User login
Rheumatology trends, research, concerns highlighted for 2016
The coming year in rheumatology brings with it a variety of trends and concerns about how rheumatologists can chart the best course for their practices and patients amid mounting fiscal and regulatory pressures.
Questions also arise as to how rheumatology can improve its attractiveness to students and residents in 2016 with the current level of effort in mentoring, outreach, and competition against higher-paying subspecialties.
There’s also high interest and expectations in the new year for studies on systemic sclerosis and microbiome research, as well as questions about what the future holds for intra-articular hyaluronic acid and over-the-counter topical nonsteroidal anti-inflammatory drugs for osteoarthritis (OA).
Rheumatology News editorial advisory board members gave their thoughts on these areas of rheumatology in 2016.

Insurance and reimbursement problems
The changing landscape in insurance plans, brought about largely by the Affordable Care Act, is having a big impact on patients and physicians, particularly in Florida, where more than 1.5 million people signed up for an ACA federal marketplace plan in 2015. Difficulty in accessing and affording care in 2016 figures to be an even greater problem, said Dr. Norman Gaylis, who is in private practice in Aventura, Fla.
In general, many policies are passing an increased burden onto patients in regard to deductibles, copayments, and costs of medications, he said. “That’s putting tremendous stress on practices. I think this is nationwide, where we’re finding that rheumatology patients are not getting access to the drugs for [several] reasons: they’ve become unaffordable, the various pharmaceutical support programs have run out of money, and the amount of work that practices are now performing in trying to get authorization for the patients far exceeds any type of revenue [it] could be generating or should be generating to cover these increased costs. So essentially there’s a reduction in reimbursement and a reduction in revenue going along at the same time.”
Dr. Gaylis noted that there is “tremendous pressure” from all sides to reduce access to rheumatology drugs, which have rising costs. For instance, the cost of a monthly supply of generic celebrex in his practice’s area is on average $120-$200, “which is almost prohibitive for many of our patients.”
“We’re finding that this year [2015] alone, 20% of patients who are on standard infusion therapy as a routine part of their management of rheumatoid arthritis have basically dropped out,” he said. “If that’s equal across the board, that means a very high number of patients are not getting optimal care.”
The trend for rheumatologists, particularly those in solo practice, to make contracts with fewer insurance companies could accelerate in 2016, Dr. Gaylis said.
Some rheumatologists are beginning to not accept insured patients with coverage from managed care companies or the lower-tier payers, and “that’s a significant trend if it starts evolving because it will create a two-tiered system in that you’ll have the more affluent patient going to one of these practices, and then you’ll have clinics where you’ll have a totally different level of care.” Whether it builds up enough to where rheumatologists begin to develop hybrid concierge practices is a fair question, he said. “It’s very difficult to conceive of a patient paying for both primary care and subspecialist concierge service. But I am starting to see signals where there may well be some integration between concierge primary care and concierge subspecialties.”

Training, mentoring more rheumatologists
Another issue going into 2016 is the lack of mentoring and assistance to medical students and residents to draw them to the subspecialty and keep them there, as well as the viability of rheumatology as an attractive subspecialty. “It’s difficult to see how we can attract medical students and residents to the specialty when the cost of their education leaves them with staggering bills to be paid. You’ve got to be extremely passionate to want to be a rheumatologist,” Dr. Gaylis said.
Dr. Elizabeth Volkmann, clinical instructor in rheumatology at the University of California, Los Angeles, agreed and said she looked forward to seeing how the future of mentoring programs in rheumatology will progress in 2016 and beyond. She noted that the American College of Rheumatology (ACR)/Childhood Arthritis and Rheumatology Research Alliance Mentoring Interest Group (AMIGO), a career-mentoring program that serves most fellows and many junior faculty in pediatric rheumatology across the United States and Canada, recently reported success in establishing mentor contact, suitability of mentor-mentee pairing, as well as benefit with respect to career development, scholarship, and work-life balance, and was especially useful to fellows, compared with junior faculty (Arthritis Care Res. 2015 Sep 28. doi: 10.1002/acr.22732).
Dr. Volkmann expressed interest in seeing how the ACR’s Choose Rheumatology! mentorship program is performing. The program seeks to pair students and residents with rheumatologist mentors who will offer advice and help to guide them. She also pointed to the European League Against Rheumatism’s working group for young rheumatologists, the Emerging EULAR Network (EMEUNET), which seeks to promote the educational, research, and mentoring needs of young clinicians and researchers in rheumatology in Europe. as a potential model for the ACR to use in the United States.
Some of the conference-based resources available to rheumatology fellows include the ACR’s 2016 State-of-the-Art Clinical Symposium, which provides a presymposium course specifically for fellows and has sessions on choosing career paths, and the Rheumatology Research Workshop, which targets rheumatologists interested in pursuing an academic research career. Fellows-in-training travel scholarships are available for both conferences.

New systemic sclerosis and microbiome research
Many new studies in systemic sclerosis will build on results obtained in earlier-phase trials or unanswered questions arising out of treatment comparison studies. “It is a very exciting time in the world of scleroderma,” said Dr. Virginia Steen, professor of medicine at Georgetown University, Washington.
At least four new trials are studying treatments for skin manifestations of systemic sclerosis, noted Dr. Steen. In patients with diffuse cutaneous systemic sclerosis, the phase II ASSET study is testing abatacept (Orencia) against placebo and another phase II study is testing riociguat (Adempas). Roche has started enrolling patients for a phase III trial of tocilizumab (Actemra) on the heels of positive findings from its phase II study. Dr. Steen and colleagues at Johns Hopkins University are also finishing up a pilot study of the effects of intravenous immunoglobulin (Privigen) on skin disease in systemic sclerosis and should have results ready for 2016.
Two additional trials will be investigating treatments for interstitial lung disease associated with systemic sclerosis: the Scleroderma Lung Study III, testing the use of mycophenolate mofetil in all patients with the addition of pirfenidone (Esbriet) or placebo, and a separate phase III study of nintedanib (Ofev) vs. placebo that just began enrolling patients.
There are also several trials of add-on therapies, including topical nitroglycerin for Raynaud’s phenomenon or riociguat for digital ulcers, and another that is testing autologous adipose–derived regenerative cells for the treatment of hand dysfunction.

In addition to these trials now underway, the expected 2016 publication of the validation of the Combined Response Index for Systemic Sclerosis will hopefully “lead to real movement forward in the treatment of systemic sclerosis,” said Dr. Daniel E. Furst, the Carl Pearson Professor of Medicine at University of California, Los Angeles.
Further examination of the Wnt signaling pathway in systemic sclerosis should also bring better insights into the disease’s pathogenesis and potential for treatment, Dr. Furst noted, as recent experimental results have shown that its activation induces fibroblast activation with subsequent myofibroblast differentiation and excessive collagen release. Small-molecule inhibitors of Wnt signaling in early clinical trials have shown promising results.
Dr. Furst and Dr. Volkmann said they also hope for studies describing better and more specific data regarding the microbiome in rheumatic disease, especially systemic sclerosis and rheumatoid arthritis, both of which have microbiome data linked with clinically meaningful outcomes (Nat Med. 2015 Aug;21[8]:895-905).
An important unanswered question, Dr. Volkmann said, is whether differences found in GI microbial composition between diseases and between different disease subtypes are clinically meaningful.

OA treatments: AAOS guidelines, OTC topical NSAIDs
The repercussions of the American Academy of Orthopaedic Surgeons 2013 clinical practice guideline’s statement on intra-articular hyaluronic acid treatment of knee OA will continue to be felt in 2016, according to Dr. Roy D. Altman, professor emeritus of medicine at University of California, Los Angeles.
The AAOS took a different stance from all other recent treatment guidelines for knee OA by stating: “Intra-articular hyaluronic acid is no longer recommended as a method of treatment for patients with symptomatic osteoarthritis of the knee.” The AAOS’s recommendation didn’t create much confusion with physicians and patients because its use continues to increase, but instead it has contributed to “a plethora of contradictory publications and increasing resistance on coverage by insurance carriers,” Dr. Altman said.
Another unresolved issue in OA treatment is “the resistance of the FDA to approve over-the-counter topical NSAIDs,” Dr. Altman said. The FDA requires a new drug application for OTC topical NSAIDs demonstrating efficacy and safety, “as if they were completely new,” when the safety exceeds presently approved OTC oral NSAIDs and has already been shown for prescription topical NSAIDs, he said.
The coming year in rheumatology brings with it a variety of trends and concerns about how rheumatologists can chart the best course for their practices and patients amid mounting fiscal and regulatory pressures.
Questions also arise as to how rheumatology can improve its attractiveness to students and residents in 2016 with the current level of effort in mentoring, outreach, and competition against higher-paying subspecialties.
There’s also high interest and expectations in the new year for studies on systemic sclerosis and microbiome research, as well as questions about what the future holds for intra-articular hyaluronic acid and over-the-counter topical nonsteroidal anti-inflammatory drugs for osteoarthritis (OA).
Rheumatology News editorial advisory board members gave their thoughts on these areas of rheumatology in 2016.
Insurance and reimbursement problems
The changing landscape in insurance plans, brought about largely by the Affordable Care Act, is having a big impact on patients and physicians, particularly in Florida, where more than 1.5 million people signed up for an ACA federal marketplace plan in 2015. Difficulty in accessing and affording care in 2016 figures to be an even greater problem, said Dr. Norman Gaylis, who is in private practice in Aventura, Fla.
In general, many policies are passing an increased burden onto patients in regard to deductibles, copayments, and costs of medications, he said. “That’s putting tremendous stress on practices. I think this is nationwide, where we’re finding that rheumatology patients are not getting access to the drugs for [several] reasons: they’ve become unaffordable, the various pharmaceutical support programs have run out of money, and the amount of work that practices are now performing in trying to get authorization for the patients far exceeds any type of revenue [it] could be generating or should be generating to cover these increased costs. So essentially there’s a reduction in reimbursement and a reduction in revenue going along at the same time.”
Dr. Gaylis noted that there is “tremendous pressure” from all sides to reduce access to rheumatology drugs, which have rising costs. For instance, the cost of a monthly supply of generic celebrex in his practice’s area is on average $120-$200, “which is almost prohibitive for many of our patients.”
“We’re finding that this year [2015] alone, 20% of patients who are on standard infusion therapy as a routine part of their management of rheumatoid arthritis have basically dropped out,” he said. “If that’s equal across the board, that means a very high number of patients are not getting optimal care.”
The trend for rheumatologists, particularly those in solo practice, to make contracts with fewer insurance companies could accelerate in 2016, Dr. Gaylis said.
Some rheumatologists are beginning to not accept insured patients with coverage from managed care companies or the lower-tier payers, and “that’s a significant trend if it starts evolving because it will create a two-tiered system in that you’ll have the more affluent patient going to one of these practices, and then you’ll have clinics where you’ll have a totally different level of care.” Whether it builds up enough to where rheumatologists begin to develop hybrid concierge practices is a fair question, he said. “It’s very difficult to conceive of a patient paying for both primary care and subspecialist concierge service. But I am starting to see signals where there may well be some integration between concierge primary care and concierge subspecialties.”
Training, mentoring more rheumatologists
Another issue going into 2016 is the lack of mentoring and assistance to medical students and residents to draw them to the subspecialty and keep them there, as well as the viability of rheumatology as an attractive subspecialty. “It’s difficult to see how we can attract medical students and residents to the specialty when the cost of their education leaves them with staggering bills to be paid. You’ve got to be extremely passionate to want to be a rheumatologist,” Dr. Gaylis said.
Dr. Elizabeth Volkmann, clinical instructor in rheumatology at the University of California, Los Angeles, agreed and said she looked forward to seeing how the future of mentoring programs in rheumatology will progress in 2016 and beyond. She noted that the American College of Rheumatology (ACR)/Childhood Arthritis and Rheumatology Research Alliance Mentoring Interest Group (AMIGO), a career-mentoring program that serves most fellows and many junior faculty in pediatric rheumatology across the United States and Canada, recently reported success in establishing mentor contact, suitability of mentor-mentee pairing, as well as benefit with respect to career development, scholarship, and work-life balance, and was especially useful to fellows, compared with junior faculty (Arthritis Care Res. 2015 Sep 28. doi: 10.1002/acr.22732).
Dr. Volkmann expressed interest in seeing how the ACR’s Choose Rheumatology! mentorship program is performing. The program seeks to pair students and residents with rheumatologist mentors who will offer advice and help to guide them. She also pointed to the European League Against Rheumatism’s working group for young rheumatologists, the Emerging EULAR Network (EMEUNET), which seeks to promote the educational, research, and mentoring needs of young clinicians and researchers in rheumatology in Europe. as a potential model for the ACR to use in the United States.
Some of the conference-based resources available to rheumatology fellows include the ACR’s 2016 State-of-the-Art Clinical Symposium, which provides a presymposium course specifically for fellows and has sessions on choosing career paths, and the Rheumatology Research Workshop, which targets rheumatologists interested in pursuing an academic research career. Fellows-in-training travel scholarships are available for both conferences.
New systemic sclerosis and microbiome research
Many new studies in systemic sclerosis will build on results obtained in earlier-phase trials or unanswered questions arising out of treatment comparison studies. “It is a very exciting time in the world of scleroderma,” said Dr. Virginia Steen, professor of medicine at Georgetown University, Washington.
At least four new trials are studying treatments for skin manifestations of systemic sclerosis, noted Dr. Steen. In patients with diffuse cutaneous systemic sclerosis, the phase II ASSET study is testing abatacept (Orencia) against placebo and another phase II study is testing riociguat (Adempas). Roche has started enrolling patients for a phase III trial of tocilizumab (Actemra) on the heels of positive findings from its phase II study. Dr. Steen and colleagues at Johns Hopkins University are also finishing up a pilot study of the effects of intravenous immunoglobulin (Privigen) on skin disease in systemic sclerosis and should have results ready for 2016.
Two additional trials will be investigating treatments for interstitial lung disease associated with systemic sclerosis: the Scleroderma Lung Study III, testing the use of mycophenolate mofetil in all patients with the addition of pirfenidone (Esbriet) or placebo, and a separate phase III study of nintedanib (Ofev) vs. placebo that just began enrolling patients.
There are also several trials of add-on therapies, including topical nitroglycerin for Raynaud’s phenomenon or riociguat for digital ulcers, and another that is testing autologous adipose–derived regenerative cells for the treatment of hand dysfunction.
In addition to these trials now underway, the expected 2016 publication of the validation of the Combined Response Index for Systemic Sclerosis will hopefully “lead to real movement forward in the treatment of systemic sclerosis,” said Dr. Daniel E. Furst, the Carl Pearson Professor of Medicine at University of California, Los Angeles.
Further examination of the Wnt signaling pathway in systemic sclerosis should also bring better insights into the disease’s pathogenesis and potential for treatment, Dr. Furst noted, as recent experimental results have shown that its activation induces fibroblast activation with subsequent myofibroblast differentiation and excessive collagen release. Small-molecule inhibitors of Wnt signaling in early clinical trials have shown promising results.
Dr. Furst and Dr. Volkmann said they also hope for studies describing better and more specific data regarding the microbiome in rheumatic disease, especially systemic sclerosis and rheumatoid arthritis, both of which have microbiome data linked with clinically meaningful outcomes (Nat Med. 2015 Aug;21[8]:895-905).
An important unanswered question, Dr. Volkmann said, is whether differences found in GI microbial composition between diseases and between different disease subtypes are clinically meaningful.
OA treatments: AAOS guidelines, OTC topical NSAIDs
The repercussions of the American Academy of Orthopaedic Surgeons 2013 clinical practice guideline’s statement on intra-articular hyaluronic acid treatment of knee OA will continue to be felt in 2016, according to Dr. Roy D. Altman, professor emeritus of medicine at University of California, Los Angeles.
The AAOS took a different stance from all other recent treatment guidelines for knee OA by stating: “Intra-articular hyaluronic acid is no longer recommended as a method of treatment for patients with symptomatic osteoarthritis of the knee.” The AAOS’s recommendation didn’t create much confusion with physicians and patients because its use continues to increase, but instead it has contributed to “a plethora of contradictory publications and increasing resistance on coverage by insurance carriers,” Dr. Altman said.
Another unresolved issue in OA treatment is “the resistance of the FDA to approve over-the-counter topical NSAIDs,” Dr. Altman said. The FDA requires a new drug application for OTC topical NSAIDs demonstrating efficacy and safety, “as if they were completely new,” when the safety exceeds presently approved OTC oral NSAIDs and has already been shown for prescription topical NSAIDs, he said.
The coming year in rheumatology brings with it a variety of trends and concerns about how rheumatologists can chart the best course for their practices and patients amid mounting fiscal and regulatory pressures.
Questions also arise as to how rheumatology can improve its attractiveness to students and residents in 2016 with the current level of effort in mentoring, outreach, and competition against higher-paying subspecialties.
There’s also high interest and expectations in the new year for studies on systemic sclerosis and microbiome research, as well as questions about what the future holds for intra-articular hyaluronic acid and over-the-counter topical nonsteroidal anti-inflammatory drugs for osteoarthritis (OA).
Rheumatology News editorial advisory board members gave their thoughts on these areas of rheumatology in 2016.
Insurance and reimbursement problems
The changing landscape in insurance plans, brought about largely by the Affordable Care Act, is having a big impact on patients and physicians, particularly in Florida, where more than 1.5 million people signed up for an ACA federal marketplace plan in 2015. Difficulty in accessing and affording care in 2016 figures to be an even greater problem, said Dr. Norman Gaylis, who is in private practice in Aventura, Fla.
In general, many policies are passing an increased burden onto patients in regard to deductibles, copayments, and costs of medications, he said. “That’s putting tremendous stress on practices. I think this is nationwide, where we’re finding that rheumatology patients are not getting access to the drugs for [several] reasons: they’ve become unaffordable, the various pharmaceutical support programs have run out of money, and the amount of work that practices are now performing in trying to get authorization for the patients far exceeds any type of revenue [it] could be generating or should be generating to cover these increased costs. So essentially there’s a reduction in reimbursement and a reduction in revenue going along at the same time.”
Dr. Gaylis noted that there is “tremendous pressure” from all sides to reduce access to rheumatology drugs, which have rising costs. For instance, the cost of a monthly supply of generic celebrex in his practice’s area is on average $120-$200, “which is almost prohibitive for many of our patients.”
“We’re finding that this year [2015] alone, 20% of patients who are on standard infusion therapy as a routine part of their management of rheumatoid arthritis have basically dropped out,” he said. “If that’s equal across the board, that means a very high number of patients are not getting optimal care.”
The trend for rheumatologists, particularly those in solo practice, to make contracts with fewer insurance companies could accelerate in 2016, Dr. Gaylis said.
Some rheumatologists are beginning to not accept insured patients with coverage from managed care companies or the lower-tier payers, and “that’s a significant trend if it starts evolving because it will create a two-tiered system in that you’ll have the more affluent patient going to one of these practices, and then you’ll have clinics where you’ll have a totally different level of care.” Whether it builds up enough to where rheumatologists begin to develop hybrid concierge practices is a fair question, he said. “It’s very difficult to conceive of a patient paying for both primary care and subspecialist concierge service. But I am starting to see signals where there may well be some integration between concierge primary care and concierge subspecialties.”
Training, mentoring more rheumatologists
Another issue going into 2016 is the lack of mentoring and assistance to medical students and residents to draw them to the subspecialty and keep them there, as well as the viability of rheumatology as an attractive subspecialty. “It’s difficult to see how we can attract medical students and residents to the specialty when the cost of their education leaves them with staggering bills to be paid. You’ve got to be extremely passionate to want to be a rheumatologist,” Dr. Gaylis said.
Dr. Elizabeth Volkmann, clinical instructor in rheumatology at the University of California, Los Angeles, agreed and said she looked forward to seeing how the future of mentoring programs in rheumatology will progress in 2016 and beyond. She noted that the American College of Rheumatology (ACR)/Childhood Arthritis and Rheumatology Research Alliance Mentoring Interest Group (AMIGO), a career-mentoring program that serves most fellows and many junior faculty in pediatric rheumatology across the United States and Canada, recently reported success in establishing mentor contact, suitability of mentor-mentee pairing, as well as benefit with respect to career development, scholarship, and work-life balance, and was especially useful to fellows, compared with junior faculty (Arthritis Care Res. 2015 Sep 28. doi: 10.1002/acr.22732).
Dr. Volkmann expressed interest in seeing how the ACR’s Choose Rheumatology! mentorship program is performing. The program seeks to pair students and residents with rheumatologist mentors who will offer advice and help to guide them. She also pointed to the European League Against Rheumatism’s working group for young rheumatologists, the Emerging EULAR Network (EMEUNET), which seeks to promote the educational, research, and mentoring needs of young clinicians and researchers in rheumatology in Europe. as a potential model for the ACR to use in the United States.
Some of the conference-based resources available to rheumatology fellows include the ACR’s 2016 State-of-the-Art Clinical Symposium, which provides a presymposium course specifically for fellows and has sessions on choosing career paths, and the Rheumatology Research Workshop, which targets rheumatologists interested in pursuing an academic research career. Fellows-in-training travel scholarships are available for both conferences.
New systemic sclerosis and microbiome research
Many new studies in systemic sclerosis will build on results obtained in earlier-phase trials or unanswered questions arising out of treatment comparison studies. “It is a very exciting time in the world of scleroderma,” said Dr. Virginia Steen, professor of medicine at Georgetown University, Washington.
At least four new trials are studying treatments for skin manifestations of systemic sclerosis, noted Dr. Steen. In patients with diffuse cutaneous systemic sclerosis, the phase II ASSET study is testing abatacept (Orencia) against placebo and another phase II study is testing riociguat (Adempas). Roche has started enrolling patients for a phase III trial of tocilizumab (Actemra) on the heels of positive findings from its phase II study. Dr. Steen and colleagues at Johns Hopkins University are also finishing up a pilot study of the effects of intravenous immunoglobulin (Privigen) on skin disease in systemic sclerosis and should have results ready for 2016.
Two additional trials will be investigating treatments for interstitial lung disease associated with systemic sclerosis: the Scleroderma Lung Study III, testing the use of mycophenolate mofetil in all patients with the addition of pirfenidone (Esbriet) or placebo, and a separate phase III study of nintedanib (Ofev) vs. placebo that just began enrolling patients.
There are also several trials of add-on therapies, including topical nitroglycerin for Raynaud’s phenomenon or riociguat for digital ulcers, and another that is testing autologous adipose–derived regenerative cells for the treatment of hand dysfunction.
In addition to these trials now underway, the expected 2016 publication of the validation of the Combined Response Index for Systemic Sclerosis will hopefully “lead to real movement forward in the treatment of systemic sclerosis,” said Dr. Daniel E. Furst, the Carl Pearson Professor of Medicine at University of California, Los Angeles.
Further examination of the Wnt signaling pathway in systemic sclerosis should also bring better insights into the disease’s pathogenesis and potential for treatment, Dr. Furst noted, as recent experimental results have shown that its activation induces fibroblast activation with subsequent myofibroblast differentiation and excessive collagen release. Small-molecule inhibitors of Wnt signaling in early clinical trials have shown promising results.
Dr. Furst and Dr. Volkmann said they also hope for studies describing better and more specific data regarding the microbiome in rheumatic disease, especially systemic sclerosis and rheumatoid arthritis, both of which have microbiome data linked with clinically meaningful outcomes (Nat Med. 2015 Aug;21[8]:895-905).
An important unanswered question, Dr. Volkmann said, is whether differences found in GI microbial composition between diseases and between different disease subtypes are clinically meaningful.
OA treatments: AAOS guidelines, OTC topical NSAIDs
The repercussions of the American Academy of Orthopaedic Surgeons 2013 clinical practice guideline’s statement on intra-articular hyaluronic acid treatment of knee OA will continue to be felt in 2016, according to Dr. Roy D. Altman, professor emeritus of medicine at University of California, Los Angeles.
The AAOS took a different stance from all other recent treatment guidelines for knee OA by stating: “Intra-articular hyaluronic acid is no longer recommended as a method of treatment for patients with symptomatic osteoarthritis of the knee.” The AAOS’s recommendation didn’t create much confusion with physicians and patients because its use continues to increase, but instead it has contributed to “a plethora of contradictory publications and increasing resistance on coverage by insurance carriers,” Dr. Altman said.
Another unresolved issue in OA treatment is “the resistance of the FDA to approve over-the-counter topical NSAIDs,” Dr. Altman said. The FDA requires a new drug application for OTC topical NSAIDs demonstrating efficacy and safety, “as if they were completely new,” when the safety exceeds presently approved OTC oral NSAIDs and has already been shown for prescription topical NSAIDs, he said.

Concerns Grow as Top Clinicians Choose Nonclinical Roles
On a spring day a couple of years ago, I met with some internal medicine residents in a “Healthcare Systems Immersion” elective. I was to provide thoughts about the nonclinical portion of my work that I spend consulting with other hospitalist groups.

I asked for their thoughts about whether the ranks of doctors providing direct bedside care were losing too many of the most talented clinicians to nonclinical roles. The most vocal resident was confident that was not the case; these doctors would ultimately have a positive impact on the care of larger numbers of patients through administrative work than through direct patient care.
I wonder if she is right.
Numerous Hospitalists Opt for Nonnclinical Work
It seems like lots of hospitalists are transitioning to nonclinical work. My experience is that most who have administrative or other nonclinical roles continue—for part of their time—to provide direct patient care. But some leave clinical work behind altogether. Some of them are very prominent people in our field, like the top physician at CMS, the current U.S. Surgeon General, and this year’s most influential physician executive as judged by Modern Healthcare. I think it is pretty cool that these people come from our specialty.
I couldn’t find published survey data on the portion of hospitalists, or doctors in any specialty, who have entirely (or almost entirely) nonclinical roles. My impression is that this was a vanishingly small number across all specialties 30 or 40 years ago, but it seems to have increased pretty dramatically in the last 10 years. At the start of my career, few hospitals had a physician in an administrative position. Now it is common.
Physician leadership roles now include information technology (CMIO), quality (CQO), leader of the employed physician group, and hospital CEO (at least two hospitalists I know are in this role). And there are lots of nonclinical roles for doctors outside of hospitals.
Pros, Cons for Healthcare
I’ve had mixed feelings watching many people leave clinical practice. Most of them, like those mentioned above, continue to make important contributions to our healthcare system; they improve the services and care patients receive. Yet it seems like some of the best clinicians are taken from active practice and are difficult to replace.
At the start of my career, the few doctors who left clinical practice for nonclinical work tended to do so late in their careers. Now many make this choice very early in their careers. Of the six or seven residents I met with above, several planned to pursue entirely nonclinical work either immediately upon completing residency or after just a few years of clinical practice. They were at one of the top internal medicine programs in the country and will, presumably, provide direct clinical care to a really small number of patients over their careers.
It makes me wonder if there is a meaningful effect of more talented people having, and exercising, the option to leave clinical practice, resulting in a tilt toward somewhat-less-talented doctors left to treat patients. I hope there is no meaningful effect in this direction, but I’m not sure.
Reasons to Move
My experience is that most doctors who have left clinical work will wax eloquent about how they really loved it and weren’t fleeing it but did so because they wanted to “try something new” or contribute to healthcare in other ways. I’m suspicious that for many of them this isn’t entirely true. Some must have been fleeing it. They were burned out, tired of being on call, and so on, and were eager to find relief from clinical work more than they were “drawn to a new career challenge.” They just don’t want to admit it.
I sometimes think about what several nationally prominent hospitalist leaders have said to me over my career. Not long ago, one said, “Wow. You’re still seeing patients and making rounds? I can’t believe it. You need to find something better.”
This doctor seemed to equate an entire career spent in clinical practice as something done mostly by those who aren’t talented enough to have other options. What a change from 30 or 40 years ago.
Several years ago, in a very moving conversation, another nationally prominent hospitalist leader told me, “It’s all about the patient and how we care for them at the bedside. There’s no better way we can spend our time.”
The Best Career
Within a few years, he left clinical practice entirely, even though he was still mid-career.
I hold in highest esteem hospitalists and other doctors who spend a full career in direct patient care and do it well. At the top of that list is my own dad, who is up there with Osler when it comes to dedicated physicians.
Of course, those who spend most or all of their time in nonclinical work really can make important contributions that help the healthcare system better serve patients, in some cases clearly making a bigger difference for more patients than they could via direct clinical care. We need talented people in both roles, but we also need to always be looking for ways to minimize the numbers of doctors who feel the need to flee a clinical career.
Like many hospitalists, I think about these things a lot when making decisions about my own career. I hope we all have the wisdom to make the best choices for ourselves, and for the patients we set out to serve when we entered medical school. TH

On a spring day a couple of years ago, I met with some internal medicine residents in a “Healthcare Systems Immersion” elective. I was to provide thoughts about the nonclinical portion of my work that I spend consulting with other hospitalist groups.
I asked for their thoughts about whether the ranks of doctors providing direct bedside care were losing too many of the most talented clinicians to nonclinical roles. The most vocal resident was confident that was not the case; these doctors would ultimately have a positive impact on the care of larger numbers of patients through administrative work than through direct patient care.
I wonder if she is right.
Numerous Hospitalists Opt for Nonnclinical Work
It seems like lots of hospitalists are transitioning to nonclinical work. My experience is that most who have administrative or other nonclinical roles continue—for part of their time—to provide direct patient care. But some leave clinical work behind altogether. Some of them are very prominent people in our field, like the top physician at CMS, the current U.S. Surgeon General, and this year’s most influential physician executive as judged by Modern Healthcare. I think it is pretty cool that these people come from our specialty.
I couldn’t find published survey data on the portion of hospitalists, or doctors in any specialty, who have entirely (or almost entirely) nonclinical roles. My impression is that this was a vanishingly small number across all specialties 30 or 40 years ago, but it seems to have increased pretty dramatically in the last 10 years. At the start of my career, few hospitals had a physician in an administrative position. Now it is common.
Physician leadership roles now include information technology (CMIO), quality (CQO), leader of the employed physician group, and hospital CEO (at least two hospitalists I know are in this role). And there are lots of nonclinical roles for doctors outside of hospitals.
Pros, Cons for Healthcare
I’ve had mixed feelings watching many people leave clinical practice. Most of them, like those mentioned above, continue to make important contributions to our healthcare system; they improve the services and care patients receive. Yet it seems like some of the best clinicians are taken from active practice and are difficult to replace.
At the start of my career, the few doctors who left clinical practice for nonclinical work tended to do so late in their careers. Now many make this choice very early in their careers. Of the six or seven residents I met with above, several planned to pursue entirely nonclinical work either immediately upon completing residency or after just a few years of clinical practice. They were at one of the top internal medicine programs in the country and will, presumably, provide direct clinical care to a really small number of patients over their careers.
It makes me wonder if there is a meaningful effect of more talented people having, and exercising, the option to leave clinical practice, resulting in a tilt toward somewhat-less-talented doctors left to treat patients. I hope there is no meaningful effect in this direction, but I’m not sure.
Reasons to Move
My experience is that most doctors who have left clinical work will wax eloquent about how they really loved it and weren’t fleeing it but did so because they wanted to “try something new” or contribute to healthcare in other ways. I’m suspicious that for many of them this isn’t entirely true. Some must have been fleeing it. They were burned out, tired of being on call, and so on, and were eager to find relief from clinical work more than they were “drawn to a new career challenge.” They just don’t want to admit it.
I sometimes think about what several nationally prominent hospitalist leaders have said to me over my career. Not long ago, one said, “Wow. You’re still seeing patients and making rounds? I can’t believe it. You need to find something better.”
This doctor seemed to equate an entire career spent in clinical practice as something done mostly by those who aren’t talented enough to have other options. What a change from 30 or 40 years ago.
Several years ago, in a very moving conversation, another nationally prominent hospitalist leader told me, “It’s all about the patient and how we care for them at the bedside. There’s no better way we can spend our time.”
The Best Career
Within a few years, he left clinical practice entirely, even though he was still mid-career.
I hold in highest esteem hospitalists and other doctors who spend a full career in direct patient care and do it well. At the top of that list is my own dad, who is up there with Osler when it comes to dedicated physicians.
Of course, those who spend most or all of their time in nonclinical work really can make important contributions that help the healthcare system better serve patients, in some cases clearly making a bigger difference for more patients than they could via direct clinical care. We need talented people in both roles, but we also need to always be looking for ways to minimize the numbers of doctors who feel the need to flee a clinical career.
Like many hospitalists, I think about these things a lot when making decisions about my own career. I hope we all have the wisdom to make the best choices for ourselves, and for the patients we set out to serve when we entered medical school. TH
On a spring day a couple of years ago, I met with some internal medicine residents in a “Healthcare Systems Immersion” elective. I was to provide thoughts about the nonclinical portion of my work that I spend consulting with other hospitalist groups.
I asked for their thoughts about whether the ranks of doctors providing direct bedside care were losing too many of the most talented clinicians to nonclinical roles. The most vocal resident was confident that was not the case; these doctors would ultimately have a positive impact on the care of larger numbers of patients through administrative work than through direct patient care.
I wonder if she is right.
Numerous Hospitalists Opt for Nonnclinical Work
It seems like lots of hospitalists are transitioning to nonclinical work. My experience is that most who have administrative or other nonclinical roles continue—for part of their time—to provide direct patient care. But some leave clinical work behind altogether. Some of them are very prominent people in our field, like the top physician at CMS, the current U.S. Surgeon General, and this year’s most influential physician executive as judged by Modern Healthcare. I think it is pretty cool that these people come from our specialty.
I couldn’t find published survey data on the portion of hospitalists, or doctors in any specialty, who have entirely (or almost entirely) nonclinical roles. My impression is that this was a vanishingly small number across all specialties 30 or 40 years ago, but it seems to have increased pretty dramatically in the last 10 years. At the start of my career, few hospitals had a physician in an administrative position. Now it is common.
Physician leadership roles now include information technology (CMIO), quality (CQO), leader of the employed physician group, and hospital CEO (at least two hospitalists I know are in this role). And there are lots of nonclinical roles for doctors outside of hospitals.
Pros, Cons for Healthcare
I’ve had mixed feelings watching many people leave clinical practice. Most of them, like those mentioned above, continue to make important contributions to our healthcare system; they improve the services and care patients receive. Yet it seems like some of the best clinicians are taken from active practice and are difficult to replace.
At the start of my career, the few doctors who left clinical practice for nonclinical work tended to do so late in their careers. Now many make this choice very early in their careers. Of the six or seven residents I met with above, several planned to pursue entirely nonclinical work either immediately upon completing residency or after just a few years of clinical practice. They were at one of the top internal medicine programs in the country and will, presumably, provide direct clinical care to a really small number of patients over their careers.
It makes me wonder if there is a meaningful effect of more talented people having, and exercising, the option to leave clinical practice, resulting in a tilt toward somewhat-less-talented doctors left to treat patients. I hope there is no meaningful effect in this direction, but I’m not sure.
Reasons to Move
My experience is that most doctors who have left clinical work will wax eloquent about how they really loved it and weren’t fleeing it but did so because they wanted to “try something new” or contribute to healthcare in other ways. I’m suspicious that for many of them this isn’t entirely true. Some must have been fleeing it. They were burned out, tired of being on call, and so on, and were eager to find relief from clinical work more than they were “drawn to a new career challenge.” They just don’t want to admit it.
I sometimes think about what several nationally prominent hospitalist leaders have said to me over my career. Not long ago, one said, “Wow. You’re still seeing patients and making rounds? I can’t believe it. You need to find something better.”
This doctor seemed to equate an entire career spent in clinical practice as something done mostly by those who aren’t talented enough to have other options. What a change from 30 or 40 years ago.
Several years ago, in a very moving conversation, another nationally prominent hospitalist leader told me, “It’s all about the patient and how we care for them at the bedside. There’s no better way we can spend our time.”
The Best Career
Within a few years, he left clinical practice entirely, even though he was still mid-career.
I hold in highest esteem hospitalists and other doctors who spend a full career in direct patient care and do it well. At the top of that list is my own dad, who is up there with Osler when it comes to dedicated physicians.
Of course, those who spend most or all of their time in nonclinical work really can make important contributions that help the healthcare system better serve patients, in some cases clearly making a bigger difference for more patients than they could via direct clinical care. We need talented people in both roles, but we also need to always be looking for ways to minimize the numbers of doctors who feel the need to flee a clinical career.
Like many hospitalists, I think about these things a lot when making decisions about my own career. I hope we all have the wisdom to make the best choices for ourselves, and for the patients we set out to serve when we entered medical school. TH
Hospitalists Can Lend Expertise, Join SHM's Campaign to Improve Antibiotic Stewardship

Many antimicrobial stewards, such as infection prevention specialists, hospital epidemiologists, pharmacists, nurses, and hospitalists, are at the center of quality improvement and seek to achieve optimal clinical outcomes related to antimicrobial use.4 These antimicrobial stewards often strive to minimize harms and other adverse events, reduce the costs of healthcare for infections, and decrease the threat of antimicrobial resistance.3
Hospitalists play a critical role in quality improvement and directly influence inpatient outcomes daily. It’s essential that hospitalists continue to make patient safety and quality care a priority while employing a multidisciplinary approach in implementing antimicrobial stewardship best practices. Although antimicrobial stewardship programs have typically been led by infectious disease physicians and pharmacists, SHM recognizes the significant value of hospitalist leadership and/or participation.5 Although most hospitalists are familiar with the adverse effects of overprescribing antibiotics, their insight and collaboration with other hospital clinicians is necessary in order to Fight the Resistance.
Fight the Resistance, a new behavior change campaign from SHM and our Center for Hospital Innovation and Improvement, is intended to encourage appropriate prescribing and use of antibiotics in the hospital. The campaign’s primary objective is to change prescribing behaviors among hospitalists and other hospital clinicians and facilitate behavior change related to antibiotic prescribing.
The campaign officially launched on Nov. 10, 2015, with a kickoff webinar presented by Scott Flanders, MD, FACP, MHM, and Melhim Bou Alwan, MD. Dr. Flanders discussed the importance of hospitalist involvement in antimicrobial stewardship and the significance of working in multidisciplinary teams in order to reduce overprescribing and the threat of antibiotic resistance.
Dr. Bou Alwan explained SHM’s efforts to fight antimicrobial resistance and informed the audience of SHM’s commitment to antibiotic stewardship. The webinar launch was a huge success, and SHM is excited to continue fighting the resistance with physicians across the country.
In order to Fight the Resistance, SHM is asking hospitalists to commit to the following actions:
- Work with your team. Physicians, nurse practitioners, physician assistants, pharmacists, and infectious disease experts need to work together to ensure that antibiotics are used appropriately. Consider the patients part of your team, too, by discussing with them why antibiotics may not be the best choice of treatment.
- Pay attention to appropriate antibiotic choice and resistance patterns, and identify mechanisms that can be used to educate providers about overprescribing in your hospital.
- Rethink your antibiotic treatment time course. Be sure to adhere to your hospital’s antibiotic treatment guidelines, track use of antibiotics, and set a stop date from when you first prescribe them.
SHM believes changing antibiotic prescription behaviors is a team effort and encourages hospitalists to get involved by visiting www.fighttheresistance.org. There you can find Fight the Resistance themed posters, resources, and educational materials to encourage enhanced stewardship and teamwork in your hospital. TH
Mobola Owolabi is senior project manager for The Center for Hospital Innovation and Improvement
References
- The White House. Office of the Press Secretary. FACT SHEET: Obama Administration releases national action plan to combat antibiotic-resistant bacteria. March 27, 2015. Available at: https://www.whitehouse.gov/the-press-office/2015/03/27/fact-sheet-obama-administration-releases-national-action-plan-combat-ant. Accessed December 3, 2015.
- CDC. Federal engagement in antimicrobial resistance. June 2015. Available at: http://www.cdc.gov/drugresistance/federal-engagement-in-ar/index.html. Accessed December 3, 2015.
- Infectious Diseases Society of America. Promoting antimicrobial stewardship in human medicine. 2015. Available at: http://www.idsociety.org/Stewardship_Policy/. Accessed December 3, 2015.
- CDC. Core elements of hospital antibiotic stewardship programs. 2015. Available at: http://www.cdc.gov/getsmart/healthcare/implementation/core-elements.html. Accessed December 3, 2015.
- Rohde JM, Jacobsen D, Rosenberg DJ. Role of the hospitalist in antimicrobial stewardship: a review of work completed and description of a multisite collaborative. Clin Ther. 2013; 35(6):751-757.
Many antimicrobial stewards, such as infection prevention specialists, hospital epidemiologists, pharmacists, nurses, and hospitalists, are at the center of quality improvement and seek to achieve optimal clinical outcomes related to antimicrobial use.4 These antimicrobial stewards often strive to minimize harms and other adverse events, reduce the costs of healthcare for infections, and decrease the threat of antimicrobial resistance.3
Hospitalists play a critical role in quality improvement and directly influence inpatient outcomes daily. It’s essential that hospitalists continue to make patient safety and quality care a priority while employing a multidisciplinary approach in implementing antimicrobial stewardship best practices. Although antimicrobial stewardship programs have typically been led by infectious disease physicians and pharmacists, SHM recognizes the significant value of hospitalist leadership and/or participation.5 Although most hospitalists are familiar with the adverse effects of overprescribing antibiotics, their insight and collaboration with other hospital clinicians is necessary in order to Fight the Resistance.
Fight the Resistance, a new behavior change campaign from SHM and our Center for Hospital Innovation and Improvement, is intended to encourage appropriate prescribing and use of antibiotics in the hospital. The campaign’s primary objective is to change prescribing behaviors among hospitalists and other hospital clinicians and facilitate behavior change related to antibiotic prescribing.
The campaign officially launched on Nov. 10, 2015, with a kickoff webinar presented by Scott Flanders, MD, FACP, MHM, and Melhim Bou Alwan, MD. Dr. Flanders discussed the importance of hospitalist involvement in antimicrobial stewardship and the significance of working in multidisciplinary teams in order to reduce overprescribing and the threat of antibiotic resistance.
Dr. Bou Alwan explained SHM’s efforts to fight antimicrobial resistance and informed the audience of SHM’s commitment to antibiotic stewardship. The webinar launch was a huge success, and SHM is excited to continue fighting the resistance with physicians across the country.
In order to Fight the Resistance, SHM is asking hospitalists to commit to the following actions:
- Work with your team. Physicians, nurse practitioners, physician assistants, pharmacists, and infectious disease experts need to work together to ensure that antibiotics are used appropriately. Consider the patients part of your team, too, by discussing with them why antibiotics may not be the best choice of treatment.
- Pay attention to appropriate antibiotic choice and resistance patterns, and identify mechanisms that can be used to educate providers about overprescribing in your hospital.
- Rethink your antibiotic treatment time course. Be sure to adhere to your hospital’s antibiotic treatment guidelines, track use of antibiotics, and set a stop date from when you first prescribe them.
SHM believes changing antibiotic prescription behaviors is a team effort and encourages hospitalists to get involved by visiting www.fighttheresistance.org. There you can find Fight the Resistance themed posters, resources, and educational materials to encourage enhanced stewardship and teamwork in your hospital. TH
Mobola Owolabi is senior project manager for The Center for Hospital Innovation and Improvement
References
- The White House. Office of the Press Secretary. FACT SHEET: Obama Administration releases national action plan to combat antibiotic-resistant bacteria. March 27, 2015. Available at: https://www.whitehouse.gov/the-press-office/2015/03/27/fact-sheet-obama-administration-releases-national-action-plan-combat-ant. Accessed December 3, 2015.
- CDC. Federal engagement in antimicrobial resistance. June 2015. Available at: http://www.cdc.gov/drugresistance/federal-engagement-in-ar/index.html. Accessed December 3, 2015.
- Infectious Diseases Society of America. Promoting antimicrobial stewardship in human medicine. 2015. Available at: http://www.idsociety.org/Stewardship_Policy/. Accessed December 3, 2015.
- CDC. Core elements of hospital antibiotic stewardship programs. 2015. Available at: http://www.cdc.gov/getsmart/healthcare/implementation/core-elements.html. Accessed December 3, 2015.
- Rohde JM, Jacobsen D, Rosenberg DJ. Role of the hospitalist in antimicrobial stewardship: a review of work completed and description of a multisite collaborative. Clin Ther. 2013; 35(6):751-757.
Many antimicrobial stewards, such as infection prevention specialists, hospital epidemiologists, pharmacists, nurses, and hospitalists, are at the center of quality improvement and seek to achieve optimal clinical outcomes related to antimicrobial use.4 These antimicrobial stewards often strive to minimize harms and other adverse events, reduce the costs of healthcare for infections, and decrease the threat of antimicrobial resistance.3
Hospitalists play a critical role in quality improvement and directly influence inpatient outcomes daily. It’s essential that hospitalists continue to make patient safety and quality care a priority while employing a multidisciplinary approach in implementing antimicrobial stewardship best practices. Although antimicrobial stewardship programs have typically been led by infectious disease physicians and pharmacists, SHM recognizes the significant value of hospitalist leadership and/or participation.5 Although most hospitalists are familiar with the adverse effects of overprescribing antibiotics, their insight and collaboration with other hospital clinicians is necessary in order to Fight the Resistance.
Fight the Resistance, a new behavior change campaign from SHM and our Center for Hospital Innovation and Improvement, is intended to encourage appropriate prescribing and use of antibiotics in the hospital. The campaign’s primary objective is to change prescribing behaviors among hospitalists and other hospital clinicians and facilitate behavior change related to antibiotic prescribing.
The campaign officially launched on Nov. 10, 2015, with a kickoff webinar presented by Scott Flanders, MD, FACP, MHM, and Melhim Bou Alwan, MD. Dr. Flanders discussed the importance of hospitalist involvement in antimicrobial stewardship and the significance of working in multidisciplinary teams in order to reduce overprescribing and the threat of antibiotic resistance.
Dr. Bou Alwan explained SHM’s efforts to fight antimicrobial resistance and informed the audience of SHM’s commitment to antibiotic stewardship. The webinar launch was a huge success, and SHM is excited to continue fighting the resistance with physicians across the country.
In order to Fight the Resistance, SHM is asking hospitalists to commit to the following actions:
- Work with your team. Physicians, nurse practitioners, physician assistants, pharmacists, and infectious disease experts need to work together to ensure that antibiotics are used appropriately. Consider the patients part of your team, too, by discussing with them why antibiotics may not be the best choice of treatment.
- Pay attention to appropriate antibiotic choice and resistance patterns, and identify mechanisms that can be used to educate providers about overprescribing in your hospital.
- Rethink your antibiotic treatment time course. Be sure to adhere to your hospital’s antibiotic treatment guidelines, track use of antibiotics, and set a stop date from when you first prescribe them.
SHM believes changing antibiotic prescription behaviors is a team effort and encourages hospitalists to get involved by visiting www.fighttheresistance.org. There you can find Fight the Resistance themed posters, resources, and educational materials to encourage enhanced stewardship and teamwork in your hospital. TH
Mobola Owolabi is senior project manager for The Center for Hospital Innovation and Improvement
References
- The White House. Office of the Press Secretary. FACT SHEET: Obama Administration releases national action plan to combat antibiotic-resistant bacteria. March 27, 2015. Available at: https://www.whitehouse.gov/the-press-office/2015/03/27/fact-sheet-obama-administration-releases-national-action-plan-combat-ant. Accessed December 3, 2015.
- CDC. Federal engagement in antimicrobial resistance. June 2015. Available at: http://www.cdc.gov/drugresistance/federal-engagement-in-ar/index.html. Accessed December 3, 2015.
- Infectious Diseases Society of America. Promoting antimicrobial stewardship in human medicine. 2015. Available at: http://www.idsociety.org/Stewardship_Policy/. Accessed December 3, 2015.
- CDC. Core elements of hospital antibiotic stewardship programs. 2015. Available at: http://www.cdc.gov/getsmart/healthcare/implementation/core-elements.html. Accessed December 3, 2015.
- Rohde JM, Jacobsen D, Rosenberg DJ. Role of the hospitalist in antimicrobial stewardship: a review of work completed and description of a multisite collaborative. Clin Ther. 2013; 35(6):751-757.

Survey reveals need to evaluate EOL discussions

Photo courtesy of the
National Cancer Institute
and Mathews Media Group
End-of-life (EOL) discussions often occur “too late” for patients with hematologic malignancies, according to a survey of US hematologists.
The researchers who conducted the survey speculate that physicians may delay EOL discussions with these patients because, unlike most solid tumors,
which are incurable when they reach an advanced stage, many advanced hematologic malignancies remain curable.
So it may not be clear that a patient has entered the EOL phase.
Oreofe O. Odejide, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, and colleagues conducted the survey and reported the results in a letter to JAMA Internal Medicine.
The researchers mailed their survey on EOL discussions to US hematologists found in the clinical directory of the American Society of Hematology. The individuals surveyed provide direct care for adults with hematologic malignancies.
Three hundred and forty-nine hematologists completed the survey. Most were men (75.4%), and they had a median age of 52. More than half (55.4%) practiced in community centers, and 42.9% practiced primarily in tertiary centers.
Three hundred and forty-five individuals answered the question about typical timing of EOL discussions, and 55.9% said these discussions occur too late.
Hematologists practicing in tertiary centers were more likely to report late EOL discussions than those practicing in community centers—64.9% and 48.7%, respectively (P=0.003). This difference was still significant in multivariable analysis, with an odds ratio of 1.92 (P=0.004).
When it comes to specific aspects of EOL care, 42.5% of the hematologists reported conducting their first conversation about resuscitation status at less than optimal times; 23.2% reported waiting until death was clearly imminent before having an initial conversation about hospice care; and 39.9% reported waiting until death was clearly imminent before having an initial conversation about the preferred site of death.
The researchers said the lack of a clear distinction between the curative and EOL phases of hematologic malignancies may explain these findings. Additionally, physicians may hesitate to have EOL discussions because they don’t want to affect a patient’s mentality or because they themselves find it difficult to “give up” on patients who might still be cured. ![]()
Photo courtesy of the
National Cancer Institute
and Mathews Media Group
End-of-life (EOL) discussions often occur “too late” for patients with hematologic malignancies, according to a survey of US hematologists.
The researchers who conducted the survey speculate that physicians may delay EOL discussions with these patients because, unlike most solid tumors,
which are incurable when they reach an advanced stage, many advanced hematologic malignancies remain curable.
So it may not be clear that a patient has entered the EOL phase.
Oreofe O. Odejide, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, and colleagues conducted the survey and reported the results in a letter to JAMA Internal Medicine.
The researchers mailed their survey on EOL discussions to US hematologists found in the clinical directory of the American Society of Hematology. The individuals surveyed provide direct care for adults with hematologic malignancies.
Three hundred and forty-nine hematologists completed the survey. Most were men (75.4%), and they had a median age of 52. More than half (55.4%) practiced in community centers, and 42.9% practiced primarily in tertiary centers.
Three hundred and forty-five individuals answered the question about typical timing of EOL discussions, and 55.9% said these discussions occur too late.
Hematologists practicing in tertiary centers were more likely to report late EOL discussions than those practicing in community centers—64.9% and 48.7%, respectively (P=0.003). This difference was still significant in multivariable analysis, with an odds ratio of 1.92 (P=0.004).
When it comes to specific aspects of EOL care, 42.5% of the hematologists reported conducting their first conversation about resuscitation status at less than optimal times; 23.2% reported waiting until death was clearly imminent before having an initial conversation about hospice care; and 39.9% reported waiting until death was clearly imminent before having an initial conversation about the preferred site of death.
The researchers said the lack of a clear distinction between the curative and EOL phases of hematologic malignancies may explain these findings. Additionally, physicians may hesitate to have EOL discussions because they don’t want to affect a patient’s mentality or because they themselves find it difficult to “give up” on patients who might still be cured. ![]()
Photo courtesy of the
National Cancer Institute
and Mathews Media Group
End-of-life (EOL) discussions often occur “too late” for patients with hematologic malignancies, according to a survey of US hematologists.
The researchers who conducted the survey speculate that physicians may delay EOL discussions with these patients because, unlike most solid tumors,
which are incurable when they reach an advanced stage, many advanced hematologic malignancies remain curable.
So it may not be clear that a patient has entered the EOL phase.
Oreofe O. Odejide, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, and colleagues conducted the survey and reported the results in a letter to JAMA Internal Medicine.
The researchers mailed their survey on EOL discussions to US hematologists found in the clinical directory of the American Society of Hematology. The individuals surveyed provide direct care for adults with hematologic malignancies.
Three hundred and forty-nine hematologists completed the survey. Most were men (75.4%), and they had a median age of 52. More than half (55.4%) practiced in community centers, and 42.9% practiced primarily in tertiary centers.
Three hundred and forty-five individuals answered the question about typical timing of EOL discussions, and 55.9% said these discussions occur too late.
Hematologists practicing in tertiary centers were more likely to report late EOL discussions than those practicing in community centers—64.9% and 48.7%, respectively (P=0.003). This difference was still significant in multivariable analysis, with an odds ratio of 1.92 (P=0.004).
When it comes to specific aspects of EOL care, 42.5% of the hematologists reported conducting their first conversation about resuscitation status at less than optimal times; 23.2% reported waiting until death was clearly imminent before having an initial conversation about hospice care; and 39.9% reported waiting until death was clearly imminent before having an initial conversation about the preferred site of death.
The researchers said the lack of a clear distinction between the curative and EOL phases of hematologic malignancies may explain these findings. Additionally, physicians may hesitate to have EOL discussions because they don’t want to affect a patient’s mentality or because they themselves find it difficult to “give up” on patients who might still be cured. ![]()
FDA changes deferral policy for MSM blood donors
Photo by Михаило Јовановић
The US Food and Drug Administration (FDA) has issued a final guidance outlining updated blood donor deferral recommendations.
As part of the guidance, the FDA is changing its recommendation that men who have sex with men (MSM) be indefinitely deferred from donating blood—a policy that has been in place for approximately 30 years.
Now, the agency is recommending that MSMs be deferred for 12 months since their last sexual contact with another man.
The FDA’s guidance also reflects a change in the rationale for deferring potential blood donors with hemophilia or related clotting disorders who have received clotting factor concentrates.
The FDA recommends that blood establishments make corresponding revisions to donor educational materials, donor history questionnaires, and accompanying materials, as well as donor requalification and product management procedures.
MSM deferral
The FDA said its recommendation regarding MSM blood donors reflects the most current scientific evidence and will help ensure continued safety of the blood supply by reducing the risk of human immunodeficiency virus (HIV) transmission by blood and blood products.
The agency also said this recommendation better aligns the deferral period for MSMs with the deferral period for other men and women at increased risk for HIV infection, such as those who had a recent blood transfusion or those who have been accidentally exposed to the blood of another individual.
Before issuing this guidance, the FDA reviewed its policies regarding HIV transmission through blood products to determine appropriate changes based on the most recent scientific evidence. The agency examined a variety of studies, epidemiologic data, and shared experiences from other countries that have made recent MSM deferral policy changes.
“In reviewing our policies to help reduce the risk of HIV transmission through blood products, we rigorously examined several alternative options, including individual risk assessment,” said Peter Marks, MD, PhD, deputy director of the FDA’s Center for Biologics Evaluation and Research.
“Ultimately, the 12-month deferral window is supported by the best available scientific evidence, at this point in time, relevant to the US population. We will continue to actively conduct research in this area and further revise our policies as new data emerge.”
Several countries, including the UK and Australia, currently have 12-month deferral policies for MSM blood donors.
During the change in Australia from an indefinite blood donor deferral policy for MSMs to a 12-month deferral, studies evaluating over 8 million units of donated blood were performed using a national blood surveillance system. These studies (CR Seed et al, Transfusion 2010; TTA Lucky et al, Transfusion 2014) show no change in risk to the blood supply with use of the 12-month deferral.
A study conducted in the UK produced similar results, although it also suggested that 3 in 10 MSMs don’t comply with the 12-month deferral policy.
And a study conducted in Canada, which recently shortened its MSM deferral period to 5 years, showed no change in risk to the blood supply with the 5-year deferral as compared to indefinite deferral. Based on these results, Canadian regulators are considering changing to a 12-month deferral period as well.
Patients with clotting disorders
The FDA’s new guidance also reflects a change in the rationale for deferring patients with hemophilia or related clotting disorders who have received clotting factor concentrates. Previously, potential donors with hemophilia or related clotting disorders were deferred due to the increased risk of HIV transmission to potential recipients.
Based on new scientific evidence, these potential donors are still deferred, but not due to the risk of HIV transmission—instead, for their own protection due to potential harm from large needles used during the donation process.
FDA policies and actions
Throughout the process of updating blood donor deferral policies over the past several years, the FDA has worked with other government agencies, considered input from external advisory committees, reviewed comments from stakeholders to its May 2015 draft guidance, and examined the most recent available scientific evidence to support the current policy revision.
The FDA has also implemented a nationally representative safety monitoring system for the blood supply with assistance from the National Heart, Lung and Blood Institute at the National Institutes of Health. This system will provide information to help inform future actions the FDA may take on blood donor policies.
The FDA said it will continue to reevaluate and update its blood donor deferral policies as new scientific information becomes available. ![]()
Photo by Михаило Јовановић
The US Food and Drug Administration (FDA) has issued a final guidance outlining updated blood donor deferral recommendations.
As part of the guidance, the FDA is changing its recommendation that men who have sex with men (MSM) be indefinitely deferred from donating blood—a policy that has been in place for approximately 30 years.
Now, the agency is recommending that MSMs be deferred for 12 months since their last sexual contact with another man.
The FDA’s guidance also reflects a change in the rationale for deferring potential blood donors with hemophilia or related clotting disorders who have received clotting factor concentrates.
The FDA recommends that blood establishments make corresponding revisions to donor educational materials, donor history questionnaires, and accompanying materials, as well as donor requalification and product management procedures.
MSM deferral
The FDA said its recommendation regarding MSM blood donors reflects the most current scientific evidence and will help ensure continued safety of the blood supply by reducing the risk of human immunodeficiency virus (HIV) transmission by blood and blood products.
The agency also said this recommendation better aligns the deferral period for MSMs with the deferral period for other men and women at increased risk for HIV infection, such as those who had a recent blood transfusion or those who have been accidentally exposed to the blood of another individual.
Before issuing this guidance, the FDA reviewed its policies regarding HIV transmission through blood products to determine appropriate changes based on the most recent scientific evidence. The agency examined a variety of studies, epidemiologic data, and shared experiences from other countries that have made recent MSM deferral policy changes.
“In reviewing our policies to help reduce the risk of HIV transmission through blood products, we rigorously examined several alternative options, including individual risk assessment,” said Peter Marks, MD, PhD, deputy director of the FDA’s Center for Biologics Evaluation and Research.
“Ultimately, the 12-month deferral window is supported by the best available scientific evidence, at this point in time, relevant to the US population. We will continue to actively conduct research in this area and further revise our policies as new data emerge.”
Several countries, including the UK and Australia, currently have 12-month deferral policies for MSM blood donors.
During the change in Australia from an indefinite blood donor deferral policy for MSMs to a 12-month deferral, studies evaluating over 8 million units of donated blood were performed using a national blood surveillance system. These studies (CR Seed et al, Transfusion 2010; TTA Lucky et al, Transfusion 2014) show no change in risk to the blood supply with use of the 12-month deferral.
A study conducted in the UK produced similar results, although it also suggested that 3 in 10 MSMs don’t comply with the 12-month deferral policy.
And a study conducted in Canada, which recently shortened its MSM deferral period to 5 years, showed no change in risk to the blood supply with the 5-year deferral as compared to indefinite deferral. Based on these results, Canadian regulators are considering changing to a 12-month deferral period as well.
Patients with clotting disorders
The FDA’s new guidance also reflects a change in the rationale for deferring patients with hemophilia or related clotting disorders who have received clotting factor concentrates. Previously, potential donors with hemophilia or related clotting disorders were deferred due to the increased risk of HIV transmission to potential recipients.
Based on new scientific evidence, these potential donors are still deferred, but not due to the risk of HIV transmission—instead, for their own protection due to potential harm from large needles used during the donation process.
FDA policies and actions
Throughout the process of updating blood donor deferral policies over the past several years, the FDA has worked with other government agencies, considered input from external advisory committees, reviewed comments from stakeholders to its May 2015 draft guidance, and examined the most recent available scientific evidence to support the current policy revision.
The FDA has also implemented a nationally representative safety monitoring system for the blood supply with assistance from the National Heart, Lung and Blood Institute at the National Institutes of Health. This system will provide information to help inform future actions the FDA may take on blood donor policies.
The FDA said it will continue to reevaluate and update its blood donor deferral policies as new scientific information becomes available. ![]()
Photo by Михаило Јовановић
The US Food and Drug Administration (FDA) has issued a final guidance outlining updated blood donor deferral recommendations.
As part of the guidance, the FDA is changing its recommendation that men who have sex with men (MSM) be indefinitely deferred from donating blood—a policy that has been in place for approximately 30 years.
Now, the agency is recommending that MSMs be deferred for 12 months since their last sexual contact with another man.
The FDA’s guidance also reflects a change in the rationale for deferring potential blood donors with hemophilia or related clotting disorders who have received clotting factor concentrates.
The FDA recommends that blood establishments make corresponding revisions to donor educational materials, donor history questionnaires, and accompanying materials, as well as donor requalification and product management procedures.
MSM deferral
The FDA said its recommendation regarding MSM blood donors reflects the most current scientific evidence and will help ensure continued safety of the blood supply by reducing the risk of human immunodeficiency virus (HIV) transmission by blood and blood products.
The agency also said this recommendation better aligns the deferral period for MSMs with the deferral period for other men and women at increased risk for HIV infection, such as those who had a recent blood transfusion or those who have been accidentally exposed to the blood of another individual.
Before issuing this guidance, the FDA reviewed its policies regarding HIV transmission through blood products to determine appropriate changes based on the most recent scientific evidence. The agency examined a variety of studies, epidemiologic data, and shared experiences from other countries that have made recent MSM deferral policy changes.
“In reviewing our policies to help reduce the risk of HIV transmission through blood products, we rigorously examined several alternative options, including individual risk assessment,” said Peter Marks, MD, PhD, deputy director of the FDA’s Center for Biologics Evaluation and Research.
“Ultimately, the 12-month deferral window is supported by the best available scientific evidence, at this point in time, relevant to the US population. We will continue to actively conduct research in this area and further revise our policies as new data emerge.”
Several countries, including the UK and Australia, currently have 12-month deferral policies for MSM blood donors.
During the change in Australia from an indefinite blood donor deferral policy for MSMs to a 12-month deferral, studies evaluating over 8 million units of donated blood were performed using a national blood surveillance system. These studies (CR Seed et al, Transfusion 2010; TTA Lucky et al, Transfusion 2014) show no change in risk to the blood supply with use of the 12-month deferral.
A study conducted in the UK produced similar results, although it also suggested that 3 in 10 MSMs don’t comply with the 12-month deferral policy.
And a study conducted in Canada, which recently shortened its MSM deferral period to 5 years, showed no change in risk to the blood supply with the 5-year deferral as compared to indefinite deferral. Based on these results, Canadian regulators are considering changing to a 12-month deferral period as well.
Patients with clotting disorders
The FDA’s new guidance also reflects a change in the rationale for deferring patients with hemophilia or related clotting disorders who have received clotting factor concentrates. Previously, potential donors with hemophilia or related clotting disorders were deferred due to the increased risk of HIV transmission to potential recipients.
Based on new scientific evidence, these potential donors are still deferred, but not due to the risk of HIV transmission—instead, for their own protection due to potential harm from large needles used during the donation process.
FDA policies and actions
Throughout the process of updating blood donor deferral policies over the past several years, the FDA has worked with other government agencies, considered input from external advisory committees, reviewed comments from stakeholders to its May 2015 draft guidance, and examined the most recent available scientific evidence to support the current policy revision.
The FDA has also implemented a nationally representative safety monitoring system for the blood supply with assistance from the National Heart, Lung and Blood Institute at the National Institutes of Health. This system will provide information to help inform future actions the FDA may take on blood donor policies.
The FDA said it will continue to reevaluate and update its blood donor deferral policies as new scientific information becomes available. ![]()
Antibody shows promise for treating CLL

Preclinical research suggests a humanized monoclonal antibody called cirmtuzumab (UC-961) might be an effective treatment for chronic lymphocytic leukemia (CLL).
Experiments revealed that the Wnt5a protein acts on the tumor-surface proteins ROR1 and ROR2 to accelerate the proliferation and spread of CLL cells.
But cirmtuzumab, which is specific for ROR1, can block the effects of Wnt5a and inhibit the growth and spread of CLL cells both in vitro and in vivo.
Investigators reported these results in The Journal of Clinical Investigation.
They noted that ROR1 and ROR2 are considered orphan receptors, which are expressed primarily during embryonic development. The expression of these proteins, particularly ROR1, becomes suppressed during fetal development and is negligible on normal adult tissues. However, CLL and many solid tissue cancers re-express these orphan receptors.
“Our findings show that ROR1 and ROR2 team up to stimulate tumor cell growth and metastasis in response to Wnt5a, which appears overexpressed in patients with CLL and can act as a survival/growth factor for leukemia cells,” said study author Thomas J. Kipps, MD, PhD, of Moores Cancer Center at the University of California, San Diego.
“By blocking the capacity of Wnt5a to stimulate tumor cells, cirmtuzumab can inhibit the growth and spread of cancer cells. We now have better insight into how cirmtuzumab works against leukemia cells. This should help find better ways to treat patients who have other cancers with cirmtuzumab, which currently is being evaluated in a phase 1 clinical trial for patients with CLL.”
The JCI paper follows a series of related findings by Dr Kipps and his colleagues in recent years.
In 2008, they reported that patients vaccinated with their own CLL cells could make antibodies against ROR1, some of which had the ability to reduce CLL cell survival. They found ROR1 on CLL cells but not on all normal adult tissues examined.
In 2012, the investigators reported finding ROR1 on many different types of cancer, particularly cancers that appear less differentiated and more likely to spread to other parts in the body. Because this protein was not found on normal adult tissues, these findings made ROR1 a new target for anticancer drug research.
In June 2013, the team linked ROR1 to a process used in early development, suggesting cancer cells hijack an embryological process called epithelial-mesenchymal transition to spread or metastasize more quickly.
In January 2014, the investigators reported expression of ROR1 resulted in a faster-developing, more aggressive form of CLL in mice.
In September 2014, the team launched a phase 1 trial of cirmtuzumab in patients with CLL. The trial is ongoing.
In November 2014, the investigators described cellular experiments indicating that cirmtuzumab might be effective against cancer stem cells, which appear responsible for the relapse and spread of cancer after conventional therapy.
The latest research more precisely defines the roles of ROR1 and ROR2 in CLL development.
Both are evolutionarily conserved proteins that are found in many species and are most active in the early stages of embryogenesis when cells are migrating to form organs and parts of the body. The lack of either during this process results in severe developmental abnormalities.
Low levels of ROR2 remain in some adult tissues, but ROR1 is found only in cancer cells. The investigators found that, in response to signaling by Wnt5a, ROR1 and ROR2 come together to signal the growth and migration of CLL cells.
But treating mice with cirmtuzumab disrupted the process, inhibiting the engraftment of CLL cells and slowing or stopping the disease from spreading. ![]()
Preclinical research suggests a humanized monoclonal antibody called cirmtuzumab (UC-961) might be an effective treatment for chronic lymphocytic leukemia (CLL).
Experiments revealed that the Wnt5a protein acts on the tumor-surface proteins ROR1 and ROR2 to accelerate the proliferation and spread of CLL cells.
But cirmtuzumab, which is specific for ROR1, can block the effects of Wnt5a and inhibit the growth and spread of CLL cells both in vitro and in vivo.
Investigators reported these results in The Journal of Clinical Investigation.
They noted that ROR1 and ROR2 are considered orphan receptors, which are expressed primarily during embryonic development. The expression of these proteins, particularly ROR1, becomes suppressed during fetal development and is negligible on normal adult tissues. However, CLL and many solid tissue cancers re-express these orphan receptors.
“Our findings show that ROR1 and ROR2 team up to stimulate tumor cell growth and metastasis in response to Wnt5a, which appears overexpressed in patients with CLL and can act as a survival/growth factor for leukemia cells,” said study author Thomas J. Kipps, MD, PhD, of Moores Cancer Center at the University of California, San Diego.
“By blocking the capacity of Wnt5a to stimulate tumor cells, cirmtuzumab can inhibit the growth and spread of cancer cells. We now have better insight into how cirmtuzumab works against leukemia cells. This should help find better ways to treat patients who have other cancers with cirmtuzumab, which currently is being evaluated in a phase 1 clinical trial for patients with CLL.”
The JCI paper follows a series of related findings by Dr Kipps and his colleagues in recent years.
In 2008, they reported that patients vaccinated with their own CLL cells could make antibodies against ROR1, some of which had the ability to reduce CLL cell survival. They found ROR1 on CLL cells but not on all normal adult tissues examined.
In 2012, the investigators reported finding ROR1 on many different types of cancer, particularly cancers that appear less differentiated and more likely to spread to other parts in the body. Because this protein was not found on normal adult tissues, these findings made ROR1 a new target for anticancer drug research.
In June 2013, the team linked ROR1 to a process used in early development, suggesting cancer cells hijack an embryological process called epithelial-mesenchymal transition to spread or metastasize more quickly.
In January 2014, the investigators reported expression of ROR1 resulted in a faster-developing, more aggressive form of CLL in mice.
In September 2014, the team launched a phase 1 trial of cirmtuzumab in patients with CLL. The trial is ongoing.
In November 2014, the investigators described cellular experiments indicating that cirmtuzumab might be effective against cancer stem cells, which appear responsible for the relapse and spread of cancer after conventional therapy.
The latest research more precisely defines the roles of ROR1 and ROR2 in CLL development.
Both are evolutionarily conserved proteins that are found in many species and are most active in the early stages of embryogenesis when cells are migrating to form organs and parts of the body. The lack of either during this process results in severe developmental abnormalities.
Low levels of ROR2 remain in some adult tissues, but ROR1 is found only in cancer cells. The investigators found that, in response to signaling by Wnt5a, ROR1 and ROR2 come together to signal the growth and migration of CLL cells.
But treating mice with cirmtuzumab disrupted the process, inhibiting the engraftment of CLL cells and slowing or stopping the disease from spreading. ![]()
Preclinical research suggests a humanized monoclonal antibody called cirmtuzumab (UC-961) might be an effective treatment for chronic lymphocytic leukemia (CLL).
Experiments revealed that the Wnt5a protein acts on the tumor-surface proteins ROR1 and ROR2 to accelerate the proliferation and spread of CLL cells.
But cirmtuzumab, which is specific for ROR1, can block the effects of Wnt5a and inhibit the growth and spread of CLL cells both in vitro and in vivo.
Investigators reported these results in The Journal of Clinical Investigation.
They noted that ROR1 and ROR2 are considered orphan receptors, which are expressed primarily during embryonic development. The expression of these proteins, particularly ROR1, becomes suppressed during fetal development and is negligible on normal adult tissues. However, CLL and many solid tissue cancers re-express these orphan receptors.
“Our findings show that ROR1 and ROR2 team up to stimulate tumor cell growth and metastasis in response to Wnt5a, which appears overexpressed in patients with CLL and can act as a survival/growth factor for leukemia cells,” said study author Thomas J. Kipps, MD, PhD, of Moores Cancer Center at the University of California, San Diego.
“By blocking the capacity of Wnt5a to stimulate tumor cells, cirmtuzumab can inhibit the growth and spread of cancer cells. We now have better insight into how cirmtuzumab works against leukemia cells. This should help find better ways to treat patients who have other cancers with cirmtuzumab, which currently is being evaluated in a phase 1 clinical trial for patients with CLL.”
The JCI paper follows a series of related findings by Dr Kipps and his colleagues in recent years.
In 2008, they reported that patients vaccinated with their own CLL cells could make antibodies against ROR1, some of which had the ability to reduce CLL cell survival. They found ROR1 on CLL cells but not on all normal adult tissues examined.
In 2012, the investigators reported finding ROR1 on many different types of cancer, particularly cancers that appear less differentiated and more likely to spread to other parts in the body. Because this protein was not found on normal adult tissues, these findings made ROR1 a new target for anticancer drug research.
In June 2013, the team linked ROR1 to a process used in early development, suggesting cancer cells hijack an embryological process called epithelial-mesenchymal transition to spread or metastasize more quickly.
In January 2014, the investigators reported expression of ROR1 resulted in a faster-developing, more aggressive form of CLL in mice.
In September 2014, the team launched a phase 1 trial of cirmtuzumab in patients with CLL. The trial is ongoing.
In November 2014, the investigators described cellular experiments indicating that cirmtuzumab might be effective against cancer stem cells, which appear responsible for the relapse and spread of cancer after conventional therapy.
The latest research more precisely defines the roles of ROR1 and ROR2 in CLL development.
Both are evolutionarily conserved proteins that are found in many species and are most active in the early stages of embryogenesis when cells are migrating to form organs and parts of the body. The lack of either during this process results in severe developmental abnormalities.
Low levels of ROR2 remain in some adult tissues, but ROR1 is found only in cancer cells. The investigators found that, in response to signaling by Wnt5a, ROR1 and ROR2 come together to signal the growth and migration of CLL cells.
But treating mice with cirmtuzumab disrupted the process, inhibiting the engraftment of CLL cells and slowing or stopping the disease from spreading. ![]()
Anemia tied to cognitive impairment

A population-based study conducted in Germany has suggested a link between anemia and mild cognitive impairment (MCI).
Researchers found that subjects with anemia, defined as hemoglobin <13 g/dL in men and <12 g/dL in women, performed worse on cognitive tests than their nonanemic peers.
And MCI occurred almost twice as often in subjects with anemia than in subjects with normal hemoglobin levels.
This study was published in the Journal of Alzheimer’s Disease.
About MCI
MCI represents an intermediate and possibly modifiable stage between normal cognitive aging and dementia. Although individuals with MCI have an increased risk of developing dementia or Alzheimer’s disease, they can also remain stable for many years or even revert to a cognitively normal state over time. This modifiable characteristic makes the concept of MCI a promising target in the prevention of dementia.
The following 4 criteria are used to diagnose MCI. First, subjects must report a decline in cognitive performance over the past 2 years. Second, they must show a cognitive impairment in objective cognitive tasks that is greater than one would expect taking their age and education into consideration.
Third, the impairment must not be as pronounced as in demented individuals since people with MCI can perform normal daily living activities or are only slightly impaired in carrying out complex instrumental functions. Fourth, the cognitive impairment has to be insufficient to fulfil criteria for dementia.
The concept of MCI distinguishes between amnestic MCI (aMCI) and non-amnestic MCI (naMCI). In the former, impairment in the memory domain is evident, most likely reflecting Alzheimer’s disease pathology. In the latter, impairment in non-memory domains is present, mainly reflecting vascular pathology but also frontotemporal dementia or dementia with Lewy bodies.
Study details
The Heinz Nixdorf Recall study is an observational, population-based, prospective study in which researchers examined 4814 subjects between 2000 and 2003. Subjects were 50 to 80 years of age and lived in the metropolitan Ruhr Area. Both genders were equally represented.
After 5 years, the researchers conducted a second examination with 92% of the subjects taking part. The publication includes cross-sectional results of the second examination.
First, 163 subjects with anemia and 3870 without anemia were included to compare their performance in all cognitive subtests.
The subjects took verbal memory tests, which were used to gauge immediate recall and delayed recall. They were also tested on executive functioning, which included problem-solving/speed of processing, verbal fluency, visual spatial organization, and the clock drawing test.
In the initial analysis, anemic subjects showed more pronounced cardiovascular risk profiles and lower cognitive performance in all administered cognitive subtests. After adjusting for age, anemic subjects showed a significantly lower performance in the immediate recall task (P=0.009) and the verbal fluency task (P=0.004).
Next, the researchers compared 579 subjects diagnosed with MCI—299 with aMCI and 280 with naMCI—to 1438 cognitively normal subjects to determine the association between anemia at follow-up and MCI.
The team found that MCI occurred more often in anemic than non-anemic subjects. The unadjusted odds ratio (OR) was 2.59 (P<0.001). The OR after adjustment for age, gender, and years of education was 2.15 (P=0.002).
In a third analysis, the researchers adjusted for the aforementioned variables as well as body mass index, high-sensitivity C-reactive protein, glomerular filtration rate, cholesterol, serum iron, hypertension, diabetes mellitus, history of coronary heart disease, history of stroke, history of cancer, APOE4, smoking, severe depressive symptoms, and use of antidepressants. The OR after adjustment for these factors was 1.92 (P=0.04).
Similar results were found for aMCI and naMCI. The researchers said this suggests that having a low hemoglobin level may contribute to cognitive impairment via different pathways.
The team believes that, overall, their study results indicate that anemia is associated with an increased risk of MCI independent of traditional cardiovascular risk factors. They said the association between anemia and MCI has important clinical relevance because—depending on etiology—anemia can be treated effectively, and this might provide means to prevent or delay cognitive decline. ![]()
A population-based study conducted in Germany has suggested a link between anemia and mild cognitive impairment (MCI).
Researchers found that subjects with anemia, defined as hemoglobin <13 g/dL in men and <12 g/dL in women, performed worse on cognitive tests than their nonanemic peers.
And MCI occurred almost twice as often in subjects with anemia than in subjects with normal hemoglobin levels.
This study was published in the Journal of Alzheimer’s Disease.
About MCI
MCI represents an intermediate and possibly modifiable stage between normal cognitive aging and dementia. Although individuals with MCI have an increased risk of developing dementia or Alzheimer’s disease, they can also remain stable for many years or even revert to a cognitively normal state over time. This modifiable characteristic makes the concept of MCI a promising target in the prevention of dementia.
The following 4 criteria are used to diagnose MCI. First, subjects must report a decline in cognitive performance over the past 2 years. Second, they must show a cognitive impairment in objective cognitive tasks that is greater than one would expect taking their age and education into consideration.
Third, the impairment must not be as pronounced as in demented individuals since people with MCI can perform normal daily living activities or are only slightly impaired in carrying out complex instrumental functions. Fourth, the cognitive impairment has to be insufficient to fulfil criteria for dementia.
The concept of MCI distinguishes between amnestic MCI (aMCI) and non-amnestic MCI (naMCI). In the former, impairment in the memory domain is evident, most likely reflecting Alzheimer’s disease pathology. In the latter, impairment in non-memory domains is present, mainly reflecting vascular pathology but also frontotemporal dementia or dementia with Lewy bodies.
Study details
The Heinz Nixdorf Recall study is an observational, population-based, prospective study in which researchers examined 4814 subjects between 2000 and 2003. Subjects were 50 to 80 years of age and lived in the metropolitan Ruhr Area. Both genders were equally represented.
After 5 years, the researchers conducted a second examination with 92% of the subjects taking part. The publication includes cross-sectional results of the second examination.
First, 163 subjects with anemia and 3870 without anemia were included to compare their performance in all cognitive subtests.
The subjects took verbal memory tests, which were used to gauge immediate recall and delayed recall. They were also tested on executive functioning, which included problem-solving/speed of processing, verbal fluency, visual spatial organization, and the clock drawing test.
In the initial analysis, anemic subjects showed more pronounced cardiovascular risk profiles and lower cognitive performance in all administered cognitive subtests. After adjusting for age, anemic subjects showed a significantly lower performance in the immediate recall task (P=0.009) and the verbal fluency task (P=0.004).
Next, the researchers compared 579 subjects diagnosed with MCI—299 with aMCI and 280 with naMCI—to 1438 cognitively normal subjects to determine the association between anemia at follow-up and MCI.
The team found that MCI occurred more often in anemic than non-anemic subjects. The unadjusted odds ratio (OR) was 2.59 (P<0.001). The OR after adjustment for age, gender, and years of education was 2.15 (P=0.002).
In a third analysis, the researchers adjusted for the aforementioned variables as well as body mass index, high-sensitivity C-reactive protein, glomerular filtration rate, cholesterol, serum iron, hypertension, diabetes mellitus, history of coronary heart disease, history of stroke, history of cancer, APOE4, smoking, severe depressive symptoms, and use of antidepressants. The OR after adjustment for these factors was 1.92 (P=0.04).
Similar results were found for aMCI and naMCI. The researchers said this suggests that having a low hemoglobin level may contribute to cognitive impairment via different pathways.
The team believes that, overall, their study results indicate that anemia is associated with an increased risk of MCI independent of traditional cardiovascular risk factors. They said the association between anemia and MCI has important clinical relevance because—depending on etiology—anemia can be treated effectively, and this might provide means to prevent or delay cognitive decline. ![]()
A population-based study conducted in Germany has suggested a link between anemia and mild cognitive impairment (MCI).
Researchers found that subjects with anemia, defined as hemoglobin <13 g/dL in men and <12 g/dL in women, performed worse on cognitive tests than their nonanemic peers.
And MCI occurred almost twice as often in subjects with anemia than in subjects with normal hemoglobin levels.
This study was published in the Journal of Alzheimer’s Disease.
About MCI
MCI represents an intermediate and possibly modifiable stage between normal cognitive aging and dementia. Although individuals with MCI have an increased risk of developing dementia or Alzheimer’s disease, they can also remain stable for many years or even revert to a cognitively normal state over time. This modifiable characteristic makes the concept of MCI a promising target in the prevention of dementia.
The following 4 criteria are used to diagnose MCI. First, subjects must report a decline in cognitive performance over the past 2 years. Second, they must show a cognitive impairment in objective cognitive tasks that is greater than one would expect taking their age and education into consideration.
Third, the impairment must not be as pronounced as in demented individuals since people with MCI can perform normal daily living activities or are only slightly impaired in carrying out complex instrumental functions. Fourth, the cognitive impairment has to be insufficient to fulfil criteria for dementia.
The concept of MCI distinguishes between amnestic MCI (aMCI) and non-amnestic MCI (naMCI). In the former, impairment in the memory domain is evident, most likely reflecting Alzheimer’s disease pathology. In the latter, impairment in non-memory domains is present, mainly reflecting vascular pathology but also frontotemporal dementia or dementia with Lewy bodies.
Study details
The Heinz Nixdorf Recall study is an observational, population-based, prospective study in which researchers examined 4814 subjects between 2000 and 2003. Subjects were 50 to 80 years of age and lived in the metropolitan Ruhr Area. Both genders were equally represented.
After 5 years, the researchers conducted a second examination with 92% of the subjects taking part. The publication includes cross-sectional results of the second examination.
First, 163 subjects with anemia and 3870 without anemia were included to compare their performance in all cognitive subtests.
The subjects took verbal memory tests, which were used to gauge immediate recall and delayed recall. They were also tested on executive functioning, which included problem-solving/speed of processing, verbal fluency, visual spatial organization, and the clock drawing test.
In the initial analysis, anemic subjects showed more pronounced cardiovascular risk profiles and lower cognitive performance in all administered cognitive subtests. After adjusting for age, anemic subjects showed a significantly lower performance in the immediate recall task (P=0.009) and the verbal fluency task (P=0.004).
Next, the researchers compared 579 subjects diagnosed with MCI—299 with aMCI and 280 with naMCI—to 1438 cognitively normal subjects to determine the association between anemia at follow-up and MCI.
The team found that MCI occurred more often in anemic than non-anemic subjects. The unadjusted odds ratio (OR) was 2.59 (P<0.001). The OR after adjustment for age, gender, and years of education was 2.15 (P=0.002).
In a third analysis, the researchers adjusted for the aforementioned variables as well as body mass index, high-sensitivity C-reactive protein, glomerular filtration rate, cholesterol, serum iron, hypertension, diabetes mellitus, history of coronary heart disease, history of stroke, history of cancer, APOE4, smoking, severe depressive symptoms, and use of antidepressants. The OR after adjustment for these factors was 1.92 (P=0.04).
Similar results were found for aMCI and naMCI. The researchers said this suggests that having a low hemoglobin level may contribute to cognitive impairment via different pathways.
The team believes that, overall, their study results indicate that anemia is associated with an increased risk of MCI independent of traditional cardiovascular risk factors. They said the association between anemia and MCI has important clinical relevance because—depending on etiology—anemia can be treated effectively, and this might provide means to prevent or delay cognitive decline. ![]()
The Social Worker’s Role in Delirium Care for Hospitalized Veterans
Delirium, or the state of mental confusion that may occur with physical or mental illness, is common, morbid, and costly; however, of the diagnosed cases, delirium is mentioned in hospital discharge summaries only 16% to 55% of the time.1-3
Social workers often coordinate care transitions for hospitalized older veterans. They serve as interdisciplinary team members who communicate with VA medical staff as well as with the patient and family. This position, in addition to their training in communication and advocacy, primes social workers for a role in delirium care and provides the needed support for veterans who experience delirium and their families.
Background
Delirium is a sudden disturbance of attention with reduced awareness of the environment. Because attention is impaired, other changes in cognition are common, including perceptual and thought disturbances. Additionally, delirium includes fluctuations in consciousness over the course of a day. The acute development of these cognitive disturbances is distinct from a preexisting chronic cognitive impairment, such as dementia. Delirium is a direct consequence of underlying medical conditions, such as infections, polypharmacy, dehydration, and surgery.4
Delirium subtypes all have inattention as a core symptom. In half of the cases, patients are hypoactive and will not awaken easily or participate in daily care plans readily.4 Hyperactive delirium occurs in a quarter of cases. In the remaining mixed delirium cases patients fluctuate between the 2 states.4
Delirium is often falsely mistaken for dementia. Although delirium and dementia can present similarly, delirium has a sudden onset, which can alert health care professionals (HCPs) to the likelihood of delirium. Another important distinction is that delirium is typically reversible. Symptom manifestations of delirium may also be confused with depression.
Related: Delirium in the Cardiac ICU
Preventing delirium is important due to its many negative health outcomes. Older adults who develop delirium are more likely to die sooner. In a Canadian study of hospitalized patients aged ≥ 65 years, 41.6% of the delirium cohort and 14.4% of the control group died within 12 months of hospital admission.5 The death rate predicted by delirium in the Canadian study was comparable to the death rate of those who experience other serious medical conditions, such as sepsis or a heart attack.6
Those who survive delirium experience other serious outcomes, such as a negative impact on function and cognition and an increase in long-term care placement.7 Even when the condition resolves quickly, lasting functional impairment may be evident without return to baseline functioning.8 Hospitalized veterans are generally older, making them susceptible to developing delirium.9
Prevalence
Delirium can result from multiple medical conditions and develops in up to 50% of patients after general surgery and up to 80% of patients in the intensive care unit.10,11 From 20% to 40% of hospitalized older adults and from 50% to 89% of patients with preexisting Alzheimer disease may develop delirium.12-15 The increasing number of aging adults who will be hospitalized may also result in an increased prevalence of delirium.1,16
Delirium is a result of various predisposing and precipitating factors.1 Predisposing vulnerabilities are intrinsic to the individual, whereas precipitating external stressors are found in the environment. External stressors may trigger delirium in an individual who is vulnerable due to predisposing risk. The primary risk factors for delirium include dementia, advanced age, sensory impairment, fracture, infection, and dehydration (Table 1).12
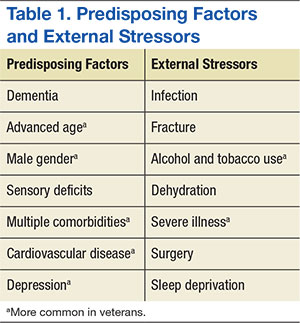
Predisposing factors for delirium, such as age and sex, lifestyle choices (alcohol, tobacco), and chronic conditions (atherosclerosis, depression, prior stroke/transient ischemic attack) are more prevalent in the veteran population.9,17-20 In 2011, the median age for male veterans was 64 and the median age for male nonveterans was 41. Of male veterans, 49.9% are aged ≥ 65 years in comparison with 10.5% of the nonveteran male population.21 Veterans also have higher rates of comorbidities; a significant risk factor for delirium.20 A study by Agha and colleagues found that veterans were 14 times more likely to have 5 or more medical conditions than that of the general population.9 In a study comparing veterans aged ≥ 65 years with their age matched nonveteran peers, the health status of the veterans was poorer overall.22 Veterans are more likely to have posttraumatic stress disorder, which can increase the risk of postsurgery delirium and dementia, a primary risk factor for delirium.23-26
Delirium Intervention
Up to 40% of delirium cases can be prevented.27 But delirium may remain undetected in older veterans because its symptoms are sometimes thought to be the unavoidable consequences of aging, dementia, preexisting mental health conditions, substance abuse, a disease process, or the hospital environment.28 Therefore, to avoid the negative consequences of delirium, prevention is critical.28
The goals of delirium treatment are to identify and reverse its underlying cause(s).29 Because delirium is typically multifactorial, an HCP must carefully consider the various sources that could have initiated a change in mental status. Delirium may be prevented if HCPs can reduce patient risk factors. The 2010 National Institute for Health and Clinical Excellence (NICE) Delirium Guideline recommended a set of prevention strategies to address delirium risk factors (Table 2).12
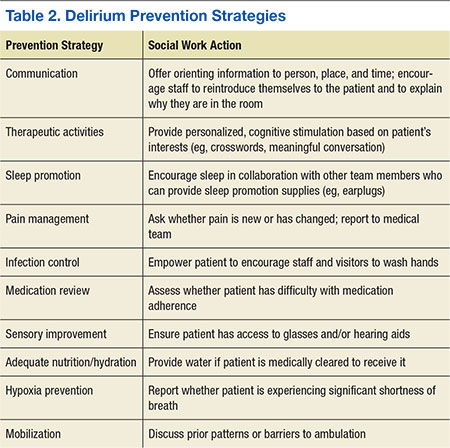
As a member of the health care team, social workers can help prevent delirium through attention to pain management, infection control, medication review, sensory improvement, adequate nutrition and hydration, hypoxia prevention, and mobilization.12No pharmacologic approach has been approved for the treatment of delirium.30 Drugs may manage symptoms associated with delirium, but they do not treat the disease and could be associated with toxicity in high-risk patients. However, there are a variety of nonpharmacologic preventative measures that have proven effective. Environmental interventions to prevent delirium include orientation, cognitive stimulation, and sensory aids. A 2013 meta-analysis of 19 delirium prevention programs found that most were effective in preventing delirium in patients at risk during hospitalization.31 This review found that the most successful programs included multidisciplinary teams providing staff education and therapeutic cognitive activities.31 Social workers can encourage and directly provide such services. The Delirium Toolbox is a delirium risk modification program that was piloted with frontline staff, including social workers, at the VA Boston Healthcare System in West Roxbury, Massachusetts, and has been associated with restraint reduction, shortened length of stay (LOS), and lower variable direct costs.32
Social Worker Role
Several studies, both national and international, have indicated that little has been done over the past 2 decades to increase the diagnosis of delirium, because only 12% to 35% of delirium cases are clinically detected within the emergency department and in acute care settings.33-37 Patients may hesitate to report their experience due to a sense of embarrassment or because of an inability to describe it.38
Social workers are skilled at helping individuals feel more at ease when disclosing distressing experiences. Delirium is relevant to HCPs and hospital social workers with care transition responsibilities, because delirium detection should impact discharge planning.1,39 Delirium education needs to be included in efforts to improve transitions from intensive care settings to lower levels of care and from lower levels of care to discharge.40 Hospital social workers are in a position to offer additional support because they see patients at a critical juncture in their care and can take steps to improve postdischarge outcomes.41
Prior to Onset
Social workers can play an important role prior to delirium onset.42 Patient education on delirium needs to be provided during the routine hospital intake assessment. Informing patients in advance that delirium is common, based on their risk factors, as well as what to expect if delirium is experienced has been found to provide comfort.38 Families who anticipated possible delirium-related confusion reported that they experienced less distress.38
Related: Baseball Reminiscence Therapy for Cognitively Impaired Veterans
During hospitalization, social workers can ascertain from families whether an alteration in mental status is a rapid change, possibly indicating delirium, or a gradual dementia onset. The social work skills of advocacy and education can be used to support delirium-risk identification to avoid adverse outcomes.43 When no family caregiver is present to provide a history of the individual’s cognitive function prior to hospitalization, the social worker may be the first to notice an acute change in cognitive status and can report this to the medical team.
During Delirium
Lack of patient responsiveness and difficulty following a conversation are possible signs of delirium. This situation should be reported to the medical team for further delirium assessment and diagnosis.4 The social worker can also attempt to determine whether a patient’s presentation is unusual by contacting the family. Social work training recognizes the important role of the family.44 Social workers often interact with families at the critical period between acute onset of delirium in the hospital and discharge.42 Studies have shown that delirium causes stress for the patient’s loved ones. Moreover, caregivers of patients who experience the syndrome are at a 12 times increased risk of meeting the criteria for generalized anxiety disorder.30 In one study, delirium was rated as more distressing for the caregivers who witnessed it than for the patients who experienced it.38 Education has been shown to reduce delirium-related distress.30
In cases where delirium is irreversible, such as during the active dying process, social workers can serve in a palliative role to ease family confusion and provide comfort.30 The presence of family and other familiar people are considered part of the nonpharmacologic management of delirium.28
Posthospitalization
Delirium complicates physical aspects of care for families, as their loved one may need direct care in areas where they were previously independent due to a loss of function. Logistic considerations such as increased supervision may be necessary due to delirium, and the patient’s condition may be upsetting and confusing for family members, triggering the need for emotional support. During the discharge process, social workers can provide support and education to family members or placement facilities.38
Social workers in the hospital setting are often responsible for discharge planning, including the reduction of extended LOS and unnecessary readmissions to the hospital.45 Increased LOS and hospital readmissions are 2 of the primary negative outcomes associated with delirium. Delirium can persist for months beyond hospitalization, making it a relevant issue at the time of discharge and beyond.46 Distress related to delirium has been documented up to 2 years after onset, due to manifestations of anxiety and depression.38
Distress impacts patients as well as caregivers who witness the delirium and provide care to the patient afterward.38 Long-term changes in mood in addition to loss of function as a result of delirium can lead to an increase in stress for both patients and their caregivers.30 The social work emphasis on counseling and family dynamics as well as the common role of coordinating post-discharge arrangements makes the profession uniquely suited for delirium care.
Barriers
Social workers can play a key role in delirium risk identification and coordination of care but face substantial barriers. Delirium assessments are complex and require training and education in the features of delirium and cognitive assessment.47 To date, social workers receive limited education about delirium and typically do not make deliberate efforts in prevention, support, and follow-up care.
Conclusion
Social workers will encounter delirium, and their training makes them particularly suited to address this health concern. An understanding of the larger ecologic system is a foundational aspect of social work and an essential component of delirium prevention and care.41 The multipathway nature of delirium as well as the importance of prevention suggests that multiple disciplines, including social work, should be involved.1 The American Delirium Society and the European Delirium Association both recognize the need for all HCPs to be engaged in delirium care.1,48
Related: Sharing Alzheimer Research, FasterSharing Alzheimer Research, Faster
Social workers in the hospital setting provide communication, advocacy, and education to other HCPs, as well as to patients and families (Figure). Because delirium directly impacts the emotional and logistic needs of patients and their families, it would be advantageous for social workers to take a more active role in delirium risk identification, prevention, and care. Fortunately, the nonpharmacologic approaches that social workers are skilled in providing (eg, education and emotional support) have been shown to benefit patients with delirium and their families.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Rudolph JL, Boustani M, Kamholz B, Shaughnessey M, Shay K; American Delirium Society. Delirium: a strategic plan to bring an ancient disease into the 21st century. J Am Geriatr Soc. 2011;59(suppl 2):S237-S240.
2. Hope C, Estrada N, Weir C, Teng CC, Damal K, Sauer BC. Documentation of delirium in the VA electronic health record. BMC Res Notes. 2014;7:208.
3. van Zyl LT, Davidson PR. Delirium in hospital: an underreported event at discharge. Can J Psychiatry. 2003;48(8):555-560.
4. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
5. McCusker J, Cole M, Abrahamowicz M, Primeau F, Belzile E. Delirium predicts 12-month mortality. Arch Intern Med. 2002;162(4):457-463.
6. Inouye SK. Delirium in older persons. N Engl J Med. 2006;354(11):1157-1165.
7. McCusker J, Cole M, Dendukuri N, Belzile E, Primeau F. Delirium in older medical inpatients and subsequent cognitive and functional status: a prospective study. CMAJ. 2001;165(5):575-583.
8. Quinlan N, Rudolph JL. Postoperative delirium and functional decline after noncardiac surgery. J Am Geriatr Soc. 2011;59(suppl 2):S301-S304.
9. Agha Z, Lofgren RP, VanRuiswyk JV, Layde PM. Are patients at Veterans Affairs medical centers sicker? A comparative analysis of health status and medical resource use. Arch Intern Med. 2000;160(21):3252-3257.
10. Marcantonio ER, Simon SE, Bergmann MA, Jones RN, Murphy KM, Morris JN. Delirium symptoms in post-acute care: prevalent, persistent, and associated with poor functional recovery. J Am Geriatr Soc. 2003;51(1):4-9.
11. McNicoll L, Pisani MA, Zhang Y, Ely EW, Siegel MD, Inouye SK. Delirium in the intensive care unit: occurrence and clinical course in older patients. J Am Geriatr Soc. 2003;51(5):591-598.
12. National Institute for Health and Clinical Excellence. Delirium: Diagnosis, Prevention and Management. National Institute for Health and Clinical Excellence Website. https://www.nice.org.uk/guidance/cg103/resources/delirium-174507018181. Published July 2010.
13. Fick D, Foreman M. Consequences of not recognizing delirium superimposed on dementia in hospitalized elderly individuals. J Gerontol Nurs. 2000;26(1):30-40.
14. Fick DM, Agostini JV, Inouye SK. Delirium superimposed on dementia: a systematic review. J Am Geriatr Soc. 2002;50(10):1723-1732.
15. Edlund A, Lundström M, Brännström B, Bucht G, Gustafson Y. Delirium before and after operation for femoral neck fracture. J Am Geriatr Soc. 2001;49(10):1335-1340.
16. Popejoy LL, Galambos C, Moylan K, Madsen R. Challenges to hospital discharge planning for older adults. Clin Nurs Res. 2012;21(4):431-449.
17. Marcantonio ER, Goldman L, Mangione CM, et al. A clinical prediction rule for delirium after elective noncardiac surgery. JAMA. 1994;271(2):134-139.
18. Rudolph JL, Jones RN, Rasmussen LS, Silverstein JH, Inouye SK, Marcantonio ER. Independent vascular and cognitive risk factors for postoperative delirium. Am J Med. 2007;120(9):807-813.
19. Rudolph JL, Babikian VL, Birjiniuk V, et al. Atherosclerosis is associated with delirium after coronary artery bypass graft surgery. J Am Geriatr Soc. 2005;53(3):462-466.
20. Rudolph JL, Jones RN, Levkoff SE, et al. Derivation and validation of a preoperative prediction rule for delirium after cardiac surgery. Circulation. 2009;119(2):229-236.
21. U.S. Department of Veterans Affairs, National Center for Veterans Analysis and Statistics. Profile of Veterans: 2013 Data from the American Community Survey. U.S. Department of Veterans Affairs Website. http://www.va.gov/vetdata/docs/SpecialReports/Profile_of_Veterans_2013.pdf. Accessed November 14, 2015.
22. Selim AJ, Berlowitz DR, Fincke G, et al. The health status of elderly veteran enrollees in the Veterans Health Administration. J Am Geriatr Soc. 2004;52(8):1271-1276.
23. McGuire JM. The incidence of and risk factors for emergence delirium in U.S. military combat veterans. J Perianesth Nurs. 2012;27(4):236-245.
24. Lepousé C, Lautner CA, Liu L, Gomis P, Leon A. Emergence delirium in adults in the post-anaesthesia care unit. Br J Anaesth. 2006;96(6):747-753.
25. Meziab O, Kirby KA, Williams B, Yaffe K, Byers AL, Barnes DE. Prisoner of war status, posttraumatic stress disorder, and dementia in older veterans. Alzheimers Dement. 2014;10(3)(suppl):S236-S241.
26. Elie M, Cole MG, Primeau FJ, Bellavance F. Delirium risk factors in elderly hospitalized patients. J Gen Intern Med. 1998;13(3):204-212.
27. Fong TG, Tulebaev SR, Inouye SK. Delirium in elderly adults: diagnosis, prevention and treatment. Nat Rev Neurol. 2009;5(4):210-220.
28. Conley DM. The gerontological clinical nurse specialist's role in prevention, early recognition, and management of delirium in hospitalized older adults. Urol Nurs. 2011;31(6):337-342.
29. Meagher DJ. Delirium: optimising management. BMJ. 2001;322(7279):144-149.
30. Irwin SA, Pirrello RD, Hirst JM, Buckholz GT, Ferris FD. Clarifying delirium management: practical, evidenced-based, expert recommendations for clinical practice. J Palliat Med. 2013;16(4):423-435.
31. Reston JT, Schoelles KM. In-facility delirium prevention programs as a patient safety strategy: a systematic review. Ann Intern Med. 2013;158(5, pt 2):375-380.
32. Rudolph JL, Archambault E, Kelly B; VA Boston Delirium Task Force. A delirium risk modification program is associated with hospital outcomes. J Am Med Dir Assoc. 2014;15(12):957.e7-957.e11.
33. Gustafson Y, Brännström B, Norberg A, Bucht G, Winblad B. Underdiagnosis and poor documentation of acute confusional states in elderly hip fracture patients. J Am Geriatr Soc. 1991;39(8):760-765.
34. Hustey FM, Meldon SW. The prevalence and documentation of impaired mental status in elderly emergency department patients. Ann Emerg Med. 2002;39(3):248-253.
35. Kales HC, Kamholz BA, Visnic SG, Blow FC. Recorded delirium in a national sample of elderly inpatients: potential implications for recognition. J Geriatr Psychiatry Neurol. 2003;16(1):32-38.
36. Lemiengre J, Nelis T, Joosten E, et al. Detection of delirium by bedside nurses using the confusion assessment method. J Am Geriatr Soc. 2006;54(4):685-689.
37. Milisen K, Foreman MD, Wouters B, et al. Documentation of delirium in elderly patients with hip fracture. J Gerontol Nurs. 2002;28(11):23-29.
38. Partridge JS, Martin FC, Harari D, Dhesi JK. The delirium experience: what is the effect on patients, relatives and staff and what can be done to modify this? Int J Geriatr Psychiatry. 2013;28(8):804-812.
39. Simons K, Connolly RP, Bonifas R, et al. Psychosocial assessment of nursing home residents via MDS 3.0: recommendations for social service training, staffing, and roles in interdisciplinary care. J Am Med Dir Assoc. 2012;13(2):190.e9-190.e15.
40. Alici Y. Interventions to improve recognition of delirium: a sine qua non for successful transitional care programs. Arch Intern Med. 2012;172(1):80-82.
41. Judd RG, Sheffield S. Hospital social work: contemporary roles and professional activities. Soc Work Health Care. 2010;49(9):856-871.
42. Duffy F, Healy JP. Social work with older people in a hospital setting. Soc Work Health Care. 2011;50(2):109-123.
43. Anderson CP, Ngo LH, Marcantonio ER. Complications in post-acute care are associated with persistent delirium. J Am Geriatr Soc. 2012;60(6):1122-1127.
44. Bauer M, Fitzgerald L, Haesler E, Manfrin M. Hospital discharge planning for frail older people and their family. Are we delivering best practice? A review of the evidence. J Clin Nurs. 2009;18(18):2539-2546.
45. Shepperd S, Lannin NA, Clemson LM, McCluskey A, Cameron ID, Barras SL. Discharge planning from hospital to home. Cochrane Database Syst Rev. 2013;1:CD000313.
46. McCusker J, Cole M, Dendukuri N, Han L, Belzile E. The course of delirium in older medical inpatients: A prospective study. J Gen Intern Med. 2003;18(9):696-704.
47. Inouye SK, Foreman MD, Mion LC, Katz KH, Cooney LM Jr. Nurses' recognition of delirium and its symptoms: comparison of nurse and researcher ratings. Arch Intern Med. 2001;161(20):2467-2473.
48. Teodorczuk A, Reynish E, Milisen K. Improving recognition of delirium in clinical practice: a call for action. BMC Geriatr. 2012;12:55.
Delirium, or the state of mental confusion that may occur with physical or mental illness, is common, morbid, and costly; however, of the diagnosed cases, delirium is mentioned in hospital discharge summaries only 16% to 55% of the time.1-3
Social workers often coordinate care transitions for hospitalized older veterans. They serve as interdisciplinary team members who communicate with VA medical staff as well as with the patient and family. This position, in addition to their training in communication and advocacy, primes social workers for a role in delirium care and provides the needed support for veterans who experience delirium and their families.
Background
Delirium is a sudden disturbance of attention with reduced awareness of the environment. Because attention is impaired, other changes in cognition are common, including perceptual and thought disturbances. Additionally, delirium includes fluctuations in consciousness over the course of a day. The acute development of these cognitive disturbances is distinct from a preexisting chronic cognitive impairment, such as dementia. Delirium is a direct consequence of underlying medical conditions, such as infections, polypharmacy, dehydration, and surgery.4
Delirium subtypes all have inattention as a core symptom. In half of the cases, patients are hypoactive and will not awaken easily or participate in daily care plans readily.4 Hyperactive delirium occurs in a quarter of cases. In the remaining mixed delirium cases patients fluctuate between the 2 states.4
Delirium is often falsely mistaken for dementia. Although delirium and dementia can present similarly, delirium has a sudden onset, which can alert health care professionals (HCPs) to the likelihood of delirium. Another important distinction is that delirium is typically reversible. Symptom manifestations of delirium may also be confused with depression.
Related: Delirium in the Cardiac ICU
Preventing delirium is important due to its many negative health outcomes. Older adults who develop delirium are more likely to die sooner. In a Canadian study of hospitalized patients aged ≥ 65 years, 41.6% of the delirium cohort and 14.4% of the control group died within 12 months of hospital admission.5 The death rate predicted by delirium in the Canadian study was comparable to the death rate of those who experience other serious medical conditions, such as sepsis or a heart attack.6
Those who survive delirium experience other serious outcomes, such as a negative impact on function and cognition and an increase in long-term care placement.7 Even when the condition resolves quickly, lasting functional impairment may be evident without return to baseline functioning.8 Hospitalized veterans are generally older, making them susceptible to developing delirium.9
Prevalence
Delirium can result from multiple medical conditions and develops in up to 50% of patients after general surgery and up to 80% of patients in the intensive care unit.10,11 From 20% to 40% of hospitalized older adults and from 50% to 89% of patients with preexisting Alzheimer disease may develop delirium.12-15 The increasing number of aging adults who will be hospitalized may also result in an increased prevalence of delirium.1,16
Delirium is a result of various predisposing and precipitating factors.1 Predisposing vulnerabilities are intrinsic to the individual, whereas precipitating external stressors are found in the environment. External stressors may trigger delirium in an individual who is vulnerable due to predisposing risk. The primary risk factors for delirium include dementia, advanced age, sensory impairment, fracture, infection, and dehydration (Table 1).12
Predisposing factors for delirium, such as age and sex, lifestyle choices (alcohol, tobacco), and chronic conditions (atherosclerosis, depression, prior stroke/transient ischemic attack) are more prevalent in the veteran population.9,17-20 In 2011, the median age for male veterans was 64 and the median age for male nonveterans was 41. Of male veterans, 49.9% are aged ≥ 65 years in comparison with 10.5% of the nonveteran male population.21 Veterans also have higher rates of comorbidities; a significant risk factor for delirium.20 A study by Agha and colleagues found that veterans were 14 times more likely to have 5 or more medical conditions than that of the general population.9 In a study comparing veterans aged ≥ 65 years with their age matched nonveteran peers, the health status of the veterans was poorer overall.22 Veterans are more likely to have posttraumatic stress disorder, which can increase the risk of postsurgery delirium and dementia, a primary risk factor for delirium.23-26
Delirium Intervention
Up to 40% of delirium cases can be prevented.27 But delirium may remain undetected in older veterans because its symptoms are sometimes thought to be the unavoidable consequences of aging, dementia, preexisting mental health conditions, substance abuse, a disease process, or the hospital environment.28 Therefore, to avoid the negative consequences of delirium, prevention is critical.28
The goals of delirium treatment are to identify and reverse its underlying cause(s).29 Because delirium is typically multifactorial, an HCP must carefully consider the various sources that could have initiated a change in mental status. Delirium may be prevented if HCPs can reduce patient risk factors. The 2010 National Institute for Health and Clinical Excellence (NICE) Delirium Guideline recommended a set of prevention strategies to address delirium risk factors (Table 2).12
As a member of the health care team, social workers can help prevent delirium through attention to pain management, infection control, medication review, sensory improvement, adequate nutrition and hydration, hypoxia prevention, and mobilization.12No pharmacologic approach has been approved for the treatment of delirium.30 Drugs may manage symptoms associated with delirium, but they do not treat the disease and could be associated with toxicity in high-risk patients. However, there are a variety of nonpharmacologic preventative measures that have proven effective. Environmental interventions to prevent delirium include orientation, cognitive stimulation, and sensory aids. A 2013 meta-analysis of 19 delirium prevention programs found that most were effective in preventing delirium in patients at risk during hospitalization.31 This review found that the most successful programs included multidisciplinary teams providing staff education and therapeutic cognitive activities.31 Social workers can encourage and directly provide such services. The Delirium Toolbox is a delirium risk modification program that was piloted with frontline staff, including social workers, at the VA Boston Healthcare System in West Roxbury, Massachusetts, and has been associated with restraint reduction, shortened length of stay (LOS), and lower variable direct costs.32
Social Worker Role
Several studies, both national and international, have indicated that little has been done over the past 2 decades to increase the diagnosis of delirium, because only 12% to 35% of delirium cases are clinically detected within the emergency department and in acute care settings.33-37 Patients may hesitate to report their experience due to a sense of embarrassment or because of an inability to describe it.38
Social workers are skilled at helping individuals feel more at ease when disclosing distressing experiences. Delirium is relevant to HCPs and hospital social workers with care transition responsibilities, because delirium detection should impact discharge planning.1,39 Delirium education needs to be included in efforts to improve transitions from intensive care settings to lower levels of care and from lower levels of care to discharge.40 Hospital social workers are in a position to offer additional support because they see patients at a critical juncture in their care and can take steps to improve postdischarge outcomes.41
Prior to Onset
Social workers can play an important role prior to delirium onset.42 Patient education on delirium needs to be provided during the routine hospital intake assessment. Informing patients in advance that delirium is common, based on their risk factors, as well as what to expect if delirium is experienced has been found to provide comfort.38 Families who anticipated possible delirium-related confusion reported that they experienced less distress.38
Related: Baseball Reminiscence Therapy for Cognitively Impaired Veterans
During hospitalization, social workers can ascertain from families whether an alteration in mental status is a rapid change, possibly indicating delirium, or a gradual dementia onset. The social work skills of advocacy and education can be used to support delirium-risk identification to avoid adverse outcomes.43 When no family caregiver is present to provide a history of the individual’s cognitive function prior to hospitalization, the social worker may be the first to notice an acute change in cognitive status and can report this to the medical team.
During Delirium
Lack of patient responsiveness and difficulty following a conversation are possible signs of delirium. This situation should be reported to the medical team for further delirium assessment and diagnosis.4 The social worker can also attempt to determine whether a patient’s presentation is unusual by contacting the family. Social work training recognizes the important role of the family.44 Social workers often interact with families at the critical period between acute onset of delirium in the hospital and discharge.42 Studies have shown that delirium causes stress for the patient’s loved ones. Moreover, caregivers of patients who experience the syndrome are at a 12 times increased risk of meeting the criteria for generalized anxiety disorder.30 In one study, delirium was rated as more distressing for the caregivers who witnessed it than for the patients who experienced it.38 Education has been shown to reduce delirium-related distress.30
In cases where delirium is irreversible, such as during the active dying process, social workers can serve in a palliative role to ease family confusion and provide comfort.30 The presence of family and other familiar people are considered part of the nonpharmacologic management of delirium.28
Posthospitalization
Delirium complicates physical aspects of care for families, as their loved one may need direct care in areas where they were previously independent due to a loss of function. Logistic considerations such as increased supervision may be necessary due to delirium, and the patient’s condition may be upsetting and confusing for family members, triggering the need for emotional support. During the discharge process, social workers can provide support and education to family members or placement facilities.38
Social workers in the hospital setting are often responsible for discharge planning, including the reduction of extended LOS and unnecessary readmissions to the hospital.45 Increased LOS and hospital readmissions are 2 of the primary negative outcomes associated with delirium. Delirium can persist for months beyond hospitalization, making it a relevant issue at the time of discharge and beyond.46 Distress related to delirium has been documented up to 2 years after onset, due to manifestations of anxiety and depression.38
Distress impacts patients as well as caregivers who witness the delirium and provide care to the patient afterward.38 Long-term changes in mood in addition to loss of function as a result of delirium can lead to an increase in stress for both patients and their caregivers.30 The social work emphasis on counseling and family dynamics as well as the common role of coordinating post-discharge arrangements makes the profession uniquely suited for delirium care.
Barriers
Social workers can play a key role in delirium risk identification and coordination of care but face substantial barriers. Delirium assessments are complex and require training and education in the features of delirium and cognitive assessment.47 To date, social workers receive limited education about delirium and typically do not make deliberate efforts in prevention, support, and follow-up care.
Conclusion
Social workers will encounter delirium, and their training makes them particularly suited to address this health concern. An understanding of the larger ecologic system is a foundational aspect of social work and an essential component of delirium prevention and care.41 The multipathway nature of delirium as well as the importance of prevention suggests that multiple disciplines, including social work, should be involved.1 The American Delirium Society and the European Delirium Association both recognize the need for all HCPs to be engaged in delirium care.1,48
Related: Sharing Alzheimer Research, FasterSharing Alzheimer Research, Faster
Social workers in the hospital setting provide communication, advocacy, and education to other HCPs, as well as to patients and families (Figure). Because delirium directly impacts the emotional and logistic needs of patients and their families, it would be advantageous for social workers to take a more active role in delirium risk identification, prevention, and care. Fortunately, the nonpharmacologic approaches that social workers are skilled in providing (eg, education and emotional support) have been shown to benefit patients with delirium and their families.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Delirium, or the state of mental confusion that may occur with physical or mental illness, is common, morbid, and costly; however, of the diagnosed cases, delirium is mentioned in hospital discharge summaries only 16% to 55% of the time.1-3
Social workers often coordinate care transitions for hospitalized older veterans. They serve as interdisciplinary team members who communicate with VA medical staff as well as with the patient and family. This position, in addition to their training in communication and advocacy, primes social workers for a role in delirium care and provides the needed support for veterans who experience delirium and their families.
Background
Delirium is a sudden disturbance of attention with reduced awareness of the environment. Because attention is impaired, other changes in cognition are common, including perceptual and thought disturbances. Additionally, delirium includes fluctuations in consciousness over the course of a day. The acute development of these cognitive disturbances is distinct from a preexisting chronic cognitive impairment, such as dementia. Delirium is a direct consequence of underlying medical conditions, such as infections, polypharmacy, dehydration, and surgery.4
Delirium subtypes all have inattention as a core symptom. In half of the cases, patients are hypoactive and will not awaken easily or participate in daily care plans readily.4 Hyperactive delirium occurs in a quarter of cases. In the remaining mixed delirium cases patients fluctuate between the 2 states.4
Delirium is often falsely mistaken for dementia. Although delirium and dementia can present similarly, delirium has a sudden onset, which can alert health care professionals (HCPs) to the likelihood of delirium. Another important distinction is that delirium is typically reversible. Symptom manifestations of delirium may also be confused with depression.
Related: Delirium in the Cardiac ICU
Preventing delirium is important due to its many negative health outcomes. Older adults who develop delirium are more likely to die sooner. In a Canadian study of hospitalized patients aged ≥ 65 years, 41.6% of the delirium cohort and 14.4% of the control group died within 12 months of hospital admission.5 The death rate predicted by delirium in the Canadian study was comparable to the death rate of those who experience other serious medical conditions, such as sepsis or a heart attack.6
Those who survive delirium experience other serious outcomes, such as a negative impact on function and cognition and an increase in long-term care placement.7 Even when the condition resolves quickly, lasting functional impairment may be evident without return to baseline functioning.8 Hospitalized veterans are generally older, making them susceptible to developing delirium.9
Prevalence
Delirium can result from multiple medical conditions and develops in up to 50% of patients after general surgery and up to 80% of patients in the intensive care unit.10,11 From 20% to 40% of hospitalized older adults and from 50% to 89% of patients with preexisting Alzheimer disease may develop delirium.12-15 The increasing number of aging adults who will be hospitalized may also result in an increased prevalence of delirium.1,16
Delirium is a result of various predisposing and precipitating factors.1 Predisposing vulnerabilities are intrinsic to the individual, whereas precipitating external stressors are found in the environment. External stressors may trigger delirium in an individual who is vulnerable due to predisposing risk. The primary risk factors for delirium include dementia, advanced age, sensory impairment, fracture, infection, and dehydration (Table 1).12
Predisposing factors for delirium, such as age and sex, lifestyle choices (alcohol, tobacco), and chronic conditions (atherosclerosis, depression, prior stroke/transient ischemic attack) are more prevalent in the veteran population.9,17-20 In 2011, the median age for male veterans was 64 and the median age for male nonveterans was 41. Of male veterans, 49.9% are aged ≥ 65 years in comparison with 10.5% of the nonveteran male population.21 Veterans also have higher rates of comorbidities; a significant risk factor for delirium.20 A study by Agha and colleagues found that veterans were 14 times more likely to have 5 or more medical conditions than that of the general population.9 In a study comparing veterans aged ≥ 65 years with their age matched nonveteran peers, the health status of the veterans was poorer overall.22 Veterans are more likely to have posttraumatic stress disorder, which can increase the risk of postsurgery delirium and dementia, a primary risk factor for delirium.23-26
Delirium Intervention
Up to 40% of delirium cases can be prevented.27 But delirium may remain undetected in older veterans because its symptoms are sometimes thought to be the unavoidable consequences of aging, dementia, preexisting mental health conditions, substance abuse, a disease process, or the hospital environment.28 Therefore, to avoid the negative consequences of delirium, prevention is critical.28
The goals of delirium treatment are to identify and reverse its underlying cause(s).29 Because delirium is typically multifactorial, an HCP must carefully consider the various sources that could have initiated a change in mental status. Delirium may be prevented if HCPs can reduce patient risk factors. The 2010 National Institute for Health and Clinical Excellence (NICE) Delirium Guideline recommended a set of prevention strategies to address delirium risk factors (Table 2).12
As a member of the health care team, social workers can help prevent delirium through attention to pain management, infection control, medication review, sensory improvement, adequate nutrition and hydration, hypoxia prevention, and mobilization.12No pharmacologic approach has been approved for the treatment of delirium.30 Drugs may manage symptoms associated with delirium, but they do not treat the disease and could be associated with toxicity in high-risk patients. However, there are a variety of nonpharmacologic preventative measures that have proven effective. Environmental interventions to prevent delirium include orientation, cognitive stimulation, and sensory aids. A 2013 meta-analysis of 19 delirium prevention programs found that most were effective in preventing delirium in patients at risk during hospitalization.31 This review found that the most successful programs included multidisciplinary teams providing staff education and therapeutic cognitive activities.31 Social workers can encourage and directly provide such services. The Delirium Toolbox is a delirium risk modification program that was piloted with frontline staff, including social workers, at the VA Boston Healthcare System in West Roxbury, Massachusetts, and has been associated with restraint reduction, shortened length of stay (LOS), and lower variable direct costs.32
Social Worker Role
Several studies, both national and international, have indicated that little has been done over the past 2 decades to increase the diagnosis of delirium, because only 12% to 35% of delirium cases are clinically detected within the emergency department and in acute care settings.33-37 Patients may hesitate to report their experience due to a sense of embarrassment or because of an inability to describe it.38
Social workers are skilled at helping individuals feel more at ease when disclosing distressing experiences. Delirium is relevant to HCPs and hospital social workers with care transition responsibilities, because delirium detection should impact discharge planning.1,39 Delirium education needs to be included in efforts to improve transitions from intensive care settings to lower levels of care and from lower levels of care to discharge.40 Hospital social workers are in a position to offer additional support because they see patients at a critical juncture in their care and can take steps to improve postdischarge outcomes.41
Prior to Onset
Social workers can play an important role prior to delirium onset.42 Patient education on delirium needs to be provided during the routine hospital intake assessment. Informing patients in advance that delirium is common, based on their risk factors, as well as what to expect if delirium is experienced has been found to provide comfort.38 Families who anticipated possible delirium-related confusion reported that they experienced less distress.38
Related: Baseball Reminiscence Therapy for Cognitively Impaired Veterans
During hospitalization, social workers can ascertain from families whether an alteration in mental status is a rapid change, possibly indicating delirium, or a gradual dementia onset. The social work skills of advocacy and education can be used to support delirium-risk identification to avoid adverse outcomes.43 When no family caregiver is present to provide a history of the individual’s cognitive function prior to hospitalization, the social worker may be the first to notice an acute change in cognitive status and can report this to the medical team.
During Delirium
Lack of patient responsiveness and difficulty following a conversation are possible signs of delirium. This situation should be reported to the medical team for further delirium assessment and diagnosis.4 The social worker can also attempt to determine whether a patient’s presentation is unusual by contacting the family. Social work training recognizes the important role of the family.44 Social workers often interact with families at the critical period between acute onset of delirium in the hospital and discharge.42 Studies have shown that delirium causes stress for the patient’s loved ones. Moreover, caregivers of patients who experience the syndrome are at a 12 times increased risk of meeting the criteria for generalized anxiety disorder.30 In one study, delirium was rated as more distressing for the caregivers who witnessed it than for the patients who experienced it.38 Education has been shown to reduce delirium-related distress.30
In cases where delirium is irreversible, such as during the active dying process, social workers can serve in a palliative role to ease family confusion and provide comfort.30 The presence of family and other familiar people are considered part of the nonpharmacologic management of delirium.28
Posthospitalization
Delirium complicates physical aspects of care for families, as their loved one may need direct care in areas where they were previously independent due to a loss of function. Logistic considerations such as increased supervision may be necessary due to delirium, and the patient’s condition may be upsetting and confusing for family members, triggering the need for emotional support. During the discharge process, social workers can provide support and education to family members or placement facilities.38
Social workers in the hospital setting are often responsible for discharge planning, including the reduction of extended LOS and unnecessary readmissions to the hospital.45 Increased LOS and hospital readmissions are 2 of the primary negative outcomes associated with delirium. Delirium can persist for months beyond hospitalization, making it a relevant issue at the time of discharge and beyond.46 Distress related to delirium has been documented up to 2 years after onset, due to manifestations of anxiety and depression.38
Distress impacts patients as well as caregivers who witness the delirium and provide care to the patient afterward.38 Long-term changes in mood in addition to loss of function as a result of delirium can lead to an increase in stress for both patients and their caregivers.30 The social work emphasis on counseling and family dynamics as well as the common role of coordinating post-discharge arrangements makes the profession uniquely suited for delirium care.
Barriers
Social workers can play a key role in delirium risk identification and coordination of care but face substantial barriers. Delirium assessments are complex and require training and education in the features of delirium and cognitive assessment.47 To date, social workers receive limited education about delirium and typically do not make deliberate efforts in prevention, support, and follow-up care.
Conclusion
Social workers will encounter delirium, and their training makes them particularly suited to address this health concern. An understanding of the larger ecologic system is a foundational aspect of social work and an essential component of delirium prevention and care.41 The multipathway nature of delirium as well as the importance of prevention suggests that multiple disciplines, including social work, should be involved.1 The American Delirium Society and the European Delirium Association both recognize the need for all HCPs to be engaged in delirium care.1,48
Related: Sharing Alzheimer Research, FasterSharing Alzheimer Research, Faster
Social workers in the hospital setting provide communication, advocacy, and education to other HCPs, as well as to patients and families (Figure). Because delirium directly impacts the emotional and logistic needs of patients and their families, it would be advantageous for social workers to take a more active role in delirium risk identification, prevention, and care. Fortunately, the nonpharmacologic approaches that social workers are skilled in providing (eg, education and emotional support) have been shown to benefit patients with delirium and their families.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Rudolph JL, Boustani M, Kamholz B, Shaughnessey M, Shay K; American Delirium Society. Delirium: a strategic plan to bring an ancient disease into the 21st century. J Am Geriatr Soc. 2011;59(suppl 2):S237-S240.
2. Hope C, Estrada N, Weir C, Teng CC, Damal K, Sauer BC. Documentation of delirium in the VA electronic health record. BMC Res Notes. 2014;7:208.
3. van Zyl LT, Davidson PR. Delirium in hospital: an underreported event at discharge. Can J Psychiatry. 2003;48(8):555-560.
4. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
5. McCusker J, Cole M, Abrahamowicz M, Primeau F, Belzile E. Delirium predicts 12-month mortality. Arch Intern Med. 2002;162(4):457-463.
6. Inouye SK. Delirium in older persons. N Engl J Med. 2006;354(11):1157-1165.
7. McCusker J, Cole M, Dendukuri N, Belzile E, Primeau F. Delirium in older medical inpatients and subsequent cognitive and functional status: a prospective study. CMAJ. 2001;165(5):575-583.
8. Quinlan N, Rudolph JL. Postoperative delirium and functional decline after noncardiac surgery. J Am Geriatr Soc. 2011;59(suppl 2):S301-S304.
9. Agha Z, Lofgren RP, VanRuiswyk JV, Layde PM. Are patients at Veterans Affairs medical centers sicker? A comparative analysis of health status and medical resource use. Arch Intern Med. 2000;160(21):3252-3257.
10. Marcantonio ER, Simon SE, Bergmann MA, Jones RN, Murphy KM, Morris JN. Delirium symptoms in post-acute care: prevalent, persistent, and associated with poor functional recovery. J Am Geriatr Soc. 2003;51(1):4-9.
11. McNicoll L, Pisani MA, Zhang Y, Ely EW, Siegel MD, Inouye SK. Delirium in the intensive care unit: occurrence and clinical course in older patients. J Am Geriatr Soc. 2003;51(5):591-598.
12. National Institute for Health and Clinical Excellence. Delirium: Diagnosis, Prevention and Management. National Institute for Health and Clinical Excellence Website. https://www.nice.org.uk/guidance/cg103/resources/delirium-174507018181. Published July 2010.
13. Fick D, Foreman M. Consequences of not recognizing delirium superimposed on dementia in hospitalized elderly individuals. J Gerontol Nurs. 2000;26(1):30-40.
14. Fick DM, Agostini JV, Inouye SK. Delirium superimposed on dementia: a systematic review. J Am Geriatr Soc. 2002;50(10):1723-1732.
15. Edlund A, Lundström M, Brännström B, Bucht G, Gustafson Y. Delirium before and after operation for femoral neck fracture. J Am Geriatr Soc. 2001;49(10):1335-1340.
16. Popejoy LL, Galambos C, Moylan K, Madsen R. Challenges to hospital discharge planning for older adults. Clin Nurs Res. 2012;21(4):431-449.
17. Marcantonio ER, Goldman L, Mangione CM, et al. A clinical prediction rule for delirium after elective noncardiac surgery. JAMA. 1994;271(2):134-139.
18. Rudolph JL, Jones RN, Rasmussen LS, Silverstein JH, Inouye SK, Marcantonio ER. Independent vascular and cognitive risk factors for postoperative delirium. Am J Med. 2007;120(9):807-813.
19. Rudolph JL, Babikian VL, Birjiniuk V, et al. Atherosclerosis is associated with delirium after coronary artery bypass graft surgery. J Am Geriatr Soc. 2005;53(3):462-466.
20. Rudolph JL, Jones RN, Levkoff SE, et al. Derivation and validation of a preoperative prediction rule for delirium after cardiac surgery. Circulation. 2009;119(2):229-236.
21. U.S. Department of Veterans Affairs, National Center for Veterans Analysis and Statistics. Profile of Veterans: 2013 Data from the American Community Survey. U.S. Department of Veterans Affairs Website. http://www.va.gov/vetdata/docs/SpecialReports/Profile_of_Veterans_2013.pdf. Accessed November 14, 2015.
22. Selim AJ, Berlowitz DR, Fincke G, et al. The health status of elderly veteran enrollees in the Veterans Health Administration. J Am Geriatr Soc. 2004;52(8):1271-1276.
23. McGuire JM. The incidence of and risk factors for emergence delirium in U.S. military combat veterans. J Perianesth Nurs. 2012;27(4):236-245.
24. Lepousé C, Lautner CA, Liu L, Gomis P, Leon A. Emergence delirium in adults in the post-anaesthesia care unit. Br J Anaesth. 2006;96(6):747-753.
25. Meziab O, Kirby KA, Williams B, Yaffe K, Byers AL, Barnes DE. Prisoner of war status, posttraumatic stress disorder, and dementia in older veterans. Alzheimers Dement. 2014;10(3)(suppl):S236-S241.
26. Elie M, Cole MG, Primeau FJ, Bellavance F. Delirium risk factors in elderly hospitalized patients. J Gen Intern Med. 1998;13(3):204-212.
27. Fong TG, Tulebaev SR, Inouye SK. Delirium in elderly adults: diagnosis, prevention and treatment. Nat Rev Neurol. 2009;5(4):210-220.
28. Conley DM. The gerontological clinical nurse specialist's role in prevention, early recognition, and management of delirium in hospitalized older adults. Urol Nurs. 2011;31(6):337-342.
29. Meagher DJ. Delirium: optimising management. BMJ. 2001;322(7279):144-149.
30. Irwin SA, Pirrello RD, Hirst JM, Buckholz GT, Ferris FD. Clarifying delirium management: practical, evidenced-based, expert recommendations for clinical practice. J Palliat Med. 2013;16(4):423-435.
31. Reston JT, Schoelles KM. In-facility delirium prevention programs as a patient safety strategy: a systematic review. Ann Intern Med. 2013;158(5, pt 2):375-380.
32. Rudolph JL, Archambault E, Kelly B; VA Boston Delirium Task Force. A delirium risk modification program is associated with hospital outcomes. J Am Med Dir Assoc. 2014;15(12):957.e7-957.e11.
33. Gustafson Y, Brännström B, Norberg A, Bucht G, Winblad B. Underdiagnosis and poor documentation of acute confusional states in elderly hip fracture patients. J Am Geriatr Soc. 1991;39(8):760-765.
34. Hustey FM, Meldon SW. The prevalence and documentation of impaired mental status in elderly emergency department patients. Ann Emerg Med. 2002;39(3):248-253.
35. Kales HC, Kamholz BA, Visnic SG, Blow FC. Recorded delirium in a national sample of elderly inpatients: potential implications for recognition. J Geriatr Psychiatry Neurol. 2003;16(1):32-38.
36. Lemiengre J, Nelis T, Joosten E, et al. Detection of delirium by bedside nurses using the confusion assessment method. J Am Geriatr Soc. 2006;54(4):685-689.
37. Milisen K, Foreman MD, Wouters B, et al. Documentation of delirium in elderly patients with hip fracture. J Gerontol Nurs. 2002;28(11):23-29.
38. Partridge JS, Martin FC, Harari D, Dhesi JK. The delirium experience: what is the effect on patients, relatives and staff and what can be done to modify this? Int J Geriatr Psychiatry. 2013;28(8):804-812.
39. Simons K, Connolly RP, Bonifas R, et al. Psychosocial assessment of nursing home residents via MDS 3.0: recommendations for social service training, staffing, and roles in interdisciplinary care. J Am Med Dir Assoc. 2012;13(2):190.e9-190.e15.
40. Alici Y. Interventions to improve recognition of delirium: a sine qua non for successful transitional care programs. Arch Intern Med. 2012;172(1):80-82.
41. Judd RG, Sheffield S. Hospital social work: contemporary roles and professional activities. Soc Work Health Care. 2010;49(9):856-871.
42. Duffy F, Healy JP. Social work with older people in a hospital setting. Soc Work Health Care. 2011;50(2):109-123.
43. Anderson CP, Ngo LH, Marcantonio ER. Complications in post-acute care are associated with persistent delirium. J Am Geriatr Soc. 2012;60(6):1122-1127.
44. Bauer M, Fitzgerald L, Haesler E, Manfrin M. Hospital discharge planning for frail older people and their family. Are we delivering best practice? A review of the evidence. J Clin Nurs. 2009;18(18):2539-2546.
45. Shepperd S, Lannin NA, Clemson LM, McCluskey A, Cameron ID, Barras SL. Discharge planning from hospital to home. Cochrane Database Syst Rev. 2013;1:CD000313.
46. McCusker J, Cole M, Dendukuri N, Han L, Belzile E. The course of delirium in older medical inpatients: A prospective study. J Gen Intern Med. 2003;18(9):696-704.
47. Inouye SK, Foreman MD, Mion LC, Katz KH, Cooney LM Jr. Nurses' recognition of delirium and its symptoms: comparison of nurse and researcher ratings. Arch Intern Med. 2001;161(20):2467-2473.
48. Teodorczuk A, Reynish E, Milisen K. Improving recognition of delirium in clinical practice: a call for action. BMC Geriatr. 2012;12:55.
1. Rudolph JL, Boustani M, Kamholz B, Shaughnessey M, Shay K; American Delirium Society. Delirium: a strategic plan to bring an ancient disease into the 21st century. J Am Geriatr Soc. 2011;59(suppl 2):S237-S240.
2. Hope C, Estrada N, Weir C, Teng CC, Damal K, Sauer BC. Documentation of delirium in the VA electronic health record. BMC Res Notes. 2014;7:208.
3. van Zyl LT, Davidson PR. Delirium in hospital: an underreported event at discharge. Can J Psychiatry. 2003;48(8):555-560.
4. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
5. McCusker J, Cole M, Abrahamowicz M, Primeau F, Belzile E. Delirium predicts 12-month mortality. Arch Intern Med. 2002;162(4):457-463.
6. Inouye SK. Delirium in older persons. N Engl J Med. 2006;354(11):1157-1165.
7. McCusker J, Cole M, Dendukuri N, Belzile E, Primeau F. Delirium in older medical inpatients and subsequent cognitive and functional status: a prospective study. CMAJ. 2001;165(5):575-583.
8. Quinlan N, Rudolph JL. Postoperative delirium and functional decline after noncardiac surgery. J Am Geriatr Soc. 2011;59(suppl 2):S301-S304.
9. Agha Z, Lofgren RP, VanRuiswyk JV, Layde PM. Are patients at Veterans Affairs medical centers sicker? A comparative analysis of health status and medical resource use. Arch Intern Med. 2000;160(21):3252-3257.
10. Marcantonio ER, Simon SE, Bergmann MA, Jones RN, Murphy KM, Morris JN. Delirium symptoms in post-acute care: prevalent, persistent, and associated with poor functional recovery. J Am Geriatr Soc. 2003;51(1):4-9.
11. McNicoll L, Pisani MA, Zhang Y, Ely EW, Siegel MD, Inouye SK. Delirium in the intensive care unit: occurrence and clinical course in older patients. J Am Geriatr Soc. 2003;51(5):591-598.
12. National Institute for Health and Clinical Excellence. Delirium: Diagnosis, Prevention and Management. National Institute for Health and Clinical Excellence Website. https://www.nice.org.uk/guidance/cg103/resources/delirium-174507018181. Published July 2010.
13. Fick D, Foreman M. Consequences of not recognizing delirium superimposed on dementia in hospitalized elderly individuals. J Gerontol Nurs. 2000;26(1):30-40.
14. Fick DM, Agostini JV, Inouye SK. Delirium superimposed on dementia: a systematic review. J Am Geriatr Soc. 2002;50(10):1723-1732.
15. Edlund A, Lundström M, Brännström B, Bucht G, Gustafson Y. Delirium before and after operation for femoral neck fracture. J Am Geriatr Soc. 2001;49(10):1335-1340.
16. Popejoy LL, Galambos C, Moylan K, Madsen R. Challenges to hospital discharge planning for older adults. Clin Nurs Res. 2012;21(4):431-449.
17. Marcantonio ER, Goldman L, Mangione CM, et al. A clinical prediction rule for delirium after elective noncardiac surgery. JAMA. 1994;271(2):134-139.
18. Rudolph JL, Jones RN, Rasmussen LS, Silverstein JH, Inouye SK, Marcantonio ER. Independent vascular and cognitive risk factors for postoperative delirium. Am J Med. 2007;120(9):807-813.
19. Rudolph JL, Babikian VL, Birjiniuk V, et al. Atherosclerosis is associated with delirium after coronary artery bypass graft surgery. J Am Geriatr Soc. 2005;53(3):462-466.
20. Rudolph JL, Jones RN, Levkoff SE, et al. Derivation and validation of a preoperative prediction rule for delirium after cardiac surgery. Circulation. 2009;119(2):229-236.
21. U.S. Department of Veterans Affairs, National Center for Veterans Analysis and Statistics. Profile of Veterans: 2013 Data from the American Community Survey. U.S. Department of Veterans Affairs Website. http://www.va.gov/vetdata/docs/SpecialReports/Profile_of_Veterans_2013.pdf. Accessed November 14, 2015.
22. Selim AJ, Berlowitz DR, Fincke G, et al. The health status of elderly veteran enrollees in the Veterans Health Administration. J Am Geriatr Soc. 2004;52(8):1271-1276.
23. McGuire JM. The incidence of and risk factors for emergence delirium in U.S. military combat veterans. J Perianesth Nurs. 2012;27(4):236-245.
24. Lepousé C, Lautner CA, Liu L, Gomis P, Leon A. Emergence delirium in adults in the post-anaesthesia care unit. Br J Anaesth. 2006;96(6):747-753.
25. Meziab O, Kirby KA, Williams B, Yaffe K, Byers AL, Barnes DE. Prisoner of war status, posttraumatic stress disorder, and dementia in older veterans. Alzheimers Dement. 2014;10(3)(suppl):S236-S241.
26. Elie M, Cole MG, Primeau FJ, Bellavance F. Delirium risk factors in elderly hospitalized patients. J Gen Intern Med. 1998;13(3):204-212.
27. Fong TG, Tulebaev SR, Inouye SK. Delirium in elderly adults: diagnosis, prevention and treatment. Nat Rev Neurol. 2009;5(4):210-220.
28. Conley DM. The gerontological clinical nurse specialist's role in prevention, early recognition, and management of delirium in hospitalized older adults. Urol Nurs. 2011;31(6):337-342.
29. Meagher DJ. Delirium: optimising management. BMJ. 2001;322(7279):144-149.
30. Irwin SA, Pirrello RD, Hirst JM, Buckholz GT, Ferris FD. Clarifying delirium management: practical, evidenced-based, expert recommendations for clinical practice. J Palliat Med. 2013;16(4):423-435.
31. Reston JT, Schoelles KM. In-facility delirium prevention programs as a patient safety strategy: a systematic review. Ann Intern Med. 2013;158(5, pt 2):375-380.
32. Rudolph JL, Archambault E, Kelly B; VA Boston Delirium Task Force. A delirium risk modification program is associated with hospital outcomes. J Am Med Dir Assoc. 2014;15(12):957.e7-957.e11.
33. Gustafson Y, Brännström B, Norberg A, Bucht G, Winblad B. Underdiagnosis and poor documentation of acute confusional states in elderly hip fracture patients. J Am Geriatr Soc. 1991;39(8):760-765.
34. Hustey FM, Meldon SW. The prevalence and documentation of impaired mental status in elderly emergency department patients. Ann Emerg Med. 2002;39(3):248-253.
35. Kales HC, Kamholz BA, Visnic SG, Blow FC. Recorded delirium in a national sample of elderly inpatients: potential implications for recognition. J Geriatr Psychiatry Neurol. 2003;16(1):32-38.
36. Lemiengre J, Nelis T, Joosten E, et al. Detection of delirium by bedside nurses using the confusion assessment method. J Am Geriatr Soc. 2006;54(4):685-689.
37. Milisen K, Foreman MD, Wouters B, et al. Documentation of delirium in elderly patients with hip fracture. J Gerontol Nurs. 2002;28(11):23-29.
38. Partridge JS, Martin FC, Harari D, Dhesi JK. The delirium experience: what is the effect on patients, relatives and staff and what can be done to modify this? Int J Geriatr Psychiatry. 2013;28(8):804-812.
39. Simons K, Connolly RP, Bonifas R, et al. Psychosocial assessment of nursing home residents via MDS 3.0: recommendations for social service training, staffing, and roles in interdisciplinary care. J Am Med Dir Assoc. 2012;13(2):190.e9-190.e15.
40. Alici Y. Interventions to improve recognition of delirium: a sine qua non for successful transitional care programs. Arch Intern Med. 2012;172(1):80-82.
41. Judd RG, Sheffield S. Hospital social work: contemporary roles and professional activities. Soc Work Health Care. 2010;49(9):856-871.
42. Duffy F, Healy JP. Social work with older people in a hospital setting. Soc Work Health Care. 2011;50(2):109-123.
43. Anderson CP, Ngo LH, Marcantonio ER. Complications in post-acute care are associated with persistent delirium. J Am Geriatr Soc. 2012;60(6):1122-1127.
44. Bauer M, Fitzgerald L, Haesler E, Manfrin M. Hospital discharge planning for frail older people and their family. Are we delivering best practice? A review of the evidence. J Clin Nurs. 2009;18(18):2539-2546.
45. Shepperd S, Lannin NA, Clemson LM, McCluskey A, Cameron ID, Barras SL. Discharge planning from hospital to home. Cochrane Database Syst Rev. 2013;1:CD000313.
46. McCusker J, Cole M, Dendukuri N, Han L, Belzile E. The course of delirium in older medical inpatients: A prospective study. J Gen Intern Med. 2003;18(9):696-704.
47. Inouye SK, Foreman MD, Mion LC, Katz KH, Cooney LM Jr. Nurses' recognition of delirium and its symptoms: comparison of nurse and researcher ratings. Arch Intern Med. 2001;161(20):2467-2473.
48. Teodorczuk A, Reynish E, Milisen K. Improving recognition of delirium in clinical practice: a call for action. BMC Geriatr. 2012;12:55.
Urinary Excretion Indices in AKI
The Things We Do for No Reason (TWDFNR) series reviews practices which have become common parts of hospital care but which may provide little value to our patients. Practices reviewed in the TWDFNR series do not represent black and white conclusions or clinical practice standards, but are meant as a starting place for research and active discussions among hospitalists and patients. We invite you to be part of that discussion. https://www.choosingwisely.org/

A 70‐year‐old woman with a history of diabetes mellitus type 2 and hypertension was admitted with abdominal pain following 2 days of nausea and diarrhea. Initial laboratory studies revealed blood urea nitrogen (BUN) 25 mg/dL and serum creatinine 1.3 mg/dL. Computed tomography of the abdomen and pelvis with nonionic, low osmolar intravenous and oral contrast demonstrated acute diverticulitis with an associated small abscess. She was administered intravenous 0.9% sodium chloride solution and antibiotics. Blood pressure on admission was 92/55 mm Hg, and 24 hours later, her BUN and serum creatinine increased to 33 mg/dL and 1.9 mg/dL, respectively. Her urine output during the preceding 24 hours was 500 mL.
In the evaluation of acute kidney injury (AKI), is the measurement of fractional excretion of sodium (FeNa) and fractional excretion of urea (FeUr) of value?
WHY YOU MIGHT THINK ORDERING FeNa AND/OR FeUr IN THE EVALUATION OF AKI IS HELPFUL
The proper maintenance of sodium balance is paramount to regulating the size of body fluid compartments. Through the interaction of multiple physiologic processes, the kidney regulates tubular reabsorption (or lack thereof) of sodium chloride to match excretion to intake. In normal health, FeNa is typically 1%, although it may vary depending on the dietary sodium intake. The corollary is that 99% of filtered sodium is reabsorbed. Acute tubular injury (ATI) that impairs the tubular resorptive capacity for sodium may increase FeNa to >3%. In addition, during states of water conservation, urea is reabsorbed from the medullary collecting duct, explaining the discrepant rise in BUN relative to creatinine in prerenal azotemia. FeUr falls progressively as water is reabsorbed and urine flow declines, and FeUr less than 35% to 40% may result during prerenal azotemia versus >50% in health or ATI. Theoretically, FeUr is largely unaffected by diuretics, whereas FeNa is increased by diuretics.
In 1976, Espinel reported on the use of FeNa in 17 oliguric patients to discriminate prerenal azotemia from ATI.[1] Establishing what are now familiar indices, FeNa <1% was deemed consistent with prerenal physiology versus >3% indicating ATI. Notably, the study excluded patients who had received diuretics or in whom chronic kidney disease (CKD), glomerulonephritis, or urinary obstruction was suspected.
Given the limitations of FeNa in the context of diuretic use, many physicians instead use FeUr to distinguish prerenal versus ATI causes of AKI. Carvounis et al. reported FeUr and FeNa in 50 patients with prerenal azotemia, 27 with prerenal azotemia receiving diuretics and 25 patients with ATI.[2] Patients with interstitial nephritis, glomerulonephritis, and obstruction were excluded. In the entire cohort, the authors reported sensitivity of 90% and specificity of 96% for FeUr <35% in identifying prerenal azotemia (Table 1). FeNa <1% was slightly less sensitive for prerenal azotemia in the entire cohort at 77%, and this fell to 48% in the presence of diuretics as compared to 89% for FeUr. Naturally, the specificity of FeNa for ATI will fall with the use of diuretics. As shown in Table 1, FeUr <35% has an excellent positive likelihood ratio (LR+) of 22 for prerenal azotemia and a moderate LR+ of 9 for FeUr 35% being consistent with ATI, regardless of the presence of diuretics. This contrasts with FeNa, which if 1% in the presence of diuretics, lacked utility in the diagnosis of ATI. Of note, diuretic use was reported only in the prerenal azotemia group and not specifically in the ATI group. Thus, these comparisons assume diuretics have no effect on test characteristics in ATI. This assumption, however, may not be valid.
| FeUr | FeNa | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sens | Spec | PPV | NPV | LR+ | LR | Sens | Spec | PPV | NPV | LR+ | LR | |||
| ||||||||||||||
| Carvounis[2] | Prerenal | Overall | 0.90 | 0.96 | 0.99 | 0.75 | 22.4 | 0.1 | 0.77 | 0.96 | 0.98 | 0.57 | 19.2 | 0.2 |
| No diuretics | 0.90 | 0.96 | 0.98 | 0.83 | 22.5 | 0.1 | 0.92 | 0.96 | 0.98 | 0.86 | 23.0 | 0.1 | ||
| Diuretics | 0.89 | 0.96 | 0.96 | 0.89 | 22.2 | 0.1 | 0.48 | 0.96 | 0.93 | 0.63 | 12.0 | 0.5 | ||
| ATI | Overall | 0.96 | 0.90 | 0.75 | 0.99 | 9.2 | 0.0 | 0.96 | 0.77 | 0.57 | 0.98 | 4.1 | 0.1 | |
| No diuretics* | 0.96 | 0.90 | 0.83 | 0.98 | 9.6 | 0.0 | 0.96 | 0.92 | 0.86 | 0.98 | 12.0 | 0.0 | ||
| Diuretics | 0.96 | 0.89 | 0.89 | 0.96 | 8.6 | 0.0 | 0.96 | 0.48 | 0.63 | 0.93 | 1.9 | 0.1 | ||
| Diskin[8] | Prerenal | Overall | 0.97 | 0.85 | 0.96 | 0.89 | 6.5 | 0.0 | 0.44 | 0.75 | 0.88 | 0.25 | 1.8 | 0.7 |
| No diuretics | 0.91 | 0.89 | 0.95 | 0.80 | 8.2 | 0.1 | 0.83 | 0.67 | 0.86 | 0.60 | 2.5 | 0.3 | ||
| Diuretics | 1.00 | 0.82 | 0.97 | 1.00 | 5.5 | 0.0 | 0.29 | 0.82 | 0.89 | 0.18 | 1.6 | 0.9 | ||
| ATI | Overall | 0.85 | 0.97 | 0.89 | 0.96 | 33.6 | 0.2 | 0.75 | 0.44 | 0.25 | 0.88 | 1.3 | 0.6 | |
| No diuretics | 0.89 | 0.91 | 0.80 | 0.95 | 10.2 | 0.1 | 0.67 | 0.83 | 0.60 | 0.86 | 3.8 | 0.4 | ||
| Diuretics | 0.82 | 1.00 | 1.00 | 0.97 | N/A | 0.2 | 0.82 | 0.29 | 0.18 | 0.89 | 1.1 | 0.6 | ||
WHY THERE IS LITTLE REASON TO ROUTINELY ORDER FeNa AND FeUr IN PATIENTS WITH AKI
The argument against FeNa and FeUr is not primarily financial. FeNa and FeUr testing on all Medicare patients discharged with AKI in 2013 would have cost US$6 million.[3] Although a tiny fraction of annual healthcare expenditure, it would nevertheless be wasteful spending, and its true harm lays in the application of flawed diagnostic reasoning.
That flaw in our conceptual approach to AKI is the broad categorization of patients into either a prerenal or intrinsic etiology of AKI. In reality, renal injury is often multifactorial, and significant prerenal injury may progress to or coexist with intrinsic disease that is commonly ATI. Measurement of a urinary index at a single point in time will often fail to capture this spectrum of causes for AKI. Unfortunately, accurately assessing volume status through physical examination is difficult.[4] Considering FeNa and FeUr may be low in both hemorrhage as well as congestive heart failure, the measurement of these variables adds little to body volume assessment.
It cannot be overemphasized that application of FeNa and FeUr is predicated on the provider already knowing the diagnosis is either prerenal azotemia or ATI. Studies have generally excluded patients >65 years old and those with CKD or notable comorbid renal processes apart from prerenal azotemia or ATI. It is important to recall that a third of kidney biopsies may yield a diagnosis different than the prebiopsy clinical diagnosis, and the gold standard for ATI in studies of FeNa and FeUr was simply a failure of kidney function to improve promptly.[5] Why send a test that is predicated on largely already knowing the answer?
Fractional Excretion of Sodium for Diagnosis
Unfortunately, FeNa is neither sensitive nor specific enough in the general inpatient population to inform important clinical decisions regarding the etiology of AKI. Miller et al. examined 30 patients with oliguric prerenal azotemia, 55 with ATI (oliguric and nonoliguric), 10 with obstructive uropathy, and 7 with glomerulonephritis.[6] None of the patients had received diuretics within 24 hours of study entry. A FeNa <1% was present in 90% of prerenal patients and 4% of oliguric ATI. Importantly, of nonoliguric patients with ATI, 10% had a false positive FeNa <1%. Many subsequent studies have similarly documented the existence of FeNa <1% or otherwise indeterminate in ATI, particularly, but not exclusively, in nonoliguric states.[7] Diskin et al. evaluated FeNa in 100 prospective oliguric AKI patients (80 with prerenal azotemia and 20 with ATI) without CKD, with FeNa <1% being consistent with prerenal azotemia, 1% to 3% indeterminate, and >3% ATI.[8] The derived LR for FeNa for both prerenal azotemia and ATI are unlikely to alter pretest probability (Table 1). In part, this may be due to Diskin et al.'s incorporation of indeterminate FeNa, consistent with clinical reality. Carvounis et al. did not account for indeterminate values, and consequently the LR were likely overinflated in that study. It is now well‐recognized that glomerulonephritis may also result in FeNa <1% despite absence of identifiable prerenal physiology, as can intravenous iodinated contrast administration and rhabdomyolysis. Moreover, diuretic administration, polyuria due to osmotic diuresis, increased excretion of anions such as ketone bodies in diabetic ketoacidosis, the presence of CKD, and increased age, among others, can produce an FeNa that is indeterminate or >3% in the absence of ATI. Regarding diuretics, although the duration of action of furosemide is approximately 6 hours, longer‐acting loop diuretics such as torsemide or thiazide diuretics such as chlorthalidone may result in natriuresis for 24 hours.
Fractional Excretion of Urea for Diagnosis
Despite the potential superiority of FeUr to FeNa in supporting a diagnosis of prerenal azotemia in the setting of diuretic administration, FeUr nevertheless will only moderately increase the post‐test probability of prerenal azotemia under ideal conditions. In the study by Diskin et al., FeUr <40% was deemed consistent with prerenal azotemia and 40% with ATI. In the diagnosis of prerenal azotemia, the LR+ were 5.5 and 8.2 in the presence and absence of diuretics, respectively. Although the LR+ for the diagnosis of ATI was impressive, this was based on only 9 patients in the ATI‐no diuretic and 11 patients in the ATI‐diuretic groups. Carvounis, moreover, demonstrated considerably lower LR+ of approximately 9 for the diagnosis of ATI, and this study was unable to account for diuretic use specifically within the ATI group. Four of the 5 prerenal patients in Diskin et al.'s study misdiagnosed by FeUr had infection, and each were properly diagnosed by FeNa. Experimental data suggest endotoxemia may downregulate urea transporters as does aging, thereby increasing FeUr in sepsis and the elderly even in times of prerenal azotemia.[9, 10] Moreover, osmotic diuresis, such as with hyperglycemia or sickle cell nephropathy with medullary injury, may result in a falsely negative FeUr during prerenal states. In summary, these data suggest FeUr less than 35% to 40%, with the noted caveats, is most applicable to an oliguric patient in whom the pretest probability of prerenal azotemia is high, and it may be superior in the context of diuretics to the use of FeNa. Nonetheless, the impact on posttest probability is marginal. Of note, the diagnostic categories lack gold standards in these studies, and in the Carvounis study, FeNa (index under study) was 1 of several criteria actually used to categorize patients as either prerenal or ATI (outcomes under study). It is important to recognize these datasets contained very small numbers of patients with ATI, limiting the strength and generalizability of the scientific evidence. Other studies have failed to consistently demonstrate any utility to FeUr, particularly in those with CKD or critical illness.[11, 12, 13, 14, 15]
WHAT YOU SHOULD DO INSTEAD: DECIDE IF VOLUME MANIPULATION IS APPROPRIATE
The gold standard for diagnosis, as in many of the above studies, is the prompt improvement of prerenal azotemia with correction of renal hypoperfusion. Ultimately, the decision to administer intravenous fluids or diuretics in the management of AKI will often be independent of both FeNa and FeUr. In considering, for example, the case described above, it is not possible to realistically dichotomize the patient into either a prerenal or ATI category; both are quite likely present. If the clinical assessment supports a component of prerenal azotemia, a low FeNa and/or FeUr will not change the intervention. An elevated FeNa and/or FeUr, however, has at best moderate and potentially no impact on the likelihood for ATI. A patient, moreover, may still require volume manipulation in the context of established ATI. As such, these indices should not alter therapeutic decisions. There may be value in utilizing and identifying new approaches to determining a priori which patients will be fluid responsive, such as inferior vena cava ultrasound.[16] Lastly, evaluation of the urine sediment is an underutilized tool that may prove more useful in discriminating prerenal azotemia from ATI. It also helps to exclude other etiologies of AKI, such as glomerulonephritis and acute interstitial nephritis, which are typical exclusion criteria in studies of FeNa and FeUr.[17, 18]
WHEN IS FeNa AND/OR FeUr USEFUL IN DEFINING THE ETIOLOGY OF AKI?
FeNa and FeUr at best only support a clinical impression of prerenal azotemia or ATI in oliguric AKI, and the accuracy of these metrics is questionable in the setting of CKD, older age, and a variety of comorbidities. There is, however, a setting in which FeNa may be helpful. In practice, FeNa is useful in the evaluation of hepatorenal syndrome, a disorder characterized by oliguria and intense renal sodium reabsorption with resultant spot urine sodium <10 mEq/L and FeNa <1%.[19]
CONCLUSION
The evidence base supporting the use of FeNa and FeUr is limited and often not generalizable to many patients with AKI. The small sample sizes of the studies do not permit adequate capture of diverse mechanisms for renal injury, and these studies are of patients referred for Nephrology consultation and may not be representative of the larger population of patients with less severe AKI. Ultimately, the true etiology will be proven by time and response to therapy. Apart from a supportive role in the diagnosis of hepatorenal syndrome, there is little practical utility to FeNa and FeUr measurement, and these indices should not alter therapeutic decisions when inconsistent with the clinical impression. The evaluation of AKI requires thoughtful clinical assessment, and the gold standard still remains the judicious decision of when to manipulate the intravascular volume status of a patient. In regard to the presented case, urine chemistries are unhelpful due to the combined vasoconstrictive and tubulotoxic effects of the administered intravenous contrast. The ongoing hypotension further contributes to both pre‐renal as well as ischemic tubular injury.
RECOMMENDATIONS
- FeNa can aid in the diagnosis of hepatorenal syndrome. Otherwise, the routine use of FeNa and FeUr in the diagnosis and management of AKI should be avoided.
- In pre‐renal azotemia, therapeutic intervention is guided by etiology of the disorder (e.g., intravenous crystalloid support based on a history of hypovolemia and ongoing hypoperfusion, diuresis and/or inotropic support in setting of decompensated heart failure, etc.), without regard to baseline FeNa and FeUr.
- In ATI, fluid administration is appropriate if hypovolemia is present. FeNa and FeUr cannot diagnose hypovolemia.
Disclosure
Nothing to report.
Do you think this is a low‐value practice? Is this truly a Thing We Do for No Reason? Share what you do in your practice and join in the conversation online by retweeting it on Twitter (#TWDFNR) and liking it on Facebook. We invite you to propose ideas for other Things We Do for No Reason topics by emailing [email protected].
- . The FENa test. Use in the differential diagnosis of acute renal failure. JAMA. 1976;236(6):579–581.
- , , . Significance of the fractional excretion of urea in the differential diagnosis of acute renal failure. Kidney Int. 2002;62(6):2223–2229.
- Centers for Medicare 275(8):630–634.
- , , , . Etiologies and outcome of acute renal insufficiency in older adults: a renal biopsy study of 259 cases. Am J Kidney Dis. 2000;35(3):433–447.
- , , , et al. Urinary diagnostic indices in acute renal failure: a prospective study. Ann Intern Med. 1978;89(1):47–50.
- , . Urinary indices and chemistries in the differential diagnosis of prerenal failure and acute tubular necrosis. Semin Nephrol. 1985;5(3):224–233.
- , , , , . The comparative benefits of the fractional excretion of urea and sodium in various azotemic oliguric states. Nephron Clin Pract. 2010;114(2):c145–c150.
- MachasNúñez JF, Cameron JS, Oreopoulos DG, eds. The Aging Kidney in Health and Disease. New York, NY: Springer Science + Business Media, LLC; 2008.
- , , . Cytokine‐mediated regulation of urea transporters during experimental endotoxemia. Am J Physiol Renal Physiol. 2007;292(5):F1479–F1489.
- , , . Urinary biochemistry and microscopy in septic acute renal failure: a systematic review. Am J Kidney Dis. 2006;48(5):695–705.
- , , , et al. Diagnostic performance of fractional excretion of urea in the evaluation of critically ill patients with acute kidney injury: a multicenter cohort study. Crit Care. 2011;15(4):R178.
- , , , et al. Fractional excretion of urea as a diagnostic index in acute kidney injury in intensive care patients. J Crit Care. 2012;27(5):505–510.
- , , , . Diagnostic performance of fractional excretion of urea and fractional excretion of sodium in the evaluations of patients with acute kidney injury with or without diuretic treatment. Am J Kidney Dis. 2007;50(4):566–573.
- , , , et al. Transient versus persistent acute kidney injury and the diagnostic performance of fractional excretion of urea in critically ill patients. Nephron Clin Pract. 2014;126(1):8–13.
- , , , , . Emergency department bedside ultrasonographic measurement of the caval index for noninvasive determination of low central venous pressure. Ann Emerg Med. 2010;55(3):290–295.
- , , , , , . Urine microscopy is associated with severity and worsening of acute kidney injury in hospitalized patients. Clin J Am Soc Nephrol. 2010;5(3):402–408.
- , , , , . Diagnostic value of urine microscopy for differential diagnosis of acute kidney injury in hospitalized patients. Clin J Am Soc Nephrol. 2008;3(6):1615–1619.
- , , , et al. Definition and diagnostic criteria of refractory ascites and hepatorenal syndrome in cirrhosis. International Ascites Club. Hepatology. 1996;23(1):164–176.
The Things We Do for No Reason (TWDFNR) series reviews practices which have become common parts of hospital care but which may provide little value to our patients. Practices reviewed in the TWDFNR series do not represent black and white conclusions or clinical practice standards, but are meant as a starting place for research and active discussions among hospitalists and patients. We invite you to be part of that discussion. https://www.choosingwisely.org/
A 70‐year‐old woman with a history of diabetes mellitus type 2 and hypertension was admitted with abdominal pain following 2 days of nausea and diarrhea. Initial laboratory studies revealed blood urea nitrogen (BUN) 25 mg/dL and serum creatinine 1.3 mg/dL. Computed tomography of the abdomen and pelvis with nonionic, low osmolar intravenous and oral contrast demonstrated acute diverticulitis with an associated small abscess. She was administered intravenous 0.9% sodium chloride solution and antibiotics. Blood pressure on admission was 92/55 mm Hg, and 24 hours later, her BUN and serum creatinine increased to 33 mg/dL and 1.9 mg/dL, respectively. Her urine output during the preceding 24 hours was 500 mL.
In the evaluation of acute kidney injury (AKI), is the measurement of fractional excretion of sodium (FeNa) and fractional excretion of urea (FeUr) of value?
WHY YOU MIGHT THINK ORDERING FeNa AND/OR FeUr IN THE EVALUATION OF AKI IS HELPFUL
The proper maintenance of sodium balance is paramount to regulating the size of body fluid compartments. Through the interaction of multiple physiologic processes, the kidney regulates tubular reabsorption (or lack thereof) of sodium chloride to match excretion to intake. In normal health, FeNa is typically 1%, although it may vary depending on the dietary sodium intake. The corollary is that 99% of filtered sodium is reabsorbed. Acute tubular injury (ATI) that impairs the tubular resorptive capacity for sodium may increase FeNa to >3%. In addition, during states of water conservation, urea is reabsorbed from the medullary collecting duct, explaining the discrepant rise in BUN relative to creatinine in prerenal azotemia. FeUr falls progressively as water is reabsorbed and urine flow declines, and FeUr less than 35% to 40% may result during prerenal azotemia versus >50% in health or ATI. Theoretically, FeUr is largely unaffected by diuretics, whereas FeNa is increased by diuretics.
In 1976, Espinel reported on the use of FeNa in 17 oliguric patients to discriminate prerenal azotemia from ATI.[1] Establishing what are now familiar indices, FeNa <1% was deemed consistent with prerenal physiology versus >3% indicating ATI. Notably, the study excluded patients who had received diuretics or in whom chronic kidney disease (CKD), glomerulonephritis, or urinary obstruction was suspected.
Given the limitations of FeNa in the context of diuretic use, many physicians instead use FeUr to distinguish prerenal versus ATI causes of AKI. Carvounis et al. reported FeUr and FeNa in 50 patients with prerenal azotemia, 27 with prerenal azotemia receiving diuretics and 25 patients with ATI.[2] Patients with interstitial nephritis, glomerulonephritis, and obstruction were excluded. In the entire cohort, the authors reported sensitivity of 90% and specificity of 96% for FeUr <35% in identifying prerenal azotemia (Table 1). FeNa <1% was slightly less sensitive for prerenal azotemia in the entire cohort at 77%, and this fell to 48% in the presence of diuretics as compared to 89% for FeUr. Naturally, the specificity of FeNa for ATI will fall with the use of diuretics. As shown in Table 1, FeUr <35% has an excellent positive likelihood ratio (LR+) of 22 for prerenal azotemia and a moderate LR+ of 9 for FeUr 35% being consistent with ATI, regardless of the presence of diuretics. This contrasts with FeNa, which if 1% in the presence of diuretics, lacked utility in the diagnosis of ATI. Of note, diuretic use was reported only in the prerenal azotemia group and not specifically in the ATI group. Thus, these comparisons assume diuretics have no effect on test characteristics in ATI. This assumption, however, may not be valid.
| FeUr | FeNa | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sens | Spec | PPV | NPV | LR+ | LR | Sens | Spec | PPV | NPV | LR+ | LR | |||
| ||||||||||||||
| Carvounis[2] | Prerenal | Overall | 0.90 | 0.96 | 0.99 | 0.75 | 22.4 | 0.1 | 0.77 | 0.96 | 0.98 | 0.57 | 19.2 | 0.2 |
| No diuretics | 0.90 | 0.96 | 0.98 | 0.83 | 22.5 | 0.1 | 0.92 | 0.96 | 0.98 | 0.86 | 23.0 | 0.1 | ||
| Diuretics | 0.89 | 0.96 | 0.96 | 0.89 | 22.2 | 0.1 | 0.48 | 0.96 | 0.93 | 0.63 | 12.0 | 0.5 | ||
| ATI | Overall | 0.96 | 0.90 | 0.75 | 0.99 | 9.2 | 0.0 | 0.96 | 0.77 | 0.57 | 0.98 | 4.1 | 0.1 | |
| No diuretics* | 0.96 | 0.90 | 0.83 | 0.98 | 9.6 | 0.0 | 0.96 | 0.92 | 0.86 | 0.98 | 12.0 | 0.0 | ||
| Diuretics | 0.96 | 0.89 | 0.89 | 0.96 | 8.6 | 0.0 | 0.96 | 0.48 | 0.63 | 0.93 | 1.9 | 0.1 | ||
| Diskin[8] | Prerenal | Overall | 0.97 | 0.85 | 0.96 | 0.89 | 6.5 | 0.0 | 0.44 | 0.75 | 0.88 | 0.25 | 1.8 | 0.7 |
| No diuretics | 0.91 | 0.89 | 0.95 | 0.80 | 8.2 | 0.1 | 0.83 | 0.67 | 0.86 | 0.60 | 2.5 | 0.3 | ||
| Diuretics | 1.00 | 0.82 | 0.97 | 1.00 | 5.5 | 0.0 | 0.29 | 0.82 | 0.89 | 0.18 | 1.6 | 0.9 | ||
| ATI | Overall | 0.85 | 0.97 | 0.89 | 0.96 | 33.6 | 0.2 | 0.75 | 0.44 | 0.25 | 0.88 | 1.3 | 0.6 | |
| No diuretics | 0.89 | 0.91 | 0.80 | 0.95 | 10.2 | 0.1 | 0.67 | 0.83 | 0.60 | 0.86 | 3.8 | 0.4 | ||
| Diuretics | 0.82 | 1.00 | 1.00 | 0.97 | N/A | 0.2 | 0.82 | 0.29 | 0.18 | 0.89 | 1.1 | 0.6 | ||
WHY THERE IS LITTLE REASON TO ROUTINELY ORDER FeNa AND FeUr IN PATIENTS WITH AKI
The argument against FeNa and FeUr is not primarily financial. FeNa and FeUr testing on all Medicare patients discharged with AKI in 2013 would have cost US$6 million.[3] Although a tiny fraction of annual healthcare expenditure, it would nevertheless be wasteful spending, and its true harm lays in the application of flawed diagnostic reasoning.
That flaw in our conceptual approach to AKI is the broad categorization of patients into either a prerenal or intrinsic etiology of AKI. In reality, renal injury is often multifactorial, and significant prerenal injury may progress to or coexist with intrinsic disease that is commonly ATI. Measurement of a urinary index at a single point in time will often fail to capture this spectrum of causes for AKI. Unfortunately, accurately assessing volume status through physical examination is difficult.[4] Considering FeNa and FeUr may be low in both hemorrhage as well as congestive heart failure, the measurement of these variables adds little to body volume assessment.
It cannot be overemphasized that application of FeNa and FeUr is predicated on the provider already knowing the diagnosis is either prerenal azotemia or ATI. Studies have generally excluded patients >65 years old and those with CKD or notable comorbid renal processes apart from prerenal azotemia or ATI. It is important to recall that a third of kidney biopsies may yield a diagnosis different than the prebiopsy clinical diagnosis, and the gold standard for ATI in studies of FeNa and FeUr was simply a failure of kidney function to improve promptly.[5] Why send a test that is predicated on largely already knowing the answer?
Fractional Excretion of Sodium for Diagnosis
Unfortunately, FeNa is neither sensitive nor specific enough in the general inpatient population to inform important clinical decisions regarding the etiology of AKI. Miller et al. examined 30 patients with oliguric prerenal azotemia, 55 with ATI (oliguric and nonoliguric), 10 with obstructive uropathy, and 7 with glomerulonephritis.[6] None of the patients had received diuretics within 24 hours of study entry. A FeNa <1% was present in 90% of prerenal patients and 4% of oliguric ATI. Importantly, of nonoliguric patients with ATI, 10% had a false positive FeNa <1%. Many subsequent studies have similarly documented the existence of FeNa <1% or otherwise indeterminate in ATI, particularly, but not exclusively, in nonoliguric states.[7] Diskin et al. evaluated FeNa in 100 prospective oliguric AKI patients (80 with prerenal azotemia and 20 with ATI) without CKD, with FeNa <1% being consistent with prerenal azotemia, 1% to 3% indeterminate, and >3% ATI.[8] The derived LR for FeNa for both prerenal azotemia and ATI are unlikely to alter pretest probability (Table 1). In part, this may be due to Diskin et al.'s incorporation of indeterminate FeNa, consistent with clinical reality. Carvounis et al. did not account for indeterminate values, and consequently the LR were likely overinflated in that study. It is now well‐recognized that glomerulonephritis may also result in FeNa <1% despite absence of identifiable prerenal physiology, as can intravenous iodinated contrast administration and rhabdomyolysis. Moreover, diuretic administration, polyuria due to osmotic diuresis, increased excretion of anions such as ketone bodies in diabetic ketoacidosis, the presence of CKD, and increased age, among others, can produce an FeNa that is indeterminate or >3% in the absence of ATI. Regarding diuretics, although the duration of action of furosemide is approximately 6 hours, longer‐acting loop diuretics such as torsemide or thiazide diuretics such as chlorthalidone may result in natriuresis for 24 hours.
Fractional Excretion of Urea for Diagnosis
Despite the potential superiority of FeUr to FeNa in supporting a diagnosis of prerenal azotemia in the setting of diuretic administration, FeUr nevertheless will only moderately increase the post‐test probability of prerenal azotemia under ideal conditions. In the study by Diskin et al., FeUr <40% was deemed consistent with prerenal azotemia and 40% with ATI. In the diagnosis of prerenal azotemia, the LR+ were 5.5 and 8.2 in the presence and absence of diuretics, respectively. Although the LR+ for the diagnosis of ATI was impressive, this was based on only 9 patients in the ATI‐no diuretic and 11 patients in the ATI‐diuretic groups. Carvounis, moreover, demonstrated considerably lower LR+ of approximately 9 for the diagnosis of ATI, and this study was unable to account for diuretic use specifically within the ATI group. Four of the 5 prerenal patients in Diskin et al.'s study misdiagnosed by FeUr had infection, and each were properly diagnosed by FeNa. Experimental data suggest endotoxemia may downregulate urea transporters as does aging, thereby increasing FeUr in sepsis and the elderly even in times of prerenal azotemia.[9, 10] Moreover, osmotic diuresis, such as with hyperglycemia or sickle cell nephropathy with medullary injury, may result in a falsely negative FeUr during prerenal states. In summary, these data suggest FeUr less than 35% to 40%, with the noted caveats, is most applicable to an oliguric patient in whom the pretest probability of prerenal azotemia is high, and it may be superior in the context of diuretics to the use of FeNa. Nonetheless, the impact on posttest probability is marginal. Of note, the diagnostic categories lack gold standards in these studies, and in the Carvounis study, FeNa (index under study) was 1 of several criteria actually used to categorize patients as either prerenal or ATI (outcomes under study). It is important to recognize these datasets contained very small numbers of patients with ATI, limiting the strength and generalizability of the scientific evidence. Other studies have failed to consistently demonstrate any utility to FeUr, particularly in those with CKD or critical illness.[11, 12, 13, 14, 15]
WHAT YOU SHOULD DO INSTEAD: DECIDE IF VOLUME MANIPULATION IS APPROPRIATE
The gold standard for diagnosis, as in many of the above studies, is the prompt improvement of prerenal azotemia with correction of renal hypoperfusion. Ultimately, the decision to administer intravenous fluids or diuretics in the management of AKI will often be independent of both FeNa and FeUr. In considering, for example, the case described above, it is not possible to realistically dichotomize the patient into either a prerenal or ATI category; both are quite likely present. If the clinical assessment supports a component of prerenal azotemia, a low FeNa and/or FeUr will not change the intervention. An elevated FeNa and/or FeUr, however, has at best moderate and potentially no impact on the likelihood for ATI. A patient, moreover, may still require volume manipulation in the context of established ATI. As such, these indices should not alter therapeutic decisions. There may be value in utilizing and identifying new approaches to determining a priori which patients will be fluid responsive, such as inferior vena cava ultrasound.[16] Lastly, evaluation of the urine sediment is an underutilized tool that may prove more useful in discriminating prerenal azotemia from ATI. It also helps to exclude other etiologies of AKI, such as glomerulonephritis and acute interstitial nephritis, which are typical exclusion criteria in studies of FeNa and FeUr.[17, 18]
WHEN IS FeNa AND/OR FeUr USEFUL IN DEFINING THE ETIOLOGY OF AKI?
FeNa and FeUr at best only support a clinical impression of prerenal azotemia or ATI in oliguric AKI, and the accuracy of these metrics is questionable in the setting of CKD, older age, and a variety of comorbidities. There is, however, a setting in which FeNa may be helpful. In practice, FeNa is useful in the evaluation of hepatorenal syndrome, a disorder characterized by oliguria and intense renal sodium reabsorption with resultant spot urine sodium <10 mEq/L and FeNa <1%.[19]
CONCLUSION
The evidence base supporting the use of FeNa and FeUr is limited and often not generalizable to many patients with AKI. The small sample sizes of the studies do not permit adequate capture of diverse mechanisms for renal injury, and these studies are of patients referred for Nephrology consultation and may not be representative of the larger population of patients with less severe AKI. Ultimately, the true etiology will be proven by time and response to therapy. Apart from a supportive role in the diagnosis of hepatorenal syndrome, there is little practical utility to FeNa and FeUr measurement, and these indices should not alter therapeutic decisions when inconsistent with the clinical impression. The evaluation of AKI requires thoughtful clinical assessment, and the gold standard still remains the judicious decision of when to manipulate the intravascular volume status of a patient. In regard to the presented case, urine chemistries are unhelpful due to the combined vasoconstrictive and tubulotoxic effects of the administered intravenous contrast. The ongoing hypotension further contributes to both pre‐renal as well as ischemic tubular injury.
RECOMMENDATIONS
- FeNa can aid in the diagnosis of hepatorenal syndrome. Otherwise, the routine use of FeNa and FeUr in the diagnosis and management of AKI should be avoided.
- In pre‐renal azotemia, therapeutic intervention is guided by etiology of the disorder (e.g., intravenous crystalloid support based on a history of hypovolemia and ongoing hypoperfusion, diuresis and/or inotropic support in setting of decompensated heart failure, etc.), without regard to baseline FeNa and FeUr.
- In ATI, fluid administration is appropriate if hypovolemia is present. FeNa and FeUr cannot diagnose hypovolemia.
Disclosure
Nothing to report.
Do you think this is a low‐value practice? Is this truly a Thing We Do for No Reason? Share what you do in your practice and join in the conversation online by retweeting it on Twitter (#TWDFNR) and liking it on Facebook. We invite you to propose ideas for other Things We Do for No Reason topics by emailing [email protected].
The Things We Do for No Reason (TWDFNR) series reviews practices which have become common parts of hospital care but which may provide little value to our patients. Practices reviewed in the TWDFNR series do not represent black and white conclusions or clinical practice standards, but are meant as a starting place for research and active discussions among hospitalists and patients. We invite you to be part of that discussion. https://www.choosingwisely.org/
A 70‐year‐old woman with a history of diabetes mellitus type 2 and hypertension was admitted with abdominal pain following 2 days of nausea and diarrhea. Initial laboratory studies revealed blood urea nitrogen (BUN) 25 mg/dL and serum creatinine 1.3 mg/dL. Computed tomography of the abdomen and pelvis with nonionic, low osmolar intravenous and oral contrast demonstrated acute diverticulitis with an associated small abscess. She was administered intravenous 0.9% sodium chloride solution and antibiotics. Blood pressure on admission was 92/55 mm Hg, and 24 hours later, her BUN and serum creatinine increased to 33 mg/dL and 1.9 mg/dL, respectively. Her urine output during the preceding 24 hours was 500 mL.
In the evaluation of acute kidney injury (AKI), is the measurement of fractional excretion of sodium (FeNa) and fractional excretion of urea (FeUr) of value?
WHY YOU MIGHT THINK ORDERING FeNa AND/OR FeUr IN THE EVALUATION OF AKI IS HELPFUL
The proper maintenance of sodium balance is paramount to regulating the size of body fluid compartments. Through the interaction of multiple physiologic processes, the kidney regulates tubular reabsorption (or lack thereof) of sodium chloride to match excretion to intake. In normal health, FeNa is typically 1%, although it may vary depending on the dietary sodium intake. The corollary is that 99% of filtered sodium is reabsorbed. Acute tubular injury (ATI) that impairs the tubular resorptive capacity for sodium may increase FeNa to >3%. In addition, during states of water conservation, urea is reabsorbed from the medullary collecting duct, explaining the discrepant rise in BUN relative to creatinine in prerenal azotemia. FeUr falls progressively as water is reabsorbed and urine flow declines, and FeUr less than 35% to 40% may result during prerenal azotemia versus >50% in health or ATI. Theoretically, FeUr is largely unaffected by diuretics, whereas FeNa is increased by diuretics.
In 1976, Espinel reported on the use of FeNa in 17 oliguric patients to discriminate prerenal azotemia from ATI.[1] Establishing what are now familiar indices, FeNa <1% was deemed consistent with prerenal physiology versus >3% indicating ATI. Notably, the study excluded patients who had received diuretics or in whom chronic kidney disease (CKD), glomerulonephritis, or urinary obstruction was suspected.
Given the limitations of FeNa in the context of diuretic use, many physicians instead use FeUr to distinguish prerenal versus ATI causes of AKI. Carvounis et al. reported FeUr and FeNa in 50 patients with prerenal azotemia, 27 with prerenal azotemia receiving diuretics and 25 patients with ATI.[2] Patients with interstitial nephritis, glomerulonephritis, and obstruction were excluded. In the entire cohort, the authors reported sensitivity of 90% and specificity of 96% for FeUr <35% in identifying prerenal azotemia (Table 1). FeNa <1% was slightly less sensitive for prerenal azotemia in the entire cohort at 77%, and this fell to 48% in the presence of diuretics as compared to 89% for FeUr. Naturally, the specificity of FeNa for ATI will fall with the use of diuretics. As shown in Table 1, FeUr <35% has an excellent positive likelihood ratio (LR+) of 22 for prerenal azotemia and a moderate LR+ of 9 for FeUr 35% being consistent with ATI, regardless of the presence of diuretics. This contrasts with FeNa, which if 1% in the presence of diuretics, lacked utility in the diagnosis of ATI. Of note, diuretic use was reported only in the prerenal azotemia group and not specifically in the ATI group. Thus, these comparisons assume diuretics have no effect on test characteristics in ATI. This assumption, however, may not be valid.
| FeUr | FeNa | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sens | Spec | PPV | NPV | LR+ | LR | Sens | Spec | PPV | NPV | LR+ | LR | |||
| ||||||||||||||
| Carvounis[2] | Prerenal | Overall | 0.90 | 0.96 | 0.99 | 0.75 | 22.4 | 0.1 | 0.77 | 0.96 | 0.98 | 0.57 | 19.2 | 0.2 |
| No diuretics | 0.90 | 0.96 | 0.98 | 0.83 | 22.5 | 0.1 | 0.92 | 0.96 | 0.98 | 0.86 | 23.0 | 0.1 | ||
| Diuretics | 0.89 | 0.96 | 0.96 | 0.89 | 22.2 | 0.1 | 0.48 | 0.96 | 0.93 | 0.63 | 12.0 | 0.5 | ||
| ATI | Overall | 0.96 | 0.90 | 0.75 | 0.99 | 9.2 | 0.0 | 0.96 | 0.77 | 0.57 | 0.98 | 4.1 | 0.1 | |
| No diuretics* | 0.96 | 0.90 | 0.83 | 0.98 | 9.6 | 0.0 | 0.96 | 0.92 | 0.86 | 0.98 | 12.0 | 0.0 | ||
| Diuretics | 0.96 | 0.89 | 0.89 | 0.96 | 8.6 | 0.0 | 0.96 | 0.48 | 0.63 | 0.93 | 1.9 | 0.1 | ||
| Diskin[8] | Prerenal | Overall | 0.97 | 0.85 | 0.96 | 0.89 | 6.5 | 0.0 | 0.44 | 0.75 | 0.88 | 0.25 | 1.8 | 0.7 |
| No diuretics | 0.91 | 0.89 | 0.95 | 0.80 | 8.2 | 0.1 | 0.83 | 0.67 | 0.86 | 0.60 | 2.5 | 0.3 | ||
| Diuretics | 1.00 | 0.82 | 0.97 | 1.00 | 5.5 | 0.0 | 0.29 | 0.82 | 0.89 | 0.18 | 1.6 | 0.9 | ||
| ATI | Overall | 0.85 | 0.97 | 0.89 | 0.96 | 33.6 | 0.2 | 0.75 | 0.44 | 0.25 | 0.88 | 1.3 | 0.6 | |
| No diuretics | 0.89 | 0.91 | 0.80 | 0.95 | 10.2 | 0.1 | 0.67 | 0.83 | 0.60 | 0.86 | 3.8 | 0.4 | ||
| Diuretics | 0.82 | 1.00 | 1.00 | 0.97 | N/A | 0.2 | 0.82 | 0.29 | 0.18 | 0.89 | 1.1 | 0.6 | ||
WHY THERE IS LITTLE REASON TO ROUTINELY ORDER FeNa AND FeUr IN PATIENTS WITH AKI
The argument against FeNa and FeUr is not primarily financial. FeNa and FeUr testing on all Medicare patients discharged with AKI in 2013 would have cost US$6 million.[3] Although a tiny fraction of annual healthcare expenditure, it would nevertheless be wasteful spending, and its true harm lays in the application of flawed diagnostic reasoning.
That flaw in our conceptual approach to AKI is the broad categorization of patients into either a prerenal or intrinsic etiology of AKI. In reality, renal injury is often multifactorial, and significant prerenal injury may progress to or coexist with intrinsic disease that is commonly ATI. Measurement of a urinary index at a single point in time will often fail to capture this spectrum of causes for AKI. Unfortunately, accurately assessing volume status through physical examination is difficult.[4] Considering FeNa and FeUr may be low in both hemorrhage as well as congestive heart failure, the measurement of these variables adds little to body volume assessment.
It cannot be overemphasized that application of FeNa and FeUr is predicated on the provider already knowing the diagnosis is either prerenal azotemia or ATI. Studies have generally excluded patients >65 years old and those with CKD or notable comorbid renal processes apart from prerenal azotemia or ATI. It is important to recall that a third of kidney biopsies may yield a diagnosis different than the prebiopsy clinical diagnosis, and the gold standard for ATI in studies of FeNa and FeUr was simply a failure of kidney function to improve promptly.[5] Why send a test that is predicated on largely already knowing the answer?
Fractional Excretion of Sodium for Diagnosis
Unfortunately, FeNa is neither sensitive nor specific enough in the general inpatient population to inform important clinical decisions regarding the etiology of AKI. Miller et al. examined 30 patients with oliguric prerenal azotemia, 55 with ATI (oliguric and nonoliguric), 10 with obstructive uropathy, and 7 with glomerulonephritis.[6] None of the patients had received diuretics within 24 hours of study entry. A FeNa <1% was present in 90% of prerenal patients and 4% of oliguric ATI. Importantly, of nonoliguric patients with ATI, 10% had a false positive FeNa <1%. Many subsequent studies have similarly documented the existence of FeNa <1% or otherwise indeterminate in ATI, particularly, but not exclusively, in nonoliguric states.[7] Diskin et al. evaluated FeNa in 100 prospective oliguric AKI patients (80 with prerenal azotemia and 20 with ATI) without CKD, with FeNa <1% being consistent with prerenal azotemia, 1% to 3% indeterminate, and >3% ATI.[8] The derived LR for FeNa for both prerenal azotemia and ATI are unlikely to alter pretest probability (Table 1). In part, this may be due to Diskin et al.'s incorporation of indeterminate FeNa, consistent with clinical reality. Carvounis et al. did not account for indeterminate values, and consequently the LR were likely overinflated in that study. It is now well‐recognized that glomerulonephritis may also result in FeNa <1% despite absence of identifiable prerenal physiology, as can intravenous iodinated contrast administration and rhabdomyolysis. Moreover, diuretic administration, polyuria due to osmotic diuresis, increased excretion of anions such as ketone bodies in diabetic ketoacidosis, the presence of CKD, and increased age, among others, can produce an FeNa that is indeterminate or >3% in the absence of ATI. Regarding diuretics, although the duration of action of furosemide is approximately 6 hours, longer‐acting loop diuretics such as torsemide or thiazide diuretics such as chlorthalidone may result in natriuresis for 24 hours.
Fractional Excretion of Urea for Diagnosis
Despite the potential superiority of FeUr to FeNa in supporting a diagnosis of prerenal azotemia in the setting of diuretic administration, FeUr nevertheless will only moderately increase the post‐test probability of prerenal azotemia under ideal conditions. In the study by Diskin et al., FeUr <40% was deemed consistent with prerenal azotemia and 40% with ATI. In the diagnosis of prerenal azotemia, the LR+ were 5.5 and 8.2 in the presence and absence of diuretics, respectively. Although the LR+ for the diagnosis of ATI was impressive, this was based on only 9 patients in the ATI‐no diuretic and 11 patients in the ATI‐diuretic groups. Carvounis, moreover, demonstrated considerably lower LR+ of approximately 9 for the diagnosis of ATI, and this study was unable to account for diuretic use specifically within the ATI group. Four of the 5 prerenal patients in Diskin et al.'s study misdiagnosed by FeUr had infection, and each were properly diagnosed by FeNa. Experimental data suggest endotoxemia may downregulate urea transporters as does aging, thereby increasing FeUr in sepsis and the elderly even in times of prerenal azotemia.[9, 10] Moreover, osmotic diuresis, such as with hyperglycemia or sickle cell nephropathy with medullary injury, may result in a falsely negative FeUr during prerenal states. In summary, these data suggest FeUr less than 35% to 40%, with the noted caveats, is most applicable to an oliguric patient in whom the pretest probability of prerenal azotemia is high, and it may be superior in the context of diuretics to the use of FeNa. Nonetheless, the impact on posttest probability is marginal. Of note, the diagnostic categories lack gold standards in these studies, and in the Carvounis study, FeNa (index under study) was 1 of several criteria actually used to categorize patients as either prerenal or ATI (outcomes under study). It is important to recognize these datasets contained very small numbers of patients with ATI, limiting the strength and generalizability of the scientific evidence. Other studies have failed to consistently demonstrate any utility to FeUr, particularly in those with CKD or critical illness.[11, 12, 13, 14, 15]
WHAT YOU SHOULD DO INSTEAD: DECIDE IF VOLUME MANIPULATION IS APPROPRIATE
The gold standard for diagnosis, as in many of the above studies, is the prompt improvement of prerenal azotemia with correction of renal hypoperfusion. Ultimately, the decision to administer intravenous fluids or diuretics in the management of AKI will often be independent of both FeNa and FeUr. In considering, for example, the case described above, it is not possible to realistically dichotomize the patient into either a prerenal or ATI category; both are quite likely present. If the clinical assessment supports a component of prerenal azotemia, a low FeNa and/or FeUr will not change the intervention. An elevated FeNa and/or FeUr, however, has at best moderate and potentially no impact on the likelihood for ATI. A patient, moreover, may still require volume manipulation in the context of established ATI. As such, these indices should not alter therapeutic decisions. There may be value in utilizing and identifying new approaches to determining a priori which patients will be fluid responsive, such as inferior vena cava ultrasound.[16] Lastly, evaluation of the urine sediment is an underutilized tool that may prove more useful in discriminating prerenal azotemia from ATI. It also helps to exclude other etiologies of AKI, such as glomerulonephritis and acute interstitial nephritis, which are typical exclusion criteria in studies of FeNa and FeUr.[17, 18]
WHEN IS FeNa AND/OR FeUr USEFUL IN DEFINING THE ETIOLOGY OF AKI?
FeNa and FeUr at best only support a clinical impression of prerenal azotemia or ATI in oliguric AKI, and the accuracy of these metrics is questionable in the setting of CKD, older age, and a variety of comorbidities. There is, however, a setting in which FeNa may be helpful. In practice, FeNa is useful in the evaluation of hepatorenal syndrome, a disorder characterized by oliguria and intense renal sodium reabsorption with resultant spot urine sodium <10 mEq/L and FeNa <1%.[19]
CONCLUSION
The evidence base supporting the use of FeNa and FeUr is limited and often not generalizable to many patients with AKI. The small sample sizes of the studies do not permit adequate capture of diverse mechanisms for renal injury, and these studies are of patients referred for Nephrology consultation and may not be representative of the larger population of patients with less severe AKI. Ultimately, the true etiology will be proven by time and response to therapy. Apart from a supportive role in the diagnosis of hepatorenal syndrome, there is little practical utility to FeNa and FeUr measurement, and these indices should not alter therapeutic decisions when inconsistent with the clinical impression. The evaluation of AKI requires thoughtful clinical assessment, and the gold standard still remains the judicious decision of when to manipulate the intravascular volume status of a patient. In regard to the presented case, urine chemistries are unhelpful due to the combined vasoconstrictive and tubulotoxic effects of the administered intravenous contrast. The ongoing hypotension further contributes to both pre‐renal as well as ischemic tubular injury.
RECOMMENDATIONS
- FeNa can aid in the diagnosis of hepatorenal syndrome. Otherwise, the routine use of FeNa and FeUr in the diagnosis and management of AKI should be avoided.
- In pre‐renal azotemia, therapeutic intervention is guided by etiology of the disorder (e.g., intravenous crystalloid support based on a history of hypovolemia and ongoing hypoperfusion, diuresis and/or inotropic support in setting of decompensated heart failure, etc.), without regard to baseline FeNa and FeUr.
- In ATI, fluid administration is appropriate if hypovolemia is present. FeNa and FeUr cannot diagnose hypovolemia.
Disclosure
Nothing to report.
Do you think this is a low‐value practice? Is this truly a Thing We Do for No Reason? Share what you do in your practice and join in the conversation online by retweeting it on Twitter (#TWDFNR) and liking it on Facebook. We invite you to propose ideas for other Things We Do for No Reason topics by emailing [email protected].
- . The FENa test. Use in the differential diagnosis of acute renal failure. JAMA. 1976;236(6):579–581.
- , , . Significance of the fractional excretion of urea in the differential diagnosis of acute renal failure. Kidney Int. 2002;62(6):2223–2229.
- Centers for Medicare 275(8):630–634.
- , , , . Etiologies and outcome of acute renal insufficiency in older adults: a renal biopsy study of 259 cases. Am J Kidney Dis. 2000;35(3):433–447.
- , , , et al. Urinary diagnostic indices in acute renal failure: a prospective study. Ann Intern Med. 1978;89(1):47–50.
- , . Urinary indices and chemistries in the differential diagnosis of prerenal failure and acute tubular necrosis. Semin Nephrol. 1985;5(3):224–233.
- , , , , . The comparative benefits of the fractional excretion of urea and sodium in various azotemic oliguric states. Nephron Clin Pract. 2010;114(2):c145–c150.
- MachasNúñez JF, Cameron JS, Oreopoulos DG, eds. The Aging Kidney in Health and Disease. New York, NY: Springer Science + Business Media, LLC; 2008.
- , , . Cytokine‐mediated regulation of urea transporters during experimental endotoxemia. Am J Physiol Renal Physiol. 2007;292(5):F1479–F1489.
- , , . Urinary biochemistry and microscopy in septic acute renal failure: a systematic review. Am J Kidney Dis. 2006;48(5):695–705.
- , , , et al. Diagnostic performance of fractional excretion of urea in the evaluation of critically ill patients with acute kidney injury: a multicenter cohort study. Crit Care. 2011;15(4):R178.
- , , , et al. Fractional excretion of urea as a diagnostic index in acute kidney injury in intensive care patients. J Crit Care. 2012;27(5):505–510.
- , , , . Diagnostic performance of fractional excretion of urea and fractional excretion of sodium in the evaluations of patients with acute kidney injury with or without diuretic treatment. Am J Kidney Dis. 2007;50(4):566–573.
- , , , et al. Transient versus persistent acute kidney injury and the diagnostic performance of fractional excretion of urea in critically ill patients. Nephron Clin Pract. 2014;126(1):8–13.
- , , , , . Emergency department bedside ultrasonographic measurement of the caval index for noninvasive determination of low central venous pressure. Ann Emerg Med. 2010;55(3):290–295.
- , , , , , . Urine microscopy is associated with severity and worsening of acute kidney injury in hospitalized patients. Clin J Am Soc Nephrol. 2010;5(3):402–408.
- , , , , . Diagnostic value of urine microscopy for differential diagnosis of acute kidney injury in hospitalized patients. Clin J Am Soc Nephrol. 2008;3(6):1615–1619.
- , , , et al. Definition and diagnostic criteria of refractory ascites and hepatorenal syndrome in cirrhosis. International Ascites Club. Hepatology. 1996;23(1):164–176.
- . The FENa test. Use in the differential diagnosis of acute renal failure. JAMA. 1976;236(6):579–581.
- , , . Significance of the fractional excretion of urea in the differential diagnosis of acute renal failure. Kidney Int. 2002;62(6):2223–2229.
- Centers for Medicare 275(8):630–634.
- , , , . Etiologies and outcome of acute renal insufficiency in older adults: a renal biopsy study of 259 cases. Am J Kidney Dis. 2000;35(3):433–447.
- , , , et al. Urinary diagnostic indices in acute renal failure: a prospective study. Ann Intern Med. 1978;89(1):47–50.
- , . Urinary indices and chemistries in the differential diagnosis of prerenal failure and acute tubular necrosis. Semin Nephrol. 1985;5(3):224–233.
- , , , , . The comparative benefits of the fractional excretion of urea and sodium in various azotemic oliguric states. Nephron Clin Pract. 2010;114(2):c145–c150.
- MachasNúñez JF, Cameron JS, Oreopoulos DG, eds. The Aging Kidney in Health and Disease. New York, NY: Springer Science + Business Media, LLC; 2008.
- , , . Cytokine‐mediated regulation of urea transporters during experimental endotoxemia. Am J Physiol Renal Physiol. 2007;292(5):F1479–F1489.
- , , . Urinary biochemistry and microscopy in septic acute renal failure: a systematic review. Am J Kidney Dis. 2006;48(5):695–705.
- , , , et al. Diagnostic performance of fractional excretion of urea in the evaluation of critically ill patients with acute kidney injury: a multicenter cohort study. Crit Care. 2011;15(4):R178.
- , , , et al. Fractional excretion of urea as a diagnostic index in acute kidney injury in intensive care patients. J Crit Care. 2012;27(5):505–510.
- , , , . Diagnostic performance of fractional excretion of urea and fractional excretion of sodium in the evaluations of patients with acute kidney injury with or without diuretic treatment. Am J Kidney Dis. 2007;50(4):566–573.
- , , , et al. Transient versus persistent acute kidney injury and the diagnostic performance of fractional excretion of urea in critically ill patients. Nephron Clin Pract. 2014;126(1):8–13.
- , , , , . Emergency department bedside ultrasonographic measurement of the caval index for noninvasive determination of low central venous pressure. Ann Emerg Med. 2010;55(3):290–295.
- , , , , , . Urine microscopy is associated with severity and worsening of acute kidney injury in hospitalized patients. Clin J Am Soc Nephrol. 2010;5(3):402–408.
- , , , , . Diagnostic value of urine microscopy for differential diagnosis of acute kidney injury in hospitalized patients. Clin J Am Soc Nephrol. 2008;3(6):1615–1619.
- , , , et al. Definition and diagnostic criteria of refractory ascites and hepatorenal syndrome in cirrhosis. International Ascites Club. Hepatology. 1996;23(1):164–176.
© 2015 Society of Hospital Medicine
Say Ahh…
Credit: These cases were adapted from Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013. You can now get the second edition of the Color Atlas of Family Medicine as an app for mobile devices by clicking this link: usatinemedia.com.
1. A painless white, thick lesion with fissuring had been on the side of this 57-year-old man’s tongue for the past 7 months. The patient drinks two to three beers in the evening and smokes one pack of cigarettes a day.

Diagnosis: This patient was given a diagnosis of leukoplakia and the biopsy indicated that the lesion was premalignant. The World Health Organization defines leukoplakia as “white plaques of questionable risk” in cases where other known diseases that don’t carry an increased risk for cancer have been excluded. For all types of leukoplakia, the risk of malignant transformation is approximately 1%, with a much higher risk associated with leukoplakias that contain red spots and/or rough spots.
For more information, read “White patch on tongue.” J Fam Pract. 2014.
For the next photograph, proceed to the next page >>
2. This nonhealing painful lesion on the side of her tongue had increased in size recently, and the patient, a 56-year-old homeless woman, was worried because her dad had died from oral cancer. She had smoked since she was 11 and acknowledged being a heavy drinker.
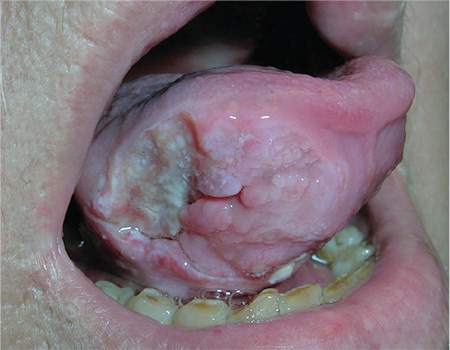
Diagnosis: A punch biopsy revealed that the patient had a squamous cell carcinoma. Fully two-thirds of oropharyngeal cancers (OPCs) will present with advanced disease at the time of diagnosis. Ninety percent of OPCs are of the squamous cell type. Reports in the literature suggest that clinicians may be missing early disease by not conducting thorough soft-tissue examinations on a routine basis. However, the fact that more than 35% of patients do not see a dentist on a routine basis likely contributes to the diagnostic delay. The 5-year survival rate for OPC is 62% for whites and 42% for blacks.
Tobacco use is the major risk factor for OPC and is implicated in approximately 75% of cases. Alcohol use is also a major risk factor. The combined use of tobacco and alcohol increases the risk of OPC far more than either alone. Human papillomavirus (especially HPV 16) is a newly recognized major risk factor for carcinomas affecting the lingual and palatine tonsils.
For more information, read “Lesion on side of tongue.” J Fam Pract. 2014.
For the next photograph, proceed to the next page >>
3. A 58-year-old man sought care for painful sores that had been in his mouth, on and off, for a year. The ulcers erupted on his tongue, gums, buccal mucosa, and inner lips, making it painful to eat. The patient was not taking any medications.
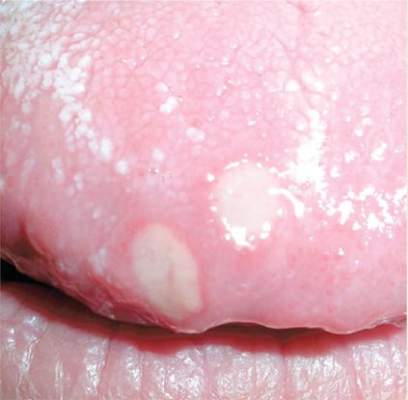
Diagnosis: The clinician recognized his condition as recurrent aphthous ulcers with giant ulcers. Aphthous ulcers are painful ulcerations in the mouth, which can be single, multiple, occasional, or recurrent. These ulcers can be small or large but are uniformly painful and may interfere with eating, speaking, and swallowing.
For more information, read “Lesions on tongue.” J Fam Pract. 2014.
For the next photograph, proceed to the next page >>
4. A 60-year-old man, smelling of alcohol and tobacco, complained of a black discoloration of his tongue and occasional gagging sensation. He smoked one to 2 packs of cigarettes and drank 6 to 8 beers daily. On physical exam, his teeth were stained and his tongue shows elongated papillae with brown discoloration.
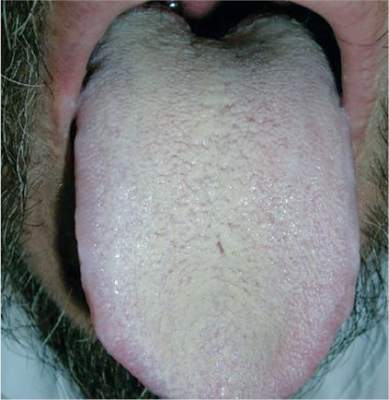
Diagnosis: This patient had a black hairy tongue (BHT), poor oral hygiene, and tobacco and alcohol addiction. BHT is a benign disorder of the tongue characterized by abnormally hypertrophied and elongated filiform papillae on the surface of the tongue. In addition, there is defective desquamation of the papillae on the dorsal tongue, resulting in a hair-like appearance. These papillae, which are normally about 1 mm in length, may become as long as 12 mm. The elongated filiform papillae can then collect debris, bacteria, fungus, or other foreign materials.
For more information, read “Discoloration of the tongue.” J Fam Pract. 2014.
For the next photograph, proceed to the next page >>
5. A 5-year-old girl had a temperature of 102.4°F, a sore throat, and a red tongue with prominent papillae. The posterior pharynx was also erythematous with slight exudate visible. The anterior cervical lymph nodes were mildly tender and somewhat enlarged, and no rashes were noted.
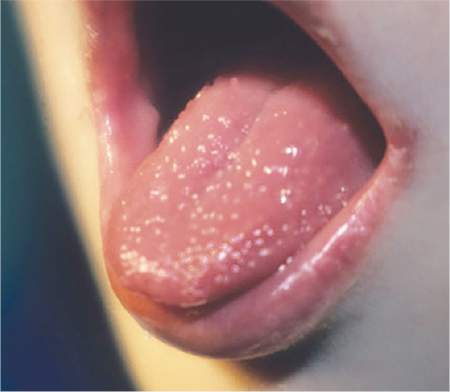
Diagnosis: The child had a strawberry tongue and scarlet fever caused by strep pharyngitis. The tongue had prominent papillae along with erythema, making it resemble a strawberry. Strawberry tongue is most commonly seen in children with scarlet fever or Kawasaki disease. Strawberry tongue usually develops within the first 2 to 3 days of illness. A white or yellowish coating usually precedes the classic red tongue with white papillae.
For more information, read “Papillae on tongue.” J Fam Pract. 2014.
Credit: These cases were adapted from Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013. You can now get the second edition of the Color Atlas of Family Medicine as an app for mobile devices by clicking this link: usatinemedia.com.
1. A painless white, thick lesion with fissuring had been on the side of this 57-year-old man’s tongue for the past 7 months. The patient drinks two to three beers in the evening and smokes one pack of cigarettes a day.
Diagnosis: This patient was given a diagnosis of leukoplakia and the biopsy indicated that the lesion was premalignant. The World Health Organization defines leukoplakia as “white plaques of questionable risk” in cases where other known diseases that don’t carry an increased risk for cancer have been excluded. For all types of leukoplakia, the risk of malignant transformation is approximately 1%, with a much higher risk associated with leukoplakias that contain red spots and/or rough spots.
For more information, read “White patch on tongue.” J Fam Pract. 2014.
For the next photograph, proceed to the next page >>
2. This nonhealing painful lesion on the side of her tongue had increased in size recently, and the patient, a 56-year-old homeless woman, was worried because her dad had died from oral cancer. She had smoked since she was 11 and acknowledged being a heavy drinker.
Diagnosis: A punch biopsy revealed that the patient had a squamous cell carcinoma. Fully two-thirds of oropharyngeal cancers (OPCs) will present with advanced disease at the time of diagnosis. Ninety percent of OPCs are of the squamous cell type. Reports in the literature suggest that clinicians may be missing early disease by not conducting thorough soft-tissue examinations on a routine basis. However, the fact that more than 35% of patients do not see a dentist on a routine basis likely contributes to the diagnostic delay. The 5-year survival rate for OPC is 62% for whites and 42% for blacks.
Tobacco use is the major risk factor for OPC and is implicated in approximately 75% of cases. Alcohol use is also a major risk factor. The combined use of tobacco and alcohol increases the risk of OPC far more than either alone. Human papillomavirus (especially HPV 16) is a newly recognized major risk factor for carcinomas affecting the lingual and palatine tonsils.
For more information, read “Lesion on side of tongue.” J Fam Pract. 2014.
For the next photograph, proceed to the next page >>
3. A 58-year-old man sought care for painful sores that had been in his mouth, on and off, for a year. The ulcers erupted on his tongue, gums, buccal mucosa, and inner lips, making it painful to eat. The patient was not taking any medications.
Diagnosis: The clinician recognized his condition as recurrent aphthous ulcers with giant ulcers. Aphthous ulcers are painful ulcerations in the mouth, which can be single, multiple, occasional, or recurrent. These ulcers can be small or large but are uniformly painful and may interfere with eating, speaking, and swallowing.
For more information, read “Lesions on tongue.” J Fam Pract. 2014.
For the next photograph, proceed to the next page >>
4. A 60-year-old man, smelling of alcohol and tobacco, complained of a black discoloration of his tongue and occasional gagging sensation. He smoked one to 2 packs of cigarettes and drank 6 to 8 beers daily. On physical exam, his teeth were stained and his tongue shows elongated papillae with brown discoloration.
Diagnosis: This patient had a black hairy tongue (BHT), poor oral hygiene, and tobacco and alcohol addiction. BHT is a benign disorder of the tongue characterized by abnormally hypertrophied and elongated filiform papillae on the surface of the tongue. In addition, there is defective desquamation of the papillae on the dorsal tongue, resulting in a hair-like appearance. These papillae, which are normally about 1 mm in length, may become as long as 12 mm. The elongated filiform papillae can then collect debris, bacteria, fungus, or other foreign materials.
For more information, read “Discoloration of the tongue.” J Fam Pract. 2014.
For the next photograph, proceed to the next page >>
5. A 5-year-old girl had a temperature of 102.4°F, a sore throat, and a red tongue with prominent papillae. The posterior pharynx was also erythematous with slight exudate visible. The anterior cervical lymph nodes were mildly tender and somewhat enlarged, and no rashes were noted.
Diagnosis: The child had a strawberry tongue and scarlet fever caused by strep pharyngitis. The tongue had prominent papillae along with erythema, making it resemble a strawberry. Strawberry tongue is most commonly seen in children with scarlet fever or Kawasaki disease. Strawberry tongue usually develops within the first 2 to 3 days of illness. A white or yellowish coating usually precedes the classic red tongue with white papillae.
For more information, read “Papillae on tongue.” J Fam Pract. 2014.
Credit: These cases were adapted from Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013. You can now get the second edition of the Color Atlas of Family Medicine as an app for mobile devices by clicking this link: usatinemedia.com.
1. A painless white, thick lesion with fissuring had been on the side of this 57-year-old man’s tongue for the past 7 months. The patient drinks two to three beers in the evening and smokes one pack of cigarettes a day.
Diagnosis: This patient was given a diagnosis of leukoplakia and the biopsy indicated that the lesion was premalignant. The World Health Organization defines leukoplakia as “white plaques of questionable risk” in cases where other known diseases that don’t carry an increased risk for cancer have been excluded. For all types of leukoplakia, the risk of malignant transformation is approximately 1%, with a much higher risk associated with leukoplakias that contain red spots and/or rough spots.
For more information, read “White patch on tongue.” J Fam Pract. 2014.
For the next photograph, proceed to the next page >>
2. This nonhealing painful lesion on the side of her tongue had increased in size recently, and the patient, a 56-year-old homeless woman, was worried because her dad had died from oral cancer. She had smoked since she was 11 and acknowledged being a heavy drinker.
Diagnosis: A punch biopsy revealed that the patient had a squamous cell carcinoma. Fully two-thirds of oropharyngeal cancers (OPCs) will present with advanced disease at the time of diagnosis. Ninety percent of OPCs are of the squamous cell type. Reports in the literature suggest that clinicians may be missing early disease by not conducting thorough soft-tissue examinations on a routine basis. However, the fact that more than 35% of patients do not see a dentist on a routine basis likely contributes to the diagnostic delay. The 5-year survival rate for OPC is 62% for whites and 42% for blacks.
Tobacco use is the major risk factor for OPC and is implicated in approximately 75% of cases. Alcohol use is also a major risk factor. The combined use of tobacco and alcohol increases the risk of OPC far more than either alone. Human papillomavirus (especially HPV 16) is a newly recognized major risk factor for carcinomas affecting the lingual and palatine tonsils.
For more information, read “Lesion on side of tongue.” J Fam Pract. 2014.
For the next photograph, proceed to the next page >>
3. A 58-year-old man sought care for painful sores that had been in his mouth, on and off, for a year. The ulcers erupted on his tongue, gums, buccal mucosa, and inner lips, making it painful to eat. The patient was not taking any medications.
Diagnosis: The clinician recognized his condition as recurrent aphthous ulcers with giant ulcers. Aphthous ulcers are painful ulcerations in the mouth, which can be single, multiple, occasional, or recurrent. These ulcers can be small or large but are uniformly painful and may interfere with eating, speaking, and swallowing.
For more information, read “Lesions on tongue.” J Fam Pract. 2014.
For the next photograph, proceed to the next page >>
4. A 60-year-old man, smelling of alcohol and tobacco, complained of a black discoloration of his tongue and occasional gagging sensation. He smoked one to 2 packs of cigarettes and drank 6 to 8 beers daily. On physical exam, his teeth were stained and his tongue shows elongated papillae with brown discoloration.
Diagnosis: This patient had a black hairy tongue (BHT), poor oral hygiene, and tobacco and alcohol addiction. BHT is a benign disorder of the tongue characterized by abnormally hypertrophied and elongated filiform papillae on the surface of the tongue. In addition, there is defective desquamation of the papillae on the dorsal tongue, resulting in a hair-like appearance. These papillae, which are normally about 1 mm in length, may become as long as 12 mm. The elongated filiform papillae can then collect debris, bacteria, fungus, or other foreign materials.
For more information, read “Discoloration of the tongue.” J Fam Pract. 2014.
For the next photograph, proceed to the next page >>
5. A 5-year-old girl had a temperature of 102.4°F, a sore throat, and a red tongue with prominent papillae. The posterior pharynx was also erythematous with slight exudate visible. The anterior cervical lymph nodes were mildly tender and somewhat enlarged, and no rashes were noted.
Diagnosis: The child had a strawberry tongue and scarlet fever caused by strep pharyngitis. The tongue had prominent papillae along with erythema, making it resemble a strawberry. Strawberry tongue is most commonly seen in children with scarlet fever or Kawasaki disease. Strawberry tongue usually develops within the first 2 to 3 days of illness. A white or yellowish coating usually precedes the classic red tongue with white papillae.
For more information, read “Papillae on tongue.” J Fam Pract. 2014.