User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Photoallergic Contact Dermatitis: No Fun in the Sun
Photoallergic contact dermatitis (PACD), a subtype of allergic contact dermatitis that occurs because of the specific combination of exposure to an exogenous chemical applied topically to the skin and UV radiation, may be more common than was once thought.1 Although the incidence in the general population is unknown, current research points to approximately 20% to 40% of patients with suspected photosensitivity having a PACD diagnosis.2 Recently, the North American Contact Dermatitis Group (NACDG) reported that 21% of 373 patients undergoing photopatch testing (PPT) were diagnosed with PACD2; however, PPT is not routinely performed, which may contribute to underdiagnosis.
Mechanism of Disease
Similar to allergic contact dermatitis, PACD is a delayed type IV hypersensitivity reaction; however, it only occurs when an exogenous chemical is applied topically to the skin with concomitant exposure to UV radiation, usually in the UVA range (315–400 nm).3,4 When exposed to UV radiation, it is thought that the exogenous chemical combines with a protein in the skin and transforms into a photoantigen. In the sensitization phase, the photoantigen is taken up by antigen-presenting cells in the epidermis and transported to local lymph nodes where antigen-specific T cells are generated.5 In the elicitation phase, the inflammatory reaction of PACD occurs upon subsequent exposure to the same chemical plus UV radiation.4 Development of PACD does not necessarily depend on the dose of the chemical or the amount of UV radiation.6 Why certain individuals may be more susceptible is unknown, though major histocompatibility complex haplotypes could be influential.7,8
Clinical Manifestations
Photoallergic contact dermatitis primarily presents in sun-exposed areas of the skin (eg, face, neck, V area of the chest, dorsal upper extremities) with sparing of naturally photoprotected sites, such as the upper eyelids and nasolabial and retroauricular folds. Other than its characteristic photodistribution, PACD often is clinically indistinguishable from routine allergic contact dermatitis. It manifests as a pruritic, poorly demarcated, eczematous or sometimes vesiculobullous eruption that develops in a delayed fashion—24 to 72 hours after sun exposure. The dermatitis may extend to other parts of the body either through spread of the chemical agent by the hands or clothing or due to the systemic nature of the immune response. The severity of the presentation can vary depending on multiple factors, such as concentration and absorption of the agent, length of exposure, intensity and duration of UV radiation exposure, and individual susceptibility.4 Chronic PACD may become lichenified. Generally, rashes resolve after discontinuation of the causative agent; however, long-term exposure may lead to development of chronic actinic dermatitis, with persistent photodistributed eczema regardless of contact with the initial inciting agent.9
Differential Diagnosis
The differential diagnosis for patients presenting with photodistributed dermatitis is broad; therefore, taking a thorough history is important. Considerations include age of onset, timing and persistence of reactions, use of topical and systemic medications (both prescription and over-the-counter [OTC]), personal care products, occupation, and hobbies, as well as a thorough review of systems.
It is important to distinguish PACD from phototoxic contact dermatitis (PTCD)(also known as photoirritant contact dermatitis)(Table). Asking about the onset and timing of the eruption may be critical for distinction, as PTCD can occur within minutes to hours of the first exposure to a chemical and UV radiation, while there is a sensitization delay in PACD.6 Phytophotodermatitis is a well-known type of PTCD caused by exposure to furocoumarin-containing plants, most commonly limes.10 Other causes of PTCD include tar products and certain medications.11 Importantly, PPT to a known phototoxic chemical should never be performed because it will cause a strong reaction in anyone tested, regardless of exposure history.

Other diagnoses to consider include photoaggravated dermatoses (eg, atopic dermatitis, lupus erythematosus, dermatomyositis) and idiopathic photodermatoses (eg, chronic actinic dermatitis, actinic prurigo, polymorphous light eruption). Although atopic dermatitis usually improves with UV light exposure, photoaggravated atopic dermatitis is suggested in eczema patients who flare with sun exposure, in a seasonal pattern, or after phototherapy; this condition is challenging to differentiate from PACD if PPT is not performed.12 The diagnosis of idiopathic photodermatoses is nuanced; however, asking about the timeline of the reaction including onset, duration, and persistence, as well as characterization of unique clinical features, can help in differentiation.13 In certain scenarios, a biopsy may be helpful. A thorough review of systems will help to assess for autoimmune connective tissue disorders, and relevant serologies should be checked as indicated.
Diagnosis
Histologically, PACD presents similarly to allergic contact dermatitis with spongiotic dermatitis; therefore, biopsy cannot be relied upon to make the diagnosis.6 Photopatch testing is required for definitive diagnosis. It is reasonable to perform PPT in any patient with chronic dermatitis primarily affecting sun-exposed areas without a clear alternative diagnosis.14,15 Of note, at present there are no North American consensus guidelines for PPT, but typically duplicate sets of photoallergens are applied to both sides of the patient’s back and one side is exposed to UVA radiation. The reactions are compared after 48 to 96 hours.15 A positive reaction only at the irradiated site is consistent with photoallergy, while a reaction of equal strength at both the irradiated and nonirradiated sites indicates regular contact allergy. The case of a reaction occurring at both sites with a stronger response at the irradiated site is known as photoaggravated contact allergy, which can be thought of as allergic contact dermatitis that worsens but does not solely occur with exposure to sunlight.
Although PPT is necessary for the accurate diagnosis of PACD, it is infrequently used. Two surveys of 112 and 117 American Contact Dermatitis Society members, respectively, have revealed that only around half performed PPT, most of them testing fewer than 20 times per year.16,17 Additionally, there was variability in the test methodology and allergens employed. Nevertheless, most respondents tested sunscreens, nonsteroidal anti-inflammatory drugs (NSAIDs), fragrances, and their patients’ own products.16,17 The most common reasons for not performing PPT were lack of equipment, insufficient skills, rare clinical suspicion, and cost. Dermatologists at academic centers performed more PPT than those in other practice settings, including multispecialty group practices and private offices.16 These findings highlight multiple factors that may contribute to reduced patient access to PPT and thus potential underdiagnosis of PACD.
Common Photoallergens
The most common photoallergens change over time in response to market trends; for example, fragrance was once a top photoallergen in the United States in the 1970s and 1980s but declined in prominence after musk ambrette—the primary allergen associated with PACD at the time—was removed as an ingredient in fragrances.18
In the largest and most recent PPT series from North America (1999-2009),2 sunscreens comprised 7 of the top 10 most common photoallergens, which is consistent with other studies showing sunscreens to be the most common North American photoallergens.19-22 The frequency of PACD due to sunscreens likely relates to their increasing use worldwide as awareness of photocarcinogenesis and photoaging grows, as well as the common use of UV filters in nonsunscreen personal care products, ranging from lip balms to perfumes and bodywashes. Chemical (organic) UV filters—in particular oxybenzone (benzophenone-3) and avobenzone (butyl methoxydibenzoylmethane)—are the most common sunscreen photoallergens.2,23 Para-aminobenzoic acid was once a common photoallergen, but it is no longer used in US sunscreens due to safety concerns.19,20 The physical (inorganic) UV filters zinc oxide and titanium dioxide are not known photosensitizers.
Methylisothiazolinone (MI) is a highly allergenic preservative commonly used in a wide array of personal care products, including sunscreens.24 In the most recent NACDG patch test data, MI was the second most common contact allergen.25 Allergic contact dermatitis caused by MI in sunscreen can mimic PACD.26 In addition, MI can cause photoaggravated contact dermatitis, with some affected patients experiencing ongoing photosensitivity even after avoiding this allergen.26-30 The European Union and Canada have introduced restrictions on the use of MI in personal care products, but no such regulatory measures have been taken in the United States to date.25,31,32
After sunscreens, another common cause of PACD are topical NSAIDs, which are frequently used for musculoskeletal pain relief. These are of particular concern in Europe, where a variety of formulations are widely available OTC.33 Ketoprofen and etofenamate are responsible for the largest number of PACD reactions in Europe.2,34,35 Meanwhile, the only OTC topical NSAID available in the United States is diclofenac gel, which was approved in 2020. Cases of PACD due to use of diclofenac gel have been reported in the literature, but testing in larger populations is needed.36-39
Notably, ketoprofen may co- or cross-react with certain UV filters—oxybenzone and octocrylene—and the lipid-lowering agent fenofibrate due to chemical similarities.40-43 Despite the relatively high number of photoallergic reactions to ketoprofen in the NACDG photopatch series, only 25% (5/20) were considered clinically relevant (ie, the allergen could not be verified as present in the known skin contactants of the patient, and the patient was not exposed to circumstances in which contact with materials known to contain the allergen would likely occur), which suggests that they likely represented cross-reactions in patients sensitized to sunscreens.2
Other agents that may cause PACD include antimicrobials, plants and plant derivatives, and pesticides.2,4,18 The antimicrobial fentichlor is a common cause of positive PPT reactions, but it rarely is clinically relevant.44
Treatment
The primary management of PACD centers on identification of the causative photoallergen to avoid future exposure. Patients should be educated on the various names by which the causative allergen can be identified on product labels and should be given a list of safe products that are free from relevant allergens and cross-reacting chemicals.45 Additionally, sun protection education should be provided. Exposure to UVA radiation can occur through windows, making the use of broad-spectrum sunscreens and protective clothing crucial. In cases of sunscreen-induced PACD, the responsible chemical UV filter(s) should be avoided, or alternatively, patients may use physical sunscreens containing only zinc oxide and/or titanium dioxide as active ingredients, as these are not known to cause PACD.4
When avoidance alone is insufficient, topical corticosteroids are the usual first-line treatment for localized PACD. When steroid-sparing treatments are preferred, topical calcineurin inhibitors such as tacrolimus and pimecrolimus may be used. If PACD is more widespread and severe, systemic therapy using steroids or steroid-sparing agents may be necessary to provide symptomatic relief.4
Final Interpretation
Photoallergic contact dermatitis is not uncommon, particularly among photosensitive patients. Most cases are due to sunscreens or topical NSAIDs. Consideration of PPT should be given in any patient with a chronic photodistributed dermatitis to evaluate for the possibility of PACD.
- Darvay A, White IR, Rycroft RJ, et al. Photoallergic contact dermatitis is uncommon. Br J Dermatol. 2001;145:597-601.
- DeLeo VA, Adler BL, Warshaw EM, et al. Photopatch test results of the North American contact dermatitis group, 1999-2009. Photodermatol Photoimmunol Photomed. 2022;38:288-291.
- Kerr A, Ferguson J. Photoallergic contact dermatitis. Photodermatol Photoimmunol Photomed. 2010;26:56-65.
- As¸kın Ö, Cesur SK, Engin B, et al. Photoallergic contact dermatitis. Curr Derm Rep. 2019;8:157-163.
- Wilm A, Berneburg M. Photoallergy. J Dtsch Dermatol Ges. 2015;13:7-13.
- DeLeo VA. Photocontact dermatitis. Dermatol Ther. 2004;17:279-288.
- Imai S, Atarashi K, Ikesue K, et al. Establishment of murine model of allergic photocontact dermatitis to ketoprofen and characterization of pathogenic T cells. J Dermatol Sci. 2006;41:127-136.
- Tokura Y, Yagi H, Satoh T, et al. Inhibitory effect of melanin pigment on sensitization and elicitation of murine contact photosensitivity: mechanism of low responsiveness in C57BL/10 background mice. J Invest Dermatol. 1993;101:673-678.
- Stein KR, Scheinfeld NS. Drug-induced photoallergic and phototoxic reactions. Expert Opin Drug Saf. 2007;6:431-443.
- Janusz SC, Schwartz RA. Botanical briefs: phytophotodermatitis is an occupational and recreational dermatosis in the limelight. Cutis. 2021;107:187-189.
- Atwal SK, Chen A, Adler BL. Phototoxic contact dermatitis from over-the-counter 8-methoxypsoralen. Cutis. 2022;109:E2-E3.
- Rutter KJ, Farrar MD, Marjanovic EJ, et al. Clinicophotobiological characterization of photoaggravated atopic dermatitis [published online July 27, 2022]. JAMA Dermatol. doi:10.1001/jamadermatol.2022.2823
- Lecha M. Idiopathic photodermatoses: clinical, diagnostic and therapeutic aspects. J Eur Acad Dermatol Venereol. 2001;15:499-505.
- Marks JG Jr, Anderson BE, DeLeo VA. Contact & Occupational Dermatology. 4th ed. Jaypee Brothers; 2016.
- Bruynzeel DP, Ferguson J, Andersen K, et al. Photopatch testing: a consensus methodology for Europe. J Eur Acad Dermatol Venereol. 2004;18:679-682.
- Kim T, Taylor JS, Maibach HI, et al. Photopatch testing among members of the American Contact Dermatitis Society. Dermatitis. 2020;31:59-67.
- Asemota E, Crawford G, Kovarik C, et al. A survey examining photopatch test and phototest methodologies of contact dermatologists in the United States: platform for developing a consensus. Dermatitis. 2017;28:265-269.
- Scalf LA, Davis MD, Rohlinger AL, et al. Photopatch testing of 182 patients: a 6-year experience at the Mayo Clinic. Dermatitis. 2009;20:44-52.
- Greenspoon J, Ahluwalia R, Juma N, et al. Allergic and photoallergic contact dermatitis: a 10-year experience. Dermatitis. 2013;24:29-32.
- Victor FC, Cohen DE, Soter NA. A 20-year analysis of previous and emerging allergens that elicit photoallergic contact dermatitis. J Am Acad Dermatol. 2010;62:605-610.
- Schauder S, Ippen H. Contact and photocontact sensitivity to sunscreens. review of a 15-year experience and of the literature. Contact Dermatitis. 1997;37:221-232.
- Collaris EJ, Frank J. Photoallergic contact dermatitis caused by ultraviolet filters in different sunscreens. Int J Dermatol. 2008;47(suppl 1):35-37.
- Heurung AR, Raju SI, Warshaw EM. Adverse reactions to sunscreen agents: epidemiology, responsible irritants and allergens, clinical characteristics, and management. Dermatitis. 2014;25:289-326.
- Reeder M, Atwater AR. Methylisothiazolinone and isothiazolinone allergy. Cutis. 2019;104:94-96.
- DeKoven JG, Silverberg JI, Warshaw EM, et al. North American Contact Dermatitis Group Patch Test Results: 2017-2018. Dermatitis. 2021;32:111-123.
- Kullberg SA, Voller LM, Warshaw EM. Methylisothiazolinone in “dermatology-recommended” sunscreens: an important mimicker of photoallergic contact dermatitis. Photodermatol Photoimmunol Photomed. 2021;37:366-370.
- Herman A, Aerts O, de Montjoye L, et al. Isothiazolinone derivatives and allergic contact dermatitis: a review and update. J Eur Acad Dermatol Venereol. 2019;33:267-276.
- Adler BL, Houle MC, Pratt M. Photoaggravated contact dermatitis to methylisothiazolinone and associated photosensitivity: a case series [published online January 25, 2022]. Dermatitis. doi:10.1097/DER.0000000000000833
- Aerts O, Goossens A, Marguery MC, et al. Photoaggravated allergic contact dermatitis and transient photosensitivity caused by methylisothiazolinone. Contact Dermatitis. 2018;78:241-245.
- Pirmez R, Fernandes AL, Melo MG. Photoaggravated contact dermatitis to Kathon CG (methylchloroisothiazolinone/methylisothiazolinone): a novel pattern of involvement in a growing epidemic?. Br J Dermatol. 2015;173:1343-1344.
- Uter W, Aalto-Korte K, Agner T, et al. The epidemic of methylisothiazolinone contact allergy in Europe: follow-up on changing exposures.J Eur Acad Dermatol Venereol. 2020;34:333-339.
- Government of Canada. Changes to the cosmetic ingredient hotlist. December 3, 2019. Updated August 26, 2022. Accessed October 20, 2022. https://www.canada.ca/en/health-canada/services/consumer-product-safety/cosmetics/cosmetic-ingredient-hotlist-prohibited-restricted-ingredients/changes.html
- Barkin RL. Topical nonsteroidal anti-inflammatory drugs: the importance of drug, delivery, and therapeutic outcome. Am J Ther. 2015;22:388-407.
- European Multicentre Photopatch Test Study (EMCPPTS) Taskforce. A European multicentre photopatch test study. Br J Dermatol. 2012;166:1002-1009.
- Ophaswongse S, Maibach H. Topical nonsteroidal antiinflammatory drugs: allergic and photoallergic contact dermatitis and phototoxicity. Contact Dermatitis. 1993;29:57-64.
- Kowalzick L, Ziegler H. Photoallergic contact dermatitis from topical diclofenac in Solaraze gel. Contact Dermatitis. 2006;54:348-349.
- Montoro J, Rodríguez M, Díaz M, et al. Photoallergic contact dermatitis due to diclofenac. Contact Dermatitis. 2003;48:115.
- Fernández-Jorge B, Goday-Buján JJ, Murga M, et al. Photoallergic contact dermatitis due to diclofenac with cross-reaction to aceclofenac: two case reports. Contact Dermatitis. 2009;61:236-237.
- Akat PB. Severe photosensitivity reaction induced by topical diclofenac. Indian J Pharmacol. 2013;45:408-409.
- Leroy D, Dompmartin A, Szczurko C, et al. Photodermatitis from ketoprofen with cross-reactivity to fenofibrate and benzophenones. Photodermatol Photoimmunol Photomed. 1997;13:93-97.
- Devleeschouwer V, Roelandts R, Garmyn M, et al. Allergic and photoallergic contact dermatitis from ketoprofen: results of (photo) patch testing and follow-up of 42 patients. Contact Dermatitis. 2008;58:159-166.
- Matsushita T, Kamide R. Five cases of photocontact dermatitisdue to topical ketoprofen: photopatch testing and cross-reaction study. Photodermatol Photoimmunol Photomed. 2001;17:26-31.
- de Groot AC, Roberts DW. Contact and photocontact allergy to octocrylene: a review. Contact Dermatitis. 2014;70:193-204.
- Wolverton JE, Soter NA, Cohen DE. Fentichlor photocontact dermatitis: a persistent enigma. Dermatitis. 2013;24:77-81.
- Mowad CM, Anderson B, Scheinman P, et al. Allergic contact dermatitis: patient management and education. J Am Acad Dermatol. 2016;74:1043-1054.
Photoallergic contact dermatitis (PACD), a subtype of allergic contact dermatitis that occurs because of the specific combination of exposure to an exogenous chemical applied topically to the skin and UV radiation, may be more common than was once thought.1 Although the incidence in the general population is unknown, current research points to approximately 20% to 40% of patients with suspected photosensitivity having a PACD diagnosis.2 Recently, the North American Contact Dermatitis Group (NACDG) reported that 21% of 373 patients undergoing photopatch testing (PPT) were diagnosed with PACD2; however, PPT is not routinely performed, which may contribute to underdiagnosis.
Mechanism of Disease
Similar to allergic contact dermatitis, PACD is a delayed type IV hypersensitivity reaction; however, it only occurs when an exogenous chemical is applied topically to the skin with concomitant exposure to UV radiation, usually in the UVA range (315–400 nm).3,4 When exposed to UV radiation, it is thought that the exogenous chemical combines with a protein in the skin and transforms into a photoantigen. In the sensitization phase, the photoantigen is taken up by antigen-presenting cells in the epidermis and transported to local lymph nodes where antigen-specific T cells are generated.5 In the elicitation phase, the inflammatory reaction of PACD occurs upon subsequent exposure to the same chemical plus UV radiation.4 Development of PACD does not necessarily depend on the dose of the chemical or the amount of UV radiation.6 Why certain individuals may be more susceptible is unknown, though major histocompatibility complex haplotypes could be influential.7,8
Clinical Manifestations
Photoallergic contact dermatitis primarily presents in sun-exposed areas of the skin (eg, face, neck, V area of the chest, dorsal upper extremities) with sparing of naturally photoprotected sites, such as the upper eyelids and nasolabial and retroauricular folds. Other than its characteristic photodistribution, PACD often is clinically indistinguishable from routine allergic contact dermatitis. It manifests as a pruritic, poorly demarcated, eczematous or sometimes vesiculobullous eruption that develops in a delayed fashion—24 to 72 hours after sun exposure. The dermatitis may extend to other parts of the body either through spread of the chemical agent by the hands or clothing or due to the systemic nature of the immune response. The severity of the presentation can vary depending on multiple factors, such as concentration and absorption of the agent, length of exposure, intensity and duration of UV radiation exposure, and individual susceptibility.4 Chronic PACD may become lichenified. Generally, rashes resolve after discontinuation of the causative agent; however, long-term exposure may lead to development of chronic actinic dermatitis, with persistent photodistributed eczema regardless of contact with the initial inciting agent.9
Differential Diagnosis
The differential diagnosis for patients presenting with photodistributed dermatitis is broad; therefore, taking a thorough history is important. Considerations include age of onset, timing and persistence of reactions, use of topical and systemic medications (both prescription and over-the-counter [OTC]), personal care products, occupation, and hobbies, as well as a thorough review of systems.
It is important to distinguish PACD from phototoxic contact dermatitis (PTCD)(also known as photoirritant contact dermatitis)(Table). Asking about the onset and timing of the eruption may be critical for distinction, as PTCD can occur within minutes to hours of the first exposure to a chemical and UV radiation, while there is a sensitization delay in PACD.6 Phytophotodermatitis is a well-known type of PTCD caused by exposure to furocoumarin-containing plants, most commonly limes.10 Other causes of PTCD include tar products and certain medications.11 Importantly, PPT to a known phototoxic chemical should never be performed because it will cause a strong reaction in anyone tested, regardless of exposure history.
Other diagnoses to consider include photoaggravated dermatoses (eg, atopic dermatitis, lupus erythematosus, dermatomyositis) and idiopathic photodermatoses (eg, chronic actinic dermatitis, actinic prurigo, polymorphous light eruption). Although atopic dermatitis usually improves with UV light exposure, photoaggravated atopic dermatitis is suggested in eczema patients who flare with sun exposure, in a seasonal pattern, or after phototherapy; this condition is challenging to differentiate from PACD if PPT is not performed.12 The diagnosis of idiopathic photodermatoses is nuanced; however, asking about the timeline of the reaction including onset, duration, and persistence, as well as characterization of unique clinical features, can help in differentiation.13 In certain scenarios, a biopsy may be helpful. A thorough review of systems will help to assess for autoimmune connective tissue disorders, and relevant serologies should be checked as indicated.
Diagnosis
Histologically, PACD presents similarly to allergic contact dermatitis with spongiotic dermatitis; therefore, biopsy cannot be relied upon to make the diagnosis.6 Photopatch testing is required for definitive diagnosis. It is reasonable to perform PPT in any patient with chronic dermatitis primarily affecting sun-exposed areas without a clear alternative diagnosis.14,15 Of note, at present there are no North American consensus guidelines for PPT, but typically duplicate sets of photoallergens are applied to both sides of the patient’s back and one side is exposed to UVA radiation. The reactions are compared after 48 to 96 hours.15 A positive reaction only at the irradiated site is consistent with photoallergy, while a reaction of equal strength at both the irradiated and nonirradiated sites indicates regular contact allergy. The case of a reaction occurring at both sites with a stronger response at the irradiated site is known as photoaggravated contact allergy, which can be thought of as allergic contact dermatitis that worsens but does not solely occur with exposure to sunlight.
Although PPT is necessary for the accurate diagnosis of PACD, it is infrequently used. Two surveys of 112 and 117 American Contact Dermatitis Society members, respectively, have revealed that only around half performed PPT, most of them testing fewer than 20 times per year.16,17 Additionally, there was variability in the test methodology and allergens employed. Nevertheless, most respondents tested sunscreens, nonsteroidal anti-inflammatory drugs (NSAIDs), fragrances, and their patients’ own products.16,17 The most common reasons for not performing PPT were lack of equipment, insufficient skills, rare clinical suspicion, and cost. Dermatologists at academic centers performed more PPT than those in other practice settings, including multispecialty group practices and private offices.16 These findings highlight multiple factors that may contribute to reduced patient access to PPT and thus potential underdiagnosis of PACD.
Common Photoallergens
The most common photoallergens change over time in response to market trends; for example, fragrance was once a top photoallergen in the United States in the 1970s and 1980s but declined in prominence after musk ambrette—the primary allergen associated with PACD at the time—was removed as an ingredient in fragrances.18
In the largest and most recent PPT series from North America (1999-2009),2 sunscreens comprised 7 of the top 10 most common photoallergens, which is consistent with other studies showing sunscreens to be the most common North American photoallergens.19-22 The frequency of PACD due to sunscreens likely relates to their increasing use worldwide as awareness of photocarcinogenesis and photoaging grows, as well as the common use of UV filters in nonsunscreen personal care products, ranging from lip balms to perfumes and bodywashes. Chemical (organic) UV filters—in particular oxybenzone (benzophenone-3) and avobenzone (butyl methoxydibenzoylmethane)—are the most common sunscreen photoallergens.2,23 Para-aminobenzoic acid was once a common photoallergen, but it is no longer used in US sunscreens due to safety concerns.19,20 The physical (inorganic) UV filters zinc oxide and titanium dioxide are not known photosensitizers.
Methylisothiazolinone (MI) is a highly allergenic preservative commonly used in a wide array of personal care products, including sunscreens.24 In the most recent NACDG patch test data, MI was the second most common contact allergen.25 Allergic contact dermatitis caused by MI in sunscreen can mimic PACD.26 In addition, MI can cause photoaggravated contact dermatitis, with some affected patients experiencing ongoing photosensitivity even after avoiding this allergen.26-30 The European Union and Canada have introduced restrictions on the use of MI in personal care products, but no such regulatory measures have been taken in the United States to date.25,31,32
After sunscreens, another common cause of PACD are topical NSAIDs, which are frequently used for musculoskeletal pain relief. These are of particular concern in Europe, where a variety of formulations are widely available OTC.33 Ketoprofen and etofenamate are responsible for the largest number of PACD reactions in Europe.2,34,35 Meanwhile, the only OTC topical NSAID available in the United States is diclofenac gel, which was approved in 2020. Cases of PACD due to use of diclofenac gel have been reported in the literature, but testing in larger populations is needed.36-39
Notably, ketoprofen may co- or cross-react with certain UV filters—oxybenzone and octocrylene—and the lipid-lowering agent fenofibrate due to chemical similarities.40-43 Despite the relatively high number of photoallergic reactions to ketoprofen in the NACDG photopatch series, only 25% (5/20) were considered clinically relevant (ie, the allergen could not be verified as present in the known skin contactants of the patient, and the patient was not exposed to circumstances in which contact with materials known to contain the allergen would likely occur), which suggests that they likely represented cross-reactions in patients sensitized to sunscreens.2
Other agents that may cause PACD include antimicrobials, plants and plant derivatives, and pesticides.2,4,18 The antimicrobial fentichlor is a common cause of positive PPT reactions, but it rarely is clinically relevant.44
Treatment
The primary management of PACD centers on identification of the causative photoallergen to avoid future exposure. Patients should be educated on the various names by which the causative allergen can be identified on product labels and should be given a list of safe products that are free from relevant allergens and cross-reacting chemicals.45 Additionally, sun protection education should be provided. Exposure to UVA radiation can occur through windows, making the use of broad-spectrum sunscreens and protective clothing crucial. In cases of sunscreen-induced PACD, the responsible chemical UV filter(s) should be avoided, or alternatively, patients may use physical sunscreens containing only zinc oxide and/or titanium dioxide as active ingredients, as these are not known to cause PACD.4
When avoidance alone is insufficient, topical corticosteroids are the usual first-line treatment for localized PACD. When steroid-sparing treatments are preferred, topical calcineurin inhibitors such as tacrolimus and pimecrolimus may be used. If PACD is more widespread and severe, systemic therapy using steroids or steroid-sparing agents may be necessary to provide symptomatic relief.4
Final Interpretation
Photoallergic contact dermatitis is not uncommon, particularly among photosensitive patients. Most cases are due to sunscreens or topical NSAIDs. Consideration of PPT should be given in any patient with a chronic photodistributed dermatitis to evaluate for the possibility of PACD.
Photoallergic contact dermatitis (PACD), a subtype of allergic contact dermatitis that occurs because of the specific combination of exposure to an exogenous chemical applied topically to the skin and UV radiation, may be more common than was once thought.1 Although the incidence in the general population is unknown, current research points to approximately 20% to 40% of patients with suspected photosensitivity having a PACD diagnosis.2 Recently, the North American Contact Dermatitis Group (NACDG) reported that 21% of 373 patients undergoing photopatch testing (PPT) were diagnosed with PACD2; however, PPT is not routinely performed, which may contribute to underdiagnosis.
Mechanism of Disease
Similar to allergic contact dermatitis, PACD is a delayed type IV hypersensitivity reaction; however, it only occurs when an exogenous chemical is applied topically to the skin with concomitant exposure to UV radiation, usually in the UVA range (315–400 nm).3,4 When exposed to UV radiation, it is thought that the exogenous chemical combines with a protein in the skin and transforms into a photoantigen. In the sensitization phase, the photoantigen is taken up by antigen-presenting cells in the epidermis and transported to local lymph nodes where antigen-specific T cells are generated.5 In the elicitation phase, the inflammatory reaction of PACD occurs upon subsequent exposure to the same chemical plus UV radiation.4 Development of PACD does not necessarily depend on the dose of the chemical or the amount of UV radiation.6 Why certain individuals may be more susceptible is unknown, though major histocompatibility complex haplotypes could be influential.7,8
Clinical Manifestations
Photoallergic contact dermatitis primarily presents in sun-exposed areas of the skin (eg, face, neck, V area of the chest, dorsal upper extremities) with sparing of naturally photoprotected sites, such as the upper eyelids and nasolabial and retroauricular folds. Other than its characteristic photodistribution, PACD often is clinically indistinguishable from routine allergic contact dermatitis. It manifests as a pruritic, poorly demarcated, eczematous or sometimes vesiculobullous eruption that develops in a delayed fashion—24 to 72 hours after sun exposure. The dermatitis may extend to other parts of the body either through spread of the chemical agent by the hands or clothing or due to the systemic nature of the immune response. The severity of the presentation can vary depending on multiple factors, such as concentration and absorption of the agent, length of exposure, intensity and duration of UV radiation exposure, and individual susceptibility.4 Chronic PACD may become lichenified. Generally, rashes resolve after discontinuation of the causative agent; however, long-term exposure may lead to development of chronic actinic dermatitis, with persistent photodistributed eczema regardless of contact with the initial inciting agent.9
Differential Diagnosis
The differential diagnosis for patients presenting with photodistributed dermatitis is broad; therefore, taking a thorough history is important. Considerations include age of onset, timing and persistence of reactions, use of topical and systemic medications (both prescription and over-the-counter [OTC]), personal care products, occupation, and hobbies, as well as a thorough review of systems.
It is important to distinguish PACD from phototoxic contact dermatitis (PTCD)(also known as photoirritant contact dermatitis)(Table). Asking about the onset and timing of the eruption may be critical for distinction, as PTCD can occur within minutes to hours of the first exposure to a chemical and UV radiation, while there is a sensitization delay in PACD.6 Phytophotodermatitis is a well-known type of PTCD caused by exposure to furocoumarin-containing plants, most commonly limes.10 Other causes of PTCD include tar products and certain medications.11 Importantly, PPT to a known phototoxic chemical should never be performed because it will cause a strong reaction in anyone tested, regardless of exposure history.
Other diagnoses to consider include photoaggravated dermatoses (eg, atopic dermatitis, lupus erythematosus, dermatomyositis) and idiopathic photodermatoses (eg, chronic actinic dermatitis, actinic prurigo, polymorphous light eruption). Although atopic dermatitis usually improves with UV light exposure, photoaggravated atopic dermatitis is suggested in eczema patients who flare with sun exposure, in a seasonal pattern, or after phototherapy; this condition is challenging to differentiate from PACD if PPT is not performed.12 The diagnosis of idiopathic photodermatoses is nuanced; however, asking about the timeline of the reaction including onset, duration, and persistence, as well as characterization of unique clinical features, can help in differentiation.13 In certain scenarios, a biopsy may be helpful. A thorough review of systems will help to assess for autoimmune connective tissue disorders, and relevant serologies should be checked as indicated.
Diagnosis
Histologically, PACD presents similarly to allergic contact dermatitis with spongiotic dermatitis; therefore, biopsy cannot be relied upon to make the diagnosis.6 Photopatch testing is required for definitive diagnosis. It is reasonable to perform PPT in any patient with chronic dermatitis primarily affecting sun-exposed areas without a clear alternative diagnosis.14,15 Of note, at present there are no North American consensus guidelines for PPT, but typically duplicate sets of photoallergens are applied to both sides of the patient’s back and one side is exposed to UVA radiation. The reactions are compared after 48 to 96 hours.15 A positive reaction only at the irradiated site is consistent with photoallergy, while a reaction of equal strength at both the irradiated and nonirradiated sites indicates regular contact allergy. The case of a reaction occurring at both sites with a stronger response at the irradiated site is known as photoaggravated contact allergy, which can be thought of as allergic contact dermatitis that worsens but does not solely occur with exposure to sunlight.
Although PPT is necessary for the accurate diagnosis of PACD, it is infrequently used. Two surveys of 112 and 117 American Contact Dermatitis Society members, respectively, have revealed that only around half performed PPT, most of them testing fewer than 20 times per year.16,17 Additionally, there was variability in the test methodology and allergens employed. Nevertheless, most respondents tested sunscreens, nonsteroidal anti-inflammatory drugs (NSAIDs), fragrances, and their patients’ own products.16,17 The most common reasons for not performing PPT were lack of equipment, insufficient skills, rare clinical suspicion, and cost. Dermatologists at academic centers performed more PPT than those in other practice settings, including multispecialty group practices and private offices.16 These findings highlight multiple factors that may contribute to reduced patient access to PPT and thus potential underdiagnosis of PACD.
Common Photoallergens
The most common photoallergens change over time in response to market trends; for example, fragrance was once a top photoallergen in the United States in the 1970s and 1980s but declined in prominence after musk ambrette—the primary allergen associated with PACD at the time—was removed as an ingredient in fragrances.18
In the largest and most recent PPT series from North America (1999-2009),2 sunscreens comprised 7 of the top 10 most common photoallergens, which is consistent with other studies showing sunscreens to be the most common North American photoallergens.19-22 The frequency of PACD due to sunscreens likely relates to their increasing use worldwide as awareness of photocarcinogenesis and photoaging grows, as well as the common use of UV filters in nonsunscreen personal care products, ranging from lip balms to perfumes and bodywashes. Chemical (organic) UV filters—in particular oxybenzone (benzophenone-3) and avobenzone (butyl methoxydibenzoylmethane)—are the most common sunscreen photoallergens.2,23 Para-aminobenzoic acid was once a common photoallergen, but it is no longer used in US sunscreens due to safety concerns.19,20 The physical (inorganic) UV filters zinc oxide and titanium dioxide are not known photosensitizers.
Methylisothiazolinone (MI) is a highly allergenic preservative commonly used in a wide array of personal care products, including sunscreens.24 In the most recent NACDG patch test data, MI was the second most common contact allergen.25 Allergic contact dermatitis caused by MI in sunscreen can mimic PACD.26 In addition, MI can cause photoaggravated contact dermatitis, with some affected patients experiencing ongoing photosensitivity even after avoiding this allergen.26-30 The European Union and Canada have introduced restrictions on the use of MI in personal care products, but no such regulatory measures have been taken in the United States to date.25,31,32
After sunscreens, another common cause of PACD are topical NSAIDs, which are frequently used for musculoskeletal pain relief. These are of particular concern in Europe, where a variety of formulations are widely available OTC.33 Ketoprofen and etofenamate are responsible for the largest number of PACD reactions in Europe.2,34,35 Meanwhile, the only OTC topical NSAID available in the United States is diclofenac gel, which was approved in 2020. Cases of PACD due to use of diclofenac gel have been reported in the literature, but testing in larger populations is needed.36-39
Notably, ketoprofen may co- or cross-react with certain UV filters—oxybenzone and octocrylene—and the lipid-lowering agent fenofibrate due to chemical similarities.40-43 Despite the relatively high number of photoallergic reactions to ketoprofen in the NACDG photopatch series, only 25% (5/20) were considered clinically relevant (ie, the allergen could not be verified as present in the known skin contactants of the patient, and the patient was not exposed to circumstances in which contact with materials known to contain the allergen would likely occur), which suggests that they likely represented cross-reactions in patients sensitized to sunscreens.2
Other agents that may cause PACD include antimicrobials, plants and plant derivatives, and pesticides.2,4,18 The antimicrobial fentichlor is a common cause of positive PPT reactions, but it rarely is clinically relevant.44
Treatment
The primary management of PACD centers on identification of the causative photoallergen to avoid future exposure. Patients should be educated on the various names by which the causative allergen can be identified on product labels and should be given a list of safe products that are free from relevant allergens and cross-reacting chemicals.45 Additionally, sun protection education should be provided. Exposure to UVA radiation can occur through windows, making the use of broad-spectrum sunscreens and protective clothing crucial. In cases of sunscreen-induced PACD, the responsible chemical UV filter(s) should be avoided, or alternatively, patients may use physical sunscreens containing only zinc oxide and/or titanium dioxide as active ingredients, as these are not known to cause PACD.4
When avoidance alone is insufficient, topical corticosteroids are the usual first-line treatment for localized PACD. When steroid-sparing treatments are preferred, topical calcineurin inhibitors such as tacrolimus and pimecrolimus may be used. If PACD is more widespread and severe, systemic therapy using steroids or steroid-sparing agents may be necessary to provide symptomatic relief.4
Final Interpretation
Photoallergic contact dermatitis is not uncommon, particularly among photosensitive patients. Most cases are due to sunscreens or topical NSAIDs. Consideration of PPT should be given in any patient with a chronic photodistributed dermatitis to evaluate for the possibility of PACD.
- Darvay A, White IR, Rycroft RJ, et al. Photoallergic contact dermatitis is uncommon. Br J Dermatol. 2001;145:597-601.
- DeLeo VA, Adler BL, Warshaw EM, et al. Photopatch test results of the North American contact dermatitis group, 1999-2009. Photodermatol Photoimmunol Photomed. 2022;38:288-291.
- Kerr A, Ferguson J. Photoallergic contact dermatitis. Photodermatol Photoimmunol Photomed. 2010;26:56-65.
- As¸kın Ö, Cesur SK, Engin B, et al. Photoallergic contact dermatitis. Curr Derm Rep. 2019;8:157-163.
- Wilm A, Berneburg M. Photoallergy. J Dtsch Dermatol Ges. 2015;13:7-13.
- DeLeo VA. Photocontact dermatitis. Dermatol Ther. 2004;17:279-288.
- Imai S, Atarashi K, Ikesue K, et al. Establishment of murine model of allergic photocontact dermatitis to ketoprofen and characterization of pathogenic T cells. J Dermatol Sci. 2006;41:127-136.
- Tokura Y, Yagi H, Satoh T, et al. Inhibitory effect of melanin pigment on sensitization and elicitation of murine contact photosensitivity: mechanism of low responsiveness in C57BL/10 background mice. J Invest Dermatol. 1993;101:673-678.
- Stein KR, Scheinfeld NS. Drug-induced photoallergic and phototoxic reactions. Expert Opin Drug Saf. 2007;6:431-443.
- Janusz SC, Schwartz RA. Botanical briefs: phytophotodermatitis is an occupational and recreational dermatosis in the limelight. Cutis. 2021;107:187-189.
- Atwal SK, Chen A, Adler BL. Phototoxic contact dermatitis from over-the-counter 8-methoxypsoralen. Cutis. 2022;109:E2-E3.
- Rutter KJ, Farrar MD, Marjanovic EJ, et al. Clinicophotobiological characterization of photoaggravated atopic dermatitis [published online July 27, 2022]. JAMA Dermatol. doi:10.1001/jamadermatol.2022.2823
- Lecha M. Idiopathic photodermatoses: clinical, diagnostic and therapeutic aspects. J Eur Acad Dermatol Venereol. 2001;15:499-505.
- Marks JG Jr, Anderson BE, DeLeo VA. Contact & Occupational Dermatology. 4th ed. Jaypee Brothers; 2016.
- Bruynzeel DP, Ferguson J, Andersen K, et al. Photopatch testing: a consensus methodology for Europe. J Eur Acad Dermatol Venereol. 2004;18:679-682.
- Kim T, Taylor JS, Maibach HI, et al. Photopatch testing among members of the American Contact Dermatitis Society. Dermatitis. 2020;31:59-67.
- Asemota E, Crawford G, Kovarik C, et al. A survey examining photopatch test and phototest methodologies of contact dermatologists in the United States: platform for developing a consensus. Dermatitis. 2017;28:265-269.
- Scalf LA, Davis MD, Rohlinger AL, et al. Photopatch testing of 182 patients: a 6-year experience at the Mayo Clinic. Dermatitis. 2009;20:44-52.
- Greenspoon J, Ahluwalia R, Juma N, et al. Allergic and photoallergic contact dermatitis: a 10-year experience. Dermatitis. 2013;24:29-32.
- Victor FC, Cohen DE, Soter NA. A 20-year analysis of previous and emerging allergens that elicit photoallergic contact dermatitis. J Am Acad Dermatol. 2010;62:605-610.
- Schauder S, Ippen H. Contact and photocontact sensitivity to sunscreens. review of a 15-year experience and of the literature. Contact Dermatitis. 1997;37:221-232.
- Collaris EJ, Frank J. Photoallergic contact dermatitis caused by ultraviolet filters in different sunscreens. Int J Dermatol. 2008;47(suppl 1):35-37.
- Heurung AR, Raju SI, Warshaw EM. Adverse reactions to sunscreen agents: epidemiology, responsible irritants and allergens, clinical characteristics, and management. Dermatitis. 2014;25:289-326.
- Reeder M, Atwater AR. Methylisothiazolinone and isothiazolinone allergy. Cutis. 2019;104:94-96.
- DeKoven JG, Silverberg JI, Warshaw EM, et al. North American Contact Dermatitis Group Patch Test Results: 2017-2018. Dermatitis. 2021;32:111-123.
- Kullberg SA, Voller LM, Warshaw EM. Methylisothiazolinone in “dermatology-recommended” sunscreens: an important mimicker of photoallergic contact dermatitis. Photodermatol Photoimmunol Photomed. 2021;37:366-370.
- Herman A, Aerts O, de Montjoye L, et al. Isothiazolinone derivatives and allergic contact dermatitis: a review and update. J Eur Acad Dermatol Venereol. 2019;33:267-276.
- Adler BL, Houle MC, Pratt M. Photoaggravated contact dermatitis to methylisothiazolinone and associated photosensitivity: a case series [published online January 25, 2022]. Dermatitis. doi:10.1097/DER.0000000000000833
- Aerts O, Goossens A, Marguery MC, et al. Photoaggravated allergic contact dermatitis and transient photosensitivity caused by methylisothiazolinone. Contact Dermatitis. 2018;78:241-245.
- Pirmez R, Fernandes AL, Melo MG. Photoaggravated contact dermatitis to Kathon CG (methylchloroisothiazolinone/methylisothiazolinone): a novel pattern of involvement in a growing epidemic?. Br J Dermatol. 2015;173:1343-1344.
- Uter W, Aalto-Korte K, Agner T, et al. The epidemic of methylisothiazolinone contact allergy in Europe: follow-up on changing exposures.J Eur Acad Dermatol Venereol. 2020;34:333-339.
- Government of Canada. Changes to the cosmetic ingredient hotlist. December 3, 2019. Updated August 26, 2022. Accessed October 20, 2022. https://www.canada.ca/en/health-canada/services/consumer-product-safety/cosmetics/cosmetic-ingredient-hotlist-prohibited-restricted-ingredients/changes.html
- Barkin RL. Topical nonsteroidal anti-inflammatory drugs: the importance of drug, delivery, and therapeutic outcome. Am J Ther. 2015;22:388-407.
- European Multicentre Photopatch Test Study (EMCPPTS) Taskforce. A European multicentre photopatch test study. Br J Dermatol. 2012;166:1002-1009.
- Ophaswongse S, Maibach H. Topical nonsteroidal antiinflammatory drugs: allergic and photoallergic contact dermatitis and phototoxicity. Contact Dermatitis. 1993;29:57-64.
- Kowalzick L, Ziegler H. Photoallergic contact dermatitis from topical diclofenac in Solaraze gel. Contact Dermatitis. 2006;54:348-349.
- Montoro J, Rodríguez M, Díaz M, et al. Photoallergic contact dermatitis due to diclofenac. Contact Dermatitis. 2003;48:115.
- Fernández-Jorge B, Goday-Buján JJ, Murga M, et al. Photoallergic contact dermatitis due to diclofenac with cross-reaction to aceclofenac: two case reports. Contact Dermatitis. 2009;61:236-237.
- Akat PB. Severe photosensitivity reaction induced by topical diclofenac. Indian J Pharmacol. 2013;45:408-409.
- Leroy D, Dompmartin A, Szczurko C, et al. Photodermatitis from ketoprofen with cross-reactivity to fenofibrate and benzophenones. Photodermatol Photoimmunol Photomed. 1997;13:93-97.
- Devleeschouwer V, Roelandts R, Garmyn M, et al. Allergic and photoallergic contact dermatitis from ketoprofen: results of (photo) patch testing and follow-up of 42 patients. Contact Dermatitis. 2008;58:159-166.
- Matsushita T, Kamide R. Five cases of photocontact dermatitisdue to topical ketoprofen: photopatch testing and cross-reaction study. Photodermatol Photoimmunol Photomed. 2001;17:26-31.
- de Groot AC, Roberts DW. Contact and photocontact allergy to octocrylene: a review. Contact Dermatitis. 2014;70:193-204.
- Wolverton JE, Soter NA, Cohen DE. Fentichlor photocontact dermatitis: a persistent enigma. Dermatitis. 2013;24:77-81.
- Mowad CM, Anderson B, Scheinman P, et al. Allergic contact dermatitis: patient management and education. J Am Acad Dermatol. 2016;74:1043-1054.
- Darvay A, White IR, Rycroft RJ, et al. Photoallergic contact dermatitis is uncommon. Br J Dermatol. 2001;145:597-601.
- DeLeo VA, Adler BL, Warshaw EM, et al. Photopatch test results of the North American contact dermatitis group, 1999-2009. Photodermatol Photoimmunol Photomed. 2022;38:288-291.
- Kerr A, Ferguson J. Photoallergic contact dermatitis. Photodermatol Photoimmunol Photomed. 2010;26:56-65.
- As¸kın Ö, Cesur SK, Engin B, et al. Photoallergic contact dermatitis. Curr Derm Rep. 2019;8:157-163.
- Wilm A, Berneburg M. Photoallergy. J Dtsch Dermatol Ges. 2015;13:7-13.
- DeLeo VA. Photocontact dermatitis. Dermatol Ther. 2004;17:279-288.
- Imai S, Atarashi K, Ikesue K, et al. Establishment of murine model of allergic photocontact dermatitis to ketoprofen and characterization of pathogenic T cells. J Dermatol Sci. 2006;41:127-136.
- Tokura Y, Yagi H, Satoh T, et al. Inhibitory effect of melanin pigment on sensitization and elicitation of murine contact photosensitivity: mechanism of low responsiveness in C57BL/10 background mice. J Invest Dermatol. 1993;101:673-678.
- Stein KR, Scheinfeld NS. Drug-induced photoallergic and phototoxic reactions. Expert Opin Drug Saf. 2007;6:431-443.
- Janusz SC, Schwartz RA. Botanical briefs: phytophotodermatitis is an occupational and recreational dermatosis in the limelight. Cutis. 2021;107:187-189.
- Atwal SK, Chen A, Adler BL. Phototoxic contact dermatitis from over-the-counter 8-methoxypsoralen. Cutis. 2022;109:E2-E3.
- Rutter KJ, Farrar MD, Marjanovic EJ, et al. Clinicophotobiological characterization of photoaggravated atopic dermatitis [published online July 27, 2022]. JAMA Dermatol. doi:10.1001/jamadermatol.2022.2823
- Lecha M. Idiopathic photodermatoses: clinical, diagnostic and therapeutic aspects. J Eur Acad Dermatol Venereol. 2001;15:499-505.
- Marks JG Jr, Anderson BE, DeLeo VA. Contact & Occupational Dermatology. 4th ed. Jaypee Brothers; 2016.
- Bruynzeel DP, Ferguson J, Andersen K, et al. Photopatch testing: a consensus methodology for Europe. J Eur Acad Dermatol Venereol. 2004;18:679-682.
- Kim T, Taylor JS, Maibach HI, et al. Photopatch testing among members of the American Contact Dermatitis Society. Dermatitis. 2020;31:59-67.
- Asemota E, Crawford G, Kovarik C, et al. A survey examining photopatch test and phototest methodologies of contact dermatologists in the United States: platform for developing a consensus. Dermatitis. 2017;28:265-269.
- Scalf LA, Davis MD, Rohlinger AL, et al. Photopatch testing of 182 patients: a 6-year experience at the Mayo Clinic. Dermatitis. 2009;20:44-52.
- Greenspoon J, Ahluwalia R, Juma N, et al. Allergic and photoallergic contact dermatitis: a 10-year experience. Dermatitis. 2013;24:29-32.
- Victor FC, Cohen DE, Soter NA. A 20-year analysis of previous and emerging allergens that elicit photoallergic contact dermatitis. J Am Acad Dermatol. 2010;62:605-610.
- Schauder S, Ippen H. Contact and photocontact sensitivity to sunscreens. review of a 15-year experience and of the literature. Contact Dermatitis. 1997;37:221-232.
- Collaris EJ, Frank J. Photoallergic contact dermatitis caused by ultraviolet filters in different sunscreens. Int J Dermatol. 2008;47(suppl 1):35-37.
- Heurung AR, Raju SI, Warshaw EM. Adverse reactions to sunscreen agents: epidemiology, responsible irritants and allergens, clinical characteristics, and management. Dermatitis. 2014;25:289-326.
- Reeder M, Atwater AR. Methylisothiazolinone and isothiazolinone allergy. Cutis. 2019;104:94-96.
- DeKoven JG, Silverberg JI, Warshaw EM, et al. North American Contact Dermatitis Group Patch Test Results: 2017-2018. Dermatitis. 2021;32:111-123.
- Kullberg SA, Voller LM, Warshaw EM. Methylisothiazolinone in “dermatology-recommended” sunscreens: an important mimicker of photoallergic contact dermatitis. Photodermatol Photoimmunol Photomed. 2021;37:366-370.
- Herman A, Aerts O, de Montjoye L, et al. Isothiazolinone derivatives and allergic contact dermatitis: a review and update. J Eur Acad Dermatol Venereol. 2019;33:267-276.
- Adler BL, Houle MC, Pratt M. Photoaggravated contact dermatitis to methylisothiazolinone and associated photosensitivity: a case series [published online January 25, 2022]. Dermatitis. doi:10.1097/DER.0000000000000833
- Aerts O, Goossens A, Marguery MC, et al. Photoaggravated allergic contact dermatitis and transient photosensitivity caused by methylisothiazolinone. Contact Dermatitis. 2018;78:241-245.
- Pirmez R, Fernandes AL, Melo MG. Photoaggravated contact dermatitis to Kathon CG (methylchloroisothiazolinone/methylisothiazolinone): a novel pattern of involvement in a growing epidemic?. Br J Dermatol. 2015;173:1343-1344.
- Uter W, Aalto-Korte K, Agner T, et al. The epidemic of methylisothiazolinone contact allergy in Europe: follow-up on changing exposures.J Eur Acad Dermatol Venereol. 2020;34:333-339.
- Government of Canada. Changes to the cosmetic ingredient hotlist. December 3, 2019. Updated August 26, 2022. Accessed October 20, 2022. https://www.canada.ca/en/health-canada/services/consumer-product-safety/cosmetics/cosmetic-ingredient-hotlist-prohibited-restricted-ingredients/changes.html
- Barkin RL. Topical nonsteroidal anti-inflammatory drugs: the importance of drug, delivery, and therapeutic outcome. Am J Ther. 2015;22:388-407.
- European Multicentre Photopatch Test Study (EMCPPTS) Taskforce. A European multicentre photopatch test study. Br J Dermatol. 2012;166:1002-1009.
- Ophaswongse S, Maibach H. Topical nonsteroidal antiinflammatory drugs: allergic and photoallergic contact dermatitis and phototoxicity. Contact Dermatitis. 1993;29:57-64.
- Kowalzick L, Ziegler H. Photoallergic contact dermatitis from topical diclofenac in Solaraze gel. Contact Dermatitis. 2006;54:348-349.
- Montoro J, Rodríguez M, Díaz M, et al. Photoallergic contact dermatitis due to diclofenac. Contact Dermatitis. 2003;48:115.
- Fernández-Jorge B, Goday-Buján JJ, Murga M, et al. Photoallergic contact dermatitis due to diclofenac with cross-reaction to aceclofenac: two case reports. Contact Dermatitis. 2009;61:236-237.
- Akat PB. Severe photosensitivity reaction induced by topical diclofenac. Indian J Pharmacol. 2013;45:408-409.
- Leroy D, Dompmartin A, Szczurko C, et al. Photodermatitis from ketoprofen with cross-reactivity to fenofibrate and benzophenones. Photodermatol Photoimmunol Photomed. 1997;13:93-97.
- Devleeschouwer V, Roelandts R, Garmyn M, et al. Allergic and photoallergic contact dermatitis from ketoprofen: results of (photo) patch testing and follow-up of 42 patients. Contact Dermatitis. 2008;58:159-166.
- Matsushita T, Kamide R. Five cases of photocontact dermatitisdue to topical ketoprofen: photopatch testing and cross-reaction study. Photodermatol Photoimmunol Photomed. 2001;17:26-31.
- de Groot AC, Roberts DW. Contact and photocontact allergy to octocrylene: a review. Contact Dermatitis. 2014;70:193-204.
- Wolverton JE, Soter NA, Cohen DE. Fentichlor photocontact dermatitis: a persistent enigma. Dermatitis. 2013;24:77-81.
- Mowad CM, Anderson B, Scheinman P, et al. Allergic contact dermatitis: patient management and education. J Am Acad Dermatol. 2016;74:1043-1054.
Practice Points
- Photoallergic contact dermatitis (PACD) presents clinically and histologically similar to allergic contact dermatitis but is concentrated in sun-exposed body sites.
- Sunscreens currently are the most common photoallergens in North America, whereas topical nonsteroidal anti-inflammatory drugs are more common culprits in Europe.
- Photopatch testing is required to diagnose PACD; however, it is infrequently performed, and there currently are no North American consensus guidelines.
Update on Tinea Capitis Diagnosis and Treatment
Tinea capitis (TC) most often is caused by Trichophyton tonsurans and Microsporum canis. The peak incidence is between 3 and 7 years of age. Noninflammatory TC typically presents as fine scaling with single or multiple scaly patches of circular alopecia (grey patches); diffuse or patchy, fine, white, adherent scaling of the scalp resembling generalized dandruff with subtle hair loss; or single or multiple patches of well-demarcated areas of alopecia with fine scale studded with broken-off hairs at the scalp surface, resulting in a black dot appearance. Inflammatory variants of TC include kerion and favus.1 Herein, updates on diagnosis, treatment, and monitoring of TC are provided, as well as a discussion of changes in the fungal microbiome associated with TC. Lastly, insights to some queries that practitioners may encounter when treating children with TC are provided.
Genetic Susceptibility
Molecular techniques have identified a number of macrophage regulator, leukocyte activation and migration, and cutaneous permeability genes associated with susceptibility to TC. These findings indicate that genetically determined deficiency in adaptive immune responses may affect the predisposition to dermatophyte infections.2
Clinical Varieties of Infection
Dermatophytes causing ringworm are capable of invading the hair shafts and can simultaneously invade smooth or glabrous skin (eg, T tonsurans, Trichophyton schoenleinii, Trichophyton violaceum). Some causative dermatophytes can even penetrate the nails (eg, Trichophyton soudanense). The clinical presentation is dependent on 3 main patterns of hair invasion3:
• Ectothrix: A mid-follicular pattern of invasion with hyphae growing down to the hair bulb that commonly is caused by Microsporum species. It clinically presents with scaling and inflammation with hair shafts breaking 2 to 3 mm above the scalp level.
• Endothrix: This pattern is nonfluorescent on Wood lamp examination, and hairs often break at the scalp level (black dot type). Trichophyton tonsurans, T soudanense, Trichophyton rubrum, and T violaceum are common causes.
• Favus: In this pattern, T schoenleinii is a common cause, and hairs grow to considerable lengths above the scalp with less damage than the other patterns. The hair shafts present with characteristic air spaces, and hyphae form clusters at the level of the epidermis.
Diagnosis
Optimal treatment of TC relies on proper identification of the causative agent. Fungal culture remains the gold standard of mycologic diagnosis regardless of its delayed results, which may take up to 4 weeks for proper identification of the fungal colonies and require ample expertise to interpret the morphologic features of the grown colonies.4
Other tests such as the potassium hydroxide preparation are nonspecific and do not identify the dermatophyte species. Although this method has been reported to have 5% to 15% false-negative results in routine practice depending on the skill of the observer and the quality of sampling, microscopic examination is essential, as it may allow the clinician to start treatment sooner pending culture results. The use of a Wood lamp is not suitable for definitive species identification, as this technique primarily is useful for observing fluorescence in ectothrix infection caused by Microsporum species, with the exception of T schoenleinii; otherwise, Trichophyton species, which cause endothrix infections, do not fluoresce.5Polymerase chain reaction is a sensitive technique that can help identify both the genus and species of common dermatophytes. Common target sequences include the ribosomal internal transcribed spacer and translation elongation factor 1α. The use of matrix-assisted laser desorption/ionization time-of-flight mass spectrometry also has become popular for dermatophyte identification.6Trichoscopic diagnosis of TC, which is simple and noninvasive, is becoming increasingly popular. Features such as short, broken, black dot, comma, corkscrew, and/or zigzag hairs, as well as perifollicular scaling, are helpful for diagnosing TC (Figure). Moreover, trichoscopy can be useful for differentiating other common causes of hair loss, such as trichotillomania and alopecia areata. It had been reported that the trichoscopic features of TC can be seen as early as 2 weeks after starting treatment and therefore this can be a reliable period in which to follow-up with the patient to evaluate progress. The disappearance of black dots and comma hairs can be appreciated from 2 weeks onwards by trichoscopic evaluation.4
, comma hairs (red arrows), corkscrew hairs (green arrows), short broken hairs (blue arrow), and perifollicular scaling (yellow arrow).")
Treatment
The common recommendation for first-line treatment of TC is the use of systemic antifungals with the use of a topical agent as an adjuvant to prevent the spread of fungal spores. For almost 6 decades, griseofulvin had been the gold-standard fungistatic used for treating TC in patients older than 2 years until the 2007 US Food and Drug Administration (FDA) approval of terbinafine fungicidal oral granules for treatment of TC in patients older than 4 years.7
Meta-analyses have demonstrated comparable efficacy for a 4-week course of terbinafine compared to 6 weeks of griseofulvin for TC based on the infectious organism. Terbinafine demonstrated superiority in treating T tonsurans and a similar efficacy in treating T violaceum, while griseofulvin was superior in treating M canis and other Microsporum species.8,9
The off-label use of fluconazole and itraconazole to treat TC is gaining popularity, with limited trials showing increased evidence of their effectiveness. There is not much clinical evidence to support the use of other oral antifungals, including the newer azoles such as voriconazole or posaconazole.9
Newer limited evidence has shown the off-label use of photodynamic therapy to be a promising alternative to systemic antifungal therapy in treating TC, pending validation by larger sample trials.10In my practice, I have found that severe cases of TC demonstrating inflammation or possible widespread id reactions are better treated with oral steroids. Ketoconazole shampoo or selenium sulfide used 2 to 3 times weekly to prevent spread in the early phases of therapy is a good adjunct to systemic treatment. Cases with kerions should be assessed for the possibility of a coexisting bacterial infection under the crusts, and if confirmed, antibiotics should be started.9The commonly used systemic antifungals generally are safe with a low side-effect profile, but there is a risk for hepatotoxicity. The FDA recommends that baseline alanine transaminase and aspartate transaminase levels should be obtained prior to beginning a terbinafine-based treatment regimen.11 The American Academy of Pediatrics has specifically stated that laboratory testing of serum hepatic enzymes is not a requirement if a griseofulvin-based regimen does not exceed 8 weeks; however, transaminase levels (alanine transaminase and aspartate transaminase) should be considered in patients using terbinafine at baseline or if treatment is prolonged beyond 4 to 6 weeks.12 In agreement with the FDA guidelines, the Canadian Pediatric Society has suggested that liver enzymes should be periodically monitored in patients being treated with terbinafine beyond 4 to 6 weeks.13
Changes in the Fungal Microbiome
Research has shown that changes in the fungal microbiome were associated with an altered bacterial community in patients with TC. During fungal infection, the relative abundances of Cutibacterium and Corynebacterium increased, and the relative abundance of Streptococcus decreased. In addition, some uncommon bacterial genera such as Herbaspirillum and Methylorubrum were detected on the scalp in TC.14
Carrier State
Carrier state is determined for those siblings and contacts of cases with a clinically normal scalp that are positive on culture. Those individuals could represent a potential reservoir responsible for contamination (or recontamination) of the patient as well as treatment failure. Opinions remain divided as to whether to use oral antifungal therapy in these carriers or maintain therapy on antifungal shampoos containing ketoconazole or povidone-iodine. Due to the paucity of available data, my experience has shown that it is sufficient to use antifungal shampoos for such carriers. In zoophilic infections, it is important to identify and treat the animal source.6-9
Final Thoughts
Successful treatment of TC requires accurate identification of the pathogen, which commonly is achieved via fungal culture. Despite its practical value, the conventional identification of dermatophytes based on morphologic features can be highly challenging due to the low positive rate and delayed results. Trichoscopy is a quick, handy, and noninvasive tool that can better indicate the diagnosis and also is helpful for follow-up on treatment progress. Due to better understanding of the immunology and genetic susceptibility associated with TC spread, the current treatment pipeline holds more insight into better control of this condition. Increased surveillance, prompt diagnosis, and early onset of systemic treatment are the key to proper prevention of spread of TC.
- Leung AKC, Hon KL, Leong KF, et al. Tinea capitis: an updated review. Recent Pat Inflamm Allergy Drug Discov. 2020;14:58-68.
- Abdel-Rahman SM, Preuett BL. Genetic predictors of susceptibility to cutaneous fungal infections: a pilot genome wide association study to refine a candidate gene search. J Dermatol Sci. 2012;67:147-152.
- Hay RJ. Tinea capitis: current status. Mycopathologia. 2017;182:87-93.
- Wahbah HR, Atallah RB, Eldahshan RM, et al. A prospective clinical and trichoscopic study of tinea capitis in children during treatment [published online May 23, 2022]. Dermatol Ther. 2022;35:E15582. doi:10.1111/dth.15582
- Salehi Z, Shams-Ghahfarokhi M, Razzaghi-Abyaneh M. Molecular epidemiology, genetic diversity, and antifungal susceptibility of major pathogenic dermatophytes isolated from human dermatophytosis. Front Microbiol. 2021;12:643509.
- Lamisil. Package insert. Novartis; 2011. Accessed October 17, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/020539s021lbl.pdf
- Gupta AK, Drummond-Main C. Meta-analysis of randomized, controlled trials comparing particular doses of griseofulvin and terbinafine for the treatment of tinea capitis. Pediatr Dermatol. 2013;30:1-6.
- Tey HL, Tan AS, Chan YC. Meta-analysis of randomized, controlled trials comparing griseofulvin and terbinafine in the treatment of tinea capitis. J Am Acad Dermatol. 2011;64:663-670.
- Gupta AK, Friedlander SF, Simkovich AJ. Tinea capitis: an update. Pediatr Dermatol. 2022;39:167-172.
- Aspiroz C, Melcon B, Cerro PA, et al. Tinea capitis caused by Microsporum canis treated with methyl-aminolevulinate daylight photodynamic therapy and ketoconazole shampooing. Photodermatol Photoimmunol Photomed. 2021;37:567-568.
- Aleohin N, Bar J, Bar-Ilan E, et al. Laboratory monitoring during antifungal treatment of paediatric tinea capitis. Mycoses. 2021;64:157-161.
- Kimberlin DW, Brady MT, Jackson MA, et al, eds. Tinea capitis. In: Red Book 2018-2021: Report of the Committee of Infectious Diseases. American Academy of Pediatrics; 2018:798-801.
- Bortolussi R, Martin S, Audcent T, et al. Antifungal agents for common outpatient paediatric infections. Canadian Paediatric Society website. Published June 20, 2019. Accessed October 4, 2022. https://www.cps.ca/en/documents/position/antifungal-agents-common-infections
- Tao R, Zhu P, Zhou Y, et al. Altered skin fungal and bacterial community compositions in tinea capitis. Mycoses. 2022;65:834-840.
Tinea capitis (TC) most often is caused by Trichophyton tonsurans and Microsporum canis. The peak incidence is between 3 and 7 years of age. Noninflammatory TC typically presents as fine scaling with single or multiple scaly patches of circular alopecia (grey patches); diffuse or patchy, fine, white, adherent scaling of the scalp resembling generalized dandruff with subtle hair loss; or single or multiple patches of well-demarcated areas of alopecia with fine scale studded with broken-off hairs at the scalp surface, resulting in a black dot appearance. Inflammatory variants of TC include kerion and favus.1 Herein, updates on diagnosis, treatment, and monitoring of TC are provided, as well as a discussion of changes in the fungal microbiome associated with TC. Lastly, insights to some queries that practitioners may encounter when treating children with TC are provided.
Genetic Susceptibility
Molecular techniques have identified a number of macrophage regulator, leukocyte activation and migration, and cutaneous permeability genes associated with susceptibility to TC. These findings indicate that genetically determined deficiency in adaptive immune responses may affect the predisposition to dermatophyte infections.2
Clinical Varieties of Infection
Dermatophytes causing ringworm are capable of invading the hair shafts and can simultaneously invade smooth or glabrous skin (eg, T tonsurans, Trichophyton schoenleinii, Trichophyton violaceum). Some causative dermatophytes can even penetrate the nails (eg, Trichophyton soudanense). The clinical presentation is dependent on 3 main patterns of hair invasion3:
• Ectothrix: A mid-follicular pattern of invasion with hyphae growing down to the hair bulb that commonly is caused by Microsporum species. It clinically presents with scaling and inflammation with hair shafts breaking 2 to 3 mm above the scalp level.
• Endothrix: This pattern is nonfluorescent on Wood lamp examination, and hairs often break at the scalp level (black dot type). Trichophyton tonsurans, T soudanense, Trichophyton rubrum, and T violaceum are common causes.
• Favus: In this pattern, T schoenleinii is a common cause, and hairs grow to considerable lengths above the scalp with less damage than the other patterns. The hair shafts present with characteristic air spaces, and hyphae form clusters at the level of the epidermis.
Diagnosis
Optimal treatment of TC relies on proper identification of the causative agent. Fungal culture remains the gold standard of mycologic diagnosis regardless of its delayed results, which may take up to 4 weeks for proper identification of the fungal colonies and require ample expertise to interpret the morphologic features of the grown colonies.4
Other tests such as the potassium hydroxide preparation are nonspecific and do not identify the dermatophyte species. Although this method has been reported to have 5% to 15% false-negative results in routine practice depending on the skill of the observer and the quality of sampling, microscopic examination is essential, as it may allow the clinician to start treatment sooner pending culture results. The use of a Wood lamp is not suitable for definitive species identification, as this technique primarily is useful for observing fluorescence in ectothrix infection caused by Microsporum species, with the exception of T schoenleinii; otherwise, Trichophyton species, which cause endothrix infections, do not fluoresce.5Polymerase chain reaction is a sensitive technique that can help identify both the genus and species of common dermatophytes. Common target sequences include the ribosomal internal transcribed spacer and translation elongation factor 1α. The use of matrix-assisted laser desorption/ionization time-of-flight mass spectrometry also has become popular for dermatophyte identification.6Trichoscopic diagnosis of TC, which is simple and noninvasive, is becoming increasingly popular. Features such as short, broken, black dot, comma, corkscrew, and/or zigzag hairs, as well as perifollicular scaling, are helpful for diagnosing TC (Figure). Moreover, trichoscopy can be useful for differentiating other common causes of hair loss, such as trichotillomania and alopecia areata. It had been reported that the trichoscopic features of TC can be seen as early as 2 weeks after starting treatment and therefore this can be a reliable period in which to follow-up with the patient to evaluate progress. The disappearance of black dots and comma hairs can be appreciated from 2 weeks onwards by trichoscopic evaluation.4
Treatment
The common recommendation for first-line treatment of TC is the use of systemic antifungals with the use of a topical agent as an adjuvant to prevent the spread of fungal spores. For almost 6 decades, griseofulvin had been the gold-standard fungistatic used for treating TC in patients older than 2 years until the 2007 US Food and Drug Administration (FDA) approval of terbinafine fungicidal oral granules for treatment of TC in patients older than 4 years.7
Meta-analyses have demonstrated comparable efficacy for a 4-week course of terbinafine compared to 6 weeks of griseofulvin for TC based on the infectious organism. Terbinafine demonstrated superiority in treating T tonsurans and a similar efficacy in treating T violaceum, while griseofulvin was superior in treating M canis and other Microsporum species.8,9
The off-label use of fluconazole and itraconazole to treat TC is gaining popularity, with limited trials showing increased evidence of their effectiveness. There is not much clinical evidence to support the use of other oral antifungals, including the newer azoles such as voriconazole or posaconazole.9
Newer limited evidence has shown the off-label use of photodynamic therapy to be a promising alternative to systemic antifungal therapy in treating TC, pending validation by larger sample trials.10In my practice, I have found that severe cases of TC demonstrating inflammation or possible widespread id reactions are better treated with oral steroids. Ketoconazole shampoo or selenium sulfide used 2 to 3 times weekly to prevent spread in the early phases of therapy is a good adjunct to systemic treatment. Cases with kerions should be assessed for the possibility of a coexisting bacterial infection under the crusts, and if confirmed, antibiotics should be started.9The commonly used systemic antifungals generally are safe with a low side-effect profile, but there is a risk for hepatotoxicity. The FDA recommends that baseline alanine transaminase and aspartate transaminase levels should be obtained prior to beginning a terbinafine-based treatment regimen.11 The American Academy of Pediatrics has specifically stated that laboratory testing of serum hepatic enzymes is not a requirement if a griseofulvin-based regimen does not exceed 8 weeks; however, transaminase levels (alanine transaminase and aspartate transaminase) should be considered in patients using terbinafine at baseline or if treatment is prolonged beyond 4 to 6 weeks.12 In agreement with the FDA guidelines, the Canadian Pediatric Society has suggested that liver enzymes should be periodically monitored in patients being treated with terbinafine beyond 4 to 6 weeks.13
Changes in the Fungal Microbiome
Research has shown that changes in the fungal microbiome were associated with an altered bacterial community in patients with TC. During fungal infection, the relative abundances of Cutibacterium and Corynebacterium increased, and the relative abundance of Streptococcus decreased. In addition, some uncommon bacterial genera such as Herbaspirillum and Methylorubrum were detected on the scalp in TC.14
Carrier State
Carrier state is determined for those siblings and contacts of cases with a clinically normal scalp that are positive on culture. Those individuals could represent a potential reservoir responsible for contamination (or recontamination) of the patient as well as treatment failure. Opinions remain divided as to whether to use oral antifungal therapy in these carriers or maintain therapy on antifungal shampoos containing ketoconazole or povidone-iodine. Due to the paucity of available data, my experience has shown that it is sufficient to use antifungal shampoos for such carriers. In zoophilic infections, it is important to identify and treat the animal source.6-9
Final Thoughts
Successful treatment of TC requires accurate identification of the pathogen, which commonly is achieved via fungal culture. Despite its practical value, the conventional identification of dermatophytes based on morphologic features can be highly challenging due to the low positive rate and delayed results. Trichoscopy is a quick, handy, and noninvasive tool that can better indicate the diagnosis and also is helpful for follow-up on treatment progress. Due to better understanding of the immunology and genetic susceptibility associated with TC spread, the current treatment pipeline holds more insight into better control of this condition. Increased surveillance, prompt diagnosis, and early onset of systemic treatment are the key to proper prevention of spread of TC.
Tinea capitis (TC) most often is caused by Trichophyton tonsurans and Microsporum canis. The peak incidence is between 3 and 7 years of age. Noninflammatory TC typically presents as fine scaling with single or multiple scaly patches of circular alopecia (grey patches); diffuse or patchy, fine, white, adherent scaling of the scalp resembling generalized dandruff with subtle hair loss; or single or multiple patches of well-demarcated areas of alopecia with fine scale studded with broken-off hairs at the scalp surface, resulting in a black dot appearance. Inflammatory variants of TC include kerion and favus.1 Herein, updates on diagnosis, treatment, and monitoring of TC are provided, as well as a discussion of changes in the fungal microbiome associated with TC. Lastly, insights to some queries that practitioners may encounter when treating children with TC are provided.
Genetic Susceptibility
Molecular techniques have identified a number of macrophage regulator, leukocyte activation and migration, and cutaneous permeability genes associated with susceptibility to TC. These findings indicate that genetically determined deficiency in adaptive immune responses may affect the predisposition to dermatophyte infections.2
Clinical Varieties of Infection
Dermatophytes causing ringworm are capable of invading the hair shafts and can simultaneously invade smooth or glabrous skin (eg, T tonsurans, Trichophyton schoenleinii, Trichophyton violaceum). Some causative dermatophytes can even penetrate the nails (eg, Trichophyton soudanense). The clinical presentation is dependent on 3 main patterns of hair invasion3:
• Ectothrix: A mid-follicular pattern of invasion with hyphae growing down to the hair bulb that commonly is caused by Microsporum species. It clinically presents with scaling and inflammation with hair shafts breaking 2 to 3 mm above the scalp level.
• Endothrix: This pattern is nonfluorescent on Wood lamp examination, and hairs often break at the scalp level (black dot type). Trichophyton tonsurans, T soudanense, Trichophyton rubrum, and T violaceum are common causes.
• Favus: In this pattern, T schoenleinii is a common cause, and hairs grow to considerable lengths above the scalp with less damage than the other patterns. The hair shafts present with characteristic air spaces, and hyphae form clusters at the level of the epidermis.
Diagnosis
Optimal treatment of TC relies on proper identification of the causative agent. Fungal culture remains the gold standard of mycologic diagnosis regardless of its delayed results, which may take up to 4 weeks for proper identification of the fungal colonies and require ample expertise to interpret the morphologic features of the grown colonies.4
Other tests such as the potassium hydroxide preparation are nonspecific and do not identify the dermatophyte species. Although this method has been reported to have 5% to 15% false-negative results in routine practice depending on the skill of the observer and the quality of sampling, microscopic examination is essential, as it may allow the clinician to start treatment sooner pending culture results. The use of a Wood lamp is not suitable for definitive species identification, as this technique primarily is useful for observing fluorescence in ectothrix infection caused by Microsporum species, with the exception of T schoenleinii; otherwise, Trichophyton species, which cause endothrix infections, do not fluoresce.5Polymerase chain reaction is a sensitive technique that can help identify both the genus and species of common dermatophytes. Common target sequences include the ribosomal internal transcribed spacer and translation elongation factor 1α. The use of matrix-assisted laser desorption/ionization time-of-flight mass spectrometry also has become popular for dermatophyte identification.6Trichoscopic diagnosis of TC, which is simple and noninvasive, is becoming increasingly popular. Features such as short, broken, black dot, comma, corkscrew, and/or zigzag hairs, as well as perifollicular scaling, are helpful for diagnosing TC (Figure). Moreover, trichoscopy can be useful for differentiating other common causes of hair loss, such as trichotillomania and alopecia areata. It had been reported that the trichoscopic features of TC can be seen as early as 2 weeks after starting treatment and therefore this can be a reliable period in which to follow-up with the patient to evaluate progress. The disappearance of black dots and comma hairs can be appreciated from 2 weeks onwards by trichoscopic evaluation.4
Treatment
The common recommendation for first-line treatment of TC is the use of systemic antifungals with the use of a topical agent as an adjuvant to prevent the spread of fungal spores. For almost 6 decades, griseofulvin had been the gold-standard fungistatic used for treating TC in patients older than 2 years until the 2007 US Food and Drug Administration (FDA) approval of terbinafine fungicidal oral granules for treatment of TC in patients older than 4 years.7
Meta-analyses have demonstrated comparable efficacy for a 4-week course of terbinafine compared to 6 weeks of griseofulvin for TC based on the infectious organism. Terbinafine demonstrated superiority in treating T tonsurans and a similar efficacy in treating T violaceum, while griseofulvin was superior in treating M canis and other Microsporum species.8,9
The off-label use of fluconazole and itraconazole to treat TC is gaining popularity, with limited trials showing increased evidence of their effectiveness. There is not much clinical evidence to support the use of other oral antifungals, including the newer azoles such as voriconazole or posaconazole.9
Newer limited evidence has shown the off-label use of photodynamic therapy to be a promising alternative to systemic antifungal therapy in treating TC, pending validation by larger sample trials.10In my practice, I have found that severe cases of TC demonstrating inflammation or possible widespread id reactions are better treated with oral steroids. Ketoconazole shampoo or selenium sulfide used 2 to 3 times weekly to prevent spread in the early phases of therapy is a good adjunct to systemic treatment. Cases with kerions should be assessed for the possibility of a coexisting bacterial infection under the crusts, and if confirmed, antibiotics should be started.9The commonly used systemic antifungals generally are safe with a low side-effect profile, but there is a risk for hepatotoxicity. The FDA recommends that baseline alanine transaminase and aspartate transaminase levels should be obtained prior to beginning a terbinafine-based treatment regimen.11 The American Academy of Pediatrics has specifically stated that laboratory testing of serum hepatic enzymes is not a requirement if a griseofulvin-based regimen does not exceed 8 weeks; however, transaminase levels (alanine transaminase and aspartate transaminase) should be considered in patients using terbinafine at baseline or if treatment is prolonged beyond 4 to 6 weeks.12 In agreement with the FDA guidelines, the Canadian Pediatric Society has suggested that liver enzymes should be periodically monitored in patients being treated with terbinafine beyond 4 to 6 weeks.13
Changes in the Fungal Microbiome
Research has shown that changes in the fungal microbiome were associated with an altered bacterial community in patients with TC. During fungal infection, the relative abundances of Cutibacterium and Corynebacterium increased, and the relative abundance of Streptococcus decreased. In addition, some uncommon bacterial genera such as Herbaspirillum and Methylorubrum were detected on the scalp in TC.14
Carrier State
Carrier state is determined for those siblings and contacts of cases with a clinically normal scalp that are positive on culture. Those individuals could represent a potential reservoir responsible for contamination (or recontamination) of the patient as well as treatment failure. Opinions remain divided as to whether to use oral antifungal therapy in these carriers or maintain therapy on antifungal shampoos containing ketoconazole or povidone-iodine. Due to the paucity of available data, my experience has shown that it is sufficient to use antifungal shampoos for such carriers. In zoophilic infections, it is important to identify and treat the animal source.6-9
Final Thoughts
Successful treatment of TC requires accurate identification of the pathogen, which commonly is achieved via fungal culture. Despite its practical value, the conventional identification of dermatophytes based on morphologic features can be highly challenging due to the low positive rate and delayed results. Trichoscopy is a quick, handy, and noninvasive tool that can better indicate the diagnosis and also is helpful for follow-up on treatment progress. Due to better understanding of the immunology and genetic susceptibility associated with TC spread, the current treatment pipeline holds more insight into better control of this condition. Increased surveillance, prompt diagnosis, and early onset of systemic treatment are the key to proper prevention of spread of TC.
- Leung AKC, Hon KL, Leong KF, et al. Tinea capitis: an updated review. Recent Pat Inflamm Allergy Drug Discov. 2020;14:58-68.
- Abdel-Rahman SM, Preuett BL. Genetic predictors of susceptibility to cutaneous fungal infections: a pilot genome wide association study to refine a candidate gene search. J Dermatol Sci. 2012;67:147-152.
- Hay RJ. Tinea capitis: current status. Mycopathologia. 2017;182:87-93.
- Wahbah HR, Atallah RB, Eldahshan RM, et al. A prospective clinical and trichoscopic study of tinea capitis in children during treatment [published online May 23, 2022]. Dermatol Ther. 2022;35:E15582. doi:10.1111/dth.15582
- Salehi Z, Shams-Ghahfarokhi M, Razzaghi-Abyaneh M. Molecular epidemiology, genetic diversity, and antifungal susceptibility of major pathogenic dermatophytes isolated from human dermatophytosis. Front Microbiol. 2021;12:643509.
- Lamisil. Package insert. Novartis; 2011. Accessed October 17, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/020539s021lbl.pdf
- Gupta AK, Drummond-Main C. Meta-analysis of randomized, controlled trials comparing particular doses of griseofulvin and terbinafine for the treatment of tinea capitis. Pediatr Dermatol. 2013;30:1-6.
- Tey HL, Tan AS, Chan YC. Meta-analysis of randomized, controlled trials comparing griseofulvin and terbinafine in the treatment of tinea capitis. J Am Acad Dermatol. 2011;64:663-670.
- Gupta AK, Friedlander SF, Simkovich AJ. Tinea capitis: an update. Pediatr Dermatol. 2022;39:167-172.
- Aspiroz C, Melcon B, Cerro PA, et al. Tinea capitis caused by Microsporum canis treated with methyl-aminolevulinate daylight photodynamic therapy and ketoconazole shampooing. Photodermatol Photoimmunol Photomed. 2021;37:567-568.
- Aleohin N, Bar J, Bar-Ilan E, et al. Laboratory monitoring during antifungal treatment of paediatric tinea capitis. Mycoses. 2021;64:157-161.
- Kimberlin DW, Brady MT, Jackson MA, et al, eds. Tinea capitis. In: Red Book 2018-2021: Report of the Committee of Infectious Diseases. American Academy of Pediatrics; 2018:798-801.
- Bortolussi R, Martin S, Audcent T, et al. Antifungal agents for common outpatient paediatric infections. Canadian Paediatric Society website. Published June 20, 2019. Accessed October 4, 2022. https://www.cps.ca/en/documents/position/antifungal-agents-common-infections
- Tao R, Zhu P, Zhou Y, et al. Altered skin fungal and bacterial community compositions in tinea capitis. Mycoses. 2022;65:834-840.
- Leung AKC, Hon KL, Leong KF, et al. Tinea capitis: an updated review. Recent Pat Inflamm Allergy Drug Discov. 2020;14:58-68.
- Abdel-Rahman SM, Preuett BL. Genetic predictors of susceptibility to cutaneous fungal infections: a pilot genome wide association study to refine a candidate gene search. J Dermatol Sci. 2012;67:147-152.
- Hay RJ. Tinea capitis: current status. Mycopathologia. 2017;182:87-93.
- Wahbah HR, Atallah RB, Eldahshan RM, et al. A prospective clinical and trichoscopic study of tinea capitis in children during treatment [published online May 23, 2022]. Dermatol Ther. 2022;35:E15582. doi:10.1111/dth.15582
- Salehi Z, Shams-Ghahfarokhi M, Razzaghi-Abyaneh M. Molecular epidemiology, genetic diversity, and antifungal susceptibility of major pathogenic dermatophytes isolated from human dermatophytosis. Front Microbiol. 2021;12:643509.
- Lamisil. Package insert. Novartis; 2011. Accessed October 17, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/020539s021lbl.pdf
- Gupta AK, Drummond-Main C. Meta-analysis of randomized, controlled trials comparing particular doses of griseofulvin and terbinafine for the treatment of tinea capitis. Pediatr Dermatol. 2013;30:1-6.
- Tey HL, Tan AS, Chan YC. Meta-analysis of randomized, controlled trials comparing griseofulvin and terbinafine in the treatment of tinea capitis. J Am Acad Dermatol. 2011;64:663-670.
- Gupta AK, Friedlander SF, Simkovich AJ. Tinea capitis: an update. Pediatr Dermatol. 2022;39:167-172.
- Aspiroz C, Melcon B, Cerro PA, et al. Tinea capitis caused by Microsporum canis treated with methyl-aminolevulinate daylight photodynamic therapy and ketoconazole shampooing. Photodermatol Photoimmunol Photomed. 2021;37:567-568.
- Aleohin N, Bar J, Bar-Ilan E, et al. Laboratory monitoring during antifungal treatment of paediatric tinea capitis. Mycoses. 2021;64:157-161.
- Kimberlin DW, Brady MT, Jackson MA, et al, eds. Tinea capitis. In: Red Book 2018-2021: Report of the Committee of Infectious Diseases. American Academy of Pediatrics; 2018:798-801.
- Bortolussi R, Martin S, Audcent T, et al. Antifungal agents for common outpatient paediatric infections. Canadian Paediatric Society website. Published June 20, 2019. Accessed October 4, 2022. https://www.cps.ca/en/documents/position/antifungal-agents-common-infections
- Tao R, Zhu P, Zhou Y, et al. Altered skin fungal and bacterial community compositions in tinea capitis. Mycoses. 2022;65:834-840.

A 95-year-old White male with hypertension presented with itchy patches and bullae on the trunk and extremities
and is associated with various predisposing factors, including HLA genes, comorbidities, aging, and trigger factors such as drugs, trauma, radiation, chemotherapy, and infections. The autoimmune reaction is mediated by a dysregulation of T cells in which IgG and IgE autoantibodies form against hemidesmosomal proteins (BP180 and BP230). These autoantibodies induce neutrophil activation, recruitment, and degradation in the basement membrane of the skin.
Typically, patients present with intense pruritus followed by an urticarial or eczematous eruption. Tense blisters and bullae occur commonly on the trunk and extremities. Drug-associated bullous pemphigoid (DABP) is a common manifestation of the disease with histologic and immunologic features similar to those of the idiopathic version. Eruptions can be triggered by systemic or topical medications, and incidence of these reactions may be related to a genetic predisposition for the disease.
Some research suggests that drug-induced changes to the antigenic properties of the epidermal basement membrane result in an augmented immune response, while others point to structural modification in these zones that stimulate the immune system. Thiol- and phenol-based drugs have been largely implicated in the development of DABP because they are capable of structural modification and disruption of the dermo-epidermal junction in the basement membrane.
DABP often presents with patients taking multiple medications. Some of the most common medications are gliptins, PD-1 inhibitors, diuretics, antibiotics, anti-inflammatory drugs, and ACE-inhibitors, and other cardiovascular drugs. DABP may present with mucosal eruptions unlike its idiopathic counterpart that is mostly contained to the skin.

On this patient, two punch biopsies were taken. Histopathology revealed an eosinophil-rich subepidermal blister with a smooth epidermal undersurface consistent with bullous pemphigoid. Direct immunofluorescence was positive with a deposition of IgG and C3 at the epidermal side of salt split basement membrane zone.
Treatment for BP includes high potency topical and systemic steroids. Tetracyclines and niacinamide have been reported to improve the condition. Treatment is tailored to allow for cutaneous healing and control pruritus, but the physician must be mindful of the patient’s comorbidities and capacity for self-care. Prognosis is often better for DABP as withdrawal of the medication greatly accelerates clearance of the lesions. Worse prognosis is related to increased number of comorbidities and older age. Our patient’s BP is controlled currently with topical steroids and oral doxycycline.
This case and photo were submitted by Lucas Shapiro, BS, Nova Southeastern University College of Osteopathic Medicine, Tampa, and Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Miyamoto D et al. An Bras Dermatol. 2019 Mar-Apr;94(2):133-46.
2. Moro et al. Biomolecules. 2020 Oct 10;10(10):1432.
3. Verheyden M et al. Acta Derm Venereol. 2020 Aug 17;100(15):adv00224.
and is associated with various predisposing factors, including HLA genes, comorbidities, aging, and trigger factors such as drugs, trauma, radiation, chemotherapy, and infections. The autoimmune reaction is mediated by a dysregulation of T cells in which IgG and IgE autoantibodies form against hemidesmosomal proteins (BP180 and BP230). These autoantibodies induce neutrophil activation, recruitment, and degradation in the basement membrane of the skin.
Typically, patients present with intense pruritus followed by an urticarial or eczematous eruption. Tense blisters and bullae occur commonly on the trunk and extremities. Drug-associated bullous pemphigoid (DABP) is a common manifestation of the disease with histologic and immunologic features similar to those of the idiopathic version. Eruptions can be triggered by systemic or topical medications, and incidence of these reactions may be related to a genetic predisposition for the disease.
Some research suggests that drug-induced changes to the antigenic properties of the epidermal basement membrane result in an augmented immune response, while others point to structural modification in these zones that stimulate the immune system. Thiol- and phenol-based drugs have been largely implicated in the development of DABP because they are capable of structural modification and disruption of the dermo-epidermal junction in the basement membrane.
DABP often presents with patients taking multiple medications. Some of the most common medications are gliptins, PD-1 inhibitors, diuretics, antibiotics, anti-inflammatory drugs, and ACE-inhibitors, and other cardiovascular drugs. DABP may present with mucosal eruptions unlike its idiopathic counterpart that is mostly contained to the skin.
On this patient, two punch biopsies were taken. Histopathology revealed an eosinophil-rich subepidermal blister with a smooth epidermal undersurface consistent with bullous pemphigoid. Direct immunofluorescence was positive with a deposition of IgG and C3 at the epidermal side of salt split basement membrane zone.
Treatment for BP includes high potency topical and systemic steroids. Tetracyclines and niacinamide have been reported to improve the condition. Treatment is tailored to allow for cutaneous healing and control pruritus, but the physician must be mindful of the patient’s comorbidities and capacity for self-care. Prognosis is often better for DABP as withdrawal of the medication greatly accelerates clearance of the lesions. Worse prognosis is related to increased number of comorbidities and older age. Our patient’s BP is controlled currently with topical steroids and oral doxycycline.
This case and photo were submitted by Lucas Shapiro, BS, Nova Southeastern University College of Osteopathic Medicine, Tampa, and Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Miyamoto D et al. An Bras Dermatol. 2019 Mar-Apr;94(2):133-46.
2. Moro et al. Biomolecules. 2020 Oct 10;10(10):1432.
3. Verheyden M et al. Acta Derm Venereol. 2020 Aug 17;100(15):adv00224.
and is associated with various predisposing factors, including HLA genes, comorbidities, aging, and trigger factors such as drugs, trauma, radiation, chemotherapy, and infections. The autoimmune reaction is mediated by a dysregulation of T cells in which IgG and IgE autoantibodies form against hemidesmosomal proteins (BP180 and BP230). These autoantibodies induce neutrophil activation, recruitment, and degradation in the basement membrane of the skin.
Typically, patients present with intense pruritus followed by an urticarial or eczematous eruption. Tense blisters and bullae occur commonly on the trunk and extremities. Drug-associated bullous pemphigoid (DABP) is a common manifestation of the disease with histologic and immunologic features similar to those of the idiopathic version. Eruptions can be triggered by systemic or topical medications, and incidence of these reactions may be related to a genetic predisposition for the disease.
Some research suggests that drug-induced changes to the antigenic properties of the epidermal basement membrane result in an augmented immune response, while others point to structural modification in these zones that stimulate the immune system. Thiol- and phenol-based drugs have been largely implicated in the development of DABP because they are capable of structural modification and disruption of the dermo-epidermal junction in the basement membrane.
DABP often presents with patients taking multiple medications. Some of the most common medications are gliptins, PD-1 inhibitors, diuretics, antibiotics, anti-inflammatory drugs, and ACE-inhibitors, and other cardiovascular drugs. DABP may present with mucosal eruptions unlike its idiopathic counterpart that is mostly contained to the skin.
On this patient, two punch biopsies were taken. Histopathology revealed an eosinophil-rich subepidermal blister with a smooth epidermal undersurface consistent with bullous pemphigoid. Direct immunofluorescence was positive with a deposition of IgG and C3 at the epidermal side of salt split basement membrane zone.
Treatment for BP includes high potency topical and systemic steroids. Tetracyclines and niacinamide have been reported to improve the condition. Treatment is tailored to allow for cutaneous healing and control pruritus, but the physician must be mindful of the patient’s comorbidities and capacity for self-care. Prognosis is often better for DABP as withdrawal of the medication greatly accelerates clearance of the lesions. Worse prognosis is related to increased number of comorbidities and older age. Our patient’s BP is controlled currently with topical steroids and oral doxycycline.
This case and photo were submitted by Lucas Shapiro, BS, Nova Southeastern University College of Osteopathic Medicine, Tampa, and Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Miyamoto D et al. An Bras Dermatol. 2019 Mar-Apr;94(2):133-46.
2. Moro et al. Biomolecules. 2020 Oct 10;10(10):1432.
3. Verheyden M et al. Acta Derm Venereol. 2020 Aug 17;100(15):adv00224.
A 95-year-old White male with hypertension presented with a history of very itchy patches and bullae on the trunk and extremities.
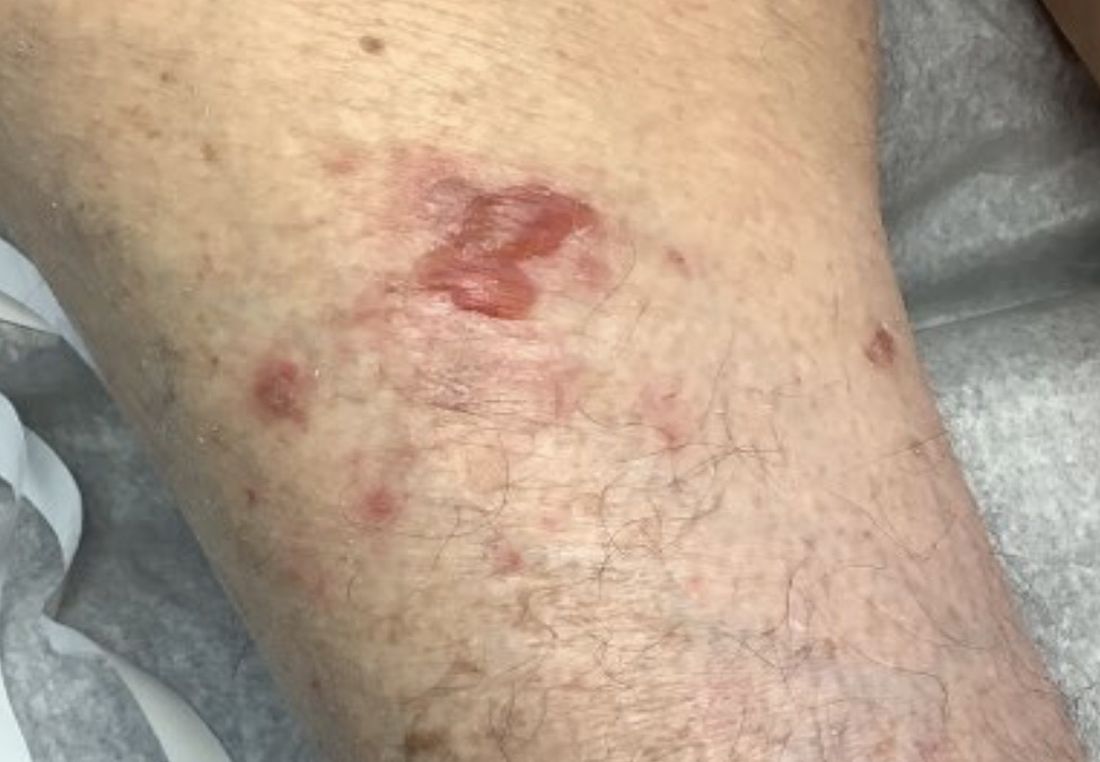
The truth of alcohol consequences
Bad drinking consequence No. 87: Joining the LOTME team
Alcohol and college students go together like peanut butter and jelly. Or peanut butter and chocolate. Or peanut butter and toothpaste. Peanut butter goes with a lot of things.
Naturally, when you combine alcohol and college students, bad decisions are sure to follow. But have you ever wondered just how many bad decisions alcohol causes? A team of researchers from Penn State University, the undisputed champion of poor drinking decisions (trust us, we know), sure has. They’ve even conducted a 4-year study of 1,700 students as they carved a drunken swath through the many fine local drinking establishments, such as East Halls or that one frat house that hosts medieval battle–style ping pong tournaments.
The students were surveyed twice a year throughout the study, and the researchers compiled a list of all the various consequences their subjects experienced. Ultimately, college students will experience an average of 102 consequences from drinking during their 4-year college careers, which is an impressive number. Try thinking up a hundred consequences for anything.
Some consequences are less common than others – we imagine “missing the Renaissance Faire because you felt drunker the morning after than while you were drinking” is pretty low on the list – but more than 96% of students reported that they’d experienced a hangover and that drinking had caused them to say or do embarrassing things. Also, more than 70% said they needed additional alcohol to feel any effect, a potential sign of alcohol use disorder.
Once they had their list, the researchers focused on 12 of the more common and severe consequences, such as blacking out, hangovers, and missing work/class, and asked the study participants how their parents would react to their drinking and those specific consequences. Students who believed their parents would disapprove of alcohol-related consequences actually experienced fewer consequences overall.
College students, it seems, really do care what their parents think, even if they don’t express it, the researchers said. That gives space for parents to offer advice about the consequences of hard drinking, making decisions while drunk, or bringing godawful Fireball whiskey to parties. Seriously, don’t do that. Stuff’s bad, and you should feel bad for bringing it. Your parents raised you better than that.
COVID ‘expert’ discusses data sharing
We interrupt our regularly scheduled programming to bring you this special news event. Elon Musk, the world’s second-most annoying human, is holding a press conference to discuss, of all things, COVID-19.
Reporter: Hey, Mr. Musketeer, what qualifies you to talk about a global pandemic?
EM: As the official king of the Twitterverse, I’m pretty much an expert on any topic.
Reporter: Okay then, Mr. Muskmelon, what can you tell us about the new study in Agricultural Economics, which looked at consumers’ knowledge of local COVID infection rates and their willingness to eat at restaurants?

EM: Well, I know that one of the investigators, Rigoberto Lopez, PhD, of the University of Connecticut, said “no news is bad news.” Restaurants located in cities where local regulations required COVID tracking recovered faster than those in areas that did not, according to data from 87 restaurants in 10 Chinese cities that were gathered between Dec. 1, 2019, and March 27, 2020. Having access to local infection rate data made customers more comfortable going out to eat, the investigators explained.
Second reporter: Interesting, Mr. Muskox, but how about this headline from CNN: “Workers flee China’s biggest iPhone factory over Covid outbreak”? Do you agree with analysts, who said that “the chaos at Zhengzhou could jeopardize Apple and Foxconn’s output in the coming weeks,” as CNN put it?
EM: I did see that a manager at Foxconn, which owns the factory and is known to its friends as Hon Hai Precision Industry, told a Chinese media outlet that “workers are panicking over the spread of the virus at the factory and lack of access to official information.” As we’ve already discussed, no news is bad news.
That’s all the time I have to chat with you today. I’m off to fire some more Twitter employees.
In case you hadn’t already guessed, Vlad Putin is officially more annoying than Elon Musk. We now return to this week’s typical LOTME shenanigans, already in progress.
The deadliest month
With climate change making the world hotter, leading to more heat stroke and organ failure, you would think the summer months would be the most deadly. In reality, though, it’s quite the opposite.

There are multiple factors that make January the most deadly month out of the year, as LiveScience discovered in a recent analysis.
Let’s go through them, shall we?
Respiratory viruses: Robert Glatter, MD, of Lenox Hill Hospital in New York, told LiveScence that winter is the time for illnesses like the flu, bacterial pneumonia, and RSV. Millions of people worldwide die from the flu, according to the CDC. And the World Health Organization reported lower respiratory infections as the fourth-leading cause of death worldwide before COVID came along.
Heart disease: Heart conditions are actually more fatal in the winter months, according to a study published in Circulation. The cold puts more stress on the heart to keep the body warm, which can be a challenge for people who already have preexisting heart conditions.
Space heaters: Dr. Glatter also told Live Science that the use of space heaters could be a factor in the cold winter months since they can lead to carbon monoxide poisoning and even fires. Silent killers.
Holiday season: A time for joy and merriment, certainly, but Christmas et al. have their downsides. By January we’re coming off a 3-month food and alcohol binge, which leads to cardiac stress. There’s also the psychological stress that comes with the season. Sometimes the most wonderful time of the year just isn’t.
So even though summer is hot, fall has hurricanes, and spring tends to have the highest suicide rate, winter still ends up being the deadliest season.
Bad drinking consequence No. 87: Joining the LOTME team
Alcohol and college students go together like peanut butter and jelly. Or peanut butter and chocolate. Or peanut butter and toothpaste. Peanut butter goes with a lot of things.
Naturally, when you combine alcohol and college students, bad decisions are sure to follow. But have you ever wondered just how many bad decisions alcohol causes? A team of researchers from Penn State University, the undisputed champion of poor drinking decisions (trust us, we know), sure has. They’ve even conducted a 4-year study of 1,700 students as they carved a drunken swath through the many fine local drinking establishments, such as East Halls or that one frat house that hosts medieval battle–style ping pong tournaments.
The students were surveyed twice a year throughout the study, and the researchers compiled a list of all the various consequences their subjects experienced. Ultimately, college students will experience an average of 102 consequences from drinking during their 4-year college careers, which is an impressive number. Try thinking up a hundred consequences for anything.
Some consequences are less common than others – we imagine “missing the Renaissance Faire because you felt drunker the morning after than while you were drinking” is pretty low on the list – but more than 96% of students reported that they’d experienced a hangover and that drinking had caused them to say or do embarrassing things. Also, more than 70% said they needed additional alcohol to feel any effect, a potential sign of alcohol use disorder.
Once they had their list, the researchers focused on 12 of the more common and severe consequences, such as blacking out, hangovers, and missing work/class, and asked the study participants how their parents would react to their drinking and those specific consequences. Students who believed their parents would disapprove of alcohol-related consequences actually experienced fewer consequences overall.
College students, it seems, really do care what their parents think, even if they don’t express it, the researchers said. That gives space for parents to offer advice about the consequences of hard drinking, making decisions while drunk, or bringing godawful Fireball whiskey to parties. Seriously, don’t do that. Stuff’s bad, and you should feel bad for bringing it. Your parents raised you better than that.
COVID ‘expert’ discusses data sharing
We interrupt our regularly scheduled programming to bring you this special news event. Elon Musk, the world’s second-most annoying human, is holding a press conference to discuss, of all things, COVID-19.
Reporter: Hey, Mr. Musketeer, what qualifies you to talk about a global pandemic?
EM: As the official king of the Twitterverse, I’m pretty much an expert on any topic.
Reporter: Okay then, Mr. Muskmelon, what can you tell us about the new study in Agricultural Economics, which looked at consumers’ knowledge of local COVID infection rates and their willingness to eat at restaurants?
EM: Well, I know that one of the investigators, Rigoberto Lopez, PhD, of the University of Connecticut, said “no news is bad news.” Restaurants located in cities where local regulations required COVID tracking recovered faster than those in areas that did not, according to data from 87 restaurants in 10 Chinese cities that were gathered between Dec. 1, 2019, and March 27, 2020. Having access to local infection rate data made customers more comfortable going out to eat, the investigators explained.
Second reporter: Interesting, Mr. Muskox, but how about this headline from CNN: “Workers flee China’s biggest iPhone factory over Covid outbreak”? Do you agree with analysts, who said that “the chaos at Zhengzhou could jeopardize Apple and Foxconn’s output in the coming weeks,” as CNN put it?
EM: I did see that a manager at Foxconn, which owns the factory and is known to its friends as Hon Hai Precision Industry, told a Chinese media outlet that “workers are panicking over the spread of the virus at the factory and lack of access to official information.” As we’ve already discussed, no news is bad news.
That’s all the time I have to chat with you today. I’m off to fire some more Twitter employees.
In case you hadn’t already guessed, Vlad Putin is officially more annoying than Elon Musk. We now return to this week’s typical LOTME shenanigans, already in progress.
The deadliest month
With climate change making the world hotter, leading to more heat stroke and organ failure, you would think the summer months would be the most deadly. In reality, though, it’s quite the opposite.
There are multiple factors that make January the most deadly month out of the year, as LiveScience discovered in a recent analysis.
Let’s go through them, shall we?
Respiratory viruses: Robert Glatter, MD, of Lenox Hill Hospital in New York, told LiveScence that winter is the time for illnesses like the flu, bacterial pneumonia, and RSV. Millions of people worldwide die from the flu, according to the CDC. And the World Health Organization reported lower respiratory infections as the fourth-leading cause of death worldwide before COVID came along.
Heart disease: Heart conditions are actually more fatal in the winter months, according to a study published in Circulation. The cold puts more stress on the heart to keep the body warm, which can be a challenge for people who already have preexisting heart conditions.
Space heaters: Dr. Glatter also told Live Science that the use of space heaters could be a factor in the cold winter months since they can lead to carbon monoxide poisoning and even fires. Silent killers.
Holiday season: A time for joy and merriment, certainly, but Christmas et al. have their downsides. By January we’re coming off a 3-month food and alcohol binge, which leads to cardiac stress. There’s also the psychological stress that comes with the season. Sometimes the most wonderful time of the year just isn’t.
So even though summer is hot, fall has hurricanes, and spring tends to have the highest suicide rate, winter still ends up being the deadliest season.
Bad drinking consequence No. 87: Joining the LOTME team
Alcohol and college students go together like peanut butter and jelly. Or peanut butter and chocolate. Or peanut butter and toothpaste. Peanut butter goes with a lot of things.
Naturally, when you combine alcohol and college students, bad decisions are sure to follow. But have you ever wondered just how many bad decisions alcohol causes? A team of researchers from Penn State University, the undisputed champion of poor drinking decisions (trust us, we know), sure has. They’ve even conducted a 4-year study of 1,700 students as they carved a drunken swath through the many fine local drinking establishments, such as East Halls or that one frat house that hosts medieval battle–style ping pong tournaments.
The students were surveyed twice a year throughout the study, and the researchers compiled a list of all the various consequences their subjects experienced. Ultimately, college students will experience an average of 102 consequences from drinking during their 4-year college careers, which is an impressive number. Try thinking up a hundred consequences for anything.
Some consequences are less common than others – we imagine “missing the Renaissance Faire because you felt drunker the morning after than while you were drinking” is pretty low on the list – but more than 96% of students reported that they’d experienced a hangover and that drinking had caused them to say or do embarrassing things. Also, more than 70% said they needed additional alcohol to feel any effect, a potential sign of alcohol use disorder.
Once they had their list, the researchers focused on 12 of the more common and severe consequences, such as blacking out, hangovers, and missing work/class, and asked the study participants how their parents would react to their drinking and those specific consequences. Students who believed their parents would disapprove of alcohol-related consequences actually experienced fewer consequences overall.
College students, it seems, really do care what their parents think, even if they don’t express it, the researchers said. That gives space for parents to offer advice about the consequences of hard drinking, making decisions while drunk, or bringing godawful Fireball whiskey to parties. Seriously, don’t do that. Stuff’s bad, and you should feel bad for bringing it. Your parents raised you better than that.
COVID ‘expert’ discusses data sharing
We interrupt our regularly scheduled programming to bring you this special news event. Elon Musk, the world’s second-most annoying human, is holding a press conference to discuss, of all things, COVID-19.
Reporter: Hey, Mr. Musketeer, what qualifies you to talk about a global pandemic?
EM: As the official king of the Twitterverse, I’m pretty much an expert on any topic.
Reporter: Okay then, Mr. Muskmelon, what can you tell us about the new study in Agricultural Economics, which looked at consumers’ knowledge of local COVID infection rates and their willingness to eat at restaurants?
EM: Well, I know that one of the investigators, Rigoberto Lopez, PhD, of the University of Connecticut, said “no news is bad news.” Restaurants located in cities where local regulations required COVID tracking recovered faster than those in areas that did not, according to data from 87 restaurants in 10 Chinese cities that were gathered between Dec. 1, 2019, and March 27, 2020. Having access to local infection rate data made customers more comfortable going out to eat, the investigators explained.
Second reporter: Interesting, Mr. Muskox, but how about this headline from CNN: “Workers flee China’s biggest iPhone factory over Covid outbreak”? Do you agree with analysts, who said that “the chaos at Zhengzhou could jeopardize Apple and Foxconn’s output in the coming weeks,” as CNN put it?
EM: I did see that a manager at Foxconn, which owns the factory and is known to its friends as Hon Hai Precision Industry, told a Chinese media outlet that “workers are panicking over the spread of the virus at the factory and lack of access to official information.” As we’ve already discussed, no news is bad news.
That’s all the time I have to chat with you today. I’m off to fire some more Twitter employees.
In case you hadn’t already guessed, Vlad Putin is officially more annoying than Elon Musk. We now return to this week’s typical LOTME shenanigans, already in progress.
The deadliest month
With climate change making the world hotter, leading to more heat stroke and organ failure, you would think the summer months would be the most deadly. In reality, though, it’s quite the opposite.
There are multiple factors that make January the most deadly month out of the year, as LiveScience discovered in a recent analysis.
Let’s go through them, shall we?
Respiratory viruses: Robert Glatter, MD, of Lenox Hill Hospital in New York, told LiveScence that winter is the time for illnesses like the flu, bacterial pneumonia, and RSV. Millions of people worldwide die from the flu, according to the CDC. And the World Health Organization reported lower respiratory infections as the fourth-leading cause of death worldwide before COVID came along.
Heart disease: Heart conditions are actually more fatal in the winter months, according to a study published in Circulation. The cold puts more stress on the heart to keep the body warm, which can be a challenge for people who already have preexisting heart conditions.
Space heaters: Dr. Glatter also told Live Science that the use of space heaters could be a factor in the cold winter months since they can lead to carbon monoxide poisoning and even fires. Silent killers.
Holiday season: A time for joy and merriment, certainly, but Christmas et al. have their downsides. By January we’re coming off a 3-month food and alcohol binge, which leads to cardiac stress. There’s also the psychological stress that comes with the season. Sometimes the most wonderful time of the year just isn’t.
So even though summer is hot, fall has hurricanes, and spring tends to have the highest suicide rate, winter still ends up being the deadliest season.
Hyaluronidase for Skin Necrosis Induced by Amiodarone
To the Editor:
Amiodarone is an oral or intravenous (IV) drug commonly used to treat supraventricular and ventricular arrhythmia as well as atrial fibrillation.1 Adverse drug reactions associated with the use of amiodarone include pulmonary, gastrointestinal, thyroid, ocular, neurologic, and cutaneous reactions.1 Long-term use of amiodarone—typically more than 4 months—can lead to slate-gray skin discoloration and photosensitivity, both of which can be reversed with drug withdrawal.2,3 Phlebitis also has been described in less than 3% of patients who receive peripheral IV administration of amiodarone.4
Amiodarone-induced skin necrosis due to extravasation is a rare complication of this antiarrhythmic medication, with only 3 reported cases in the literature according to a PubMed search of articles indexed for MEDLINE using the search terms amiodarone and skin and (necrosis or ischemia or extravasation or reaction).5–7 Although hyaluronidase is a known therapy for extravasation of fluids, including parenteral nutrition and chemotherapy, its use for the treatment of extravasation from amiodarone is not well documented.6 We report a case of skin necrosis of the left dorsal forearm and the left dorsal and ventral hand following infusion of amiodarone through a peripheral IV line, which was treated with injections of hyaluronidase.
A 77-year-old man was admitted to the emergency department for sepsis secondary to cholangitis in the setting of an obstructive gallbladder stone. His medical history was notable for multivessel coronary artery disease and atrial flutter treated with ablation. One day after admission, endoscopic retrograde cholangiopancreatography was attempted and aborted due to atrial fibrillation with rapid ventricular response. A second endoscopic retrograde cholangiopancreatography attempt was made 4 days later, during which the patient underwent cardiac arrest. During this event, amiodarone was administered in a 200-mL solution (1.8 mg/mL) in 5% dextrose through a peripheral IV line in the left forearm. The patient was stabilized and transferred to the intensive care unit.
Twenty-four hours after amiodarone administration, erythema was noted on the left dorsal forearm. Within hours, the digits of the hand became a dark, dusky color, which spread to involve the forearm. Surgical debridement was not deemed necessary; the left arm was elevated, and warm compresses were applied regularly. Within the next week, the skin of the left hand and dorsal forearm had progressively worsened and took on a well-demarcated, dusky blue hue surrounded by an erythematous border involving the proximal forearm and upper arm (Figure 1A). The skin was fragile and had overlying bullae (Figure 1B).

Hyaluronidase (1000 U) was injected into the surrounding areas of erythema, which resolved from the left proximal forearm to the elbow within 2 days after injection (Figure 2). The dusky violaceous patches were persistent, and the necrotic bullae were unchanged. Hyaluronidase (1000 U) was injected into necrotic skin of the left dorsal forearm and dorsal and ventral hand. No improvement was noted on subsequent evaluations of this area. While still an inpatient, he received wound care and twice-daily Doppler ultrasounds in the areas of necrosis. The patient lost sensation in the left hand with increased soft tissue necrosis and developed an eschar on the left dorsal forearm. Due to the progressive loss of function and necrosis, a partial forearm amputation was performed that healed well, and the patient experienced improvement in range of motion of the left upper extremity.

Well-known adverse reactions of amiodarone treatment include pulmonary fibrosis, hepatic dysfunction, hypothyroidism and hyperthyroidism, peripheral neuropathy, and corneal deposits.1 Cutaneous adverse reactions include photosensitivity (phototoxic and photoallergic reactions), hyperpigmentation, pseudoporphyria, and linear IgA bullous dermatosis. Less commonly, it also can cause urticaria, pruritus, erythema nodosum, purpura, and toxic epidermal necrolysis.3 Amiodarone-induced skin necrosis is rare, first described by Russell and Saltissi5 in 2006 in a 60-year-old man who developed dark discoloration and edema of the forearm 24 hours after initiation of an amiodarone peripheral IV. The patient was treated with hot or cold packs and steroid cream per the pharmaceutical company’s recommendations; however, patient outcomes were not discussed.5 A 77-year-old man who received subcutaneous amiodarone due to misplaced vascular access developed edema and bullae of the forearm followed by tissue necrosis, resulting in notably reduced mobility.6 Fox et al7 described a 60-year-old man who developed atrial fibrillation after emergent spinal fusion and laminectomy. He received intradermal hyaluronidase administration within 24 hours of developing severe pain from extravasation induced by amiodarone with no adverse outcomes and full recovery.7
There are numerous properties of amiodarone that may have resulted in the skin necrosis seen in these cases. The acidic pH (3.5–4.5) of amiodarone can contribute to coagulative necrosis, cellular desiccation, eschar formation, and edema.8 It also can contain additives such as polysorbate and benzyl alcohol, which may contribute to the drug’s vesicant properties.9
Current recommendations for IV administration of amiodarone include delivery through a central vein with high concentrations (>2 mg/mL) because peripheral infusion is slower and may cause phlebitis.4 In-line filters also may be a potential method of preventing phlebitis with peripheral IV administration of amiodarone.10 Extravasation of amiodarone can be treated nonpharmacologically with limb elevation and warm compresses, as these methods may promote vasodilation and enhance drug removal.5-7 However, when extravasation leads to progressive erythema and skin necrosis or is refractory to these therapies, intradermal injection of hyaluronidase should be considered. Hyaluronidase mediates the degradation of hyaluronic acid in the extracellular matrix, allowing for increased permeability of injected fluids into tissues and diluting the concentration of toxins at the site of exposure.9,11 It has been used to treat extravasation of fluids such as parenteral nutrition, electrolyte infusion, antibiotics, aminophylline, mannitol, and chemotherapy.11 Although hyaluronidase has been recognized as therapeutic for extravasation, there is no established consistent dosing or proper technique. In the setting of infiltration of chemotherapy, doses of hyaluronidase ranging from 150 to 1500 U/mL can be subcutaneously or intradermally injected into the site within 1 hour of extravasation. Side effects of using hyaluronidase are rare, including local pruritus, allergic reactions, urticaria, and angioedema.12
The patient described by Fox et al7 who fully recovered from amiodarone extravasation after hyaluronidase injections likely benefited from quick intervention, as he received amiodarone within 24 hours of the care team identifying initial erythema. Although our patient did have improvement of the areas of erythema on the forearm, evidence of skin and subcutaneous tissue necrosis on the left hand and proximal forearm was already apparent and not reversible, most likely caused by late intervention of intradermal hyaluronidase almost a week after the extravasation event. It is important to identify amiodarone as the source of extravasation and administer intradermal hyaluronidase in a timely fashion for extravasation refractory to conventional measurements to prevent progression to severe tissue damage.
Our case draws attention to the risk for skin necrosis with peripheral IV administration of amiodarone. Interventions include limb elevation, warm compresses, and consideration of intradermal hyaluronidase within 24 hours of extravasation, as this may reduce the severity of subsequent tissue damage with minimal side effects.
- Epstein AE, Olshansky B, Naccarelli GV, et al. Practical management guide for clinicians who treat patients with amiodarone. Am J Med. 2016;129:468-475. doi:10.1016/j.amjmed.2015.08.039
- Harris L, McKenna WJ, Rowland E, et al. Side effects of long-term amiodarone therapy. Circulation. 1983;67:45-51. doi:10.1161/01.cir.67.1.45
- Jaworski K, Walecka I, Rudnicka L, et al. Cutaneous adverse reactions of amiodarone. Med Sci Monit. 2014;20:2369-2372. doi:10.12659/MSM.890881
- Kowey Peter R, Marinchak Roger A, Rials Seth J, et al. Intravenous amiodarone. J Am Coll Cardiol. 1997;29:1190-1198. doi:10.1016/S0735-1097(97)00069-7
- Russell SJ, Saltissi S. Amiodarone induced skin necrosis. Heart. 2006;92:1395. doi:10.1136/hrt.2005.086157
- Grove EL. Skin necrosis and consequences of accidental subcutaneous administration of amiodarone. Ugeskr Laeger. 2015;177:V66928.
- Fox AN, Villanueva R, Miller JL. Management of amiodarone extravasation with intradermal hyaluronidase. Am J Health Syst Pharm. 2017;74:1545-1548. doi:10.2146/ajhp160737
- Reynolds PM, MacLaren R, Mueller SW, et al. Management of extravasation injuries: a focused evaluation of noncytotoxic medications. Pharmacotherapy. 2014;34:617-632. doi:https://doi.org/10.1002/phar.1396
- Le A, Patel S. Extravasation of noncytotoxic drugs: a review of the literature. Ann Pharmacother. 2014;48:870-886. doi:10.1177/1060028014527820
- Slim AM, Roth JE, Duffy B, et al. The incidence of phlebitis with intravenous amiodarone at guideline dose recommendations. Mil Med. 2007;172:1279-1283.
- Girish KS, Kemparaju K. The magic glue hyaluronan and its eraser hyaluronidase: a biological overview. Life Sci. 2007;80:1921-1943. doi:10.1016/j.lfs.2007.02.037
- Jung H. Hyaluronidase: an overview of its properties, applications, and side effects. Arch Plast Surg. 2020;47:297-300. doi:10.5999/aps.2020.00752
To the Editor:
Amiodarone is an oral or intravenous (IV) drug commonly used to treat supraventricular and ventricular arrhythmia as well as atrial fibrillation.1 Adverse drug reactions associated with the use of amiodarone include pulmonary, gastrointestinal, thyroid, ocular, neurologic, and cutaneous reactions.1 Long-term use of amiodarone—typically more than 4 months—can lead to slate-gray skin discoloration and photosensitivity, both of which can be reversed with drug withdrawal.2,3 Phlebitis also has been described in less than 3% of patients who receive peripheral IV administration of amiodarone.4
Amiodarone-induced skin necrosis due to extravasation is a rare complication of this antiarrhythmic medication, with only 3 reported cases in the literature according to a PubMed search of articles indexed for MEDLINE using the search terms amiodarone and skin and (necrosis or ischemia or extravasation or reaction).5–7 Although hyaluronidase is a known therapy for extravasation of fluids, including parenteral nutrition and chemotherapy, its use for the treatment of extravasation from amiodarone is not well documented.6 We report a case of skin necrosis of the left dorsal forearm and the left dorsal and ventral hand following infusion of amiodarone through a peripheral IV line, which was treated with injections of hyaluronidase.
A 77-year-old man was admitted to the emergency department for sepsis secondary to cholangitis in the setting of an obstructive gallbladder stone. His medical history was notable for multivessel coronary artery disease and atrial flutter treated with ablation. One day after admission, endoscopic retrograde cholangiopancreatography was attempted and aborted due to atrial fibrillation with rapid ventricular response. A second endoscopic retrograde cholangiopancreatography attempt was made 4 days later, during which the patient underwent cardiac arrest. During this event, amiodarone was administered in a 200-mL solution (1.8 mg/mL) in 5% dextrose through a peripheral IV line in the left forearm. The patient was stabilized and transferred to the intensive care unit.
Twenty-four hours after amiodarone administration, erythema was noted on the left dorsal forearm. Within hours, the digits of the hand became a dark, dusky color, which spread to involve the forearm. Surgical debridement was not deemed necessary; the left arm was elevated, and warm compresses were applied regularly. Within the next week, the skin of the left hand and dorsal forearm had progressively worsened and took on a well-demarcated, dusky blue hue surrounded by an erythematous border involving the proximal forearm and upper arm (Figure 1A). The skin was fragile and had overlying bullae (Figure 1B).
Hyaluronidase (1000 U) was injected into the surrounding areas of erythema, which resolved from the left proximal forearm to the elbow within 2 days after injection (Figure 2). The dusky violaceous patches were persistent, and the necrotic bullae were unchanged. Hyaluronidase (1000 U) was injected into necrotic skin of the left dorsal forearm and dorsal and ventral hand. No improvement was noted on subsequent evaluations of this area. While still an inpatient, he received wound care and twice-daily Doppler ultrasounds in the areas of necrosis. The patient lost sensation in the left hand with increased soft tissue necrosis and developed an eschar on the left dorsal forearm. Due to the progressive loss of function and necrosis, a partial forearm amputation was performed that healed well, and the patient experienced improvement in range of motion of the left upper extremity.
Well-known adverse reactions of amiodarone treatment include pulmonary fibrosis, hepatic dysfunction, hypothyroidism and hyperthyroidism, peripheral neuropathy, and corneal deposits.1 Cutaneous adverse reactions include photosensitivity (phototoxic and photoallergic reactions), hyperpigmentation, pseudoporphyria, and linear IgA bullous dermatosis. Less commonly, it also can cause urticaria, pruritus, erythema nodosum, purpura, and toxic epidermal necrolysis.3 Amiodarone-induced skin necrosis is rare, first described by Russell and Saltissi5 in 2006 in a 60-year-old man who developed dark discoloration and edema of the forearm 24 hours after initiation of an amiodarone peripheral IV. The patient was treated with hot or cold packs and steroid cream per the pharmaceutical company’s recommendations; however, patient outcomes were not discussed.5 A 77-year-old man who received subcutaneous amiodarone due to misplaced vascular access developed edema and bullae of the forearm followed by tissue necrosis, resulting in notably reduced mobility.6 Fox et al7 described a 60-year-old man who developed atrial fibrillation after emergent spinal fusion and laminectomy. He received intradermal hyaluronidase administration within 24 hours of developing severe pain from extravasation induced by amiodarone with no adverse outcomes and full recovery.7
There are numerous properties of amiodarone that may have resulted in the skin necrosis seen in these cases. The acidic pH (3.5–4.5) of amiodarone can contribute to coagulative necrosis, cellular desiccation, eschar formation, and edema.8 It also can contain additives such as polysorbate and benzyl alcohol, which may contribute to the drug’s vesicant properties.9
Current recommendations for IV administration of amiodarone include delivery through a central vein with high concentrations (>2 mg/mL) because peripheral infusion is slower and may cause phlebitis.4 In-line filters also may be a potential method of preventing phlebitis with peripheral IV administration of amiodarone.10 Extravasation of amiodarone can be treated nonpharmacologically with limb elevation and warm compresses, as these methods may promote vasodilation and enhance drug removal.5-7 However, when extravasation leads to progressive erythema and skin necrosis or is refractory to these therapies, intradermal injection of hyaluronidase should be considered. Hyaluronidase mediates the degradation of hyaluronic acid in the extracellular matrix, allowing for increased permeability of injected fluids into tissues and diluting the concentration of toxins at the site of exposure.9,11 It has been used to treat extravasation of fluids such as parenteral nutrition, electrolyte infusion, antibiotics, aminophylline, mannitol, and chemotherapy.11 Although hyaluronidase has been recognized as therapeutic for extravasation, there is no established consistent dosing or proper technique. In the setting of infiltration of chemotherapy, doses of hyaluronidase ranging from 150 to 1500 U/mL can be subcutaneously or intradermally injected into the site within 1 hour of extravasation. Side effects of using hyaluronidase are rare, including local pruritus, allergic reactions, urticaria, and angioedema.12
The patient described by Fox et al7 who fully recovered from amiodarone extravasation after hyaluronidase injections likely benefited from quick intervention, as he received amiodarone within 24 hours of the care team identifying initial erythema. Although our patient did have improvement of the areas of erythema on the forearm, evidence of skin and subcutaneous tissue necrosis on the left hand and proximal forearm was already apparent and not reversible, most likely caused by late intervention of intradermal hyaluronidase almost a week after the extravasation event. It is important to identify amiodarone as the source of extravasation and administer intradermal hyaluronidase in a timely fashion for extravasation refractory to conventional measurements to prevent progression to severe tissue damage.
Our case draws attention to the risk for skin necrosis with peripheral IV administration of amiodarone. Interventions include limb elevation, warm compresses, and consideration of intradermal hyaluronidase within 24 hours of extravasation, as this may reduce the severity of subsequent tissue damage with minimal side effects.
To the Editor:
Amiodarone is an oral or intravenous (IV) drug commonly used to treat supraventricular and ventricular arrhythmia as well as atrial fibrillation.1 Adverse drug reactions associated with the use of amiodarone include pulmonary, gastrointestinal, thyroid, ocular, neurologic, and cutaneous reactions.1 Long-term use of amiodarone—typically more than 4 months—can lead to slate-gray skin discoloration and photosensitivity, both of which can be reversed with drug withdrawal.2,3 Phlebitis also has been described in less than 3% of patients who receive peripheral IV administration of amiodarone.4
Amiodarone-induced skin necrosis due to extravasation is a rare complication of this antiarrhythmic medication, with only 3 reported cases in the literature according to a PubMed search of articles indexed for MEDLINE using the search terms amiodarone and skin and (necrosis or ischemia or extravasation or reaction).5–7 Although hyaluronidase is a known therapy for extravasation of fluids, including parenteral nutrition and chemotherapy, its use for the treatment of extravasation from amiodarone is not well documented.6 We report a case of skin necrosis of the left dorsal forearm and the left dorsal and ventral hand following infusion of amiodarone through a peripheral IV line, which was treated with injections of hyaluronidase.
A 77-year-old man was admitted to the emergency department for sepsis secondary to cholangitis in the setting of an obstructive gallbladder stone. His medical history was notable for multivessel coronary artery disease and atrial flutter treated with ablation. One day after admission, endoscopic retrograde cholangiopancreatography was attempted and aborted due to atrial fibrillation with rapid ventricular response. A second endoscopic retrograde cholangiopancreatography attempt was made 4 days later, during which the patient underwent cardiac arrest. During this event, amiodarone was administered in a 200-mL solution (1.8 mg/mL) in 5% dextrose through a peripheral IV line in the left forearm. The patient was stabilized and transferred to the intensive care unit.
Twenty-four hours after amiodarone administration, erythema was noted on the left dorsal forearm. Within hours, the digits of the hand became a dark, dusky color, which spread to involve the forearm. Surgical debridement was not deemed necessary; the left arm was elevated, and warm compresses were applied regularly. Within the next week, the skin of the left hand and dorsal forearm had progressively worsened and took on a well-demarcated, dusky blue hue surrounded by an erythematous border involving the proximal forearm and upper arm (Figure 1A). The skin was fragile and had overlying bullae (Figure 1B).
Hyaluronidase (1000 U) was injected into the surrounding areas of erythema, which resolved from the left proximal forearm to the elbow within 2 days after injection (Figure 2). The dusky violaceous patches were persistent, and the necrotic bullae were unchanged. Hyaluronidase (1000 U) was injected into necrotic skin of the left dorsal forearm and dorsal and ventral hand. No improvement was noted on subsequent evaluations of this area. While still an inpatient, he received wound care and twice-daily Doppler ultrasounds in the areas of necrosis. The patient lost sensation in the left hand with increased soft tissue necrosis and developed an eschar on the left dorsal forearm. Due to the progressive loss of function and necrosis, a partial forearm amputation was performed that healed well, and the patient experienced improvement in range of motion of the left upper extremity.
Well-known adverse reactions of amiodarone treatment include pulmonary fibrosis, hepatic dysfunction, hypothyroidism and hyperthyroidism, peripheral neuropathy, and corneal deposits.1 Cutaneous adverse reactions include photosensitivity (phototoxic and photoallergic reactions), hyperpigmentation, pseudoporphyria, and linear IgA bullous dermatosis. Less commonly, it also can cause urticaria, pruritus, erythema nodosum, purpura, and toxic epidermal necrolysis.3 Amiodarone-induced skin necrosis is rare, first described by Russell and Saltissi5 in 2006 in a 60-year-old man who developed dark discoloration and edema of the forearm 24 hours after initiation of an amiodarone peripheral IV. The patient was treated with hot or cold packs and steroid cream per the pharmaceutical company’s recommendations; however, patient outcomes were not discussed.5 A 77-year-old man who received subcutaneous amiodarone due to misplaced vascular access developed edema and bullae of the forearm followed by tissue necrosis, resulting in notably reduced mobility.6 Fox et al7 described a 60-year-old man who developed atrial fibrillation after emergent spinal fusion and laminectomy. He received intradermal hyaluronidase administration within 24 hours of developing severe pain from extravasation induced by amiodarone with no adverse outcomes and full recovery.7
There are numerous properties of amiodarone that may have resulted in the skin necrosis seen in these cases. The acidic pH (3.5–4.5) of amiodarone can contribute to coagulative necrosis, cellular desiccation, eschar formation, and edema.8 It also can contain additives such as polysorbate and benzyl alcohol, which may contribute to the drug’s vesicant properties.9
Current recommendations for IV administration of amiodarone include delivery through a central vein with high concentrations (>2 mg/mL) because peripheral infusion is slower and may cause phlebitis.4 In-line filters also may be a potential method of preventing phlebitis with peripheral IV administration of amiodarone.10 Extravasation of amiodarone can be treated nonpharmacologically with limb elevation and warm compresses, as these methods may promote vasodilation and enhance drug removal.5-7 However, when extravasation leads to progressive erythema and skin necrosis or is refractory to these therapies, intradermal injection of hyaluronidase should be considered. Hyaluronidase mediates the degradation of hyaluronic acid in the extracellular matrix, allowing for increased permeability of injected fluids into tissues and diluting the concentration of toxins at the site of exposure.9,11 It has been used to treat extravasation of fluids such as parenteral nutrition, electrolyte infusion, antibiotics, aminophylline, mannitol, and chemotherapy.11 Although hyaluronidase has been recognized as therapeutic for extravasation, there is no established consistent dosing or proper technique. In the setting of infiltration of chemotherapy, doses of hyaluronidase ranging from 150 to 1500 U/mL can be subcutaneously or intradermally injected into the site within 1 hour of extravasation. Side effects of using hyaluronidase are rare, including local pruritus, allergic reactions, urticaria, and angioedema.12
The patient described by Fox et al7 who fully recovered from amiodarone extravasation after hyaluronidase injections likely benefited from quick intervention, as he received amiodarone within 24 hours of the care team identifying initial erythema. Although our patient did have improvement of the areas of erythema on the forearm, evidence of skin and subcutaneous tissue necrosis on the left hand and proximal forearm was already apparent and not reversible, most likely caused by late intervention of intradermal hyaluronidase almost a week after the extravasation event. It is important to identify amiodarone as the source of extravasation and administer intradermal hyaluronidase in a timely fashion for extravasation refractory to conventional measurements to prevent progression to severe tissue damage.
Our case draws attention to the risk for skin necrosis with peripheral IV administration of amiodarone. Interventions include limb elevation, warm compresses, and consideration of intradermal hyaluronidase within 24 hours of extravasation, as this may reduce the severity of subsequent tissue damage with minimal side effects.
- Epstein AE, Olshansky B, Naccarelli GV, et al. Practical management guide for clinicians who treat patients with amiodarone. Am J Med. 2016;129:468-475. doi:10.1016/j.amjmed.2015.08.039
- Harris L, McKenna WJ, Rowland E, et al. Side effects of long-term amiodarone therapy. Circulation. 1983;67:45-51. doi:10.1161/01.cir.67.1.45
- Jaworski K, Walecka I, Rudnicka L, et al. Cutaneous adverse reactions of amiodarone. Med Sci Monit. 2014;20:2369-2372. doi:10.12659/MSM.890881
- Kowey Peter R, Marinchak Roger A, Rials Seth J, et al. Intravenous amiodarone. J Am Coll Cardiol. 1997;29:1190-1198. doi:10.1016/S0735-1097(97)00069-7
- Russell SJ, Saltissi S. Amiodarone induced skin necrosis. Heart. 2006;92:1395. doi:10.1136/hrt.2005.086157
- Grove EL. Skin necrosis and consequences of accidental subcutaneous administration of amiodarone. Ugeskr Laeger. 2015;177:V66928.
- Fox AN, Villanueva R, Miller JL. Management of amiodarone extravasation with intradermal hyaluronidase. Am J Health Syst Pharm. 2017;74:1545-1548. doi:10.2146/ajhp160737
- Reynolds PM, MacLaren R, Mueller SW, et al. Management of extravasation injuries: a focused evaluation of noncytotoxic medications. Pharmacotherapy. 2014;34:617-632. doi:https://doi.org/10.1002/phar.1396
- Le A, Patel S. Extravasation of noncytotoxic drugs: a review of the literature. Ann Pharmacother. 2014;48:870-886. doi:10.1177/1060028014527820
- Slim AM, Roth JE, Duffy B, et al. The incidence of phlebitis with intravenous amiodarone at guideline dose recommendations. Mil Med. 2007;172:1279-1283.
- Girish KS, Kemparaju K. The magic glue hyaluronan and its eraser hyaluronidase: a biological overview. Life Sci. 2007;80:1921-1943. doi:10.1016/j.lfs.2007.02.037
- Jung H. Hyaluronidase: an overview of its properties, applications, and side effects. Arch Plast Surg. 2020;47:297-300. doi:10.5999/aps.2020.00752
- Epstein AE, Olshansky B, Naccarelli GV, et al. Practical management guide for clinicians who treat patients with amiodarone. Am J Med. 2016;129:468-475. doi:10.1016/j.amjmed.2015.08.039
- Harris L, McKenna WJ, Rowland E, et al. Side effects of long-term amiodarone therapy. Circulation. 1983;67:45-51. doi:10.1161/01.cir.67.1.45
- Jaworski K, Walecka I, Rudnicka L, et al. Cutaneous adverse reactions of amiodarone. Med Sci Monit. 2014;20:2369-2372. doi:10.12659/MSM.890881
- Kowey Peter R, Marinchak Roger A, Rials Seth J, et al. Intravenous amiodarone. J Am Coll Cardiol. 1997;29:1190-1198. doi:10.1016/S0735-1097(97)00069-7
- Russell SJ, Saltissi S. Amiodarone induced skin necrosis. Heart. 2006;92:1395. doi:10.1136/hrt.2005.086157
- Grove EL. Skin necrosis and consequences of accidental subcutaneous administration of amiodarone. Ugeskr Laeger. 2015;177:V66928.
- Fox AN, Villanueva R, Miller JL. Management of amiodarone extravasation with intradermal hyaluronidase. Am J Health Syst Pharm. 2017;74:1545-1548. doi:10.2146/ajhp160737
- Reynolds PM, MacLaren R, Mueller SW, et al. Management of extravasation injuries: a focused evaluation of noncytotoxic medications. Pharmacotherapy. 2014;34:617-632. doi:https://doi.org/10.1002/phar.1396
- Le A, Patel S. Extravasation of noncytotoxic drugs: a review of the literature. Ann Pharmacother. 2014;48:870-886. doi:10.1177/1060028014527820
- Slim AM, Roth JE, Duffy B, et al. The incidence of phlebitis with intravenous amiodarone at guideline dose recommendations. Mil Med. 2007;172:1279-1283.
- Girish KS, Kemparaju K. The magic glue hyaluronan and its eraser hyaluronidase: a biological overview. Life Sci. 2007;80:1921-1943. doi:10.1016/j.lfs.2007.02.037
- Jung H. Hyaluronidase: an overview of its properties, applications, and side effects. Arch Plast Surg. 2020;47:297-300. doi:10.5999/aps.2020.00752
Practice Points
- Intravenous amiodarone administered peripherally can induce skin extravasation, leading to necrosis.
- Dermatologists should be aware that early intervention with intradermal hyaluronidase may reduce the severity of tissue damage caused by amiodarone-induced skin necrosis.

Genital Lentiginosis: A Benign Pigmentary Abnormality Often Raising Concern for Melanoma
To the Editor:
Genital lentiginosis (also known as mucosal melanotic macules, vulvar melanosis, penile melanosis, and penile lentigines) occurs in men and women.1 Lesions present in adult life as multifocal, asymmetrical, pigmented patches that can have a mottled appearance or exhibit skip areas. The irregular appearance of the pigmented areas often raises concern for melanoma. Biopsy reveals increased pigmentation along the basal layer of the epidermis; the irregular distribution of single melanocytes and pagetoid spread typical of melanoma in situ is not identified.

Genital lentiginosis usually occurs as an isolated finding; however, the condition can be a manifestation of Laugier-Hunziker syndrome, Carney complex, and Bannayan-Riley-Ruvalcaba syndrome.1-3 When it occurs as an isolated finding, the patient can be reassured and treatment is unnecessary. Because genital lentiginosis may mimic the appearance of melanoma, it is important for physicians to differentiate the two and make a correct diagnosis. We present a case of genital lentiginosis that mimicked vulvar melanoma.
.")
A 64-year-old woman was referred by her gynecologist to dermatology to rule out vulvar melanoma. The patient had a history of hypothyroidism and hypercholesterolemia but was otherwise in good health. Genital examination revealed asymptomatic pigmented macules and patches of unknown duration (Figure 1). Specimens were taken from 3 areas by punch biopsy to clarify the diagnosis. All 3 specimens showed identical features including basilar pigmentation, occasional melanophages in the papillary dermis, and no evidence of nests or pagetoid spread of atypical melanocytes (Figures 2 and 3). Histologic findings were diagnostic for genital lentiginosis. The patient was reassured, and no treatment was provided. At 6-month follow-up there was no change in clinical appearance.
.")
Genital lentiginosis is characterized by brown lesions that can have a mottled appearance and often are associated with skip areas.1 Lesions can be strikingly irregular and darkly pigmented.
Although the lesions of genital lentiginosis most often are isolated findings, they can be a clue to several uncommon syndromes such as autosomal-dominant Bannayan-Riley-Ruvalcaba syndrome, which is associated with genital lentiginosis, intestinal polyposis, and macrocephaly.3 Vascular malformations, lipomatosis, verrucal keratoses, and acrochordons can occur. Bannayan-Riley-Ruvalcaba syndrome and Cowden syndrome may share genetic linkage; mutations in the tumor suppressor PTEN (phosphatase and tensin homolog deleted on chromosome ten) has been implicated in both syndromes.4 Underlying Carney complex should be excluded when genital lentiginosis is encountered.
Genital lentiginosis is idiopathic in most instances, but reports of lesions occurring after annular lichen planus suggest a possible mechanism.5 The disappearance of lentigines after imatinib therapy suggests a role for c-kit, a receptor tyrosine kinase that is involved in intracellular signaling, in some cases.6 At times, lesions can simulate trichrome vitiligo or have a reticulate pattern.7
Men and women present at different points in the course of disease. Men often present with penile lesions 14 years after onset, on average; they notice a gradual increase in the size of lesions. Because women can have greater difficulty self-examining the genital region, they tend to present much later in the course but often within a few months after initial inspection.1,8
Genital lentiginosis can mimic melanoma with nonhomogeneous pigmentation, asymmetry, and unilateral distribution, which makes dermoscopic assessment of colors helpful in narrowing the differential diagnosis. Melanoma is associated with combinations of gray, red, blue, and white, which are not found in genital lentiginosis.9
Biopsy of a genital lentigo is diagnostic, distinguishing the lesion from melanoma—failing to reveal the atypical melanocytes and pagetoid spread characteristic of melanoma in situ. Histologic findings can cause diagnostic difficulties when concurrent lichen sclerosus is associated with genital lentigines or nevi.10
Lentigines on sun-damaged skin or in the setting of xeroderma pigmentosum have been associated with melanoma,11-13 but genital lentigines are not considered a form of precancerous melanosis. In women, early diagnosis is important when there is concern for melanoma because the prognosis for vulvar melanoma is improved in thin lesions.14
Other entities in the differential include secondary syphilis, which commonly presents as macules and scaly papules and can be found on mucosal surfaces such as the oral cavity,15 as well as Kaposi sarcoma, which is characterized by purplish, brown, or black macules, plaques, and nodules, more commonly in immunosuppressed patients.16
To avoid unwarranted concern and unnecessary surgery, dermatologists should be aware of genital lentigines and their characteristic presentation in adults.
- Hwang L, Wilson H, Orengo I. Off-center fold: irregular, pigmented genital macules. Arch Dermatol. 2000;136:1559-1564. doi:10.1001/archderm.136.12.1559-b
- Rhodes AR, Silverman RA, Harrist TJ, et al. Mucocutaneous lentigines, cardiomucocutaneous myxomas, and multiple blue nevi: the “LAMB” syndrome. J Am Acad Dermatol. 1984;10:72-82. doi:10.1016/s0190-9622(84)80047-x
- Erkek E, Hizel S, Sanl C, et al. Clinical and histopathological findings in Bannayan-Riley-Ruvalcaba syndrome. J Am Acad Dermatol. 2005;53:639-643. doi:10.1016/j.jaad.2005.06.022
- Blum RR, Rahimizadeh A, Kardon N, et al. Genital lentigines in a 6-year-old boy with a family history of Cowden’s disease: clinical and genetic evidence of the linkage between Bannayan-Riley-Ruvalcaba syndrome and Cowden’s disease. J Cutan Med Surg. 2001;5:228-230. doi:10.1177/120347540100500307
- Isbary G, Dyall-Smith D, Coras-Stepanek B, et al. Penile lentigo (genital mucosal macule) following annular lichen planus: a possible association? Australas J Dermatol. 2014;55:159-161. doi:10.1111/ajd.12169
- Campbell T, Felsten L, Moore J. Disappearance of lentigines in a patient receiving imatinib treatment for familial gastrointestinal stromal tumor syndrome. Arch Dermatol. 2009;145:1313-1316. doi:10.1001/archdermatol.2009.263
- Romero- A, R, , et al. Reticulate genital pigmentation associated with localized vitiligo. Arch Dermatol. 2010; 146:574-575. doi:10.1001/archdermatol.2010.69
- Barnhill RL, Albert LS, Shama SK, et al. Genital lentiginosis: a clinical and histopathologic study. J Am Acad Dermatol. 1990;22:453-460. doi:10.1016/0190-9622(90)70064-o
- De Giorgi V, Gori A, Salvati L, et al. Clinical and dermoscopic features of vulvar melanosis over the last 20 years. JAMA Dermatol. 2020;156:1185–1191. doi:10.1001/jamadermatol.2020.2528
- El Shabrawi-Caelen L, Soyer HP, Schaeppi H, et al. Genital lentigines and melanocytic nevi with superimposed lichen sclerosus: a diagnostic challenge. J Am Acad Dermatol. 2004;50:690-694. doi:10.1016/j.jaad.2003.09.034
- Shatkin M, Helm MF, Muhlbauer A, et al. Solar lentigo evolving into fatal metastatic melanoma in a patient who initially refused surgery. N A J Med Sci. 2020;1:28-31. doi:10.7156/najms.2020.1301028
- Stern JB, Peck GL, Haupt HM, et al. Malignant melanoma in xeroderma pigmentosum: search for a precursor lesion. J Am Acad Dermatol. 1993;28:591-594. doi:10.1016/0190-9622(93)70079-9
- Byrom L, Barksdale S, Weedon D, et al. Unstable solar lentigo: a defined separate entity. Australas J Dermatol. 2016;57:229-234. doi:10.1111/ajd.12447
- Panizzon RG. Vulvar melanoma. Semin Dermatol. 1996;15:67-70. doi:10.1016/s1085-5629(96)80021-6
- Chapel TA. The signs and symptoms of secondary syphilis. Sex Transm Dis. 1980;7:161-164. doi:10.1097/00007435-198010000-00002
- Schwartz RA. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151. doi:10.1002/jso.20090
To the Editor:
Genital lentiginosis (also known as mucosal melanotic macules, vulvar melanosis, penile melanosis, and penile lentigines) occurs in men and women.1 Lesions present in adult life as multifocal, asymmetrical, pigmented patches that can have a mottled appearance or exhibit skip areas. The irregular appearance of the pigmented areas often raises concern for melanoma. Biopsy reveals increased pigmentation along the basal layer of the epidermis; the irregular distribution of single melanocytes and pagetoid spread typical of melanoma in situ is not identified.
Genital lentiginosis usually occurs as an isolated finding; however, the condition can be a manifestation of Laugier-Hunziker syndrome, Carney complex, and Bannayan-Riley-Ruvalcaba syndrome.1-3 When it occurs as an isolated finding, the patient can be reassured and treatment is unnecessary. Because genital lentiginosis may mimic the appearance of melanoma, it is important for physicians to differentiate the two and make a correct diagnosis. We present a case of genital lentiginosis that mimicked vulvar melanoma.
A 64-year-old woman was referred by her gynecologist to dermatology to rule out vulvar melanoma. The patient had a history of hypothyroidism and hypercholesterolemia but was otherwise in good health. Genital examination revealed asymptomatic pigmented macules and patches of unknown duration (Figure 1). Specimens were taken from 3 areas by punch biopsy to clarify the diagnosis. All 3 specimens showed identical features including basilar pigmentation, occasional melanophages in the papillary dermis, and no evidence of nests or pagetoid spread of atypical melanocytes (Figures 2 and 3). Histologic findings were diagnostic for genital lentiginosis. The patient was reassured, and no treatment was provided. At 6-month follow-up there was no change in clinical appearance.
Genital lentiginosis is characterized by brown lesions that can have a mottled appearance and often are associated with skip areas.1 Lesions can be strikingly irregular and darkly pigmented.
Although the lesions of genital lentiginosis most often are isolated findings, they can be a clue to several uncommon syndromes such as autosomal-dominant Bannayan-Riley-Ruvalcaba syndrome, which is associated with genital lentiginosis, intestinal polyposis, and macrocephaly.3 Vascular malformations, lipomatosis, verrucal keratoses, and acrochordons can occur. Bannayan-Riley-Ruvalcaba syndrome and Cowden syndrome may share genetic linkage; mutations in the tumor suppressor PTEN (phosphatase and tensin homolog deleted on chromosome ten) has been implicated in both syndromes.4 Underlying Carney complex should be excluded when genital lentiginosis is encountered.
Genital lentiginosis is idiopathic in most instances, but reports of lesions occurring after annular lichen planus suggest a possible mechanism.5 The disappearance of lentigines after imatinib therapy suggests a role for c-kit, a receptor tyrosine kinase that is involved in intracellular signaling, in some cases.6 At times, lesions can simulate trichrome vitiligo or have a reticulate pattern.7
Men and women present at different points in the course of disease. Men often present with penile lesions 14 years after onset, on average; they notice a gradual increase in the size of lesions. Because women can have greater difficulty self-examining the genital region, they tend to present much later in the course but often within a few months after initial inspection.1,8
Genital lentiginosis can mimic melanoma with nonhomogeneous pigmentation, asymmetry, and unilateral distribution, which makes dermoscopic assessment of colors helpful in narrowing the differential diagnosis. Melanoma is associated with combinations of gray, red, blue, and white, which are not found in genital lentiginosis.9
Biopsy of a genital lentigo is diagnostic, distinguishing the lesion from melanoma—failing to reveal the atypical melanocytes and pagetoid spread characteristic of melanoma in situ. Histologic findings can cause diagnostic difficulties when concurrent lichen sclerosus is associated with genital lentigines or nevi.10
Lentigines on sun-damaged skin or in the setting of xeroderma pigmentosum have been associated with melanoma,11-13 but genital lentigines are not considered a form of precancerous melanosis. In women, early diagnosis is important when there is concern for melanoma because the prognosis for vulvar melanoma is improved in thin lesions.14
Other entities in the differential include secondary syphilis, which commonly presents as macules and scaly papules and can be found on mucosal surfaces such as the oral cavity,15 as well as Kaposi sarcoma, which is characterized by purplish, brown, or black macules, plaques, and nodules, more commonly in immunosuppressed patients.16
To avoid unwarranted concern and unnecessary surgery, dermatologists should be aware of genital lentigines and their characteristic presentation in adults.
To the Editor:
Genital lentiginosis (also known as mucosal melanotic macules, vulvar melanosis, penile melanosis, and penile lentigines) occurs in men and women.1 Lesions present in adult life as multifocal, asymmetrical, pigmented patches that can have a mottled appearance or exhibit skip areas. The irregular appearance of the pigmented areas often raises concern for melanoma. Biopsy reveals increased pigmentation along the basal layer of the epidermis; the irregular distribution of single melanocytes and pagetoid spread typical of melanoma in situ is not identified.
Genital lentiginosis usually occurs as an isolated finding; however, the condition can be a manifestation of Laugier-Hunziker syndrome, Carney complex, and Bannayan-Riley-Ruvalcaba syndrome.1-3 When it occurs as an isolated finding, the patient can be reassured and treatment is unnecessary. Because genital lentiginosis may mimic the appearance of melanoma, it is important for physicians to differentiate the two and make a correct diagnosis. We present a case of genital lentiginosis that mimicked vulvar melanoma.
A 64-year-old woman was referred by her gynecologist to dermatology to rule out vulvar melanoma. The patient had a history of hypothyroidism and hypercholesterolemia but was otherwise in good health. Genital examination revealed asymptomatic pigmented macules and patches of unknown duration (Figure 1). Specimens were taken from 3 areas by punch biopsy to clarify the diagnosis. All 3 specimens showed identical features including basilar pigmentation, occasional melanophages in the papillary dermis, and no evidence of nests or pagetoid spread of atypical melanocytes (Figures 2 and 3). Histologic findings were diagnostic for genital lentiginosis. The patient was reassured, and no treatment was provided. At 6-month follow-up there was no change in clinical appearance.
Genital lentiginosis is characterized by brown lesions that can have a mottled appearance and often are associated with skip areas.1 Lesions can be strikingly irregular and darkly pigmented.
Although the lesions of genital lentiginosis most often are isolated findings, they can be a clue to several uncommon syndromes such as autosomal-dominant Bannayan-Riley-Ruvalcaba syndrome, which is associated with genital lentiginosis, intestinal polyposis, and macrocephaly.3 Vascular malformations, lipomatosis, verrucal keratoses, and acrochordons can occur. Bannayan-Riley-Ruvalcaba syndrome and Cowden syndrome may share genetic linkage; mutations in the tumor suppressor PTEN (phosphatase and tensin homolog deleted on chromosome ten) has been implicated in both syndromes.4 Underlying Carney complex should be excluded when genital lentiginosis is encountered.
Genital lentiginosis is idiopathic in most instances, but reports of lesions occurring after annular lichen planus suggest a possible mechanism.5 The disappearance of lentigines after imatinib therapy suggests a role for c-kit, a receptor tyrosine kinase that is involved in intracellular signaling, in some cases.6 At times, lesions can simulate trichrome vitiligo or have a reticulate pattern.7
Men and women present at different points in the course of disease. Men often present with penile lesions 14 years after onset, on average; they notice a gradual increase in the size of lesions. Because women can have greater difficulty self-examining the genital region, they tend to present much later in the course but often within a few months after initial inspection.1,8
Genital lentiginosis can mimic melanoma with nonhomogeneous pigmentation, asymmetry, and unilateral distribution, which makes dermoscopic assessment of colors helpful in narrowing the differential diagnosis. Melanoma is associated with combinations of gray, red, blue, and white, which are not found in genital lentiginosis.9
Biopsy of a genital lentigo is diagnostic, distinguishing the lesion from melanoma—failing to reveal the atypical melanocytes and pagetoid spread characteristic of melanoma in situ. Histologic findings can cause diagnostic difficulties when concurrent lichen sclerosus is associated with genital lentigines or nevi.10
Lentigines on sun-damaged skin or in the setting of xeroderma pigmentosum have been associated with melanoma,11-13 but genital lentigines are not considered a form of precancerous melanosis. In women, early diagnosis is important when there is concern for melanoma because the prognosis for vulvar melanoma is improved in thin lesions.14
Other entities in the differential include secondary syphilis, which commonly presents as macules and scaly papules and can be found on mucosal surfaces such as the oral cavity,15 as well as Kaposi sarcoma, which is characterized by purplish, brown, or black macules, plaques, and nodules, more commonly in immunosuppressed patients.16
To avoid unwarranted concern and unnecessary surgery, dermatologists should be aware of genital lentigines and their characteristic presentation in adults.
- Hwang L, Wilson H, Orengo I. Off-center fold: irregular, pigmented genital macules. Arch Dermatol. 2000;136:1559-1564. doi:10.1001/archderm.136.12.1559-b
- Rhodes AR, Silverman RA, Harrist TJ, et al. Mucocutaneous lentigines, cardiomucocutaneous myxomas, and multiple blue nevi: the “LAMB” syndrome. J Am Acad Dermatol. 1984;10:72-82. doi:10.1016/s0190-9622(84)80047-x
- Erkek E, Hizel S, Sanl C, et al. Clinical and histopathological findings in Bannayan-Riley-Ruvalcaba syndrome. J Am Acad Dermatol. 2005;53:639-643. doi:10.1016/j.jaad.2005.06.022
- Blum RR, Rahimizadeh A, Kardon N, et al. Genital lentigines in a 6-year-old boy with a family history of Cowden’s disease: clinical and genetic evidence of the linkage between Bannayan-Riley-Ruvalcaba syndrome and Cowden’s disease. J Cutan Med Surg. 2001;5:228-230. doi:10.1177/120347540100500307
- Isbary G, Dyall-Smith D, Coras-Stepanek B, et al. Penile lentigo (genital mucosal macule) following annular lichen planus: a possible association? Australas J Dermatol. 2014;55:159-161. doi:10.1111/ajd.12169
- Campbell T, Felsten L, Moore J. Disappearance of lentigines in a patient receiving imatinib treatment for familial gastrointestinal stromal tumor syndrome. Arch Dermatol. 2009;145:1313-1316. doi:10.1001/archdermatol.2009.263
- Romero- A, R, , et al. Reticulate genital pigmentation associated with localized vitiligo. Arch Dermatol. 2010; 146:574-575. doi:10.1001/archdermatol.2010.69
- Barnhill RL, Albert LS, Shama SK, et al. Genital lentiginosis: a clinical and histopathologic study. J Am Acad Dermatol. 1990;22:453-460. doi:10.1016/0190-9622(90)70064-o
- De Giorgi V, Gori A, Salvati L, et al. Clinical and dermoscopic features of vulvar melanosis over the last 20 years. JAMA Dermatol. 2020;156:1185–1191. doi:10.1001/jamadermatol.2020.2528
- El Shabrawi-Caelen L, Soyer HP, Schaeppi H, et al. Genital lentigines and melanocytic nevi with superimposed lichen sclerosus: a diagnostic challenge. J Am Acad Dermatol. 2004;50:690-694. doi:10.1016/j.jaad.2003.09.034
- Shatkin M, Helm MF, Muhlbauer A, et al. Solar lentigo evolving into fatal metastatic melanoma in a patient who initially refused surgery. N A J Med Sci. 2020;1:28-31. doi:10.7156/najms.2020.1301028
- Stern JB, Peck GL, Haupt HM, et al. Malignant melanoma in xeroderma pigmentosum: search for a precursor lesion. J Am Acad Dermatol. 1993;28:591-594. doi:10.1016/0190-9622(93)70079-9
- Byrom L, Barksdale S, Weedon D, et al. Unstable solar lentigo: a defined separate entity. Australas J Dermatol. 2016;57:229-234. doi:10.1111/ajd.12447
- Panizzon RG. Vulvar melanoma. Semin Dermatol. 1996;15:67-70. doi:10.1016/s1085-5629(96)80021-6
- Chapel TA. The signs and symptoms of secondary syphilis. Sex Transm Dis. 1980;7:161-164. doi:10.1097/00007435-198010000-00002
- Schwartz RA. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151. doi:10.1002/jso.20090
- Hwang L, Wilson H, Orengo I. Off-center fold: irregular, pigmented genital macules. Arch Dermatol. 2000;136:1559-1564. doi:10.1001/archderm.136.12.1559-b
- Rhodes AR, Silverman RA, Harrist TJ, et al. Mucocutaneous lentigines, cardiomucocutaneous myxomas, and multiple blue nevi: the “LAMB” syndrome. J Am Acad Dermatol. 1984;10:72-82. doi:10.1016/s0190-9622(84)80047-x
- Erkek E, Hizel S, Sanl C, et al. Clinical and histopathological findings in Bannayan-Riley-Ruvalcaba syndrome. J Am Acad Dermatol. 2005;53:639-643. doi:10.1016/j.jaad.2005.06.022
- Blum RR, Rahimizadeh A, Kardon N, et al. Genital lentigines in a 6-year-old boy with a family history of Cowden’s disease: clinical and genetic evidence of the linkage between Bannayan-Riley-Ruvalcaba syndrome and Cowden’s disease. J Cutan Med Surg. 2001;5:228-230. doi:10.1177/120347540100500307
- Isbary G, Dyall-Smith D, Coras-Stepanek B, et al. Penile lentigo (genital mucosal macule) following annular lichen planus: a possible association? Australas J Dermatol. 2014;55:159-161. doi:10.1111/ajd.12169
- Campbell T, Felsten L, Moore J. Disappearance of lentigines in a patient receiving imatinib treatment for familial gastrointestinal stromal tumor syndrome. Arch Dermatol. 2009;145:1313-1316. doi:10.1001/archdermatol.2009.263
- Romero- A, R, , et al. Reticulate genital pigmentation associated with localized vitiligo. Arch Dermatol. 2010; 146:574-575. doi:10.1001/archdermatol.2010.69
- Barnhill RL, Albert LS, Shama SK, et al. Genital lentiginosis: a clinical and histopathologic study. J Am Acad Dermatol. 1990;22:453-460. doi:10.1016/0190-9622(90)70064-o
- De Giorgi V, Gori A, Salvati L, et al. Clinical and dermoscopic features of vulvar melanosis over the last 20 years. JAMA Dermatol. 2020;156:1185–1191. doi:10.1001/jamadermatol.2020.2528
- El Shabrawi-Caelen L, Soyer HP, Schaeppi H, et al. Genital lentigines and melanocytic nevi with superimposed lichen sclerosus: a diagnostic challenge. J Am Acad Dermatol. 2004;50:690-694. doi:10.1016/j.jaad.2003.09.034
- Shatkin M, Helm MF, Muhlbauer A, et al. Solar lentigo evolving into fatal metastatic melanoma in a patient who initially refused surgery. N A J Med Sci. 2020;1:28-31. doi:10.7156/najms.2020.1301028
- Stern JB, Peck GL, Haupt HM, et al. Malignant melanoma in xeroderma pigmentosum: search for a precursor lesion. J Am Acad Dermatol. 1993;28:591-594. doi:10.1016/0190-9622(93)70079-9
- Byrom L, Barksdale S, Weedon D, et al. Unstable solar lentigo: a defined separate entity. Australas J Dermatol. 2016;57:229-234. doi:10.1111/ajd.12447
- Panizzon RG. Vulvar melanoma. Semin Dermatol. 1996;15:67-70. doi:10.1016/s1085-5629(96)80021-6
- Chapel TA. The signs and symptoms of secondary syphilis. Sex Transm Dis. 1980;7:161-164. doi:10.1097/00007435-198010000-00002
- Schwartz RA. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151. doi:10.1002/jso.20090
Practice Points
- The irregular appearance of genital lentiginosis—multifocal, asymmetric, irregular, and darkly pigmented patches—often raises concern for melanoma but is benign.
- Certain genetic conditions can present with genital lentiginosis.
- Dermoscopic assessment of the lesion color is highly helpful in narrowing the differential diagnosis; seeing no gray, red, blue, or white makes melanoma less likely.
- Be aware of genital lentigines and their characteristic presentation in adulthood to avoid unwarranted concern and unneeded surgery.

Mycetomalike Skin Infection Due to Gordonia bronchialis in an Immunocompetent Patient
Mycetoma is a chronic subcutaneous infection due to fungal (eumycetoma) or aerobic actinomycetes (actinomycetoma) organisms. Clinical lesions develop from a granulomatous infiltrate organizing around the infectious organism. Patients can present with extensive subcutaneous nodularity and draining sinuses that can lead to deformation of the affected extremity. These infections are rare in developed countries, and the prevalence and incidence remain unknown. It has been reported that actinomycetes represent 60% of mycetoma cases worldwide, with the majority of cases in Central America from Nocardia (86%) and Actinomadura madurae (10%). 1Gordonia species are aerobic, partially acid-fast, gram-positive actinobacteria that may comprise a notable minority of actinomycete isolates. 2 The species Gordonia bronchialis is of particular interest as a human pathogen because of increasing reports of nosocomial infections. 3,4 We describe a case of a mycetomalike infection due to G bronchialis in an immunocompetent patient with complete resolution after 3 months of antibiotics.

Case Report
An 86-year-old man presented to the emergency department with a pruritic rash on the right forearm. He had a history of chronic kidney disease, hypertension, and inverse psoriasis complicated by steroid atrophy. He reported trauma to the right antecubital fossa approximately 1 to 2 months prior from a car door; he received wound care over several weeks at an outside hospital. The initial wound healed completely, but he subsequently noticed erythema spreading down the forearm. At the current presentation, he was empirically treated with mid-potency topical steroids and cefuroxime for 7 days. Initial laboratory results were notable for a white blood cell count of 5.7×103 cells/μL (reference range,3.7–8.4×103 cells/μL) and a creatinine level of 1.5 mg/dL (reference range, 0.57–1.25 mg/dL). The patient returned to the emergency department 2 weeks later with spreading of the initial rash and worsening pruritus. Dermatologic evaluation revealed the patient was afebrile and had violaceous papules and nodules that coalesced into plaques on the right arm, with the largest measuring approximately 15 cm. Areas of superficial erosion and crusting were noted (Figure 1A). The patient denied constitutional symptoms and had no axillary or cervical lymphadenopathy. The differential initially included an atypical infection vs a neoplasm. Two 5-mm punch biopsies were performed, which demonstrated a suppurative granulomatous infiltrate in the dermis with extension into the subcutis (Figure 2A). Focal vacuolations within the dermis demonstrated aggregates of gram-positive pseudofilamentous organisms (Figures 2B and 2C). Aerobic tissue cultures grew G bronchialis that was susceptible to all antibiotics tested and Staphylococcus epidermidis. Fungal and mycobacterial cultures were negative. The patient was placed on amoxicillin 875 mg–clavulanate 125 mg twice daily for 3 weeks. However, he demonstrated progression of the rash, with increased induration and confluence of plaques on the forearm (Figure 1B). A repeat excisional biopsy was performed, and a tissue sample was sent for 16S ribosomal RNA sequencing identification. However, neither conventional cultures nor sequencing demonstrated evidence of G bronchialis or any other pathogen. Additionally, bacterial, fungal, and mycobacterial blood cultures were negative. Amoxicillin-clavulanate was stopped, and he was placed on trimethoprim-sulfamethoxazole for 2 weeks, then changed to linezolid (600 mg twice daily) due to continued lack of improvement of the rash. After 2 weeks of linezolid, the rash was slightly improved, but the patient had notable side effects (eg, nausea, mucositis). Therefore, he was switched back to trimethoprim-sulfamethoxazole for another 6 weeks. Antibiotic therapy was discontinued after there was notable regression of indurated plaques (Figure 1C); he received more than 3 months of antibiotics in all. At 1 month after completion of antibiotic therapy, the patient had no evidence of recurrence.
. B, Clumped pseudofilamento")
Comment
Microbiology of Gordonia Species—Gordonia bronchialis originally was isolated in 1971 by Tsukamura et al5 from the sputum of patients with cavitary tuberculosis and bronchiectasis in Japan. Other Gordonia species (formerly Rhodococcus or Gordona) later were identified in soil, seawater, sediment, and wastewater. Gordonia bronchialis is a gram-positive aerobic actinomycete short rod that organizes in cordlike compact groups. It is weakly acid fast, nonmotile, and nonsporulating. Colonies exhibit pinkish-brown pigmentation. Our understanding of the clinical significance of this organism continues to evolve, and it is not always clearly pathogenic. Because Gordonia isolates may be dismissed as commensals or misidentified as Nocardia or Rhodococcus by routine biochemical tests, it is possible that infections may go undetected. Speciation requires gene sequencing; as our utilization of molecular methods has increased, the identification of clinically relevant aerobic actinomycetes, including Gordonia, has improved,6 and the following species have been recognized as pathogens: Gordonia araii, G bronchialis, Gordonia effusa, Gordonia otitidis, Gordonia polyisoprenivorans, Gordonia rubirpertincta, Gordonia sputi, and Gordonia terrae.7
Cases Reported in the Literature—A PubMed search of articles indexed for MEDLINE using the term Gordonia bronchialis yielded 35 previously reported human cases of G bronchialis infection, most often associated with medical devices or procedures.8-31 Eighteen of these cases were sternal surgical site infections in patients with a history of cardiac surgery,3,4,12-16,30 including 2 outbreaks following coronary artery bypass grafting that were thought to be related to intraoperative transmission from a nurse.3,4 Of the remaining cases, 12 were linked to a procedure or an indwelling catheter: 4 cases of peritonitis in the setting of continuous ambulatory peritoneal dialysis17,18,26,27; 3 cases of skin and soft tissue infection (1 at the site of a prior needle injection,10 1 after acupuncture,11 and 1 after breast reduction surgery29); 1 case of ventriculitis in a premature neonate with an underlying intraventricular shunt19; 2 cases of pacemaker-induced endocarditis20,28; 1 case of tibial osteomyelitis related to a bioresorbable polymer screw21; and 1 case of chronic endophthalmitis with underlying intraocular lens implants.22 The Table lists all cases of G bronchialis skin or surgical site infections encountered in our literature search as well as the treatment provided in each case.


Only 4 of these 35 cases of G bronchialis infections were skin and soft tissue infections. All 4 occurred in immunocompetent hosts, and 3 were associated with needle punctures or surgery. The fourth case involved a recurrent breast abscess that occurred in a patient without known risk factors or recent procedures.23 Other Gordonia species have been associated with cutaneous infections, including Gordonia amicalis, G terrae, and recently Gordonia westfalica, with the latter 2 demonstrating actinomycetoma formation.32-34 Our case is remarkable in that it represents actinomycetoma due to G bronchialis. Of note, our patient was immunocompetent and did not have any radiation or chronic lymphedema involving the affected extremity. However, his history of steroid-induced skin atrophy may have predisposed him to this rare infection.
Clinical Presentation—Classic mycetoma demonstrate organismal granules within the dermis, surrounded by a neutrophilic infiltrate, which is in turn surrounded by histiocytes and multinucleated giant cells. Periodic acid–Schiff and silver stains can identify fungal organisms, while Gram stain helps to elucidate bacterial etiologies.1 In our patient, a biopsy revealed several dermal aggregates of pseudofilamentous gram-positive organisms surrounded by a neutrophilic and histiocytic infiltrate.8 Because this case presented over weeks to months rather than months to years, it progressed more rapidly than a classic mycetoma. However, the dermatologic and histologic features were consistent with mycetoma.
Management—General treatment of actinomycetoma requires identification of the causative organism and prolonged administration of antibiotics, typically in combination.35-37 Most G bronchialis infections associated with surgical intervention or implants in the literature required surgical debridement and removal of contaminated material for clinical cure, with the exception of 3 cases of sternal wound infection and 1 case of peritonitis that recovered with antimicrobial therapy alone.3,17 Combination therapy often was used, but monotherapy, particularly with a fluoroquinolone, has been reported. Susceptibility data are limited, but in general, Gordonia species appear susceptible to imipenem, ciprofloxacin, amikacin, gentamicin, and linezolid, with variable susceptibility to vancomycin (89% of isolates), third-generation cephalosporins (80%–90% of isolates), tetracyclines (≤85% of isolates), penicillin (≤70% of isolates), and trimethoprim-sulfamethoxazole (≤65% of isolates).7,10,19,38-40 Although there are no standardized recommendations for the treatment of these infections, the most commonly used drugs to treat Gordonia are carbapenems and fluoroquinolones, with or without an aminoglycoside, followed by third-generation cephalosporins and vancomycin, depending on susceptibilities. Additional antibiotics (alone or in combination) that have previously been used with favorable outcomes include amoxicillin or amoxicillin-clavulanate, piperacillin-tazobactam, rifampicin, trimethoprim-sulfamethoxazole, minocycline, doxycycline, and daptomycin.
Our patient received amoxicillin-clavulanate, trimethoprim-sulfamethoxazole, and linezolid. We considered combination therapy but decided against it due to concern for toxicity, given his age and poor renal function. The antibiotic that was most important to his recovery was unclear; the patient insisted that his body, not antibiotics, deserved most of the credit for healing his arm. Although cultures and polymerase chain reaction assays were negative after 3 weeks of amoxicillin-clavulanate, the patient did not show clinical improvement—reasons could be because the antibiotic reduced but did not eliminate the bacterial burden, sampling error of the biopsy, or it takes much longer for the body to heal than it takes to kill the bacteria. Most likely a combination of factors was at play.
Conclusion
Gordonia bronchialis is an emerging cause of human infections typically occurring after trauma, inoculation, or surgery. Most infections are localized; however, the present case highlights the ability of this species to form a massive cutaneous infection. Treatment should be tailored to susceptibility, with close follow-up to ensure improvement and resolution. For clinicians encountering a similar case, we encourage biopsy prior to empiric antibiotics, as antibiotic therapy can decrease the yield of subsequent testing. Treatment should be guided by the clinical course and may need to last weeks to months. Combination therapy for Gordonia infections should be considered in severe cases, in cases presenting as actinomycetoma, in those not responding to therapy, or when the susceptibility profile is unknown or unreliable.
Acknowledgments—The authors thank this veteran for allowing us to participate in his care and to learn from his experience. He gave his consent for us to share his story and the photographs of the arm.
- Arenas R, Fernandez Martinez RF, Torres-Guerrero E, et al. Actinomycetoma: an update on diagnosis and treatment. Cutis. 2017;99:E11-E15.
- Poonwan N, Mekha N, Yazawa K, et al. Characterization of clinical isolates of pathogenic Nocardia strains and related actinomycetes in Thailand from 1996 to 2003. Mycopathologia. 2005;159:361-368.
- Richet HM, Craven PC, Brown JM, et al. A cluster of Rhodococcus (Gordona) bronchialis sternal-wound infections after coronary-artery bypass surgery. N Engl J Med. 1991;324:104-109.
- Wright SN, Gerry JS, Busowski MT, et al. Gordonia bronchialis sternal wound infection in 3 patients following open heart surgery: intraoperative transmission from a healthcare worker. Infect Control Hosp Epidemiol. 2012;33:1238-1241.
- Tsukamura M. Proposal of a new genus, Gordona, for slightly acid-fast organisms occurring in sputa of patients with pulmonary disease and in soil. J Gen Microbiol. 1971;68:15-26.
- Wang T, Kong F, Chen S, et al. Improved identification of Gordonia, Rhodococcus and Tsukamurella species by 5′-end 16s rRNA gene sequencing. Pathology. 2011;43:58-63.
- Aoyama K, Kang Y, Yazawa K, et al. Characterization of clinical isolates of Gordonia species in Japanese clinical samples during 1998-2008. Mycopathologia. 2009;168:175-183.
- Ivanova N, Sikorski J, Jando M, et al. Complete genome sequence of Gordonia bronchialis type strain (3410 T). Stand Genomic Sci. 2010;2:19-28.
- Johnson JA, Onderdonk AB, Cosimi LA, et al. Gordonia bronchialis bacteremia and pleural infection: case report and review of the literature. J Clin Microbiol. 2011;49:1662-1666.
- Bartolomé-Álvarez J, Sáez-Nieto JA, Escudero-Jiménez A, et al. Cutaneous abscess due to Gordonia bronchialis: case report and literature review. Rev Esp Quimioter. 2016;29:170-173.
- Choi ME, Jung CJ, Won CH, et al. Case report of cutaneous nodule caused by Gordonia bronchialis in an immunocompetent patient after receiving acupuncture. J Dermatol. 2019;46:343-346.
- Nguyen DB, Gupta N, Abou-Daoud A, et al. A polymicrobial outbreak of surgical site infections following cardiac surgery at a community hospital in Florida, 2011-2012. Am J Infect Control. 2014;42:432-435.
- Chang JH, Ji M, Hong HL, et al. Sternal osteomyelitis caused byGordonia bronchialis after open-heart surgery. Infect Chemother. 2014;46:110-114.
- Rodriguez-Lozano J, Pérez-Llantada E, Agüero J, et al. Sternal wound infection caused by Gordonia bronchialis: identification by MALDI-TOF MS. JMM Case Rep. 2016;3:e005067.
- Akrami K, Coletta J, Mehta S, et al. Gordonia sternal wound infection treated with ceftaroline: case report and literature review. JMM Case Rep. 2017;4:e005113.
- Ambesh P, Kapoor A, Kazmi D, et al. Sternal osteomyelitis by Gordonia bronchialis in an immunocompetent patient after open heart surgery. Ann Card Anaesth. 2019;22:221-224.
- Ma TKW, Chow KM, Kwan BCH, et al. Peritoneal-dialysis related peritonitis caused by Gordonia species: report of four cases and literature review. Nephrology. 2014;19:379-383.
- Lam JYW, Wu AKL, Leung WS, et al. Gordonia species as emerging causes of continuous-ambulatory-peritoneal-dialysis-related peritonitis identified by 16S rRNA and secA1 gene sequencing and matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS). J Clin Microbiol. 2015;53:671-676.
- Blaschke AJ, Bender J, Byington CL, et al. Gordonia species: emerging pathogens in pediatric patients that are identified by 16S ribosomal RNA gene sequencing. Clin Infect Dis. 2007;45:483-486.
- Titécat M, Loïez C, Courcol RJ, et al. Difficulty with Gordonia bronchialis identification by Microflex mass spectrometer in a pacemaker‐induced endocarditis. JMM Case Rep. 2014;1:E003681.
- Siddiqui N, Toumeh A, Georgescu C. Tibial osteomyelitis caused by Gordonia bronchialis in an immunocompetent patient. J Clin Microbiol. 2012;50:3119-3121.
- Choi R, Strnad L, Flaxel CJ, et al. Gordonia bronchialis–associated endophthalmitis. Emerg Infect Dis. 2019;25:1017-1019.
- Werno AM, Anderson TP, Chambers ST, et al. Recurrent breast abscess caused by Gordonia bronchialis in an immunocompetent patient. J Clin Microbiol. 2005;43:3009-3010.
- Sng LH, Koh TH, Toney SR, et al. Bacteremia caused by Gordonia bronchialis in a patient with sequestrated lung. J Clin Microbiol. 2004;42:2870-2871.
- Ramanan P, Deziel PJ, Wengenack NL. Gordonia bacteremia. J Clin Microbiol. 2013;51:3443-3447.
- Sukackiene D, Rimsevicius L, Kiveryte S, et al. A case of successfully treated relapsing peritoneal dialysis-associated peritonitis caused by Gordonia bronchialis in a farmer. Nephrol Ther. 2018;14:109-111.
- Bruno V, Tjon J, Lin S, et al. Peritoneal dialysis-related peritonitis caused by Gordonia bronchialis: first pediatric report. Pediatr Nephrol. 2022;37:217-220. doi: 10.1007/s00467-021-05313-3
- Mormeneo Bayo S, Palacián Ruíz MP, Asin Samper U, et al. Pacemaker-induced endocarditis by Gordonia bronchialis. Enferm Infecc Microbiol Clin (Engl Ed). 2022;40:255-257.
- Davidson AL, Driscoll CR, Luther VP, et al. Recurrent skin and soft tissue infection following breast reduction surgery caused by Gordonia bronchialis: a case report. Plast Reconstr Surg Glob Open. 2022;10:E4395.
- Nwaedozie S, Mojarrab JN, Gopinath P, et al. Sternal osteomyelitis caused by Gordonia bronchialis in an immunocompetent patient following coronary artery bypass surgery. IDCases. 2022;29:E01548.
- Nakahama H, Hanada S, Takada K, et al. Obstructive pneumonia caused by Gordonia bronchialis with a bronchial foreign body. Int J Infect Dis. 2022;124:157-158. doi:10.1016/j.ijid.2022.09.028
- Lai CC, Hsieh JH, Tsai HY, et al. Cutaneous infection caused by Gordonia amicalis after a traumatic injury. J Clin Microbiol. 2012;50:1821-1822.
- Bakker XR, Spauwen PHM, Dolmans WMV. Mycetoma of the hand caused by Gordona terrae: a case report. J Hand Surg Am. 2004;29:188-190.
- Gueneau R, Blanchet D, Rodriguez-Nava V, et al. Actinomycetoma caused by Gordonia westfalica: first reported case of human infection. New Microbes New Infect. 2020;34:100658.
- Auwaerter PG, ed. The Johns Hopkins POC-IT ABX Guide. Johns Hopkins Medicine; 2021.
- Welsh O, Sauceda E, Gonzalez J, et al. Amikacin alone andin combination with trimethoprim-sulfamethoxazole in the treatment of actinomycotic mycetoma. J Am Acad Dermatol. 1987;17:443-448.
- Zijlstra EE, van de Sande WWJ, Welsh O, et al. Mycetoma: a unique neglected tropical disease. Lancet Infect Dis. 2016;16:100-112.
- Pham AS, Dé I, Rolston KV, et al. Catheter-related bacteremia caused by the nocardioform actinomycete Gordonia terrae. Clin Infect Dis. 2003;36:524-527.
- Renvoise A, Harle JR, Raoult D, et al. Gordonia sputi bacteremia. Emerg Infect Dis. 2009;15:1535-1537.
- Moser BD, Pellegrini GJ, Lasker BA, et al. Pattern of antimicrobial susceptibility obtained from blood isolates of a rare but emerging human pathogen, Gordonia polyisoprenivorans. Antimicrob Agents Chemother. 2012;56:4991-4993.
Mycetoma is a chronic subcutaneous infection due to fungal (eumycetoma) or aerobic actinomycetes (actinomycetoma) organisms. Clinical lesions develop from a granulomatous infiltrate organizing around the infectious organism. Patients can present with extensive subcutaneous nodularity and draining sinuses that can lead to deformation of the affected extremity. These infections are rare in developed countries, and the prevalence and incidence remain unknown. It has been reported that actinomycetes represent 60% of mycetoma cases worldwide, with the majority of cases in Central America from Nocardia (86%) and Actinomadura madurae (10%). 1Gordonia species are aerobic, partially acid-fast, gram-positive actinobacteria that may comprise a notable minority of actinomycete isolates. 2 The species Gordonia bronchialis is of particular interest as a human pathogen because of increasing reports of nosocomial infections. 3,4 We describe a case of a mycetomalike infection due to G bronchialis in an immunocompetent patient with complete resolution after 3 months of antibiotics.
Case Report
An 86-year-old man presented to the emergency department with a pruritic rash on the right forearm. He had a history of chronic kidney disease, hypertension, and inverse psoriasis complicated by steroid atrophy. He reported trauma to the right antecubital fossa approximately 1 to 2 months prior from a car door; he received wound care over several weeks at an outside hospital. The initial wound healed completely, but he subsequently noticed erythema spreading down the forearm. At the current presentation, he was empirically treated with mid-potency topical steroids and cefuroxime for 7 days. Initial laboratory results were notable for a white blood cell count of 5.7×103 cells/μL (reference range,3.7–8.4×103 cells/μL) and a creatinine level of 1.5 mg/dL (reference range, 0.57–1.25 mg/dL). The patient returned to the emergency department 2 weeks later with spreading of the initial rash and worsening pruritus. Dermatologic evaluation revealed the patient was afebrile and had violaceous papules and nodules that coalesced into plaques on the right arm, with the largest measuring approximately 15 cm. Areas of superficial erosion and crusting were noted (Figure 1A). The patient denied constitutional symptoms and had no axillary or cervical lymphadenopathy. The differential initially included an atypical infection vs a neoplasm. Two 5-mm punch biopsies were performed, which demonstrated a suppurative granulomatous infiltrate in the dermis with extension into the subcutis (Figure 2A). Focal vacuolations within the dermis demonstrated aggregates of gram-positive pseudofilamentous organisms (Figures 2B and 2C). Aerobic tissue cultures grew G bronchialis that was susceptible to all antibiotics tested and Staphylococcus epidermidis. Fungal and mycobacterial cultures were negative. The patient was placed on amoxicillin 875 mg–clavulanate 125 mg twice daily for 3 weeks. However, he demonstrated progression of the rash, with increased induration and confluence of plaques on the forearm (Figure 1B). A repeat excisional biopsy was performed, and a tissue sample was sent for 16S ribosomal RNA sequencing identification. However, neither conventional cultures nor sequencing demonstrated evidence of G bronchialis or any other pathogen. Additionally, bacterial, fungal, and mycobacterial blood cultures were negative. Amoxicillin-clavulanate was stopped, and he was placed on trimethoprim-sulfamethoxazole for 2 weeks, then changed to linezolid (600 mg twice daily) due to continued lack of improvement of the rash. After 2 weeks of linezolid, the rash was slightly improved, but the patient had notable side effects (eg, nausea, mucositis). Therefore, he was switched back to trimethoprim-sulfamethoxazole for another 6 weeks. Antibiotic therapy was discontinued after there was notable regression of indurated plaques (Figure 1C); he received more than 3 months of antibiotics in all. At 1 month after completion of antibiotic therapy, the patient had no evidence of recurrence.
Comment
Microbiology of Gordonia Species—Gordonia bronchialis originally was isolated in 1971 by Tsukamura et al5 from the sputum of patients with cavitary tuberculosis and bronchiectasis in Japan. Other Gordonia species (formerly Rhodococcus or Gordona) later were identified in soil, seawater, sediment, and wastewater. Gordonia bronchialis is a gram-positive aerobic actinomycete short rod that organizes in cordlike compact groups. It is weakly acid fast, nonmotile, and nonsporulating. Colonies exhibit pinkish-brown pigmentation. Our understanding of the clinical significance of this organism continues to evolve, and it is not always clearly pathogenic. Because Gordonia isolates may be dismissed as commensals or misidentified as Nocardia or Rhodococcus by routine biochemical tests, it is possible that infections may go undetected. Speciation requires gene sequencing; as our utilization of molecular methods has increased, the identification of clinically relevant aerobic actinomycetes, including Gordonia, has improved,6 and the following species have been recognized as pathogens: Gordonia araii, G bronchialis, Gordonia effusa, Gordonia otitidis, Gordonia polyisoprenivorans, Gordonia rubirpertincta, Gordonia sputi, and Gordonia terrae.7
Cases Reported in the Literature—A PubMed search of articles indexed for MEDLINE using the term Gordonia bronchialis yielded 35 previously reported human cases of G bronchialis infection, most often associated with medical devices or procedures.8-31 Eighteen of these cases were sternal surgical site infections in patients with a history of cardiac surgery,3,4,12-16,30 including 2 outbreaks following coronary artery bypass grafting that were thought to be related to intraoperative transmission from a nurse.3,4 Of the remaining cases, 12 were linked to a procedure or an indwelling catheter: 4 cases of peritonitis in the setting of continuous ambulatory peritoneal dialysis17,18,26,27; 3 cases of skin and soft tissue infection (1 at the site of a prior needle injection,10 1 after acupuncture,11 and 1 after breast reduction surgery29); 1 case of ventriculitis in a premature neonate with an underlying intraventricular shunt19; 2 cases of pacemaker-induced endocarditis20,28; 1 case of tibial osteomyelitis related to a bioresorbable polymer screw21; and 1 case of chronic endophthalmitis with underlying intraocular lens implants.22 The Table lists all cases of G bronchialis skin or surgical site infections encountered in our literature search as well as the treatment provided in each case.
Only 4 of these 35 cases of G bronchialis infections were skin and soft tissue infections. All 4 occurred in immunocompetent hosts, and 3 were associated with needle punctures or surgery. The fourth case involved a recurrent breast abscess that occurred in a patient without known risk factors or recent procedures.23 Other Gordonia species have been associated with cutaneous infections, including Gordonia amicalis, G terrae, and recently Gordonia westfalica, with the latter 2 demonstrating actinomycetoma formation.32-34 Our case is remarkable in that it represents actinomycetoma due to G bronchialis. Of note, our patient was immunocompetent and did not have any radiation or chronic lymphedema involving the affected extremity. However, his history of steroid-induced skin atrophy may have predisposed him to this rare infection.
Clinical Presentation—Classic mycetoma demonstrate organismal granules within the dermis, surrounded by a neutrophilic infiltrate, which is in turn surrounded by histiocytes and multinucleated giant cells. Periodic acid–Schiff and silver stains can identify fungal organisms, while Gram stain helps to elucidate bacterial etiologies.1 In our patient, a biopsy revealed several dermal aggregates of pseudofilamentous gram-positive organisms surrounded by a neutrophilic and histiocytic infiltrate.8 Because this case presented over weeks to months rather than months to years, it progressed more rapidly than a classic mycetoma. However, the dermatologic and histologic features were consistent with mycetoma.
Management—General treatment of actinomycetoma requires identification of the causative organism and prolonged administration of antibiotics, typically in combination.35-37 Most G bronchialis infections associated with surgical intervention or implants in the literature required surgical debridement and removal of contaminated material for clinical cure, with the exception of 3 cases of sternal wound infection and 1 case of peritonitis that recovered with antimicrobial therapy alone.3,17 Combination therapy often was used, but monotherapy, particularly with a fluoroquinolone, has been reported. Susceptibility data are limited, but in general, Gordonia species appear susceptible to imipenem, ciprofloxacin, amikacin, gentamicin, and linezolid, with variable susceptibility to vancomycin (89% of isolates), third-generation cephalosporins (80%–90% of isolates), tetracyclines (≤85% of isolates), penicillin (≤70% of isolates), and trimethoprim-sulfamethoxazole (≤65% of isolates).7,10,19,38-40 Although there are no standardized recommendations for the treatment of these infections, the most commonly used drugs to treat Gordonia are carbapenems and fluoroquinolones, with or without an aminoglycoside, followed by third-generation cephalosporins and vancomycin, depending on susceptibilities. Additional antibiotics (alone or in combination) that have previously been used with favorable outcomes include amoxicillin or amoxicillin-clavulanate, piperacillin-tazobactam, rifampicin, trimethoprim-sulfamethoxazole, minocycline, doxycycline, and daptomycin.
Our patient received amoxicillin-clavulanate, trimethoprim-sulfamethoxazole, and linezolid. We considered combination therapy but decided against it due to concern for toxicity, given his age and poor renal function. The antibiotic that was most important to his recovery was unclear; the patient insisted that his body, not antibiotics, deserved most of the credit for healing his arm. Although cultures and polymerase chain reaction assays were negative after 3 weeks of amoxicillin-clavulanate, the patient did not show clinical improvement—reasons could be because the antibiotic reduced but did not eliminate the bacterial burden, sampling error of the biopsy, or it takes much longer for the body to heal than it takes to kill the bacteria. Most likely a combination of factors was at play.
Conclusion
Gordonia bronchialis is an emerging cause of human infections typically occurring after trauma, inoculation, or surgery. Most infections are localized; however, the present case highlights the ability of this species to form a massive cutaneous infection. Treatment should be tailored to susceptibility, with close follow-up to ensure improvement and resolution. For clinicians encountering a similar case, we encourage biopsy prior to empiric antibiotics, as antibiotic therapy can decrease the yield of subsequent testing. Treatment should be guided by the clinical course and may need to last weeks to months. Combination therapy for Gordonia infections should be considered in severe cases, in cases presenting as actinomycetoma, in those not responding to therapy, or when the susceptibility profile is unknown or unreliable.
Acknowledgments—The authors thank this veteran for allowing us to participate in his care and to learn from his experience. He gave his consent for us to share his story and the photographs of the arm.
Mycetoma is a chronic subcutaneous infection due to fungal (eumycetoma) or aerobic actinomycetes (actinomycetoma) organisms. Clinical lesions develop from a granulomatous infiltrate organizing around the infectious organism. Patients can present with extensive subcutaneous nodularity and draining sinuses that can lead to deformation of the affected extremity. These infections are rare in developed countries, and the prevalence and incidence remain unknown. It has been reported that actinomycetes represent 60% of mycetoma cases worldwide, with the majority of cases in Central America from Nocardia (86%) and Actinomadura madurae (10%). 1Gordonia species are aerobic, partially acid-fast, gram-positive actinobacteria that may comprise a notable minority of actinomycete isolates. 2 The species Gordonia bronchialis is of particular interest as a human pathogen because of increasing reports of nosocomial infections. 3,4 We describe a case of a mycetomalike infection due to G bronchialis in an immunocompetent patient with complete resolution after 3 months of antibiotics.
Case Report
An 86-year-old man presented to the emergency department with a pruritic rash on the right forearm. He had a history of chronic kidney disease, hypertension, and inverse psoriasis complicated by steroid atrophy. He reported trauma to the right antecubital fossa approximately 1 to 2 months prior from a car door; he received wound care over several weeks at an outside hospital. The initial wound healed completely, but he subsequently noticed erythema spreading down the forearm. At the current presentation, he was empirically treated with mid-potency topical steroids and cefuroxime for 7 days. Initial laboratory results were notable for a white blood cell count of 5.7×103 cells/μL (reference range,3.7–8.4×103 cells/μL) and a creatinine level of 1.5 mg/dL (reference range, 0.57–1.25 mg/dL). The patient returned to the emergency department 2 weeks later with spreading of the initial rash and worsening pruritus. Dermatologic evaluation revealed the patient was afebrile and had violaceous papules and nodules that coalesced into plaques on the right arm, with the largest measuring approximately 15 cm. Areas of superficial erosion and crusting were noted (Figure 1A). The patient denied constitutional symptoms and had no axillary or cervical lymphadenopathy. The differential initially included an atypical infection vs a neoplasm. Two 5-mm punch biopsies were performed, which demonstrated a suppurative granulomatous infiltrate in the dermis with extension into the subcutis (Figure 2A). Focal vacuolations within the dermis demonstrated aggregates of gram-positive pseudofilamentous organisms (Figures 2B and 2C). Aerobic tissue cultures grew G bronchialis that was susceptible to all antibiotics tested and Staphylococcus epidermidis. Fungal and mycobacterial cultures were negative. The patient was placed on amoxicillin 875 mg–clavulanate 125 mg twice daily for 3 weeks. However, he demonstrated progression of the rash, with increased induration and confluence of plaques on the forearm (Figure 1B). A repeat excisional biopsy was performed, and a tissue sample was sent for 16S ribosomal RNA sequencing identification. However, neither conventional cultures nor sequencing demonstrated evidence of G bronchialis or any other pathogen. Additionally, bacterial, fungal, and mycobacterial blood cultures were negative. Amoxicillin-clavulanate was stopped, and he was placed on trimethoprim-sulfamethoxazole for 2 weeks, then changed to linezolid (600 mg twice daily) due to continued lack of improvement of the rash. After 2 weeks of linezolid, the rash was slightly improved, but the patient had notable side effects (eg, nausea, mucositis). Therefore, he was switched back to trimethoprim-sulfamethoxazole for another 6 weeks. Antibiotic therapy was discontinued after there was notable regression of indurated plaques (Figure 1C); he received more than 3 months of antibiotics in all. At 1 month after completion of antibiotic therapy, the patient had no evidence of recurrence.
Comment
Microbiology of Gordonia Species—Gordonia bronchialis originally was isolated in 1971 by Tsukamura et al5 from the sputum of patients with cavitary tuberculosis and bronchiectasis in Japan. Other Gordonia species (formerly Rhodococcus or Gordona) later were identified in soil, seawater, sediment, and wastewater. Gordonia bronchialis is a gram-positive aerobic actinomycete short rod that organizes in cordlike compact groups. It is weakly acid fast, nonmotile, and nonsporulating. Colonies exhibit pinkish-brown pigmentation. Our understanding of the clinical significance of this organism continues to evolve, and it is not always clearly pathogenic. Because Gordonia isolates may be dismissed as commensals or misidentified as Nocardia or Rhodococcus by routine biochemical tests, it is possible that infections may go undetected. Speciation requires gene sequencing; as our utilization of molecular methods has increased, the identification of clinically relevant aerobic actinomycetes, including Gordonia, has improved,6 and the following species have been recognized as pathogens: Gordonia araii, G bronchialis, Gordonia effusa, Gordonia otitidis, Gordonia polyisoprenivorans, Gordonia rubirpertincta, Gordonia sputi, and Gordonia terrae.7
Cases Reported in the Literature—A PubMed search of articles indexed for MEDLINE using the term Gordonia bronchialis yielded 35 previously reported human cases of G bronchialis infection, most often associated with medical devices or procedures.8-31 Eighteen of these cases were sternal surgical site infections in patients with a history of cardiac surgery,3,4,12-16,30 including 2 outbreaks following coronary artery bypass grafting that were thought to be related to intraoperative transmission from a nurse.3,4 Of the remaining cases, 12 were linked to a procedure or an indwelling catheter: 4 cases of peritonitis in the setting of continuous ambulatory peritoneal dialysis17,18,26,27; 3 cases of skin and soft tissue infection (1 at the site of a prior needle injection,10 1 after acupuncture,11 and 1 after breast reduction surgery29); 1 case of ventriculitis in a premature neonate with an underlying intraventricular shunt19; 2 cases of pacemaker-induced endocarditis20,28; 1 case of tibial osteomyelitis related to a bioresorbable polymer screw21; and 1 case of chronic endophthalmitis with underlying intraocular lens implants.22 The Table lists all cases of G bronchialis skin or surgical site infections encountered in our literature search as well as the treatment provided in each case.
Only 4 of these 35 cases of G bronchialis infections were skin and soft tissue infections. All 4 occurred in immunocompetent hosts, and 3 were associated with needle punctures or surgery. The fourth case involved a recurrent breast abscess that occurred in a patient without known risk factors or recent procedures.23 Other Gordonia species have been associated with cutaneous infections, including Gordonia amicalis, G terrae, and recently Gordonia westfalica, with the latter 2 demonstrating actinomycetoma formation.32-34 Our case is remarkable in that it represents actinomycetoma due to G bronchialis. Of note, our patient was immunocompetent and did not have any radiation or chronic lymphedema involving the affected extremity. However, his history of steroid-induced skin atrophy may have predisposed him to this rare infection.
Clinical Presentation—Classic mycetoma demonstrate organismal granules within the dermis, surrounded by a neutrophilic infiltrate, which is in turn surrounded by histiocytes and multinucleated giant cells. Periodic acid–Schiff and silver stains can identify fungal organisms, while Gram stain helps to elucidate bacterial etiologies.1 In our patient, a biopsy revealed several dermal aggregates of pseudofilamentous gram-positive organisms surrounded by a neutrophilic and histiocytic infiltrate.8 Because this case presented over weeks to months rather than months to years, it progressed more rapidly than a classic mycetoma. However, the dermatologic and histologic features were consistent with mycetoma.
Management—General treatment of actinomycetoma requires identification of the causative organism and prolonged administration of antibiotics, typically in combination.35-37 Most G bronchialis infections associated with surgical intervention or implants in the literature required surgical debridement and removal of contaminated material for clinical cure, with the exception of 3 cases of sternal wound infection and 1 case of peritonitis that recovered with antimicrobial therapy alone.3,17 Combination therapy often was used, but monotherapy, particularly with a fluoroquinolone, has been reported. Susceptibility data are limited, but in general, Gordonia species appear susceptible to imipenem, ciprofloxacin, amikacin, gentamicin, and linezolid, with variable susceptibility to vancomycin (89% of isolates), third-generation cephalosporins (80%–90% of isolates), tetracyclines (≤85% of isolates), penicillin (≤70% of isolates), and trimethoprim-sulfamethoxazole (≤65% of isolates).7,10,19,38-40 Although there are no standardized recommendations for the treatment of these infections, the most commonly used drugs to treat Gordonia are carbapenems and fluoroquinolones, with or without an aminoglycoside, followed by third-generation cephalosporins and vancomycin, depending on susceptibilities. Additional antibiotics (alone or in combination) that have previously been used with favorable outcomes include amoxicillin or amoxicillin-clavulanate, piperacillin-tazobactam, rifampicin, trimethoprim-sulfamethoxazole, minocycline, doxycycline, and daptomycin.
Our patient received amoxicillin-clavulanate, trimethoprim-sulfamethoxazole, and linezolid. We considered combination therapy but decided against it due to concern for toxicity, given his age and poor renal function. The antibiotic that was most important to his recovery was unclear; the patient insisted that his body, not antibiotics, deserved most of the credit for healing his arm. Although cultures and polymerase chain reaction assays were negative after 3 weeks of amoxicillin-clavulanate, the patient did not show clinical improvement—reasons could be because the antibiotic reduced but did not eliminate the bacterial burden, sampling error of the biopsy, or it takes much longer for the body to heal than it takes to kill the bacteria. Most likely a combination of factors was at play.
Conclusion
Gordonia bronchialis is an emerging cause of human infections typically occurring after trauma, inoculation, or surgery. Most infections are localized; however, the present case highlights the ability of this species to form a massive cutaneous infection. Treatment should be tailored to susceptibility, with close follow-up to ensure improvement and resolution. For clinicians encountering a similar case, we encourage biopsy prior to empiric antibiotics, as antibiotic therapy can decrease the yield of subsequent testing. Treatment should be guided by the clinical course and may need to last weeks to months. Combination therapy for Gordonia infections should be considered in severe cases, in cases presenting as actinomycetoma, in those not responding to therapy, or when the susceptibility profile is unknown or unreliable.
Acknowledgments—The authors thank this veteran for allowing us to participate in his care and to learn from his experience. He gave his consent for us to share his story and the photographs of the arm.
- Arenas R, Fernandez Martinez RF, Torres-Guerrero E, et al. Actinomycetoma: an update on diagnosis and treatment. Cutis. 2017;99:E11-E15.
- Poonwan N, Mekha N, Yazawa K, et al. Characterization of clinical isolates of pathogenic Nocardia strains and related actinomycetes in Thailand from 1996 to 2003. Mycopathologia. 2005;159:361-368.
- Richet HM, Craven PC, Brown JM, et al. A cluster of Rhodococcus (Gordona) bronchialis sternal-wound infections after coronary-artery bypass surgery. N Engl J Med. 1991;324:104-109.
- Wright SN, Gerry JS, Busowski MT, et al. Gordonia bronchialis sternal wound infection in 3 patients following open heart surgery: intraoperative transmission from a healthcare worker. Infect Control Hosp Epidemiol. 2012;33:1238-1241.
- Tsukamura M. Proposal of a new genus, Gordona, for slightly acid-fast organisms occurring in sputa of patients with pulmonary disease and in soil. J Gen Microbiol. 1971;68:15-26.
- Wang T, Kong F, Chen S, et al. Improved identification of Gordonia, Rhodococcus and Tsukamurella species by 5′-end 16s rRNA gene sequencing. Pathology. 2011;43:58-63.
- Aoyama K, Kang Y, Yazawa K, et al. Characterization of clinical isolates of Gordonia species in Japanese clinical samples during 1998-2008. Mycopathologia. 2009;168:175-183.
- Ivanova N, Sikorski J, Jando M, et al. Complete genome sequence of Gordonia bronchialis type strain (3410 T). Stand Genomic Sci. 2010;2:19-28.
- Johnson JA, Onderdonk AB, Cosimi LA, et al. Gordonia bronchialis bacteremia and pleural infection: case report and review of the literature. J Clin Microbiol. 2011;49:1662-1666.
- Bartolomé-Álvarez J, Sáez-Nieto JA, Escudero-Jiménez A, et al. Cutaneous abscess due to Gordonia bronchialis: case report and literature review. Rev Esp Quimioter. 2016;29:170-173.
- Choi ME, Jung CJ, Won CH, et al. Case report of cutaneous nodule caused by Gordonia bronchialis in an immunocompetent patient after receiving acupuncture. J Dermatol. 2019;46:343-346.
- Nguyen DB, Gupta N, Abou-Daoud A, et al. A polymicrobial outbreak of surgical site infections following cardiac surgery at a community hospital in Florida, 2011-2012. Am J Infect Control. 2014;42:432-435.
- Chang JH, Ji M, Hong HL, et al. Sternal osteomyelitis caused byGordonia bronchialis after open-heart surgery. Infect Chemother. 2014;46:110-114.
- Rodriguez-Lozano J, Pérez-Llantada E, Agüero J, et al. Sternal wound infection caused by Gordonia bronchialis: identification by MALDI-TOF MS. JMM Case Rep. 2016;3:e005067.
- Akrami K, Coletta J, Mehta S, et al. Gordonia sternal wound infection treated with ceftaroline: case report and literature review. JMM Case Rep. 2017;4:e005113.
- Ambesh P, Kapoor A, Kazmi D, et al. Sternal osteomyelitis by Gordonia bronchialis in an immunocompetent patient after open heart surgery. Ann Card Anaesth. 2019;22:221-224.
- Ma TKW, Chow KM, Kwan BCH, et al. Peritoneal-dialysis related peritonitis caused by Gordonia species: report of four cases and literature review. Nephrology. 2014;19:379-383.
- Lam JYW, Wu AKL, Leung WS, et al. Gordonia species as emerging causes of continuous-ambulatory-peritoneal-dialysis-related peritonitis identified by 16S rRNA and secA1 gene sequencing and matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS). J Clin Microbiol. 2015;53:671-676.
- Blaschke AJ, Bender J, Byington CL, et al. Gordonia species: emerging pathogens in pediatric patients that are identified by 16S ribosomal RNA gene sequencing. Clin Infect Dis. 2007;45:483-486.
- Titécat M, Loïez C, Courcol RJ, et al. Difficulty with Gordonia bronchialis identification by Microflex mass spectrometer in a pacemaker‐induced endocarditis. JMM Case Rep. 2014;1:E003681.
- Siddiqui N, Toumeh A, Georgescu C. Tibial osteomyelitis caused by Gordonia bronchialis in an immunocompetent patient. J Clin Microbiol. 2012;50:3119-3121.
- Choi R, Strnad L, Flaxel CJ, et al. Gordonia bronchialis–associated endophthalmitis. Emerg Infect Dis. 2019;25:1017-1019.
- Werno AM, Anderson TP, Chambers ST, et al. Recurrent breast abscess caused by Gordonia bronchialis in an immunocompetent patient. J Clin Microbiol. 2005;43:3009-3010.
- Sng LH, Koh TH, Toney SR, et al. Bacteremia caused by Gordonia bronchialis in a patient with sequestrated lung. J Clin Microbiol. 2004;42:2870-2871.
- Ramanan P, Deziel PJ, Wengenack NL. Gordonia bacteremia. J Clin Microbiol. 2013;51:3443-3447.
- Sukackiene D, Rimsevicius L, Kiveryte S, et al. A case of successfully treated relapsing peritoneal dialysis-associated peritonitis caused by Gordonia bronchialis in a farmer. Nephrol Ther. 2018;14:109-111.
- Bruno V, Tjon J, Lin S, et al. Peritoneal dialysis-related peritonitis caused by Gordonia bronchialis: first pediatric report. Pediatr Nephrol. 2022;37:217-220. doi: 10.1007/s00467-021-05313-3
- Mormeneo Bayo S, Palacián Ruíz MP, Asin Samper U, et al. Pacemaker-induced endocarditis by Gordonia bronchialis. Enferm Infecc Microbiol Clin (Engl Ed). 2022;40:255-257.
- Davidson AL, Driscoll CR, Luther VP, et al. Recurrent skin and soft tissue infection following breast reduction surgery caused by Gordonia bronchialis: a case report. Plast Reconstr Surg Glob Open. 2022;10:E4395.
- Nwaedozie S, Mojarrab JN, Gopinath P, et al. Sternal osteomyelitis caused by Gordonia bronchialis in an immunocompetent patient following coronary artery bypass surgery. IDCases. 2022;29:E01548.
- Nakahama H, Hanada S, Takada K, et al. Obstructive pneumonia caused by Gordonia bronchialis with a bronchial foreign body. Int J Infect Dis. 2022;124:157-158. doi:10.1016/j.ijid.2022.09.028
- Lai CC, Hsieh JH, Tsai HY, et al. Cutaneous infection caused by Gordonia amicalis after a traumatic injury. J Clin Microbiol. 2012;50:1821-1822.
- Bakker XR, Spauwen PHM, Dolmans WMV. Mycetoma of the hand caused by Gordona terrae: a case report. J Hand Surg Am. 2004;29:188-190.
- Gueneau R, Blanchet D, Rodriguez-Nava V, et al. Actinomycetoma caused by Gordonia westfalica: first reported case of human infection. New Microbes New Infect. 2020;34:100658.
- Auwaerter PG, ed. The Johns Hopkins POC-IT ABX Guide. Johns Hopkins Medicine; 2021.
- Welsh O, Sauceda E, Gonzalez J, et al. Amikacin alone andin combination with trimethoprim-sulfamethoxazole in the treatment of actinomycotic mycetoma. J Am Acad Dermatol. 1987;17:443-448.
- Zijlstra EE, van de Sande WWJ, Welsh O, et al. Mycetoma: a unique neglected tropical disease. Lancet Infect Dis. 2016;16:100-112.
- Pham AS, Dé I, Rolston KV, et al. Catheter-related bacteremia caused by the nocardioform actinomycete Gordonia terrae. Clin Infect Dis. 2003;36:524-527.
- Renvoise A, Harle JR, Raoult D, et al. Gordonia sputi bacteremia. Emerg Infect Dis. 2009;15:1535-1537.
- Moser BD, Pellegrini GJ, Lasker BA, et al. Pattern of antimicrobial susceptibility obtained from blood isolates of a rare but emerging human pathogen, Gordonia polyisoprenivorans. Antimicrob Agents Chemother. 2012;56:4991-4993.
- Arenas R, Fernandez Martinez RF, Torres-Guerrero E, et al. Actinomycetoma: an update on diagnosis and treatment. Cutis. 2017;99:E11-E15.
- Poonwan N, Mekha N, Yazawa K, et al. Characterization of clinical isolates of pathogenic Nocardia strains and related actinomycetes in Thailand from 1996 to 2003. Mycopathologia. 2005;159:361-368.
- Richet HM, Craven PC, Brown JM, et al. A cluster of Rhodococcus (Gordona) bronchialis sternal-wound infections after coronary-artery bypass surgery. N Engl J Med. 1991;324:104-109.
- Wright SN, Gerry JS, Busowski MT, et al. Gordonia bronchialis sternal wound infection in 3 patients following open heart surgery: intraoperative transmission from a healthcare worker. Infect Control Hosp Epidemiol. 2012;33:1238-1241.
- Tsukamura M. Proposal of a new genus, Gordona, for slightly acid-fast organisms occurring in sputa of patients with pulmonary disease and in soil. J Gen Microbiol. 1971;68:15-26.
- Wang T, Kong F, Chen S, et al. Improved identification of Gordonia, Rhodococcus and Tsukamurella species by 5′-end 16s rRNA gene sequencing. Pathology. 2011;43:58-63.
- Aoyama K, Kang Y, Yazawa K, et al. Characterization of clinical isolates of Gordonia species in Japanese clinical samples during 1998-2008. Mycopathologia. 2009;168:175-183.
- Ivanova N, Sikorski J, Jando M, et al. Complete genome sequence of Gordonia bronchialis type strain (3410 T). Stand Genomic Sci. 2010;2:19-28.
- Johnson JA, Onderdonk AB, Cosimi LA, et al. Gordonia bronchialis bacteremia and pleural infection: case report and review of the literature. J Clin Microbiol. 2011;49:1662-1666.
- Bartolomé-Álvarez J, Sáez-Nieto JA, Escudero-Jiménez A, et al. Cutaneous abscess due to Gordonia bronchialis: case report and literature review. Rev Esp Quimioter. 2016;29:170-173.
- Choi ME, Jung CJ, Won CH, et al. Case report of cutaneous nodule caused by Gordonia bronchialis in an immunocompetent patient after receiving acupuncture. J Dermatol. 2019;46:343-346.
- Nguyen DB, Gupta N, Abou-Daoud A, et al. A polymicrobial outbreak of surgical site infections following cardiac surgery at a community hospital in Florida, 2011-2012. Am J Infect Control. 2014;42:432-435.
- Chang JH, Ji M, Hong HL, et al. Sternal osteomyelitis caused byGordonia bronchialis after open-heart surgery. Infect Chemother. 2014;46:110-114.
- Rodriguez-Lozano J, Pérez-Llantada E, Agüero J, et al. Sternal wound infection caused by Gordonia bronchialis: identification by MALDI-TOF MS. JMM Case Rep. 2016;3:e005067.
- Akrami K, Coletta J, Mehta S, et al. Gordonia sternal wound infection treated with ceftaroline: case report and literature review. JMM Case Rep. 2017;4:e005113.
- Ambesh P, Kapoor A, Kazmi D, et al. Sternal osteomyelitis by Gordonia bronchialis in an immunocompetent patient after open heart surgery. Ann Card Anaesth. 2019;22:221-224.
- Ma TKW, Chow KM, Kwan BCH, et al. Peritoneal-dialysis related peritonitis caused by Gordonia species: report of four cases and literature review. Nephrology. 2014;19:379-383.
- Lam JYW, Wu AKL, Leung WS, et al. Gordonia species as emerging causes of continuous-ambulatory-peritoneal-dialysis-related peritonitis identified by 16S rRNA and secA1 gene sequencing and matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS). J Clin Microbiol. 2015;53:671-676.
- Blaschke AJ, Bender J, Byington CL, et al. Gordonia species: emerging pathogens in pediatric patients that are identified by 16S ribosomal RNA gene sequencing. Clin Infect Dis. 2007;45:483-486.
- Titécat M, Loïez C, Courcol RJ, et al. Difficulty with Gordonia bronchialis identification by Microflex mass spectrometer in a pacemaker‐induced endocarditis. JMM Case Rep. 2014;1:E003681.
- Siddiqui N, Toumeh A, Georgescu C. Tibial osteomyelitis caused by Gordonia bronchialis in an immunocompetent patient. J Clin Microbiol. 2012;50:3119-3121.
- Choi R, Strnad L, Flaxel CJ, et al. Gordonia bronchialis–associated endophthalmitis. Emerg Infect Dis. 2019;25:1017-1019.
- Werno AM, Anderson TP, Chambers ST, et al. Recurrent breast abscess caused by Gordonia bronchialis in an immunocompetent patient. J Clin Microbiol. 2005;43:3009-3010.
- Sng LH, Koh TH, Toney SR, et al. Bacteremia caused by Gordonia bronchialis in a patient with sequestrated lung. J Clin Microbiol. 2004;42:2870-2871.
- Ramanan P, Deziel PJ, Wengenack NL. Gordonia bacteremia. J Clin Microbiol. 2013;51:3443-3447.
- Sukackiene D, Rimsevicius L, Kiveryte S, et al. A case of successfully treated relapsing peritoneal dialysis-associated peritonitis caused by Gordonia bronchialis in a farmer. Nephrol Ther. 2018;14:109-111.
- Bruno V, Tjon J, Lin S, et al. Peritoneal dialysis-related peritonitis caused by Gordonia bronchialis: first pediatric report. Pediatr Nephrol. 2022;37:217-220. doi: 10.1007/s00467-021-05313-3
- Mormeneo Bayo S, Palacián Ruíz MP, Asin Samper U, et al. Pacemaker-induced endocarditis by Gordonia bronchialis. Enferm Infecc Microbiol Clin (Engl Ed). 2022;40:255-257.
- Davidson AL, Driscoll CR, Luther VP, et al. Recurrent skin and soft tissue infection following breast reduction surgery caused by Gordonia bronchialis: a case report. Plast Reconstr Surg Glob Open. 2022;10:E4395.
- Nwaedozie S, Mojarrab JN, Gopinath P, et al. Sternal osteomyelitis caused by Gordonia bronchialis in an immunocompetent patient following coronary artery bypass surgery. IDCases. 2022;29:E01548.
- Nakahama H, Hanada S, Takada K, et al. Obstructive pneumonia caused by Gordonia bronchialis with a bronchial foreign body. Int J Infect Dis. 2022;124:157-158. doi:10.1016/j.ijid.2022.09.028
- Lai CC, Hsieh JH, Tsai HY, et al. Cutaneous infection caused by Gordonia amicalis after a traumatic injury. J Clin Microbiol. 2012;50:1821-1822.
- Bakker XR, Spauwen PHM, Dolmans WMV. Mycetoma of the hand caused by Gordona terrae: a case report. J Hand Surg Am. 2004;29:188-190.
- Gueneau R, Blanchet D, Rodriguez-Nava V, et al. Actinomycetoma caused by Gordonia westfalica: first reported case of human infection. New Microbes New Infect. 2020;34:100658.
- Auwaerter PG, ed. The Johns Hopkins POC-IT ABX Guide. Johns Hopkins Medicine; 2021.
- Welsh O, Sauceda E, Gonzalez J, et al. Amikacin alone andin combination with trimethoprim-sulfamethoxazole in the treatment of actinomycotic mycetoma. J Am Acad Dermatol. 1987;17:443-448.
- Zijlstra EE, van de Sande WWJ, Welsh O, et al. Mycetoma: a unique neglected tropical disease. Lancet Infect Dis. 2016;16:100-112.
- Pham AS, Dé I, Rolston KV, et al. Catheter-related bacteremia caused by the nocardioform actinomycete Gordonia terrae. Clin Infect Dis. 2003;36:524-527.
- Renvoise A, Harle JR, Raoult D, et al. Gordonia sputi bacteremia. Emerg Infect Dis. 2009;15:1535-1537.
- Moser BD, Pellegrini GJ, Lasker BA, et al. Pattern of antimicrobial susceptibility obtained from blood isolates of a rare but emerging human pathogen, Gordonia polyisoprenivorans. Antimicrob Agents Chemother. 2012;56:4991-4993.
Practice Points
- Gordonia bronchialis is an emerging cause of human skin and soft tissue infection, typically occurring after trauma, inoculation, or surgery.
- Gordonia species can cause a mycetomalike skin infection.
- Increasing use of molecular methods to identify bacteria has improved identification of clinically relevant actinomycetes, such as Helvetica Neue LT StdGordonia, and increases the likelihood that clinicians will see these organisms on culture results.

Iron Screening in Alopecia Areata Patients May Catch Hereditary Hemochromatosis Early
The role of micronutrients in the hair follicle cycle is not fully understood; thus deficiency and/or excess of certain micronutrients may be a modifiable risk factor associated with the development and/or treatment of some types of hair loss and therefore may be included in the workup during an alopecia consultation.
Hereditary hemochromatosis (HHC) is the most common genetic disorder identified in White individuals, with a worldwide prevalence of 1 in 220 to 1 in 250 individuals for a homozygous mutation. It most commonly affects individuals of Northern European descent.1 Men usually present in the fourth to sixth decades of life, while women usually develop symptoms after menopause, as pregnancy and menstruation delay the onset of the disease.2 Early symptoms of HHC include fatigue, joint pain, abdominal pain, and weight loss. Men are more likely to develop complications; in fact, 1 in 10 men with HHC will develop severe liver disease.3 As the disease progresses, affected individuals can present with cardiomyopathy (restrictive and dilated), cirrhosis, hypogonadism (usually hypogonadotrophic), arthropathy, diabetes mellitus, hepatomegaly, hepatic cirrhosis, and primary liver cancer (eg, hepatocellular carcinoma, cholangiocarcinoma).2 Approximately 90% of patients with HHC present with hyperpigmentation at the time of diagnosis.4 Thinning or loss of hair is another finding in HHC, primarily reported in the axillae and pubic regions, and is ascribed to hepatotesticular insufficiency.5
Alopecia areata (AA) is the most common cause of autoimmune, inflammation-induced hair loss, with a calculated lifetime risk of 2%.6 This disease manifests as loss of hair in well-circumscribed patches of skin, most commonly on the scalp; AA also may affect other hair-bearing sites on the body. It is associated with an increased risk for other autoimmune disorders, such as psoriasis, thyroid disease, rheumatoid arthritis, systemic lupus erythematosus, and vitiligo.7
Alopecia areata is induced by an inflammatory infiltrate of CD4+ and CD8+ T lymphocytes around hair follicles in the anagen stage, the active growth phase.8 Although the diagnosis is clinical, some clinicians order laboratory thyroid studies to investigate conditions that may be associated with AA. Common treatments include topical, intralesional, and/or systemic corticosteroids; contact immunotherapy; topical and more recently oral minoxidil; phototherapy; and topical and systemic JAK inhibitors, including tofacitinib.4,9
We reviewed the medical records of 533 patients who were seen in The University of Texas Southwestern (Dallas, Texas) dermatology clinic from January 2015 through January 2020 and were diagnosed with AA. We examined their demographic data and medical history. We sought to determine any relationship between various types of alopecia and certain micronutrient levels through laboratory test results. Ferritin and iron saturation studies were evaluated. We report 4 cases of HHC concurrent with AA, of which 2 HHC diagnoses were uncovered through iron studies as part of the alopecia evaluation.
Case Reports
Patient 1—A 55-year-old White woman presented to the clinic for an alopecia consultation. She had a medical history of hypothyroidism and AA that was treated unsuccessfully with triamcinolone acetonide steroid injections; topical minoxidil; topical steroids; and systemic steroids, specifically oral prednisone. Following evaluation, she successfully transitioned to treatment with oral tofacitinib and continued to do well on tofacitinib.
The patient’s alopecia workup revealed a ferritin level of 245 ng/mL (reference range, 13–150 ng/mL) and iron saturation of 60% (reference range, 20%–50%). She was referred to the hematology department for further evaluation and was diagnosed with HHC. Genetic testing revealed a heterozygous H63D mutation; therapeutic phlebotomy was recommended. Her sister also was recently diagnosed with HHC.
Patient 2—A 55-year-old White man was referred for evaluation and treatment of alopecia universalis. He had a medical history of skin cancer and vitiligo. He attempted contact immunotherapy with diphenylcyclopropenone scalp treatment but stopped due to intolerable inflammation. Intervention with a topical steroid and topical minoxidil was unsuccessful, but use of triamcinolone acetonide steroid injection on the scalp and topical bimatoprost 0.03% on the eyebrows produced satisfactory results.
The patient’s alopecia workup revealed a ferritin level of 422 ng/mL (reference range, 30–400 ng/mL), which prompted a hematology consultation for further evaluation. Notably, the patient ate red meat several times a week, used iron skillets, and denied receiving blood transfusions. His social habits included 3 alcoholic beverages a night, 5 days a week. Ultrasonography of the liver was recommended to assess potential damage from iron overload and alcohol consumption; the results suggested chronic liver disease, not definitive for cirrhosis, and no evidence of hepatocellular carcinoma. Genetic analysis later revealed the heterozygous H63D variant; therapeutic phlebotomy was recommended.
Patient 3—A 22-year-old White man presented with AA involving his facial beard. He had a medical history of vitiligo and psoriasis and a family history of AA as well as other autoimmune diseases including Hashimoto thyroiditis, psoriasis, eczema, and autoimmune hepatitis. Diphenylcyclopropenone treatment was not successful.
Laboratory studies revealed mildly elevated transaminase and ferritin levels. The patient also presented to the gastroenterologist for evaluation of abdominal pain. Subsequent hematology evaluation confirmed the presence of compound heterozygous C282Y and H63D mutations in the HFE gene, and the patient’s mother was later determined to be homozygous for the C282Y mutation with no elevated ferritin level. The patient’s ferritin level at diagnosis was approximately 500 ng/mL (reference range, 22–322 ng/mL); he required a modest number of therapeutic phlebotomies to normalize his ferritin level.
Patient 4—A 62-year-old White woman presented for evaluation and treatment of patchy hair loss on the scalp of 7 months’ duration. She was subsequently diagnosed with AA. After unsuccessful treatment with a triamcinolone acetonide steroid injection, topical immunotherapy with diphenylcyclopropenone was recommended. The patient achieved full hair regrowth after 35 treatments administered at 3-week intervals.
The patient had a medical history of HHC, including homozygosity for the C282Y mutation, and a family history of HHC in 1 sister. Treatment was therapeutic phlebotomy.
Comment
HHC in the Setting of AA—We presented 4 White patients with both HHC and AA. A PubMed search of articles indexed for MEDLINE using the terms HHC and AA yielded only 1 other reported case of newly identified HHC in a 56-year-old man who presented with pigmented purpuric dermatitis and AA that affected the beard.10 Because HHC is the most common genetic disorder identified in White individuals and has a varied clinical presentation, the documentation of AA may be an important cutaneous clue to help clinicians diagnose HHC early.
Iron Overload in Patients With HHC—The genetic association between HHC and AA, if any, is unknown. What is known is that iron overload can catalyze reactive oxygen species, which can overwhelm cellular antioxidant capacities at particular levels and cause injury to its constituents.11 Data show that the levels of oxidative stress are elevated in the scalp of patients with AA compared to controls and increased 2-fold during the early phase of disease vs late-phase disease.12 Thus, it is possible that increased iron levels in HHC may contribute to AA in genetically susceptible individuals by direct toxicity that ultimately results in the AA hair disorder that is CD8+ T-cell mediated.
Data show that 78% (31/40) of men and 36% (14/39) of women identified with homozygous C282Y mutations determined from family genetic analyses exhibited iron overload.13 In general, a normal life expectancy is possible for patients promptly treated with appropriate therapeutic phlebotomies.14 Thus, early diagnosis and appropriate therapy can prevent consequences of iron overload, which include cirrhosis, diabetes mellitus, and cardiomyopathy.13Iron Screening in the Alopecia Workup—Our cases illustrate how iron screening tests as part of the alopecia workup identified a cohort of White patients with iron overload and subsequently led to an early diagnosis of HHC. The calculated 2% lifetime risk for developing AA highlights the importance of evaluating iron status as part of the AA workup, particularly for White men, and the potential health benefit from early diagnosis of HHC. Limitations of this case series included its retrospective nature and small patient number.
- Bacon BR, Adams PC, Kowdley KV, et al. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54:328-343.
- Barton JC, Edwards CQ. HFE hemochromatosis. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews® [Internet]. University of Washington, Seattle; 1993-2020.
- Centers for Disease Control and Prevention. Hereditary hemochromatosis. Accessed September 13, 2022. https://www.cdc.gov/genomics/disease/hemochromatosis.htm
- Ibrahim O, Bayart CB, Hogan S, et al. Treatment of alopecia areata with tofacitinib. JAMA Dermatol. 2017;153:600-602.
- Tweed MJ, Roland JM. Haemochromatosis as an endocrine cause of subfertility. BMJ. 1998;316:915-916. doi:10.1136/bmj.316.7135.915
- Gilhar A, Etzioni A, Paus R. Alopecia areata. N Engl J Med. 2012;366:1515-1525.
- Barahmani N, Schabath MB, Duvic M, et al. History of atopy or autoimmunity increases risk of alopecia areata. J Am Acad Dermatol. 2009;61:581-591.
- McElwee KJ, Freyschmidt-Paul P, Hoffmann R, et al. Transfer of CD8(+) cells induces localized hair loss whereas CD4(+)/CD25(−) cells promote systemic alopecia areata and CD4(+)/CD25(+) cells blockade disease onset in the C3H/HeJ mouse model. J Invest Dermatol. 2005;124:947-957.
- MacDonald Hull SP, Wood ML, Hutchinson PE, et al. Guidelines for the management of alopecia areata. Br J Dermatol. 2003;149:692-699.
- Sredoja Tišma V, Bulimbašic´ S, Jaganjac M, et al. Progressive pigmented purpuric dermatitis and alopecia areata as unusual skin manifestations in recognizing hereditary hemochromatosis. Acta Dermatovenerol Croat. 2012;20:181-186.
- Cabantchik ZI. Labile iron in cells and body fluids: physiology, pathology, and pharmacology. Front Pharmacol. 2014;5:45.
- Akar A, Arca E, Erbil H, et al. Antioxidant enzymes and lipid peroxidation in the scalp of patients with alopecia areata. J Dermatol Sci. 2002;29:85-90.
- Ryan E, Byrnes V, Coughlan B, et al. Underdiagnosis of hereditary haemochromatosis: lack of presentation or penetration? Gut. 2002;51:108-112.
- Niederau C, Strohmeyer G. Strategies for early diagnosis of haemochromatosis. Eur J Gastroenterol Hepatol. 2002;14:217-221.
The role of micronutrients in the hair follicle cycle is not fully understood; thus deficiency and/or excess of certain micronutrients may be a modifiable risk factor associated with the development and/or treatment of some types of hair loss and therefore may be included in the workup during an alopecia consultation.
Hereditary hemochromatosis (HHC) is the most common genetic disorder identified in White individuals, with a worldwide prevalence of 1 in 220 to 1 in 250 individuals for a homozygous mutation. It most commonly affects individuals of Northern European descent.1 Men usually present in the fourth to sixth decades of life, while women usually develop symptoms after menopause, as pregnancy and menstruation delay the onset of the disease.2 Early symptoms of HHC include fatigue, joint pain, abdominal pain, and weight loss. Men are more likely to develop complications; in fact, 1 in 10 men with HHC will develop severe liver disease.3 As the disease progresses, affected individuals can present with cardiomyopathy (restrictive and dilated), cirrhosis, hypogonadism (usually hypogonadotrophic), arthropathy, diabetes mellitus, hepatomegaly, hepatic cirrhosis, and primary liver cancer (eg, hepatocellular carcinoma, cholangiocarcinoma).2 Approximately 90% of patients with HHC present with hyperpigmentation at the time of diagnosis.4 Thinning or loss of hair is another finding in HHC, primarily reported in the axillae and pubic regions, and is ascribed to hepatotesticular insufficiency.5
Alopecia areata (AA) is the most common cause of autoimmune, inflammation-induced hair loss, with a calculated lifetime risk of 2%.6 This disease manifests as loss of hair in well-circumscribed patches of skin, most commonly on the scalp; AA also may affect other hair-bearing sites on the body. It is associated with an increased risk for other autoimmune disorders, such as psoriasis, thyroid disease, rheumatoid arthritis, systemic lupus erythematosus, and vitiligo.7
Alopecia areata is induced by an inflammatory infiltrate of CD4+ and CD8+ T lymphocytes around hair follicles in the anagen stage, the active growth phase.8 Although the diagnosis is clinical, some clinicians order laboratory thyroid studies to investigate conditions that may be associated with AA. Common treatments include topical, intralesional, and/or systemic corticosteroids; contact immunotherapy; topical and more recently oral minoxidil; phototherapy; and topical and systemic JAK inhibitors, including tofacitinib.4,9
We reviewed the medical records of 533 patients who were seen in The University of Texas Southwestern (Dallas, Texas) dermatology clinic from January 2015 through January 2020 and were diagnosed with AA. We examined their demographic data and medical history. We sought to determine any relationship between various types of alopecia and certain micronutrient levels through laboratory test results. Ferritin and iron saturation studies were evaluated. We report 4 cases of HHC concurrent with AA, of which 2 HHC diagnoses were uncovered through iron studies as part of the alopecia evaluation.
Case Reports
Patient 1—A 55-year-old White woman presented to the clinic for an alopecia consultation. She had a medical history of hypothyroidism and AA that was treated unsuccessfully with triamcinolone acetonide steroid injections; topical minoxidil; topical steroids; and systemic steroids, specifically oral prednisone. Following evaluation, she successfully transitioned to treatment with oral tofacitinib and continued to do well on tofacitinib.
The patient’s alopecia workup revealed a ferritin level of 245 ng/mL (reference range, 13–150 ng/mL) and iron saturation of 60% (reference range, 20%–50%). She was referred to the hematology department for further evaluation and was diagnosed with HHC. Genetic testing revealed a heterozygous H63D mutation; therapeutic phlebotomy was recommended. Her sister also was recently diagnosed with HHC.
Patient 2—A 55-year-old White man was referred for evaluation and treatment of alopecia universalis. He had a medical history of skin cancer and vitiligo. He attempted contact immunotherapy with diphenylcyclopropenone scalp treatment but stopped due to intolerable inflammation. Intervention with a topical steroid and topical minoxidil was unsuccessful, but use of triamcinolone acetonide steroid injection on the scalp and topical bimatoprost 0.03% on the eyebrows produced satisfactory results.
The patient’s alopecia workup revealed a ferritin level of 422 ng/mL (reference range, 30–400 ng/mL), which prompted a hematology consultation for further evaluation. Notably, the patient ate red meat several times a week, used iron skillets, and denied receiving blood transfusions. His social habits included 3 alcoholic beverages a night, 5 days a week. Ultrasonography of the liver was recommended to assess potential damage from iron overload and alcohol consumption; the results suggested chronic liver disease, not definitive for cirrhosis, and no evidence of hepatocellular carcinoma. Genetic analysis later revealed the heterozygous H63D variant; therapeutic phlebotomy was recommended.
Patient 3—A 22-year-old White man presented with AA involving his facial beard. He had a medical history of vitiligo and psoriasis and a family history of AA as well as other autoimmune diseases including Hashimoto thyroiditis, psoriasis, eczema, and autoimmune hepatitis. Diphenylcyclopropenone treatment was not successful.
Laboratory studies revealed mildly elevated transaminase and ferritin levels. The patient also presented to the gastroenterologist for evaluation of abdominal pain. Subsequent hematology evaluation confirmed the presence of compound heterozygous C282Y and H63D mutations in the HFE gene, and the patient’s mother was later determined to be homozygous for the C282Y mutation with no elevated ferritin level. The patient’s ferritin level at diagnosis was approximately 500 ng/mL (reference range, 22–322 ng/mL); he required a modest number of therapeutic phlebotomies to normalize his ferritin level.
Patient 4—A 62-year-old White woman presented for evaluation and treatment of patchy hair loss on the scalp of 7 months’ duration. She was subsequently diagnosed with AA. After unsuccessful treatment with a triamcinolone acetonide steroid injection, topical immunotherapy with diphenylcyclopropenone was recommended. The patient achieved full hair regrowth after 35 treatments administered at 3-week intervals.
The patient had a medical history of HHC, including homozygosity for the C282Y mutation, and a family history of HHC in 1 sister. Treatment was therapeutic phlebotomy.
Comment
HHC in the Setting of AA—We presented 4 White patients with both HHC and AA. A PubMed search of articles indexed for MEDLINE using the terms HHC and AA yielded only 1 other reported case of newly identified HHC in a 56-year-old man who presented with pigmented purpuric dermatitis and AA that affected the beard.10 Because HHC is the most common genetic disorder identified in White individuals and has a varied clinical presentation, the documentation of AA may be an important cutaneous clue to help clinicians diagnose HHC early.
Iron Overload in Patients With HHC—The genetic association between HHC and AA, if any, is unknown. What is known is that iron overload can catalyze reactive oxygen species, which can overwhelm cellular antioxidant capacities at particular levels and cause injury to its constituents.11 Data show that the levels of oxidative stress are elevated in the scalp of patients with AA compared to controls and increased 2-fold during the early phase of disease vs late-phase disease.12 Thus, it is possible that increased iron levels in HHC may contribute to AA in genetically susceptible individuals by direct toxicity that ultimately results in the AA hair disorder that is CD8+ T-cell mediated.
Data show that 78% (31/40) of men and 36% (14/39) of women identified with homozygous C282Y mutations determined from family genetic analyses exhibited iron overload.13 In general, a normal life expectancy is possible for patients promptly treated with appropriate therapeutic phlebotomies.14 Thus, early diagnosis and appropriate therapy can prevent consequences of iron overload, which include cirrhosis, diabetes mellitus, and cardiomyopathy.13Iron Screening in the Alopecia Workup—Our cases illustrate how iron screening tests as part of the alopecia workup identified a cohort of White patients with iron overload and subsequently led to an early diagnosis of HHC. The calculated 2% lifetime risk for developing AA highlights the importance of evaluating iron status as part of the AA workup, particularly for White men, and the potential health benefit from early diagnosis of HHC. Limitations of this case series included its retrospective nature and small patient number.
The role of micronutrients in the hair follicle cycle is not fully understood; thus deficiency and/or excess of certain micronutrients may be a modifiable risk factor associated with the development and/or treatment of some types of hair loss and therefore may be included in the workup during an alopecia consultation.
Hereditary hemochromatosis (HHC) is the most common genetic disorder identified in White individuals, with a worldwide prevalence of 1 in 220 to 1 in 250 individuals for a homozygous mutation. It most commonly affects individuals of Northern European descent.1 Men usually present in the fourth to sixth decades of life, while women usually develop symptoms after menopause, as pregnancy and menstruation delay the onset of the disease.2 Early symptoms of HHC include fatigue, joint pain, abdominal pain, and weight loss. Men are more likely to develop complications; in fact, 1 in 10 men with HHC will develop severe liver disease.3 As the disease progresses, affected individuals can present with cardiomyopathy (restrictive and dilated), cirrhosis, hypogonadism (usually hypogonadotrophic), arthropathy, diabetes mellitus, hepatomegaly, hepatic cirrhosis, and primary liver cancer (eg, hepatocellular carcinoma, cholangiocarcinoma).2 Approximately 90% of patients with HHC present with hyperpigmentation at the time of diagnosis.4 Thinning or loss of hair is another finding in HHC, primarily reported in the axillae and pubic regions, and is ascribed to hepatotesticular insufficiency.5
Alopecia areata (AA) is the most common cause of autoimmune, inflammation-induced hair loss, with a calculated lifetime risk of 2%.6 This disease manifests as loss of hair in well-circumscribed patches of skin, most commonly on the scalp; AA also may affect other hair-bearing sites on the body. It is associated with an increased risk for other autoimmune disorders, such as psoriasis, thyroid disease, rheumatoid arthritis, systemic lupus erythematosus, and vitiligo.7
Alopecia areata is induced by an inflammatory infiltrate of CD4+ and CD8+ T lymphocytes around hair follicles in the anagen stage, the active growth phase.8 Although the diagnosis is clinical, some clinicians order laboratory thyroid studies to investigate conditions that may be associated with AA. Common treatments include topical, intralesional, and/or systemic corticosteroids; contact immunotherapy; topical and more recently oral minoxidil; phototherapy; and topical and systemic JAK inhibitors, including tofacitinib.4,9
We reviewed the medical records of 533 patients who were seen in The University of Texas Southwestern (Dallas, Texas) dermatology clinic from January 2015 through January 2020 and were diagnosed with AA. We examined their demographic data and medical history. We sought to determine any relationship between various types of alopecia and certain micronutrient levels through laboratory test results. Ferritin and iron saturation studies were evaluated. We report 4 cases of HHC concurrent with AA, of which 2 HHC diagnoses were uncovered through iron studies as part of the alopecia evaluation.
Case Reports
Patient 1—A 55-year-old White woman presented to the clinic for an alopecia consultation. She had a medical history of hypothyroidism and AA that was treated unsuccessfully with triamcinolone acetonide steroid injections; topical minoxidil; topical steroids; and systemic steroids, specifically oral prednisone. Following evaluation, she successfully transitioned to treatment with oral tofacitinib and continued to do well on tofacitinib.
The patient’s alopecia workup revealed a ferritin level of 245 ng/mL (reference range, 13–150 ng/mL) and iron saturation of 60% (reference range, 20%–50%). She was referred to the hematology department for further evaluation and was diagnosed with HHC. Genetic testing revealed a heterozygous H63D mutation; therapeutic phlebotomy was recommended. Her sister also was recently diagnosed with HHC.
Patient 2—A 55-year-old White man was referred for evaluation and treatment of alopecia universalis. He had a medical history of skin cancer and vitiligo. He attempted contact immunotherapy with diphenylcyclopropenone scalp treatment but stopped due to intolerable inflammation. Intervention with a topical steroid and topical minoxidil was unsuccessful, but use of triamcinolone acetonide steroid injection on the scalp and topical bimatoprost 0.03% on the eyebrows produced satisfactory results.
The patient’s alopecia workup revealed a ferritin level of 422 ng/mL (reference range, 30–400 ng/mL), which prompted a hematology consultation for further evaluation. Notably, the patient ate red meat several times a week, used iron skillets, and denied receiving blood transfusions. His social habits included 3 alcoholic beverages a night, 5 days a week. Ultrasonography of the liver was recommended to assess potential damage from iron overload and alcohol consumption; the results suggested chronic liver disease, not definitive for cirrhosis, and no evidence of hepatocellular carcinoma. Genetic analysis later revealed the heterozygous H63D variant; therapeutic phlebotomy was recommended.
Patient 3—A 22-year-old White man presented with AA involving his facial beard. He had a medical history of vitiligo and psoriasis and a family history of AA as well as other autoimmune diseases including Hashimoto thyroiditis, psoriasis, eczema, and autoimmune hepatitis. Diphenylcyclopropenone treatment was not successful.
Laboratory studies revealed mildly elevated transaminase and ferritin levels. The patient also presented to the gastroenterologist for evaluation of abdominal pain. Subsequent hematology evaluation confirmed the presence of compound heterozygous C282Y and H63D mutations in the HFE gene, and the patient’s mother was later determined to be homozygous for the C282Y mutation with no elevated ferritin level. The patient’s ferritin level at diagnosis was approximately 500 ng/mL (reference range, 22–322 ng/mL); he required a modest number of therapeutic phlebotomies to normalize his ferritin level.
Patient 4—A 62-year-old White woman presented for evaluation and treatment of patchy hair loss on the scalp of 7 months’ duration. She was subsequently diagnosed with AA. After unsuccessful treatment with a triamcinolone acetonide steroid injection, topical immunotherapy with diphenylcyclopropenone was recommended. The patient achieved full hair regrowth after 35 treatments administered at 3-week intervals.
The patient had a medical history of HHC, including homozygosity for the C282Y mutation, and a family history of HHC in 1 sister. Treatment was therapeutic phlebotomy.
Comment
HHC in the Setting of AA—We presented 4 White patients with both HHC and AA. A PubMed search of articles indexed for MEDLINE using the terms HHC and AA yielded only 1 other reported case of newly identified HHC in a 56-year-old man who presented with pigmented purpuric dermatitis and AA that affected the beard.10 Because HHC is the most common genetic disorder identified in White individuals and has a varied clinical presentation, the documentation of AA may be an important cutaneous clue to help clinicians diagnose HHC early.
Iron Overload in Patients With HHC—The genetic association between HHC and AA, if any, is unknown. What is known is that iron overload can catalyze reactive oxygen species, which can overwhelm cellular antioxidant capacities at particular levels and cause injury to its constituents.11 Data show that the levels of oxidative stress are elevated in the scalp of patients with AA compared to controls and increased 2-fold during the early phase of disease vs late-phase disease.12 Thus, it is possible that increased iron levels in HHC may contribute to AA in genetically susceptible individuals by direct toxicity that ultimately results in the AA hair disorder that is CD8+ T-cell mediated.
Data show that 78% (31/40) of men and 36% (14/39) of women identified with homozygous C282Y mutations determined from family genetic analyses exhibited iron overload.13 In general, a normal life expectancy is possible for patients promptly treated with appropriate therapeutic phlebotomies.14 Thus, early diagnosis and appropriate therapy can prevent consequences of iron overload, which include cirrhosis, diabetes mellitus, and cardiomyopathy.13Iron Screening in the Alopecia Workup—Our cases illustrate how iron screening tests as part of the alopecia workup identified a cohort of White patients with iron overload and subsequently led to an early diagnosis of HHC. The calculated 2% lifetime risk for developing AA highlights the importance of evaluating iron status as part of the AA workup, particularly for White men, and the potential health benefit from early diagnosis of HHC. Limitations of this case series included its retrospective nature and small patient number.
- Bacon BR, Adams PC, Kowdley KV, et al. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54:328-343.
- Barton JC, Edwards CQ. HFE hemochromatosis. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews® [Internet]. University of Washington, Seattle; 1993-2020.
- Centers for Disease Control and Prevention. Hereditary hemochromatosis. Accessed September 13, 2022. https://www.cdc.gov/genomics/disease/hemochromatosis.htm
- Ibrahim O, Bayart CB, Hogan S, et al. Treatment of alopecia areata with tofacitinib. JAMA Dermatol. 2017;153:600-602.
- Tweed MJ, Roland JM. Haemochromatosis as an endocrine cause of subfertility. BMJ. 1998;316:915-916. doi:10.1136/bmj.316.7135.915
- Gilhar A, Etzioni A, Paus R. Alopecia areata. N Engl J Med. 2012;366:1515-1525.
- Barahmani N, Schabath MB, Duvic M, et al. History of atopy or autoimmunity increases risk of alopecia areata. J Am Acad Dermatol. 2009;61:581-591.
- McElwee KJ, Freyschmidt-Paul P, Hoffmann R, et al. Transfer of CD8(+) cells induces localized hair loss whereas CD4(+)/CD25(−) cells promote systemic alopecia areata and CD4(+)/CD25(+) cells blockade disease onset in the C3H/HeJ mouse model. J Invest Dermatol. 2005;124:947-957.
- MacDonald Hull SP, Wood ML, Hutchinson PE, et al. Guidelines for the management of alopecia areata. Br J Dermatol. 2003;149:692-699.
- Sredoja Tišma V, Bulimbašic´ S, Jaganjac M, et al. Progressive pigmented purpuric dermatitis and alopecia areata as unusual skin manifestations in recognizing hereditary hemochromatosis. Acta Dermatovenerol Croat. 2012;20:181-186.
- Cabantchik ZI. Labile iron in cells and body fluids: physiology, pathology, and pharmacology. Front Pharmacol. 2014;5:45.
- Akar A, Arca E, Erbil H, et al. Antioxidant enzymes and lipid peroxidation in the scalp of patients with alopecia areata. J Dermatol Sci. 2002;29:85-90.
- Ryan E, Byrnes V, Coughlan B, et al. Underdiagnosis of hereditary haemochromatosis: lack of presentation or penetration? Gut. 2002;51:108-112.
- Niederau C, Strohmeyer G. Strategies for early diagnosis of haemochromatosis. Eur J Gastroenterol Hepatol. 2002;14:217-221.
- Bacon BR, Adams PC, Kowdley KV, et al. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54:328-343.
- Barton JC, Edwards CQ. HFE hemochromatosis. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews® [Internet]. University of Washington, Seattle; 1993-2020.
- Centers for Disease Control and Prevention. Hereditary hemochromatosis. Accessed September 13, 2022. https://www.cdc.gov/genomics/disease/hemochromatosis.htm
- Ibrahim O, Bayart CB, Hogan S, et al. Treatment of alopecia areata with tofacitinib. JAMA Dermatol. 2017;153:600-602.
- Tweed MJ, Roland JM. Haemochromatosis as an endocrine cause of subfertility. BMJ. 1998;316:915-916. doi:10.1136/bmj.316.7135.915
- Gilhar A, Etzioni A, Paus R. Alopecia areata. N Engl J Med. 2012;366:1515-1525.
- Barahmani N, Schabath MB, Duvic M, et al. History of atopy or autoimmunity increases risk of alopecia areata. J Am Acad Dermatol. 2009;61:581-591.
- McElwee KJ, Freyschmidt-Paul P, Hoffmann R, et al. Transfer of CD8(+) cells induces localized hair loss whereas CD4(+)/CD25(−) cells promote systemic alopecia areata and CD4(+)/CD25(+) cells blockade disease onset in the C3H/HeJ mouse model. J Invest Dermatol. 2005;124:947-957.
- MacDonald Hull SP, Wood ML, Hutchinson PE, et al. Guidelines for the management of alopecia areata. Br J Dermatol. 2003;149:692-699.
- Sredoja Tišma V, Bulimbašic´ S, Jaganjac M, et al. Progressive pigmented purpuric dermatitis and alopecia areata as unusual skin manifestations in recognizing hereditary hemochromatosis. Acta Dermatovenerol Croat. 2012;20:181-186.
- Cabantchik ZI. Labile iron in cells and body fluids: physiology, pathology, and pharmacology. Front Pharmacol. 2014;5:45.
- Akar A, Arca E, Erbil H, et al. Antioxidant enzymes and lipid peroxidation in the scalp of patients with alopecia areata. J Dermatol Sci. 2002;29:85-90.
- Ryan E, Byrnes V, Coughlan B, et al. Underdiagnosis of hereditary haemochromatosis: lack of presentation or penetration? Gut. 2002;51:108-112.
- Niederau C, Strohmeyer G. Strategies for early diagnosis of haemochromatosis. Eur J Gastroenterol Hepatol. 2002;14:217-221.
Practice Points
- Hereditary hemochromatosis (HHC) is a disorder of iron overload that presents with clinical phenotypic heterogeneity. Complications can be mitigated with early intervention.
- Alopecia areata (AA) may be a rare early cutaneous manifestation of HHC in individuals with a predisposition for autoimmunity; therefore, it is important to evaluate iron status as part of the AA workup.
Genital HSV shedding declines rapidly in first year post infection
Shedding of genital herpes simplex virus was frequent soon after first-time infection but declined significantly during the first year, based on data from 82 individuals.
Genital herpes simplex virus (HSV) infections remain common and incurable; consequently, the population with residual infection continues to rise, Christine Johnston, MD, of the University of Washington, Seattle, and colleagues wrote. However, data on the viral shedding trajectory of genital HSV-1 are limited, although HSV-1 accounts for an increasing number of infections.
In a study published in JAMA the researchers recruited 82 women with first-episode genital HSV-1 infections from sexual health and primary care clinics in Seattle, between 2013 and 2018. The participants supplied self-collected oral and genital swabs for daily HSV polymerase chain reaction testing for two 30-day periods at 2 months and 11 months after their initial symptoms. The study population was not pregnant and did not have HIV infection. The median age of the participants was 26 years, 54 were women, and 42 had primary HSV-1 infections. Primary HSV-1 infection was defined as the lack of HSV antibody at baseline or an evolving antibody profile, based on the University of Washington HSV Western Blot.
The primary outcome was the rates of genital and oral HSV shedding and lesions at 2 and 11 months and up to 2 years after an initial HSV-1 infection.
At 2 months, approximately two-thirds (64.6%) of the participants had HSV-1 in the genital tract and 29.3% had virus in the mouth. Genital shedding of HSV-1 was detected in 12.1% of 2,264 total testing days at 2 months, but this rate declined to 7.1% of 1,719 testing days at 11 months (relative risk, 0.52).
The researchers identified oral HSV-1 shedding on 3.9% of 2,247 testing days at 2 months, with a slight increase to 5.1% of 1,714 testing days at 11 months.
Both genital and oral lesions were rare, with reports of 2.6% and 0.4%, respectively, at 2 months and 3.8% and 0.5%, respectively, at 11 months.
The risk of genital shedding was significantly higher in individuals with primary HSV-1, compared with those with nonprimary infections (7.9% vs. 2.9%; RR, 2.75). The overall rate of genital shedding was 17.2% for those with primary HSV-1, of which 15.2% was asymptomatic. Oral shedding was similar for individuals with primary and nonprimary HSV in a multivariate analysis.
In addition, HSV-specific CD4+ and CD8+ T-cell responses were identified in all participants, and these remained stable during the study period. No association appeared between rates of genital and oral shedding and the proportion of cells that expressed two, three, or four cytokines.
The current study is the first known to comprehensively assess genital and oral HSV-1 viral shedding using polymerase chain reaction, the researchers wrote. “Characterizing shedding rates is clinically important because patients with genital herpes are often concerned about transmission to sexual partners, which usually occurs in the absence of lesions.”
The study findings were limited by several factors including the 22% loss of participants to follow-up by the end of the first year, and the use of data from a single location with a primarily White population, the researchers noted. Another limitation was reliance on self-reports and the potential underestimation of recurrences because of the possible use of antiviral medications between swabbing periods.
However, the results indicate the early frequency of HSV-1 shedding and suggest that suppressive therapy might benefit individuals with primary HSV-1 during their first year of infection, the researchers said.
Findings may improve HSV management
The current study helps fill a knowledge gap regarding the natural history of genital HSV-1 infections, Richard J. Whitley, MD, and Edward W. Hook III, MD, both of the University of Alabama at Birmingham, wrote in an accompanying editorial. Despite the small study population, the data represent the largest cohort to date of individuals with first-episode infection and up to 2 years’ follow-up.
Although HSV-2 shedding is greater and associated with more symptoms, seroprevalence of HSV-2 in the United States is declining, they noted. Therefore, the findings can inform patient counseling and recommendations for antiviral therapy that may extend to managing HSV-1 in pregnant women as well, although no pregnant women were included in the study.
“For clinicians, these data emphasize the importance of determining the HSV viral type in persons presenting with initial episodes of genital herpes to accurately counsel patients regarding risk of clinical recurrence, the likelihood of asymptomatic shedding of virus and hence transmission, and antiviral prophylaxis,” the editorialists emphasized. For investigators, the results should prompt additional studies of the host defense against HSV and improved serological testing.
Study supports need for attention to HSV-1
“Genital herpes is an extremely common sexually transmitted infection, and often only HSV-2 is measured,” Sarah W. Prager, MD, of the University of Washington, Seattle, said in an interview. “This study shows that HSV-1 also accounts for a significant amount of genital disease, and should also be considered when determining prevalence of genital herpes.
“I was not surprised to see that viral shedding decreased significantly over the first year after diagnosis, and similarly not surprised that lesions were rare after the initial infection,” said Dr. Prager, who was not involved in the study. “I was somewhat surprised to see that genital HSV-1 shedding was more common than oral shedding.”
Dr. Prager said that she would advise clinicians against serum HSV testing unless someone has an active genital lesion. “Testing after a lesion will often reveal HSV-1, and patients should be counseled that shedding will decrease over the first year. Subsequent genital lesions are uncommon, but certainly possible, and oral lesions and shedding are both rare.” ]
More research is needed in a more diverse population, Dr. Prager emphasized. Following patients for more than a year and learning more about the use of antiviral medications also would be useful.
The study was supported in part by the National Institutes of Health/National Institute of Allergy and Infectious Diseases through grants to several authors, including lead author Dr. Johnston. Dr. Johnston also disclosed personal fees from AbbVie, grants from Gilead, royalties from UpToDate, and personal fees from GlaxoSmithKline unrelated to the current study. Dr. Whitley disclosed personal fees from Virios Therapeutics as a board member and shareholder during the conduct of the study, royalties from Aettis unrelated to the submitted work, and serving on an advisory board for Visby Diagnostics. Dr. Hook disclosed serving on an advisory board for Visby Diagnostics unrelated to the submitted work. Dr. Prager had no conflicts to disclose and serves on the editorial advisory board of Ob.Gyn News.
Shedding of genital herpes simplex virus was frequent soon after first-time infection but declined significantly during the first year, based on data from 82 individuals.
Genital herpes simplex virus (HSV) infections remain common and incurable; consequently, the population with residual infection continues to rise, Christine Johnston, MD, of the University of Washington, Seattle, and colleagues wrote. However, data on the viral shedding trajectory of genital HSV-1 are limited, although HSV-1 accounts for an increasing number of infections.
In a study published in JAMA the researchers recruited 82 women with first-episode genital HSV-1 infections from sexual health and primary care clinics in Seattle, between 2013 and 2018. The participants supplied self-collected oral and genital swabs for daily HSV polymerase chain reaction testing for two 30-day periods at 2 months and 11 months after their initial symptoms. The study population was not pregnant and did not have HIV infection. The median age of the participants was 26 years, 54 were women, and 42 had primary HSV-1 infections. Primary HSV-1 infection was defined as the lack of HSV antibody at baseline or an evolving antibody profile, based on the University of Washington HSV Western Blot.
The primary outcome was the rates of genital and oral HSV shedding and lesions at 2 and 11 months and up to 2 years after an initial HSV-1 infection.
At 2 months, approximately two-thirds (64.6%) of the participants had HSV-1 in the genital tract and 29.3% had virus in the mouth. Genital shedding of HSV-1 was detected in 12.1% of 2,264 total testing days at 2 months, but this rate declined to 7.1% of 1,719 testing days at 11 months (relative risk, 0.52).
The researchers identified oral HSV-1 shedding on 3.9% of 2,247 testing days at 2 months, with a slight increase to 5.1% of 1,714 testing days at 11 months.
Both genital and oral lesions were rare, with reports of 2.6% and 0.4%, respectively, at 2 months and 3.8% and 0.5%, respectively, at 11 months.
The risk of genital shedding was significantly higher in individuals with primary HSV-1, compared with those with nonprimary infections (7.9% vs. 2.9%; RR, 2.75). The overall rate of genital shedding was 17.2% for those with primary HSV-1, of which 15.2% was asymptomatic. Oral shedding was similar for individuals with primary and nonprimary HSV in a multivariate analysis.
In addition, HSV-specific CD4+ and CD8+ T-cell responses were identified in all participants, and these remained stable during the study period. No association appeared between rates of genital and oral shedding and the proportion of cells that expressed two, three, or four cytokines.
The current study is the first known to comprehensively assess genital and oral HSV-1 viral shedding using polymerase chain reaction, the researchers wrote. “Characterizing shedding rates is clinically important because patients with genital herpes are often concerned about transmission to sexual partners, which usually occurs in the absence of lesions.”
The study findings were limited by several factors including the 22% loss of participants to follow-up by the end of the first year, and the use of data from a single location with a primarily White population, the researchers noted. Another limitation was reliance on self-reports and the potential underestimation of recurrences because of the possible use of antiviral medications between swabbing periods.
However, the results indicate the early frequency of HSV-1 shedding and suggest that suppressive therapy might benefit individuals with primary HSV-1 during their first year of infection, the researchers said.
Findings may improve HSV management
The current study helps fill a knowledge gap regarding the natural history of genital HSV-1 infections, Richard J. Whitley, MD, and Edward W. Hook III, MD, both of the University of Alabama at Birmingham, wrote in an accompanying editorial. Despite the small study population, the data represent the largest cohort to date of individuals with first-episode infection and up to 2 years’ follow-up.
Although HSV-2 shedding is greater and associated with more symptoms, seroprevalence of HSV-2 in the United States is declining, they noted. Therefore, the findings can inform patient counseling and recommendations for antiviral therapy that may extend to managing HSV-1 in pregnant women as well, although no pregnant women were included in the study.
“For clinicians, these data emphasize the importance of determining the HSV viral type in persons presenting with initial episodes of genital herpes to accurately counsel patients regarding risk of clinical recurrence, the likelihood of asymptomatic shedding of virus and hence transmission, and antiviral prophylaxis,” the editorialists emphasized. For investigators, the results should prompt additional studies of the host defense against HSV and improved serological testing.
Study supports need for attention to HSV-1
“Genital herpes is an extremely common sexually transmitted infection, and often only HSV-2 is measured,” Sarah W. Prager, MD, of the University of Washington, Seattle, said in an interview. “This study shows that HSV-1 also accounts for a significant amount of genital disease, and should also be considered when determining prevalence of genital herpes.
“I was not surprised to see that viral shedding decreased significantly over the first year after diagnosis, and similarly not surprised that lesions were rare after the initial infection,” said Dr. Prager, who was not involved in the study. “I was somewhat surprised to see that genital HSV-1 shedding was more common than oral shedding.”
Dr. Prager said that she would advise clinicians against serum HSV testing unless someone has an active genital lesion. “Testing after a lesion will often reveal HSV-1, and patients should be counseled that shedding will decrease over the first year. Subsequent genital lesions are uncommon, but certainly possible, and oral lesions and shedding are both rare.” ]
More research is needed in a more diverse population, Dr. Prager emphasized. Following patients for more than a year and learning more about the use of antiviral medications also would be useful.
The study was supported in part by the National Institutes of Health/National Institute of Allergy and Infectious Diseases through grants to several authors, including lead author Dr. Johnston. Dr. Johnston also disclosed personal fees from AbbVie, grants from Gilead, royalties from UpToDate, and personal fees from GlaxoSmithKline unrelated to the current study. Dr. Whitley disclosed personal fees from Virios Therapeutics as a board member and shareholder during the conduct of the study, royalties from Aettis unrelated to the submitted work, and serving on an advisory board for Visby Diagnostics. Dr. Hook disclosed serving on an advisory board for Visby Diagnostics unrelated to the submitted work. Dr. Prager had no conflicts to disclose and serves on the editorial advisory board of Ob.Gyn News.
Shedding of genital herpes simplex virus was frequent soon after first-time infection but declined significantly during the first year, based on data from 82 individuals.
Genital herpes simplex virus (HSV) infections remain common and incurable; consequently, the population with residual infection continues to rise, Christine Johnston, MD, of the University of Washington, Seattle, and colleagues wrote. However, data on the viral shedding trajectory of genital HSV-1 are limited, although HSV-1 accounts for an increasing number of infections.
In a study published in JAMA the researchers recruited 82 women with first-episode genital HSV-1 infections from sexual health and primary care clinics in Seattle, between 2013 and 2018. The participants supplied self-collected oral and genital swabs for daily HSV polymerase chain reaction testing for two 30-day periods at 2 months and 11 months after their initial symptoms. The study population was not pregnant and did not have HIV infection. The median age of the participants was 26 years, 54 were women, and 42 had primary HSV-1 infections. Primary HSV-1 infection was defined as the lack of HSV antibody at baseline or an evolving antibody profile, based on the University of Washington HSV Western Blot.
The primary outcome was the rates of genital and oral HSV shedding and lesions at 2 and 11 months and up to 2 years after an initial HSV-1 infection.
At 2 months, approximately two-thirds (64.6%) of the participants had HSV-1 in the genital tract and 29.3% had virus in the mouth. Genital shedding of HSV-1 was detected in 12.1% of 2,264 total testing days at 2 months, but this rate declined to 7.1% of 1,719 testing days at 11 months (relative risk, 0.52).
The researchers identified oral HSV-1 shedding on 3.9% of 2,247 testing days at 2 months, with a slight increase to 5.1% of 1,714 testing days at 11 months.
Both genital and oral lesions were rare, with reports of 2.6% and 0.4%, respectively, at 2 months and 3.8% and 0.5%, respectively, at 11 months.
The risk of genital shedding was significantly higher in individuals with primary HSV-1, compared with those with nonprimary infections (7.9% vs. 2.9%; RR, 2.75). The overall rate of genital shedding was 17.2% for those with primary HSV-1, of which 15.2% was asymptomatic. Oral shedding was similar for individuals with primary and nonprimary HSV in a multivariate analysis.
In addition, HSV-specific CD4+ and CD8+ T-cell responses were identified in all participants, and these remained stable during the study period. No association appeared between rates of genital and oral shedding and the proportion of cells that expressed two, three, or four cytokines.
The current study is the first known to comprehensively assess genital and oral HSV-1 viral shedding using polymerase chain reaction, the researchers wrote. “Characterizing shedding rates is clinically important because patients with genital herpes are often concerned about transmission to sexual partners, which usually occurs in the absence of lesions.”
The study findings were limited by several factors including the 22% loss of participants to follow-up by the end of the first year, and the use of data from a single location with a primarily White population, the researchers noted. Another limitation was reliance on self-reports and the potential underestimation of recurrences because of the possible use of antiviral medications between swabbing periods.
However, the results indicate the early frequency of HSV-1 shedding and suggest that suppressive therapy might benefit individuals with primary HSV-1 during their first year of infection, the researchers said.
Findings may improve HSV management
The current study helps fill a knowledge gap regarding the natural history of genital HSV-1 infections, Richard J. Whitley, MD, and Edward W. Hook III, MD, both of the University of Alabama at Birmingham, wrote in an accompanying editorial. Despite the small study population, the data represent the largest cohort to date of individuals with first-episode infection and up to 2 years’ follow-up.
Although HSV-2 shedding is greater and associated with more symptoms, seroprevalence of HSV-2 in the United States is declining, they noted. Therefore, the findings can inform patient counseling and recommendations for antiviral therapy that may extend to managing HSV-1 in pregnant women as well, although no pregnant women were included in the study.
“For clinicians, these data emphasize the importance of determining the HSV viral type in persons presenting with initial episodes of genital herpes to accurately counsel patients regarding risk of clinical recurrence, the likelihood of asymptomatic shedding of virus and hence transmission, and antiviral prophylaxis,” the editorialists emphasized. For investigators, the results should prompt additional studies of the host defense against HSV and improved serological testing.
Study supports need for attention to HSV-1
“Genital herpes is an extremely common sexually transmitted infection, and often only HSV-2 is measured,” Sarah W. Prager, MD, of the University of Washington, Seattle, said in an interview. “This study shows that HSV-1 also accounts for a significant amount of genital disease, and should also be considered when determining prevalence of genital herpes.
“I was not surprised to see that viral shedding decreased significantly over the first year after diagnosis, and similarly not surprised that lesions were rare after the initial infection,” said Dr. Prager, who was not involved in the study. “I was somewhat surprised to see that genital HSV-1 shedding was more common than oral shedding.”
Dr. Prager said that she would advise clinicians against serum HSV testing unless someone has an active genital lesion. “Testing after a lesion will often reveal HSV-1, and patients should be counseled that shedding will decrease over the first year. Subsequent genital lesions are uncommon, but certainly possible, and oral lesions and shedding are both rare.” ]
More research is needed in a more diverse population, Dr. Prager emphasized. Following patients for more than a year and learning more about the use of antiviral medications also would be useful.
The study was supported in part by the National Institutes of Health/National Institute of Allergy and Infectious Diseases through grants to several authors, including lead author Dr. Johnston. Dr. Johnston also disclosed personal fees from AbbVie, grants from Gilead, royalties from UpToDate, and personal fees from GlaxoSmithKline unrelated to the current study. Dr. Whitley disclosed personal fees from Virios Therapeutics as a board member and shareholder during the conduct of the study, royalties from Aettis unrelated to the submitted work, and serving on an advisory board for Visby Diagnostics. Dr. Hook disclosed serving on an advisory board for Visby Diagnostics unrelated to the submitted work. Dr. Prager had no conflicts to disclose and serves on the editorial advisory board of Ob.Gyn News.
FROM JAMA
Rapid action or sustained effect? Methotrexate vs. ciclosporin for pediatric AD
MONTREAL – in the TREAT study, investigators reported at the annual meeting of the International Society of Atopic Dermatitis.
The findings are important, since many regulatory bodies require patients to have tried such first-line conventional systemic therapies before moving on to novel therapeutics, explained Carsten Flohr, MD, PhD, research and development lead at St John’s Institute of Dermatology, Guy’s and St Thomas’ NHS Foundation Trust London.
“We don’t really have much pediatric trial data; very often the pediatric data that we have is buried in adult trials and when it comes to an adequately powered randomized controlled trial with conventional systemic medication in pediatric patients, we don’t have one – so we’re lacking that gold standard,” said Dr. Flohr, chair in dermatology and population health sciences at King’s College London.
In the TREAT trial, 103 patients with AD (mean age, 10 years) who had not responded to topical treatment, were randomly assigned to oral ciclosporin (4 mg/kg daily) or methotrexate (0.4 mg/kg weekly) for 36 weeks and then followed for another 24 weeks off therapy for the co-primary outcomes of change in objective Scoring Atopic Dermatitis (o-SCORAD) at 12 weeks, as well as time to first significant flare after treatment cessation, defined as returning to baseline o-SCORAD, or restarting a systemic treatment.
Secondary outcomes included disease severity and quality of life (QOL) measures, as well as safety. At baseline, the mean o-SCORAD was 46.81, with mean Eczema Area and Severity Index (EASI) and Patient Oriented Eczema Measure (POEM) scores of 28.05 and 20.62 respectively. The mean Children’s Dermatology Life Quality Index (CDLQI) score was 14.96.
Looking at change in eczema severity measured by o-SCORAD at 12 weeks, ciclosporin was superior to methotrexate, with a mean difference in o-SCORAD change of -5.69 (P =.01). For the co-primary endpoint of time to first significant flare during the 24 weeks after treatment cessation, “there was a trend toward more flare activity in the ciclosporin group, although with a hazard ratio of 1.55, this was statistically not significant,” Dr. Flohr said.
On a graph showing mean EASI scores from baseline through the 60-week study period, Dr. Flohr explained how the score first dropped more precipitously in patients treated with ciclosporin compared with those treated with methotrexate, reaching a statistically significant difference between the groups by 12 weeks (–3.13, P = .0145).
However, after that time, while the EASI score among those on methotrexate continued to drop, the ciclosporin score evened out, so that by 20 weeks, methotrexate EASI scores were better, and remained so until the end of treatment and further, out to 60 weeks (mean difference -6.36, P < .001). “The most interesting bit of this graph is [that] the curve is pointing downwards for methotrexate up to the 9-month point, suggesting these people had not reached their full therapeutic potential yet, whereas if you’re on ciclosporin you plateau and there’s not much additional improvement, if at all, and then people [on ciclosporin] start going up in their disease activity off therapy,” he said.
The same pattern was seen with all the other outcome measures, including o-SCORAD and POEM.
Quality of life significantly improved by about 8 points in both treatment groups, with no significant differences between groups, and this improvement was sustained through the 24 weeks following cessation of therapy. However, during this treatment-free phase, patients on methotrexate had fewer parent-reported flares compared with those on ciclosporin (mean 6.19 vs 5.40 flares, P =.0251), although there was no difference between groups in time to first flare.
Describing the treatment safety as “overall reassuring,” Dr. Flohr said there were slightly more nonserious adverse events in the methotrexate arm (407 vs. 369), with nausea occurring more often in this group (43.1% vs. 17.6%).
“I think we were seeing this clinically, but to see it in a clinical trial gives us more confidence in discussing with parents,” said session moderator Melinda Gooderham, MD, assistant professor at Queens University, Kingston, Ont., and medical director at the SKiN Centre for Dermatology in Peterborough.
What she also took away from the study was safety of these treatments. “The discontinuation rate was not different with either drug, so it’s not like ciclosporin works fast but all these people have problems and discontinue,” Dr. Gooderham told this news organization. “That’s also reassuring.”
Asked which treatment she prefers, Dr. Gooderham, a consultant physician at Peterborough Regional Health Centre, picked methotrexate “because of the lasting effect. But there are times when you may need more rapid control ... where I might choose ciclosporin first, but for me it’s maybe 90% methotrexate first, 10% ciclosporin.”
Dr. Flohr and Dr. Gooderham report no relevant financial relationships. The study was funded by the National Institute for Health and Care Research.
A version of this article first appeared on Medscape.com.
MONTREAL – in the TREAT study, investigators reported at the annual meeting of the International Society of Atopic Dermatitis.
The findings are important, since many regulatory bodies require patients to have tried such first-line conventional systemic therapies before moving on to novel therapeutics, explained Carsten Flohr, MD, PhD, research and development lead at St John’s Institute of Dermatology, Guy’s and St Thomas’ NHS Foundation Trust London.
“We don’t really have much pediatric trial data; very often the pediatric data that we have is buried in adult trials and when it comes to an adequately powered randomized controlled trial with conventional systemic medication in pediatric patients, we don’t have one – so we’re lacking that gold standard,” said Dr. Flohr, chair in dermatology and population health sciences at King’s College London.
In the TREAT trial, 103 patients with AD (mean age, 10 years) who had not responded to topical treatment, were randomly assigned to oral ciclosporin (4 mg/kg daily) or methotrexate (0.4 mg/kg weekly) for 36 weeks and then followed for another 24 weeks off therapy for the co-primary outcomes of change in objective Scoring Atopic Dermatitis (o-SCORAD) at 12 weeks, as well as time to first significant flare after treatment cessation, defined as returning to baseline o-SCORAD, or restarting a systemic treatment.
Secondary outcomes included disease severity and quality of life (QOL) measures, as well as safety. At baseline, the mean o-SCORAD was 46.81, with mean Eczema Area and Severity Index (EASI) and Patient Oriented Eczema Measure (POEM) scores of 28.05 and 20.62 respectively. The mean Children’s Dermatology Life Quality Index (CDLQI) score was 14.96.
Looking at change in eczema severity measured by o-SCORAD at 12 weeks, ciclosporin was superior to methotrexate, with a mean difference in o-SCORAD change of -5.69 (P =.01). For the co-primary endpoint of time to first significant flare during the 24 weeks after treatment cessation, “there was a trend toward more flare activity in the ciclosporin group, although with a hazard ratio of 1.55, this was statistically not significant,” Dr. Flohr said.
On a graph showing mean EASI scores from baseline through the 60-week study period, Dr. Flohr explained how the score first dropped more precipitously in patients treated with ciclosporin compared with those treated with methotrexate, reaching a statistically significant difference between the groups by 12 weeks (–3.13, P = .0145).
However, after that time, while the EASI score among those on methotrexate continued to drop, the ciclosporin score evened out, so that by 20 weeks, methotrexate EASI scores were better, and remained so until the end of treatment and further, out to 60 weeks (mean difference -6.36, P < .001). “The most interesting bit of this graph is [that] the curve is pointing downwards for methotrexate up to the 9-month point, suggesting these people had not reached their full therapeutic potential yet, whereas if you’re on ciclosporin you plateau and there’s not much additional improvement, if at all, and then people [on ciclosporin] start going up in their disease activity off therapy,” he said.
The same pattern was seen with all the other outcome measures, including o-SCORAD and POEM.
Quality of life significantly improved by about 8 points in both treatment groups, with no significant differences between groups, and this improvement was sustained through the 24 weeks following cessation of therapy. However, during this treatment-free phase, patients on methotrexate had fewer parent-reported flares compared with those on ciclosporin (mean 6.19 vs 5.40 flares, P =.0251), although there was no difference between groups in time to first flare.
Describing the treatment safety as “overall reassuring,” Dr. Flohr said there were slightly more nonserious adverse events in the methotrexate arm (407 vs. 369), with nausea occurring more often in this group (43.1% vs. 17.6%).
“I think we were seeing this clinically, but to see it in a clinical trial gives us more confidence in discussing with parents,” said session moderator Melinda Gooderham, MD, assistant professor at Queens University, Kingston, Ont., and medical director at the SKiN Centre for Dermatology in Peterborough.
What she also took away from the study was safety of these treatments. “The discontinuation rate was not different with either drug, so it’s not like ciclosporin works fast but all these people have problems and discontinue,” Dr. Gooderham told this news organization. “That’s also reassuring.”
Asked which treatment she prefers, Dr. Gooderham, a consultant physician at Peterborough Regional Health Centre, picked methotrexate “because of the lasting effect. But there are times when you may need more rapid control ... where I might choose ciclosporin first, but for me it’s maybe 90% methotrexate first, 10% ciclosporin.”
Dr. Flohr and Dr. Gooderham report no relevant financial relationships. The study was funded by the National Institute for Health and Care Research.
A version of this article first appeared on Medscape.com.
MONTREAL – in the TREAT study, investigators reported at the annual meeting of the International Society of Atopic Dermatitis.
The findings are important, since many regulatory bodies require patients to have tried such first-line conventional systemic therapies before moving on to novel therapeutics, explained Carsten Flohr, MD, PhD, research and development lead at St John’s Institute of Dermatology, Guy’s and St Thomas’ NHS Foundation Trust London.
“We don’t really have much pediatric trial data; very often the pediatric data that we have is buried in adult trials and when it comes to an adequately powered randomized controlled trial with conventional systemic medication in pediatric patients, we don’t have one – so we’re lacking that gold standard,” said Dr. Flohr, chair in dermatology and population health sciences at King’s College London.
In the TREAT trial, 103 patients with AD (mean age, 10 years) who had not responded to topical treatment, were randomly assigned to oral ciclosporin (4 mg/kg daily) or methotrexate (0.4 mg/kg weekly) for 36 weeks and then followed for another 24 weeks off therapy for the co-primary outcomes of change in objective Scoring Atopic Dermatitis (o-SCORAD) at 12 weeks, as well as time to first significant flare after treatment cessation, defined as returning to baseline o-SCORAD, or restarting a systemic treatment.
Secondary outcomes included disease severity and quality of life (QOL) measures, as well as safety. At baseline, the mean o-SCORAD was 46.81, with mean Eczema Area and Severity Index (EASI) and Patient Oriented Eczema Measure (POEM) scores of 28.05 and 20.62 respectively. The mean Children’s Dermatology Life Quality Index (CDLQI) score was 14.96.
Looking at change in eczema severity measured by o-SCORAD at 12 weeks, ciclosporin was superior to methotrexate, with a mean difference in o-SCORAD change of -5.69 (P =.01). For the co-primary endpoint of time to first significant flare during the 24 weeks after treatment cessation, “there was a trend toward more flare activity in the ciclosporin group, although with a hazard ratio of 1.55, this was statistically not significant,” Dr. Flohr said.
On a graph showing mean EASI scores from baseline through the 60-week study period, Dr. Flohr explained how the score first dropped more precipitously in patients treated with ciclosporin compared with those treated with methotrexate, reaching a statistically significant difference between the groups by 12 weeks (–3.13, P = .0145).
However, after that time, while the EASI score among those on methotrexate continued to drop, the ciclosporin score evened out, so that by 20 weeks, methotrexate EASI scores were better, and remained so until the end of treatment and further, out to 60 weeks (mean difference -6.36, P < .001). “The most interesting bit of this graph is [that] the curve is pointing downwards for methotrexate up to the 9-month point, suggesting these people had not reached their full therapeutic potential yet, whereas if you’re on ciclosporin you plateau and there’s not much additional improvement, if at all, and then people [on ciclosporin] start going up in their disease activity off therapy,” he said.
The same pattern was seen with all the other outcome measures, including o-SCORAD and POEM.
Quality of life significantly improved by about 8 points in both treatment groups, with no significant differences between groups, and this improvement was sustained through the 24 weeks following cessation of therapy. However, during this treatment-free phase, patients on methotrexate had fewer parent-reported flares compared with those on ciclosporin (mean 6.19 vs 5.40 flares, P =.0251), although there was no difference between groups in time to first flare.
Describing the treatment safety as “overall reassuring,” Dr. Flohr said there were slightly more nonserious adverse events in the methotrexate arm (407 vs. 369), with nausea occurring more often in this group (43.1% vs. 17.6%).
“I think we were seeing this clinically, but to see it in a clinical trial gives us more confidence in discussing with parents,” said session moderator Melinda Gooderham, MD, assistant professor at Queens University, Kingston, Ont., and medical director at the SKiN Centre for Dermatology in Peterborough.
What she also took away from the study was safety of these treatments. “The discontinuation rate was not different with either drug, so it’s not like ciclosporin works fast but all these people have problems and discontinue,” Dr. Gooderham told this news organization. “That’s also reassuring.”
Asked which treatment she prefers, Dr. Gooderham, a consultant physician at Peterborough Regional Health Centre, picked methotrexate “because of the lasting effect. But there are times when you may need more rapid control ... where I might choose ciclosporin first, but for me it’s maybe 90% methotrexate first, 10% ciclosporin.”
Dr. Flohr and Dr. Gooderham report no relevant financial relationships. The study was funded by the National Institute for Health and Care Research.
A version of this article first appeared on Medscape.com.
AT ISAD 2022