User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Clinical Accuracy of Skin Cancer Diagnosis: Investigation of Keratinocyte Carcinoma Mismatch Rates
Clinical Accuracy of Skin Cancer Diagnosis: Investigation of Keratinocyte Carcinoma Mismatch Rates
To the Editor:
The incidence of nonmelanoma skin cancer (NMSC) is rapidly increasing worldwide. Due to its highly curable nature when treated early, accurate diagnosis is the cornerstone to good patient outcomes.1 Accurate diagnosis of skin cancer and subsequent treatment decisions rely heavily on the congruence between clinical observations and histopathologic assessments. Clinical misdiagnosis of a malignant lesion can lead to delayed and suboptimal treatment, which may contribute to serious complications such as metastasis or even mortality. In this study, data from clinically diagnosed basal cell carcinomas (BCCs) and squamous cell carcinomas (SCCs) were compared to their identified histopathologic subtype classifications. The accuracy of the clinical diagnosis of these NMSCs was assessed by determining the rate of misdiagnosis and the respective positive predictive value (PPV).
A retrospective review of medical records from a private dermatology practice in Lubbock, Texas, was conducted to identify patients diagnosed with NMSC from January 1, 2017, through December 31, 2021. A total of 11,229 NMSCs were diagnosed and treated in 5877 patients. Of the NMSCs diagnosed, 11,145 were identified as keratinocyte carcinomas and were classified as BCCs or SCCs. The accuracy of the clinical diagnoses was determined by comparison to the histologic subtype identified via biopsy of the lesion. Although the use of a dermatoscope during the clinical encounter was not formally recorded, reports from the examining dermatologists indicated it was not used in the majority of cases.
If a lesion was clinically diagnosed as a BCC but was identified as a subtype of SCC on histology (or vice versa), the lesion was considered to be mismatched. The number of mismatched lesions and the mismatch rate for each lesion type/subtype is recorded in the Table. Of the total 11,145 keratinocyte carcinomas included in our study, there was an overall 10.63% mismatch rate, with 1185 of the malignancies having a differing clinical diagnosis (eg, BCC vs SCC) from the histologic findings. The clinical mismatch rate was notably higher for SCC compared to BCC (15.83% vs 7.03%, respectively).
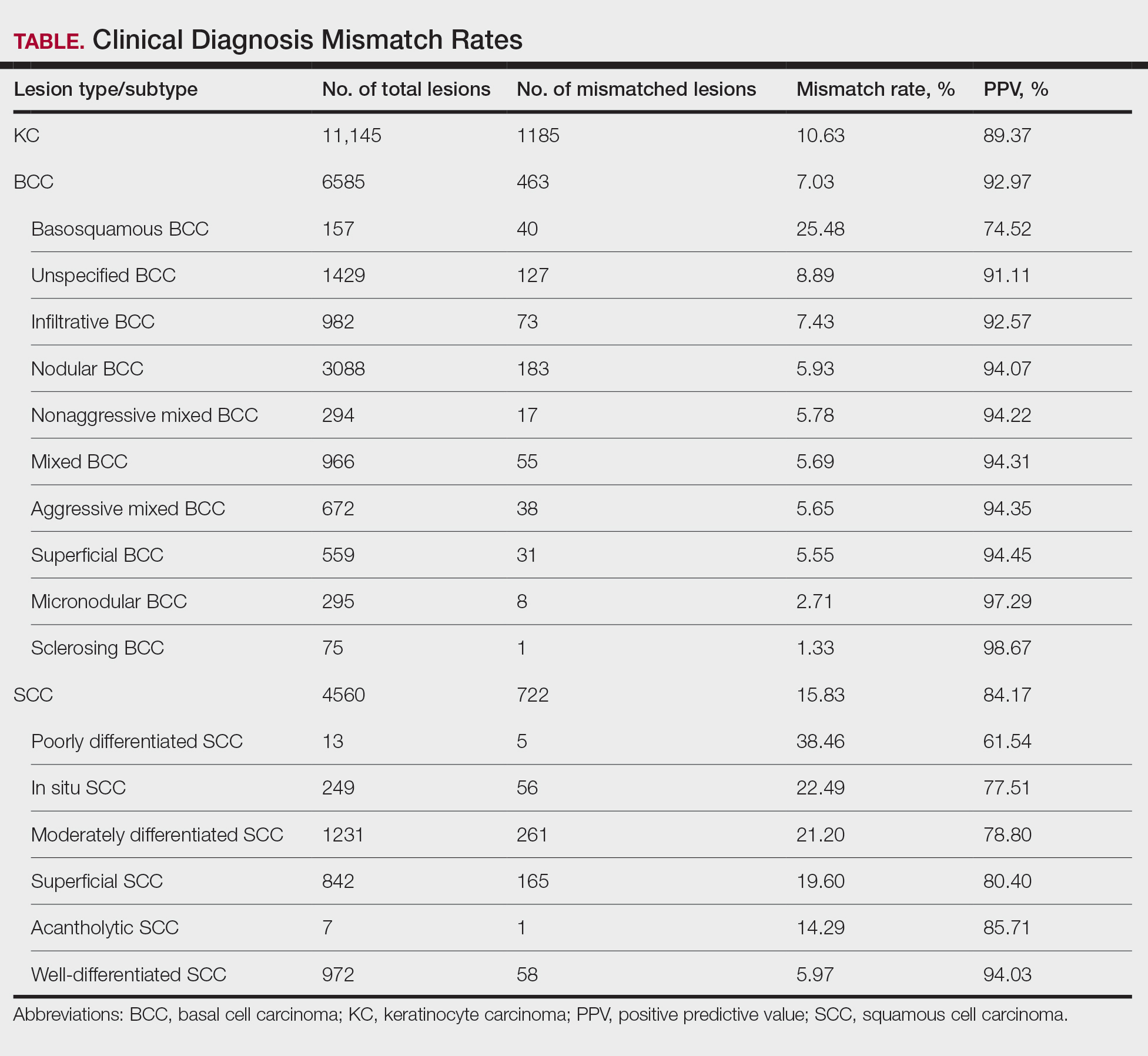
The Table provides a breakdown of the BCC subtypes identified by histology with their computed mismatch rate and PPV. It is worth clarifying that lesions classified as more than one BCC subtype per the histologic findings were diagnosed as mixed BCC; these were further classified as mixed-aggressive BCC (if at least one aggressive BCC subtype was present) and mixed nonaggressive BCC (if no aggressive BCC subtype was present). Overall, BCCs were less likely to be misdiagnosed, with an average PPV of 92.97% compared to 84.17% for SCCs. Basosquamous BCC was the BCC subtype with the highest mismatch rate (25.48%), while sclerosing BCC has the lowest overall mismatch rate (1.33%). The most common malignancy was BCC, with nodular BCC being the most common subtype.
The Table also breaks down the SCC subtypes, reporting the most commonly misdiagnosed of any BCC or SCC subtype to be poorly differentiated SCC (mismatch rate, 38.46%). The lowest mismatch rate of the SCC subtypes was 5.97% for well-differentiated SCC.
There was an overall PPV of 89.37% in clinically evaluated malignancies and their respective histologic subtypes. Basal cell carcinoma had a lower overall mismatch rate of 7.03% compared to 15.83% in SCC. The most common misdiagnosis was attributed to poorly differentiated SCC (mismatch rate, 38.46%), while the least common misdiagnosed malignancy was sclerosing BCC (1.33%). The high mismatch rate of poorly differentiated SCC may be due to its diverging presentation from a typical SCC as a flat lesion with the absence of scaling, keratin, or bleeding, leading to the misdiagnosis of BCC.2
Accurate clinical diagnosis of NMSCs is the basis for further evaluation and treatment that should ensue in a timely manner; however, accurately identifying BCCs vs SCCs solely based on clinical examination can be challenging due to variable manifestations and overlapping features. Basal cell carcinoma commonly presents as a shiny pink/flesh-colored nodule, macule, or patch with surface telangiectasia, sometimes appearing with ulceration or crusting.3 Alternatively, SCC typically appears as a firm, sharply demarcated, red nodule with a thick overlying scale.4 Definitive diagnoses can be difficult upon clinical examination since these features can be shared between the 2 subtypes. To aid in these uncertainties, a growing number of clinicians are implementing the use of dermoscopy in their everyday practice.
Dermoscopy is an extremely useful tool in improving the diagnostic accuracy of skin cancers compared to examination with the naked eye, as it provides detailed visualization of specific structures and patterns in skin cancer lesions.5 The dermoscopic appearance of BCC is characterized by pearly blue-gray or translucent globules with arborizing vessels, spoke-wheel structures, and leaflike areas.5,6 Conversely, dermoscopic features of SCC may include a milky-red globule with a scaly, sharply demarcated, crusted lesion with polymorphous vasculature, sometimes resembling a persistent sore or nonhealing wound.4,5 Though the use of dermoscopy can aid in diagnosis upon initial examination, certain factors such as trauma, ulceration, and previous treatments that distorted the lesion’s architecture may lead to misdiagnosis. Furthermore, the distinct vascular patterns found in BCC and SCC may be mistaken for each other and therefore lead to misdiagnosis upon examination.7 Other variables that may complicate diagnosis include the location of the lesion, its size, and the presence of other skin conditions or nearby lesions.
The primary limitation of the current study was the limited scope of the data, as they were derived from patients seen at one private dermatology practice, preventing the generalizability of our findings. However, our results show trends similar to those observed in other studies analyzing the clinical accuracy of skin cancer diagnoses, with higher PPVs for BCC compared to SCC. A study by Ahnlide and Bjellerup8 was based in a hospital dermatology department and demonstrated a PPV of 85.5% for BCC compared to 92.97% in our study; for SCC, the PPV was 67.3% compared to 84.17% in our study. In another study by Heal et al,9 data were collected from an Australian registry that included records of all histologically confirmed skin cancers from December 1996 to October 1999 from 202 general practitioners and 42 specialists, including 1 dermatologist. The PPVs for BCC and SCC were 72.7% and 49.4%, respectively. Although our results indicated higher PPVs compared to these 2 studies, some of the discrepancies can be accounted for by the differences in clinical setting as well as the lack of expertise of nondermatologist physicians in identifying skin malignancies in the study by Heal et al.9
The current study was further limited by the lack of data quantifying the number of lesions clinically suspected to be malignant but found to be histologically benign. It is typical for clinicians to have a low threshold to biopsy a suspicious lesion with atypical features (eg, rapid evolution and growth, bleeding, crusting). Furthermore, the identification of risk factors in the patient’s medical and family history (eg, exposure to radiation, personal or family history of skin cancers) can heavily influence a clinician’s decision to biopsy a lesion with an atypical appearance.10 Many benign lesions are biopsied to avoid missing a diagnosis of malignancy. Consequently, our results suggest a high degree of clinical misdiagnosis of BCCs and SCCs. Obtaining data on the number of lesions suspected to be BCC or SCC that were found to be histologically benign would be a valuable addition to our study, as it would provide a measurable insight into the sensitivity of clinicians’ decision-making to identify a lesion as suspicious and warranting biopsy.
While clinical diagnosis plays a vital role in identifying suspected NMSCs such as BCC and SCC, its accuracy can be limited even with the use of dermoscopy. Overall, our data have shown a high rate of diagnostic accuracy upon suspicion of malignancy, but the different variables that affect clinical presentation promote histologic diagnosis to prevail as the gold standard.
- Seyed Ahadi M, Firooz A, Rahimi H, et al. Clinical diagnosis has a high negative predictive value in evaluation of malignant skin lesions. Dermatol Res Pract. 2021;2021:6618990. doi:10.1155/2021/6618990
- Lallas A, Pyne J, Kyrgidis A, et al. The clinical and dermoscopic features of invasive cutaneous squamous cell carcinoma depend on the histopathological grade of differentiation. Br J Dermatol. 2015;172:1308- 1315. doi:10.1111/bjd.13510
- McDaniel B, Badri T, Steele RB. Basal cell carcinoma. September 19, 2022. In: StatPearls. StatPearls Publishing; 2023.
- Suárez AL, Louis P, Kitts J, et al. Clinical and dermoscopic features of combined cutaneous squamous cell carcinoma (SCC)/neuroendocrine [Merkel cell] carcinoma (MCC). J Am Acad Dermatol. 2015;73:968-975. doi:10.1016/j.jaad.2015.08.041
- Wolner ZJ, Yélamos O, Liopyris K, et al. Enhancing skin cancer diagnosis with dermoscopy. Dermatol Clin. 2017;35:417-437. doi:10.1016/j.det.2017.06.003
- Reiter O, Mimouni I, Dusza S, et al. Dermoscopic features of basal cell carcinoma and its subtypes: a systematic review. J Am Acad Dermatol. 2021;85:653-664. doi:10.1016/j.jaad.2019.11.008
- Pruneda C, Ramesh M, Hope L, et al. Nonmelanoma skin cancers: diagnostic accuracy of midlevel providers versus dermatologists. The Dermatologist. March 2023. Accessed March 18, 2025. https://www.hmpgloballearningnetwork.com/site/thederm/feature-story/nonmelanoma-skin-cancers-diagnostic-accuracy-midlevel-providers-vs
- Ahnlide I, Bjellerup M. Accuracy of clinical skin tumour diagnosis in a dermatological setting. Acta Derm Venereol. 2013;93:305-308. doi:10.2340/00015555-1560
- Heal CF, Raasch BA, Buettner PG, et al. Accuracy of clinical diagnosis of skin lesions. Br J Dermatol. 2008;159:661-668.
- Fu S, Kim S, Wasko C. Dermatological guide for primary care physicians: full body skin checks, skin cancer detection, and patient education on self-skin checks and sun protection. Proc (Bayl Univ Med Cent). 2024;37:647-654. doi:10.1080/08998280.2024.2351751
To the Editor:
The incidence of nonmelanoma skin cancer (NMSC) is rapidly increasing worldwide. Due to its highly curable nature when treated early, accurate diagnosis is the cornerstone to good patient outcomes.1 Accurate diagnosis of skin cancer and subsequent treatment decisions rely heavily on the congruence between clinical observations and histopathologic assessments. Clinical misdiagnosis of a malignant lesion can lead to delayed and suboptimal treatment, which may contribute to serious complications such as metastasis or even mortality. In this study, data from clinically diagnosed basal cell carcinomas (BCCs) and squamous cell carcinomas (SCCs) were compared to their identified histopathologic subtype classifications. The accuracy of the clinical diagnosis of these NMSCs was assessed by determining the rate of misdiagnosis and the respective positive predictive value (PPV).
A retrospective review of medical records from a private dermatology practice in Lubbock, Texas, was conducted to identify patients diagnosed with NMSC from January 1, 2017, through December 31, 2021. A total of 11,229 NMSCs were diagnosed and treated in 5877 patients. Of the NMSCs diagnosed, 11,145 were identified as keratinocyte carcinomas and were classified as BCCs or SCCs. The accuracy of the clinical diagnoses was determined by comparison to the histologic subtype identified via biopsy of the lesion. Although the use of a dermatoscope during the clinical encounter was not formally recorded, reports from the examining dermatologists indicated it was not used in the majority of cases.
If a lesion was clinically diagnosed as a BCC but was identified as a subtype of SCC on histology (or vice versa), the lesion was considered to be mismatched. The number of mismatched lesions and the mismatch rate for each lesion type/subtype is recorded in the Table. Of the total 11,145 keratinocyte carcinomas included in our study, there was an overall 10.63% mismatch rate, with 1185 of the malignancies having a differing clinical diagnosis (eg, BCC vs SCC) from the histologic findings. The clinical mismatch rate was notably higher for SCC compared to BCC (15.83% vs 7.03%, respectively).
The Table provides a breakdown of the BCC subtypes identified by histology with their computed mismatch rate and PPV. It is worth clarifying that lesions classified as more than one BCC subtype per the histologic findings were diagnosed as mixed BCC; these were further classified as mixed-aggressive BCC (if at least one aggressive BCC subtype was present) and mixed nonaggressive BCC (if no aggressive BCC subtype was present). Overall, BCCs were less likely to be misdiagnosed, with an average PPV of 92.97% compared to 84.17% for SCCs. Basosquamous BCC was the BCC subtype with the highest mismatch rate (25.48%), while sclerosing BCC has the lowest overall mismatch rate (1.33%). The most common malignancy was BCC, with nodular BCC being the most common subtype.
The Table also breaks down the SCC subtypes, reporting the most commonly misdiagnosed of any BCC or SCC subtype to be poorly differentiated SCC (mismatch rate, 38.46%). The lowest mismatch rate of the SCC subtypes was 5.97% for well-differentiated SCC.
There was an overall PPV of 89.37% in clinically evaluated malignancies and their respective histologic subtypes. Basal cell carcinoma had a lower overall mismatch rate of 7.03% compared to 15.83% in SCC. The most common misdiagnosis was attributed to poorly differentiated SCC (mismatch rate, 38.46%), while the least common misdiagnosed malignancy was sclerosing BCC (1.33%). The high mismatch rate of poorly differentiated SCC may be due to its diverging presentation from a typical SCC as a flat lesion with the absence of scaling, keratin, or bleeding, leading to the misdiagnosis of BCC.2
Accurate clinical diagnosis of NMSCs is the basis for further evaluation and treatment that should ensue in a timely manner; however, accurately identifying BCCs vs SCCs solely based on clinical examination can be challenging due to variable manifestations and overlapping features. Basal cell carcinoma commonly presents as a shiny pink/flesh-colored nodule, macule, or patch with surface telangiectasia, sometimes appearing with ulceration or crusting.3 Alternatively, SCC typically appears as a firm, sharply demarcated, red nodule with a thick overlying scale.4 Definitive diagnoses can be difficult upon clinical examination since these features can be shared between the 2 subtypes. To aid in these uncertainties, a growing number of clinicians are implementing the use of dermoscopy in their everyday practice.
Dermoscopy is an extremely useful tool in improving the diagnostic accuracy of skin cancers compared to examination with the naked eye, as it provides detailed visualization of specific structures and patterns in skin cancer lesions.5 The dermoscopic appearance of BCC is characterized by pearly blue-gray or translucent globules with arborizing vessels, spoke-wheel structures, and leaflike areas.5,6 Conversely, dermoscopic features of SCC may include a milky-red globule with a scaly, sharply demarcated, crusted lesion with polymorphous vasculature, sometimes resembling a persistent sore or nonhealing wound.4,5 Though the use of dermoscopy can aid in diagnosis upon initial examination, certain factors such as trauma, ulceration, and previous treatments that distorted the lesion’s architecture may lead to misdiagnosis. Furthermore, the distinct vascular patterns found in BCC and SCC may be mistaken for each other and therefore lead to misdiagnosis upon examination.7 Other variables that may complicate diagnosis include the location of the lesion, its size, and the presence of other skin conditions or nearby lesions.
The primary limitation of the current study was the limited scope of the data, as they were derived from patients seen at one private dermatology practice, preventing the generalizability of our findings. However, our results show trends similar to those observed in other studies analyzing the clinical accuracy of skin cancer diagnoses, with higher PPVs for BCC compared to SCC. A study by Ahnlide and Bjellerup8 was based in a hospital dermatology department and demonstrated a PPV of 85.5% for BCC compared to 92.97% in our study; for SCC, the PPV was 67.3% compared to 84.17% in our study. In another study by Heal et al,9 data were collected from an Australian registry that included records of all histologically confirmed skin cancers from December 1996 to October 1999 from 202 general practitioners and 42 specialists, including 1 dermatologist. The PPVs for BCC and SCC were 72.7% and 49.4%, respectively. Although our results indicated higher PPVs compared to these 2 studies, some of the discrepancies can be accounted for by the differences in clinical setting as well as the lack of expertise of nondermatologist physicians in identifying skin malignancies in the study by Heal et al.9
The current study was further limited by the lack of data quantifying the number of lesions clinically suspected to be malignant but found to be histologically benign. It is typical for clinicians to have a low threshold to biopsy a suspicious lesion with atypical features (eg, rapid evolution and growth, bleeding, crusting). Furthermore, the identification of risk factors in the patient’s medical and family history (eg, exposure to radiation, personal or family history of skin cancers) can heavily influence a clinician’s decision to biopsy a lesion with an atypical appearance.10 Many benign lesions are biopsied to avoid missing a diagnosis of malignancy. Consequently, our results suggest a high degree of clinical misdiagnosis of BCCs and SCCs. Obtaining data on the number of lesions suspected to be BCC or SCC that were found to be histologically benign would be a valuable addition to our study, as it would provide a measurable insight into the sensitivity of clinicians’ decision-making to identify a lesion as suspicious and warranting biopsy.
While clinical diagnosis plays a vital role in identifying suspected NMSCs such as BCC and SCC, its accuracy can be limited even with the use of dermoscopy. Overall, our data have shown a high rate of diagnostic accuracy upon suspicion of malignancy, but the different variables that affect clinical presentation promote histologic diagnosis to prevail as the gold standard.
To the Editor:
The incidence of nonmelanoma skin cancer (NMSC) is rapidly increasing worldwide. Due to its highly curable nature when treated early, accurate diagnosis is the cornerstone to good patient outcomes.1 Accurate diagnosis of skin cancer and subsequent treatment decisions rely heavily on the congruence between clinical observations and histopathologic assessments. Clinical misdiagnosis of a malignant lesion can lead to delayed and suboptimal treatment, which may contribute to serious complications such as metastasis or even mortality. In this study, data from clinically diagnosed basal cell carcinomas (BCCs) and squamous cell carcinomas (SCCs) were compared to their identified histopathologic subtype classifications. The accuracy of the clinical diagnosis of these NMSCs was assessed by determining the rate of misdiagnosis and the respective positive predictive value (PPV).
A retrospective review of medical records from a private dermatology practice in Lubbock, Texas, was conducted to identify patients diagnosed with NMSC from January 1, 2017, through December 31, 2021. A total of 11,229 NMSCs were diagnosed and treated in 5877 patients. Of the NMSCs diagnosed, 11,145 were identified as keratinocyte carcinomas and were classified as BCCs or SCCs. The accuracy of the clinical diagnoses was determined by comparison to the histologic subtype identified via biopsy of the lesion. Although the use of a dermatoscope during the clinical encounter was not formally recorded, reports from the examining dermatologists indicated it was not used in the majority of cases.
If a lesion was clinically diagnosed as a BCC but was identified as a subtype of SCC on histology (or vice versa), the lesion was considered to be mismatched. The number of mismatched lesions and the mismatch rate for each lesion type/subtype is recorded in the Table. Of the total 11,145 keratinocyte carcinomas included in our study, there was an overall 10.63% mismatch rate, with 1185 of the malignancies having a differing clinical diagnosis (eg, BCC vs SCC) from the histologic findings. The clinical mismatch rate was notably higher for SCC compared to BCC (15.83% vs 7.03%, respectively).
The Table provides a breakdown of the BCC subtypes identified by histology with their computed mismatch rate and PPV. It is worth clarifying that lesions classified as more than one BCC subtype per the histologic findings were diagnosed as mixed BCC; these were further classified as mixed-aggressive BCC (if at least one aggressive BCC subtype was present) and mixed nonaggressive BCC (if no aggressive BCC subtype was present). Overall, BCCs were less likely to be misdiagnosed, with an average PPV of 92.97% compared to 84.17% for SCCs. Basosquamous BCC was the BCC subtype with the highest mismatch rate (25.48%), while sclerosing BCC has the lowest overall mismatch rate (1.33%). The most common malignancy was BCC, with nodular BCC being the most common subtype.
The Table also breaks down the SCC subtypes, reporting the most commonly misdiagnosed of any BCC or SCC subtype to be poorly differentiated SCC (mismatch rate, 38.46%). The lowest mismatch rate of the SCC subtypes was 5.97% for well-differentiated SCC.
There was an overall PPV of 89.37% in clinically evaluated malignancies and their respective histologic subtypes. Basal cell carcinoma had a lower overall mismatch rate of 7.03% compared to 15.83% in SCC. The most common misdiagnosis was attributed to poorly differentiated SCC (mismatch rate, 38.46%), while the least common misdiagnosed malignancy was sclerosing BCC (1.33%). The high mismatch rate of poorly differentiated SCC may be due to its diverging presentation from a typical SCC as a flat lesion with the absence of scaling, keratin, or bleeding, leading to the misdiagnosis of BCC.2
Accurate clinical diagnosis of NMSCs is the basis for further evaluation and treatment that should ensue in a timely manner; however, accurately identifying BCCs vs SCCs solely based on clinical examination can be challenging due to variable manifestations and overlapping features. Basal cell carcinoma commonly presents as a shiny pink/flesh-colored nodule, macule, or patch with surface telangiectasia, sometimes appearing with ulceration or crusting.3 Alternatively, SCC typically appears as a firm, sharply demarcated, red nodule with a thick overlying scale.4 Definitive diagnoses can be difficult upon clinical examination since these features can be shared between the 2 subtypes. To aid in these uncertainties, a growing number of clinicians are implementing the use of dermoscopy in their everyday practice.
Dermoscopy is an extremely useful tool in improving the diagnostic accuracy of skin cancers compared to examination with the naked eye, as it provides detailed visualization of specific structures and patterns in skin cancer lesions.5 The dermoscopic appearance of BCC is characterized by pearly blue-gray or translucent globules with arborizing vessels, spoke-wheel structures, and leaflike areas.5,6 Conversely, dermoscopic features of SCC may include a milky-red globule with a scaly, sharply demarcated, crusted lesion with polymorphous vasculature, sometimes resembling a persistent sore or nonhealing wound.4,5 Though the use of dermoscopy can aid in diagnosis upon initial examination, certain factors such as trauma, ulceration, and previous treatments that distorted the lesion’s architecture may lead to misdiagnosis. Furthermore, the distinct vascular patterns found in BCC and SCC may be mistaken for each other and therefore lead to misdiagnosis upon examination.7 Other variables that may complicate diagnosis include the location of the lesion, its size, and the presence of other skin conditions or nearby lesions.
The primary limitation of the current study was the limited scope of the data, as they were derived from patients seen at one private dermatology practice, preventing the generalizability of our findings. However, our results show trends similar to those observed in other studies analyzing the clinical accuracy of skin cancer diagnoses, with higher PPVs for BCC compared to SCC. A study by Ahnlide and Bjellerup8 was based in a hospital dermatology department and demonstrated a PPV of 85.5% for BCC compared to 92.97% in our study; for SCC, the PPV was 67.3% compared to 84.17% in our study. In another study by Heal et al,9 data were collected from an Australian registry that included records of all histologically confirmed skin cancers from December 1996 to October 1999 from 202 general practitioners and 42 specialists, including 1 dermatologist. The PPVs for BCC and SCC were 72.7% and 49.4%, respectively. Although our results indicated higher PPVs compared to these 2 studies, some of the discrepancies can be accounted for by the differences in clinical setting as well as the lack of expertise of nondermatologist physicians in identifying skin malignancies in the study by Heal et al.9
The current study was further limited by the lack of data quantifying the number of lesions clinically suspected to be malignant but found to be histologically benign. It is typical for clinicians to have a low threshold to biopsy a suspicious lesion with atypical features (eg, rapid evolution and growth, bleeding, crusting). Furthermore, the identification of risk factors in the patient’s medical and family history (eg, exposure to radiation, personal or family history of skin cancers) can heavily influence a clinician’s decision to biopsy a lesion with an atypical appearance.10 Many benign lesions are biopsied to avoid missing a diagnosis of malignancy. Consequently, our results suggest a high degree of clinical misdiagnosis of BCCs and SCCs. Obtaining data on the number of lesions suspected to be BCC or SCC that were found to be histologically benign would be a valuable addition to our study, as it would provide a measurable insight into the sensitivity of clinicians’ decision-making to identify a lesion as suspicious and warranting biopsy.
While clinical diagnosis plays a vital role in identifying suspected NMSCs such as BCC and SCC, its accuracy can be limited even with the use of dermoscopy. Overall, our data have shown a high rate of diagnostic accuracy upon suspicion of malignancy, but the different variables that affect clinical presentation promote histologic diagnosis to prevail as the gold standard.
- Seyed Ahadi M, Firooz A, Rahimi H, et al. Clinical diagnosis has a high negative predictive value in evaluation of malignant skin lesions. Dermatol Res Pract. 2021;2021:6618990. doi:10.1155/2021/6618990
- Lallas A, Pyne J, Kyrgidis A, et al. The clinical and dermoscopic features of invasive cutaneous squamous cell carcinoma depend on the histopathological grade of differentiation. Br J Dermatol. 2015;172:1308- 1315. doi:10.1111/bjd.13510
- McDaniel B, Badri T, Steele RB. Basal cell carcinoma. September 19, 2022. In: StatPearls. StatPearls Publishing; 2023.
- Suárez AL, Louis P, Kitts J, et al. Clinical and dermoscopic features of combined cutaneous squamous cell carcinoma (SCC)/neuroendocrine [Merkel cell] carcinoma (MCC). J Am Acad Dermatol. 2015;73:968-975. doi:10.1016/j.jaad.2015.08.041
- Wolner ZJ, Yélamos O, Liopyris K, et al. Enhancing skin cancer diagnosis with dermoscopy. Dermatol Clin. 2017;35:417-437. doi:10.1016/j.det.2017.06.003
- Reiter O, Mimouni I, Dusza S, et al. Dermoscopic features of basal cell carcinoma and its subtypes: a systematic review. J Am Acad Dermatol. 2021;85:653-664. doi:10.1016/j.jaad.2019.11.008
- Pruneda C, Ramesh M, Hope L, et al. Nonmelanoma skin cancers: diagnostic accuracy of midlevel providers versus dermatologists. The Dermatologist. March 2023. Accessed March 18, 2025. https://www.hmpgloballearningnetwork.com/site/thederm/feature-story/nonmelanoma-skin-cancers-diagnostic-accuracy-midlevel-providers-vs
- Ahnlide I, Bjellerup M. Accuracy of clinical skin tumour diagnosis in a dermatological setting. Acta Derm Venereol. 2013;93:305-308. doi:10.2340/00015555-1560
- Heal CF, Raasch BA, Buettner PG, et al. Accuracy of clinical diagnosis of skin lesions. Br J Dermatol. 2008;159:661-668.
- Fu S, Kim S, Wasko C. Dermatological guide for primary care physicians: full body skin checks, skin cancer detection, and patient education on self-skin checks and sun protection. Proc (Bayl Univ Med Cent). 2024;37:647-654. doi:10.1080/08998280.2024.2351751
- Seyed Ahadi M, Firooz A, Rahimi H, et al. Clinical diagnosis has a high negative predictive value in evaluation of malignant skin lesions. Dermatol Res Pract. 2021;2021:6618990. doi:10.1155/2021/6618990
- Lallas A, Pyne J, Kyrgidis A, et al. The clinical and dermoscopic features of invasive cutaneous squamous cell carcinoma depend on the histopathological grade of differentiation. Br J Dermatol. 2015;172:1308- 1315. doi:10.1111/bjd.13510
- McDaniel B, Badri T, Steele RB. Basal cell carcinoma. September 19, 2022. In: StatPearls. StatPearls Publishing; 2023.
- Suárez AL, Louis P, Kitts J, et al. Clinical and dermoscopic features of combined cutaneous squamous cell carcinoma (SCC)/neuroendocrine [Merkel cell] carcinoma (MCC). J Am Acad Dermatol. 2015;73:968-975. doi:10.1016/j.jaad.2015.08.041
- Wolner ZJ, Yélamos O, Liopyris K, et al. Enhancing skin cancer diagnosis with dermoscopy. Dermatol Clin. 2017;35:417-437. doi:10.1016/j.det.2017.06.003
- Reiter O, Mimouni I, Dusza S, et al. Dermoscopic features of basal cell carcinoma and its subtypes: a systematic review. J Am Acad Dermatol. 2021;85:653-664. doi:10.1016/j.jaad.2019.11.008
- Pruneda C, Ramesh M, Hope L, et al. Nonmelanoma skin cancers: diagnostic accuracy of midlevel providers versus dermatologists. The Dermatologist. March 2023. Accessed March 18, 2025. https://www.hmpgloballearningnetwork.com/site/thederm/feature-story/nonmelanoma-skin-cancers-diagnostic-accuracy-midlevel-providers-vs
- Ahnlide I, Bjellerup M. Accuracy of clinical skin tumour diagnosis in a dermatological setting. Acta Derm Venereol. 2013;93:305-308. doi:10.2340/00015555-1560
- Heal CF, Raasch BA, Buettner PG, et al. Accuracy of clinical diagnosis of skin lesions. Br J Dermatol. 2008;159:661-668.
- Fu S, Kim S, Wasko C. Dermatological guide for primary care physicians: full body skin checks, skin cancer detection, and patient education on self-skin checks and sun protection. Proc (Bayl Univ Med Cent). 2024;37:647-654. doi:10.1080/08998280.2024.2351751
Clinical Accuracy of Skin Cancer Diagnosis: Investigation of Keratinocyte Carcinoma Mismatch Rates
Clinical Accuracy of Skin Cancer Diagnosis: Investigation of Keratinocyte Carcinoma Mismatch Rates
PRACTICE POINTS
- Malignant lesions may be misdiagnosed when assessments are guided by clinical features that align with typical presentations of other lesion types, potentially leading to diagnostic errors among experienced clinicians.
- Although dermoscopy is a beneficial tool in examining potential skin cancers, clinical observations should not bypass the gold standard of histopathologic examination.
Repair of a Large Full-Thickness Conchal Bowl Defect
Repair of a Large Full-Thickness Conchal Bowl Defect
Practice Gap
Large full-thickness conchal bowl defects often pose a reconstructive challenge. Maintaining the shape and structural integrity of the concha is fundamental for optimal cosmetic and functional outcomes. Prior reports have suggested wedge excisions, composite grafts, interpolation flaps with or without cartilage struts, and hinge flaps as possible options for reconstruction.1-3 However, patients with large defects who prefer single-stage reconstruction procedures present a unique challenge. Herein, we describe a single-stage full-thickness hinge flap technique for a large conchal bowl defect.
The Technique
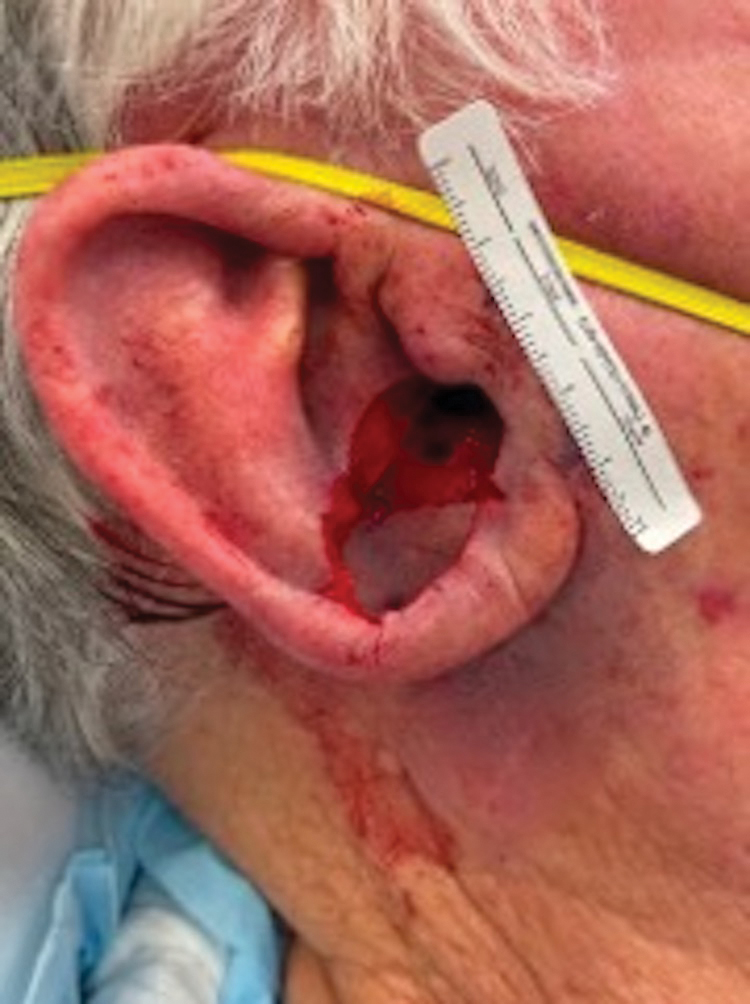
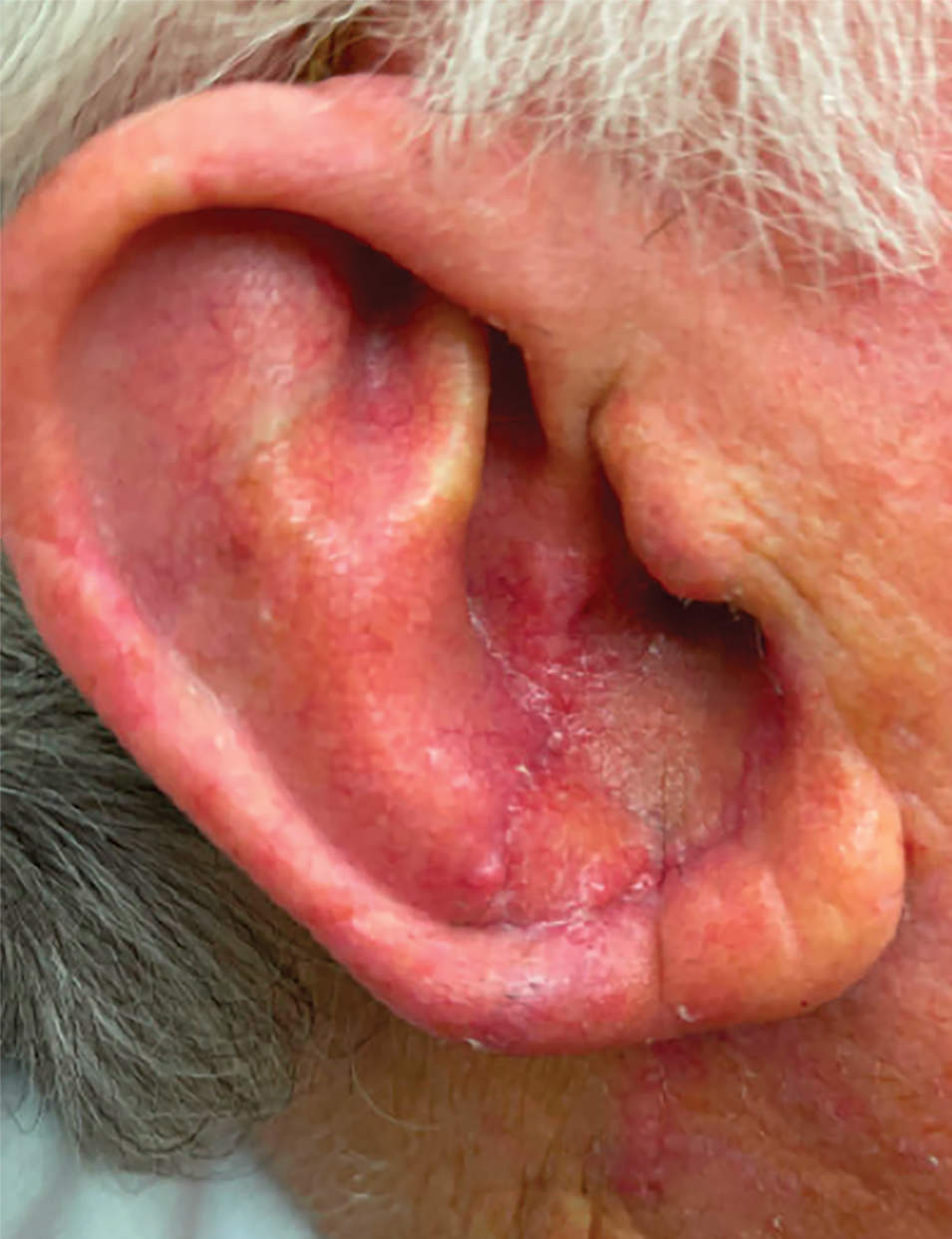
A 77-year-old man was referred to our dermatology clinic by an outside dermatologist for Mohs micrographic surgery of a biopsy-proven cutaneous squamous cell carcinoma on the right conchal bowl measuring 1.1×2.1 cm and extending to the edge of the external auditory canal (EAC). The excision was performed that same day and was completed in 2 stages, achieving negative margins and resulting in a full-thickness defect measuring 2.0×3.6 cm that included the posterior auricular sulcus, cavum, antitragus, and proximal EAC (Figure 1). The patient requested a single-stage procedure but emphasized that his main priority was an optimal cosmetic outcome.
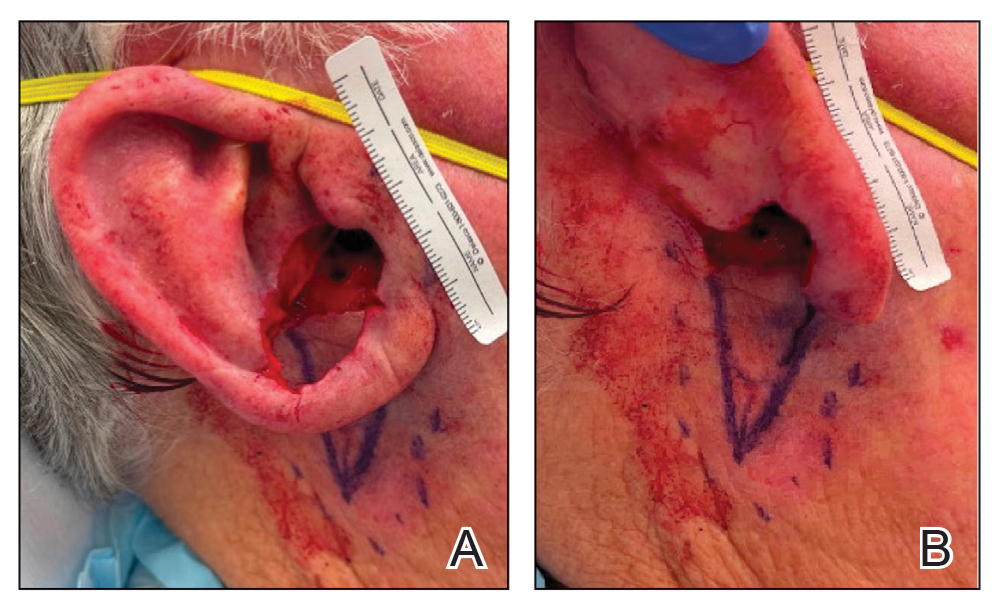
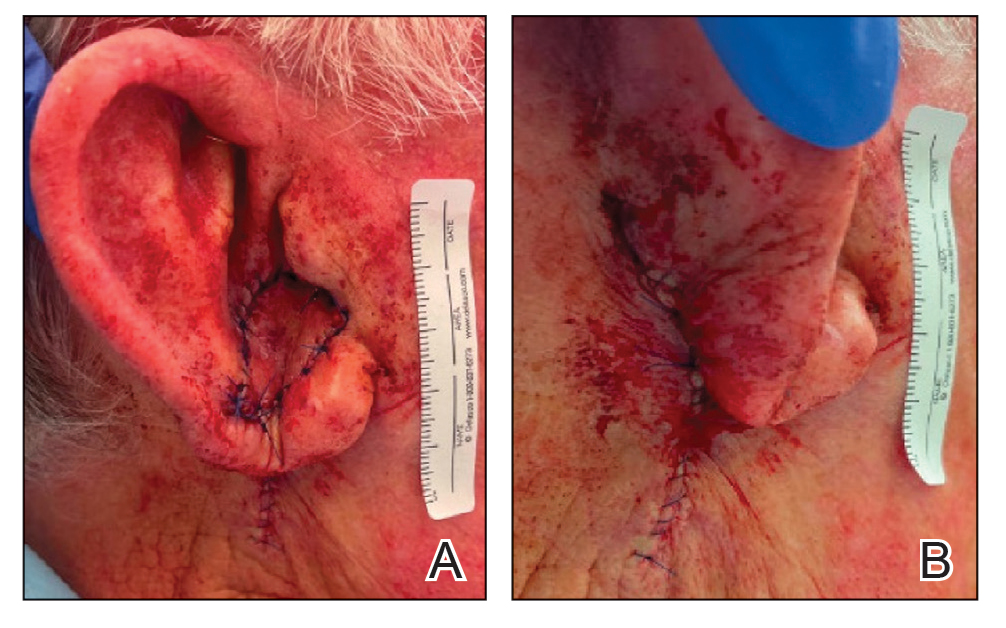
To repair this large defect, a full-thickness hinge flap with Burow graft was performed. The hinge-type flap was designed in a triangular fashion emanating at the posterior auricular sulcus adjacent to the posterior aspect of the defect and extending down the lateral neck (Figure 2). The flap was incised and the surrounding tissue was undermined, maintaining a robust pedicle in the center of its body on the superolateral neck. The flap was passed through the posterior aspect of the full-thickness defect and was secured in place with 4-0 polyglactin sutures in a buried interrupted fashion, thereby recreating the anterior portion of the defect. The superficial skin edges were reapproximated using 4-0 and 5-0 polypropylene sutures in a running interrupted fashion. The distal Burow triangle created from closure of the flap’s secondary defect was aggressively thinned and was utilized as a full-thickness graft for the residual postauricular groove defect (Figure 3). At 2 weeks’ follow-up, the patient was healing well with no postoperative issues and the sutures were removed (Figure 4).
Practice Implications
There are many different reconstructive options for conchal bowl defects, including primary repair, wedge excision, composite graft and interpolation flaps with or without cartilage struts, and hinge flaps. Structural support, EAC patency, auricle symmetry, overall auricle size, and re-creation of natural contours were considered when designing the reconstruction of the defect in our patient; however, his main priority was achieving the greatest cosmetic outcome in a single-stage procedure, therefore limiting our reconstruction options.
Wedge excision, in which the residual lobule and inferior helical rim are removed, could have been considered in our patient but would have drastically altered the symmetry of the size of the ears. A folded postauricular flap, as described in the otolaryngology literature, is an interpolation flap based on the posterior auricular artery that was designed for full-thickness defects of the auricle to prevent any posterior pinning.1 This technique may have worked well in our case, but the patient preferred to avoid a multistage procedure. Additionally, the positional symmetry of the ears was maintained despite utilizing a hinge flap, which does not involve takedown of the pedicle. A composite graft from the contralateral ear could be considered for smaller conchal bowl defects but likely would have resulted in graft failure in our patient’s large defect due to its need for rich blood supply to heal and dependence on lateral wound edges. Cartilage struts in conjunction with a flap could have been considered in this scenario for greater structural support, but in our patient’s case, by maintaining the robust pedicle of our flap and having residual superior cartilage, further structural support was not necessary.
A prior case report described a partial and full-thickness defect in a similar location that was repaired with a retroauricular hinge flap, in which a portion of the flap was extensively de-epithelialized to address the varied thicknesses of the surgical defect.2 In our patient, the defect abutted the skin reservoir on the superolateral neck, and therefore no de-epithelialization was required as the entire epithelialized portion was utilized to recreate the anterior aspect of the defect. Postauricular hinge-type flaps are a reliable, single-stage surgical alternative to the 2-stage folded postauricular interpolation flap when reconstructing large conchal bowl defects. For small full-thickness defects of the ear, a composite graft may be considered; however, blood supply and other nutritional requirements limit this option for large full-thickness defects.
- Roche AM, Griffin M, Shelton R, et al. The folded postauricular flap: a novel approach to reconstruction of large full thickness defects of the conchal bowl. Am J Otolaryngol. 2017;38:706-709. doi:10.1016 /j.amjoto.2017.09.006
- Klein JC, Nijhawan RI. Retroauricular hinge flaps for full-thickness conchal bowl defects. J Am Acad Dermatol. 2024;90:E71-E72. doi:10.1016/j.jaad.2022.10.056
- Pickrell BB, Hughes CD, Maricevich RS. Partial ear defects. Semin Plast Surg. 2017 Aug;31:134-140. doi:10.1055/s-0037-1603968.
Practice Gap
Large full-thickness conchal bowl defects often pose a reconstructive challenge. Maintaining the shape and structural integrity of the concha is fundamental for optimal cosmetic and functional outcomes. Prior reports have suggested wedge excisions, composite grafts, interpolation flaps with or without cartilage struts, and hinge flaps as possible options for reconstruction.1-3 However, patients with large defects who prefer single-stage reconstruction procedures present a unique challenge. Herein, we describe a single-stage full-thickness hinge flap technique for a large conchal bowl defect.
The Technique
A 77-year-old man was referred to our dermatology clinic by an outside dermatologist for Mohs micrographic surgery of a biopsy-proven cutaneous squamous cell carcinoma on the right conchal bowl measuring 1.1×2.1 cm and extending to the edge of the external auditory canal (EAC). The excision was performed that same day and was completed in 2 stages, achieving negative margins and resulting in a full-thickness defect measuring 2.0×3.6 cm that included the posterior auricular sulcus, cavum, antitragus, and proximal EAC (Figure 1). The patient requested a single-stage procedure but emphasized that his main priority was an optimal cosmetic outcome.
To repair this large defect, a full-thickness hinge flap with Burow graft was performed. The hinge-type flap was designed in a triangular fashion emanating at the posterior auricular sulcus adjacent to the posterior aspect of the defect and extending down the lateral neck (Figure 2). The flap was incised and the surrounding tissue was undermined, maintaining a robust pedicle in the center of its body on the superolateral neck. The flap was passed through the posterior aspect of the full-thickness defect and was secured in place with 4-0 polyglactin sutures in a buried interrupted fashion, thereby recreating the anterior portion of the defect. The superficial skin edges were reapproximated using 4-0 and 5-0 polypropylene sutures in a running interrupted fashion. The distal Burow triangle created from closure of the flap’s secondary defect was aggressively thinned and was utilized as a full-thickness graft for the residual postauricular groove defect (Figure 3). At 2 weeks’ follow-up, the patient was healing well with no postoperative issues and the sutures were removed (Figure 4).
Practice Implications
There are many different reconstructive options for conchal bowl defects, including primary repair, wedge excision, composite graft and interpolation flaps with or without cartilage struts, and hinge flaps. Structural support, EAC patency, auricle symmetry, overall auricle size, and re-creation of natural contours were considered when designing the reconstruction of the defect in our patient; however, his main priority was achieving the greatest cosmetic outcome in a single-stage procedure, therefore limiting our reconstruction options.
Wedge excision, in which the residual lobule and inferior helical rim are removed, could have been considered in our patient but would have drastically altered the symmetry of the size of the ears. A folded postauricular flap, as described in the otolaryngology literature, is an interpolation flap based on the posterior auricular artery that was designed for full-thickness defects of the auricle to prevent any posterior pinning.1 This technique may have worked well in our case, but the patient preferred to avoid a multistage procedure. Additionally, the positional symmetry of the ears was maintained despite utilizing a hinge flap, which does not involve takedown of the pedicle. A composite graft from the contralateral ear could be considered for smaller conchal bowl defects but likely would have resulted in graft failure in our patient’s large defect due to its need for rich blood supply to heal and dependence on lateral wound edges. Cartilage struts in conjunction with a flap could have been considered in this scenario for greater structural support, but in our patient’s case, by maintaining the robust pedicle of our flap and having residual superior cartilage, further structural support was not necessary.
A prior case report described a partial and full-thickness defect in a similar location that was repaired with a retroauricular hinge flap, in which a portion of the flap was extensively de-epithelialized to address the varied thicknesses of the surgical defect.2 In our patient, the defect abutted the skin reservoir on the superolateral neck, and therefore no de-epithelialization was required as the entire epithelialized portion was utilized to recreate the anterior aspect of the defect. Postauricular hinge-type flaps are a reliable, single-stage surgical alternative to the 2-stage folded postauricular interpolation flap when reconstructing large conchal bowl defects. For small full-thickness defects of the ear, a composite graft may be considered; however, blood supply and other nutritional requirements limit this option for large full-thickness defects.
Practice Gap
Large full-thickness conchal bowl defects often pose a reconstructive challenge. Maintaining the shape and structural integrity of the concha is fundamental for optimal cosmetic and functional outcomes. Prior reports have suggested wedge excisions, composite grafts, interpolation flaps with or without cartilage struts, and hinge flaps as possible options for reconstruction.1-3 However, patients with large defects who prefer single-stage reconstruction procedures present a unique challenge. Herein, we describe a single-stage full-thickness hinge flap technique for a large conchal bowl defect.
The Technique
A 77-year-old man was referred to our dermatology clinic by an outside dermatologist for Mohs micrographic surgery of a biopsy-proven cutaneous squamous cell carcinoma on the right conchal bowl measuring 1.1×2.1 cm and extending to the edge of the external auditory canal (EAC). The excision was performed that same day and was completed in 2 stages, achieving negative margins and resulting in a full-thickness defect measuring 2.0×3.6 cm that included the posterior auricular sulcus, cavum, antitragus, and proximal EAC (Figure 1). The patient requested a single-stage procedure but emphasized that his main priority was an optimal cosmetic outcome.
To repair this large defect, a full-thickness hinge flap with Burow graft was performed. The hinge-type flap was designed in a triangular fashion emanating at the posterior auricular sulcus adjacent to the posterior aspect of the defect and extending down the lateral neck (Figure 2). The flap was incised and the surrounding tissue was undermined, maintaining a robust pedicle in the center of its body on the superolateral neck. The flap was passed through the posterior aspect of the full-thickness defect and was secured in place with 4-0 polyglactin sutures in a buried interrupted fashion, thereby recreating the anterior portion of the defect. The superficial skin edges were reapproximated using 4-0 and 5-0 polypropylene sutures in a running interrupted fashion. The distal Burow triangle created from closure of the flap’s secondary defect was aggressively thinned and was utilized as a full-thickness graft for the residual postauricular groove defect (Figure 3). At 2 weeks’ follow-up, the patient was healing well with no postoperative issues and the sutures were removed (Figure 4).
Practice Implications
There are many different reconstructive options for conchal bowl defects, including primary repair, wedge excision, composite graft and interpolation flaps with or without cartilage struts, and hinge flaps. Structural support, EAC patency, auricle symmetry, overall auricle size, and re-creation of natural contours were considered when designing the reconstruction of the defect in our patient; however, his main priority was achieving the greatest cosmetic outcome in a single-stage procedure, therefore limiting our reconstruction options.
Wedge excision, in which the residual lobule and inferior helical rim are removed, could have been considered in our patient but would have drastically altered the symmetry of the size of the ears. A folded postauricular flap, as described in the otolaryngology literature, is an interpolation flap based on the posterior auricular artery that was designed for full-thickness defects of the auricle to prevent any posterior pinning.1 This technique may have worked well in our case, but the patient preferred to avoid a multistage procedure. Additionally, the positional symmetry of the ears was maintained despite utilizing a hinge flap, which does not involve takedown of the pedicle. A composite graft from the contralateral ear could be considered for smaller conchal bowl defects but likely would have resulted in graft failure in our patient’s large defect due to its need for rich blood supply to heal and dependence on lateral wound edges. Cartilage struts in conjunction with a flap could have been considered in this scenario for greater structural support, but in our patient’s case, by maintaining the robust pedicle of our flap and having residual superior cartilage, further structural support was not necessary.
A prior case report described a partial and full-thickness defect in a similar location that was repaired with a retroauricular hinge flap, in which a portion of the flap was extensively de-epithelialized to address the varied thicknesses of the surgical defect.2 In our patient, the defect abutted the skin reservoir on the superolateral neck, and therefore no de-epithelialization was required as the entire epithelialized portion was utilized to recreate the anterior aspect of the defect. Postauricular hinge-type flaps are a reliable, single-stage surgical alternative to the 2-stage folded postauricular interpolation flap when reconstructing large conchal bowl defects. For small full-thickness defects of the ear, a composite graft may be considered; however, blood supply and other nutritional requirements limit this option for large full-thickness defects.
- Roche AM, Griffin M, Shelton R, et al. The folded postauricular flap: a novel approach to reconstruction of large full thickness defects of the conchal bowl. Am J Otolaryngol. 2017;38:706-709. doi:10.1016 /j.amjoto.2017.09.006
- Klein JC, Nijhawan RI. Retroauricular hinge flaps for full-thickness conchal bowl defects. J Am Acad Dermatol. 2024;90:E71-E72. doi:10.1016/j.jaad.2022.10.056
- Pickrell BB, Hughes CD, Maricevich RS. Partial ear defects. Semin Plast Surg. 2017 Aug;31:134-140. doi:10.1055/s-0037-1603968.
- Roche AM, Griffin M, Shelton R, et al. The folded postauricular flap: a novel approach to reconstruction of large full thickness defects of the conchal bowl. Am J Otolaryngol. 2017;38:706-709. doi:10.1016 /j.amjoto.2017.09.006
- Klein JC, Nijhawan RI. Retroauricular hinge flaps for full-thickness conchal bowl defects. J Am Acad Dermatol. 2024;90:E71-E72. doi:10.1016/j.jaad.2022.10.056
- Pickrell BB, Hughes CD, Maricevich RS. Partial ear defects. Semin Plast Surg. 2017 Aug;31:134-140. doi:10.1055/s-0037-1603968.
Repair of a Large Full-Thickness Conchal Bowl Defect
Repair of a Large Full-Thickness Conchal Bowl Defect

Papulonodules on the Ankle in a Patient with Lung Cancer
Papulonodules on the Ankle in a Patient with Lung Cancer
THE DIAGNOSIS: Pembrolizumab-Induced Eruptive Squamous Proliferation
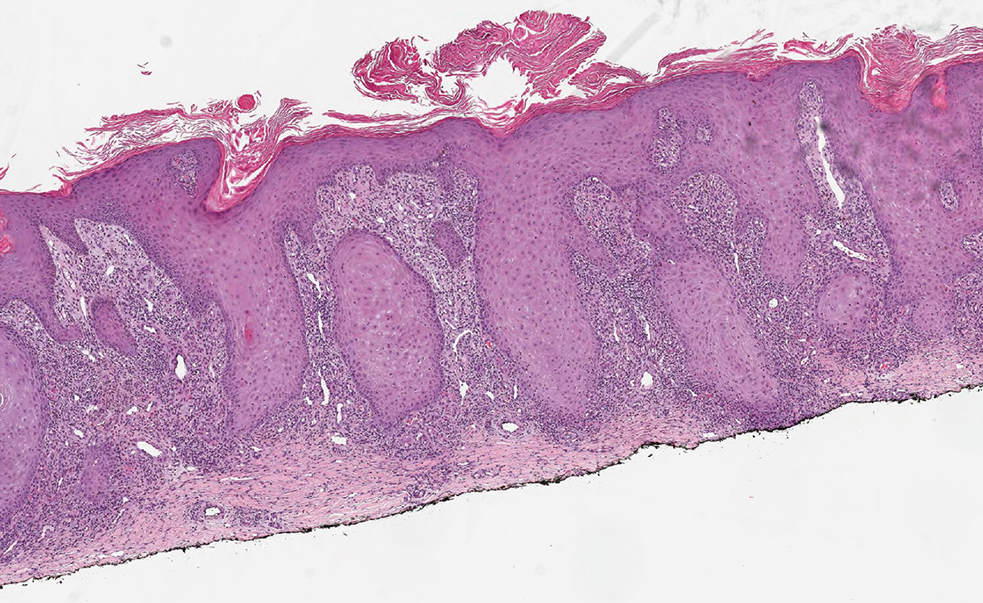
Histopathology showed a broad squamous proliferation with acanthosis of the epidermis. Large glassy keratinocytes were seen with scattered necrotic keratinocytes (Figure), and a dense lichenoid band of inflammation was present subjacent to the proliferation. Notably, no hypergranulosis, remarkable keratinocyte atypia, or increased mitotic figures were seen. Based on the patient’s medical history and biopsy results, a diagnosis of pembrolizumab-induced eruptive squamous proliferation was made. The diagnosis was supported by a growing body of evidence of this type of reaction in patients taking programmed death 1 (PD-1) inhibitors.1,2 Conservative treatment with clobetasol ointment 0.05% was initiated with complete resolution of the lesions at the 2-month follow-up appointment. Other common treatments include topical steroids, injected corticosteroids, or cryosurgery to locally control the inflammation and atypical proliferation of cells.3
Pembrolizumab is a humanized IgG4 monoclonal antibody targeting the PD-1 receptor that has been utilized for its antitumor activity against various cancers, including unresectable and metastatic melanoma, head and neck cancers, and non–small cell lung cancer (NSCLC).1,4,5 While this drug has extended the lives of many patients with cancer, there are adverse reactions associated with PD-1 inhibitors (eg, pembrolizumab, nivolumab). Skin toxicity to PD-1 inhibitors is the one of most common immune-mediated reactions worldwide, occurring in approximately 30% of patients.6,7 Reactions can occur while a patient is taking the inciting drug and can continue up to 2 months after treatment discontinuation.8 Skin reactions associated with PD-1 inhibitors vary from lichenoid reactions and vitiligolike patches to psoriasis or eczema flares and are organized into 4 categories: inflammatory, immunobullous, alteration of keratinocytes, and alteration of melanocytes.9 Our patient demonstrated alteration of keratinocytes, which is characterized by overlapping features of hypertrophic lichen planus and early keratoacanthoma.
The differential diagnoses for pembrolizumab-induced eruptive squamous proliferation include squamous cell carcinoma (SCC), psoriasis, hypertrophic lichen planus, and cutaneous metastasis of NSCLC. Hypertrophic lupus erythematosus also is a well-documented reaction to use of immune-checkpoint inhibitors.10 Direct immunofluorescence could have helped differentiate hypertrophic lupus erythematosus from an eruptive squamous proliferation in our patient; however, due to her response to treatment, no additional workup was done.
Squamous cell carcinoma, which is the most common type of skin cancer in Black patients in the United States,11 has been shown to manifest after a PD-1 inhibitor is taken.12 Although it typically has a more chronic persistent course, the clinical appearance of SCC can be similar to the findings seen in our patient. Histologically, SCC may demonstrate necrosis, but the atypical proliferations will invade the dermis—a feature not seen in our patient’s histopathology.13
Lichen planus (LP) is an eruptive immune reaction of violaceous polygonal papules and plaques commonly seen on the ankles14 that has been shown to be an adverse effect of pembrolizumab.15 There are several subtypes of LP including hypertrophic versions, which can appear clinically similar to the findings seen in our patient. On dermoscopy, the classic finding of white lines, known as Wickham striae, is seen in all subtypes and can help diagnose this pathologic process. Under the microscope, LP can manifest with hyperkeratosis without parakeratosis, irregular thickening of the stratum granulosum, sawtooth rete ridges, and destruction of the basal layer.14
Psoriasis also has been shown to be exacerbated by anti–PD-1 therapy, although the majority of patients diagnosed with PD-1–induced psoriasis have a personal or family history of the disease.6 Clinically, psoriasis can have a hyperpigmented or violaceous appearance in patients with skin of color.16 The histopathology of psoriasis typically reveals confluent parakeratosis, neutrophils in the stratum corneum, regular acanthosis, thinning of the suprapapillary plates, and vessels in the dermal papillae.17
Although cutaneous metastasis of NSCLC may appear clinically similar to the current case, it is one of the rarer organ sites of metastasis for lung cancer.18 In our patient, biopsy quickly ruled out this diagnosis. If it had been a site of metastasis, histopathology would have shown a dermal-based proliferation of dysplastic cells without epidermal connection.19
It is important for dermatologists to recognize eruptive squamous proliferations associated with pembrolizumab, as they often respond to conservative treatment and typically do not require dose reduction or discontinuation of the inciting drug.
- Freshwater T, Kondic A, Ahamadi M, et al. Evaluation of dosing strategy for pembrolizumab for oncology indications. J Immunother Cancer. 2017;5:43. doi:10.1186/s40425-017-0242-5
- Preti BTB, Pencz A, Cowger JJM, et al. Skin deep: a fascinating case report of immunotherapy-triggered, treatment-refractory autoimmune lichen planus and keratoacanthoma. Case Rep Oncol. 2021;14: 1189-1193. doi:10.1159/000518313
- Fradet M, Sibaud V, Tournier E, et al. Multiple keratoacanthoma-like lesions in a patient treated with pembrolizumab. Acta Derm Venereol. 2019;99:1301-1302. doi:10.2340/00015555-3301
- Flynn JP, Gerriets V. Pembrolizumab. StatPearls [Internet]. StatPearls Publishing; 2023. Updated June 26, 2023. Accessed April 2, 2025.
- Antonov NK, Nair KG, Halasz CL. Transient eruptive keratoacanthomas associated with Nivolumab. JAAD Case Rep. 2019;5:342-345. doi:10.1016/j.jdcr.2019.01.025
- Voudouri D, Nikolaou V, Laschos K, et al. Anti-Pd1/Pdl1 induced psoriasis. Curr Probl Cancer. 2017;41:407-412. doi:10.1016 /j.currproblcancer.2017.10.003
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25. doi:10.1016/j.ejca.2016.02.010
- Coscarart A, Martel J, Lee MP, et al. Pembrolizumab-induced pseudoepitheliomatous eruption consistent with hypertrophic lichen planus. J Cutan Pathol. 2020;47:275-279. doi:10.1111/cup.13587
- Curry JL, Tetzlaff MT, Nagarajan P, et al. Diverse types of dermatologic toxicities from immune checkpoint blockade therapy. J Cutan Pathol. 2017;44:158-176. doi:10.1111/cup.12858
- Vitzthum von Eckstaedt H, Singh A, Reid P, et al. Immune checkpoint inhibitors and lupus erythematosus. Pharmaceuticals (Basel). 2024;2:15;17. doi:10.3390/ph17020252
- Halder RM, Bridgeman-Shah S. Skin cancer in African Americans. Cancer. 1995;75:667-673.
- Vu M, Chapman S, Lenz B, et al. Squamous cell carcinoma or squamous proliferation associated with nivolumab treatment for metastatic melanoma. Dermatol Online J. 2022;6:28. doi:10.5070/d328357786
- Howell JY, Ramsey ML. Squamous cell skin cancer. StatPearls [Internet]. StatPearls Publishing; 2024. Updated July 2, 2024. Accessed April 2, 2025.
- Arnold DL, Krishnamurthy K. Lichen planus. StatPearls [Internet]. StatPearls Publishing; 2024. Updated October 29, 2024. Accessed April 2, 2025.
- Yamashita A, Akasaka E, Nakano H, et al. Pembrolizumab-induced lichen planus on the scalp of a patient with non-small-cell lung carcinoma. Case Rep Dermatol. 2021;13:487-491. doi:10.1159/000519486
- Alexis AF, Blackcloud P. Psoriasis in skin of color: epidemiology, genetics, clinical presentation, and treatment nuances. J Clin Aesthet Dermatol. 2014;7:16-24.
- Murphy M, Kerr P, Grant-Kels JM. The histopathologic spectrum of psoriasis. Clin Dermatol. 2007;25:524-528. doi:10.1016 /j.clindermatol.2007.08.005.
- Hidaka T, Ishii Y, Kitamura S. Clinical features of skin metastasis from lung cancer. Intern Med. 1996;35:459-462. doi:10.2169 /internalmedicine.35.459.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613–620. doi:10.1001/archderm.143.5.613
THE DIAGNOSIS: Pembrolizumab-Induced Eruptive Squamous Proliferation
Histopathology showed a broad squamous proliferation with acanthosis of the epidermis. Large glassy keratinocytes were seen with scattered necrotic keratinocytes (Figure), and a dense lichenoid band of inflammation was present subjacent to the proliferation. Notably, no hypergranulosis, remarkable keratinocyte atypia, or increased mitotic figures were seen. Based on the patient’s medical history and biopsy results, a diagnosis of pembrolizumab-induced eruptive squamous proliferation was made. The diagnosis was supported by a growing body of evidence of this type of reaction in patients taking programmed death 1 (PD-1) inhibitors.1,2 Conservative treatment with clobetasol ointment 0.05% was initiated with complete resolution of the lesions at the 2-month follow-up appointment. Other common treatments include topical steroids, injected corticosteroids, or cryosurgery to locally control the inflammation and atypical proliferation of cells.3
Pembrolizumab is a humanized IgG4 monoclonal antibody targeting the PD-1 receptor that has been utilized for its antitumor activity against various cancers, including unresectable and metastatic melanoma, head and neck cancers, and non–small cell lung cancer (NSCLC).1,4,5 While this drug has extended the lives of many patients with cancer, there are adverse reactions associated with PD-1 inhibitors (eg, pembrolizumab, nivolumab). Skin toxicity to PD-1 inhibitors is the one of most common immune-mediated reactions worldwide, occurring in approximately 30% of patients.6,7 Reactions can occur while a patient is taking the inciting drug and can continue up to 2 months after treatment discontinuation.8 Skin reactions associated with PD-1 inhibitors vary from lichenoid reactions and vitiligolike patches to psoriasis or eczema flares and are organized into 4 categories: inflammatory, immunobullous, alteration of keratinocytes, and alteration of melanocytes.9 Our patient demonstrated alteration of keratinocytes, which is characterized by overlapping features of hypertrophic lichen planus and early keratoacanthoma.
The differential diagnoses for pembrolizumab-induced eruptive squamous proliferation include squamous cell carcinoma (SCC), psoriasis, hypertrophic lichen planus, and cutaneous metastasis of NSCLC. Hypertrophic lupus erythematosus also is a well-documented reaction to use of immune-checkpoint inhibitors.10 Direct immunofluorescence could have helped differentiate hypertrophic lupus erythematosus from an eruptive squamous proliferation in our patient; however, due to her response to treatment, no additional workup was done.
Squamous cell carcinoma, which is the most common type of skin cancer in Black patients in the United States,11 has been shown to manifest after a PD-1 inhibitor is taken.12 Although it typically has a more chronic persistent course, the clinical appearance of SCC can be similar to the findings seen in our patient. Histologically, SCC may demonstrate necrosis, but the atypical proliferations will invade the dermis—a feature not seen in our patient’s histopathology.13
Lichen planus (LP) is an eruptive immune reaction of violaceous polygonal papules and plaques commonly seen on the ankles14 that has been shown to be an adverse effect of pembrolizumab.15 There are several subtypes of LP including hypertrophic versions, which can appear clinically similar to the findings seen in our patient. On dermoscopy, the classic finding of white lines, known as Wickham striae, is seen in all subtypes and can help diagnose this pathologic process. Under the microscope, LP can manifest with hyperkeratosis without parakeratosis, irregular thickening of the stratum granulosum, sawtooth rete ridges, and destruction of the basal layer.14
Psoriasis also has been shown to be exacerbated by anti–PD-1 therapy, although the majority of patients diagnosed with PD-1–induced psoriasis have a personal or family history of the disease.6 Clinically, psoriasis can have a hyperpigmented or violaceous appearance in patients with skin of color.16 The histopathology of psoriasis typically reveals confluent parakeratosis, neutrophils in the stratum corneum, regular acanthosis, thinning of the suprapapillary plates, and vessels in the dermal papillae.17
Although cutaneous metastasis of NSCLC may appear clinically similar to the current case, it is one of the rarer organ sites of metastasis for lung cancer.18 In our patient, biopsy quickly ruled out this diagnosis. If it had been a site of metastasis, histopathology would have shown a dermal-based proliferation of dysplastic cells without epidermal connection.19
It is important for dermatologists to recognize eruptive squamous proliferations associated with pembrolizumab, as they often respond to conservative treatment and typically do not require dose reduction or discontinuation of the inciting drug.
THE DIAGNOSIS: Pembrolizumab-Induced Eruptive Squamous Proliferation
Histopathology showed a broad squamous proliferation with acanthosis of the epidermis. Large glassy keratinocytes were seen with scattered necrotic keratinocytes (Figure), and a dense lichenoid band of inflammation was present subjacent to the proliferation. Notably, no hypergranulosis, remarkable keratinocyte atypia, or increased mitotic figures were seen. Based on the patient’s medical history and biopsy results, a diagnosis of pembrolizumab-induced eruptive squamous proliferation was made. The diagnosis was supported by a growing body of evidence of this type of reaction in patients taking programmed death 1 (PD-1) inhibitors.1,2 Conservative treatment with clobetasol ointment 0.05% was initiated with complete resolution of the lesions at the 2-month follow-up appointment. Other common treatments include topical steroids, injected corticosteroids, or cryosurgery to locally control the inflammation and atypical proliferation of cells.3
Pembrolizumab is a humanized IgG4 monoclonal antibody targeting the PD-1 receptor that has been utilized for its antitumor activity against various cancers, including unresectable and metastatic melanoma, head and neck cancers, and non–small cell lung cancer (NSCLC).1,4,5 While this drug has extended the lives of many patients with cancer, there are adverse reactions associated with PD-1 inhibitors (eg, pembrolizumab, nivolumab). Skin toxicity to PD-1 inhibitors is the one of most common immune-mediated reactions worldwide, occurring in approximately 30% of patients.6,7 Reactions can occur while a patient is taking the inciting drug and can continue up to 2 months after treatment discontinuation.8 Skin reactions associated with PD-1 inhibitors vary from lichenoid reactions and vitiligolike patches to psoriasis or eczema flares and are organized into 4 categories: inflammatory, immunobullous, alteration of keratinocytes, and alteration of melanocytes.9 Our patient demonstrated alteration of keratinocytes, which is characterized by overlapping features of hypertrophic lichen planus and early keratoacanthoma.
The differential diagnoses for pembrolizumab-induced eruptive squamous proliferation include squamous cell carcinoma (SCC), psoriasis, hypertrophic lichen planus, and cutaneous metastasis of NSCLC. Hypertrophic lupus erythematosus also is a well-documented reaction to use of immune-checkpoint inhibitors.10 Direct immunofluorescence could have helped differentiate hypertrophic lupus erythematosus from an eruptive squamous proliferation in our patient; however, due to her response to treatment, no additional workup was done.
Squamous cell carcinoma, which is the most common type of skin cancer in Black patients in the United States,11 has been shown to manifest after a PD-1 inhibitor is taken.12 Although it typically has a more chronic persistent course, the clinical appearance of SCC can be similar to the findings seen in our patient. Histologically, SCC may demonstrate necrosis, but the atypical proliferations will invade the dermis—a feature not seen in our patient’s histopathology.13
Lichen planus (LP) is an eruptive immune reaction of violaceous polygonal papules and plaques commonly seen on the ankles14 that has been shown to be an adverse effect of pembrolizumab.15 There are several subtypes of LP including hypertrophic versions, which can appear clinically similar to the findings seen in our patient. On dermoscopy, the classic finding of white lines, known as Wickham striae, is seen in all subtypes and can help diagnose this pathologic process. Under the microscope, LP can manifest with hyperkeratosis without parakeratosis, irregular thickening of the stratum granulosum, sawtooth rete ridges, and destruction of the basal layer.14
Psoriasis also has been shown to be exacerbated by anti–PD-1 therapy, although the majority of patients diagnosed with PD-1–induced psoriasis have a personal or family history of the disease.6 Clinically, psoriasis can have a hyperpigmented or violaceous appearance in patients with skin of color.16 The histopathology of psoriasis typically reveals confluent parakeratosis, neutrophils in the stratum corneum, regular acanthosis, thinning of the suprapapillary plates, and vessels in the dermal papillae.17
Although cutaneous metastasis of NSCLC may appear clinically similar to the current case, it is one of the rarer organ sites of metastasis for lung cancer.18 In our patient, biopsy quickly ruled out this diagnosis. If it had been a site of metastasis, histopathology would have shown a dermal-based proliferation of dysplastic cells without epidermal connection.19
It is important for dermatologists to recognize eruptive squamous proliferations associated with pembrolizumab, as they often respond to conservative treatment and typically do not require dose reduction or discontinuation of the inciting drug.
- Freshwater T, Kondic A, Ahamadi M, et al. Evaluation of dosing strategy for pembrolizumab for oncology indications. J Immunother Cancer. 2017;5:43. doi:10.1186/s40425-017-0242-5
- Preti BTB, Pencz A, Cowger JJM, et al. Skin deep: a fascinating case report of immunotherapy-triggered, treatment-refractory autoimmune lichen planus and keratoacanthoma. Case Rep Oncol. 2021;14: 1189-1193. doi:10.1159/000518313
- Fradet M, Sibaud V, Tournier E, et al. Multiple keratoacanthoma-like lesions in a patient treated with pembrolizumab. Acta Derm Venereol. 2019;99:1301-1302. doi:10.2340/00015555-3301
- Flynn JP, Gerriets V. Pembrolizumab. StatPearls [Internet]. StatPearls Publishing; 2023. Updated June 26, 2023. Accessed April 2, 2025.
- Antonov NK, Nair KG, Halasz CL. Transient eruptive keratoacanthomas associated with Nivolumab. JAAD Case Rep. 2019;5:342-345. doi:10.1016/j.jdcr.2019.01.025
- Voudouri D, Nikolaou V, Laschos K, et al. Anti-Pd1/Pdl1 induced psoriasis. Curr Probl Cancer. 2017;41:407-412. doi:10.1016 /j.currproblcancer.2017.10.003
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25. doi:10.1016/j.ejca.2016.02.010
- Coscarart A, Martel J, Lee MP, et al. Pembrolizumab-induced pseudoepitheliomatous eruption consistent with hypertrophic lichen planus. J Cutan Pathol. 2020;47:275-279. doi:10.1111/cup.13587
- Curry JL, Tetzlaff MT, Nagarajan P, et al. Diverse types of dermatologic toxicities from immune checkpoint blockade therapy. J Cutan Pathol. 2017;44:158-176. doi:10.1111/cup.12858
- Vitzthum von Eckstaedt H, Singh A, Reid P, et al. Immune checkpoint inhibitors and lupus erythematosus. Pharmaceuticals (Basel). 2024;2:15;17. doi:10.3390/ph17020252
- Halder RM, Bridgeman-Shah S. Skin cancer in African Americans. Cancer. 1995;75:667-673.
- Vu M, Chapman S, Lenz B, et al. Squamous cell carcinoma or squamous proliferation associated with nivolumab treatment for metastatic melanoma. Dermatol Online J. 2022;6:28. doi:10.5070/d328357786
- Howell JY, Ramsey ML. Squamous cell skin cancer. StatPearls [Internet]. StatPearls Publishing; 2024. Updated July 2, 2024. Accessed April 2, 2025.
- Arnold DL, Krishnamurthy K. Lichen planus. StatPearls [Internet]. StatPearls Publishing; 2024. Updated October 29, 2024. Accessed April 2, 2025.
- Yamashita A, Akasaka E, Nakano H, et al. Pembrolizumab-induced lichen planus on the scalp of a patient with non-small-cell lung carcinoma. Case Rep Dermatol. 2021;13:487-491. doi:10.1159/000519486
- Alexis AF, Blackcloud P. Psoriasis in skin of color: epidemiology, genetics, clinical presentation, and treatment nuances. J Clin Aesthet Dermatol. 2014;7:16-24.
- Murphy M, Kerr P, Grant-Kels JM. The histopathologic spectrum of psoriasis. Clin Dermatol. 2007;25:524-528. doi:10.1016 /j.clindermatol.2007.08.005.
- Hidaka T, Ishii Y, Kitamura S. Clinical features of skin metastasis from lung cancer. Intern Med. 1996;35:459-462. doi:10.2169 /internalmedicine.35.459.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613–620. doi:10.1001/archderm.143.5.613
- Freshwater T, Kondic A, Ahamadi M, et al. Evaluation of dosing strategy for pembrolizumab for oncology indications. J Immunother Cancer. 2017;5:43. doi:10.1186/s40425-017-0242-5
- Preti BTB, Pencz A, Cowger JJM, et al. Skin deep: a fascinating case report of immunotherapy-triggered, treatment-refractory autoimmune lichen planus and keratoacanthoma. Case Rep Oncol. 2021;14: 1189-1193. doi:10.1159/000518313
- Fradet M, Sibaud V, Tournier E, et al. Multiple keratoacanthoma-like lesions in a patient treated with pembrolizumab. Acta Derm Venereol. 2019;99:1301-1302. doi:10.2340/00015555-3301
- Flynn JP, Gerriets V. Pembrolizumab. StatPearls [Internet]. StatPearls Publishing; 2023. Updated June 26, 2023. Accessed April 2, 2025.
- Antonov NK, Nair KG, Halasz CL. Transient eruptive keratoacanthomas associated with Nivolumab. JAAD Case Rep. 2019;5:342-345. doi:10.1016/j.jdcr.2019.01.025
- Voudouri D, Nikolaou V, Laschos K, et al. Anti-Pd1/Pdl1 induced psoriasis. Curr Probl Cancer. 2017;41:407-412. doi:10.1016 /j.currproblcancer.2017.10.003
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25. doi:10.1016/j.ejca.2016.02.010
- Coscarart A, Martel J, Lee MP, et al. Pembrolizumab-induced pseudoepitheliomatous eruption consistent with hypertrophic lichen planus. J Cutan Pathol. 2020;47:275-279. doi:10.1111/cup.13587
- Curry JL, Tetzlaff MT, Nagarajan P, et al. Diverse types of dermatologic toxicities from immune checkpoint blockade therapy. J Cutan Pathol. 2017;44:158-176. doi:10.1111/cup.12858
- Vitzthum von Eckstaedt H, Singh A, Reid P, et al. Immune checkpoint inhibitors and lupus erythematosus. Pharmaceuticals (Basel). 2024;2:15;17. doi:10.3390/ph17020252
- Halder RM, Bridgeman-Shah S. Skin cancer in African Americans. Cancer. 1995;75:667-673.
- Vu M, Chapman S, Lenz B, et al. Squamous cell carcinoma or squamous proliferation associated with nivolumab treatment for metastatic melanoma. Dermatol Online J. 2022;6:28. doi:10.5070/d328357786
- Howell JY, Ramsey ML. Squamous cell skin cancer. StatPearls [Internet]. StatPearls Publishing; 2024. Updated July 2, 2024. Accessed April 2, 2025.
- Arnold DL, Krishnamurthy K. Lichen planus. StatPearls [Internet]. StatPearls Publishing; 2024. Updated October 29, 2024. Accessed April 2, 2025.
- Yamashita A, Akasaka E, Nakano H, et al. Pembrolizumab-induced lichen planus on the scalp of a patient with non-small-cell lung carcinoma. Case Rep Dermatol. 2021;13:487-491. doi:10.1159/000519486
- Alexis AF, Blackcloud P. Psoriasis in skin of color: epidemiology, genetics, clinical presentation, and treatment nuances. J Clin Aesthet Dermatol. 2014;7:16-24.
- Murphy M, Kerr P, Grant-Kels JM. The histopathologic spectrum of psoriasis. Clin Dermatol. 2007;25:524-528. doi:10.1016 /j.clindermatol.2007.08.005.
- Hidaka T, Ishii Y, Kitamura S. Clinical features of skin metastasis from lung cancer. Intern Med. 1996;35:459-462. doi:10.2169 /internalmedicine.35.459.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613–620. doi:10.1001/archderm.143.5.613
Papulonodules on the Ankle in a Patient with Lung Cancer
Papulonodules on the Ankle in a Patient with Lung Cancer
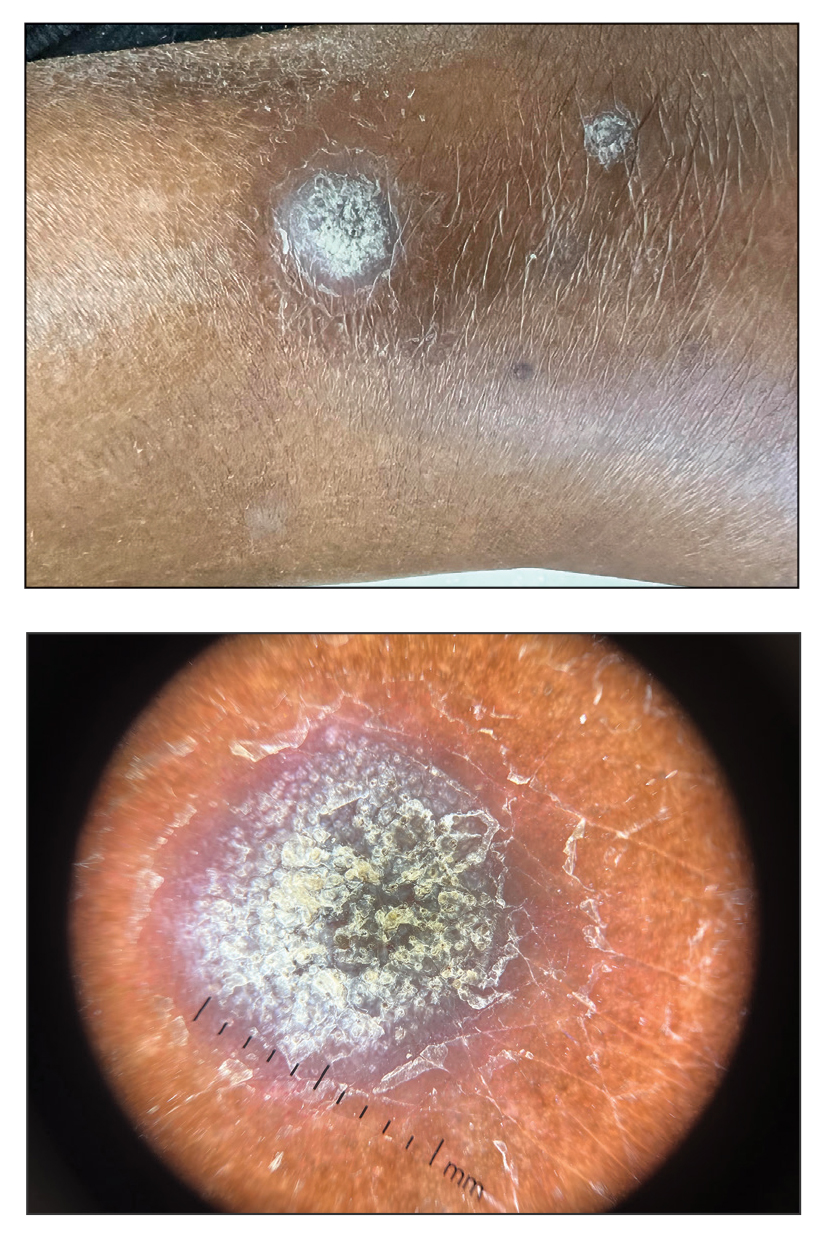
A 75-year-old woman presented to the dermatology department with well-circumscribed, round, hyperkeratotic papulonodules on the ankle of 3 months’ duration (top). The papulonodules also were evaluated by dermoscopy, which highlighted in greater detail the hyperkeratosis seen grossly (bottom). The patient had a history of chronic obstructive pulmonary disease and metastatic lung cancer and had been taking pembrolizumab for the past 2 years. The lesions initially appeared on the medial right foot and slowly spread proximally. Most of the lesions resolved spontaneously except for 2 on the right ankle. At the current presentation, one lesion was slightly tender to palpation, but both were otherwise asymptomatic. A lesion was biopsied and sent for dermatopathologic evaluation.

Acral Erythema, Edema, and Scaly Plaques in a Patient With Polyneuropathy
Acral Erythema, Edema, and Scaly Plaques in a Patient With Polyneuropathy
THE DIAGNOSIS: Borderline-Borderline Leprosy With Type 1 Lepra Reaction
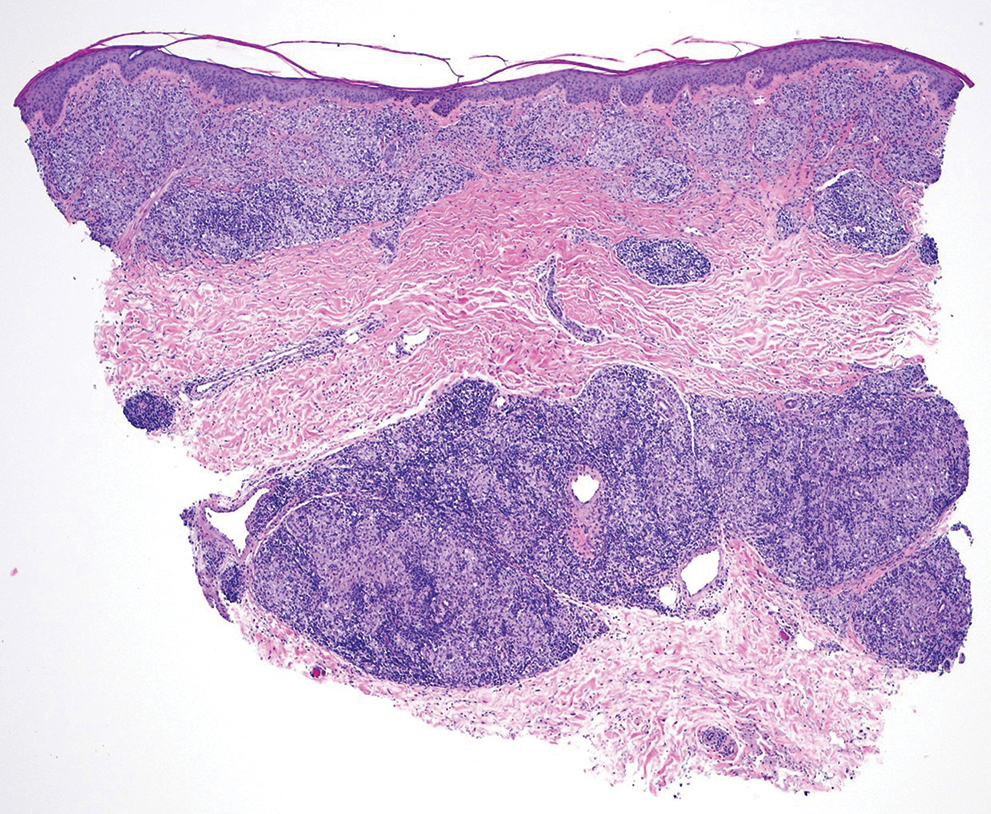
Punch biopsies from plaques on the right elbow and right shin revealed diffuse granulomatous dermatitis (Figure 1) with a narrow Grenz zone in the superficial dermis. The upper dermis contained a dense bandlike infiltrate of histiocytes with abundant foamy-gray cytoplasm and a moderate admixture of lymphocytes. The mid and deep dermis contained a nodular, perivascular, periadnexal, and perineural infiltrate of histiocytes and a dense admixture of lymphocytes. Periodic acid-Schiff and Gram stains were negative for microorganisms. Fite stain was positive for numerous organisms in histiocytes and small dermal nerves (Figure 2). These findings and the clinical examination confirmed a diagnosis of borderline-borderline leprosy with type 1 lepra reaction. The patient was started on dapsone 100 mg, rifampin 600 mg, and clofazimine 100 mg once daily and experienced clinical improvement within 6 months.
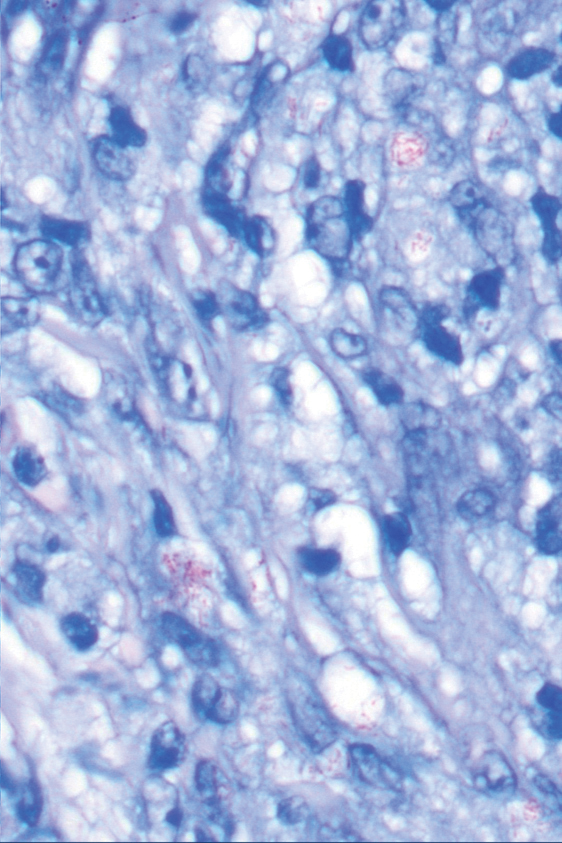
The World Health Organization reported more than 200,000 new leprosy cases globally in 2019, with most occurring in India, Brazil, and Indonesia.1 About 150 to 250 new cases are detected in the United States annually.1 The Ridley-Jopling classification of leprosy divides the condition into 5 categories: tuberculoid, borderline tuberculoid, borderline-borderline (BB), borderline lepromatous, and lepromatous. At one end of the spectrum, tuberculoid leprosy—a predominant Th1 immune response mediated by CD4 lymphocytes, interleukin (IL) 2, and interferon gamma2—is characterized by sharply demarcated erythematous and hypopigmented plaques with raised borders and an annular appearance.2,3 Lesions typically have atrophic and hypopigmented centers that often appear in an asymmetric distribution on the arms and legs.2,3 Histologic features include dermal tuberculoid granulomas with epithelioid cells—some located directly beneath the epidermis and others around deep vessels and nerves3—multinucleated Langerhans giant cells, thickened peripheral nerves with intraneural lymphocytic infiltrates, and granulomas with central necrosis. Fite-Faraco staining exhibits few bacteria.2
Lepromatous leprosy occurs in individuals with impaired T-cell immunity, leading to multiple red-brown nodular infiltrates in the skin and mucous membranes.2,3 Lesions typically are symmetric and favor the face and auricle of the ear.2,3 Histologically, there are bluish-gray foamy macrophages that form diffuse or nodular infiltrates with few lymphocytes,2 with a Grenz zone between the epidermis and dermis. Nerves may show lamination of the perineurium resembling an onion skin.2,3 Immunohistochemistry shows predominant CD8-positive infiltrates with a Th2 response and positive IL-4 and IL-10. Fite-Faraco stain shows numerous mycobacteria arranged in clusters and in histiocytes.2
Tuberculoid leprosy is treated with dapsone 100 mg and rifampin 600 mg once daily for 6 months,4 and lepromatous leprosy is treated with dapsone 100 mg, rifampin 600 mg, and clofazimine 50 mg once daily for 12 months.4 The prognosis for both is good with treatment; erythema and induration of skin lesions may improve within a few months, but residual nerve damage is common, especially in those with advanced disease prior to treatment.2 For direct contacts, a single dose of rifampin may be given.4
Borderline-borderline leprosy manifests with numerous asymmetric annular plaques, as seen in our patient (Figure 3). Histology findings can be variable and often overlap with other forms of leprosy. There can be epithelioid granulomas and only a few acid-fast bacilli (AFB) or diffuse histiocytic aggregates with foamy histiocytes containing large numbers of AFB.3 Nerve involvement is variable but can be severe in the setting of type 1 lepra reaction, which was present in our patient. Type 1 lepra reaction—a type IV cell-mediated allergic hypersensitivity reaction to Mycobacterium leprae antigens—manifests clinically with hyperesthesia, erythema, edema, and subsequent scaling.2 It occurs in up to 30% of patients with borderline leprosy, usually within 12 months of treatment initiation.2 Our patient had considerable edema and erythema of the hands and feet (Figure 4) along with extensive polyneuropathy prior to starting therapy.
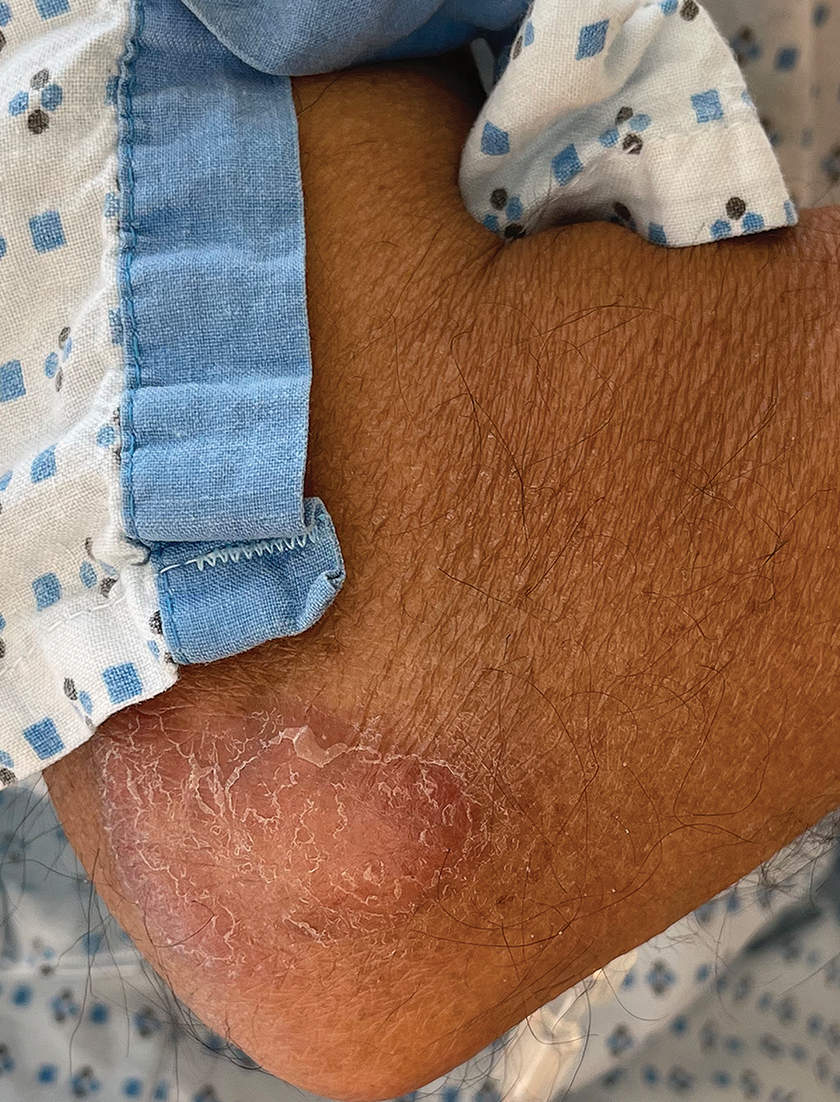
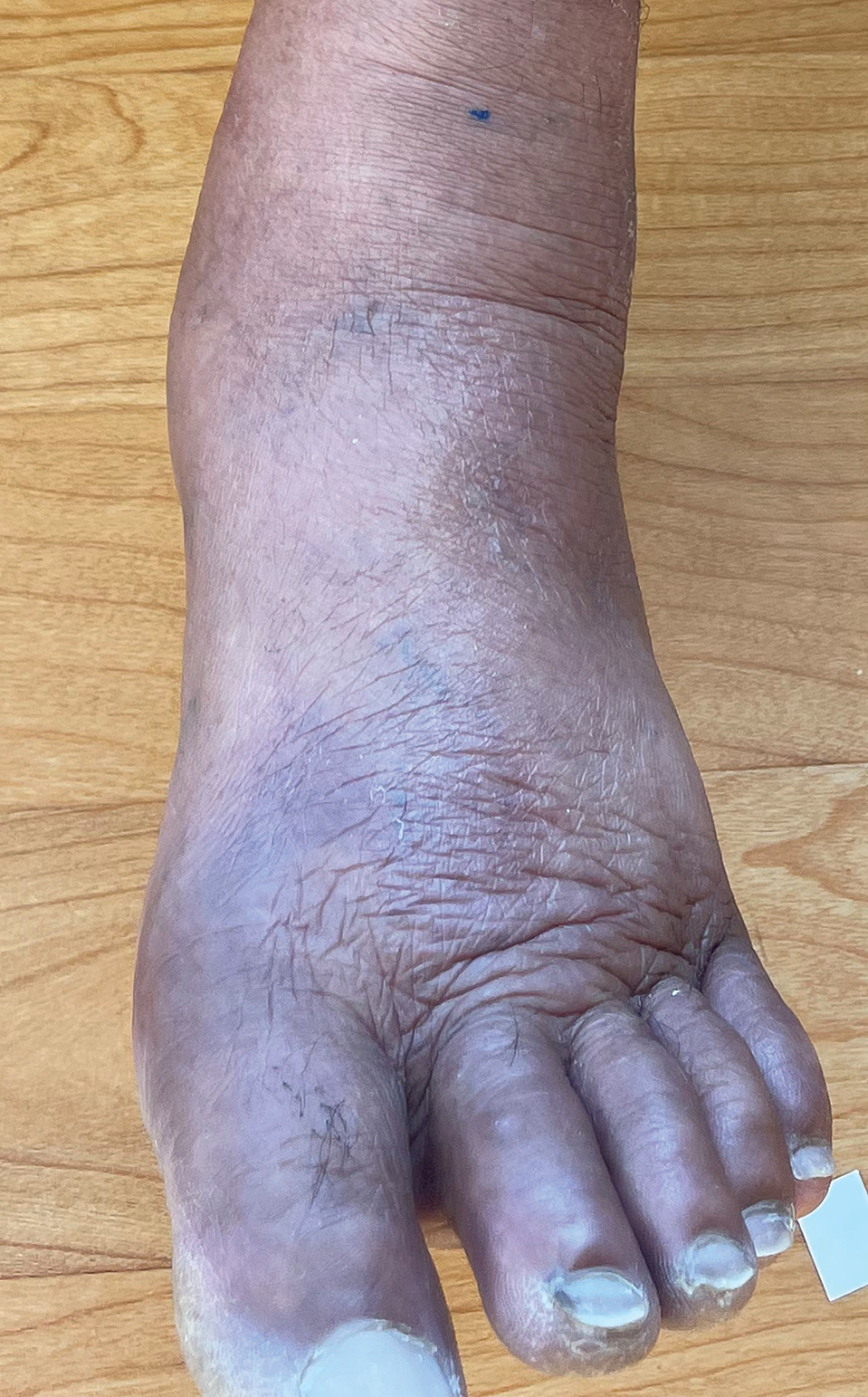
Lucio phenomenon is a rare leprosy reaction found in patients with untreated lepromatous leprosy characterized by erythematous to violaceous macules that lead to ulceronecrotic lesions.5 Histologically, there are many AFB in the vascular endothelium, leukocytoclastic vasculitis, and ischemic epidermal necrosis.5 Our patient did not have ulcerative or necrotic lesions.
The classic skin lesions of psoriasis vulgaris can be described as well-demarcated pink plaques with white or silvery scales that usually are distributed symmetrically and often are found on extensor surfaces.6 Rapidly progressive lesions can be annular with normal skin in the center, mimicking the lesions seen in tuberculoid leprosy. Clinically, both psoriasis and tuberculoid forms of leprosy are sharply demarcated; however, psoriatic lesions often have micaceous overlying scale that is not present in leprosy. Characteristic histologic findings of psoriasis are hyperkeratosis, parakeratosis, and acanthosis of the epidermis with dilated blood vessels and a lymphocytic infiltrate, predominantly into the dermis.7 Psoriatic arthritis has a variable clinical course but tends to emerge 5 to 12 years after initial skin manifestation.8 Classic clinical symptoms include swelling, tenderness, stiffness, and pain in joints and surrounding tissues.8 Other than edema, our patient did not exhibit signs of psoriatic arthritis.
Sarcoidosis is a systemic autoimmune disease characterized by noncaseating epithelioid granulomas affecting various organs, with cutaneous manifestations present in approximately 30% of all cases. Cutaneous manifestations can be variable, including maculopapular lesions, plaques, and nodules.9 Differentiating between cutaneous sarcoidosis and tuberculoid leprosy can be challenging, as both are granulomatous processes; however, histology of sarcoidosis demonstrates noncaseating granulomas in the dermis and/or subcutaneous tissues without AFB9 compared to granulomas with necrotic centers in tuberculoid leprosy.
Cutaneous tuberculosis has variable morphologies. One subtype, lupus vulgaris, can manifest with violaceous, scaly, eroded plaques that could be confused for leprosy. Lupus vulgaris usually results from hematogenous or lymphatic seeding in individuals with high or moderate immunity to M tuberculosis.10
Histologically, the dermis has tuberculoid granulomas containing multinucleated giant cells,10 which can mimic those seen in BB leprosy. Tuberculin skin test results often are positive10; while this test was not performed in our patient, chest radiography was unremarkable, making this diagnosis less likely.
Mycobacterium leprae infections should be considered in a patient with a worsening rash and progressive polyneuropathy. Clinical diagnosis can be challenging due to similarities with other diseases; however, histopathologic findings can help differentiate M leprae from other conditions. This infection is treatable, and early detection can minimize long-term patient morbidity.
- CDC. Hansen’s disease (leprosy). Accessed April 23, 2025. https://www.cdc.gov/leprosy/about/index.html
- Fischer M. Leprosy—an overview of clinical features, diagnosis, and treatment. J Dtsch Dermatol Ges. 2017;15:801-827.
- Maymone MBC, Laughter M, Venkatesh S, et al. Leprosy: clinical aspects and diagnostic techniques. J Am Acad Dermatol. 2020; 83:1-14.
- World Health Organization. Guidelines for the diagnosis, treatment and prevention of leprosy. October 6, 2018. Accessed April 2, 2025. https://www.who.int/publications/i/item/9789290226383
- Frade MAC, Coltro PS, Filho FB, et al. Lucio’s phenomenon: a systematic literature review of definition, clinical features, histopathogenesis and management. Indian J Dermatol Venereol Leprol. 2022;88:464-477.
- Kimmel GW, Lebwohl M. Psoriasis: overview and diagnosis. In: Evidence-Based Psoriasis. Springer International Publishing; 2018:1-16.
- Griffiths CE, Barker JN. Pathogenesis and clinical features of psoriasis. Lancet. 2007;370:263-271.
- Menter A. Psoriasis and psoriatic arthritis overview. Am J Manag Care. 2016;22(8 suppl):S216-S224.
- Wu JH, Imadojemu S, Caplan AS. The evolving landscape of cutaneous sarcoidosis: pathogenic insight, clinical challenges, and new frontiers in therapy. Am J Clin Dermatol. 2022;23:499-514.
- Hill MK, Sanders CV. Cutaneous tuberculosis. Microbiol Spectr. 2017;5:1-6.
THE DIAGNOSIS: Borderline-Borderline Leprosy With Type 1 Lepra Reaction
Punch biopsies from plaques on the right elbow and right shin revealed diffuse granulomatous dermatitis (Figure 1) with a narrow Grenz zone in the superficial dermis. The upper dermis contained a dense bandlike infiltrate of histiocytes with abundant foamy-gray cytoplasm and a moderate admixture of lymphocytes. The mid and deep dermis contained a nodular, perivascular, periadnexal, and perineural infiltrate of histiocytes and a dense admixture of lymphocytes. Periodic acid-Schiff and Gram stains were negative for microorganisms. Fite stain was positive for numerous organisms in histiocytes and small dermal nerves (Figure 2). These findings and the clinical examination confirmed a diagnosis of borderline-borderline leprosy with type 1 lepra reaction. The patient was started on dapsone 100 mg, rifampin 600 mg, and clofazimine 100 mg once daily and experienced clinical improvement within 6 months.
The World Health Organization reported more than 200,000 new leprosy cases globally in 2019, with most occurring in India, Brazil, and Indonesia.1 About 150 to 250 new cases are detected in the United States annually.1 The Ridley-Jopling classification of leprosy divides the condition into 5 categories: tuberculoid, borderline tuberculoid, borderline-borderline (BB), borderline lepromatous, and lepromatous. At one end of the spectrum, tuberculoid leprosy—a predominant Th1 immune response mediated by CD4 lymphocytes, interleukin (IL) 2, and interferon gamma2—is characterized by sharply demarcated erythematous and hypopigmented plaques with raised borders and an annular appearance.2,3 Lesions typically have atrophic and hypopigmented centers that often appear in an asymmetric distribution on the arms and legs.2,3 Histologic features include dermal tuberculoid granulomas with epithelioid cells—some located directly beneath the epidermis and others around deep vessels and nerves3—multinucleated Langerhans giant cells, thickened peripheral nerves with intraneural lymphocytic infiltrates, and granulomas with central necrosis. Fite-Faraco staining exhibits few bacteria.2
Lepromatous leprosy occurs in individuals with impaired T-cell immunity, leading to multiple red-brown nodular infiltrates in the skin and mucous membranes.2,3 Lesions typically are symmetric and favor the face and auricle of the ear.2,3 Histologically, there are bluish-gray foamy macrophages that form diffuse or nodular infiltrates with few lymphocytes,2 with a Grenz zone between the epidermis and dermis. Nerves may show lamination of the perineurium resembling an onion skin.2,3 Immunohistochemistry shows predominant CD8-positive infiltrates with a Th2 response and positive IL-4 and IL-10. Fite-Faraco stain shows numerous mycobacteria arranged in clusters and in histiocytes.2
Tuberculoid leprosy is treated with dapsone 100 mg and rifampin 600 mg once daily for 6 months,4 and lepromatous leprosy is treated with dapsone 100 mg, rifampin 600 mg, and clofazimine 50 mg once daily for 12 months.4 The prognosis for both is good with treatment; erythema and induration of skin lesions may improve within a few months, but residual nerve damage is common, especially in those with advanced disease prior to treatment.2 For direct contacts, a single dose of rifampin may be given.4
Borderline-borderline leprosy manifests with numerous asymmetric annular plaques, as seen in our patient (Figure 3). Histology findings can be variable and often overlap with other forms of leprosy. There can be epithelioid granulomas and only a few acid-fast bacilli (AFB) or diffuse histiocytic aggregates with foamy histiocytes containing large numbers of AFB.3 Nerve involvement is variable but can be severe in the setting of type 1 lepra reaction, which was present in our patient. Type 1 lepra reaction—a type IV cell-mediated allergic hypersensitivity reaction to Mycobacterium leprae antigens—manifests clinically with hyperesthesia, erythema, edema, and subsequent scaling.2 It occurs in up to 30% of patients with borderline leprosy, usually within 12 months of treatment initiation.2 Our patient had considerable edema and erythema of the hands and feet (Figure 4) along with extensive polyneuropathy prior to starting therapy.
Lucio phenomenon is a rare leprosy reaction found in patients with untreated lepromatous leprosy characterized by erythematous to violaceous macules that lead to ulceronecrotic lesions.5 Histologically, there are many AFB in the vascular endothelium, leukocytoclastic vasculitis, and ischemic epidermal necrosis.5 Our patient did not have ulcerative or necrotic lesions.
The classic skin lesions of psoriasis vulgaris can be described as well-demarcated pink plaques with white or silvery scales that usually are distributed symmetrically and often are found on extensor surfaces.6 Rapidly progressive lesions can be annular with normal skin in the center, mimicking the lesions seen in tuberculoid leprosy. Clinically, both psoriasis and tuberculoid forms of leprosy are sharply demarcated; however, psoriatic lesions often have micaceous overlying scale that is not present in leprosy. Characteristic histologic findings of psoriasis are hyperkeratosis, parakeratosis, and acanthosis of the epidermis with dilated blood vessels and a lymphocytic infiltrate, predominantly into the dermis.7 Psoriatic arthritis has a variable clinical course but tends to emerge 5 to 12 years after initial skin manifestation.8 Classic clinical symptoms include swelling, tenderness, stiffness, and pain in joints and surrounding tissues.8 Other than edema, our patient did not exhibit signs of psoriatic arthritis.
Sarcoidosis is a systemic autoimmune disease characterized by noncaseating epithelioid granulomas affecting various organs, with cutaneous manifestations present in approximately 30% of all cases. Cutaneous manifestations can be variable, including maculopapular lesions, plaques, and nodules.9 Differentiating between cutaneous sarcoidosis and tuberculoid leprosy can be challenging, as both are granulomatous processes; however, histology of sarcoidosis demonstrates noncaseating granulomas in the dermis and/or subcutaneous tissues without AFB9 compared to granulomas with necrotic centers in tuberculoid leprosy.
Cutaneous tuberculosis has variable morphologies. One subtype, lupus vulgaris, can manifest with violaceous, scaly, eroded plaques that could be confused for leprosy. Lupus vulgaris usually results from hematogenous or lymphatic seeding in individuals with high or moderate immunity to M tuberculosis.10
Histologically, the dermis has tuberculoid granulomas containing multinucleated giant cells,10 which can mimic those seen in BB leprosy. Tuberculin skin test results often are positive10; while this test was not performed in our patient, chest radiography was unremarkable, making this diagnosis less likely.
Mycobacterium leprae infections should be considered in a patient with a worsening rash and progressive polyneuropathy. Clinical diagnosis can be challenging due to similarities with other diseases; however, histopathologic findings can help differentiate M leprae from other conditions. This infection is treatable, and early detection can minimize long-term patient morbidity.
THE DIAGNOSIS: Borderline-Borderline Leprosy With Type 1 Lepra Reaction
Punch biopsies from plaques on the right elbow and right shin revealed diffuse granulomatous dermatitis (Figure 1) with a narrow Grenz zone in the superficial dermis. The upper dermis contained a dense bandlike infiltrate of histiocytes with abundant foamy-gray cytoplasm and a moderate admixture of lymphocytes. The mid and deep dermis contained a nodular, perivascular, periadnexal, and perineural infiltrate of histiocytes and a dense admixture of lymphocytes. Periodic acid-Schiff and Gram stains were negative for microorganisms. Fite stain was positive for numerous organisms in histiocytes and small dermal nerves (Figure 2). These findings and the clinical examination confirmed a diagnosis of borderline-borderline leprosy with type 1 lepra reaction. The patient was started on dapsone 100 mg, rifampin 600 mg, and clofazimine 100 mg once daily and experienced clinical improvement within 6 months.
The World Health Organization reported more than 200,000 new leprosy cases globally in 2019, with most occurring in India, Brazil, and Indonesia.1 About 150 to 250 new cases are detected in the United States annually.1 The Ridley-Jopling classification of leprosy divides the condition into 5 categories: tuberculoid, borderline tuberculoid, borderline-borderline (BB), borderline lepromatous, and lepromatous. At one end of the spectrum, tuberculoid leprosy—a predominant Th1 immune response mediated by CD4 lymphocytes, interleukin (IL) 2, and interferon gamma2—is characterized by sharply demarcated erythematous and hypopigmented plaques with raised borders and an annular appearance.2,3 Lesions typically have atrophic and hypopigmented centers that often appear in an asymmetric distribution on the arms and legs.2,3 Histologic features include dermal tuberculoid granulomas with epithelioid cells—some located directly beneath the epidermis and others around deep vessels and nerves3—multinucleated Langerhans giant cells, thickened peripheral nerves with intraneural lymphocytic infiltrates, and granulomas with central necrosis. Fite-Faraco staining exhibits few bacteria.2
Lepromatous leprosy occurs in individuals with impaired T-cell immunity, leading to multiple red-brown nodular infiltrates in the skin and mucous membranes.2,3 Lesions typically are symmetric and favor the face and auricle of the ear.2,3 Histologically, there are bluish-gray foamy macrophages that form diffuse or nodular infiltrates with few lymphocytes,2 with a Grenz zone between the epidermis and dermis. Nerves may show lamination of the perineurium resembling an onion skin.2,3 Immunohistochemistry shows predominant CD8-positive infiltrates with a Th2 response and positive IL-4 and IL-10. Fite-Faraco stain shows numerous mycobacteria arranged in clusters and in histiocytes.2
Tuberculoid leprosy is treated with dapsone 100 mg and rifampin 600 mg once daily for 6 months,4 and lepromatous leprosy is treated with dapsone 100 mg, rifampin 600 mg, and clofazimine 50 mg once daily for 12 months.4 The prognosis for both is good with treatment; erythema and induration of skin lesions may improve within a few months, but residual nerve damage is common, especially in those with advanced disease prior to treatment.2 For direct contacts, a single dose of rifampin may be given.4
Borderline-borderline leprosy manifests with numerous asymmetric annular plaques, as seen in our patient (Figure 3). Histology findings can be variable and often overlap with other forms of leprosy. There can be epithelioid granulomas and only a few acid-fast bacilli (AFB) or diffuse histiocytic aggregates with foamy histiocytes containing large numbers of AFB.3 Nerve involvement is variable but can be severe in the setting of type 1 lepra reaction, which was present in our patient. Type 1 lepra reaction—a type IV cell-mediated allergic hypersensitivity reaction to Mycobacterium leprae antigens—manifests clinically with hyperesthesia, erythema, edema, and subsequent scaling.2 It occurs in up to 30% of patients with borderline leprosy, usually within 12 months of treatment initiation.2 Our patient had considerable edema and erythema of the hands and feet (Figure 4) along with extensive polyneuropathy prior to starting therapy.
Lucio phenomenon is a rare leprosy reaction found in patients with untreated lepromatous leprosy characterized by erythematous to violaceous macules that lead to ulceronecrotic lesions.5 Histologically, there are many AFB in the vascular endothelium, leukocytoclastic vasculitis, and ischemic epidermal necrosis.5 Our patient did not have ulcerative or necrotic lesions.
The classic skin lesions of psoriasis vulgaris can be described as well-demarcated pink plaques with white or silvery scales that usually are distributed symmetrically and often are found on extensor surfaces.6 Rapidly progressive lesions can be annular with normal skin in the center, mimicking the lesions seen in tuberculoid leprosy. Clinically, both psoriasis and tuberculoid forms of leprosy are sharply demarcated; however, psoriatic lesions often have micaceous overlying scale that is not present in leprosy. Characteristic histologic findings of psoriasis are hyperkeratosis, parakeratosis, and acanthosis of the epidermis with dilated blood vessels and a lymphocytic infiltrate, predominantly into the dermis.7 Psoriatic arthritis has a variable clinical course but tends to emerge 5 to 12 years after initial skin manifestation.8 Classic clinical symptoms include swelling, tenderness, stiffness, and pain in joints and surrounding tissues.8 Other than edema, our patient did not exhibit signs of psoriatic arthritis.
Sarcoidosis is a systemic autoimmune disease characterized by noncaseating epithelioid granulomas affecting various organs, with cutaneous manifestations present in approximately 30% of all cases. Cutaneous manifestations can be variable, including maculopapular lesions, plaques, and nodules.9 Differentiating between cutaneous sarcoidosis and tuberculoid leprosy can be challenging, as both are granulomatous processes; however, histology of sarcoidosis demonstrates noncaseating granulomas in the dermis and/or subcutaneous tissues without AFB9 compared to granulomas with necrotic centers in tuberculoid leprosy.
Cutaneous tuberculosis has variable morphologies. One subtype, lupus vulgaris, can manifest with violaceous, scaly, eroded plaques that could be confused for leprosy. Lupus vulgaris usually results from hematogenous or lymphatic seeding in individuals with high or moderate immunity to M tuberculosis.10
Histologically, the dermis has tuberculoid granulomas containing multinucleated giant cells,10 which can mimic those seen in BB leprosy. Tuberculin skin test results often are positive10; while this test was not performed in our patient, chest radiography was unremarkable, making this diagnosis less likely.
Mycobacterium leprae infections should be considered in a patient with a worsening rash and progressive polyneuropathy. Clinical diagnosis can be challenging due to similarities with other diseases; however, histopathologic findings can help differentiate M leprae from other conditions. This infection is treatable, and early detection can minimize long-term patient morbidity.
- CDC. Hansen’s disease (leprosy). Accessed April 23, 2025. https://www.cdc.gov/leprosy/about/index.html
- Fischer M. Leprosy—an overview of clinical features, diagnosis, and treatment. J Dtsch Dermatol Ges. 2017;15:801-827.
- Maymone MBC, Laughter M, Venkatesh S, et al. Leprosy: clinical aspects and diagnostic techniques. J Am Acad Dermatol. 2020; 83:1-14.
- World Health Organization. Guidelines for the diagnosis, treatment and prevention of leprosy. October 6, 2018. Accessed April 2, 2025. https://www.who.int/publications/i/item/9789290226383
- Frade MAC, Coltro PS, Filho FB, et al. Lucio’s phenomenon: a systematic literature review of definition, clinical features, histopathogenesis and management. Indian J Dermatol Venereol Leprol. 2022;88:464-477.
- Kimmel GW, Lebwohl M. Psoriasis: overview and diagnosis. In: Evidence-Based Psoriasis. Springer International Publishing; 2018:1-16.
- Griffiths CE, Barker JN. Pathogenesis and clinical features of psoriasis. Lancet. 2007;370:263-271.
- Menter A. Psoriasis and psoriatic arthritis overview. Am J Manag Care. 2016;22(8 suppl):S216-S224.
- Wu JH, Imadojemu S, Caplan AS. The evolving landscape of cutaneous sarcoidosis: pathogenic insight, clinical challenges, and new frontiers in therapy. Am J Clin Dermatol. 2022;23:499-514.
- Hill MK, Sanders CV. Cutaneous tuberculosis. Microbiol Spectr. 2017;5:1-6.
- CDC. Hansen’s disease (leprosy). Accessed April 23, 2025. https://www.cdc.gov/leprosy/about/index.html
- Fischer M. Leprosy—an overview of clinical features, diagnosis, and treatment. J Dtsch Dermatol Ges. 2017;15:801-827.
- Maymone MBC, Laughter M, Venkatesh S, et al. Leprosy: clinical aspects and diagnostic techniques. J Am Acad Dermatol. 2020; 83:1-14.
- World Health Organization. Guidelines for the diagnosis, treatment and prevention of leprosy. October 6, 2018. Accessed April 2, 2025. https://www.who.int/publications/i/item/9789290226383
- Frade MAC, Coltro PS, Filho FB, et al. Lucio’s phenomenon: a systematic literature review of definition, clinical features, histopathogenesis and management. Indian J Dermatol Venereol Leprol. 2022;88:464-477.
- Kimmel GW, Lebwohl M. Psoriasis: overview and diagnosis. In: Evidence-Based Psoriasis. Springer International Publishing; 2018:1-16.
- Griffiths CE, Barker JN. Pathogenesis and clinical features of psoriasis. Lancet. 2007;370:263-271.
- Menter A. Psoriasis and psoriatic arthritis overview. Am J Manag Care. 2016;22(8 suppl):S216-S224.
- Wu JH, Imadojemu S, Caplan AS. The evolving landscape of cutaneous sarcoidosis: pathogenic insight, clinical challenges, and new frontiers in therapy. Am J Clin Dermatol. 2022;23:499-514.
- Hill MK, Sanders CV. Cutaneous tuberculosis. Microbiol Spectr. 2017;5:1-6.
Acral Erythema, Edema, and Scaly Plaques in a Patient With Polyneuropathy
Acral Erythema, Edema, and Scaly Plaques in a Patient With Polyneuropathy
A 67-year-old man presented to his primary care physician with scaly plaques on the extensor surfaces along with distal neuropathy that had been slowly worsening over the past 6 months. The patient was prescribed triamcinolone cream 0.1% twice daily for presumed atopic dermatitis. Three months later, his symptoms rapidly worsened and he developed edema of the hands and feet. He was seen by neurology, and electromyography revealed severe distal sensorimotor neuropathy, prompting hospital admission for further evaluation of a potential rapidly progressive autoimmune disease. Laboratory workup and imaging were ordered, and the patient began an intravenous course of methylprednisolone. Minimal improvement in his symptoms was noted after 1 day, at which time dermatology was consulted.
Physical examination by dermatology revealed well-defined plaques with annular scale on extensor surfaces of the arms and legs, and edema on the hands and feet as well as distal sensorimotor neuropathy. The patient reported associated unspecified weight loss but denied any chest pain, shortness of breath, fevers, chills, cough, night sweats, exposure to chemicals, or recent travel. He reported that he had immigrated from India 37 years prior; his last visit to India was 6 years ago. He currently was taking famotidine for gastrointestinal reflux disease and losartan for hypertension. There was no personal or family history of autoimmune diseases. A complete workup for hematologic, thyroid, liver, and renal function was unremarkable. Initial autoimmune workup was negative for antinuclear antibodies and rheumatoid factor. Serum protein electrophoresis was normal. Results of testing for HIV, hepatitis B, and hepatitis C were negative. Chest radiography was unremarkable. Erythrocyte sedimentation rate and C-reactive protein level were elevated.
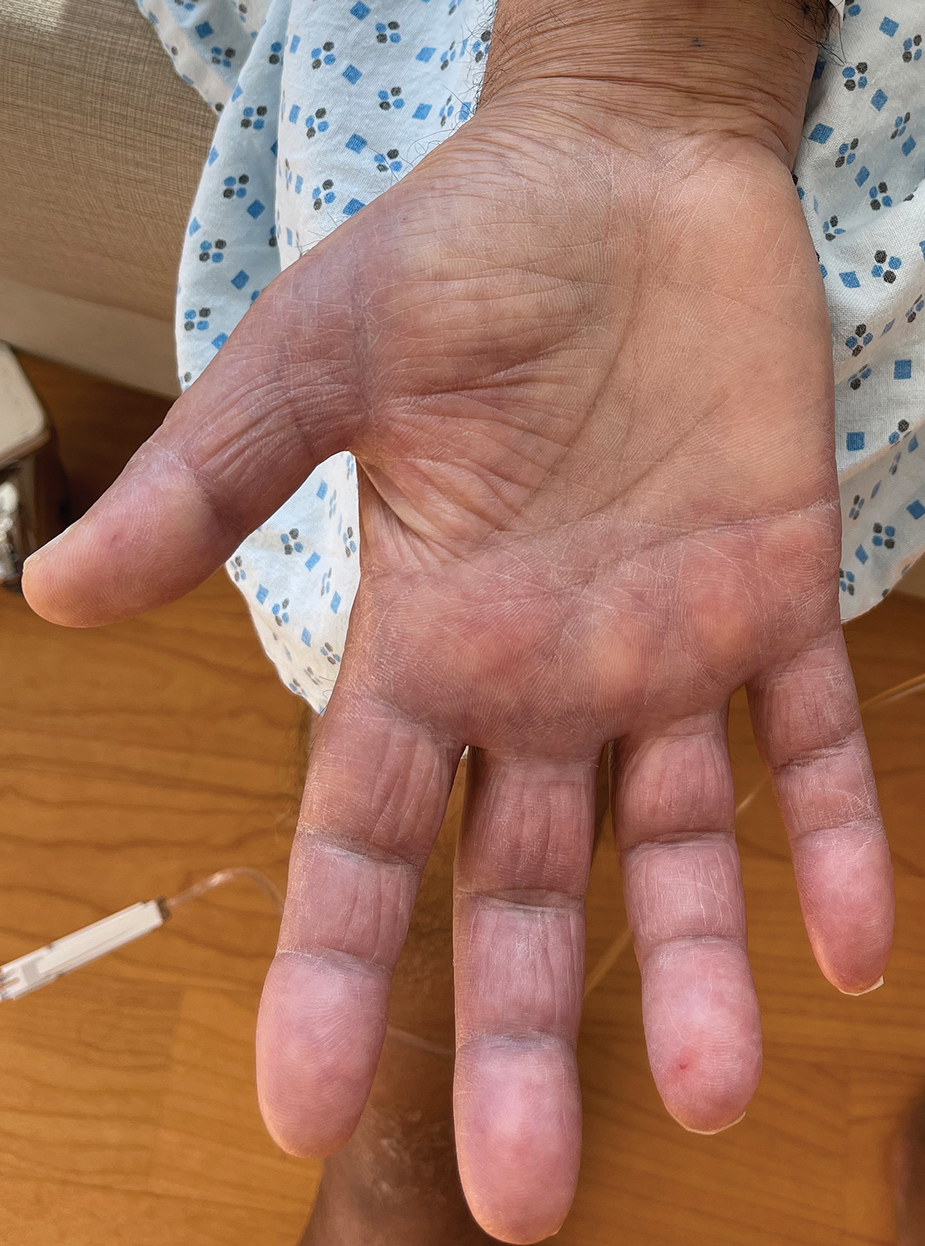
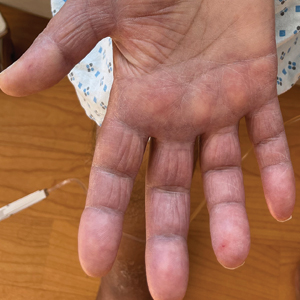
Exploring the Relationship Between Psoriasis and Mobility Among US Adults
Exploring the Relationship Between Psoriasis and Mobility Among US Adults
To the Editor:
Psoriasis is a chronic inflammatory condition that affects individuals in various extracutaneous ways.1 Prior studies have documented a decrease in exercise intensity among patients with psoriasis2; however, few studies have specifically investigated baseline mobility in this population. Baseline mobility denotes an individual’s fundamental ability to walk or move around without assistance of any kind. Impaired mobility—when baseline mobility is compromised—is an aspect of the wider diversity, equity, and inclusion framework that underscores the significance of recognizing challenges and promoting inclusive measures, both at the point of care and in research.3 study sought to analyze the relationship between psoriasis and baseline mobility among US adults (aged 45 to 80 years) utilizing the latest data from the National Health and Nutrition Examination Survey (NHANES) database for psoriasis.4 We used three 2-year cycles of NHANES data to create a 2009-2014 dataset.
The overall NHANES response rate among adults aged 45 to 80 years between 2009 and 2014 was 67.9%. Patients were categorized as having impaired mobility if they responded “yes” to the following question: “Because of a health problem, do you have difficulty walking without using any special equipment?” Psoriasis status was assessed by the following question: “Have you ever been told by a doctor or other health professional that you had psoriasis?” Multivariable logistic regression analyses were performed using Stata/SE 18.0 software (StataCorp LLC) to assess the relationship between psoriasis and impaired mobility. Age, income, education, sex, race, tobacco use, diabetes status, body mass index, and arthritis status were controlled for in our models.
Our analysis initially included 9982 participants; 14 did not respond to questions assessing psoriasis and impaired mobility and were excluded. The prevalence of impaired mobility in patients with psoriasis was 17.1% compared with 10.9% among those without psoriasis (Table 1). There was a significant association between psoriasis and impaired mobility among patients aged 45 to 80 years after adjusting for potential confounding variables (adjusted odds ratio [AOR], 1.54; 95% CI, 1.04- 2.29; P=.032)(Table 2). Analyses of subgroups yielded no statistically significant results.
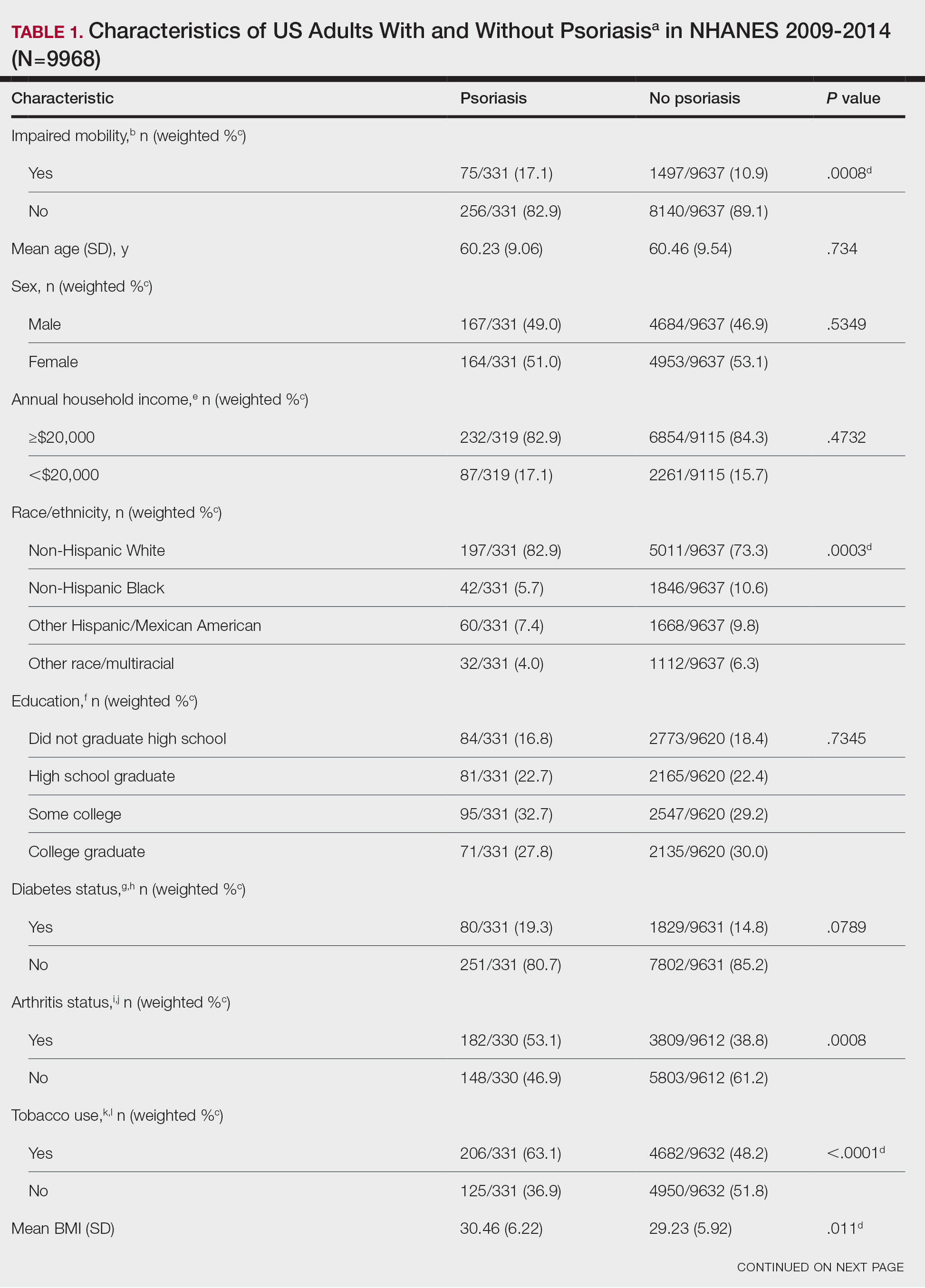

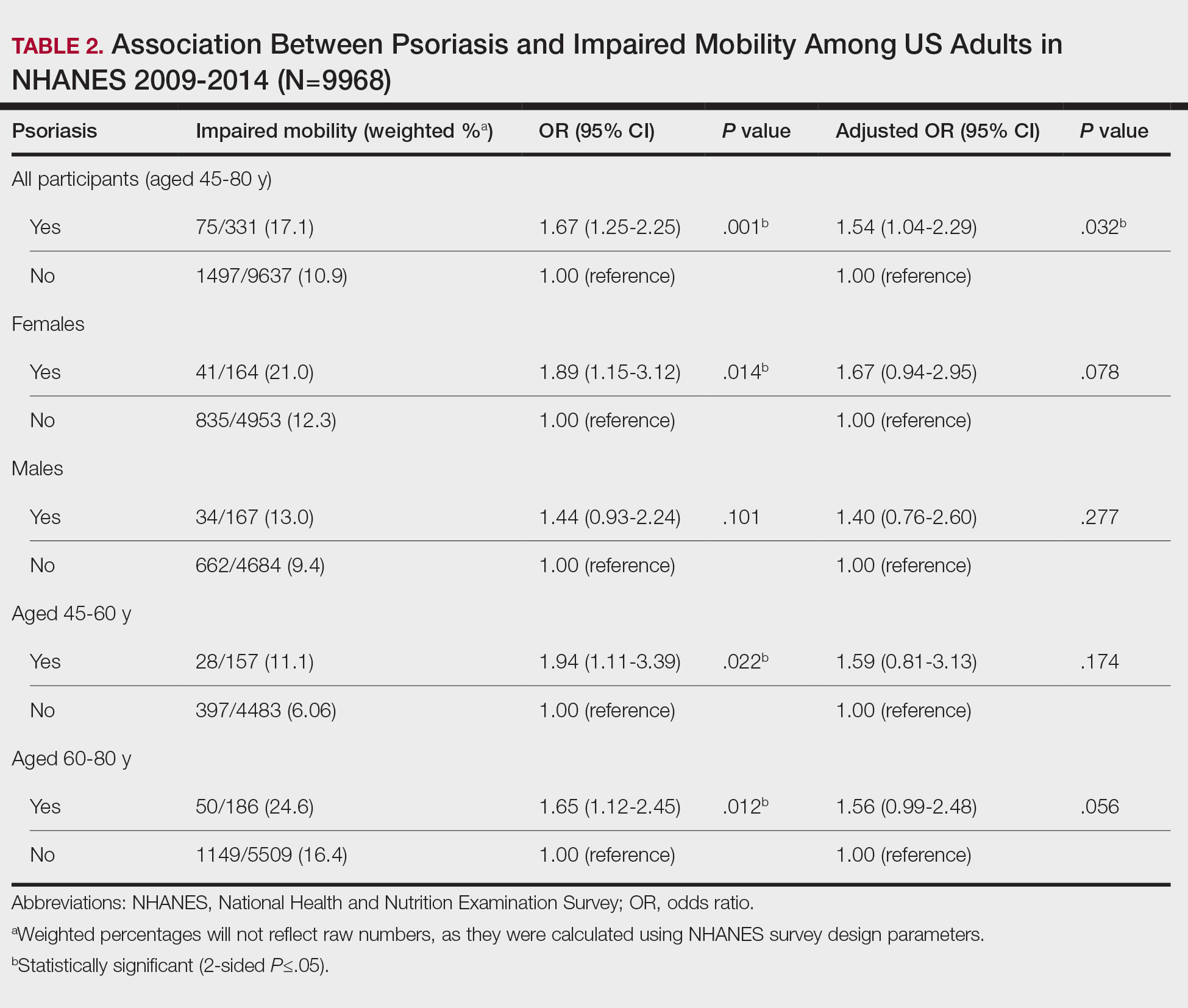
Our study demonstrated a statistically significant difference in mobility between individuals with psoriasis compared with the general population, which remained significant when controlling for arthritis, obesity, and diabetes (P=.032). This may be the result of several influences. First, the location of the psoriasis may impact mobility. Plantar psoriasis—a manifestation on the soles of the feet—can cause discomfort and pain, which can hinder walking and standing.5 Second, a study by Lasselin et al6 found that systemic inflammation contributes to mobility impairment through alterations in gait and posture, which suggests that the inflammatory processes inherent in psoriasis could intrinsically modify walking speed and stride, potentially exacerbating mobility difficulties independent of other comorbid conditions. These findings suggest that psoriasis may disproportionately affect individuals with impaired mobility, independent of comorbid arthritis, obesity, and diabetes.
These findings have broad implications for diversity, equity, and inclusion. They should prompt us to consider the practical challenges faced by this patient population and the ways that we can address barriers to care. Offering telehealth appointments, making primary care referrals for impaired mobility workups, and advising patients of direct-to-home delivery of prescriptions are good places to start.
Limitations to our study include the lack of specificity in the survey question, self-reporting bias, and the inability to control for the psoriasis location. Further investigations are warranted in large, representative US adult populations to assess the implications of impaired mobility in patients with psoriasis.
- Elmets CA, Leonardi CL, Davis DMR, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with awareness and attention to comorbidities. J Am Acad Dermatol. 2019;80:1073-1113. doi: 10.1016/j.jaad.2018.11.058
- Zheng Q, Sun XY, Miao X, et al. Association between physical activity and risk of prevalent psoriasis: A MOOSE-compliant meta-analysis. Medicine (Baltimore). 2018;97:e11394. doi: 10.1097 /MD.0000000000011394
- Mullin AE, Coe IR, Gooden EA, et al. Inclusion, diversity, equity, and accessibility: from organizational responsibility to leadership competency. Healthc Manage Forum. 2021;34311-315. doi: 10.1177/08404704211038232
- Centers for Disease Control and Prevention. National Health and Nutrition Examination Survey. NHANES questionnaires, datasets, and related documentation. Accessed October 21, 2023. https://wwwn.cdc.gov/nchs/nhanes/
- Romani M, Biela G, Farr K, et al. Plantar psoriasis: a review of the literature. Clin Podiatr Med Surg. 2021;38:541-552. doi: 10.1016 /j.cpm.2021.06.009
- Lasselin J, Sundelin T, Wayne PM, et al. Biological motion during inflammation in humans. Brain Behav Immun. 2020;84:147-153. doi: 10.1016/j.bbi.2019.11.019
To the Editor:
Psoriasis is a chronic inflammatory condition that affects individuals in various extracutaneous ways.1 Prior studies have documented a decrease in exercise intensity among patients with psoriasis2; however, few studies have specifically investigated baseline mobility in this population. Baseline mobility denotes an individual’s fundamental ability to walk or move around without assistance of any kind. Impaired mobility—when baseline mobility is compromised—is an aspect of the wider diversity, equity, and inclusion framework that underscores the significance of recognizing challenges and promoting inclusive measures, both at the point of care and in research.3 study sought to analyze the relationship between psoriasis and baseline mobility among US adults (aged 45 to 80 years) utilizing the latest data from the National Health and Nutrition Examination Survey (NHANES) database for psoriasis.4 We used three 2-year cycles of NHANES data to create a 2009-2014 dataset.
The overall NHANES response rate among adults aged 45 to 80 years between 2009 and 2014 was 67.9%. Patients were categorized as having impaired mobility if they responded “yes” to the following question: “Because of a health problem, do you have difficulty walking without using any special equipment?” Psoriasis status was assessed by the following question: “Have you ever been told by a doctor or other health professional that you had psoriasis?” Multivariable logistic regression analyses were performed using Stata/SE 18.0 software (StataCorp LLC) to assess the relationship between psoriasis and impaired mobility. Age, income, education, sex, race, tobacco use, diabetes status, body mass index, and arthritis status were controlled for in our models.
Our analysis initially included 9982 participants; 14 did not respond to questions assessing psoriasis and impaired mobility and were excluded. The prevalence of impaired mobility in patients with psoriasis was 17.1% compared with 10.9% among those without psoriasis (Table 1). There was a significant association between psoriasis and impaired mobility among patients aged 45 to 80 years after adjusting for potential confounding variables (adjusted odds ratio [AOR], 1.54; 95% CI, 1.04- 2.29; P=.032)(Table 2). Analyses of subgroups yielded no statistically significant results.
Our study demonstrated a statistically significant difference in mobility between individuals with psoriasis compared with the general population, which remained significant when controlling for arthritis, obesity, and diabetes (P=.032). This may be the result of several influences. First, the location of the psoriasis may impact mobility. Plantar psoriasis—a manifestation on the soles of the feet—can cause discomfort and pain, which can hinder walking and standing.5 Second, a study by Lasselin et al6 found that systemic inflammation contributes to mobility impairment through alterations in gait and posture, which suggests that the inflammatory processes inherent in psoriasis could intrinsically modify walking speed and stride, potentially exacerbating mobility difficulties independent of other comorbid conditions. These findings suggest that psoriasis may disproportionately affect individuals with impaired mobility, independent of comorbid arthritis, obesity, and diabetes.
These findings have broad implications for diversity, equity, and inclusion. They should prompt us to consider the practical challenges faced by this patient population and the ways that we can address barriers to care. Offering telehealth appointments, making primary care referrals for impaired mobility workups, and advising patients of direct-to-home delivery of prescriptions are good places to start.
Limitations to our study include the lack of specificity in the survey question, self-reporting bias, and the inability to control for the psoriasis location. Further investigations are warranted in large, representative US adult populations to assess the implications of impaired mobility in patients with psoriasis.
To the Editor:
Psoriasis is a chronic inflammatory condition that affects individuals in various extracutaneous ways.1 Prior studies have documented a decrease in exercise intensity among patients with psoriasis2; however, few studies have specifically investigated baseline mobility in this population. Baseline mobility denotes an individual’s fundamental ability to walk or move around without assistance of any kind. Impaired mobility—when baseline mobility is compromised—is an aspect of the wider diversity, equity, and inclusion framework that underscores the significance of recognizing challenges and promoting inclusive measures, both at the point of care and in research.3 study sought to analyze the relationship between psoriasis and baseline mobility among US adults (aged 45 to 80 years) utilizing the latest data from the National Health and Nutrition Examination Survey (NHANES) database for psoriasis.4 We used three 2-year cycles of NHANES data to create a 2009-2014 dataset.
The overall NHANES response rate among adults aged 45 to 80 years between 2009 and 2014 was 67.9%. Patients were categorized as having impaired mobility if they responded “yes” to the following question: “Because of a health problem, do you have difficulty walking without using any special equipment?” Psoriasis status was assessed by the following question: “Have you ever been told by a doctor or other health professional that you had psoriasis?” Multivariable logistic regression analyses were performed using Stata/SE 18.0 software (StataCorp LLC) to assess the relationship between psoriasis and impaired mobility. Age, income, education, sex, race, tobacco use, diabetes status, body mass index, and arthritis status were controlled for in our models.
Our analysis initially included 9982 participants; 14 did not respond to questions assessing psoriasis and impaired mobility and were excluded. The prevalence of impaired mobility in patients with psoriasis was 17.1% compared with 10.9% among those without psoriasis (Table 1). There was a significant association between psoriasis and impaired mobility among patients aged 45 to 80 years after adjusting for potential confounding variables (adjusted odds ratio [AOR], 1.54; 95% CI, 1.04- 2.29; P=.032)(Table 2). Analyses of subgroups yielded no statistically significant results.
Our study demonstrated a statistically significant difference in mobility between individuals with psoriasis compared with the general population, which remained significant when controlling for arthritis, obesity, and diabetes (P=.032). This may be the result of several influences. First, the location of the psoriasis may impact mobility. Plantar psoriasis—a manifestation on the soles of the feet—can cause discomfort and pain, which can hinder walking and standing.5 Second, a study by Lasselin et al6 found that systemic inflammation contributes to mobility impairment through alterations in gait and posture, which suggests that the inflammatory processes inherent in psoriasis could intrinsically modify walking speed and stride, potentially exacerbating mobility difficulties independent of other comorbid conditions. These findings suggest that psoriasis may disproportionately affect individuals with impaired mobility, independent of comorbid arthritis, obesity, and diabetes.
These findings have broad implications for diversity, equity, and inclusion. They should prompt us to consider the practical challenges faced by this patient population and the ways that we can address barriers to care. Offering telehealth appointments, making primary care referrals for impaired mobility workups, and advising patients of direct-to-home delivery of prescriptions are good places to start.
Limitations to our study include the lack of specificity in the survey question, self-reporting bias, and the inability to control for the psoriasis location. Further investigations are warranted in large, representative US adult populations to assess the implications of impaired mobility in patients with psoriasis.
- Elmets CA, Leonardi CL, Davis DMR, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with awareness and attention to comorbidities. J Am Acad Dermatol. 2019;80:1073-1113. doi: 10.1016/j.jaad.2018.11.058
- Zheng Q, Sun XY, Miao X, et al. Association between physical activity and risk of prevalent psoriasis: A MOOSE-compliant meta-analysis. Medicine (Baltimore). 2018;97:e11394. doi: 10.1097 /MD.0000000000011394
- Mullin AE, Coe IR, Gooden EA, et al. Inclusion, diversity, equity, and accessibility: from organizational responsibility to leadership competency. Healthc Manage Forum. 2021;34311-315. doi: 10.1177/08404704211038232
- Centers for Disease Control and Prevention. National Health and Nutrition Examination Survey. NHANES questionnaires, datasets, and related documentation. Accessed October 21, 2023. https://wwwn.cdc.gov/nchs/nhanes/
- Romani M, Biela G, Farr K, et al. Plantar psoriasis: a review of the literature. Clin Podiatr Med Surg. 2021;38:541-552. doi: 10.1016 /j.cpm.2021.06.009
- Lasselin J, Sundelin T, Wayne PM, et al. Biological motion during inflammation in humans. Brain Behav Immun. 2020;84:147-153. doi: 10.1016/j.bbi.2019.11.019
- Elmets CA, Leonardi CL, Davis DMR, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with awareness and attention to comorbidities. J Am Acad Dermatol. 2019;80:1073-1113. doi: 10.1016/j.jaad.2018.11.058
- Zheng Q, Sun XY, Miao X, et al. Association between physical activity and risk of prevalent psoriasis: A MOOSE-compliant meta-analysis. Medicine (Baltimore). 2018;97:e11394. doi: 10.1097 /MD.0000000000011394
- Mullin AE, Coe IR, Gooden EA, et al. Inclusion, diversity, equity, and accessibility: from organizational responsibility to leadership competency. Healthc Manage Forum. 2021;34311-315. doi: 10.1177/08404704211038232
- Centers for Disease Control and Prevention. National Health and Nutrition Examination Survey. NHANES questionnaires, datasets, and related documentation. Accessed October 21, 2023. https://wwwn.cdc.gov/nchs/nhanes/
- Romani M, Biela G, Farr K, et al. Plantar psoriasis: a review of the literature. Clin Podiatr Med Surg. 2021;38:541-552. doi: 10.1016 /j.cpm.2021.06.009
- Lasselin J, Sundelin T, Wayne PM, et al. Biological motion during inflammation in humans. Brain Behav Immun. 2020;84:147-153. doi: 10.1016/j.bbi.2019.11.019
Exploring the Relationship Between Psoriasis and Mobility Among US Adults
Exploring the Relationship Between Psoriasis and Mobility Among US Adults
PRACTICE POINTS
- Mobility issues are more common in patients who have psoriasis than in those who do not.
- It is important to assess patients with psoriasis for mobility issues regardless of age or comorbid conditions such as arthritis, obesity, and diabetes.
- Dermatologists can help patients with psoriasis and impaired mobility overcome potential barriers to care by incorporating telehealth services into their practices and informing patients of direct-to-home delivery of prescriptions.
Dermatologists’ Perspectives Toward Disability Assessment: A Nationwide Survey Report
Dermatologists’ Perspectives Toward Disability Assessment: A Nationwide Survey Report
To the Editor:
Cutaneous medical conditions can have a substantial impact on patients’ functioning and quality of life. Many patients with severe skin disease are eligible to receive disability assistance that can provide them with essential income and health care. Previous research has highlighted disability assessment as one of the most important ways physicians can help mitigate the health consequences of poverty.1 Dermatologists can play an important role in the disability assessment process by documenting the facts associated with patients’ skin conditions.
Although skin conditions have a relatively high prevalence, they remain underrepresented in disability claims. Between 1997 and 2004, occupational skin diseases accounted for 12% to 17% of nonfatal work-related illnesses; however, during that same period, skin conditions comprised only 0.21% of disability claims in the United States.2,3 Historically, there has been hesitancy among dermatologists to complete disability paperwork; a 1976 survey of dermatologists cited extensive paperwork, “troublesome patients,” and fee schedule issues as reasons.4 The lack of training regarding disability assessment in medical school and residency also has been noted.5
To characterize modern attitudes toward disability assessments, we conducted a survey of dermatologists across the United States. Our study was reviewed and declared exempt by the institutional review board of the Lundquist Institute for Biomedical Innovation at Harbor-UCLA Medical Center (Torrance, California)(approval #18CR-32242-01). Using convenience sampling, we emailed dermatologists from the Association of Professors of Dermatology and dermatology state societies in all 50 states inviting them to participate in our voluntary and anonymous survey, which was administered using SurveyMonkey. The use of all society mailing lists was approved by the respective owners. The 15-question survey included multiple choice, Likert scale, and free response sections. Summary and descriptive statistics were used to describe respondent demographics and identify any patterns in responses.
For each Likert-based question, participants ranked their degree of agreement with a statement as: 1=strongly disagree, 2=somewhat disagree, 3=neither agree nor disagree/neutral, 4=somewhat agree, and 5=strongly agree. The mean response and standard deviation were reported for each Likert scale prompt. Preplanned 1-sample t testing was used to analyze Likert scale data, in which the mean response for each prompt was compared to a baseline response of 3 (neutral). A P value <.05 was considered statistically significant. Statistical analyses were performed using SPSS Statistics for MacOS, version 27 (IBM).
Seventy-eight dermatologists agreed to participate, and 70 completed the survey, for a response rate of 89.7% (Table 1). The dermatologists we surveyed practiced in a variety of clinical settings, including academic public hospitals (46.2% [36/78]), academic private hospitals (33.3% [26/78]), and private practices (32.1% [25/78]), and 60.3% (47/78) reported providing disability documentation at some point. Most of the respondents (64.3% [45/70]) did not perform assessments in an average month (Table 2). Medical assessment documentation was provided most frequently for workers’ compensation (50.0% [35/70]), private insurance (27.1% [19/70]), and Social Security Disability Insurance (25.7% [18/70]). Dermatologists overwhelmingly reported no formal training for disability assessment in medical school (94.3% [66/70]), residency (97.1% [68/70]), or clinical practice (81.4% [57/70]).


In the Likert scale prompts, respondents agreed that they were uncertain of their role in disability assessment (mean response, 3.6; P<.001). Moreover, they were uncomfortable providing assessments (mean response, 3.5; P<.001) and felt that they did not have sufficient time to perform them (mean response, 3.6; P<.001). Dermatologists disagreed that they received adequate compensation for performing assessments (mean response, 2.2; P<.001) and felt that they did not have enough time to participate in assessments (mean response, 3.6; P<.001). Respondents generally did not feel distrustful of patients seeking disability assessment (mean response, 2.8; P=.043). Dermatologists neither agreed nor disagreed when asked if they thought that physicians can determine disability status (mean response, 3.2; P=.118). The details of the Likert scale responses are described in Table 3. Respondents also were uncertain as to which dermatologic conditions were eligible for disability. When asked to select which conditions from a list of 10 were eligible per the Social Security Administration listing of disability impairments, only 15.4% (12/70) of respondents correctly identified that all the conditions qualified; these included ichthyosis, pemphigus vulgaris, allergic contact dermatitis, hidradenitis suppurativa, systemic lupus erythematosus, chromoblastomycosis, xeroderma pigmentosum, burns, malignant melanoma, and scleroderma.6

In the free-response prompts, respondents frequently described extensive paperwork, inadequate time, and lack of reimbursement as barriers to providing documentation. Often, dermatologists found that the forms were not well matched to the skin conditions they were evaluating and rather had a musculoskeletal focus. Multiple individuals commented on the challenge in assessing the percentage of disability and functional/psychosocial impairment in skin conditions. One respondent noted that workers’ compensation forms ask if the patient is “…permanent and stationary…for most conditions this has no meaning in dermatology.” Some felt hesitant to provide documentation because they had insufficient patient history, especially regarding employment, and opted to defer to primary care providers who might be more familiar with the full patient history.
A dermatologist described their perspective as follows:
“…As a specialist I feel that I don’t have a complete look into all the factors that could contribute to a patient[’]s need to go on disability, and I don’t have experience with filling out disability requests. That being said, if a patient[’]s request for disability was due to a skin disease that I know way more about than [a] primary care [physician] would, I would do the disability assessment.”
Another respondent noted the complexity in “establishing causality” for workers’ compensation. Another dermatologist reported,
“The most frequent challenging situation I encounter is being asked to evaluate for maximum medical improvement after patch testing. If the patient is not fully avoiding contact allergens either at home or at work, then I typically document that they are not at [maximum medical improvement]. The reality is that most frequently it is due to exposure to allergens at home so the line between what is a legitimate worker’s comp[ensation] issue and what is a home life choice is blurry.”
Nevertheless, respondents expressed interest in learning more about disability assessment procedures. Summary guides, lectures, and prefilled paperwork were the most popular initiatives that respondents agreed would be beneficial toward becoming educated regarding disability assessment (78.6%, 58.6%, and 58.6%, respectively)(Table 2). One respondent noted that “previous [internal medicine] history help[ed]” them in performing cutaneous disability assessments.
As with any survey, our study did have some inherent limitations. Only a relatively small sample size was willing to complete the survey. There was a predominance of respondents from California (34.6% [27/78]), as well as those practicing for less than 15 years (58.9% [46/78])(Figure). This could limit generalizability to the national population of dermatologists. In addition, there was potential for recall bias and errors in responding given the self-reported nature of the study. Different individuals may interpret the Likert scale options in various ways, which could skew results unintentionally. However, the survey was largely qualitative in nature, making it a legitimate tool for answering our research questions. Moreover, we were able to hear the perspectives of dermatologists across diverse practice settings, with free response prompts to increase the depth of the survey.
Almost 50 years later, our survey echoes common themes from Adams’ 1976 survey.4 Inadequate compensation, limited time, and burdensome paperwork all continue to hinder dermatologists’ ability to perform disability assessments. Our participants frequently commented that the current disability forms are not congruent with the nature of skin conditions, making it challenging to accurately document the facts.
Moreover, respondents felt uncertain in their role in disability assessment and occasionally noted distrust of patients or insufficient patient history as barriers to completing assessments. They also were unsure if physicians can grant disability status. This is a common misconception among physicians that leads to discomfort in helping with disability assessment.7 The role of physicians in disability assessment is to document the facts of a patient’s illness, not to determine whether they are eligible for benefits. We discovered uncertainty in our respondents’ ability to identify conditions eligible for disability, highlighting an area in need of greater education for physicians.
Despite these obstacles, respondents were interested in learning more about disability assessment and highlighted several practical approaches that could help them better perform this task. As skin specialists, dermatologists are the best-equipped physicians to assess cutaneous conditions and should play a greater role in performing disability assessments, which could be achieved through increased educational initiatives and individual physician motivation.7 We call for greater collaboration and reflection on the importance of disability assistance among dermatologists to increase participation in the disability-assessment process.
- O’Connell JJ, Zevin BD, Quick PD, et al. Documenting disability: simple strategies for medical providers. Health Care for the Homeless Clinicians’ Network. September 2007. Accessed March 31, 2025. https://nhchc.org/wp-content/uploads/2019/08/DocumentingDisability2007.pdf
- US Bureau of Labor Statistics. Injuries, illnesses, and fatalities. Accessed March 31, 2025. https://www.bls.gov/iif/
- Meseguer J. Outcome variation in the Social Security Disability Insurance Program: the role of primary diagnoses. Soc Secur Bull. 2013;73:39-75.
- Adams RM. Attitudes of California dermatologists toward Worker’s Compensation: results of a survey. West J Med. 1976;125:169-175.
- Talmage J, Melhorn J, Hyman M. AMA Guides to the Evaluation of Work Ability and Return to Work. 2nd ed. American Medical Association; 2011.
- Social Security Administration. Disability evaluation under Social Security. 8.00 skin disorders - adult. March 31, 2025. https://www.ssa.gov/disability/professionals/bluebook/8.00-Skin-Adult.htm
- Dawson J, Smogorzewski J. Demystifying disability assessments for dermatologists—a call to action. JAMA Dermatol. 2021;157:903-904. doi:10.1001/jamadermatol.2021.1767
To the Editor:
Cutaneous medical conditions can have a substantial impact on patients’ functioning and quality of life. Many patients with severe skin disease are eligible to receive disability assistance that can provide them with essential income and health care. Previous research has highlighted disability assessment as one of the most important ways physicians can help mitigate the health consequences of poverty.1 Dermatologists can play an important role in the disability assessment process by documenting the facts associated with patients’ skin conditions.
Although skin conditions have a relatively high prevalence, they remain underrepresented in disability claims. Between 1997 and 2004, occupational skin diseases accounted for 12% to 17% of nonfatal work-related illnesses; however, during that same period, skin conditions comprised only 0.21% of disability claims in the United States.2,3 Historically, there has been hesitancy among dermatologists to complete disability paperwork; a 1976 survey of dermatologists cited extensive paperwork, “troublesome patients,” and fee schedule issues as reasons.4 The lack of training regarding disability assessment in medical school and residency also has been noted.5
To characterize modern attitudes toward disability assessments, we conducted a survey of dermatologists across the United States. Our study was reviewed and declared exempt by the institutional review board of the Lundquist Institute for Biomedical Innovation at Harbor-UCLA Medical Center (Torrance, California)(approval #18CR-32242-01). Using convenience sampling, we emailed dermatologists from the Association of Professors of Dermatology and dermatology state societies in all 50 states inviting them to participate in our voluntary and anonymous survey, which was administered using SurveyMonkey. The use of all society mailing lists was approved by the respective owners. The 15-question survey included multiple choice, Likert scale, and free response sections. Summary and descriptive statistics were used to describe respondent demographics and identify any patterns in responses.
For each Likert-based question, participants ranked their degree of agreement with a statement as: 1=strongly disagree, 2=somewhat disagree, 3=neither agree nor disagree/neutral, 4=somewhat agree, and 5=strongly agree. The mean response and standard deviation were reported for each Likert scale prompt. Preplanned 1-sample t testing was used to analyze Likert scale data, in which the mean response for each prompt was compared to a baseline response of 3 (neutral). A P value <.05 was considered statistically significant. Statistical analyses were performed using SPSS Statistics for MacOS, version 27 (IBM).
Seventy-eight dermatologists agreed to participate, and 70 completed the survey, for a response rate of 89.7% (Table 1). The dermatologists we surveyed practiced in a variety of clinical settings, including academic public hospitals (46.2% [36/78]), academic private hospitals (33.3% [26/78]), and private practices (32.1% [25/78]), and 60.3% (47/78) reported providing disability documentation at some point. Most of the respondents (64.3% [45/70]) did not perform assessments in an average month (Table 2). Medical assessment documentation was provided most frequently for workers’ compensation (50.0% [35/70]), private insurance (27.1% [19/70]), and Social Security Disability Insurance (25.7% [18/70]). Dermatologists overwhelmingly reported no formal training for disability assessment in medical school (94.3% [66/70]), residency (97.1% [68/70]), or clinical practice (81.4% [57/70]).
In the Likert scale prompts, respondents agreed that they were uncertain of their role in disability assessment (mean response, 3.6; P<.001). Moreover, they were uncomfortable providing assessments (mean response, 3.5; P<.001) and felt that they did not have sufficient time to perform them (mean response, 3.6; P<.001). Dermatologists disagreed that they received adequate compensation for performing assessments (mean response, 2.2; P<.001) and felt that they did not have enough time to participate in assessments (mean response, 3.6; P<.001). Respondents generally did not feel distrustful of patients seeking disability assessment (mean response, 2.8; P=.043). Dermatologists neither agreed nor disagreed when asked if they thought that physicians can determine disability status (mean response, 3.2; P=.118). The details of the Likert scale responses are described in Table 3. Respondents also were uncertain as to which dermatologic conditions were eligible for disability. When asked to select which conditions from a list of 10 were eligible per the Social Security Administration listing of disability impairments, only 15.4% (12/70) of respondents correctly identified that all the conditions qualified; these included ichthyosis, pemphigus vulgaris, allergic contact dermatitis, hidradenitis suppurativa, systemic lupus erythematosus, chromoblastomycosis, xeroderma pigmentosum, burns, malignant melanoma, and scleroderma.6
In the free-response prompts, respondents frequently described extensive paperwork, inadequate time, and lack of reimbursement as barriers to providing documentation. Often, dermatologists found that the forms were not well matched to the skin conditions they were evaluating and rather had a musculoskeletal focus. Multiple individuals commented on the challenge in assessing the percentage of disability and functional/psychosocial impairment in skin conditions. One respondent noted that workers’ compensation forms ask if the patient is “…permanent and stationary…for most conditions this has no meaning in dermatology.” Some felt hesitant to provide documentation because they had insufficient patient history, especially regarding employment, and opted to defer to primary care providers who might be more familiar with the full patient history.
A dermatologist described their perspective as follows:
“…As a specialist I feel that I don’t have a complete look into all the factors that could contribute to a patient[’]s need to go on disability, and I don’t have experience with filling out disability requests. That being said, if a patient[’]s request for disability was due to a skin disease that I know way more about than [a] primary care [physician] would, I would do the disability assessment.”
Another respondent noted the complexity in “establishing causality” for workers’ compensation. Another dermatologist reported,
“The most frequent challenging situation I encounter is being asked to evaluate for maximum medical improvement after patch testing. If the patient is not fully avoiding contact allergens either at home or at work, then I typically document that they are not at [maximum medical improvement]. The reality is that most frequently it is due to exposure to allergens at home so the line between what is a legitimate worker’s comp[ensation] issue and what is a home life choice is blurry.”
Nevertheless, respondents expressed interest in learning more about disability assessment procedures. Summary guides, lectures, and prefilled paperwork were the most popular initiatives that respondents agreed would be beneficial toward becoming educated regarding disability assessment (78.6%, 58.6%, and 58.6%, respectively)(Table 2). One respondent noted that “previous [internal medicine] history help[ed]” them in performing cutaneous disability assessments.
As with any survey, our study did have some inherent limitations. Only a relatively small sample size was willing to complete the survey. There was a predominance of respondents from California (34.6% [27/78]), as well as those practicing for less than 15 years (58.9% [46/78])(Figure). This could limit generalizability to the national population of dermatologists. In addition, there was potential for recall bias and errors in responding given the self-reported nature of the study. Different individuals may interpret the Likert scale options in various ways, which could skew results unintentionally. However, the survey was largely qualitative in nature, making it a legitimate tool for answering our research questions. Moreover, we were able to hear the perspectives of dermatologists across diverse practice settings, with free response prompts to increase the depth of the survey.
Almost 50 years later, our survey echoes common themes from Adams’ 1976 survey.4 Inadequate compensation, limited time, and burdensome paperwork all continue to hinder dermatologists’ ability to perform disability assessments. Our participants frequently commented that the current disability forms are not congruent with the nature of skin conditions, making it challenging to accurately document the facts.
Moreover, respondents felt uncertain in their role in disability assessment and occasionally noted distrust of patients or insufficient patient history as barriers to completing assessments. They also were unsure if physicians can grant disability status. This is a common misconception among physicians that leads to discomfort in helping with disability assessment.7 The role of physicians in disability assessment is to document the facts of a patient’s illness, not to determine whether they are eligible for benefits. We discovered uncertainty in our respondents’ ability to identify conditions eligible for disability, highlighting an area in need of greater education for physicians.
Despite these obstacles, respondents were interested in learning more about disability assessment and highlighted several practical approaches that could help them better perform this task. As skin specialists, dermatologists are the best-equipped physicians to assess cutaneous conditions and should play a greater role in performing disability assessments, which could be achieved through increased educational initiatives and individual physician motivation.7 We call for greater collaboration and reflection on the importance of disability assistance among dermatologists to increase participation in the disability-assessment process.
To the Editor:
Cutaneous medical conditions can have a substantial impact on patients’ functioning and quality of life. Many patients with severe skin disease are eligible to receive disability assistance that can provide them with essential income and health care. Previous research has highlighted disability assessment as one of the most important ways physicians can help mitigate the health consequences of poverty.1 Dermatologists can play an important role in the disability assessment process by documenting the facts associated with patients’ skin conditions.
Although skin conditions have a relatively high prevalence, they remain underrepresented in disability claims. Between 1997 and 2004, occupational skin diseases accounted for 12% to 17% of nonfatal work-related illnesses; however, during that same period, skin conditions comprised only 0.21% of disability claims in the United States.2,3 Historically, there has been hesitancy among dermatologists to complete disability paperwork; a 1976 survey of dermatologists cited extensive paperwork, “troublesome patients,” and fee schedule issues as reasons.4 The lack of training regarding disability assessment in medical school and residency also has been noted.5
To characterize modern attitudes toward disability assessments, we conducted a survey of dermatologists across the United States. Our study was reviewed and declared exempt by the institutional review board of the Lundquist Institute for Biomedical Innovation at Harbor-UCLA Medical Center (Torrance, California)(approval #18CR-32242-01). Using convenience sampling, we emailed dermatologists from the Association of Professors of Dermatology and dermatology state societies in all 50 states inviting them to participate in our voluntary and anonymous survey, which was administered using SurveyMonkey. The use of all society mailing lists was approved by the respective owners. The 15-question survey included multiple choice, Likert scale, and free response sections. Summary and descriptive statistics were used to describe respondent demographics and identify any patterns in responses.
For each Likert-based question, participants ranked their degree of agreement with a statement as: 1=strongly disagree, 2=somewhat disagree, 3=neither agree nor disagree/neutral, 4=somewhat agree, and 5=strongly agree. The mean response and standard deviation were reported for each Likert scale prompt. Preplanned 1-sample t testing was used to analyze Likert scale data, in which the mean response for each prompt was compared to a baseline response of 3 (neutral). A P value <.05 was considered statistically significant. Statistical analyses were performed using SPSS Statistics for MacOS, version 27 (IBM).
Seventy-eight dermatologists agreed to participate, and 70 completed the survey, for a response rate of 89.7% (Table 1). The dermatologists we surveyed practiced in a variety of clinical settings, including academic public hospitals (46.2% [36/78]), academic private hospitals (33.3% [26/78]), and private practices (32.1% [25/78]), and 60.3% (47/78) reported providing disability documentation at some point. Most of the respondents (64.3% [45/70]) did not perform assessments in an average month (Table 2). Medical assessment documentation was provided most frequently for workers’ compensation (50.0% [35/70]), private insurance (27.1% [19/70]), and Social Security Disability Insurance (25.7% [18/70]). Dermatologists overwhelmingly reported no formal training for disability assessment in medical school (94.3% [66/70]), residency (97.1% [68/70]), or clinical practice (81.4% [57/70]).
In the Likert scale prompts, respondents agreed that they were uncertain of their role in disability assessment (mean response, 3.6; P<.001). Moreover, they were uncomfortable providing assessments (mean response, 3.5; P<.001) and felt that they did not have sufficient time to perform them (mean response, 3.6; P<.001). Dermatologists disagreed that they received adequate compensation for performing assessments (mean response, 2.2; P<.001) and felt that they did not have enough time to participate in assessments (mean response, 3.6; P<.001). Respondents generally did not feel distrustful of patients seeking disability assessment (mean response, 2.8; P=.043). Dermatologists neither agreed nor disagreed when asked if they thought that physicians can determine disability status (mean response, 3.2; P=.118). The details of the Likert scale responses are described in Table 3. Respondents also were uncertain as to which dermatologic conditions were eligible for disability. When asked to select which conditions from a list of 10 were eligible per the Social Security Administration listing of disability impairments, only 15.4% (12/70) of respondents correctly identified that all the conditions qualified; these included ichthyosis, pemphigus vulgaris, allergic contact dermatitis, hidradenitis suppurativa, systemic lupus erythematosus, chromoblastomycosis, xeroderma pigmentosum, burns, malignant melanoma, and scleroderma.6
In the free-response prompts, respondents frequently described extensive paperwork, inadequate time, and lack of reimbursement as barriers to providing documentation. Often, dermatologists found that the forms were not well matched to the skin conditions they were evaluating and rather had a musculoskeletal focus. Multiple individuals commented on the challenge in assessing the percentage of disability and functional/psychosocial impairment in skin conditions. One respondent noted that workers’ compensation forms ask if the patient is “…permanent and stationary…for most conditions this has no meaning in dermatology.” Some felt hesitant to provide documentation because they had insufficient patient history, especially regarding employment, and opted to defer to primary care providers who might be more familiar with the full patient history.
A dermatologist described their perspective as follows:
“…As a specialist I feel that I don’t have a complete look into all the factors that could contribute to a patient[’]s need to go on disability, and I don’t have experience with filling out disability requests. That being said, if a patient[’]s request for disability was due to a skin disease that I know way more about than [a] primary care [physician] would, I would do the disability assessment.”
Another respondent noted the complexity in “establishing causality” for workers’ compensation. Another dermatologist reported,
“The most frequent challenging situation I encounter is being asked to evaluate for maximum medical improvement after patch testing. If the patient is not fully avoiding contact allergens either at home or at work, then I typically document that they are not at [maximum medical improvement]. The reality is that most frequently it is due to exposure to allergens at home so the line between what is a legitimate worker’s comp[ensation] issue and what is a home life choice is blurry.”
Nevertheless, respondents expressed interest in learning more about disability assessment procedures. Summary guides, lectures, and prefilled paperwork were the most popular initiatives that respondents agreed would be beneficial toward becoming educated regarding disability assessment (78.6%, 58.6%, and 58.6%, respectively)(Table 2). One respondent noted that “previous [internal medicine] history help[ed]” them in performing cutaneous disability assessments.
As with any survey, our study did have some inherent limitations. Only a relatively small sample size was willing to complete the survey. There was a predominance of respondents from California (34.6% [27/78]), as well as those practicing for less than 15 years (58.9% [46/78])(Figure). This could limit generalizability to the national population of dermatologists. In addition, there was potential for recall bias and errors in responding given the self-reported nature of the study. Different individuals may interpret the Likert scale options in various ways, which could skew results unintentionally. However, the survey was largely qualitative in nature, making it a legitimate tool for answering our research questions. Moreover, we were able to hear the perspectives of dermatologists across diverse practice settings, with free response prompts to increase the depth of the survey.
Almost 50 years later, our survey echoes common themes from Adams’ 1976 survey.4 Inadequate compensation, limited time, and burdensome paperwork all continue to hinder dermatologists’ ability to perform disability assessments. Our participants frequently commented that the current disability forms are not congruent with the nature of skin conditions, making it challenging to accurately document the facts.
Moreover, respondents felt uncertain in their role in disability assessment and occasionally noted distrust of patients or insufficient patient history as barriers to completing assessments. They also were unsure if physicians can grant disability status. This is a common misconception among physicians that leads to discomfort in helping with disability assessment.7 The role of physicians in disability assessment is to document the facts of a patient’s illness, not to determine whether they are eligible for benefits. We discovered uncertainty in our respondents’ ability to identify conditions eligible for disability, highlighting an area in need of greater education for physicians.
Despite these obstacles, respondents were interested in learning more about disability assessment and highlighted several practical approaches that could help them better perform this task. As skin specialists, dermatologists are the best-equipped physicians to assess cutaneous conditions and should play a greater role in performing disability assessments, which could be achieved through increased educational initiatives and individual physician motivation.7 We call for greater collaboration and reflection on the importance of disability assistance among dermatologists to increase participation in the disability-assessment process.
- O’Connell JJ, Zevin BD, Quick PD, et al. Documenting disability: simple strategies for medical providers. Health Care for the Homeless Clinicians’ Network. September 2007. Accessed March 31, 2025. https://nhchc.org/wp-content/uploads/2019/08/DocumentingDisability2007.pdf
- US Bureau of Labor Statistics. Injuries, illnesses, and fatalities. Accessed March 31, 2025. https://www.bls.gov/iif/
- Meseguer J. Outcome variation in the Social Security Disability Insurance Program: the role of primary diagnoses. Soc Secur Bull. 2013;73:39-75.
- Adams RM. Attitudes of California dermatologists toward Worker’s Compensation: results of a survey. West J Med. 1976;125:169-175.
- Talmage J, Melhorn J, Hyman M. AMA Guides to the Evaluation of Work Ability and Return to Work. 2nd ed. American Medical Association; 2011.
- Social Security Administration. Disability evaluation under Social Security. 8.00 skin disorders - adult. March 31, 2025. https://www.ssa.gov/disability/professionals/bluebook/8.00-Skin-Adult.htm
- Dawson J, Smogorzewski J. Demystifying disability assessments for dermatologists—a call to action. JAMA Dermatol. 2021;157:903-904. doi:10.1001/jamadermatol.2021.1767
- O’Connell JJ, Zevin BD, Quick PD, et al. Documenting disability: simple strategies for medical providers. Health Care for the Homeless Clinicians’ Network. September 2007. Accessed March 31, 2025. https://nhchc.org/wp-content/uploads/2019/08/DocumentingDisability2007.pdf
- US Bureau of Labor Statistics. Injuries, illnesses, and fatalities. Accessed March 31, 2025. https://www.bls.gov/iif/
- Meseguer J. Outcome variation in the Social Security Disability Insurance Program: the role of primary diagnoses. Soc Secur Bull. 2013;73:39-75.
- Adams RM. Attitudes of California dermatologists toward Worker’s Compensation: results of a survey. West J Med. 1976;125:169-175.
- Talmage J, Melhorn J, Hyman M. AMA Guides to the Evaluation of Work Ability and Return to Work. 2nd ed. American Medical Association; 2011.
- Social Security Administration. Disability evaluation under Social Security. 8.00 skin disorders - adult. March 31, 2025. https://www.ssa.gov/disability/professionals/bluebook/8.00-Skin-Adult.htm
- Dawson J, Smogorzewski J. Demystifying disability assessments for dermatologists—a call to action. JAMA Dermatol. 2021;157:903-904. doi:10.1001/jamadermatol.2021.1767
Dermatologists’ Perspectives Toward Disability Assessment: A Nationwide Survey Report
Dermatologists’ Perspectives Toward Disability Assessment: A Nationwide Survey Report
PRACTICE POINTS
- As experts in skin conditions, dermatologists are most qualified to assist with disability assessment for dermatologic concerns.
- There are several barriers to dermatologists participating in the disability assessment process, including lack of time, compensation, and education on the subject.
- Many dermatologists may be interested in learning more about disability assessment, and education could be provided in the form of summary guides, lectures, and prefilled paperwork.
Analysis of Errors in the Management of Cutaneous Disorders
Analysis of Errors in the Management of Cutaneous Disorders
Humans are inherently prone to errors. The extent and consequences of medical errors were documented in the 2000 publication of To Err is Human: Building a Safer Health System.1 Published research on medical errors in dermatology has emphasized the heuristic issues involved in diagnosis,2-6 essentially approaching the “why?” and “how?” of such errors. By contrast, the current study aimed to elucidate the “what?”—what are the dermatologic conditions most prone to diagnostic and/or management errors? One study published in 1987 approached this question by analyzing patterns of errors for dermatologic conditions in patients referred for specialty care by primary care physicians.7 The current study aimed to update and expand on the findings of this 1987 report by comparing more recent data on the errors made by providers and patients regarding skin conditions.
Methods
Data were collected prospectively from March 18, 2021, through July 25, 2023. Prospective data were obtained by recording the nature of errors noted for all patients seen by a board-certified dermatologist (R.J.P.) during routine outpatient practice in Norfolk, Virginia. This practice is limited to medical dermatology and accepts patients of any age from any referral source, with or without medical insurance. Retrospective data were obtained by review of electronic medical records for all patients seen by the same board-certified dermatologist from June 5, 2020, through March 12, 2021, who previously had been seen by an outside provider or were self-referred. In this study, the term diagnosis is used to describe providers’ explicit or imputed conclusions as to the nature of a dermatosis, and the term interpretation is used to describe patients' conclusions about their own condition. For this study, the patients’ self-made interpretations of their dermatoses were deemed to be correct when they agreed with those made by the dermatologist using standard clinicopathologic criteria supplemented by rapid bedside diagnostic techniques, as detailed in the 1987 study.7
Cases in which diagnostic or therapeutic errors were noted were entered into a spreadsheet that excluded patients’ names or other identifiers. For each noted case of diagnostic or therapeutic error, the following data were entered: patient’s age and sex; the name of the incorrect diagnosis, interpretation, or treatment; and the name of the correct (missed) diagnosis, along with the source of the error (provider or patient). Provider diagnoses were determined from medical records or patient statements or were imputed from the generally accepted indications for prescribed treatments. A provider was deemed to be any practitioner with prescriptive authority. Patients’ interpretations of their conditions were determined by patient statements or were imputed based on the indications for treatments being used. A treatment error was recorded when a diagnosis or interpretation was deemed to be correct, but treatment was deemed to be inappropriate. The same dermatologist (R.J.P) made all determinations as to the nature of the errors and their source.
Diagnostic errors were determined in several situations: (1) if the interpretation made by the patient of their dermatosis differed from the correct diagnosis in the absence of any additional diagnostic documentation, the correct diagnosis was scored as a missed diagnosis and the incorrect interpretation was scored as such; (2) if the provider’s diagnosis in the patient’s medical record differed from the correct diagnosis, both the correct (missed) and incorrect diagnoses were recorded; and (3) if the indication(s) of the medication(s) prescribed by the provider or used by the patient for their condition differed from the correct diagnosis, an imputed diagnosis based on this indication was scored as the incorrect diagnosis and the correct (missed) diagnosis was recorded; for example, an error would be entered into the spreadsheet for a patient using terbinafine cream for what was actually psoriasis. For a medication with multiple active agents, an error would be entered into the spreadsheet only if none of its indications matched the correct diagnosis; for example, if the patient had been prescribed a betamethasone/clotrimazole product, no error would be scored if the correct diagnosis was a steroid-responsive dermatosis, dermatophytosis, candidiasis, or tinea versicolor. For a single medication with multiple indications, no error would be recorded if the correct diagnosis was any of these indications; for example, in a patient who had been prescribed topical ketoconazole, no error would be scored if the correct diagnosis was dermatophytosis, candidiasis, tinea versicolor, or seborrheic dermatitis. Additionally, no error would be recorded if the correct diagnosis was uncertain at the time of initial patient evaluation or during chart review.
Standard spreadsheet functions and the pandas package8 from the Python programming language9 were used to extract relevant data from the spreadsheet (Tables 1-4).




Results
A total of 446 patient visits (182 males, 264 females) were included in the study, in which a total of 486 errors were found in the combined prospective and retrospective portions of the study. These errors involved 1.4% of all patient visits for the study period—specifically, all in routine practice as well as all patient records retrospectively reviewed. The age of the patients ranged from 4 to 95 years; the mean age was 51.5 years for males and 50.8 years for females.
The study results are outlined in Tables 1 through 4. To minimize the amount of data provided with no appreciable effect on the results, cases in which an incorrect or missed diagnosis/interpretation occurred only once (ie, unique case errors) were excluded from the tables. Tables 1 and 2 indicate the numbers and types of incorrect and missed diagnoses.
In the combined patient and provider cases, there were 434 instances in which provider diagnoses and patient interpretations were incorrect, 320 (73.7%) of which involved infectious disorders. By contrast, of the 413 instances of provider and patient missed diagnoses 289 (70.0%) were inflammatory dermatoses. The pattern was similar for patients’ incorrect interpretations compared to the incorrect diagnoses of the medical providers. Patients incorrectly interpreted their dermatoses as infectious in 79.5% (101/127) of cases. Similarly, providers incorrectly diagnosed their patients’ dermatoses as infectious in 75.4% (211/280) of cases (Table 3). For patients’ missed diagnoses, 70.7% (82/116) involved inflammatory dermatoses. For providers’ missed diagnoses, 63.9% (179/280) involved inflammatory dermatoses (Table 4).
Treatment errors in the context of correct diagnoses were uncommon. Fifteen (3.4%) such cases were noted in the 446 error-containing patient visits. In 4 (26.7%) of the 15 cases, potent topical corticosteroids were used long term on inappropriate cutaneous sites (eg, genital, facial, or intertriginous areas). Another 4 (26.7%) cases involved fungal infections: nystatin used for tinea versicolor in 1 case and for dermatophytosis in another, widespread dermatophytosis treated topically, and use of a nonindicated topical antifungal for onychomycosis. Other examples involved inadequate dosing of systemic corticosteroids for extensive acute contact dermatitis, psoriasis treated with systemic corticosteroids, inadequate dosing of medication for seborrheic dermatitis, and treatment with valacyclovir based solely on serologic testing.
Comment
The results of our study indicate that errors in management of cutaneous disorders are overwhelmingly diagnostic in nature, while treatment errors appear to be unusual when the correct diagnosis is made. Both the current study and the 1987 study indicated a notable tendency of providers to incorrectly diagnose infectious disorders and to miss the diagnosis of inflammatory dermatoses.7 The current study extends this finding to include patients’ interpretive errors.
It is notable that many of the incorrect and missed diagnoses can be confirmed or ruled out by rapid bedside techniques, namely potassium hydroxide (KOH) preparation for dermatophytes, candidiasis, and tinea versicolor; wet preparation for scabies and pediculosis; Tzanck preparation for herpes simplex and herpes zoster; and crush preparation for molluscum contagiosum. Notably, 57.8% (281/486) of cases in which error was noted involved disorders for which the use of one of these bedside diagnostic tests could have correctly established a diagnosis or ruled out an incorrect one; thus in an ideal world in which these tests were performed perfectly in all appropriate cases, more than half of the errors detected in this study could have been avoided. Dermatophytosis was involved in 35.8% (174/486) of the error-containing patient encounters in this study; therefore, if only the KOH preparation is considered, more than one-third of all errors documented in this study could have been avoided. Unfortunately, surveys have suggested that among dermatologists in the United States and some other countries, KOH preparations are used infrequently.10-12
Certain limitations were inherent to this study. The data were derived from a single dermatology practice by one physician in one geographic region over a short period of time. These factors may limit the generalizability of the results. Although the goal was to identify all errors made for the patients seen, some errors likely were missed due to incomplete patient history or inaccurate medication listings. There is no absolute way to determine if the diagnoses or the treatments deemed correct by the dermatologist were, in fact, correct. For cases in which a patient’s interpretation or a provider’s diagnosis was imputed from the indication(s) associated with the medication(s) being used, one cannot exclude the possibility that a medication was used appropriately for a nonlabeled or nonstandard indication. The designation of treatment errors may be subject to different interpretations by different clinicians. Despite these limitations, it is likely that the results of this study can be extrapolated to reasonably similar dermatology practices. The apparently persistent and consistent tendency of clinicians to incorrectly diagnose infectious dermatoses and to miss inflammatory conditions has implications for teaching of medical dermatology in the academic and clinical settings. In particular, given that dermatophytosis is the diagnosis involved in the highest number of errors, special emphasis should be placed on this infection in clinician education.
Acknowledgement—The authors would like to acknowledge the essential contributions to this study by Urvi Jain (Virginia Beach, Virginia), particularly for analysis and interpretation of data and for suggestions to improve the manuscript.
- Institute of Medicine (US) Committee on Quality of Health Care in America. To Err is Human: Building a Safer Health System. Kohn LT, Corrigan JM, Donaldson MS, eds. National Academies Press; 2000.
- Lowenstein EJ, Sidlow R, Ko CJ. Visual perception, cognition, and error in dermatologic diagnosis: diagnosis and error. J Am Acad Dermatol. 2019;81:1237-1245.
- Ko CJ, Braverman I, Sidlow R, et al. Visual perception, cognition, and error in dermatologic diagnosis: key cognitive principles. J Am Acad Dermatol. 2019;81:1227-1234.
- Lowenstein EJ. Dermatology and its unique diagnostic heuristics. J Am Acad Dermatol. 2018;78:1239-1240.
- Elston DM. Cognitive bias and medical errors. J Am Acad Dermatol. 2019;81:1249.
- Costa Filho GB, Moura AS, Brandão PR, et al. Effects of deliberate reflection on diagnostic accuracy, confidence and diagnostic calibration in dermatology. Perspect Med Educ. 2019;8:230-236.
- Pariser RJ, Pariser DM. Primary physicians’ errors in handling cutaneous disorders. J Am Acad Dermatol. 1987;17:239-245.
- van Rossum G, Drake FL Jr. Python Reference Manual. Centrum voor Wiskunde en Informatica; 1995.
- The pandas development team. pandas-dev/pandas: Pandas. Zenodo. February 2020. doi:10.5281/zenodo.3509134
- Murphy EC, Friedman AJ. Use of in-office preparations by dermatologists for the diagnosis of cutaneous fungal infections. J Drugs Dermatol. 2019;18:798-802.
- Dhafiri MA, Alhamed AS, Aljughayman MA. Use of potassium hydroxide in dermatology daily practice: a local study from Saudi Arabia. Cureus. 2022;14:E30612. doi:10.7759/cureus .30612.eCollection
- Chandler JD, Yamamoto R, Hay RJ. Use of direct microscopy to diagnose superficial mycoses: a survey of UK dermatology practice. Br J Dermatol. 2023;189:480-481.
Humans are inherently prone to errors. The extent and consequences of medical errors were documented in the 2000 publication of To Err is Human: Building a Safer Health System.1 Published research on medical errors in dermatology has emphasized the heuristic issues involved in diagnosis,2-6 essentially approaching the “why?” and “how?” of such errors. By contrast, the current study aimed to elucidate the “what?”—what are the dermatologic conditions most prone to diagnostic and/or management errors? One study published in 1987 approached this question by analyzing patterns of errors for dermatologic conditions in patients referred for specialty care by primary care physicians.7 The current study aimed to update and expand on the findings of this 1987 report by comparing more recent data on the errors made by providers and patients regarding skin conditions.
Methods
Data were collected prospectively from March 18, 2021, through July 25, 2023. Prospective data were obtained by recording the nature of errors noted for all patients seen by a board-certified dermatologist (R.J.P.) during routine outpatient practice in Norfolk, Virginia. This practice is limited to medical dermatology and accepts patients of any age from any referral source, with or without medical insurance. Retrospective data were obtained by review of electronic medical records for all patients seen by the same board-certified dermatologist from June 5, 2020, through March 12, 2021, who previously had been seen by an outside provider or were self-referred. In this study, the term diagnosis is used to describe providers’ explicit or imputed conclusions as to the nature of a dermatosis, and the term interpretation is used to describe patients' conclusions about their own condition. For this study, the patients’ self-made interpretations of their dermatoses were deemed to be correct when they agreed with those made by the dermatologist using standard clinicopathologic criteria supplemented by rapid bedside diagnostic techniques, as detailed in the 1987 study.7
Cases in which diagnostic or therapeutic errors were noted were entered into a spreadsheet that excluded patients’ names or other identifiers. For each noted case of diagnostic or therapeutic error, the following data were entered: patient’s age and sex; the name of the incorrect diagnosis, interpretation, or treatment; and the name of the correct (missed) diagnosis, along with the source of the error (provider or patient). Provider diagnoses were determined from medical records or patient statements or were imputed from the generally accepted indications for prescribed treatments. A provider was deemed to be any practitioner with prescriptive authority. Patients’ interpretations of their conditions were determined by patient statements or were imputed based on the indications for treatments being used. A treatment error was recorded when a diagnosis or interpretation was deemed to be correct, but treatment was deemed to be inappropriate. The same dermatologist (R.J.P) made all determinations as to the nature of the errors and their source.
Diagnostic errors were determined in several situations: (1) if the interpretation made by the patient of their dermatosis differed from the correct diagnosis in the absence of any additional diagnostic documentation, the correct diagnosis was scored as a missed diagnosis and the incorrect interpretation was scored as such; (2) if the provider’s diagnosis in the patient’s medical record differed from the correct diagnosis, both the correct (missed) and incorrect diagnoses were recorded; and (3) if the indication(s) of the medication(s) prescribed by the provider or used by the patient for their condition differed from the correct diagnosis, an imputed diagnosis based on this indication was scored as the incorrect diagnosis and the correct (missed) diagnosis was recorded; for example, an error would be entered into the spreadsheet for a patient using terbinafine cream for what was actually psoriasis. For a medication with multiple active agents, an error would be entered into the spreadsheet only if none of its indications matched the correct diagnosis; for example, if the patient had been prescribed a betamethasone/clotrimazole product, no error would be scored if the correct diagnosis was a steroid-responsive dermatosis, dermatophytosis, candidiasis, or tinea versicolor. For a single medication with multiple indications, no error would be recorded if the correct diagnosis was any of these indications; for example, in a patient who had been prescribed topical ketoconazole, no error would be scored if the correct diagnosis was dermatophytosis, candidiasis, tinea versicolor, or seborrheic dermatitis. Additionally, no error would be recorded if the correct diagnosis was uncertain at the time of initial patient evaluation or during chart review.
Standard spreadsheet functions and the pandas package8 from the Python programming language9 were used to extract relevant data from the spreadsheet (Tables 1-4).
Results
A total of 446 patient visits (182 males, 264 females) were included in the study, in which a total of 486 errors were found in the combined prospective and retrospective portions of the study. These errors involved 1.4% of all patient visits for the study period—specifically, all in routine practice as well as all patient records retrospectively reviewed. The age of the patients ranged from 4 to 95 years; the mean age was 51.5 years for males and 50.8 years for females.
The study results are outlined in Tables 1 through 4. To minimize the amount of data provided with no appreciable effect on the results, cases in which an incorrect or missed diagnosis/interpretation occurred only once (ie, unique case errors) were excluded from the tables. Tables 1 and 2 indicate the numbers and types of incorrect and missed diagnoses.
In the combined patient and provider cases, there were 434 instances in which provider diagnoses and patient interpretations were incorrect, 320 (73.7%) of which involved infectious disorders. By contrast, of the 413 instances of provider and patient missed diagnoses 289 (70.0%) were inflammatory dermatoses. The pattern was similar for patients’ incorrect interpretations compared to the incorrect diagnoses of the medical providers. Patients incorrectly interpreted their dermatoses as infectious in 79.5% (101/127) of cases. Similarly, providers incorrectly diagnosed their patients’ dermatoses as infectious in 75.4% (211/280) of cases (Table 3). For patients’ missed diagnoses, 70.7% (82/116) involved inflammatory dermatoses. For providers’ missed diagnoses, 63.9% (179/280) involved inflammatory dermatoses (Table 4).
Treatment errors in the context of correct diagnoses were uncommon. Fifteen (3.4%) such cases were noted in the 446 error-containing patient visits. In 4 (26.7%) of the 15 cases, potent topical corticosteroids were used long term on inappropriate cutaneous sites (eg, genital, facial, or intertriginous areas). Another 4 (26.7%) cases involved fungal infections: nystatin used for tinea versicolor in 1 case and for dermatophytosis in another, widespread dermatophytosis treated topically, and use of a nonindicated topical antifungal for onychomycosis. Other examples involved inadequate dosing of systemic corticosteroids for extensive acute contact dermatitis, psoriasis treated with systemic corticosteroids, inadequate dosing of medication for seborrheic dermatitis, and treatment with valacyclovir based solely on serologic testing.
Comment
The results of our study indicate that errors in management of cutaneous disorders are overwhelmingly diagnostic in nature, while treatment errors appear to be unusual when the correct diagnosis is made. Both the current study and the 1987 study indicated a notable tendency of providers to incorrectly diagnose infectious disorders and to miss the diagnosis of inflammatory dermatoses.7 The current study extends this finding to include patients’ interpretive errors.
It is notable that many of the incorrect and missed diagnoses can be confirmed or ruled out by rapid bedside techniques, namely potassium hydroxide (KOH) preparation for dermatophytes, candidiasis, and tinea versicolor; wet preparation for scabies and pediculosis; Tzanck preparation for herpes simplex and herpes zoster; and crush preparation for molluscum contagiosum. Notably, 57.8% (281/486) of cases in which error was noted involved disorders for which the use of one of these bedside diagnostic tests could have correctly established a diagnosis or ruled out an incorrect one; thus in an ideal world in which these tests were performed perfectly in all appropriate cases, more than half of the errors detected in this study could have been avoided. Dermatophytosis was involved in 35.8% (174/486) of the error-containing patient encounters in this study; therefore, if only the KOH preparation is considered, more than one-third of all errors documented in this study could have been avoided. Unfortunately, surveys have suggested that among dermatologists in the United States and some other countries, KOH preparations are used infrequently.10-12
Certain limitations were inherent to this study. The data were derived from a single dermatology practice by one physician in one geographic region over a short period of time. These factors may limit the generalizability of the results. Although the goal was to identify all errors made for the patients seen, some errors likely were missed due to incomplete patient history or inaccurate medication listings. There is no absolute way to determine if the diagnoses or the treatments deemed correct by the dermatologist were, in fact, correct. For cases in which a patient’s interpretation or a provider’s diagnosis was imputed from the indication(s) associated with the medication(s) being used, one cannot exclude the possibility that a medication was used appropriately for a nonlabeled or nonstandard indication. The designation of treatment errors may be subject to different interpretations by different clinicians. Despite these limitations, it is likely that the results of this study can be extrapolated to reasonably similar dermatology practices. The apparently persistent and consistent tendency of clinicians to incorrectly diagnose infectious dermatoses and to miss inflammatory conditions has implications for teaching of medical dermatology in the academic and clinical settings. In particular, given that dermatophytosis is the diagnosis involved in the highest number of errors, special emphasis should be placed on this infection in clinician education.
Acknowledgement—The authors would like to acknowledge the essential contributions to this study by Urvi Jain (Virginia Beach, Virginia), particularly for analysis and interpretation of data and for suggestions to improve the manuscript.
Humans are inherently prone to errors. The extent and consequences of medical errors were documented in the 2000 publication of To Err is Human: Building a Safer Health System.1 Published research on medical errors in dermatology has emphasized the heuristic issues involved in diagnosis,2-6 essentially approaching the “why?” and “how?” of such errors. By contrast, the current study aimed to elucidate the “what?”—what are the dermatologic conditions most prone to diagnostic and/or management errors? One study published in 1987 approached this question by analyzing patterns of errors for dermatologic conditions in patients referred for specialty care by primary care physicians.7 The current study aimed to update and expand on the findings of this 1987 report by comparing more recent data on the errors made by providers and patients regarding skin conditions.
Methods
Data were collected prospectively from March 18, 2021, through July 25, 2023. Prospective data were obtained by recording the nature of errors noted for all patients seen by a board-certified dermatologist (R.J.P.) during routine outpatient practice in Norfolk, Virginia. This practice is limited to medical dermatology and accepts patients of any age from any referral source, with or without medical insurance. Retrospective data were obtained by review of electronic medical records for all patients seen by the same board-certified dermatologist from June 5, 2020, through March 12, 2021, who previously had been seen by an outside provider or were self-referred. In this study, the term diagnosis is used to describe providers’ explicit or imputed conclusions as to the nature of a dermatosis, and the term interpretation is used to describe patients' conclusions about their own condition. For this study, the patients’ self-made interpretations of their dermatoses were deemed to be correct when they agreed with those made by the dermatologist using standard clinicopathologic criteria supplemented by rapid bedside diagnostic techniques, as detailed in the 1987 study.7
Cases in which diagnostic or therapeutic errors were noted were entered into a spreadsheet that excluded patients’ names or other identifiers. For each noted case of diagnostic or therapeutic error, the following data were entered: patient’s age and sex; the name of the incorrect diagnosis, interpretation, or treatment; and the name of the correct (missed) diagnosis, along with the source of the error (provider or patient). Provider diagnoses were determined from medical records or patient statements or were imputed from the generally accepted indications for prescribed treatments. A provider was deemed to be any practitioner with prescriptive authority. Patients’ interpretations of their conditions were determined by patient statements or were imputed based on the indications for treatments being used. A treatment error was recorded when a diagnosis or interpretation was deemed to be correct, but treatment was deemed to be inappropriate. The same dermatologist (R.J.P) made all determinations as to the nature of the errors and their source.
Diagnostic errors were determined in several situations: (1) if the interpretation made by the patient of their dermatosis differed from the correct diagnosis in the absence of any additional diagnostic documentation, the correct diagnosis was scored as a missed diagnosis and the incorrect interpretation was scored as such; (2) if the provider’s diagnosis in the patient’s medical record differed from the correct diagnosis, both the correct (missed) and incorrect diagnoses were recorded; and (3) if the indication(s) of the medication(s) prescribed by the provider or used by the patient for their condition differed from the correct diagnosis, an imputed diagnosis based on this indication was scored as the incorrect diagnosis and the correct (missed) diagnosis was recorded; for example, an error would be entered into the spreadsheet for a patient using terbinafine cream for what was actually psoriasis. For a medication with multiple active agents, an error would be entered into the spreadsheet only if none of its indications matched the correct diagnosis; for example, if the patient had been prescribed a betamethasone/clotrimazole product, no error would be scored if the correct diagnosis was a steroid-responsive dermatosis, dermatophytosis, candidiasis, or tinea versicolor. For a single medication with multiple indications, no error would be recorded if the correct diagnosis was any of these indications; for example, in a patient who had been prescribed topical ketoconazole, no error would be scored if the correct diagnosis was dermatophytosis, candidiasis, tinea versicolor, or seborrheic dermatitis. Additionally, no error would be recorded if the correct diagnosis was uncertain at the time of initial patient evaluation or during chart review.
Standard spreadsheet functions and the pandas package8 from the Python programming language9 were used to extract relevant data from the spreadsheet (Tables 1-4).
Results
A total of 446 patient visits (182 males, 264 females) were included in the study, in which a total of 486 errors were found in the combined prospective and retrospective portions of the study. These errors involved 1.4% of all patient visits for the study period—specifically, all in routine practice as well as all patient records retrospectively reviewed. The age of the patients ranged from 4 to 95 years; the mean age was 51.5 years for males and 50.8 years for females.
The study results are outlined in Tables 1 through 4. To minimize the amount of data provided with no appreciable effect on the results, cases in which an incorrect or missed diagnosis/interpretation occurred only once (ie, unique case errors) were excluded from the tables. Tables 1 and 2 indicate the numbers and types of incorrect and missed diagnoses.
In the combined patient and provider cases, there were 434 instances in which provider diagnoses and patient interpretations were incorrect, 320 (73.7%) of which involved infectious disorders. By contrast, of the 413 instances of provider and patient missed diagnoses 289 (70.0%) were inflammatory dermatoses. The pattern was similar for patients’ incorrect interpretations compared to the incorrect diagnoses of the medical providers. Patients incorrectly interpreted their dermatoses as infectious in 79.5% (101/127) of cases. Similarly, providers incorrectly diagnosed their patients’ dermatoses as infectious in 75.4% (211/280) of cases (Table 3). For patients’ missed diagnoses, 70.7% (82/116) involved inflammatory dermatoses. For providers’ missed diagnoses, 63.9% (179/280) involved inflammatory dermatoses (Table 4).
Treatment errors in the context of correct diagnoses were uncommon. Fifteen (3.4%) such cases were noted in the 446 error-containing patient visits. In 4 (26.7%) of the 15 cases, potent topical corticosteroids were used long term on inappropriate cutaneous sites (eg, genital, facial, or intertriginous areas). Another 4 (26.7%) cases involved fungal infections: nystatin used for tinea versicolor in 1 case and for dermatophytosis in another, widespread dermatophytosis treated topically, and use of a nonindicated topical antifungal for onychomycosis. Other examples involved inadequate dosing of systemic corticosteroids for extensive acute contact dermatitis, psoriasis treated with systemic corticosteroids, inadequate dosing of medication for seborrheic dermatitis, and treatment with valacyclovir based solely on serologic testing.
Comment
The results of our study indicate that errors in management of cutaneous disorders are overwhelmingly diagnostic in nature, while treatment errors appear to be unusual when the correct diagnosis is made. Both the current study and the 1987 study indicated a notable tendency of providers to incorrectly diagnose infectious disorders and to miss the diagnosis of inflammatory dermatoses.7 The current study extends this finding to include patients’ interpretive errors.
It is notable that many of the incorrect and missed diagnoses can be confirmed or ruled out by rapid bedside techniques, namely potassium hydroxide (KOH) preparation for dermatophytes, candidiasis, and tinea versicolor; wet preparation for scabies and pediculosis; Tzanck preparation for herpes simplex and herpes zoster; and crush preparation for molluscum contagiosum. Notably, 57.8% (281/486) of cases in which error was noted involved disorders for which the use of one of these bedside diagnostic tests could have correctly established a diagnosis or ruled out an incorrect one; thus in an ideal world in which these tests were performed perfectly in all appropriate cases, more than half of the errors detected in this study could have been avoided. Dermatophytosis was involved in 35.8% (174/486) of the error-containing patient encounters in this study; therefore, if only the KOH preparation is considered, more than one-third of all errors documented in this study could have been avoided. Unfortunately, surveys have suggested that among dermatologists in the United States and some other countries, KOH preparations are used infrequently.10-12
Certain limitations were inherent to this study. The data were derived from a single dermatology practice by one physician in one geographic region over a short period of time. These factors may limit the generalizability of the results. Although the goal was to identify all errors made for the patients seen, some errors likely were missed due to incomplete patient history or inaccurate medication listings. There is no absolute way to determine if the diagnoses or the treatments deemed correct by the dermatologist were, in fact, correct. For cases in which a patient’s interpretation or a provider’s diagnosis was imputed from the indication(s) associated with the medication(s) being used, one cannot exclude the possibility that a medication was used appropriately for a nonlabeled or nonstandard indication. The designation of treatment errors may be subject to different interpretations by different clinicians. Despite these limitations, it is likely that the results of this study can be extrapolated to reasonably similar dermatology practices. The apparently persistent and consistent tendency of clinicians to incorrectly diagnose infectious dermatoses and to miss inflammatory conditions has implications for teaching of medical dermatology in the academic and clinical settings. In particular, given that dermatophytosis is the diagnosis involved in the highest number of errors, special emphasis should be placed on this infection in clinician education.
Acknowledgement—The authors would like to acknowledge the essential contributions to this study by Urvi Jain (Virginia Beach, Virginia), particularly for analysis and interpretation of data and for suggestions to improve the manuscript.
- Institute of Medicine (US) Committee on Quality of Health Care in America. To Err is Human: Building a Safer Health System. Kohn LT, Corrigan JM, Donaldson MS, eds. National Academies Press; 2000.
- Lowenstein EJ, Sidlow R, Ko CJ. Visual perception, cognition, and error in dermatologic diagnosis: diagnosis and error. J Am Acad Dermatol. 2019;81:1237-1245.
- Ko CJ, Braverman I, Sidlow R, et al. Visual perception, cognition, and error in dermatologic diagnosis: key cognitive principles. J Am Acad Dermatol. 2019;81:1227-1234.
- Lowenstein EJ. Dermatology and its unique diagnostic heuristics. J Am Acad Dermatol. 2018;78:1239-1240.
- Elston DM. Cognitive bias and medical errors. J Am Acad Dermatol. 2019;81:1249.
- Costa Filho GB, Moura AS, Brandão PR, et al. Effects of deliberate reflection on diagnostic accuracy, confidence and diagnostic calibration in dermatology. Perspect Med Educ. 2019;8:230-236.
- Pariser RJ, Pariser DM. Primary physicians’ errors in handling cutaneous disorders. J Am Acad Dermatol. 1987;17:239-245.
- van Rossum G, Drake FL Jr. Python Reference Manual. Centrum voor Wiskunde en Informatica; 1995.
- The pandas development team. pandas-dev/pandas: Pandas. Zenodo. February 2020. doi:10.5281/zenodo.3509134
- Murphy EC, Friedman AJ. Use of in-office preparations by dermatologists for the diagnosis of cutaneous fungal infections. J Drugs Dermatol. 2019;18:798-802.
- Dhafiri MA, Alhamed AS, Aljughayman MA. Use of potassium hydroxide in dermatology daily practice: a local study from Saudi Arabia. Cureus. 2022;14:E30612. doi:10.7759/cureus .30612.eCollection
- Chandler JD, Yamamoto R, Hay RJ. Use of direct microscopy to diagnose superficial mycoses: a survey of UK dermatology practice. Br J Dermatol. 2023;189:480-481.
- Institute of Medicine (US) Committee on Quality of Health Care in America. To Err is Human: Building a Safer Health System. Kohn LT, Corrigan JM, Donaldson MS, eds. National Academies Press; 2000.
- Lowenstein EJ, Sidlow R, Ko CJ. Visual perception, cognition, and error in dermatologic diagnosis: diagnosis and error. J Am Acad Dermatol. 2019;81:1237-1245.
- Ko CJ, Braverman I, Sidlow R, et al. Visual perception, cognition, and error in dermatologic diagnosis: key cognitive principles. J Am Acad Dermatol. 2019;81:1227-1234.
- Lowenstein EJ. Dermatology and its unique diagnostic heuristics. J Am Acad Dermatol. 2018;78:1239-1240.
- Elston DM. Cognitive bias and medical errors. J Am Acad Dermatol. 2019;81:1249.
- Costa Filho GB, Moura AS, Brandão PR, et al. Effects of deliberate reflection on diagnostic accuracy, confidence and diagnostic calibration in dermatology. Perspect Med Educ. 2019;8:230-236.
- Pariser RJ, Pariser DM. Primary physicians’ errors in handling cutaneous disorders. J Am Acad Dermatol. 1987;17:239-245.
- van Rossum G, Drake FL Jr. Python Reference Manual. Centrum voor Wiskunde en Informatica; 1995.
- The pandas development team. pandas-dev/pandas: Pandas. Zenodo. February 2020. doi:10.5281/zenodo.3509134
- Murphy EC, Friedman AJ. Use of in-office preparations by dermatologists for the diagnosis of cutaneous fungal infections. J Drugs Dermatol. 2019;18:798-802.
- Dhafiri MA, Alhamed AS, Aljughayman MA. Use of potassium hydroxide in dermatology daily practice: a local study from Saudi Arabia. Cureus. 2022;14:E30612. doi:10.7759/cureus .30612.eCollection
- Chandler JD, Yamamoto R, Hay RJ. Use of direct microscopy to diagnose superficial mycoses: a survey of UK dermatology practice. Br J Dermatol. 2023;189:480-481.
Analysis of Errors in the Management of Cutaneous Disorders
Analysis of Errors in the Management of Cutaneous Disorders
PRACTICE POINTS
- Errors in the management of cutaneous disorders predominantly are due to misdiagnosis rather than treatment oversights.
- There is a tendency among medical providers to incorrectly diagnose dermatoses as infectious disorders and to miss the diagnosis of inflammatory dermatoses.
- A similar pattern of errors occurs for patients’ interpretations of their own skin conditions.
- Use of available rapid bedside diagnostic techniques can reduce the likelihood of errors made by medical providers.
White Atrophic Plaques on the Thighs
White Atrophic Plaques on the Thighs
THE DIAGNOSIS: Lichen Sclerosus
Given the clinical appearance of white atrophic plaques with characteristic wrinkling of the skin, a diagnosis of lichen sclerosus was strongly suspected. At the initial office visit, the patient was prescribed clobetasol 0.05% ointment twice daily for 6 weeks. Histopathology revealed hyperkeratosis, follicular plugging, papillary dermal pallor, and adjacent lymphocytic inflammation, confirming the clinical diagnosis of lichen sclerosus (Figure). The patient then was lost to follow-up.
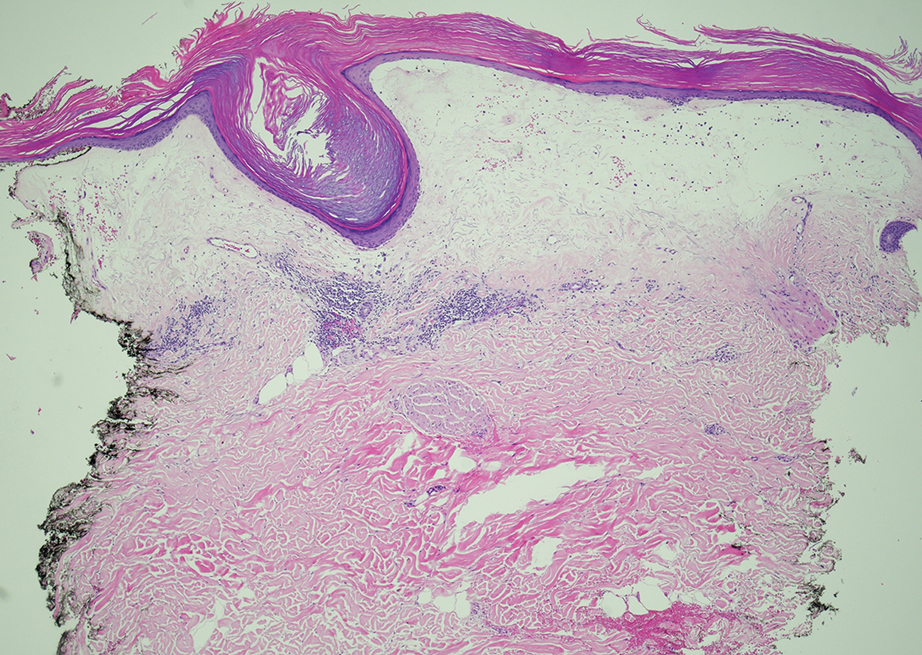
Lichen sclerosus is a chronic benign dermatologic condition of unknown etiology that is characterized by epidermal atrophy and inflammation and is common in postmenopausal women. It features pale, ivory-colored lesions with partially atrophic skin and a wrinkled cigarette paper appearance.1 The differential for lichen sclerosus is broad, and definitive diagnosis is made via biopsy to rule out potential malignancy and other inflammatory skin diseases.1 Lichen sclerosus is an immune-mediated disorder driven by type 1 T helper cells and regulated by miR-155. There has been an association with extracellular matrix protein 1, a glycoprotein that is found in the dermal-epidermal basement membrane zone, which provides structural integrity to the skin. Autoantibodies against extracellular matrix protein 1 and other antigens in the basement membrane generally are found in anogenital lichen sclerosus; however, their precise roles in the pathogenesis of lichen sclerosus remains unclear.1
The differential diagnoses for lichen sclerosus include psoriasis, tinea corporis, lichen simplex chronicus, and atopic dermatitis. Psoriasis typically manifests as pink plaques with silver scales on the elbows, knees, and scalp in adult patients.2 Our patient’s white plaques may have suggested psoriasis, but the partially atrophic skin with a wrinkled cigarette paper appearance was not compatible with that diagnosis.
Tinea corporis, a superficial fungal infection of the skin, manifests as circular or ovoid lesions with raised erythematous scaly borders, often with central clearing resembling a ring, that can occur anywhere on the body other than the feet, groin, face, scalp, or beard area.3 The fact that our patient previously had tried topical antifungal medications with no relief and that the skin lesions were atrophic rather than ring shaped made the diagnosis of tinea corporis unlikely.
Lichen simplex chronicus is a chronic condition caused by friction or scratching that is characterized by dry, patchy, scaly, and thickened areas of the skin. Typically affecting the head, arms, neck, scalp, and genital region, lichen simplex chronicus manifests with violaceous or hyperpigmented lesions.4 The nonpruritic atrophic plaques on the inner thighs and the presence of white patches on the vaginal area were not indicative of lichen simplex chronicus in our patient.
Atopic dermatitis manifests as pruritic erythematous scaly papules and plaques with secondary excoriation and possible lichenification. In adults, atopic dermatitis commonly appears on flexural surfaces.2 Atopic dermatitis does not manifest with atrophy and skin wrinkling as seen in our patient.
In the management of lichen sclerosus, the standard treatment is potent topical corticosteroids. Alternatively, topical calcineurin inhibitors can be employed; however, due to the unknown nature of the condition’s underlying cause, targeted treatment is challenging. Our case underscores how lichen sclerosus can be misdiagnosed, highlighting the need for more frequent reporting in the literature to enhance early recognition and reduce delays in patient treatment.
- De Luca DA, Papara C, Vorobyev A, et al. Lichen sclerosus: the 2023 update. Front Med (Lausanne). 2023;10:1106318. doi:10.3389 /fmed.2023.1106318
- Chovatiya R, Silverberg JI. Pathophysiology of atopic dermatitis and psoriasis: implications for management in children. Children (Basel). 2019;6:108. doi:10.3390/children6100108
- Trayes KP, Savage K, Studdiford JS. Annular lesions: diagnosis and treatment. Am Fam Physician. 2018;98:283-291.
- Ju T, Vander Does A, Mohsin N, et al. Lichen simplex chronicus itch: an update. Acta Derm Venereol. 2022;102:adv00796. doi:10.2340 /actadv.v102.4367
THE DIAGNOSIS: Lichen Sclerosus
Given the clinical appearance of white atrophic plaques with characteristic wrinkling of the skin, a diagnosis of lichen sclerosus was strongly suspected. At the initial office visit, the patient was prescribed clobetasol 0.05% ointment twice daily for 6 weeks. Histopathology revealed hyperkeratosis, follicular plugging, papillary dermal pallor, and adjacent lymphocytic inflammation, confirming the clinical diagnosis of lichen sclerosus (Figure). The patient then was lost to follow-up.
Lichen sclerosus is a chronic benign dermatologic condition of unknown etiology that is characterized by epidermal atrophy and inflammation and is common in postmenopausal women. It features pale, ivory-colored lesions with partially atrophic skin and a wrinkled cigarette paper appearance.1 The differential for lichen sclerosus is broad, and definitive diagnosis is made via biopsy to rule out potential malignancy and other inflammatory skin diseases.1 Lichen sclerosus is an immune-mediated disorder driven by type 1 T helper cells and regulated by miR-155. There has been an association with extracellular matrix protein 1, a glycoprotein that is found in the dermal-epidermal basement membrane zone, which provides structural integrity to the skin. Autoantibodies against extracellular matrix protein 1 and other antigens in the basement membrane generally are found in anogenital lichen sclerosus; however, their precise roles in the pathogenesis of lichen sclerosus remains unclear.1
The differential diagnoses for lichen sclerosus include psoriasis, tinea corporis, lichen simplex chronicus, and atopic dermatitis. Psoriasis typically manifests as pink plaques with silver scales on the elbows, knees, and scalp in adult patients.2 Our patient’s white plaques may have suggested psoriasis, but the partially atrophic skin with a wrinkled cigarette paper appearance was not compatible with that diagnosis.
Tinea corporis, a superficial fungal infection of the skin, manifests as circular or ovoid lesions with raised erythematous scaly borders, often with central clearing resembling a ring, that can occur anywhere on the body other than the feet, groin, face, scalp, or beard area.3 The fact that our patient previously had tried topical antifungal medications with no relief and that the skin lesions were atrophic rather than ring shaped made the diagnosis of tinea corporis unlikely.
Lichen simplex chronicus is a chronic condition caused by friction or scratching that is characterized by dry, patchy, scaly, and thickened areas of the skin. Typically affecting the head, arms, neck, scalp, and genital region, lichen simplex chronicus manifests with violaceous or hyperpigmented lesions.4 The nonpruritic atrophic plaques on the inner thighs and the presence of white patches on the vaginal area were not indicative of lichen simplex chronicus in our patient.
Atopic dermatitis manifests as pruritic erythematous scaly papules and plaques with secondary excoriation and possible lichenification. In adults, atopic dermatitis commonly appears on flexural surfaces.2 Atopic dermatitis does not manifest with atrophy and skin wrinkling as seen in our patient.
In the management of lichen sclerosus, the standard treatment is potent topical corticosteroids. Alternatively, topical calcineurin inhibitors can be employed; however, due to the unknown nature of the condition’s underlying cause, targeted treatment is challenging. Our case underscores how lichen sclerosus can be misdiagnosed, highlighting the need for more frequent reporting in the literature to enhance early recognition and reduce delays in patient treatment.
THE DIAGNOSIS: Lichen Sclerosus
Given the clinical appearance of white atrophic plaques with characteristic wrinkling of the skin, a diagnosis of lichen sclerosus was strongly suspected. At the initial office visit, the patient was prescribed clobetasol 0.05% ointment twice daily for 6 weeks. Histopathology revealed hyperkeratosis, follicular plugging, papillary dermal pallor, and adjacent lymphocytic inflammation, confirming the clinical diagnosis of lichen sclerosus (Figure). The patient then was lost to follow-up.
Lichen sclerosus is a chronic benign dermatologic condition of unknown etiology that is characterized by epidermal atrophy and inflammation and is common in postmenopausal women. It features pale, ivory-colored lesions with partially atrophic skin and a wrinkled cigarette paper appearance.1 The differential for lichen sclerosus is broad, and definitive diagnosis is made via biopsy to rule out potential malignancy and other inflammatory skin diseases.1 Lichen sclerosus is an immune-mediated disorder driven by type 1 T helper cells and regulated by miR-155. There has been an association with extracellular matrix protein 1, a glycoprotein that is found in the dermal-epidermal basement membrane zone, which provides structural integrity to the skin. Autoantibodies against extracellular matrix protein 1 and other antigens in the basement membrane generally are found in anogenital lichen sclerosus; however, their precise roles in the pathogenesis of lichen sclerosus remains unclear.1
The differential diagnoses for lichen sclerosus include psoriasis, tinea corporis, lichen simplex chronicus, and atopic dermatitis. Psoriasis typically manifests as pink plaques with silver scales on the elbows, knees, and scalp in adult patients.2 Our patient’s white plaques may have suggested psoriasis, but the partially atrophic skin with a wrinkled cigarette paper appearance was not compatible with that diagnosis.
Tinea corporis, a superficial fungal infection of the skin, manifests as circular or ovoid lesions with raised erythematous scaly borders, often with central clearing resembling a ring, that can occur anywhere on the body other than the feet, groin, face, scalp, or beard area.3 The fact that our patient previously had tried topical antifungal medications with no relief and that the skin lesions were atrophic rather than ring shaped made the diagnosis of tinea corporis unlikely.
Lichen simplex chronicus is a chronic condition caused by friction or scratching that is characterized by dry, patchy, scaly, and thickened areas of the skin. Typically affecting the head, arms, neck, scalp, and genital region, lichen simplex chronicus manifests with violaceous or hyperpigmented lesions.4 The nonpruritic atrophic plaques on the inner thighs and the presence of white patches on the vaginal area were not indicative of lichen simplex chronicus in our patient.
Atopic dermatitis manifests as pruritic erythematous scaly papules and plaques with secondary excoriation and possible lichenification. In adults, atopic dermatitis commonly appears on flexural surfaces.2 Atopic dermatitis does not manifest with atrophy and skin wrinkling as seen in our patient.
In the management of lichen sclerosus, the standard treatment is potent topical corticosteroids. Alternatively, topical calcineurin inhibitors can be employed; however, due to the unknown nature of the condition’s underlying cause, targeted treatment is challenging. Our case underscores how lichen sclerosus can be misdiagnosed, highlighting the need for more frequent reporting in the literature to enhance early recognition and reduce delays in patient treatment.
- De Luca DA, Papara C, Vorobyev A, et al. Lichen sclerosus: the 2023 update. Front Med (Lausanne). 2023;10:1106318. doi:10.3389 /fmed.2023.1106318
- Chovatiya R, Silverberg JI. Pathophysiology of atopic dermatitis and psoriasis: implications for management in children. Children (Basel). 2019;6:108. doi:10.3390/children6100108
- Trayes KP, Savage K, Studdiford JS. Annular lesions: diagnosis and treatment. Am Fam Physician. 2018;98:283-291.
- Ju T, Vander Does A, Mohsin N, et al. Lichen simplex chronicus itch: an update. Acta Derm Venereol. 2022;102:adv00796. doi:10.2340 /actadv.v102.4367
- De Luca DA, Papara C, Vorobyev A, et al. Lichen sclerosus: the 2023 update. Front Med (Lausanne). 2023;10:1106318. doi:10.3389 /fmed.2023.1106318
- Chovatiya R, Silverberg JI. Pathophysiology of atopic dermatitis and psoriasis: implications for management in children. Children (Basel). 2019;6:108. doi:10.3390/children6100108
- Trayes KP, Savage K, Studdiford JS. Annular lesions: diagnosis and treatment. Am Fam Physician. 2018;98:283-291.
- Ju T, Vander Does A, Mohsin N, et al. Lichen simplex chronicus itch: an update. Acta Derm Venereol. 2022;102:adv00796. doi:10.2340 /actadv.v102.4367
White Atrophic Plaques on the Thighs
White Atrophic Plaques on the Thighs
A 71-year-old woman presented to the dermatology clinic for evaluation of intense pruritus of the vaginal region and a nonpruritic rash on the inner thighs of 7 months’ duration. Physical examination revealed white atrophic plaques with scaling and a wrinkled appearance on the inner thighs. White atrophic patches also were noted on the vulva. The patient reported that she had tried over-the-counter antifungals with no improvement. A punch biopsy was performed.
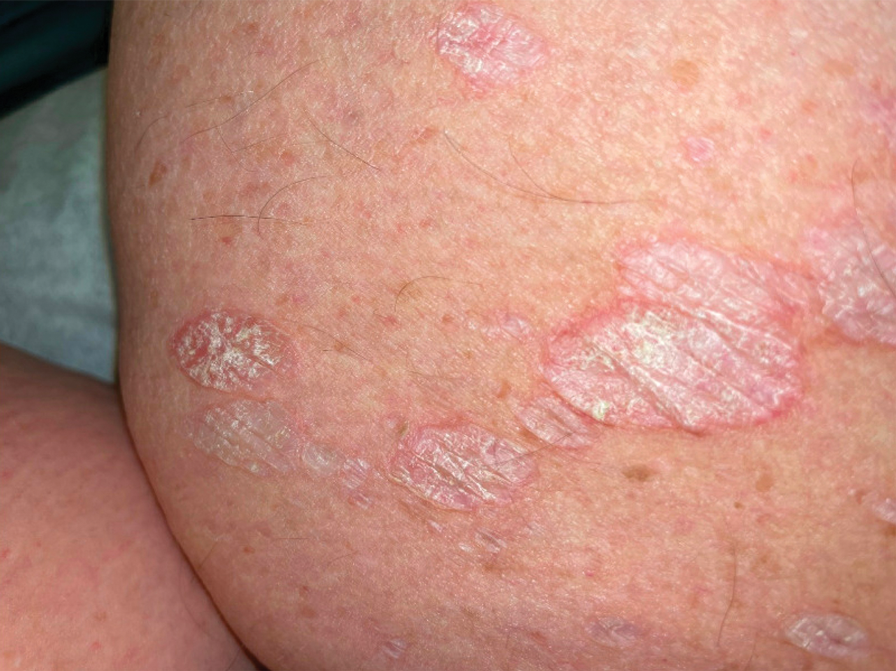

Beyond the Razor: Managing Pseudofolliculitis Barbae in Skin of Color
Beyond the Razor: Managing Pseudofolliculitis Barbae in Skin of Color
THE COMPARISON
- A. Pustules, erythematous to violaceous nodules, and hyperpigmented patches on the lower cheek and chin.
- B. Brown papules, pink keloidal papules and nodules, pustules, and hyperpigmented papules on the mandibular area and neck.
- C. Coarse hairs, pustules, and pink papules on the mandibular area and neck.
Pseudofolliculitis barbae (PFB), also known as razor bumps, is a common inflammatory condition characterized by papules and pustules that typically appear in the beard and cheek regions. It occurs when shaved hair regrows and penetrates the skin, leading to irritation and inflammation. While anyone who shaves can develop PFB, it is more prevalent and severe in individuals with naturally tightly coiled, coarse-textured hair.1,2 Pseudofolliculitis barbae is common in individuals who shave frequently due to personal choice or profession, such as members of the US military3,4 and firefighters, who are required to remain clean shaven for safety (eg, ensuring proper fit of a respirator mask).5 Early diagnosis and treatment of PFB are essential to prevent long-term complications such as scarring or hyperpigmentation, which may be more severe in those with darker skin tones.
Epidemiology
Pseudofolliculitis barbae is most common in Black men, affecting 45% to 83% of men of African ancestry.1,2 This condition also can affect individuals of various ethnicities with coarse or curly hair. The spiral shape of the hair increases the likelihood that it will regrow into the skin after shaving.6 Women with hirsutism who shave also can develop PFB.
Key Clinical Features
The papules and pustules seen in PFB may be flesh colored, erythematous, hyperpigmented, brown, or violaceous. Erythema may be less pronounced in darker vs lighter skin tones. Persistent and severe postinflammatory hyperpigmentation may occur, and hypertrophic or keloidal scars may develop in affected areas. Dermoscopy may reveal extrafollicular hair penetration as well as follicular or perifollicular pustules accompanied by hyperkeratosis.
Worth Noting
The most effective management for PFB is to discontinue shaving.1 If shaving is desired or necessary, it is recommended that patients apply lukewarm water to the affected area followed by a generous amount of shaving foam or gel to create a protective antifriction layer that allows the razor to glide more smoothly over the skin and reduces subsequent irritation.2 Using the right razor technology also may help alleviate symptoms. Research has shown that multiblade razors used in conjunction with preshave hair hydration and postshave moisturization do not worsen PFB.2 A recent study found that multiblade razor technology paired with use of a shave foam or gel actually improved skin appearance in patients with PFB.7
It is important to direct patients to shave in the direction of hair growth; however, this may not be possible for individuals with curly or coarse hair, as the hair may grow in many directions.8,9 Patients also should avoid pulling the skin taut while shaving, as doing so allows the hair to be clipped below the surface, where it can repenetrate the skin and cause further irritation. As an alternative to shaving with a razor, patients can use hair clippers to trim beard hair, which leaves behind stubble and interrupts the cycle of retracted hairs under the skin. Nd:YAG laser therapy has demonstrated efficacy in reduction of PFB papules and pustules.9-12 Greater mean improvement in inflammatory papules and reduction in hair density was noted in participants who received Nd:YAG laser plus eflornithine compared with those who received the laser or eflornithine alone.11 Patients should not pluck or dig into the skin to remove any ingrown hairs. If a tweezer is used, the patient should gently lift the tip of the ingrown hair with the tweezer to dislodge it from the skin and prevent plucking out the hair completely.
To help manage inflammation after shaving, topical treatments such as benzoyl peroxide 5%/clindamycin 1% gel can be used.3,13 A low-potency steroid such as topical hydrocortisone 2.5% applied once or twice daily for up to 2 to 3 days may be helpful.1,14 Adjunctive treatments including keratolytics (eg, topical retinoids, hydroxy acids) reduce perifollicular hyperkeratosis.14,15 Agents containing alpha hydroxy acids (eg, glycolic acid) also can decrease the curvature of the hair itself by reducing the sulfhydryl bonds.6 If secondary bacterial infections occur, oral antibiotics (eg, doxycycline) may be necessary.
Health Disparity Highlight
Individuals with darker skin tones are at higher risk for PFB and associated complications. Limited access to dermatology services may further exacerbate these challenges. Individuals with PFB may not seek medical treatment until the condition becomes severe. Clinicians also may underestimate the severity of PFB—particularly in those with darker skin tones—based on erythema alone because it may be less pronounced in darker vs lighter skin tones.16
While permanent hair reduction with laser therapy is a treatment option for PFB, it may be inaccessible to some patients because it can be expensive and is coded as a cosmetic procedure. Additionally, patients may not have access to specialists who are experienced in performing the procedure in those with darker skin tones.9 Some patients also may not want to permanently reduce the amount of hair that grows in the beard area for personal or religious reasons.17
Pseudofolliculitis barbae also has been linked to professional disparities. One study found that members of the US Air Force who had medical shaving waivers experienced longer times to promotion than those with no waiver.18 Delays in promotion may be linked to perceptions of unprofessionalism, exclusion from high-profile duties, and concerns about career progression. While this delay was similar for individuals of all races, the majority of those in the waiver group were Black/African American. In 2021, 4 Black firefighters with PFB were unsuccessful in their bid to get a medical accommodation regarding a New York City Fire Department policy requiring them to be clean shaven where the oxygen mask seals against the skin.5 More research is needed on mask safety and efficiency relative to the length of facial hair. Accommodations or tailored masks for facial hair conditions also are necessary so individuals with PFB can meet job requirements while managing their condition.
- Alexis A, Heath CR, Halder RM. Folliculitis keloidalis nuchae and pseudofolliculitis barbae: are prevention and effective treatment within reach? em>Dermatol Clin. 2014;32:183-191.
- Gray J, McMichael AJ. Pseudofolliculitis barbae: understanding the condition and the role of facial grooming. Int J Cosmet Sci. 2016;38 (suppl 1):24-27.
- Tshudy MT, Cho S. Pseudofolliculitis barbae in the U.S. military, a review. Mil Med. 2021;186:E52-E57.
- Jung I, Lannan FM, Weiss A, et al. Treatment and current policies on pseudofolliculitis barbae in the US military. Cutis. 2023;112:299-302.
- Jiang YR. Reasonable accommodation and disparate impact: clean shave policy discrimination in today’s workplace. J Law Med Ethics. 2023;51:185-195.
- Taylor SC, Barbosa V, Burgess C, et al. Hair and scalp disorders in adult and pediatric patients with skin of color. Cutis. 2017;100:31-35.
- Moran E, McMichael A, De Souza B, et al. New razor technology improves appearance and quality of life in men with pseudofolliculitis barbae. Cutis. 2022;110:329-334.
- Maurer M, Rietzler M, Burghardt R, et al. The male beard hair and facial skin—challenges for shaving. Int J Cosmet Sci. 2016;38 (suppl 1):3-9.
- Ross EV. How would you treat this patient with lasers & EBDs? casebased panel. Presented at: Skin of Color Update; September 13, 2024; New York, NY.
- Ross EV, Cooke LM, Timko AL, et al. Treatment of pseudofolliculitis barbae in skin types IV, V, and VI with a long-pulsed neodymium:yttrium aluminum garnet laser. J Am Acad Dermatol. 2002;47:263-270.
- Shokeir H, Samy N, Taymour M. Pseudofolliculitis barbae treatment: efficacy of topical eflornithine, long-pulsed Nd-YAG laser versus their combination. J Cosmet Dermatol. 2021;20:3517-3525.
- Amer A, Elsayed A, Gharib K. Evaluation of efficacy and safety of chemical peeling and long-pulse Nd:YAG laser in treatment of pseudofolliculitis barbae. Dermatol Ther. 2021;34:E14859.
- Cook-Bolden FE, Barba A, Halder R, et al. Twice-daily applications of benzoyl peroxide 5%/clindamycin 1% gel versus vehicle in the treatment of pseudofolliculitis barbae. Cutis. 2004;73(6 suppl):18-24.
- Nussbaum D, Friedman A. Pseudofolliculitis barbae: a review of current treatment options. J Drugs Dermatol. 2019;18:246-250.
- Quarles FN, Brody H, Johnson BA, et al. Pseudofolliculitis barbae. Dermatol Ther. 2007;20:133-136.
- McMichael AJ, Frey C. Challenging the tools used to measure cutaneous lupus severity in patients of all skin types. JAMA Dermatol. 2025;161:9-10.
- Okonkwo E, Neal B, Harper HL. Pseudofolliculitis barbae in the military and the need for social awareness. Mil Med. 2021;186:143-144.
- Ritchie S, Park J, Banta J, et al. Shaving waivers in the United States Air Force and their impact on promotions of Black/African-American members. Mil Med. 2023;188:E242-E247.
THE COMPARISON
- A. Pustules, erythematous to violaceous nodules, and hyperpigmented patches on the lower cheek and chin.
- B. Brown papules, pink keloidal papules and nodules, pustules, and hyperpigmented papules on the mandibular area and neck.
- C. Coarse hairs, pustules, and pink papules on the mandibular area and neck.
Pseudofolliculitis barbae (PFB), also known as razor bumps, is a common inflammatory condition characterized by papules and pustules that typically appear in the beard and cheek regions. It occurs when shaved hair regrows and penetrates the skin, leading to irritation and inflammation. While anyone who shaves can develop PFB, it is more prevalent and severe in individuals with naturally tightly coiled, coarse-textured hair.1,2 Pseudofolliculitis barbae is common in individuals who shave frequently due to personal choice or profession, such as members of the US military3,4 and firefighters, who are required to remain clean shaven for safety (eg, ensuring proper fit of a respirator mask).5 Early diagnosis and treatment of PFB are essential to prevent long-term complications such as scarring or hyperpigmentation, which may be more severe in those with darker skin tones.
Epidemiology
Pseudofolliculitis barbae is most common in Black men, affecting 45% to 83% of men of African ancestry.1,2 This condition also can affect individuals of various ethnicities with coarse or curly hair. The spiral shape of the hair increases the likelihood that it will regrow into the skin after shaving.6 Women with hirsutism who shave also can develop PFB.
Key Clinical Features
The papules and pustules seen in PFB may be flesh colored, erythematous, hyperpigmented, brown, or violaceous. Erythema may be less pronounced in darker vs lighter skin tones. Persistent and severe postinflammatory hyperpigmentation may occur, and hypertrophic or keloidal scars may develop in affected areas. Dermoscopy may reveal extrafollicular hair penetration as well as follicular or perifollicular pustules accompanied by hyperkeratosis.
Worth Noting
The most effective management for PFB is to discontinue shaving.1 If shaving is desired or necessary, it is recommended that patients apply lukewarm water to the affected area followed by a generous amount of shaving foam or gel to create a protective antifriction layer that allows the razor to glide more smoothly over the skin and reduces subsequent irritation.2 Using the right razor technology also may help alleviate symptoms. Research has shown that multiblade razors used in conjunction with preshave hair hydration and postshave moisturization do not worsen PFB.2 A recent study found that multiblade razor technology paired with use of a shave foam or gel actually improved skin appearance in patients with PFB.7
It is important to direct patients to shave in the direction of hair growth; however, this may not be possible for individuals with curly or coarse hair, as the hair may grow in many directions.8,9 Patients also should avoid pulling the skin taut while shaving, as doing so allows the hair to be clipped below the surface, where it can repenetrate the skin and cause further irritation. As an alternative to shaving with a razor, patients can use hair clippers to trim beard hair, which leaves behind stubble and interrupts the cycle of retracted hairs under the skin. Nd:YAG laser therapy has demonstrated efficacy in reduction of PFB papules and pustules.9-12 Greater mean improvement in inflammatory papules and reduction in hair density was noted in participants who received Nd:YAG laser plus eflornithine compared with those who received the laser or eflornithine alone.11 Patients should not pluck or dig into the skin to remove any ingrown hairs. If a tweezer is used, the patient should gently lift the tip of the ingrown hair with the tweezer to dislodge it from the skin and prevent plucking out the hair completely.
To help manage inflammation after shaving, topical treatments such as benzoyl peroxide 5%/clindamycin 1% gel can be used.3,13 A low-potency steroid such as topical hydrocortisone 2.5% applied once or twice daily for up to 2 to 3 days may be helpful.1,14 Adjunctive treatments including keratolytics (eg, topical retinoids, hydroxy acids) reduce perifollicular hyperkeratosis.14,15 Agents containing alpha hydroxy acids (eg, glycolic acid) also can decrease the curvature of the hair itself by reducing the sulfhydryl bonds.6 If secondary bacterial infections occur, oral antibiotics (eg, doxycycline) may be necessary.
Health Disparity Highlight
Individuals with darker skin tones are at higher risk for PFB and associated complications. Limited access to dermatology services may further exacerbate these challenges. Individuals with PFB may not seek medical treatment until the condition becomes severe. Clinicians also may underestimate the severity of PFB—particularly in those with darker skin tones—based on erythema alone because it may be less pronounced in darker vs lighter skin tones.16
While permanent hair reduction with laser therapy is a treatment option for PFB, it may be inaccessible to some patients because it can be expensive and is coded as a cosmetic procedure. Additionally, patients may not have access to specialists who are experienced in performing the procedure in those with darker skin tones.9 Some patients also may not want to permanently reduce the amount of hair that grows in the beard area for personal or religious reasons.17
Pseudofolliculitis barbae also has been linked to professional disparities. One study found that members of the US Air Force who had medical shaving waivers experienced longer times to promotion than those with no waiver.18 Delays in promotion may be linked to perceptions of unprofessionalism, exclusion from high-profile duties, and concerns about career progression. While this delay was similar for individuals of all races, the majority of those in the waiver group were Black/African American. In 2021, 4 Black firefighters with PFB were unsuccessful in their bid to get a medical accommodation regarding a New York City Fire Department policy requiring them to be clean shaven where the oxygen mask seals against the skin.5 More research is needed on mask safety and efficiency relative to the length of facial hair. Accommodations or tailored masks for facial hair conditions also are necessary so individuals with PFB can meet job requirements while managing their condition.
THE COMPARISON
- A. Pustules, erythematous to violaceous nodules, and hyperpigmented patches on the lower cheek and chin.
- B. Brown papules, pink keloidal papules and nodules, pustules, and hyperpigmented papules on the mandibular area and neck.
- C. Coarse hairs, pustules, and pink papules on the mandibular area and neck.
Pseudofolliculitis barbae (PFB), also known as razor bumps, is a common inflammatory condition characterized by papules and pustules that typically appear in the beard and cheek regions. It occurs when shaved hair regrows and penetrates the skin, leading to irritation and inflammation. While anyone who shaves can develop PFB, it is more prevalent and severe in individuals with naturally tightly coiled, coarse-textured hair.1,2 Pseudofolliculitis barbae is common in individuals who shave frequently due to personal choice or profession, such as members of the US military3,4 and firefighters, who are required to remain clean shaven for safety (eg, ensuring proper fit of a respirator mask).5 Early diagnosis and treatment of PFB are essential to prevent long-term complications such as scarring or hyperpigmentation, which may be more severe in those with darker skin tones.
Epidemiology
Pseudofolliculitis barbae is most common in Black men, affecting 45% to 83% of men of African ancestry.1,2 This condition also can affect individuals of various ethnicities with coarse or curly hair. The spiral shape of the hair increases the likelihood that it will regrow into the skin after shaving.6 Women with hirsutism who shave also can develop PFB.
Key Clinical Features
The papules and pustules seen in PFB may be flesh colored, erythematous, hyperpigmented, brown, or violaceous. Erythema may be less pronounced in darker vs lighter skin tones. Persistent and severe postinflammatory hyperpigmentation may occur, and hypertrophic or keloidal scars may develop in affected areas. Dermoscopy may reveal extrafollicular hair penetration as well as follicular or perifollicular pustules accompanied by hyperkeratosis.
Worth Noting
The most effective management for PFB is to discontinue shaving.1 If shaving is desired or necessary, it is recommended that patients apply lukewarm water to the affected area followed by a generous amount of shaving foam or gel to create a protective antifriction layer that allows the razor to glide more smoothly over the skin and reduces subsequent irritation.2 Using the right razor technology also may help alleviate symptoms. Research has shown that multiblade razors used in conjunction with preshave hair hydration and postshave moisturization do not worsen PFB.2 A recent study found that multiblade razor technology paired with use of a shave foam or gel actually improved skin appearance in patients with PFB.7
It is important to direct patients to shave in the direction of hair growth; however, this may not be possible for individuals with curly or coarse hair, as the hair may grow in many directions.8,9 Patients also should avoid pulling the skin taut while shaving, as doing so allows the hair to be clipped below the surface, where it can repenetrate the skin and cause further irritation. As an alternative to shaving with a razor, patients can use hair clippers to trim beard hair, which leaves behind stubble and interrupts the cycle of retracted hairs under the skin. Nd:YAG laser therapy has demonstrated efficacy in reduction of PFB papules and pustules.9-12 Greater mean improvement in inflammatory papules and reduction in hair density was noted in participants who received Nd:YAG laser plus eflornithine compared with those who received the laser or eflornithine alone.11 Patients should not pluck or dig into the skin to remove any ingrown hairs. If a tweezer is used, the patient should gently lift the tip of the ingrown hair with the tweezer to dislodge it from the skin and prevent plucking out the hair completely.
To help manage inflammation after shaving, topical treatments such as benzoyl peroxide 5%/clindamycin 1% gel can be used.3,13 A low-potency steroid such as topical hydrocortisone 2.5% applied once or twice daily for up to 2 to 3 days may be helpful.1,14 Adjunctive treatments including keratolytics (eg, topical retinoids, hydroxy acids) reduce perifollicular hyperkeratosis.14,15 Agents containing alpha hydroxy acids (eg, glycolic acid) also can decrease the curvature of the hair itself by reducing the sulfhydryl bonds.6 If secondary bacterial infections occur, oral antibiotics (eg, doxycycline) may be necessary.
Health Disparity Highlight
Individuals with darker skin tones are at higher risk for PFB and associated complications. Limited access to dermatology services may further exacerbate these challenges. Individuals with PFB may not seek medical treatment until the condition becomes severe. Clinicians also may underestimate the severity of PFB—particularly in those with darker skin tones—based on erythema alone because it may be less pronounced in darker vs lighter skin tones.16
While permanent hair reduction with laser therapy is a treatment option for PFB, it may be inaccessible to some patients because it can be expensive and is coded as a cosmetic procedure. Additionally, patients may not have access to specialists who are experienced in performing the procedure in those with darker skin tones.9 Some patients also may not want to permanently reduce the amount of hair that grows in the beard area for personal or religious reasons.17
Pseudofolliculitis barbae also has been linked to professional disparities. One study found that members of the US Air Force who had medical shaving waivers experienced longer times to promotion than those with no waiver.18 Delays in promotion may be linked to perceptions of unprofessionalism, exclusion from high-profile duties, and concerns about career progression. While this delay was similar for individuals of all races, the majority of those in the waiver group were Black/African American. In 2021, 4 Black firefighters with PFB were unsuccessful in their bid to get a medical accommodation regarding a New York City Fire Department policy requiring them to be clean shaven where the oxygen mask seals against the skin.5 More research is needed on mask safety and efficiency relative to the length of facial hair. Accommodations or tailored masks for facial hair conditions also are necessary so individuals with PFB can meet job requirements while managing their condition.
- Alexis A, Heath CR, Halder RM. Folliculitis keloidalis nuchae and pseudofolliculitis barbae: are prevention and effective treatment within reach? em>Dermatol Clin. 2014;32:183-191.
- Gray J, McMichael AJ. Pseudofolliculitis barbae: understanding the condition and the role of facial grooming. Int J Cosmet Sci. 2016;38 (suppl 1):24-27.
- Tshudy MT, Cho S. Pseudofolliculitis barbae in the U.S. military, a review. Mil Med. 2021;186:E52-E57.
- Jung I, Lannan FM, Weiss A, et al. Treatment and current policies on pseudofolliculitis barbae in the US military. Cutis. 2023;112:299-302.
- Jiang YR. Reasonable accommodation and disparate impact: clean shave policy discrimination in today’s workplace. J Law Med Ethics. 2023;51:185-195.
- Taylor SC, Barbosa V, Burgess C, et al. Hair and scalp disorders in adult and pediatric patients with skin of color. Cutis. 2017;100:31-35.
- Moran E, McMichael A, De Souza B, et al. New razor technology improves appearance and quality of life in men with pseudofolliculitis barbae. Cutis. 2022;110:329-334.
- Maurer M, Rietzler M, Burghardt R, et al. The male beard hair and facial skin—challenges for shaving. Int J Cosmet Sci. 2016;38 (suppl 1):3-9.
- Ross EV. How would you treat this patient with lasers & EBDs? casebased panel. Presented at: Skin of Color Update; September 13, 2024; New York, NY.
- Ross EV, Cooke LM, Timko AL, et al. Treatment of pseudofolliculitis barbae in skin types IV, V, and VI with a long-pulsed neodymium:yttrium aluminum garnet laser. J Am Acad Dermatol. 2002;47:263-270.
- Shokeir H, Samy N, Taymour M. Pseudofolliculitis barbae treatment: efficacy of topical eflornithine, long-pulsed Nd-YAG laser versus their combination. J Cosmet Dermatol. 2021;20:3517-3525.
- Amer A, Elsayed A, Gharib K. Evaluation of efficacy and safety of chemical peeling and long-pulse Nd:YAG laser in treatment of pseudofolliculitis barbae. Dermatol Ther. 2021;34:E14859.
- Cook-Bolden FE, Barba A, Halder R, et al. Twice-daily applications of benzoyl peroxide 5%/clindamycin 1% gel versus vehicle in the treatment of pseudofolliculitis barbae. Cutis. 2004;73(6 suppl):18-24.
- Nussbaum D, Friedman A. Pseudofolliculitis barbae: a review of current treatment options. J Drugs Dermatol. 2019;18:246-250.
- Quarles FN, Brody H, Johnson BA, et al. Pseudofolliculitis barbae. Dermatol Ther. 2007;20:133-136.
- McMichael AJ, Frey C. Challenging the tools used to measure cutaneous lupus severity in patients of all skin types. JAMA Dermatol. 2025;161:9-10.
- Okonkwo E, Neal B, Harper HL. Pseudofolliculitis barbae in the military and the need for social awareness. Mil Med. 2021;186:143-144.
- Ritchie S, Park J, Banta J, et al. Shaving waivers in the United States Air Force and their impact on promotions of Black/African-American members. Mil Med. 2023;188:E242-E247.
- Alexis A, Heath CR, Halder RM. Folliculitis keloidalis nuchae and pseudofolliculitis barbae: are prevention and effective treatment within reach? em>Dermatol Clin. 2014;32:183-191.
- Gray J, McMichael AJ. Pseudofolliculitis barbae: understanding the condition and the role of facial grooming. Int J Cosmet Sci. 2016;38 (suppl 1):24-27.
- Tshudy MT, Cho S. Pseudofolliculitis barbae in the U.S. military, a review. Mil Med. 2021;186:E52-E57.
- Jung I, Lannan FM, Weiss A, et al. Treatment and current policies on pseudofolliculitis barbae in the US military. Cutis. 2023;112:299-302.
- Jiang YR. Reasonable accommodation and disparate impact: clean shave policy discrimination in today’s workplace. J Law Med Ethics. 2023;51:185-195.
- Taylor SC, Barbosa V, Burgess C, et al. Hair and scalp disorders in adult and pediatric patients with skin of color. Cutis. 2017;100:31-35.
- Moran E, McMichael A, De Souza B, et al. New razor technology improves appearance and quality of life in men with pseudofolliculitis barbae. Cutis. 2022;110:329-334.
- Maurer M, Rietzler M, Burghardt R, et al. The male beard hair and facial skin—challenges for shaving. Int J Cosmet Sci. 2016;38 (suppl 1):3-9.
- Ross EV. How would you treat this patient with lasers & EBDs? casebased panel. Presented at: Skin of Color Update; September 13, 2024; New York, NY.
- Ross EV, Cooke LM, Timko AL, et al. Treatment of pseudofolliculitis barbae in skin types IV, V, and VI with a long-pulsed neodymium:yttrium aluminum garnet laser. J Am Acad Dermatol. 2002;47:263-270.
- Shokeir H, Samy N, Taymour M. Pseudofolliculitis barbae treatment: efficacy of topical eflornithine, long-pulsed Nd-YAG laser versus their combination. J Cosmet Dermatol. 2021;20:3517-3525.
- Amer A, Elsayed A, Gharib K. Evaluation of efficacy and safety of chemical peeling and long-pulse Nd:YAG laser in treatment of pseudofolliculitis barbae. Dermatol Ther. 2021;34:E14859.
- Cook-Bolden FE, Barba A, Halder R, et al. Twice-daily applications of benzoyl peroxide 5%/clindamycin 1% gel versus vehicle in the treatment of pseudofolliculitis barbae. Cutis. 2004;73(6 suppl):18-24.
- Nussbaum D, Friedman A. Pseudofolliculitis barbae: a review of current treatment options. J Drugs Dermatol. 2019;18:246-250.
- Quarles FN, Brody H, Johnson BA, et al. Pseudofolliculitis barbae. Dermatol Ther. 2007;20:133-136.
- McMichael AJ, Frey C. Challenging the tools used to measure cutaneous lupus severity in patients of all skin types. JAMA Dermatol. 2025;161:9-10.
- Okonkwo E, Neal B, Harper HL. Pseudofolliculitis barbae in the military and the need for social awareness. Mil Med. 2021;186:143-144.
- Ritchie S, Park J, Banta J, et al. Shaving waivers in the United States Air Force and their impact on promotions of Black/African-American members. Mil Med. 2023;188:E242-E247.
Beyond the Razor: Managing Pseudofolliculitis Barbae in Skin of Color
Beyond the Razor: Managing Pseudofolliculitis Barbae in Skin of Color

Baricitinib-Induced Trichilemmal Cyst Reactivation in a Woman With Alopecia Areata
Baricitinib-Induced Trichilemmal Cyst Reactivation in a Woman With Alopecia Areata
To the Editor:
Alopecia areata (AA), an autoimmune disease characterized by inflammatory and nonscarring hair loss, can have a considerable impact on quality of life.1 Baricitinib is a Janus kinase inhibitor that recently was approved by the US Food and Drug Administration for treatment of severe AA in adult patients, becoming the only on-label treatment available.2 So far, the most common adverse effects reported in phase 3 trials have been acne, upper respiratory tract infections, headaches, urinary tract infections, and elevated creatine kinase levels.3
At our trichology unit in the dermatology department of a Spanish tertiary-care hospital in Seville, we have successfully used baricitinib to treat 18 patients with severe, therapy-resistant AA. Herein, we present a case of trichilemmal cyst reactivation in one of our patients following successful treatment with baricitinib.
A 53-year-old woman with a history of trichilemmal cysts presented to the dermatology department with total body hair loss of 5 years' duration that was diagnosed as AA universalis (Figure, A). The patient reported that the trichilemmal cysts had shrunk drastically 1 month after complete loss of body hair (Severity of Alopecia Tool [SALT] score, 100)(Figure, B). The largest cyst was surgically removed, and the diagnosis was histologically confirmed by a pathologist. Her mother and sister also had a history of multiple trichilemmal cysts.
The patient previously had failed treatment with oral prednisone 50 mg/d, oral cyclosporine 4 mg/kg/d, oral dexamethasone 4 mg twice weekly, and oral azathioprine 300 mg/wk. Due to the new indication of baricitinib for AA, we opted to start the patient on oral baricitinib 4 mg/d. By week 8 of treatment, she had achieved total hair regrowth (SALT score, 0). This rapid response might indicate a quick-responder phenotype, referring to a subset of patients who exhibit a fast and robust response to treatment (SALT90), generally before week 16, although more evidence is needed.
Notably, we observed the reactivation of 4 trichilemmal cysts on the scalp 6 weeks after starting baricitinib. To our knowledge, this side effect has not previously been reported. We hypothesize that reactivation of the cysts may have been due to the inhibition of the Janus kinase/signal transducer and activator of transcription pathway, which reduces the effects of cytokines and leads to reactivation of hair follicles that were inactive because of inflammation.4 As a result, the outer root sheath of the hair follicle can once again be filled with keratin, thereby reactivating the trichilemmal cysts. Based on our experience with this case, it may be relevant to consider personal and family history of trichilemmal cysts before starting treatment with baricitinib for AA and advise the patient about the possibility of this adverse effect.
- Freitas E, Guttman-Yassky E, Torres T. Baricitinib for the treatment of alopecia areata. Drugs. 2023;83:761-770. doi:10.1007 /s40265-023-01873-w
- US Food and Drug Administration. FDA approves first systemic treatment for alopecia areata [news release]. July 13, 2022. Accessed March 17, 2025. https://www.prnewswire.com/news-releases/fda-approves-first-systemic-treatment-for-alopecia-areata-301566884.html
- King B, Ohyama M, Kwon O, et al. Two phase 3 trials of baricitinib for alopecia areata. N Engl J Med. 2022;386:1687-1699. doi:10.1056 /NEJMoa2110343
- Lensing M, Jabbari A. An overview of JAK/STAT pathways and JAK inhibition in alopecia areata. Front Immunol. 2022;13:955035. doi:10.3389/fimmu.2022.955035
To the Editor:
Alopecia areata (AA), an autoimmune disease characterized by inflammatory and nonscarring hair loss, can have a considerable impact on quality of life.1 Baricitinib is a Janus kinase inhibitor that recently was approved by the US Food and Drug Administration for treatment of severe AA in adult patients, becoming the only on-label treatment available.2 So far, the most common adverse effects reported in phase 3 trials have been acne, upper respiratory tract infections, headaches, urinary tract infections, and elevated creatine kinase levels.3
At our trichology unit in the dermatology department of a Spanish tertiary-care hospital in Seville, we have successfully used baricitinib to treat 18 patients with severe, therapy-resistant AA. Herein, we present a case of trichilemmal cyst reactivation in one of our patients following successful treatment with baricitinib.
A 53-year-old woman with a history of trichilemmal cysts presented to the dermatology department with total body hair loss of 5 years' duration that was diagnosed as AA universalis (Figure, A). The patient reported that the trichilemmal cysts had shrunk drastically 1 month after complete loss of body hair (Severity of Alopecia Tool [SALT] score, 100)(Figure, B). The largest cyst was surgically removed, and the diagnosis was histologically confirmed by a pathologist. Her mother and sister also had a history of multiple trichilemmal cysts.
The patient previously had failed treatment with oral prednisone 50 mg/d, oral cyclosporine 4 mg/kg/d, oral dexamethasone 4 mg twice weekly, and oral azathioprine 300 mg/wk. Due to the new indication of baricitinib for AA, we opted to start the patient on oral baricitinib 4 mg/d. By week 8 of treatment, she had achieved total hair regrowth (SALT score, 0). This rapid response might indicate a quick-responder phenotype, referring to a subset of patients who exhibit a fast and robust response to treatment (SALT90), generally before week 16, although more evidence is needed.
Notably, we observed the reactivation of 4 trichilemmal cysts on the scalp 6 weeks after starting baricitinib. To our knowledge, this side effect has not previously been reported. We hypothesize that reactivation of the cysts may have been due to the inhibition of the Janus kinase/signal transducer and activator of transcription pathway, which reduces the effects of cytokines and leads to reactivation of hair follicles that were inactive because of inflammation.4 As a result, the outer root sheath of the hair follicle can once again be filled with keratin, thereby reactivating the trichilemmal cysts. Based on our experience with this case, it may be relevant to consider personal and family history of trichilemmal cysts before starting treatment with baricitinib for AA and advise the patient about the possibility of this adverse effect.
To the Editor:
Alopecia areata (AA), an autoimmune disease characterized by inflammatory and nonscarring hair loss, can have a considerable impact on quality of life.1 Baricitinib is a Janus kinase inhibitor that recently was approved by the US Food and Drug Administration for treatment of severe AA in adult patients, becoming the only on-label treatment available.2 So far, the most common adverse effects reported in phase 3 trials have been acne, upper respiratory tract infections, headaches, urinary tract infections, and elevated creatine kinase levels.3
At our trichology unit in the dermatology department of a Spanish tertiary-care hospital in Seville, we have successfully used baricitinib to treat 18 patients with severe, therapy-resistant AA. Herein, we present a case of trichilemmal cyst reactivation in one of our patients following successful treatment with baricitinib.
A 53-year-old woman with a history of trichilemmal cysts presented to the dermatology department with total body hair loss of 5 years' duration that was diagnosed as AA universalis (Figure, A). The patient reported that the trichilemmal cysts had shrunk drastically 1 month after complete loss of body hair (Severity of Alopecia Tool [SALT] score, 100)(Figure, B). The largest cyst was surgically removed, and the diagnosis was histologically confirmed by a pathologist. Her mother and sister also had a history of multiple trichilemmal cysts.
The patient previously had failed treatment with oral prednisone 50 mg/d, oral cyclosporine 4 mg/kg/d, oral dexamethasone 4 mg twice weekly, and oral azathioprine 300 mg/wk. Due to the new indication of baricitinib for AA, we opted to start the patient on oral baricitinib 4 mg/d. By week 8 of treatment, she had achieved total hair regrowth (SALT score, 0). This rapid response might indicate a quick-responder phenotype, referring to a subset of patients who exhibit a fast and robust response to treatment (SALT90), generally before week 16, although more evidence is needed.
Notably, we observed the reactivation of 4 trichilemmal cysts on the scalp 6 weeks after starting baricitinib. To our knowledge, this side effect has not previously been reported. We hypothesize that reactivation of the cysts may have been due to the inhibition of the Janus kinase/signal transducer and activator of transcription pathway, which reduces the effects of cytokines and leads to reactivation of hair follicles that were inactive because of inflammation.4 As a result, the outer root sheath of the hair follicle can once again be filled with keratin, thereby reactivating the trichilemmal cysts. Based on our experience with this case, it may be relevant to consider personal and family history of trichilemmal cysts before starting treatment with baricitinib for AA and advise the patient about the possibility of this adverse effect.
- Freitas E, Guttman-Yassky E, Torres T. Baricitinib for the treatment of alopecia areata. Drugs. 2023;83:761-770. doi:10.1007 /s40265-023-01873-w
- US Food and Drug Administration. FDA approves first systemic treatment for alopecia areata [news release]. July 13, 2022. Accessed March 17, 2025. https://www.prnewswire.com/news-releases/fda-approves-first-systemic-treatment-for-alopecia-areata-301566884.html
- King B, Ohyama M, Kwon O, et al. Two phase 3 trials of baricitinib for alopecia areata. N Engl J Med. 2022;386:1687-1699. doi:10.1056 /NEJMoa2110343
- Lensing M, Jabbari A. An overview of JAK/STAT pathways and JAK inhibition in alopecia areata. Front Immunol. 2022;13:955035. doi:10.3389/fimmu.2022.955035
- Freitas E, Guttman-Yassky E, Torres T. Baricitinib for the treatment of alopecia areata. Drugs. 2023;83:761-770. doi:10.1007 /s40265-023-01873-w
- US Food and Drug Administration. FDA approves first systemic treatment for alopecia areata [news release]. July 13, 2022. Accessed March 17, 2025. https://www.prnewswire.com/news-releases/fda-approves-first-systemic-treatment-for-alopecia-areata-301566884.html
- King B, Ohyama M, Kwon O, et al. Two phase 3 trials of baricitinib for alopecia areata. N Engl J Med. 2022;386:1687-1699. doi:10.1056 /NEJMoa2110343
- Lensing M, Jabbari A. An overview of JAK/STAT pathways and JAK inhibition in alopecia areata. Front Immunol. 2022;13:955035. doi:10.3389/fimmu.2022.955035
Baricitinib-Induced Trichilemmal Cyst Reactivation in a Woman With Alopecia Areata
Baricitinib-Induced Trichilemmal Cyst Reactivation in a Woman With Alopecia Areata
PRACTICE POINTS
- The rapid growth of trichilemmal cysts may serve as an indicator of a quick-responder phenotype to baricitinib in cases of alopecia areata (AA), although more evidence is needed.
- It is imperative to consider personal and family history of trichilemmal cysts prior to initiating baricitinib treatment for AA.
