ba D, et al. Positron emission tomography costs less to patients than conventional screening for malignancy in dermatomyositis. Semin Arthritis Rheum. 2019;49:140-144. doi:10.1016/j.semarthrit.2018.10.021
- Satoh M, Tanaka S, Ceribelli A, et al. A comprehensive overview on myositis-specific antibodies: new and old biomarkers in idiopathic inflammatory myopathy. Clin Rev Allergy Immunol. 2017;52:1-19. doi:10.1007/s12016-015-8510-y
- Vaughan H, Rugo HS, Haemel A. Risk-based screening for cancer in patients with dermatomyositis: toward a more individualized approach. JAMA Dermatol. 2022;158:244-247. doi:10.1001/jamadermatol.2021.5841
- Khanna U, Galimberti F, Li Y, et al. Dermatomyositis and malignancy: should all patients with dermatomyositis undergo malignancy screening? Ann Transl Med. 2021;9:432. doi:10.21037/atm-20-5215
- Oldroyd AGS, Allard AB, Callen JP, et al. Corrigendum to: A systematic review and meta-analysis to inform cancer screening guidelines in idiopathic inflammatory myopathies. Rheumatology (Oxford). 2021;60:5483. doi:10.1093/rheumatology/keab616
- Tchuenche M, Haté V, McPherson D, et al. Estimating client out-of-pocket costs for accessing voluntary medical male circumcision in South Africa. PLoS One. 2016;11:E0164147. doi:10.1371/journal.pone.0164147
- Teni FS, Gebresillassie BM, Birru EM, et al. Costs incurred by outpatients at a university hospital in northwestern Ethiopia: a cross-sectional study. BMC Health Serv Res. 2018;18:842. doi:10.1186/s12913-018-3628-2
- Harris PA, Taylor R, Thielke R, et al. Research electronic data capture (REDCap)—a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377-381. doi:10.1016/j.jbi.2008.08.010
- Bala MV, Mauskopf JA, Wood LL. Willingness to pay as a measure of health benefits. Pharmacoeconomics. 1999;15:9-18. doi:10.2165/00019053-199915010-00002
- Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971-982. doi:10.1016/S0140-6736(03)14368-1
- Schmidt J. Current classification and management of inflammatory myopathies. J Neuromuscul Dis. 2018;5:109-129. doi:10.3233/JND-180308
- Lazarou IN, Guerne PA. Classification, diagnosis, and management of idiopathic inflammatory myopathies. J Rheumatol. 201;40:550-564. doi:10.3899/jrheum.120682
- Wang J, Guo G, Chen G, et al. Meta-analysis of the association of dermatomyositis and polymyositis with cancer. Br J Dermatol. 2013;169:838-847. doi:10.1111/bjd.12564
- Zampieri S, Valente M, Adami N, et al. Polymyositis, dermatomyositis and malignancy: a further intriguing link. Autoimmun Rev. 2010;9:449-453. doi:10.1016/j.autrev.2009.12.005
- Sigurgeirsson B, Lindelöf B, Edhag O, et al. Risk of cancer in patients with dermatomyositis or polymyositis. a population-based study. N Engl J Med. 1992;326:363-367. doi:10.1056/nejm199202063260602
- Chen YJ, Wu CY, Huang YL, et al. Cancer risks of dermatomyositis and polymyositis: a nationwide cohort study in Taiwan. Arthritis Res Ther. 2010;12:R70. doi:10.1186/ar2987
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case-control study. Br J Dermatol. 2001;144:825-831. doi:10.1046/j.1365-2133.2001.04140.x
- Targoff IN, Mamyrova G, Trieu EP, et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum. 2006;54:3682-3689. doi:10.1002/art.22164
- Chow WH, Gridley G, Mellemkjær L, et al. Cancer risk following polymyositis and dermatomyositis: a nationwide cohort study in Denmark. Cancer Causes Control. 1995;6:9-13. doi:10.1007/BF00051675
- Buchbinder R, Forbes A, Hall S, et al. Incidence of malignant disease in biopsy-proven inflammatory myopathy: a population-based cohort study. Ann Intern Med. 2001;134:1087-1095. doi:10.7326/0003-4819-134-12-200106190-00008
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100. doi:10.1016/S0140-6736(00)03540-6
- Leatham H, Schadt C, Chisolm S, et al. Evidence supports blind screening for internal malignancy in dermatomyositis: data from 2 large US dermatology cohorts. Medicine (Baltimore). 2018;97:E9639. doi:10.1097/MD.0000000000009639
- Sparsa A, Liozon E, Herrmann F, et al. Routine vs extensive malignancy search for adult dermatomyositis and polymyositis: a study of 40 patients. Arch Dermatol. 2002;138:885-890.
- Dutton K, Soden M. Malignancy screening in autoimmune myositis among Australian rheumatologists. Intern Med J. 2017;47:1367-1375. doi:10.1111/imj.13556
- Selva-O’Callaghan A, Martinez-Gómez X, Trallero-Araguás E, et al. The diagnostic work-up of cancer-associated myositis. Curr Opin Rheumatol. 2018;30:630-636. doi:10.1097/BOR.0000000000000535
- Selva-O’Callaghan A, Grau JM, Gámez-Cenzano C, et al. Conventional cancer screening versus PET/CT in dermatomyositis/polymyositis. Am J Med. 2010;123:558-562. doi:10.1016/j.amjmed.2009.11.012
- Kundrick A, Kirby J, Ba D, et al. Positron emission tomography costs less to patients than conventional screening for malignancy in dermatomyositis. Semin Arthritis Rheum. 2019;49:140-144. doi:10.1016/j.semarthrit.2018.10.021
- Satoh M, Tanaka S, Ceribelli A, et al. A comprehensive overview on myositis-specific antibodies: new and old biomarkers in idiopathic inflammatory myopathy. Clin Rev Allergy Immunol. 2017;52:1-19. doi:10.1007/s12016-015-8510-y
- Vaughan H, Rugo HS, Haemel A. Risk-based screening for cancer in patients with dermatomyositis: toward a more individualized approach. JAMA Dermatol. 2022;158:244-247. doi:10.1001/jamadermatol.2021.5841
- Khanna U, Galimberti F, Li Y, et al. Dermatomyositis and malignancy: should all patients with dermatomyositis undergo malignancy screening? Ann Transl Med. 2021;9:432. doi:10.21037/atm-20-5215
- Oldroyd AGS, Allard AB, Callen JP, et al. Corrigendum to: A systematic review and meta-analysis to inform cancer screening guidelines in idiopathic inflammatory myopathies. Rheumatology (Oxford). 2021;60:5483. doi:10.1093/rheumatology/keab616
- Tchuenche M, Haté V, McPherson D, et al. Estimating client out-of-pocket costs for accessing voluntary medical male circumcision in South Africa. PLoS One. 2016;11:E0164147. doi:10.1371/journal.pone.0164147
- Teni FS, Gebresillassie BM, Birru EM, et al. Costs incurred by outpatients at a university hospital in northwestern Ethiopia: a cross-sectional study. BMC Health Serv Res. 2018;18:842. doi:10.1186/s12913-018-3628-2
- Harris PA, Taylor R, Thielke R, et al. Research electronic data capture (REDCap)—a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377-381. doi:10.1016/j.jbi.2008.08.010
- Bala MV, Mauskopf JA, Wood LL. Willingness to pay as a measure of health benefits. Pharmacoeconomics. 1999;15:9-18. doi:10.2165/00019053-199915010-00002
Practice Points
- Dermatomyositis (DM) is associated with an increased risk for malignancy. Patient perspective needs to be considered in developing cancer screening guidelines for patients with DM, particularly given the similar efficacy of available screening modalities.
- Current modalities for cancer screening in DM include whole-body positron emission tomography/computed tomography (PET/CT) and a conventional cancer screening panel (CSP), which includes a battery of tests typically requiring multiple visits. Patients may find the simplicity of PET/CT more preferrable than the more complex CSP.
- Indirect costs of cancer screening include missed work, travel and childcare expenses, and lost wages. Conventional cancer screening has greater indirect costs than PET/CT.

Extensive Erosions and Ulcerations on the Trunk and Extremities in a Neonate
The Diagnosis: Dominant Dystrophic Epidermolysis Bullosa
Blisters in a neonate may be caused by infectious, traumatic, autoimmune, or congenital etiologies. Biopsy findings correlated with clinical findings usually can establish a prompt diagnosis when the clinical diagnosis is uncertain. Direct immunofluorescence (DIF) as well as indirect immunofluorescence studies are useful when autoimmune blistering disease or congenital or heritable disorders of skin fragility are in the differential diagnosis. Many genetic abnormalities of skin fragility are associated with marked morbidity and mortality, and prompt diagnosis is essential to provide proper care. Our patient’s parents had no history of skin disorders, and there was no known family history of blistering disease or traumatic birth. A heritable disorder of skin fragility was still a top consideration because of the extensive blistering in the absence of any other symptoms.
Although dystrophic epidermolysis bullosa (DEB) is an uncommon cause of skin fragility in neonates, our patient’s presentation was typical because of the extensive blistering and increased fragility of the skin at pressure points. Dystrophic epidermolysis bullosa has both dominant and recessive presentations that span a spectrum from mild and focal skin blistering to extensive blistering with esophageal involvement.1 Early diagnosis and treatment can mitigate potential failure to thrive or premature death. Inherited mutations in the type VII collagen gene, COL7A1, are causative.2 Dominant DEB may be associated with dental caries, swallowing problems secondary to esophageal scarring, and constipation, as well as dystrophic or absent nails. Immunomapping studies of the skin often reveal type VII collagen cytoplasmic granules in the epidermis and weaker reaction in the roof of the subepidermal separation (quiz image).3 Abnormalities in type VII collagen impact the production of anchoring fibrils. Blister cleavage occurs in the sublamina densa with type VII collagen staining evident on the blister roof (quiz image).4 Patients with severe generalized recessive DEB may have barely detectable type VII collagen. In our patient, the cytoplasmic staining and weak staining in the epidermal roof of the separation confirmed the clinical impression of dominant DEB.
Autoimmune blistering disease should be considered in the histologic differential diagnosis, but it usually is associated with obvious disease in the mother. Direct immunofluorescence of pemphigoid gestationis reveals linear deposition of C3 at the basement membrane zone, which also can be associated with IgG (Figure 1). Neonates receiving passive transfer of antibodies may develop annular erythema, vesicles, and even dyshidroticlike changes on the soles.5

Suction blisters are subepithelial.6,7 When they occur in the neonatal period, they often are localized and are thought to be the result of vigorous sucking in utero.6 They quickly resolve without treatment and do not reveal abnormalities on DIF. If immunomapping is done for type VII collagen, it will be located at the floor of the suction blister (Figure 2).

Bullous pemphigoid is associated with deposition of linear IgG along the dermoepidermal junction—IgG4 is most common—and/or C3 (Figure 3). Direct immunofluorescence on split-skin biopsy reveals IgG on the epidermal side of the blister in bullous pemphigoid in contrast to epidermolysis bullosa acquisita, where the immune deposits are found on the dermal side of the split.8,9 Linear IgA bullous disease is associated with IgA deposition (Figure 4).10,11 Secretory IgA derived from breast milk can be causative.11 Neonatal linear IgA bullous disease is a serious condition associated with marked mucosal involvement that can eventuate in respiratory compromise. Prompt recognition is important; breastfeeding must be stopped and supportive therapy must be provided.

Other types of vesicular or pustular eruptions in the newborn usually are easily diagnosed by their typical clinical presentation without biopsy. Erythema toxicum neonatorum usually presents within 1 to 2 days of birth. It is self-limited and often resembles acne, but it also occurs on the trunk and extremities. Transient neonatal pustular melanosis may be present at birth and predominantly is seen in newborns with skin of color. Lesions easily rupture and usually resolve within 1 to 2 days. Infectious causes of blistering often can be identified on clinical examination and confirmed by culture. Herpes simplex virus infection is associated with characteristic multinucleated giant cells as well as steel grey nuclei evident on routine histologic evaluation. Bullous impetigo reveals superficial acantholysis and will have negative findings on DIF.12

When a neonate presents with widespread blistering, both genetic disorders of skin fragility as well as passive transfer of antibodies from maternal autoimmune disease need to be considered. Direct immunofluorescence and indirect immunofluorescence immunomapping findings can be useful in clarifying the diagnosis when heritable disorders of skin fragility or autoimmune blistering diseases are a clinical consideration.
- Has C, Bauer JW, Bodemer C, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183:614-627. doi:10.1111/bjd.18921
- Dang N, Murrell DF. Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp Dermatol. 2008;17:553-568. doi:10.1111/j.1600-0625.2008.00723.x
- Has C, He Y. Research techniques made simple: immunofluorescence antigen mapping in epidermolysis bullosa. J Invest Dermatol. 2016;136:E65-E71. doi:10.1016/j.jid.2016.05.093
- Rao R, Mellerio J, Bhogal BS, et al. Immunofluorescence antigen mapping for hereditary epidermolysis bullosa. Indian J Dermatol Venereol Leprol. 2012;78:692-697.
- Aoyama Y, Asai K, Hioki K, et al. Herpes gestationis in a mother and newborn: immunoclinical perspectives based on a weekly followup of the enzyme-linked immunosorbent assay index of a bullous pemphigoid antigen noncollagenous domain. Arch Dermatol. 2007;143:1168-1172. doi:10.1001/archderm.143.9.1168
- Afsar FS, Cun S, Seremet S. Neonatal sucking blister [published online November 15, 2019]. Dermatol Online J. 2019;25:13030 /qt33b1w59j.
- Yu WY, Wei ML. Suction blisters. JAMA Dermatol. 2019;155:237. doi:10.1001/jamadermatol.2018.3277
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Reis-Filho EG, Silva Tde A, Aguirre LH, et al. Bullous pemphigoid in a 3-month-old infant: case report and literature review of this dermatosis in childhood. An Bras Dermatol. 2013;88:961-965. doi:10.1590/abd1806-4841.20132378
- Hruza LL, Mallory SB, Fitzgibbons J, et al. Linear IgA bullous dermatosis in a neonate. Pediatr Dermatol. 1993;10:171-176. doi:10.1111/j.1525-1470
- Egami S, Suzuki C, Kurihara Y, et al. Neonatal linear IgA bullous dermatosis mediated by breast milk–borne maternal IgA. JAMA Dermatol. 2021;157:1107-1111. doi:10.1001/jamadermatol.2021.2392
- Ligtenberg KG, Hu JK, Panse G, et al. Bullous impetigo masquerading as pemphigus foliaceus in an adult patient. JAAD Case Rep. 2020; 6:428-430. doi:10.1016/j.jdcr.2020.02.040
The Diagnosis: Dominant Dystrophic Epidermolysis Bullosa
Blisters in a neonate may be caused by infectious, traumatic, autoimmune, or congenital etiologies. Biopsy findings correlated with clinical findings usually can establish a prompt diagnosis when the clinical diagnosis is uncertain. Direct immunofluorescence (DIF) as well as indirect immunofluorescence studies are useful when autoimmune blistering disease or congenital or heritable disorders of skin fragility are in the differential diagnosis. Many genetic abnormalities of skin fragility are associated with marked morbidity and mortality, and prompt diagnosis is essential to provide proper care. Our patient’s parents had no history of skin disorders, and there was no known family history of blistering disease or traumatic birth. A heritable disorder of skin fragility was still a top consideration because of the extensive blistering in the absence of any other symptoms.
Although dystrophic epidermolysis bullosa (DEB) is an uncommon cause of skin fragility in neonates, our patient’s presentation was typical because of the extensive blistering and increased fragility of the skin at pressure points. Dystrophic epidermolysis bullosa has both dominant and recessive presentations that span a spectrum from mild and focal skin blistering to extensive blistering with esophageal involvement.1 Early diagnosis and treatment can mitigate potential failure to thrive or premature death. Inherited mutations in the type VII collagen gene, COL7A1, are causative.2 Dominant DEB may be associated with dental caries, swallowing problems secondary to esophageal scarring, and constipation, as well as dystrophic or absent nails. Immunomapping studies of the skin often reveal type VII collagen cytoplasmic granules in the epidermis and weaker reaction in the roof of the subepidermal separation (quiz image).3 Abnormalities in type VII collagen impact the production of anchoring fibrils. Blister cleavage occurs in the sublamina densa with type VII collagen staining evident on the blister roof (quiz image).4 Patients with severe generalized recessive DEB may have barely detectable type VII collagen. In our patient, the cytoplasmic staining and weak staining in the epidermal roof of the separation confirmed the clinical impression of dominant DEB.
Autoimmune blistering disease should be considered in the histologic differential diagnosis, but it usually is associated with obvious disease in the mother. Direct immunofluorescence of pemphigoid gestationis reveals linear deposition of C3 at the basement membrane zone, which also can be associated with IgG (Figure 1). Neonates receiving passive transfer of antibodies may develop annular erythema, vesicles, and even dyshidroticlike changes on the soles.5
Suction blisters are subepithelial.6,7 When they occur in the neonatal period, they often are localized and are thought to be the result of vigorous sucking in utero.6 They quickly resolve without treatment and do not reveal abnormalities on DIF. If immunomapping is done for type VII collagen, it will be located at the floor of the suction blister (Figure 2).
Bullous pemphigoid is associated with deposition of linear IgG along the dermoepidermal junction—IgG4 is most common—and/or C3 (Figure 3). Direct immunofluorescence on split-skin biopsy reveals IgG on the epidermal side of the blister in bullous pemphigoid in contrast to epidermolysis bullosa acquisita, where the immune deposits are found on the dermal side of the split.8,9 Linear IgA bullous disease is associated with IgA deposition (Figure 4).10,11 Secretory IgA derived from breast milk can be causative.11 Neonatal linear IgA bullous disease is a serious condition associated with marked mucosal involvement that can eventuate in respiratory compromise. Prompt recognition is important; breastfeeding must be stopped and supportive therapy must be provided.
Other types of vesicular or pustular eruptions in the newborn usually are easily diagnosed by their typical clinical presentation without biopsy. Erythema toxicum neonatorum usually presents within 1 to 2 days of birth. It is self-limited and often resembles acne, but it also occurs on the trunk and extremities. Transient neonatal pustular melanosis may be present at birth and predominantly is seen in newborns with skin of color. Lesions easily rupture and usually resolve within 1 to 2 days. Infectious causes of blistering often can be identified on clinical examination and confirmed by culture. Herpes simplex virus infection is associated with characteristic multinucleated giant cells as well as steel grey nuclei evident on routine histologic evaluation. Bullous impetigo reveals superficial acantholysis and will have negative findings on DIF.12
When a neonate presents with widespread blistering, both genetic disorders of skin fragility as well as passive transfer of antibodies from maternal autoimmune disease need to be considered. Direct immunofluorescence and indirect immunofluorescence immunomapping findings can be useful in clarifying the diagnosis when heritable disorders of skin fragility or autoimmune blistering diseases are a clinical consideration.
The Diagnosis: Dominant Dystrophic Epidermolysis Bullosa
Blisters in a neonate may be caused by infectious, traumatic, autoimmune, or congenital etiologies. Biopsy findings correlated with clinical findings usually can establish a prompt diagnosis when the clinical diagnosis is uncertain. Direct immunofluorescence (DIF) as well as indirect immunofluorescence studies are useful when autoimmune blistering disease or congenital or heritable disorders of skin fragility are in the differential diagnosis. Many genetic abnormalities of skin fragility are associated with marked morbidity and mortality, and prompt diagnosis is essential to provide proper care. Our patient’s parents had no history of skin disorders, and there was no known family history of blistering disease or traumatic birth. A heritable disorder of skin fragility was still a top consideration because of the extensive blistering in the absence of any other symptoms.
Although dystrophic epidermolysis bullosa (DEB) is an uncommon cause of skin fragility in neonates, our patient’s presentation was typical because of the extensive blistering and increased fragility of the skin at pressure points. Dystrophic epidermolysis bullosa has both dominant and recessive presentations that span a spectrum from mild and focal skin blistering to extensive blistering with esophageal involvement.1 Early diagnosis and treatment can mitigate potential failure to thrive or premature death. Inherited mutations in the type VII collagen gene, COL7A1, are causative.2 Dominant DEB may be associated with dental caries, swallowing problems secondary to esophageal scarring, and constipation, as well as dystrophic or absent nails. Immunomapping studies of the skin often reveal type VII collagen cytoplasmic granules in the epidermis and weaker reaction in the roof of the subepidermal separation (quiz image).3 Abnormalities in type VII collagen impact the production of anchoring fibrils. Blister cleavage occurs in the sublamina densa with type VII collagen staining evident on the blister roof (quiz image).4 Patients with severe generalized recessive DEB may have barely detectable type VII collagen. In our patient, the cytoplasmic staining and weak staining in the epidermal roof of the separation confirmed the clinical impression of dominant DEB.
Autoimmune blistering disease should be considered in the histologic differential diagnosis, but it usually is associated with obvious disease in the mother. Direct immunofluorescence of pemphigoid gestationis reveals linear deposition of C3 at the basement membrane zone, which also can be associated with IgG (Figure 1). Neonates receiving passive transfer of antibodies may develop annular erythema, vesicles, and even dyshidroticlike changes on the soles.5
Suction blisters are subepithelial.6,7 When they occur in the neonatal period, they often are localized and are thought to be the result of vigorous sucking in utero.6 They quickly resolve without treatment and do not reveal abnormalities on DIF. If immunomapping is done for type VII collagen, it will be located at the floor of the suction blister (Figure 2).
Bullous pemphigoid is associated with deposition of linear IgG along the dermoepidermal junction—IgG4 is most common—and/or C3 (Figure 3). Direct immunofluorescence on split-skin biopsy reveals IgG on the epidermal side of the blister in bullous pemphigoid in contrast to epidermolysis bullosa acquisita, where the immune deposits are found on the dermal side of the split.8,9 Linear IgA bullous disease is associated with IgA deposition (Figure 4).10,11 Secretory IgA derived from breast milk can be causative.11 Neonatal linear IgA bullous disease is a serious condition associated with marked mucosal involvement that can eventuate in respiratory compromise. Prompt recognition is important; breastfeeding must be stopped and supportive therapy must be provided.
Other types of vesicular or pustular eruptions in the newborn usually are easily diagnosed by their typical clinical presentation without biopsy. Erythema toxicum neonatorum usually presents within 1 to 2 days of birth. It is self-limited and often resembles acne, but it also occurs on the trunk and extremities. Transient neonatal pustular melanosis may be present at birth and predominantly is seen in newborns with skin of color. Lesions easily rupture and usually resolve within 1 to 2 days. Infectious causes of blistering often can be identified on clinical examination and confirmed by culture. Herpes simplex virus infection is associated with characteristic multinucleated giant cells as well as steel grey nuclei evident on routine histologic evaluation. Bullous impetigo reveals superficial acantholysis and will have negative findings on DIF.12
When a neonate presents with widespread blistering, both genetic disorders of skin fragility as well as passive transfer of antibodies from maternal autoimmune disease need to be considered. Direct immunofluorescence and indirect immunofluorescence immunomapping findings can be useful in clarifying the diagnosis when heritable disorders of skin fragility or autoimmune blistering diseases are a clinical consideration.
- Has C, Bauer JW, Bodemer C, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183:614-627. doi:10.1111/bjd.18921
- Dang N, Murrell DF. Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp Dermatol. 2008;17:553-568. doi:10.1111/j.1600-0625.2008.00723.x
- Has C, He Y. Research techniques made simple: immunofluorescence antigen mapping in epidermolysis bullosa. J Invest Dermatol. 2016;136:E65-E71. doi:10.1016/j.jid.2016.05.093
- Rao R, Mellerio J, Bhogal BS, et al. Immunofluorescence antigen mapping for hereditary epidermolysis bullosa. Indian J Dermatol Venereol Leprol. 2012;78:692-697.
- Aoyama Y, Asai K, Hioki K, et al. Herpes gestationis in a mother and newborn: immunoclinical perspectives based on a weekly followup of the enzyme-linked immunosorbent assay index of a bullous pemphigoid antigen noncollagenous domain. Arch Dermatol. 2007;143:1168-1172. doi:10.1001/archderm.143.9.1168
- Afsar FS, Cun S, Seremet S. Neonatal sucking blister [published online November 15, 2019]. Dermatol Online J. 2019;25:13030 /qt33b1w59j.
- Yu WY, Wei ML. Suction blisters. JAMA Dermatol. 2019;155:237. doi:10.1001/jamadermatol.2018.3277
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Reis-Filho EG, Silva Tde A, Aguirre LH, et al. Bullous pemphigoid in a 3-month-old infant: case report and literature review of this dermatosis in childhood. An Bras Dermatol. 2013;88:961-965. doi:10.1590/abd1806-4841.20132378
- Hruza LL, Mallory SB, Fitzgibbons J, et al. Linear IgA bullous dermatosis in a neonate. Pediatr Dermatol. 1993;10:171-176. doi:10.1111/j.1525-1470
- Egami S, Suzuki C, Kurihara Y, et al. Neonatal linear IgA bullous dermatosis mediated by breast milk–borne maternal IgA. JAMA Dermatol. 2021;157:1107-1111. doi:10.1001/jamadermatol.2021.2392
- Ligtenberg KG, Hu JK, Panse G, et al. Bullous impetigo masquerading as pemphigus foliaceus in an adult patient. JAAD Case Rep. 2020; 6:428-430. doi:10.1016/j.jdcr.2020.02.040
- Has C, Bauer JW, Bodemer C, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183:614-627. doi:10.1111/bjd.18921
- Dang N, Murrell DF. Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp Dermatol. 2008;17:553-568. doi:10.1111/j.1600-0625.2008.00723.x
- Has C, He Y. Research techniques made simple: immunofluorescence antigen mapping in epidermolysis bullosa. J Invest Dermatol. 2016;136:E65-E71. doi:10.1016/j.jid.2016.05.093
- Rao R, Mellerio J, Bhogal BS, et al. Immunofluorescence antigen mapping for hereditary epidermolysis bullosa. Indian J Dermatol Venereol Leprol. 2012;78:692-697.
- Aoyama Y, Asai K, Hioki K, et al. Herpes gestationis in a mother and newborn: immunoclinical perspectives based on a weekly followup of the enzyme-linked immunosorbent assay index of a bullous pemphigoid antigen noncollagenous domain. Arch Dermatol. 2007;143:1168-1172. doi:10.1001/archderm.143.9.1168
- Afsar FS, Cun S, Seremet S. Neonatal sucking blister [published online November 15, 2019]. Dermatol Online J. 2019;25:13030 /qt33b1w59j.
- Yu WY, Wei ML. Suction blisters. JAMA Dermatol. 2019;155:237. doi:10.1001/jamadermatol.2018.3277
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Reis-Filho EG, Silva Tde A, Aguirre LH, et al. Bullous pemphigoid in a 3-month-old infant: case report and literature review of this dermatosis in childhood. An Bras Dermatol. 2013;88:961-965. doi:10.1590/abd1806-4841.20132378
- Hruza LL, Mallory SB, Fitzgibbons J, et al. Linear IgA bullous dermatosis in a neonate. Pediatr Dermatol. 1993;10:171-176. doi:10.1111/j.1525-1470
- Egami S, Suzuki C, Kurihara Y, et al. Neonatal linear IgA bullous dermatosis mediated by breast milk–borne maternal IgA. JAMA Dermatol. 2021;157:1107-1111. doi:10.1001/jamadermatol.2021.2392
- Ligtenberg KG, Hu JK, Panse G, et al. Bullous impetigo masquerading as pemphigus foliaceus in an adult patient. JAAD Case Rep. 2020; 6:428-430. doi:10.1016/j.jdcr.2020.02.040
A neonate was born with extensive erosions and ulcerations on the trunk and extremities. The eroded areas had a beefy red appearance. A biopsy taken from a small blister was stained for type VII collagen by indirect immunofluorescence.
.")
.")
Genital Lentiginosis: A Benign Pigmentary Abnormality Often Raising Concern for Melanoma
To the Editor:
Genital lentiginosis (also known as mucosal melanotic macules, vulvar melanosis, penile melanosis, and penile lentigines) occurs in men and women.1 Lesions present in adult life as multifocal, asymmetrical, pigmented patches that can have a mottled appearance or exhibit skip areas. The irregular appearance of the pigmented areas often raises concern for melanoma. Biopsy reveals increased pigmentation along the basal layer of the epidermis; the irregular distribution of single melanocytes and pagetoid spread typical of melanoma in situ is not identified.

Genital lentiginosis usually occurs as an isolated finding; however, the condition can be a manifestation of Laugier-Hunziker syndrome, Carney complex, and Bannayan-Riley-Ruvalcaba syndrome.1-3 When it occurs as an isolated finding, the patient can be reassured and treatment is unnecessary. Because genital lentiginosis may mimic the appearance of melanoma, it is important for physicians to differentiate the two and make a correct diagnosis. We present a case of genital lentiginosis that mimicked vulvar melanoma.
.")
A 64-year-old woman was referred by her gynecologist to dermatology to rule out vulvar melanoma. The patient had a history of hypothyroidism and hypercholesterolemia but was otherwise in good health. Genital examination revealed asymptomatic pigmented macules and patches of unknown duration (Figure 1). Specimens were taken from 3 areas by punch biopsy to clarify the diagnosis. All 3 specimens showed identical features including basilar pigmentation, occasional melanophages in the papillary dermis, and no evidence of nests or pagetoid spread of atypical melanocytes (Figures 2 and 3). Histologic findings were diagnostic for genital lentiginosis. The patient was reassured, and no treatment was provided. At 6-month follow-up there was no change in clinical appearance.
.")
Genital lentiginosis is characterized by brown lesions that can have a mottled appearance and often are associated with skip areas.1 Lesions can be strikingly irregular and darkly pigmented.
Although the lesions of genital lentiginosis most often are isolated findings, they can be a clue to several uncommon syndromes such as autosomal-dominant Bannayan-Riley-Ruvalcaba syndrome, which is associated with genital lentiginosis, intestinal polyposis, and macrocephaly.3 Vascular malformations, lipomatosis, verrucal keratoses, and acrochordons can occur. Bannayan-Riley-Ruvalcaba syndrome and Cowden syndrome may share genetic linkage; mutations in the tumor suppressor PTEN (phosphatase and tensin homolog deleted on chromosome ten) has been implicated in both syndromes.4 Underlying Carney complex should be excluded when genital lentiginosis is encountered.
Genital lentiginosis is idiopathic in most instances, but reports of lesions occurring after annular lichen planus suggest a possible mechanism.5 The disappearance of lentigines after imatinib therapy suggests a role for c-kit, a receptor tyrosine kinase that is involved in intracellular signaling, in some cases.6 At times, lesions can simulate trichrome vitiligo or have a reticulate pattern.7
Men and women present at different points in the course of disease. Men often present with penile lesions 14 years after onset, on average; they notice a gradual increase in the size of lesions. Because women can have greater difficulty self-examining the genital region, they tend to present much later in the course but often within a few months after initial inspection.1,8
Genital lentiginosis can mimic melanoma with nonhomogeneous pigmentation, asymmetry, and unilateral distribution, which makes dermoscopic assessment of colors helpful in narrowing the differential diagnosis. Melanoma is associated with combinations of gray, red, blue, and white, which are not found in genital lentiginosis.9
Biopsy of a genital lentigo is diagnostic, distinguishing the lesion from melanoma—failing to reveal the atypical melanocytes and pagetoid spread characteristic of melanoma in situ. Histologic findings can cause diagnostic difficulties when concurrent lichen sclerosus is associated with genital lentigines or nevi.10
Lentigines on sun-damaged skin or in the setting of xeroderma pigmentosum have been associated with melanoma,11-13 but genital lentigines are not considered a form of precancerous melanosis. In women, early diagnosis is important when there is concern for melanoma because the prognosis for vulvar melanoma is improved in thin lesions.14
Other entities in the differential include secondary syphilis, which commonly presents as macules and scaly papules and can be found on mucosal surfaces such as the oral cavity,15 as well as Kaposi sarcoma, which is characterized by purplish, brown, or black macules, plaques, and nodules, more commonly in immunosuppressed patients.16
To avoid unwarranted concern and unnecessary surgery, dermatologists should be aware of genital lentigines and their characteristic presentation in adults.
- Hwang L, Wilson H, Orengo I. Off-center fold: irregular, pigmented genital macules. Arch Dermatol. 2000;136:1559-1564. doi:10.1001/archderm.136.12.1559-b
- Rhodes AR, Silverman RA, Harrist TJ, et al. Mucocutaneous lentigines, cardiomucocutaneous myxomas, and multiple blue nevi: the “LAMB” syndrome. J Am Acad Dermatol. 1984;10:72-82. doi:10.1016/s0190-9622(84)80047-x
- Erkek E, Hizel S, Sanl C, et al. Clinical and histopathological findings in Bannayan-Riley-Ruvalcaba syndrome. J Am Acad Dermatol. 2005;53:639-643. doi:10.1016/j.jaad.2005.06.022
- Blum RR, Rahimizadeh A, Kardon N, et al. Genital lentigines in a 6-year-old boy with a family history of Cowden’s disease: clinical and genetic evidence of the linkage between Bannayan-Riley-Ruvalcaba syndrome and Cowden’s disease. J Cutan Med Surg. 2001;5:228-230. doi:10.1177/120347540100500307
- Isbary G, Dyall-Smith D, Coras-Stepanek B, et al. Penile lentigo (genital mucosal macule) following annular lichen planus: a possible association? Australas J Dermatol. 2014;55:159-161. doi:10.1111/ajd.12169
- Campbell T, Felsten L, Moore J. Disappearance of lentigines in a patient receiving imatinib treatment for familial gastrointestinal stromal tumor syndrome. Arch Dermatol. 2009;145:1313-1316. doi:10.1001/archdermatol.2009.263
- Romero- A, R, , et al. Reticulate genital pigmentation associated with localized vitiligo. Arch Dermatol. 2010; 146:574-575. doi:10.1001/archdermatol.2010.69
- Barnhill RL, Albert LS, Shama SK, et al. Genital lentiginosis: a clinical and histopathologic study. J Am Acad Dermatol. 1990;22:453-460. doi:10.1016/0190-9622(90)70064-o
- De Giorgi V, Gori A, Salvati L, et al. Clinical and dermoscopic features of vulvar melanosis over the last 20 years. JAMA Dermatol. 2020;156:1185–1191. doi:10.1001/jamadermatol.2020.2528
- El Shabrawi-Caelen L, Soyer HP, Schaeppi H, et al. Genital lentigines and melanocytic nevi with superimposed lichen sclerosus: a diagnostic challenge. J Am Acad Dermatol. 2004;50:690-694. doi:10.1016/j.jaad.2003.09.034
- Shatkin M, Helm MF, Muhlbauer A, et al. Solar lentigo evolving into fatal metastatic melanoma in a patient who initially refused surgery. N A J Med Sci. 2020;1:28-31. doi:10.7156/najms.2020.1301028
- Stern JB, Peck GL, Haupt HM, et al. Malignant melanoma in xeroderma pigmentosum: search for a precursor lesion. J Am Acad Dermatol. 1993;28:591-594. doi:10.1016/0190-9622(93)70079-9
- Byrom L, Barksdale S, Weedon D, et al. Unstable solar lentigo: a defined separate entity. Australas J Dermatol. 2016;57:229-234. doi:10.1111/ajd.12447
- Panizzon RG. Vulvar melanoma. Semin Dermatol. 1996;15:67-70. doi:10.1016/s1085-5629(96)80021-6
- Chapel TA. The signs and symptoms of secondary syphilis. Sex Transm Dis. 1980;7:161-164. doi:10.1097/00007435-198010000-00002
- Schwartz RA. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151. doi:10.1002/jso.20090
To the Editor:
Genital lentiginosis (also known as mucosal melanotic macules, vulvar melanosis, penile melanosis, and penile lentigines) occurs in men and women.1 Lesions present in adult life as multifocal, asymmetrical, pigmented patches that can have a mottled appearance or exhibit skip areas. The irregular appearance of the pigmented areas often raises concern for melanoma. Biopsy reveals increased pigmentation along the basal layer of the epidermis; the irregular distribution of single melanocytes and pagetoid spread typical of melanoma in situ is not identified.
Genital lentiginosis usually occurs as an isolated finding; however, the condition can be a manifestation of Laugier-Hunziker syndrome, Carney complex, and Bannayan-Riley-Ruvalcaba syndrome.1-3 When it occurs as an isolated finding, the patient can be reassured and treatment is unnecessary. Because genital lentiginosis may mimic the appearance of melanoma, it is important for physicians to differentiate the two and make a correct diagnosis. We present a case of genital lentiginosis that mimicked vulvar melanoma.
A 64-year-old woman was referred by her gynecologist to dermatology to rule out vulvar melanoma. The patient had a history of hypothyroidism and hypercholesterolemia but was otherwise in good health. Genital examination revealed asymptomatic pigmented macules and patches of unknown duration (Figure 1). Specimens were taken from 3 areas by punch biopsy to clarify the diagnosis. All 3 specimens showed identical features including basilar pigmentation, occasional melanophages in the papillary dermis, and no evidence of nests or pagetoid spread of atypical melanocytes (Figures 2 and 3). Histologic findings were diagnostic for genital lentiginosis. The patient was reassured, and no treatment was provided. At 6-month follow-up there was no change in clinical appearance.
Genital lentiginosis is characterized by brown lesions that can have a mottled appearance and often are associated with skip areas.1 Lesions can be strikingly irregular and darkly pigmented.
Although the lesions of genital lentiginosis most often are isolated findings, they can be a clue to several uncommon syndromes such as autosomal-dominant Bannayan-Riley-Ruvalcaba syndrome, which is associated with genital lentiginosis, intestinal polyposis, and macrocephaly.3 Vascular malformations, lipomatosis, verrucal keratoses, and acrochordons can occur. Bannayan-Riley-Ruvalcaba syndrome and Cowden syndrome may share genetic linkage; mutations in the tumor suppressor PTEN (phosphatase and tensin homolog deleted on chromosome ten) has been implicated in both syndromes.4 Underlying Carney complex should be excluded when genital lentiginosis is encountered.
Genital lentiginosis is idiopathic in most instances, but reports of lesions occurring after annular lichen planus suggest a possible mechanism.5 The disappearance of lentigines after imatinib therapy suggests a role for c-kit, a receptor tyrosine kinase that is involved in intracellular signaling, in some cases.6 At times, lesions can simulate trichrome vitiligo or have a reticulate pattern.7
Men and women present at different points in the course of disease. Men often present with penile lesions 14 years after onset, on average; they notice a gradual increase in the size of lesions. Because women can have greater difficulty self-examining the genital region, they tend to present much later in the course but often within a few months after initial inspection.1,8
Genital lentiginosis can mimic melanoma with nonhomogeneous pigmentation, asymmetry, and unilateral distribution, which makes dermoscopic assessment of colors helpful in narrowing the differential diagnosis. Melanoma is associated with combinations of gray, red, blue, and white, which are not found in genital lentiginosis.9
Biopsy of a genital lentigo is diagnostic, distinguishing the lesion from melanoma—failing to reveal the atypical melanocytes and pagetoid spread characteristic of melanoma in situ. Histologic findings can cause diagnostic difficulties when concurrent lichen sclerosus is associated with genital lentigines or nevi.10
Lentigines on sun-damaged skin or in the setting of xeroderma pigmentosum have been associated with melanoma,11-13 but genital lentigines are not considered a form of precancerous melanosis. In women, early diagnosis is important when there is concern for melanoma because the prognosis for vulvar melanoma is improved in thin lesions.14
Other entities in the differential include secondary syphilis, which commonly presents as macules and scaly papules and can be found on mucosal surfaces such as the oral cavity,15 as well as Kaposi sarcoma, which is characterized by purplish, brown, or black macules, plaques, and nodules, more commonly in immunosuppressed patients.16
To avoid unwarranted concern and unnecessary surgery, dermatologists should be aware of genital lentigines and their characteristic presentation in adults.
To the Editor:
Genital lentiginosis (also known as mucosal melanotic macules, vulvar melanosis, penile melanosis, and penile lentigines) occurs in men and women.1 Lesions present in adult life as multifocal, asymmetrical, pigmented patches that can have a mottled appearance or exhibit skip areas. The irregular appearance of the pigmented areas often raises concern for melanoma. Biopsy reveals increased pigmentation along the basal layer of the epidermis; the irregular distribution of single melanocytes and pagetoid spread typical of melanoma in situ is not identified.
Genital lentiginosis usually occurs as an isolated finding; however, the condition can be a manifestation of Laugier-Hunziker syndrome, Carney complex, and Bannayan-Riley-Ruvalcaba syndrome.1-3 When it occurs as an isolated finding, the patient can be reassured and treatment is unnecessary. Because genital lentiginosis may mimic the appearance of melanoma, it is important for physicians to differentiate the two and make a correct diagnosis. We present a case of genital lentiginosis that mimicked vulvar melanoma.
A 64-year-old woman was referred by her gynecologist to dermatology to rule out vulvar melanoma. The patient had a history of hypothyroidism and hypercholesterolemia but was otherwise in good health. Genital examination revealed asymptomatic pigmented macules and patches of unknown duration (Figure 1). Specimens were taken from 3 areas by punch biopsy to clarify the diagnosis. All 3 specimens showed identical features including basilar pigmentation, occasional melanophages in the papillary dermis, and no evidence of nests or pagetoid spread of atypical melanocytes (Figures 2 and 3). Histologic findings were diagnostic for genital lentiginosis. The patient was reassured, and no treatment was provided. At 6-month follow-up there was no change in clinical appearance.
Genital lentiginosis is characterized by brown lesions that can have a mottled appearance and often are associated with skip areas.1 Lesions can be strikingly irregular and darkly pigmented.
Although the lesions of genital lentiginosis most often are isolated findings, they can be a clue to several uncommon syndromes such as autosomal-dominant Bannayan-Riley-Ruvalcaba syndrome, which is associated with genital lentiginosis, intestinal polyposis, and macrocephaly.3 Vascular malformations, lipomatosis, verrucal keratoses, and acrochordons can occur. Bannayan-Riley-Ruvalcaba syndrome and Cowden syndrome may share genetic linkage; mutations in the tumor suppressor PTEN (phosphatase and tensin homolog deleted on chromosome ten) has been implicated in both syndromes.4 Underlying Carney complex should be excluded when genital lentiginosis is encountered.
Genital lentiginosis is idiopathic in most instances, but reports of lesions occurring after annular lichen planus suggest a possible mechanism.5 The disappearance of lentigines after imatinib therapy suggests a role for c-kit, a receptor tyrosine kinase that is involved in intracellular signaling, in some cases.6 At times, lesions can simulate trichrome vitiligo or have a reticulate pattern.7
Men and women present at different points in the course of disease. Men often present with penile lesions 14 years after onset, on average; they notice a gradual increase in the size of lesions. Because women can have greater difficulty self-examining the genital region, they tend to present much later in the course but often within a few months after initial inspection.1,8
Genital lentiginosis can mimic melanoma with nonhomogeneous pigmentation, asymmetry, and unilateral distribution, which makes dermoscopic assessment of colors helpful in narrowing the differential diagnosis. Melanoma is associated with combinations of gray, red, blue, and white, which are not found in genital lentiginosis.9
Biopsy of a genital lentigo is diagnostic, distinguishing the lesion from melanoma—failing to reveal the atypical melanocytes and pagetoid spread characteristic of melanoma in situ. Histologic findings can cause diagnostic difficulties when concurrent lichen sclerosus is associated with genital lentigines or nevi.10
Lentigines on sun-damaged skin or in the setting of xeroderma pigmentosum have been associated with melanoma,11-13 but genital lentigines are not considered a form of precancerous melanosis. In women, early diagnosis is important when there is concern for melanoma because the prognosis for vulvar melanoma is improved in thin lesions.14
Other entities in the differential include secondary syphilis, which commonly presents as macules and scaly papules and can be found on mucosal surfaces such as the oral cavity,15 as well as Kaposi sarcoma, which is characterized by purplish, brown, or black macules, plaques, and nodules, more commonly in immunosuppressed patients.16
To avoid unwarranted concern and unnecessary surgery, dermatologists should be aware of genital lentigines and their characteristic presentation in adults.
- Hwang L, Wilson H, Orengo I. Off-center fold: irregular, pigmented genital macules. Arch Dermatol. 2000;136:1559-1564. doi:10.1001/archderm.136.12.1559-b
- Rhodes AR, Silverman RA, Harrist TJ, et al. Mucocutaneous lentigines, cardiomucocutaneous myxomas, and multiple blue nevi: the “LAMB” syndrome. J Am Acad Dermatol. 1984;10:72-82. doi:10.1016/s0190-9622(84)80047-x
- Erkek E, Hizel S, Sanl C, et al. Clinical and histopathological findings in Bannayan-Riley-Ruvalcaba syndrome. J Am Acad Dermatol. 2005;53:639-643. doi:10.1016/j.jaad.2005.06.022
- Blum RR, Rahimizadeh A, Kardon N, et al. Genital lentigines in a 6-year-old boy with a family history of Cowden’s disease: clinical and genetic evidence of the linkage between Bannayan-Riley-Ruvalcaba syndrome and Cowden’s disease. J Cutan Med Surg. 2001;5:228-230. doi:10.1177/120347540100500307
- Isbary G, Dyall-Smith D, Coras-Stepanek B, et al. Penile lentigo (genital mucosal macule) following annular lichen planus: a possible association? Australas J Dermatol. 2014;55:159-161. doi:10.1111/ajd.12169
- Campbell T, Felsten L, Moore J. Disappearance of lentigines in a patient receiving imatinib treatment for familial gastrointestinal stromal tumor syndrome. Arch Dermatol. 2009;145:1313-1316. doi:10.1001/archdermatol.2009.263
- Romero- A, R, , et al. Reticulate genital pigmentation associated with localized vitiligo. Arch Dermatol. 2010; 146:574-575. doi:10.1001/archdermatol.2010.69
- Barnhill RL, Albert LS, Shama SK, et al. Genital lentiginosis: a clinical and histopathologic study. J Am Acad Dermatol. 1990;22:453-460. doi:10.1016/0190-9622(90)70064-o
- De Giorgi V, Gori A, Salvati L, et al. Clinical and dermoscopic features of vulvar melanosis over the last 20 years. JAMA Dermatol. 2020;156:1185–1191. doi:10.1001/jamadermatol.2020.2528
- El Shabrawi-Caelen L, Soyer HP, Schaeppi H, et al. Genital lentigines and melanocytic nevi with superimposed lichen sclerosus: a diagnostic challenge. J Am Acad Dermatol. 2004;50:690-694. doi:10.1016/j.jaad.2003.09.034
- Shatkin M, Helm MF, Muhlbauer A, et al. Solar lentigo evolving into fatal metastatic melanoma in a patient who initially refused surgery. N A J Med Sci. 2020;1:28-31. doi:10.7156/najms.2020.1301028
- Stern JB, Peck GL, Haupt HM, et al. Malignant melanoma in xeroderma pigmentosum: search for a precursor lesion. J Am Acad Dermatol. 1993;28:591-594. doi:10.1016/0190-9622(93)70079-9
- Byrom L, Barksdale S, Weedon D, et al. Unstable solar lentigo: a defined separate entity. Australas J Dermatol. 2016;57:229-234. doi:10.1111/ajd.12447
- Panizzon RG. Vulvar melanoma. Semin Dermatol. 1996;15:67-70. doi:10.1016/s1085-5629(96)80021-6
- Chapel TA. The signs and symptoms of secondary syphilis. Sex Transm Dis. 1980;7:161-164. doi:10.1097/00007435-198010000-00002
- Schwartz RA. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151. doi:10.1002/jso.20090
- Hwang L, Wilson H, Orengo I. Off-center fold: irregular, pigmented genital macules. Arch Dermatol. 2000;136:1559-1564. doi:10.1001/archderm.136.12.1559-b
- Rhodes AR, Silverman RA, Harrist TJ, et al. Mucocutaneous lentigines, cardiomucocutaneous myxomas, and multiple blue nevi: the “LAMB” syndrome. J Am Acad Dermatol. 1984;10:72-82. doi:10.1016/s0190-9622(84)80047-x
- Erkek E, Hizel S, Sanl C, et al. Clinical and histopathological findings in Bannayan-Riley-Ruvalcaba syndrome. J Am Acad Dermatol. 2005;53:639-643. doi:10.1016/j.jaad.2005.06.022
- Blum RR, Rahimizadeh A, Kardon N, et al. Genital lentigines in a 6-year-old boy with a family history of Cowden’s disease: clinical and genetic evidence of the linkage between Bannayan-Riley-Ruvalcaba syndrome and Cowden’s disease. J Cutan Med Surg. 2001;5:228-230. doi:10.1177/120347540100500307
- Isbary G, Dyall-Smith D, Coras-Stepanek B, et al. Penile lentigo (genital mucosal macule) following annular lichen planus: a possible association? Australas J Dermatol. 2014;55:159-161. doi:10.1111/ajd.12169
- Campbell T, Felsten L, Moore J. Disappearance of lentigines in a patient receiving imatinib treatment for familial gastrointestinal stromal tumor syndrome. Arch Dermatol. 2009;145:1313-1316. doi:10.1001/archdermatol.2009.263
- Romero- A, R, , et al. Reticulate genital pigmentation associated with localized vitiligo. Arch Dermatol. 2010; 146:574-575. doi:10.1001/archdermatol.2010.69
- Barnhill RL, Albert LS, Shama SK, et al. Genital lentiginosis: a clinical and histopathologic study. J Am Acad Dermatol. 1990;22:453-460. doi:10.1016/0190-9622(90)70064-o
- De Giorgi V, Gori A, Salvati L, et al. Clinical and dermoscopic features of vulvar melanosis over the last 20 years. JAMA Dermatol. 2020;156:1185–1191. doi:10.1001/jamadermatol.2020.2528
- El Shabrawi-Caelen L, Soyer HP, Schaeppi H, et al. Genital lentigines and melanocytic nevi with superimposed lichen sclerosus: a diagnostic challenge. J Am Acad Dermatol. 2004;50:690-694. doi:10.1016/j.jaad.2003.09.034
- Shatkin M, Helm MF, Muhlbauer A, et al. Solar lentigo evolving into fatal metastatic melanoma in a patient who initially refused surgery. N A J Med Sci. 2020;1:28-31. doi:10.7156/najms.2020.1301028
- Stern JB, Peck GL, Haupt HM, et al. Malignant melanoma in xeroderma pigmentosum: search for a precursor lesion. J Am Acad Dermatol. 1993;28:591-594. doi:10.1016/0190-9622(93)70079-9
- Byrom L, Barksdale S, Weedon D, et al. Unstable solar lentigo: a defined separate entity. Australas J Dermatol. 2016;57:229-234. doi:10.1111/ajd.12447
- Panizzon RG. Vulvar melanoma. Semin Dermatol. 1996;15:67-70. doi:10.1016/s1085-5629(96)80021-6
- Chapel TA. The signs and symptoms of secondary syphilis. Sex Transm Dis. 1980;7:161-164. doi:10.1097/00007435-198010000-00002
- Schwartz RA. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151. doi:10.1002/jso.20090
Practice Points
- The irregular appearance of genital lentiginosis—multifocal, asymmetric, irregular, and darkly pigmented patches—often raises concern for melanoma but is benign.
- Certain genetic conditions can present with genital lentiginosis.
- Dermoscopic assessment of the lesion color is highly helpful in narrowing the differential diagnosis; seeing no gray, red, blue, or white makes melanoma less likely.
- Be aware of genital lentigines and their characteristic presentation in adulthood to avoid unwarranted concern and unneeded surgery.

Surgical Specimens and Margins
We have attended grand rounds presentations at which students announce that Mohs micrographic surgery evaluates 100% of the surgical margin, whereas standard excision samples 1% to 2% of the margin; we have even fielded questions from neighbors who have come across this information on the internet.1-5 This statement describes a best-case scenario for Mohs surgery and a worst-case scenario for standard excision. We believe that it is important for clinicians to have a more nuanced understanding of how simple excisions are processed so that they can have pertinent discussions with patients, especially now that there is increasing access to personal health information along with increased agency in patient decision-making.
Margins for Mohs Surgery
Theoretically, Mohs surgery should sample all true surgical margins by complete circumferential, peripheral, and deep-margin assessment. Unfortunately, some sections are not cut full face—sections may not always sample a complete surface—when technicians make an error or lack expertise. Some sections may have small tissue folds or small gaps that prevent complete visualization. We estimate that the Mohs sections we review in consultation that are prepared by private practice Mohs surgeons in our communities visualize approximately 98% of surgical margins on average. Incomplete sections contribute to the rare tumor recurrences after Mohs surgery of approximately 2% to 3%.6
Standard Excision Margins
When we obtained the references cited in articles asserting that
Here is a simple example to show that more margin is accessed in some cases. Consider this hypothetical situation: If a tumor can be readily visualized grossly and housed entirely within an imaginary cuboid (rectangular) prism that is removed in an elliptical specimen with a length of 6 cm, a width of 2 cm, and a height of 1 cm (Figure), then standard sectioning assesses a greater margin.

Bread-loaf sectioning would be expected to examine the complete surface of 2 sides (faces) of the cuboid. Assessing 2 of the 5 clinically relevant sides provides information for approximately 50% of the margins, as sections in the next parallel plane can be expected to be clear after the first clear section is identified. The clinically useful information is not limited to the sum of the widths of sections. Encountering a clear plane typically indicates that there will be no tumor in more distal parallel planes. Warne et al6 developed a formula that can accurately predict the percentage of the margin evaluated by proxy that considers the curvature of the ellipse.
Comparing Standard Excision and Mohs Surgery
Mohs surgery consistently results in the best outcomes, but standard excision is effective, too. Standard excision is relatively simple, requires less equipment, is less time consuming, and can provide good value when resources are finite. Data on recurrence of basal cell carcinoma after simple excision are limited, but the recurrence rate is reported to be approximately 3%.7,8 A meta-analysis found that the recurrence rate of basal cell carcinoma treated with standard excision was 0.4%, 1.6%, 2.6%, and 4% with 5-mm, 4-mm, 3-mm, and 2-mm surgical margins, respectively.9
Mohs surgery is the best, most effective, and most tissue-sparing technique for certain nonmelanoma skin cancers. This observation is reflected in guidelines worldwide.10 The adequacy of standard approaches to margin evaluation depends on the capabilities and focus of the laboratory team. Dermatopathologists often are called to the laboratory to decide which technique will be best for a particular case.11 Technicians are trained to take more sections in areas where abnormalities are seen, and some laboratories take photographs of specimens or provide sketches for correlation. Dermatopathologists also routinely request additional sections in areas where visible tumor extends close to surgical margins on microscopic examination.
It is not simply a matter of knowing how much of the margin is sampled but if the most pertinent areas are adequately sampled. Simple sectioning can work well and be cost effective. Many clinicians are unaware of how tissue processing can vary from laboratory to laboratory. There are no uniformly accepted standards for how tissue should be processed. Assiduous and thoughtful evaluation of specimens can affect results. As with any service, some laboratories provide more detailed and conscientious care while others focus more on immediate costs. Clinicians should understand how their specimens are processed by discussing margin evaluation with their dermatopathologist.
Final Thoughts
Used appropriately, Mohs surgery is an excellent technique that can provide outstanding results. Standard excision also has an important place in the dermatologist’s armamentarium and typically provides information about more than 1% to 2% of the margin. Understanding the techniques used to process specimens is critical to delivering the best possible care.
- Tolkachjov SN, Brodland DG, Coldiron BM, et al. Understanding Mohs micrographic surgery: a review and practical guide for the nondermatologist. Mayo Clin Proc. 2017;92:1261-1271. doi:10.1016/j.mayocp.2017.04.009
- Thomas RM, Amonette RA. Mohs micrographic surgery. Am Fam Physician. 1988;37:135-142.
- Buker JL, Amonette RA. Micrographic surgery. Clin Dermatol. 1992:10:309-315. doi:10.1016/0738-081x(92)90074-9
- Kauvar ANB. Mohs: the gold standard. The Skin Cancer Foundation website. Updated March 9, 2021. Accessed June 15, 2022. https://www.skincancer.org/treatment-resources/mohs-surgery/mohs-the-gold-standard/
- van Delft LCJ, Nelemans PJ, van Loo E, et al. The illusion of conventional histological resection margin control. Br J Dermatol. 2019;180:1240-1241. doi:10.1111/bjd.17510
- Warne MM, Klawonn MM, Brodell RT. Bread loaf sections provide useful information on more than 0.5% of surgical margins [published July 5, 2022]. Br J Dermatol. doi:10.1111/bjd.21740
- Mehrany K, Weenig RH, Pittelkow MR, et al. High recurrence rates of basal cell carcinoma after Mohs surgery in patients with chronic lymphocytic leukemia. Arch Dermatol. 2004;140:985-988. doi:10.1001/archderm.140.8.985
- Smeets NWJ, Krekels GAM, Ostertag JU, et al. Surgical excision vs Mohs’ micrographic surgery for basal-cell carcinoma of the face: randomised controlled trial. Lancet. 2004;364:1766-1772. doi:10.1016/S0140-6736(04)17399-6
- Gulleth Y, Goldberg N, Silverman RP, et al. What is the best surgical margin for a basal cell carcinoma: a meta-analysis of theliterature. Plast Reconstr Surg. 2010;126:1222-1231. doi:10.1097/PRS.0b013e3181ea450d
- Nahhas AF, Scarbrough CA, Trotter S. A review of the global guidelines on surgical margins for nonmelanoma skin cancers. J Clin Aesthet Dermatol. 2017;10:37-46.
- Rapini RP. Comparison of methods for checking surgical margins. J Am Acad Dermatol. 1990; 23:288-294. doi:10.1016/0190-9622(90)70212-z
We have attended grand rounds presentations at which students announce that Mohs micrographic surgery evaluates 100% of the surgical margin, whereas standard excision samples 1% to 2% of the margin; we have even fielded questions from neighbors who have come across this information on the internet.1-5 This statement describes a best-case scenario for Mohs surgery and a worst-case scenario for standard excision. We believe that it is important for clinicians to have a more nuanced understanding of how simple excisions are processed so that they can have pertinent discussions with patients, especially now that there is increasing access to personal health information along with increased agency in patient decision-making.
Margins for Mohs Surgery
Theoretically, Mohs surgery should sample all true surgical margins by complete circumferential, peripheral, and deep-margin assessment. Unfortunately, some sections are not cut full face—sections may not always sample a complete surface—when technicians make an error or lack expertise. Some sections may have small tissue folds or small gaps that prevent complete visualization. We estimate that the Mohs sections we review in consultation that are prepared by private practice Mohs surgeons in our communities visualize approximately 98% of surgical margins on average. Incomplete sections contribute to the rare tumor recurrences after Mohs surgery of approximately 2% to 3%.6
Standard Excision Margins
When we obtained the references cited in articles asserting that
Here is a simple example to show that more margin is accessed in some cases. Consider this hypothetical situation: If a tumor can be readily visualized grossly and housed entirely within an imaginary cuboid (rectangular) prism that is removed in an elliptical specimen with a length of 6 cm, a width of 2 cm, and a height of 1 cm (Figure), then standard sectioning assesses a greater margin.
Bread-loaf sectioning would be expected to examine the complete surface of 2 sides (faces) of the cuboid. Assessing 2 of the 5 clinically relevant sides provides information for approximately 50% of the margins, as sections in the next parallel plane can be expected to be clear after the first clear section is identified. The clinically useful information is not limited to the sum of the widths of sections. Encountering a clear plane typically indicates that there will be no tumor in more distal parallel planes. Warne et al6 developed a formula that can accurately predict the percentage of the margin evaluated by proxy that considers the curvature of the ellipse.
Comparing Standard Excision and Mohs Surgery
Mohs surgery consistently results in the best outcomes, but standard excision is effective, too. Standard excision is relatively simple, requires less equipment, is less time consuming, and can provide good value when resources are finite. Data on recurrence of basal cell carcinoma after simple excision are limited, but the recurrence rate is reported to be approximately 3%.7,8 A meta-analysis found that the recurrence rate of basal cell carcinoma treated with standard excision was 0.4%, 1.6%, 2.6%, and 4% with 5-mm, 4-mm, 3-mm, and 2-mm surgical margins, respectively.9
Mohs surgery is the best, most effective, and most tissue-sparing technique for certain nonmelanoma skin cancers. This observation is reflected in guidelines worldwide.10 The adequacy of standard approaches to margin evaluation depends on the capabilities and focus of the laboratory team. Dermatopathologists often are called to the laboratory to decide which technique will be best for a particular case.11 Technicians are trained to take more sections in areas where abnormalities are seen, and some laboratories take photographs of specimens or provide sketches for correlation. Dermatopathologists also routinely request additional sections in areas where visible tumor extends close to surgical margins on microscopic examination.
It is not simply a matter of knowing how much of the margin is sampled but if the most pertinent areas are adequately sampled. Simple sectioning can work well and be cost effective. Many clinicians are unaware of how tissue processing can vary from laboratory to laboratory. There are no uniformly accepted standards for how tissue should be processed. Assiduous and thoughtful evaluation of specimens can affect results. As with any service, some laboratories provide more detailed and conscientious care while others focus more on immediate costs. Clinicians should understand how their specimens are processed by discussing margin evaluation with their dermatopathologist.
Final Thoughts
Used appropriately, Mohs surgery is an excellent technique that can provide outstanding results. Standard excision also has an important place in the dermatologist’s armamentarium and typically provides information about more than 1% to 2% of the margin. Understanding the techniques used to process specimens is critical to delivering the best possible care.
We have attended grand rounds presentations at which students announce that Mohs micrographic surgery evaluates 100% of the surgical margin, whereas standard excision samples 1% to 2% of the margin; we have even fielded questions from neighbors who have come across this information on the internet.1-5 This statement describes a best-case scenario for Mohs surgery and a worst-case scenario for standard excision. We believe that it is important for clinicians to have a more nuanced understanding of how simple excisions are processed so that they can have pertinent discussions with patients, especially now that there is increasing access to personal health information along with increased agency in patient decision-making.
Margins for Mohs Surgery
Theoretically, Mohs surgery should sample all true surgical margins by complete circumferential, peripheral, and deep-margin assessment. Unfortunately, some sections are not cut full face—sections may not always sample a complete surface—when technicians make an error or lack expertise. Some sections may have small tissue folds or small gaps that prevent complete visualization. We estimate that the Mohs sections we review in consultation that are prepared by private practice Mohs surgeons in our communities visualize approximately 98% of surgical margins on average. Incomplete sections contribute to the rare tumor recurrences after Mohs surgery of approximately 2% to 3%.6
Standard Excision Margins
When we obtained the references cited in articles asserting that
Here is a simple example to show that more margin is accessed in some cases. Consider this hypothetical situation: If a tumor can be readily visualized grossly and housed entirely within an imaginary cuboid (rectangular) prism that is removed in an elliptical specimen with a length of 6 cm, a width of 2 cm, and a height of 1 cm (Figure), then standard sectioning assesses a greater margin.
Bread-loaf sectioning would be expected to examine the complete surface of 2 sides (faces) of the cuboid. Assessing 2 of the 5 clinically relevant sides provides information for approximately 50% of the margins, as sections in the next parallel plane can be expected to be clear after the first clear section is identified. The clinically useful information is not limited to the sum of the widths of sections. Encountering a clear plane typically indicates that there will be no tumor in more distal parallel planes. Warne et al6 developed a formula that can accurately predict the percentage of the margin evaluated by proxy that considers the curvature of the ellipse.
Comparing Standard Excision and Mohs Surgery
Mohs surgery consistently results in the best outcomes, but standard excision is effective, too. Standard excision is relatively simple, requires less equipment, is less time consuming, and can provide good value when resources are finite. Data on recurrence of basal cell carcinoma after simple excision are limited, but the recurrence rate is reported to be approximately 3%.7,8 A meta-analysis found that the recurrence rate of basal cell carcinoma treated with standard excision was 0.4%, 1.6%, 2.6%, and 4% with 5-mm, 4-mm, 3-mm, and 2-mm surgical margins, respectively.9
Mohs surgery is the best, most effective, and most tissue-sparing technique for certain nonmelanoma skin cancers. This observation is reflected in guidelines worldwide.10 The adequacy of standard approaches to margin evaluation depends on the capabilities and focus of the laboratory team. Dermatopathologists often are called to the laboratory to decide which technique will be best for a particular case.11 Technicians are trained to take more sections in areas where abnormalities are seen, and some laboratories take photographs of specimens or provide sketches for correlation. Dermatopathologists also routinely request additional sections in areas where visible tumor extends close to surgical margins on microscopic examination.
It is not simply a matter of knowing how much of the margin is sampled but if the most pertinent areas are adequately sampled. Simple sectioning can work well and be cost effective. Many clinicians are unaware of how tissue processing can vary from laboratory to laboratory. There are no uniformly accepted standards for how tissue should be processed. Assiduous and thoughtful evaluation of specimens can affect results. As with any service, some laboratories provide more detailed and conscientious care while others focus more on immediate costs. Clinicians should understand how their specimens are processed by discussing margin evaluation with their dermatopathologist.
Final Thoughts
Used appropriately, Mohs surgery is an excellent technique that can provide outstanding results. Standard excision also has an important place in the dermatologist’s armamentarium and typically provides information about more than 1% to 2% of the margin. Understanding the techniques used to process specimens is critical to delivering the best possible care.
- Tolkachjov SN, Brodland DG, Coldiron BM, et al. Understanding Mohs micrographic surgery: a review and practical guide for the nondermatologist. Mayo Clin Proc. 2017;92:1261-1271. doi:10.1016/j.mayocp.2017.04.009
- Thomas RM, Amonette RA. Mohs micrographic surgery. Am Fam Physician. 1988;37:135-142.
- Buker JL, Amonette RA. Micrographic surgery. Clin Dermatol. 1992:10:309-315. doi:10.1016/0738-081x(92)90074-9
- Kauvar ANB. Mohs: the gold standard. The Skin Cancer Foundation website. Updated March 9, 2021. Accessed June 15, 2022. https://www.skincancer.org/treatment-resources/mohs-surgery/mohs-the-gold-standard/
- van Delft LCJ, Nelemans PJ, van Loo E, et al. The illusion of conventional histological resection margin control. Br J Dermatol. 2019;180:1240-1241. doi:10.1111/bjd.17510
- Warne MM, Klawonn MM, Brodell RT. Bread loaf sections provide useful information on more than 0.5% of surgical margins [published July 5, 2022]. Br J Dermatol. doi:10.1111/bjd.21740
- Mehrany K, Weenig RH, Pittelkow MR, et al. High recurrence rates of basal cell carcinoma after Mohs surgery in patients with chronic lymphocytic leukemia. Arch Dermatol. 2004;140:985-988. doi:10.1001/archderm.140.8.985
- Smeets NWJ, Krekels GAM, Ostertag JU, et al. Surgical excision vs Mohs’ micrographic surgery for basal-cell carcinoma of the face: randomised controlled trial. Lancet. 2004;364:1766-1772. doi:10.1016/S0140-6736(04)17399-6
- Gulleth Y, Goldberg N, Silverman RP, et al. What is the best surgical margin for a basal cell carcinoma: a meta-analysis of theliterature. Plast Reconstr Surg. 2010;126:1222-1231. doi:10.1097/PRS.0b013e3181ea450d
- Nahhas AF, Scarbrough CA, Trotter S. A review of the global guidelines on surgical margins for nonmelanoma skin cancers. J Clin Aesthet Dermatol. 2017;10:37-46.
- Rapini RP. Comparison of methods for checking surgical margins. J Am Acad Dermatol. 1990; 23:288-294. doi:10.1016/0190-9622(90)70212-z
- Tolkachjov SN, Brodland DG, Coldiron BM, et al. Understanding Mohs micrographic surgery: a review and practical guide for the nondermatologist. Mayo Clin Proc. 2017;92:1261-1271. doi:10.1016/j.mayocp.2017.04.009
- Thomas RM, Amonette RA. Mohs micrographic surgery. Am Fam Physician. 1988;37:135-142.
- Buker JL, Amonette RA. Micrographic surgery. Clin Dermatol. 1992:10:309-315. doi:10.1016/0738-081x(92)90074-9
- Kauvar ANB. Mohs: the gold standard. The Skin Cancer Foundation website. Updated March 9, 2021. Accessed June 15, 2022. https://www.skincancer.org/treatment-resources/mohs-surgery/mohs-the-gold-standard/
- van Delft LCJ, Nelemans PJ, van Loo E, et al. The illusion of conventional histological resection margin control. Br J Dermatol. 2019;180:1240-1241. doi:10.1111/bjd.17510
- Warne MM, Klawonn MM, Brodell RT. Bread loaf sections provide useful information on more than 0.5% of surgical margins [published July 5, 2022]. Br J Dermatol. doi:10.1111/bjd.21740
- Mehrany K, Weenig RH, Pittelkow MR, et al. High recurrence rates of basal cell carcinoma after Mohs surgery in patients with chronic lymphocytic leukemia. Arch Dermatol. 2004;140:985-988. doi:10.1001/archderm.140.8.985
- Smeets NWJ, Krekels GAM, Ostertag JU, et al. Surgical excision vs Mohs’ micrographic surgery for basal-cell carcinoma of the face: randomised controlled trial. Lancet. 2004;364:1766-1772. doi:10.1016/S0140-6736(04)17399-6
- Gulleth Y, Goldberg N, Silverman RP, et al. What is the best surgical margin for a basal cell carcinoma: a meta-analysis of theliterature. Plast Reconstr Surg. 2010;126:1222-1231. doi:10.1097/PRS.0b013e3181ea450d
- Nahhas AF, Scarbrough CA, Trotter S. A review of the global guidelines on surgical margins for nonmelanoma skin cancers. J Clin Aesthet Dermatol. 2017;10:37-46.
- Rapini RP. Comparison of methods for checking surgical margins. J Am Acad Dermatol. 1990; 23:288-294. doi:10.1016/0190-9622(90)70212-z
Practice Points
- Margin analysis in simple excisions can provide useful information by proxy about more than the 1% of the margin often quoted in the literature.
- Simple excisions of uncomplicated keratinocytic carcinomas are associated with high cure rates.
Rapid Onset of Widespread Nodules and Lymphadenopathy
The Diagnosis: Primary Cutaneous γδ T-cell Lymphoma
Primary cutaneous γδ T-cell lymphoma (PCGDTL) is a distinct entity that can be confused with other types of cutaneous T-cell lymphomas. Often rapidly fatal, PCGDTL has a broad clinical spectrum that may include indolent variants—subcutaneous, epidermotropic, and dermal.1 Primary cutaneous γδ T-cell lymphoma represents less than 1% of all cutaneous T-cell lymphomas.2 Diagnosis and treatment remain challenging. Patients typically present with nodular lesions that progress to ulceration and necrosis. Early lesions can be confused with erythema nodosum, mycosis fungoides, or infection on clinical examination; biopsy establishes the diagnosis. Typical findings include a cytotoxic phenotype, variable epidermotropism, dermal and subcutaneous involvement, and loss of CD4 and often CD8 expression. Testing for Epstein-Barr virus expression yields negative results. The neoplastic lymphocytes in dermal and subcutaneous PCGDTL typically are T-cell intracellular antigen-1 (TIA-1) and granzyme positive.1
Immunohistochemistry failed to reveal CD8, CD56, granzyme, or T-cell intracellular antigen-1 staining of neoplastic cells in our patient but stained diffusely positive with CD3 and CD4. A CD20 stain decorated only a few dermal cells. The patient’s skin lesions continued to enlarge, and the massive lymphadenopathy made breathing difficult. Computed tomography revealed diffuse systemic involvement. An axillary lymph node biopsy revealed sinusoids with complete diffuse effacement of architecture as well as frequent mitotic figures and karyorrhectic debris (Figure 1A). Negative staining for T-cell receptor beta-F1 of the axillary lymph node biopsy and clonal rearrangement of the T-cell receptor gamma chain supported the diagnosis of PCGDTL. Nuclear staining for Epstein-Barr virus–encoded RNA was negative. Human T-cell leukemia virus type 1 antibodies and polymerase chain reaction also were negative. Flow cytometry demonstrated an atypical population of CD3+, CD4+, and CD7− γδ T lymphocytes, further supporting the diagnosis of lymphoma.
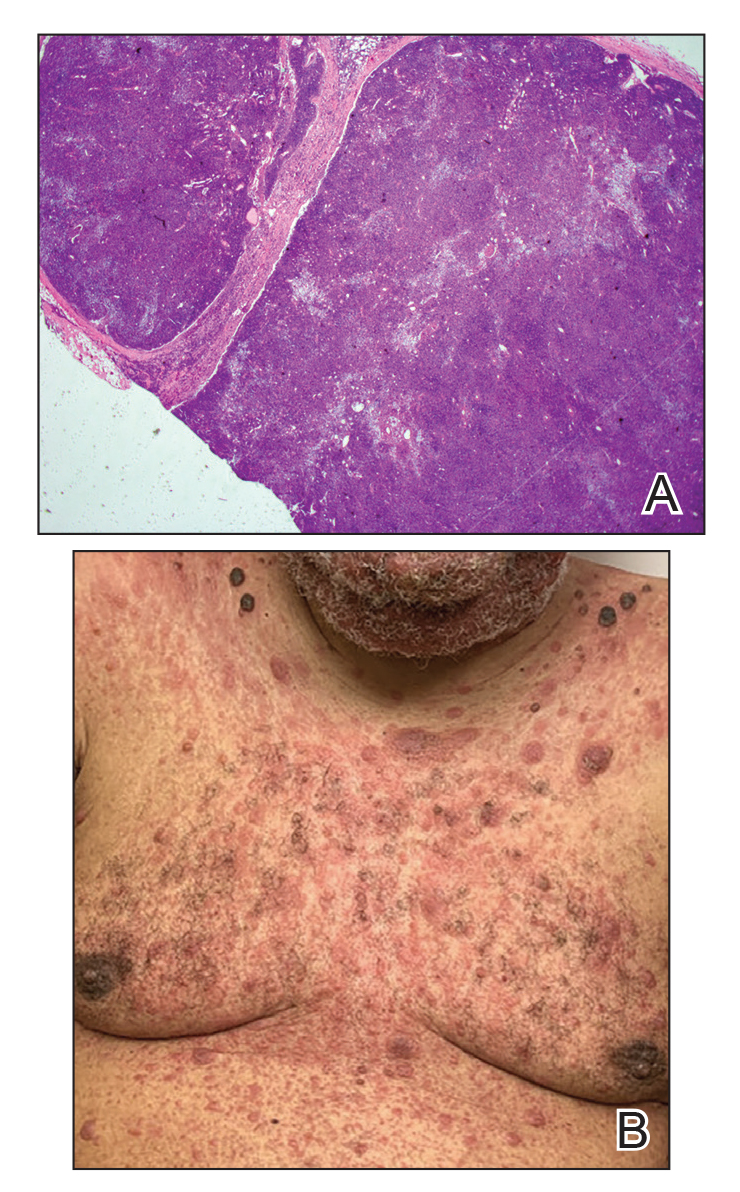
The median life expectancy for patients with dermal or subcutaneous PCGDTL is 10 to 15 months after diagnosis.3 The 5-year life expectancy for PCGDTL is approximately 11%.2 Limited treatment options contribute to the poor outcome. Chemotherapy regimens such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisolone) and EPOCH (etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) have yielded inconsistent results. Stem cell transplant has been tried in progressive disease and also has yielded mixed results.2 Brentuximab is indicated for individuals whose tumors express CD30.4 Associated hemophagic lymphohistiocytosis portends a poor prognosis.5
Despite treatment with etoposide, vincristine, doxorubicin, and high-dose oral steroids, our patient developed progressive difficulty breathing, stridor, kidney injury, and anemia. Our patient died less than 1 month after diagnosis—after only 1 round of chemotherapy—secondary to progressive disease and an uncontrollable gastrointestinal tract bleed. The leonine facies (Figure 1B) encountered in our patient can raise a differential diagnosis that includes infectious as well as neoplastic etiologies; however, most infectious etiologies associated with leonine facies manifest in a chronic fashion rather than with a sudden eruption, as noted in our patient.
Leprosy is caused by Mycobacterium leprae, a grampositive bacillus. The condition manifests across a spectrum, with the poles being tuberculoid and lepromatous, and borderline variants in between.6-8 Lepromatous leprosy arises in individuals who are unable to mount cellular immunity against M leprae secondary to anergy.6 Lepromatous leprosy often presents with numerous papules and nodules. Aside from cutaneous manifestations, lepromatous leprosy has a predilection for peripheral nerves and specifically Schwann cells. Histologically, biopsy reveals a flat epidermis and a cell-free subepidermal grenz zone. Within the dermis, there is a diffuse histiocytic infiltrate that typically is not centered around nerves (Figure 2).6,7 Mycobacterium leprae can appear scattered throughout or clustered in globi. Mycobacterium leprae stains red with Ziehl-Neelsen or Wade-Fite stains.6,7 Immunohistochemistry reveals a CD4+ helper T cell (TH2) predominance, supported by the increased expression of type 2 reaction cytokines such as IL-4, IL-5, IL-10, and IL-13.8

Diffuse large B-cell lymphoma (DLBCL) embodies 10% to 20% of all primary cutaneous lymphomas; it is more prevalent in older adults (age range, 70–82 years) and women. Clinically, DLBCL presents as either single or multiple rapidly progressing nodules or plaques, usually violaceous or blue-red in color.9,10 The most common area of presentation is on the legs, though it also can surface at other sites.9 On histology, DLBCL has clearly malignant features including frequent mitotic figures, large immunoblasts, and involvement throughout the dermis as well as perivascularly (Figure 3). Spindle-shaped cells and anaplastic features can be present. Immunohistochemically, DLBCL stains strongly positive for CD20 and B-cell lymphoma 2 (Bcl-2) along with other pan–B-cell markers.9-11 The aggressive leg type of DLBCL stains positively for multiple myeloma oncogene 1 (MUM-1).9,11
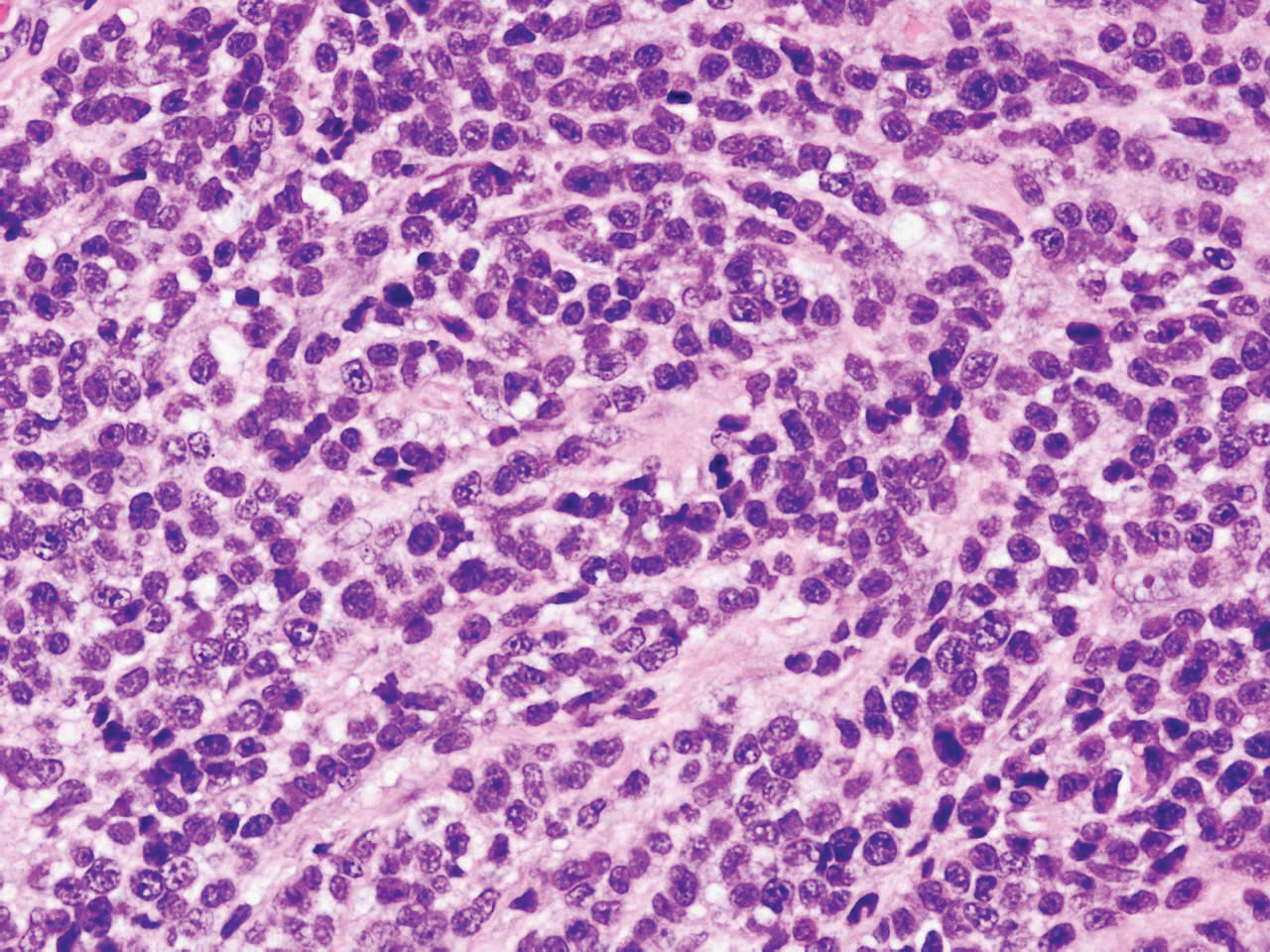
Cutaneous metastatic adenocarcinoma from internal malignancies occurs in approximately 5% of cancer patients with metastatic spread.12 Most of these cutaneous lesions develop in close proximity to the primary tumor such as on the trunk, head, or neck. All cutaneous metastases carry a poor prognosis. Clinical presentation can vary greatly, ranging from painless, firm, or elastic nodules to lesions that mimic inflammatory skin conditions such as erysipelas or scleroderma. The majority of these metastases develop as painless firm nodules that are flesh colored, pink, red-brown, or purple.12,13 The histopathology of metastatic adenocarcinoma demonstrates an infiltrative nodular appearance, though there rarely are well-circumscribed nodules found.13 The lesion originates in the dermis or subcutaneous tissue. It is a glandulartype lesion that may reflect the tissue of the primary tumor (Figure 4).12,14 Immunohistochemical stains likely will remain consistent with those of the primary tumor, which is not always the case.14

Merkel cell carcinoma (MCC) is an aggressive cutaneous malignancy of epithelial and neuroendocrine origin, first described as trabecular carcinoma due to the arrangement of tumor resembling cancellous bone.15,16 Merkel cells are mechanoreceptors found near nerve terminals.17 Approximately 80% of MCCs are associated with Merkel cell polyomavirus, which is a small, double-stranded DNA virus with an icosahedral capsid.17,18 Merkel cell polyomavirus–positive cases of MCC tend to have a better prognosis. In Merkel cell polyomavirus–negative MCC, there is an association with UV damage and increased chromosomal aberrations.18 Merkel cell carcinoma is known for its high rate of recurrence as well as local and distant metastasis. Nodal involvement is the most important prognostic indicator.15 Clinically, MCC is associated with the AEIOU mnemonic (asymptomatic, expanding rapidly, immunosuppression, older than 50 years, UV exposed/fair skin).15-17 Lesions appear as red-blue papules on sun-exposed skin and usually are smaller than 2 cm by their greatest dimension. On histopathology, MCC demonstrates small, round, blue cells arranged in sheets or nests originating in the dermis and occasionally can infiltrate the subcutis and lymphovascular surroundings (Figure 5).16-19 Cells have scant eosinophilic cytoplasm and may have fine granular chromatin. Numerous mitotic figures and apoptotic cells also are present. On immunohistochemistry, these cells will stain positive for cytokeratin AE1/AE3, anticytokeratin (CAM 5.2), CK20, and CD56. Due to their neuroendocrine derivation, they also are commonly synaptophysin, neuron-specific enolase, and chromogranin A positive. Notably, MCC will stain negative for leukocyte common antigen, CD20, CD3, CD34, and thyroid transcription factor 1 (TTF-1).16,17
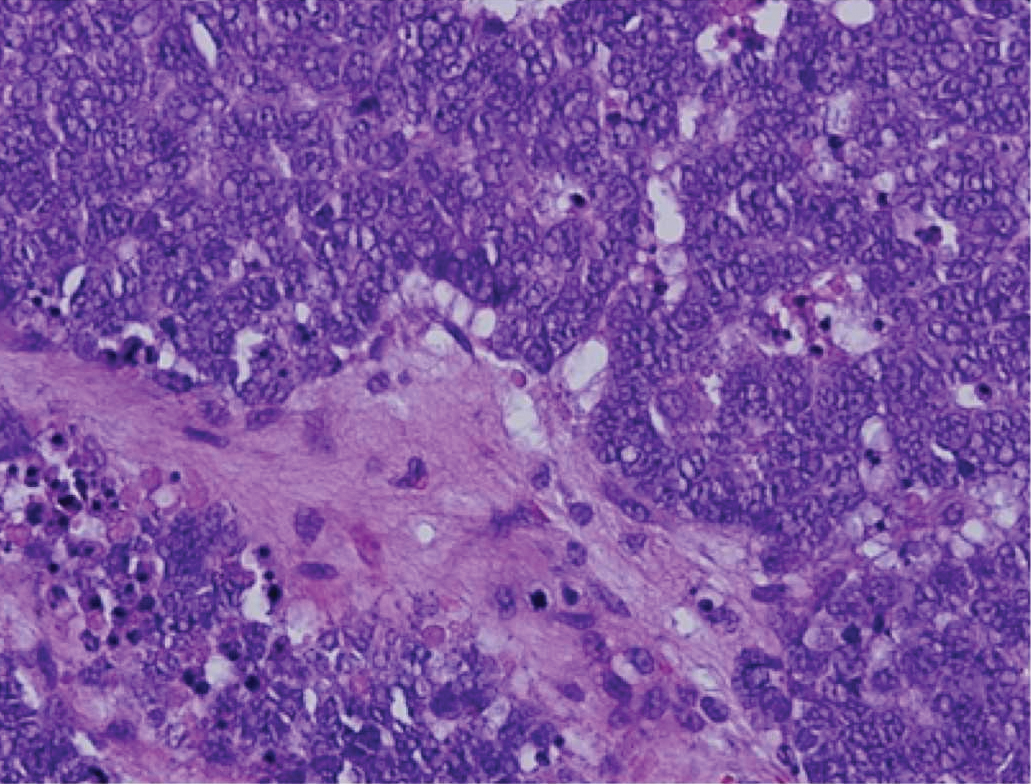
Primary cutaneous γδ T-cell lymphoma can be difficult to diagnose and requires urgent treatment. Clinicians and dermatopathologists need to work together to establish the diagnosis. There is a high mortality rate associated with PCGDTL, making prompt recognition and timely treatment critical. Acknowledgments—Thank you to our colleagues with the Penn State Health Hematology/Oncology Department (Hershey, Pennsylvania) for comanagement of this patient.
Acknowledgments
Thank you to our colleagues with the Penn State Health Hematology/Oncology Department (Hershey, Pennsylvania) for comanagement of this patient.
- Merrill ED, Agbay R, Miranda RN, et al. Primary cutaneous T-cell lymphomas showing gamma-delta (γδ) phenotype and predominantly epidermotropic pattern are clinicopathologically distinct from classic primary cutaneous γδ T-cell lymphomas. Am J Surg Pathol. 2017;41:204-215.
- Foppoli M, Ferreri AJ. Gamma‐delta T‐cell lymphomas. Eur J Haematol. 2015;94:206-218.
- Toro JR, Liewehr DJ, Pabby N, et al. Gamma-delta T-cell phenotype is associated with significantly decreased survival in cutaneous T-cell lymphoma. Blood. 2003;101:3407-3412.
- Rubio-Gonzalez B, Zain J, Garcia L, et al. Cutaneous gamma-delta T-cell lymphoma successfully treated with brentuximab vedotin. JAMA Dermatol. 2016;152:1388-1390.
- Tong H, Ren Y, Liu H, et al. Clinical characteristics of T-cell lymphoma associated with hemophagocytic syndrome: comparison of T-cell lymphoma with and without hemophagocytic syndrome. Leuk Lymphoma. 2008;49:81-87.
- Brehmer-Andersson E. Leprosy. Dermatopathology. New York, NY: Springer; 2006:110-113.
- Massone C, Belachew WA, Schettini A. Histopathology of the lepromatous skin biopsy. Clin Dermatol. 2015;33:38-45.
- Naafs B, Noto S. Reactions in leprosy. In: Nunzi E, Massone C, eds. Leprosy: A Practical Guide. Milan, Italy: Springer; 2012:219-239.
- Hope CB, Pincus LB. Primary cutaneous B-cell lymphomas. Clin Lab Med. 2017;37:547-574.
- Billero VL, LaSenna CE, Romanelli M, et al. Primary cutaneous diffuse large B-cell lymphoma presenting as chronic non-healing ulcer. Int Wound J. 2017;14:830-832.
- Testo N, Olson L, Subramaniyam S, et al. Primary cutaneous diffuse large B-cell lymphoma with a MYC-IGH rearrangement and gain of BCL2: expanding the spectrum of MYC/BCL2 double hit lymphomas. Am J Dermatopathol. 2016;38:769-774.
- Boyd AS. Pulmonary signet-ring cell adenocarcinoma metastatic to the skin. Am J Dermatopathol. 2017;39:E66-E68.
- Guanziroli E, Coggi A, Venegoni L, et al. Cutaneous metastases of internal malignancies: an experience from a single institution. Eur J Dermatol. 2017;27:609-614.
- Fernandez-Flores A, Cassarino DS. Cutaneous metastasis of adenocarcinoma of the ampulla of Vater. Am J Dermatopathol. 2018;40:758-761.
- Trinidad CM, Torres-Cabala CA, Prieto VG, et. Al. Update on eighth edition American Joint Committee on Cancer classification for Merkel Cell carcinoma and histopathological parameters that determine prognosis. J Clin Pathol. 2017;72:337-340.
- Bandino JP, Purvis CG, Shaffer BR, et al. A comparison of the histopathologic growth patterns between non-Merkel cell small round blue cell tumors and Merkel cell carcinoma. Am J Dermatopathol. 2018;40:815-818.
- Mauzo SH, Rerrarotto R, Bell D, et al. Molecular characteristics and potential therapeutic targets in Merkel cell carcinoma. J Clin Pathol. 2016;69:382-390.
- Lowe G, Brewer J, Bordeaux J. Epidemiology and genetics. In: Alam M, Bordeaux JS, Yu SS, eds. Merkel Cell Carcinoma. New York, NY: Springer; 2013:26-28.
- North J, McCalmont T. Histopathologic diagnosis. In: Alam M, Bordeaux JS, Yu SS, eds. Merkel Cell Carcinoma. New York, NY: Springer; 2013:66-69.
The Diagnosis: Primary Cutaneous γδ T-cell Lymphoma
Primary cutaneous γδ T-cell lymphoma (PCGDTL) is a distinct entity that can be confused with other types of cutaneous T-cell lymphomas. Often rapidly fatal, PCGDTL has a broad clinical spectrum that may include indolent variants—subcutaneous, epidermotropic, and dermal.1 Primary cutaneous γδ T-cell lymphoma represents less than 1% of all cutaneous T-cell lymphomas.2 Diagnosis and treatment remain challenging. Patients typically present with nodular lesions that progress to ulceration and necrosis. Early lesions can be confused with erythema nodosum, mycosis fungoides, or infection on clinical examination; biopsy establishes the diagnosis. Typical findings include a cytotoxic phenotype, variable epidermotropism, dermal and subcutaneous involvement, and loss of CD4 and often CD8 expression. Testing for Epstein-Barr virus expression yields negative results. The neoplastic lymphocytes in dermal and subcutaneous PCGDTL typically are T-cell intracellular antigen-1 (TIA-1) and granzyme positive.1
Immunohistochemistry failed to reveal CD8, CD56, granzyme, or T-cell intracellular antigen-1 staining of neoplastic cells in our patient but stained diffusely positive with CD3 and CD4. A CD20 stain decorated only a few dermal cells. The patient’s skin lesions continued to enlarge, and the massive lymphadenopathy made breathing difficult. Computed tomography revealed diffuse systemic involvement. An axillary lymph node biopsy revealed sinusoids with complete diffuse effacement of architecture as well as frequent mitotic figures and karyorrhectic debris (Figure 1A). Negative staining for T-cell receptor beta-F1 of the axillary lymph node biopsy and clonal rearrangement of the T-cell receptor gamma chain supported the diagnosis of PCGDTL. Nuclear staining for Epstein-Barr virus–encoded RNA was negative. Human T-cell leukemia virus type 1 antibodies and polymerase chain reaction also were negative. Flow cytometry demonstrated an atypical population of CD3+, CD4+, and CD7− γδ T lymphocytes, further supporting the diagnosis of lymphoma.
The median life expectancy for patients with dermal or subcutaneous PCGDTL is 10 to 15 months after diagnosis.3 The 5-year life expectancy for PCGDTL is approximately 11%.2 Limited treatment options contribute to the poor outcome. Chemotherapy regimens such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisolone) and EPOCH (etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) have yielded inconsistent results. Stem cell transplant has been tried in progressive disease and also has yielded mixed results.2 Brentuximab is indicated for individuals whose tumors express CD30.4 Associated hemophagic lymphohistiocytosis portends a poor prognosis.5
Despite treatment with etoposide, vincristine, doxorubicin, and high-dose oral steroids, our patient developed progressive difficulty breathing, stridor, kidney injury, and anemia. Our patient died less than 1 month after diagnosis—after only 1 round of chemotherapy—secondary to progressive disease and an uncontrollable gastrointestinal tract bleed. The leonine facies (Figure 1B) encountered in our patient can raise a differential diagnosis that includes infectious as well as neoplastic etiologies; however, most infectious etiologies associated with leonine facies manifest in a chronic fashion rather than with a sudden eruption, as noted in our patient.
Leprosy is caused by Mycobacterium leprae, a grampositive bacillus. The condition manifests across a spectrum, with the poles being tuberculoid and lepromatous, and borderline variants in between.6-8 Lepromatous leprosy arises in individuals who are unable to mount cellular immunity against M leprae secondary to anergy.6 Lepromatous leprosy often presents with numerous papules and nodules. Aside from cutaneous manifestations, lepromatous leprosy has a predilection for peripheral nerves and specifically Schwann cells. Histologically, biopsy reveals a flat epidermis and a cell-free subepidermal grenz zone. Within the dermis, there is a diffuse histiocytic infiltrate that typically is not centered around nerves (Figure 2).6,7 Mycobacterium leprae can appear scattered throughout or clustered in globi. Mycobacterium leprae stains red with Ziehl-Neelsen or Wade-Fite stains.6,7 Immunohistochemistry reveals a CD4+ helper T cell (TH2) predominance, supported by the increased expression of type 2 reaction cytokines such as IL-4, IL-5, IL-10, and IL-13.8
Diffuse large B-cell lymphoma (DLBCL) embodies 10% to 20% of all primary cutaneous lymphomas; it is more prevalent in older adults (age range, 70–82 years) and women. Clinically, DLBCL presents as either single or multiple rapidly progressing nodules or plaques, usually violaceous or blue-red in color.9,10 The most common area of presentation is on the legs, though it also can surface at other sites.9 On histology, DLBCL has clearly malignant features including frequent mitotic figures, large immunoblasts, and involvement throughout the dermis as well as perivascularly (Figure 3). Spindle-shaped cells and anaplastic features can be present. Immunohistochemically, DLBCL stains strongly positive for CD20 and B-cell lymphoma 2 (Bcl-2) along with other pan–B-cell markers.9-11 The aggressive leg type of DLBCL stains positively for multiple myeloma oncogene 1 (MUM-1).9,11
Cutaneous metastatic adenocarcinoma from internal malignancies occurs in approximately 5% of cancer patients with metastatic spread.12 Most of these cutaneous lesions develop in close proximity to the primary tumor such as on the trunk, head, or neck. All cutaneous metastases carry a poor prognosis. Clinical presentation can vary greatly, ranging from painless, firm, or elastic nodules to lesions that mimic inflammatory skin conditions such as erysipelas or scleroderma. The majority of these metastases develop as painless firm nodules that are flesh colored, pink, red-brown, or purple.12,13 The histopathology of metastatic adenocarcinoma demonstrates an infiltrative nodular appearance, though there rarely are well-circumscribed nodules found.13 The lesion originates in the dermis or subcutaneous tissue. It is a glandulartype lesion that may reflect the tissue of the primary tumor (Figure 4).12,14 Immunohistochemical stains likely will remain consistent with those of the primary tumor, which is not always the case.14
Merkel cell carcinoma (MCC) is an aggressive cutaneous malignancy of epithelial and neuroendocrine origin, first described as trabecular carcinoma due to the arrangement of tumor resembling cancellous bone.15,16 Merkel cells are mechanoreceptors found near nerve terminals.17 Approximately 80% of MCCs are associated with Merkel cell polyomavirus, which is a small, double-stranded DNA virus with an icosahedral capsid.17,18 Merkel cell polyomavirus–positive cases of MCC tend to have a better prognosis. In Merkel cell polyomavirus–negative MCC, there is an association with UV damage and increased chromosomal aberrations.18 Merkel cell carcinoma is known for its high rate of recurrence as well as local and distant metastasis. Nodal involvement is the most important prognostic indicator.15 Clinically, MCC is associated with the AEIOU mnemonic (asymptomatic, expanding rapidly, immunosuppression, older than 50 years, UV exposed/fair skin).15-17 Lesions appear as red-blue papules on sun-exposed skin and usually are smaller than 2 cm by their greatest dimension. On histopathology, MCC demonstrates small, round, blue cells arranged in sheets or nests originating in the dermis and occasionally can infiltrate the subcutis and lymphovascular surroundings (Figure 5).16-19 Cells have scant eosinophilic cytoplasm and may have fine granular chromatin. Numerous mitotic figures and apoptotic cells also are present. On immunohistochemistry, these cells will stain positive for cytokeratin AE1/AE3, anticytokeratin (CAM 5.2), CK20, and CD56. Due to their neuroendocrine derivation, they also are commonly synaptophysin, neuron-specific enolase, and chromogranin A positive. Notably, MCC will stain negative for leukocyte common antigen, CD20, CD3, CD34, and thyroid transcription factor 1 (TTF-1).16,17
Primary cutaneous γδ T-cell lymphoma can be difficult to diagnose and requires urgent treatment. Clinicians and dermatopathologists need to work together to establish the diagnosis. There is a high mortality rate associated with PCGDTL, making prompt recognition and timely treatment critical. Acknowledgments—Thank you to our colleagues with the Penn State Health Hematology/Oncology Department (Hershey, Pennsylvania) for comanagement of this patient.
Acknowledgments
Thank you to our colleagues with the Penn State Health Hematology/Oncology Department (Hershey, Pennsylvania) for comanagement of this patient.
The Diagnosis: Primary Cutaneous γδ T-cell Lymphoma
Primary cutaneous γδ T-cell lymphoma (PCGDTL) is a distinct entity that can be confused with other types of cutaneous T-cell lymphomas. Often rapidly fatal, PCGDTL has a broad clinical spectrum that may include indolent variants—subcutaneous, epidermotropic, and dermal.1 Primary cutaneous γδ T-cell lymphoma represents less than 1% of all cutaneous T-cell lymphomas.2 Diagnosis and treatment remain challenging. Patients typically present with nodular lesions that progress to ulceration and necrosis. Early lesions can be confused with erythema nodosum, mycosis fungoides, or infection on clinical examination; biopsy establishes the diagnosis. Typical findings include a cytotoxic phenotype, variable epidermotropism, dermal and subcutaneous involvement, and loss of CD4 and often CD8 expression. Testing for Epstein-Barr virus expression yields negative results. The neoplastic lymphocytes in dermal and subcutaneous PCGDTL typically are T-cell intracellular antigen-1 (TIA-1) and granzyme positive.1
Immunohistochemistry failed to reveal CD8, CD56, granzyme, or T-cell intracellular antigen-1 staining of neoplastic cells in our patient but stained diffusely positive with CD3 and CD4. A CD20 stain decorated only a few dermal cells. The patient’s skin lesions continued to enlarge, and the massive lymphadenopathy made breathing difficult. Computed tomography revealed diffuse systemic involvement. An axillary lymph node biopsy revealed sinusoids with complete diffuse effacement of architecture as well as frequent mitotic figures and karyorrhectic debris (Figure 1A). Negative staining for T-cell receptor beta-F1 of the axillary lymph node biopsy and clonal rearrangement of the T-cell receptor gamma chain supported the diagnosis of PCGDTL. Nuclear staining for Epstein-Barr virus–encoded RNA was negative. Human T-cell leukemia virus type 1 antibodies and polymerase chain reaction also were negative. Flow cytometry demonstrated an atypical population of CD3+, CD4+, and CD7− γδ T lymphocytes, further supporting the diagnosis of lymphoma.
The median life expectancy for patients with dermal or subcutaneous PCGDTL is 10 to 15 months after diagnosis.3 The 5-year life expectancy for PCGDTL is approximately 11%.2 Limited treatment options contribute to the poor outcome. Chemotherapy regimens such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisolone) and EPOCH (etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) have yielded inconsistent results. Stem cell transplant has been tried in progressive disease and also has yielded mixed results.2 Brentuximab is indicated for individuals whose tumors express CD30.4 Associated hemophagic lymphohistiocytosis portends a poor prognosis.5
Despite treatment with etoposide, vincristine, doxorubicin, and high-dose oral steroids, our patient developed progressive difficulty breathing, stridor, kidney injury, and anemia. Our patient died less than 1 month after diagnosis—after only 1 round of chemotherapy—secondary to progressive disease and an uncontrollable gastrointestinal tract bleed. The leonine facies (Figure 1B) encountered in our patient can raise a differential diagnosis that includes infectious as well as neoplastic etiologies; however, most infectious etiologies associated with leonine facies manifest in a chronic fashion rather than with a sudden eruption, as noted in our patient.
Leprosy is caused by Mycobacterium leprae, a grampositive bacillus. The condition manifests across a spectrum, with the poles being tuberculoid and lepromatous, and borderline variants in between.6-8 Lepromatous leprosy arises in individuals who are unable to mount cellular immunity against M leprae secondary to anergy.6 Lepromatous leprosy often presents with numerous papules and nodules. Aside from cutaneous manifestations, lepromatous leprosy has a predilection for peripheral nerves and specifically Schwann cells. Histologically, biopsy reveals a flat epidermis and a cell-free subepidermal grenz zone. Within the dermis, there is a diffuse histiocytic infiltrate that typically is not centered around nerves (Figure 2).6,7 Mycobacterium leprae can appear scattered throughout or clustered in globi. Mycobacterium leprae stains red with Ziehl-Neelsen or Wade-Fite stains.6,7 Immunohistochemistry reveals a CD4+ helper T cell (TH2) predominance, supported by the increased expression of type 2 reaction cytokines such as IL-4, IL-5, IL-10, and IL-13.8
Diffuse large B-cell lymphoma (DLBCL) embodies 10% to 20% of all primary cutaneous lymphomas; it is more prevalent in older adults (age range, 70–82 years) and women. Clinically, DLBCL presents as either single or multiple rapidly progressing nodules or plaques, usually violaceous or blue-red in color.9,10 The most common area of presentation is on the legs, though it also can surface at other sites.9 On histology, DLBCL has clearly malignant features including frequent mitotic figures, large immunoblasts, and involvement throughout the dermis as well as perivascularly (Figure 3). Spindle-shaped cells and anaplastic features can be present. Immunohistochemically, DLBCL stains strongly positive for CD20 and B-cell lymphoma 2 (Bcl-2) along with other pan–B-cell markers.9-11 The aggressive leg type of DLBCL stains positively for multiple myeloma oncogene 1 (MUM-1).9,11
Cutaneous metastatic adenocarcinoma from internal malignancies occurs in approximately 5% of cancer patients with metastatic spread.12 Most of these cutaneous lesions develop in close proximity to the primary tumor such as on the trunk, head, or neck. All cutaneous metastases carry a poor prognosis. Clinical presentation can vary greatly, ranging from painless, firm, or elastic nodules to lesions that mimic inflammatory skin conditions such as erysipelas or scleroderma. The majority of these metastases develop as painless firm nodules that are flesh colored, pink, red-brown, or purple.12,13 The histopathology of metastatic adenocarcinoma demonstrates an infiltrative nodular appearance, though there rarely are well-circumscribed nodules found.13 The lesion originates in the dermis or subcutaneous tissue. It is a glandulartype lesion that may reflect the tissue of the primary tumor (Figure 4).12,14 Immunohistochemical stains likely will remain consistent with those of the primary tumor, which is not always the case.14
Merkel cell carcinoma (MCC) is an aggressive cutaneous malignancy of epithelial and neuroendocrine origin, first described as trabecular carcinoma due to the arrangement of tumor resembling cancellous bone.15,16 Merkel cells are mechanoreceptors found near nerve terminals.17 Approximately 80% of MCCs are associated with Merkel cell polyomavirus, which is a small, double-stranded DNA virus with an icosahedral capsid.17,18 Merkel cell polyomavirus–positive cases of MCC tend to have a better prognosis. In Merkel cell polyomavirus–negative MCC, there is an association with UV damage and increased chromosomal aberrations.18 Merkel cell carcinoma is known for its high rate of recurrence as well as local and distant metastasis. Nodal involvement is the most important prognostic indicator.15 Clinically, MCC is associated with the AEIOU mnemonic (asymptomatic, expanding rapidly, immunosuppression, older than 50 years, UV exposed/fair skin).15-17 Lesions appear as red-blue papules on sun-exposed skin and usually are smaller than 2 cm by their greatest dimension. On histopathology, MCC demonstrates small, round, blue cells arranged in sheets or nests originating in the dermis and occasionally can infiltrate the subcutis and lymphovascular surroundings (Figure 5).16-19 Cells have scant eosinophilic cytoplasm and may have fine granular chromatin. Numerous mitotic figures and apoptotic cells also are present. On immunohistochemistry, these cells will stain positive for cytokeratin AE1/AE3, anticytokeratin (CAM 5.2), CK20, and CD56. Due to their neuroendocrine derivation, they also are commonly synaptophysin, neuron-specific enolase, and chromogranin A positive. Notably, MCC will stain negative for leukocyte common antigen, CD20, CD3, CD34, and thyroid transcription factor 1 (TTF-1).16,17
Primary cutaneous γδ T-cell lymphoma can be difficult to diagnose and requires urgent treatment. Clinicians and dermatopathologists need to work together to establish the diagnosis. There is a high mortality rate associated with PCGDTL, making prompt recognition and timely treatment critical. Acknowledgments—Thank you to our colleagues with the Penn State Health Hematology/Oncology Department (Hershey, Pennsylvania) for comanagement of this patient.
Acknowledgments
Thank you to our colleagues with the Penn State Health Hematology/Oncology Department (Hershey, Pennsylvania) for comanagement of this patient.
- Merrill ED, Agbay R, Miranda RN, et al. Primary cutaneous T-cell lymphomas showing gamma-delta (γδ) phenotype and predominantly epidermotropic pattern are clinicopathologically distinct from classic primary cutaneous γδ T-cell lymphomas. Am J Surg Pathol. 2017;41:204-215.
- Foppoli M, Ferreri AJ. Gamma‐delta T‐cell lymphomas. Eur J Haematol. 2015;94:206-218.
- Toro JR, Liewehr DJ, Pabby N, et al. Gamma-delta T-cell phenotype is associated with significantly decreased survival in cutaneous T-cell lymphoma. Blood. 2003;101:3407-3412.
- Rubio-Gonzalez B, Zain J, Garcia L, et al. Cutaneous gamma-delta T-cell lymphoma successfully treated with brentuximab vedotin. JAMA Dermatol. 2016;152:1388-1390.
- Tong H, Ren Y, Liu H, et al. Clinical characteristics of T-cell lymphoma associated with hemophagocytic syndrome: comparison of T-cell lymphoma with and without hemophagocytic syndrome. Leuk Lymphoma. 2008;49:81-87.
- Brehmer-Andersson E. Leprosy. Dermatopathology. New York, NY: Springer; 2006:110-113.
- Massone C, Belachew WA, Schettini A. Histopathology of the lepromatous skin biopsy. Clin Dermatol. 2015;33:38-45.
- Naafs B, Noto S. Reactions in leprosy. In: Nunzi E, Massone C, eds. Leprosy: A Practical Guide. Milan, Italy: Springer; 2012:219-239.
- Hope CB, Pincus LB. Primary cutaneous B-cell lymphomas. Clin Lab Med. 2017;37:547-574.
- Billero VL, LaSenna CE, Romanelli M, et al. Primary cutaneous diffuse large B-cell lymphoma presenting as chronic non-healing ulcer. Int Wound J. 2017;14:830-832.
- Testo N, Olson L, Subramaniyam S, et al. Primary cutaneous diffuse large B-cell lymphoma with a MYC-IGH rearrangement and gain of BCL2: expanding the spectrum of MYC/BCL2 double hit lymphomas. Am J Dermatopathol. 2016;38:769-774.
- Boyd AS. Pulmonary signet-ring cell adenocarcinoma metastatic to the skin. Am J Dermatopathol. 2017;39:E66-E68.
- Guanziroli E, Coggi A, Venegoni L, et al. Cutaneous metastases of internal malignancies: an experience from a single institution. Eur J Dermatol. 2017;27:609-614.
- Fernandez-Flores A, Cassarino DS. Cutaneous metastasis of adenocarcinoma of the ampulla of Vater. Am J Dermatopathol. 2018;40:758-761.
- Trinidad CM, Torres-Cabala CA, Prieto VG, et. Al. Update on eighth edition American Joint Committee on Cancer classification for Merkel Cell carcinoma and histopathological parameters that determine prognosis. J Clin Pathol. 2017;72:337-340.
- Bandino JP, Purvis CG, Shaffer BR, et al. A comparison of the histopathologic growth patterns between non-Merkel cell small round blue cell tumors and Merkel cell carcinoma. Am J Dermatopathol. 2018;40:815-818.
- Mauzo SH, Rerrarotto R, Bell D, et al. Molecular characteristics and potential therapeutic targets in Merkel cell carcinoma. J Clin Pathol. 2016;69:382-390.
- Lowe G, Brewer J, Bordeaux J. Epidemiology and genetics. In: Alam M, Bordeaux JS, Yu SS, eds. Merkel Cell Carcinoma. New York, NY: Springer; 2013:26-28.
- North J, McCalmont T. Histopathologic diagnosis. In: Alam M, Bordeaux JS, Yu SS, eds. Merkel Cell Carcinoma. New York, NY: Springer; 2013:66-69.
- Merrill ED, Agbay R, Miranda RN, et al. Primary cutaneous T-cell lymphomas showing gamma-delta (γδ) phenotype and predominantly epidermotropic pattern are clinicopathologically distinct from classic primary cutaneous γδ T-cell lymphomas. Am J Surg Pathol. 2017;41:204-215.
- Foppoli M, Ferreri AJ. Gamma‐delta T‐cell lymphomas. Eur J Haematol. 2015;94:206-218.
- Toro JR, Liewehr DJ, Pabby N, et al. Gamma-delta T-cell phenotype is associated with significantly decreased survival in cutaneous T-cell lymphoma. Blood. 2003;101:3407-3412.
- Rubio-Gonzalez B, Zain J, Garcia L, et al. Cutaneous gamma-delta T-cell lymphoma successfully treated with brentuximab vedotin. JAMA Dermatol. 2016;152:1388-1390.
- Tong H, Ren Y, Liu H, et al. Clinical characteristics of T-cell lymphoma associated with hemophagocytic syndrome: comparison of T-cell lymphoma with and without hemophagocytic syndrome. Leuk Lymphoma. 2008;49:81-87.
- Brehmer-Andersson E. Leprosy. Dermatopathology. New York, NY: Springer; 2006:110-113.
- Massone C, Belachew WA, Schettini A. Histopathology of the lepromatous skin biopsy. Clin Dermatol. 2015;33:38-45.
- Naafs B, Noto S. Reactions in leprosy. In: Nunzi E, Massone C, eds. Leprosy: A Practical Guide. Milan, Italy: Springer; 2012:219-239.
- Hope CB, Pincus LB. Primary cutaneous B-cell lymphomas. Clin Lab Med. 2017;37:547-574.
- Billero VL, LaSenna CE, Romanelli M, et al. Primary cutaneous diffuse large B-cell lymphoma presenting as chronic non-healing ulcer. Int Wound J. 2017;14:830-832.
- Testo N, Olson L, Subramaniyam S, et al. Primary cutaneous diffuse large B-cell lymphoma with a MYC-IGH rearrangement and gain of BCL2: expanding the spectrum of MYC/BCL2 double hit lymphomas. Am J Dermatopathol. 2016;38:769-774.
- Boyd AS. Pulmonary signet-ring cell adenocarcinoma metastatic to the skin. Am J Dermatopathol. 2017;39:E66-E68.
- Guanziroli E, Coggi A, Venegoni L, et al. Cutaneous metastases of internal malignancies: an experience from a single institution. Eur J Dermatol. 2017;27:609-614.
- Fernandez-Flores A, Cassarino DS. Cutaneous metastasis of adenocarcinoma of the ampulla of Vater. Am J Dermatopathol. 2018;40:758-761.
- Trinidad CM, Torres-Cabala CA, Prieto VG, et. Al. Update on eighth edition American Joint Committee on Cancer classification for Merkel Cell carcinoma and histopathological parameters that determine prognosis. J Clin Pathol. 2017;72:337-340.
- Bandino JP, Purvis CG, Shaffer BR, et al. A comparison of the histopathologic growth patterns between non-Merkel cell small round blue cell tumors and Merkel cell carcinoma. Am J Dermatopathol. 2018;40:815-818.
- Mauzo SH, Rerrarotto R, Bell D, et al. Molecular characteristics and potential therapeutic targets in Merkel cell carcinoma. J Clin Pathol. 2016;69:382-390.
- Lowe G, Brewer J, Bordeaux J. Epidemiology and genetics. In: Alam M, Bordeaux JS, Yu SS, eds. Merkel Cell Carcinoma. New York, NY: Springer; 2013:26-28.
- North J, McCalmont T. Histopathologic diagnosis. In: Alam M, Bordeaux JS, Yu SS, eds. Merkel Cell Carcinoma. New York, NY: Springer; 2013:66-69.
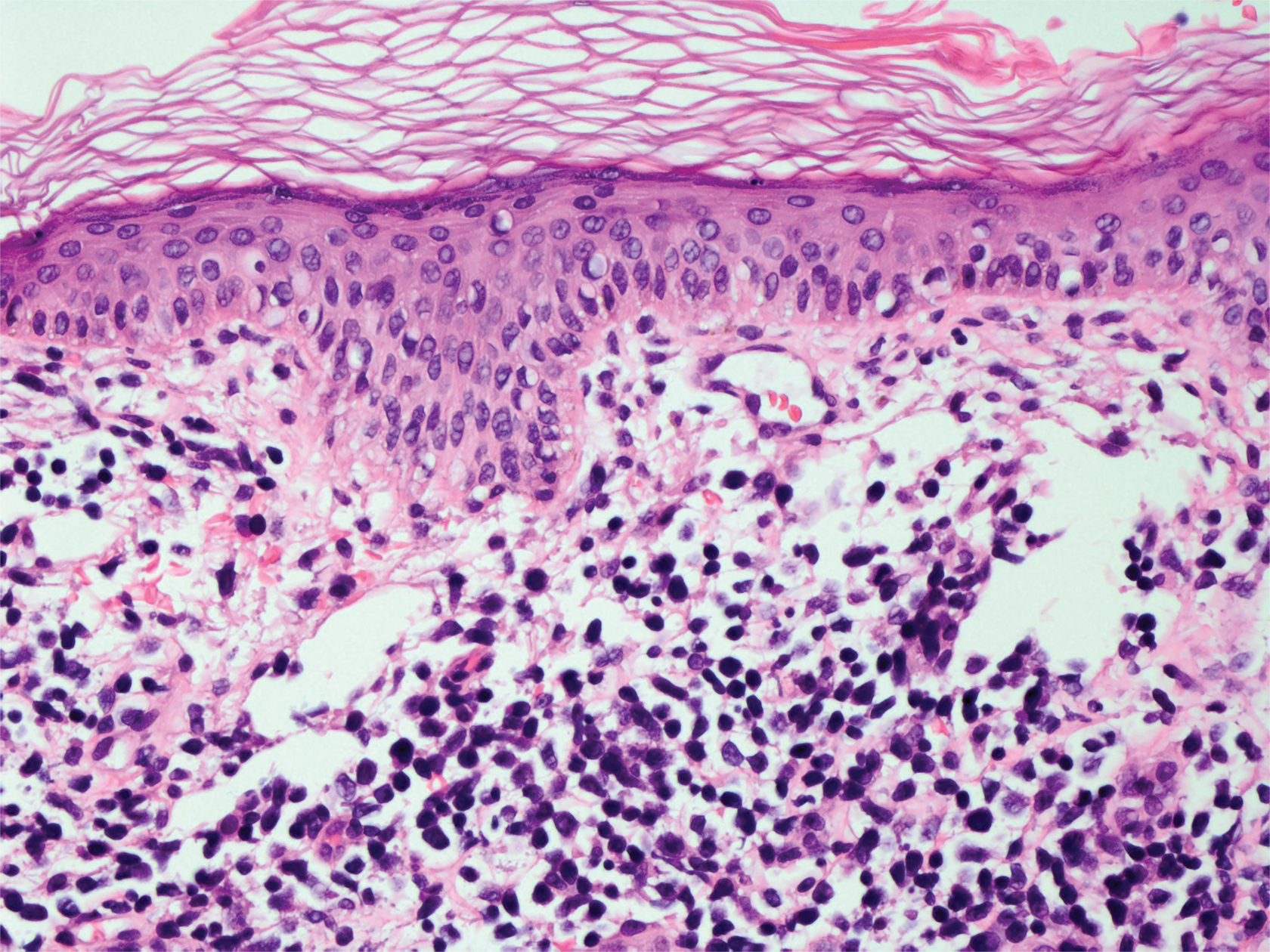
A 71-year-old man presented with an eruption on the face, shoulders, upper back, and arms of 3 weeks’ duration. The lesions were asymptomatic, and he denied fever, chills, or weight loss. He had a history of type 2 diabetes mellitus, hypertension, and hypercholesterolemia. Physical examination revealed coarse facial features with purple-pink nodules on the face and trunk and ulcerated nodules on the upper extremities. Mucous membrane involvement was noted, and there was marked occipital and submandibular lymphadenopathy. A biopsy of an arm nodule revealed a superficial and deep dermal and periadnexal lymphocytic infiltrate of atypical CD3+ cells.
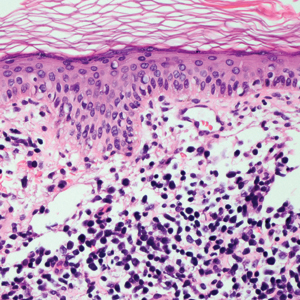
Bullae and Hyperpigmented Patches on the Legs
The Diagnosis: Lichen Planus Pemphigoides
A skin biopsy from the right thigh demonstrated subepidermal blisters containing neutrophils (Figure 1). Direct immunofluorescence revealed linear basement membrane zone staining with C3 and trace staining with IgG (Figure 2), supporting a diagnosis of lichen planus pemphigoides (LPP). Oral prednisone—starting at 60 mg daily and tapered to 40 mg for a week, 20 mg for a week, then 10 mg for a month—along with triamcinolone ointment 0.1% to affected areas led to improvement. Hydrochlorothiazide and UV light therapy were discontinued. Doxycycline 100 mg twice daily and nicotinamide 500 mg twice daily prescribed as adjunctive therapy also led to improvement. The patient achieved remission with doxycycline and was doing well without prednisone; however, he experienced a flare of his disease about 6 months later and was started on mycophenolate mofetil 1 g twice daily after clearance from his gastroenterologist, given his history of hepatitis B. He has been doing well since starting mycophenolate mofetil.
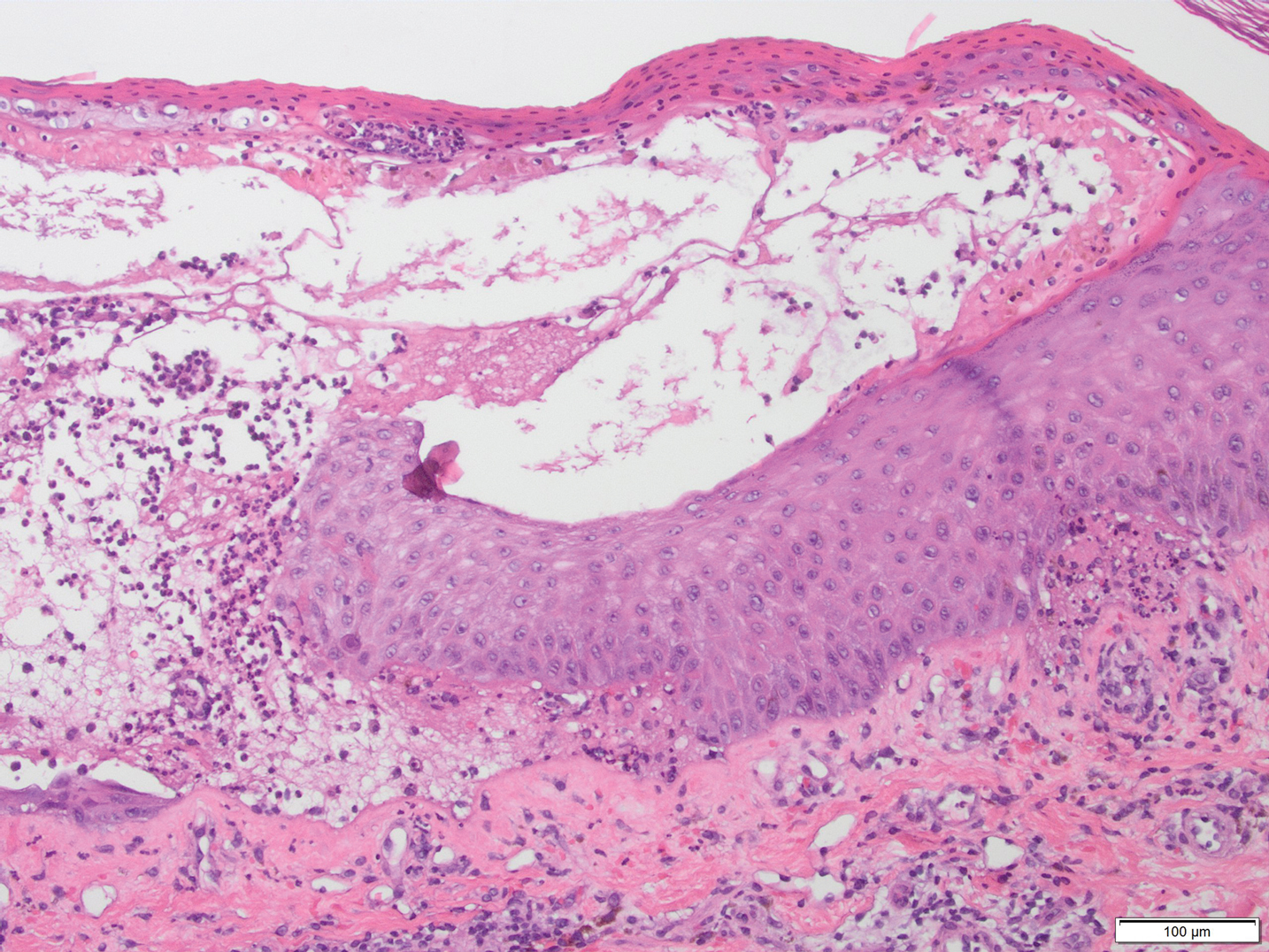
Lichen planus pemphigoides is a rare autoimmune bullous dermatosis with features of both lichen planus and bullous pemphigoid.1 Violaceous papules and tense bullae may be superimposed or arise independently. The chest, abdomen, back, and upper and lower extremities typically are involved.2 Oral mucosal involvement with white reticular streaks or erosions and nail involvement have been reported.2 Histopathologic and immunologic findings establish the diagnosis. Lichen planus pemphigoides is associated with subepidermal bullae and linear deposits of IgG and C3 on the basement membrane zone.1 Autoantibodies to bullous pemphigoid (BP) antigens BP180 and BP230 are associated with LPP.3 The pathogenesis of LPP remains unclear, but there are associations with chronic diseases, medications, and certain therapies.1,4-6 Several case reports have linked LPP to chronic viral hepatitis infections, as well as malignant tumors of the skin, mucosa, and gastrointestinal tract.2 Lichen planus pemphigoides has been reported in a patient on entecavir for hepatitis B as well as in a patient treated for hepatitis C with interferon and ribavirin.1 Lichen planus pemphigoides has been described in patients treated with the angiotensin-converting enzyme inhibitors enalapril, captopril, and ramipril.4,5,7 UV phototherapy also has been associated with the development of LPP.6 Hydrochlorothiazide previously has been reported as a cause of drug-induced lichen planus.8 A PubMed search of articles indexed for MEDLINE using the terms lichen planus pemphigoides and hydrochlorothiazide revealed no reports of hydrochlorothiazide-induced LPP.
Lichen planus pemphigoides demonstrates overlap with other blistering dermatoses, such as BP, bullous lupus erythematosus, and bullous lichen planus. Although histologically and immunologically similar to BP, LPP can be differentiated clinically by the presence of violaceous papules or plaques typical of lichen planus.9 Bullous pemphigoid is more common in individuals older than 70 years, whereas LPP tends to occur in middle-aged adults.2 Bullous lupus erythematosus usually is associated with manifestations of systemic lupus erythematosus and autoantibodies to collagen type VII.10 Salt-split skin studies demonstrate immunofluorescence on the dermal side of the split. Individuals affected by bullous lupus erythematosus typically have a history of photosensitivity.10 Blisters in LPP may form de novo from unaffected skin, whereas the bullae in bullous lichen planus are limited to existing lichenoid papules.9 The autoantibodies typical of LPP are absent in bullous lichen planus. Lichen planus actinicus is a variant of lichen planus that presents with annular, dyschromic, or violaceous plaques in a photodistributed pattern without bullous lesions.9
Lichen planus pemphigoides most commonly is treated with systemic corticosteroids. Topical steroids, dapsone, erythromycin, tetracycline and nicotinamide, azathioprine, and mycophenolate mofetil have been reported as adjuncts to systemic steroid therapy.2,11 Most reports describe treatment success with resolution of blistering lesions.
- Jang SH, Yun SJ, Lee SC, et al. Lichen planus pemphigoides associated with chronic hepatitis B virus infection. Clin Exp Dermatol. 2015;40:868-871.
- Zaraa I, Mahfoudh A, Sellami MK, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol. 2013;52:406-412.
- Harting MS, Hsu S. Lichen planus pemphigoides: a case report and review of the literature. Dermatol Online J. 2006;12:10.
- Onprasert W, Chanprapaph K. Lichen planus pemphigoides induced by enalapril: a case report and a review of literature. Case Rep Dermatol. 2017;9:217-224.
- Ben Salem C, Chengeul L, Ghariani N, et al. Captopril-induced lichen planus pemphigoides. Pharmacoepidemiol Drug Saf. 2008;17:722-724.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Zhu YI, Fitzpatrick JE, Kornfield BW. Lichen planus pemphigoides associated with Ramipril. Int J Dermatol. 2006;45:1453-1455.
- Sin B, Miller M, Chew E. Hydrochlorothiazide induced lichen planus in the emergency department. J Pharm Pract. 2017;30:266-269.
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Women Dermatol. 2015;1:140-149.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Fivenson DP, Kimbrough TL. Lichen planus pemphigoides: combination therapy with tetracycline and nicotinamide. J Am Acad Dermatol. 1997;36:638-640.
The Diagnosis: Lichen Planus Pemphigoides
A skin biopsy from the right thigh demonstrated subepidermal blisters containing neutrophils (Figure 1). Direct immunofluorescence revealed linear basement membrane zone staining with C3 and trace staining with IgG (Figure 2), supporting a diagnosis of lichen planus pemphigoides (LPP). Oral prednisone—starting at 60 mg daily and tapered to 40 mg for a week, 20 mg for a week, then 10 mg for a month—along with triamcinolone ointment 0.1% to affected areas led to improvement. Hydrochlorothiazide and UV light therapy were discontinued. Doxycycline 100 mg twice daily and nicotinamide 500 mg twice daily prescribed as adjunctive therapy also led to improvement. The patient achieved remission with doxycycline and was doing well without prednisone; however, he experienced a flare of his disease about 6 months later and was started on mycophenolate mofetil 1 g twice daily after clearance from his gastroenterologist, given his history of hepatitis B. He has been doing well since starting mycophenolate mofetil.
Lichen planus pemphigoides is a rare autoimmune bullous dermatosis with features of both lichen planus and bullous pemphigoid.1 Violaceous papules and tense bullae may be superimposed or arise independently. The chest, abdomen, back, and upper and lower extremities typically are involved.2 Oral mucosal involvement with white reticular streaks or erosions and nail involvement have been reported.2 Histopathologic and immunologic findings establish the diagnosis. Lichen planus pemphigoides is associated with subepidermal bullae and linear deposits of IgG and C3 on the basement membrane zone.1 Autoantibodies to bullous pemphigoid (BP) antigens BP180 and BP230 are associated with LPP.3 The pathogenesis of LPP remains unclear, but there are associations with chronic diseases, medications, and certain therapies.1,4-6 Several case reports have linked LPP to chronic viral hepatitis infections, as well as malignant tumors of the skin, mucosa, and gastrointestinal tract.2 Lichen planus pemphigoides has been reported in a patient on entecavir for hepatitis B as well as in a patient treated for hepatitis C with interferon and ribavirin.1 Lichen planus pemphigoides has been described in patients treated with the angiotensin-converting enzyme inhibitors enalapril, captopril, and ramipril.4,5,7 UV phototherapy also has been associated with the development of LPP.6 Hydrochlorothiazide previously has been reported as a cause of drug-induced lichen planus.8 A PubMed search of articles indexed for MEDLINE using the terms lichen planus pemphigoides and hydrochlorothiazide revealed no reports of hydrochlorothiazide-induced LPP.
Lichen planus pemphigoides demonstrates overlap with other blistering dermatoses, such as BP, bullous lupus erythematosus, and bullous lichen planus. Although histologically and immunologically similar to BP, LPP can be differentiated clinically by the presence of violaceous papules or plaques typical of lichen planus.9 Bullous pemphigoid is more common in individuals older than 70 years, whereas LPP tends to occur in middle-aged adults.2 Bullous lupus erythematosus usually is associated with manifestations of systemic lupus erythematosus and autoantibodies to collagen type VII.10 Salt-split skin studies demonstrate immunofluorescence on the dermal side of the split. Individuals affected by bullous lupus erythematosus typically have a history of photosensitivity.10 Blisters in LPP may form de novo from unaffected skin, whereas the bullae in bullous lichen planus are limited to existing lichenoid papules.9 The autoantibodies typical of LPP are absent in bullous lichen planus. Lichen planus actinicus is a variant of lichen planus that presents with annular, dyschromic, or violaceous plaques in a photodistributed pattern without bullous lesions.9
Lichen planus pemphigoides most commonly is treated with systemic corticosteroids. Topical steroids, dapsone, erythromycin, tetracycline and nicotinamide, azathioprine, and mycophenolate mofetil have been reported as adjuncts to systemic steroid therapy.2,11 Most reports describe treatment success with resolution of blistering lesions.
The Diagnosis: Lichen Planus Pemphigoides
A skin biopsy from the right thigh demonstrated subepidermal blisters containing neutrophils (Figure 1). Direct immunofluorescence revealed linear basement membrane zone staining with C3 and trace staining with IgG (Figure 2), supporting a diagnosis of lichen planus pemphigoides (LPP). Oral prednisone—starting at 60 mg daily and tapered to 40 mg for a week, 20 mg for a week, then 10 mg for a month—along with triamcinolone ointment 0.1% to affected areas led to improvement. Hydrochlorothiazide and UV light therapy were discontinued. Doxycycline 100 mg twice daily and nicotinamide 500 mg twice daily prescribed as adjunctive therapy also led to improvement. The patient achieved remission with doxycycline and was doing well without prednisone; however, he experienced a flare of his disease about 6 months later and was started on mycophenolate mofetil 1 g twice daily after clearance from his gastroenterologist, given his history of hepatitis B. He has been doing well since starting mycophenolate mofetil.
Lichen planus pemphigoides is a rare autoimmune bullous dermatosis with features of both lichen planus and bullous pemphigoid.1 Violaceous papules and tense bullae may be superimposed or arise independently. The chest, abdomen, back, and upper and lower extremities typically are involved.2 Oral mucosal involvement with white reticular streaks or erosions and nail involvement have been reported.2 Histopathologic and immunologic findings establish the diagnosis. Lichen planus pemphigoides is associated with subepidermal bullae and linear deposits of IgG and C3 on the basement membrane zone.1 Autoantibodies to bullous pemphigoid (BP) antigens BP180 and BP230 are associated with LPP.3 The pathogenesis of LPP remains unclear, but there are associations with chronic diseases, medications, and certain therapies.1,4-6 Several case reports have linked LPP to chronic viral hepatitis infections, as well as malignant tumors of the skin, mucosa, and gastrointestinal tract.2 Lichen planus pemphigoides has been reported in a patient on entecavir for hepatitis B as well as in a patient treated for hepatitis C with interferon and ribavirin.1 Lichen planus pemphigoides has been described in patients treated with the angiotensin-converting enzyme inhibitors enalapril, captopril, and ramipril.4,5,7 UV phototherapy also has been associated with the development of LPP.6 Hydrochlorothiazide previously has been reported as a cause of drug-induced lichen planus.8 A PubMed search of articles indexed for MEDLINE using the terms lichen planus pemphigoides and hydrochlorothiazide revealed no reports of hydrochlorothiazide-induced LPP.
Lichen planus pemphigoides demonstrates overlap with other blistering dermatoses, such as BP, bullous lupus erythematosus, and bullous lichen planus. Although histologically and immunologically similar to BP, LPP can be differentiated clinically by the presence of violaceous papules or plaques typical of lichen planus.9 Bullous pemphigoid is more common in individuals older than 70 years, whereas LPP tends to occur in middle-aged adults.2 Bullous lupus erythematosus usually is associated with manifestations of systemic lupus erythematosus and autoantibodies to collagen type VII.10 Salt-split skin studies demonstrate immunofluorescence on the dermal side of the split. Individuals affected by bullous lupus erythematosus typically have a history of photosensitivity.10 Blisters in LPP may form de novo from unaffected skin, whereas the bullae in bullous lichen planus are limited to existing lichenoid papules.9 The autoantibodies typical of LPP are absent in bullous lichen planus. Lichen planus actinicus is a variant of lichen planus that presents with annular, dyschromic, or violaceous plaques in a photodistributed pattern without bullous lesions.9
Lichen planus pemphigoides most commonly is treated with systemic corticosteroids. Topical steroids, dapsone, erythromycin, tetracycline and nicotinamide, azathioprine, and mycophenolate mofetil have been reported as adjuncts to systemic steroid therapy.2,11 Most reports describe treatment success with resolution of blistering lesions.
- Jang SH, Yun SJ, Lee SC, et al. Lichen planus pemphigoides associated with chronic hepatitis B virus infection. Clin Exp Dermatol. 2015;40:868-871.
- Zaraa I, Mahfoudh A, Sellami MK, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol. 2013;52:406-412.
- Harting MS, Hsu S. Lichen planus pemphigoides: a case report and review of the literature. Dermatol Online J. 2006;12:10.
- Onprasert W, Chanprapaph K. Lichen planus pemphigoides induced by enalapril: a case report and a review of literature. Case Rep Dermatol. 2017;9:217-224.
- Ben Salem C, Chengeul L, Ghariani N, et al. Captopril-induced lichen planus pemphigoides. Pharmacoepidemiol Drug Saf. 2008;17:722-724.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Zhu YI, Fitzpatrick JE, Kornfield BW. Lichen planus pemphigoides associated with Ramipril. Int J Dermatol. 2006;45:1453-1455.
- Sin B, Miller M, Chew E. Hydrochlorothiazide induced lichen planus in the emergency department. J Pharm Pract. 2017;30:266-269.
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Women Dermatol. 2015;1:140-149.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Fivenson DP, Kimbrough TL. Lichen planus pemphigoides: combination therapy with tetracycline and nicotinamide. J Am Acad Dermatol. 1997;36:638-640.
- Jang SH, Yun SJ, Lee SC, et al. Lichen planus pemphigoides associated with chronic hepatitis B virus infection. Clin Exp Dermatol. 2015;40:868-871.
- Zaraa I, Mahfoudh A, Sellami MK, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol. 2013;52:406-412.
- Harting MS, Hsu S. Lichen planus pemphigoides: a case report and review of the literature. Dermatol Online J. 2006;12:10.
- Onprasert W, Chanprapaph K. Lichen planus pemphigoides induced by enalapril: a case report and a review of literature. Case Rep Dermatol. 2017;9:217-224.
- Ben Salem C, Chengeul L, Ghariani N, et al. Captopril-induced lichen planus pemphigoides. Pharmacoepidemiol Drug Saf. 2008;17:722-724.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Zhu YI, Fitzpatrick JE, Kornfield BW. Lichen planus pemphigoides associated with Ramipril. Int J Dermatol. 2006;45:1453-1455.
- Sin B, Miller M, Chew E. Hydrochlorothiazide induced lichen planus in the emergency department. J Pharm Pract. 2017;30:266-269.
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Women Dermatol. 2015;1:140-149.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Fivenson DP, Kimbrough TL. Lichen planus pemphigoides: combination therapy with tetracycline and nicotinamide. J Am Acad Dermatol. 1997;36:638-640.
A 50-year-old man presented with a pruritic bullous dermatosis on the lower legs, arms, and back of 1 month’s duration. He had an 8-year history of lichen planus, and the lesions recently had worsened despite the addition of UVB phototherapy. His medical history was remarkable for hepatitis B treated with entecavir and the addition of hydrochlorothiazide for essential hypertension 2 weeks prior to the dramatic worsening of the rash. Physical examination revealed multiple bullae on the lower legs associated with violaceous and hyperpigmented papules and patches. He also had violaceous papules on the lower back and eroded lesions on the oral mucosa. Shave biopsies were obtained from the right thigh and mid back, and histopathologic analysis was performed for both routine histology and direct immunofluorescence.
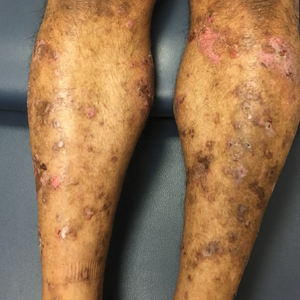
The many variants of psoriasis
The “heartbreak of psoriasis,” coined by an advertiser in the 1960s, conveyed the notion that this disease was a cosmetic disorder mainly limited to skin involvement. John Updike’s article in the September 1985 issue of The New Yorker, “At War With My Skin,” detailed Mr. Updike’s feelings of isolation and stress related to his condition, helping to reframe the popular concept of psoriasis.1 Updike’s eloquent account describing his struggles to find effective treatment increased public awareness about psoriasis, which in fact affects other body systems as well.
The overall prevalence of psoriasis is 1.5% to 3.1% in the United States and United Kingdom.2,3 More than 6.5 million adults in the United States > 20 years of age are affected.3 The most commonly affected demographic group is non-Hispanic Caucasians.
Our expanding knowledge of pathogenesis
Studies of genetic linkage have identified genes and single nucleotide polymorphisms associated with psoriasis.4 The interaction between environmental triggers and the innate and adaptive immune systems leads to keratinocyte hyperproliferation. Tumor necrosis factor (TNF), interleukin (IL) 23, and IL-17 are important cytokines associated with psoriatic inflammation.4 There are common pathways of inflammation in both psoriasis and cardiovascular disease resulting in oxidative stress and endothelial cell dysfunction.4 Ninety percent of early-onset psoriasis is associated with human leukocyte antigen (HLA)-Cw6.4 And alterations in the microbiome of the skin may contribute, as reduced microbial diversity has been found in psoriatic lesions.5
Comorbidities are common
Psoriasis is an independent risk factor for diabetes and major adverse cardiovascular events.6 Hypertension, dyslipidemia, inflammatory bowel disease, nonalcoholic fatty liver disease, chronic kidney disease, and lymphoma (particularly cutaneous T-cell lymphoma) are also associated with psoriasis.6 Psoriatic arthritis is frequently encountered with cutaneous psoriasis; however, it is often not recognized until late in the disease course.
There also appears to be an association among psoriasis, dietary factors, and celiac disease.7-9 Positive testing for IgA anti-endomysial antibodies and IgA tissue transglutaminase antibodies should prompt consideration of starting a gluten-free diet, which has been shown to improve psoriatic
The different types of psoriasis
The classic presentation of psoriasis involves stubborn plaques with silvery scale on extensor surfaces such as the elbows and knees. The severity of the disease corresponds with the amount of body surface area affected. While plaque-type psoriasis is the most common form, other patterns exist. Individuals may exhibit 1 dominant pattern or multiple psoriatic variants simultaneously. Most types of psoriasis have 3 characteristic features: erythema, skin thickening, and scales.
Certain history and physical clues can aid in diagnosing psoriasis; these include the Koebner phenomenon, the Auspitz sign, and the Woronoff ring. The Koebner phenomenon refers to the development of psoriatic lesions in an area of trauma (FIGURE 1), frequently resulting in a linear streak-like appearance. The Auspitz sign describes the pinpoint bleeding that may be encountered with the removal of a psoriatic plaque. The Woronoff ring is a pale blanching ring that may surround a psoriatic lesion.

Continue to: Chronic plaque-type psoriasis
Chronic plaque-type psoriasis (Figures 2A and 2B), the most common variant, is characterized by sharply demarcated pink papules and plaques with a silvery scale in a symmetric distribution on the extensor surfaces, scalp, trunk, and lumbosacral areas.

Guttate psoriasis (FIGURE 3) features small (often < 1 cm) pink scaly papules that appear suddenly. It is more commonly seen in children and is usually preceded by an upper respiratory tract infection, often with Streptococcus.10 If strep testing is positive, guttate psoriasis may improve after appropriate antibiotic treatment.

Erythrodermic psoriasis (FIGUREs 4A and 4B) involves at least 75% of the body with erythema and scaling.11 Erythroderma can be caused by many other conditions such as atopic dermatitis, a drug reaction, Sezary syndrome, seborrheic dermatitis, and pityriasis rubra pilaris. Treatments for other conditions in the differential diagnosis can potentially make psoriasis worse. Unfortunately, findings on a skin biopsy are often nonspecific, making careful clinical observation crucial to arriving at an accurate diagnosis.

Pustular psoriasis is characterized by bright erythema and sterile pustules. Pustular psoriasis can be triggered by pregnancy, sudden tapering of corticosteroids, hypocalcemia, and infection. Involvement of the palms and soles with severe desquamation can drastically impact daily functioning and quality of life.
Inverse or flexural psoriasis (FIGUREs 5A and 5B) is characterized by shiny, pink-to-red sharply demarcated plaques involving intertriginous areas, typically the groin, inguinal crease, axilla, inframammary regions, and intergluteal cleft.

Continue to: Geographic tongue
Geographic tongue describes psoriasis of the tongue. The mucosa of the tongue has white plaques with a geographic border. Instead of scale, the moisture on the tongue causes areas of hyperkeratosis that appear white.
Nail psoriasis can manifest as nail pitting (FIGURE 6), oil staining, onycholysis (distal lifting of the nail), and subungual hyperkeratosis. Nail psoriasis is often quite distressing for patients and can be difficult to treat.

Palmoplantar psoriasis (FIGUREs 7A and 7B) can be painful due to the involvement of the palms of the hands and soles of the feet. Lesions will either be similar to other psoriatic plaques with well-demarcated erythematous scaling lesions or involve thickening and scale without associated erythema.

Psoriatic arthritis can cause significant joint damage and disability. Most affected individuals with psoriatic arthritis have a history of preceding skin disease.12 There are no specific lab tests for psoriasis; radiologic studies can show bulky syndesmophytes, central and marginal erosions, and periostitis. Patterns of joint involvement are variable. Psoriatic arthritis is more likely to affect the distal interphalangeal joints than rheumatoid arthritis and is more likely to affect the metacarpophalangeal joints than osteoarthritis.13
Psoriatic arthritis often progresses insidiously and is commonly described as causing discomfort rather than acute pain. Enthesitis, inflammation at the site where tendons or ligaments insert into the bone, is often present. Joint destruction may lead to the telescoping “opera glass” digit (FIGURE 8).

Continue to: Drug-provoked psoriasis
Drug-provoked psoriasis is divided into 2 groups: drug-induced and drug-aggravated. Drug-induced psoriasis will improve after discontinuation of the causative drug and tends to occur in patients without a personal or family history of psoriasis. Drug-aggravated psoriasis continues to progress after the discontinuation of the offending drug and is more often seen in patients with a history of psoriasis.14 Drugs that most commonly provoke psoriasis are beta-blockers, lithium, and antimalarials.10 Other potentially aggravating agents include antibiotics, digoxin, and nonsteroidal anti-inflammatory drugs.10
Consider these skin disorders in the differential diagnosis
The diagnosis of psoriasis is usually clinical, and a skin biopsy is rarely needed. However, a range of other skin disorders should be kept in mind when considering the differential diagnosis.
Mycosis fungoides is a type of cutaneous T-cell lymphoma that forms erythematous plaques that may show wrinkling and epidermal atrophy in sun-protected sites. Onset usually occurs among the elderly.
Pityriasis rubra pilaris is characterized by salmon-colored patches that may have small areas of normal skin (“islands of sparing”), hyperkeratotic follicular papules, and hyperkeratosis of the palms and soles.
Seborrheic dermatitis, dandruff of the skin, usually involves the scalp and nasolabial areas and the T-zone of the face.
Continue to: Lichen planus
Lichen planus usually appears slightly more purple than psoriasis and typically involves the mouth, flexural surfaces of the wrists, genitals, and ankles.
Other conditions in the differential include pityriasis lichenoides chronica, which may be identified on skin biopsy. Inverse psoriasis can be difficult to differentiate from candida intertrigo, erythrasma, or tinea cruris.
A potassium hydroxide (KOH) preparation can help differentiate psoriasis from candida or tinea. In psoriasis, a KOH test will be negative for fungal elements. Mycology culture on skin scrapings may be performed to rule out fungal infection. Erythrasma may exhibit a coral red appearance under Wood lamp examination.
If a lesion fails to respond to appropriate treatment, a careful drug history and biopsy can help clarify the diagnosis.
Document disease
It’s important to thoroughly document the extent and severity of the psoriasis and to monitor the impact of treatment. The Psoriasis Area and Severity Index is a commonly used method that calculates a score based on the area (extent) of involvement surrounding 4 major anatomical regions (head, upper extremities, trunk, and lower extremities), as well as the degree of erythema, induration, and scaling of lesions. The average redness, thickness, and scaling are graded on a scale of 0 to 4 and the extent of involvement is calculated to form a total numerical score ranging from 0 (no disease) to 72 (maximal disease).
Continue to: Many options in the treatment arsenal
Many options in the treatment arsenal
Many treatments can improve psoriasis.9,15-19 Most affected individuals discover that emollients and exposure to natural sunlight can be effective, as are soothing baths (balneotherapy) or topical coal tar application. More persistent disease requires prescription therapy. Individualize therapy according to the severity of disease, location of the lesions, involvement of joints, and comorbidities (FIGURE 9).15

If ≤ 10% of the body surface area is involved, treatment options generally are explored in a stepwise progression from safest and most affordable to more involved therapies as needed: moisturization and avoidance of repetitive trauma, topical corticosteroids (TCS), vitamin D analogs, topical calcineurin inhibitors, and vitamin A creams. Recalcitrant disease will likely require ultraviolet (UV) light treatment or a systemic agent.15
If > 10% of the body surface area is involved, but joints are not involved, consider UV light treatment or a combination of alcitretin and TCS. If the joints are involved, likely initial options would be methotrexate, cyclosporine, or TNF-α inhibitor. Additional options to consider are anti-IL-17 or anti-IL-23 agents.15
If there’s joint involvement. In individuals with mild peripheral arthritis involving fewer than 4 joints without evidence of joint damage on imaging, nonsteroidal anti-inflammatory drugs are the mainstay of treatment. If the peripheral arthritis persists, or if it is associated with moderate-to-severe erosions or with substantial functional limitations, initiate treatment with a conventional disease-modifying antirheumatic drug. If the disease remains active, consider biologic agents.
Case studies
Mild-to-moderate psoriasis
Patient A is a 19-year-old woman presenting for evaluation of a persistently dry, flaking scalp. She has had the itchy scalp for years, as well as several small “patches” across her elbows, legs, knees, and abdomen. Over-the-counter emollients have not helped. The patient also says she has had brittle nails on several of her fingers, which she keeps covered with thick polish.
Continue to: The condition exemplified...
The condition exemplified by Patient A can typically be managed with topical products.
Topical steroids may be classified by different delivery vehicles, active ingredients, and potencies. The National Psoriasis Foundation's Topical Steroids Potency Chart can provide guidance (visit www.psoriasis.org/about-psoriasis/treatments/topicals/steroids/potency-chart and scroll down). Prescribing an appropriate amount is important; the standard 30-g prescription tube is generally required to cover the entire skin surface. Ointments have a greasy consistency (typically a petroleum base), which enhances potency and hydrates the skin. Creams and lotions are easier to rub on and spread. Gels are alcohol based and readily absorbed.16 Solutions, foams, and shampoos are particularly useful to treat psoriasis in hairy areas such as the scalp.
Corticosteroid potency ranges from Class I to Class VII, with the former being the most potent. While TCS products are typically effective with minimal systemic absorption, it is important to counsel patients on the risk of skin atrophy, impaired wound healing, and skin pigmentation changes with chronic use. With nail psoriasis, a potent topical steroid (including flurandrenolide [Cordran] tape) applied to the proximal nail fold has shown benefit.20
Topical calcineurin inhibitors (TCIs; eg, tacrolimus ointment and pimecrolimus cream) are anti-inflammatory agents often used in conjunction with topical steroids to minimize steroid use and associated adverse effects.15 A possible steroid-sparing regimen includes using a TCI Monday through Friday and a topical steroid on the weekend.
Topical vitamin D analogs (calcipotriene, calcipotriol, calcitriol) inhibit proliferation of keratinocytes and decrease the production of inflammatory mediators.15,17-19,21 Application of a vitamin D analog in combination with a high-potency TCS, systemic treatment, or phototherapy can provide greater efficacy, a more rapid onset of action, and less irritation than can the vitamin D analog used alone.21 If used in combination with UV light, apply topical vitamin D after the light therapy to prevent degradation.
Continue to: UV light therapy
UV light therapy is often used in cases refractory to topical therapy. Patients are typically prescribed 2 to 3 treatments per week with narrowband UVB (311-313 nm), the excimer laser (308 nm), or, less commonly, PUVA (UV treatment with psoralens). Treatment begins with a minimal erythema dose—the lowest dose to achieve minimal erythema of the skin before burning. When that is determined, exposure is increased as needed—depending on the response. If this is impractical or too time-consuming for the patient, an alternative recommendation would be increased exposure to natural sunlight or even use of a tanning booth. However, patients must then be cautioned about the increased risk of skin cancer.
Refractory/severe psoriasis
Patient B is a 35-year-old man with a longstanding history of psoriasis affecting his scalp and nails. Over the past 10 years, psoriatic lesions have also appeared and grown across his lower back, gluteal fold, legs, abdomen, and arms. He is now being evaluated by a rheumatologist for worsening symmetric joint pain that includes his lower back.
Methotrexate has been used to treat psoriasis and psoriatic arthritis since the 1950s. Methotrexate is a competitive inhibitor of dihydrofolate reductase and is typically given as an oral medication dosed once weekly with folic acid supplementation on the other 6 days.17 The most common adverse effects encountered with methotrexate are gastrointestinal upset and oral ulcers; however, routine monitoring for myelosuppression and hepatotoxicity is required.
Biologic therapy. When conventional therapies fail, immune-targeted treatment with “biologics” may be initiated. As knowledge of signaling pathways and the immunopathogenesis of psoriasis has increased, so has the number of biologic agents, which are generally well tolerated and effective in managing plaque psoriasis and psoriatic arthritis. Although their use, which requires monitoring, is handled primarily by specialists, familiarizing yourself with available agents can be helpful (TABLE).22

Nutritional modification and supplementation in treating skin disease still requires further investigation. Fish oil has shown benefit for cutaneous psoriasis in randomized controlled trials.7,8 Oral vitamin D supplementation requires further study, whereas selenium and B12 supplementation have not conferred consistent benefit.7 Given that several studies have demonstrated a relationship between body mass index and psoriatic disease severity, weight loss may be helpful in the management of psoriasis as well as psoriatic arthritis.8
Continue to: Other systemic agents
Other systemic agents—for individuals who cannot tolerate the biologic agents—include acitretin, azathioprine, mycophenolate mofetil, and cyclosporine.15,17
Paradoxical psoriatic reactions
When a psoriatic condition develops during biologic drug therapy, it is known as a paradoxical psoriatic reaction. The onset of de novo psoriasis has been documented during TNF-α inhibitor therapy for individuals with underlying rheumatoid arthritis.23 Skin biopsy reveals the same findings as common plaque psoriasis.
Using immunosuppressive Tx? Screen for tuberculosis
Testing to exclude a diagnosis of latent or undiagnosed tuberculosis must be performed prior to initiating immunosuppressive therapy with methotrexate or a biologic agent. Tuberculin skin testing, QuantiFERON-TB gold test, and the T-SPOT.TB test are accepted screening modalities. Discordance between tuberculin skin tests and the interferon gamma release assays in latent TB highlights the need for further study using the available QuantiFERON-TB gold test and the T-SPOT.TB test.24
CORRESPONDENCE
Karl T. Clebak, MD, FAAFP, Penn State Health Milton S. Hershey Medical Center, Department of Family and Community Medicine, 500 University Drive, Hershey, PA 17033; [email protected].
1. Jackson R. John Updike on psoriasis. At war with my skin, from the journal of a leper. J Cutan Med Surg. 2000;4:113-115.
2. Gelfand JM, Weinstein R, Porter SB, et al. Prevalence and treatment of psoriasis in the United Kingdom: a population-based study. Arch Dermatol. 2005;141:1537-1541.
3. Helmick CG, Lee-Han H, Hirsch SC, et al. Prevalence of psoriasis among adults in the U.S.: 2003-2006 and 2009-2010 National Health and Nutrition Examination Surveys. Am J Prev Med. 2014;47:37-45.
4. Alexander H, Nestle FO. Pathogenesis and immunotherapy in cutaneous psoriasis: what can rheumatologists learn? Curr Opin Rheumatol. 2017;29:71-78.
5. Fahlén A, Engstrand L, Baker BS, et al. Comparison of bacterial microbiota in skin biopsies from normal and psoriatic skin. Arch Dermatol Res. 2012;304:15-22.
6. Takeshita J, Grewal S, Langan SM, et al. Psoriasis and comorbid diseases: epidemiology. J Am Acad Dermatol. 2017;76:377-390.
7. Millsop JW, Bhatia BK, Debbaneh M, et al. Diet and psoriasis, part III: role of nutritional supplements. J Am Acad Dermatol. 2014;71:561-569.
8. Debbaneh M, Millsop JW, Bhatia BK, et al. Diet and psoriasis, part I: impact of weight loss interventions. J Am Acad Dermatol. 2014;71:133-140.
9. Bhatia BK, Millsop JW, Debbaneh M, et al. Diet and psoriasis, part II: celiac disease and role of a gluten-free diet. J Am Acad Dermatol. 2014;71:350-358.
10. Fry L, Baker BS. Triggering psoriasis: the role of infections and medications. Clin Dermatol. 2007;25:606-615.
11. Singh RK, Lee KM, Ucmak D, et al. Erythrodermic psoriasis: pathophysiology and current treatment perspectives. Psoriasis (Aukl). 2016;6:93-104.
12. Garg A, Gladman D. Recognizing psoriatic arthritis in the dermatology clinic. J Am Acad Dermatol. 2010;63:733-748.
13. McGonagle D, Hermann KG, Tan AL. Differentiation between osteoarthritis and psoriatic arthritis: implications for pathogenesis and treatment in the biologic therapy era. Rheumatology. 2015;54:29-38.
14. Kim GK, Del Rosso JQ. Drug-provoked psoriasis: is it drug induced or drug aggravated?: understanding pathophysiology and clinical relevance. J Clin Aesthet Dermatol. 2010;3:32-38.
15. Kupetsky EA, Keller M. Psoriasis vulgaris: an evidence-based guide for primary care. J Am Board Fam Med. 2013;26:787-801.
16. Helm MF, Farah JB, Carvalho M, et al. Compounded topical medications for diseases of the skin: a long tradition still relevant today. N Am J Med Sci. 2017;10:116-118.
17. Weigle N, McBane S. Psoriasis. Am Fam Physician. 2013;87:626-633.
18. Helfrich YR, Sachs DL, Kang S. Topical vitamin D3. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 2nd ed. Philadelphia, PA: Saunders; 2007:691-695.
19. Lebwohl M, Siskin SB, Epinette W, et al. A multicenter trial of calcipotriene ointment and halobetasol ointment compared with either agent alone for the treatment of psoriasis. J Am Acad Dermatol. 1996;35:268-269.
20. Pasch MC. Nail psoriasis: a review of treatment options. Drugs. 2016;76:675-705.
21. Bagel J, Gold LS. Combining topical psoriasis treatment to enhance systemic and phototherapy: a review of the literature. J Drugs Dermatol. 2017;16:1209-1222.
22. Rønholt K, Iversen L. Old and new biological therapies for psoriasis. Int J Mol Sci. 2017;18:e2297.
23. Toussirot É, Aubin F. Paradoxical reactions under TNF-alpha blocking agents and other biologic agents given for chronic immune-mediated diseases: an analytical and comprehensive overview. RMD Open. 2016;2:e000239.
24. Connell TG, Ritz N, Paxton GA, et al. A three-way comparison of tuberculin skin testing, QuantiFERON-TB gold and T-SPOT.TB in children. PLoS One. 2008;3:e2624.
The “heartbreak of psoriasis,” coined by an advertiser in the 1960s, conveyed the notion that this disease was a cosmetic disorder mainly limited to skin involvement. John Updike’s article in the September 1985 issue of The New Yorker, “At War With My Skin,” detailed Mr. Updike’s feelings of isolation and stress related to his condition, helping to reframe the popular concept of psoriasis.1 Updike’s eloquent account describing his struggles to find effective treatment increased public awareness about psoriasis, which in fact affects other body systems as well.
The overall prevalence of psoriasis is 1.5% to 3.1% in the United States and United Kingdom.2,3 More than 6.5 million adults in the United States > 20 years of age are affected.3 The most commonly affected demographic group is non-Hispanic Caucasians.
Our expanding knowledge of pathogenesis
Studies of genetic linkage have identified genes and single nucleotide polymorphisms associated with psoriasis.4 The interaction between environmental triggers and the innate and adaptive immune systems leads to keratinocyte hyperproliferation. Tumor necrosis factor (TNF), interleukin (IL) 23, and IL-17 are important cytokines associated with psoriatic inflammation.4 There are common pathways of inflammation in both psoriasis and cardiovascular disease resulting in oxidative stress and endothelial cell dysfunction.4 Ninety percent of early-onset psoriasis is associated with human leukocyte antigen (HLA)-Cw6.4 And alterations in the microbiome of the skin may contribute, as reduced microbial diversity has been found in psoriatic lesions.5
Comorbidities are common
Psoriasis is an independent risk factor for diabetes and major adverse cardiovascular events.6 Hypertension, dyslipidemia, inflammatory bowel disease, nonalcoholic fatty liver disease, chronic kidney disease, and lymphoma (particularly cutaneous T-cell lymphoma) are also associated with psoriasis.6 Psoriatic arthritis is frequently encountered with cutaneous psoriasis; however, it is often not recognized until late in the disease course.
There also appears to be an association among psoriasis, dietary factors, and celiac disease.7-9 Positive testing for IgA anti-endomysial antibodies and IgA tissue transglutaminase antibodies should prompt consideration of starting a gluten-free diet, which has been shown to improve psoriatic
The different types of psoriasis
The classic presentation of psoriasis involves stubborn plaques with silvery scale on extensor surfaces such as the elbows and knees. The severity of the disease corresponds with the amount of body surface area affected. While plaque-type psoriasis is the most common form, other patterns exist. Individuals may exhibit 1 dominant pattern or multiple psoriatic variants simultaneously. Most types of psoriasis have 3 characteristic features: erythema, skin thickening, and scales.
Certain history and physical clues can aid in diagnosing psoriasis; these include the Koebner phenomenon, the Auspitz sign, and the Woronoff ring. The Koebner phenomenon refers to the development of psoriatic lesions in an area of trauma (FIGURE 1), frequently resulting in a linear streak-like appearance. The Auspitz sign describes the pinpoint bleeding that may be encountered with the removal of a psoriatic plaque. The Woronoff ring is a pale blanching ring that may surround a psoriatic lesion.
Continue to: Chronic plaque-type psoriasis
Chronic plaque-type psoriasis (Figures 2A and 2B), the most common variant, is characterized by sharply demarcated pink papules and plaques with a silvery scale in a symmetric distribution on the extensor surfaces, scalp, trunk, and lumbosacral areas.
Guttate psoriasis (FIGURE 3) features small (often < 1 cm) pink scaly papules that appear suddenly. It is more commonly seen in children and is usually preceded by an upper respiratory tract infection, often with Streptococcus.10 If strep testing is positive, guttate psoriasis may improve after appropriate antibiotic treatment.
Erythrodermic psoriasis (FIGUREs 4A and 4B) involves at least 75% of the body with erythema and scaling.11 Erythroderma can be caused by many other conditions such as atopic dermatitis, a drug reaction, Sezary syndrome, seborrheic dermatitis, and pityriasis rubra pilaris. Treatments for other conditions in the differential diagnosis can potentially make psoriasis worse. Unfortunately, findings on a skin biopsy are often nonspecific, making careful clinical observation crucial to arriving at an accurate diagnosis.
Pustular psoriasis is characterized by bright erythema and sterile pustules. Pustular psoriasis can be triggered by pregnancy, sudden tapering of corticosteroids, hypocalcemia, and infection. Involvement of the palms and soles with severe desquamation can drastically impact daily functioning and quality of life.
Inverse or flexural psoriasis (FIGUREs 5A and 5B) is characterized by shiny, pink-to-red sharply demarcated plaques involving intertriginous areas, typically the groin, inguinal crease, axilla, inframammary regions, and intergluteal cleft.
Continue to: Geographic tongue
Geographic tongue describes psoriasis of the tongue. The mucosa of the tongue has white plaques with a geographic border. Instead of scale, the moisture on the tongue causes areas of hyperkeratosis that appear white.
Nail psoriasis can manifest as nail pitting (FIGURE 6), oil staining, onycholysis (distal lifting of the nail), and subungual hyperkeratosis. Nail psoriasis is often quite distressing for patients and can be difficult to treat.
Palmoplantar psoriasis (FIGUREs 7A and 7B) can be painful due to the involvement of the palms of the hands and soles of the feet. Lesions will either be similar to other psoriatic plaques with well-demarcated erythematous scaling lesions or involve thickening and scale without associated erythema.
Psoriatic arthritis can cause significant joint damage and disability. Most affected individuals with psoriatic arthritis have a history of preceding skin disease.12 There are no specific lab tests for psoriasis; radiologic studies can show bulky syndesmophytes, central and marginal erosions, and periostitis. Patterns of joint involvement are variable. Psoriatic arthritis is more likely to affect the distal interphalangeal joints than rheumatoid arthritis and is more likely to affect the metacarpophalangeal joints than osteoarthritis.13
Psoriatic arthritis often progresses insidiously and is commonly described as causing discomfort rather than acute pain. Enthesitis, inflammation at the site where tendons or ligaments insert into the bone, is often present. Joint destruction may lead to the telescoping “opera glass” digit (FIGURE 8).
Continue to: Drug-provoked psoriasis
Drug-provoked psoriasis is divided into 2 groups: drug-induced and drug-aggravated. Drug-induced psoriasis will improve after discontinuation of the causative drug and tends to occur in patients without a personal or family history of psoriasis. Drug-aggravated psoriasis continues to progress after the discontinuation of the offending drug and is more often seen in patients with a history of psoriasis.14 Drugs that most commonly provoke psoriasis are beta-blockers, lithium, and antimalarials.10 Other potentially aggravating agents include antibiotics, digoxin, and nonsteroidal anti-inflammatory drugs.10
Consider these skin disorders in the differential diagnosis
The diagnosis of psoriasis is usually clinical, and a skin biopsy is rarely needed. However, a range of other skin disorders should be kept in mind when considering the differential diagnosis.
Mycosis fungoides is a type of cutaneous T-cell lymphoma that forms erythematous plaques that may show wrinkling and epidermal atrophy in sun-protected sites. Onset usually occurs among the elderly.
Pityriasis rubra pilaris is characterized by salmon-colored patches that may have small areas of normal skin (“islands of sparing”), hyperkeratotic follicular papules, and hyperkeratosis of the palms and soles.
Seborrheic dermatitis, dandruff of the skin, usually involves the scalp and nasolabial areas and the T-zone of the face.
Continue to: Lichen planus
Lichen planus usually appears slightly more purple than psoriasis and typically involves the mouth, flexural surfaces of the wrists, genitals, and ankles.
Other conditions in the differential include pityriasis lichenoides chronica, which may be identified on skin biopsy. Inverse psoriasis can be difficult to differentiate from candida intertrigo, erythrasma, or tinea cruris.
A potassium hydroxide (KOH) preparation can help differentiate psoriasis from candida or tinea. In psoriasis, a KOH test will be negative for fungal elements. Mycology culture on skin scrapings may be performed to rule out fungal infection. Erythrasma may exhibit a coral red appearance under Wood lamp examination.
If a lesion fails to respond to appropriate treatment, a careful drug history and biopsy can help clarify the diagnosis.
Document disease
It’s important to thoroughly document the extent and severity of the psoriasis and to monitor the impact of treatment. The Psoriasis Area and Severity Index is a commonly used method that calculates a score based on the area (extent) of involvement surrounding 4 major anatomical regions (head, upper extremities, trunk, and lower extremities), as well as the degree of erythema, induration, and scaling of lesions. The average redness, thickness, and scaling are graded on a scale of 0 to 4 and the extent of involvement is calculated to form a total numerical score ranging from 0 (no disease) to 72 (maximal disease).
Continue to: Many options in the treatment arsenal
Many options in the treatment arsenal
Many treatments can improve psoriasis.9,15-19 Most affected individuals discover that emollients and exposure to natural sunlight can be effective, as are soothing baths (balneotherapy) or topical coal tar application. More persistent disease requires prescription therapy. Individualize therapy according to the severity of disease, location of the lesions, involvement of joints, and comorbidities (FIGURE 9).15
If ≤ 10% of the body surface area is involved, treatment options generally are explored in a stepwise progression from safest and most affordable to more involved therapies as needed: moisturization and avoidance of repetitive trauma, topical corticosteroids (TCS), vitamin D analogs, topical calcineurin inhibitors, and vitamin A creams. Recalcitrant disease will likely require ultraviolet (UV) light treatment or a systemic agent.15
If > 10% of the body surface area is involved, but joints are not involved, consider UV light treatment or a combination of alcitretin and TCS. If the joints are involved, likely initial options would be methotrexate, cyclosporine, or TNF-α inhibitor. Additional options to consider are anti-IL-17 or anti-IL-23 agents.15
If there’s joint involvement. In individuals with mild peripheral arthritis involving fewer than 4 joints without evidence of joint damage on imaging, nonsteroidal anti-inflammatory drugs are the mainstay of treatment. If the peripheral arthritis persists, or if it is associated with moderate-to-severe erosions or with substantial functional limitations, initiate treatment with a conventional disease-modifying antirheumatic drug. If the disease remains active, consider biologic agents.
Case studies
Mild-to-moderate psoriasis
Patient A is a 19-year-old woman presenting for evaluation of a persistently dry, flaking scalp. She has had the itchy scalp for years, as well as several small “patches” across her elbows, legs, knees, and abdomen. Over-the-counter emollients have not helped. The patient also says she has had brittle nails on several of her fingers, which she keeps covered with thick polish.
Continue to: The condition exemplified...
The condition exemplified by Patient A can typically be managed with topical products.
Topical steroids may be classified by different delivery vehicles, active ingredients, and potencies. The National Psoriasis Foundation's Topical Steroids Potency Chart can provide guidance (visit www.psoriasis.org/about-psoriasis/treatments/topicals/steroids/potency-chart and scroll down). Prescribing an appropriate amount is important; the standard 30-g prescription tube is generally required to cover the entire skin surface. Ointments have a greasy consistency (typically a petroleum base), which enhances potency and hydrates the skin. Creams and lotions are easier to rub on and spread. Gels are alcohol based and readily absorbed.16 Solutions, foams, and shampoos are particularly useful to treat psoriasis in hairy areas such as the scalp.
Corticosteroid potency ranges from Class I to Class VII, with the former being the most potent. While TCS products are typically effective with minimal systemic absorption, it is important to counsel patients on the risk of skin atrophy, impaired wound healing, and skin pigmentation changes with chronic use. With nail psoriasis, a potent topical steroid (including flurandrenolide [Cordran] tape) applied to the proximal nail fold has shown benefit.20
Topical calcineurin inhibitors (TCIs; eg, tacrolimus ointment and pimecrolimus cream) are anti-inflammatory agents often used in conjunction with topical steroids to minimize steroid use and associated adverse effects.15 A possible steroid-sparing regimen includes using a TCI Monday through Friday and a topical steroid on the weekend.
Topical vitamin D analogs (calcipotriene, calcipotriol, calcitriol) inhibit proliferation of keratinocytes and decrease the production of inflammatory mediators.15,17-19,21 Application of a vitamin D analog in combination with a high-potency TCS, systemic treatment, or phototherapy can provide greater efficacy, a more rapid onset of action, and less irritation than can the vitamin D analog used alone.21 If used in combination with UV light, apply topical vitamin D after the light therapy to prevent degradation.
Continue to: UV light therapy
UV light therapy is often used in cases refractory to topical therapy. Patients are typically prescribed 2 to 3 treatments per week with narrowband UVB (311-313 nm), the excimer laser (308 nm), or, less commonly, PUVA (UV treatment with psoralens). Treatment begins with a minimal erythema dose—the lowest dose to achieve minimal erythema of the skin before burning. When that is determined, exposure is increased as needed—depending on the response. If this is impractical or too time-consuming for the patient, an alternative recommendation would be increased exposure to natural sunlight or even use of a tanning booth. However, patients must then be cautioned about the increased risk of skin cancer.
Refractory/severe psoriasis
Patient B is a 35-year-old man with a longstanding history of psoriasis affecting his scalp and nails. Over the past 10 years, psoriatic lesions have also appeared and grown across his lower back, gluteal fold, legs, abdomen, and arms. He is now being evaluated by a rheumatologist for worsening symmetric joint pain that includes his lower back.
Methotrexate has been used to treat psoriasis and psoriatic arthritis since the 1950s. Methotrexate is a competitive inhibitor of dihydrofolate reductase and is typically given as an oral medication dosed once weekly with folic acid supplementation on the other 6 days.17 The most common adverse effects encountered with methotrexate are gastrointestinal upset and oral ulcers; however, routine monitoring for myelosuppression and hepatotoxicity is required.
Biologic therapy. When conventional therapies fail, immune-targeted treatment with “biologics” may be initiated. As knowledge of signaling pathways and the immunopathogenesis of psoriasis has increased, so has the number of biologic agents, which are generally well tolerated and effective in managing plaque psoriasis and psoriatic arthritis. Although their use, which requires monitoring, is handled primarily by specialists, familiarizing yourself with available agents can be helpful (TABLE).22
Nutritional modification and supplementation in treating skin disease still requires further investigation. Fish oil has shown benefit for cutaneous psoriasis in randomized controlled trials.7,8 Oral vitamin D supplementation requires further study, whereas selenium and B12 supplementation have not conferred consistent benefit.7 Given that several studies have demonstrated a relationship between body mass index and psoriatic disease severity, weight loss may be helpful in the management of psoriasis as well as psoriatic arthritis.8
Continue to: Other systemic agents
Other systemic agents—for individuals who cannot tolerate the biologic agents—include acitretin, azathioprine, mycophenolate mofetil, and cyclosporine.15,17
Paradoxical psoriatic reactions
When a psoriatic condition develops during biologic drug therapy, it is known as a paradoxical psoriatic reaction. The onset of de novo psoriasis has been documented during TNF-α inhibitor therapy for individuals with underlying rheumatoid arthritis.23 Skin biopsy reveals the same findings as common plaque psoriasis.
Using immunosuppressive Tx? Screen for tuberculosis
Testing to exclude a diagnosis of latent or undiagnosed tuberculosis must be performed prior to initiating immunosuppressive therapy with methotrexate or a biologic agent. Tuberculin skin testing, QuantiFERON-TB gold test, and the T-SPOT.TB test are accepted screening modalities. Discordance between tuberculin skin tests and the interferon gamma release assays in latent TB highlights the need for further study using the available QuantiFERON-TB gold test and the T-SPOT.TB test.24
CORRESPONDENCE
Karl T. Clebak, MD, FAAFP, Penn State Health Milton S. Hershey Medical Center, Department of Family and Community Medicine, 500 University Drive, Hershey, PA 17033; [email protected].
The “heartbreak of psoriasis,” coined by an advertiser in the 1960s, conveyed the notion that this disease was a cosmetic disorder mainly limited to skin involvement. John Updike’s article in the September 1985 issue of The New Yorker, “At War With My Skin,” detailed Mr. Updike’s feelings of isolation and stress related to his condition, helping to reframe the popular concept of psoriasis.1 Updike’s eloquent account describing his struggles to find effective treatment increased public awareness about psoriasis, which in fact affects other body systems as well.
The overall prevalence of psoriasis is 1.5% to 3.1% in the United States and United Kingdom.2,3 More than 6.5 million adults in the United States > 20 years of age are affected.3 The most commonly affected demographic group is non-Hispanic Caucasians.
Our expanding knowledge of pathogenesis
Studies of genetic linkage have identified genes and single nucleotide polymorphisms associated with psoriasis.4 The interaction between environmental triggers and the innate and adaptive immune systems leads to keratinocyte hyperproliferation. Tumor necrosis factor (TNF), interleukin (IL) 23, and IL-17 are important cytokines associated with psoriatic inflammation.4 There are common pathways of inflammation in both psoriasis and cardiovascular disease resulting in oxidative stress and endothelial cell dysfunction.4 Ninety percent of early-onset psoriasis is associated with human leukocyte antigen (HLA)-Cw6.4 And alterations in the microbiome of the skin may contribute, as reduced microbial diversity has been found in psoriatic lesions.5
Comorbidities are common
Psoriasis is an independent risk factor for diabetes and major adverse cardiovascular events.6 Hypertension, dyslipidemia, inflammatory bowel disease, nonalcoholic fatty liver disease, chronic kidney disease, and lymphoma (particularly cutaneous T-cell lymphoma) are also associated with psoriasis.6 Psoriatic arthritis is frequently encountered with cutaneous psoriasis; however, it is often not recognized until late in the disease course.
There also appears to be an association among psoriasis, dietary factors, and celiac disease.7-9 Positive testing for IgA anti-endomysial antibodies and IgA tissue transglutaminase antibodies should prompt consideration of starting a gluten-free diet, which has been shown to improve psoriatic
The different types of psoriasis
The classic presentation of psoriasis involves stubborn plaques with silvery scale on extensor surfaces such as the elbows and knees. The severity of the disease corresponds with the amount of body surface area affected. While plaque-type psoriasis is the most common form, other patterns exist. Individuals may exhibit 1 dominant pattern or multiple psoriatic variants simultaneously. Most types of psoriasis have 3 characteristic features: erythema, skin thickening, and scales.
Certain history and physical clues can aid in diagnosing psoriasis; these include the Koebner phenomenon, the Auspitz sign, and the Woronoff ring. The Koebner phenomenon refers to the development of psoriatic lesions in an area of trauma (FIGURE 1), frequently resulting in a linear streak-like appearance. The Auspitz sign describes the pinpoint bleeding that may be encountered with the removal of a psoriatic plaque. The Woronoff ring is a pale blanching ring that may surround a psoriatic lesion.
Continue to: Chronic plaque-type psoriasis
Chronic plaque-type psoriasis (Figures 2A and 2B), the most common variant, is characterized by sharply demarcated pink papules and plaques with a silvery scale in a symmetric distribution on the extensor surfaces, scalp, trunk, and lumbosacral areas.
Guttate psoriasis (FIGURE 3) features small (often < 1 cm) pink scaly papules that appear suddenly. It is more commonly seen in children and is usually preceded by an upper respiratory tract infection, often with Streptococcus.10 If strep testing is positive, guttate psoriasis may improve after appropriate antibiotic treatment.
Erythrodermic psoriasis (FIGUREs 4A and 4B) involves at least 75% of the body with erythema and scaling.11 Erythroderma can be caused by many other conditions such as atopic dermatitis, a drug reaction, Sezary syndrome, seborrheic dermatitis, and pityriasis rubra pilaris. Treatments for other conditions in the differential diagnosis can potentially make psoriasis worse. Unfortunately, findings on a skin biopsy are often nonspecific, making careful clinical observation crucial to arriving at an accurate diagnosis.
Pustular psoriasis is characterized by bright erythema and sterile pustules. Pustular psoriasis can be triggered by pregnancy, sudden tapering of corticosteroids, hypocalcemia, and infection. Involvement of the palms and soles with severe desquamation can drastically impact daily functioning and quality of life.
Inverse or flexural psoriasis (FIGUREs 5A and 5B) is characterized by shiny, pink-to-red sharply demarcated plaques involving intertriginous areas, typically the groin, inguinal crease, axilla, inframammary regions, and intergluteal cleft.
Continue to: Geographic tongue
Geographic tongue describes psoriasis of the tongue. The mucosa of the tongue has white plaques with a geographic border. Instead of scale, the moisture on the tongue causes areas of hyperkeratosis that appear white.
Nail psoriasis can manifest as nail pitting (FIGURE 6), oil staining, onycholysis (distal lifting of the nail), and subungual hyperkeratosis. Nail psoriasis is often quite distressing for patients and can be difficult to treat.
Palmoplantar psoriasis (FIGUREs 7A and 7B) can be painful due to the involvement of the palms of the hands and soles of the feet. Lesions will either be similar to other psoriatic plaques with well-demarcated erythematous scaling lesions or involve thickening and scale without associated erythema.
Psoriatic arthritis can cause significant joint damage and disability. Most affected individuals with psoriatic arthritis have a history of preceding skin disease.12 There are no specific lab tests for psoriasis; radiologic studies can show bulky syndesmophytes, central and marginal erosions, and periostitis. Patterns of joint involvement are variable. Psoriatic arthritis is more likely to affect the distal interphalangeal joints than rheumatoid arthritis and is more likely to affect the metacarpophalangeal joints than osteoarthritis.13
Psoriatic arthritis often progresses insidiously and is commonly described as causing discomfort rather than acute pain. Enthesitis, inflammation at the site where tendons or ligaments insert into the bone, is often present. Joint destruction may lead to the telescoping “opera glass” digit (FIGURE 8).
Continue to: Drug-provoked psoriasis
Drug-provoked psoriasis is divided into 2 groups: drug-induced and drug-aggravated. Drug-induced psoriasis will improve after discontinuation of the causative drug and tends to occur in patients without a personal or family history of psoriasis. Drug-aggravated psoriasis continues to progress after the discontinuation of the offending drug and is more often seen in patients with a history of psoriasis.14 Drugs that most commonly provoke psoriasis are beta-blockers, lithium, and antimalarials.10 Other potentially aggravating agents include antibiotics, digoxin, and nonsteroidal anti-inflammatory drugs.10
Consider these skin disorders in the differential diagnosis
The diagnosis of psoriasis is usually clinical, and a skin biopsy is rarely needed. However, a range of other skin disorders should be kept in mind when considering the differential diagnosis.
Mycosis fungoides is a type of cutaneous T-cell lymphoma that forms erythematous plaques that may show wrinkling and epidermal atrophy in sun-protected sites. Onset usually occurs among the elderly.
Pityriasis rubra pilaris is characterized by salmon-colored patches that may have small areas of normal skin (“islands of sparing”), hyperkeratotic follicular papules, and hyperkeratosis of the palms and soles.
Seborrheic dermatitis, dandruff of the skin, usually involves the scalp and nasolabial areas and the T-zone of the face.
Continue to: Lichen planus
Lichen planus usually appears slightly more purple than psoriasis and typically involves the mouth, flexural surfaces of the wrists, genitals, and ankles.
Other conditions in the differential include pityriasis lichenoides chronica, which may be identified on skin biopsy. Inverse psoriasis can be difficult to differentiate from candida intertrigo, erythrasma, or tinea cruris.
A potassium hydroxide (KOH) preparation can help differentiate psoriasis from candida or tinea. In psoriasis, a KOH test will be negative for fungal elements. Mycology culture on skin scrapings may be performed to rule out fungal infection. Erythrasma may exhibit a coral red appearance under Wood lamp examination.
If a lesion fails to respond to appropriate treatment, a careful drug history and biopsy can help clarify the diagnosis.
Document disease
It’s important to thoroughly document the extent and severity of the psoriasis and to monitor the impact of treatment. The Psoriasis Area and Severity Index is a commonly used method that calculates a score based on the area (extent) of involvement surrounding 4 major anatomical regions (head, upper extremities, trunk, and lower extremities), as well as the degree of erythema, induration, and scaling of lesions. The average redness, thickness, and scaling are graded on a scale of 0 to 4 and the extent of involvement is calculated to form a total numerical score ranging from 0 (no disease) to 72 (maximal disease).
Continue to: Many options in the treatment arsenal
Many options in the treatment arsenal
Many treatments can improve psoriasis.9,15-19 Most affected individuals discover that emollients and exposure to natural sunlight can be effective, as are soothing baths (balneotherapy) or topical coal tar application. More persistent disease requires prescription therapy. Individualize therapy according to the severity of disease, location of the lesions, involvement of joints, and comorbidities (FIGURE 9).15
If ≤ 10% of the body surface area is involved, treatment options generally are explored in a stepwise progression from safest and most affordable to more involved therapies as needed: moisturization and avoidance of repetitive trauma, topical corticosteroids (TCS), vitamin D analogs, topical calcineurin inhibitors, and vitamin A creams. Recalcitrant disease will likely require ultraviolet (UV) light treatment or a systemic agent.15
If > 10% of the body surface area is involved, but joints are not involved, consider UV light treatment or a combination of alcitretin and TCS. If the joints are involved, likely initial options would be methotrexate, cyclosporine, or TNF-α inhibitor. Additional options to consider are anti-IL-17 or anti-IL-23 agents.15
If there’s joint involvement. In individuals with mild peripheral arthritis involving fewer than 4 joints without evidence of joint damage on imaging, nonsteroidal anti-inflammatory drugs are the mainstay of treatment. If the peripheral arthritis persists, or if it is associated with moderate-to-severe erosions or with substantial functional limitations, initiate treatment with a conventional disease-modifying antirheumatic drug. If the disease remains active, consider biologic agents.
Case studies
Mild-to-moderate psoriasis
Patient A is a 19-year-old woman presenting for evaluation of a persistently dry, flaking scalp. She has had the itchy scalp for years, as well as several small “patches” across her elbows, legs, knees, and abdomen. Over-the-counter emollients have not helped. The patient also says she has had brittle nails on several of her fingers, which she keeps covered with thick polish.
Continue to: The condition exemplified...
The condition exemplified by Patient A can typically be managed with topical products.
Topical steroids may be classified by different delivery vehicles, active ingredients, and potencies. The National Psoriasis Foundation's Topical Steroids Potency Chart can provide guidance (visit www.psoriasis.org/about-psoriasis/treatments/topicals/steroids/potency-chart and scroll down). Prescribing an appropriate amount is important; the standard 30-g prescription tube is generally required to cover the entire skin surface. Ointments have a greasy consistency (typically a petroleum base), which enhances potency and hydrates the skin. Creams and lotions are easier to rub on and spread. Gels are alcohol based and readily absorbed.16 Solutions, foams, and shampoos are particularly useful to treat psoriasis in hairy areas such as the scalp.
Corticosteroid potency ranges from Class I to Class VII, with the former being the most potent. While TCS products are typically effective with minimal systemic absorption, it is important to counsel patients on the risk of skin atrophy, impaired wound healing, and skin pigmentation changes with chronic use. With nail psoriasis, a potent topical steroid (including flurandrenolide [Cordran] tape) applied to the proximal nail fold has shown benefit.20
Topical calcineurin inhibitors (TCIs; eg, tacrolimus ointment and pimecrolimus cream) are anti-inflammatory agents often used in conjunction with topical steroids to minimize steroid use and associated adverse effects.15 A possible steroid-sparing regimen includes using a TCI Monday through Friday and a topical steroid on the weekend.
Topical vitamin D analogs (calcipotriene, calcipotriol, calcitriol) inhibit proliferation of keratinocytes and decrease the production of inflammatory mediators.15,17-19,21 Application of a vitamin D analog in combination with a high-potency TCS, systemic treatment, or phototherapy can provide greater efficacy, a more rapid onset of action, and less irritation than can the vitamin D analog used alone.21 If used in combination with UV light, apply topical vitamin D after the light therapy to prevent degradation.
Continue to: UV light therapy
UV light therapy is often used in cases refractory to topical therapy. Patients are typically prescribed 2 to 3 treatments per week with narrowband UVB (311-313 nm), the excimer laser (308 nm), or, less commonly, PUVA (UV treatment with psoralens). Treatment begins with a minimal erythema dose—the lowest dose to achieve minimal erythema of the skin before burning. When that is determined, exposure is increased as needed—depending on the response. If this is impractical or too time-consuming for the patient, an alternative recommendation would be increased exposure to natural sunlight or even use of a tanning booth. However, patients must then be cautioned about the increased risk of skin cancer.
Refractory/severe psoriasis
Patient B is a 35-year-old man with a longstanding history of psoriasis affecting his scalp and nails. Over the past 10 years, psoriatic lesions have also appeared and grown across his lower back, gluteal fold, legs, abdomen, and arms. He is now being evaluated by a rheumatologist for worsening symmetric joint pain that includes his lower back.
Methotrexate has been used to treat psoriasis and psoriatic arthritis since the 1950s. Methotrexate is a competitive inhibitor of dihydrofolate reductase and is typically given as an oral medication dosed once weekly with folic acid supplementation on the other 6 days.17 The most common adverse effects encountered with methotrexate are gastrointestinal upset and oral ulcers; however, routine monitoring for myelosuppression and hepatotoxicity is required.
Biologic therapy. When conventional therapies fail, immune-targeted treatment with “biologics” may be initiated. As knowledge of signaling pathways and the immunopathogenesis of psoriasis has increased, so has the number of biologic agents, which are generally well tolerated and effective in managing plaque psoriasis and psoriatic arthritis. Although their use, which requires monitoring, is handled primarily by specialists, familiarizing yourself with available agents can be helpful (TABLE).22
Nutritional modification and supplementation in treating skin disease still requires further investigation. Fish oil has shown benefit for cutaneous psoriasis in randomized controlled trials.7,8 Oral vitamin D supplementation requires further study, whereas selenium and B12 supplementation have not conferred consistent benefit.7 Given that several studies have demonstrated a relationship between body mass index and psoriatic disease severity, weight loss may be helpful in the management of psoriasis as well as psoriatic arthritis.8
Continue to: Other systemic agents
Other systemic agents—for individuals who cannot tolerate the biologic agents—include acitretin, azathioprine, mycophenolate mofetil, and cyclosporine.15,17
Paradoxical psoriatic reactions
When a psoriatic condition develops during biologic drug therapy, it is known as a paradoxical psoriatic reaction. The onset of de novo psoriasis has been documented during TNF-α inhibitor therapy for individuals with underlying rheumatoid arthritis.23 Skin biopsy reveals the same findings as common plaque psoriasis.
Using immunosuppressive Tx? Screen for tuberculosis
Testing to exclude a diagnosis of latent or undiagnosed tuberculosis must be performed prior to initiating immunosuppressive therapy with methotrexate or a biologic agent. Tuberculin skin testing, QuantiFERON-TB gold test, and the T-SPOT.TB test are accepted screening modalities. Discordance between tuberculin skin tests and the interferon gamma release assays in latent TB highlights the need for further study using the available QuantiFERON-TB gold test and the T-SPOT.TB test.24
CORRESPONDENCE
Karl T. Clebak, MD, FAAFP, Penn State Health Milton S. Hershey Medical Center, Department of Family and Community Medicine, 500 University Drive, Hershey, PA 17033; [email protected].
1. Jackson R. John Updike on psoriasis. At war with my skin, from the journal of a leper. J Cutan Med Surg. 2000;4:113-115.
2. Gelfand JM, Weinstein R, Porter SB, et al. Prevalence and treatment of psoriasis in the United Kingdom: a population-based study. Arch Dermatol. 2005;141:1537-1541.
3. Helmick CG, Lee-Han H, Hirsch SC, et al. Prevalence of psoriasis among adults in the U.S.: 2003-2006 and 2009-2010 National Health and Nutrition Examination Surveys. Am J Prev Med. 2014;47:37-45.
4. Alexander H, Nestle FO. Pathogenesis and immunotherapy in cutaneous psoriasis: what can rheumatologists learn? Curr Opin Rheumatol. 2017;29:71-78.
5. Fahlén A, Engstrand L, Baker BS, et al. Comparison of bacterial microbiota in skin biopsies from normal and psoriatic skin. Arch Dermatol Res. 2012;304:15-22.
6. Takeshita J, Grewal S, Langan SM, et al. Psoriasis and comorbid diseases: epidemiology. J Am Acad Dermatol. 2017;76:377-390.
7. Millsop JW, Bhatia BK, Debbaneh M, et al. Diet and psoriasis, part III: role of nutritional supplements. J Am Acad Dermatol. 2014;71:561-569.
8. Debbaneh M, Millsop JW, Bhatia BK, et al. Diet and psoriasis, part I: impact of weight loss interventions. J Am Acad Dermatol. 2014;71:133-140.
9. Bhatia BK, Millsop JW, Debbaneh M, et al. Diet and psoriasis, part II: celiac disease and role of a gluten-free diet. J Am Acad Dermatol. 2014;71:350-358.
10. Fry L, Baker BS. Triggering psoriasis: the role of infections and medications. Clin Dermatol. 2007;25:606-615.
11. Singh RK, Lee KM, Ucmak D, et al. Erythrodermic psoriasis: pathophysiology and current treatment perspectives. Psoriasis (Aukl). 2016;6:93-104.
12. Garg A, Gladman D. Recognizing psoriatic arthritis in the dermatology clinic. J Am Acad Dermatol. 2010;63:733-748.
13. McGonagle D, Hermann KG, Tan AL. Differentiation between osteoarthritis and psoriatic arthritis: implications for pathogenesis and treatment in the biologic therapy era. Rheumatology. 2015;54:29-38.
14. Kim GK, Del Rosso JQ. Drug-provoked psoriasis: is it drug induced or drug aggravated?: understanding pathophysiology and clinical relevance. J Clin Aesthet Dermatol. 2010;3:32-38.
15. Kupetsky EA, Keller M. Psoriasis vulgaris: an evidence-based guide for primary care. J Am Board Fam Med. 2013;26:787-801.
16. Helm MF, Farah JB, Carvalho M, et al. Compounded topical medications for diseases of the skin: a long tradition still relevant today. N Am J Med Sci. 2017;10:116-118.
17. Weigle N, McBane S. Psoriasis. Am Fam Physician. 2013;87:626-633.
18. Helfrich YR, Sachs DL, Kang S. Topical vitamin D3. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 2nd ed. Philadelphia, PA: Saunders; 2007:691-695.
19. Lebwohl M, Siskin SB, Epinette W, et al. A multicenter trial of calcipotriene ointment and halobetasol ointment compared with either agent alone for the treatment of psoriasis. J Am Acad Dermatol. 1996;35:268-269.
20. Pasch MC. Nail psoriasis: a review of treatment options. Drugs. 2016;76:675-705.
21. Bagel J, Gold LS. Combining topical psoriasis treatment to enhance systemic and phototherapy: a review of the literature. J Drugs Dermatol. 2017;16:1209-1222.
22. Rønholt K, Iversen L. Old and new biological therapies for psoriasis. Int J Mol Sci. 2017;18:e2297.
23. Toussirot É, Aubin F. Paradoxical reactions under TNF-alpha blocking agents and other biologic agents given for chronic immune-mediated diseases: an analytical and comprehensive overview. RMD Open. 2016;2:e000239.
24. Connell TG, Ritz N, Paxton GA, et al. A three-way comparison of tuberculin skin testing, QuantiFERON-TB gold and T-SPOT.TB in children. PLoS One. 2008;3:e2624.
1. Jackson R. John Updike on psoriasis. At war with my skin, from the journal of a leper. J Cutan Med Surg. 2000;4:113-115.
2. Gelfand JM, Weinstein R, Porter SB, et al. Prevalence and treatment of psoriasis in the United Kingdom: a population-based study. Arch Dermatol. 2005;141:1537-1541.
3. Helmick CG, Lee-Han H, Hirsch SC, et al. Prevalence of psoriasis among adults in the U.S.: 2003-2006 and 2009-2010 National Health and Nutrition Examination Surveys. Am J Prev Med. 2014;47:37-45.
4. Alexander H, Nestle FO. Pathogenesis and immunotherapy in cutaneous psoriasis: what can rheumatologists learn? Curr Opin Rheumatol. 2017;29:71-78.
5. Fahlén A, Engstrand L, Baker BS, et al. Comparison of bacterial microbiota in skin biopsies from normal and psoriatic skin. Arch Dermatol Res. 2012;304:15-22.
6. Takeshita J, Grewal S, Langan SM, et al. Psoriasis and comorbid diseases: epidemiology. J Am Acad Dermatol. 2017;76:377-390.
7. Millsop JW, Bhatia BK, Debbaneh M, et al. Diet and psoriasis, part III: role of nutritional supplements. J Am Acad Dermatol. 2014;71:561-569.
8. Debbaneh M, Millsop JW, Bhatia BK, et al. Diet and psoriasis, part I: impact of weight loss interventions. J Am Acad Dermatol. 2014;71:133-140.
9. Bhatia BK, Millsop JW, Debbaneh M, et al. Diet and psoriasis, part II: celiac disease and role of a gluten-free diet. J Am Acad Dermatol. 2014;71:350-358.
10. Fry L, Baker BS. Triggering psoriasis: the role of infections and medications. Clin Dermatol. 2007;25:606-615.
11. Singh RK, Lee KM, Ucmak D, et al. Erythrodermic psoriasis: pathophysiology and current treatment perspectives. Psoriasis (Aukl). 2016;6:93-104.
12. Garg A, Gladman D. Recognizing psoriatic arthritis in the dermatology clinic. J Am Acad Dermatol. 2010;63:733-748.
13. McGonagle D, Hermann KG, Tan AL. Differentiation between osteoarthritis and psoriatic arthritis: implications for pathogenesis and treatment in the biologic therapy era. Rheumatology. 2015;54:29-38.
14. Kim GK, Del Rosso JQ. Drug-provoked psoriasis: is it drug induced or drug aggravated?: understanding pathophysiology and clinical relevance. J Clin Aesthet Dermatol. 2010;3:32-38.
15. Kupetsky EA, Keller M. Psoriasis vulgaris: an evidence-based guide for primary care. J Am Board Fam Med. 2013;26:787-801.
16. Helm MF, Farah JB, Carvalho M, et al. Compounded topical medications for diseases of the skin: a long tradition still relevant today. N Am J Med Sci. 2017;10:116-118.
17. Weigle N, McBane S. Psoriasis. Am Fam Physician. 2013;87:626-633.
18. Helfrich YR, Sachs DL, Kang S. Topical vitamin D3. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 2nd ed. Philadelphia, PA: Saunders; 2007:691-695.
19. Lebwohl M, Siskin SB, Epinette W, et al. A multicenter trial of calcipotriene ointment and halobetasol ointment compared with either agent alone for the treatment of psoriasis. J Am Acad Dermatol. 1996;35:268-269.
20. Pasch MC. Nail psoriasis: a review of treatment options. Drugs. 2016;76:675-705.
21. Bagel J, Gold LS. Combining topical psoriasis treatment to enhance systemic and phototherapy: a review of the literature. J Drugs Dermatol. 2017;16:1209-1222.
22. Rønholt K, Iversen L. Old and new biological therapies for psoriasis. Int J Mol Sci. 2017;18:e2297.
23. Toussirot É, Aubin F. Paradoxical reactions under TNF-alpha blocking agents and other biologic agents given for chronic immune-mediated diseases: an analytical and comprehensive overview. RMD Open. 2016;2:e000239.
24. Connell TG, Ritz N, Paxton GA, et al. A three-way comparison of tuberculin skin testing, QuantiFERON-TB gold and T-SPOT.TB in children. PLoS One. 2008;3:e2624.
PRACTICE RECOMMENDATIONS
› Consider guttate psoriasis if small (often < 1 cm) pink scaly papules appear suddenly, particularly in a child who has an upper respiratory tract infection. C
› Document extent of disease using a tool such as the Psoriasis Area and Severity Index, which calculates a score based on the area (extent) of involvement surrounding 4 major anatomical regions. C
› Consider prescribing UV light treatment or a combination of alcitretin and topical corticosteroid if > 10% of the body surface area is involved but joints are not affected. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series

Chronic blistering rash on hands
A 60-year-old man presented to our dermatology clinic with a chronic, recurrent pruritic rash on his hands and neck. He noted that the rash developed into blisters, which he would pick until they scabbed over. The rash only manifested on sun-exposed areas.
The patient did not take any medications. He admitted to drinking alcohol (4 beers/d on average) and had roughly a 50-pack year history of smoking. There was no family history of similar symptoms.
On physical exam, we noted erosions and ulcerations with hemorrhagic crust on the dorsal aspect of his hands, along with milia on the knuckle pads (FIGURE 1A). Further skin examination revealed hypopigmented scars on his shoulders and lower extremities bilaterally, with hypertrichosis of the cheeks (FIGURE 1B).

WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Porphyria cutanea tarda
Based on the clinical presentation and the patient’s history of smoking and alcohol consumption, we suspected that this was a case of porphyria cutanea tarda (PCT). Laboratory studies, including a complete blood count, basic metabolic panel, iron studies, and liver function tests, were ordered. These revealed elevated levels of serum alanine transaminase (116 IU/L; reference range, 20-60 IU/L), aspartate aminotransferase (184 IU/L; reference range, 6-34 IU/L in men), and ferritin (1594 ng/mL; reference range, 12-300 ng/mL in men), consistent with PCT. Total porphyrins were then measured and found to be elevated (128.5 mcg/dL; reference range, 0 to 1 mcg/dL), which confirmed the diagnosis. Further testing revealed that the patient was positive for both hepatitis C virus (HCV) and hepatitis B virus infection.
While PCT is the most common porphyria worldwide, it is nonetheless a rare disorder that results from deficient activity (< 20% of normal) of uroporphyrinogen decarboxylase (UROD), the fifth enzyme in the heme synthetic pathway.1,2 It is typically (~75% cases) an acquired disorder of mid- to late adulthood and more commonly affects males.1 In the remainder of cases, patients have a genetic predisposition—a mutation of the UROD or HFE gene. Patients with a genetic predisposition may present earlier.2,3 Susceptibility factors for both forms of PCT include chronic alcohol use, HCV and/or human immunodeficiency virus (HIV) infection, estrogen therapy, and a history of chronic/heavy smoking.1,4
Cutaneous manifestations of PCT are caused by the accumulation of porphyrins, which are photo-oxidized in the skin.1 Findings include photosensitivity, skin fragility, blistering, scarring, hypo- or hyperpigmentation, and milia in sun-exposed areas, such as the dorsum of the hands, forearms, face, ears, neck, and feet.1,2 Hypertrichosis can occur, particularly on the cheeks and forearms.1 Elevated transaminases often accompany cutaneous findings, due to porphyrin accumulation in hepatocytes and the hepatotoxic effects of alcohol, HCV infection, or iron overload.5 Iron overload, in part due to dysregulation of hepcidin, can lead to increased serum ferritin, iron, and transferrin saturation.1
Differential includes autoimmune and autosomal conditions
Diseases that manifest with blistering, elevated porphyrins or porphyrin precursors, and iron overload should be included in the differential diagnosis.
Bullous pemphigoid is an autoimmune subepithelial blistering disorder that occurs when antibodies attack hemidesmosomes in the epidermis. It commonly manifests in the elderly and classically presents with tense bullae, typically on the trunk, abdomen, and proximal extremities. Serologic testing and biopsy can confirm the diagnosis.6
Continue to: Pseudoporphyria...
Pseudoporphyria has a similar presentation to PCT but with no abnormalities in porphyrin metabolism. Risk factors include UV radiation exposure; use of medications such as nonsteroidal anti-inflammatory drugs, diuretics, and retinoids; chronic renal failure; and hemodialysis.7
Acute intermittent porphyria is an autosomal dominant disorder due to deficiency of porphobilinogen deaminase, a heme biosynthetic enzyme. Clinical manifestations usually arise in adulthood and include neurovisceral attacks (eg, abdominal pain, vomiting, muscle weakness). Diagnosis during an acute attack can be made by measuring urinary 5-aminolaevulinc acid and porphobilinogen.1
Hereditary hemochromatosis is an autosomal recessive disorder most commonly due to mutations in the HFE gene. Patients typically have iron overload and abnormal liver function test results. The main cutaneous finding is skin hyperpigmentation. Patients also may develop diabetes mellitus, arthropathy, cardiac disease, and hypopituitarism, although most are diagnosed with asymptomatic disease following routine laboratory studies.8
Confirm the diagnosis with total porphyrin measurement
The preferred initial test to confirm the diagnosis of PCT is measurement of plasma or urine total porphyrins, which will be elevated.1 Further testing is then performed to discern PCT from the other, less common cutaneous porphyrias.1 If needed, biopsy can be done to exclude other diagnoses. Testing for HIV and viral hepatitis infection may be performed when clinical suspicion is high.1 Testing for UROD and HFE mutations may also be advised.1
Treatment choice is guided by iron levels
For patients with normal iron levels, low-dose hydroxychloroquine 100 mg or chloroquine 125 mg twice per week can be used until restoration of normal plasma or urine porphyrin levels has been achieved for several months.1 For those with iron excess (serum ferritin > 600 ng/dL), repeat phlebotomy is the preferred treatment; a unit of blood (350-500 mL) is typically removed, as tolerated, until iron stores return to normal.1 In severe cases of PCT, these therapies can be used in combination.1 Clinical remission with these methods can be expected within 6 to 9 months.9
Continue to: In addition...
In addition, it is important to provide patient education regarding proper sun protection and risk factor modification.1 Underlying HIV and viral hepatitis infection should be managed appropriately by the relevant specialists.
Our patient was counseled on proper sun protection and encouraged to cease alcohol consumption and smoking. We subsequently referred him to Hepatology for the treatment of his liver disease. Given that the patient’s ferritin level was so high (1594 ng/mL), serial phlebotomy was initiated twice monthly until levels reached the lower limit of normal. He was also started on direct-acting antiviral therapy with Epclusa (sofosbuvir/velpatasvir) for 12 weeks for treatment of his HCV and is currently in remission.
CORRESPONDENCE
Christopher G. Bazewicz, MD, Department of Dermatology, Penn State Milton S. Hershey Medical Center, 500 University Drive, Hershey, PA 17033; [email protected]
1. Puy H, Gouya L, Deybach JC. Porphyrias. Lancet. 2010;375:924-937.
2. Méndez M, Poblete-Gutiérrez P, García-Bravo M, et al. Molecular heterogeneity of familial porphyria cutanea tarda in Spain: characterization of 10 novel mutations in the UROD gene. Br J Dermatol. 2007;157:501-507.
3. Brady JJ, Jackson HA, Roberts AG, et al. Co-inheritance of mutations in the uroporphyrinogen decarboxylase and hemochromatosis genes accelerates the onset of porphyria cutanea tarda. J Invest Dermatol. 2000;115:868-874.
4. Jalil S, Grady JJ, Lee C, et al. Associations among behavior-related susceptibility factors in porphyria cutanea tarda. Clin Gastroenterol Hepatol. 2010;8:297-302, 302.e1.
5. Gisbert JP, García-Buey L, Alonso A, et al. Hepatocellular carcinoma risk in patients with porphyria cutanea tarda. Eur J Gastroenterol Hepatol. 2004;16:689-692.
6. Di Zenzo G, Della Torre R, Zambruno G, et al. Bullous pemphigoid: from the clinic to the bench. Clin Dermatol. 2012;30:3-16.
7. Green JJ, Manders SM. Pseudoporphyria. J Am Acad Dermatol. 2001;44:100-108.
8. Crownover BK, Covey CJ. Hereditary hemochromatosis. Am Fam Physician. 2013;87:183-190.
9. Sarkany RP. The management of porphyria cutanea tarda. Clin Exp Dermatol. 2001;26:225-232.
A 60-year-old man presented to our dermatology clinic with a chronic, recurrent pruritic rash on his hands and neck. He noted that the rash developed into blisters, which he would pick until they scabbed over. The rash only manifested on sun-exposed areas.
The patient did not take any medications. He admitted to drinking alcohol (4 beers/d on average) and had roughly a 50-pack year history of smoking. There was no family history of similar symptoms.
On physical exam, we noted erosions and ulcerations with hemorrhagic crust on the dorsal aspect of his hands, along with milia on the knuckle pads (FIGURE 1A). Further skin examination revealed hypopigmented scars on his shoulders and lower extremities bilaterally, with hypertrichosis of the cheeks (FIGURE 1B).
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Porphyria cutanea tarda
Based on the clinical presentation and the patient’s history of smoking and alcohol consumption, we suspected that this was a case of porphyria cutanea tarda (PCT). Laboratory studies, including a complete blood count, basic metabolic panel, iron studies, and liver function tests, were ordered. These revealed elevated levels of serum alanine transaminase (116 IU/L; reference range, 20-60 IU/L), aspartate aminotransferase (184 IU/L; reference range, 6-34 IU/L in men), and ferritin (1594 ng/mL; reference range, 12-300 ng/mL in men), consistent with PCT. Total porphyrins were then measured and found to be elevated (128.5 mcg/dL; reference range, 0 to 1 mcg/dL), which confirmed the diagnosis. Further testing revealed that the patient was positive for both hepatitis C virus (HCV) and hepatitis B virus infection.
While PCT is the most common porphyria worldwide, it is nonetheless a rare disorder that results from deficient activity (< 20% of normal) of uroporphyrinogen decarboxylase (UROD), the fifth enzyme in the heme synthetic pathway.1,2 It is typically (~75% cases) an acquired disorder of mid- to late adulthood and more commonly affects males.1 In the remainder of cases, patients have a genetic predisposition—a mutation of the UROD or HFE gene. Patients with a genetic predisposition may present earlier.2,3 Susceptibility factors for both forms of PCT include chronic alcohol use, HCV and/or human immunodeficiency virus (HIV) infection, estrogen therapy, and a history of chronic/heavy smoking.1,4
Cutaneous manifestations of PCT are caused by the accumulation of porphyrins, which are photo-oxidized in the skin.1 Findings include photosensitivity, skin fragility, blistering, scarring, hypo- or hyperpigmentation, and milia in sun-exposed areas, such as the dorsum of the hands, forearms, face, ears, neck, and feet.1,2 Hypertrichosis can occur, particularly on the cheeks and forearms.1 Elevated transaminases often accompany cutaneous findings, due to porphyrin accumulation in hepatocytes and the hepatotoxic effects of alcohol, HCV infection, or iron overload.5 Iron overload, in part due to dysregulation of hepcidin, can lead to increased serum ferritin, iron, and transferrin saturation.1
Differential includes autoimmune and autosomal conditions
Diseases that manifest with blistering, elevated porphyrins or porphyrin precursors, and iron overload should be included in the differential diagnosis.
Bullous pemphigoid is an autoimmune subepithelial blistering disorder that occurs when antibodies attack hemidesmosomes in the epidermis. It commonly manifests in the elderly and classically presents with tense bullae, typically on the trunk, abdomen, and proximal extremities. Serologic testing and biopsy can confirm the diagnosis.6
Continue to: Pseudoporphyria...
Pseudoporphyria has a similar presentation to PCT but with no abnormalities in porphyrin metabolism. Risk factors include UV radiation exposure; use of medications such as nonsteroidal anti-inflammatory drugs, diuretics, and retinoids; chronic renal failure; and hemodialysis.7
Acute intermittent porphyria is an autosomal dominant disorder due to deficiency of porphobilinogen deaminase, a heme biosynthetic enzyme. Clinical manifestations usually arise in adulthood and include neurovisceral attacks (eg, abdominal pain, vomiting, muscle weakness). Diagnosis during an acute attack can be made by measuring urinary 5-aminolaevulinc acid and porphobilinogen.1
Hereditary hemochromatosis is an autosomal recessive disorder most commonly due to mutations in the HFE gene. Patients typically have iron overload and abnormal liver function test results. The main cutaneous finding is skin hyperpigmentation. Patients also may develop diabetes mellitus, arthropathy, cardiac disease, and hypopituitarism, although most are diagnosed with asymptomatic disease following routine laboratory studies.8
Confirm the diagnosis with total porphyrin measurement
The preferred initial test to confirm the diagnosis of PCT is measurement of plasma or urine total porphyrins, which will be elevated.1 Further testing is then performed to discern PCT from the other, less common cutaneous porphyrias.1 If needed, biopsy can be done to exclude other diagnoses. Testing for HIV and viral hepatitis infection may be performed when clinical suspicion is high.1 Testing for UROD and HFE mutations may also be advised.1
Treatment choice is guided by iron levels
For patients with normal iron levels, low-dose hydroxychloroquine 100 mg or chloroquine 125 mg twice per week can be used until restoration of normal plasma or urine porphyrin levels has been achieved for several months.1 For those with iron excess (serum ferritin > 600 ng/dL), repeat phlebotomy is the preferred treatment; a unit of blood (350-500 mL) is typically removed, as tolerated, until iron stores return to normal.1 In severe cases of PCT, these therapies can be used in combination.1 Clinical remission with these methods can be expected within 6 to 9 months.9
Continue to: In addition...
In addition, it is important to provide patient education regarding proper sun protection and risk factor modification.1 Underlying HIV and viral hepatitis infection should be managed appropriately by the relevant specialists.
Our patient was counseled on proper sun protection and encouraged to cease alcohol consumption and smoking. We subsequently referred him to Hepatology for the treatment of his liver disease. Given that the patient’s ferritin level was so high (1594 ng/mL), serial phlebotomy was initiated twice monthly until levels reached the lower limit of normal. He was also started on direct-acting antiviral therapy with Epclusa (sofosbuvir/velpatasvir) for 12 weeks for treatment of his HCV and is currently in remission.
CORRESPONDENCE
Christopher G. Bazewicz, MD, Department of Dermatology, Penn State Milton S. Hershey Medical Center, 500 University Drive, Hershey, PA 17033; [email protected]
A 60-year-old man presented to our dermatology clinic with a chronic, recurrent pruritic rash on his hands and neck. He noted that the rash developed into blisters, which he would pick until they scabbed over. The rash only manifested on sun-exposed areas.
The patient did not take any medications. He admitted to drinking alcohol (4 beers/d on average) and had roughly a 50-pack year history of smoking. There was no family history of similar symptoms.
On physical exam, we noted erosions and ulcerations with hemorrhagic crust on the dorsal aspect of his hands, along with milia on the knuckle pads (FIGURE 1A). Further skin examination revealed hypopigmented scars on his shoulders and lower extremities bilaterally, with hypertrichosis of the cheeks (FIGURE 1B).
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Porphyria cutanea tarda
Based on the clinical presentation and the patient’s history of smoking and alcohol consumption, we suspected that this was a case of porphyria cutanea tarda (PCT). Laboratory studies, including a complete blood count, basic metabolic panel, iron studies, and liver function tests, were ordered. These revealed elevated levels of serum alanine transaminase (116 IU/L; reference range, 20-60 IU/L), aspartate aminotransferase (184 IU/L; reference range, 6-34 IU/L in men), and ferritin (1594 ng/mL; reference range, 12-300 ng/mL in men), consistent with PCT. Total porphyrins were then measured and found to be elevated (128.5 mcg/dL; reference range, 0 to 1 mcg/dL), which confirmed the diagnosis. Further testing revealed that the patient was positive for both hepatitis C virus (HCV) and hepatitis B virus infection.
While PCT is the most common porphyria worldwide, it is nonetheless a rare disorder that results from deficient activity (< 20% of normal) of uroporphyrinogen decarboxylase (UROD), the fifth enzyme in the heme synthetic pathway.1,2 It is typically (~75% cases) an acquired disorder of mid- to late adulthood and more commonly affects males.1 In the remainder of cases, patients have a genetic predisposition—a mutation of the UROD or HFE gene. Patients with a genetic predisposition may present earlier.2,3 Susceptibility factors for both forms of PCT include chronic alcohol use, HCV and/or human immunodeficiency virus (HIV) infection, estrogen therapy, and a history of chronic/heavy smoking.1,4
Cutaneous manifestations of PCT are caused by the accumulation of porphyrins, which are photo-oxidized in the skin.1 Findings include photosensitivity, skin fragility, blistering, scarring, hypo- or hyperpigmentation, and milia in sun-exposed areas, such as the dorsum of the hands, forearms, face, ears, neck, and feet.1,2 Hypertrichosis can occur, particularly on the cheeks and forearms.1 Elevated transaminases often accompany cutaneous findings, due to porphyrin accumulation in hepatocytes and the hepatotoxic effects of alcohol, HCV infection, or iron overload.5 Iron overload, in part due to dysregulation of hepcidin, can lead to increased serum ferritin, iron, and transferrin saturation.1
Differential includes autoimmune and autosomal conditions
Diseases that manifest with blistering, elevated porphyrins or porphyrin precursors, and iron overload should be included in the differential diagnosis.
Bullous pemphigoid is an autoimmune subepithelial blistering disorder that occurs when antibodies attack hemidesmosomes in the epidermis. It commonly manifests in the elderly and classically presents with tense bullae, typically on the trunk, abdomen, and proximal extremities. Serologic testing and biopsy can confirm the diagnosis.6
Continue to: Pseudoporphyria...
Pseudoporphyria has a similar presentation to PCT but with no abnormalities in porphyrin metabolism. Risk factors include UV radiation exposure; use of medications such as nonsteroidal anti-inflammatory drugs, diuretics, and retinoids; chronic renal failure; and hemodialysis.7
Acute intermittent porphyria is an autosomal dominant disorder due to deficiency of porphobilinogen deaminase, a heme biosynthetic enzyme. Clinical manifestations usually arise in adulthood and include neurovisceral attacks (eg, abdominal pain, vomiting, muscle weakness). Diagnosis during an acute attack can be made by measuring urinary 5-aminolaevulinc acid and porphobilinogen.1
Hereditary hemochromatosis is an autosomal recessive disorder most commonly due to mutations in the HFE gene. Patients typically have iron overload and abnormal liver function test results. The main cutaneous finding is skin hyperpigmentation. Patients also may develop diabetes mellitus, arthropathy, cardiac disease, and hypopituitarism, although most are diagnosed with asymptomatic disease following routine laboratory studies.8
Confirm the diagnosis with total porphyrin measurement
The preferred initial test to confirm the diagnosis of PCT is measurement of plasma or urine total porphyrins, which will be elevated.1 Further testing is then performed to discern PCT from the other, less common cutaneous porphyrias.1 If needed, biopsy can be done to exclude other diagnoses. Testing for HIV and viral hepatitis infection may be performed when clinical suspicion is high.1 Testing for UROD and HFE mutations may also be advised.1
Treatment choice is guided by iron levels
For patients with normal iron levels, low-dose hydroxychloroquine 100 mg or chloroquine 125 mg twice per week can be used until restoration of normal plasma or urine porphyrin levels has been achieved for several months.1 For those with iron excess (serum ferritin > 600 ng/dL), repeat phlebotomy is the preferred treatment; a unit of blood (350-500 mL) is typically removed, as tolerated, until iron stores return to normal.1 In severe cases of PCT, these therapies can be used in combination.1 Clinical remission with these methods can be expected within 6 to 9 months.9
Continue to: In addition...
In addition, it is important to provide patient education regarding proper sun protection and risk factor modification.1 Underlying HIV and viral hepatitis infection should be managed appropriately by the relevant specialists.
Our patient was counseled on proper sun protection and encouraged to cease alcohol consumption and smoking. We subsequently referred him to Hepatology for the treatment of his liver disease. Given that the patient’s ferritin level was so high (1594 ng/mL), serial phlebotomy was initiated twice monthly until levels reached the lower limit of normal. He was also started on direct-acting antiviral therapy with Epclusa (sofosbuvir/velpatasvir) for 12 weeks for treatment of his HCV and is currently in remission.
CORRESPONDENCE
Christopher G. Bazewicz, MD, Department of Dermatology, Penn State Milton S. Hershey Medical Center, 500 University Drive, Hershey, PA 17033; [email protected]
1. Puy H, Gouya L, Deybach JC. Porphyrias. Lancet. 2010;375:924-937.
2. Méndez M, Poblete-Gutiérrez P, García-Bravo M, et al. Molecular heterogeneity of familial porphyria cutanea tarda in Spain: characterization of 10 novel mutations in the UROD gene. Br J Dermatol. 2007;157:501-507.
3. Brady JJ, Jackson HA, Roberts AG, et al. Co-inheritance of mutations in the uroporphyrinogen decarboxylase and hemochromatosis genes accelerates the onset of porphyria cutanea tarda. J Invest Dermatol. 2000;115:868-874.
4. Jalil S, Grady JJ, Lee C, et al. Associations among behavior-related susceptibility factors in porphyria cutanea tarda. Clin Gastroenterol Hepatol. 2010;8:297-302, 302.e1.
5. Gisbert JP, García-Buey L, Alonso A, et al. Hepatocellular carcinoma risk in patients with porphyria cutanea tarda. Eur J Gastroenterol Hepatol. 2004;16:689-692.
6. Di Zenzo G, Della Torre R, Zambruno G, et al. Bullous pemphigoid: from the clinic to the bench. Clin Dermatol. 2012;30:3-16.
7. Green JJ, Manders SM. Pseudoporphyria. J Am Acad Dermatol. 2001;44:100-108.
8. Crownover BK, Covey CJ. Hereditary hemochromatosis. Am Fam Physician. 2013;87:183-190.
9. Sarkany RP. The management of porphyria cutanea tarda. Clin Exp Dermatol. 2001;26:225-232.
1. Puy H, Gouya L, Deybach JC. Porphyrias. Lancet. 2010;375:924-937.
2. Méndez M, Poblete-Gutiérrez P, García-Bravo M, et al. Molecular heterogeneity of familial porphyria cutanea tarda in Spain: characterization of 10 novel mutations in the UROD gene. Br J Dermatol. 2007;157:501-507.
3. Brady JJ, Jackson HA, Roberts AG, et al. Co-inheritance of mutations in the uroporphyrinogen decarboxylase and hemochromatosis genes accelerates the onset of porphyria cutanea tarda. J Invest Dermatol. 2000;115:868-874.
4. Jalil S, Grady JJ, Lee C, et al. Associations among behavior-related susceptibility factors in porphyria cutanea tarda. Clin Gastroenterol Hepatol. 2010;8:297-302, 302.e1.
5. Gisbert JP, García-Buey L, Alonso A, et al. Hepatocellular carcinoma risk in patients with porphyria cutanea tarda. Eur J Gastroenterol Hepatol. 2004;16:689-692.
6. Di Zenzo G, Della Torre R, Zambruno G, et al. Bullous pemphigoid: from the clinic to the bench. Clin Dermatol. 2012;30:3-16.
7. Green JJ, Manders SM. Pseudoporphyria. J Am Acad Dermatol. 2001;44:100-108.
8. Crownover BK, Covey CJ. Hereditary hemochromatosis. Am Fam Physician. 2013;87:183-190.
9. Sarkany RP. The management of porphyria cutanea tarda. Clin Exp Dermatol. 2001;26:225-232.

Erythematous swollen ear
A 25-year-old woman presented with an exceedingly tender right ear. She’d had the helix of her ear pierced 3 days prior to presentation and 2 days after that, the ear had become tender. The tenderness was progressively worsening and associated with throbbing pain. The patient, who’d had her ears pierced before, was otherwise in good health and denied fever, chills, or travel outside of the country. She had been going to the gym regularly and took frequent showers. Physical examination revealed an erythematous swollen ear that was tender to the touch (FIGURE). The entire auricle was swollen except for the earlobe. The patient also reported purulent material draining from the helical piercing site.

WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Auricular perichondritis
Auricular perichondritis is an inflammation of the connective tissue surrounding the cartilage of the ear. Infectious and autoimmune factors may play a role. The underlying cartilage also may become involved. A useful clinical clue to the diagnosis of auricular perichondritis is sparing of the earlobe, which does not contain cartilage. Autoimmune causes typically have bilateral involvement. Infectious causes are usually associated with trauma and purulent drainage at the wound site. Ear piercings are an increasingly common cause, but perichondritis due to minor trauma, as a surgical complication, or in the absence of an obvious inciting trigger can occur. A careful history usually will reveal the cause.
In this case, the patient indicated that an open piercing gun at a shopping mall kiosk had been used to pierce her ear. Piercing with a sterile straight needle would have been preferable and less likely to be associated with secondary infection, as the shearing trauma to the perichondrium experienced with a piercing gun is thought to predispose to infection.1 Exposure to fresh water from the shower could have been a source for Pseudomonas infection.1
Differential: Pinpointing the diagnosis early is vital
A red and tender ear can raise a differential diagnosis that includes erysipelas, relapsing polychondritis, and auricular perichondritis. Erysipelas is a bacterial infection that spreads through the lymphatic system and is associated with intense and well-demarcated erythema. Erysipelas typically involves the face or lower legs. Infection after piercing or traumatic injury should raise suspicion of pseudomonal infection.2-5 Untreated infection can spread quickly and lead to permanent ear deformity. Although the same pattern of inflammation can be seen in relapsing polychondritis, relapsing polychondritis typically involves both ears as well as the eyes and joints.
Prompt treatment is necessary to avoid cosmetic disfigurement
The timing of the reaction in our patient made infection obvious because Pseudomonas aeruginosa seems to have a particular affinity for damaged cartilage.2
Ciprofloxacin 500 mg twice daily is the treatment of choice. Although many skin infections can be empirically treated with oral cephalosporin, penicillin, or erythromycin, it is important to recognize that infected piercing sites and auricular perichondritis due to pseudomonal infection will not respond to these agents. That’s because these agents do not provide as good coverage for Pseudomonas as they do for Staphylococci or other bacteria more often associated with skin infection. Treatment with an agent such as amoxicillin and clavulanic acid or oral cephalexin can mean the loss of valuable time and subsequent cosmetic disfigurement.6
Continue to: When fluctuance is present...
When fluctuance is present, incision and drainage, or even debridement, may be necessary. When extensive infection leads to cartilage necrosis and liquefaction, treatment is difficult and may result in lasting disfigurement. Prompt empiric treatment currently is considered the best option.6
Our patient was prescribed a course of ciprofloxacin 500 mg every 12 hours for 10 days. She noted improvement within 2 days, and the infection resolved without complication.
CORRESPONDENCE
Matthew F. Helm, MD, Penn State Health Hershey Medical Center, 500 University Dr, HU14, Hershey, PA 17033; [email protected]
1. Sandhu A, Gross M, Wylie J, et al. Pseudomonas aeruginosa necrotizing chondritis complicating high helical ear piercing case report: clinical and public health perspectives. Can J Public Health. 2007;98:74-77.
2. Prasad HK, Sreedharan S, Prasad HS, et al. Perichondritis of the auricle and its management. J Laryngol Otol. 2007;121:530-534.
3. Fisher CG, Kacica MA, Bennett NM. Risk factors for cartilage infections of the ear. Am J Prev Med. 2005;29:204-209.
4. Lee TC, Gold WL. Necrotizing Pseudomonas chondritis after piercing of the upper ear. CMAJ. 2011;183:819-821.
5. Rowshan HH, Keith K, Baur D, et al. Pseudomonas aeruginosa infection of the auricular cartilage caused by “high ear piercing”: a case report and review of the literature. J Oral Maxillofac Surg. 2008;66:543-546.
6. Liu ZW, Chokkalingam P. Piercing associated perichondritis of the pinna: are we treating it correctly? J Laryngol Otol. 2013;127:505-508.
A 25-year-old woman presented with an exceedingly tender right ear. She’d had the helix of her ear pierced 3 days prior to presentation and 2 days after that, the ear had become tender. The tenderness was progressively worsening and associated with throbbing pain. The patient, who’d had her ears pierced before, was otherwise in good health and denied fever, chills, or travel outside of the country. She had been going to the gym regularly and took frequent showers. Physical examination revealed an erythematous swollen ear that was tender to the touch (FIGURE). The entire auricle was swollen except for the earlobe. The patient also reported purulent material draining from the helical piercing site.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Auricular perichondritis
Auricular perichondritis is an inflammation of the connective tissue surrounding the cartilage of the ear. Infectious and autoimmune factors may play a role. The underlying cartilage also may become involved. A useful clinical clue to the diagnosis of auricular perichondritis is sparing of the earlobe, which does not contain cartilage. Autoimmune causes typically have bilateral involvement. Infectious causes are usually associated with trauma and purulent drainage at the wound site. Ear piercings are an increasingly common cause, but perichondritis due to minor trauma, as a surgical complication, or in the absence of an obvious inciting trigger can occur. A careful history usually will reveal the cause.
In this case, the patient indicated that an open piercing gun at a shopping mall kiosk had been used to pierce her ear. Piercing with a sterile straight needle would have been preferable and less likely to be associated with secondary infection, as the shearing trauma to the perichondrium experienced with a piercing gun is thought to predispose to infection.1 Exposure to fresh water from the shower could have been a source for Pseudomonas infection.1
Differential: Pinpointing the diagnosis early is vital
A red and tender ear can raise a differential diagnosis that includes erysipelas, relapsing polychondritis, and auricular perichondritis. Erysipelas is a bacterial infection that spreads through the lymphatic system and is associated with intense and well-demarcated erythema. Erysipelas typically involves the face or lower legs. Infection after piercing or traumatic injury should raise suspicion of pseudomonal infection.2-5 Untreated infection can spread quickly and lead to permanent ear deformity. Although the same pattern of inflammation can be seen in relapsing polychondritis, relapsing polychondritis typically involves both ears as well as the eyes and joints.
Prompt treatment is necessary to avoid cosmetic disfigurement
The timing of the reaction in our patient made infection obvious because Pseudomonas aeruginosa seems to have a particular affinity for damaged cartilage.2
Ciprofloxacin 500 mg twice daily is the treatment of choice. Although many skin infections can be empirically treated with oral cephalosporin, penicillin, or erythromycin, it is important to recognize that infected piercing sites and auricular perichondritis due to pseudomonal infection will not respond to these agents. That’s because these agents do not provide as good coverage for Pseudomonas as they do for Staphylococci or other bacteria more often associated with skin infection. Treatment with an agent such as amoxicillin and clavulanic acid or oral cephalexin can mean the loss of valuable time and subsequent cosmetic disfigurement.6
Continue to: When fluctuance is present...
When fluctuance is present, incision and drainage, or even debridement, may be necessary. When extensive infection leads to cartilage necrosis and liquefaction, treatment is difficult and may result in lasting disfigurement. Prompt empiric treatment currently is considered the best option.6
Our patient was prescribed a course of ciprofloxacin 500 mg every 12 hours for 10 days. She noted improvement within 2 days, and the infection resolved without complication.
CORRESPONDENCE
Matthew F. Helm, MD, Penn State Health Hershey Medical Center, 500 University Dr, HU14, Hershey, PA 17033; [email protected]
A 25-year-old woman presented with an exceedingly tender right ear. She’d had the helix of her ear pierced 3 days prior to presentation and 2 days after that, the ear had become tender. The tenderness was progressively worsening and associated with throbbing pain. The patient, who’d had her ears pierced before, was otherwise in good health and denied fever, chills, or travel outside of the country. She had been going to the gym regularly and took frequent showers. Physical examination revealed an erythematous swollen ear that was tender to the touch (FIGURE). The entire auricle was swollen except for the earlobe. The patient also reported purulent material draining from the helical piercing site.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Auricular perichondritis
Auricular perichondritis is an inflammation of the connective tissue surrounding the cartilage of the ear. Infectious and autoimmune factors may play a role. The underlying cartilage also may become involved. A useful clinical clue to the diagnosis of auricular perichondritis is sparing of the earlobe, which does not contain cartilage. Autoimmune causes typically have bilateral involvement. Infectious causes are usually associated with trauma and purulent drainage at the wound site. Ear piercings are an increasingly common cause, but perichondritis due to minor trauma, as a surgical complication, or in the absence of an obvious inciting trigger can occur. A careful history usually will reveal the cause.
In this case, the patient indicated that an open piercing gun at a shopping mall kiosk had been used to pierce her ear. Piercing with a sterile straight needle would have been preferable and less likely to be associated with secondary infection, as the shearing trauma to the perichondrium experienced with a piercing gun is thought to predispose to infection.1 Exposure to fresh water from the shower could have been a source for Pseudomonas infection.1
Differential: Pinpointing the diagnosis early is vital
A red and tender ear can raise a differential diagnosis that includes erysipelas, relapsing polychondritis, and auricular perichondritis. Erysipelas is a bacterial infection that spreads through the lymphatic system and is associated with intense and well-demarcated erythema. Erysipelas typically involves the face or lower legs. Infection after piercing or traumatic injury should raise suspicion of pseudomonal infection.2-5 Untreated infection can spread quickly and lead to permanent ear deformity. Although the same pattern of inflammation can be seen in relapsing polychondritis, relapsing polychondritis typically involves both ears as well as the eyes and joints.
Prompt treatment is necessary to avoid cosmetic disfigurement
The timing of the reaction in our patient made infection obvious because Pseudomonas aeruginosa seems to have a particular affinity for damaged cartilage.2
Ciprofloxacin 500 mg twice daily is the treatment of choice. Although many skin infections can be empirically treated with oral cephalosporin, penicillin, or erythromycin, it is important to recognize that infected piercing sites and auricular perichondritis due to pseudomonal infection will not respond to these agents. That’s because these agents do not provide as good coverage for Pseudomonas as they do for Staphylococci or other bacteria more often associated with skin infection. Treatment with an agent such as amoxicillin and clavulanic acid or oral cephalexin can mean the loss of valuable time and subsequent cosmetic disfigurement.6
Continue to: When fluctuance is present...
When fluctuance is present, incision and drainage, or even debridement, may be necessary. When extensive infection leads to cartilage necrosis and liquefaction, treatment is difficult and may result in lasting disfigurement. Prompt empiric treatment currently is considered the best option.6
Our patient was prescribed a course of ciprofloxacin 500 mg every 12 hours for 10 days. She noted improvement within 2 days, and the infection resolved without complication.
CORRESPONDENCE
Matthew F. Helm, MD, Penn State Health Hershey Medical Center, 500 University Dr, HU14, Hershey, PA 17033; [email protected]
1. Sandhu A, Gross M, Wylie J, et al. Pseudomonas aeruginosa necrotizing chondritis complicating high helical ear piercing case report: clinical and public health perspectives. Can J Public Health. 2007;98:74-77.
2. Prasad HK, Sreedharan S, Prasad HS, et al. Perichondritis of the auricle and its management. J Laryngol Otol. 2007;121:530-534.
3. Fisher CG, Kacica MA, Bennett NM. Risk factors for cartilage infections of the ear. Am J Prev Med. 2005;29:204-209.
4. Lee TC, Gold WL. Necrotizing Pseudomonas chondritis after piercing of the upper ear. CMAJ. 2011;183:819-821.
5. Rowshan HH, Keith K, Baur D, et al. Pseudomonas aeruginosa infection of the auricular cartilage caused by “high ear piercing”: a case report and review of the literature. J Oral Maxillofac Surg. 2008;66:543-546.
6. Liu ZW, Chokkalingam P. Piercing associated perichondritis of the pinna: are we treating it correctly? J Laryngol Otol. 2013;127:505-508.
1. Sandhu A, Gross M, Wylie J, et al. Pseudomonas aeruginosa necrotizing chondritis complicating high helical ear piercing case report: clinical and public health perspectives. Can J Public Health. 2007;98:74-77.
2. Prasad HK, Sreedharan S, Prasad HS, et al. Perichondritis of the auricle and its management. J Laryngol Otol. 2007;121:530-534.
3. Fisher CG, Kacica MA, Bennett NM. Risk factors for cartilage infections of the ear. Am J Prev Med. 2005;29:204-209.
4. Lee TC, Gold WL. Necrotizing Pseudomonas chondritis after piercing of the upper ear. CMAJ. 2011;183:819-821.
5. Rowshan HH, Keith K, Baur D, et al. Pseudomonas aeruginosa infection of the auricular cartilage caused by “high ear piercing”: a case report and review of the literature. J Oral Maxillofac Surg. 2008;66:543-546.
6. Liu ZW, Chokkalingam P. Piercing associated perichondritis of the pinna: are we treating it correctly? J Laryngol Otol. 2013;127:505-508.
