User login
Methacrylate Polymer Powder Dressing for a Lower Leg Surgical Defect
To the Editor:
Surgical wounds on the lower leg are challenging to manage because venous stasis, bacterial colonization, and high tension may contribute to protracted healing. Advances in technology led to the development of novel, polymer-based wound-healing modalities that hold promise for the management of these wounds.
A 75-year-old man presented with a well-differentiated squamous cell carcinoma with a 3-mm depth of invasion on the left pretibial region. His comorbidities were notable for hypertension, hypercholesterolemia, varicose veins, myocardial infarction, peripheral vascular disease, and a 32 pack-year cigarette smoking history. Current medications included clopidogrel bisulfate and warfarin sodium to manage a recently placed coronary artery stent.
The tumor was cleared after 2 stages of Mohs micrographic surgery with excision down to tibialis anterior fascia (Figure 1A). The resultant defect measured 43×33 mm in area and 9 mm in depth (wound size, 12,771 mm3). Reconstructive options were discussed, including random-pattern flap repair and skin graft. Given the patient’s risk of bleeding, the decision was made to forego a flap repair. Additionally, the patient was a heavy smoker and could not comply with the wound care and elevation and ambulation restrictions required for optimal skin graft care. Therefore, a decision was made to proceed with secondary intention healing using a methacrylate polymer powder dressing.
After achieving hemostasis, a novel 10-mg sterile, biologically inert methacrylate polymer powder dressing was poured over the wound in a uniform layer to fill and seal the entire wound surface (Figure 1B). Sterile normal saline 0.1 mL was sprayed onto the powder to activate particle aggregation. No secondary dressing was used, and the patient was permitted to get the dressing wet after 48 hours.
The dressing was changed in a similar fashion 4 weeks after application, following gentle debridement with gauze and normal saline. Eight weeks after surgery, the wound exhibited healthy granulation tissue and measured 5×6 mm in area and 2 mm in depth (wound size, 60 mm3), which represented a 99.5% reduction in wound size (Figure 1C). The dressing was not painful, and there were no reported adverse effects. The patient continued to smoke and ambulate fully throughout this period. No antibiotics were used.

Methacrylate polymer powder dressings are a novel and sophisticated dressing modality with great promise for the management of surgical wounds on the lower limb. The dressing is a sterile powder consisting of 84.8% poly-2-hydroxyethylmethacrylate, 14.9% poly-2-hydroxypropylmethacrylate, and 0.3% sodium deoxycholate. These hydrophilic polymers have a covalent methacrylate backbone with a hydroxyl aliphatic side chain. When saline or wound exudate contacts the powder, the spheres hydrate and nonreversibly aggregate to form a moist, flexible dressing that conforms to the topography of the wound and seals it (Figure 2).1

Once the spheres have aggregated, they are designed to orient in a honeycomb formation with 4- to 10-nm openings that serve as capillary channels (Figure 3). This porous architecture of the polymer is essential for adequate moisture management. It allows for vapor transpiration at a rate of 12 L/m2 per day, which ensures the capillary flow from the moist wound surface is evenly distributed through the dressing, contributing to its 68% water content. Notably, this approximately three-fifths water composition is similar to the water makeup of human skin. Optimized moisture management is theorized to enhance epithelial migration, stimulate angiogenesis, retain growth factors, promote autolytic debridement, and maintain ideal voltage and oxygen gradients for wound healing. The risk for infection is not increased by the existence of these pores, as their small size does not allow for bacterial migration.1

This case demonstrates the effectiveness of using a methacrylate polymer powder dressing to promote timely wound healing in a poorly vascularized lower leg surgical wound. The low maintenance, user-friendly dressing was changed at monthly intervals, which spared the patient the inconvenience and pain associated with the repeated application of more conventional primary and secondary dressings. The dressing was well tolerated and resulted in a 99.5% reduction in wound size. Further studies are needed to investigate the utility of this promising technology.
1. Fitzgerald RH, Bharara M, Mills JL, et al. Use of a nanoflex powder dressing for wound management following debridement for necrotising fasciitis in the diabetic foot. Int Wound J. 2009;6:133-139.
To the Editor:
Surgical wounds on the lower leg are challenging to manage because venous stasis, bacterial colonization, and high tension may contribute to protracted healing. Advances in technology led to the development of novel, polymer-based wound-healing modalities that hold promise for the management of these wounds.
A 75-year-old man presented with a well-differentiated squamous cell carcinoma with a 3-mm depth of invasion on the left pretibial region. His comorbidities were notable for hypertension, hypercholesterolemia, varicose veins, myocardial infarction, peripheral vascular disease, and a 32 pack-year cigarette smoking history. Current medications included clopidogrel bisulfate and warfarin sodium to manage a recently placed coronary artery stent.
The tumor was cleared after 2 stages of Mohs micrographic surgery with excision down to tibialis anterior fascia (Figure 1A). The resultant defect measured 43×33 mm in area and 9 mm in depth (wound size, 12,771 mm3). Reconstructive options were discussed, including random-pattern flap repair and skin graft. Given the patient’s risk of bleeding, the decision was made to forego a flap repair. Additionally, the patient was a heavy smoker and could not comply with the wound care and elevation and ambulation restrictions required for optimal skin graft care. Therefore, a decision was made to proceed with secondary intention healing using a methacrylate polymer powder dressing.
After achieving hemostasis, a novel 10-mg sterile, biologically inert methacrylate polymer powder dressing was poured over the wound in a uniform layer to fill and seal the entire wound surface (Figure 1B). Sterile normal saline 0.1 mL was sprayed onto the powder to activate particle aggregation. No secondary dressing was used, and the patient was permitted to get the dressing wet after 48 hours.
The dressing was changed in a similar fashion 4 weeks after application, following gentle debridement with gauze and normal saline. Eight weeks after surgery, the wound exhibited healthy granulation tissue and measured 5×6 mm in area and 2 mm in depth (wound size, 60 mm3), which represented a 99.5% reduction in wound size (Figure 1C). The dressing was not painful, and there were no reported adverse effects. The patient continued to smoke and ambulate fully throughout this period. No antibiotics were used.
Methacrylate polymer powder dressings are a novel and sophisticated dressing modality with great promise for the management of surgical wounds on the lower limb. The dressing is a sterile powder consisting of 84.8% poly-2-hydroxyethylmethacrylate, 14.9% poly-2-hydroxypropylmethacrylate, and 0.3% sodium deoxycholate. These hydrophilic polymers have a covalent methacrylate backbone with a hydroxyl aliphatic side chain. When saline or wound exudate contacts the powder, the spheres hydrate and nonreversibly aggregate to form a moist, flexible dressing that conforms to the topography of the wound and seals it (Figure 2).1
Once the spheres have aggregated, they are designed to orient in a honeycomb formation with 4- to 10-nm openings that serve as capillary channels (Figure 3). This porous architecture of the polymer is essential for adequate moisture management. It allows for vapor transpiration at a rate of 12 L/m2 per day, which ensures the capillary flow from the moist wound surface is evenly distributed through the dressing, contributing to its 68% water content. Notably, this approximately three-fifths water composition is similar to the water makeup of human skin. Optimized moisture management is theorized to enhance epithelial migration, stimulate angiogenesis, retain growth factors, promote autolytic debridement, and maintain ideal voltage and oxygen gradients for wound healing. The risk for infection is not increased by the existence of these pores, as their small size does not allow for bacterial migration.1
This case demonstrates the effectiveness of using a methacrylate polymer powder dressing to promote timely wound healing in a poorly vascularized lower leg surgical wound. The low maintenance, user-friendly dressing was changed at monthly intervals, which spared the patient the inconvenience and pain associated with the repeated application of more conventional primary and secondary dressings. The dressing was well tolerated and resulted in a 99.5% reduction in wound size. Further studies are needed to investigate the utility of this promising technology.
To the Editor:
Surgical wounds on the lower leg are challenging to manage because venous stasis, bacterial colonization, and high tension may contribute to protracted healing. Advances in technology led to the development of novel, polymer-based wound-healing modalities that hold promise for the management of these wounds.
A 75-year-old man presented with a well-differentiated squamous cell carcinoma with a 3-mm depth of invasion on the left pretibial region. His comorbidities were notable for hypertension, hypercholesterolemia, varicose veins, myocardial infarction, peripheral vascular disease, and a 32 pack-year cigarette smoking history. Current medications included clopidogrel bisulfate and warfarin sodium to manage a recently placed coronary artery stent.
The tumor was cleared after 2 stages of Mohs micrographic surgery with excision down to tibialis anterior fascia (Figure 1A). The resultant defect measured 43×33 mm in area and 9 mm in depth (wound size, 12,771 mm3). Reconstructive options were discussed, including random-pattern flap repair and skin graft. Given the patient’s risk of bleeding, the decision was made to forego a flap repair. Additionally, the patient was a heavy smoker and could not comply with the wound care and elevation and ambulation restrictions required for optimal skin graft care. Therefore, a decision was made to proceed with secondary intention healing using a methacrylate polymer powder dressing.
After achieving hemostasis, a novel 10-mg sterile, biologically inert methacrylate polymer powder dressing was poured over the wound in a uniform layer to fill and seal the entire wound surface (Figure 1B). Sterile normal saline 0.1 mL was sprayed onto the powder to activate particle aggregation. No secondary dressing was used, and the patient was permitted to get the dressing wet after 48 hours.
The dressing was changed in a similar fashion 4 weeks after application, following gentle debridement with gauze and normal saline. Eight weeks after surgery, the wound exhibited healthy granulation tissue and measured 5×6 mm in area and 2 mm in depth (wound size, 60 mm3), which represented a 99.5% reduction in wound size (Figure 1C). The dressing was not painful, and there were no reported adverse effects. The patient continued to smoke and ambulate fully throughout this period. No antibiotics were used.
Methacrylate polymer powder dressings are a novel and sophisticated dressing modality with great promise for the management of surgical wounds on the lower limb. The dressing is a sterile powder consisting of 84.8% poly-2-hydroxyethylmethacrylate, 14.9% poly-2-hydroxypropylmethacrylate, and 0.3% sodium deoxycholate. These hydrophilic polymers have a covalent methacrylate backbone with a hydroxyl aliphatic side chain. When saline or wound exudate contacts the powder, the spheres hydrate and nonreversibly aggregate to form a moist, flexible dressing that conforms to the topography of the wound and seals it (Figure 2).1
Once the spheres have aggregated, they are designed to orient in a honeycomb formation with 4- to 10-nm openings that serve as capillary channels (Figure 3). This porous architecture of the polymer is essential for adequate moisture management. It allows for vapor transpiration at a rate of 12 L/m2 per day, which ensures the capillary flow from the moist wound surface is evenly distributed through the dressing, contributing to its 68% water content. Notably, this approximately three-fifths water composition is similar to the water makeup of human skin. Optimized moisture management is theorized to enhance epithelial migration, stimulate angiogenesis, retain growth factors, promote autolytic debridement, and maintain ideal voltage and oxygen gradients for wound healing. The risk for infection is not increased by the existence of these pores, as their small size does not allow for bacterial migration.1
This case demonstrates the effectiveness of using a methacrylate polymer powder dressing to promote timely wound healing in a poorly vascularized lower leg surgical wound. The low maintenance, user-friendly dressing was changed at monthly intervals, which spared the patient the inconvenience and pain associated with the repeated application of more conventional primary and secondary dressings. The dressing was well tolerated and resulted in a 99.5% reduction in wound size. Further studies are needed to investigate the utility of this promising technology.
1. Fitzgerald RH, Bharara M, Mills JL, et al. Use of a nanoflex powder dressing for wound management following debridement for necrotising fasciitis in the diabetic foot. Int Wound J. 2009;6:133-139.
1. Fitzgerald RH, Bharara M, Mills JL, et al. Use of a nanoflex powder dressing for wound management following debridement for necrotising fasciitis in the diabetic foot. Int Wound J. 2009;6:133-139.
PRACTICE POINTS
- Lower leg surgical wounds are difficult to manage, as venous stasis, bacterial colonization, and high tension may contribute to protracted healing.
- A methacrylate polymer powder dressing is user friendly and facilitates granulation and reduction in size of difficult lower leg wounds.

Kaposi’s sarcoma: Antiretroviral-related improvements in survival measured
than their uninfected counterparts, based on the first such analysis of the American College of Surgeons’ National Cancer Database.
One-year overall survival for all patients with Kaposi’s sarcoma (KS), 74.9% in 2004-2007, rose by 6.4 percentage points to 81.3% in 2016-2018, with the use of ART for HIV starting in 2008. Two-year survival was up by an even larger 8.3 percentage points: 68.0% to 76.3%, said Amar D. Desai of New Jersey Medical School, Newark, and Shari R. Lipner, MD, of Weill Cornell Medicine, New York.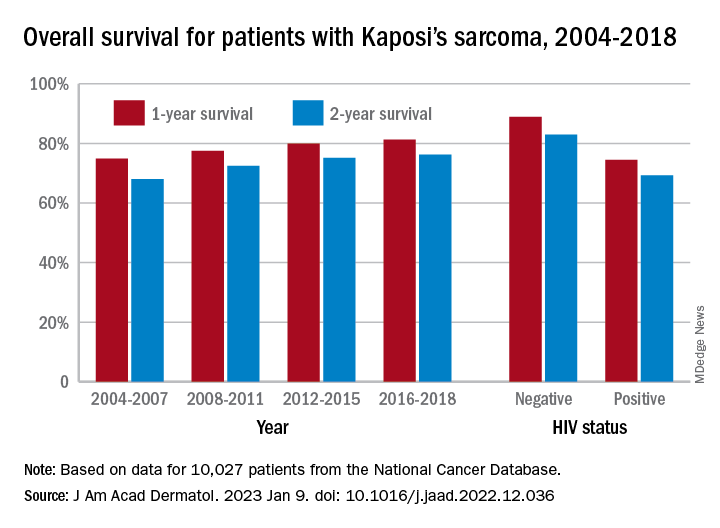
Since HIV-infected patients represented a much lower 46.7% of the Kaposi’s population in 2016-2018 than in 2004-2007 (70.5%), “better outcomes for all KS patients likely reflects advancements in ART, preventing many HIV+ patients from progressing to AIDS, changes in clinical practice with earlier treatment start, and more off-label treatments,” they wrote in the Journal of the American Academy of Dermatology.
Overall survival rates for the 10,027 patients with KS with data available in the National Cancer Database were 77.9% at 1 year and 72.4% at 2 years. HIV status had a significant (P < .0074) effect over the entire study period: One-year survival rates were 88.9% for HIV-negative and 74.5% for HIV-positive patients, and 2-year rates were 83.0% (HIV-negative) and 69.3% (HIV-positive), the investigators reported in what they called “the largest analysis since the advent of antiretroviral therapy for HIV in 2008.”
The improvement in overall survival, along with the continued differences in survival between HIV infected and noninfected patients, indicate that “dermatologists, as part of a multidisciplinary team including oncologists and infectious disease physicians, can play significant roles in early KS diagnosis,” Mr. Desai and Dr. Lipner said.
Mr. Desai had no conflicts of interest to report. Dr. Lipner has served as a consultant for Ortho-Dermatologics, Hoth Therapeutics, and BelleTorus Corporation.
than their uninfected counterparts, based on the first such analysis of the American College of Surgeons’ National Cancer Database.
One-year overall survival for all patients with Kaposi’s sarcoma (KS), 74.9% in 2004-2007, rose by 6.4 percentage points to 81.3% in 2016-2018, with the use of ART for HIV starting in 2008. Two-year survival was up by an even larger 8.3 percentage points: 68.0% to 76.3%, said Amar D. Desai of New Jersey Medical School, Newark, and Shari R. Lipner, MD, of Weill Cornell Medicine, New York.
Since HIV-infected patients represented a much lower 46.7% of the Kaposi’s population in 2016-2018 than in 2004-2007 (70.5%), “better outcomes for all KS patients likely reflects advancements in ART, preventing many HIV+ patients from progressing to AIDS, changes in clinical practice with earlier treatment start, and more off-label treatments,” they wrote in the Journal of the American Academy of Dermatology.
Overall survival rates for the 10,027 patients with KS with data available in the National Cancer Database were 77.9% at 1 year and 72.4% at 2 years. HIV status had a significant (P < .0074) effect over the entire study period: One-year survival rates were 88.9% for HIV-negative and 74.5% for HIV-positive patients, and 2-year rates were 83.0% (HIV-negative) and 69.3% (HIV-positive), the investigators reported in what they called “the largest analysis since the advent of antiretroviral therapy for HIV in 2008.”
The improvement in overall survival, along with the continued differences in survival between HIV infected and noninfected patients, indicate that “dermatologists, as part of a multidisciplinary team including oncologists and infectious disease physicians, can play significant roles in early KS diagnosis,” Mr. Desai and Dr. Lipner said.
Mr. Desai had no conflicts of interest to report. Dr. Lipner has served as a consultant for Ortho-Dermatologics, Hoth Therapeutics, and BelleTorus Corporation.
than their uninfected counterparts, based on the first such analysis of the American College of Surgeons’ National Cancer Database.
One-year overall survival for all patients with Kaposi’s sarcoma (KS), 74.9% in 2004-2007, rose by 6.4 percentage points to 81.3% in 2016-2018, with the use of ART for HIV starting in 2008. Two-year survival was up by an even larger 8.3 percentage points: 68.0% to 76.3%, said Amar D. Desai of New Jersey Medical School, Newark, and Shari R. Lipner, MD, of Weill Cornell Medicine, New York.
Since HIV-infected patients represented a much lower 46.7% of the Kaposi’s population in 2016-2018 than in 2004-2007 (70.5%), “better outcomes for all KS patients likely reflects advancements in ART, preventing many HIV+ patients from progressing to AIDS, changes in clinical practice with earlier treatment start, and more off-label treatments,” they wrote in the Journal of the American Academy of Dermatology.
Overall survival rates for the 10,027 patients with KS with data available in the National Cancer Database were 77.9% at 1 year and 72.4% at 2 years. HIV status had a significant (P < .0074) effect over the entire study period: One-year survival rates were 88.9% for HIV-negative and 74.5% for HIV-positive patients, and 2-year rates were 83.0% (HIV-negative) and 69.3% (HIV-positive), the investigators reported in what they called “the largest analysis since the advent of antiretroviral therapy for HIV in 2008.”
The improvement in overall survival, along with the continued differences in survival between HIV infected and noninfected patients, indicate that “dermatologists, as part of a multidisciplinary team including oncologists and infectious disease physicians, can play significant roles in early KS diagnosis,” Mr. Desai and Dr. Lipner said.
Mr. Desai had no conflicts of interest to report. Dr. Lipner has served as a consultant for Ortho-Dermatologics, Hoth Therapeutics, and BelleTorus Corporation.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Oral Propranolol Used as Adjunct Therapy in Cutaneous Angiosarcoma
To the Editor:
Angiosarcoma is a malignancy of the vascular endothelium that most commonly presents on the skin.1 Patients diagnosed with cutaneous angiosarcoma, which is a rare and aggressive malignancy, have a 5-year survival rate of approximately 30%.2,3 Angiosarcoma can be seen in the setting of chronic lymphedema; radiation therapy; and sporadically in elderly patients, where it is commonly seen on the head and neck. Presentation on the head and neck has been associated with worse outcomes, with a projected overall 10-year survival rate of 13.8%; the survival rate is lower if the tumor is surgically unresectable or larger in size. Metastasis can occur via both lymphatic and hematogenous routes, with pulmonary and hepatic metastases most frequently observed.1 Prognostications of poor outcomes for patients with head and neck cutaneous angiosarcoma via a 5-year survival rate were identified in a meta-analysis and included the following: patient age older than 70 years, larger tumors, tumor location of scalp vs face, nonsurgical treatments, and lack of clear margins on histology.2
Treatment of angiosarcoma historically has encompassed both surgical resection and adjuvant radiation therapy with suboptimal success. Evidence supporting various treatment regimens remains sparse due to the low incidence of the neoplasm. Although surgical resection is the only documented curative treatment, cutaneous angiosarcomas frequently are found to have positive surgical margins and require adjuvant radiation. Use of high-dose radiation (>50 Gy) with application over a wide treatment area such as total scalp irradiation is recommended.4 Although radiation has been found to diminish local recurrence rates, it has not substantially affected rates of distant disease recurrence.1 Cytotoxic chemotherapy has clinical utility in minimizing progression, but standard regimens afford a progression-free survival of only months.3 Adjuvant treatment with paclitaxel has been shown to have improved efficacy in scalp angiosarcoma vs other visceral sites, showing a nonprogression rate of 42% at 4 months after treatment.5 More recently, targeted chemotherapeutics, including the vascular endothelial growth factor inhibitor bevacizumab and tyrosine kinase inhibitor sorafenib, have shown some survival benefit, but it is unclear if these agents are superior to traditional cytotoxic agents.4,6-10 A phase 2 study of paclitaxel administered weekly with or without bevacizumab showed similar progression-free survival and overall survival, albeit at the expense of added toxicity experienced by participants in the combined group.10
The addition of the nonselective β-adrenergic blocker propranolol to the treatment armamentarium, which was pursued due to its utility in the treatment of benign infantile hemangioma and demonstrated ability to limit the expression of adrenergic receptors in angiosarcoma, has gained clinical attention for possible augmentation of cutaneous angiosarcoma therapy.11-14 Propranolol has been shown to reduce metastasis in other neoplasms—both vascular and nonvascular—and may play a role as an adjuvant treatment to current therapies in angiosarcoma.15-20 We report a patient with cutaneous angiosarcoma (T2 classification) with disease-free survival of nearly 6 years without evidence of recurrence in the setting of continuous propranolol use supplementary to chemotherapy and radiation.

A 78-year-old man with a history of multiple basal cell carcinomas, hypertension, and remote smoking history presented to the dermatology clinic with an enlarging red-brown plaque on the scalp of 2 months’ duration. The lesion had grown rapidly to involve the forehead, right temple, preauricular region, and parietal scalp. At presentation, the tumor measured more than 20 cm in diameter at its greatest point (Figure 1). Physical examination revealed a 6-mm purple nodule within the lesion on the patient’s right parietal scalp. No clinical lymphadenopathy was appreciated at the time of diagnosis. Punch biopsies of the right parietal scalp nodule and right temple patch showed findings consistent with angiosarcoma with diffuse cytoplasmic staining of CD31 in atypical endothelial cells and no staining for human herpesvirus 8 (Figure 2). Concurrent computed tomography of the head showed thickening of the right epidermis, dermis, and deeper scalp tissues, but there was no evidence of skull involvement. Computed tomography of the thorax, abdomen, and pelvis showed no evidence of metastatic disease. After a diagnostic workup, the patient was diagnosed with T2bN0M0 angiosarcoma.
. B, Cytoplasmic CD31 staining of endothelial lining of slit-like atypical")
The lesion was determined to be nonresectable due to the extent of the patient’s cutaneous disease. The patient was started on a regimen of paclitaxel, scalp radiation, and oral propranolol. Propranolol 40 mg twice daily was initiated at the time of diagnosis with a plan to continue indefinitely. Starting 1 month after staging, the patient completed 10 weekly cycles of paclitaxel, and he was treated with 60 Gy of scalp radiation in 30 fractions, starting with the second cycle of paclitaxel. He tolerated both well with no reported adverse events. Repeat computed tomography performed 1 month after completion of chemotherapy and radiation showed no evidence of a mass or fluid collection in subcutaneous scalp tissues and no evidence of metastatic disease. This correlated with an observed clinical regression at 1 month and complete clinical response at 5 months with residual hemosiderin and radiation changes. The area of prior disease involvement subsequently evolved from violet to dusky gray in appearance to an eventual complete resolution 26 months after diagnosis, accompanied by atrophic radiation-induced sequelae (Figure 3).

The patient’s postchemotherapy course was complicated by hospitalization for a suspected malignant pleural effusion. Analysis revealed growing ground-glass opacities and nodules in the right lower lung lobe. A thoracentesis with cytology studies was negative for malignancy. Continued monitoring over 19 months demonstrated eventual resolution of those findings. He experienced notable complication from local radiation therapy to the scalp with chronic cutaneous ulceration refractory to wound care and surgical intervention. The patient did not exhibit additional signs or symptoms concerning for recurrence or metastasis and was followed by dermatology and oncology until he died nearly 5 years after initial diagnosis due to complications from acute hypoxic respiratory failure secondary to COVID-19. The last imaging obtained showed no convincing evidence of metastasis, though spinal imaging within a month of his death showed lesions favored to represent benign angiomatous growths. His survival after diagnosis ultimately reached 57 months without confirmed disease recurrence and cause of death unrelated to malignancy history, which is a markedly long documented survival for this extent of disease.
Cutaneous angiosarcoma is an aggressive yet rare malignancy without effective treatments for prolonging survival or eradicating disease. Cutaneous angiosarcoma of the head and neck has a reported 10-year survival rate of 13.8%.1 Although angiosarcoma in any location holds a bleak prognosis, cutaneous angiosarcoma of the scalp with a T2 classification has a 2-year survival rate of 0%. Moreover, even if remission is achieved, disease is highly recurrent, typically within months with the current standard of care.3,21,22
Emerging evidence for the possible role of β-adrenergic receptor blockade in the treatment of malignant vascular neoplasms is promising. Microarrays from a host of vascular growths have demonstrated expression of β-adrenergic receptors in 77% of sampled angiosarcoma specimens in addition to strong expression in infantile hemangiomas, hemangiomas, hemangioendotheliomas, and vascular malformations.19 Research findings have further verified the validity of this approach with the demonstration of b1-, b2-, and b3- adrenergic receptor expression by angiosarcoma cell lines. Propranolol subsequently was shown to effectively target proliferation of these cells and induce apoptosis in a dose-dependent manner and moreover be synergistic in effect with other chemotherapies.15 Several genes have exhibited differential expression between control tumor cells and propranolol-treated cells. Specifically, target genes including AXL (a receptor tyrosine kinase associated with cell adhesion, proliferation, and apoptosis and found to upregulated in melanoma and leukemia) and ERBB receptor feedback inhibitor 1 (receptor tyrosine kinase, with ERBB family members commonly overexpressed or mutated in the setting malignancy) have been posited as possible explanatory factors in the observed angiosarcoma response to propranolol.23
Several cases describing propranolol use as an adjunctive therapy for angiosarcoma suggest a beneficial role in clinical medicine. One case report described propranolol monotherapy for lesion to our patient, with a resultant reduction in Ki-67 as a measure of proliferative index within 1 week of initiating propranolol therapy.13 Propranolol also has been shown to halt or slow progression of metastatic disease in visceral and metastatic angiosarcomas.12-14 In combination with oral etoposide and cyclophosphamide, maintenance propranolol therapy in 7 cases of advanced cutaneous angiosarcoma resulted in 1 complete response and 3 very good partial responses, with a median progression-free survival of 11 months.11 Larger-scale studies have not been published, but the growing number of case reports and case series warrants further investigation of the utility of propranolol as an adjunct to current therapies in advanced angiosarcoma.
- Abraham JA, Hornicek FJ, Kaufman AM, et al. Treatment and outcome of 82 patients with angiosarcoma. Ann Surg Oncol. 2007;14:1953-1967.
- Shin JY, Roh SG, Lee NH, et al. Predisposing factors for poor prognosis of angiosarcoma of the scalp and face: systematic review and meta-analysis. Head Neck. 2017;39:380-386.
- Fury MG, Antonescu CR, Zee KJV, et al. A 14-year retrospective review of angiosarcoma: clinical characteristics, prognostic factors, and treatment outcomes with surgery and chemotherapy. Cancer. 2005;11:241-247.
- Dossett LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- Penel N, Bui BN, Bay JO, et al. Phase II trial of weekly paclitaxel for unresectable angiosarcoma: the ANGIOTAX study. J Clin Oncol. 2008;26:5269-5274.
- Agulnik M, Yarber JL, Okuno SH, et al. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann Oncol. 2013;24:257-263.
- Maki RG, D’Adamo DR, Keohan ML, et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J Clin Oncol. 2009;27:3133-3140.
- Ishida Y, Otsuka A, Kabashima K. Cutaneous angiosarcoma: update on biology and latest treatment. Curr Opin Oncol. 2018;30:107-112.
- Ray-Coquard I, Italiano A, Bompas E, et al. Sorafenib for patients with advanced angiosarcoma: a phase II trial from the French Sarcoma Group (GSF/GETO). Oncologist. 2012;17:260-266.
- Ray-Coquard IL, Domont J, Tresch-Bruneel E, et al. Paclitaxel given once per week with or without bevacizumab in patients with advanced angiosarcoma: a randomized phase II trial. J Clin Oncol. 2015;33:2797-2802.
- Pasquier E, Andre N, Street J, et al. Effective management of advanced angiosarcoma by the synergistic combination of propranolol and vinblastine-based metronomic chemotherapy: a bench to bedside study. EBioMedicine. 2016;6:87-95.
- Banavali S, Pasquier E, Andre N. Targeted therapy with propranolol and metronomic chemotherapy combination: sustained complete response of a relapsing metastatic angiosarcoma. Ecancermedicalscience. 2015;9:499.
- Chow W, Amaya CN, Rains S, et al. Growth attenuation of cutaneous angiosarcoma with propranolol-mediated beta-blockade. JAMA Dermatol. 2015;151:1226-1229.
- Daguze J, Saint-Jean M, Peuvrel L, et al. Visceral metastatic angiosarcoma treated effectively with oral cyclophosphamide combined with propranolol. JAAD Case Rep. 2016;2:497-499.
- Stiles JM, Amaya C, Rains S, et al. Targeting of beta adrenergic receptors results in therapeutic efficacy against models of hemangioendothelioma and angiosarcoma. PLoS One. 2013;8:e60021.
- Chang PY, Chung CH, Chang WC, et al. The effect of propranolol on the prognosis of hepatocellular carcinoma: a nationwide population-based study. PLoS One. 2019;14:e0216828.
- De Giorgi V, Grazzini M, Benemei S, et al. Propranolol for off-label treatment of patients with melanoma: results from a cohort study. JAMA Oncol. 2018;4:e172908.
- Rico M, Baglioni M, Bondarenko M, et al. Metformin and propranolol combination prevents cancer progression and metastasis in different breast cancer models. Oncotarget. 2017;8:2874-2889.
- Chisholm KM, Chang KW, Truong MT, et al. β-Adrenergic receptor expression in vascular tumors. Mod Pathol. 2012;25:1446-1451.
- Leaute-Labreze C, Dumas de la Roque E, Hubiche T, et al. Propranolol for severe hemangiomas of infancy. N Engl J Med. 2008;358:2649-2651.
- Maddox JC, Evans HL. Angiosarcoma of skin and soft tissue: a study of forty-four cases. Cancer. 1981;48:1907-1921.
- Morgan MB, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Zhou S, Liu P, Jiang W, et al. Identification of potential target genes associated with the effect of propranolol on angiosarcoma via microarray analysis. Oncol Lett. 2017;13:4267-4275.
To the Editor:
Angiosarcoma is a malignancy of the vascular endothelium that most commonly presents on the skin.1 Patients diagnosed with cutaneous angiosarcoma, which is a rare and aggressive malignancy, have a 5-year survival rate of approximately 30%.2,3 Angiosarcoma can be seen in the setting of chronic lymphedema; radiation therapy; and sporadically in elderly patients, where it is commonly seen on the head and neck. Presentation on the head and neck has been associated with worse outcomes, with a projected overall 10-year survival rate of 13.8%; the survival rate is lower if the tumor is surgically unresectable or larger in size. Metastasis can occur via both lymphatic and hematogenous routes, with pulmonary and hepatic metastases most frequently observed.1 Prognostications of poor outcomes for patients with head and neck cutaneous angiosarcoma via a 5-year survival rate were identified in a meta-analysis and included the following: patient age older than 70 years, larger tumors, tumor location of scalp vs face, nonsurgical treatments, and lack of clear margins on histology.2
Treatment of angiosarcoma historically has encompassed both surgical resection and adjuvant radiation therapy with suboptimal success. Evidence supporting various treatment regimens remains sparse due to the low incidence of the neoplasm. Although surgical resection is the only documented curative treatment, cutaneous angiosarcomas frequently are found to have positive surgical margins and require adjuvant radiation. Use of high-dose radiation (>50 Gy) with application over a wide treatment area such as total scalp irradiation is recommended.4 Although radiation has been found to diminish local recurrence rates, it has not substantially affected rates of distant disease recurrence.1 Cytotoxic chemotherapy has clinical utility in minimizing progression, but standard regimens afford a progression-free survival of only months.3 Adjuvant treatment with paclitaxel has been shown to have improved efficacy in scalp angiosarcoma vs other visceral sites, showing a nonprogression rate of 42% at 4 months after treatment.5 More recently, targeted chemotherapeutics, including the vascular endothelial growth factor inhibitor bevacizumab and tyrosine kinase inhibitor sorafenib, have shown some survival benefit, but it is unclear if these agents are superior to traditional cytotoxic agents.4,6-10 A phase 2 study of paclitaxel administered weekly with or without bevacizumab showed similar progression-free survival and overall survival, albeit at the expense of added toxicity experienced by participants in the combined group.10
The addition of the nonselective β-adrenergic blocker propranolol to the treatment armamentarium, which was pursued due to its utility in the treatment of benign infantile hemangioma and demonstrated ability to limit the expression of adrenergic receptors in angiosarcoma, has gained clinical attention for possible augmentation of cutaneous angiosarcoma therapy.11-14 Propranolol has been shown to reduce metastasis in other neoplasms—both vascular and nonvascular—and may play a role as an adjuvant treatment to current therapies in angiosarcoma.15-20 We report a patient with cutaneous angiosarcoma (T2 classification) with disease-free survival of nearly 6 years without evidence of recurrence in the setting of continuous propranolol use supplementary to chemotherapy and radiation.
A 78-year-old man with a history of multiple basal cell carcinomas, hypertension, and remote smoking history presented to the dermatology clinic with an enlarging red-brown plaque on the scalp of 2 months’ duration. The lesion had grown rapidly to involve the forehead, right temple, preauricular region, and parietal scalp. At presentation, the tumor measured more than 20 cm in diameter at its greatest point (Figure 1). Physical examination revealed a 6-mm purple nodule within the lesion on the patient’s right parietal scalp. No clinical lymphadenopathy was appreciated at the time of diagnosis. Punch biopsies of the right parietal scalp nodule and right temple patch showed findings consistent with angiosarcoma with diffuse cytoplasmic staining of CD31 in atypical endothelial cells and no staining for human herpesvirus 8 (Figure 2). Concurrent computed tomography of the head showed thickening of the right epidermis, dermis, and deeper scalp tissues, but there was no evidence of skull involvement. Computed tomography of the thorax, abdomen, and pelvis showed no evidence of metastatic disease. After a diagnostic workup, the patient was diagnosed with T2bN0M0 angiosarcoma.
The lesion was determined to be nonresectable due to the extent of the patient’s cutaneous disease. The patient was started on a regimen of paclitaxel, scalp radiation, and oral propranolol. Propranolol 40 mg twice daily was initiated at the time of diagnosis with a plan to continue indefinitely. Starting 1 month after staging, the patient completed 10 weekly cycles of paclitaxel, and he was treated with 60 Gy of scalp radiation in 30 fractions, starting with the second cycle of paclitaxel. He tolerated both well with no reported adverse events. Repeat computed tomography performed 1 month after completion of chemotherapy and radiation showed no evidence of a mass or fluid collection in subcutaneous scalp tissues and no evidence of metastatic disease. This correlated with an observed clinical regression at 1 month and complete clinical response at 5 months with residual hemosiderin and radiation changes. The area of prior disease involvement subsequently evolved from violet to dusky gray in appearance to an eventual complete resolution 26 months after diagnosis, accompanied by atrophic radiation-induced sequelae (Figure 3).
The patient’s postchemotherapy course was complicated by hospitalization for a suspected malignant pleural effusion. Analysis revealed growing ground-glass opacities and nodules in the right lower lung lobe. A thoracentesis with cytology studies was negative for malignancy. Continued monitoring over 19 months demonstrated eventual resolution of those findings. He experienced notable complication from local radiation therapy to the scalp with chronic cutaneous ulceration refractory to wound care and surgical intervention. The patient did not exhibit additional signs or symptoms concerning for recurrence or metastasis and was followed by dermatology and oncology until he died nearly 5 years after initial diagnosis due to complications from acute hypoxic respiratory failure secondary to COVID-19. The last imaging obtained showed no convincing evidence of metastasis, though spinal imaging within a month of his death showed lesions favored to represent benign angiomatous growths. His survival after diagnosis ultimately reached 57 months without confirmed disease recurrence and cause of death unrelated to malignancy history, which is a markedly long documented survival for this extent of disease.
Cutaneous angiosarcoma is an aggressive yet rare malignancy without effective treatments for prolonging survival or eradicating disease. Cutaneous angiosarcoma of the head and neck has a reported 10-year survival rate of 13.8%.1 Although angiosarcoma in any location holds a bleak prognosis, cutaneous angiosarcoma of the scalp with a T2 classification has a 2-year survival rate of 0%. Moreover, even if remission is achieved, disease is highly recurrent, typically within months with the current standard of care.3,21,22
Emerging evidence for the possible role of β-adrenergic receptor blockade in the treatment of malignant vascular neoplasms is promising. Microarrays from a host of vascular growths have demonstrated expression of β-adrenergic receptors in 77% of sampled angiosarcoma specimens in addition to strong expression in infantile hemangiomas, hemangiomas, hemangioendotheliomas, and vascular malformations.19 Research findings have further verified the validity of this approach with the demonstration of b1-, b2-, and b3- adrenergic receptor expression by angiosarcoma cell lines. Propranolol subsequently was shown to effectively target proliferation of these cells and induce apoptosis in a dose-dependent manner and moreover be synergistic in effect with other chemotherapies.15 Several genes have exhibited differential expression between control tumor cells and propranolol-treated cells. Specifically, target genes including AXL (a receptor tyrosine kinase associated with cell adhesion, proliferation, and apoptosis and found to upregulated in melanoma and leukemia) and ERBB receptor feedback inhibitor 1 (receptor tyrosine kinase, with ERBB family members commonly overexpressed or mutated in the setting malignancy) have been posited as possible explanatory factors in the observed angiosarcoma response to propranolol.23
Several cases describing propranolol use as an adjunctive therapy for angiosarcoma suggest a beneficial role in clinical medicine. One case report described propranolol monotherapy for lesion to our patient, with a resultant reduction in Ki-67 as a measure of proliferative index within 1 week of initiating propranolol therapy.13 Propranolol also has been shown to halt or slow progression of metastatic disease in visceral and metastatic angiosarcomas.12-14 In combination with oral etoposide and cyclophosphamide, maintenance propranolol therapy in 7 cases of advanced cutaneous angiosarcoma resulted in 1 complete response and 3 very good partial responses, with a median progression-free survival of 11 months.11 Larger-scale studies have not been published, but the growing number of case reports and case series warrants further investigation of the utility of propranolol as an adjunct to current therapies in advanced angiosarcoma.
To the Editor:
Angiosarcoma is a malignancy of the vascular endothelium that most commonly presents on the skin.1 Patients diagnosed with cutaneous angiosarcoma, which is a rare and aggressive malignancy, have a 5-year survival rate of approximately 30%.2,3 Angiosarcoma can be seen in the setting of chronic lymphedema; radiation therapy; and sporadically in elderly patients, where it is commonly seen on the head and neck. Presentation on the head and neck has been associated with worse outcomes, with a projected overall 10-year survival rate of 13.8%; the survival rate is lower if the tumor is surgically unresectable or larger in size. Metastasis can occur via both lymphatic and hematogenous routes, with pulmonary and hepatic metastases most frequently observed.1 Prognostications of poor outcomes for patients with head and neck cutaneous angiosarcoma via a 5-year survival rate were identified in a meta-analysis and included the following: patient age older than 70 years, larger tumors, tumor location of scalp vs face, nonsurgical treatments, and lack of clear margins on histology.2
Treatment of angiosarcoma historically has encompassed both surgical resection and adjuvant radiation therapy with suboptimal success. Evidence supporting various treatment regimens remains sparse due to the low incidence of the neoplasm. Although surgical resection is the only documented curative treatment, cutaneous angiosarcomas frequently are found to have positive surgical margins and require adjuvant radiation. Use of high-dose radiation (>50 Gy) with application over a wide treatment area such as total scalp irradiation is recommended.4 Although radiation has been found to diminish local recurrence rates, it has not substantially affected rates of distant disease recurrence.1 Cytotoxic chemotherapy has clinical utility in minimizing progression, but standard regimens afford a progression-free survival of only months.3 Adjuvant treatment with paclitaxel has been shown to have improved efficacy in scalp angiosarcoma vs other visceral sites, showing a nonprogression rate of 42% at 4 months after treatment.5 More recently, targeted chemotherapeutics, including the vascular endothelial growth factor inhibitor bevacizumab and tyrosine kinase inhibitor sorafenib, have shown some survival benefit, but it is unclear if these agents are superior to traditional cytotoxic agents.4,6-10 A phase 2 study of paclitaxel administered weekly with or without bevacizumab showed similar progression-free survival and overall survival, albeit at the expense of added toxicity experienced by participants in the combined group.10
The addition of the nonselective β-adrenergic blocker propranolol to the treatment armamentarium, which was pursued due to its utility in the treatment of benign infantile hemangioma and demonstrated ability to limit the expression of adrenergic receptors in angiosarcoma, has gained clinical attention for possible augmentation of cutaneous angiosarcoma therapy.11-14 Propranolol has been shown to reduce metastasis in other neoplasms—both vascular and nonvascular—and may play a role as an adjuvant treatment to current therapies in angiosarcoma.15-20 We report a patient with cutaneous angiosarcoma (T2 classification) with disease-free survival of nearly 6 years without evidence of recurrence in the setting of continuous propranolol use supplementary to chemotherapy and radiation.
A 78-year-old man with a history of multiple basal cell carcinomas, hypertension, and remote smoking history presented to the dermatology clinic with an enlarging red-brown plaque on the scalp of 2 months’ duration. The lesion had grown rapidly to involve the forehead, right temple, preauricular region, and parietal scalp. At presentation, the tumor measured more than 20 cm in diameter at its greatest point (Figure 1). Physical examination revealed a 6-mm purple nodule within the lesion on the patient’s right parietal scalp. No clinical lymphadenopathy was appreciated at the time of diagnosis. Punch biopsies of the right parietal scalp nodule and right temple patch showed findings consistent with angiosarcoma with diffuse cytoplasmic staining of CD31 in atypical endothelial cells and no staining for human herpesvirus 8 (Figure 2). Concurrent computed tomography of the head showed thickening of the right epidermis, dermis, and deeper scalp tissues, but there was no evidence of skull involvement. Computed tomography of the thorax, abdomen, and pelvis showed no evidence of metastatic disease. After a diagnostic workup, the patient was diagnosed with T2bN0M0 angiosarcoma.
The lesion was determined to be nonresectable due to the extent of the patient’s cutaneous disease. The patient was started on a regimen of paclitaxel, scalp radiation, and oral propranolol. Propranolol 40 mg twice daily was initiated at the time of diagnosis with a plan to continue indefinitely. Starting 1 month after staging, the patient completed 10 weekly cycles of paclitaxel, and he was treated with 60 Gy of scalp radiation in 30 fractions, starting with the second cycle of paclitaxel. He tolerated both well with no reported adverse events. Repeat computed tomography performed 1 month after completion of chemotherapy and radiation showed no evidence of a mass or fluid collection in subcutaneous scalp tissues and no evidence of metastatic disease. This correlated with an observed clinical regression at 1 month and complete clinical response at 5 months with residual hemosiderin and radiation changes. The area of prior disease involvement subsequently evolved from violet to dusky gray in appearance to an eventual complete resolution 26 months after diagnosis, accompanied by atrophic radiation-induced sequelae (Figure 3).
The patient’s postchemotherapy course was complicated by hospitalization for a suspected malignant pleural effusion. Analysis revealed growing ground-glass opacities and nodules in the right lower lung lobe. A thoracentesis with cytology studies was negative for malignancy. Continued monitoring over 19 months demonstrated eventual resolution of those findings. He experienced notable complication from local radiation therapy to the scalp with chronic cutaneous ulceration refractory to wound care and surgical intervention. The patient did not exhibit additional signs or symptoms concerning for recurrence or metastasis and was followed by dermatology and oncology until he died nearly 5 years after initial diagnosis due to complications from acute hypoxic respiratory failure secondary to COVID-19. The last imaging obtained showed no convincing evidence of metastasis, though spinal imaging within a month of his death showed lesions favored to represent benign angiomatous growths. His survival after diagnosis ultimately reached 57 months without confirmed disease recurrence and cause of death unrelated to malignancy history, which is a markedly long documented survival for this extent of disease.
Cutaneous angiosarcoma is an aggressive yet rare malignancy without effective treatments for prolonging survival or eradicating disease. Cutaneous angiosarcoma of the head and neck has a reported 10-year survival rate of 13.8%.1 Although angiosarcoma in any location holds a bleak prognosis, cutaneous angiosarcoma of the scalp with a T2 classification has a 2-year survival rate of 0%. Moreover, even if remission is achieved, disease is highly recurrent, typically within months with the current standard of care.3,21,22
Emerging evidence for the possible role of β-adrenergic receptor blockade in the treatment of malignant vascular neoplasms is promising. Microarrays from a host of vascular growths have demonstrated expression of β-adrenergic receptors in 77% of sampled angiosarcoma specimens in addition to strong expression in infantile hemangiomas, hemangiomas, hemangioendotheliomas, and vascular malformations.19 Research findings have further verified the validity of this approach with the demonstration of b1-, b2-, and b3- adrenergic receptor expression by angiosarcoma cell lines. Propranolol subsequently was shown to effectively target proliferation of these cells and induce apoptosis in a dose-dependent manner and moreover be synergistic in effect with other chemotherapies.15 Several genes have exhibited differential expression between control tumor cells and propranolol-treated cells. Specifically, target genes including AXL (a receptor tyrosine kinase associated with cell adhesion, proliferation, and apoptosis and found to upregulated in melanoma and leukemia) and ERBB receptor feedback inhibitor 1 (receptor tyrosine kinase, with ERBB family members commonly overexpressed or mutated in the setting malignancy) have been posited as possible explanatory factors in the observed angiosarcoma response to propranolol.23
Several cases describing propranolol use as an adjunctive therapy for angiosarcoma suggest a beneficial role in clinical medicine. One case report described propranolol monotherapy for lesion to our patient, with a resultant reduction in Ki-67 as a measure of proliferative index within 1 week of initiating propranolol therapy.13 Propranolol also has been shown to halt or slow progression of metastatic disease in visceral and metastatic angiosarcomas.12-14 In combination with oral etoposide and cyclophosphamide, maintenance propranolol therapy in 7 cases of advanced cutaneous angiosarcoma resulted in 1 complete response and 3 very good partial responses, with a median progression-free survival of 11 months.11 Larger-scale studies have not been published, but the growing number of case reports and case series warrants further investigation of the utility of propranolol as an adjunct to current therapies in advanced angiosarcoma.
- Abraham JA, Hornicek FJ, Kaufman AM, et al. Treatment and outcome of 82 patients with angiosarcoma. Ann Surg Oncol. 2007;14:1953-1967.
- Shin JY, Roh SG, Lee NH, et al. Predisposing factors for poor prognosis of angiosarcoma of the scalp and face: systematic review and meta-analysis. Head Neck. 2017;39:380-386.
- Fury MG, Antonescu CR, Zee KJV, et al. A 14-year retrospective review of angiosarcoma: clinical characteristics, prognostic factors, and treatment outcomes with surgery and chemotherapy. Cancer. 2005;11:241-247.
- Dossett LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- Penel N, Bui BN, Bay JO, et al. Phase II trial of weekly paclitaxel for unresectable angiosarcoma: the ANGIOTAX study. J Clin Oncol. 2008;26:5269-5274.
- Agulnik M, Yarber JL, Okuno SH, et al. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann Oncol. 2013;24:257-263.
- Maki RG, D’Adamo DR, Keohan ML, et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J Clin Oncol. 2009;27:3133-3140.
- Ishida Y, Otsuka A, Kabashima K. Cutaneous angiosarcoma: update on biology and latest treatment. Curr Opin Oncol. 2018;30:107-112.
- Ray-Coquard I, Italiano A, Bompas E, et al. Sorafenib for patients with advanced angiosarcoma: a phase II trial from the French Sarcoma Group (GSF/GETO). Oncologist. 2012;17:260-266.
- Ray-Coquard IL, Domont J, Tresch-Bruneel E, et al. Paclitaxel given once per week with or without bevacizumab in patients with advanced angiosarcoma: a randomized phase II trial. J Clin Oncol. 2015;33:2797-2802.
- Pasquier E, Andre N, Street J, et al. Effective management of advanced angiosarcoma by the synergistic combination of propranolol and vinblastine-based metronomic chemotherapy: a bench to bedside study. EBioMedicine. 2016;6:87-95.
- Banavali S, Pasquier E, Andre N. Targeted therapy with propranolol and metronomic chemotherapy combination: sustained complete response of a relapsing metastatic angiosarcoma. Ecancermedicalscience. 2015;9:499.
- Chow W, Amaya CN, Rains S, et al. Growth attenuation of cutaneous angiosarcoma with propranolol-mediated beta-blockade. JAMA Dermatol. 2015;151:1226-1229.
- Daguze J, Saint-Jean M, Peuvrel L, et al. Visceral metastatic angiosarcoma treated effectively with oral cyclophosphamide combined with propranolol. JAAD Case Rep. 2016;2:497-499.
- Stiles JM, Amaya C, Rains S, et al. Targeting of beta adrenergic receptors results in therapeutic efficacy against models of hemangioendothelioma and angiosarcoma. PLoS One. 2013;8:e60021.
- Chang PY, Chung CH, Chang WC, et al. The effect of propranolol on the prognosis of hepatocellular carcinoma: a nationwide population-based study. PLoS One. 2019;14:e0216828.
- De Giorgi V, Grazzini M, Benemei S, et al. Propranolol for off-label treatment of patients with melanoma: results from a cohort study. JAMA Oncol. 2018;4:e172908.
- Rico M, Baglioni M, Bondarenko M, et al. Metformin and propranolol combination prevents cancer progression and metastasis in different breast cancer models. Oncotarget. 2017;8:2874-2889.
- Chisholm KM, Chang KW, Truong MT, et al. β-Adrenergic receptor expression in vascular tumors. Mod Pathol. 2012;25:1446-1451.
- Leaute-Labreze C, Dumas de la Roque E, Hubiche T, et al. Propranolol for severe hemangiomas of infancy. N Engl J Med. 2008;358:2649-2651.
- Maddox JC, Evans HL. Angiosarcoma of skin and soft tissue: a study of forty-four cases. Cancer. 1981;48:1907-1921.
- Morgan MB, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Zhou S, Liu P, Jiang W, et al. Identification of potential target genes associated with the effect of propranolol on angiosarcoma via microarray analysis. Oncol Lett. 2017;13:4267-4275.
- Abraham JA, Hornicek FJ, Kaufman AM, et al. Treatment and outcome of 82 patients with angiosarcoma. Ann Surg Oncol. 2007;14:1953-1967.
- Shin JY, Roh SG, Lee NH, et al. Predisposing factors for poor prognosis of angiosarcoma of the scalp and face: systematic review and meta-analysis. Head Neck. 2017;39:380-386.
- Fury MG, Antonescu CR, Zee KJV, et al. A 14-year retrospective review of angiosarcoma: clinical characteristics, prognostic factors, and treatment outcomes with surgery and chemotherapy. Cancer. 2005;11:241-247.
- Dossett LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- Penel N, Bui BN, Bay JO, et al. Phase II trial of weekly paclitaxel for unresectable angiosarcoma: the ANGIOTAX study. J Clin Oncol. 2008;26:5269-5274.
- Agulnik M, Yarber JL, Okuno SH, et al. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann Oncol. 2013;24:257-263.
- Maki RG, D’Adamo DR, Keohan ML, et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J Clin Oncol. 2009;27:3133-3140.
- Ishida Y, Otsuka A, Kabashima K. Cutaneous angiosarcoma: update on biology and latest treatment. Curr Opin Oncol. 2018;30:107-112.
- Ray-Coquard I, Italiano A, Bompas E, et al. Sorafenib for patients with advanced angiosarcoma: a phase II trial from the French Sarcoma Group (GSF/GETO). Oncologist. 2012;17:260-266.
- Ray-Coquard IL, Domont J, Tresch-Bruneel E, et al. Paclitaxel given once per week with or without bevacizumab in patients with advanced angiosarcoma: a randomized phase II trial. J Clin Oncol. 2015;33:2797-2802.
- Pasquier E, Andre N, Street J, et al. Effective management of advanced angiosarcoma by the synergistic combination of propranolol and vinblastine-based metronomic chemotherapy: a bench to bedside study. EBioMedicine. 2016;6:87-95.
- Banavali S, Pasquier E, Andre N. Targeted therapy with propranolol and metronomic chemotherapy combination: sustained complete response of a relapsing metastatic angiosarcoma. Ecancermedicalscience. 2015;9:499.
- Chow W, Amaya CN, Rains S, et al. Growth attenuation of cutaneous angiosarcoma with propranolol-mediated beta-blockade. JAMA Dermatol. 2015;151:1226-1229.
- Daguze J, Saint-Jean M, Peuvrel L, et al. Visceral metastatic angiosarcoma treated effectively with oral cyclophosphamide combined with propranolol. JAAD Case Rep. 2016;2:497-499.
- Stiles JM, Amaya C, Rains S, et al. Targeting of beta adrenergic receptors results in therapeutic efficacy against models of hemangioendothelioma and angiosarcoma. PLoS One. 2013;8:e60021.
- Chang PY, Chung CH, Chang WC, et al. The effect of propranolol on the prognosis of hepatocellular carcinoma: a nationwide population-based study. PLoS One. 2019;14:e0216828.
- De Giorgi V, Grazzini M, Benemei S, et al. Propranolol for off-label treatment of patients with melanoma: results from a cohort study. JAMA Oncol. 2018;4:e172908.
- Rico M, Baglioni M, Bondarenko M, et al. Metformin and propranolol combination prevents cancer progression and metastasis in different breast cancer models. Oncotarget. 2017;8:2874-2889.
- Chisholm KM, Chang KW, Truong MT, et al. β-Adrenergic receptor expression in vascular tumors. Mod Pathol. 2012;25:1446-1451.
- Leaute-Labreze C, Dumas de la Roque E, Hubiche T, et al. Propranolol for severe hemangiomas of infancy. N Engl J Med. 2008;358:2649-2651.
- Maddox JC, Evans HL. Angiosarcoma of skin and soft tissue: a study of forty-four cases. Cancer. 1981;48:1907-1921.
- Morgan MB, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Zhou S, Liu P, Jiang W, et al. Identification of potential target genes associated with the effect of propranolol on angiosarcoma via microarray analysis. Oncol Lett. 2017;13:4267-4275.
PRACTICE POINTS
- In one classic presentation, cutaneous angiosarcoma characteristically appears as a bruiselike patch on the head and neck of an elderly gentleman.
- Although cutaneous angiosarcoma typically portends a poor prognosis at the time of diagnosis, adjunctive oral propranolol may be a promising and relatively benign therapy, posited to afford benefit in a manner similar to its efficacy in the treatment of infantile hemangiomas.

Rapidly Growing Nodule Within a Previously Radiated Area of the Scalp
The Diagnosis: Pseudoangiomatous Squamous Cell Carcinoma
Pseudoangiomatous squamous cell carcinoma (PSCC), a variant of acantholytic squamous cell carcinoma (SCC), is a rare epithelial neoplasm that can mimic angiosarcoma.1 Clinically, PSCC presents as a white-gray ulcer or nodular pink tumor on sun-exposed areas, typically on the head and neck. Due to its increased potential for metastasis, this variant of SCC is considered particularly aggressive. Histologically, PSCC shows nests of acantholytic atypical keratinocytes arranged in anastomosing arrays that form pseudovascular or pseudoglandular structures.2 Acantholytic spaces frequently are filled with erythrocytes. Immunohistochemically, PSCC tumor cells express classic squamous markers such as cytokeratin (CK) 5 and p63 but not vascular markers such as CD31, CD34, and von Willebrand factor.3 In our patient, histopathology of the lesion revealed invasive nests, lobules, and interconnected columns of well-differentiated squamous tumor cells that emanated from the base of the epidermis. The tumor exhibited acantholysis forming ectatic and slitlike spaces, some of which contained erythrocytes. The neoplastic cells, including those lining pseudovascular spaces, positively stained for CK5 (Figure 1A) and nuclear p63 but lacked reactivity to CD31 (Figure 1B) and CD34, corroborating squamous and not vascular differentiation. Current treatment guidelines include Mohs micrographic surgery, excisional surgery, or radiation.4 Our patient’s lesion was completely removed by Mohs micrographic surgery. Three months later, there was no evidence of recurrence.
 squamous cell carcinoma.")
Angiosarcoma is an aggressive neoplasm associated with a poor prognosis and 5-year survival rate of 30% to 40%. The etiology of angiosarcoma still is unclear, but identified risk factors include prior radiation therapy, lymphedema (Stewart-Treves syndrome), and genetic predisposition.5 In the skin, angiosarcoma often occurs in the head and neck region, accounting for 60% of cutaneous cases.5,6 Early in the disease, most patients present with a bruiselike lesion on the scalp or forehead, often delaying the diagnosis.6 As the cancer progresses, tissue infiltration, edema, and hemorrhage contribute to the formation of violaceous nodules, which eventually prompt for biopsy. Angiosarcoma spans a broad histologic spectrum depending on the cytology of malignant cells (eg, spindle, small round, epithelioid) and their capacity for vasoformation. Welldifferentiated angiosarcoma shows retiform slitlike spaces in between collagen bundles that are lined by hyperchromatic hobnailing endothelial cells (Figure 2).7 Epithelioid angiosarcoma can be mistaken for SCC.8 Immunohistochemically, angiosarcoma stains positively for CD31, CD34, ETS-related gene 1, D2-40, and factor VIII.9 In our patient, the neoplasm was negative for vascular markers CD31 and CD34.

Bacillary angiomatosis (BA), caused by Bartonella henselae, is a rare disease that first was identified in HIV patients with diminished CD4+ T-cell counts. In the skin, BA often manifests as centrally ulcerated, single or clustered, reddish-purple nodules.10 Histologically, it is characterized by highly vascularized, histiocyterich infiltrates with admixed neutrophils and plasma cells (Figure 3). Capillaries often proliferate in a lobular fashion.11 Atypical cytology with areas of necrosis may mimic angiosarcoma.12 The pathognomonic feature of BA is the presence of enlarged histiocytes with pink-purplish cytoplasm corresponding to intracytoplasmic aggregates of bacteria, which can be revealed by Warthin-Starry or Grocott-Gomori methenamine-silver staining. Immunohistochemically, proliferative benign capillaries are highlighted by CD34 and CD31, and histiocytes are decorated by CD68.12 This diagnosis was excluded based on the patient’s history, clinical presentation, and positive staining for CK5 and p63.

Squamoid eccrine ductal carcinoma is an exceedingly rare subtype of eccrine carcinoma that mimics SCC both clinically and histologically.13 It most often occurs on the head and neck of elderly patients. This neoplasm can look similar to SCC and its variants, including PSCC. Histologically, squamoid eccrine ductal carcinoma exhibits a biphasic growth pattern.14 Well-differentiated squamous dysplasia transitions to carcinoma with eccrine duct formation as the tumor percolates deep into the dermis (Figure 4). As a result, superficial skin biopsies often lead to an incorrect diagnosis.15 Unlike SCC, the risk for locoregional and widespread metastasis is elevated. Identifying ducts in the deep aspect of the tumor is critical, thus immunohistochemical staining for carcinoembryonic antigen and epithelial membrane antigen is paramount for the diagnosis.15 Pseudoangiomatous SCC will stain negative for carcinoembryonic antigen, as was the case in our patient.

Pseudoepitheliomatous hyperplasia is a benign histologic reaction that can result from trauma, chronic inflammation (ie, pyoderma gangrenosum), tattoo placement, underlying neoplasia or fungal infection, or a spider bite reaction.14,15 It most commonly is seen as a well-demarcated nodule or plaque associated with scaling or crusting. Papules vary in size from less than 1 cm to several centimeters. Histologically, it is defined by an acanthotic proliferation of the adnexal epithelium and epidermis (Figure 5).16,17 Irregular strands, cords, and nests of squamoid cells can extend into the dermis.18 It can closely mimic SCC, but there are a few key differences. Pseudoepitheliomatous hyperplasia will not display atypical mitotic figures or atypical nuclei and will never invade lymphatics or vascular systems.19 Pseudoepitheliomatous hyperplasia shows identical histology to well-differentiated SCC, and thus clinicopathologic correlation and mindful histologic evaluation are crucial. The presence of an increased influx of neutrophils and histiocytes should prompt for microbial stains or deeper sectioning. A superficial biopsy should be followed by a deep biopsy. In our patient, microorganismal stains were negative.

- Kiyohara T, Miyamoto M, Shijimaya T, et al. Pseudovascular squamous cell carcinoma: a review of the published work and reassessment of prognosis. J Dermatol. 2018;45:1448-1451.
- Nagore E, Sánchez-Motilla JM, Pérez-Vallés A, et al. Pseudovascular squamous cell carcinoma of the skin. Clin Exp Dermatol. 2000;25:206-208.
- Han X, Lin X, Shao X. Pseudovascular adenoid squamous cell carcinoma of the tongue: a case report and literature review. Int J Clin Exp Pathol. 2020;13:1086-1089.
- Singh S, Bisht N, Purkayastha A, et al. Acantholytic squamous cell carcinoma of the scalp in an elderly patient treated with radical radiotherapy. J Cancer Res Pract. 2018;5:165-168.
- Cao J, Wang J, He C, et al. Angiosarcoma: a review of diagnosis and current treatment. Am J Cancer Res. 2019;9:2303-2313.
- Buehler D, Rice SR, Moody JS, et al. Angiosarcoma outcomes and prognostic factors: a 25-year single institution experience. Am J Clin Oncol. 2014;37:473-479.
- Ronen S, Ivan D, Torres-Cabala CA, et al. Post‐radiation vascular lesions of the breast. J Cutan Pathol. 2019;46:52-58.
- Shilpa K, Leelavathy B, Gorur D, et al. Early-onset epithelioid angiosarcoma: diagnostic enigma, a rare case report. Indian J Dermatopathol Diagn Dermatol. 2019;6:36-38.
- Gaballah AH, Jensen CT, Palmquist S, et al. Angiosarcoma: clinical and imaging features from head to toe [published online May 4, 2017]. Br J Radiol. 2017;90:20170039. doi:10.1259/bjr.20170039
- Hoffman CF, Papadopoulos D, Palmer DM, et al. A case report of bacillary angiomatosis in a patient infected with human immunodeficiency virus. Cutis. 2002;69:175-178.
- Biwer E, Uerlich M, Wimheuer R, et al. Bacillary angiomatosis: an important differential diagnosis in patients with HIV. Am J Dermatopathol. 1994;16:110.
- Medeiros LJ, Miranda RN. Bacillary angiomatosis. In: Medeiros LJ, Miranda RN, eds. Diagnostic Pathology: Lymph Nodes and Extranodal Lymphomas. 2nd ed. Elsevier; 2018:58-63.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Mckissack S, Wohltmann W, Dalton S, et al. Squamoid eccrine ductal carcinoma: an aggressive mimicker of squamous cell carcinoma. Am J Dermatopathol. 2019;41:140-143.
- Wollina U. Pyoderma gangrenosum—a review. Orphanet J Rare Dis. 2007;2:19
- Chow P, Goddard L, Greenway H, et al. Squamoid eccrine ductal carcinoma: the Scripps experience. Dermatol Surg. 2021;47:1115-1117.
- Zayour M, Lazova R. Pseudoepitheliomatous hyperplasia: a review. Am J Dermatopathol. 2011;33:112-122; quiz 123-126.
- Lynch JM. Understanding pseudoepitheliomatous hyperplasia. Pathol Case Rev. 2004;9:36-45.
- Goel R, Wallace ML. Pseudoepitheliomatous hyperplasia secondary to cutaneous aspergillus. Am J Dermatopathol. 2001;23:224-226.
The Diagnosis: Pseudoangiomatous Squamous Cell Carcinoma
Pseudoangiomatous squamous cell carcinoma (PSCC), a variant of acantholytic squamous cell carcinoma (SCC), is a rare epithelial neoplasm that can mimic angiosarcoma.1 Clinically, PSCC presents as a white-gray ulcer or nodular pink tumor on sun-exposed areas, typically on the head and neck. Due to its increased potential for metastasis, this variant of SCC is considered particularly aggressive. Histologically, PSCC shows nests of acantholytic atypical keratinocytes arranged in anastomosing arrays that form pseudovascular or pseudoglandular structures.2 Acantholytic spaces frequently are filled with erythrocytes. Immunohistochemically, PSCC tumor cells express classic squamous markers such as cytokeratin (CK) 5 and p63 but not vascular markers such as CD31, CD34, and von Willebrand factor.3 In our patient, histopathology of the lesion revealed invasive nests, lobules, and interconnected columns of well-differentiated squamous tumor cells that emanated from the base of the epidermis. The tumor exhibited acantholysis forming ectatic and slitlike spaces, some of which contained erythrocytes. The neoplastic cells, including those lining pseudovascular spaces, positively stained for CK5 (Figure 1A) and nuclear p63 but lacked reactivity to CD31 (Figure 1B) and CD34, corroborating squamous and not vascular differentiation. Current treatment guidelines include Mohs micrographic surgery, excisional surgery, or radiation.4 Our patient’s lesion was completely removed by Mohs micrographic surgery. Three months later, there was no evidence of recurrence.
Angiosarcoma is an aggressive neoplasm associated with a poor prognosis and 5-year survival rate of 30% to 40%. The etiology of angiosarcoma still is unclear, but identified risk factors include prior radiation therapy, lymphedema (Stewart-Treves syndrome), and genetic predisposition.5 In the skin, angiosarcoma often occurs in the head and neck region, accounting for 60% of cutaneous cases.5,6 Early in the disease, most patients present with a bruiselike lesion on the scalp or forehead, often delaying the diagnosis.6 As the cancer progresses, tissue infiltration, edema, and hemorrhage contribute to the formation of violaceous nodules, which eventually prompt for biopsy. Angiosarcoma spans a broad histologic spectrum depending on the cytology of malignant cells (eg, spindle, small round, epithelioid) and their capacity for vasoformation. Welldifferentiated angiosarcoma shows retiform slitlike spaces in between collagen bundles that are lined by hyperchromatic hobnailing endothelial cells (Figure 2).7 Epithelioid angiosarcoma can be mistaken for SCC.8 Immunohistochemically, angiosarcoma stains positively for CD31, CD34, ETS-related gene 1, D2-40, and factor VIII.9 In our patient, the neoplasm was negative for vascular markers CD31 and CD34.
Bacillary angiomatosis (BA), caused by Bartonella henselae, is a rare disease that first was identified in HIV patients with diminished CD4+ T-cell counts. In the skin, BA often manifests as centrally ulcerated, single or clustered, reddish-purple nodules.10 Histologically, it is characterized by highly vascularized, histiocyterich infiltrates with admixed neutrophils and plasma cells (Figure 3). Capillaries often proliferate in a lobular fashion.11 Atypical cytology with areas of necrosis may mimic angiosarcoma.12 The pathognomonic feature of BA is the presence of enlarged histiocytes with pink-purplish cytoplasm corresponding to intracytoplasmic aggregates of bacteria, which can be revealed by Warthin-Starry or Grocott-Gomori methenamine-silver staining. Immunohistochemically, proliferative benign capillaries are highlighted by CD34 and CD31, and histiocytes are decorated by CD68.12 This diagnosis was excluded based on the patient’s history, clinical presentation, and positive staining for CK5 and p63.
Squamoid eccrine ductal carcinoma is an exceedingly rare subtype of eccrine carcinoma that mimics SCC both clinically and histologically.13 It most often occurs on the head and neck of elderly patients. This neoplasm can look similar to SCC and its variants, including PSCC. Histologically, squamoid eccrine ductal carcinoma exhibits a biphasic growth pattern.14 Well-differentiated squamous dysplasia transitions to carcinoma with eccrine duct formation as the tumor percolates deep into the dermis (Figure 4). As a result, superficial skin biopsies often lead to an incorrect diagnosis.15 Unlike SCC, the risk for locoregional and widespread metastasis is elevated. Identifying ducts in the deep aspect of the tumor is critical, thus immunohistochemical staining for carcinoembryonic antigen and epithelial membrane antigen is paramount for the diagnosis.15 Pseudoangiomatous SCC will stain negative for carcinoembryonic antigen, as was the case in our patient.
Pseudoepitheliomatous hyperplasia is a benign histologic reaction that can result from trauma, chronic inflammation (ie, pyoderma gangrenosum), tattoo placement, underlying neoplasia or fungal infection, or a spider bite reaction.14,15 It most commonly is seen as a well-demarcated nodule or plaque associated with scaling or crusting. Papules vary in size from less than 1 cm to several centimeters. Histologically, it is defined by an acanthotic proliferation of the adnexal epithelium and epidermis (Figure 5).16,17 Irregular strands, cords, and nests of squamoid cells can extend into the dermis.18 It can closely mimic SCC, but there are a few key differences. Pseudoepitheliomatous hyperplasia will not display atypical mitotic figures or atypical nuclei and will never invade lymphatics or vascular systems.19 Pseudoepitheliomatous hyperplasia shows identical histology to well-differentiated SCC, and thus clinicopathologic correlation and mindful histologic evaluation are crucial. The presence of an increased influx of neutrophils and histiocytes should prompt for microbial stains or deeper sectioning. A superficial biopsy should be followed by a deep biopsy. In our patient, microorganismal stains were negative.
The Diagnosis: Pseudoangiomatous Squamous Cell Carcinoma
Pseudoangiomatous squamous cell carcinoma (PSCC), a variant of acantholytic squamous cell carcinoma (SCC), is a rare epithelial neoplasm that can mimic angiosarcoma.1 Clinically, PSCC presents as a white-gray ulcer or nodular pink tumor on sun-exposed areas, typically on the head and neck. Due to its increased potential for metastasis, this variant of SCC is considered particularly aggressive. Histologically, PSCC shows nests of acantholytic atypical keratinocytes arranged in anastomosing arrays that form pseudovascular or pseudoglandular structures.2 Acantholytic spaces frequently are filled with erythrocytes. Immunohistochemically, PSCC tumor cells express classic squamous markers such as cytokeratin (CK) 5 and p63 but not vascular markers such as CD31, CD34, and von Willebrand factor.3 In our patient, histopathology of the lesion revealed invasive nests, lobules, and interconnected columns of well-differentiated squamous tumor cells that emanated from the base of the epidermis. The tumor exhibited acantholysis forming ectatic and slitlike spaces, some of which contained erythrocytes. The neoplastic cells, including those lining pseudovascular spaces, positively stained for CK5 (Figure 1A) and nuclear p63 but lacked reactivity to CD31 (Figure 1B) and CD34, corroborating squamous and not vascular differentiation. Current treatment guidelines include Mohs micrographic surgery, excisional surgery, or radiation.4 Our patient’s lesion was completely removed by Mohs micrographic surgery. Three months later, there was no evidence of recurrence.
Angiosarcoma is an aggressive neoplasm associated with a poor prognosis and 5-year survival rate of 30% to 40%. The etiology of angiosarcoma still is unclear, but identified risk factors include prior radiation therapy, lymphedema (Stewart-Treves syndrome), and genetic predisposition.5 In the skin, angiosarcoma often occurs in the head and neck region, accounting for 60% of cutaneous cases.5,6 Early in the disease, most patients present with a bruiselike lesion on the scalp or forehead, often delaying the diagnosis.6 As the cancer progresses, tissue infiltration, edema, and hemorrhage contribute to the formation of violaceous nodules, which eventually prompt for biopsy. Angiosarcoma spans a broad histologic spectrum depending on the cytology of malignant cells (eg, spindle, small round, epithelioid) and their capacity for vasoformation. Welldifferentiated angiosarcoma shows retiform slitlike spaces in between collagen bundles that are lined by hyperchromatic hobnailing endothelial cells (Figure 2).7 Epithelioid angiosarcoma can be mistaken for SCC.8 Immunohistochemically, angiosarcoma stains positively for CD31, CD34, ETS-related gene 1, D2-40, and factor VIII.9 In our patient, the neoplasm was negative for vascular markers CD31 and CD34.
Bacillary angiomatosis (BA), caused by Bartonella henselae, is a rare disease that first was identified in HIV patients with diminished CD4+ T-cell counts. In the skin, BA often manifests as centrally ulcerated, single or clustered, reddish-purple nodules.10 Histologically, it is characterized by highly vascularized, histiocyterich infiltrates with admixed neutrophils and plasma cells (Figure 3). Capillaries often proliferate in a lobular fashion.11 Atypical cytology with areas of necrosis may mimic angiosarcoma.12 The pathognomonic feature of BA is the presence of enlarged histiocytes with pink-purplish cytoplasm corresponding to intracytoplasmic aggregates of bacteria, which can be revealed by Warthin-Starry or Grocott-Gomori methenamine-silver staining. Immunohistochemically, proliferative benign capillaries are highlighted by CD34 and CD31, and histiocytes are decorated by CD68.12 This diagnosis was excluded based on the patient’s history, clinical presentation, and positive staining for CK5 and p63.
Squamoid eccrine ductal carcinoma is an exceedingly rare subtype of eccrine carcinoma that mimics SCC both clinically and histologically.13 It most often occurs on the head and neck of elderly patients. This neoplasm can look similar to SCC and its variants, including PSCC. Histologically, squamoid eccrine ductal carcinoma exhibits a biphasic growth pattern.14 Well-differentiated squamous dysplasia transitions to carcinoma with eccrine duct formation as the tumor percolates deep into the dermis (Figure 4). As a result, superficial skin biopsies often lead to an incorrect diagnosis.15 Unlike SCC, the risk for locoregional and widespread metastasis is elevated. Identifying ducts in the deep aspect of the tumor is critical, thus immunohistochemical staining for carcinoembryonic antigen and epithelial membrane antigen is paramount for the diagnosis.15 Pseudoangiomatous SCC will stain negative for carcinoembryonic antigen, as was the case in our patient.
Pseudoepitheliomatous hyperplasia is a benign histologic reaction that can result from trauma, chronic inflammation (ie, pyoderma gangrenosum), tattoo placement, underlying neoplasia or fungal infection, or a spider bite reaction.14,15 It most commonly is seen as a well-demarcated nodule or plaque associated with scaling or crusting. Papules vary in size from less than 1 cm to several centimeters. Histologically, it is defined by an acanthotic proliferation of the adnexal epithelium and epidermis (Figure 5).16,17 Irregular strands, cords, and nests of squamoid cells can extend into the dermis.18 It can closely mimic SCC, but there are a few key differences. Pseudoepitheliomatous hyperplasia will not display atypical mitotic figures or atypical nuclei and will never invade lymphatics or vascular systems.19 Pseudoepitheliomatous hyperplasia shows identical histology to well-differentiated SCC, and thus clinicopathologic correlation and mindful histologic evaluation are crucial. The presence of an increased influx of neutrophils and histiocytes should prompt for microbial stains or deeper sectioning. A superficial biopsy should be followed by a deep biopsy. In our patient, microorganismal stains were negative.
- Kiyohara T, Miyamoto M, Shijimaya T, et al. Pseudovascular squamous cell carcinoma: a review of the published work and reassessment of prognosis. J Dermatol. 2018;45:1448-1451.
- Nagore E, Sánchez-Motilla JM, Pérez-Vallés A, et al. Pseudovascular squamous cell carcinoma of the skin. Clin Exp Dermatol. 2000;25:206-208.
- Han X, Lin X, Shao X. Pseudovascular adenoid squamous cell carcinoma of the tongue: a case report and literature review. Int J Clin Exp Pathol. 2020;13:1086-1089.
- Singh S, Bisht N, Purkayastha A, et al. Acantholytic squamous cell carcinoma of the scalp in an elderly patient treated with radical radiotherapy. J Cancer Res Pract. 2018;5:165-168.
- Cao J, Wang J, He C, et al. Angiosarcoma: a review of diagnosis and current treatment. Am J Cancer Res. 2019;9:2303-2313.
- Buehler D, Rice SR, Moody JS, et al. Angiosarcoma outcomes and prognostic factors: a 25-year single institution experience. Am J Clin Oncol. 2014;37:473-479.
- Ronen S, Ivan D, Torres-Cabala CA, et al. Post‐radiation vascular lesions of the breast. J Cutan Pathol. 2019;46:52-58.
- Shilpa K, Leelavathy B, Gorur D, et al. Early-onset epithelioid angiosarcoma: diagnostic enigma, a rare case report. Indian J Dermatopathol Diagn Dermatol. 2019;6:36-38.
- Gaballah AH, Jensen CT, Palmquist S, et al. Angiosarcoma: clinical and imaging features from head to toe [published online May 4, 2017]. Br J Radiol. 2017;90:20170039. doi:10.1259/bjr.20170039
- Hoffman CF, Papadopoulos D, Palmer DM, et al. A case report of bacillary angiomatosis in a patient infected with human immunodeficiency virus. Cutis. 2002;69:175-178.
- Biwer E, Uerlich M, Wimheuer R, et al. Bacillary angiomatosis: an important differential diagnosis in patients with HIV. Am J Dermatopathol. 1994;16:110.
- Medeiros LJ, Miranda RN. Bacillary angiomatosis. In: Medeiros LJ, Miranda RN, eds. Diagnostic Pathology: Lymph Nodes and Extranodal Lymphomas. 2nd ed. Elsevier; 2018:58-63.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Mckissack S, Wohltmann W, Dalton S, et al. Squamoid eccrine ductal carcinoma: an aggressive mimicker of squamous cell carcinoma. Am J Dermatopathol. 2019;41:140-143.
- Wollina U. Pyoderma gangrenosum—a review. Orphanet J Rare Dis. 2007;2:19
- Chow P, Goddard L, Greenway H, et al. Squamoid eccrine ductal carcinoma: the Scripps experience. Dermatol Surg. 2021;47:1115-1117.
- Zayour M, Lazova R. Pseudoepitheliomatous hyperplasia: a review. Am J Dermatopathol. 2011;33:112-122; quiz 123-126.
- Lynch JM. Understanding pseudoepitheliomatous hyperplasia. Pathol Case Rev. 2004;9:36-45.
- Goel R, Wallace ML. Pseudoepitheliomatous hyperplasia secondary to cutaneous aspergillus. Am J Dermatopathol. 2001;23:224-226.
- Kiyohara T, Miyamoto M, Shijimaya T, et al. Pseudovascular squamous cell carcinoma: a review of the published work and reassessment of prognosis. J Dermatol. 2018;45:1448-1451.
- Nagore E, Sánchez-Motilla JM, Pérez-Vallés A, et al. Pseudovascular squamous cell carcinoma of the skin. Clin Exp Dermatol. 2000;25:206-208.
- Han X, Lin X, Shao X. Pseudovascular adenoid squamous cell carcinoma of the tongue: a case report and literature review. Int J Clin Exp Pathol. 2020;13:1086-1089.
- Singh S, Bisht N, Purkayastha A, et al. Acantholytic squamous cell carcinoma of the scalp in an elderly patient treated with radical radiotherapy. J Cancer Res Pract. 2018;5:165-168.
- Cao J, Wang J, He C, et al. Angiosarcoma: a review of diagnosis and current treatment. Am J Cancer Res. 2019;9:2303-2313.
- Buehler D, Rice SR, Moody JS, et al. Angiosarcoma outcomes and prognostic factors: a 25-year single institution experience. Am J Clin Oncol. 2014;37:473-479.
- Ronen S, Ivan D, Torres-Cabala CA, et al. Post‐radiation vascular lesions of the breast. J Cutan Pathol. 2019;46:52-58.
- Shilpa K, Leelavathy B, Gorur D, et al. Early-onset epithelioid angiosarcoma: diagnostic enigma, a rare case report. Indian J Dermatopathol Diagn Dermatol. 2019;6:36-38.
- Gaballah AH, Jensen CT, Palmquist S, et al. Angiosarcoma: clinical and imaging features from head to toe [published online May 4, 2017]. Br J Radiol. 2017;90:20170039. doi:10.1259/bjr.20170039
- Hoffman CF, Papadopoulos D, Palmer DM, et al. A case report of bacillary angiomatosis in a patient infected with human immunodeficiency virus. Cutis. 2002;69:175-178.
- Biwer E, Uerlich M, Wimheuer R, et al. Bacillary angiomatosis: an important differential diagnosis in patients with HIV. Am J Dermatopathol. 1994;16:110.
- Medeiros LJ, Miranda RN. Bacillary angiomatosis. In: Medeiros LJ, Miranda RN, eds. Diagnostic Pathology: Lymph Nodes and Extranodal Lymphomas. 2nd ed. Elsevier; 2018:58-63.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Mckissack S, Wohltmann W, Dalton S, et al. Squamoid eccrine ductal carcinoma: an aggressive mimicker of squamous cell carcinoma. Am J Dermatopathol. 2019;41:140-143.
- Wollina U. Pyoderma gangrenosum—a review. Orphanet J Rare Dis. 2007;2:19
- Chow P, Goddard L, Greenway H, et al. Squamoid eccrine ductal carcinoma: the Scripps experience. Dermatol Surg. 2021;47:1115-1117.
- Zayour M, Lazova R. Pseudoepitheliomatous hyperplasia: a review. Am J Dermatopathol. 2011;33:112-122; quiz 123-126.
- Lynch JM. Understanding pseudoepitheliomatous hyperplasia. Pathol Case Rev. 2004;9:36-45.
- Goel R, Wallace ML. Pseudoepitheliomatous hyperplasia secondary to cutaneous aspergillus. Am J Dermatopathol. 2001;23:224-226.
An 84-year-old man with a history of nonmelanoma skin cancer presented to our clinic with a 1.6×1.5-cm exophytic lesion on the left posterior parietal scalp. The lesion nearly doubled in size over the last 4 months. The patient received radiation therapy in this area for the treatment of basal cell carcinoma 7 years prior to presentation. A shave biopsy was performed.


The Role of Dietary Antioxidants in Melanoma and Nonmelanoma Skin Cancer
Nonmelanoma skin cancer (NMSC) is the most common cancer in the United States, and cutaneous melanoma is projected to be the fifth most common form of cancer in 2022, with increasing incidence and high potential for mortality.1-3 Estimates indicate that 35% to 45% of all cancers in White patients are cutaneous, with 4% to 5% occurring in Hispanic patients, 2% to 4% in Asian patients, and 1% to 2% in Black patients.4 Of the keratinocyte carcinomas, basal cell carcinoma (BCC) is the most prevalent, projected to affect approximately 33% to 39% of White males and 23% to 28% of White females in the United States during their lifetimes. Squamous cell carcinoma (SCC) is the second most common skin malignancy, with a lifetime risk of 9% to 14% for White males and 4% to 9% for White females in the United States.5 The incidence of melanoma continues to increase, with approximately 99,780 new cases expected in the United States in 2022.1
UV-induced DNA damage plays a key role in the pathogenesis and development of various skin malignancies.6 UV radiation from sunlight or tanning devices causes photocarcinogenesis due to molecular and cellular effects, including the generation of reactive oxygen species, DNA damage due to the formation of cyclobutane pyrimidine dimers and pyrimidine-pyrimidone, melanogenesis, apoptosis, and the increased expression of harmful genes and proteins.6 The summation of this damage can result in skin malignancies, including NMSC and melanoma.6,7 Dietary antioxidants theoretically help prevent oxidative reactions from occurring within the body, and it has been suggested that intake of dietary antioxidants may decrease DNA damage and prevent tumorigenesis secondary to UV radiation.8 Antioxidants exist naturally in the body but can be acquired exogenously. Investigators have studied dietary antioxidants in preventing skin cancer formation with promising results in the laboratory setting.8-11 Recently, more robust human studies have been initiated to further delineate this relationship. We present clinical evidence of several frequently utilized antioxidant vitamins and their effects on melanoma and NMSC.
Antioxidants
Vitamin A—Vitamin A is a fat-soluble vitamin found in animal sources, including fish, liver, and eggs. Carotenoids, such as beta carotene, are provitamin A plant derivatives found in fruits and vegetables that are converted into biologically active retinol and retinoic acid.12 Retinols play a key role in cellular growth and differentiation and are thought to be protective against skin cancer via the inactivation of free radicals and immunologic enhancement due to their antiproliferative, antioxidative, and antiapoptotic effects.13-16 Animal studies have demonstrated this protective effect and the ability of retinoids to suppress carcinogenesis; however, human studies reveal conflicting results.17,18
Greenberg et al19 investigated the use of beta carotene in preventing the formation of NMSC. Patients (N=1805) were randomized to receive 50 mg of beta carotene daily or placebo. Over a 5-year period, there was no significant reduction in the occurrence of NMSC (relative risk [RR], 1.05; 95% CI, 0.91-1.22).19 Frieling et al20 conducted a similar randomized, double-blind, placebo-controlled trial investigating beta carotene for primary prevention of NMSC in 22,071 healthy male physicians. The study group received 50 mg of beta carotene every other day for 12 years’ duration, and there was no significant effect on the incidence of first NMSC development (RR, 0.98; 95% CI, 0.92-1.05).20
A case-control study by Naldi et al21 found an inverse association between vitamin A intake and development of melanoma. Study participants were stratified into quartiles based on level of dietary intake and found an odds ratio (OR) of 0.71 for beta carotene (95% CI, 0.50-1.02), 0.57 for retinol (95% CI, 0.39-0.83), and 0.51 for total vitamin A (95% CI, 0.35-0.75) when comparing the upper quartile of vitamin A intake to the lower quartile. Upper-quartile cutoff values of vitamin A intake were 214 µg/d for beta carotene, 149 µg/d for retinol, and 359 µg/d for total vitamin A.21 More recently, a meta-analysis by Zhang et al22 pooled data from 8 case-control studies and 2 prospective studies. Intake of retinol but not total vitamin A or beta carotene was associated with a reduced risk for development of melanoma (retinol: OR, 0.80; 95% CI, 0.69-0.92; total vitamin A: OR, 0.86; 95% CI, 0.59-1.25; beta carotene: OR, 0.87; 95% CI, 0.62-1.20).22 Feskanich et al23 demonstrated similar findings with use of food-frequency questionnaires in White women, suggesting that retinol intake from food combined with supplements may be protective for women who were otherwise at a low risk for melanoma based on nondietary factors. These factors included painful or blistering sunburns during childhood, history of more than 6 sunburns, more than 3 moles on the left arm, having red or blonde hair, and having a parent or sibling with melanoma (P=.01). However, this relationship did not hold true when looking at women at an intermediate or high risk for melanoma (P=.16 and P=.46).23
When looking at high-risk patients, such as transplant patients, oral retinoids have been beneficial in preventing NMSC.24-27 Bavinck et al24 investigated 44 renal transplant patients with a history of more than 10 NMSCs treated with 30 mg of acitretin daily vs placebo. Patients receiving oral retinoid supplementation developed fewer NMSCs over a 6-month treatment period (P=.01).24 Similarly, George et al25 investigated acitretin in renal transplant patients and found a statistically significant decrease in number of SCCs in patients on supplementation (P=.002). Solomon-Cohen et al26 performed a retrospective case-crossover study in solid organ transplant recipients and found that those treated with 10 mg of acitretin daily for 2 years had a significant reduction in the number of new keratinocyte carcinomas (P=.002). Other investigators have demonstrated similar results, and in 2006, Otley et al27 proposed standardized dosing of acitretin for chemoprevention in high-risk patients, including patients developing 5 to 10 NMSCs per year, solid organ transplant recipients, and those with syndromes associated with the development of NMSC.28,29 Overall, in the general population, vitamin A and related compounds have not demonstrated a significant association with decreased development of NMSC; however, oral retinoids have proven useful for high-risk patients. Furthermore, several studies have suggested a negative association between vitamin A levels and the incidence of melanoma, specifically in the retinol formulation.
Vitamin B3—Nicotinamide (also known as niacinamide) is a water-soluble form of vitamin B3 and is obtained from animal-based and plant-based foods, such as meat, fish, and legumes.30 Nicotinamide plays a key role in cellular metabolism, cellular signaling, and DNA repair, including protection from UV damage within keratinocytes.31,32 Early mouse models demonstrated decreased formation of skin tumors in mice treated with topical or oral nicotinamide.32,33 A number of human studies have revealed similar results.34-36
Chen et al34 conducted the ONTRAC study, a phase 3, double-blind, randomized controlled trial (RCT) looking at 386 participants with a history of at least 2 NMSCs in the preceding 5 years. At 12 months, those treated with 500 mg of nicotinamide twice daily demonstrated a statistically significant decreased rate of SCC formation (P=.05). A decreased incidence of BCC development was noted; however, this trend did not reach statistical significance (P=.12). Precancerous skin lesions also were found to be decreased in the treatment group, with 20% lower incidence of actinic keratoses (AKs) after 9 months of treatment (P<.001).34 Drago et al35 specifically studied the incidence of AKs in 38 transplant recipients—8 liver and 30 kidney—and found that previously noted AKs had decreased in size for 18 of 19 patients taking 500 mg of nicotinamide daily when originally photographed AKs were remeasured at 6-month follow-up, with 7 of these 18 patients demonstrating complete clinical regression. Of those on nicotinamide supplementation, no new AKs developed compared to the control group, which demonstrated increased size of AKs or development of new AKs in 91% of patients, with 7 AKs progressing into SCC.35
Nicotinamide has been demonstrated to be useful in preventing skin cancer in high-risk populations, such as transplant patients or those with a high incidence of NMSC.34,36 Despite promising results within the laboratory setting, nicotinamide’s effects on melanoma in humans remains less clear.31,37 Studies suggest that nicotinamide enhances tumor-infiltrating lymphocytes and DNA repair mechanisms in melanocytes, which may translate into nicotinamide, providing chemoprevention for melanoma, but research in human patients is limited.31,37
Vitamin B9—Folate, the natural form of vitamin B9, is a water-soluble compound that is found in many foods, especially green leafy vegetables, and often is supplemented because of its health benefits.38,39 In the skin, folic acid plays a key role in cellular replication and proliferation.38 Controversy exists regarding folate’s effects on cellular growth and turnover with respect to cancer incidence.38,40 Donnenfeld et al41 conducted a prospective study assessing dietary folic acid intake and development of NMSC. A total of 5880 participants completed dietary records throughout the first 2 years of the study. After an average follow-up period of 12.6 years, there was an overall increased incidence of skin cancer in those with increased dietary folate (P=.03). Furthermore, when striating by skin cancer type, there was an increased incidence of NMSC overall as well as BCC when analyzing by type of NMSC (P=.03 for NMSC; P=.05 for BCC). However, when stratifying by gender, these findings only held true for women.41 Similar effects were observed by Fung et al,42 who prospectively studied the intake of various vitamins in relationship to the development of BCC in women. During 12 years of follow-up, a positive association was observed between folate intake and BCC development (OR, 1.2; 95% CI, 1.10-1.31).42 Fung et al43 also investigated the role of several vitamins in the development of SCC and found that folate showed a negative association, which did not reach statistical significance (RR, 0.79; 95% CI, 0.56-1.11). Furthermore, Vollset et al40 conducted a meta-analysis comparing folic acid to placebo in the incidence of various types of cancer. The study excluded NMSC but reported no significant association between the development of melanoma and folic acid supplementation.40 In summary, the effects of folate have diverse consequences, potentially promoting the formation of NMSC, but studies suggest that an individual’s gender and other genetic and environmental factors also may play a role.
Vitamin C—Vitamin C (also known as ascorbic acid) is a water-soluble vitamin with antioxidant immune-mediating effects. It is found in various fruits and vegetables and serves as a cofactor for enzymes within the body playing a key role in immune function and collagen formation.44,45 It has been postulated that ascorbic acid can provide protection from UV radiation damage via its intracellular activity but conversely can contribute to oxidative damage.44 Multiple in vitro laboratory studies and animal models have demonstrated photoprotective effects of ascorbic acid.46-48 Despite these findings, minimal photoprotective effects have been found in the human population.
Kune et al49 performed a case-control study of 88 males with previously diagnosed NMSC undergoing surgical removal and investigated patients’ prior dietary habits. Patients with NMSC had a statistically significantly lower level of vitamin C–containing food in their diet than those without NMSC (P=.004).49 In addition, Vural et al50 analyzed plasma samples and blood cells of patients with AK and BCC and found a significant decrease in ascorbic acid levels in both the AK (P<.001) and BCC (P<.001) groups compared with controls. However, studies have found that consumption of certain dietary compounds can rapidly increase plasma concentration levels, which may serve as a major confounding variable in this study. Plasma concentrations of ascorbic acid and beta carotene were found to be significantly increased following consumption of a high-antioxidant diet for as short a duration as 2 weeks (P<.05).51 More recently, Heinen et al52 performed a prospective study on 1001 adults. In patients without a history of skin cancer, they found that vitamin C from food sources plus dietary supplements was positively associated with the development of BCC (P=.03).52 Similarly, Fung et al42 performed a study in women and found a positive association between vitamin C intake and the development of BCC (OR, 1.13; 95% CI, 1.03-1.23).
The relationship between vitamin C intake—either in dietary or supplemental form—and melanoma remains controversial. Mice-based studies found that high concentrations of orally administered vitamin C induce cytotoxicity in melanoma cell lines, but at low concentrations they promote tumor growth of malignant melanoma.53 Feskanich et al23 examined the relationship between vitamin C intake and melanoma development via food frequency questionnaires in White women and found that vitamin C was associated with a higher risk for melanoma (P=.05), and furthermore, a positive dose response with frequency of orange juice intake was observed (P=.008). Overall, despite promising laboratory studies, there is a lack of RCTs investigating the use of vitamin C supplementation for prevention of NMSC and melanoma in humans, and the oral benefits of vitamin C for chemoprevention remain unclear.
Vitamin D—Vitamin D is a fat-soluble vitamin that is found in fish, liver, egg, and cheese, and is endogenously produced when UV radiation from sun exposure interacts with the skin, triggering the synthesis of vitamin D.54 Vitamin D is biologically inactive and must be converted to its active form 1,25-dihydroxyvitamin D after entering the body. Vitamin D modulates many genes involved in cellular proliferation and differentiation.54 Vitamin D receptors are expressed on keratinocytes and melanocytes.55 Animal studies have demonstrated a potentially protective effect of vitamin D in the development of NMSC.56 In a mouse model, Ellison et al56 found that mice without vitamin D receptors developed skin tumors more rapidly than those with vitamin D receptors.
Unfortunately, these findings have not been demonstrated in humans, and studies have even reported an increased risk for development of NMSC in patients with normal or increased vitamin D levels compared with those with low levels of vitamin D.57-60 Eide et al57 studied 3223 patients seeking advice for low bone density by recording their vitamin D levels at the time of presentation and monitoring development of NMSC. Vitamin D levels greater than 15 ng/mL were positively associated with the development of NMSC (OR, 1.7; 95% CI, 1.04-2.7). This association held true for both SCC and BCC, with a higher risk estimated for SCC (OR, 3.2; 95% CI, 0.4-24.0 for SCC; OR, 1.7; 95% CI, 0.5-5.8 for BCC).57 An increased vitamin D serum level also was found to be significantly associated with a higher risk for BCC and melanoma by van der Pols et al.58 This prospective study looked at the incidence of skin cancer over 11 years. Study participants with vitamin D levels over 75 nmol/L more frequently developed BCC (P=.01) and melanoma (P=.05). In contrast, SCC was less frequently observed in participants with these high levels of vitamin D (P=.07).58 Furthermore, Park et al60 looked at vitamin D and skin cancer risk for men and women in the United States and found no association with risk for SCC or melanoma but a positive association with BCC (P=.05 for total vitamin D; P<.01 for dietary vitamin D). Additional studies have been performed with inconsistent results, and multiple authors suggest the possible confounding relationship between vitamin D levels and UV radiation exposure.59-62 Furthermore, some studies have even demonstrated a negative association between vitamin D and NMSC. Tang et al63 performed a retrospective case-control study in elderly males, investigating serum levels of vitamin D and patients’ self-reported history of NMSC, which demonstrated that higher levels of vitamin D were associated with a decreased risk for NMSC. Overall, the relationship between vitamin D and skin cancer development remains unclear for both melanoma and NMSC.
Vitamin E—Vitamin E is a fat-soluble vitamin that is found in plant-based oils, nuts, seeds, fruits, and vegetables.64 It works as an antioxidant to protect against free radicals and heighten immune function, and it also serves as a pro-oxidant.65,66 Vitamin E naturally exists in 8 chemical forms, of which gamma-tocopherol is the most frequently obtained form in the diet, and alpha-tocopherol is the most abundant form found in the body.64,65
Early animal studies demonstrated the inhibition of UV-induced damage in mice receiving vitamin E supplementation.67,68 Human studies have not consistently shown these effects. Vural et al50 investigated plasma samples and blood cells of patients with AKs and BCCs and reported a significant decrease in alpha-tocopherol levels in both the AK (P<.05) and BCC (P<.001) groups compared with controls. However, studies also have demonstrated a positive association between vitamin E intake and the development of BCC, including one by Fung et al,42 which found a significant association in women (OR, 1.15; 95% CI, 1.06-1.26).
Vitamin E has been found to inhibit melanin synthesis in the laboratory, suggesting a potentially protective effect in melanoma.69,70 However, in the study performed by Feskanich et al23 examining vitamin intake and melanoma incidence via food-frequency questionnaires, vitamin E was not associated with a lower risk for melanoma. Despite promising laboratory studies, the data surrounding the use of a vitamin E supplement for prevention of melanoma and NMSC in humans remains unclear.
Selenium—Selenium is a trace mineral found in plants, meat, and fish. It plays a key role in reproduction, hormone metabolism, DNA synthesis, and protection from oxidative damage.71 In mice studies, lack of selenium-containing proteins resulted in skin abnormalities, including the development of a hyperplastic epidermis and aberrant hair follicle morphogenesis with alopecia after birth, and numerous experimental studies have demonstrated a negative association between selenium intake and cancer.72,73 However, human studies have yielded alternative results.
The Nutritional Prevention of Cancer Study Group analyzed 1312 dermatology patients with a history of NMSC.74 The study population was obtained from 7 dermatology clinics with randomization to control for confounding variables. Study participants received either 200 μg of selenium daily or placebo.74 Baseline characteristics of each study group were overall balanced. Selenium intake was found to have no effect on the development of BCC (hazard ratio [HR], 1.09; 95% CI, 0.94-1.26) but an increased risk for developing SCC (HR, 1.25; 95% CI, 1.03-1.51) and total NMSC (HR, 1.17; 95% CI, 1.02-1.34).74,75 Similarly, Reid et al76 performed an RCT comparing patients treated with 400 μg/d of selenium to those treated with 200 μg/d of selenium. When compared with placebo, those treated with 200 μg/d of selenium had a statistically significantly increased incidence of NMSC (P=.006); however, those treated with 400 μg/d of selenium had no significant change in total incidence of NMSC (P=.51).76 Furthermore, Vinceti et al77 performed a review of 83 studies from the literature investigating the effect of dietary selenium, and from the RCTs, there was no beneficial effect of selenium in reducing cancer risk in general; however, some studies demonstrated an increased incidence of other types of cancer, including melanoma. Of the RCTs included in the study investigating NMSC incidence specifically, it was found that the incidence was not affected by selenium administration (RR, 1.16; 95% CI, 0.30-4.42; 2 studies, 2027 participants).77 Despite data from several studies demonstrating an increased risk for NMSC, the effects of selenium on the risk for NMSC and melanoma remain unclear.
Combination Antioxidant Studies
In addition to investigating the use of single antioxidants in skin cancer prevention, studies utilizing the combination of various antioxidants or other dietary minerals have been conducted. Hercberg et al78 performed a randomized, double-blinded, placebo-controlled trial of 13,017 adults (7876 women and 5141 men) receiving a combination of 120 mg vitamin C, 30 mg vitamin E, 100 μg selenium, 6 mg beta carotene, and 20 mg zinc. Study participants were followed for an average of 7.5 years, and the development of skin cancers were recorded. Overall, the incidence rate of skin cancer did not differ between the 2 treatment groups; however, when segregated by gender, the study found that there was an increased risk for developing skin cancer in women taking the antioxidant supplement combination compared with placebo (P=.03). This difference was not observed in the 2 treatment groups of male patients (P=.11). When looking specifically at NMSC, there was no difference between treatment groups for male or female patients (P=.39 for males; P=.15 for females). In contrast, there was a higher incidence of melanoma identified in female patients taking the combination antioxidant supplement (P=.01), but this was not seen within the male study population (P=.51).78 In addition, Chang et al79 performed a meta-analysis of 10 previously published RCTs. Analysis revealed that treatment with a variety of supplements, including vitamins A, C, E, and beta carotene, were found to have no preventative effects on the incidence of skin cancer development (RR, 0.98; CI, 0.98-1.03). Notable limitations to this study included the variability in protocols of the studies included in this meta-analysis, the limited number of RCTs investigating vitamin supplementation and the risk for skin cancer development, and the influence of dietary intake on study outcomes.79
Other Dietary Agents
Furocoumarins—Furocoumarins are botanical substances found in various fruits and plants, including many citrus products. Furocoumarins are activated by UV light radiation and can lead to development of a phototoxic eruption. Several studies have suggested a pharmacogenetic effect of furocoumarins.80 Sun et al80 collected dietary data from 47,453 men and 75,291 women on furocoumarin intake and correlation with the development of NMSC. Overall, the study suggested that the intake of furocoumarins may lead to an increase in the development of BCC (HR, 1.16; 95% CI, 1.11-1.21; P=.002); however, there was no significant association identified between total intake of furocoumarins in the risk for SCC or melanoma.80 Furthermore, Sakaki et al81 conducted a survey study looking at the consumption of citrus products and the development of NMSC. The group found that there was an increased risk for NMSC in those consuming an increased amount of citrus products (P=.007).81
Conclusion
Dietary antioxidants have been investigated for their potential role in the prevention of tumorigenesis. Specific antioxidant vitamins, such as vitamin A derivatives and niacinamide, have demonstrated clinical utility in the prevention of NMSC in high-risk populations. Retinol also has been associated with a reduced incidence of melanoma. Numerous antioxidants have demonstrated promising data within the laboratory setting; however, inconsistent results have been appreciated in humans. Furthermore, several research studies suggest that folate, vitamin D, and furocoumarins may be associated with an increased risk for skin cancer development; however, these studies are inconclusive, and dietary studies are challenging to conduct. Overall, RCTs investigating the role of antioxidants for chemoprevention are limited. Moreover, the study of dietary antioxidants and vitamins may be affected by various confounding variables that can be difficult to account for because of patients’ potentially poor recall of dietary intake and the effect of dietary intake in supplemental studies. Given the increasing prevalence of skin cancer worldwide, further research into the clinical utility of antioxidants in skin cancer prevention is warranted.
- Siegel RL, Miller KD, Fuchs HE, et al. Cancer statistics, 2022. CA Cancer J Clin. 2022;72:7-33.
- Global Burden of Disease Cancer Collaboration; Fitzmaurice C, Abate D, Abbasi N, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 1990 to 2017: a systematic analysis for the Global Burden of Disease Study. JAMA Oncol. 2019;5:1749-1768.
- Leiter U, Keim U, Garbe C. Epidemiology of skin cancer: update 2019. In: Reichrath J, ed. Sunlight, Vitamin D and Skin Cancer. Springer International Publishing; 2020:123-139.
- Bradford PT. Skin cancer in skin of color. Dermatol Nurs. 2009;21:170-177, 206; quiz 178.
- Miller DL, Weinstock MA. Nonmelanoma skin cancer in the United States: incidence. J Am Acad Dermatol. 1994;30:774-778.
- Young AR, Claveau J, Rossi AB. Ultraviolet radiation and the skin: photobiology and sunscreen photoprotection. J Am Acad Dermatol. 2017;76(3S1):S100-S109.
- Pleasance ED, Cheetham RK, Stephens PJ, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191-196.
- Baek J, Lee MG. Oxidative stress and antioxidant strategies in dermatology. Redox Rep. 2016;21:164-169.
- Katta R, Brown DN. Diet and skin cancer: the potential role of dietary antioxidants in nonmelanoma skin cancer prevention. J Skin Cancer. 2015;2015:893149.
- Stoj V, Shahriari N, Shao K, et al. Nutrition and nonmelanoma skin cancers. Clin Dermatol. 2022;40:173-185.
- O’Connor EA, Evans CV, Ivlev I, et al. Vitamin and mineral supplements for the primary prevention of cardiovascular disease and cancer: updated evidence report and systematic review for the US Preventive Services Task Force. JAMA. 2022;327:2334-2347.
- National Institutes of Health Office of Dietary Supplements. Vitamin A and carotenoids. fact sheet for health professionals. Updated June 15, 2022. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/VitaminA-HealthProfessional/
- Keller KL, Fenske NA. Uses of vitamins A, C, and E and related compounds in dermatology: a review. J Am Acad Dermatol. 1998;39:611-625.
- Wright TI, Spencer JM, Flowers FP. Chemoprevention of nonmelanoma skin cancer. J Am Acad Dermatol. 2006;54:933-946; quiz 947-950.
- Bushue N, Wan YJY. Retinoid pathway and cancer therapeutics. Adv Drug Deliv Rev. 2010;62:1285-1298.
- Stahl W, Sies H. β-Carotene and other carotenoids in protection from sunlight. Am J Clin Nutr. 2012;96:1179S-1184S.
- Bukhari MH, Qureshi SS, Niazi S, et al. Chemotherapeutic/chemopreventive role of retinoids in chemically induced skin carcinogenesis in albino mice. Int J Dermatol. 2007;46:1160-1165.
- Lambert LA, Wamer WG, Wei RR, et al. The protective but nonsynergistic effect of dietary beta-carotene and vitamin E on skin tumorigenesis in Skh mice. Nutr Cancer. 1994;21:1-12.
- Greenberg ER, Baron JA, Stukel TA, et al. A clinical trial of beta carotene to prevent basal-cell and squamous-cell cancers of the skin. The Skin Cancer Prevention Study Group. N Engl J Med. 1990;323:789-795.
- Frieling UM, Schaumberg DA, Kupper TS, et al. A randomized, 12-year primary-prevention trial of beta carotene supplementation for nonmelanoma skin cancer in the physician’s health study. Arch Dermatol. 2000;136:179-184.
- Naldi L, Gallus S, Tavani A, et al; Oncology Study Group of the Italian Group for Epidemiologic Research in Dermatology. Risk of melanoma and vitamin A, coffee and alcohol: a case-control study from Italy. Eur J Cancer Prev. 2004;13:503-508.
- Zhang YP, Chu RX, Liu H. Vitamin A intake and risk of melanoma: a meta-analysis. PloS One. 2014;9:e102527.
- Feskanich D, Willett WC, Hunter DJ, et al. Dietary intakes of vitamins A, C, and E and risk of melanoma in two cohorts of women. Br J Cancer. 2003;88:1381-1387.
- Bavinck JN, Tieben LM, Van der Woude FJ, et al. Prevention of skin cancer and reduction of keratotic skin lesions during acitretin therapy in renal transplant recipients: a double-blind, placebo-controlled study. J Clin Oncol. 1995;13:1933-1938.
- George R, Weightman W, Russ GR, et al. Acitretin for chemoprevention of non-melanoma skin cancers in renal transplant recipients. Australas J Dermatol. 2002;43:269-273.
- Solomon-Cohen E, Reiss-Huss S, Hodak E, et al. Low-dose acitretin for secondary prevention of keratinocyte carcinomas in solid-organ transplant recipients. Dermatology. 2022;238:161-166.
- Otley CC, Stasko T, Tope WD, et al. Chemoprevention of nonmelanoma skin cancer with systemic retinoids: practical dosing and management of adverse effects. Dermatol Surg. 2006;32:562-568.
- Kadakia KC, Barton DL, Loprinzi CL, et al. Randomized controlled trial of acitretin versus placebo in patients at high-risk for basal cell or squamous cell carcinoma of the skin (North Central Cancer Treatment Group Study 969251). Cancer. 2012;118:2128-2137.
- McKenna DB, Murphy GM. Skin cancer chemoprophylaxis in renal transplant recipients: 5 years of experience using low-dose acitretin. Br J Dermatol. 1999;140:656-660.
- National Institutes of Health Office of Dietary Supplements. Niacin: fact sheet for health professionals. Updated August 23, 2022. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/Niacin-HealthProfessional/
- Malesu R, Martin AJ, Lyons JG, et al. Nicotinamide for skin cancer chemoprevention: effects of nicotinamide on melanoma in vitro and in vivo. Photochem Photobiol Sci. 2020;19:171-179.
- Gensler HL. Prevention of photoimmunosuppression and photocarcinogenesis by topical nicotinamide. Nutr Cancer. 1997;29:157-162.
- Gensler HL, Williams T, Huang AC, et al. Oral niacin prevents photocarcinogenesis and photoimmunosuppression in mice. Nutr Cancer. 1999;34:36-41.
- Chen AC, Martin AJ, Choy B, et al. A phase 3 randomized trial of nicotinamide for skin-cancer chemoprevention. N Engl J Med. 2015;373:1618-1626.
- Drago F, Ciccarese G, Cogorno L, et al. Prevention of non-melanoma skin cancers with nicotinamide in transplant recipients: a case-control study. Eur J Dermatol. 2017;27:382-385.
- Yélamos O, Halpern AC, Weinstock MA. Reply to “A phase II randomized controlled trial of nicotinamide for skin cancer chemoprevention in renal transplant recipients.” Br J Dermatol. 2017;176:551-552.
- Scatozza F, Moschella F, D’Arcangelo D, et al. Nicotinamide inhibits melanoma in vitro and in vivo. J Exp Clin Cancer Res. 2020;39:211.
- National Institutes of Health Office of Dietary Supplements. Folate: fact sheet for health professionals. Updated November 1, 2022. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/Folate-HealthProfessional/
- Butzbach K, Epe B. Photogenotoxicity of folic acid. Free Radic Biol Med. 2013;65:821-827.
- Vollset SE, Clarke R, Lewington S, et al. Effects of folic acid supplementation on overall and site-specific cancer incidence during the randomised trials: meta-analyses of data on 50,000 individuals. Lancet. 2013;381:1029-1036.
- Donnenfeld M, Deschasaux M, Latino-Martel P, et al. Prospective association between dietary folate intake and skin cancer risk: results from the Supplémentation en Vitamines et Minéraux Antioxydants cohort. Am J Clin Nutr. 2015;102:471-478.
- Fung TT, Hunter DJ, Spiegelman D, et al. Vitamins and carotenoids intake and the risk of basal cell carcinoma of the skin in women (United States). Cancer Causes Control. 2002;13:221-230.
- Fung TT, Spiegelman D, Egan KM, et al. Vitamin and carotenoid intake and risk of squamous cell carcinoma of the skin. Int J Cancer. 2003;103:110-115.
- National Institutes of Health Office of Dietary Supplements. Vitamin C: fact sheet for health professionals. Updated March 26, 2021. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/VitaminC-HealthProfessional/
- Spoelstra-de Man AME, Elbers PWG, Oudemans-Van Straaten HM. Vitamin C: should we supplement? Curr Opin Crit Care. 2018;24:248-255.
- Moison RMW, Beijersbergen van Henegouwen GMJ. Topical antioxidant vitamins C and E prevent UVB-radiation-induced peroxidation of eicosapentaenoic acid in pig skin. Radiat Res. 2002;157:402-409.
- Lin JY, Selim MA, Shea CR, et al. UV photoprotection by combination topical antioxidants vitamin C and vitamin E. J Am Acad Dermatol. 2003;48:866-874.
- Pauling L, Willoughby R, Reynolds R, et al. Incidence of squamous cell carcinoma in hairless mice irradiated with ultraviolet light in relation to intake of ascorbic acid (vitamin C) and of D, L-alpha-tocopheryl acetate (vitamin E). Int J Vitam Nutr Res Suppl. 1982;23:53-82.
- Kune GA, Bannerman S, Field B, et al. Diet, alcohol, smoking, serum beta-carotene, and vitamin A in male nonmelanocytic skin cancer patients and controls. Nutr Cancer. 1992;18:237-244.
- Vural P, Canbaz M, Selçuki D. Plasma antioxidant defense in actinic keratosis and basal cell carcinoma. J Eur Acad Dermatol Venereol. 1999;13:96-101.
- Record IR, Dreosti IE, McInerney JK. Changes in plasma antioxidant status following consumption of diets high or low in fruit and vegetables or following dietary supplementation with an antioxidant mixture. Br J Nutr. 2001;85:459-464.
- Heinen MM, Hughes MC, Ibiebele TI, et al. Intake of antioxidant nutrients and the risk of skin cancer. Eur J Cancer. 2007;43:2707-2716.
- Yang G, Yan Y, Ma Y, et al. Vitamin C at high concentrations induces cytotoxicity in malignant melanoma but promotes tumor growth at low concentrations. Mol Carcinog. 2017;56:1965-1976.
- National Institutes of Health Office of Dietary Supplements. Vitamin D: fact sheet for health professionals. Updated August 12, 2022. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/VitaminD-HealthProfessional/
- Reichrath J, Saternus R, Vogt T. Endocrine actions of vitamin D in skin: relevance for photocarcinogenesis of non-melanoma skin cancer, and beyond. Mol Cell Endocrinol. 2017;453:96-102.
- Ellison TI, Smith MK, Gilliam AC, et al. Inactivation of the vitamin D receptor enhances susceptibility of murine skin to UV-induced tumorigenesis. J Invest Dermatol. 2008;128:2508-2517.
- Eide MJ, Johnson DA, Jacobsen GR, et al. Vitamin D and nonmelanoma skin cancer in a health maintenance organization cohort. Arch Dermatol. 2011;147:1379-1384.
- van der Pols JC, Russell A, Bauer U, et al. Vitamin D status and skin cancer risk independent of time outdoors: 11-year prospective study in an Australian community. J Invest Dermatol. 2013;133:637-641.
- Caini S, Gnagnarella P, Stanganelli I, et al. Vitamin D and the risk of non-melanoma skin cancer: a systematic literature review and meta-analysis on behalf of the Italian Melanoma Intergroup. Cancers (Basel). 2021;13:4815.
- Park SM, Li T, Wu S, et al. Vitamin D intake and risk of skin cancer in US women and men. PLoS One. 2016;11:e0160308.
- Afzal S, Nordestgaard BG, Bojesen SE. Plasma 25-hydroxyvitamin D and risk of non-melanoma and melanoma skin cancer: a prospective cohort study. J Invest Dermatol. 2013;133:629-636.
- Asgari MM, Tang J, Warton ME, et al. Association of prediagnostic serum vitamin D levels with the development of basal cell carcinoma. J Invest Dermatol. 2010;130:1438-1443.
- Tang JY, Parimi N, Wu A, et al. Inverse association between serum 25(OH) vitamin D levels and non-melanoma skin cancer in elderly men. Cancer Causes Control. 2010;21:387-391.
- Keen MA, Hassan I. Vitamin E in dermatology. Indian Dermatol Online J. 2016;7:311-315.
- National Institutes of Health Office of Dietary Supplements. Vitamin E: fact sheet for health professionals. Updated March 26, 2021. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/VitaminE-HealthProfessional/
- Pearson P, Lewis SA, Britton J, et al. The pro-oxidant activity of high-dose vitamin E supplements in vivo. BioDrugs. 2006;20:271-273.
- Gerrish KE, Gensler HL. Prevention of photocarcinogenesis by dietary vitamin E. Nutr Cancer. 1993;19:125-133.
- McVean M, Liebler DC. Prevention of DNA photodamage by vitamin E compounds and sunscreens: roles of ultraviolet absorbance and cellular uptake. Mol Carcinog. 1999;24:169-176.
- Prasad KN, Cohrs RJ, Sharma OK. Decreased expressions of c-myc and H-ras oncogenes in vitamin E succinate induced morphologically differentiated murine B-16 melanoma cells in culture. Biochem Cell Biol. 1990;68:1250-1255.
- Funasaka Y, Komoto M, Ichihashi M. Depigmenting effect of alpha-tocopheryl ferulate on normal human melanocytes. Pigment Cell Res. 2000;13(suppl 8):170-174.
- National Institutes of Health Office of Dietary Supplements. Selenium: fact sheet for health professionals. Updated March 26, 2021. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/Selenium-HealthProfessional/
- Sengupta A, Lichti UF, Carlson BA, et al. Selenoproteins are essential for proper keratinocyte function and skin development. PLoS One. 2010;5:e12249.
- Das RK, Hossain SKU, Bhattacharya S. Diphenylmethyl selenocyanate inhibits DMBA-croton oil induced two-stage mouse skin carcinogenesis by inducing apoptosis and inhibiting cutaneous cell proliferation. Cancer Lett. 2005;230:90-101.
- Clark LC, Combs GF Jr, Turnbull BW, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA. 1996;276:1957-1963.
- Duffield-Lillico AJ, Slate EH, Reid ME, et al. Selenium supplementation and secondary prevention of nonmelanoma skin cancer in a randomized trial. J Natl Cancer Inst. 2003;95:1477-1481.
- Reid ME, Duffield-Lillico AJ, Slate E, et al. The nutritional prevention of cancer: 400 mcg per day selenium treatment. Nutr Cancer. 2008;60:155-163.
- Vinceti M, Filippini T, Del Giovane C, et al. Selenium for preventing cancer. Cochrane Database Syst Rev. 2018;1:CD005195.
- Hercberg S, Ezzedine K, Guinot C, et al. Antioxidant supplementation increases the risk of skin cancers in women but not in men. J Nutr. 2007;137:2098-2105.
- Chang YJ, Myung SK, Chung ST, et al. Effects of vitamin treatment or supplements with purported antioxidant properties on skin cancer prevention: a meta-analysis of randomized controlled trials. Dermatology. 2011;223:36-44.
- Sun W, Rice MS, Park MK, et al. Intake of furocoumarins and risk of skin cancer in 2 prospective US cohort studies. J Nutr. 2020;150:1535-1544.
- Sakaki JR, Melough MM, Roberts MB, et al. Citrus consumption and the risk of non-melanoma skin cancer in the Women’s Health Initiative. Cancers (Basel). 2021;13:2173.
Nonmelanoma skin cancer (NMSC) is the most common cancer in the United States, and cutaneous melanoma is projected to be the fifth most common form of cancer in 2022, with increasing incidence and high potential for mortality.1-3 Estimates indicate that 35% to 45% of all cancers in White patients are cutaneous, with 4% to 5% occurring in Hispanic patients, 2% to 4% in Asian patients, and 1% to 2% in Black patients.4 Of the keratinocyte carcinomas, basal cell carcinoma (BCC) is the most prevalent, projected to affect approximately 33% to 39% of White males and 23% to 28% of White females in the United States during their lifetimes. Squamous cell carcinoma (SCC) is the second most common skin malignancy, with a lifetime risk of 9% to 14% for White males and 4% to 9% for White females in the United States.5 The incidence of melanoma continues to increase, with approximately 99,780 new cases expected in the United States in 2022.1
UV-induced DNA damage plays a key role in the pathogenesis and development of various skin malignancies.6 UV radiation from sunlight or tanning devices causes photocarcinogenesis due to molecular and cellular effects, including the generation of reactive oxygen species, DNA damage due to the formation of cyclobutane pyrimidine dimers and pyrimidine-pyrimidone, melanogenesis, apoptosis, and the increased expression of harmful genes and proteins.6 The summation of this damage can result in skin malignancies, including NMSC and melanoma.6,7 Dietary antioxidants theoretically help prevent oxidative reactions from occurring within the body, and it has been suggested that intake of dietary antioxidants may decrease DNA damage and prevent tumorigenesis secondary to UV radiation.8 Antioxidants exist naturally in the body but can be acquired exogenously. Investigators have studied dietary antioxidants in preventing skin cancer formation with promising results in the laboratory setting.8-11 Recently, more robust human studies have been initiated to further delineate this relationship. We present clinical evidence of several frequently utilized antioxidant vitamins and their effects on melanoma and NMSC.
Antioxidants
Vitamin A—Vitamin A is a fat-soluble vitamin found in animal sources, including fish, liver, and eggs. Carotenoids, such as beta carotene, are provitamin A plant derivatives found in fruits and vegetables that are converted into biologically active retinol and retinoic acid.12 Retinols play a key role in cellular growth and differentiation and are thought to be protective against skin cancer via the inactivation of free radicals and immunologic enhancement due to their antiproliferative, antioxidative, and antiapoptotic effects.13-16 Animal studies have demonstrated this protective effect and the ability of retinoids to suppress carcinogenesis; however, human studies reveal conflicting results.17,18
Greenberg et al19 investigated the use of beta carotene in preventing the formation of NMSC. Patients (N=1805) were randomized to receive 50 mg of beta carotene daily or placebo. Over a 5-year period, there was no significant reduction in the occurrence of NMSC (relative risk [RR], 1.05; 95% CI, 0.91-1.22).19 Frieling et al20 conducted a similar randomized, double-blind, placebo-controlled trial investigating beta carotene for primary prevention of NMSC in 22,071 healthy male physicians. The study group received 50 mg of beta carotene every other day for 12 years’ duration, and there was no significant effect on the incidence of first NMSC development (RR, 0.98; 95% CI, 0.92-1.05).20
A case-control study by Naldi et al21 found an inverse association between vitamin A intake and development of melanoma. Study participants were stratified into quartiles based on level of dietary intake and found an odds ratio (OR) of 0.71 for beta carotene (95% CI, 0.50-1.02), 0.57 for retinol (95% CI, 0.39-0.83), and 0.51 for total vitamin A (95% CI, 0.35-0.75) when comparing the upper quartile of vitamin A intake to the lower quartile. Upper-quartile cutoff values of vitamin A intake were 214 µg/d for beta carotene, 149 µg/d for retinol, and 359 µg/d for total vitamin A.21 More recently, a meta-analysis by Zhang et al22 pooled data from 8 case-control studies and 2 prospective studies. Intake of retinol but not total vitamin A or beta carotene was associated with a reduced risk for development of melanoma (retinol: OR, 0.80; 95% CI, 0.69-0.92; total vitamin A: OR, 0.86; 95% CI, 0.59-1.25; beta carotene: OR, 0.87; 95% CI, 0.62-1.20).22 Feskanich et al23 demonstrated similar findings with use of food-frequency questionnaires in White women, suggesting that retinol intake from food combined with supplements may be protective for women who were otherwise at a low risk for melanoma based on nondietary factors. These factors included painful or blistering sunburns during childhood, history of more than 6 sunburns, more than 3 moles on the left arm, having red or blonde hair, and having a parent or sibling with melanoma (P=.01). However, this relationship did not hold true when looking at women at an intermediate or high risk for melanoma (P=.16 and P=.46).23
When looking at high-risk patients, such as transplant patients, oral retinoids have been beneficial in preventing NMSC.24-27 Bavinck et al24 investigated 44 renal transplant patients with a history of more than 10 NMSCs treated with 30 mg of acitretin daily vs placebo. Patients receiving oral retinoid supplementation developed fewer NMSCs over a 6-month treatment period (P=.01).24 Similarly, George et al25 investigated acitretin in renal transplant patients and found a statistically significant decrease in number of SCCs in patients on supplementation (P=.002). Solomon-Cohen et al26 performed a retrospective case-crossover study in solid organ transplant recipients and found that those treated with 10 mg of acitretin daily for 2 years had a significant reduction in the number of new keratinocyte carcinomas (P=.002). Other investigators have demonstrated similar results, and in 2006, Otley et al27 proposed standardized dosing of acitretin for chemoprevention in high-risk patients, including patients developing 5 to 10 NMSCs per year, solid organ transplant recipients, and those with syndromes associated with the development of NMSC.28,29 Overall, in the general population, vitamin A and related compounds have not demonstrated a significant association with decreased development of NMSC; however, oral retinoids have proven useful for high-risk patients. Furthermore, several studies have suggested a negative association between vitamin A levels and the incidence of melanoma, specifically in the retinol formulation.
Vitamin B3—Nicotinamide (also known as niacinamide) is a water-soluble form of vitamin B3 and is obtained from animal-based and plant-based foods, such as meat, fish, and legumes.30 Nicotinamide plays a key role in cellular metabolism, cellular signaling, and DNA repair, including protection from UV damage within keratinocytes.31,32 Early mouse models demonstrated decreased formation of skin tumors in mice treated with topical or oral nicotinamide.32,33 A number of human studies have revealed similar results.34-36
Chen et al34 conducted the ONTRAC study, a phase 3, double-blind, randomized controlled trial (RCT) looking at 386 participants with a history of at least 2 NMSCs in the preceding 5 years. At 12 months, those treated with 500 mg of nicotinamide twice daily demonstrated a statistically significant decreased rate of SCC formation (P=.05). A decreased incidence of BCC development was noted; however, this trend did not reach statistical significance (P=.12). Precancerous skin lesions also were found to be decreased in the treatment group, with 20% lower incidence of actinic keratoses (AKs) after 9 months of treatment (P<.001).34 Drago et al35 specifically studied the incidence of AKs in 38 transplant recipients—8 liver and 30 kidney—and found that previously noted AKs had decreased in size for 18 of 19 patients taking 500 mg of nicotinamide daily when originally photographed AKs were remeasured at 6-month follow-up, with 7 of these 18 patients demonstrating complete clinical regression. Of those on nicotinamide supplementation, no new AKs developed compared to the control group, which demonstrated increased size of AKs or development of new AKs in 91% of patients, with 7 AKs progressing into SCC.35
Nicotinamide has been demonstrated to be useful in preventing skin cancer in high-risk populations, such as transplant patients or those with a high incidence of NMSC.34,36 Despite promising results within the laboratory setting, nicotinamide’s effects on melanoma in humans remains less clear.31,37 Studies suggest that nicotinamide enhances tumor-infiltrating lymphocytes and DNA repair mechanisms in melanocytes, which may translate into nicotinamide, providing chemoprevention for melanoma, but research in human patients is limited.31,37
Vitamin B9—Folate, the natural form of vitamin B9, is a water-soluble compound that is found in many foods, especially green leafy vegetables, and often is supplemented because of its health benefits.38,39 In the skin, folic acid plays a key role in cellular replication and proliferation.38 Controversy exists regarding folate’s effects on cellular growth and turnover with respect to cancer incidence.38,40 Donnenfeld et al41 conducted a prospective study assessing dietary folic acid intake and development of NMSC. A total of 5880 participants completed dietary records throughout the first 2 years of the study. After an average follow-up period of 12.6 years, there was an overall increased incidence of skin cancer in those with increased dietary folate (P=.03). Furthermore, when striating by skin cancer type, there was an increased incidence of NMSC overall as well as BCC when analyzing by type of NMSC (P=.03 for NMSC; P=.05 for BCC). However, when stratifying by gender, these findings only held true for women.41 Similar effects were observed by Fung et al,42 who prospectively studied the intake of various vitamins in relationship to the development of BCC in women. During 12 years of follow-up, a positive association was observed between folate intake and BCC development (OR, 1.2; 95% CI, 1.10-1.31).42 Fung et al43 also investigated the role of several vitamins in the development of SCC and found that folate showed a negative association, which did not reach statistical significance (RR, 0.79; 95% CI, 0.56-1.11). Furthermore, Vollset et al40 conducted a meta-analysis comparing folic acid to placebo in the incidence of various types of cancer. The study excluded NMSC but reported no significant association between the development of melanoma and folic acid supplementation.40 In summary, the effects of folate have diverse consequences, potentially promoting the formation of NMSC, but studies suggest that an individual’s gender and other genetic and environmental factors also may play a role.
Vitamin C—Vitamin C (also known as ascorbic acid) is a water-soluble vitamin with antioxidant immune-mediating effects. It is found in various fruits and vegetables and serves as a cofactor for enzymes within the body playing a key role in immune function and collagen formation.44,45 It has been postulated that ascorbic acid can provide protection from UV radiation damage via its intracellular activity but conversely can contribute to oxidative damage.44 Multiple in vitro laboratory studies and animal models have demonstrated photoprotective effects of ascorbic acid.46-48 Despite these findings, minimal photoprotective effects have been found in the human population.
Kune et al49 performed a case-control study of 88 males with previously diagnosed NMSC undergoing surgical removal and investigated patients’ prior dietary habits. Patients with NMSC had a statistically significantly lower level of vitamin C–containing food in their diet than those without NMSC (P=.004).49 In addition, Vural et al50 analyzed plasma samples and blood cells of patients with AK and BCC and found a significant decrease in ascorbic acid levels in both the AK (P<.001) and BCC (P<.001) groups compared with controls. However, studies have found that consumption of certain dietary compounds can rapidly increase plasma concentration levels, which may serve as a major confounding variable in this study. Plasma concentrations of ascorbic acid and beta carotene were found to be significantly increased following consumption of a high-antioxidant diet for as short a duration as 2 weeks (P<.05).51 More recently, Heinen et al52 performed a prospective study on 1001 adults. In patients without a history of skin cancer, they found that vitamin C from food sources plus dietary supplements was positively associated with the development of BCC (P=.03).52 Similarly, Fung et al42 performed a study in women and found a positive association between vitamin C intake and the development of BCC (OR, 1.13; 95% CI, 1.03-1.23).
The relationship between vitamin C intake—either in dietary or supplemental form—and melanoma remains controversial. Mice-based studies found that high concentrations of orally administered vitamin C induce cytotoxicity in melanoma cell lines, but at low concentrations they promote tumor growth of malignant melanoma.53 Feskanich et al23 examined the relationship between vitamin C intake and melanoma development via food frequency questionnaires in White women and found that vitamin C was associated with a higher risk for melanoma (P=.05), and furthermore, a positive dose response with frequency of orange juice intake was observed (P=.008). Overall, despite promising laboratory studies, there is a lack of RCTs investigating the use of vitamin C supplementation for prevention of NMSC and melanoma in humans, and the oral benefits of vitamin C for chemoprevention remain unclear.
Vitamin D—Vitamin D is a fat-soluble vitamin that is found in fish, liver, egg, and cheese, and is endogenously produced when UV radiation from sun exposure interacts with the skin, triggering the synthesis of vitamin D.54 Vitamin D is biologically inactive and must be converted to its active form 1,25-dihydroxyvitamin D after entering the body. Vitamin D modulates many genes involved in cellular proliferation and differentiation.54 Vitamin D receptors are expressed on keratinocytes and melanocytes.55 Animal studies have demonstrated a potentially protective effect of vitamin D in the development of NMSC.56 In a mouse model, Ellison et al56 found that mice without vitamin D receptors developed skin tumors more rapidly than those with vitamin D receptors.
Unfortunately, these findings have not been demonstrated in humans, and studies have even reported an increased risk for development of NMSC in patients with normal or increased vitamin D levels compared with those with low levels of vitamin D.57-60 Eide et al57 studied 3223 patients seeking advice for low bone density by recording their vitamin D levels at the time of presentation and monitoring development of NMSC. Vitamin D levels greater than 15 ng/mL were positively associated with the development of NMSC (OR, 1.7; 95% CI, 1.04-2.7). This association held true for both SCC and BCC, with a higher risk estimated for SCC (OR, 3.2; 95% CI, 0.4-24.0 for SCC; OR, 1.7; 95% CI, 0.5-5.8 for BCC).57 An increased vitamin D serum level also was found to be significantly associated with a higher risk for BCC and melanoma by van der Pols et al.58 This prospective study looked at the incidence of skin cancer over 11 years. Study participants with vitamin D levels over 75 nmol/L more frequently developed BCC (P=.01) and melanoma (P=.05). In contrast, SCC was less frequently observed in participants with these high levels of vitamin D (P=.07).58 Furthermore, Park et al60 looked at vitamin D and skin cancer risk for men and women in the United States and found no association with risk for SCC or melanoma but a positive association with BCC (P=.05 for total vitamin D; P<.01 for dietary vitamin D). Additional studies have been performed with inconsistent results, and multiple authors suggest the possible confounding relationship between vitamin D levels and UV radiation exposure.59-62 Furthermore, some studies have even demonstrated a negative association between vitamin D and NMSC. Tang et al63 performed a retrospective case-control study in elderly males, investigating serum levels of vitamin D and patients’ self-reported history of NMSC, which demonstrated that higher levels of vitamin D were associated with a decreased risk for NMSC. Overall, the relationship between vitamin D and skin cancer development remains unclear for both melanoma and NMSC.
Vitamin E—Vitamin E is a fat-soluble vitamin that is found in plant-based oils, nuts, seeds, fruits, and vegetables.64 It works as an antioxidant to protect against free radicals and heighten immune function, and it also serves as a pro-oxidant.65,66 Vitamin E naturally exists in 8 chemical forms, of which gamma-tocopherol is the most frequently obtained form in the diet, and alpha-tocopherol is the most abundant form found in the body.64,65
Early animal studies demonstrated the inhibition of UV-induced damage in mice receiving vitamin E supplementation.67,68 Human studies have not consistently shown these effects. Vural et al50 investigated plasma samples and blood cells of patients with AKs and BCCs and reported a significant decrease in alpha-tocopherol levels in both the AK (P<.05) and BCC (P<.001) groups compared with controls. However, studies also have demonstrated a positive association between vitamin E intake and the development of BCC, including one by Fung et al,42 which found a significant association in women (OR, 1.15; 95% CI, 1.06-1.26).
Vitamin E has been found to inhibit melanin synthesis in the laboratory, suggesting a potentially protective effect in melanoma.69,70 However, in the study performed by Feskanich et al23 examining vitamin intake and melanoma incidence via food-frequency questionnaires, vitamin E was not associated with a lower risk for melanoma. Despite promising laboratory studies, the data surrounding the use of a vitamin E supplement for prevention of melanoma and NMSC in humans remains unclear.
Selenium—Selenium is a trace mineral found in plants, meat, and fish. It plays a key role in reproduction, hormone metabolism, DNA synthesis, and protection from oxidative damage.71 In mice studies, lack of selenium-containing proteins resulted in skin abnormalities, including the development of a hyperplastic epidermis and aberrant hair follicle morphogenesis with alopecia after birth, and numerous experimental studies have demonstrated a negative association between selenium intake and cancer.72,73 However, human studies have yielded alternative results.
The Nutritional Prevention of Cancer Study Group analyzed 1312 dermatology patients with a history of NMSC.74 The study population was obtained from 7 dermatology clinics with randomization to control for confounding variables. Study participants received either 200 μg of selenium daily or placebo.74 Baseline characteristics of each study group were overall balanced. Selenium intake was found to have no effect on the development of BCC (hazard ratio [HR], 1.09; 95% CI, 0.94-1.26) but an increased risk for developing SCC (HR, 1.25; 95% CI, 1.03-1.51) and total NMSC (HR, 1.17; 95% CI, 1.02-1.34).74,75 Similarly, Reid et al76 performed an RCT comparing patients treated with 400 μg/d of selenium to those treated with 200 μg/d of selenium. When compared with placebo, those treated with 200 μg/d of selenium had a statistically significantly increased incidence of NMSC (P=.006); however, those treated with 400 μg/d of selenium had no significant change in total incidence of NMSC (P=.51).76 Furthermore, Vinceti et al77 performed a review of 83 studies from the literature investigating the effect of dietary selenium, and from the RCTs, there was no beneficial effect of selenium in reducing cancer risk in general; however, some studies demonstrated an increased incidence of other types of cancer, including melanoma. Of the RCTs included in the study investigating NMSC incidence specifically, it was found that the incidence was not affected by selenium administration (RR, 1.16; 95% CI, 0.30-4.42; 2 studies, 2027 participants).77 Despite data from several studies demonstrating an increased risk for NMSC, the effects of selenium on the risk for NMSC and melanoma remain unclear.
Combination Antioxidant Studies
In addition to investigating the use of single antioxidants in skin cancer prevention, studies utilizing the combination of various antioxidants or other dietary minerals have been conducted. Hercberg et al78 performed a randomized, double-blinded, placebo-controlled trial of 13,017 adults (7876 women and 5141 men) receiving a combination of 120 mg vitamin C, 30 mg vitamin E, 100 μg selenium, 6 mg beta carotene, and 20 mg zinc. Study participants were followed for an average of 7.5 years, and the development of skin cancers were recorded. Overall, the incidence rate of skin cancer did not differ between the 2 treatment groups; however, when segregated by gender, the study found that there was an increased risk for developing skin cancer in women taking the antioxidant supplement combination compared with placebo (P=.03). This difference was not observed in the 2 treatment groups of male patients (P=.11). When looking specifically at NMSC, there was no difference between treatment groups for male or female patients (P=.39 for males; P=.15 for females). In contrast, there was a higher incidence of melanoma identified in female patients taking the combination antioxidant supplement (P=.01), but this was not seen within the male study population (P=.51).78 In addition, Chang et al79 performed a meta-analysis of 10 previously published RCTs. Analysis revealed that treatment with a variety of supplements, including vitamins A, C, E, and beta carotene, were found to have no preventative effects on the incidence of skin cancer development (RR, 0.98; CI, 0.98-1.03). Notable limitations to this study included the variability in protocols of the studies included in this meta-analysis, the limited number of RCTs investigating vitamin supplementation and the risk for skin cancer development, and the influence of dietary intake on study outcomes.79
Other Dietary Agents
Furocoumarins—Furocoumarins are botanical substances found in various fruits and plants, including many citrus products. Furocoumarins are activated by UV light radiation and can lead to development of a phototoxic eruption. Several studies have suggested a pharmacogenetic effect of furocoumarins.80 Sun et al80 collected dietary data from 47,453 men and 75,291 women on furocoumarin intake and correlation with the development of NMSC. Overall, the study suggested that the intake of furocoumarins may lead to an increase in the development of BCC (HR, 1.16; 95% CI, 1.11-1.21; P=.002); however, there was no significant association identified between total intake of furocoumarins in the risk for SCC or melanoma.80 Furthermore, Sakaki et al81 conducted a survey study looking at the consumption of citrus products and the development of NMSC. The group found that there was an increased risk for NMSC in those consuming an increased amount of citrus products (P=.007).81
Conclusion
Dietary antioxidants have been investigated for their potential role in the prevention of tumorigenesis. Specific antioxidant vitamins, such as vitamin A derivatives and niacinamide, have demonstrated clinical utility in the prevention of NMSC in high-risk populations. Retinol also has been associated with a reduced incidence of melanoma. Numerous antioxidants have demonstrated promising data within the laboratory setting; however, inconsistent results have been appreciated in humans. Furthermore, several research studies suggest that folate, vitamin D, and furocoumarins may be associated with an increased risk for skin cancer development; however, these studies are inconclusive, and dietary studies are challenging to conduct. Overall, RCTs investigating the role of antioxidants for chemoprevention are limited. Moreover, the study of dietary antioxidants and vitamins may be affected by various confounding variables that can be difficult to account for because of patients’ potentially poor recall of dietary intake and the effect of dietary intake in supplemental studies. Given the increasing prevalence of skin cancer worldwide, further research into the clinical utility of antioxidants in skin cancer prevention is warranted.
Nonmelanoma skin cancer (NMSC) is the most common cancer in the United States, and cutaneous melanoma is projected to be the fifth most common form of cancer in 2022, with increasing incidence and high potential for mortality.1-3 Estimates indicate that 35% to 45% of all cancers in White patients are cutaneous, with 4% to 5% occurring in Hispanic patients, 2% to 4% in Asian patients, and 1% to 2% in Black patients.4 Of the keratinocyte carcinomas, basal cell carcinoma (BCC) is the most prevalent, projected to affect approximately 33% to 39% of White males and 23% to 28% of White females in the United States during their lifetimes. Squamous cell carcinoma (SCC) is the second most common skin malignancy, with a lifetime risk of 9% to 14% for White males and 4% to 9% for White females in the United States.5 The incidence of melanoma continues to increase, with approximately 99,780 new cases expected in the United States in 2022.1
UV-induced DNA damage plays a key role in the pathogenesis and development of various skin malignancies.6 UV radiation from sunlight or tanning devices causes photocarcinogenesis due to molecular and cellular effects, including the generation of reactive oxygen species, DNA damage due to the formation of cyclobutane pyrimidine dimers and pyrimidine-pyrimidone, melanogenesis, apoptosis, and the increased expression of harmful genes and proteins.6 The summation of this damage can result in skin malignancies, including NMSC and melanoma.6,7 Dietary antioxidants theoretically help prevent oxidative reactions from occurring within the body, and it has been suggested that intake of dietary antioxidants may decrease DNA damage and prevent tumorigenesis secondary to UV radiation.8 Antioxidants exist naturally in the body but can be acquired exogenously. Investigators have studied dietary antioxidants in preventing skin cancer formation with promising results in the laboratory setting.8-11 Recently, more robust human studies have been initiated to further delineate this relationship. We present clinical evidence of several frequently utilized antioxidant vitamins and their effects on melanoma and NMSC.
Antioxidants
Vitamin A—Vitamin A is a fat-soluble vitamin found in animal sources, including fish, liver, and eggs. Carotenoids, such as beta carotene, are provitamin A plant derivatives found in fruits and vegetables that are converted into biologically active retinol and retinoic acid.12 Retinols play a key role in cellular growth and differentiation and are thought to be protective against skin cancer via the inactivation of free radicals and immunologic enhancement due to their antiproliferative, antioxidative, and antiapoptotic effects.13-16 Animal studies have demonstrated this protective effect and the ability of retinoids to suppress carcinogenesis; however, human studies reveal conflicting results.17,18
Greenberg et al19 investigated the use of beta carotene in preventing the formation of NMSC. Patients (N=1805) were randomized to receive 50 mg of beta carotene daily or placebo. Over a 5-year period, there was no significant reduction in the occurrence of NMSC (relative risk [RR], 1.05; 95% CI, 0.91-1.22).19 Frieling et al20 conducted a similar randomized, double-blind, placebo-controlled trial investigating beta carotene for primary prevention of NMSC in 22,071 healthy male physicians. The study group received 50 mg of beta carotene every other day for 12 years’ duration, and there was no significant effect on the incidence of first NMSC development (RR, 0.98; 95% CI, 0.92-1.05).20
A case-control study by Naldi et al21 found an inverse association between vitamin A intake and development of melanoma. Study participants were stratified into quartiles based on level of dietary intake and found an odds ratio (OR) of 0.71 for beta carotene (95% CI, 0.50-1.02), 0.57 for retinol (95% CI, 0.39-0.83), and 0.51 for total vitamin A (95% CI, 0.35-0.75) when comparing the upper quartile of vitamin A intake to the lower quartile. Upper-quartile cutoff values of vitamin A intake were 214 µg/d for beta carotene, 149 µg/d for retinol, and 359 µg/d for total vitamin A.21 More recently, a meta-analysis by Zhang et al22 pooled data from 8 case-control studies and 2 prospective studies. Intake of retinol but not total vitamin A or beta carotene was associated with a reduced risk for development of melanoma (retinol: OR, 0.80; 95% CI, 0.69-0.92; total vitamin A: OR, 0.86; 95% CI, 0.59-1.25; beta carotene: OR, 0.87; 95% CI, 0.62-1.20).22 Feskanich et al23 demonstrated similar findings with use of food-frequency questionnaires in White women, suggesting that retinol intake from food combined with supplements may be protective for women who were otherwise at a low risk for melanoma based on nondietary factors. These factors included painful or blistering sunburns during childhood, history of more than 6 sunburns, more than 3 moles on the left arm, having red or blonde hair, and having a parent or sibling with melanoma (P=.01). However, this relationship did not hold true when looking at women at an intermediate or high risk for melanoma (P=.16 and P=.46).23
When looking at high-risk patients, such as transplant patients, oral retinoids have been beneficial in preventing NMSC.24-27 Bavinck et al24 investigated 44 renal transplant patients with a history of more than 10 NMSCs treated with 30 mg of acitretin daily vs placebo. Patients receiving oral retinoid supplementation developed fewer NMSCs over a 6-month treatment period (P=.01).24 Similarly, George et al25 investigated acitretin in renal transplant patients and found a statistically significant decrease in number of SCCs in patients on supplementation (P=.002). Solomon-Cohen et al26 performed a retrospective case-crossover study in solid organ transplant recipients and found that those treated with 10 mg of acitretin daily for 2 years had a significant reduction in the number of new keratinocyte carcinomas (P=.002). Other investigators have demonstrated similar results, and in 2006, Otley et al27 proposed standardized dosing of acitretin for chemoprevention in high-risk patients, including patients developing 5 to 10 NMSCs per year, solid organ transplant recipients, and those with syndromes associated with the development of NMSC.28,29 Overall, in the general population, vitamin A and related compounds have not demonstrated a significant association with decreased development of NMSC; however, oral retinoids have proven useful for high-risk patients. Furthermore, several studies have suggested a negative association between vitamin A levels and the incidence of melanoma, specifically in the retinol formulation.
Vitamin B3—Nicotinamide (also known as niacinamide) is a water-soluble form of vitamin B3 and is obtained from animal-based and plant-based foods, such as meat, fish, and legumes.30 Nicotinamide plays a key role in cellular metabolism, cellular signaling, and DNA repair, including protection from UV damage within keratinocytes.31,32 Early mouse models demonstrated decreased formation of skin tumors in mice treated with topical or oral nicotinamide.32,33 A number of human studies have revealed similar results.34-36
Chen et al34 conducted the ONTRAC study, a phase 3, double-blind, randomized controlled trial (RCT) looking at 386 participants with a history of at least 2 NMSCs in the preceding 5 years. At 12 months, those treated with 500 mg of nicotinamide twice daily demonstrated a statistically significant decreased rate of SCC formation (P=.05). A decreased incidence of BCC development was noted; however, this trend did not reach statistical significance (P=.12). Precancerous skin lesions also were found to be decreased in the treatment group, with 20% lower incidence of actinic keratoses (AKs) after 9 months of treatment (P<.001).34 Drago et al35 specifically studied the incidence of AKs in 38 transplant recipients—8 liver and 30 kidney—and found that previously noted AKs had decreased in size for 18 of 19 patients taking 500 mg of nicotinamide daily when originally photographed AKs were remeasured at 6-month follow-up, with 7 of these 18 patients demonstrating complete clinical regression. Of those on nicotinamide supplementation, no new AKs developed compared to the control group, which demonstrated increased size of AKs or development of new AKs in 91% of patients, with 7 AKs progressing into SCC.35
Nicotinamide has been demonstrated to be useful in preventing skin cancer in high-risk populations, such as transplant patients or those with a high incidence of NMSC.34,36 Despite promising results within the laboratory setting, nicotinamide’s effects on melanoma in humans remains less clear.31,37 Studies suggest that nicotinamide enhances tumor-infiltrating lymphocytes and DNA repair mechanisms in melanocytes, which may translate into nicotinamide, providing chemoprevention for melanoma, but research in human patients is limited.31,37
Vitamin B9—Folate, the natural form of vitamin B9, is a water-soluble compound that is found in many foods, especially green leafy vegetables, and often is supplemented because of its health benefits.38,39 In the skin, folic acid plays a key role in cellular replication and proliferation.38 Controversy exists regarding folate’s effects on cellular growth and turnover with respect to cancer incidence.38,40 Donnenfeld et al41 conducted a prospective study assessing dietary folic acid intake and development of NMSC. A total of 5880 participants completed dietary records throughout the first 2 years of the study. After an average follow-up period of 12.6 years, there was an overall increased incidence of skin cancer in those with increased dietary folate (P=.03). Furthermore, when striating by skin cancer type, there was an increased incidence of NMSC overall as well as BCC when analyzing by type of NMSC (P=.03 for NMSC; P=.05 for BCC). However, when stratifying by gender, these findings only held true for women.41 Similar effects were observed by Fung et al,42 who prospectively studied the intake of various vitamins in relationship to the development of BCC in women. During 12 years of follow-up, a positive association was observed between folate intake and BCC development (OR, 1.2; 95% CI, 1.10-1.31).42 Fung et al43 also investigated the role of several vitamins in the development of SCC and found that folate showed a negative association, which did not reach statistical significance (RR, 0.79; 95% CI, 0.56-1.11). Furthermore, Vollset et al40 conducted a meta-analysis comparing folic acid to placebo in the incidence of various types of cancer. The study excluded NMSC but reported no significant association between the development of melanoma and folic acid supplementation.40 In summary, the effects of folate have diverse consequences, potentially promoting the formation of NMSC, but studies suggest that an individual’s gender and other genetic and environmental factors also may play a role.
Vitamin C—Vitamin C (also known as ascorbic acid) is a water-soluble vitamin with antioxidant immune-mediating effects. It is found in various fruits and vegetables and serves as a cofactor for enzymes within the body playing a key role in immune function and collagen formation.44,45 It has been postulated that ascorbic acid can provide protection from UV radiation damage via its intracellular activity but conversely can contribute to oxidative damage.44 Multiple in vitro laboratory studies and animal models have demonstrated photoprotective effects of ascorbic acid.46-48 Despite these findings, minimal photoprotective effects have been found in the human population.
Kune et al49 performed a case-control study of 88 males with previously diagnosed NMSC undergoing surgical removal and investigated patients’ prior dietary habits. Patients with NMSC had a statistically significantly lower level of vitamin C–containing food in their diet than those without NMSC (P=.004).49 In addition, Vural et al50 analyzed plasma samples and blood cells of patients with AK and BCC and found a significant decrease in ascorbic acid levels in both the AK (P<.001) and BCC (P<.001) groups compared with controls. However, studies have found that consumption of certain dietary compounds can rapidly increase plasma concentration levels, which may serve as a major confounding variable in this study. Plasma concentrations of ascorbic acid and beta carotene were found to be significantly increased following consumption of a high-antioxidant diet for as short a duration as 2 weeks (P<.05).51 More recently, Heinen et al52 performed a prospective study on 1001 adults. In patients without a history of skin cancer, they found that vitamin C from food sources plus dietary supplements was positively associated with the development of BCC (P=.03).52 Similarly, Fung et al42 performed a study in women and found a positive association between vitamin C intake and the development of BCC (OR, 1.13; 95% CI, 1.03-1.23).
The relationship between vitamin C intake—either in dietary or supplemental form—and melanoma remains controversial. Mice-based studies found that high concentrations of orally administered vitamin C induce cytotoxicity in melanoma cell lines, but at low concentrations they promote tumor growth of malignant melanoma.53 Feskanich et al23 examined the relationship between vitamin C intake and melanoma development via food frequency questionnaires in White women and found that vitamin C was associated with a higher risk for melanoma (P=.05), and furthermore, a positive dose response with frequency of orange juice intake was observed (P=.008). Overall, despite promising laboratory studies, there is a lack of RCTs investigating the use of vitamin C supplementation for prevention of NMSC and melanoma in humans, and the oral benefits of vitamin C for chemoprevention remain unclear.
Vitamin D—Vitamin D is a fat-soluble vitamin that is found in fish, liver, egg, and cheese, and is endogenously produced when UV radiation from sun exposure interacts with the skin, triggering the synthesis of vitamin D.54 Vitamin D is biologically inactive and must be converted to its active form 1,25-dihydroxyvitamin D after entering the body. Vitamin D modulates many genes involved in cellular proliferation and differentiation.54 Vitamin D receptors are expressed on keratinocytes and melanocytes.55 Animal studies have demonstrated a potentially protective effect of vitamin D in the development of NMSC.56 In a mouse model, Ellison et al56 found that mice without vitamin D receptors developed skin tumors more rapidly than those with vitamin D receptors.
Unfortunately, these findings have not been demonstrated in humans, and studies have even reported an increased risk for development of NMSC in patients with normal or increased vitamin D levels compared with those with low levels of vitamin D.57-60 Eide et al57 studied 3223 patients seeking advice for low bone density by recording their vitamin D levels at the time of presentation and monitoring development of NMSC. Vitamin D levels greater than 15 ng/mL were positively associated with the development of NMSC (OR, 1.7; 95% CI, 1.04-2.7). This association held true for both SCC and BCC, with a higher risk estimated for SCC (OR, 3.2; 95% CI, 0.4-24.0 for SCC; OR, 1.7; 95% CI, 0.5-5.8 for BCC).57 An increased vitamin D serum level also was found to be significantly associated with a higher risk for BCC and melanoma by van der Pols et al.58 This prospective study looked at the incidence of skin cancer over 11 years. Study participants with vitamin D levels over 75 nmol/L more frequently developed BCC (P=.01) and melanoma (P=.05). In contrast, SCC was less frequently observed in participants with these high levels of vitamin D (P=.07).58 Furthermore, Park et al60 looked at vitamin D and skin cancer risk for men and women in the United States and found no association with risk for SCC or melanoma but a positive association with BCC (P=.05 for total vitamin D; P<.01 for dietary vitamin D). Additional studies have been performed with inconsistent results, and multiple authors suggest the possible confounding relationship between vitamin D levels and UV radiation exposure.59-62 Furthermore, some studies have even demonstrated a negative association between vitamin D and NMSC. Tang et al63 performed a retrospective case-control study in elderly males, investigating serum levels of vitamin D and patients’ self-reported history of NMSC, which demonstrated that higher levels of vitamin D were associated with a decreased risk for NMSC. Overall, the relationship between vitamin D and skin cancer development remains unclear for both melanoma and NMSC.
Vitamin E—Vitamin E is a fat-soluble vitamin that is found in plant-based oils, nuts, seeds, fruits, and vegetables.64 It works as an antioxidant to protect against free radicals and heighten immune function, and it also serves as a pro-oxidant.65,66 Vitamin E naturally exists in 8 chemical forms, of which gamma-tocopherol is the most frequently obtained form in the diet, and alpha-tocopherol is the most abundant form found in the body.64,65
Early animal studies demonstrated the inhibition of UV-induced damage in mice receiving vitamin E supplementation.67,68 Human studies have not consistently shown these effects. Vural et al50 investigated plasma samples and blood cells of patients with AKs and BCCs and reported a significant decrease in alpha-tocopherol levels in both the AK (P<.05) and BCC (P<.001) groups compared with controls. However, studies also have demonstrated a positive association between vitamin E intake and the development of BCC, including one by Fung et al,42 which found a significant association in women (OR, 1.15; 95% CI, 1.06-1.26).
Vitamin E has been found to inhibit melanin synthesis in the laboratory, suggesting a potentially protective effect in melanoma.69,70 However, in the study performed by Feskanich et al23 examining vitamin intake and melanoma incidence via food-frequency questionnaires, vitamin E was not associated with a lower risk for melanoma. Despite promising laboratory studies, the data surrounding the use of a vitamin E supplement for prevention of melanoma and NMSC in humans remains unclear.
Selenium—Selenium is a trace mineral found in plants, meat, and fish. It plays a key role in reproduction, hormone metabolism, DNA synthesis, and protection from oxidative damage.71 In mice studies, lack of selenium-containing proteins resulted in skin abnormalities, including the development of a hyperplastic epidermis and aberrant hair follicle morphogenesis with alopecia after birth, and numerous experimental studies have demonstrated a negative association between selenium intake and cancer.72,73 However, human studies have yielded alternative results.
The Nutritional Prevention of Cancer Study Group analyzed 1312 dermatology patients with a history of NMSC.74 The study population was obtained from 7 dermatology clinics with randomization to control for confounding variables. Study participants received either 200 μg of selenium daily or placebo.74 Baseline characteristics of each study group were overall balanced. Selenium intake was found to have no effect on the development of BCC (hazard ratio [HR], 1.09; 95% CI, 0.94-1.26) but an increased risk for developing SCC (HR, 1.25; 95% CI, 1.03-1.51) and total NMSC (HR, 1.17; 95% CI, 1.02-1.34).74,75 Similarly, Reid et al76 performed an RCT comparing patients treated with 400 μg/d of selenium to those treated with 200 μg/d of selenium. When compared with placebo, those treated with 200 μg/d of selenium had a statistically significantly increased incidence of NMSC (P=.006); however, those treated with 400 μg/d of selenium had no significant change in total incidence of NMSC (P=.51).76 Furthermore, Vinceti et al77 performed a review of 83 studies from the literature investigating the effect of dietary selenium, and from the RCTs, there was no beneficial effect of selenium in reducing cancer risk in general; however, some studies demonstrated an increased incidence of other types of cancer, including melanoma. Of the RCTs included in the study investigating NMSC incidence specifically, it was found that the incidence was not affected by selenium administration (RR, 1.16; 95% CI, 0.30-4.42; 2 studies, 2027 participants).77 Despite data from several studies demonstrating an increased risk for NMSC, the effects of selenium on the risk for NMSC and melanoma remain unclear.
Combination Antioxidant Studies
In addition to investigating the use of single antioxidants in skin cancer prevention, studies utilizing the combination of various antioxidants or other dietary minerals have been conducted. Hercberg et al78 performed a randomized, double-blinded, placebo-controlled trial of 13,017 adults (7876 women and 5141 men) receiving a combination of 120 mg vitamin C, 30 mg vitamin E, 100 μg selenium, 6 mg beta carotene, and 20 mg zinc. Study participants were followed for an average of 7.5 years, and the development of skin cancers were recorded. Overall, the incidence rate of skin cancer did not differ between the 2 treatment groups; however, when segregated by gender, the study found that there was an increased risk for developing skin cancer in women taking the antioxidant supplement combination compared with placebo (P=.03). This difference was not observed in the 2 treatment groups of male patients (P=.11). When looking specifically at NMSC, there was no difference between treatment groups for male or female patients (P=.39 for males; P=.15 for females). In contrast, there was a higher incidence of melanoma identified in female patients taking the combination antioxidant supplement (P=.01), but this was not seen within the male study population (P=.51).78 In addition, Chang et al79 performed a meta-analysis of 10 previously published RCTs. Analysis revealed that treatment with a variety of supplements, including vitamins A, C, E, and beta carotene, were found to have no preventative effects on the incidence of skin cancer development (RR, 0.98; CI, 0.98-1.03). Notable limitations to this study included the variability in protocols of the studies included in this meta-analysis, the limited number of RCTs investigating vitamin supplementation and the risk for skin cancer development, and the influence of dietary intake on study outcomes.79
Other Dietary Agents
Furocoumarins—Furocoumarins are botanical substances found in various fruits and plants, including many citrus products. Furocoumarins are activated by UV light radiation and can lead to development of a phototoxic eruption. Several studies have suggested a pharmacogenetic effect of furocoumarins.80 Sun et al80 collected dietary data from 47,453 men and 75,291 women on furocoumarin intake and correlation with the development of NMSC. Overall, the study suggested that the intake of furocoumarins may lead to an increase in the development of BCC (HR, 1.16; 95% CI, 1.11-1.21; P=.002); however, there was no significant association identified between total intake of furocoumarins in the risk for SCC or melanoma.80 Furthermore, Sakaki et al81 conducted a survey study looking at the consumption of citrus products and the development of NMSC. The group found that there was an increased risk for NMSC in those consuming an increased amount of citrus products (P=.007).81
Conclusion
Dietary antioxidants have been investigated for their potential role in the prevention of tumorigenesis. Specific antioxidant vitamins, such as vitamin A derivatives and niacinamide, have demonstrated clinical utility in the prevention of NMSC in high-risk populations. Retinol also has been associated with a reduced incidence of melanoma. Numerous antioxidants have demonstrated promising data within the laboratory setting; however, inconsistent results have been appreciated in humans. Furthermore, several research studies suggest that folate, vitamin D, and furocoumarins may be associated with an increased risk for skin cancer development; however, these studies are inconclusive, and dietary studies are challenging to conduct. Overall, RCTs investigating the role of antioxidants for chemoprevention are limited. Moreover, the study of dietary antioxidants and vitamins may be affected by various confounding variables that can be difficult to account for because of patients’ potentially poor recall of dietary intake and the effect of dietary intake in supplemental studies. Given the increasing prevalence of skin cancer worldwide, further research into the clinical utility of antioxidants in skin cancer prevention is warranted.
- Siegel RL, Miller KD, Fuchs HE, et al. Cancer statistics, 2022. CA Cancer J Clin. 2022;72:7-33.
- Global Burden of Disease Cancer Collaboration; Fitzmaurice C, Abate D, Abbasi N, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 1990 to 2017: a systematic analysis for the Global Burden of Disease Study. JAMA Oncol. 2019;5:1749-1768.
- Leiter U, Keim U, Garbe C. Epidemiology of skin cancer: update 2019. In: Reichrath J, ed. Sunlight, Vitamin D and Skin Cancer. Springer International Publishing; 2020:123-139.
- Bradford PT. Skin cancer in skin of color. Dermatol Nurs. 2009;21:170-177, 206; quiz 178.
- Miller DL, Weinstock MA. Nonmelanoma skin cancer in the United States: incidence. J Am Acad Dermatol. 1994;30:774-778.
- Young AR, Claveau J, Rossi AB. Ultraviolet radiation and the skin: photobiology and sunscreen photoprotection. J Am Acad Dermatol. 2017;76(3S1):S100-S109.
- Pleasance ED, Cheetham RK, Stephens PJ, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191-196.
- Baek J, Lee MG. Oxidative stress and antioxidant strategies in dermatology. Redox Rep. 2016;21:164-169.
- Katta R, Brown DN. Diet and skin cancer: the potential role of dietary antioxidants in nonmelanoma skin cancer prevention. J Skin Cancer. 2015;2015:893149.
- Stoj V, Shahriari N, Shao K, et al. Nutrition and nonmelanoma skin cancers. Clin Dermatol. 2022;40:173-185.
- O’Connor EA, Evans CV, Ivlev I, et al. Vitamin and mineral supplements for the primary prevention of cardiovascular disease and cancer: updated evidence report and systematic review for the US Preventive Services Task Force. JAMA. 2022;327:2334-2347.
- National Institutes of Health Office of Dietary Supplements. Vitamin A and carotenoids. fact sheet for health professionals. Updated June 15, 2022. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/VitaminA-HealthProfessional/
- Keller KL, Fenske NA. Uses of vitamins A, C, and E and related compounds in dermatology: a review. J Am Acad Dermatol. 1998;39:611-625.
- Wright TI, Spencer JM, Flowers FP. Chemoprevention of nonmelanoma skin cancer. J Am Acad Dermatol. 2006;54:933-946; quiz 947-950.
- Bushue N, Wan YJY. Retinoid pathway and cancer therapeutics. Adv Drug Deliv Rev. 2010;62:1285-1298.
- Stahl W, Sies H. β-Carotene and other carotenoids in protection from sunlight. Am J Clin Nutr. 2012;96:1179S-1184S.
- Bukhari MH, Qureshi SS, Niazi S, et al. Chemotherapeutic/chemopreventive role of retinoids in chemically induced skin carcinogenesis in albino mice. Int J Dermatol. 2007;46:1160-1165.
- Lambert LA, Wamer WG, Wei RR, et al. The protective but nonsynergistic effect of dietary beta-carotene and vitamin E on skin tumorigenesis in Skh mice. Nutr Cancer. 1994;21:1-12.
- Greenberg ER, Baron JA, Stukel TA, et al. A clinical trial of beta carotene to prevent basal-cell and squamous-cell cancers of the skin. The Skin Cancer Prevention Study Group. N Engl J Med. 1990;323:789-795.
- Frieling UM, Schaumberg DA, Kupper TS, et al. A randomized, 12-year primary-prevention trial of beta carotene supplementation for nonmelanoma skin cancer in the physician’s health study. Arch Dermatol. 2000;136:179-184.
- Naldi L, Gallus S, Tavani A, et al; Oncology Study Group of the Italian Group for Epidemiologic Research in Dermatology. Risk of melanoma and vitamin A, coffee and alcohol: a case-control study from Italy. Eur J Cancer Prev. 2004;13:503-508.
- Zhang YP, Chu RX, Liu H. Vitamin A intake and risk of melanoma: a meta-analysis. PloS One. 2014;9:e102527.
- Feskanich D, Willett WC, Hunter DJ, et al. Dietary intakes of vitamins A, C, and E and risk of melanoma in two cohorts of women. Br J Cancer. 2003;88:1381-1387.
- Bavinck JN, Tieben LM, Van der Woude FJ, et al. Prevention of skin cancer and reduction of keratotic skin lesions during acitretin therapy in renal transplant recipients: a double-blind, placebo-controlled study. J Clin Oncol. 1995;13:1933-1938.
- George R, Weightman W, Russ GR, et al. Acitretin for chemoprevention of non-melanoma skin cancers in renal transplant recipients. Australas J Dermatol. 2002;43:269-273.
- Solomon-Cohen E, Reiss-Huss S, Hodak E, et al. Low-dose acitretin for secondary prevention of keratinocyte carcinomas in solid-organ transplant recipients. Dermatology. 2022;238:161-166.
- Otley CC, Stasko T, Tope WD, et al. Chemoprevention of nonmelanoma skin cancer with systemic retinoids: practical dosing and management of adverse effects. Dermatol Surg. 2006;32:562-568.
- Kadakia KC, Barton DL, Loprinzi CL, et al. Randomized controlled trial of acitretin versus placebo in patients at high-risk for basal cell or squamous cell carcinoma of the skin (North Central Cancer Treatment Group Study 969251). Cancer. 2012;118:2128-2137.
- McKenna DB, Murphy GM. Skin cancer chemoprophylaxis in renal transplant recipients: 5 years of experience using low-dose acitretin. Br J Dermatol. 1999;140:656-660.
- National Institutes of Health Office of Dietary Supplements. Niacin: fact sheet for health professionals. Updated August 23, 2022. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/Niacin-HealthProfessional/
- Malesu R, Martin AJ, Lyons JG, et al. Nicotinamide for skin cancer chemoprevention: effects of nicotinamide on melanoma in vitro and in vivo. Photochem Photobiol Sci. 2020;19:171-179.
- Gensler HL. Prevention of photoimmunosuppression and photocarcinogenesis by topical nicotinamide. Nutr Cancer. 1997;29:157-162.
- Gensler HL, Williams T, Huang AC, et al. Oral niacin prevents photocarcinogenesis and photoimmunosuppression in mice. Nutr Cancer. 1999;34:36-41.
- Chen AC, Martin AJ, Choy B, et al. A phase 3 randomized trial of nicotinamide for skin-cancer chemoprevention. N Engl J Med. 2015;373:1618-1626.
- Drago F, Ciccarese G, Cogorno L, et al. Prevention of non-melanoma skin cancers with nicotinamide in transplant recipients: a case-control study. Eur J Dermatol. 2017;27:382-385.
- Yélamos O, Halpern AC, Weinstock MA. Reply to “A phase II randomized controlled trial of nicotinamide for skin cancer chemoprevention in renal transplant recipients.” Br J Dermatol. 2017;176:551-552.
- Scatozza F, Moschella F, D’Arcangelo D, et al. Nicotinamide inhibits melanoma in vitro and in vivo. J Exp Clin Cancer Res. 2020;39:211.
- National Institutes of Health Office of Dietary Supplements. Folate: fact sheet for health professionals. Updated November 1, 2022. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/Folate-HealthProfessional/
- Butzbach K, Epe B. Photogenotoxicity of folic acid. Free Radic Biol Med. 2013;65:821-827.
- Vollset SE, Clarke R, Lewington S, et al. Effects of folic acid supplementation on overall and site-specific cancer incidence during the randomised trials: meta-analyses of data on 50,000 individuals. Lancet. 2013;381:1029-1036.
- Donnenfeld M, Deschasaux M, Latino-Martel P, et al. Prospective association between dietary folate intake and skin cancer risk: results from the Supplémentation en Vitamines et Minéraux Antioxydants cohort. Am J Clin Nutr. 2015;102:471-478.
- Fung TT, Hunter DJ, Spiegelman D, et al. Vitamins and carotenoids intake and the risk of basal cell carcinoma of the skin in women (United States). Cancer Causes Control. 2002;13:221-230.
- Fung TT, Spiegelman D, Egan KM, et al. Vitamin and carotenoid intake and risk of squamous cell carcinoma of the skin. Int J Cancer. 2003;103:110-115.
- National Institutes of Health Office of Dietary Supplements. Vitamin C: fact sheet for health professionals. Updated March 26, 2021. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/VitaminC-HealthProfessional/
- Spoelstra-de Man AME, Elbers PWG, Oudemans-Van Straaten HM. Vitamin C: should we supplement? Curr Opin Crit Care. 2018;24:248-255.
- Moison RMW, Beijersbergen van Henegouwen GMJ. Topical antioxidant vitamins C and E prevent UVB-radiation-induced peroxidation of eicosapentaenoic acid in pig skin. Radiat Res. 2002;157:402-409.
- Lin JY, Selim MA, Shea CR, et al. UV photoprotection by combination topical antioxidants vitamin C and vitamin E. J Am Acad Dermatol. 2003;48:866-874.
- Pauling L, Willoughby R, Reynolds R, et al. Incidence of squamous cell carcinoma in hairless mice irradiated with ultraviolet light in relation to intake of ascorbic acid (vitamin C) and of D, L-alpha-tocopheryl acetate (vitamin E). Int J Vitam Nutr Res Suppl. 1982;23:53-82.
- Kune GA, Bannerman S, Field B, et al. Diet, alcohol, smoking, serum beta-carotene, and vitamin A in male nonmelanocytic skin cancer patients and controls. Nutr Cancer. 1992;18:237-244.
- Vural P, Canbaz M, Selçuki D. Plasma antioxidant defense in actinic keratosis and basal cell carcinoma. J Eur Acad Dermatol Venereol. 1999;13:96-101.
- Record IR, Dreosti IE, McInerney JK. Changes in plasma antioxidant status following consumption of diets high or low in fruit and vegetables or following dietary supplementation with an antioxidant mixture. Br J Nutr. 2001;85:459-464.
- Heinen MM, Hughes MC, Ibiebele TI, et al. Intake of antioxidant nutrients and the risk of skin cancer. Eur J Cancer. 2007;43:2707-2716.
- Yang G, Yan Y, Ma Y, et al. Vitamin C at high concentrations induces cytotoxicity in malignant melanoma but promotes tumor growth at low concentrations. Mol Carcinog. 2017;56:1965-1976.
- National Institutes of Health Office of Dietary Supplements. Vitamin D: fact sheet for health professionals. Updated August 12, 2022. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/VitaminD-HealthProfessional/
- Reichrath J, Saternus R, Vogt T. Endocrine actions of vitamin D in skin: relevance for photocarcinogenesis of non-melanoma skin cancer, and beyond. Mol Cell Endocrinol. 2017;453:96-102.
- Ellison TI, Smith MK, Gilliam AC, et al. Inactivation of the vitamin D receptor enhances susceptibility of murine skin to UV-induced tumorigenesis. J Invest Dermatol. 2008;128:2508-2517.
- Eide MJ, Johnson DA, Jacobsen GR, et al. Vitamin D and nonmelanoma skin cancer in a health maintenance organization cohort. Arch Dermatol. 2011;147:1379-1384.
- van der Pols JC, Russell A, Bauer U, et al. Vitamin D status and skin cancer risk independent of time outdoors: 11-year prospective study in an Australian community. J Invest Dermatol. 2013;133:637-641.
- Caini S, Gnagnarella P, Stanganelli I, et al. Vitamin D and the risk of non-melanoma skin cancer: a systematic literature review and meta-analysis on behalf of the Italian Melanoma Intergroup. Cancers (Basel). 2021;13:4815.
- Park SM, Li T, Wu S, et al. Vitamin D intake and risk of skin cancer in US women and men. PLoS One. 2016;11:e0160308.
- Afzal S, Nordestgaard BG, Bojesen SE. Plasma 25-hydroxyvitamin D and risk of non-melanoma and melanoma skin cancer: a prospective cohort study. J Invest Dermatol. 2013;133:629-636.
- Asgari MM, Tang J, Warton ME, et al. Association of prediagnostic serum vitamin D levels with the development of basal cell carcinoma. J Invest Dermatol. 2010;130:1438-1443.
- Tang JY, Parimi N, Wu A, et al. Inverse association between serum 25(OH) vitamin D levels and non-melanoma skin cancer in elderly men. Cancer Causes Control. 2010;21:387-391.
- Keen MA, Hassan I. Vitamin E in dermatology. Indian Dermatol Online J. 2016;7:311-315.
- National Institutes of Health Office of Dietary Supplements. Vitamin E: fact sheet for health professionals. Updated March 26, 2021. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/VitaminE-HealthProfessional/
- Pearson P, Lewis SA, Britton J, et al. The pro-oxidant activity of high-dose vitamin E supplements in vivo. BioDrugs. 2006;20:271-273.
- Gerrish KE, Gensler HL. Prevention of photocarcinogenesis by dietary vitamin E. Nutr Cancer. 1993;19:125-133.
- McVean M, Liebler DC. Prevention of DNA photodamage by vitamin E compounds and sunscreens: roles of ultraviolet absorbance and cellular uptake. Mol Carcinog. 1999;24:169-176.
- Prasad KN, Cohrs RJ, Sharma OK. Decreased expressions of c-myc and H-ras oncogenes in vitamin E succinate induced morphologically differentiated murine B-16 melanoma cells in culture. Biochem Cell Biol. 1990;68:1250-1255.
- Funasaka Y, Komoto M, Ichihashi M. Depigmenting effect of alpha-tocopheryl ferulate on normal human melanocytes. Pigment Cell Res. 2000;13(suppl 8):170-174.
- National Institutes of Health Office of Dietary Supplements. Selenium: fact sheet for health professionals. Updated March 26, 2021. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/Selenium-HealthProfessional/
- Sengupta A, Lichti UF, Carlson BA, et al. Selenoproteins are essential for proper keratinocyte function and skin development. PLoS One. 2010;5:e12249.
- Das RK, Hossain SKU, Bhattacharya S. Diphenylmethyl selenocyanate inhibits DMBA-croton oil induced two-stage mouse skin carcinogenesis by inducing apoptosis and inhibiting cutaneous cell proliferation. Cancer Lett. 2005;230:90-101.
- Clark LC, Combs GF Jr, Turnbull BW, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA. 1996;276:1957-1963.
- Duffield-Lillico AJ, Slate EH, Reid ME, et al. Selenium supplementation and secondary prevention of nonmelanoma skin cancer in a randomized trial. J Natl Cancer Inst. 2003;95:1477-1481.
- Reid ME, Duffield-Lillico AJ, Slate E, et al. The nutritional prevention of cancer: 400 mcg per day selenium treatment. Nutr Cancer. 2008;60:155-163.
- Vinceti M, Filippini T, Del Giovane C, et al. Selenium for preventing cancer. Cochrane Database Syst Rev. 2018;1:CD005195.
- Hercberg S, Ezzedine K, Guinot C, et al. Antioxidant supplementation increases the risk of skin cancers in women but not in men. J Nutr. 2007;137:2098-2105.
- Chang YJ, Myung SK, Chung ST, et al. Effects of vitamin treatment or supplements with purported antioxidant properties on skin cancer prevention: a meta-analysis of randomized controlled trials. Dermatology. 2011;223:36-44.
- Sun W, Rice MS, Park MK, et al. Intake of furocoumarins and risk of skin cancer in 2 prospective US cohort studies. J Nutr. 2020;150:1535-1544.
- Sakaki JR, Melough MM, Roberts MB, et al. Citrus consumption and the risk of non-melanoma skin cancer in the Women’s Health Initiative. Cancers (Basel). 2021;13:2173.
- Siegel RL, Miller KD, Fuchs HE, et al. Cancer statistics, 2022. CA Cancer J Clin. 2022;72:7-33.
- Global Burden of Disease Cancer Collaboration; Fitzmaurice C, Abate D, Abbasi N, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 1990 to 2017: a systematic analysis for the Global Burden of Disease Study. JAMA Oncol. 2019;5:1749-1768.
- Leiter U, Keim U, Garbe C. Epidemiology of skin cancer: update 2019. In: Reichrath J, ed. Sunlight, Vitamin D and Skin Cancer. Springer International Publishing; 2020:123-139.
- Bradford PT. Skin cancer in skin of color. Dermatol Nurs. 2009;21:170-177, 206; quiz 178.
- Miller DL, Weinstock MA. Nonmelanoma skin cancer in the United States: incidence. J Am Acad Dermatol. 1994;30:774-778.
- Young AR, Claveau J, Rossi AB. Ultraviolet radiation and the skin: photobiology and sunscreen photoprotection. J Am Acad Dermatol. 2017;76(3S1):S100-S109.
- Pleasance ED, Cheetham RK, Stephens PJ, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191-196.
- Baek J, Lee MG. Oxidative stress and antioxidant strategies in dermatology. Redox Rep. 2016;21:164-169.
- Katta R, Brown DN. Diet and skin cancer: the potential role of dietary antioxidants in nonmelanoma skin cancer prevention. J Skin Cancer. 2015;2015:893149.
- Stoj V, Shahriari N, Shao K, et al. Nutrition and nonmelanoma skin cancers. Clin Dermatol. 2022;40:173-185.
- O’Connor EA, Evans CV, Ivlev I, et al. Vitamin and mineral supplements for the primary prevention of cardiovascular disease and cancer: updated evidence report and systematic review for the US Preventive Services Task Force. JAMA. 2022;327:2334-2347.
- National Institutes of Health Office of Dietary Supplements. Vitamin A and carotenoids. fact sheet for health professionals. Updated June 15, 2022. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/VitaminA-HealthProfessional/
- Keller KL, Fenske NA. Uses of vitamins A, C, and E and related compounds in dermatology: a review. J Am Acad Dermatol. 1998;39:611-625.
- Wright TI, Spencer JM, Flowers FP. Chemoprevention of nonmelanoma skin cancer. J Am Acad Dermatol. 2006;54:933-946; quiz 947-950.
- Bushue N, Wan YJY. Retinoid pathway and cancer therapeutics. Adv Drug Deliv Rev. 2010;62:1285-1298.
- Stahl W, Sies H. β-Carotene and other carotenoids in protection from sunlight. Am J Clin Nutr. 2012;96:1179S-1184S.
- Bukhari MH, Qureshi SS, Niazi S, et al. Chemotherapeutic/chemopreventive role of retinoids in chemically induced skin carcinogenesis in albino mice. Int J Dermatol. 2007;46:1160-1165.
- Lambert LA, Wamer WG, Wei RR, et al. The protective but nonsynergistic effect of dietary beta-carotene and vitamin E on skin tumorigenesis in Skh mice. Nutr Cancer. 1994;21:1-12.
- Greenberg ER, Baron JA, Stukel TA, et al. A clinical trial of beta carotene to prevent basal-cell and squamous-cell cancers of the skin. The Skin Cancer Prevention Study Group. N Engl J Med. 1990;323:789-795.
- Frieling UM, Schaumberg DA, Kupper TS, et al. A randomized, 12-year primary-prevention trial of beta carotene supplementation for nonmelanoma skin cancer in the physician’s health study. Arch Dermatol. 2000;136:179-184.
- Naldi L, Gallus S, Tavani A, et al; Oncology Study Group of the Italian Group for Epidemiologic Research in Dermatology. Risk of melanoma and vitamin A, coffee and alcohol: a case-control study from Italy. Eur J Cancer Prev. 2004;13:503-508.
- Zhang YP, Chu RX, Liu H. Vitamin A intake and risk of melanoma: a meta-analysis. PloS One. 2014;9:e102527.
- Feskanich D, Willett WC, Hunter DJ, et al. Dietary intakes of vitamins A, C, and E and risk of melanoma in two cohorts of women. Br J Cancer. 2003;88:1381-1387.
- Bavinck JN, Tieben LM, Van der Woude FJ, et al. Prevention of skin cancer and reduction of keratotic skin lesions during acitretin therapy in renal transplant recipients: a double-blind, placebo-controlled study. J Clin Oncol. 1995;13:1933-1938.
- George R, Weightman W, Russ GR, et al. Acitretin for chemoprevention of non-melanoma skin cancers in renal transplant recipients. Australas J Dermatol. 2002;43:269-273.
- Solomon-Cohen E, Reiss-Huss S, Hodak E, et al. Low-dose acitretin for secondary prevention of keratinocyte carcinomas in solid-organ transplant recipients. Dermatology. 2022;238:161-166.
- Otley CC, Stasko T, Tope WD, et al. Chemoprevention of nonmelanoma skin cancer with systemic retinoids: practical dosing and management of adverse effects. Dermatol Surg. 2006;32:562-568.
- Kadakia KC, Barton DL, Loprinzi CL, et al. Randomized controlled trial of acitretin versus placebo in patients at high-risk for basal cell or squamous cell carcinoma of the skin (North Central Cancer Treatment Group Study 969251). Cancer. 2012;118:2128-2137.
- McKenna DB, Murphy GM. Skin cancer chemoprophylaxis in renal transplant recipients: 5 years of experience using low-dose acitretin. Br J Dermatol. 1999;140:656-660.
- National Institutes of Health Office of Dietary Supplements. Niacin: fact sheet for health professionals. Updated August 23, 2022. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/Niacin-HealthProfessional/
- Malesu R, Martin AJ, Lyons JG, et al. Nicotinamide for skin cancer chemoprevention: effects of nicotinamide on melanoma in vitro and in vivo. Photochem Photobiol Sci. 2020;19:171-179.
- Gensler HL. Prevention of photoimmunosuppression and photocarcinogenesis by topical nicotinamide. Nutr Cancer. 1997;29:157-162.
- Gensler HL, Williams T, Huang AC, et al. Oral niacin prevents photocarcinogenesis and photoimmunosuppression in mice. Nutr Cancer. 1999;34:36-41.
- Chen AC, Martin AJ, Choy B, et al. A phase 3 randomized trial of nicotinamide for skin-cancer chemoprevention. N Engl J Med. 2015;373:1618-1626.
- Drago F, Ciccarese G, Cogorno L, et al. Prevention of non-melanoma skin cancers with nicotinamide in transplant recipients: a case-control study. Eur J Dermatol. 2017;27:382-385.
- Yélamos O, Halpern AC, Weinstock MA. Reply to “A phase II randomized controlled trial of nicotinamide for skin cancer chemoprevention in renal transplant recipients.” Br J Dermatol. 2017;176:551-552.
- Scatozza F, Moschella F, D’Arcangelo D, et al. Nicotinamide inhibits melanoma in vitro and in vivo. J Exp Clin Cancer Res. 2020;39:211.
- National Institutes of Health Office of Dietary Supplements. Folate: fact sheet for health professionals. Updated November 1, 2022. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/Folate-HealthProfessional/
- Butzbach K, Epe B. Photogenotoxicity of folic acid. Free Radic Biol Med. 2013;65:821-827.
- Vollset SE, Clarke R, Lewington S, et al. Effects of folic acid supplementation on overall and site-specific cancer incidence during the randomised trials: meta-analyses of data on 50,000 individuals. Lancet. 2013;381:1029-1036.
- Donnenfeld M, Deschasaux M, Latino-Martel P, et al. Prospective association between dietary folate intake and skin cancer risk: results from the Supplémentation en Vitamines et Minéraux Antioxydants cohort. Am J Clin Nutr. 2015;102:471-478.
- Fung TT, Hunter DJ, Spiegelman D, et al. Vitamins and carotenoids intake and the risk of basal cell carcinoma of the skin in women (United States). Cancer Causes Control. 2002;13:221-230.
- Fung TT, Spiegelman D, Egan KM, et al. Vitamin and carotenoid intake and risk of squamous cell carcinoma of the skin. Int J Cancer. 2003;103:110-115.
- National Institutes of Health Office of Dietary Supplements. Vitamin C: fact sheet for health professionals. Updated March 26, 2021. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/VitaminC-HealthProfessional/
- Spoelstra-de Man AME, Elbers PWG, Oudemans-Van Straaten HM. Vitamin C: should we supplement? Curr Opin Crit Care. 2018;24:248-255.
- Moison RMW, Beijersbergen van Henegouwen GMJ. Topical antioxidant vitamins C and E prevent UVB-radiation-induced peroxidation of eicosapentaenoic acid in pig skin. Radiat Res. 2002;157:402-409.
- Lin JY, Selim MA, Shea CR, et al. UV photoprotection by combination topical antioxidants vitamin C and vitamin E. J Am Acad Dermatol. 2003;48:866-874.
- Pauling L, Willoughby R, Reynolds R, et al. Incidence of squamous cell carcinoma in hairless mice irradiated with ultraviolet light in relation to intake of ascorbic acid (vitamin C) and of D, L-alpha-tocopheryl acetate (vitamin E). Int J Vitam Nutr Res Suppl. 1982;23:53-82.
- Kune GA, Bannerman S, Field B, et al. Diet, alcohol, smoking, serum beta-carotene, and vitamin A in male nonmelanocytic skin cancer patients and controls. Nutr Cancer. 1992;18:237-244.
- Vural P, Canbaz M, Selçuki D. Plasma antioxidant defense in actinic keratosis and basal cell carcinoma. J Eur Acad Dermatol Venereol. 1999;13:96-101.
- Record IR, Dreosti IE, McInerney JK. Changes in plasma antioxidant status following consumption of diets high or low in fruit and vegetables or following dietary supplementation with an antioxidant mixture. Br J Nutr. 2001;85:459-464.
- Heinen MM, Hughes MC, Ibiebele TI, et al. Intake of antioxidant nutrients and the risk of skin cancer. Eur J Cancer. 2007;43:2707-2716.
- Yang G, Yan Y, Ma Y, et al. Vitamin C at high concentrations induces cytotoxicity in malignant melanoma but promotes tumor growth at low concentrations. Mol Carcinog. 2017;56:1965-1976.
- National Institutes of Health Office of Dietary Supplements. Vitamin D: fact sheet for health professionals. Updated August 12, 2022. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/VitaminD-HealthProfessional/
- Reichrath J, Saternus R, Vogt T. Endocrine actions of vitamin D in skin: relevance for photocarcinogenesis of non-melanoma skin cancer, and beyond. Mol Cell Endocrinol. 2017;453:96-102.
- Ellison TI, Smith MK, Gilliam AC, et al. Inactivation of the vitamin D receptor enhances susceptibility of murine skin to UV-induced tumorigenesis. J Invest Dermatol. 2008;128:2508-2517.
- Eide MJ, Johnson DA, Jacobsen GR, et al. Vitamin D and nonmelanoma skin cancer in a health maintenance organization cohort. Arch Dermatol. 2011;147:1379-1384.
- van der Pols JC, Russell A, Bauer U, et al. Vitamin D status and skin cancer risk independent of time outdoors: 11-year prospective study in an Australian community. J Invest Dermatol. 2013;133:637-641.
- Caini S, Gnagnarella P, Stanganelli I, et al. Vitamin D and the risk of non-melanoma skin cancer: a systematic literature review and meta-analysis on behalf of the Italian Melanoma Intergroup. Cancers (Basel). 2021;13:4815.
- Park SM, Li T, Wu S, et al. Vitamin D intake and risk of skin cancer in US women and men. PLoS One. 2016;11:e0160308.
- Afzal S, Nordestgaard BG, Bojesen SE. Plasma 25-hydroxyvitamin D and risk of non-melanoma and melanoma skin cancer: a prospective cohort study. J Invest Dermatol. 2013;133:629-636.
- Asgari MM, Tang J, Warton ME, et al. Association of prediagnostic serum vitamin D levels with the development of basal cell carcinoma. J Invest Dermatol. 2010;130:1438-1443.
- Tang JY, Parimi N, Wu A, et al. Inverse association between serum 25(OH) vitamin D levels and non-melanoma skin cancer in elderly men. Cancer Causes Control. 2010;21:387-391.
- Keen MA, Hassan I. Vitamin E in dermatology. Indian Dermatol Online J. 2016;7:311-315.
- National Institutes of Health Office of Dietary Supplements. Vitamin E: fact sheet for health professionals. Updated March 26, 2021. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/VitaminE-HealthProfessional/
- Pearson P, Lewis SA, Britton J, et al. The pro-oxidant activity of high-dose vitamin E supplements in vivo. BioDrugs. 2006;20:271-273.
- Gerrish KE, Gensler HL. Prevention of photocarcinogenesis by dietary vitamin E. Nutr Cancer. 1993;19:125-133.
- McVean M, Liebler DC. Prevention of DNA photodamage by vitamin E compounds and sunscreens: roles of ultraviolet absorbance and cellular uptake. Mol Carcinog. 1999;24:169-176.
- Prasad KN, Cohrs RJ, Sharma OK. Decreased expressions of c-myc and H-ras oncogenes in vitamin E succinate induced morphologically differentiated murine B-16 melanoma cells in culture. Biochem Cell Biol. 1990;68:1250-1255.
- Funasaka Y, Komoto M, Ichihashi M. Depigmenting effect of alpha-tocopheryl ferulate on normal human melanocytes. Pigment Cell Res. 2000;13(suppl 8):170-174.
- National Institutes of Health Office of Dietary Supplements. Selenium: fact sheet for health professionals. Updated March 26, 2021. Accessed November 14, 2022. https://ods.od.nih.gov/factsheets/Selenium-HealthProfessional/
- Sengupta A, Lichti UF, Carlson BA, et al. Selenoproteins are essential for proper keratinocyte function and skin development. PLoS One. 2010;5:e12249.
- Das RK, Hossain SKU, Bhattacharya S. Diphenylmethyl selenocyanate inhibits DMBA-croton oil induced two-stage mouse skin carcinogenesis by inducing apoptosis and inhibiting cutaneous cell proliferation. Cancer Lett. 2005;230:90-101.
- Clark LC, Combs GF Jr, Turnbull BW, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA. 1996;276:1957-1963.
- Duffield-Lillico AJ, Slate EH, Reid ME, et al. Selenium supplementation and secondary prevention of nonmelanoma skin cancer in a randomized trial. J Natl Cancer Inst. 2003;95:1477-1481.
- Reid ME, Duffield-Lillico AJ, Slate E, et al. The nutritional prevention of cancer: 400 mcg per day selenium treatment. Nutr Cancer. 2008;60:155-163.
- Vinceti M, Filippini T, Del Giovane C, et al. Selenium for preventing cancer. Cochrane Database Syst Rev. 2018;1:CD005195.
- Hercberg S, Ezzedine K, Guinot C, et al. Antioxidant supplementation increases the risk of skin cancers in women but not in men. J Nutr. 2007;137:2098-2105.
- Chang YJ, Myung SK, Chung ST, et al. Effects of vitamin treatment or supplements with purported antioxidant properties on skin cancer prevention: a meta-analysis of randomized controlled trials. Dermatology. 2011;223:36-44.
- Sun W, Rice MS, Park MK, et al. Intake of furocoumarins and risk of skin cancer in 2 prospective US cohort studies. J Nutr. 2020;150:1535-1544.
- Sakaki JR, Melough MM, Roberts MB, et al. Citrus consumption and the risk of non-melanoma skin cancer in the Women’s Health Initiative. Cancers (Basel). 2021;13:2173.
Practice Points
- Melanoma and nonmelanoma skin cancer (NMSC) are 2 of the most frequently diagnosed cancers in the United States. UV radiation plays a key role in the pathogenesis of both.
- Dietary antioxidants may mechanistically decrease DNA damage caused by UV radiation and could play a potential role in the prevention or development of melanoma and NMSC.
Epidermal Growth Factor Receptor Inhibitor–Induced Symmetrical Drug-Related Intertriginous and Flexural Exanthema: Should You Discontinue the Offending Agent?
Epidermal growth factor receptor (EGFR) inhibitors cause numerous cutaneous adverse events (AEs), including papulopustular eruptions, paronychia, acral fissures, xerosis, alopecia, and trichomegaly.1 Symmetrical drug-related intertriginous and flexural exanthema (SDRIFE) is an uncommon type IV hypersensitivity reaction reported most commonly in association with β-lactam antibiotics and other medications.2 Treatment of SDRIFE generally involves withdrawing the inciting medication; however, in SDRIFE secondary to oncologic therapies, medication withdrawal may not be feasible or desirable. We present 2 cases of SDRIFE secondary to EGFR inhibitors in which treatment was continued alongside supportive skin-directed therapies. We also review the literature.
Case Reports
Patient 1—A 65-year-old man with stage IV non–small cell lung cancer presented to the dermatology clinic with an eruption of 2 months’ duration that began in the periumbilical area and spread to the perianal area within 2 weeks of starting treatment with lazertinib and amivantamab. Physical examination was notable for Common Terminology Criteria for Adverse Events (CTCAE) Grade 2 periumbilical erythema and erosions as well as symmetric red-brown patches with linear erosions in the gluteal cleft (Figure 1) and Grade 2 facial papulopustular rash. Herpes simplex virus polymerase chain reaction and bacterial culture were negative. A skin biopsy from the left buttock revealed dermal edema and a perivascular lymphocytic infiltrate compatible with SDRIFE. Triamcinolone ointment 0.1% twice daily was initiated, then uptitrated to betamethasone ointment 0.05% twice daily with moderate improvement. The patient had a treatment interruption due to malignancy complications, at which time his skin improved, with recurrence of the eruption after treatment re-initiation. He resumed skin-directed treatment and was maintained on betamethasone ointment 0.05% and tacrolimus ointment 0.1% twice daily on alternating days. This treatment was continued for 4 months before the patient died from complications of the malignancy.

Patient 2—A 68-year-old woman with stage IV lung adenocarcinoma presented to the dermatology clinic with a rash of 3 weeks’ duration. Treatment with osimertinib was initiated 8 months prior to presentation, and there were no recent medication changes. Physical examination revealed CTCAE Grade 2 erythematous patches in the inguinal folds (Figure 2A), inframammary folds (Figure 2B), and on the nasal tip, as well as Grade 2 paronychia. The patient was managed with hydrocortisone cream 1% twice daily, and osimertinib was continued. At follow-up 4 weeks later, the erythema had faded to hyperpigmentation in affected areas with resolution of symptoms. No further treatment was required.

Comment
Supportive oncodermatologists and dermatology hospitalists should be aware of SDRIFE as an uncommon but increasingly recognized cutaneous AE of EGFR inhibitors. Other cases of SDRIFE secondary to EGFR inhibition are described in the Table.2-5 Although SDRIFE typically is treated by discontinuation of the offending agent, in all reported cases of EGFR inhibitor–associated SDRIFE the rash was CTCAE Grade 2, meaning that it did not interfere with instrumental activities of daily living. In 5 of 6 cases, EGFR therapy was continued while skin-directed therapies were used for symptom management.

Presentation of SDRIFE—Symmetrical drug-related intertriginous and flexural exanthema is characterized by a symmetric, sharply demarcated erythema in the inguinal, gluteal, or perianal area with at least 1 other flexural localization involved in the absence of systemic signs. It is observed most frequently at initial exposure or re-exposure to a medication. Onset typically is within a few hours to a few days after exposure to a medication.6 Interestingly, in this case series, half of reported SDRIFE cases developed 8 months or more after EGFR inhibitor initiation.
Pathophysiology of SDRIFE—The mechanism of SDRIFE has not been clearly elucidated; it generally is accepted to be a delayed-type hypersensitivity drug reaction, though other proposed pathophysiologic mechanisms for the distribution of SDRIFE include recall phenomenon or predisposing anatomic factors such as temperature, humidity, and apocrine or eccrine gland density.6,7 Epidermal growth factor receptor plays a critical role in regulating differentiation and proliferation of epidermal keratinocytes, hair follicles, and the sweat gland apparatus. Additionally, it has been hypothesized that EGFR inhibitor use may affect the microflora of the skin and that EGFR inhibitors directly affect the immune system, as demonstrated in an experiment showing EGFR inhibitor–treated mice had enhanced skin inflammation and contact hypersensitivity responses.8 How these disparate mechanisms may interact to produce SDRIFE and the reason for the notably delayed presentation of SDRIFE in half of the cases we reviewed is not known. Other delayed cutaneous AEs of EGFR inhibitor therapy, such as paronychia, are thought to be secondary to development of skin fragility and decreased keratinocyte proliferation with secondary infection.1 It is conceivable that a combination of proliferative, immunologic, and microbiome-related factors may each be playing a role in EGFR inhibitor–related SDRIFE.
Dermatology Inpatient Considerations—As seen in our cases, dermatologists can play a valuable role in diagnosing, grading, and managing cutaneous AEs associated with the administration of oncologic therapies. The array of cutaneous AEs has grown as cancer treatment options have expanded from conventional antimetabolite agents to kinase inhibitors and immune checkpoint inhibitors. Dermatologists may play an important role in differentiating the etiology of a skin finding (eg, infectious vs inflammatory) and can identify serious or dose-limiting reactions, such as Stevens-Johnson syndrome or drug reaction with eosinophilia and systemic symptoms (DRESS). If cutaneous AEs appear to occur secondary to administration of a chemotherapeutic agent, use of the National Cancer Institute CTCAE should be employed. For certain AEs (eg, alopecia, acneiform rashes, bullous dermatitis), specific grading has been developed based on a combination of body surface area involved, psychosocial impact, symptoms, and other associated morbidity.9
In management of chemotherapy-associated cutaneous AEs, dermatologists are likely to be the members of the health care team most comfortable with prescribing high-potency anti-inflammatory topical medications. Dermatologic consultation for management of cutaneous AEs has been shown to both reduce the need for systemic immunosuppression and limit interruptions in oncologic treatment.10
Conclusion
Epidermal growth factor receptor inhibitors commonly are prescribed for colorectal cancer, non–small cell lung cancer, and squamous cell carcinoma of the head and neck. They are associated with a variety of cutaneous AEs, including acneiform eruptions, paronychia, and xerosis, which rarely necessitate stopping EGFR inhibitor therapy. Our cases support an approach to managing EGFR inhibitor–related SDRIFE that does not involve discontinuation of the offending agent. Further studies are needed on the best supportive topical and systemic regimens for EGFR inhibitor–associated SDRIFE.
- Hu JC, Sadeghi P, Pinter-Brown LC, et al. Cutaneous side effects of epidermal growth factor receptor inhibitors: clinical presentation, pathogenesis, and management. J Am Acad Dermatol. 2007;56:317-326.
- Coppola R, Santo B, Silipigni S, et al. Symmetrical drug-related intertriginous and flexural exanthema and acneiform eruption in a patient with metastatic colorectal cancer treated with cetuximab. Clin Cancer Investig J. 2021;10:331-332.
- Yalici-Armagan B, Ayanoglu BT, Demirdag HG. Targeted tumour therapy induced papulopustular rash and other dermatologic side effects: a retrospective study. Cutan Ocul Toxicol. 2019;38:261-266.
- Copps B, Lacroix JP, Sasseville D. Symmetrical drug-related intertriginous and flexural exanthema secondary to epidermal growth factor receptor inhibitor gefitinib. JAAD Case Rep. 2020;6:172-175.
- Coppola R, Santo B, Ramella S, et al. Novel skin toxicity of epidermal growth factor receptor inhibitors: a case of intertrigo-like eruption in a patient with metastatic colorectal cancer treated with cetuximab. Clin Cancer Investig J. 2021;10:91-92.
- Häusermann P, Harr T, Bircher AJ. Baboon syndrome resulting from systemic drugs: is there strife between SDRIFE and allergic contact dermatitis syndrome? Contact Dermatitis. 2004;51:297-310.
- Wolf R, Orion E, Matz H. The baboon syndrome or intertriginous drug eruption: a report of eleven cases and a second look at its pathomechanism. Dermatol Online J. 2003;9:2.
- Mascia F, Mariani V, Girolomoni G, et al. Blockade of the EGF receptor induces a deranged chemokine expression in keratinocytes leading to enhanced skin inflammation. Am J Pathol. 2003;163:303-312.
- National Cancer Institute (U.S.). Common Terminology Criteria for Adverse Events: (CTCAE), Version 5.0. US Department of Health and Human Services; 2017. Accessed December 16, 2022. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf
- Chen ST, Molina GE, Lo JA, et al. Dermatology consultation reduces interruption of oncologic management among hospitalized patients with immune-related adverse events: a retrospective cohort study. J Am Acad Dermatol. 2020;82:994-996.
Epidermal growth factor receptor (EGFR) inhibitors cause numerous cutaneous adverse events (AEs), including papulopustular eruptions, paronychia, acral fissures, xerosis, alopecia, and trichomegaly.1 Symmetrical drug-related intertriginous and flexural exanthema (SDRIFE) is an uncommon type IV hypersensitivity reaction reported most commonly in association with β-lactam antibiotics and other medications.2 Treatment of SDRIFE generally involves withdrawing the inciting medication; however, in SDRIFE secondary to oncologic therapies, medication withdrawal may not be feasible or desirable. We present 2 cases of SDRIFE secondary to EGFR inhibitors in which treatment was continued alongside supportive skin-directed therapies. We also review the literature.
Case Reports
Patient 1—A 65-year-old man with stage IV non–small cell lung cancer presented to the dermatology clinic with an eruption of 2 months’ duration that began in the periumbilical area and spread to the perianal area within 2 weeks of starting treatment with lazertinib and amivantamab. Physical examination was notable for Common Terminology Criteria for Adverse Events (CTCAE) Grade 2 periumbilical erythema and erosions as well as symmetric red-brown patches with linear erosions in the gluteal cleft (Figure 1) and Grade 2 facial papulopustular rash. Herpes simplex virus polymerase chain reaction and bacterial culture were negative. A skin biopsy from the left buttock revealed dermal edema and a perivascular lymphocytic infiltrate compatible with SDRIFE. Triamcinolone ointment 0.1% twice daily was initiated, then uptitrated to betamethasone ointment 0.05% twice daily with moderate improvement. The patient had a treatment interruption due to malignancy complications, at which time his skin improved, with recurrence of the eruption after treatment re-initiation. He resumed skin-directed treatment and was maintained on betamethasone ointment 0.05% and tacrolimus ointment 0.1% twice daily on alternating days. This treatment was continued for 4 months before the patient died from complications of the malignancy.
Patient 2—A 68-year-old woman with stage IV lung adenocarcinoma presented to the dermatology clinic with a rash of 3 weeks’ duration. Treatment with osimertinib was initiated 8 months prior to presentation, and there were no recent medication changes. Physical examination revealed CTCAE Grade 2 erythematous patches in the inguinal folds (Figure 2A), inframammary folds (Figure 2B), and on the nasal tip, as well as Grade 2 paronychia. The patient was managed with hydrocortisone cream 1% twice daily, and osimertinib was continued. At follow-up 4 weeks later, the erythema had faded to hyperpigmentation in affected areas with resolution of symptoms. No further treatment was required.
Comment
Supportive oncodermatologists and dermatology hospitalists should be aware of SDRIFE as an uncommon but increasingly recognized cutaneous AE of EGFR inhibitors. Other cases of SDRIFE secondary to EGFR inhibition are described in the Table.2-5 Although SDRIFE typically is treated by discontinuation of the offending agent, in all reported cases of EGFR inhibitor–associated SDRIFE the rash was CTCAE Grade 2, meaning that it did not interfere with instrumental activities of daily living. In 5 of 6 cases, EGFR therapy was continued while skin-directed therapies were used for symptom management.
Presentation of SDRIFE—Symmetrical drug-related intertriginous and flexural exanthema is characterized by a symmetric, sharply demarcated erythema in the inguinal, gluteal, or perianal area with at least 1 other flexural localization involved in the absence of systemic signs. It is observed most frequently at initial exposure or re-exposure to a medication. Onset typically is within a few hours to a few days after exposure to a medication.6 Interestingly, in this case series, half of reported SDRIFE cases developed 8 months or more after EGFR inhibitor initiation.
Pathophysiology of SDRIFE—The mechanism of SDRIFE has not been clearly elucidated; it generally is accepted to be a delayed-type hypersensitivity drug reaction, though other proposed pathophysiologic mechanisms for the distribution of SDRIFE include recall phenomenon or predisposing anatomic factors such as temperature, humidity, and apocrine or eccrine gland density.6,7 Epidermal growth factor receptor plays a critical role in regulating differentiation and proliferation of epidermal keratinocytes, hair follicles, and the sweat gland apparatus. Additionally, it has been hypothesized that EGFR inhibitor use may affect the microflora of the skin and that EGFR inhibitors directly affect the immune system, as demonstrated in an experiment showing EGFR inhibitor–treated mice had enhanced skin inflammation and contact hypersensitivity responses.8 How these disparate mechanisms may interact to produce SDRIFE and the reason for the notably delayed presentation of SDRIFE in half of the cases we reviewed is not known. Other delayed cutaneous AEs of EGFR inhibitor therapy, such as paronychia, are thought to be secondary to development of skin fragility and decreased keratinocyte proliferation with secondary infection.1 It is conceivable that a combination of proliferative, immunologic, and microbiome-related factors may each be playing a role in EGFR inhibitor–related SDRIFE.
Dermatology Inpatient Considerations—As seen in our cases, dermatologists can play a valuable role in diagnosing, grading, and managing cutaneous AEs associated with the administration of oncologic therapies. The array of cutaneous AEs has grown as cancer treatment options have expanded from conventional antimetabolite agents to kinase inhibitors and immune checkpoint inhibitors. Dermatologists may play an important role in differentiating the etiology of a skin finding (eg, infectious vs inflammatory) and can identify serious or dose-limiting reactions, such as Stevens-Johnson syndrome or drug reaction with eosinophilia and systemic symptoms (DRESS). If cutaneous AEs appear to occur secondary to administration of a chemotherapeutic agent, use of the National Cancer Institute CTCAE should be employed. For certain AEs (eg, alopecia, acneiform rashes, bullous dermatitis), specific grading has been developed based on a combination of body surface area involved, psychosocial impact, symptoms, and other associated morbidity.9
In management of chemotherapy-associated cutaneous AEs, dermatologists are likely to be the members of the health care team most comfortable with prescribing high-potency anti-inflammatory topical medications. Dermatologic consultation for management of cutaneous AEs has been shown to both reduce the need for systemic immunosuppression and limit interruptions in oncologic treatment.10
Conclusion
Epidermal growth factor receptor inhibitors commonly are prescribed for colorectal cancer, non–small cell lung cancer, and squamous cell carcinoma of the head and neck. They are associated with a variety of cutaneous AEs, including acneiform eruptions, paronychia, and xerosis, which rarely necessitate stopping EGFR inhibitor therapy. Our cases support an approach to managing EGFR inhibitor–related SDRIFE that does not involve discontinuation of the offending agent. Further studies are needed on the best supportive topical and systemic regimens for EGFR inhibitor–associated SDRIFE.
Epidermal growth factor receptor (EGFR) inhibitors cause numerous cutaneous adverse events (AEs), including papulopustular eruptions, paronychia, acral fissures, xerosis, alopecia, and trichomegaly.1 Symmetrical drug-related intertriginous and flexural exanthema (SDRIFE) is an uncommon type IV hypersensitivity reaction reported most commonly in association with β-lactam antibiotics and other medications.2 Treatment of SDRIFE generally involves withdrawing the inciting medication; however, in SDRIFE secondary to oncologic therapies, medication withdrawal may not be feasible or desirable. We present 2 cases of SDRIFE secondary to EGFR inhibitors in which treatment was continued alongside supportive skin-directed therapies. We also review the literature.
Case Reports
Patient 1—A 65-year-old man with stage IV non–small cell lung cancer presented to the dermatology clinic with an eruption of 2 months’ duration that began in the periumbilical area and spread to the perianal area within 2 weeks of starting treatment with lazertinib and amivantamab. Physical examination was notable for Common Terminology Criteria for Adverse Events (CTCAE) Grade 2 periumbilical erythema and erosions as well as symmetric red-brown patches with linear erosions in the gluteal cleft (Figure 1) and Grade 2 facial papulopustular rash. Herpes simplex virus polymerase chain reaction and bacterial culture were negative. A skin biopsy from the left buttock revealed dermal edema and a perivascular lymphocytic infiltrate compatible with SDRIFE. Triamcinolone ointment 0.1% twice daily was initiated, then uptitrated to betamethasone ointment 0.05% twice daily with moderate improvement. The patient had a treatment interruption due to malignancy complications, at which time his skin improved, with recurrence of the eruption after treatment re-initiation. He resumed skin-directed treatment and was maintained on betamethasone ointment 0.05% and tacrolimus ointment 0.1% twice daily on alternating days. This treatment was continued for 4 months before the patient died from complications of the malignancy.
Patient 2—A 68-year-old woman with stage IV lung adenocarcinoma presented to the dermatology clinic with a rash of 3 weeks’ duration. Treatment with osimertinib was initiated 8 months prior to presentation, and there were no recent medication changes. Physical examination revealed CTCAE Grade 2 erythematous patches in the inguinal folds (Figure 2A), inframammary folds (Figure 2B), and on the nasal tip, as well as Grade 2 paronychia. The patient was managed with hydrocortisone cream 1% twice daily, and osimertinib was continued. At follow-up 4 weeks later, the erythema had faded to hyperpigmentation in affected areas with resolution of symptoms. No further treatment was required.
Comment
Supportive oncodermatologists and dermatology hospitalists should be aware of SDRIFE as an uncommon but increasingly recognized cutaneous AE of EGFR inhibitors. Other cases of SDRIFE secondary to EGFR inhibition are described in the Table.2-5 Although SDRIFE typically is treated by discontinuation of the offending agent, in all reported cases of EGFR inhibitor–associated SDRIFE the rash was CTCAE Grade 2, meaning that it did not interfere with instrumental activities of daily living. In 5 of 6 cases, EGFR therapy was continued while skin-directed therapies were used for symptom management.
Presentation of SDRIFE—Symmetrical drug-related intertriginous and flexural exanthema is characterized by a symmetric, sharply demarcated erythema in the inguinal, gluteal, or perianal area with at least 1 other flexural localization involved in the absence of systemic signs. It is observed most frequently at initial exposure or re-exposure to a medication. Onset typically is within a few hours to a few days after exposure to a medication.6 Interestingly, in this case series, half of reported SDRIFE cases developed 8 months or more after EGFR inhibitor initiation.
Pathophysiology of SDRIFE—The mechanism of SDRIFE has not been clearly elucidated; it generally is accepted to be a delayed-type hypersensitivity drug reaction, though other proposed pathophysiologic mechanisms for the distribution of SDRIFE include recall phenomenon or predisposing anatomic factors such as temperature, humidity, and apocrine or eccrine gland density.6,7 Epidermal growth factor receptor plays a critical role in regulating differentiation and proliferation of epidermal keratinocytes, hair follicles, and the sweat gland apparatus. Additionally, it has been hypothesized that EGFR inhibitor use may affect the microflora of the skin and that EGFR inhibitors directly affect the immune system, as demonstrated in an experiment showing EGFR inhibitor–treated mice had enhanced skin inflammation and contact hypersensitivity responses.8 How these disparate mechanisms may interact to produce SDRIFE and the reason for the notably delayed presentation of SDRIFE in half of the cases we reviewed is not known. Other delayed cutaneous AEs of EGFR inhibitor therapy, such as paronychia, are thought to be secondary to development of skin fragility and decreased keratinocyte proliferation with secondary infection.1 It is conceivable that a combination of proliferative, immunologic, and microbiome-related factors may each be playing a role in EGFR inhibitor–related SDRIFE.
Dermatology Inpatient Considerations—As seen in our cases, dermatologists can play a valuable role in diagnosing, grading, and managing cutaneous AEs associated with the administration of oncologic therapies. The array of cutaneous AEs has grown as cancer treatment options have expanded from conventional antimetabolite agents to kinase inhibitors and immune checkpoint inhibitors. Dermatologists may play an important role in differentiating the etiology of a skin finding (eg, infectious vs inflammatory) and can identify serious or dose-limiting reactions, such as Stevens-Johnson syndrome or drug reaction with eosinophilia and systemic symptoms (DRESS). If cutaneous AEs appear to occur secondary to administration of a chemotherapeutic agent, use of the National Cancer Institute CTCAE should be employed. For certain AEs (eg, alopecia, acneiform rashes, bullous dermatitis), specific grading has been developed based on a combination of body surface area involved, psychosocial impact, symptoms, and other associated morbidity.9
In management of chemotherapy-associated cutaneous AEs, dermatologists are likely to be the members of the health care team most comfortable with prescribing high-potency anti-inflammatory topical medications. Dermatologic consultation for management of cutaneous AEs has been shown to both reduce the need for systemic immunosuppression and limit interruptions in oncologic treatment.10
Conclusion
Epidermal growth factor receptor inhibitors commonly are prescribed for colorectal cancer, non–small cell lung cancer, and squamous cell carcinoma of the head and neck. They are associated with a variety of cutaneous AEs, including acneiform eruptions, paronychia, and xerosis, which rarely necessitate stopping EGFR inhibitor therapy. Our cases support an approach to managing EGFR inhibitor–related SDRIFE that does not involve discontinuation of the offending agent. Further studies are needed on the best supportive topical and systemic regimens for EGFR inhibitor–associated SDRIFE.
- Hu JC, Sadeghi P, Pinter-Brown LC, et al. Cutaneous side effects of epidermal growth factor receptor inhibitors: clinical presentation, pathogenesis, and management. J Am Acad Dermatol. 2007;56:317-326.
- Coppola R, Santo B, Silipigni S, et al. Symmetrical drug-related intertriginous and flexural exanthema and acneiform eruption in a patient with metastatic colorectal cancer treated with cetuximab. Clin Cancer Investig J. 2021;10:331-332.
- Yalici-Armagan B, Ayanoglu BT, Demirdag HG. Targeted tumour therapy induced papulopustular rash and other dermatologic side effects: a retrospective study. Cutan Ocul Toxicol. 2019;38:261-266.
- Copps B, Lacroix JP, Sasseville D. Symmetrical drug-related intertriginous and flexural exanthema secondary to epidermal growth factor receptor inhibitor gefitinib. JAAD Case Rep. 2020;6:172-175.
- Coppola R, Santo B, Ramella S, et al. Novel skin toxicity of epidermal growth factor receptor inhibitors: a case of intertrigo-like eruption in a patient with metastatic colorectal cancer treated with cetuximab. Clin Cancer Investig J. 2021;10:91-92.
- Häusermann P, Harr T, Bircher AJ. Baboon syndrome resulting from systemic drugs: is there strife between SDRIFE and allergic contact dermatitis syndrome? Contact Dermatitis. 2004;51:297-310.
- Wolf R, Orion E, Matz H. The baboon syndrome or intertriginous drug eruption: a report of eleven cases and a second look at its pathomechanism. Dermatol Online J. 2003;9:2.
- Mascia F, Mariani V, Girolomoni G, et al. Blockade of the EGF receptor induces a deranged chemokine expression in keratinocytes leading to enhanced skin inflammation. Am J Pathol. 2003;163:303-312.
- National Cancer Institute (U.S.). Common Terminology Criteria for Adverse Events: (CTCAE), Version 5.0. US Department of Health and Human Services; 2017. Accessed December 16, 2022. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf
- Chen ST, Molina GE, Lo JA, et al. Dermatology consultation reduces interruption of oncologic management among hospitalized patients with immune-related adverse events: a retrospective cohort study. J Am Acad Dermatol. 2020;82:994-996.
- Hu JC, Sadeghi P, Pinter-Brown LC, et al. Cutaneous side effects of epidermal growth factor receptor inhibitors: clinical presentation, pathogenesis, and management. J Am Acad Dermatol. 2007;56:317-326.
- Coppola R, Santo B, Silipigni S, et al. Symmetrical drug-related intertriginous and flexural exanthema and acneiform eruption in a patient with metastatic colorectal cancer treated with cetuximab. Clin Cancer Investig J. 2021;10:331-332.
- Yalici-Armagan B, Ayanoglu BT, Demirdag HG. Targeted tumour therapy induced papulopustular rash and other dermatologic side effects: a retrospective study. Cutan Ocul Toxicol. 2019;38:261-266.
- Copps B, Lacroix JP, Sasseville D. Symmetrical drug-related intertriginous and flexural exanthema secondary to epidermal growth factor receptor inhibitor gefitinib. JAAD Case Rep. 2020;6:172-175.
- Coppola R, Santo B, Ramella S, et al. Novel skin toxicity of epidermal growth factor receptor inhibitors: a case of intertrigo-like eruption in a patient with metastatic colorectal cancer treated with cetuximab. Clin Cancer Investig J. 2021;10:91-92.
- Häusermann P, Harr T, Bircher AJ. Baboon syndrome resulting from systemic drugs: is there strife between SDRIFE and allergic contact dermatitis syndrome? Contact Dermatitis. 2004;51:297-310.
- Wolf R, Orion E, Matz H. The baboon syndrome or intertriginous drug eruption: a report of eleven cases and a second look at its pathomechanism. Dermatol Online J. 2003;9:2.
- Mascia F, Mariani V, Girolomoni G, et al. Blockade of the EGF receptor induces a deranged chemokine expression in keratinocytes leading to enhanced skin inflammation. Am J Pathol. 2003;163:303-312.
- National Cancer Institute (U.S.). Common Terminology Criteria for Adverse Events: (CTCAE), Version 5.0. US Department of Health and Human Services; 2017. Accessed December 16, 2022. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf
- Chen ST, Molina GE, Lo JA, et al. Dermatology consultation reduces interruption of oncologic management among hospitalized patients with immune-related adverse events: a retrospective cohort study. J Am Acad Dermatol. 2020;82:994-996.
Practice Points
- Symmetrical drug-related intertriginous and flexural exanthema (SDRIFE) is an uncommon but increasingly recognized cutaneous adverse event (AE) of epidermal growth factor receptor (EGFR) inhibitors.
- Epidermal growth factor receptor inhibitor–associated SDRIFE may be approached similarly to other EGFR inhibitor–related cutaneous AEs in that it may not require discontinuation of the offending agent.

Atypical Keratotic Nodule on the Knuckle
The Diagnosis: Atypical Mycobacterial Infection
The history of rapid growth followed by shrinkage as well as the craterlike clinical appearance of our patient’s lesion were suspicious for the keratoacanthoma variant of squamous cell carcinoma (SCC). Periodic acid–Schiff green staining was negative for fungal or bacterial organisms, and the biopsy findings of keratinocyte atypia and irregular epidermal proliferation seemed to confirm our suspicion for well-differentiated SCC (Figure 1). Our patient subsequently was scheduled for Mohs micrographic surgery. Fortunately, a sample of tissue had been sent for panculture—bacterial, fungal, and mycobacterial—to rule out infectious etiologies, given the history of possible traumatic inoculation, and returned positive for Mycobacterium marinum infection prior to the surgery. Mohs surgery was canceled, and he was referred to an infectious disease specialist who started antibiotic treatment with azithromycin, ethambutol, and rifabutin. After 1 month of treatment the lesion substantially improved (Figure 2), further supporting the diagnosis of M marinum infection over SCC.

The differential diagnosis also included sporotrichosis, leishmaniasis, and chromoblastomycosis. Sporotrichosis lesions typically develop as multiple nodules and ulcers along a path of lymphatic drainage and can exhibit asteroid bodies and cigar-shaped yeast forms on histology. Chromoblastomycosis may display pseudoepitheliomatous hyperplasia and granulomatous inflammation; however, pathognomonic pigmented Medlar bodies also likely would be present.1 Leishmaniasis has a wide variety of presentations; however, it typically occurs in patients with exposure to endemic areas outside of the United States. Although leishmaniasis may demonstrate pseudoepitheliomatous hyperplasia, ulceration, and mixed inflammation on histology, it also likely would show amastigotes within dermal macrophages.2

Atypical mycobacterial infections initially may be misdiagnosed as SCC due to their tendency to induce irregular acanthosis in the form of pseudoepitheliomatous hyperplasia as well as mild keratinocyte atypia secondary to inflammation.3,4 Our case is unique because it occurred with M marinum infection specifically. The histopathologic findings of M marinum infections are variable and may additionally include granulomas, most commonly suppurative; intraepithelial abscesses; small vessel proliferation; dermal fibrosis; multinucleated giant cells; and transepidermal elimination.4,5 Periodic acid–Schiff, Ziehl-Neelsen (acid-fast bacilli), and Fite staining may be used to distinguish M marinum infection from SCC but have low sensitivities (approximately 30%). Culture remains the most reliable test, with a sensitivity of nearly 80%.5-7 In our patient, a Periodic acid–Schiff stain was obtained prior to receiving culture results, and acid-fast bacilli and Fite staining were added after the culture returned positive; however, all 3 stains failed to highlight any mycobacteria.
The primary risk factor for infection with M marinum is contact with aquatic environments or marine animals, and most cases involve the fingers or the hand.6 After we reached the diagnosis and further discussed the patient’s history, he recalled fishing for and cleaning raw shrimp around the time that he had a splinter. The Infectious Diseases Society of America recommends a treatment course extending 1 to 2 months after clinical symptoms resolve with ethambutol in addition to clarithromycin or azithromycin.8 If the infection is near a joint, rifampin should be empirically added to account for a potentially deeper infection. Imaging should be obtained to evaluate for joint space involvement, with magnetic resonance imaging being the preferred modality. If joint space involvement is confirmed, surgical debridement is indicated. Surgical debridement also is indicated for infections that fail to respond to antibiotic therapy.8
This case highlights M marinum infection as a potential mimicker of SCC, particularly if the biopsy is relatively superficial, as often occurs when obtained via the common shave technique. The distinction is critical, as M marinum infection is highly treatable and inappropriate surgery on the typical hand and finger locations may subject patients to substantial morbidity, such as the need for a skin graft, reduced mobility from scarring, or risk for serious wound infection.9 For superficial biopsies of an atypical squamous process, pathologists also may consider routinely recommending tissue culture, especially for hand and finger locations or when a history of local trauma is reported, instead of recommending complete excision or repeat biopsy alone.
- Elewski BE, Hughey LC, Hunt KM, et al. Fungal diseases. In: Bolognia J, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:1329-1363.
- Bravo FG. Protozoa and worms. In: Bolognia J, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:1470-1502.
- Zayour M, Lazova R. Pseudoepitheliomatous hyperplasia: a review. Am J Dermatopathol. 2011;33:112-122; quiz 123-126. doi:10.1097 /DAD.0b013e3181fcfb47
- Li JJ, Beresford R, Fyfe J, et al. Clinical and histopathological features of cutaneous nontuberculous mycobacterial infection: a review of 13 cases. J Cutan Pathol. 2017;44:433-443. doi:10.1111/cup.12903
- Abbas O, Marrouch N, Kattar MM, et al. Cutaneous non-tuberculous mycobacterial infections: a clinical and histopathological study of 17 cases from Lebanon. J Eur Acad Dermatol Venereol. 2011;25:33-42. doi:10.1111/j.1468-3083.2010.03684.x
- Johnson MG, Stout JE. Twenty-eight cases of Mycobacterium marinum infection: retrospective case series and literature review. Infection. 2015;43:655-662. doi:10.1007/s15010-015-0776-8
- Aubry A, Mougari F, Reibel F, et al. Mycobacterium marinum. Microbiol Spectr. 2017;5. doi:10.1128/microbiolspec.TNMI7-0038-2016
- Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416. doi:10.1164/rccm.200604-571ST
- Alam M, Ibrahim O, Nodzenski M, et al. Adverse events associated with Mohs micrographic surgery: multicenter prospective cohort study of 20,821 cases at 23 centers. JAMA Dermatol. 2013;149:1378-1385. doi:10.1001/jamadermatol.2013.6255
The Diagnosis: Atypical Mycobacterial Infection
The history of rapid growth followed by shrinkage as well as the craterlike clinical appearance of our patient’s lesion were suspicious for the keratoacanthoma variant of squamous cell carcinoma (SCC). Periodic acid–Schiff green staining was negative for fungal or bacterial organisms, and the biopsy findings of keratinocyte atypia and irregular epidermal proliferation seemed to confirm our suspicion for well-differentiated SCC (Figure 1). Our patient subsequently was scheduled for Mohs micrographic surgery. Fortunately, a sample of tissue had been sent for panculture—bacterial, fungal, and mycobacterial—to rule out infectious etiologies, given the history of possible traumatic inoculation, and returned positive for Mycobacterium marinum infection prior to the surgery. Mohs surgery was canceled, and he was referred to an infectious disease specialist who started antibiotic treatment with azithromycin, ethambutol, and rifabutin. After 1 month of treatment the lesion substantially improved (Figure 2), further supporting the diagnosis of M marinum infection over SCC.
The differential diagnosis also included sporotrichosis, leishmaniasis, and chromoblastomycosis. Sporotrichosis lesions typically develop as multiple nodules and ulcers along a path of lymphatic drainage and can exhibit asteroid bodies and cigar-shaped yeast forms on histology. Chromoblastomycosis may display pseudoepitheliomatous hyperplasia and granulomatous inflammation; however, pathognomonic pigmented Medlar bodies also likely would be present.1 Leishmaniasis has a wide variety of presentations; however, it typically occurs in patients with exposure to endemic areas outside of the United States. Although leishmaniasis may demonstrate pseudoepitheliomatous hyperplasia, ulceration, and mixed inflammation on histology, it also likely would show amastigotes within dermal macrophages.2
Atypical mycobacterial infections initially may be misdiagnosed as SCC due to their tendency to induce irregular acanthosis in the form of pseudoepitheliomatous hyperplasia as well as mild keratinocyte atypia secondary to inflammation.3,4 Our case is unique because it occurred with M marinum infection specifically. The histopathologic findings of M marinum infections are variable and may additionally include granulomas, most commonly suppurative; intraepithelial abscesses; small vessel proliferation; dermal fibrosis; multinucleated giant cells; and transepidermal elimination.4,5 Periodic acid–Schiff, Ziehl-Neelsen (acid-fast bacilli), and Fite staining may be used to distinguish M marinum infection from SCC but have low sensitivities (approximately 30%). Culture remains the most reliable test, with a sensitivity of nearly 80%.5-7 In our patient, a Periodic acid–Schiff stain was obtained prior to receiving culture results, and acid-fast bacilli and Fite staining were added after the culture returned positive; however, all 3 stains failed to highlight any mycobacteria.
The primary risk factor for infection with M marinum is contact with aquatic environments or marine animals, and most cases involve the fingers or the hand.6 After we reached the diagnosis and further discussed the patient’s history, he recalled fishing for and cleaning raw shrimp around the time that he had a splinter. The Infectious Diseases Society of America recommends a treatment course extending 1 to 2 months after clinical symptoms resolve with ethambutol in addition to clarithromycin or azithromycin.8 If the infection is near a joint, rifampin should be empirically added to account for a potentially deeper infection. Imaging should be obtained to evaluate for joint space involvement, with magnetic resonance imaging being the preferred modality. If joint space involvement is confirmed, surgical debridement is indicated. Surgical debridement also is indicated for infections that fail to respond to antibiotic therapy.8
This case highlights M marinum infection as a potential mimicker of SCC, particularly if the biopsy is relatively superficial, as often occurs when obtained via the common shave technique. The distinction is critical, as M marinum infection is highly treatable and inappropriate surgery on the typical hand and finger locations may subject patients to substantial morbidity, such as the need for a skin graft, reduced mobility from scarring, or risk for serious wound infection.9 For superficial biopsies of an atypical squamous process, pathologists also may consider routinely recommending tissue culture, especially for hand and finger locations or when a history of local trauma is reported, instead of recommending complete excision or repeat biopsy alone.
The Diagnosis: Atypical Mycobacterial Infection
The history of rapid growth followed by shrinkage as well as the craterlike clinical appearance of our patient’s lesion were suspicious for the keratoacanthoma variant of squamous cell carcinoma (SCC). Periodic acid–Schiff green staining was negative for fungal or bacterial organisms, and the biopsy findings of keratinocyte atypia and irregular epidermal proliferation seemed to confirm our suspicion for well-differentiated SCC (Figure 1). Our patient subsequently was scheduled for Mohs micrographic surgery. Fortunately, a sample of tissue had been sent for panculture—bacterial, fungal, and mycobacterial—to rule out infectious etiologies, given the history of possible traumatic inoculation, and returned positive for Mycobacterium marinum infection prior to the surgery. Mohs surgery was canceled, and he was referred to an infectious disease specialist who started antibiotic treatment with azithromycin, ethambutol, and rifabutin. After 1 month of treatment the lesion substantially improved (Figure 2), further supporting the diagnosis of M marinum infection over SCC.
The differential diagnosis also included sporotrichosis, leishmaniasis, and chromoblastomycosis. Sporotrichosis lesions typically develop as multiple nodules and ulcers along a path of lymphatic drainage and can exhibit asteroid bodies and cigar-shaped yeast forms on histology. Chromoblastomycosis may display pseudoepitheliomatous hyperplasia and granulomatous inflammation; however, pathognomonic pigmented Medlar bodies also likely would be present.1 Leishmaniasis has a wide variety of presentations; however, it typically occurs in patients with exposure to endemic areas outside of the United States. Although leishmaniasis may demonstrate pseudoepitheliomatous hyperplasia, ulceration, and mixed inflammation on histology, it also likely would show amastigotes within dermal macrophages.2
Atypical mycobacterial infections initially may be misdiagnosed as SCC due to their tendency to induce irregular acanthosis in the form of pseudoepitheliomatous hyperplasia as well as mild keratinocyte atypia secondary to inflammation.3,4 Our case is unique because it occurred with M marinum infection specifically. The histopathologic findings of M marinum infections are variable and may additionally include granulomas, most commonly suppurative; intraepithelial abscesses; small vessel proliferation; dermal fibrosis; multinucleated giant cells; and transepidermal elimination.4,5 Periodic acid–Schiff, Ziehl-Neelsen (acid-fast bacilli), and Fite staining may be used to distinguish M marinum infection from SCC but have low sensitivities (approximately 30%). Culture remains the most reliable test, with a sensitivity of nearly 80%.5-7 In our patient, a Periodic acid–Schiff stain was obtained prior to receiving culture results, and acid-fast bacilli and Fite staining were added after the culture returned positive; however, all 3 stains failed to highlight any mycobacteria.
The primary risk factor for infection with M marinum is contact with aquatic environments or marine animals, and most cases involve the fingers or the hand.6 After we reached the diagnosis and further discussed the patient’s history, he recalled fishing for and cleaning raw shrimp around the time that he had a splinter. The Infectious Diseases Society of America recommends a treatment course extending 1 to 2 months after clinical symptoms resolve with ethambutol in addition to clarithromycin or azithromycin.8 If the infection is near a joint, rifampin should be empirically added to account for a potentially deeper infection. Imaging should be obtained to evaluate for joint space involvement, with magnetic resonance imaging being the preferred modality. If joint space involvement is confirmed, surgical debridement is indicated. Surgical debridement also is indicated for infections that fail to respond to antibiotic therapy.8
This case highlights M marinum infection as a potential mimicker of SCC, particularly if the biopsy is relatively superficial, as often occurs when obtained via the common shave technique. The distinction is critical, as M marinum infection is highly treatable and inappropriate surgery on the typical hand and finger locations may subject patients to substantial morbidity, such as the need for a skin graft, reduced mobility from scarring, or risk for serious wound infection.9 For superficial biopsies of an atypical squamous process, pathologists also may consider routinely recommending tissue culture, especially for hand and finger locations or when a history of local trauma is reported, instead of recommending complete excision or repeat biopsy alone.
- Elewski BE, Hughey LC, Hunt KM, et al. Fungal diseases. In: Bolognia J, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:1329-1363.
- Bravo FG. Protozoa and worms. In: Bolognia J, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:1470-1502.
- Zayour M, Lazova R. Pseudoepitheliomatous hyperplasia: a review. Am J Dermatopathol. 2011;33:112-122; quiz 123-126. doi:10.1097 /DAD.0b013e3181fcfb47
- Li JJ, Beresford R, Fyfe J, et al. Clinical and histopathological features of cutaneous nontuberculous mycobacterial infection: a review of 13 cases. J Cutan Pathol. 2017;44:433-443. doi:10.1111/cup.12903
- Abbas O, Marrouch N, Kattar MM, et al. Cutaneous non-tuberculous mycobacterial infections: a clinical and histopathological study of 17 cases from Lebanon. J Eur Acad Dermatol Venereol. 2011;25:33-42. doi:10.1111/j.1468-3083.2010.03684.x
- Johnson MG, Stout JE. Twenty-eight cases of Mycobacterium marinum infection: retrospective case series and literature review. Infection. 2015;43:655-662. doi:10.1007/s15010-015-0776-8
- Aubry A, Mougari F, Reibel F, et al. Mycobacterium marinum. Microbiol Spectr. 2017;5. doi:10.1128/microbiolspec.TNMI7-0038-2016
- Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416. doi:10.1164/rccm.200604-571ST
- Alam M, Ibrahim O, Nodzenski M, et al. Adverse events associated with Mohs micrographic surgery: multicenter prospective cohort study of 20,821 cases at 23 centers. JAMA Dermatol. 2013;149:1378-1385. doi:10.1001/jamadermatol.2013.6255
- Elewski BE, Hughey LC, Hunt KM, et al. Fungal diseases. In: Bolognia J, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:1329-1363.
- Bravo FG. Protozoa and worms. In: Bolognia J, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:1470-1502.
- Zayour M, Lazova R. Pseudoepitheliomatous hyperplasia: a review. Am J Dermatopathol. 2011;33:112-122; quiz 123-126. doi:10.1097 /DAD.0b013e3181fcfb47
- Li JJ, Beresford R, Fyfe J, et al. Clinical and histopathological features of cutaneous nontuberculous mycobacterial infection: a review of 13 cases. J Cutan Pathol. 2017;44:433-443. doi:10.1111/cup.12903
- Abbas O, Marrouch N, Kattar MM, et al. Cutaneous non-tuberculous mycobacterial infections: a clinical and histopathological study of 17 cases from Lebanon. J Eur Acad Dermatol Venereol. 2011;25:33-42. doi:10.1111/j.1468-3083.2010.03684.x
- Johnson MG, Stout JE. Twenty-eight cases of Mycobacterium marinum infection: retrospective case series and literature review. Infection. 2015;43:655-662. doi:10.1007/s15010-015-0776-8
- Aubry A, Mougari F, Reibel F, et al. Mycobacterium marinum. Microbiol Spectr. 2017;5. doi:10.1128/microbiolspec.TNMI7-0038-2016
- Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416. doi:10.1164/rccm.200604-571ST
- Alam M, Ibrahim O, Nodzenski M, et al. Adverse events associated with Mohs micrographic surgery: multicenter prospective cohort study of 20,821 cases at 23 centers. JAMA Dermatol. 2013;149:1378-1385. doi:10.1001/jamadermatol.2013.6255
A 75-year-old man presented with a lesion on the knuckle of 5 months’ duration. He reported that the lesion initially grew very quickly before shrinking down to its current size. He denied any bleeding or pain but thought he may have had a splinter in the area around the time the lesion appeared. He reported spending a lot of time outdoors and noted several recent insect and tick bites. He also owned a boat and frequently went fishing. He previously had been treated for actinic keratoses but had no history of skin cancer and no family history of melanoma. Physical examination revealed a 2-cm erythematous nodule with central hyperkeratosis overlying the metacarpophalangeal joint of the right index finger. A shave biopsy was performed.


Study eyes sunscreens marketed to individuals with skin of color
, and more than 40% contain a UV blocker that may create a white cast.

Those are among the findings from a study by Michelle Xiong, a medical student at Brown University, Providence, R.I., and Erin M. Warshaw, MD, of the department of dermatology at Park Nicollet/Health Partners Health Services, Minneapolis, which was published online in the Journal of the American Academy of Dermatology.
“There is increasing awareness of the negative effects of ultraviolet (UV) light in individuals with skin of color (SOC), especially in regards to pigmentation disorders induced and/or exacerbated by UV exposure,” the authors wrote. “As a result, there has been a surge in sunscreens marketed to this population. We aimed to characterize cost, marketing claims, and potential allergenic ingredients in sunscreens marketed to individuals with SOC.”
Between December 2021 and October 2022, the researchers used the following search terms on Google: “sunscreen” plus “skin of 36 color,” “dark skin,” “brown skin,” “LatinX skin,” and/or “Black skin.” They extracted price, marketing claims, and ingredients from manufacturers’ websites and used 90 allergens contained in the American Contact Dermatitis Society 2020 Core series to identify potential allergens. Next, they combined cross-reactors/synonyms into allergen categories based on ACDS Contact Allergen Management Plan (CAMP) cross-reactor classification. If multiple ingredients in a sunscreen were represented by a single allergen category, it was counted only once. A similar approach was utilized for marketing categories.
A total of 12 sunscreens were included in the analysis: Absolute Joi, Black Girl Sunscreen, Black Girl Sunscreen Make It Matte, Bolden SPF Brightening Moisturizer, Eleven on the Defense Unrivaled Sun Serum, Kinlo Golden Rays Sunscreen, Live Tinted Hueguard 3-in-1 Mineral Sunscreen, Mele Dew The Most Sheer Moisturizer SPF30 Broad Spectrum Sunscreen, Mele No Shade Sunscreen Oil, Specific Beauty Active Radiance Day Moi, Unsun Mineral Sunscreen, and Urban Skin Rx Complexion Protection. Their average cost was $19.30 per ounce (range, $6.33-$50.00) and common marketing claims for these products were “no white cast” (91.7%), being free of an ingredient (83.3%), and “moisturizing” (75%).
Of the 12 sunscreens, 7 (58.3%) contained a chemical sunscreen agent, 5 (41.7%) contained a physical UV blocker, and all contained at least one allergen. The average number of allergens per product was 4.7, most commonly fragrance/botanicals (83.3%), tocopherol (83.3%), sodium benzoates/derivatives (58.3%), and sorbitan sesquiolate/derivatives (58.3%).
“Average cost of sunscreens marketed to individuals with SOC was $19.30/oz, much higher than the median price of $3.32/oz reported in a separate study of 65 popular sunscreens,” the study authors wrote. “As many of the sunscreens in our study were sold by smaller businesses, higher prices may be due to higher production costs or a perceived smaller market.”
The authors expressed surprise that five sunscreens marketed to individuals with SOC contained a physical UV blocker which may create a white cast. They contacted the manufacturers of these five sunscreens and confirmed that three used micronized formulations. “While ingested/inhaled nanoparticles of titanium dioxide may cause tissue effects, most studies of topical products show excellent safety,” they wrote.
They also noted that the average of 4.7 allergens per product observed in the analysis was similar to the average of 4.9 seen in a separate study of 52 popular sunscreens. “However, that study only included 34 allergens while this study evaluated 90 allergens,” the authors wrote. “Consumers and providers should be aware sunscreens marketed to individuals with SOC may cause allergic contact dermatitis,” they commented.

“It is interesting to see how costly these products are now compared to store bought and general commercially available sunscreens several years ago,” said Lawrence J. Green, clinical professor of dermatology at George Washington University, Washington, who was asked to comment on the study. “However, to me that is not surprising as products marketed and targeted to specific populations are often priced at a premium. It wasn’t clear to me how many of these specialized online SOC sunscreens are tinted. I wish the authors had compared the cost of tinted sunscreens in general to nontinted sunscreens because tinted ones are more useful for SOC, because when rubbed in, they can readily match SOC and can also offer protection in the visible light spectrum.”
The authors reported having no financial disclosures; the study had no funding source. Dr. Green disclosed that he is a speaker, consultant, or investigator for many pharmaceutical companies.
, and more than 40% contain a UV blocker that may create a white cast.
Those are among the findings from a study by Michelle Xiong, a medical student at Brown University, Providence, R.I., and Erin M. Warshaw, MD, of the department of dermatology at Park Nicollet/Health Partners Health Services, Minneapolis, which was published online in the Journal of the American Academy of Dermatology.
“There is increasing awareness of the negative effects of ultraviolet (UV) light in individuals with skin of color (SOC), especially in regards to pigmentation disorders induced and/or exacerbated by UV exposure,” the authors wrote. “As a result, there has been a surge in sunscreens marketed to this population. We aimed to characterize cost, marketing claims, and potential allergenic ingredients in sunscreens marketed to individuals with SOC.”
Between December 2021 and October 2022, the researchers used the following search terms on Google: “sunscreen” plus “skin of 36 color,” “dark skin,” “brown skin,” “LatinX skin,” and/or “Black skin.” They extracted price, marketing claims, and ingredients from manufacturers’ websites and used 90 allergens contained in the American Contact Dermatitis Society 2020 Core series to identify potential allergens. Next, they combined cross-reactors/synonyms into allergen categories based on ACDS Contact Allergen Management Plan (CAMP) cross-reactor classification. If multiple ingredients in a sunscreen were represented by a single allergen category, it was counted only once. A similar approach was utilized for marketing categories.
A total of 12 sunscreens were included in the analysis: Absolute Joi, Black Girl Sunscreen, Black Girl Sunscreen Make It Matte, Bolden SPF Brightening Moisturizer, Eleven on the Defense Unrivaled Sun Serum, Kinlo Golden Rays Sunscreen, Live Tinted Hueguard 3-in-1 Mineral Sunscreen, Mele Dew The Most Sheer Moisturizer SPF30 Broad Spectrum Sunscreen, Mele No Shade Sunscreen Oil, Specific Beauty Active Radiance Day Moi, Unsun Mineral Sunscreen, and Urban Skin Rx Complexion Protection. Their average cost was $19.30 per ounce (range, $6.33-$50.00) and common marketing claims for these products were “no white cast” (91.7%), being free of an ingredient (83.3%), and “moisturizing” (75%).
Of the 12 sunscreens, 7 (58.3%) contained a chemical sunscreen agent, 5 (41.7%) contained a physical UV blocker, and all contained at least one allergen. The average number of allergens per product was 4.7, most commonly fragrance/botanicals (83.3%), tocopherol (83.3%), sodium benzoates/derivatives (58.3%), and sorbitan sesquiolate/derivatives (58.3%).
“Average cost of sunscreens marketed to individuals with SOC was $19.30/oz, much higher than the median price of $3.32/oz reported in a separate study of 65 popular sunscreens,” the study authors wrote. “As many of the sunscreens in our study were sold by smaller businesses, higher prices may be due to higher production costs or a perceived smaller market.”
The authors expressed surprise that five sunscreens marketed to individuals with SOC contained a physical UV blocker which may create a white cast. They contacted the manufacturers of these five sunscreens and confirmed that three used micronized formulations. “While ingested/inhaled nanoparticles of titanium dioxide may cause tissue effects, most studies of topical products show excellent safety,” they wrote.
They also noted that the average of 4.7 allergens per product observed in the analysis was similar to the average of 4.9 seen in a separate study of 52 popular sunscreens. “However, that study only included 34 allergens while this study evaluated 90 allergens,” the authors wrote. “Consumers and providers should be aware sunscreens marketed to individuals with SOC may cause allergic contact dermatitis,” they commented.
“It is interesting to see how costly these products are now compared to store bought and general commercially available sunscreens several years ago,” said Lawrence J. Green, clinical professor of dermatology at George Washington University, Washington, who was asked to comment on the study. “However, to me that is not surprising as products marketed and targeted to specific populations are often priced at a premium. It wasn’t clear to me how many of these specialized online SOC sunscreens are tinted. I wish the authors had compared the cost of tinted sunscreens in general to nontinted sunscreens because tinted ones are more useful for SOC, because when rubbed in, they can readily match SOC and can also offer protection in the visible light spectrum.”
The authors reported having no financial disclosures; the study had no funding source. Dr. Green disclosed that he is a speaker, consultant, or investigator for many pharmaceutical companies.
, and more than 40% contain a UV blocker that may create a white cast.
Those are among the findings from a study by Michelle Xiong, a medical student at Brown University, Providence, R.I., and Erin M. Warshaw, MD, of the department of dermatology at Park Nicollet/Health Partners Health Services, Minneapolis, which was published online in the Journal of the American Academy of Dermatology.
“There is increasing awareness of the negative effects of ultraviolet (UV) light in individuals with skin of color (SOC), especially in regards to pigmentation disorders induced and/or exacerbated by UV exposure,” the authors wrote. “As a result, there has been a surge in sunscreens marketed to this population. We aimed to characterize cost, marketing claims, and potential allergenic ingredients in sunscreens marketed to individuals with SOC.”
Between December 2021 and October 2022, the researchers used the following search terms on Google: “sunscreen” plus “skin of 36 color,” “dark skin,” “brown skin,” “LatinX skin,” and/or “Black skin.” They extracted price, marketing claims, and ingredients from manufacturers’ websites and used 90 allergens contained in the American Contact Dermatitis Society 2020 Core series to identify potential allergens. Next, they combined cross-reactors/synonyms into allergen categories based on ACDS Contact Allergen Management Plan (CAMP) cross-reactor classification. If multiple ingredients in a sunscreen were represented by a single allergen category, it was counted only once. A similar approach was utilized for marketing categories.
A total of 12 sunscreens were included in the analysis: Absolute Joi, Black Girl Sunscreen, Black Girl Sunscreen Make It Matte, Bolden SPF Brightening Moisturizer, Eleven on the Defense Unrivaled Sun Serum, Kinlo Golden Rays Sunscreen, Live Tinted Hueguard 3-in-1 Mineral Sunscreen, Mele Dew The Most Sheer Moisturizer SPF30 Broad Spectrum Sunscreen, Mele No Shade Sunscreen Oil, Specific Beauty Active Radiance Day Moi, Unsun Mineral Sunscreen, and Urban Skin Rx Complexion Protection. Their average cost was $19.30 per ounce (range, $6.33-$50.00) and common marketing claims for these products were “no white cast” (91.7%), being free of an ingredient (83.3%), and “moisturizing” (75%).
Of the 12 sunscreens, 7 (58.3%) contained a chemical sunscreen agent, 5 (41.7%) contained a physical UV blocker, and all contained at least one allergen. The average number of allergens per product was 4.7, most commonly fragrance/botanicals (83.3%), tocopherol (83.3%), sodium benzoates/derivatives (58.3%), and sorbitan sesquiolate/derivatives (58.3%).
“Average cost of sunscreens marketed to individuals with SOC was $19.30/oz, much higher than the median price of $3.32/oz reported in a separate study of 65 popular sunscreens,” the study authors wrote. “As many of the sunscreens in our study were sold by smaller businesses, higher prices may be due to higher production costs or a perceived smaller market.”
The authors expressed surprise that five sunscreens marketed to individuals with SOC contained a physical UV blocker which may create a white cast. They contacted the manufacturers of these five sunscreens and confirmed that three used micronized formulations. “While ingested/inhaled nanoparticles of titanium dioxide may cause tissue effects, most studies of topical products show excellent safety,” they wrote.
They also noted that the average of 4.7 allergens per product observed in the analysis was similar to the average of 4.9 seen in a separate study of 52 popular sunscreens. “However, that study only included 34 allergens while this study evaluated 90 allergens,” the authors wrote. “Consumers and providers should be aware sunscreens marketed to individuals with SOC may cause allergic contact dermatitis,” they commented.
“It is interesting to see how costly these products are now compared to store bought and general commercially available sunscreens several years ago,” said Lawrence J. Green, clinical professor of dermatology at George Washington University, Washington, who was asked to comment on the study. “However, to me that is not surprising as products marketed and targeted to specific populations are often priced at a premium. It wasn’t clear to me how many of these specialized online SOC sunscreens are tinted. I wish the authors had compared the cost of tinted sunscreens in general to nontinted sunscreens because tinted ones are more useful for SOC, because when rubbed in, they can readily match SOC and can also offer protection in the visible light spectrum.”
The authors reported having no financial disclosures; the study had no funding source. Dr. Green disclosed that he is a speaker, consultant, or investigator for many pharmaceutical companies.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
What are the risk factors for Mohs surgery–related anxiety?
confirmed by a health care provider (HCP), results from a single-center survey demonstrated.
“Higher patient-reported anxiety in hospital settings is significantly linked to lower patient satisfaction with the quality of care and higher patient-reported postoperative pain,” corresponding author Ally-Khan Somani, MD, PhD, and colleagues wrote in the study, which was published online in Dermatologic Surgery. “Identifying factors associated with perioperative patient anxiety could improve outcomes and patient satisfaction.”
Dr. Somani, director of dermatologic surgery and cutaneous oncology in the department of dermatology at the University of Indiana, Indianapolis, and coauthors surveyed 145 patients who underwent Mohs micrographic surgery (MMS) at the university from February 2018 to March 2020. They collected patient self-reported demographics, medical history, and administered a 10-point visual analog scale assessment of anxiety at multiple stages. They also sought HCP-perceived assessments of anxiety and used a stepwise regression mode to explore factors that potentially contributed to anxiety outcomes. The mean age of the 145 patients was 63 years, 60% were female, and 77% had no self-reported anxiety confirmed by a prior HCP’s diagnosis.
Two-thirds of patients (66%) received a pre-MMS consultation with the surgeon, 59% had a history of skin cancer removal surgery, and 86% had 1-2 layers removed during the current MMS.
Prior to MMS, the researchers found that significant risk factors for increased anxiety included younger age, female sex, and self-reported history of anxiety confirmed by an HCP (P < .05), while intraoperatively, HCP-perceived patient anxiety increased with younger patient age and more layers removed. Following MMS, patient anxiety increased significantly with more layers removed and higher self-reported preoperative anxiety levels. “Although existing research is divided regarding the efficacy of pre-MMS consultation for anxiety reduction, these findings suggest that patient-reported and HCP-perceived anxiety were not significantly affected by in-person pre-MMS consultation with the surgeon,” Dr. Somani and colleagues wrote. “Thus, routinely recommending consultations may not be the best approach for improving anxiety outcomes.”
They acknowledged certain limitations of their analysis, including its single-center design, enrollment of demographically similar patients, and the fact that no objective measurements of anxiety such as heart rate or blood pressure were taken.
“One of the main benefits of Mohs surgery is that we are able to operate under local anesthesia, but this also means that our patients are acutely aware of everything going on around them,” said Patricia M. Richey, MD, who practices Mohs surgery and cosmetic dermatology in Washington, D.C., and was asked to comment on the study.
“I think it is so important that this study is primarily focusing on the patient experience,” she said. “While this study did not find that a pre-op consult impacted patient anxiety levels, I do think we can infer that it is critical to connect with your patients on some level prior to surgery, as it helps you tailor your process to make the day more tolerable for them [such as] playing music, determining the need for an oral anxiolytic, etc.”
Neither the researchers nor Dr. Richey reported having financial disclosures.
confirmed by a health care provider (HCP), results from a single-center survey demonstrated.
“Higher patient-reported anxiety in hospital settings is significantly linked to lower patient satisfaction with the quality of care and higher patient-reported postoperative pain,” corresponding author Ally-Khan Somani, MD, PhD, and colleagues wrote in the study, which was published online in Dermatologic Surgery. “Identifying factors associated with perioperative patient anxiety could improve outcomes and patient satisfaction.”
Dr. Somani, director of dermatologic surgery and cutaneous oncology in the department of dermatology at the University of Indiana, Indianapolis, and coauthors surveyed 145 patients who underwent Mohs micrographic surgery (MMS) at the university from February 2018 to March 2020. They collected patient self-reported demographics, medical history, and administered a 10-point visual analog scale assessment of anxiety at multiple stages. They also sought HCP-perceived assessments of anxiety and used a stepwise regression mode to explore factors that potentially contributed to anxiety outcomes. The mean age of the 145 patients was 63 years, 60% were female, and 77% had no self-reported anxiety confirmed by a prior HCP’s diagnosis.
Two-thirds of patients (66%) received a pre-MMS consultation with the surgeon, 59% had a history of skin cancer removal surgery, and 86% had 1-2 layers removed during the current MMS.
Prior to MMS, the researchers found that significant risk factors for increased anxiety included younger age, female sex, and self-reported history of anxiety confirmed by an HCP (P < .05), while intraoperatively, HCP-perceived patient anxiety increased with younger patient age and more layers removed. Following MMS, patient anxiety increased significantly with more layers removed and higher self-reported preoperative anxiety levels. “Although existing research is divided regarding the efficacy of pre-MMS consultation for anxiety reduction, these findings suggest that patient-reported and HCP-perceived anxiety were not significantly affected by in-person pre-MMS consultation with the surgeon,” Dr. Somani and colleagues wrote. “Thus, routinely recommending consultations may not be the best approach for improving anxiety outcomes.”
They acknowledged certain limitations of their analysis, including its single-center design, enrollment of demographically similar patients, and the fact that no objective measurements of anxiety such as heart rate or blood pressure were taken.
“One of the main benefits of Mohs surgery is that we are able to operate under local anesthesia, but this also means that our patients are acutely aware of everything going on around them,” said Patricia M. Richey, MD, who practices Mohs surgery and cosmetic dermatology in Washington, D.C., and was asked to comment on the study.
“I think it is so important that this study is primarily focusing on the patient experience,” she said. “While this study did not find that a pre-op consult impacted patient anxiety levels, I do think we can infer that it is critical to connect with your patients on some level prior to surgery, as it helps you tailor your process to make the day more tolerable for them [such as] playing music, determining the need for an oral anxiolytic, etc.”
Neither the researchers nor Dr. Richey reported having financial disclosures.
confirmed by a health care provider (HCP), results from a single-center survey demonstrated.
“Higher patient-reported anxiety in hospital settings is significantly linked to lower patient satisfaction with the quality of care and higher patient-reported postoperative pain,” corresponding author Ally-Khan Somani, MD, PhD, and colleagues wrote in the study, which was published online in Dermatologic Surgery. “Identifying factors associated with perioperative patient anxiety could improve outcomes and patient satisfaction.”
Dr. Somani, director of dermatologic surgery and cutaneous oncology in the department of dermatology at the University of Indiana, Indianapolis, and coauthors surveyed 145 patients who underwent Mohs micrographic surgery (MMS) at the university from February 2018 to March 2020. They collected patient self-reported demographics, medical history, and administered a 10-point visual analog scale assessment of anxiety at multiple stages. They also sought HCP-perceived assessments of anxiety and used a stepwise regression mode to explore factors that potentially contributed to anxiety outcomes. The mean age of the 145 patients was 63 years, 60% were female, and 77% had no self-reported anxiety confirmed by a prior HCP’s diagnosis.
Two-thirds of patients (66%) received a pre-MMS consultation with the surgeon, 59% had a history of skin cancer removal surgery, and 86% had 1-2 layers removed during the current MMS.
Prior to MMS, the researchers found that significant risk factors for increased anxiety included younger age, female sex, and self-reported history of anxiety confirmed by an HCP (P < .05), while intraoperatively, HCP-perceived patient anxiety increased with younger patient age and more layers removed. Following MMS, patient anxiety increased significantly with more layers removed and higher self-reported preoperative anxiety levels. “Although existing research is divided regarding the efficacy of pre-MMS consultation for anxiety reduction, these findings suggest that patient-reported and HCP-perceived anxiety were not significantly affected by in-person pre-MMS consultation with the surgeon,” Dr. Somani and colleagues wrote. “Thus, routinely recommending consultations may not be the best approach for improving anxiety outcomes.”
They acknowledged certain limitations of their analysis, including its single-center design, enrollment of demographically similar patients, and the fact that no objective measurements of anxiety such as heart rate or blood pressure were taken.
“One of the main benefits of Mohs surgery is that we are able to operate under local anesthesia, but this also means that our patients are acutely aware of everything going on around them,” said Patricia M. Richey, MD, who practices Mohs surgery and cosmetic dermatology in Washington, D.C., and was asked to comment on the study.
“I think it is so important that this study is primarily focusing on the patient experience,” she said. “While this study did not find that a pre-op consult impacted patient anxiety levels, I do think we can infer that it is critical to connect with your patients on some level prior to surgery, as it helps you tailor your process to make the day more tolerable for them [such as] playing music, determining the need for an oral anxiolytic, etc.”
Neither the researchers nor Dr. Richey reported having financial disclosures.
FROM DERMATOLOGIC SURGERY
Consider radiologic imaging for high-risk cutaneous SCC, expert advises
DENVER –
In a study published in 2020, Emily Ruiz, MD, MPH, and colleagues identified 87 CSCC tumors in 83 patients who underwent baseline or surveillance imaging primary at the Brigham and Women’s Hospital Mohs Surgery Clinic and the Dana-Farber Cancer Institute High-Risk Skin Cancer Clinic, both in Boston, from Jan. 1, 2017, to June 1, 2019. Of the 87 primary CSCCs, 48 (58%) underwent surveillance imaging. The researchers found that imaging detected additional disease in 26 patients, or 30% of cases, “whether that be nodal metastasis, local invasion beyond what was clinically accepted, or in-transit disease,” Dr. Ruiz, academic director of the Mohs and Dermatologic Surgery Center at Brigham and Women’s, said during the annual meeting of the American Society for Dermatologic Surgery. “But if you look at the 16 nodal metastases in this cohort, all were picked up on imaging and not on clinical exam.”

Since publication of these results, Dr. Ruiz routinely considers baseline radiologic imaging in T2b and T3 tumors; borderline T2a tumors (which she said they are now calling “T2a high,” for those who have one risk factor plus another intermediate risk factor),” and T2a tumors in patients who are profoundly immunosuppressed.
“My preference is to always do [the imaging] before treatment unless I’m up-staging them during surgery,” said Dr. Ruiz, who also directs the High-Risk Skin Cancer Clinic at Dana Farber. “We have picked up nodal metastases before surgery, which enables us to create a good therapeutic plan for our patients before we start operating. Then we image them every 6 months or so for about 2 years. Sometimes we will extend that out to 3 years.”
Some clinicians use sentinel lymph node biopsy (SLNB) as a diagnostic test, but there are mixed results about its prognostic significance. A retrospective observational study of 720 patients with CSCC found that SLNB provided no benefit regarding further metastasis or tumor-specific survival, compared with those who received routine observation and follow-up, “but head and neck surgeons in the U.S. are putting together some prospective data from multiple centers,” Dr. Ruiz said. “I think in the coming years, you will have more multicenter data to inform us as to whether to do SLNB or not.”
Surgery may be the mainstay of treatment for resectable SCC, but the emerging role of neoadjuvant therapeutics is changing the way oncologists treat these tumors. For example, in a phase 2 trial recently published in the New England Journal of Medicine, 79 patients with stage II-IV CSCC received up to four doses of immunotherapy with the programmed death receptor–1 (PD-1) blocker cemiplimab administered every 3 weeks. The primary endpoint was a pathologic complete response, defined as the absence of viable tumor cells in the surgical specimen at a central laboratory. The researchers observed that 68% of patients had an objective response.
“These were patients with localized tumors that were either very aggressive or had nodal metastases,” said Dr, Ruiz, who was the site primary investigator at Dana Farber and a coauthor of the NEJM study. “This has altered the way we approach treating our larger tumors that could be resectable but have a lot of disease either locally or in the nodal basin. We think that we can shrink down the tumor and make it easier to resect, but also there is the possibility or improving outcomes.”
At Brigham and Women’s and the Dana Farber, she and her colleagues consider immunotherapy for multiple recurrent tumors that have been previously irradiated; cases of large tumor burden locally or in the nodal basin; tumors that have a complex surgical plan; cases where there is a low likelihood of achieving clear surgical margins; and cases of in-transit disease.
“We use two to four doses of immunotherapy prior to surgery and assess the tumor response after two doses both clinically and radiologically,” she said. “If the tumor continues to grow, we would do surgery sooner.”
The side-effect profile of immunotherapy is another consideration. “Some patients are not appropriate for a neoadjuvant immunotherapy approach, such as transplant patients,” she said.
According to the latest National Comprehensive Cancer Network guidelines, surgery with or without adjuvant radiation is the current standard of care for treating CSCC. These guidelines were developed without much data to support the use of radiation, but a 20-year retrospective cohort study at Brigham and Women’s Hospital and the Cleveland Clinic Foundation found that adjuvant radiation following margin resection in high T-stage CSCC cut the risk of local and locoregional recurrence in half.
“This is something that radiation oncologists have told us for years, but there was no data to support it, so it was nice to see that borne out in clinical data,” said Dr. Ruiz, the study’s lead author. The 10% risk of local recurrence observed in the study “may not be high enough for some of our older patients, so we wanted to see if we could identify a group of high tumors that had higher risk of local recurrence,” she said. They found that patients who had a greater than 20% risk of poor outcome were those with recurrent tumors, those with tumors 6 cm or greater in size, and those with all four BWH risk factors (tumor diameter ≥ 2 cm, poorly differentiated histology, perineural invasion ≥ 0.1 mm, or tumor invasion beyond fat excluding bone invasion).
“Those risks were also cut in half if you added radiation,” she said. “So, the way I now approach counseling patients is, I try to estimate their baseline risk as best I can based on the tumor itself. I tell them that if they want to do adjuvant radiation it would cut the risk in half. Some patients are too frail and want to pass on it, while others are very interested.”
Of patients who did not receive radiation but had a disease recurrence, just under half of tumors were salvageable, about 25% died of their disease, and 23% had persistent disease. “I think this does support using radiation earlier on for the appropriate patient,” Dr. Ruiz said. “I consider the baseline risks [and] balance that with the patient’s comorbidities.”
Limited data exists on adjuvant immunotherapy for CSCC, but two ongoing randomized prospective clinical trials underway are studying the PD-1 inhibitors cemiplimab and pembrolizumab versus placebo. “We don’t have data yet, but prior to randomization, patients undergo surgery with macroscopic gross resection of all disease,” Dr. Ruiz said. “All tumors receive ART [adjuvant radiation therapy] prior to randomization”
Dr. Ruiz disclosed that she is a consultant for Sanofi, Regeneron, Genentech, and Jaunce Therapeutics. She is also a member of the advisory board for Checkpoint Therapeutics and is an investigator for Merck, Sanofi, and Regeneron.
DENVER –
In a study published in 2020, Emily Ruiz, MD, MPH, and colleagues identified 87 CSCC tumors in 83 patients who underwent baseline or surveillance imaging primary at the Brigham and Women’s Hospital Mohs Surgery Clinic and the Dana-Farber Cancer Institute High-Risk Skin Cancer Clinic, both in Boston, from Jan. 1, 2017, to June 1, 2019. Of the 87 primary CSCCs, 48 (58%) underwent surveillance imaging. The researchers found that imaging detected additional disease in 26 patients, or 30% of cases, “whether that be nodal metastasis, local invasion beyond what was clinically accepted, or in-transit disease,” Dr. Ruiz, academic director of the Mohs and Dermatologic Surgery Center at Brigham and Women’s, said during the annual meeting of the American Society for Dermatologic Surgery. “But if you look at the 16 nodal metastases in this cohort, all were picked up on imaging and not on clinical exam.”
Since publication of these results, Dr. Ruiz routinely considers baseline radiologic imaging in T2b and T3 tumors; borderline T2a tumors (which she said they are now calling “T2a high,” for those who have one risk factor plus another intermediate risk factor),” and T2a tumors in patients who are profoundly immunosuppressed.
“My preference is to always do [the imaging] before treatment unless I’m up-staging them during surgery,” said Dr. Ruiz, who also directs the High-Risk Skin Cancer Clinic at Dana Farber. “We have picked up nodal metastases before surgery, which enables us to create a good therapeutic plan for our patients before we start operating. Then we image them every 6 months or so for about 2 years. Sometimes we will extend that out to 3 years.”
Some clinicians use sentinel lymph node biopsy (SLNB) as a diagnostic test, but there are mixed results about its prognostic significance. A retrospective observational study of 720 patients with CSCC found that SLNB provided no benefit regarding further metastasis or tumor-specific survival, compared with those who received routine observation and follow-up, “but head and neck surgeons in the U.S. are putting together some prospective data from multiple centers,” Dr. Ruiz said. “I think in the coming years, you will have more multicenter data to inform us as to whether to do SLNB or not.”
Surgery may be the mainstay of treatment for resectable SCC, but the emerging role of neoadjuvant therapeutics is changing the way oncologists treat these tumors. For example, in a phase 2 trial recently published in the New England Journal of Medicine, 79 patients with stage II-IV CSCC received up to four doses of immunotherapy with the programmed death receptor–1 (PD-1) blocker cemiplimab administered every 3 weeks. The primary endpoint was a pathologic complete response, defined as the absence of viable tumor cells in the surgical specimen at a central laboratory. The researchers observed that 68% of patients had an objective response.
“These were patients with localized tumors that were either very aggressive or had nodal metastases,” said Dr, Ruiz, who was the site primary investigator at Dana Farber and a coauthor of the NEJM study. “This has altered the way we approach treating our larger tumors that could be resectable but have a lot of disease either locally or in the nodal basin. We think that we can shrink down the tumor and make it easier to resect, but also there is the possibility or improving outcomes.”
At Brigham and Women’s and the Dana Farber, she and her colleagues consider immunotherapy for multiple recurrent tumors that have been previously irradiated; cases of large tumor burden locally or in the nodal basin; tumors that have a complex surgical plan; cases where there is a low likelihood of achieving clear surgical margins; and cases of in-transit disease.
“We use two to four doses of immunotherapy prior to surgery and assess the tumor response after two doses both clinically and radiologically,” she said. “If the tumor continues to grow, we would do surgery sooner.”
The side-effect profile of immunotherapy is another consideration. “Some patients are not appropriate for a neoadjuvant immunotherapy approach, such as transplant patients,” she said.
According to the latest National Comprehensive Cancer Network guidelines, surgery with or without adjuvant radiation is the current standard of care for treating CSCC. These guidelines were developed without much data to support the use of radiation, but a 20-year retrospective cohort study at Brigham and Women’s Hospital and the Cleveland Clinic Foundation found that adjuvant radiation following margin resection in high T-stage CSCC cut the risk of local and locoregional recurrence in half.
“This is something that radiation oncologists have told us for years, but there was no data to support it, so it was nice to see that borne out in clinical data,” said Dr. Ruiz, the study’s lead author. The 10% risk of local recurrence observed in the study “may not be high enough for some of our older patients, so we wanted to see if we could identify a group of high tumors that had higher risk of local recurrence,” she said. They found that patients who had a greater than 20% risk of poor outcome were those with recurrent tumors, those with tumors 6 cm or greater in size, and those with all four BWH risk factors (tumor diameter ≥ 2 cm, poorly differentiated histology, perineural invasion ≥ 0.1 mm, or tumor invasion beyond fat excluding bone invasion).
“Those risks were also cut in half if you added radiation,” she said. “So, the way I now approach counseling patients is, I try to estimate their baseline risk as best I can based on the tumor itself. I tell them that if they want to do adjuvant radiation it would cut the risk in half. Some patients are too frail and want to pass on it, while others are very interested.”
Of patients who did not receive radiation but had a disease recurrence, just under half of tumors were salvageable, about 25% died of their disease, and 23% had persistent disease. “I think this does support using radiation earlier on for the appropriate patient,” Dr. Ruiz said. “I consider the baseline risks [and] balance that with the patient’s comorbidities.”
Limited data exists on adjuvant immunotherapy for CSCC, but two ongoing randomized prospective clinical trials underway are studying the PD-1 inhibitors cemiplimab and pembrolizumab versus placebo. “We don’t have data yet, but prior to randomization, patients undergo surgery with macroscopic gross resection of all disease,” Dr. Ruiz said. “All tumors receive ART [adjuvant radiation therapy] prior to randomization”
Dr. Ruiz disclosed that she is a consultant for Sanofi, Regeneron, Genentech, and Jaunce Therapeutics. She is also a member of the advisory board for Checkpoint Therapeutics and is an investigator for Merck, Sanofi, and Regeneron.
DENVER –
In a study published in 2020, Emily Ruiz, MD, MPH, and colleagues identified 87 CSCC tumors in 83 patients who underwent baseline or surveillance imaging primary at the Brigham and Women’s Hospital Mohs Surgery Clinic and the Dana-Farber Cancer Institute High-Risk Skin Cancer Clinic, both in Boston, from Jan. 1, 2017, to June 1, 2019. Of the 87 primary CSCCs, 48 (58%) underwent surveillance imaging. The researchers found that imaging detected additional disease in 26 patients, or 30% of cases, “whether that be nodal metastasis, local invasion beyond what was clinically accepted, or in-transit disease,” Dr. Ruiz, academic director of the Mohs and Dermatologic Surgery Center at Brigham and Women’s, said during the annual meeting of the American Society for Dermatologic Surgery. “But if you look at the 16 nodal metastases in this cohort, all were picked up on imaging and not on clinical exam.”
Since publication of these results, Dr. Ruiz routinely considers baseline radiologic imaging in T2b and T3 tumors; borderline T2a tumors (which she said they are now calling “T2a high,” for those who have one risk factor plus another intermediate risk factor),” and T2a tumors in patients who are profoundly immunosuppressed.
“My preference is to always do [the imaging] before treatment unless I’m up-staging them during surgery,” said Dr. Ruiz, who also directs the High-Risk Skin Cancer Clinic at Dana Farber. “We have picked up nodal metastases before surgery, which enables us to create a good therapeutic plan for our patients before we start operating. Then we image them every 6 months or so for about 2 years. Sometimes we will extend that out to 3 years.”
Some clinicians use sentinel lymph node biopsy (SLNB) as a diagnostic test, but there are mixed results about its prognostic significance. A retrospective observational study of 720 patients with CSCC found that SLNB provided no benefit regarding further metastasis or tumor-specific survival, compared with those who received routine observation and follow-up, “but head and neck surgeons in the U.S. are putting together some prospective data from multiple centers,” Dr. Ruiz said. “I think in the coming years, you will have more multicenter data to inform us as to whether to do SLNB or not.”
Surgery may be the mainstay of treatment for resectable SCC, but the emerging role of neoadjuvant therapeutics is changing the way oncologists treat these tumors. For example, in a phase 2 trial recently published in the New England Journal of Medicine, 79 patients with stage II-IV CSCC received up to four doses of immunotherapy with the programmed death receptor–1 (PD-1) blocker cemiplimab administered every 3 weeks. The primary endpoint was a pathologic complete response, defined as the absence of viable tumor cells in the surgical specimen at a central laboratory. The researchers observed that 68% of patients had an objective response.
“These were patients with localized tumors that were either very aggressive or had nodal metastases,” said Dr, Ruiz, who was the site primary investigator at Dana Farber and a coauthor of the NEJM study. “This has altered the way we approach treating our larger tumors that could be resectable but have a lot of disease either locally or in the nodal basin. We think that we can shrink down the tumor and make it easier to resect, but also there is the possibility or improving outcomes.”
At Brigham and Women’s and the Dana Farber, she and her colleagues consider immunotherapy for multiple recurrent tumors that have been previously irradiated; cases of large tumor burden locally or in the nodal basin; tumors that have a complex surgical plan; cases where there is a low likelihood of achieving clear surgical margins; and cases of in-transit disease.
“We use two to four doses of immunotherapy prior to surgery and assess the tumor response after two doses both clinically and radiologically,” she said. “If the tumor continues to grow, we would do surgery sooner.”
The side-effect profile of immunotherapy is another consideration. “Some patients are not appropriate for a neoadjuvant immunotherapy approach, such as transplant patients,” she said.
According to the latest National Comprehensive Cancer Network guidelines, surgery with or without adjuvant radiation is the current standard of care for treating CSCC. These guidelines were developed without much data to support the use of radiation, but a 20-year retrospective cohort study at Brigham and Women’s Hospital and the Cleveland Clinic Foundation found that adjuvant radiation following margin resection in high T-stage CSCC cut the risk of local and locoregional recurrence in half.
“This is something that radiation oncologists have told us for years, but there was no data to support it, so it was nice to see that borne out in clinical data,” said Dr. Ruiz, the study’s lead author. The 10% risk of local recurrence observed in the study “may not be high enough for some of our older patients, so we wanted to see if we could identify a group of high tumors that had higher risk of local recurrence,” she said. They found that patients who had a greater than 20% risk of poor outcome were those with recurrent tumors, those with tumors 6 cm or greater in size, and those with all four BWH risk factors (tumor diameter ≥ 2 cm, poorly differentiated histology, perineural invasion ≥ 0.1 mm, or tumor invasion beyond fat excluding bone invasion).
“Those risks were also cut in half if you added radiation,” she said. “So, the way I now approach counseling patients is, I try to estimate their baseline risk as best I can based on the tumor itself. I tell them that if they want to do adjuvant radiation it would cut the risk in half. Some patients are too frail and want to pass on it, while others are very interested.”
Of patients who did not receive radiation but had a disease recurrence, just under half of tumors were salvageable, about 25% died of their disease, and 23% had persistent disease. “I think this does support using radiation earlier on for the appropriate patient,” Dr. Ruiz said. “I consider the baseline risks [and] balance that with the patient’s comorbidities.”
Limited data exists on adjuvant immunotherapy for CSCC, but two ongoing randomized prospective clinical trials underway are studying the PD-1 inhibitors cemiplimab and pembrolizumab versus placebo. “We don’t have data yet, but prior to randomization, patients undergo surgery with macroscopic gross resection of all disease,” Dr. Ruiz said. “All tumors receive ART [adjuvant radiation therapy] prior to randomization”
Dr. Ruiz disclosed that she is a consultant for Sanofi, Regeneron, Genentech, and Jaunce Therapeutics. She is also a member of the advisory board for Checkpoint Therapeutics and is an investigator for Merck, Sanofi, and Regeneron.
AT ASDS 2022