User login
CDC updates advice on preventing sexual transmission of Zika virus
Men potentially exposed to Zika virus should use a condom during all sex or abstain from sex for at least 8 weeks, according to new recommendations from the Centers for Disease Control and Prevention on reducing the risk of sexual transmission of the virus.
Men with confirmed infections or clinical symptoms of Zika should similarly abstain or use a condom for at least 6 months, the CDC recommends in the Morbidity and Mortality Weekly Report released on March 25 (MMWR 2016. Mar 25. doi: http://dx.doi.org/10.15585/mmwr.mm6512e3er).
These recommendations update and replace those issued by the CDC on Feb. 5 and include new guidance for men who live in, or have traveled to, an area with active Zika virus transmission. The recommendations apply to all types of sexual activity involving the penis, including vaginal intercourse, anal intercourse, or fellatio.
“The previous recommendations focused on women who were already pregnant,” Dr. Denise J. Jamieson, co-lead of the Pregnancy and Birth Defects Team of the CDC Zika Virus Response Team, said during a press briefing. “What’s new is that we are now concerned about the periconceptional period, around the time the woman conceives.”
For men with pregnant sex partners, the agency recommends consistent and accurate use of condoms during any type of sex, or abstinence during the length of the pregnancy.
“This course is the best way to avoid even a minimal risk of sexual transmission of Zika virus, which could have adverse fetal effects when contracted during pregnancy,” the CDC report states, adding that pregnant women should ask their male sex partners about recent travel to areas with currently circulating Zika virus.
For couples not expecting a child, but concerned about sexual transmission of Zika, men with a confirmed Zika infection or clinical symptoms of Zika infection should consider using condoms or abstaining from sex for at least 6 months after their symptoms appear. This recommendation is based on tripling 62 days – the longest time interval after infection during which the virus was successfully isolated from semen.
If men have traveled to areas with active Zika transmission but have not developed symptoms, the CDC recommends condom use or abstinence for at least 8 weeks after leaving the area. Those living in areas with active transmission should also consider condom use during sex or abstaining from sex until active transmission has ceased.
These recommendations come as more evidence points to a link between Zika infection and fetal abnormalities, including microcephaly and fetal mortality.
“I think we’re learning more every day, and I think the evidence of a link between Zika and a range of poor pregnancy outcomes is becoming stronger and stronger,” Dr. Jamieson said. “At this point, we’re not using causal language, but the evidence is mounting.”
The CDC also released two other reports focusing on the need to increase access to contraception for residents of Puerto Rico and interim guidance for health care providers of women of childbearing age who have been potentially exposed to Zika virus.
As of March 25, the CDC has reported 273 U.S. cases of Zika virus infections from 35 states and Washington, D.C. All of these – except six sexually transmitted cases – are travel related.
Additionally, Puerto Rico’s most recent case total is 261, all locally transmitted by mosquitoes, except for three travel-associated cases. American Samoa has 14 cases, and the U.S. Virgin Islands have 11 cases, all thought to be locally transmitted.
“Long-acting contraception methods are not readily available in Puerto Rico, and from our health care provider colleagues in Puerto Rico, there is a desire to provide a more broad range of contraception options to women in Puerto Rico,” Dr. Jamieson said.
She said the CDC is developing a plan to make long-acting contraceptive methods more available in Puerto Rico.
When advising couples who wish to become pregnant after the man has had confirmed or suspected Zika infection, the CDC recommends waiting at least 6 months after the man’s onset of Zika symptoms or confirmed infection before attempting to conceive.
Although no evidence suggests that Zika virus will cause congenital infections in pregnancies conceived after a woman’s infection has resolved, data on the virus’s incubation period is limited, according to the CDC.
“Women with Zika virus disease should wait until at least 8 weeks after symptom onset before attempting conception,” wrote Dr. Emily E. Petersen and her colleagues in the guidance on caring for women of reproductive age with possible Zika virus exposure. “No data are available regarding the risk for congenital infection among pregnant women with asymptomatic infection.”
Similarly, asymptomatic women potentially exposed to Zika virus should also wait at least 8 weeks after the possible exposure date before trying to conceive.
Men potentially exposed to Zika virus should use a condom during all sex or abstain from sex for at least 8 weeks, according to new recommendations from the Centers for Disease Control and Prevention on reducing the risk of sexual transmission of the virus.
Men with confirmed infections or clinical symptoms of Zika should similarly abstain or use a condom for at least 6 months, the CDC recommends in the Morbidity and Mortality Weekly Report released on March 25 (MMWR 2016. Mar 25. doi: http://dx.doi.org/10.15585/mmwr.mm6512e3er).
These recommendations update and replace those issued by the CDC on Feb. 5 and include new guidance for men who live in, or have traveled to, an area with active Zika virus transmission. The recommendations apply to all types of sexual activity involving the penis, including vaginal intercourse, anal intercourse, or fellatio.
“The previous recommendations focused on women who were already pregnant,” Dr. Denise J. Jamieson, co-lead of the Pregnancy and Birth Defects Team of the CDC Zika Virus Response Team, said during a press briefing. “What’s new is that we are now concerned about the periconceptional period, around the time the woman conceives.”
For men with pregnant sex partners, the agency recommends consistent and accurate use of condoms during any type of sex, or abstinence during the length of the pregnancy.
“This course is the best way to avoid even a minimal risk of sexual transmission of Zika virus, which could have adverse fetal effects when contracted during pregnancy,” the CDC report states, adding that pregnant women should ask their male sex partners about recent travel to areas with currently circulating Zika virus.
For couples not expecting a child, but concerned about sexual transmission of Zika, men with a confirmed Zika infection or clinical symptoms of Zika infection should consider using condoms or abstaining from sex for at least 6 months after their symptoms appear. This recommendation is based on tripling 62 days – the longest time interval after infection during which the virus was successfully isolated from semen.
If men have traveled to areas with active Zika transmission but have not developed symptoms, the CDC recommends condom use or abstinence for at least 8 weeks after leaving the area. Those living in areas with active transmission should also consider condom use during sex or abstaining from sex until active transmission has ceased.
These recommendations come as more evidence points to a link between Zika infection and fetal abnormalities, including microcephaly and fetal mortality.
“I think we’re learning more every day, and I think the evidence of a link between Zika and a range of poor pregnancy outcomes is becoming stronger and stronger,” Dr. Jamieson said. “At this point, we’re not using causal language, but the evidence is mounting.”
The CDC also released two other reports focusing on the need to increase access to contraception for residents of Puerto Rico and interim guidance for health care providers of women of childbearing age who have been potentially exposed to Zika virus.
As of March 25, the CDC has reported 273 U.S. cases of Zika virus infections from 35 states and Washington, D.C. All of these – except six sexually transmitted cases – are travel related.
Additionally, Puerto Rico’s most recent case total is 261, all locally transmitted by mosquitoes, except for three travel-associated cases. American Samoa has 14 cases, and the U.S. Virgin Islands have 11 cases, all thought to be locally transmitted.
“Long-acting contraception methods are not readily available in Puerto Rico, and from our health care provider colleagues in Puerto Rico, there is a desire to provide a more broad range of contraception options to women in Puerto Rico,” Dr. Jamieson said.
She said the CDC is developing a plan to make long-acting contraceptive methods more available in Puerto Rico.
When advising couples who wish to become pregnant after the man has had confirmed or suspected Zika infection, the CDC recommends waiting at least 6 months after the man’s onset of Zika symptoms or confirmed infection before attempting to conceive.
Although no evidence suggests that Zika virus will cause congenital infections in pregnancies conceived after a woman’s infection has resolved, data on the virus’s incubation period is limited, according to the CDC.
“Women with Zika virus disease should wait until at least 8 weeks after symptom onset before attempting conception,” wrote Dr. Emily E. Petersen and her colleagues in the guidance on caring for women of reproductive age with possible Zika virus exposure. “No data are available regarding the risk for congenital infection among pregnant women with asymptomatic infection.”
Similarly, asymptomatic women potentially exposed to Zika virus should also wait at least 8 weeks after the possible exposure date before trying to conceive.
Men potentially exposed to Zika virus should use a condom during all sex or abstain from sex for at least 8 weeks, according to new recommendations from the Centers for Disease Control and Prevention on reducing the risk of sexual transmission of the virus.
Men with confirmed infections or clinical symptoms of Zika should similarly abstain or use a condom for at least 6 months, the CDC recommends in the Morbidity and Mortality Weekly Report released on March 25 (MMWR 2016. Mar 25. doi: http://dx.doi.org/10.15585/mmwr.mm6512e3er).
These recommendations update and replace those issued by the CDC on Feb. 5 and include new guidance for men who live in, or have traveled to, an area with active Zika virus transmission. The recommendations apply to all types of sexual activity involving the penis, including vaginal intercourse, anal intercourse, or fellatio.
“The previous recommendations focused on women who were already pregnant,” Dr. Denise J. Jamieson, co-lead of the Pregnancy and Birth Defects Team of the CDC Zika Virus Response Team, said during a press briefing. “What’s new is that we are now concerned about the periconceptional period, around the time the woman conceives.”
For men with pregnant sex partners, the agency recommends consistent and accurate use of condoms during any type of sex, or abstinence during the length of the pregnancy.
“This course is the best way to avoid even a minimal risk of sexual transmission of Zika virus, which could have adverse fetal effects when contracted during pregnancy,” the CDC report states, adding that pregnant women should ask their male sex partners about recent travel to areas with currently circulating Zika virus.
For couples not expecting a child, but concerned about sexual transmission of Zika, men with a confirmed Zika infection or clinical symptoms of Zika infection should consider using condoms or abstaining from sex for at least 6 months after their symptoms appear. This recommendation is based on tripling 62 days – the longest time interval after infection during which the virus was successfully isolated from semen.
If men have traveled to areas with active Zika transmission but have not developed symptoms, the CDC recommends condom use or abstinence for at least 8 weeks after leaving the area. Those living in areas with active transmission should also consider condom use during sex or abstaining from sex until active transmission has ceased.
These recommendations come as more evidence points to a link between Zika infection and fetal abnormalities, including microcephaly and fetal mortality.
“I think we’re learning more every day, and I think the evidence of a link between Zika and a range of poor pregnancy outcomes is becoming stronger and stronger,” Dr. Jamieson said. “At this point, we’re not using causal language, but the evidence is mounting.”
The CDC also released two other reports focusing on the need to increase access to contraception for residents of Puerto Rico and interim guidance for health care providers of women of childbearing age who have been potentially exposed to Zika virus.
As of March 25, the CDC has reported 273 U.S. cases of Zika virus infections from 35 states and Washington, D.C. All of these – except six sexually transmitted cases – are travel related.
Additionally, Puerto Rico’s most recent case total is 261, all locally transmitted by mosquitoes, except for three travel-associated cases. American Samoa has 14 cases, and the U.S. Virgin Islands have 11 cases, all thought to be locally transmitted.
“Long-acting contraception methods are not readily available in Puerto Rico, and from our health care provider colleagues in Puerto Rico, there is a desire to provide a more broad range of contraception options to women in Puerto Rico,” Dr. Jamieson said.
She said the CDC is developing a plan to make long-acting contraceptive methods more available in Puerto Rico.
When advising couples who wish to become pregnant after the man has had confirmed or suspected Zika infection, the CDC recommends waiting at least 6 months after the man’s onset of Zika symptoms or confirmed infection before attempting to conceive.
Although no evidence suggests that Zika virus will cause congenital infections in pregnancies conceived after a woman’s infection has resolved, data on the virus’s incubation period is limited, according to the CDC.
“Women with Zika virus disease should wait until at least 8 weeks after symptom onset before attempting conception,” wrote Dr. Emily E. Petersen and her colleagues in the guidance on caring for women of reproductive age with possible Zika virus exposure. “No data are available regarding the risk for congenital infection among pregnant women with asymptomatic infection.”
Similarly, asymptomatic women potentially exposed to Zika virus should also wait at least 8 weeks after the possible exposure date before trying to conceive.
ECCMID 2016: Antimicrobial resistance, the microbiome and systems vaccinology
The global infectious disease and clinical microbiology community meets every year at the European Congress of Clinical Microbiology and Infectious Diseases (ECCMID), the world’s largest congress on infectious diseases and medical microbiology, to present and discuss recent research results and to offer solutions to the most pressing infection problems.
The 2016 ECCMID annual conference, organized by the European Society of Clinical Microbiology and Infectious Diseases (ESCMID), will take place April 9-12 in Amsterdam. Discussions at this event not only help translate research findings into diagnostic tools, guidelines, best practices, and international policies; they also raise awareness of emerging health care challenges.

At ECCMID 2016, researchers will present more than 3,000 abstracts with the latest findings and recommendations to help improve diagnosis, prevention, and the clinical care given to patients. The Congress offers more than 150 oral presentations, including keynote lectures, symposia, oral sessions, educational workshops, and meet-the-experts sessions, as well as more than 2,000 poster presentations.
The main topics this year are strategies to detect and tackle antimicrobial resistance in various settings, approaches for prevention involving vaccines and infection control, as well as descriptions of novel diagnostic technologies. Always among the most popular sessions are lectures by winners of the ESCMID Award for Excellence and the Young Investigator Awards, as well as oral presentations on groundbreaking research, and late-breaking abstracts.
Also included will be mini oral “e-poster” presentations. Printed posters will be presented, but they will also be available at e-poster viewing stations, where visitors can scroll through abstracts of paper presentations.
Keynote speeches this year will feature innovative approaches to vaccines; microbiome and tuberculosis therapies; lectures on how nonhuman antibiotics affect public health; and an economic perspective on antimicrobial resistance.
Special topics
This year, the ECCMID Program Committee has decided to offer two special tracks for the late-breaking abstract sessions, focused on topics requiring a coordinated response from infection specialists across all disciplines.
The first topic is refugee and migrant health. The thousands of people who are currently migrating challenge public health systems in transition and the host countries. Clinicians and public health specialists need to develop strategies for the screening, prevention, and treatment of infectious diseases that were largely eradicated in Europe but are now gradually being reintroduced.
The second focus of the late-breaking abstracts is on emerging colistin resistance. Reports about the emergence of plasmid-borne resistance to this last-resort antibiotic have come from China, Canada, the United Kingdom, and most countries in continental Europe. Colistin resistance can spread easily between different types of bacteria, says Dr. Murat Akova, current ESCMID president and professor of medicine at Hacettepe University in Ankara, Turkey, and the world needs to wake up and take note.
In terms of viral infections, experts at the Congress will evaluate HIV and hepatitis C treatments in several interesting sessions. Researchers will also present results on emerging infections, including those caused by the Zika virus. Dr. Jean Paul Stahl, vice chairman of the ESCMID Study Group for Infectious Diseases of the Brain and professor of infectious diseases at University Hospital in Grenoble, France, says the current Zika virus epidemic is an important example of the great need we have for new evidence-based approaches on how to best manage emerging infections.
The outbreaks of Zika and Ebola in the last few years have seen the international community mobilize on infectious disease issues in a more collaborative manner than ever before, which should help reduce the severity of future outbreaks. But viral infections extend far beyond the recent outbreaks of unusual pathologies, and there are a number of important developments taking place among some of the more common viruses.
For more information on ECCMID 2016, visit http://www.eccmid.org/.
Dr. Winfried V. Kern is professor of medicine at the Albert Ludwigs University of Freiburg and head of the division of infectious diseases, department of medicine, and Centre for Infectious Diseases and Travel Medicine, University Hospital, Freiburg, Germany. His professional interests include bacterial multidrug resistance mechanisms and epidemiology, hospital antibiotic stewardship programs, health care–associated infections including infections in the immunocompromised host.
The global infectious disease and clinical microbiology community meets every year at the European Congress of Clinical Microbiology and Infectious Diseases (ECCMID), the world’s largest congress on infectious diseases and medical microbiology, to present and discuss recent research results and to offer solutions to the most pressing infection problems.
The 2016 ECCMID annual conference, organized by the European Society of Clinical Microbiology and Infectious Diseases (ESCMID), will take place April 9-12 in Amsterdam. Discussions at this event not only help translate research findings into diagnostic tools, guidelines, best practices, and international policies; they also raise awareness of emerging health care challenges.
At ECCMID 2016, researchers will present more than 3,000 abstracts with the latest findings and recommendations to help improve diagnosis, prevention, and the clinical care given to patients. The Congress offers more than 150 oral presentations, including keynote lectures, symposia, oral sessions, educational workshops, and meet-the-experts sessions, as well as more than 2,000 poster presentations.
The main topics this year are strategies to detect and tackle antimicrobial resistance in various settings, approaches for prevention involving vaccines and infection control, as well as descriptions of novel diagnostic technologies. Always among the most popular sessions are lectures by winners of the ESCMID Award for Excellence and the Young Investigator Awards, as well as oral presentations on groundbreaking research, and late-breaking abstracts.
Also included will be mini oral “e-poster” presentations. Printed posters will be presented, but they will also be available at e-poster viewing stations, where visitors can scroll through abstracts of paper presentations.
Keynote speeches this year will feature innovative approaches to vaccines; microbiome and tuberculosis therapies; lectures on how nonhuman antibiotics affect public health; and an economic perspective on antimicrobial resistance.
Special topics
This year, the ECCMID Program Committee has decided to offer two special tracks for the late-breaking abstract sessions, focused on topics requiring a coordinated response from infection specialists across all disciplines.
The first topic is refugee and migrant health. The thousands of people who are currently migrating challenge public health systems in transition and the host countries. Clinicians and public health specialists need to develop strategies for the screening, prevention, and treatment of infectious diseases that were largely eradicated in Europe but are now gradually being reintroduced.
The second focus of the late-breaking abstracts is on emerging colistin resistance. Reports about the emergence of plasmid-borne resistance to this last-resort antibiotic have come from China, Canada, the United Kingdom, and most countries in continental Europe. Colistin resistance can spread easily between different types of bacteria, says Dr. Murat Akova, current ESCMID president and professor of medicine at Hacettepe University in Ankara, Turkey, and the world needs to wake up and take note.
In terms of viral infections, experts at the Congress will evaluate HIV and hepatitis C treatments in several interesting sessions. Researchers will also present results on emerging infections, including those caused by the Zika virus. Dr. Jean Paul Stahl, vice chairman of the ESCMID Study Group for Infectious Diseases of the Brain and professor of infectious diseases at University Hospital in Grenoble, France, says the current Zika virus epidemic is an important example of the great need we have for new evidence-based approaches on how to best manage emerging infections.
The outbreaks of Zika and Ebola in the last few years have seen the international community mobilize on infectious disease issues in a more collaborative manner than ever before, which should help reduce the severity of future outbreaks. But viral infections extend far beyond the recent outbreaks of unusual pathologies, and there are a number of important developments taking place among some of the more common viruses.
For more information on ECCMID 2016, visit http://www.eccmid.org/.
Dr. Winfried V. Kern is professor of medicine at the Albert Ludwigs University of Freiburg and head of the division of infectious diseases, department of medicine, and Centre for Infectious Diseases and Travel Medicine, University Hospital, Freiburg, Germany. His professional interests include bacterial multidrug resistance mechanisms and epidemiology, hospital antibiotic stewardship programs, health care–associated infections including infections in the immunocompromised host.
The global infectious disease and clinical microbiology community meets every year at the European Congress of Clinical Microbiology and Infectious Diseases (ECCMID), the world’s largest congress on infectious diseases and medical microbiology, to present and discuss recent research results and to offer solutions to the most pressing infection problems.
The 2016 ECCMID annual conference, organized by the European Society of Clinical Microbiology and Infectious Diseases (ESCMID), will take place April 9-12 in Amsterdam. Discussions at this event not only help translate research findings into diagnostic tools, guidelines, best practices, and international policies; they also raise awareness of emerging health care challenges.
At ECCMID 2016, researchers will present more than 3,000 abstracts with the latest findings and recommendations to help improve diagnosis, prevention, and the clinical care given to patients. The Congress offers more than 150 oral presentations, including keynote lectures, symposia, oral sessions, educational workshops, and meet-the-experts sessions, as well as more than 2,000 poster presentations.
The main topics this year are strategies to detect and tackle antimicrobial resistance in various settings, approaches for prevention involving vaccines and infection control, as well as descriptions of novel diagnostic technologies. Always among the most popular sessions are lectures by winners of the ESCMID Award for Excellence and the Young Investigator Awards, as well as oral presentations on groundbreaking research, and late-breaking abstracts.
Also included will be mini oral “e-poster” presentations. Printed posters will be presented, but they will also be available at e-poster viewing stations, where visitors can scroll through abstracts of paper presentations.
Keynote speeches this year will feature innovative approaches to vaccines; microbiome and tuberculosis therapies; lectures on how nonhuman antibiotics affect public health; and an economic perspective on antimicrobial resistance.
Special topics
This year, the ECCMID Program Committee has decided to offer two special tracks for the late-breaking abstract sessions, focused on topics requiring a coordinated response from infection specialists across all disciplines.
The first topic is refugee and migrant health. The thousands of people who are currently migrating challenge public health systems in transition and the host countries. Clinicians and public health specialists need to develop strategies for the screening, prevention, and treatment of infectious diseases that were largely eradicated in Europe but are now gradually being reintroduced.
The second focus of the late-breaking abstracts is on emerging colistin resistance. Reports about the emergence of plasmid-borne resistance to this last-resort antibiotic have come from China, Canada, the United Kingdom, and most countries in continental Europe. Colistin resistance can spread easily between different types of bacteria, says Dr. Murat Akova, current ESCMID president and professor of medicine at Hacettepe University in Ankara, Turkey, and the world needs to wake up and take note.
In terms of viral infections, experts at the Congress will evaluate HIV and hepatitis C treatments in several interesting sessions. Researchers will also present results on emerging infections, including those caused by the Zika virus. Dr. Jean Paul Stahl, vice chairman of the ESCMID Study Group for Infectious Diseases of the Brain and professor of infectious diseases at University Hospital in Grenoble, France, says the current Zika virus epidemic is an important example of the great need we have for new evidence-based approaches on how to best manage emerging infections.
The outbreaks of Zika and Ebola in the last few years have seen the international community mobilize on infectious disease issues in a more collaborative manner than ever before, which should help reduce the severity of future outbreaks. But viral infections extend far beyond the recent outbreaks of unusual pathologies, and there are a number of important developments taking place among some of the more common viruses.
For more information on ECCMID 2016, visit http://www.eccmid.org/.
Dr. Winfried V. Kern is professor of medicine at the Albert Ludwigs University of Freiburg and head of the division of infectious diseases, department of medicine, and Centre for Infectious Diseases and Travel Medicine, University Hospital, Freiburg, Germany. His professional interests include bacterial multidrug resistance mechanisms and epidemiology, hospital antibiotic stewardship programs, health care–associated infections including infections in the immunocompromised host.

David Henry's JCSO podcast, March 2016
In the March podcast for The Journal of Community and Supportive Oncology, Dr David Henry discusses a number of articles that center on patient quality of life and overall quality of care, among them, an article on opioid risk assessment in palliative care and three Original Reports, one on the impact of trimodality treatment on arm function and QoL in patients with superior sulcus tumors, a second on patient perceptions and the challenges of oral anticancer therapy, and a third on the use of voluntary reporting to assess symptom burden in cancer patients. A Review article by Jose de Souza and colleagues on financial toxicity in cancer care spans the QoL and quality of care spectrum as it details the implications of the increasing cost of cancer care for the delivery of high-quality, patient-centered care and discusses potential predictors of financial toxicity and instruments that could help quantify financial burden. Also in the line-up are articles on encapsulated irinotecan for hard-to-treat cancer and Case Reports on an uncommon presentation of lung cancer and on acute promyelocytic leukemia presenting as a paraspinal mass.
Listen to the podcast below.
In the March podcast for The Journal of Community and Supportive Oncology, Dr David Henry discusses a number of articles that center on patient quality of life and overall quality of care, among them, an article on opioid risk assessment in palliative care and three Original Reports, one on the impact of trimodality treatment on arm function and QoL in patients with superior sulcus tumors, a second on patient perceptions and the challenges of oral anticancer therapy, and a third on the use of voluntary reporting to assess symptom burden in cancer patients. A Review article by Jose de Souza and colleagues on financial toxicity in cancer care spans the QoL and quality of care spectrum as it details the implications of the increasing cost of cancer care for the delivery of high-quality, patient-centered care and discusses potential predictors of financial toxicity and instruments that could help quantify financial burden. Also in the line-up are articles on encapsulated irinotecan for hard-to-treat cancer and Case Reports on an uncommon presentation of lung cancer and on acute promyelocytic leukemia presenting as a paraspinal mass.
Listen to the podcast below.
In the March podcast for The Journal of Community and Supportive Oncology, Dr David Henry discusses a number of articles that center on patient quality of life and overall quality of care, among them, an article on opioid risk assessment in palliative care and three Original Reports, one on the impact of trimodality treatment on arm function and QoL in patients with superior sulcus tumors, a second on patient perceptions and the challenges of oral anticancer therapy, and a third on the use of voluntary reporting to assess symptom burden in cancer patients. A Review article by Jose de Souza and colleagues on financial toxicity in cancer care spans the QoL and quality of care spectrum as it details the implications of the increasing cost of cancer care for the delivery of high-quality, patient-centered care and discusses potential predictors of financial toxicity and instruments that could help quantify financial burden. Also in the line-up are articles on encapsulated irinotecan for hard-to-treat cancer and Case Reports on an uncommon presentation of lung cancer and on acute promyelocytic leukemia presenting as a paraspinal mass.
Listen to the podcast below.
A line-up of new therapies and expanded combinations
Click on the PDF icon at the top of this introduction to read the full article.
Click on the PDF icon at the top of this introduction to read the full article.
Click on the PDF icon at the top of this introduction to read the full article.
Sertraline improves Cryptococcus clearance in HIV meningitis
An open-label pilot study has found adjunct treatment with the SSRI antidepressant sertraline is associated with accelerated clearance of Cryptococcus from the cerebrospinal fluid in HIV-infected individuals with cryptococcal meningitis.
The dose-finding study of 172 HIV-infected Ugandan adults with cryptococcal meningitis showed any dose of sertraline, in combination with standard amphotericin B and high-dose fluconazole therapy, was associated with an average clearance rate of –0.37 log10 colony-forming U/mL CSF per day, compared with –0.30 observed in a previous study in individuals taking standard therapy without sertraline.
According to a paper published online in Lancet Infectious Diseases, the incidence of paradoxical immune reconstitution inflammatory syndrome was 5%, compared with rates of 17%-30% observed in other studies.
The sertraline doses used in the study ranged from 100 mg/day to 400 mg/day but researchers did not observe a significant difference in serious adverse events between the lower and higher doses (Lancet Infect Dis. 2016 Mar 9. doi: 10.1016/S1473-3099[16]00074-8).
Sertraline has previously shown potent fungicidal activity against Cryptococcus in vitro and in mice, suggesting it could be a promising therapeutic option for cryptococcal meningitis, a disease that accounts for 15%-20% of AIDS-related deaths in Africa.
“The current standard therapy for cryptococcal meningitis is based on antifungal regimens that are a half-century old, are associated with a range of toxic effects, and are largely inaccessible in areas of the world where they are needed most,” wrote Dr. Joshua Rhein of the University of Minnesota, Minneapolis, and his coauthors. “For this reason, the discovery of a widely available, nontoxic, and affordable drug effective against Cryptococcus would represent a substantial advance in preventing deaths.”
The study was supported by the National Institutes of Health, Fogarty International Center, Grand Challenges Canada, and the Doris Duke Charitable Foundation. No conflicts of interest were declared.
Cryptococcal meningitis remains a major clinical challenge in Africa. The number of cases of cryptococcal meningitis remains high despite more than a decade of antiretroviral therapy rollout, but the profile of the population developing the disease is changing.
Novel treatments are urgently needed, and Dr. Rhein and his colleagues should be commended for their work to develop practical, affordable, effective new antifungal regimens for HIV-associated cryptococcal meningitis.
Joseph N. Jarvis, Ph.D., is from the Botswana-UPenn Partnership at the University of Pennsylvania, Philadelphia, and Dr. Thomas S. Harrison is from the Institute of Infection and Immunity at St. George’s University of London. These comments are taken from an accompanying editorial (Lancet Infect Dis. 2016 Mar 9. doi: 10.1016/S1473-3099[16]00128-6). Dr. Jarvis declared grants from Gilead Sciences and the National Institutes of Health outside of the submitted work, and Dr. Harrison declared grants from Gilead Sciences and personal fees from Viamet Pharmaceuticals, outside of the submitted work.
Cryptococcal meningitis remains a major clinical challenge in Africa. The number of cases of cryptococcal meningitis remains high despite more than a decade of antiretroviral therapy rollout, but the profile of the population developing the disease is changing.
Novel treatments are urgently needed, and Dr. Rhein and his colleagues should be commended for their work to develop practical, affordable, effective new antifungal regimens for HIV-associated cryptococcal meningitis.
Joseph N. Jarvis, Ph.D., is from the Botswana-UPenn Partnership at the University of Pennsylvania, Philadelphia, and Dr. Thomas S. Harrison is from the Institute of Infection and Immunity at St. George’s University of London. These comments are taken from an accompanying editorial (Lancet Infect Dis. 2016 Mar 9. doi: 10.1016/S1473-3099[16]00128-6). Dr. Jarvis declared grants from Gilead Sciences and the National Institutes of Health outside of the submitted work, and Dr. Harrison declared grants from Gilead Sciences and personal fees from Viamet Pharmaceuticals, outside of the submitted work.
Cryptococcal meningitis remains a major clinical challenge in Africa. The number of cases of cryptococcal meningitis remains high despite more than a decade of antiretroviral therapy rollout, but the profile of the population developing the disease is changing.
Novel treatments are urgently needed, and Dr. Rhein and his colleagues should be commended for their work to develop practical, affordable, effective new antifungal regimens for HIV-associated cryptococcal meningitis.
Joseph N. Jarvis, Ph.D., is from the Botswana-UPenn Partnership at the University of Pennsylvania, Philadelphia, and Dr. Thomas S. Harrison is from the Institute of Infection and Immunity at St. George’s University of London. These comments are taken from an accompanying editorial (Lancet Infect Dis. 2016 Mar 9. doi: 10.1016/S1473-3099[16]00128-6). Dr. Jarvis declared grants from Gilead Sciences and the National Institutes of Health outside of the submitted work, and Dr. Harrison declared grants from Gilead Sciences and personal fees from Viamet Pharmaceuticals, outside of the submitted work.
An open-label pilot study has found adjunct treatment with the SSRI antidepressant sertraline is associated with accelerated clearance of Cryptococcus from the cerebrospinal fluid in HIV-infected individuals with cryptococcal meningitis.
The dose-finding study of 172 HIV-infected Ugandan adults with cryptococcal meningitis showed any dose of sertraline, in combination with standard amphotericin B and high-dose fluconazole therapy, was associated with an average clearance rate of –0.37 log10 colony-forming U/mL CSF per day, compared with –0.30 observed in a previous study in individuals taking standard therapy without sertraline.
According to a paper published online in Lancet Infectious Diseases, the incidence of paradoxical immune reconstitution inflammatory syndrome was 5%, compared with rates of 17%-30% observed in other studies.
The sertraline doses used in the study ranged from 100 mg/day to 400 mg/day but researchers did not observe a significant difference in serious adverse events between the lower and higher doses (Lancet Infect Dis. 2016 Mar 9. doi: 10.1016/S1473-3099[16]00074-8).
Sertraline has previously shown potent fungicidal activity against Cryptococcus in vitro and in mice, suggesting it could be a promising therapeutic option for cryptococcal meningitis, a disease that accounts for 15%-20% of AIDS-related deaths in Africa.
“The current standard therapy for cryptococcal meningitis is based on antifungal regimens that are a half-century old, are associated with a range of toxic effects, and are largely inaccessible in areas of the world where they are needed most,” wrote Dr. Joshua Rhein of the University of Minnesota, Minneapolis, and his coauthors. “For this reason, the discovery of a widely available, nontoxic, and affordable drug effective against Cryptococcus would represent a substantial advance in preventing deaths.”
The study was supported by the National Institutes of Health, Fogarty International Center, Grand Challenges Canada, and the Doris Duke Charitable Foundation. No conflicts of interest were declared.
An open-label pilot study has found adjunct treatment with the SSRI antidepressant sertraline is associated with accelerated clearance of Cryptococcus from the cerebrospinal fluid in HIV-infected individuals with cryptococcal meningitis.
The dose-finding study of 172 HIV-infected Ugandan adults with cryptococcal meningitis showed any dose of sertraline, in combination with standard amphotericin B and high-dose fluconazole therapy, was associated with an average clearance rate of –0.37 log10 colony-forming U/mL CSF per day, compared with –0.30 observed in a previous study in individuals taking standard therapy without sertraline.
According to a paper published online in Lancet Infectious Diseases, the incidence of paradoxical immune reconstitution inflammatory syndrome was 5%, compared with rates of 17%-30% observed in other studies.
The sertraline doses used in the study ranged from 100 mg/day to 400 mg/day but researchers did not observe a significant difference in serious adverse events between the lower and higher doses (Lancet Infect Dis. 2016 Mar 9. doi: 10.1016/S1473-3099[16]00074-8).
Sertraline has previously shown potent fungicidal activity against Cryptococcus in vitro and in mice, suggesting it could be a promising therapeutic option for cryptococcal meningitis, a disease that accounts for 15%-20% of AIDS-related deaths in Africa.
“The current standard therapy for cryptococcal meningitis is based on antifungal regimens that are a half-century old, are associated with a range of toxic effects, and are largely inaccessible in areas of the world where they are needed most,” wrote Dr. Joshua Rhein of the University of Minnesota, Minneapolis, and his coauthors. “For this reason, the discovery of a widely available, nontoxic, and affordable drug effective against Cryptococcus would represent a substantial advance in preventing deaths.”
The study was supported by the National Institutes of Health, Fogarty International Center, Grand Challenges Canada, and the Doris Duke Charitable Foundation. No conflicts of interest were declared.
FROM LANCET INFECTIOUS DISEASES
Key clinical point: Sertraline may improve clearance of Cryptococcus in HIV-infected individuals with cryptococcal meningitis.
Major finding: Adjunct treatment with sertraline was associated with accelerated clearance of Cryptococcus.
Data source: Open-label, randomized, dose-finding pilot study in 172 HIV-infected individuals with cryptococcal meningitis.
Disclosures: The study was supported by the National Institutes of Health, Fogarty International Center, Grand Challenges Canada, and the Doris Duke Charitable Foundation.
Voluntary reporting to assess symptom burden among Yemeni cancer patients: common symptoms are frequently missed
Objective To assess the symptom burden experienced by Yemeni cancer patients by using VR and systematic assessment (SA).
Methods 50 cancer patients were asked an open question to voluntarily report their symptoms. This was followed by an SA of a list of 20 common physical symptoms that was drawn up based on the literature.
Results From 375 symptom entries related to the 20 symptoms, VR accounted for 66 entries (18%) and SA for 309 (82%). The mean number of VR symptoms/patient was 1.3, and the mean number of VR plus SA symptoms was 7.5 (P < .001). In all, 74% of VR symptoms and 57% of SA symptoms were moderate or severe. For each symptom, the percentage of patients who experienced it and did not report it voluntarily (missed) was 100% for bleeding, constipation, early satiety, hoarseness, taste changes, and weight loss. These were followed by anorexia (97%), skin symptoms (92%), dry mouth (91%), edema (89%), dyspnea (88%), sore mouth (88%), fatigue/weakness (85%), diarrhea (80%), dysphagia (80%), nausea (76%), cough (75%), urinary symptoms (75%), vomiting (62%), and pain (18%). Pain was the most common voluntarily reported symptom (56% of patients), the most commonly distressing (42%), and the least under-reported (18%).
Limitations Relatively small sample size; the SA included only 20 symptoms.
Conclusions SA of symptoms yields a more accurate estimation of symptom burden than does VR. As with many developing countries where the majority of cancer patients present at an incurable disease stage, Yemeni cancer patients suffer a high symptom burden, especially pain.
Click on the PDF icon at the top of this introduction to read the full article.
Objective To assess the symptom burden experienced by Yemeni cancer patients by using VR and systematic assessment (SA).
Methods 50 cancer patients were asked an open question to voluntarily report their symptoms. This was followed by an SA of a list of 20 common physical symptoms that was drawn up based on the literature.
Results From 375 symptom entries related to the 20 symptoms, VR accounted for 66 entries (18%) and SA for 309 (82%). The mean number of VR symptoms/patient was 1.3, and the mean number of VR plus SA symptoms was 7.5 (P < .001). In all, 74% of VR symptoms and 57% of SA symptoms were moderate or severe. For each symptom, the percentage of patients who experienced it and did not report it voluntarily (missed) was 100% for bleeding, constipation, early satiety, hoarseness, taste changes, and weight loss. These were followed by anorexia (97%), skin symptoms (92%), dry mouth (91%), edema (89%), dyspnea (88%), sore mouth (88%), fatigue/weakness (85%), diarrhea (80%), dysphagia (80%), nausea (76%), cough (75%), urinary symptoms (75%), vomiting (62%), and pain (18%). Pain was the most common voluntarily reported symptom (56% of patients), the most commonly distressing (42%), and the least under-reported (18%).
Limitations Relatively small sample size; the SA included only 20 symptoms.
Conclusions SA of symptoms yields a more accurate estimation of symptom burden than does VR. As with many developing countries where the majority of cancer patients present at an incurable disease stage, Yemeni cancer patients suffer a high symptom burden, especially pain.
Click on the PDF icon at the top of this introduction to read the full article.
Objective To assess the symptom burden experienced by Yemeni cancer patients by using VR and systematic assessment (SA).
Methods 50 cancer patients were asked an open question to voluntarily report their symptoms. This was followed by an SA of a list of 20 common physical symptoms that was drawn up based on the literature.
Results From 375 symptom entries related to the 20 symptoms, VR accounted for 66 entries (18%) and SA for 309 (82%). The mean number of VR symptoms/patient was 1.3, and the mean number of VR plus SA symptoms was 7.5 (P < .001). In all, 74% of VR symptoms and 57% of SA symptoms were moderate or severe. For each symptom, the percentage of patients who experienced it and did not report it voluntarily (missed) was 100% for bleeding, constipation, early satiety, hoarseness, taste changes, and weight loss. These were followed by anorexia (97%), skin symptoms (92%), dry mouth (91%), edema (89%), dyspnea (88%), sore mouth (88%), fatigue/weakness (85%), diarrhea (80%), dysphagia (80%), nausea (76%), cough (75%), urinary symptoms (75%), vomiting (62%), and pain (18%). Pain was the most common voluntarily reported symptom (56% of patients), the most commonly distressing (42%), and the least under-reported (18%).
Limitations Relatively small sample size; the SA included only 20 symptoms.
Conclusions SA of symptoms yields a more accurate estimation of symptom burden than does VR. As with many developing countries where the majority of cancer patients present at an incurable disease stage, Yemeni cancer patients suffer a high symptom burden, especially pain.
Click on the PDF icon at the top of this introduction to read the full article.
Oral anticancer therapy: a comprehensive assessment of patient perceptions and challenges
Background Oral anticancer agents are more convenient to use and better tolerated than traditional intravenous therapy but come with significant concerns about patient noncompliance, adverse effects, and high cost. Identifying areas for improvement in the medication use process may help ensure optimal use of these agents.
Methods 30 patients who were receiving oral anticancer therapy were administered a brief survey during their visits to an ambulatory Department of Veterans’ Affairs oncology clinic where pharmacists are heavily involved in providing initial and follow-up medication use education. Veterans aged 18 years or older were considered for inclusion into the study if they were currently being treated with an oral anticancer medication from a specified list for at least 1 month. Topics addressed included drug information sources, regimen compliance, management of side effects, and cost. The results were results were analyzed using univariate descriptive statistics.
Results Most of the patients were satisfied with their oral treatment, reporting ease of use with minimal side effect occurrence. Oncologists and pharmacists were equally named as sources of drug information.
Limitations Sample size was small and patients were overwhelmingly male. Response bias may be partially responsible for the observed results for regimen management, side effect occurrence, missed doses, and overall treatment satisfaction.
Conclusion Oral anticancer therapy represents a significant therapeutic advance for many types of cancer. Pharmacists can serve as vital informational resources to these patients. Further studies examining the role of pharmacist-led educational programs in terms of overall patient outcomes are warranted.
Background Oral anticancer agents are more convenient to use and better tolerated than traditional intravenous therapy but come with significant concerns about patient noncompliance, adverse effects, and high cost. Identifying areas for improvement in the medication use process may help ensure optimal use of these agents.
Methods 30 patients who were receiving oral anticancer therapy were administered a brief survey during their visits to an ambulatory Department of Veterans’ Affairs oncology clinic where pharmacists are heavily involved in providing initial and follow-up medication use education. Veterans aged 18 years or older were considered for inclusion into the study if they were currently being treated with an oral anticancer medication from a specified list for at least 1 month. Topics addressed included drug information sources, regimen compliance, management of side effects, and cost. The results were results were analyzed using univariate descriptive statistics.
Results Most of the patients were satisfied with their oral treatment, reporting ease of use with minimal side effect occurrence. Oncologists and pharmacists were equally named as sources of drug information.
Limitations Sample size was small and patients were overwhelmingly male. Response bias may be partially responsible for the observed results for regimen management, side effect occurrence, missed doses, and overall treatment satisfaction.
Conclusion Oral anticancer therapy represents a significant therapeutic advance for many types of cancer. Pharmacists can serve as vital informational resources to these patients. Further studies examining the role of pharmacist-led educational programs in terms of overall patient outcomes are warranted.
Background Oral anticancer agents are more convenient to use and better tolerated than traditional intravenous therapy but come with significant concerns about patient noncompliance, adverse effects, and high cost. Identifying areas for improvement in the medication use process may help ensure optimal use of these agents.
Methods 30 patients who were receiving oral anticancer therapy were administered a brief survey during their visits to an ambulatory Department of Veterans’ Affairs oncology clinic where pharmacists are heavily involved in providing initial and follow-up medication use education. Veterans aged 18 years or older were considered for inclusion into the study if they were currently being treated with an oral anticancer medication from a specified list for at least 1 month. Topics addressed included drug information sources, regimen compliance, management of side effects, and cost. The results were results were analyzed using univariate descriptive statistics.
Results Most of the patients were satisfied with their oral treatment, reporting ease of use with minimal side effect occurrence. Oncologists and pharmacists were equally named as sources of drug information.
Limitations Sample size was small and patients were overwhelmingly male. Response bias may be partially responsible for the observed results for regimen management, side effect occurrence, missed doses, and overall treatment satisfaction.
Conclusion Oral anticancer therapy represents a significant therapeutic advance for many types of cancer. Pharmacists can serve as vital informational resources to these patients. Further studies examining the role of pharmacist-led educational programs in terms of overall patient outcomes are warranted.
Risk score predicts rehospitalization after heart surgery
PHOENIX – A simple, five-element formula can help identify the patients undergoing heart surgery who face the greatest risk for a hospital readmission within 30 days following discharge from their index hospitalization.
The surgeons who developed this formula hope to use it in an investigational program that will target intensified management resources in postsurgical patients who face the highest readmission risk, to cut rehospitalizations and better improve their clinical status and quality of life.

The analysis that produced this formula also documented that the worst offender for triggering rehospitalizations following heart surgery is fluid overload, the proximate readmission cause for 23% of postsurgical patients, Dr. Arman Kilic said at the annual meeting of the Society of Thoracic Surgeons. The next most common cause was infection, which led to 20% of readmissions, followed by arrhythmias, responsible for 8% of readmissions, said Dr. Kilic, a thoracic surgeon at the University of Pennsylvania in Philadelphia.
Because fluid overload, often in the form of pleural effusion, is such an important driver of rehospitalizations, a more targeted management program would include better titration of diuretic treatment to patients following heart surgery, thoracentesis, and closer monitoring of clinical features that flag fluid overload such as weight.
“The volume overload issue is where the money is. If we can reduce that, it could really impact readmissions,” Dr. Kilic said in an interview.
An investigational program to target rehospitalization risk in heart surgery patients is planned at Johns Hopkins Hospital in Baltimore, where Dr. Kilic worked when he performed this analysis. Surgeons at Johns Hopkins are now in the process of getting funding for this pilot program, said Dr. John V. Conte Jr., professor of surgery and director of mechanical circulatory support at Johns Hopkins and a collaborator with Dr. Kilic on developing the risk formula.
“We’ll tailor postoperative follow-up. We’ll get high-risk patients back to the clinic sooner, and we’ll send nurse practitioners to see them to make sure they’re taking their medications and are getting weighed daily,” Dr. Conte said in an interview. “When a patient has heart surgery, they typically retain about 5-10 pounds of fluid. Patients with good renal function give up that fluid easily, but others are difficult to diurese. Many patients go home before they have been fully diuresed, and we need to follow these patients and transition them better to out-of-hospital care.”
He noted that other situations also come up that unnecessarily drive patients back to the hospital when an alternative and less expensive intervention might be equally effective. For example, some patients return to the hospital out of concern for how their chest wound is healing. Instead of being rehospitalized, such patients could be reassured by having them send a nurse a photo of their wound or by coming to an outpatient clinic.

“We need to engage more often with recently discharged patients,” Dr. Conte said in an interview. “Discharging them doesn’t mean separating them from the health care system; it should mean interacting with patients in a different way” that produces better outcomes and patient satisfaction for less money. Developing improved ways to manage recent heart surgery patients following discharge becomes even more critical later this year when, in July, the Centers for Medicare & Medicaid Services adds 30-day readmissions following coronary artery bypass grafting (CABG) to its list of procedures that can generate a penalty to hospitals if they exceed U.S. norms for readmission rates.
The risk model developed by Dr. Kilic, Dr. Conte, and their associates used data collected from 5,360 heart surgery patients treated at Johns Hopkins during 2008-2013. Nearly half the patients underwent isolated CABG, and 20% had isolated valve surgery. Overall, 585 patients (11%) had a hospital readmission within 30 days of their index discharge. One limitation of the analysis was it used data only on readmissions back to Johns Hopkins Hospital.
The researchers used data from three-quarters of the database to derive the risk formula, and from the remaining 25% of the database to validate the formula. A multivariate analysis of demographic and clinical characteristics that significantly linked with an elevated risk for readmissions identified five factors that independently made a significant contribution to readmission risk. The researchers assigned each of these five factors points depending on its relative contribution to readmission risk in the adjusted model: Severe chronic lung disease received 6 points; placement of a ventricular assist device received 5 points, while other types of heart surgery that was not CABG or valve surgery received 4 points (isolated CABG, isolated valve, or combined CABG and valve surgery received 0 points); development of acute renal failure postoperatively but before index discharge received 4 points; an index length of stay beyond 7 days received 4 points; and African American race received 3 points. The maximum number of points a patient could receive was 22.
Patients with a score of 0 had a 6% rate of a 30-day readmission; those with a score of 22 had a 63% readmission rate. For simplicity, Dr. Kilic suggested dividing patients into three categories based on their readmission risk score: Low-risk patients with a score of 0 had a readmission risk of 6%, medium-risk patients with a score of 1-10 had a readmission risk of 12%, and high-risk patients with a score of 11 or more had a readmission risk of 31%. The researchers found a 96% correlation when comparing these predicted readmission risk rates based on the derivation-subgroup analysis with the actual readmission rates seen in the validation subgroup of their database. The targeted risk-management program planned by Dr. Conte would primarily focus on high-risk patients.
Dr. Kilic and Dr. Conte said they had no relevant financial disclosures.
[email protected]
On Twitter @mitchelzoler
Dr. Kilic's data illustrates common factors resulting in rehospitalization after cardiac surgery. Fastidious fluid management in these patients and others is critical to reduce hospital readmissions. A further point to consider is that many pleural effusions, especially those on the left side, are due to retained hemothorax rather than fluid overload. In those instances, early surgical intervention with video-assisted thoracoscopic surgery, rather than prolonged diuresis, would be optimal.
Dr. Francis J. Podbielski, FCCP, serves on the editorial advisory board for CHEST Physician.
Dr. Kilic's data illustrates common factors resulting in rehospitalization after cardiac surgery. Fastidious fluid management in these patients and others is critical to reduce hospital readmissions. A further point to consider is that many pleural effusions, especially those on the left side, are due to retained hemothorax rather than fluid overload. In those instances, early surgical intervention with video-assisted thoracoscopic surgery, rather than prolonged diuresis, would be optimal.
Dr. Francis J. Podbielski, FCCP, serves on the editorial advisory board for CHEST Physician.
Dr. Kilic's data illustrates common factors resulting in rehospitalization after cardiac surgery. Fastidious fluid management in these patients and others is critical to reduce hospital readmissions. A further point to consider is that many pleural effusions, especially those on the left side, are due to retained hemothorax rather than fluid overload. In those instances, early surgical intervention with video-assisted thoracoscopic surgery, rather than prolonged diuresis, would be optimal.
Dr. Francis J. Podbielski, FCCP, serves on the editorial advisory board for CHEST Physician.
PHOENIX – A simple, five-element formula can help identify the patients undergoing heart surgery who face the greatest risk for a hospital readmission within 30 days following discharge from their index hospitalization.
The surgeons who developed this formula hope to use it in an investigational program that will target intensified management resources in postsurgical patients who face the highest readmission risk, to cut rehospitalizations and better improve their clinical status and quality of life.
The analysis that produced this formula also documented that the worst offender for triggering rehospitalizations following heart surgery is fluid overload, the proximate readmission cause for 23% of postsurgical patients, Dr. Arman Kilic said at the annual meeting of the Society of Thoracic Surgeons. The next most common cause was infection, which led to 20% of readmissions, followed by arrhythmias, responsible for 8% of readmissions, said Dr. Kilic, a thoracic surgeon at the University of Pennsylvania in Philadelphia.
Because fluid overload, often in the form of pleural effusion, is such an important driver of rehospitalizations, a more targeted management program would include better titration of diuretic treatment to patients following heart surgery, thoracentesis, and closer monitoring of clinical features that flag fluid overload such as weight.
“The volume overload issue is where the money is. If we can reduce that, it could really impact readmissions,” Dr. Kilic said in an interview.
An investigational program to target rehospitalization risk in heart surgery patients is planned at Johns Hopkins Hospital in Baltimore, where Dr. Kilic worked when he performed this analysis. Surgeons at Johns Hopkins are now in the process of getting funding for this pilot program, said Dr. John V. Conte Jr., professor of surgery and director of mechanical circulatory support at Johns Hopkins and a collaborator with Dr. Kilic on developing the risk formula.
“We’ll tailor postoperative follow-up. We’ll get high-risk patients back to the clinic sooner, and we’ll send nurse practitioners to see them to make sure they’re taking their medications and are getting weighed daily,” Dr. Conte said in an interview. “When a patient has heart surgery, they typically retain about 5-10 pounds of fluid. Patients with good renal function give up that fluid easily, but others are difficult to diurese. Many patients go home before they have been fully diuresed, and we need to follow these patients and transition them better to out-of-hospital care.”
He noted that other situations also come up that unnecessarily drive patients back to the hospital when an alternative and less expensive intervention might be equally effective. For example, some patients return to the hospital out of concern for how their chest wound is healing. Instead of being rehospitalized, such patients could be reassured by having them send a nurse a photo of their wound or by coming to an outpatient clinic.
“We need to engage more often with recently discharged patients,” Dr. Conte said in an interview. “Discharging them doesn’t mean separating them from the health care system; it should mean interacting with patients in a different way” that produces better outcomes and patient satisfaction for less money. Developing improved ways to manage recent heart surgery patients following discharge becomes even more critical later this year when, in July, the Centers for Medicare & Medicaid Services adds 30-day readmissions following coronary artery bypass grafting (CABG) to its list of procedures that can generate a penalty to hospitals if they exceed U.S. norms for readmission rates.
The risk model developed by Dr. Kilic, Dr. Conte, and their associates used data collected from 5,360 heart surgery patients treated at Johns Hopkins during 2008-2013. Nearly half the patients underwent isolated CABG, and 20% had isolated valve surgery. Overall, 585 patients (11%) had a hospital readmission within 30 days of their index discharge. One limitation of the analysis was it used data only on readmissions back to Johns Hopkins Hospital.
The researchers used data from three-quarters of the database to derive the risk formula, and from the remaining 25% of the database to validate the formula. A multivariate analysis of demographic and clinical characteristics that significantly linked with an elevated risk for readmissions identified five factors that independently made a significant contribution to readmission risk. The researchers assigned each of these five factors points depending on its relative contribution to readmission risk in the adjusted model: Severe chronic lung disease received 6 points; placement of a ventricular assist device received 5 points, while other types of heart surgery that was not CABG or valve surgery received 4 points (isolated CABG, isolated valve, or combined CABG and valve surgery received 0 points); development of acute renal failure postoperatively but before index discharge received 4 points; an index length of stay beyond 7 days received 4 points; and African American race received 3 points. The maximum number of points a patient could receive was 22.
Patients with a score of 0 had a 6% rate of a 30-day readmission; those with a score of 22 had a 63% readmission rate. For simplicity, Dr. Kilic suggested dividing patients into three categories based on their readmission risk score: Low-risk patients with a score of 0 had a readmission risk of 6%, medium-risk patients with a score of 1-10 had a readmission risk of 12%, and high-risk patients with a score of 11 or more had a readmission risk of 31%. The researchers found a 96% correlation when comparing these predicted readmission risk rates based on the derivation-subgroup analysis with the actual readmission rates seen in the validation subgroup of their database. The targeted risk-management program planned by Dr. Conte would primarily focus on high-risk patients.
Dr. Kilic and Dr. Conte said they had no relevant financial disclosures.
[email protected]
On Twitter @mitchelzoler
PHOENIX – A simple, five-element formula can help identify the patients undergoing heart surgery who face the greatest risk for a hospital readmission within 30 days following discharge from their index hospitalization.
The surgeons who developed this formula hope to use it in an investigational program that will target intensified management resources in postsurgical patients who face the highest readmission risk, to cut rehospitalizations and better improve their clinical status and quality of life.
The analysis that produced this formula also documented that the worst offender for triggering rehospitalizations following heart surgery is fluid overload, the proximate readmission cause for 23% of postsurgical patients, Dr. Arman Kilic said at the annual meeting of the Society of Thoracic Surgeons. The next most common cause was infection, which led to 20% of readmissions, followed by arrhythmias, responsible for 8% of readmissions, said Dr. Kilic, a thoracic surgeon at the University of Pennsylvania in Philadelphia.
Because fluid overload, often in the form of pleural effusion, is such an important driver of rehospitalizations, a more targeted management program would include better titration of diuretic treatment to patients following heart surgery, thoracentesis, and closer monitoring of clinical features that flag fluid overload such as weight.
“The volume overload issue is where the money is. If we can reduce that, it could really impact readmissions,” Dr. Kilic said in an interview.
An investigational program to target rehospitalization risk in heart surgery patients is planned at Johns Hopkins Hospital in Baltimore, where Dr. Kilic worked when he performed this analysis. Surgeons at Johns Hopkins are now in the process of getting funding for this pilot program, said Dr. John V. Conte Jr., professor of surgery and director of mechanical circulatory support at Johns Hopkins and a collaborator with Dr. Kilic on developing the risk formula.
“We’ll tailor postoperative follow-up. We’ll get high-risk patients back to the clinic sooner, and we’ll send nurse practitioners to see them to make sure they’re taking their medications and are getting weighed daily,” Dr. Conte said in an interview. “When a patient has heart surgery, they typically retain about 5-10 pounds of fluid. Patients with good renal function give up that fluid easily, but others are difficult to diurese. Many patients go home before they have been fully diuresed, and we need to follow these patients and transition them better to out-of-hospital care.”
He noted that other situations also come up that unnecessarily drive patients back to the hospital when an alternative and less expensive intervention might be equally effective. For example, some patients return to the hospital out of concern for how their chest wound is healing. Instead of being rehospitalized, such patients could be reassured by having them send a nurse a photo of their wound or by coming to an outpatient clinic.
“We need to engage more often with recently discharged patients,” Dr. Conte said in an interview. “Discharging them doesn’t mean separating them from the health care system; it should mean interacting with patients in a different way” that produces better outcomes and patient satisfaction for less money. Developing improved ways to manage recent heart surgery patients following discharge becomes even more critical later this year when, in July, the Centers for Medicare & Medicaid Services adds 30-day readmissions following coronary artery bypass grafting (CABG) to its list of procedures that can generate a penalty to hospitals if they exceed U.S. norms for readmission rates.
The risk model developed by Dr. Kilic, Dr. Conte, and their associates used data collected from 5,360 heart surgery patients treated at Johns Hopkins during 2008-2013. Nearly half the patients underwent isolated CABG, and 20% had isolated valve surgery. Overall, 585 patients (11%) had a hospital readmission within 30 days of their index discharge. One limitation of the analysis was it used data only on readmissions back to Johns Hopkins Hospital.
The researchers used data from three-quarters of the database to derive the risk formula, and from the remaining 25% of the database to validate the formula. A multivariate analysis of demographic and clinical characteristics that significantly linked with an elevated risk for readmissions identified five factors that independently made a significant contribution to readmission risk. The researchers assigned each of these five factors points depending on its relative contribution to readmission risk in the adjusted model: Severe chronic lung disease received 6 points; placement of a ventricular assist device received 5 points, while other types of heart surgery that was not CABG or valve surgery received 4 points (isolated CABG, isolated valve, or combined CABG and valve surgery received 0 points); development of acute renal failure postoperatively but before index discharge received 4 points; an index length of stay beyond 7 days received 4 points; and African American race received 3 points. The maximum number of points a patient could receive was 22.
Patients with a score of 0 had a 6% rate of a 30-day readmission; those with a score of 22 had a 63% readmission rate. For simplicity, Dr. Kilic suggested dividing patients into three categories based on their readmission risk score: Low-risk patients with a score of 0 had a readmission risk of 6%, medium-risk patients with a score of 1-10 had a readmission risk of 12%, and high-risk patients with a score of 11 or more had a readmission risk of 31%. The researchers found a 96% correlation when comparing these predicted readmission risk rates based on the derivation-subgroup analysis with the actual readmission rates seen in the validation subgroup of their database. The targeted risk-management program planned by Dr. Conte would primarily focus on high-risk patients.
Dr. Kilic and Dr. Conte said they had no relevant financial disclosures.
[email protected]
On Twitter @mitchelzoler
Key clinical point: A risk score predicted which heart surgery patients faced the greatest risk for hospital readmission within 30 days of their index discharge.
Major finding: Patients with a 0 score had a 6% 30-day readmission rate; a high score of 22 linked with a 63% rate.
Data source: A review of 5,360 heart surgery patients treated at one U.S. center.
Disclosures: Dr. Kilic and Dr. Conte said they had no relevant financial disclosures.

A Case of Nerves
1. After several years of progressively worsening symptoms, starting with intermittent muscular rigidity and spasms in the lower back, this patient now has stiff leg muscles (more pronounced on the right side). On exam, she demonstrates a slow, stiff manner of walking and lordosis of the lower spine.

Diagnosis: Moersch-Woltman syndrome, or stiff person syndrome, is a rare acquired neurologic disorder, affecting both men and women of any age; most, however, are women. Thought to be an autoimmune disorder, this syndrome may localized to one area of the body or may be widespread, and rapidly progressive, including the body, brain stem, and spinal cord. If untreated, the patient may have difficulty walking and performing activities of daily living.
For more information, see “Stiff Person Syndrome.”
For the next photograph, proceed to the next page >>
2. This patient lost control of her eye and eyelid rather suddenly and then noticed difficulty swallowing and slurred speech. She is now experiencing limb weakness that worsens as the day progresses.
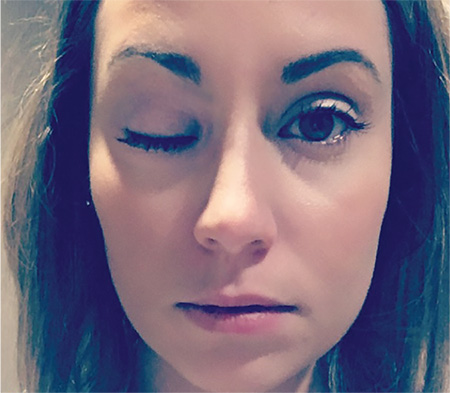
Diagnosis: Myasthenia gravis is a chronic autoimmune disease characterized by varying degrees of weakness of the skeletal muscles of the body. Muscles that control eye and eyelid movement, facial expression, chewing, talking, and swallowing are often, but not always, involved in the disorder. The muscles that control breathing and neck and limb movements may also be affected.
For more information, see “Test Accurately Diagnoses Myasthenia Gravis.” Neurology Reviews. 2016;24(3):36.
For the next photograph, proceed to the next page >>
3. After a night out on the town, this patient is concerned that something is wrong, and it’s not the splitting headache: He is unable to extend his right wrist, thumb, or fingers in the pronated position. Furthermore, the radial and dorsal skin is numb. During the history, he reports that he fell asleep sitting upright with his arm over the back of a chair.
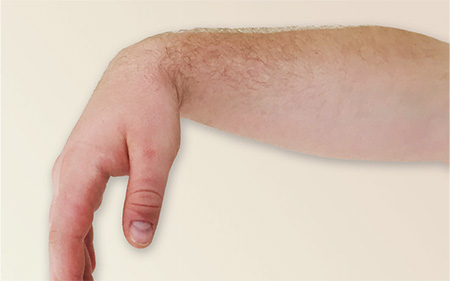
Diagnosis: Radial neuropathy, or Saturday night palsy, is typically produced by direct, prolonged pressure on the radial nerve. The nerve is especially susceptible as it spirals around the midportion of the humerus, in this case by allowing the hard edge of a chair back to compress it all night long. Although alcohol and drugs are usually implicated, radial palsy can also result from fracturing the humerus, leaning on crutches, or from long-term constriction of the wrist (tight watch or bracelet).
For more information, see “9.19 Radial Neuropathy (Saturday Night Palsy).”
For the next photograph, proceed to the next page >>
4. The patient reports spontaneous severe pain near his right shoulder, unrelated to any trauma. Two weeks later, the shoulder protrudes in a winglike position on the upper back, made more apparent when the same side arm pushes against resistance. He has limited ability to lift that arm above his head.

Diagnosis: Winged scapula injury is due to dysfunction of the anterior serratus muscle, which is supplied by the injury-prone long thoracic nerve.
Damage can be caused by a contusion or blunt trauma of the shoulder, traction of the neck, and sometimes a viral illness. Paralysis of the serratus anterior muscle has been well documented among athletes and laborers. To identify an injury, the patient stands facing a wall from about two feet away and then pushes against the wall with flat palms at waist level. Conservative treatment is often recommended; if after 6-24 months if there is no recovery, patients become candidates for corrective surgery.
For more information, see “Scapular winging: anatomical review, diagnosis, and treatments.” Curr Rev Musculoskelet Med. 2008;1(1):1-11.
Related article
For more on peripheral neuropathies, see “An easy approach to evaluating peripheral neuropathy.” J Fam Pract. 2006 October;55(10):853-861.
Photographs courtesy of Ashley Owens and Alexander Orban. They are included for illustration only and do not represent actual cases.
1. After several years of progressively worsening symptoms, starting with intermittent muscular rigidity and spasms in the lower back, this patient now has stiff leg muscles (more pronounced on the right side). On exam, she demonstrates a slow, stiff manner of walking and lordosis of the lower spine.
Diagnosis: Moersch-Woltman syndrome, or stiff person syndrome, is a rare acquired neurologic disorder, affecting both men and women of any age; most, however, are women. Thought to be an autoimmune disorder, this syndrome may localized to one area of the body or may be widespread, and rapidly progressive, including the body, brain stem, and spinal cord. If untreated, the patient may have difficulty walking and performing activities of daily living.
For more information, see “Stiff Person Syndrome.”
For the next photograph, proceed to the next page >>
2. This patient lost control of her eye and eyelid rather suddenly and then noticed difficulty swallowing and slurred speech. She is now experiencing limb weakness that worsens as the day progresses.
Diagnosis: Myasthenia gravis is a chronic autoimmune disease characterized by varying degrees of weakness of the skeletal muscles of the body. Muscles that control eye and eyelid movement, facial expression, chewing, talking, and swallowing are often, but not always, involved in the disorder. The muscles that control breathing and neck and limb movements may also be affected.
For more information, see “Test Accurately Diagnoses Myasthenia Gravis.” Neurology Reviews. 2016;24(3):36.
For the next photograph, proceed to the next page >>
3. After a night out on the town, this patient is concerned that something is wrong, and it’s not the splitting headache: He is unable to extend his right wrist, thumb, or fingers in the pronated position. Furthermore, the radial and dorsal skin is numb. During the history, he reports that he fell asleep sitting upright with his arm over the back of a chair.
Diagnosis: Radial neuropathy, or Saturday night palsy, is typically produced by direct, prolonged pressure on the radial nerve. The nerve is especially susceptible as it spirals around the midportion of the humerus, in this case by allowing the hard edge of a chair back to compress it all night long. Although alcohol and drugs are usually implicated, radial palsy can also result from fracturing the humerus, leaning on crutches, or from long-term constriction of the wrist (tight watch or bracelet).
For more information, see “9.19 Radial Neuropathy (Saturday Night Palsy).”
For the next photograph, proceed to the next page >>
4. The patient reports spontaneous severe pain near his right shoulder, unrelated to any trauma. Two weeks later, the shoulder protrudes in a winglike position on the upper back, made more apparent when the same side arm pushes against resistance. He has limited ability to lift that arm above his head.
Diagnosis: Winged scapula injury is due to dysfunction of the anterior serratus muscle, which is supplied by the injury-prone long thoracic nerve.
Damage can be caused by a contusion or blunt trauma of the shoulder, traction of the neck, and sometimes a viral illness. Paralysis of the serratus anterior muscle has been well documented among athletes and laborers. To identify an injury, the patient stands facing a wall from about two feet away and then pushes against the wall with flat palms at waist level. Conservative treatment is often recommended; if after 6-24 months if there is no recovery, patients become candidates for corrective surgery.
For more information, see “Scapular winging: anatomical review, diagnosis, and treatments.” Curr Rev Musculoskelet Med. 2008;1(1):1-11.
Related article
For more on peripheral neuropathies, see “An easy approach to evaluating peripheral neuropathy.” J Fam Pract. 2006 October;55(10):853-861.
Photographs courtesy of Ashley Owens and Alexander Orban. They are included for illustration only and do not represent actual cases.
1. After several years of progressively worsening symptoms, starting with intermittent muscular rigidity and spasms in the lower back, this patient now has stiff leg muscles (more pronounced on the right side). On exam, she demonstrates a slow, stiff manner of walking and lordosis of the lower spine.
Diagnosis: Moersch-Woltman syndrome, or stiff person syndrome, is a rare acquired neurologic disorder, affecting both men and women of any age; most, however, are women. Thought to be an autoimmune disorder, this syndrome may localized to one area of the body or may be widespread, and rapidly progressive, including the body, brain stem, and spinal cord. If untreated, the patient may have difficulty walking and performing activities of daily living.
For more information, see “Stiff Person Syndrome.”
For the next photograph, proceed to the next page >>
2. This patient lost control of her eye and eyelid rather suddenly and then noticed difficulty swallowing and slurred speech. She is now experiencing limb weakness that worsens as the day progresses.
Diagnosis: Myasthenia gravis is a chronic autoimmune disease characterized by varying degrees of weakness of the skeletal muscles of the body. Muscles that control eye and eyelid movement, facial expression, chewing, talking, and swallowing are often, but not always, involved in the disorder. The muscles that control breathing and neck and limb movements may also be affected.
For more information, see “Test Accurately Diagnoses Myasthenia Gravis.” Neurology Reviews. 2016;24(3):36.
For the next photograph, proceed to the next page >>
3. After a night out on the town, this patient is concerned that something is wrong, and it’s not the splitting headache: He is unable to extend his right wrist, thumb, or fingers in the pronated position. Furthermore, the radial and dorsal skin is numb. During the history, he reports that he fell asleep sitting upright with his arm over the back of a chair.
Diagnosis: Radial neuropathy, or Saturday night palsy, is typically produced by direct, prolonged pressure on the radial nerve. The nerve is especially susceptible as it spirals around the midportion of the humerus, in this case by allowing the hard edge of a chair back to compress it all night long. Although alcohol and drugs are usually implicated, radial palsy can also result from fracturing the humerus, leaning on crutches, or from long-term constriction of the wrist (tight watch or bracelet).
For more information, see “9.19 Radial Neuropathy (Saturday Night Palsy).”
For the next photograph, proceed to the next page >>
4. The patient reports spontaneous severe pain near his right shoulder, unrelated to any trauma. Two weeks later, the shoulder protrudes in a winglike position on the upper back, made more apparent when the same side arm pushes against resistance. He has limited ability to lift that arm above his head.
Diagnosis: Winged scapula injury is due to dysfunction of the anterior serratus muscle, which is supplied by the injury-prone long thoracic nerve.
Damage can be caused by a contusion or blunt trauma of the shoulder, traction of the neck, and sometimes a viral illness. Paralysis of the serratus anterior muscle has been well documented among athletes and laborers. To identify an injury, the patient stands facing a wall from about two feet away and then pushes against the wall with flat palms at waist level. Conservative treatment is often recommended; if after 6-24 months if there is no recovery, patients become candidates for corrective surgery.
For more information, see “Scapular winging: anatomical review, diagnosis, and treatments.” Curr Rev Musculoskelet Med. 2008;1(1):1-11.
Related article
For more on peripheral neuropathies, see “An easy approach to evaluating peripheral neuropathy.” J Fam Pract. 2006 October;55(10):853-861.
Photographs courtesy of Ashley Owens and Alexander Orban. They are included for illustration only and do not represent actual cases.
Impact of trimodality treatment on patient quality of life and arm function for superior sulcus tumors
Background Trimodality treatment leads to improved survival for superior sulcus tumor (SST) patients. Not much is known about the impact of this treatment on arm function and patient quality of life.
Objective To analyze arm function and quality of life in SST patients undergoing trimodality treatment.
Methods This was a prospective cohort study of consecutive SST patients treated with trimodality treatment that was conducted between April 1, 2010 and October 31, 2012. We obtained informed consent for 20 of 22 eligible patients. The 36-item Short Form Health Survey (SF-36) and disabilities of the arm, shoulder, and hand (DASH) questionnaires were used to asses patient quality of life and subjective arm function at 0 (preoperative day), 3, and 12 months after trimodality treatment.
Results DASH scores were significantly lower at 3 and 12 months (P = .024 and P = .011) compared with preoperative scores. Significantly lower scores were reported for the SF-36 domains of physical functioning at 12 months (P = .020) and of physical role functioning at 3 months (P = .041), and significantly more pain was reported at 3 and 12 months (P = .006 and P = .019, respectively). Patients who underwent T1 nerve root resection had lower scores for the SF-36 domain health change at 3 months (P = .037) compared with those in whom the T1 root was spared. For all other domains no differences were found.
Limitations Small sample size; patient pre-chemoradiation function and quality of life unknown.
Conclusion Subjective arm function and patient quality of life is reduced following trimodality treatment. Resection of the T1 nerve root has no significant long-term effect on the subjective arm function and quality of life.
Click on the PDF icon at the top of this introduction to read the full article.
Background Trimodality treatment leads to improved survival for superior sulcus tumor (SST) patients. Not much is known about the impact of this treatment on arm function and patient quality of life.
Objective To analyze arm function and quality of life in SST patients undergoing trimodality treatment.
Methods This was a prospective cohort study of consecutive SST patients treated with trimodality treatment that was conducted between April 1, 2010 and October 31, 2012. We obtained informed consent for 20 of 22 eligible patients. The 36-item Short Form Health Survey (SF-36) and disabilities of the arm, shoulder, and hand (DASH) questionnaires were used to asses patient quality of life and subjective arm function at 0 (preoperative day), 3, and 12 months after trimodality treatment.
Results DASH scores were significantly lower at 3 and 12 months (P = .024 and P = .011) compared with preoperative scores. Significantly lower scores were reported for the SF-36 domains of physical functioning at 12 months (P = .020) and of physical role functioning at 3 months (P = .041), and significantly more pain was reported at 3 and 12 months (P = .006 and P = .019, respectively). Patients who underwent T1 nerve root resection had lower scores for the SF-36 domain health change at 3 months (P = .037) compared with those in whom the T1 root was spared. For all other domains no differences were found.
Limitations Small sample size; patient pre-chemoradiation function and quality of life unknown.
Conclusion Subjective arm function and patient quality of life is reduced following trimodality treatment. Resection of the T1 nerve root has no significant long-term effect on the subjective arm function and quality of life.
Click on the PDF icon at the top of this introduction to read the full article.
Background Trimodality treatment leads to improved survival for superior sulcus tumor (SST) patients. Not much is known about the impact of this treatment on arm function and patient quality of life.
Objective To analyze arm function and quality of life in SST patients undergoing trimodality treatment.
Methods This was a prospective cohort study of consecutive SST patients treated with trimodality treatment that was conducted between April 1, 2010 and October 31, 2012. We obtained informed consent for 20 of 22 eligible patients. The 36-item Short Form Health Survey (SF-36) and disabilities of the arm, shoulder, and hand (DASH) questionnaires were used to asses patient quality of life and subjective arm function at 0 (preoperative day), 3, and 12 months after trimodality treatment.
Results DASH scores were significantly lower at 3 and 12 months (P = .024 and P = .011) compared with preoperative scores. Significantly lower scores were reported for the SF-36 domains of physical functioning at 12 months (P = .020) and of physical role functioning at 3 months (P = .041), and significantly more pain was reported at 3 and 12 months (P = .006 and P = .019, respectively). Patients who underwent T1 nerve root resection had lower scores for the SF-36 domain health change at 3 months (P = .037) compared with those in whom the T1 root was spared. For all other domains no differences were found.
Limitations Small sample size; patient pre-chemoradiation function and quality of life unknown.
Conclusion Subjective arm function and patient quality of life is reduced following trimodality treatment. Resection of the T1 nerve root has no significant long-term effect on the subjective arm function and quality of life.
Click on the PDF icon at the top of this introduction to read the full article.